

Bone marrow neoplastic
Bone marrow - neoplastic myeloid
Myeloproliferative neoplasms (MPN)
Polycythemia vera
Resident / Fellow Advisory Board: Frido Bruehl, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 4 November 2024
Last staff update: 4 November 2024
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: Polycythemia vera

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Positive stains | Negative stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2 | Board review style question #3 | Board review style answer #3Cite this page: Niyazi S, Patel R, Zhang L. Polycythemia vera. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/myeloproliferativepv.html. Accessed December 4th, 2024.
Definition / general
- BCR::ABL1 negative myeloproliferative neoplasm (MPN) characterized by peripheral erythrocytosis frequently accompanied by leukocytosis or thrombocytosis
Essential features
- Relatively common MPN characterized by increased red blood cell (RBC) production with elevated red blood cell count and hemoglobin level, which is not proportional to erythropoietin (EPO) production (usually has markedly decreased EPO level)
- Diagnosis requires all 3 major criteria or 2 major plus the minor criterion (see 2016 and 2022 WHO classification in Diagnosis below)
- JAK2 V617F (exon 14, valine to phenylalanine) and JAK2 exon 12 mutations are identified in > 95% and ~5% of patients, respectively (Blood 2006;108:1865, Hum Pathol 2006;37:1458, N Engl J Med 2007;356:459)
- Occurs in 3 phases
- Prodromal / prepolycythemic
- Overt polycythemic
- Postpolycythemic myelofibrosis / spent
- < 20% undergo blastic transformation within 15 years
Terminology
- Polycythemia vera (PV)
- Not accepted: Vaquez disease, Osler-Vaquez disease, polycythemia rubra vera, primary polycythemia, polycythemia
Epidemiology
- Annual incidence increases with advanced age, varies from 0.7 to 2.6 per 100,000 people in Europe and North America (Semin Thromb Hemost 2006;32:171)
- Slight male predominance; M:F = 2:1 (Leukemia 2013;27:1874)
- Median age at diagnosis is 61 years
Sites
- Peripheral blood and bone marrow are major sites of involvement
- Spleen and liver are common sites of extramedullary hematopoiesis in later stages (Blood 2016;128:5491)
Pathophysiology
- Clonal hematopoietic stem cell disorder (Blood 2003;102:3793)
- Point mutation in Janus kinase 2 (JAK2) gene at 9p24 results in constitutional activation of transcription factors of the STAT family, which promote growth factor independent proliferation and survival (Blood 2006;108:1865)
- Mechanisms of thrombosis and bleeding are due to increased circulating red blood cells and elevated platelets (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- 3 phases
- Prodromal / prepolycythemic: characterized by borderline to mild erythrocytosis
- Overt polycythemic: associated with significantly increased red cell mass
- Postpolycythemic myelofibrosis / spent: cytopenias, including anemia, are associated with ineffective hematopoiesis, bone marrow fibrosis, extramedullary hematopoiesis and hypersplenism
Etiology
- Underlying cause is unknown
- Presence of JAK2 V617F mutation is detected in over 95% of patients with PV (Blood 2008;111:1686)
- Abnormal karyotype in hematopoietic progenitor cells has been found in ~34% of patients with PV (J Clin Oncol 2011;29:761)
Clinical features
- Major symptoms are related to hypertension or vascular abnormalities caused by increased red cell mass
- Plethora can occur due to increased RBC mass
- Common symptoms include fatigue, headache, dizziness, tinnitus, vision changes, insomnia, claudication, pruritus, gastritis and early satiety (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Thrombotic events and thrombosis have been observed in up to 41% of patients with PV (Ann Intern Med 1995;123:656)
- 66% of thrombosis events tend to be arterial in nature (Rodgers: The Bethesda Handbook of Clinical Hematology, 4th Edition, 2018)
- Pruritis that occurs during or after a hot shower (aquagenic pruritus) is found in 40% of patients (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Gastrointestinal (GI) discomfort and peptic ulcer disease are common (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Early satiety may occur from impaired gastric filling due to splenomegaly (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Median survival of untreated symptomatic PV is 6 - 18 months from diagnosis, 3.5 years for PV treated with phlebotomy and 7 - 12 years for PV treated with cytoreduction (Rodgers: The Bethesda Handbook of Clinical Hematology, 4th Edition, 2018)
Diagnosis
- Diagnosis criteria (WHO 5th edition) for polycythemia vera requires meeting either all 3 major criteria or the first 2 major criteria and the minor criterion
- Major criteria
- Hemoglobin > 16.5 g/dL in men, > 16.0 g/dL in women; hematocrit (HCT) > 49% in men, > 48% in women
- Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis), including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size)
- Presence of JAK2 V617F or JAK2 exon 12 mutation
- Minor criterion
- Subnormal serum erythropoietin level (normal range of EPO in adults: 4.1 - 19.5 mU/mL)
- Major criterion 2 may not be required in cases with sustained absolute erythrocytosis
- Sustained absolute erythrocytosis is defined as hemoglobin levels greater than 18.5 g/dL in men or 16.5 g/dL in women or hematocrit greater than 55.5% in men and 49.5% in women
- Major criterion 3 (presence of JAK2 V617F or JAK2 exon 12 mutation) plus the minor criterion must also be present
- Accelerated phase: presence of 10 - 19% blasts in bone marrow or peripheral blood (Best Pract Res Clin Haematol 2022;35:101379)
- Blast phase: presence of ≥ 20% blasts in bone marrow or peripheral blood (Best Pract Res Clin Haematol 2022;35:101379)
- Major criteria
- Diagnostic criteria for postpolycythemic myelofibrosis (post-PV MF)
- Criteria based on International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and WHO (Blood 2016;127:2391, Am J Hematol 2017;92:94)
- Required criteria
- Documentation of a previous diagnosis of PV according to 2016 or 2022 WHO criteria
- Presence of grade 2 or 3 bone marrow fibrosis, based on either 2016 or 2022 WHO classification or the European consensus on grading of bone marrow fibrosis (Haematologica 2005;90:1128)
- Additional criteria (2 required)
- Anemia
- Leukoerythroblastosis
- Splenomegaly - either development of newly palpable splenomegaly or increased splenomegaly of > 5 cm from baseline
- Development of any 2 (or all 3) of the following constitutional symptoms
- Night sweats
- > 10% weight loss within past 6 months
- Unexplained fevers greater than 37.5 °C
- Required criteria
- Criteria based on International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and WHO (Blood 2016;127:2391, Am J Hematol 2017;92:94)
Laboratory
- Laboratory studies usually demonstrate erythrocytosis
- May also present with asymptomatic leukocytosis or thrombocytosis
- Bone marrow biopsy may show hypercellularity and panmyelosis
- Iron deficiency can be seen in many cases (Hematology 2023;28:2204621)
- JAK2 V617F mutation is detectable in over 95% of patients with PV (Blood 2008;111:1686)
- JAK2 exon 12 mutations may be seen in 2 - 5% of patients who are negative for the JAK2 V617F mutation (Ann Hematol 2020;99:983)
Prognostic factors
- Leukocytosis may predict inferior survival and leukemic transformation
- Risk of PV progressing to postpolycythemic myelofibrosis is 4.9% at 10 years and 9.4% at 15 years (Hematol J 2003;4:198)
- 15% develop spent phase with marrow fibrosis and marked cytopenias after 10 years; death in months if no treatment
- JAK2 V617F mutant allele burden of > 50% has been associated with fibrotic transformation (Am J Hematol 2017;92:94)
- Leukemic transformation rates at 20 years are estimated at < 10% for PV (Am J Hematol 2017;92:94)
- Risk factors for transformation include advanced age, leukocytosis and abnormal karyotype
- Leukemic transformation has been associated with treatment exposure to pipobroman or P32 / chlorambucil (Leukemia 2013;27:1874)
- Recent study showed older age, leukocytosis, venous thrombosis and abnormal karyotype are associated with adverse clinical outcomes (Leukemia 2013;27:1874)
- 3 risk groups are proposed, including low risk (0 points), intermediate risk (1 or 2 points) and high risk (3 points)
- Adverse points are assigned to age 67 years and older (5 points), age 57 - 66 years (2 points), leukocyte count 15 x 109/L (1 point) and venous thrombosis (1 point) with median survival of 28 years (low risk), 19 years (intermediate risk) and 11 years (high risk) (Leukemia 2013;27:1874)
- Next generation sequencing (NGS) study indicated that ASXL1, SRSF2 and IDH2 mutations are negative prognostic parameters associated with shorter overall, leukemia free and fibrosis free survival (Am J Hematol 2017;92:94)
- Cytogenetic abnormalities can be detected in up to 20% of patients at the time of initial diagnosis (Haematologica 2017;102:1511)
- Abnormalities may vary in different phases of disease
- Isolated del(20q), +8 and +9 are shown to be the most common abnormalities in polycythemic phase
- Del(20q) and +1q have been shown to be the most common abnormalities in postpolycythemic myelofibrosis
- Complex karyotypes have been shown to be the most common in accelerated and blast phases
- Abnormalities may vary in different phases of disease
Case reports
- 15 year old boy with cooperation of germline JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia (Blood 2016;128:1418)
- 40 year old woman who presented with subdural hematoma was found to have polycythemia vera (Neurol India 2022;70:1717)
- 51 year old woman with PV treated with intermittent phlebotomy and oral hydroxyurea irregularly for 2 years developed acute promyelocytic leukemia (Medicine (Baltimore) 2022;101:e30064)
- 56 year old woman with progression of primary myelofibrosis to polycythemia vera (Medicine (Baltimore) 2017;96:e7464)
- 62 year old woman with postpolycythemic myelofibrosis with tumor lysis syndrome after the administration of ruxolitinib (Intern Med 2017;56:2335)
- 66 year old man with PV and Erdheim-Chester disease with multiorgan involvement (Medicine (Baltimore) 2016;95:e3697)
Treatment
- Risk of PV progressing to postpolycythemic myelofibrosis is 4.9% at 10 years and 9.4% at 15 years (Hematol J 2003;4:198)
- Due to increased red blood cell production independent of mechanisms that typically regulate erythropoiesis
- 15% develop spent phase with marrow fibrosis and marked cytopenias after 10 years; death in months if no treatment
- Leukemic transformation rates at 20 years are estimated at < 10% for PV (Am J Hematol 2017;92:94)
- 2% develop acute myeloid leukemia with phlebotomy treatment but 15% with alkylating agents or radioactive phosphorus (no longer used)
- Risk of leukemic transformation (ALL) is rare, except in patients treated with cytotoxic regimens
- Therapeutic goals
- Decrease thrombosis without increasing bleeding
- Prevent progression
- Ameliorate symptoms
- Treatment guidelines (Ann Oncol 2015;26:v85, Blood 2014;124:3212, N Engl J Med 2004;350:114)
- Low / intermediate risk for thrombosis
- First line: phlebotomy
- Low dose aspirin
- Intensive management of cardiovascular risk factors
- High risk for thrombosis
- Cytoreductive therapy (hydroxyurea)
- Adjunct phlebotomy
- Low dose aspirin
- Pregnant women
- First line: phlebotomy
- Low dose aspirin
- Low molecular weight heparin in postpartum period
- Low / intermediate risk for thrombosis
- European collaboration on low dose aspirin in polycythemia vera (ECLAP) trial (2004): multicenter project that investigated the benefits and risks of low dose aspirin in patients with PV
- Study showed that daily aspirin (100 mg) lowered the risk of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke and total mortality
- Treatment also was known to nonsignificantly increase the risk of major bleeding (Am Heart J 2004;148:1068)
- Randomized study of efficacy and safety in polycythemia vera with JAK inhibitor ruxolitinib versus best available care (RESPONSE) trial (2020): longterm efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (Lancet Haematol 2020;7:e226)
- 5 year follow up of a phase 3 trial comparing patients with failed response to hydroxyurea
- Study results showed that ruxolitinib is a safe and effective longterm treatment option for patients with polycythemia vera who are resistant to or intolerant of hydroxyurea
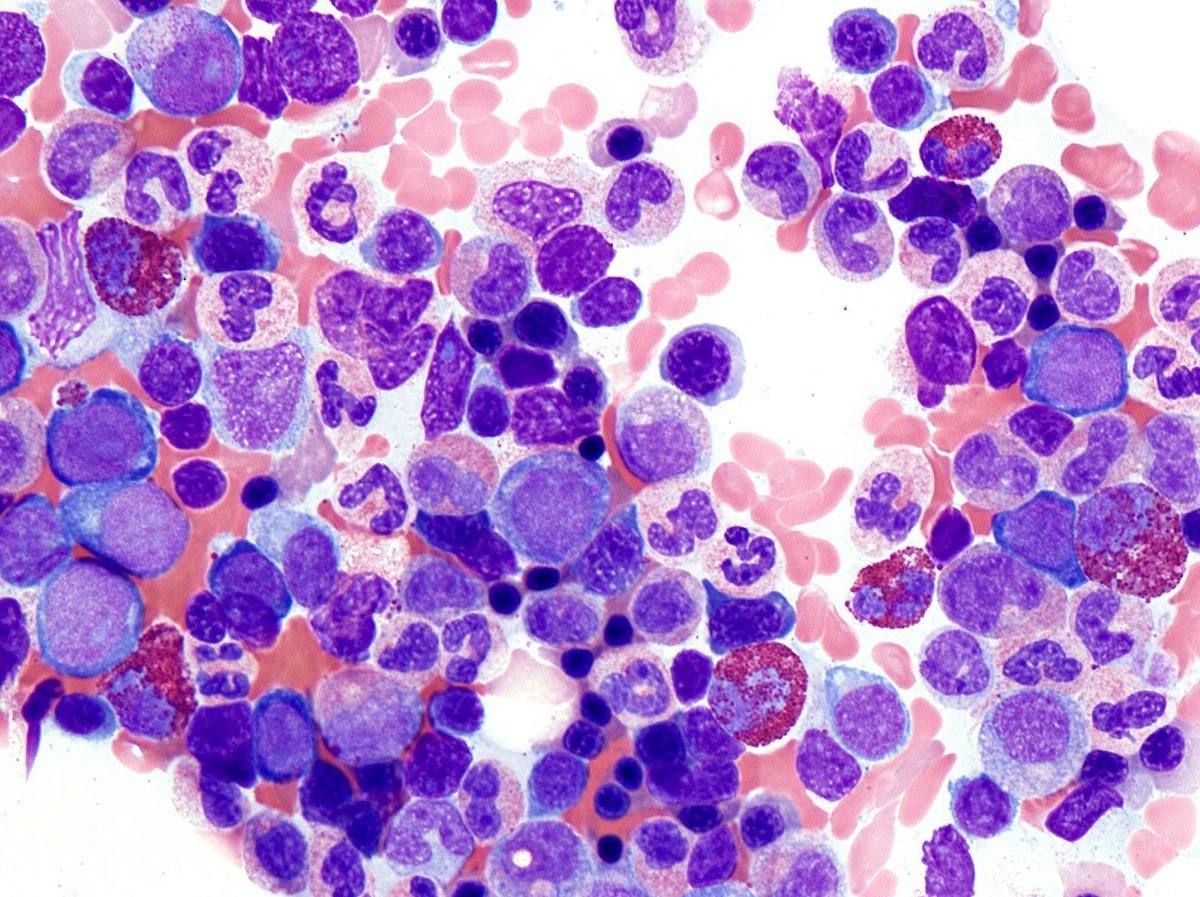
Microscopic (histologic) description
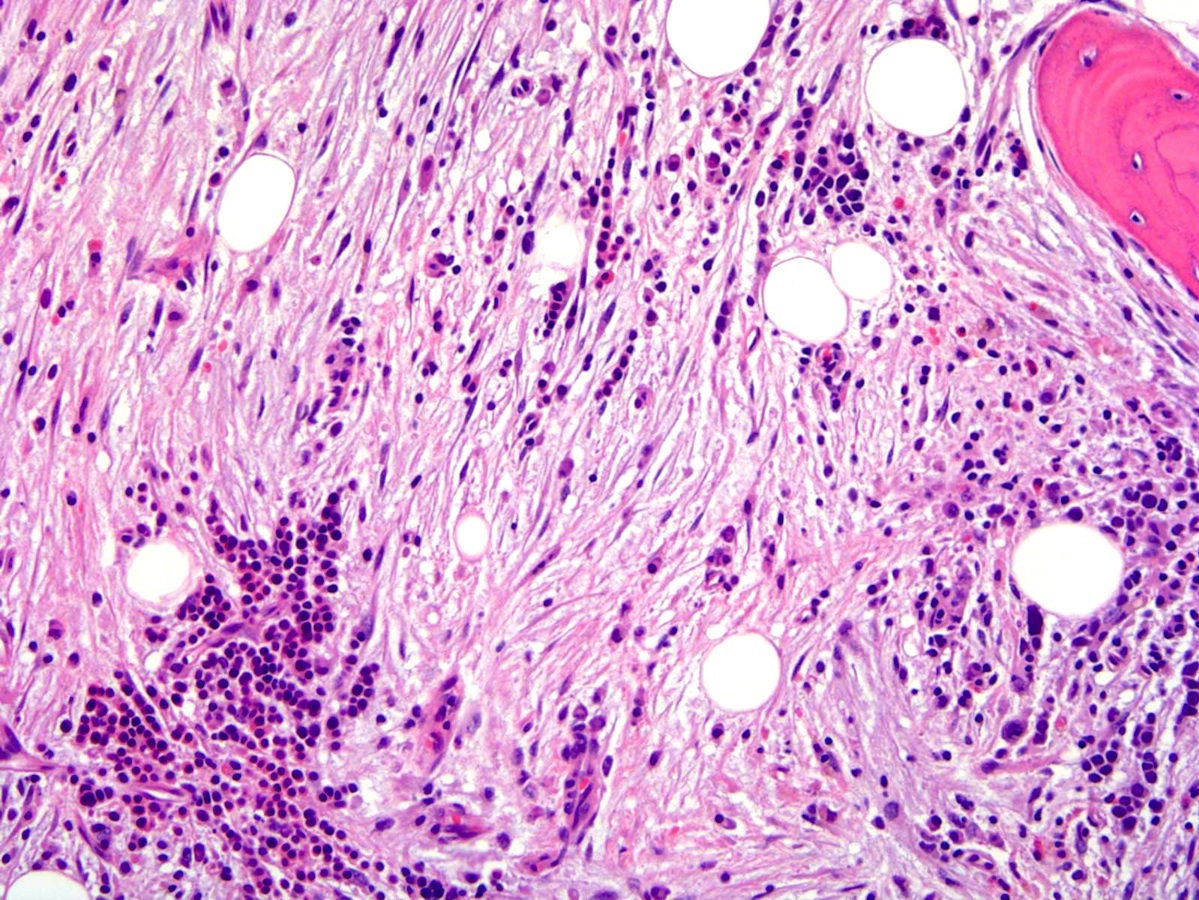
- Bone marrow (prepolycythemic and overt polycythemic phases) (see Microscopic (histologic) images 1, 4 & 7)
- Hypercellular with panmyelosis; notable predominance of erythroid and megakaryocytic lineages
- Complete and progressive maturation of all 3 hematopoietic lineages
- Abnormal megakaryocyte morphology and architecture is a prominent feature
- Mekagaryocytes are variably hyperlobulated and often seen in loose clusters; frequently seen close to bony trabeculae
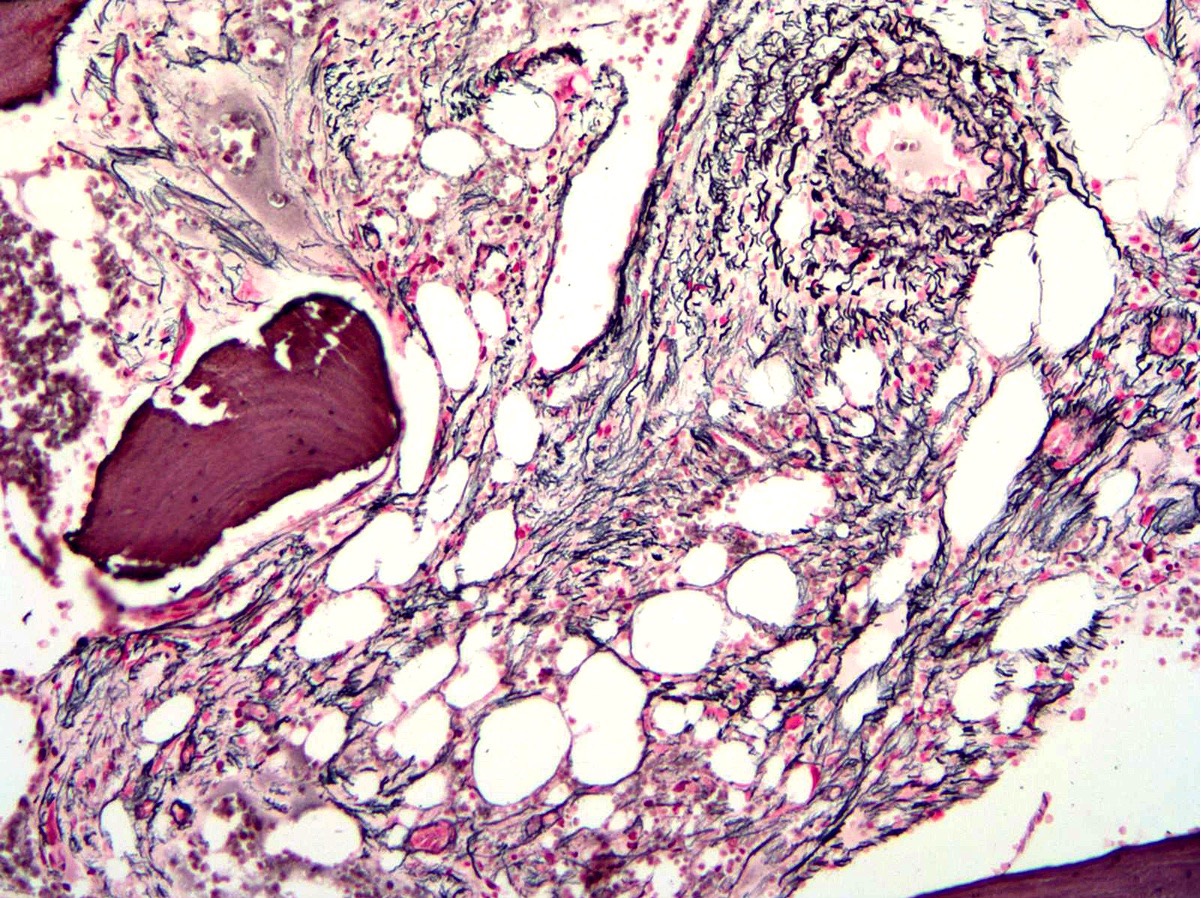
- Minority of cases (< 20%) may show mild increase in reticulin fibrosis (Blood 2012;119:2239)
- Iron stores are decreased, often absent
- Reactive lymphoid aggregates may be seen in up to 20% of cases (Histol Histopathol 2005;20:317)
- Blasts can be seen during accelerated or blast phase
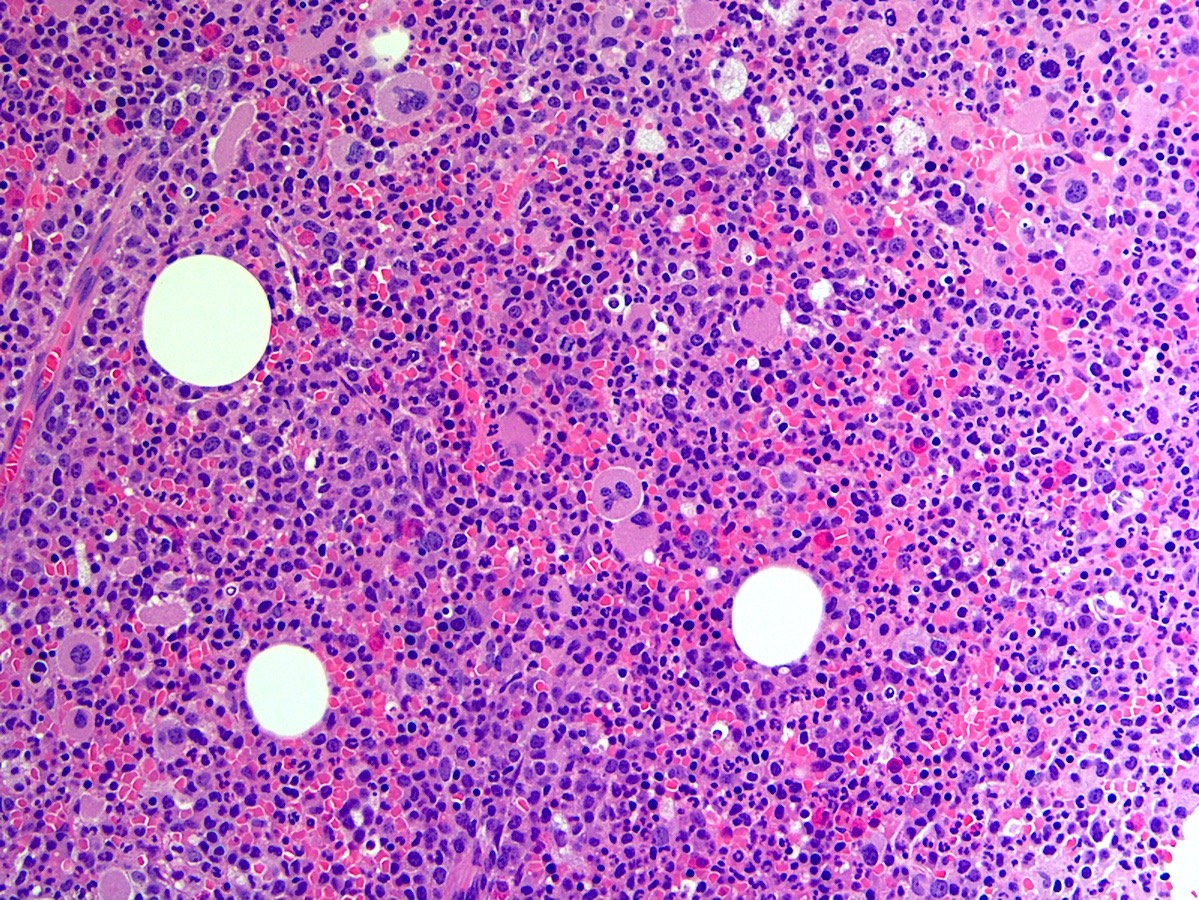
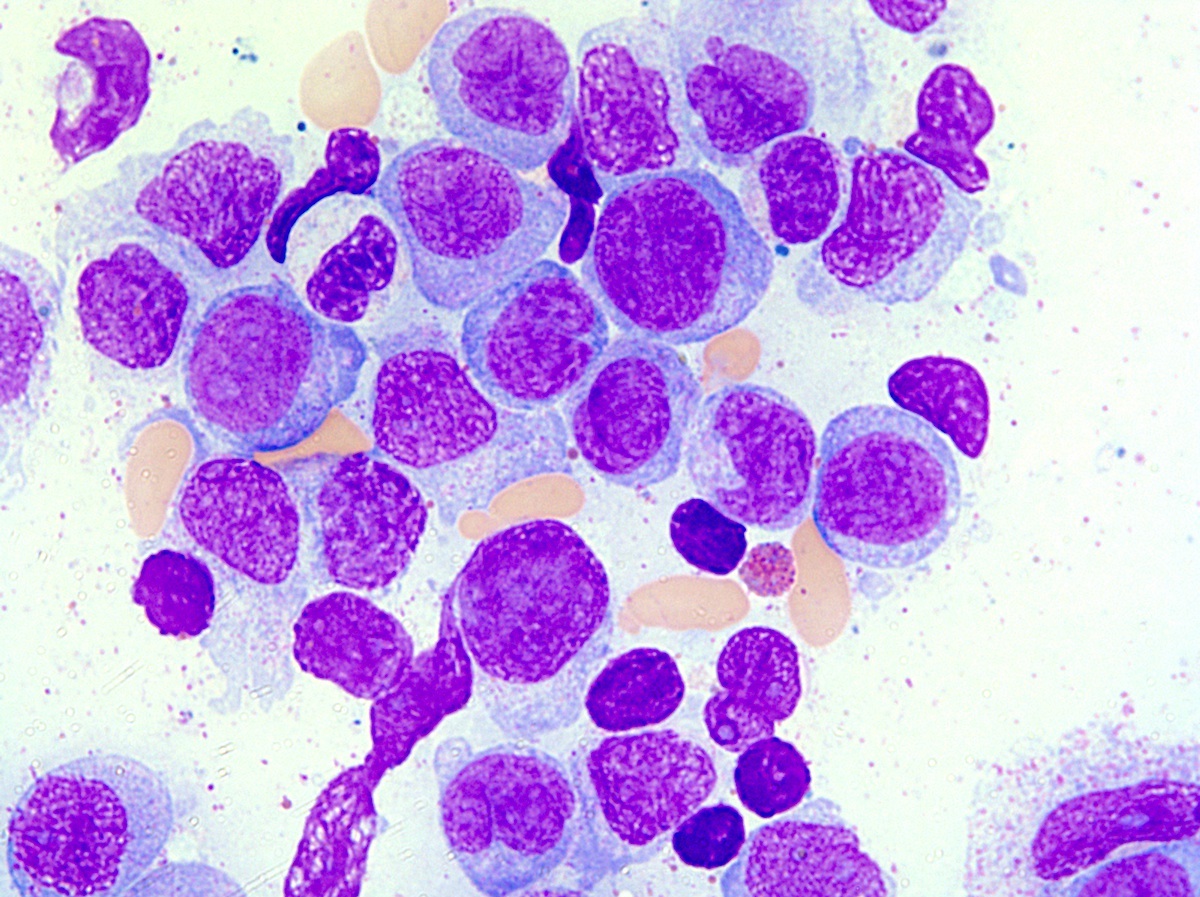
- Post-PV / spent phase (see Peripheral smear image and Microscopic (histologic) images 2, 5 & 6)
- Leukoerythroblastosis (Leukemia 2021;35:3339)
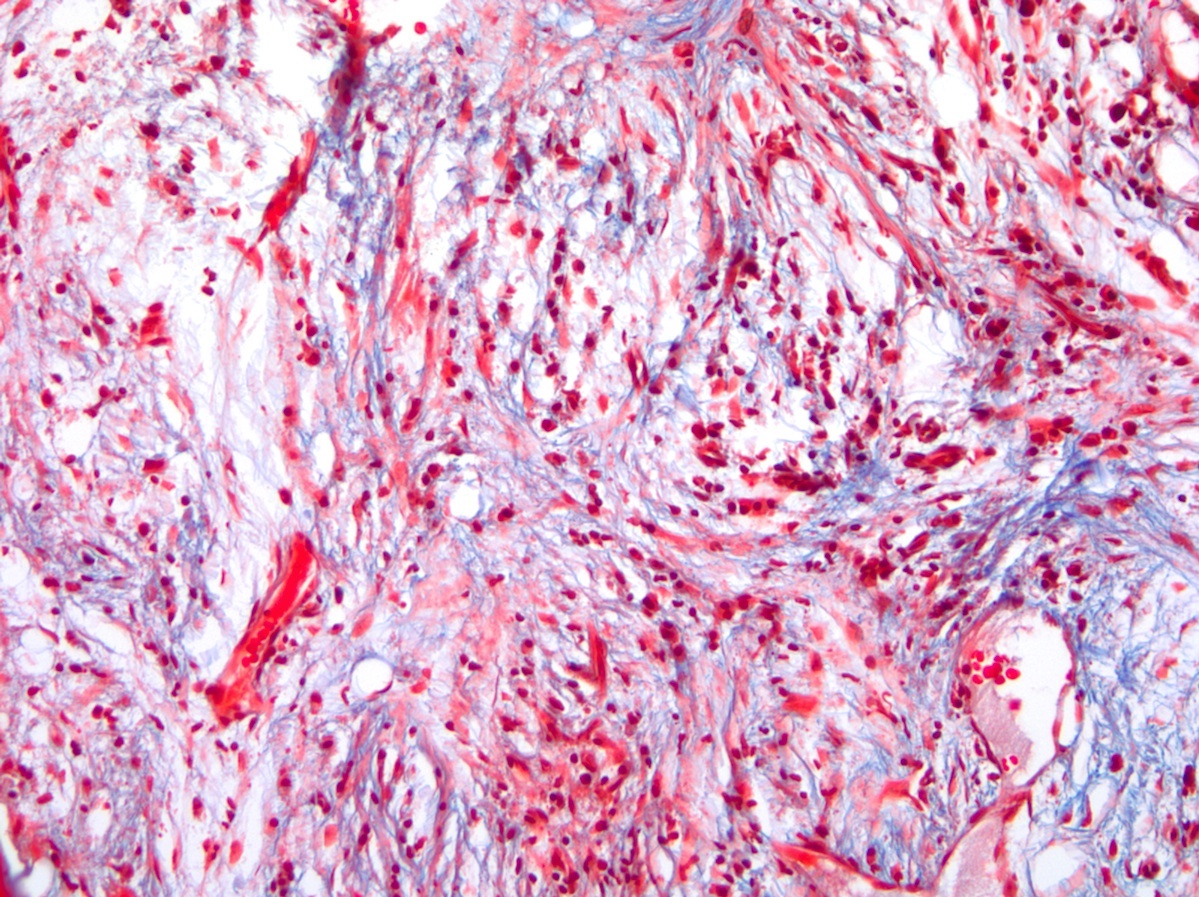
- Overt bone marrow reticulin and collagen fibrosis; osteosclerosis often prominent (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Dilated sinuses with intrasinusoidal hematopoiesis
- Decline in hematopoietic cells
Microscopic (histologic) images

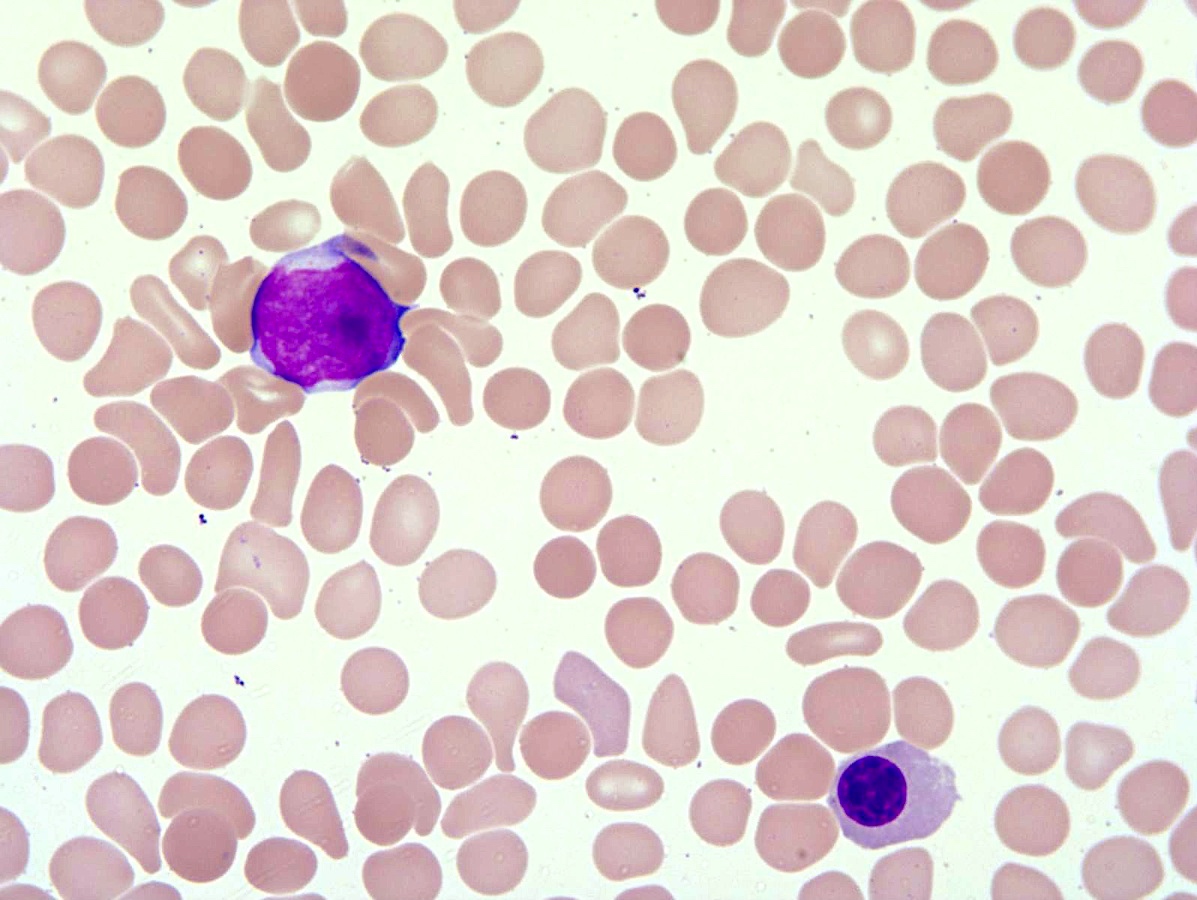
Peripheral smear description
- Peripheral blood (prepolycythemic and overt polycythemic phases)
- Normochromic, normocytic red blood cells or microcytic, hypochromatic when iron deficiency is present
- Erythrocytosis with increased hemoglobin or hematocrit (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Red blood cell morphology is unremarkable except in cases with concomitant iron deficiency
- Deeply basophilic reticulocytes or rare normoblasts may be present
- Mild neutrophilic leukocytosis with rare left shift (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Mild basophilia may be seen
- Thrombocytosis; may be particularly prominent in prodromal phase or exaggerated by concurrent iron deficiency (Am J Hematol 2020;95:1599)
- Post-PV / spent phase: myeloid metaplasia characterized by leukoerythroblastosis (see Peripheral smear image)
- Poikilocytosis with teardrop shaped red blood cells (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Findings of > 10% blasts in the peripheral blood or bone marrow or the presence of significant myelodysplasia is unusual and most likely signals transformation to an accelerated phase
- Cases in which 20% or more blasts are found are considered acute myeloid leukemia (AML) (see Microscopic (histologic) images 3 & 8) (J Clin Med 2021;10:436)
Peripheral smear images
Positive stains
- High expression of VEGF but not used for diagnosis (Am J Clin Pathol 2007;128:966)
- Glycophorin, spectrin or hemoglobin highlight increased number of erythroid precursors
- CD34 highlights increased myeloblasts when transformed
- CD61, CD42b and factor VIII highlight megakaryocytic hyperplasia
Negative stains
- MPL (thrombopoietin receptor) on megakaryocytes is usually moderate to strong in normal controls and secondary erythrocytosis but is not used for diagnosis (Blood 2000;96:771)
Molecular / cytogenetics description
- Most frequent somatic activating mutations of JAK2 are located in exon 14 (JAK2 V617F) (> 95%) followed by exon 12 (~5%)
- JAK2 V617F is not specific and is found at a lower frequency in other MPN
- Patients with JAK2 V617F are usually elderly with higher level of hemoglobin, white blood cell (WBC) count and lower level of platelets, while patients with JAK2 exon 12 mutation are frequently younger with erythroid hyperplasia but low erythropoietin production (low EPO level) (Am J Hematol 2017;92:94)
- Cytogenetic abnormalities are detectable in about 20% of patients and most commonly include del(20q), +8, +9 and +1q (Haematologica 2017;102:1511)
- Cytogenetic abnormalities increase in frequency when patients with PV transform to acute myeloid leukemia (AML), myelodysplastic syndrome (MDS) or myelofibrosis (MF) (Leuk Lymphoma 2013;54:2667)
- Next generation sequencing identified several gene mutations in PV in addition to JAK2, which includes most frequently TET2 and ASXL1 followed by SRSF2 and IDH2, with combined prevalence of 15% (Blood Adv 2016;1:21)
Sample pathology report
- Bone marrow, left posterior iliac crest, aspirate and biopsy:
- Myeloproliferative neoplasm (see comment)
- Comment: The patient has a history of persistent erythrocytosis with a decreased erythropoietin level. A JAK2 V617F mutation was identified in the peripheral blood (VAF: 10%). The combined morphologic and ancillary laboratory findings are diagnostic of polycythemia vera.
- Hypercellular bone marrow (80 - 90%) with panmyelosis
- Microscopic examination
- Peripheral blood
- Erythrocytosis
- Normal WBCs and platelets
- Peripheral smear: normocytic erythrocytosis
- Cytopenias present: none
- Red cell morphology: normocytic erythrocytosis
- Reticulocytosis: not increased
- Other significant findings: none
- Bone marrow aspirate smear
- Adequacy: spicular, cellular
- M:E ratio: 2:1
- Bone marrow differential count: blasts: 2.1 %, promyelocytes: 1.0%, myelocytes: 14.2%, metamyelocytes: 15.8%, band neutrophils: 3.5%, segmented neutrophils: 9.8%, eosinophils: 11.8%, basophils: 1.2%, monocytes: 2.0%, lymphocytes: 10.0%, early normoblasts: 2.5%, late normoblasts: 24.0%
- Significant findings: maturing trilineage hematopoiesis, no overt dysplasia, megakaryocytes are relatively increased and show variable morphology, including small forms and large hyperlobulated forms
- Bone marrow clot section and core biopsy
- Bony trabeculae: unremarkable
- Cellularity: 80 - 90%
- Megakaryocytes: increased, with occasional loose clustering
- Estimated M:E ratio: 3:1
- Iron stores: decreased
- Reticulin fibrosis: none (marrow fibrosis 0/3)
- Significant findings: hypercellular bone marrow with panmyelosis
- Peripheral blood
Differential diagnosis
- Chronic myelogenous leukemia (CML):
- Leukocytosis (WBC ranges from 12 k/L to 1,000 k/L; median 100 k/L), basophilia, thrombocytosis, monocytosis (often accounts for < 2 - 3% of WBC)
- No evidence of true erythrocytosis
- Positive for BCR::ABL1 gene rearrangement
- Negative for JAK2 V617F or JAK2 exon 12 or 13 mutation
- Essential thrombocythemia (ET) and early phase of primary myelofibrosis (PMF):
- Following the adoption of the updated 2016 WHO classification of PV or when a patient presents with thrombocytosis, it is particularly important to differentiate masked / prodromal polycythemia from JAK2 positive ET by careful examination of bone marrow morphology
- In contrast to PV, ET and PMF show distinct clinical and pathologic features (bone marrow biopsy of PV shows panmyelopoiesis with more atypical or pleomorphic appearing mature megakaryocytes) (Am J Hematol 2017;92:1062, Blood 2016;127:2391)
- ET: peripheral thrombocytosis but no evidence of erythrocytosis; bone marrow with essentially normocellular marrow, hyperlobated megakaryocytes
- PMF, early phase: cytosis but no evidence of erythrocytosis; bone marrow with atypical megakaryocytic proliferation, often associated with clustering, accompanied by myeloid hyperplasia
- PMF fibrotic phase: peripheral cytopenia; bone marrow with atypical megakaryopoiesis, syncytial clustering of megakaryocytes, reticulin fibrosis (marrow fibrosis 2 - 3/3)
- Following the adoption of the updated 2016 WHO classification of PV or when a patient presents with thrombocytosis, it is particularly important to differentiate masked / prodromal polycythemia from JAK2 positive ET by careful examination of bone marrow morphology
- Secondary erythrocytosis / polycythemia:
- In contrast to primary polycythemia vera, EPO levels are elevated, usually as a secondary response to chronic hypoxemia
- May be compensatory to various physiologic or environmental processes
- Lung disease, high altitude living, cyanotic heart disease, heavy smoking, carbon monoxide poisoning, body builder or hormone replacement (e.g., testolone)
- May be caused by EPO secreting tumors (i.e., renal cell carcinoma, hepatocellular carcinoma, cerebellar hemangioblastoma)
- May be caused by an inherited defect that stabilizes HIF1A (prolyl hydroxylase mutations, homozygous VHL mutations, etc.)
- Negative JAK2 mutation and normal serum EPO level help to differentiate from PV
- Gaisbock syndrome: normal megakaryocytes, less cellular marrow, no increase in reticulin, more normal iron stores, normal or high erythropoietin levels (Angiology 1978;29:520)
Additional references
Board review style question #1
Which of the following is the most helpful when differentiating between polycythemia vera (PV) and secondary erythrocytosis?
- Grade of marrow fibrosis
- Increased erythroid precursors seen in the bone marrow
- JAK2 V617F mutation status
- Presence of BCR::ABL1 gene fusion
- Red blood cell morphology
Board review style answer #1
C. JAK2 V617F mutation status. Detection of the JAK2 V617F mutation is a key criterion for diagnosing PV; therefore, out of the answer choices provided, JAK2 V617F mutation status would be the most helpful in differentiating PV from secondary erythrocytosis. Keep in mind the mutation can also be associated with essential thrombocythemia (ET) and primary myelofibrosis or other myeloid neoplasms. Answer A is incorrect because bone marrow fibrosis is a nonspecific response to chronic stimulation of hematopoiesis and cannot be used to differentiate between PV and secondary erythrocytosis. Answer B is incorrect because both PV and secondary erythrocytosis will show an increase in erythroid precursors on bone marrow biopsy. Answer E is incorrect because both PV and secondary erythrocytosis can exhibit variations in red blood cell morphology; there is no specific parameter for distinguishing between these entities on morphology alone. Answer D is incorrect because BCR::ABL1 gene fusion products are usually not detected in PV but have been rarely reported in certain cases of PV; however, it is not specific and does not help to differentiate PV from secondary erythrocytosis.
Comment Here
Reference: Polycythemia vera
Comment Here
Reference: Polycythemia vera
Board review style question #2
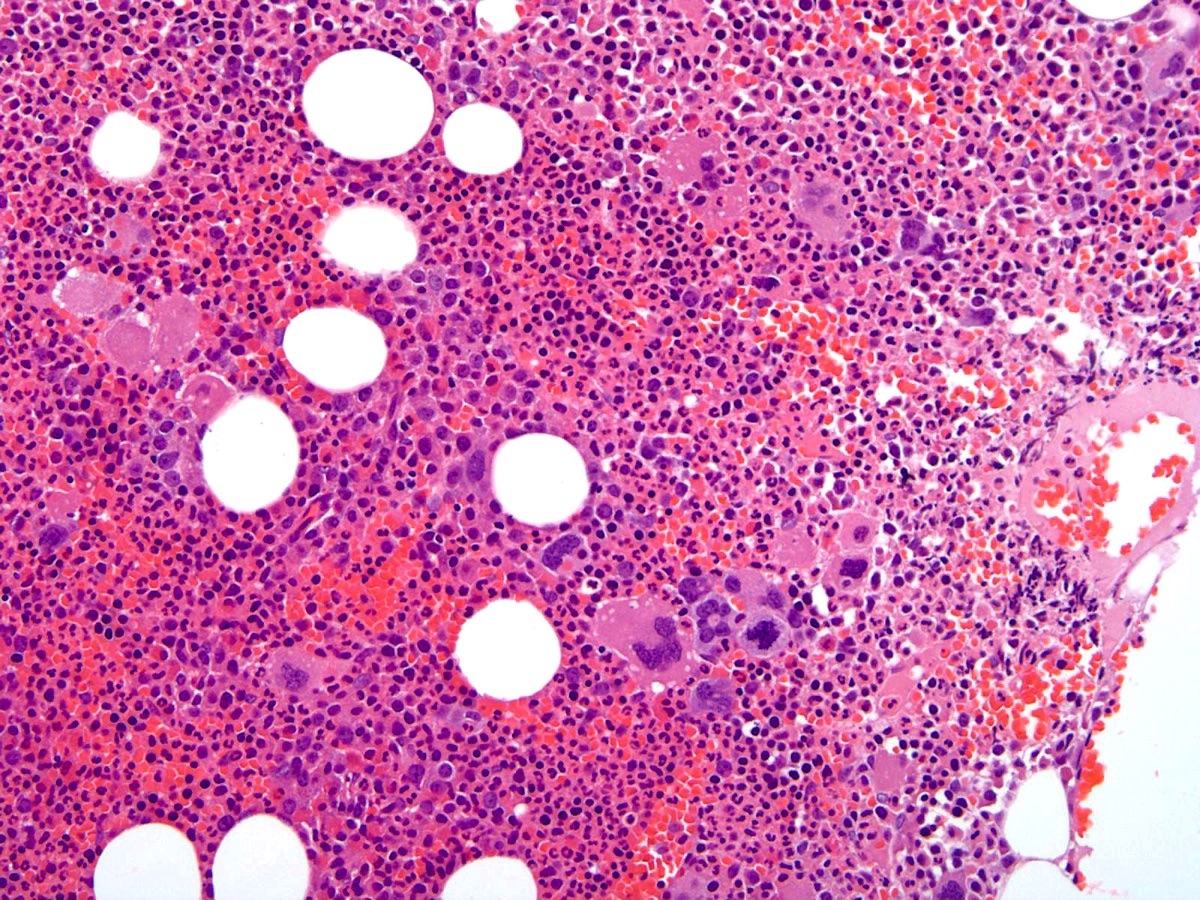
A patient underwent bone marrow biopsy for unexplainable pruritus and erythromelalgia. The bone marrow findings are depicted in the image above. What is a notable microscopic feature of polycythemia vera (PV) that can be seen in the bone marrow during the prepolycythemic and overt polycythemic phases?
- Decreased erythroid precursors
- Hypercellularity with panmyelosis
- Hypocellularity with sparse megakaryocytes
- Increased reticulin fibrosis
- Presence of collagen fibrosis highlighted by trichrome stain
Board review style answer #2
B. Hypercellularity with panmyelosis. During the prepolycythemic and overt polycythemic phases of PV, the bone marrow is hypercellular with notable panmyelosis. Answer C is incorrect because the bone marrow is hypercellular, rather than hypocellular, in prepolycythemic and overt polycythemic phases of PV and megakaryocytes are often increased, rather than sparse. Answer A is incorrect because PV involves an increase in erythroid production; therefore, in both prepolycythemic and overt polycythemic phases of PV, there are increased (rather than decreased) erythroid precursor cells seen in the bone marrow. Answer D is incorrect because fibrosis is typically a feature of the postpolycythemic myelofibrosis phase of PV and not the prepolycythemic or overt polycythemic phase. Answer E is incorrect because the presence of trichrome positive collagen fibrosis is usually not observed in prefibrotic PV bone marrow and is more often seen in the postpolycythemic myelofibrosis phase.
Comment Here
Reference: Polycythemia vera
Comment Here
Reference: Polycythemia vera
Board review style question #3
A 55 year old man is found to have a significantly elevated hemoglobin level. He has no significant medical history and no significant surgical history. He has a body mass index (BMI) of 21 and runs 40 minutes every day. He takes no medications. Vital signs are within normal limits. Physical exam is notable for a palpable spleen. Laboratory data shows a hematocrit of 53%, hemoglobin of 18 g/dL, leukocyte count of 8 x 109/L and platelet count of 800 x 1009/L. Genetic testing is positive for JAK2 V617F mutation. Presence of the JAK2 V617F mutation is one of the major criteria for diagnosing polycythemia vera (PV). Which of the following is also a major criteria requirement for diagnosis based on WHO classification?
- Bone marrow hypocellularity for age
- Hemoglobin greater than 16.5 g/dL
- Platelet count less than 100 x 103/μL
- Presence of extramedullary hematopoiesis
Board review style answer #3
B. Hemoglobin greater than 16.5 g/dL. One of the major criteria for diagnosing PV includes a hemoglobin greater than 16.5 g/dL in men and greater than 16.0 g/dL in women (see Diagnosis). Answer A is incorrect because bone marrow hypercellularity (not hypocellularity) is also a major criterion. Answer D is incorrect because, while extramedullary hematopoiesis may occur in PV, it is not a major criterion for diagnosis. Answer C is incorrect because decreased platelet count (thrombocytopenia) is not a major criterion for diagnosis and is seen less frequently than increased platelet count (thrombocytosis) in PV.
Comment Here
Reference: Polycythemia vera
Comment Here
Reference: Polycythemia vera

