
Thyroid & parathyroid
Congenital / metabolic anomalies
Lysosomal storage diseases
Author: Andrey Bychkov, M.D., Ph.D.

Last author update: 1 May 2016
Last staff update: 28 July 2023
Copyright: 2017-2025, PathologyOutlines.com, Inc.
PubMed search: Lysosomal storage diseases thyroid

Table of Contents
Definition / general | Terminology | Epidemiology | Sites | Pathophysiology / etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Virtual slides | Cytology images | Peripheral smear description | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Videos | Additional referencesCite this page: Bychkov A. Lysosomal storage diseases. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/thyroidlsd.html. Accessed April 2nd, 2025.
Definition / general
- Lysosomal storage disease (LSD) is a subgroup of inherited metabolic disorders, caused by mutations in genes encoding lysosomal enzymes, which results in cell damage due to excessive storage of undegraded substrates
- Main target organ / systems are nervous, muscular, reticuloendothelial and liver
- Thyroid involvement is uncommon
- Major LSDs were first described in the late 19th (Tay-Sachs, Gaucher and Fabry diseases) and early 20th centuries (Niemann-Pick and Pompe diseases); however the lysosomal concept was developed only in the 1960s (Pediatr Endocrinol Rev 2013;11:59)
Terminology
- Synonyms: lysosomal storage disease / disorder, lysosomal disease, thesaurismosis, storage disease
- Broader category of inherited metabolic disorders is equivalent to metabolic storage disorder or inborn errors of metabolism (Ann Endocrinol (Paris) 2009;70:14)
- Approximately 50 diseases are included in the LSD group, similar to the number of different enzymes within a lysosome
- Almost all names are eponymous
- Classification is based on the nature of the primary accumulated material:
- Lipid storage diseases, including sphingolipidoses (Fabry, Gaucher, Niemann-Pick) and gangliosidosis (Tay-Sachs)
- Mucopolysacharidoses (Hunter, Hurler, Morquio)
- Glycoproteinoses
- Mucolipidoses
- Glycogen storage disease (Pompe)
- Cystinosis
Epidemiology
- Although individual LSDs are rare, the aggregate prevalence is 1:5,000 births, which is comparable to that of the most common genetic diseases (Nat Rev Neurol 2013;9:583)
- LSDs are the second most common group of inborn errors of metabolism (1:1,500 births) after small molecule diseases, e.g. phenylketonuria and galactosemia (Pediatrics 2000;105:e10)
- Some LSDs have ethnic or geographical predilections (Madame Curie Bioscience Database: Lysosomal Storage Disorders [Accessed 30 October 2017]):
- Ashkenazi Jews: Tay-Sachs and Gaucher disease
- French Canadian: Tay-Sachs
- Finnish: aspartylglucosaminuria and Salla disease
- Russian: Hurler syndrome
Sites
- Distribution of affected organs in LSDs is dependent on the nature of the substrates that accumulate and their specific cellular distribution, as well as the cell turnover rate in each tissue (Nat Rev Neurol 2013;9:583):
- Sphingolipids: brain and nerves
- Mucopolysaccharides: cardiovascular system and connective tissue, including bones
- Glycogen: muscles
- Cystine: kidney
Pathophysiology / etiology
- LSDs are caused by mutations in genes encoding soluble acidic hydrolases, activator proteins, transporter proteins, integral membrane proteins or nonlysosomal proteins that are necessary for lysosomal function (Annu Rev Med 2015;66:471)
- Main genes in LSDs: glucosylceramidase beta / GBA (Gaucher), galactosidase alpha / GLA (Fabry), sphingomyelin phosphodiesterase 1 / SMPD1 (Niemann-Pick), hexosaminidase subunit alpha / HEXA (Tay-Sachs), iduronate 2-sulfatase / IDS (Hunter), glucosidase alpha, acid / GAA (Pompe), cystinosin, lysosomal cystine transporter / CTNS (cystinosis)
- As a result, metabolic machinery of the cell is impaired by defects in degradative and synthetic enzymes, lysosomal membrane defects, disorders of lysosome biogenesis and endosome lysosome traffic
- Pathogenetic cascade leads to intralysosomal accumulation of undegraded substrates in multiple tissues and organs
- Excessive storage of a substrate triggers cell damage via several mechanisms: activation of apoptosis, alterations of plasma membrane lipid content (affect receptor responses and downstream signaling), prolonged inflammation, calcium imbalance, dysregulation of autophagy, etc. (Nat Rev Neurol 2013;9:583)
- All LSDs are inherited in an autosomal recessive manner, except X linked recessive Fabry, Hunter and Danon diseases
- Severity and age of onset in LSDs depend on a range of factors: residual enzyme activity (20% is still enough for normal function), mutant protein size, location of the mutation with respect to catalytic site, distribution of tissue specific and cell specific substrates, cell turnover rate, defective protein expression and other mechanisms that influence the life span of the affected cells (Nat Rev Neurol 2013;9:583)
Diagrams / tables
Images hosted on other servers:

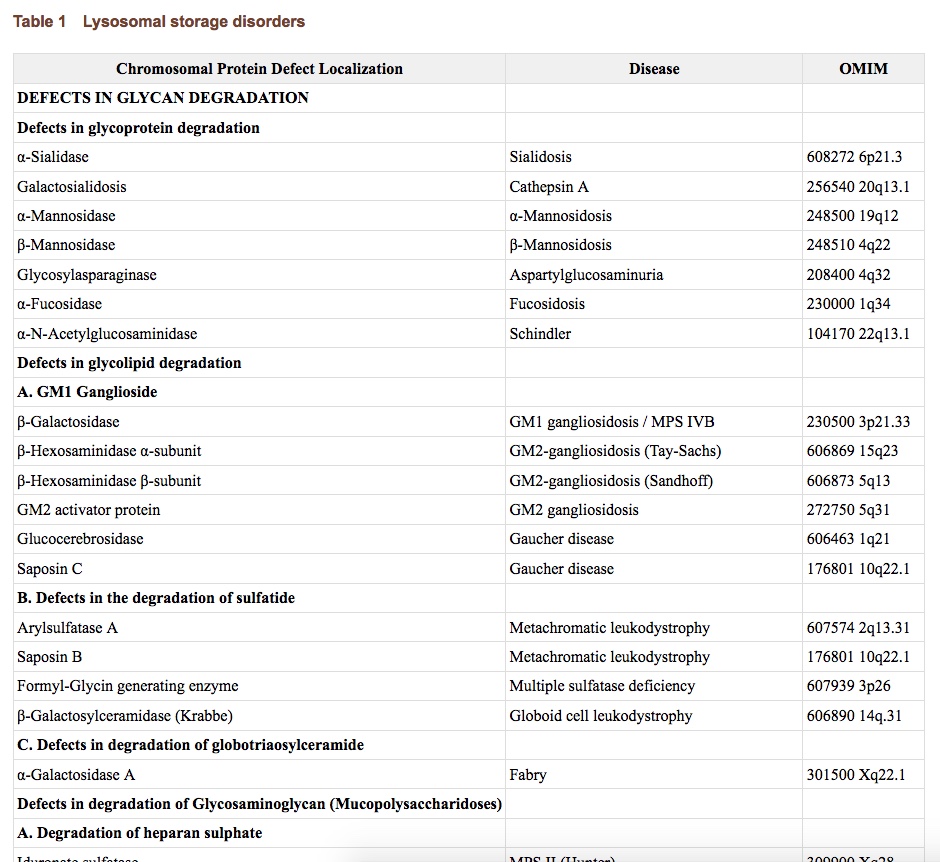
Classification
Clinical features
- In LSDs, buildup of material may begin in utero; disease progressively evolves over time, often becoming evident during the newborn period (i.e. infantile form)
- Up to 15% of nonimmune hydrops fetalis are due to LSD (Stocker: Stocker and Dehner's Pediatric Pathology, 3rd Edition, 2010)
- Presence of residual enzyme activity can result in mild and late onset forms, i.e. juvenile and adult variants (Madame Curie Bioscience Database: Lysosomal Storage Disorders [Accessed 30 October 2017])
- Involvement of multiple organs and systems is typical, with variable association of visceral, ocular, hematological, skeletal and neurological manifestations and there is partial phenotypic overlap among different disorders (Nat Rev Neurol 2013;9:583)
- About two thirds of patients with LSDs display a significant neurological component, which ranges from progressive neurodegeneration and severe cognitive impairment to epileptic, behavioral and psychiatric disorders (Annu Rev Med 2015;66:471)
- Abnormal thyroid function is mainly represented by primary hypothyroidism (Orphanet J Rare Dis 2012;7:11):
- Hypothyroidism due to cystine accumulation in follicles occurs in 50 - 75% of patients with cystinosis, both children and adults (JAMA 1993;270:2200, Orphanet J Rare Dis 2016;11:47)
- Frequent subclinical hypothyroidism was recently reported in Fabry disease (J Inherit Metab Dis 2005;28:715)
Diagnosis
- Current guidelines recommend that diagnosis must be established by specific enzyme assays and by mutational analysis (Genet Med 2011;13:457)
- Early detection of LSDs via genetic screening programs, prenatal diagnosis and screening of newborns in at risk families has major impact for the long term course and outcome of the disease (National Organization for Rare Disorders: Lysosomal Storage Disorders [Accessed 30 October 2017])
Laboratory
- Accumulated substrate can be detected in various clinical samples, such as urine, blood, amniotic fluid and tissue biopsies (Khong: Keeling's Fetal and Neonatal Pathology, 5th Edition, 2015)
- Lysosomal enzyme activities are usually determined by a fluorometric assay in cultured fibroblasts and leukocytes (Mehta: Fabry Disease - Perspectives from 5 Years of FOS, 2006)
Radiology images
Images hosted on other servers:

Skeletal deformities in mucopoly-saccharidoses
Prognostic factors
- Most early onset (diagnosed in infants) LSDs have a rather predictable clinical course, when neurologic deterioration continues until the death of the child within 5 - 10 years, usually by infection
- Predicting the clinical course in later onset patients, especially adolescents and adults, is nearly impossible (Arch Neurol 2003;60:322)
Case reports
- Sphingolipidoses
- 20 year old woman with Gaucher disease and symptoms suspicious for thyroid carcinoma (Pol Arch Med Wewn 1998;100:337)
- 48 year old woman with hypothyroidism in Fabry disease (Intern Med 1994;33:172)
- Two patients with thyroid peroxidase deficiency in Batten disease (Arch Pathol 1975;99:430)
- Two patients with Gaucher disease and papillary thyroid cancer (Am J Hematol 2010;85:340)
- Mucopolysacharidoses
- 19 year old man with Hunter syndrome and thyroid deposits (Acta Pathol Jpn 1976;26:115)
- 20 year old man with thyroid cell vacuolation in Morquio A syndrome (Mol Genet Metab 2013;109:301)
- Glycogenoses
- Two infants with Pompe disease (Arch Pathol Lab Med 1985;109:921)
- Two 15 month old girls with minimal thyroid inclusions in Pompe disease (Am J Dis Child 1964;108:503)
- Family case with Pompe disease and thyroxine binding globulin deficiency (Funct Neurol 1996;11:105)
- Cystinosis
- 23 year old man with cystinosis and atrophic thyroid (Acta Pathol Jpn 1985;35:145)
- 23 year old man with cystinotic kidney transplant and hyperthyroidism after radiotherapy for Hodgkin lymphoma (Pediatr Transplant 2011;15:e56)
Treatment
- There is no cure for most LSDs; symptomatic treatment is usually provided by a multidisciplinary team (neurologic, orthopedic, cardiologic, etc.)
- Early diagnosis with prompt initiation of presymptomatic therapy for defective soluble enzymes in the neonatal period or as early as possible can improve outcome (Nat Rev Neurol 2013;9:583)
- Cystinosis is treated with cysteamine, which reduces intracellular cystine
- Enzyme replacement therapy is a promising treatment; normal enzyme can be provided by intravenous injection or as a precursor secreted into the circulation by engineered cells from the patient or by an allograft of transplanted cells (Annu Rev Med 2015;66:471)
- Other advanced treatment strategies: stem cell therapies, substrate reduction and chaperone mediated delivery (Nat Rev Neurol 2013;9:583)
Clinical images
Images hosted on other servers:
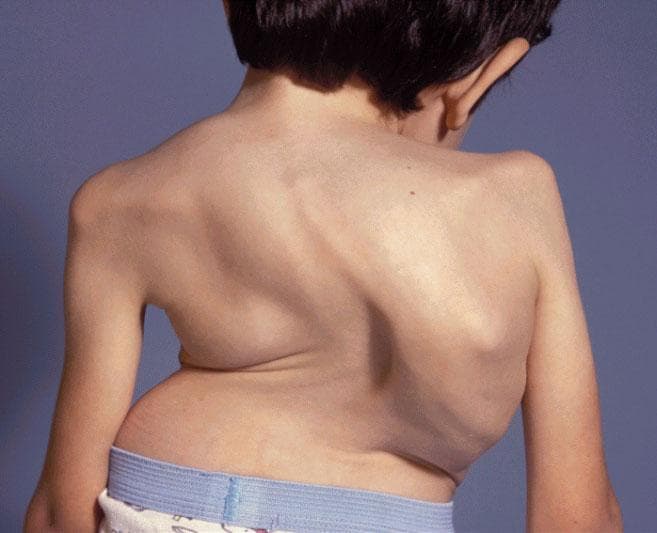
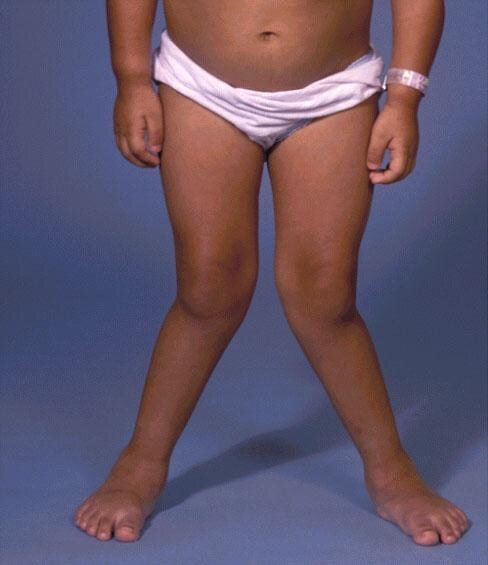
Skeletal deformities in mucopolysaccharidoses
Gross description
- Unremarkable thyroid gland
- In cystinosis thyroid may be atrophied (Acta Pathol Jpn 1985;35:145), occasionally to degree when it cannot be identified (Am J Med 1970;48:678)
Microscopic (histologic) description
- LSDs are characterized by progressive intracellular (rarely extracellular, e.g. mucopolysacharidoses) accumulation of a substrate
- Most prominent thyroid abnormalities are observed in cystinosis (Am J Med 1970;48:678, J Pediatr 1977;91:204):
- Destruction and infiltration of thyroid epithelial cells by cystine crystals
- Focal areas of papillary hyperplasia with dilated follicles and focal acute inflammation
- Follicular atrophy with fibrosis and decreased amounts of colloid
- Frozen sections show abundance of birefringent crystals within follicular cells and macrophages but these are usually lost in fixed tissue sections; to preserve crystals, specimens should be fixed in absolute alcohol, not formalin, otherwise cystine may dissolve
- Minor changes represented by intracellular deposits are found in thyroid in other LSDs:
- Tay-Sachs: follicular epithelial cells contain yellow, autofluorescent pigmented granules with lipofuscin (Am J Dis Child 1964;108:503)
- Batten: lipofuscin deposits (Lancet 1970;2:296)
- Hunter: cytoplasmic vacuolation (Acta Pathol Jpn 1976;26:115)
- Morquio: vacuolation of follicular and parafollicular cells (Mol Genet Metab 2013;109:301)
- Pompe: intralysosomal glycogen (Arch Pathol Lab Med 1985;109:921)
Microscopic (histologic) images
Case #164
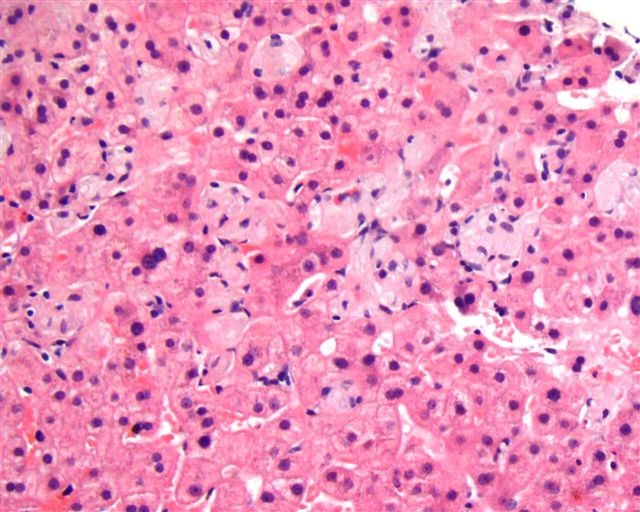
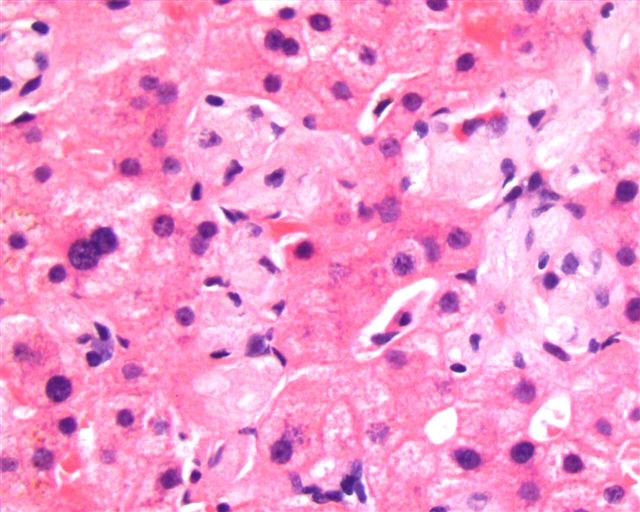
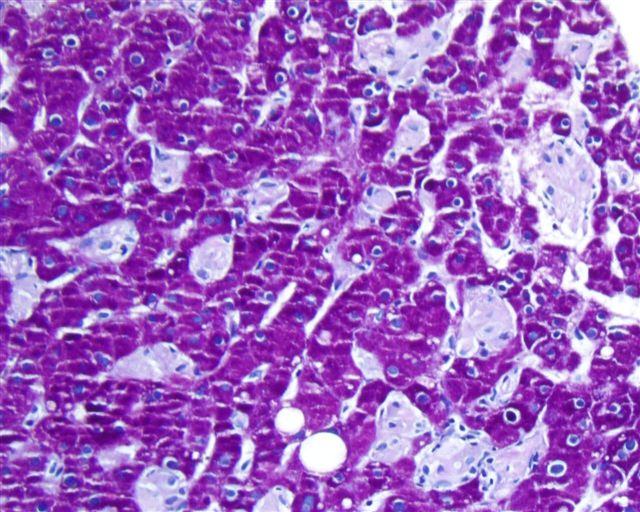
Liver biopsy in Gaucher disease (H&E, PAS)
Images hosted on other servers:






Hepatic glycogenosis (H&E, PAS, Masson)



Niemann-Pick disease (hepatic steatosis)
Virtual slides
Images hosted on other servers:
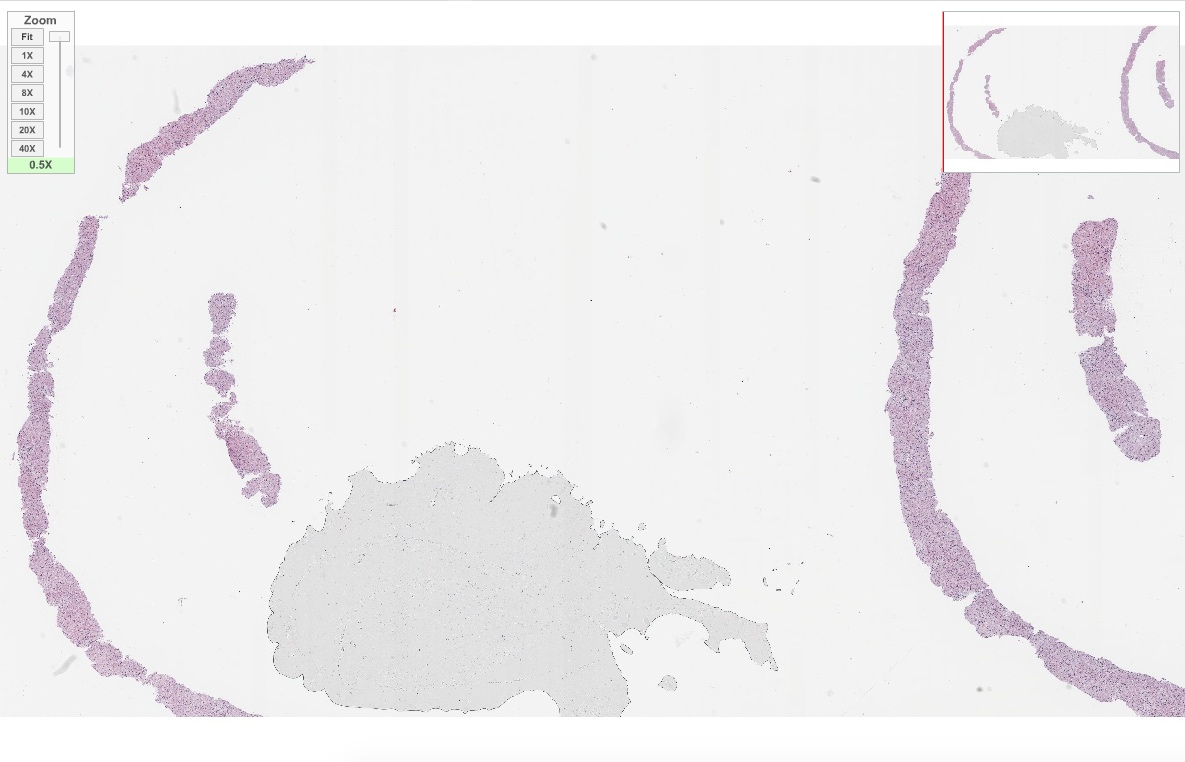
Niemann-Pick disease (liver biopsy)
Cytology images
Images hosted on other servers:



Gaucher cell in bone marrow
Peripheral smear description
- Presence of vacuolated lymphocytes is a simple screening test for storage disorders (Gilbert-Barness: Potter's Pathology of the Fetus, Infant and Child, 2nd Edition, 2007)
Positive stains
- Accumulated substrates can be confirmed by histochemistry, e.g. PAS stain for glycogen and lipofuscin (Stocker: Stocker and Dehner's Pediatric Pathology, 3rd Edition, 2010)
Negative stains
- CD107b helps to differentiate cardiomyopathy due to Danon disease
Electron microscopy description
- Cystinosis: numerous square, rectangular or lozenge shaped cystine crystals in the cytoplasm of foam cells, with smaller crystals located within osmiophilic dense bodies (Acta Pathol Jpn 1985;35:145)
- Hunter: clear cytoplasmic inclusion bodies containing various amounts of flocculent material representing glycosaminoglycans (Acta Pathol Jpn 1988;38:1175)
- Tay-Sachs: osmophilic deposits in cytoplasm of follicular cells is seen in brain, muscle, other tissues (Lab Invest 1974;30:102)
- Ultrastructural studies may be used as a screening tool or if a storage disease is suspected (Stocker: Stocker and Dehner's Pediatric Pathology, 3rd Edition, 2010)
Electron microscopy images
Images hosted on other servers:

Lamellated "zebra" bodies in Fabry disease (fig 10)
Leukocyte in mannosidosis
Videos
Histopathology of glycogen storage disease (heart, liver)
Primer in inherited metabolic disorders (2013)
Mechanism of LSDs (2014)
Lecture series on LSDs by Excellence in Pediatrics Institute (2014 - 2016)
Additional references
- Web sources: Wikipedia: Lysosomal Storage Disease [Accessed 30 October 2017], eMedicine: Lysosomal Storage Disease [Accessed 30 October 2017], Neuropathology: Inherited Metabolic Disorders [Accessed 30 October 2017]
- Books: Barranger: Lysosomal Storage Disorders, 1st Edition, 2007, Gilbert-Barness: Metabolic Diseases - Foundations of Clinical Management, Genetics and Pathology, 1st Edition, 2000, Mehta: Lysosomal Storage Disorders - A Practical Guide, 1st Edition, 2013, Pastores: Lysosomal Storage Disorders - Principles and Practice, 1st Edition, 2009, Saudubray: Inborn Metabolic Diseases - Diagnosis and Treatment, 5th Edition, 2012
- Societies: SSIEM, Cystinosis Research Network
