
Pancreas
Neuroendocrine neoplasms
MEN1 syndrome
Author: Sabrina C. Sopha, M.D.

Last author update: 1 October 2017
Last staff update: 27 February 2025 (update in progress)
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: MEN1 pancreas


Table of Contents
Definition / general | Essential features | Epidemiology | Sites | Pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Gross images | Microscopic (histologic) images | Molecular / cytogenetics description | Board review style question #1 | Board review style answer #1Cite this page: Sopha S. MEN1 syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/pancreasMEN1.html. Accessed April 3rd, 2025.
Definition / general
- Syndrome characterized by mutations in MEN1 gene and development of multiple endocrine tumors (and other tumor types)
- Formerly known as Wermer syndrome
- Autosomal dominant inheritance
Essential features
- Defined by MEN1 mutation (chromosome 11q13, menin, tumor suppressor gene, 100% penetrance)
- Classic constellation of pituitary, parathyroid and pancreatic tumors (the 3 P's)
- Variety of nonendocrine tumors are also part of MEN1 (meningioma, ependymoma, angiofibroma)
Epidemiology
- Prevalence of MEN1 is 1:30,000 to 1:500,000 (geographic variability due to familial clustering) (F1000Res 2017;6:73)
- MEN1 cases are 90% familial and 10% sporadic (F1000Res 2017;6:73)
- 100% penetrance (100% with parathyroid adenomas by age 50) (F1000Res 2017;6:73)
- Most frequent cause of MEN1 related death is complications secondary to pancreatic / gastrointestinal neuroendocrine tumors (J Pediatr Genet 2016;5:89)
Sites
- Variety of neuroendocrine and nonneuroendocrine tumors (J Pediatr Genet 2016;5:89, Cancer Genet 2016;209:36, Endocr Relat Cancer 2017;24:T119)
- Parathyroid (90% of patients)
- Anterior pituitary (30 - 40% of patients)
- Pancreas / gastrointestinal tract (30 - 70% of patients)
- Adrenal cortex
- Thymus
- Facial skin (angiofibromas, collagenomas)
- Testis (epididymomas)
- Meninges (meningiomas)
-
Of note, nonsyndromic tumors have somatic MEN1 mutations at the following frequencies (Endocr Relat Cancer 2017;24:T119):
- Glucagonoma 60%
- VIPoma 57%
- Nonfunctioning PanNETs 44%
- Gastrinoma 38%
- Bronchial carcinoid 35%
- Parathyroid adenoma 35%
- Lipoma 28%
- Insulinoma 2 - 19%
- Angiofibroma 10%
- Anterior pituitary tumor 3.5%
- Adrenocortical tumor 2%
Pathophysiology
- MEN1 gene first identified in 1997, found on chromosome 11q13 (Science 1997;276:404)
- Menin, the protein product of MEN1, is a tumor suppressor gene
- Twelve mutations were initially identified and more than 1,800 mutations have been identified to date
- Most of these are private (family specific)
- Seventy five percent are inactivating (F1000Res 2017;6:73)
- 5 - 20% may not harbor mutations in the MEN1 gene coding region (F1000Res 2017;6:73, Cancer Genet 2016;209:36)
- There is no genotype phenotype correlation with the type of MEN1 mutation and the type of tumors which develop (F1000Res 2017;6:73)
- Menin does not show homology with any other known protein but is conserved across species from Drosophila to humans (F1000Res 2017;6:73, Cancer Genet 2016;209:36)
- Normal functions of menin: cell proliferation, apoptosis and genome integrity (F1000Res 2017;6:73)
- Mechanism by which a nonfunctional menin protein causes tumor development remains unknown
Diagrams / tables
Images hosted on other servers:
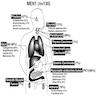
Location of tumors in patients with MEN1
Clinical features
- Endocrine tumors found in parathyroid, anterior pituitary, pancreas / gastrointestinal tract, adrenal cortex and thymus
- Non endocrine tumors: angiofibromas, collagenomas, epididymomas, meningiomas
- Increased frequency of renal stones compared with the general population
- Penetrance is 100%, with all patients developing parathyroid adenomas by age 50
- Patients with gastrinomas, typically found in the duodenum, may have Zollinger-Ellison syndrome
- Primary gastrinomas may be microadenomas but the lymph node metastases can be much larger than the primary (J Pediatr Genet 2016;5:89)
- Patients with parathyroid adenomas have hypersecretion of parathyroid hormone (PTH) with resultant symptoms of hypercalcemia (GI pain, renal stones, clouded mental functioning)
- Cushing disease, Nelson syndrome, acromegaly or hyperprolactinemia in cases of functional pituitary adenomas
- Pituitary adenomas can also cause a number of visual field defects due to compression of the optic chiasm or extraocular muscle compression
- Syndromes of other functioning pancreatic and gastrointestinal neuroendocrine neoplasms can be found on Neuroendocrine Neoplasms - General
Diagnosis
- Identification of a MEN1 mutation by PCR amplification of exons and splicing sites
- Notably, this fails to identify the subset of MEN1 patients that do not have the mutation within the coding region (World J Exp Med 2015;5:124)
- Next generation sequencing would bypass these limitations
- Mutational analysis should be offered to:
- Asymptomatic first degree relatives of a known MEN1 patient
- Patients with at least two typical MEN1 tumors or one MEN1 tumor and a positive family history
- Patients with early onset (< 30 years) of a typical MEN1 manifestation (hyperparathyroidism, Zollinger-Ellison syndrome) (Endocr Relat Cancer 2017;24:T209)
Laboratory
- Relevant to the type of tumor present and only functional tumors
Radiology description
- Osteoporosis, terminal tuft erosion (distal phalanges) and nephrolithiasis in patients with parathyroid adenomas and hypercalcemia
- Calvarial thickening, frontal bossing, enlarged sinuses (especially frontal sinuses), enlarged sella turcica, prognathic mandible and increased vertebral fractures in patients with acromegaly due to pituitary adenoma
Prognostic factors
- As described for pancreatic and gastrointestinal tumors in the WHO classification
Case reports
- 18 year old woman with hyperprolactinemia and hypercalcemia since age 13 (Clinical Chemistry 2015;61:1328)
- 36 year old man with pancreatic neuroendocrine tumor, pituitary adenoma and acromegaly (W V Med J 2012;108:26)
- 51 year old man with urolithiasis and 2 jejunal perforations 9 months apart (Korean J Gastroenterol 2013;61:333)
Treatment
- Surgical resection of neuroendocrine tumors
- Minimally invasive trans sphenoidal approach for pituitary adenomas
- Increased screening and surveillance in known kindreds: insulinoma starting age 5, pituitary adenoma starting age 5, parathyroid adenoma starting age 8, gastrinoma starting age 20, adrenal gland starting before age 10, thymic tumors starting before age 15 (World J Exp Med 2015;5:124)
Gross images
AFIP images
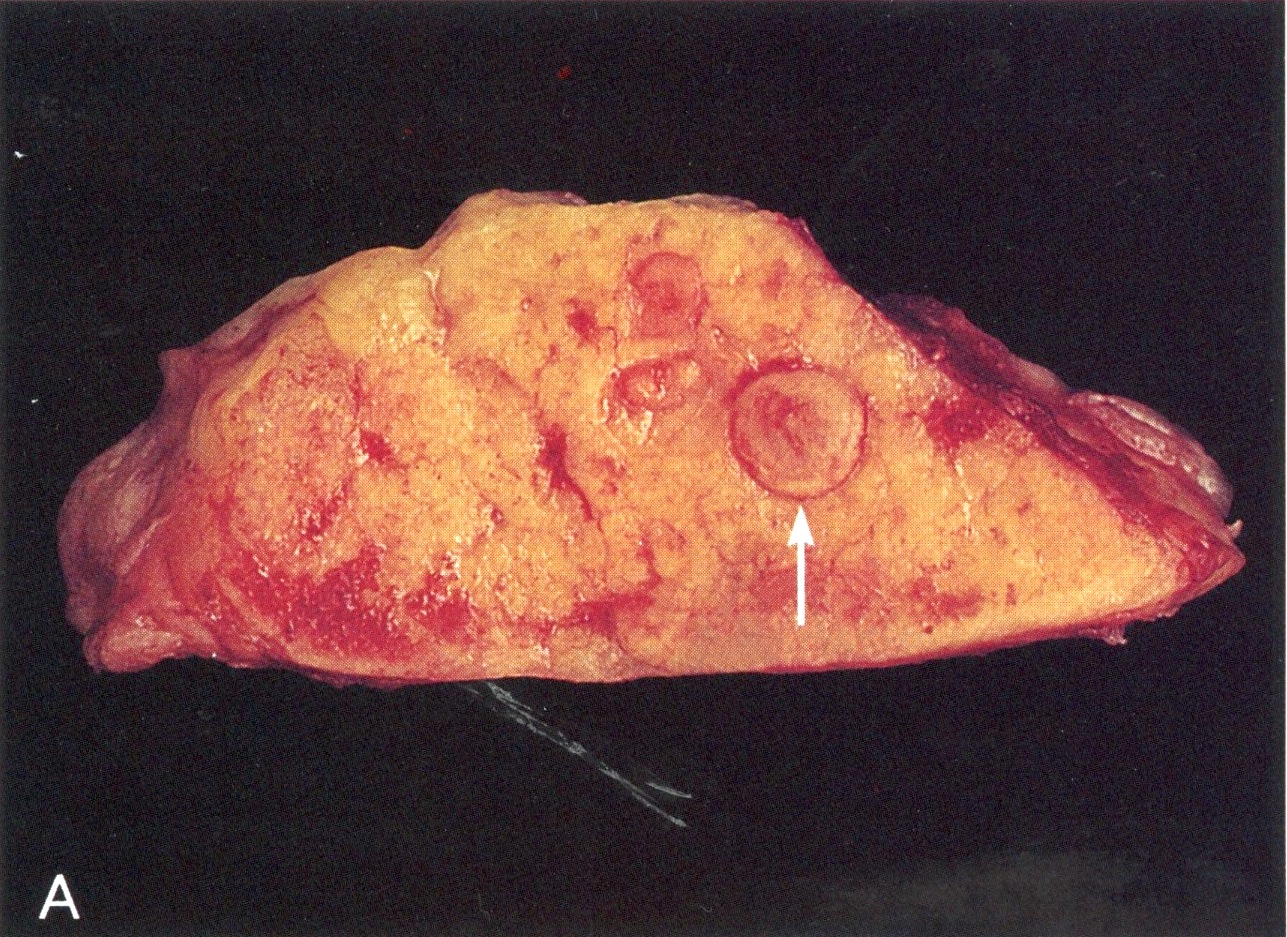
Intramucosal tumor nodules
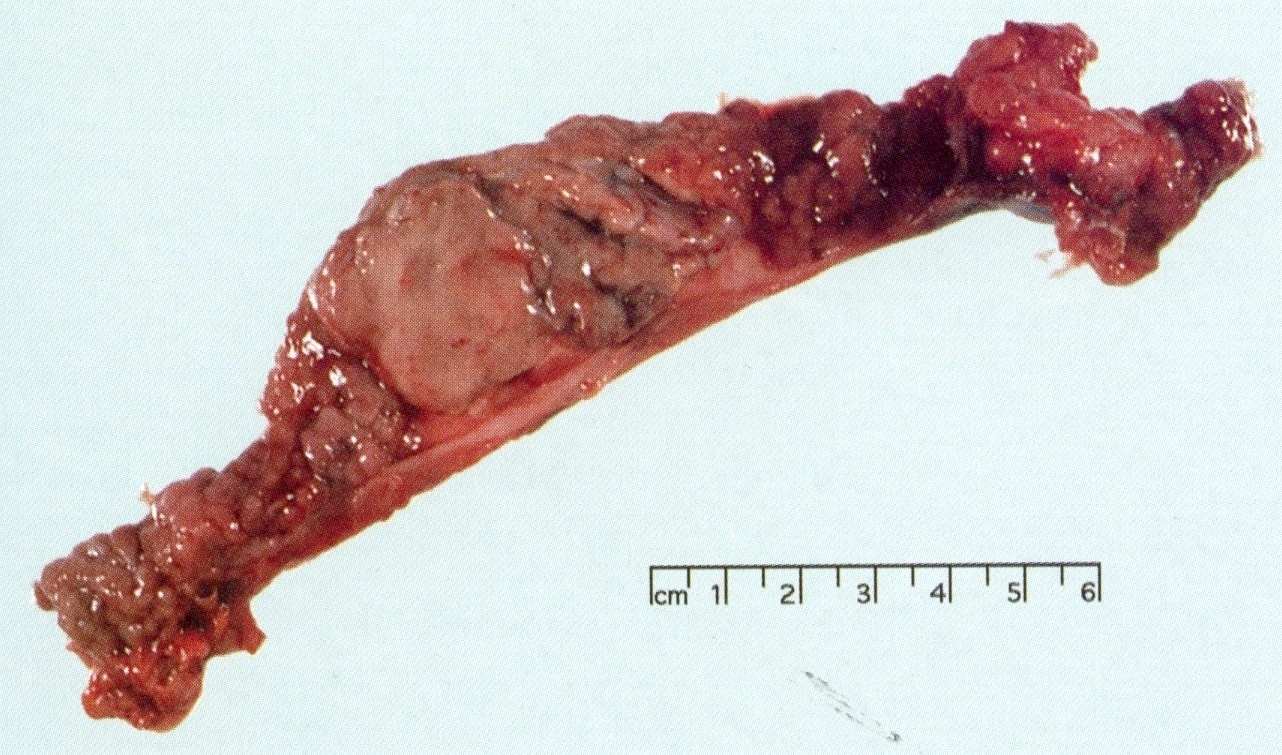
Zollinger-Ellison syndrome
Microscopic (histologic) images
AFIP images
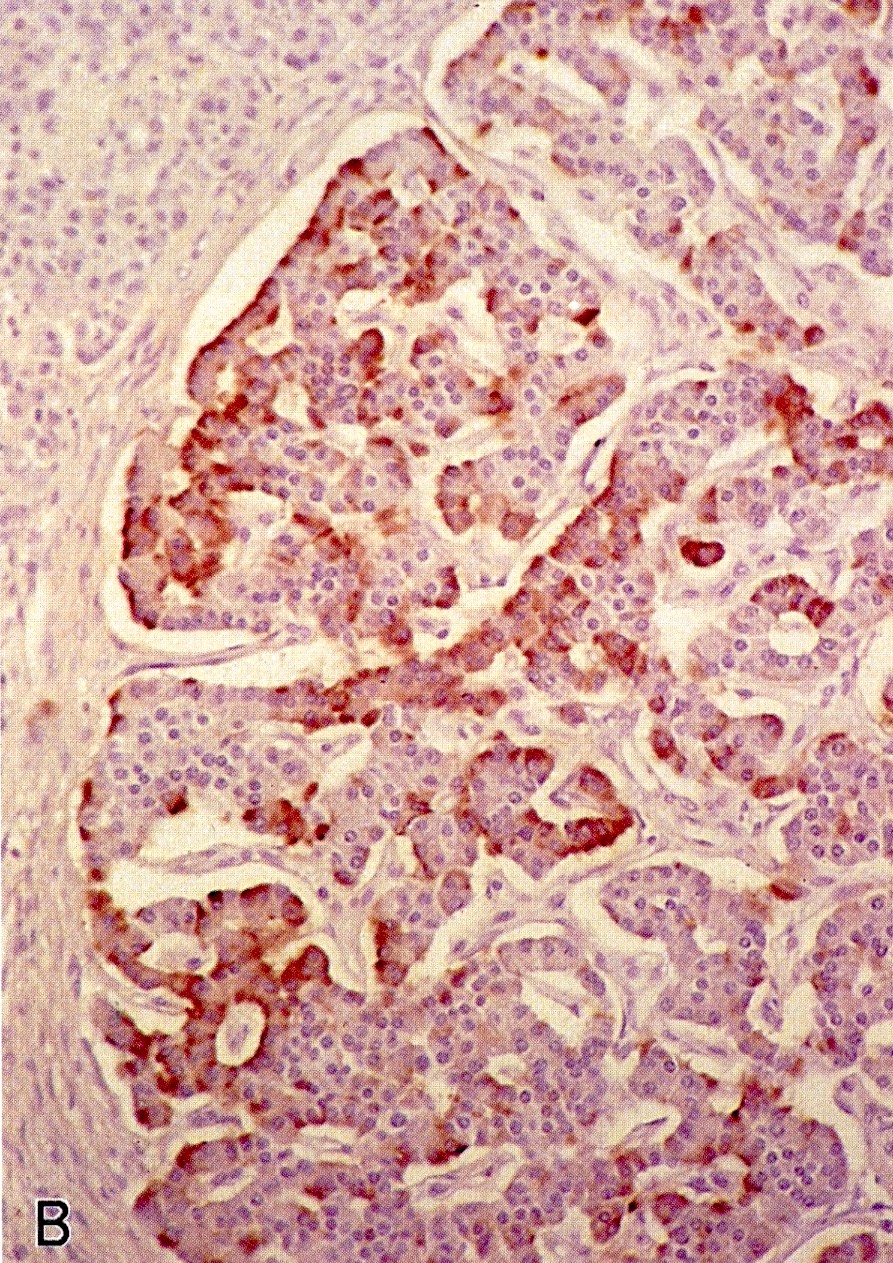
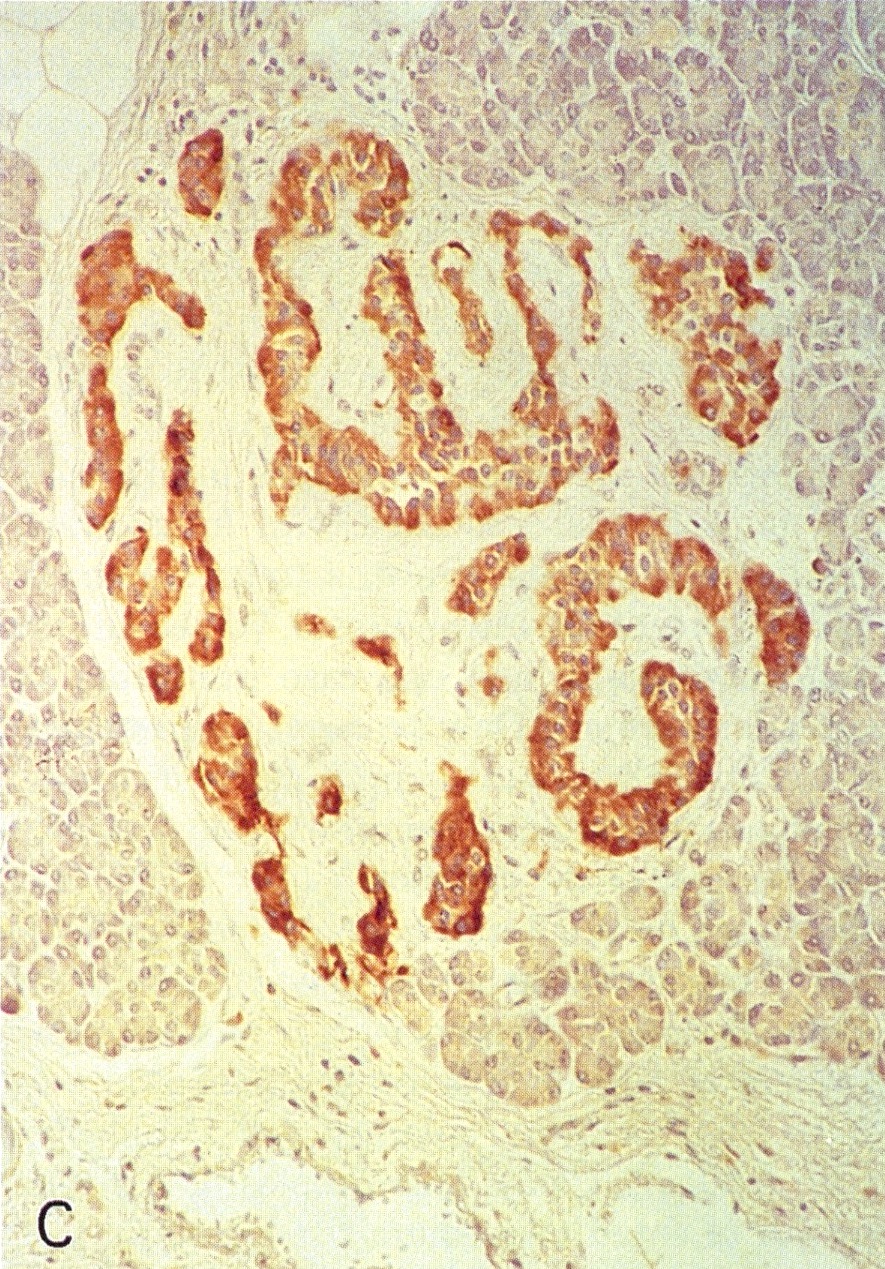
Pancreatic microadenomas in patients with MEN1
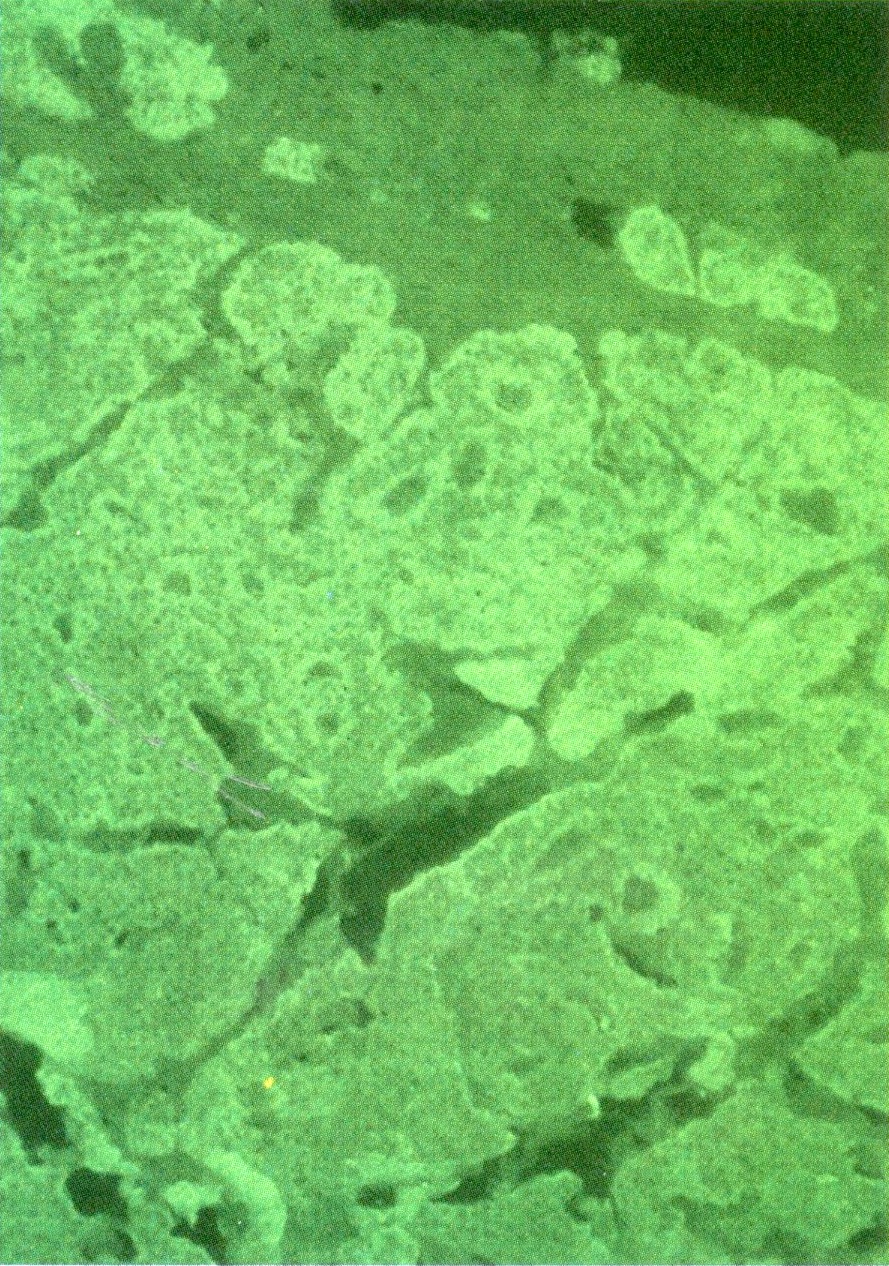
Duodenal gastrinoma in the mucosa and submucosa
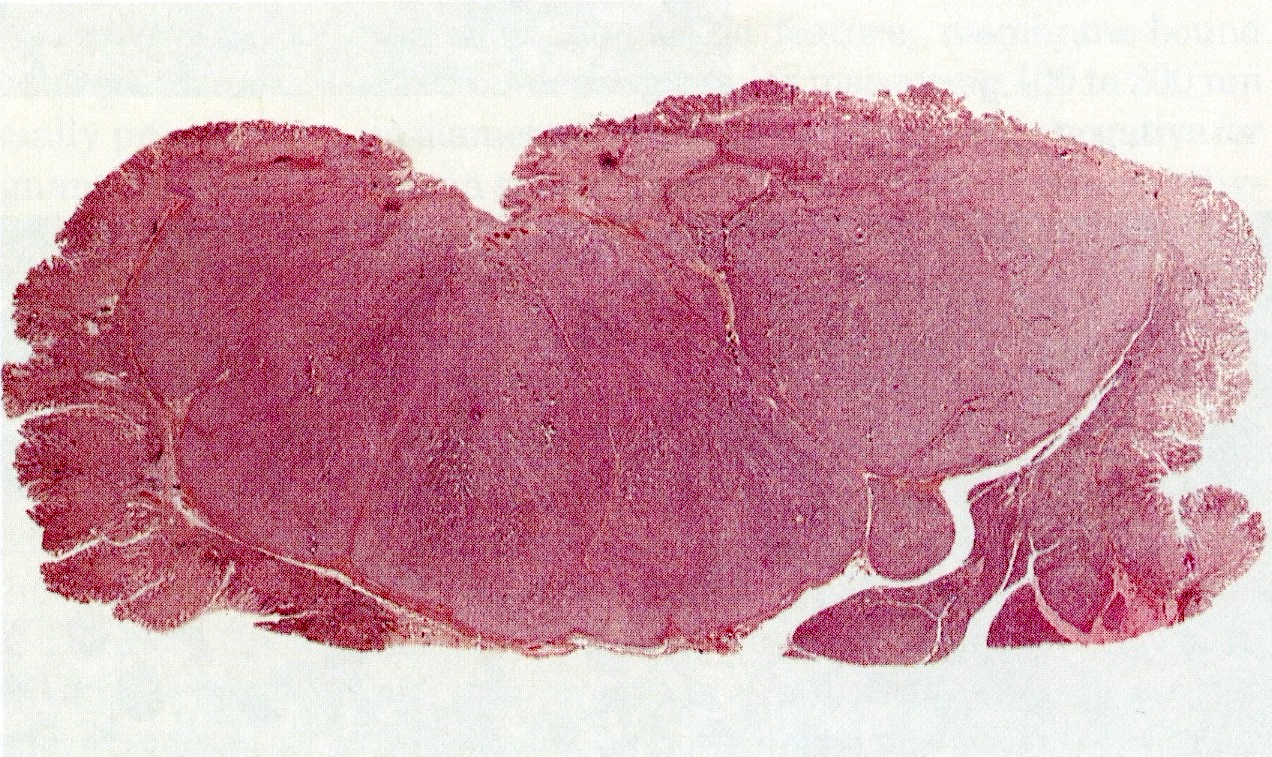
Hypertrophic and eroded mucosa
Molecular / cytogenetics description
- MEN1 mutation (chromosome 11q13)
Board review style question #1
- Which of the following patients would NOT be representative of a patient with MEN1 syndrome?
- A patient with facial angiofibromas, trichodiscomas and achrochordons
- A patient with MEN1 mutation and no tumor development at time of evaluation
- A patient with parathyroid adenoma and nephrolithiasis
- A patient with parathyroid adenoma, meningioma and epididymoma
- A patient with parathyroid adenoma, pituitary adenoma and pancreatic neuroendocrine tumor
Board review style answer #1
A. The patient NOT representative of a patient with MEN1 syndrome is the patient with angiofibromas, trichodiscomas and achrochordons. Angiofibromas are a part of MEN1 syndrome but trichodiscomas and achrochordons are not (they are part of Birt-Hogg-Dubé syndrome).
Comment Here
Reference: MEN1 syndrome
Comment Here
Reference: MEN1 syndrome
