
Soft tissue
Uncertain differentiation
Extrarenal rhabdoid tumor
Editorial Board Member: Borislav A. Alexiev, M.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 29 September 2020
Last staff update: 26 April 2023
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Rhabdoid tumor [title] soft tissue
See Also: Rhabdoid tumor of the kidney

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Clinical features | Diagnosis | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Frozen section description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Cytology images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Chundriger Q, Ud Din N. Extrarenal rhabdoid tumor. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/softtissuerhabdoidtumor.html. Accessed April 1st, 2025.
Definition / general
- Heterogeneous group of neoplasms unified by the presence of globular cytoplasmic inclusions, vesicular nuclei with prominent nucleoli and aggressive behavior
- Response to therapy is poor
- Metastasizes to lung, liver and lymph nodes early in the course of disease
Essential features
- Abundant eosinophilic cytoplasm (due to hyaline globular inclusions of intermediate filaments) with eccentric vesicular nuclei and prominent nucleoli
- Skeletal muscle markers and S100 negative
- Karyotypic rearrangements of 22q11.2 resulting in homozygous inactivation of SMARCB1 (hSNF5 / INI1), with subsequent loss of INI1 nuclear expression by immunohistochemistry
Terminology
- Malignant rhabdoid tumor (synonym)
ICD coding
- ICD-O: 8963/3 - rhabdoid tumor, NOS
- ICD-11: 2B5F.2 & XH3RF3 - sarcoma, not elsewhere classified of other specified sites & malignant rhabdoid tumor
Epidemiology
- Genuine rhabdoid tumors exclusively belong to the pediatric age group and are very rare
- Adult cases represent expression of a particular phenotype, which can occur in any type of sarcoma or even carcinoma but it is virtually universally associated with an aggressive course
- Mean age at diagnosis < 1 year for tumors with germ line SMARCB1 alterations and 1.5 years for sporadic tumors (Crit Rev Oncog 2015;20:199)
- M = F
Sites
- Deep soft tissue including limb girdles, retroperitoneum, abdominal cavity, liver, pelvis, neck, paraspinal and perineal regions
- Rare unusual sites include:
- Intraocular (J Pediatr Ophthalmol Strabismus 2018;55:e7, J Pediatr Hematol Oncol 2020;42:228)
- Paratesticular (Cureus 2020;12:e8273)
- Intradural (Childs Nerv Syst 2018;34:165)
Pathophysiology
- Mutations in 22q11.2, which cause homozygous inactivation of the SMARCB1 gene (Genes Chromosomes Cancer 2011;50:379)
- More recently, has been described as one of the many neoplasms belonging to a group of SW1 / SNF deficient tumors (Semin Diagn Pathol 2018;35:193)
Clinical features
- Clinical presentation is related to the site of involvement
- Intraocular tumors present with proptosis (J Pediatr Hematol Oncol 2020;42:228)
- Deep / visceral involvement causes local pain and pressure symptoms and may result in loss of function of an anatomic site if local neural bundle / plexus is compromised (Brain Dev 2017;39:717)
Diagnosis
- Rhabdoid cell morphology in a pediatric tumor along with loss of INI1
- Requires thorough sampling and immunohistochemical studies to rule out rhabdoid phenotype in another underlying tumor, particularly in adult cases (Mod Pathol 2016;29:1232)
- Demonstration of 22q11.2 mutations in a tumor with well developed rhabdoid morphology (Genes Chromosomes Cancer 2011;50:379)
Radiology description
- Heterogenous appearance on CT and MRI (J Pediatr Surg 2018;53:567)
- May show cystic areas
Radiology images
Images hosted on other servers:

Intratumoral hemorrhage, bone erosion

Lobulated mediastinal mass
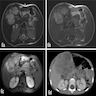
Multilobulated hyperintense mass
Prognostic factors
- Rhabdoid phenotype itself is a poor prognostic factor
- Advanced stage and presentation at < 1 year of age is associated with poorer survival (Oncologist 2019;24:e551)
- 5 year survival rate is < 15% (Cancer 2007;110:2061, Pediatr Blood Cancer 2008;51:363)
- Rhabdoid tumors of liver behave more aggressively (Blood Cancer 2011;57:423)
Case reports
- Infant 36 weeks with bulging parieto-occipital mass (Head Neck Pathol 2017;11:224)
- Infant 37 weeks with echogenic bladder mass (Urology 2019;123:221)
- 1 year old boy presenting with an immobile arm (Brain Dev 2017;39:717)
- 15 year old girl with inguinal mass (Pediatr Blood Cancer 2019;66:e27784)
- Teenager with primary intraocular tumor (J Pediatr Ophthalmol Strabismus 2018;55:e7)
- 65 year old man with tumor of colon and disease free survival (J Med Case Rep 2018;12:39)
Treatment
- No definite chemotherapy regimen is available; some cases report successful therapy using alkaloids, platinum agents and combinations of chemotherapy, surgery and radiation (Crit Rev Oncog 2015;20:199)
- Targeted therapy is under investigation, utilizing various epigenetic pathways including DNA and histone methylation, histone deacetylation, cell cycle arrest and antimitotic mechanisms (Crit Rev Oncog 2015;20:199)
Clinical images
Images hosted on other servers:
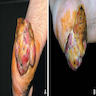
Ulcerating tumor at heel
Gross description
- Tumor is infiltrative with a tan-white solid appearance
- Calcifications and hemorrhage may be seen (Urology 2020;137:164)
Gross images
Contributed by Mark R. Wick, M.D.
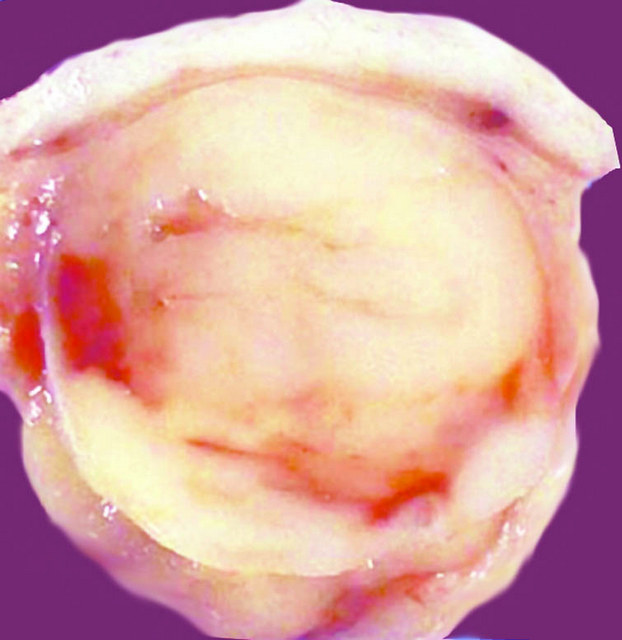
Superficial, subcuticular
Images hosted on other servers:

Polypoid mass with hemorrhage
Frozen section description
- Frozen sections are rarely performed and show sheets of cells with typical rhabdoid appearance and vesicular nuclei with nucleoli
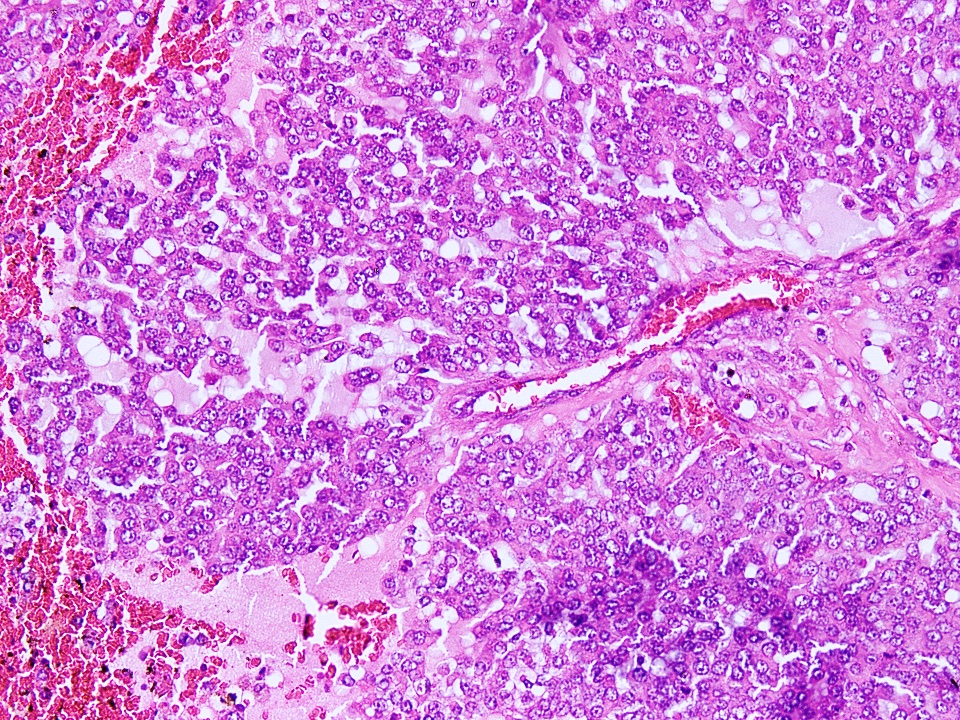
Microscopic (histologic) description
- Tumor cells have classic rhabdoid or skeletal-like profile, comprised of abundant cytoplasm with eosinophilic hyaline globules and vesicular nuclei with prominent nucleoli (Anticancer Res 2005;25:4573, Ann Diagn Pathol 2012;16:504)
- Some cases may show areas of spindling of tumor cells
- Nuclear pleomorphism can be prominent
- Background stroma may be variably myxoid to fibromyxoid
- Tumor necrosis and mitotic activity can be variable
Microscopic (histologic) images
Contributed by Nasir Ud Din, M.B.B.S.
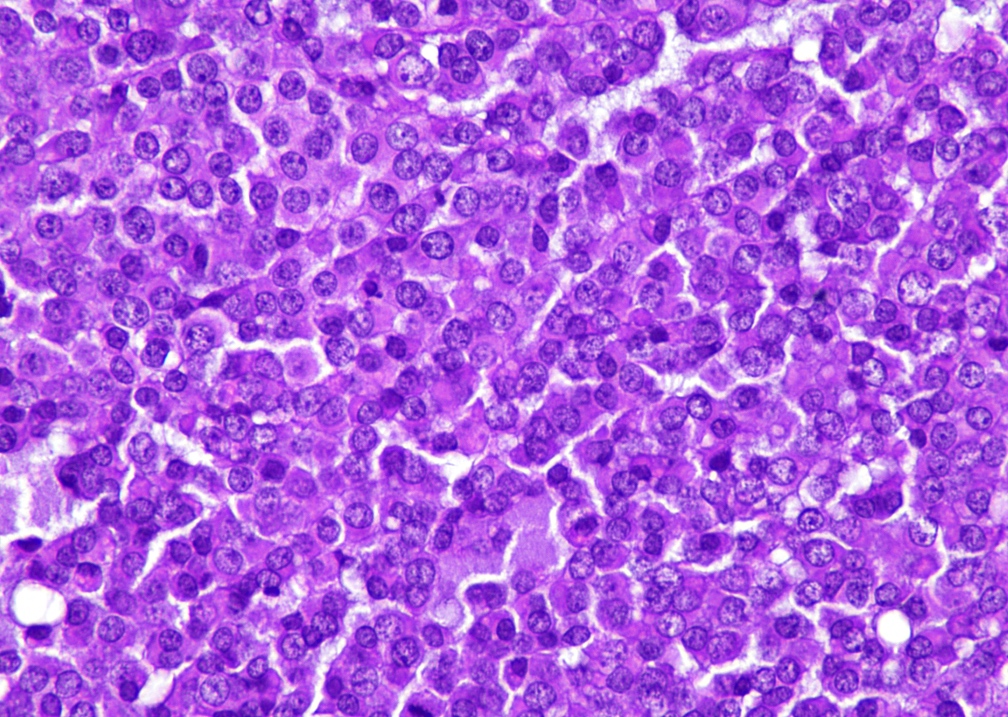
Rhabdoid cells

Prominent nucleoli
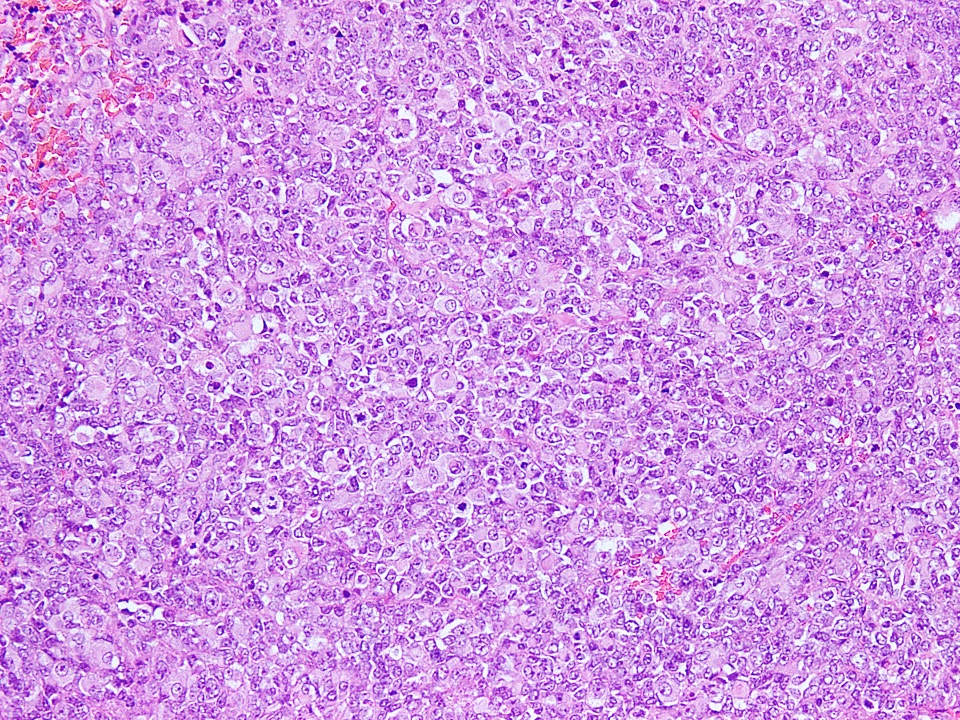
Sheet-like growth
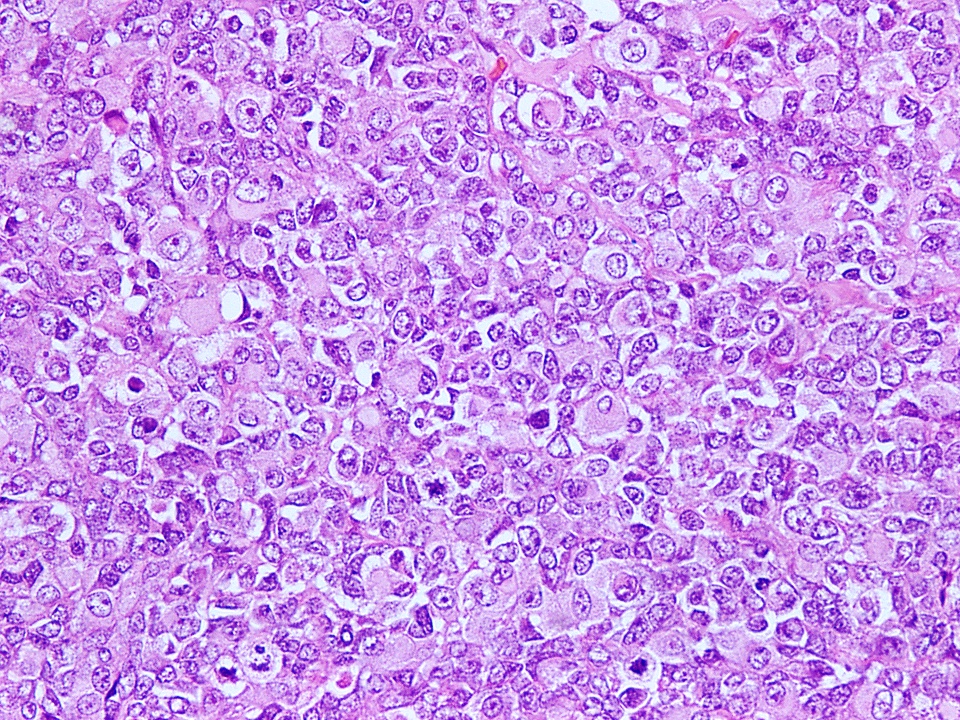
Nuclear pleomorphism
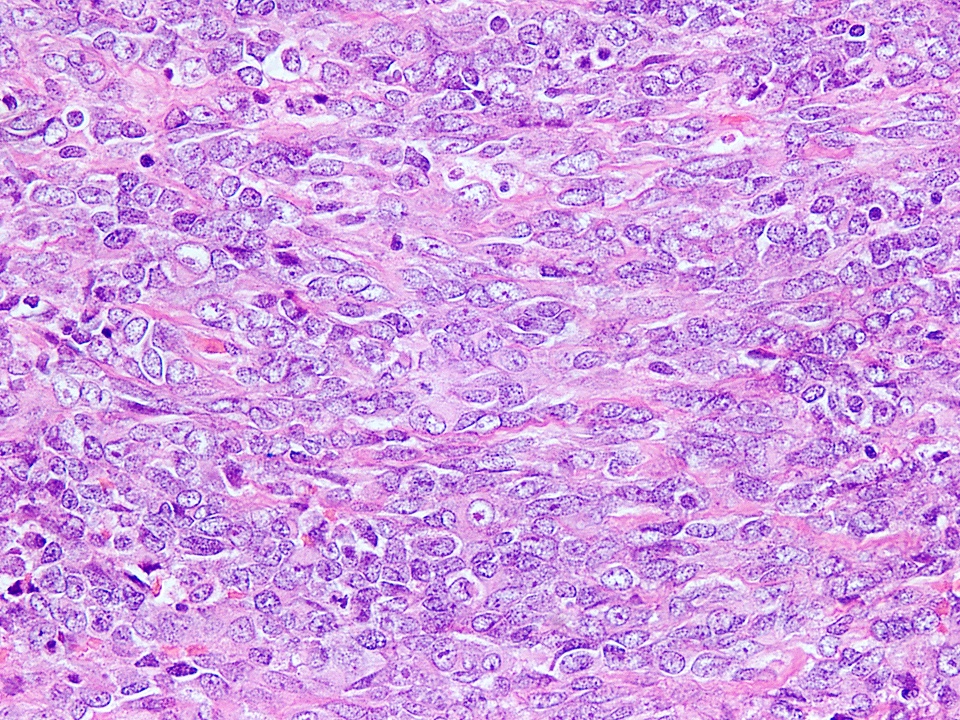
Spindling of tumor cells
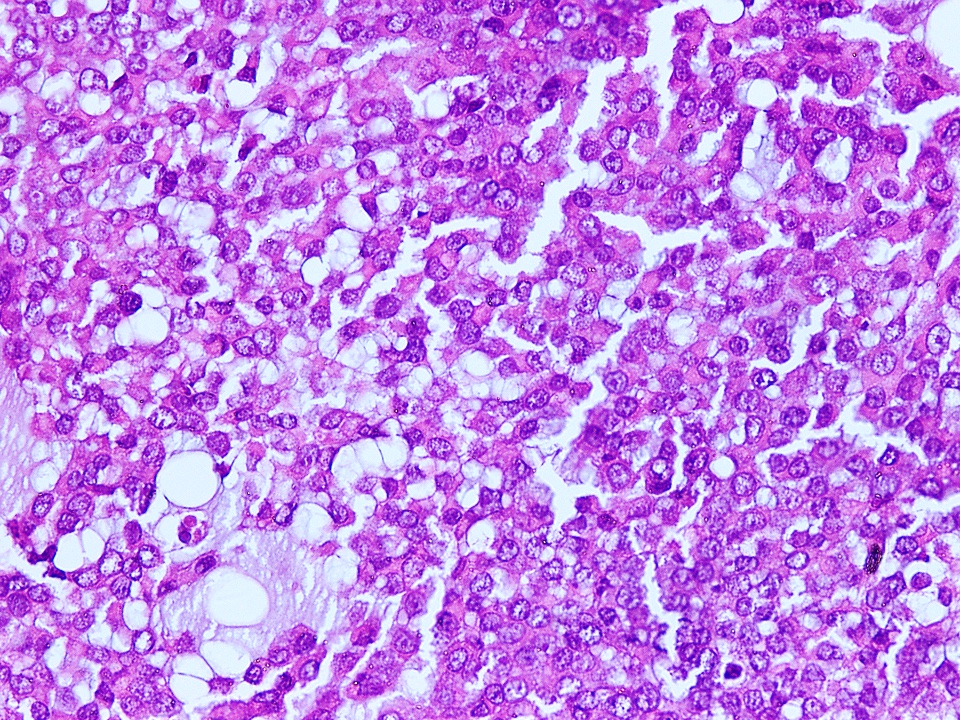
Myxoid stroma
Fibromyxoid stroma

Tumor necrosis
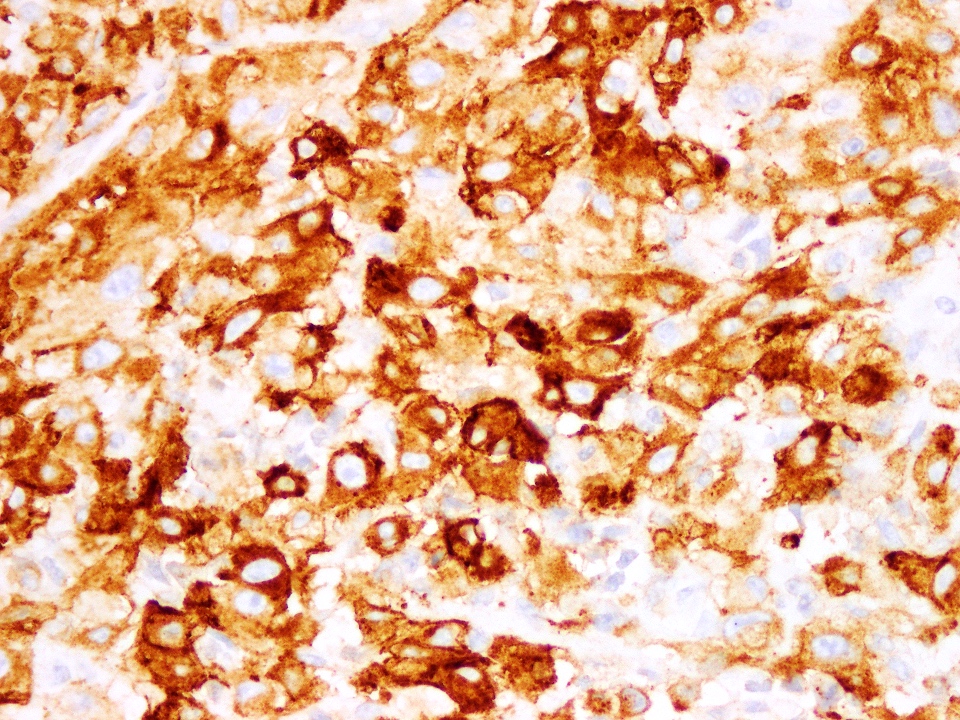
EMA
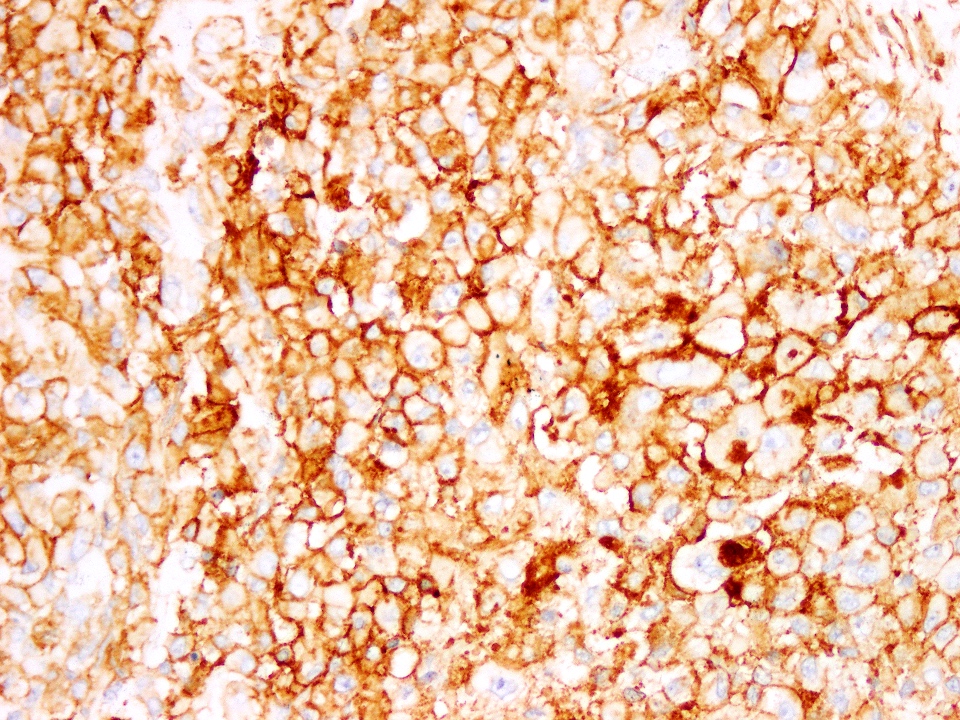
CD99
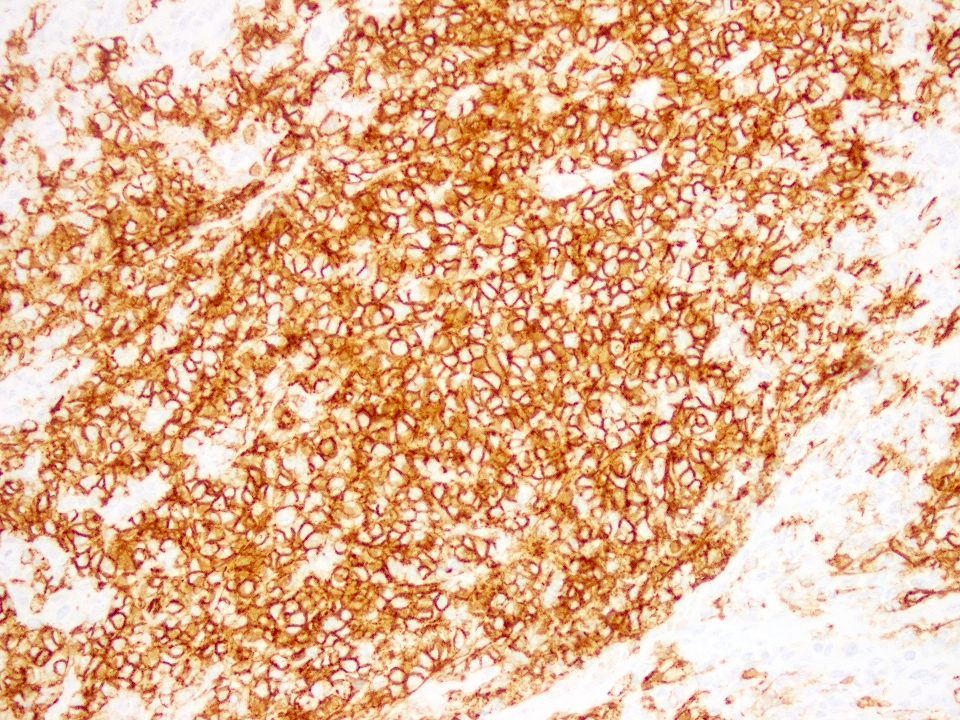
CD56
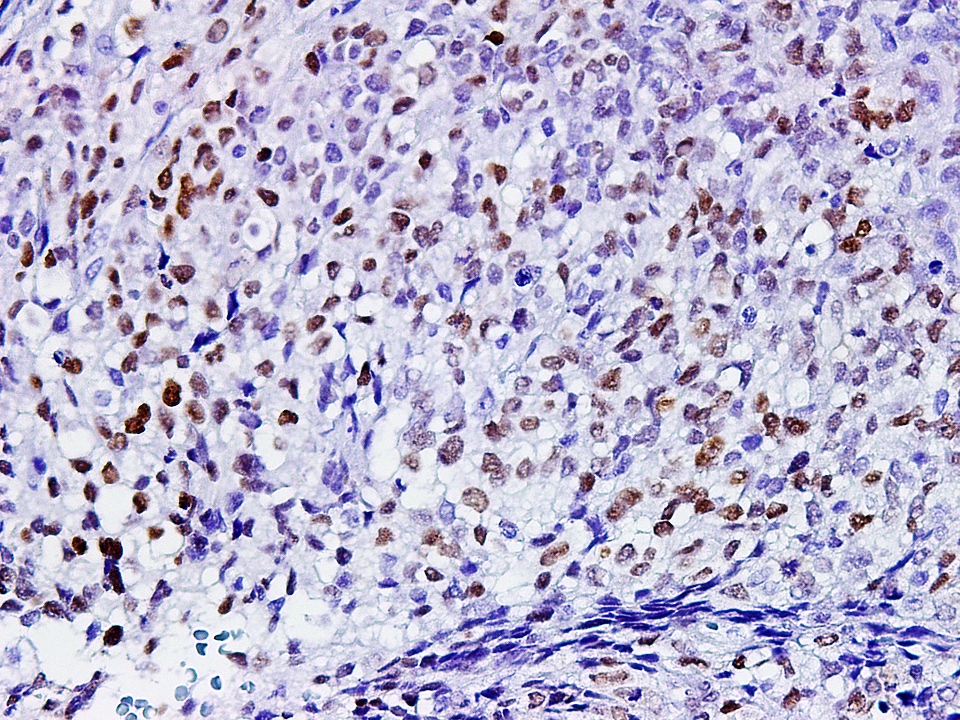
SALL4
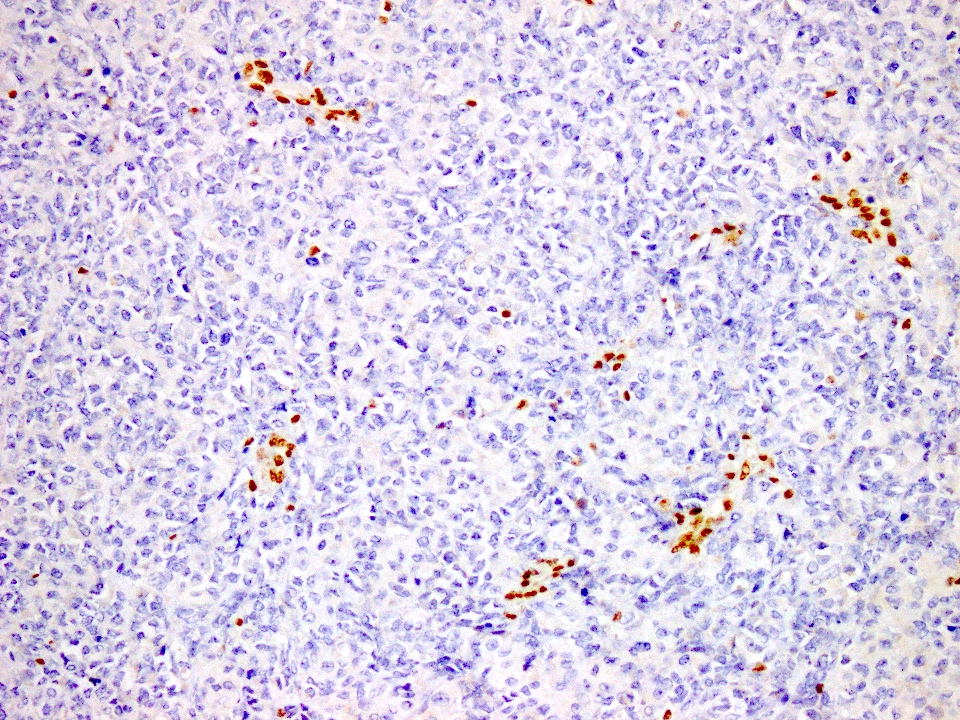

Loss of INI1
Cytology description
- Variably cellular smears with individual cells and structureless clusters of rhabdoid cells, spindle cells or round cells
- Nuclei show either prominent nucleoli in rhabdoid cells or nuclei with homogenous chromatin in round cells (Cancer Cytopathol 2011;119:49)
- Cytoplasmic inclusions are eosinophilic on Giemsa and pale gray on Papanicolaou stain (Indian J Pathol Microbiol 2011;54:819)
- Differentials on cytology include extrarenal Wilms tumor, rhabdomyosarcoma, spindle cell sarcoma and round blue cell tumors including lymphoma (Indian J Pathol Microbiol 2011;54:819)
Cytology images
Contributed by Mark R. Wick, M.D.
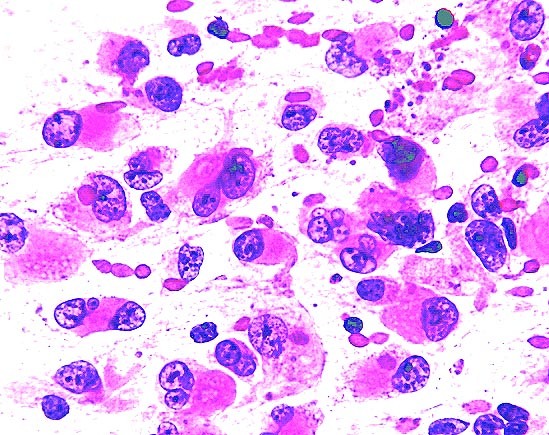
Rhabdoid cells in smears
Images hosted on other servers:

Giemsa and Papanicolaou stains
Positive stains
- EMA, cytokeratin (variable positivity) and vimentin
- CD99, CD56 and synaptophysin in some cases
- SALL4 and glypican 3 also frequently positive (Hum Pathol 2015;46:225, Histopathology 2015;66:252, Hum Pathol 2013;44:526)
Negative stains
Electron microscopy description
- Eosinophilic hyaline inclusions are composed of clusters of intermediate filaments, which can form cytoplasmic whorls (J Pediatr Ophthalmol Strabismus 2013;50:e18)
Electron microscopy images
Images hosted on other servers:
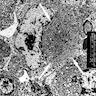
Cytoplasmic whorls
Molecular / cytogenetics description
- Mutations in 22q11.2 (deletion of SMARCB1)
- Mutations in any of the genes encoding SWI / SNF complex (Semin Diagn Pathol 2018;35:193, Pediatr Hematol Oncol 2013;30:587)
Molecular / cytogenetics images
Images hosted on other servers:

Homozygous deletion on MLPA
Sample pathology report
- Right arm, excision:
- Extrarenal rhabdoid tumor (see comment)
- Comment: Tumor cells are rhabdoid in appearance with eccentric nuclei. Positive cytokeratin, negative skeletal markers and loss of INI1 are seen. 22q11.2 mutation confirms the diagnosis.
Differential diagnosis
In children:
In adults:
- Rhabdomyosarcoma:
- Embryonal rhabdomyosarcoma:
- Common in children < 10 years old
- M > F
- Most common sites: head and neck region, genitourinary region
- May have rhabdomyoblasts / strap cells with cross striations
- May show eccentric cytoplasm similar to extrarenal rhabdoid tumor
- Shows positive staining for markers of skeletal differentiation (desmin, myogenin and MyoD1)
- Retained nuclear INI1
- Alveolar rhabdomyosarcoma:
- Occurs in adolescents and young adults
- Grows as alveoli-like spaces formed by loss of cohesion centrally
- Monomorphic round cells with hyperchromatic nuclei and scant cytoplasm
- Strong and diffuse desmin and myogenin positivity
- May express focal / weak cytokeratins (5 - 50% cases)
- Intact nuclear INI1
- Embryonal rhabdomyosarcoma:
- Ewing sarcoma:
- Shows sheet-like growth of round blue cells with scant cytoplasm and hyperchromatic nuclei
- Diffuse CD99 and NKX2.2 expression
- Retained nuclear INI1
- May show cytokeratin positivity in up to 20% cases
- EWSR1 mutations are diagnostic
- Desmoplastic round blue cell tumor:
- Mean age 25 years
- M > F
- Abdominal cavity is the most common site
- Contains prominent desmoplastic stroma
- Round cells grow in nodules separated by stroma
- May show polyphenotypic differentiation with positive keratins, vimentin, desmin, CD56, NSE and nuclear WT1
- Retained nuclear INI1
- EWSR1-WT1 gene fusion is consistent
In adults:
- Extrarenal rhabdoid tumor should be considered only after other tumors with a rhabdoid appearance have been ruled out
- Carcinoma:
- Age over 50, a history of carcinoma and areas of cohesive epithelial differentiation help in reaching correct diagnosis
- Malignant melanoma:
- Nonpigmented melanoma mimics rhabdoid tumor
- Occurs in older age group
- Positive melanocytic markers HMB45, MelanA and negative cytokeratins
- Anaplastic large cell lymphoma:
- May occasionally exhibit rhabdoid features
- Positive for CD30, weak / focal positive for CD45 / LCA and CD3 and negative for keratins
- Proximal type epithelioid sarcoma:
Board review style question #1
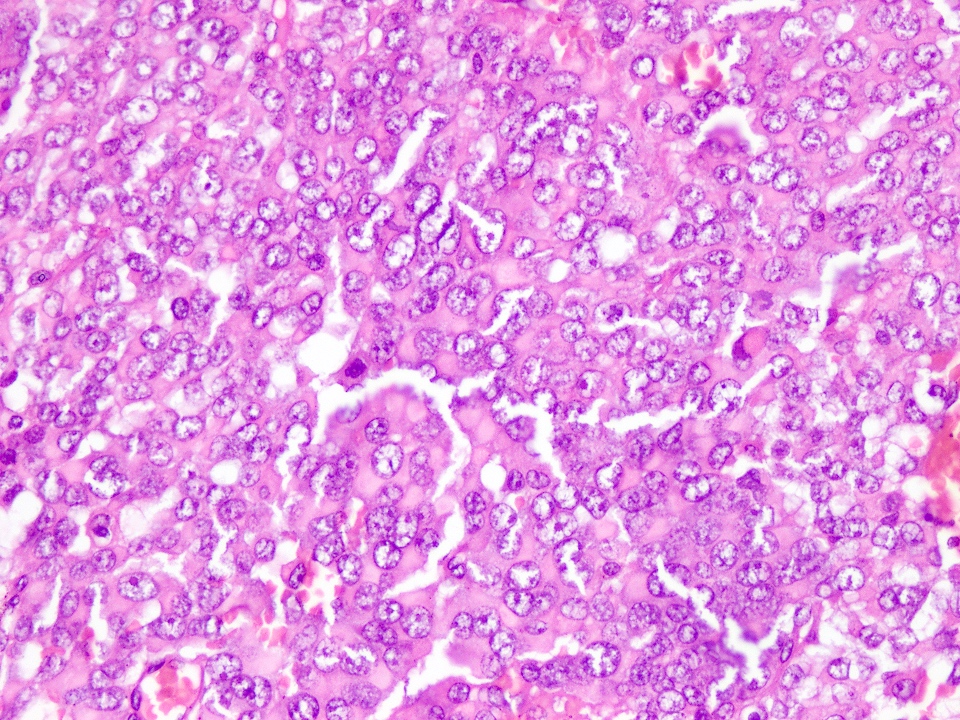
A 2 month old baby presented with a progressively enlarging soft tissue mass in the right axillary region. Microscopic examination of the resected tumor showed morphology depicted in the picture. Immunohistochemical stains to help reach a diagnosis are
- Negative desmin and nuclear INI1
- Negative vimentin and positive S100
- Positive cytokeratin and CD34
- Positive cytokeratin and desmin
- Positive cytokeratin and negative vimentin
Board review style answer #1
A. Negative desmin and nuclear INI1. This is an extrarenal rhabdoid tumor.
Comment Here
Reference: Extrarenal rhabdoid tumor
Comment Here
Reference: Extrarenal rhabdoid tumor
Board review style question #2
Homozygous inactivation of SMARCB1 seen in extrarenal rhabdoid tumors are due to mutations in
- 9q22
- 11p13
- 11q24
- 21q12
- 22q11
Board review style answer #2
