

Transfusion medicine
Transfusion therapy
Other therapy
Sickle cell disease
Advisory Board: Kyle Annen, D.O.
Deputy Editor-in-Chief: Patricia Tsang, M.D., M.B.A.

Last author update: 25 January 2023
Last staff update: 4 October 2023
Copyright: 2022-2024, PathologyOutlines.com, Inc.
PubMed Search: Sickle cell disease

Table of Contents
Definition / general | Essential features | Terminology | Pathophysiology | Clinical features | Transmission | Symptoms | Screening | Blood donor screening | Laboratory | Laboratory images | Case reports | Treatment | Microscopic (histologic) description | Peripheral smear images | Positive stains | Sample assessment & plan | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Tanhehco Y. Sickle cell disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/transfusionmedsicklecell.html. Accessed December 18th, 2024.
Definition / general
- Sickle cell disease is a hemoglobinopathy caused by a point mutation in the β globin gene that leads to the production of hemoglobin S, which polymerizes under deoxygenated conditions and causes red blood cells (RBCs) to form a sickle shape
- Sickle RBCs result in hemolysis, vaso-occlusive pain crises and endothelial damage
Essential features
- Sickle cell disease is caused by a single point mutation in the β globin gene that results in the production of hemoglobin S, which polymerizes under deoxygenated conditions and causes RBCs to sickle (Lancet 2010;376:2018)
- Sickle RBCs are rigid and less deformable, which causes vaso-occlusive pain crises, hemolytic anemia, endothelial injury and inflammation (Lancet 2010;376:2018)
- Complications of sickle cell disease include stroke, acute chest syndrome, priapism, multiorgan failure, splenic / hepatic sequestration, intrahepatic cholestasis, aplastic crisis, pulmonary hypertension and avascular necrosis
- Current treatment options include simple and exchange RBC transfusions, disease modifying drug therapies and hematopoietic stem cell transplantation (Lancet 2017;390:311)
- Sickle cell trait is protective against the parasite Plasmodium falciparum
Terminology
- Sickle cell anemia
Pathophysiology
- Single point mutation (A to T substitution) in the first exon of the β globin gene, converting glutamic acid into valine
- Defective hemoglobin tetramers are formed (Hemoglobin S, Hgb S) that polymerize under deoxygenated conditions
- Polymerized Hgb S causes RBCs to form a sickle shape
- Sickle RBCs are rigid, less deformable and have a shortened lifespan of 10 - 20 days
- Rigid sickle RBCs lead to hemolysis, vascular occlusion, endothelial injury and inflammation
- Patients have hemolytic anemia and experience vaso-occlusive pain crises
- Sickle RBCs and leukocytes adhere to the vascular endothelium, causing vascular obstruction and tissue ischemia
- Chronic inflammation with an increased risk of thrombosis especially during vasooclussive event (J Thromb Thrombolysis 2013;35:352)
- Immunocompromised due to autoinfarction of spleen or surgical splenectomy (Birth Defects Orig Artic Ser 1975;11:322)
- Have hypercoagulable state based on thermoelastographic profiles (Arch Pathol Lab Med 2005;129:760, J Clin Pathol 1980;33:622)
Clinical features
- Stroke
- Acute chest syndrome
- Priapism
- Multiorgan failure
- Splenic / hepatic sequestration
- Intrahepatic cholestasis
- Aplastic crisis
- Pulmonary hypertension
- Avascular necrosis
- Reference: J Clin Med 2021;10:4232
Transmission
- Autosomal recessive gene inheritance pattern
- Variable genotypes: SS, SC, S/β0 thalassemia, S/β+ thalassemia, SD, SE are common ones
- Reference: Lancet 2010;376:2018
Symptoms
- Jaundice
- Fatigue
- Dyspnea
- Infection
- Pain and swelling
- Organ damage
- Reference: Lancet 2010;376:2018
Screening
- Newborn screening is performed as early as 24 - 48 hours after birth
- Methods used to detect the presence of Hgb S include gel electrophoresis and high performance liquid chromatography (HPLC) (Syst Rev 2020;9:250)
- Confirmatory testing is performed for positive screen results
Blood donor screening
- Donors with sickle cell disease are not eligible to donate blood; donors with sickle cell trait may be eligible
- Blood with sickle cell trait often causes failure of leukocyte reduction filters and may be deferred due to this
Laboratory
- Positive for anemia: low hemoglobin, low hematocrit, low RBC, high reticulocyte
- Positive for hemolysis: low haptoglobin, high lactate dehydrogenase, high total bilirubin, high indirect bilirubin
- Positive for Hgb S: high hemoglobin S
- Positive for inflammation: high C reactive protein, high erythrocyte sedimentation rate
- Positive for iron studies if chronically transfused and iron overloaded: high serum ferritin, high serum iron, high serum transferrin saturation, low total iron binding capacity
- Associated with increases in thrombin generation, fibrinolytic activation, platelet activation, increased antiphospholipid antibodies, decreased levels of circulating anticoagulants and contact factors
- Also increased circulating levels of tissue factor and endothelial cells expressing a tissue factor phenotype
- Reference: Ann Intern Med 2021;174:ITC1
Laboratory images
Contributed by Patricia Tsang, M.D., M.B.A.
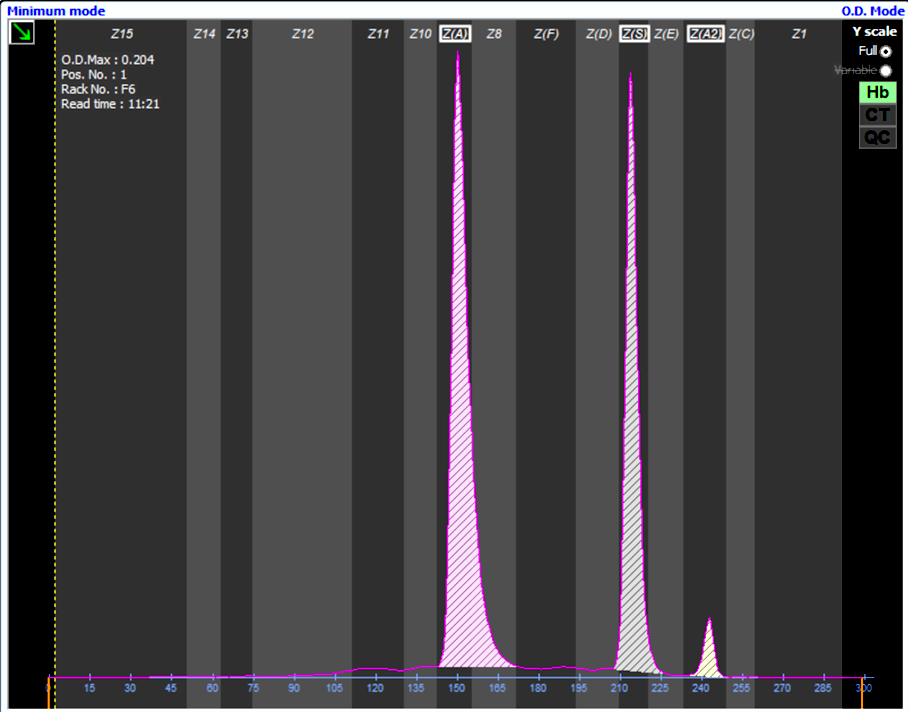
Sickle cell trait
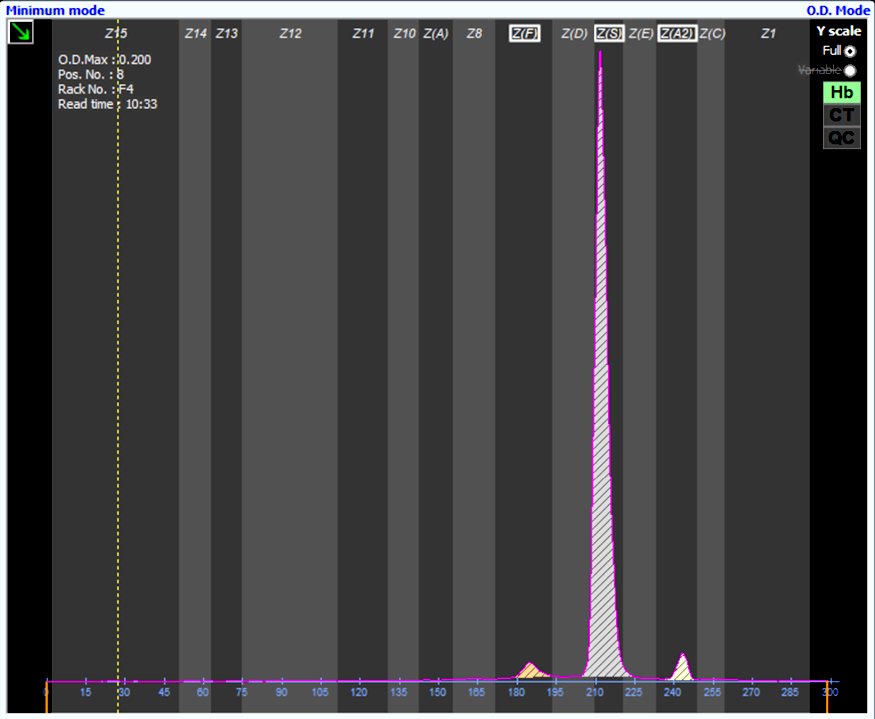
SS hemoglobin
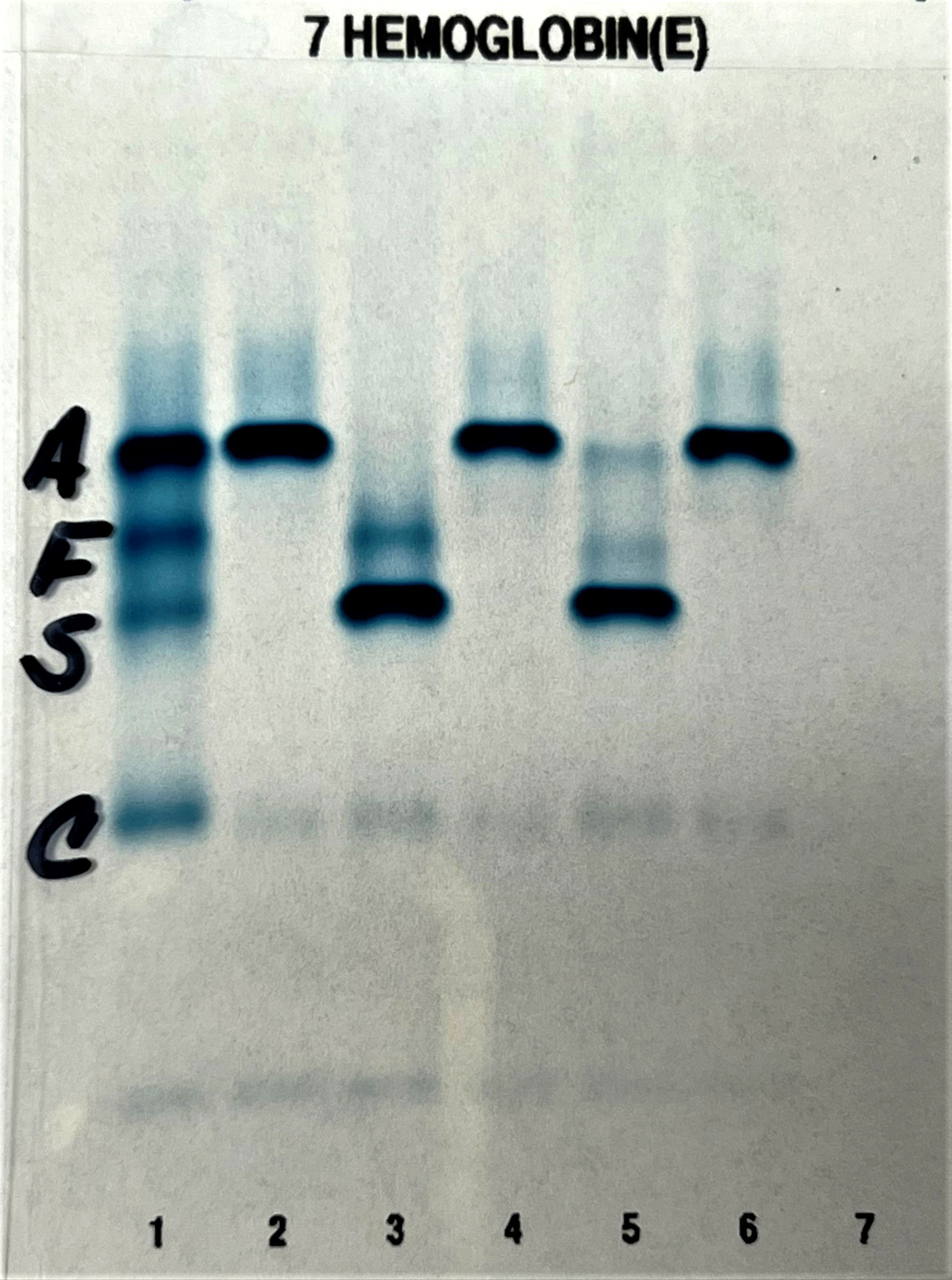
Alkaline gel hemoglobin electrophoresis
Case reports
- 8 year old boy with a rare hemoglobinopathy (hemoglobin S / hemoglobin Quebec-Chori) who presented with complications of sickle cell disease characterized by pain crises, osteomyelitis and priapism (J Pediatr Hematol Oncol 2020;42:e775)
- 16 year old girl with concurrent sickle cell anemia and ulcerative colitis (Paediatr Int Child Health 2021 Sep 3 [Epub ahead of print])
- 20 year old woman with sickle cell anemia (Hgb SS) and no history of alloantibodies, who developed a severe delayed hemolytic transfusion reaction 5 days after an elective preoperative packed RBC transfusion that rapidly progressed to multiorgan failure and death (J Pediatr Hematol Oncol 2019;41:624)
- 10 patients with sickle cell disease who developed COVID-19 infections in a developing country (Acta Haematol 2021 Sep 17 [Epub ahead of print])
Treatment
- RBC transfusions: simple or exchange (N Engl J Med 1998;339:5)
- Monitor for iron overload as a potential side effect of chronic transfusion therapy
- Prophylactic red cell antigen matching for Rh (C, E, K) recommended by American Society of Hematology (ASH) (Blood Adv 2020;4:327)
- Alloimmunization rate as high as 50% without prophylactic red cell antigen matching, 5 - 24% with limited prophylactic matching and ~7% with extended prophylactic matching (Pediatr Blood Cancer 2011;57:294, Transfusion 2001;41:1086, Pediatr Blood Cancer 2013;60:1487, Transfus Med Rev 2019;33:12, Transfusion 2011;51:1732)
- Extended red cell antigen matching (Jka / Jkb, Fya / Fyb, S / s) may further prevent alloimmunization (Blood Adv 2020;4:327)
- Disease modifying drug therapies: hydroxyurea, L glutamine, crizanlizumab, voxelotor (N Engl J Med 2017;376:429, Blood 2019;133:1865)
- Hematopoietic stem cell transplantation: allogeneic donor, autologous donor with gene therapy (clinical trials)
- Sickle cell patients should avoid handling reptiles and perhaps should avoid pet chickens due to an increased risk of salmonella infection (Pediatr Hematol Oncol 2002;24:75, CDC: Salmonella Outbreaks Linked to Backyard Poultry [Accessed 6 January 2022])
Microscopic (histologic) description
- May have increased normoblasts and megaloblastic changes due to folate deficiency
- Increased perivascular fibrosis in small and medium sized vessels (Arch Pathol Lab Med 2004;128:634)
- Occasional aplastic crisis
Peripheral smear images
Contributed by Patricia Tsang, M.D., M.B.A.
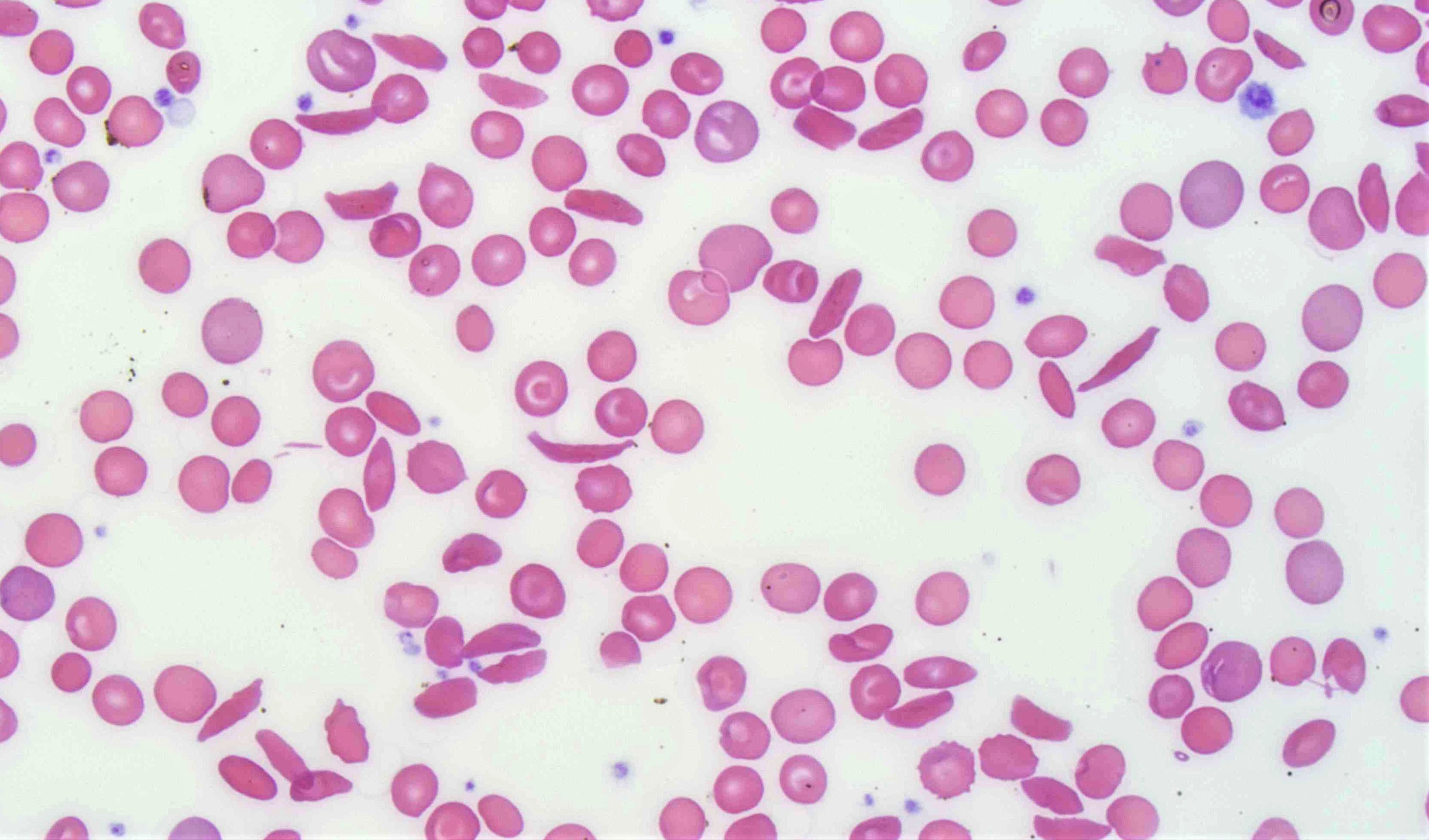
Sickle cell blood smear
Positive stains
- CD1a, CD1b and CD1c in monocytes (Hum Immunol 2004;65:1370)
Sample assessment & plan
- Patient with sickle cell disease (Hgb SS) and a history of stroke, priapism and vaso-occlusive pain crisis presented with shortness of breath and was subsequently diagnosed with acute chest syndrome. Hemoglobin S level found to be 80%. Patient to undergo red blood cell exchange with 8 units of ABO compatible and crossmatch compatible packed RBCs that are Rh and K antigen matched, leukocyte reduced, irradiated and Hgb S negative. Post procedure target Hgb S is less than 30% and target hematocrit is 27%.
Additional references
Board review style question #1
A 12 year old girl with sickle cell disease presents to the emergency department with fever (102 °F), difficulty breathing, cough and chest pain. Her mother reported that the patient had a cold 2 weeks ago. On physical exam, her vitals were T 102 °F, BP 100/60, P 80, RR 25 and SaO2 80%. She was coughing and appeared to be in pain. No other abnormal findings were noted. A chest Xray was performed and showed a new pulmonary opacity. Her hematocrit was 15% and Hgb S level was 75%.
What is the most likely diagnosis for the patient?
What is the most likely diagnosis for the patient?
- Acute chest syndrome
- Asthma
- Influenza
- Pneumonia
Board review style answer #1
Board review style question #2
A 12 year old girl with sickle cell disease presents to the emergency department with fever (102 °F), difficulty breathing, cough and chest pain. Her mother reported that the patient had a cold 2 weeks ago. On physical exam, her vitals were T 102 °F, BP 100/60, P 80, RR 25 and SaO2 80%. She was coughing and appeared to be in pain. No other abnormal findings were noted. A chest Xray was performed and showed a new pulmonary opacity. Her hematocrit was 15% and Hgb S level was 75%.
The patient is being managed with red blood cell exchange. What should the target hematocrit (Hct) and Hgb S levels be?
The patient is being managed with red blood cell exchange. What should the target hematocrit (Hct) and Hgb S levels be?
- Hct > 30%, Hgb S < 30%
- Hct > 30%, Hgb S > 30%
- Hct < 30%, Hgb S < 30%
- Hct < 30%, Hgb S > 30%
Board review style answer #2

