
Soft tissue
Peripheral nerve
Malignant
Malignant peripheral nerve sheath tumor (MPNST)
Authors: Erica Kao, M.D., Jose G. Mantilla, M.D.
Resident / Fellow Advisory Board: Farres Obeidin, M.D.
Editorial Board Member: Borislav A. Alexiev, M.D.


Last author update: 20 January 2021
Last staff update: 19 September 2023
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Malignant peripheral nerve sheath tumor

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Frozen section description | Microscopic (histologic) description | Microscopic (histologic) images | Virtual slides | Cytology description | Cytology images | Positive stains | Negative stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Kao E, Mantilla JG. Malignant peripheral nerve sheath tumor (MPNST). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/softtissuempnst.html. Accessed March 30th, 2025.
Definition / general
- Malignant neoplasm arising from peripheral nerve
- May arise from a preexisting nerve sheath tumor in neurofibromatosis type 1 (NF1) or in the setting of prior radiation therapy
- In the absence of association with NF1 or radiation, diagnosis is challenging and based on histologic and immunohistochemical features suggesting Schwannian differentiation (Am J Surg Pathol 2016;40:896)
- Variants:
- Epithelioid MPNST
- Rare subtype, usually not associated with NF1 (Am J Surg Pathol 2019;43:835)
- Rare cases may arise in epithelioid Schwannoma
- Typically have diffuse and strong expression of S100 and SOX10 (Am J Surg Pathol 2019;43:835)
- Distinct molecular features from conventional MPNSTs and harbor SMARCB1 gene inactivation and INI1 loss in up to 40 - 67% of cases (Am J Surg Pathol 2019;43:835, Histopathology 2016;68:286, Am J Surg Pathol 2015;39:673)
- MPNST with heterologous rhabdomyoblastic differentiation (Malignant Triton tumor)
- Has been associated with adverse clinical behavior (Eur J Surg Oncol 2013;39:46)
- Epithelioid MPNST
Essential features
- Sarcoma with peripheral nerve sheath differentiation with typically aggressive behavior
- Can occur in the following settings:
- Sporadic (~ 50%)
- In neurofibromatosis type 1 (40 - 50%)
- In the setting of prior radiation therapy (10%) (World Neurosurg 2017;105:961)
- Morphology: marbling at low magnification (alternating areas of hypocellularity and hypercellularity) with perivascular accentuation, uniform cytologic features
- Heterologous differentiation in 10 - 15% of cases
- Rhabdomyoblastic differentiation associated with adverse clinical behavior (Eur J Surg Oncol 2013;39:46)
- SOX10 and S100 IHC only seen in 50% of cases
Terminology
- Malignant peripheral nerve sheath tumor (MPNST)
- Obsolete: neurofibrosarcoma, malignant schwannoma, neurogenic sarcoma
ICD coding
- ICD-O: 9540/3 - malignant peripheral nerve sheath tumor
- ICD-11:
- 2B5E & XH2XP8 - malignant nerve sheath tumor of peripheral nerves or autonomic nervous system, primary site, malignant peripheral nerve sheath tumor
- 2B5E & XH4V81 - malignant nerve sheath tumor of peripheral nerves or autonomic nervous system, primary site, malignant peripheral nerve sheath tumor, epithelioid
- 2B5E & XH2VV8 - malignant nerve sheath tumor of peripheral nerves or autonomic nervous system, primary site, malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation
Epidemiology
- MPNST can occur in the following settings:
- Sporadic (approximately 50%)
- In neurofibromatosis type 1 (40 - 50%)
- In the setting of prior radiation therapy (10%)
- Plexiform neurofibroma is a common precursor lesion in patients with NF1
- Patients with NF1 have an 8 - 13% lifetime risk of developing MPNST
- Plexiform neurofibromas are seen in approximately 50% of patients (Sarcoma 2017;2017:7429697)
- 10 - 15% of plexiform neurofibromas transform to MPNST (Hum Mutat 2008;29:E103)
- Other nerve sheath tumors (schwannoma, ganglioneuroma) may rarely give rise to secondary MPNST (Am J Surg Pathol 1994;18:882, Am J Surg Pathol 2001;25:13, Pathology 2015;47:595, Histopathology 1988;12:445)
Sites
- Can arise in virtually any anatomic location
- Most common sites are the trunk and extremities, followed by head and neck (Am J Surg Pathol 2016;40:896)
Pathophysiology
- Germline mutations in NF1 predispose to the development of peripheral nerve sheath neoplasms in patients with type 1 neurofibromatosis (Acta Neuropathol 2012;123:349)
- In the setting of NF1, lesions often arise from plexiform neurofibroma (Acta Neuropathol 2012;123:349)
- Transformation process is accompanied by progressive genomic changes involving NF1, CDKN2A / CDKN2B and PRC2 (Hum Pathol 2017;67:1)
- Radiation therapy predisposes to the development of secondary sarcomas through repeated DNA damage and defective repair
Etiology
- See Epidemiology
Clinical features
- No sex predilection
- Patients with NF1 are typically younger than their sporadic and radiation associated counterparts (J Med Genet 2002;39:311)
Diagnosis
- Histologic evaluation is necessary but not always specific and requires correlation with clinical and radiologic findings
- Helpful features include close association with peripheral nerves and history of NF1 or precursor lesions
Radiology description
- Association with a large peripheral nerve or neurofibroma on MRI
- FDG PET avid; not specific but can help distinguish MPNST from neurofibroma (Skeletal Radiol 2016;45:1097, Neurology 2019;93:e1076)
Radiology images
Contributed by Jose G. Mantilla, M.D.
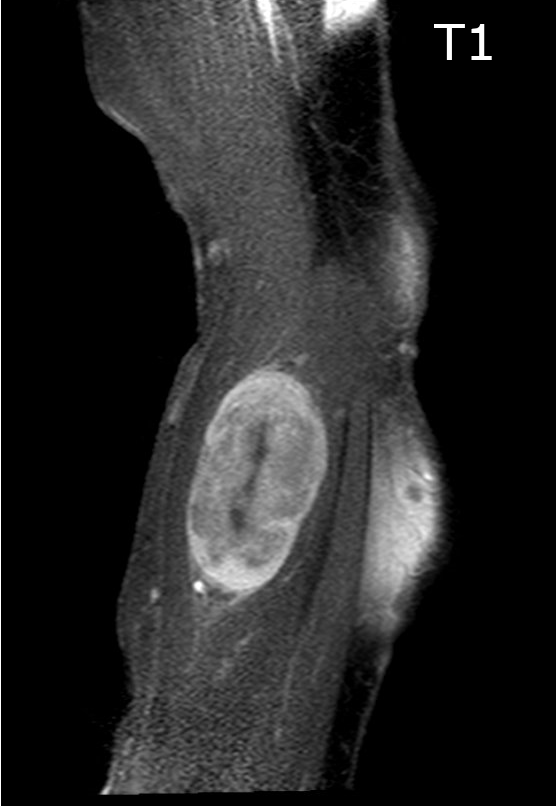
Forearm lesion T1
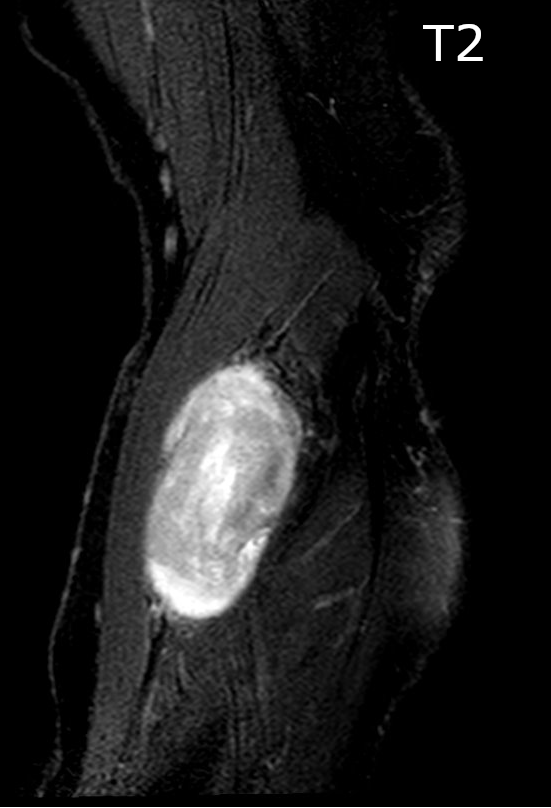
Forearm lesion T2
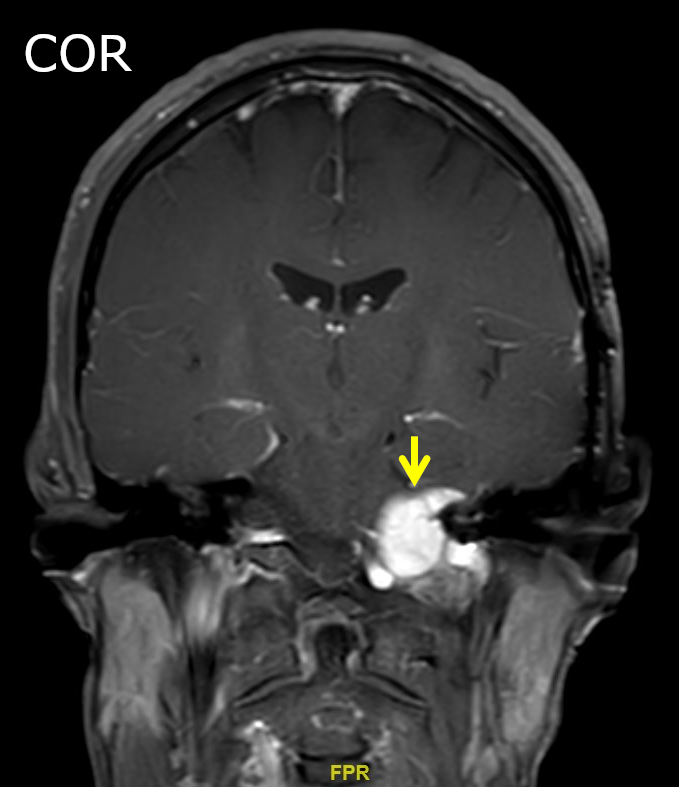
Cerebellopontine angle mass coronal view
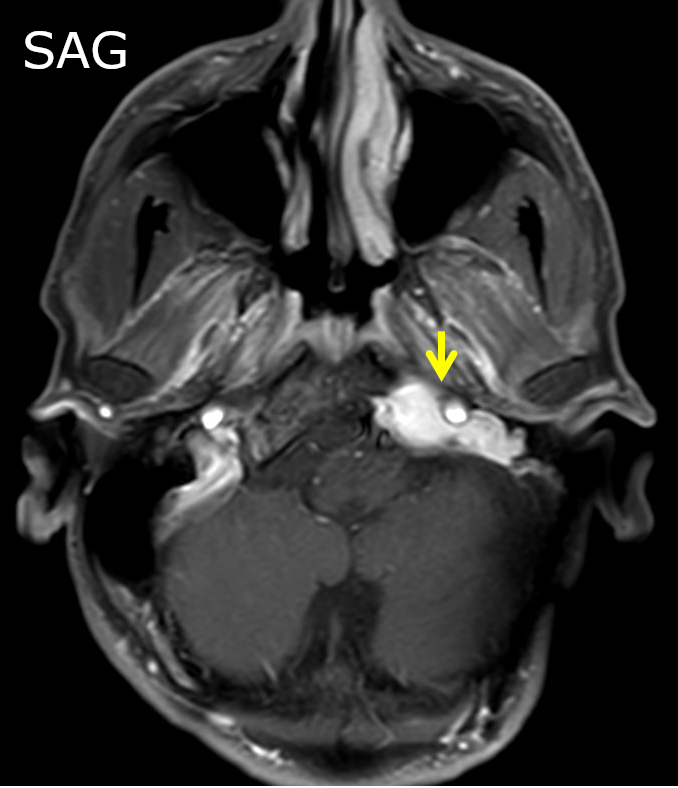
Cerebellopontine angle mass sagittal view
Prognostic factors
- Generally aggressive behavior with frequent metastatic disease and local recurrence
- FNCLCC grading controversial, high grade may be associated with aggressive behavior (Eur J Cancer 2016;56:77, Radiother Oncol 2019;137:61)
- Trunk has worse prognosis than extremities (Radiother Oncol 2019;137:61, J Neurosurg 2017;126:319)
- NF1 and radiation associated associated MPNST have worse prognosis (Am J Surg Pathol 2016;40:896, Radiother Oncol 2019;137:61)
- Rhabdomyoblastic differentiation (malignant triton tumor) associated with aggressive behavior (Eur J Surg Oncol 2013;39:46)
Case reports
- 3 year old girl with spinal intradural malignant peripheral nerve sheath tumor (Turk Neurosurg 2018;28:317)
- 26 year old man with malignant triton tumor of the gluteal region (Acta Orthop Traumatol Turc 2018;52:236)
- 29 year old woman with transformation of vestibular schwannoma (Asian J Neurosurg 2018;13:810)
- 45 year old man with right parotid swelling (Indian J Surg Oncol 2018;9:629)
Treatment
- Aggressive surgical resection followed by radiation therapy to achieve local control (World J Surg Oncol 2006;4:55, Int J Radiat Oncol Biol Phys 1998;42:351)
- Therapeutic options for metastatic MPNST are limited; conventional chemotherapy is usually limited to patients with metastatic disease (Surg Oncol Clin N Am 2016;25:789)
- Dramatic response to vemurafenib has been reported in a single case harboring BRAF V600E mutation (J Natl Compr Canc Netw 2013;11:1466)
Gross description
- Fusiform to globoid, pseudoencapsulated tumor often with gross evidence of necrosis
- If the tumor is arising from a nerve, an attached medium or large nerve is evident
Gross images
Contributed by Jose G. Mantilla, M.D.
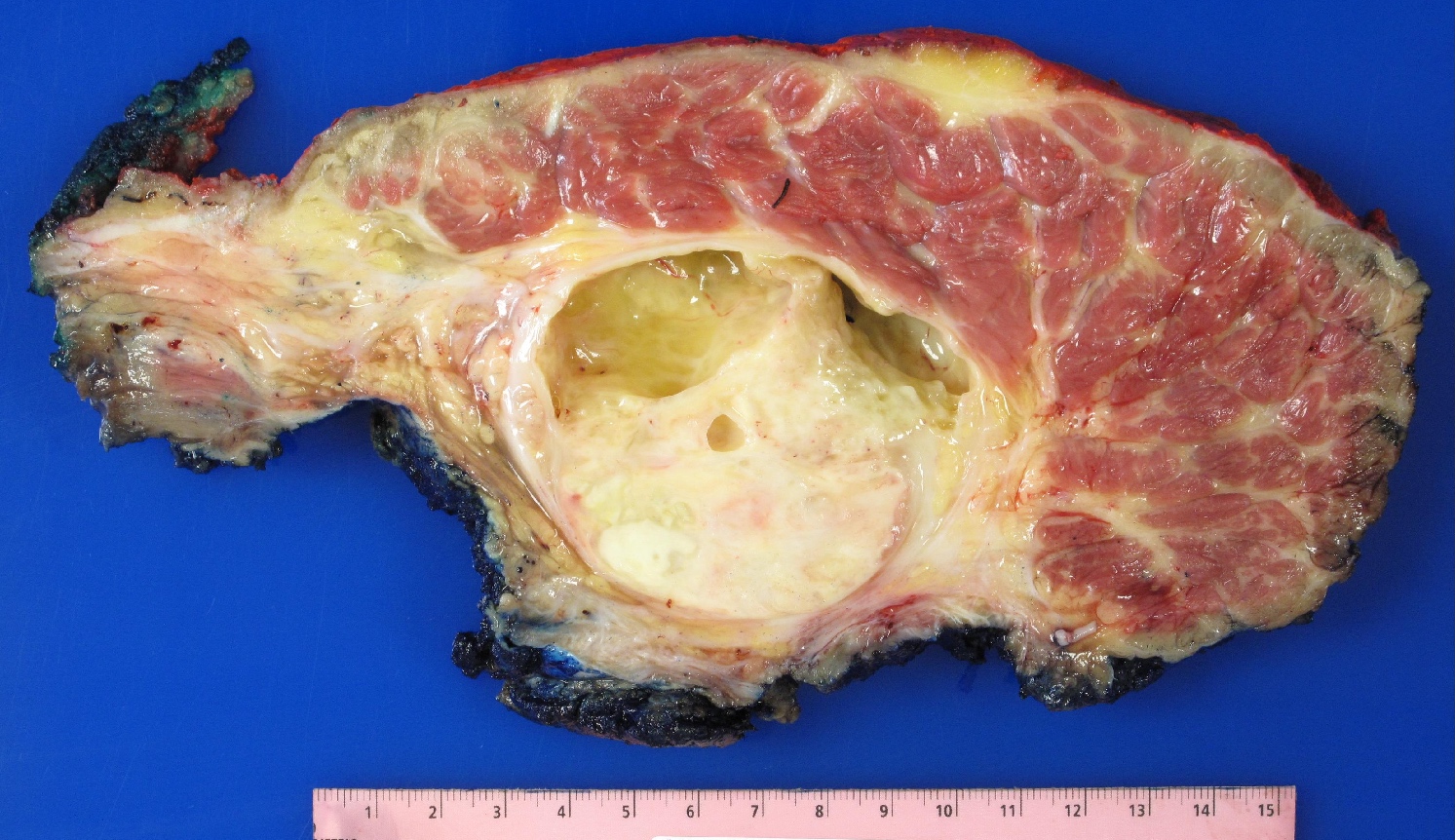
Gross findings
Frozen section description
- In high grade tumors, overt features of malignancy are readily identified, including nuclear pleomorphism, brisk mitotic activity and areas of geographic necrosis
- Diagnosis of low grade lesions is difficult in frozen sections
- In NF1, mitotic activity, increased cellularity and nuclear atypia within a neurofibroma raise concern for MPNST
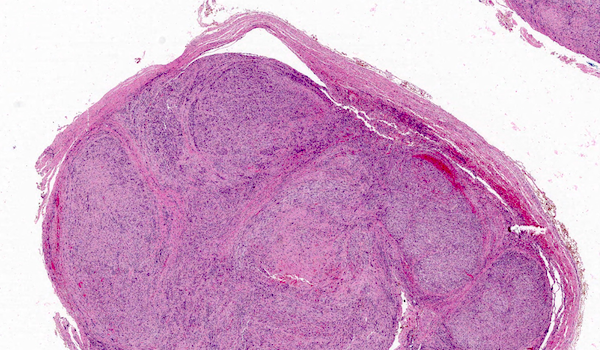
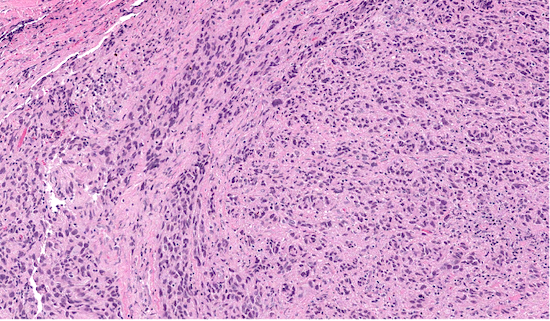
Microscopic (histologic) description
- Low power: marbled appearance due to alternating hypocellular and hypercellular areas with perivascular accentuation
- Uniform spindle cells with hyperchromatic, thin, wavy, or focally buckled nuclei
- May have uniform cellularity with fibrosarcoma-like fascicular growth, raising the differential diagnosis of synovial sarcoma
- Can have foci of myxoid stroma and hyalinization
- Epithelioid morphology can be present
- Precursor lesion, such as neurofibroma, may be identifiable
- Nuclear palisading is uncommon
- Heterologous differentiation may include chondrosarcomatous, osteosarcomatous and rhabdomyosarcomatous components (malignant triton tumor)
- Glandular elements are exceedingly rare (Int J Surg Pathol 2017;25:310)
- Proposed nomenclature for the spectrum of NF1 associated nerve sheath tumors:
- Atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP):
- At least 2 of the following features: cytologic atypia, loss of neurofibroma architecture, hypercellularity, > 1/50 HPF and < 3/10 HPF
- MPNST, low grade: features of ANNUBP but with mitotic index of 3 - 9/10 HPF and no necrosis
- MPNST, high grade: MPNST with at least 10 mits/10 HPF or 3 - 9 mits/10 HPF combined with necrosis (Hum Pathol 2017;67:1)
- Atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP):
Microscopic (histologic) images
Contributed by Jose G. Mantilla, M.D.
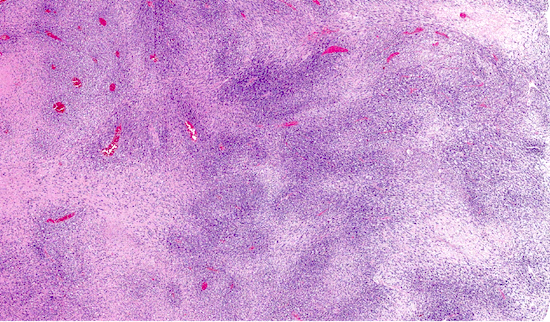
Marbling
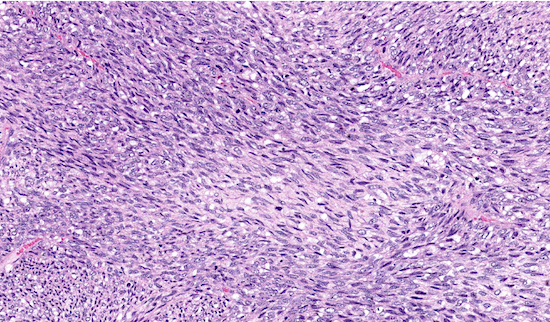
Fascicular growth
Rhabdomyo-
sarcomatous elements
Epithelioid type
Epithelioid type
Contributed by @MirunaPopescu13 and @JMGardnerMD on Twitter
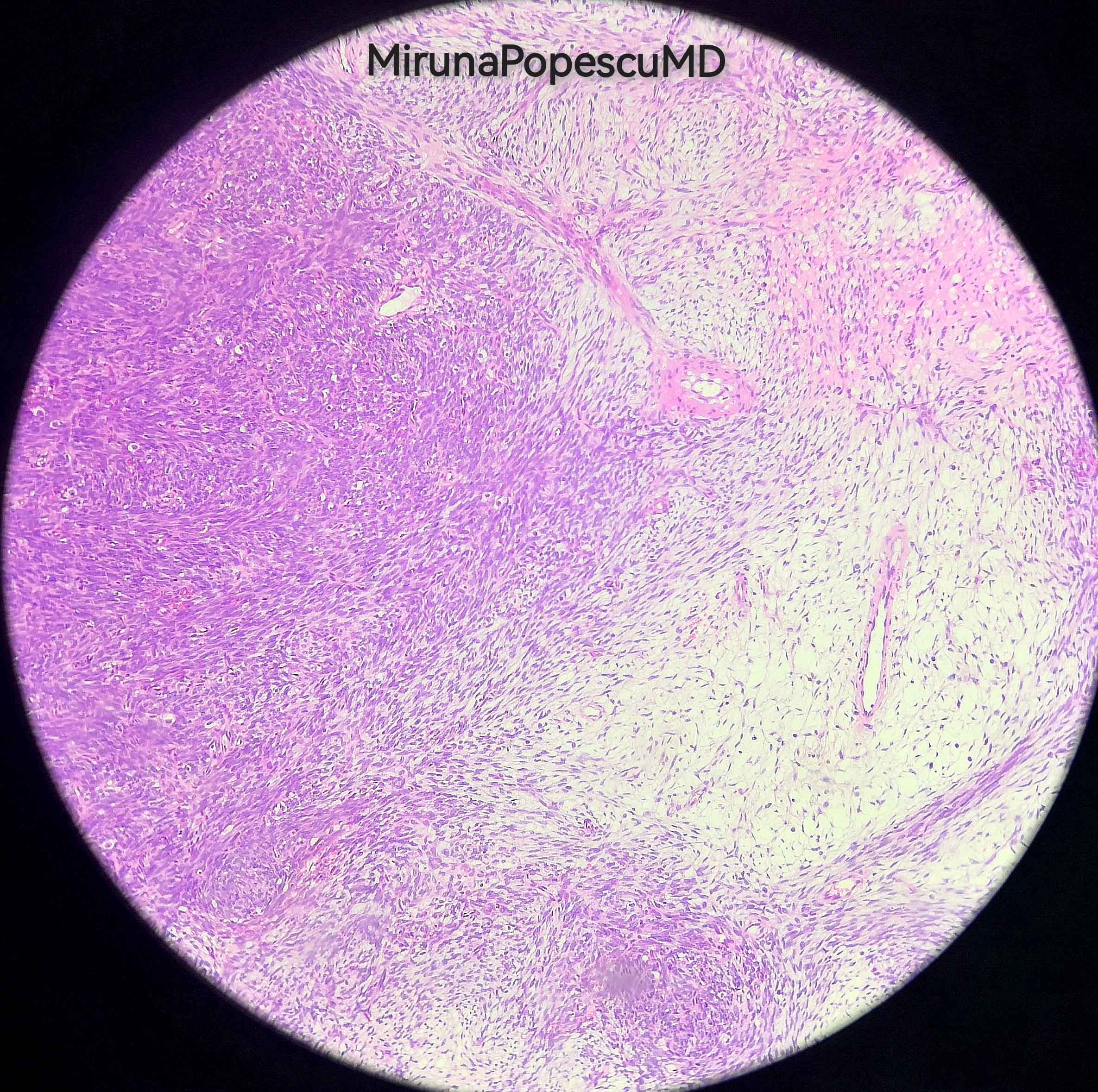
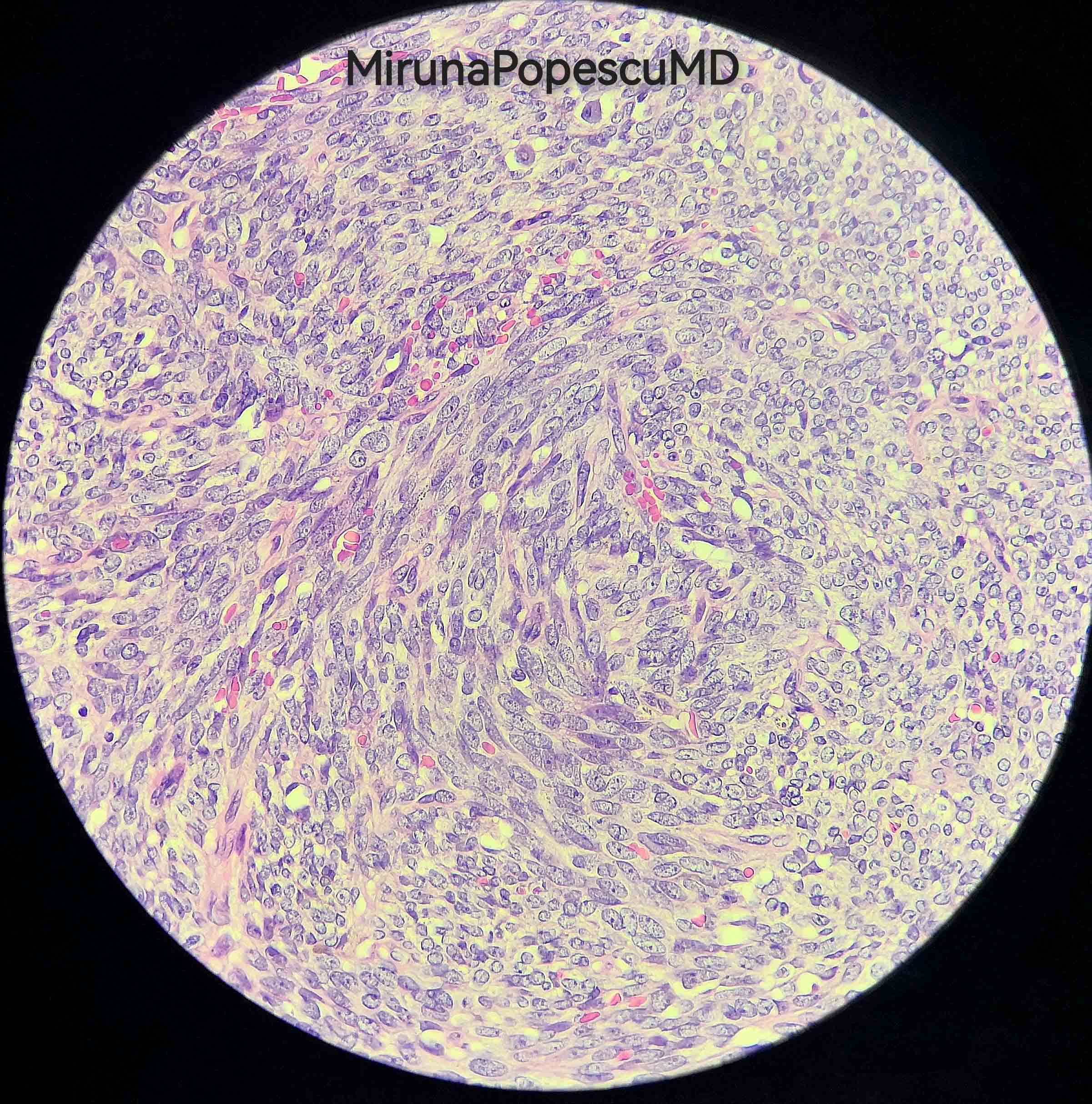
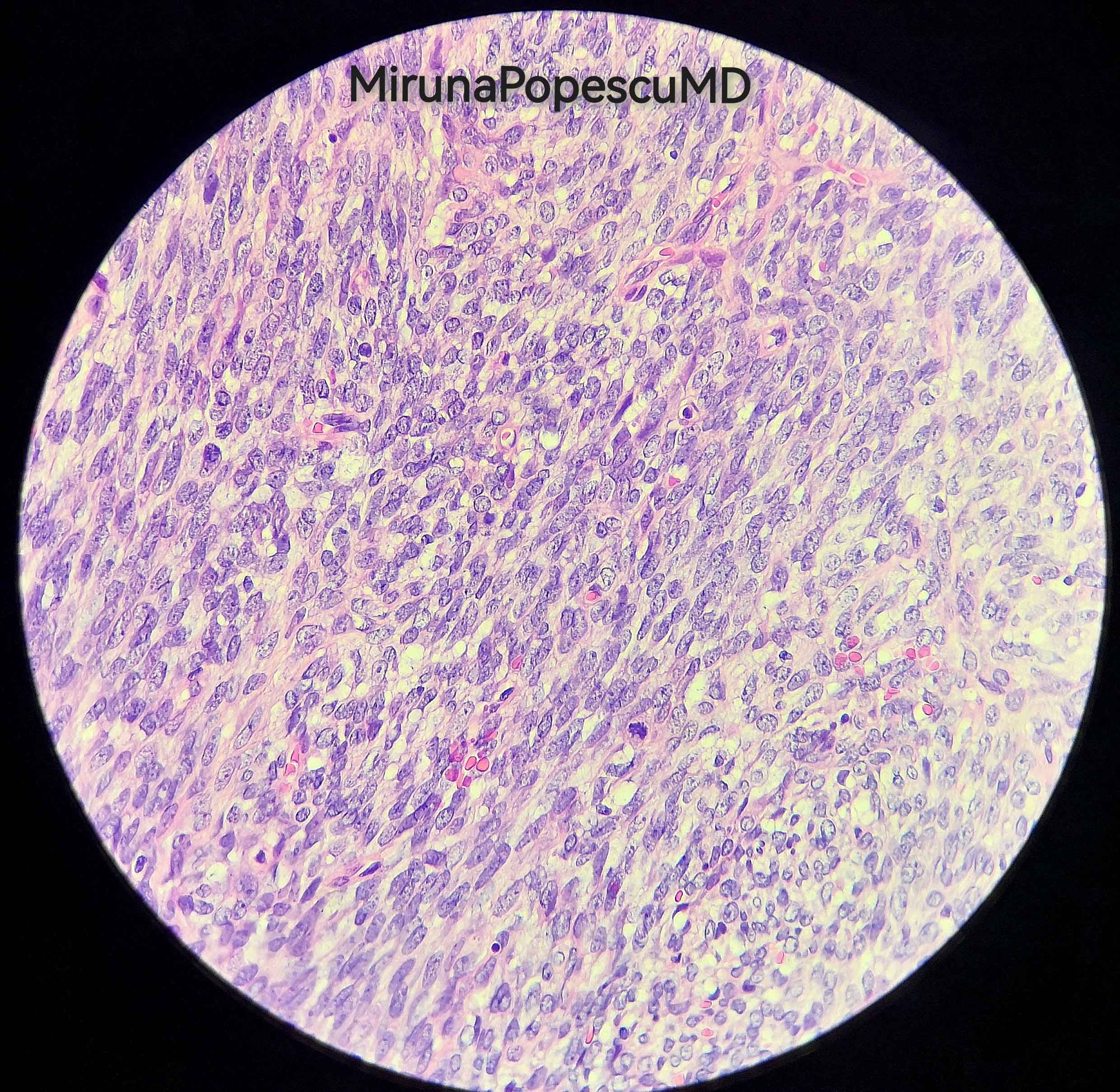
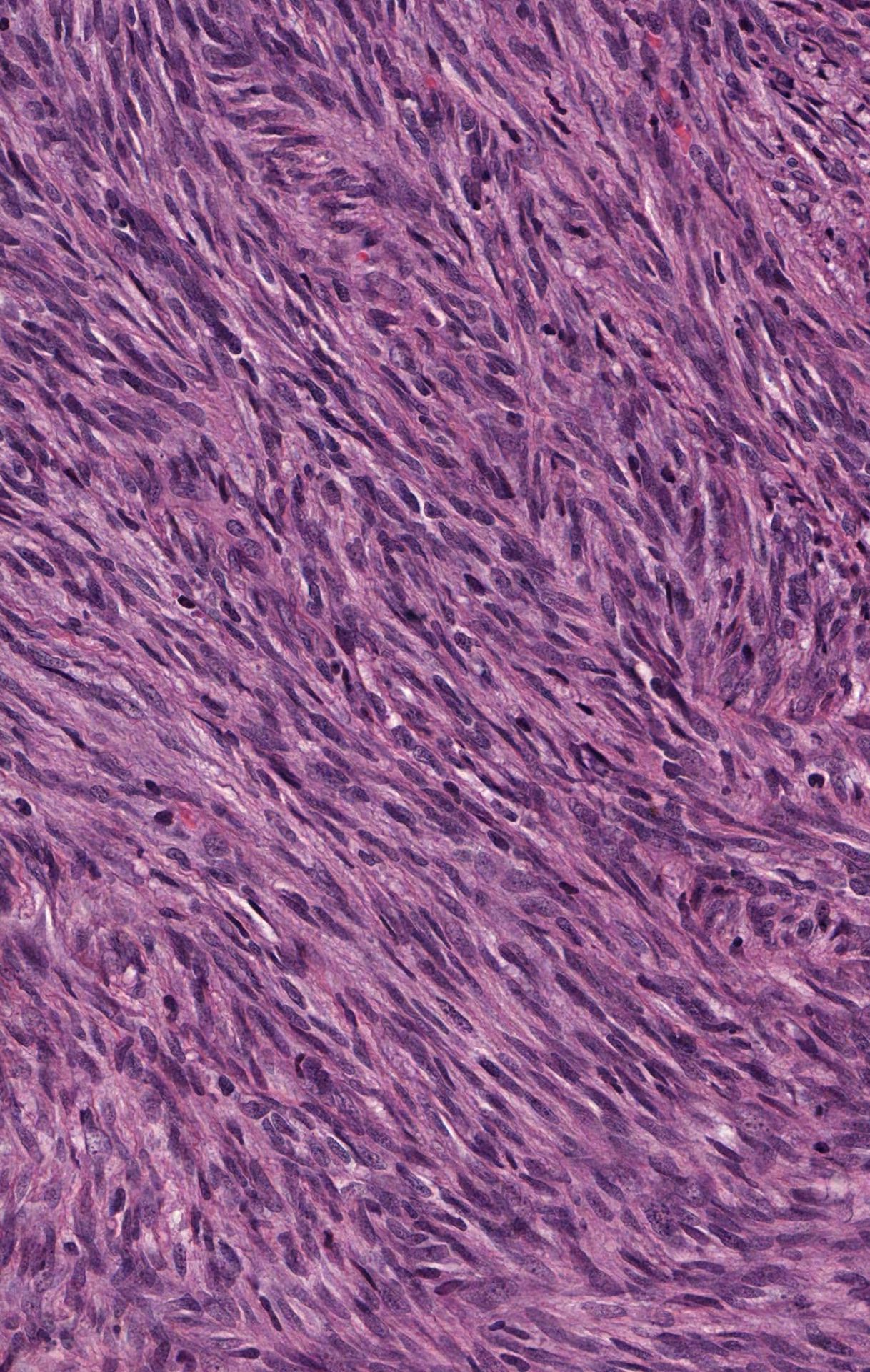
Malignant peripheral nerve sheath tumor (MPNST)
Virtual slides
Images hosted on other servers:
Marbling, hyperchromatic nuclei

Perivascular accentuation
Cytology description
- Highly cellular smears of uniform spindled cells singly and in clusters (Cancer Cytopathol 2012;120:334)
- Cytomorphologic overlap with other sarcomas is significant
Cytology images
Contributed by Jose G. Mantilla, M.D.

Uniform spindle cells
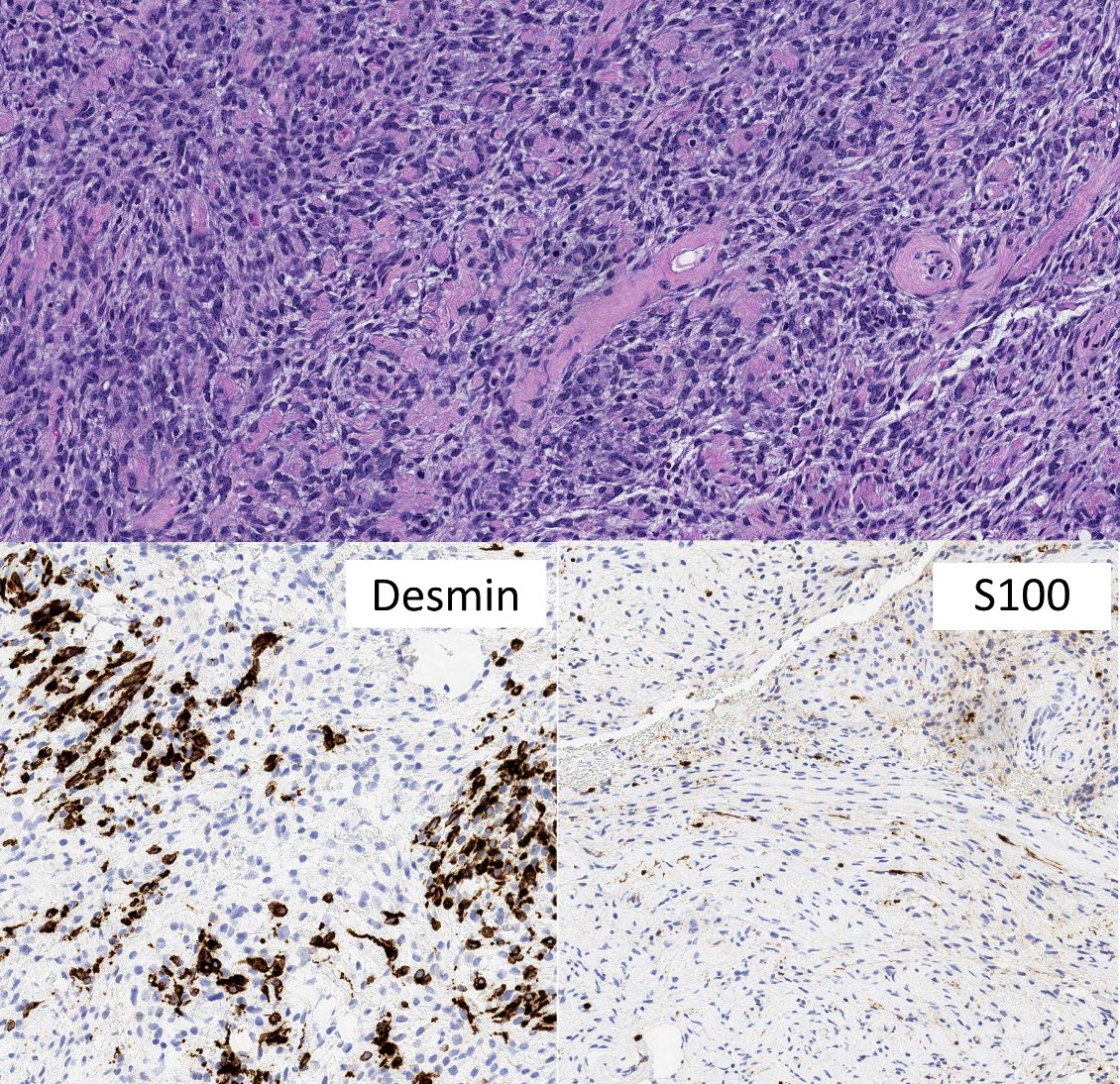
Positive stains
Negative stains
- Loss of nuclear H3K27me3 in high grade sporadic and radiation associated tumors
- Sensitivity is lower in low grade and NF1 associated MPNST (complete loss in 88/122 (72%) and mosaic / partial loss in 23/122 (19%), loss of expression in 47/68 tumors (69%)) (Mod Pathol 2017;30:1677, Am J Surg Pathol 2016;40:479)
- Loss of nuclear INI1 immunoreactivity in a large subset of epithelioid MPNST (3/6 (50%) tumors, 35/52 tumors (67%)) (Histopathology 2016;68:286, Am J Surg Pathol 2015;39:673)
Molecular / cytogenetics description
- Cytogenetically complex sarcomas
- Inactivating mutations in CDKN2A / CDKN2B and PRC2 (Clin Cancer Res 2011;17:3771, Nat Genet 2014;46:1227)
- Germline NF1 mutations in the setting of NF1 (see Pathophysiology)
- As precursor lesions transform, they tend to acquire additional mutations
- Point mutations in BRAF V600 in a subset of cases (Histopathology 2013;62:499, J Natl Compr Canc Netw 2018;16:967, J Neurooncol 2014;120:267)
Sample pathology report
- Right hand mass, excision (consult case):
- Spindle cell sarcoma, consistent with malignant peripheral nerve sheath tumor (see comment)
- Tumor size: 3.8 cm in greatest dimension, per outside report
- Inked tissue edges involved by sarcoma
- Comment: Histologic sections contain a dermal based neoplasm with a plexiform architectural pattern. It is composed of variably pleomorphic spindle cells forming fascicles and storiform structures with a marbling pattern. Mitotic activity is present in up to 7/10 high power fields and no necrosis is identified. The periphery of the lesion is relatively hypocellular with a single area composed of cytologically bland spindle cells, which morphologically resembles neurofibroma (slide A3). This portion of the lesion shows immunoreactivity for S100 and CD34. The remainder of the lesion is has focal S100 reactivity. Negative stains include EMA, CK AE1 / AE3, desmin, SMA, MART1, MUC4, CD56, neurofilament and ER. Overall, the findings are consistent with malignant peripheral nerve sheath tumor.
Differential diagnosis
- Atypical neurofibroma:
- Occurs in the context of NF1
- Distinguished from low grade MPNST based on lower mitotic activity (< 3/10 high power fields), lack of prominent cytologic atypia, preserved architecture and lack of necrosis (Hum Pathol 2017;67:1)
- Monophasic synovial sarcoma:
- Monotonous cytologic features
- Patchy expression of keratin or EMA
- SYT gene rearrangements
- Dedifferentiated liposarcoma:
- MDM2 amplification
- Spindle cell melanoma:
- Diffuse expression of S100, often with expression of mature melanocytic markers
- Leiomyosarcoma:
- Blunt ended cigar shaped nuclei with bright pink eosinophilic cytoplasm
- Positive for SMA, desmin and h-caldesmon
- Neurofibroma:
- No mitoses, less cellularity, no nuclear atypia
- Low grade fibromyxoid sarcoma:
- Low grade cytologic features
- MUC4 positive
- FUS-CREB gene rearrangement
- Cellular schwannoma:
- Foamy macrophages and hyalinized vessel walls
- Intense S100 protein expression
Board review style question #1
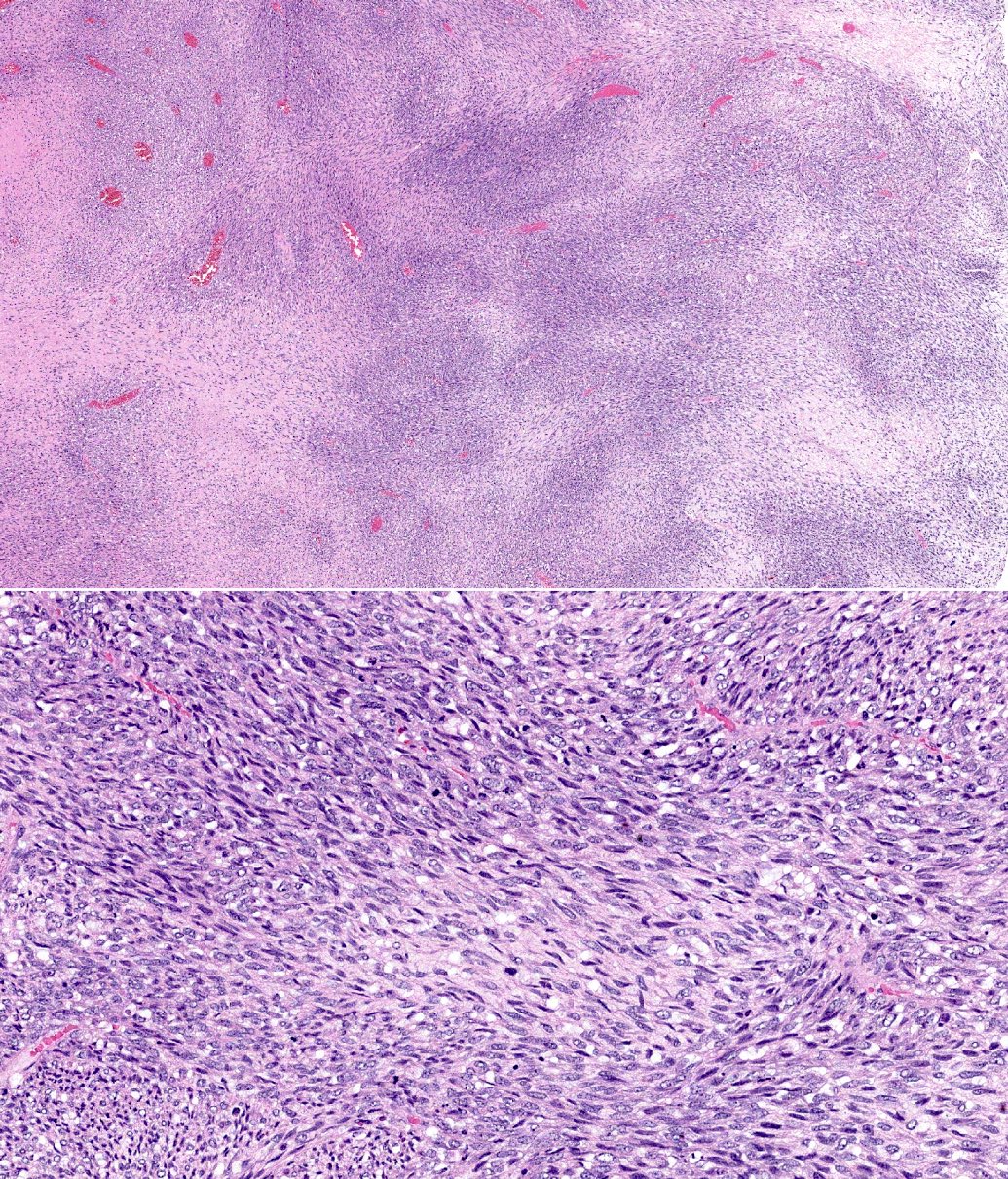
- Which of the following is true about malignant peripheral nerve sheath tumors?
- Characteristic histology is of a marbled appearance at low power with alternating hypocellular and hypercellular areas
- Glandular elements are never seen in this entity, which can help distinguish it from synovial sarcoma
- Lesion always has diffuse and strong S100 positivity due to its nerve sheath origin
- Most of these tumors are radiation associated as opposed to arising de novo or in a preexisting neurofibroma
Board review style answer #1
A. Characteristic histology is of a marbled appearance at low power with alternating hypocellular and hypercellular areas
Comment Here
Reference: MPNST
Comment Here
Reference: MPNST
Board review style question #2
- Which of the following is true about the immunophenotypic and molecular characteristics of MPNST?
- Loss of expression of H3K27me3 has been described in high grade appearing tumors
- All cases have some degree of SOX10 expression
- Germline mutations have no influence on the development of MPNST
- MPNST is defined by recurrent cytogenetic alterations
Board review style answer #2
A. Loss of expression of H3K27me3 has been described in high grade appearing tumors
Comment Here
Reference: MPNST
Comment Here
Reference: MPNST
