
Bone marrow neoplastic
Bone marrow - neoplastic myeloid
Myeloproliferative neoplasms (MPN)
Primary myelofibrosis
Resident / Fellow Advisory Board: Frido Bruehl, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last staff update: 20 March 2025 (update in progress)
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Primary myelofibrosis

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Positive stains | Flow cytometry description | Flow cytometry images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Kaseb H, Hudnall SD. Primary myelofibrosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/myeloproliferativemyelofibrosis.html. Accessed April 2nd, 2025.
Definition / general
- Primary myelofibrosis is an uncommon myeloproliferative neoplasm characterized by a proliferation of predominantly abnormal megakaryocytes and granulocytes in the bone marrow, which in fully developed disease is associated with reactive deposition of fibrous connective tissue and extramedullary hematopoiesis
Essential features
- Shows a stepwise evolution from an initial prefibrotic / early stage to overt fibrotic stage
- Is a clonal stem cell defect characterized by
- Impaired medullary hematopoiesis
- Granulocytic and megakaryocytic proliferation in the bone marrow
- Gradual increase in bone marrow reticulin and collagen fibrosis
- Extramedullary hematopoiesis in the spleen, liver and other organs
- Impaired medullary hematopoiesis
- The International Consensus Classification (ICC) 2022 distinguishes prefibrotic from overtly fibrotic PMF (Blood 2022;140:1200)
Terminology
- Myelofibrosis refers to the increase in the amount and density of reticulin fibers in the bone marrow (can be caused by infections, inflammation, neoplasms, etc.)
ICD coding
- ICD-10: D75.81 - myelofibrosis
Epidemiology
- Estimated annual incidence of overt phase is 0.5 - 1.5 cases per 100,000 population
- Prefibrotic / early phase accounts for 30 - 50% of all cases
- Increasing prevalence due to earlier diagnosis (preprimary myelofibrosis)
- Equal gender predisposition (Am J Hematol 2014;89:915)
- Disease of older adults (mean age: 60 years)
- Extremely rare in children; however, some childhood cases may be inherited and associated with other anomalies (Curr Hematol Malig Rep 2020;15:141, Jaffe: Hematopathology, 2nd Edition, 2016)
- Increased incidence in individuals of Ashkenazi Jewish descent (Leuk Lymphoma 1992;7:251, Hsi: Hematopathology - Foundations in Diagnostic Pathology, 3rd Edition, 2017)
Sites
- Mainly blood and bone marrow
- Other variably included sites: liver, spleen
Pathophysiology
- Clonal stem cell defect
- Fibrosis is due to neoplastic megakaryocytes releasing platelet derived growth factor, basic fibroblast growth factor, transforming growth factor beta or other cytokines, which causes nonneoplastic fibroblasts in marrow to deposit collagen
- Myelofibrosis and osteosclerosis are secondary changes due to the abnormal release of growth factors and fibrogenic cytokines
- Prominent angiogenesis in the bone marrow and spleen due to increased serum levels of vascular endothelial growth factor
- Teardrop red blood cells and leukoerythroblastosis are caused by extramedullary hematopoiesis
- References: Int J Lab Hematol 2023:45:59, Leukemia 2022;36:1703
Clinical features
- Presentation
- Asymptomatic (30%)
- Symptomatic (70%)
- Splenomegaly (90%): often considered a hallmark
- Hepatomegaly (50%): portal hypertension can develop as a complication of hepatomegaly and may precede the onset of the disease (Gastroenterology 1988;94:1063)
- Constitutional symptoms such as fatigue, dyspnea, weight loss, night sweats, low grade fever and cachexia
- Fatigue is the most common presenting symptom
- Anemia
- Leukocytosis or thrombocytosis is common; leukopenia or thrombocytopenia is less common
- Gouty arthritis and renal stones
- Marked thrombocytosis may lead to thrombosis or hemorrhagic episodes
- Uncommon symptoms include pruritus and pulmonary hypertension (Am J Hematol 2012;87:136, Leukemia 2008;22:646)
- 2 phases
- Prefibrotic
- Usually presents with thrombocytosis
- No blasts
- No / borderline anemia
- No / borderline splenomegaly
- Normal or borderline increased lactate dehydrogenase
- Misdiagnosis of essential thrombocythemia is possible if the bone marrow biopsy is not carefully examined
- Overt / fibrotic
- Thrombocytopenia
- Leukoerythroblastosis
- Anemia
- Splenomegaly
- Increased lactate dehydrogenase
- Increased myeloblasts can be seen (< 20%); 10 - 19% blasts indicates progression to accelerated phase
- Transformation to acute myeloid leukemia occurs in ~5 - 20% at a median of 3 years after the diagnosis of the overt phase (Jaffe: Hematopathology, 2nd Edition, 2016)
- 2 factors at the time of diagnosis are considered predictors of leukemic transformation: circulating blasts ≥ 3% and platelet count < 100,000/μL (Cancer 2008;112:2726)
- Prefibrotic
- Extramedullary hematopoiesis can form masses
- Survival: 3.5 - 5.5 years
Diagnosis
| Diagnostic criteria for prefibrotic primary myelofibrosis | Diagnostic criteria for primary myelofibrosis, fibrotic stage |
| Diagnosis of prefibrotic primary myelofibrosis requires that all 3 major criteria and at least 1 minor criterion are met | Diagnosis of overt primary myelofibrosis requires that all 3 major criteria and at least 1 minor criterion are met |
| Major criteria | |
|
|
| Minor criteria | |
Presence of at least 1 of the following, confirmed in 2 consecutive determinations
|
Presence of at least 1 of the following, confirmed in 2 consecutive determinations
|
Laboratory
- Complete blood count / peripheral blood smear
- Prefibrotic
- Initially normal or increased blood counts
- Mild neutrophilia with a left shift
- Thrombocytosis (mild / moderate)
- No / borderline anemia
- No myeloblasts
- No leukoerythroblastosis
- Initially normal or increased blood counts
- Overt
- Thrombocytopenia with bizarre abnormal large platelets with altered granulation; in addition, fragmented megakaryocytes can be seen on the peripheral smear
- Leukoerythroblastosis and anemia
- Myeloblasts (usually > 5%)
- Prefibrotic
- Lactate dehydrogenase (LDH)
- Prefibrotic: normal or borderline increased lactate dehydrogenase
- Overt: increased lactate dehydrogenase
- Alkaline phosphatase, uric acid, leukocyte alkaline phosphatase and vitamin B12 are increased (Leuk Lymphoma 1996;22:303)
- Bone marrow aspirate / biopsy
- Prefibrotic
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Usually morphologically more variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Erythropoiesis is reduced
- Absent or only slight reticulin fibrosis
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Overt
- Hypocellular bone marrow with usually alternating cellular and hypocellular regions
- Atypical megakaryocytes can form clusters or sheets
- More variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Marked reticulin (MF2 / 3) or collagen fibrosis
- New bone formation and osteosclerosis
- Bone marrow aspirate is often difficult (dry tap)
- Prefibrotic
- See also Molecular description
Prognostic factors
- Has the least favorable prognosis among the myeloproliferative neoplasms
- Patients are at risk of death due to disease progression, leukemic transformation, thrombohemorrhagic complications and infections
- Survival
- Prefibrotic: 10 - 15 years
- Overt: 3 - 5 years
- Dynamic International Prognostic Scoring System (DIPSS plus)
- Includes 8 predictors of inferior survival
- Patient age > 65 years
- Red blood cell transfusion dependency
- Unfavorable karyotype
- Hemoglobin < 10 g/dL
- White blood cells > 25 × 109/L
- Constitutional symptoms (fever, night sweats, weight loss)
- Circulating blasts ≥ 1%
- Platelet count < 100 × 109/L
- Risk status is defined by the number of adverse prognostic factors present
- 0 (low risk), 1 (intermediate - 1 risk), 2 or 3 (intermediate - 2 risk) or > 4 (high risk)
- Includes 8 predictors of inferior survival
- Genetics inspired International Prognostic Scoring System (Leukemia 2018;32:1631)
- Points are scored as follows
- Very high risk karyotype f (2 points)
- Unfavorable karyotype g (1 point)
- ASXL1 mutation (1 point)
- SRSF2 mutation (1 point)
- U2AF1 Q157 mutation (1 point)
- Type 1-like CALR absent (1 point)
- Risk status is defined by the points
- 0 (low), 1 (intermediate - 1), 2 (intermediate - 2), > 3 (high)
- Points are scored as follows
- Absolute monocyte count ≥ 1 × 109/L confers an unfavorable outcome
- Unfavorable karyotype
- Complex karyotype, sole abnormality
- 2 abnormalities that include +8, -7 / 7q-, i(17q), inv(3), -5 / del(5q), 12p- or 11q23.3 rearrangement
- CALR mutation confers a better prognosis compared to other mutations
Case reports
- 51 year old Korean man with tumor lysis syndrome (Am J Case Rep 2019;20:146)
- 54 year old woman with massive hemothorax due to primary extramedullary hematopoiesis of the pleura (Cureus 2018;10:e3675)
- 71 year old patient treated with ruxolitinib (Drugs Context 2019;8:212569)
- 80 year old man with a JAK2 V617F mutation transforming into Philadelphia chromosome positive acute myeloid leukemia (Biomark Res 2018;6:33)
- 86 year old woman presented with chronic lymphocytic lymphoma (Case Rep Hematol 2018;2018:7426739)
Treatment
- Management based on the risk stratification by
- GIPSS (Genetically Inspired Prognostic Scoring System): uses karyotyping, driver mutations and HMA mutations
- MIPSS70+ v2.0 (Mutation and Karyotype Enhanced International Prognostic Scoring System) criteria: uses clinical information, karyotyping and mutations
- Higher risk
- Eligible for transplantation: allogeneic hematopoietic cell transplantation
- Ineligible for transplantation: enrollment in a clinical trial or treatment with ruxolitinib or hydroxyurea for relief of symptoms (N Engl J Med 2012;366:787)
- Lower risk
- Asymptomatic: observation
- Symptomatic: treatment with ruxolitinib or hydroxyurea
Microscopic (histologic) description
- Bone marrow
- Prefibrotic stage
- Hypercellular with large, dysplastic, clustered (loose or tight) megakaryocytes and excess granulocytes
- Increased reticulin is present around clusters of megakaryocytes; megakaryocytes have aberrant N:C ratios and hyperchromatic, bulbous or irregularly folded nuclei
- Often bare megakaryocytic nuclei
- Megakaryocytic features are most useful to distinguish this stage of primary myelofibrosis from essential thrombocythemia
- Fibrotic phase
- Hypocellular and diffusely fibrotic bone marrow with atypical streaming megakaryocytes
- Marrow osteosclerosis with irregular, broad bony trabeculae
- Markedly dilated sinuses; associated with dry bone marrow taps
- Prefibrotic stage
- Spleen: red pulp sinuses contain megakaryocytes, granulocyte precursors, nucleated red cells; may be nodules of extramedullary hematopoiesis
- References: Int J Lab Hematol 2023:45:59, Leukemia 2022;36:1703
Microscopic (histologic) images
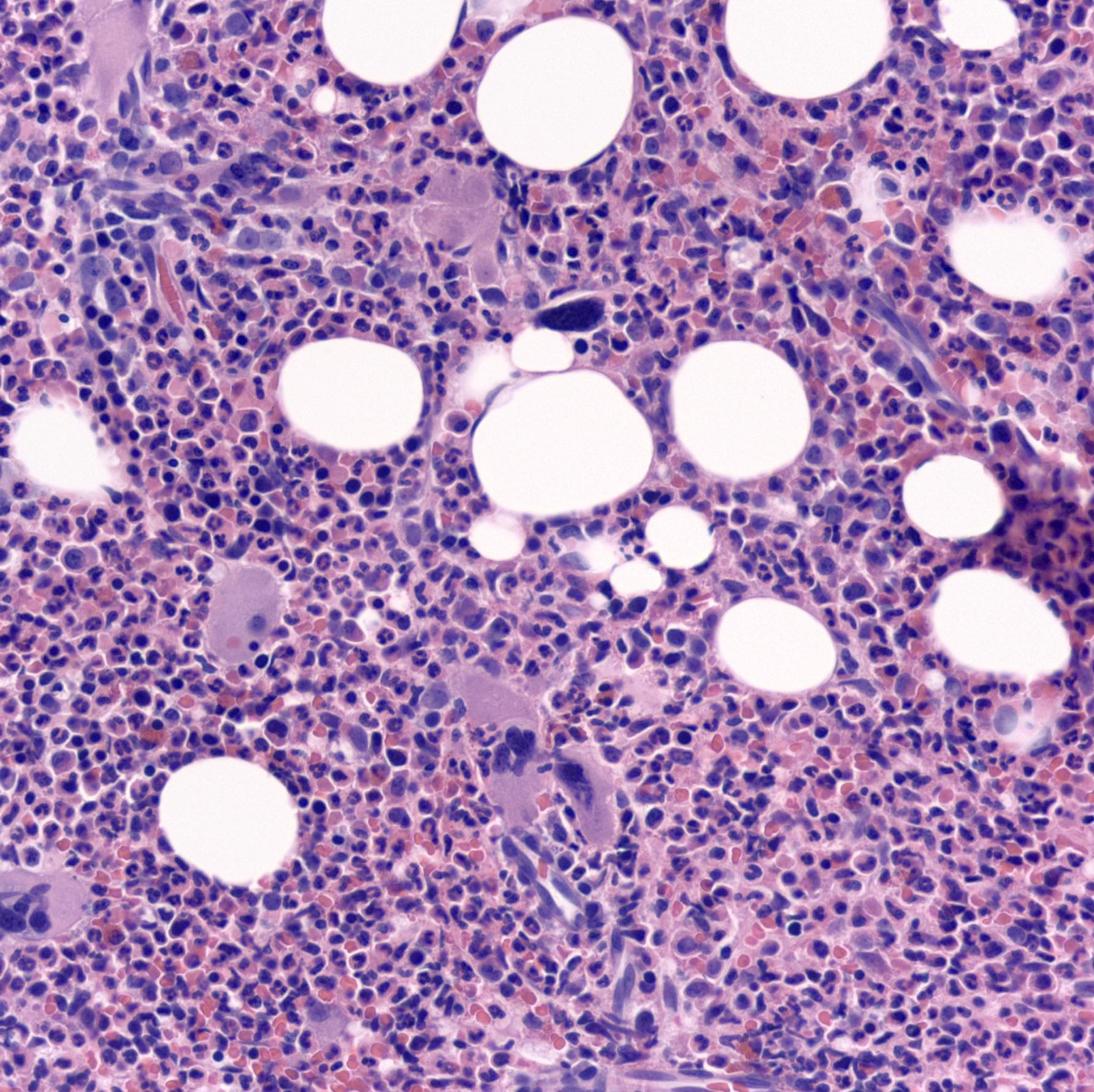
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H. and AFIP

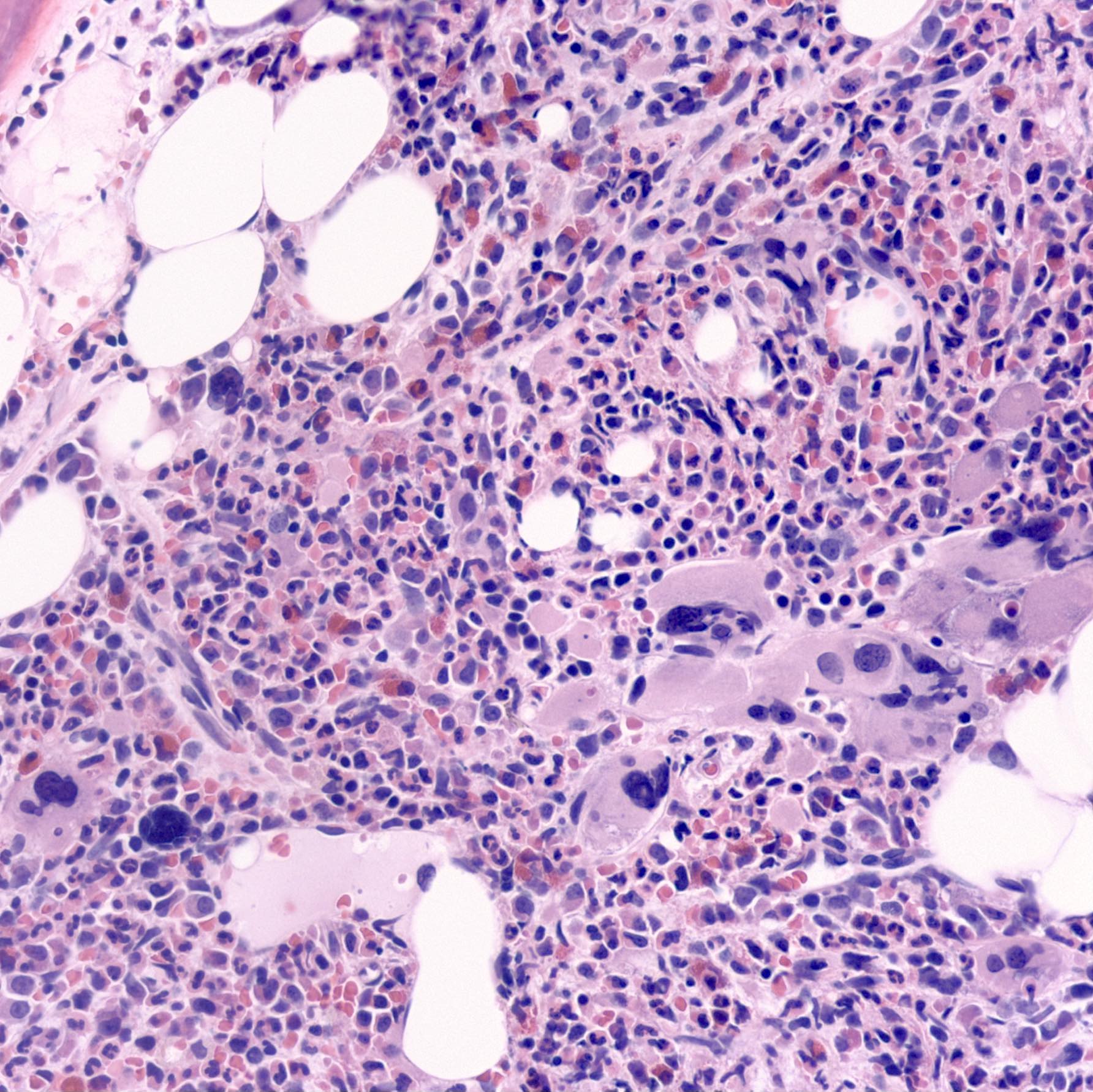
Atypical megakaryocytes
Blast transformation

Cell intermediate
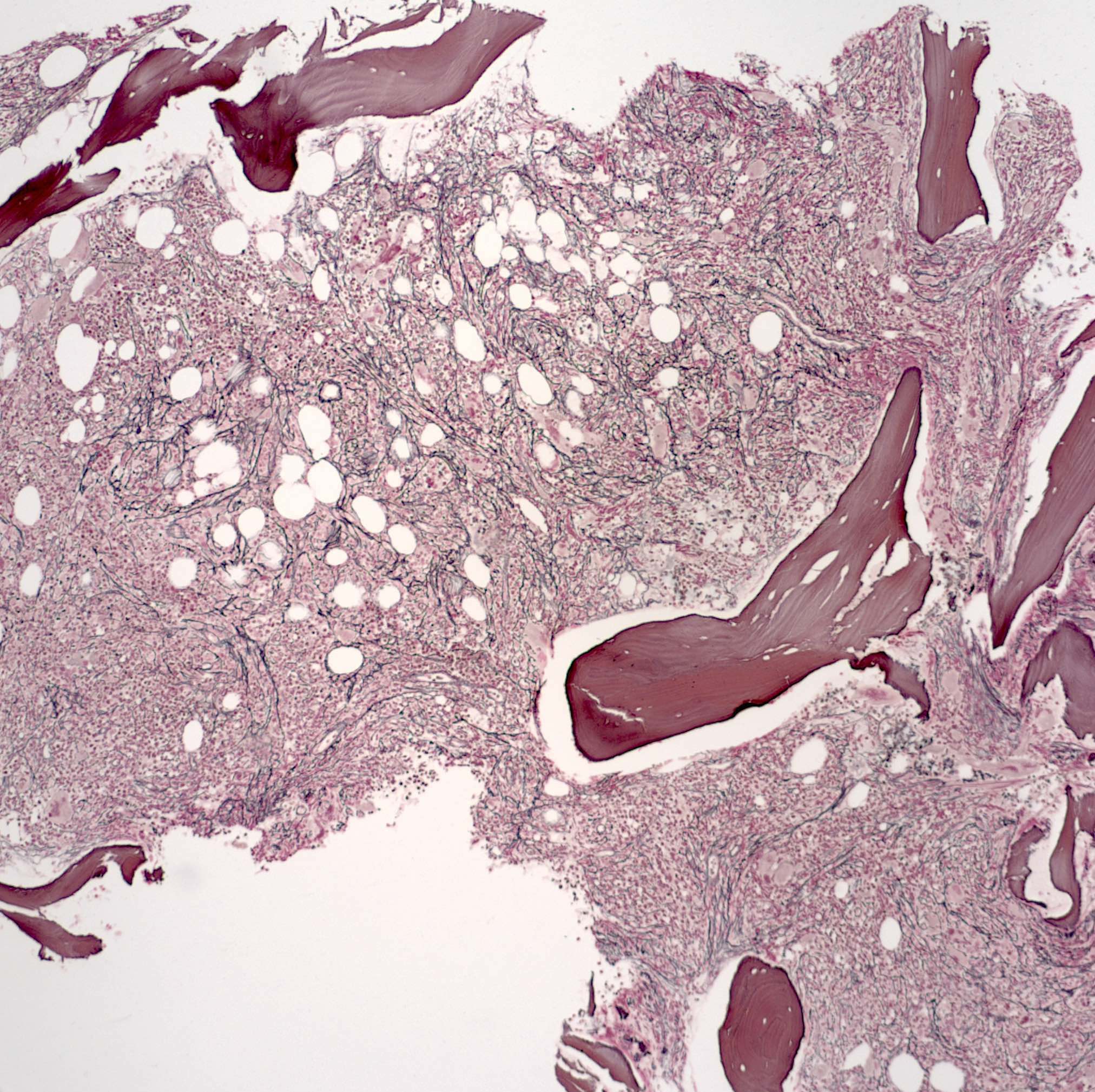
Fibrotic bone marrow
Peripheral smear description
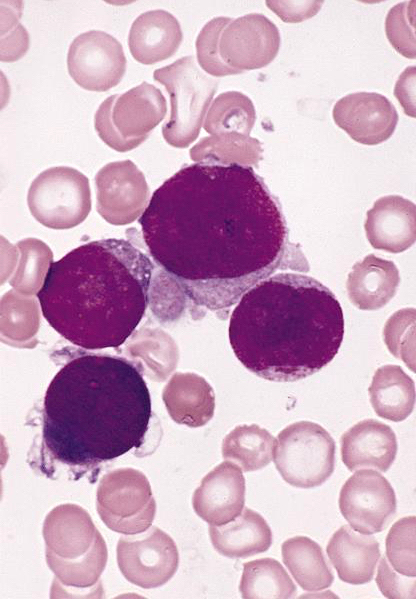
- Leukoerythroblastosis (immature granulocytes and normoblasts in peripheral blood) is common in later phases; also dacrocytes (teardrop erythrocytes)
Peripheral smear images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
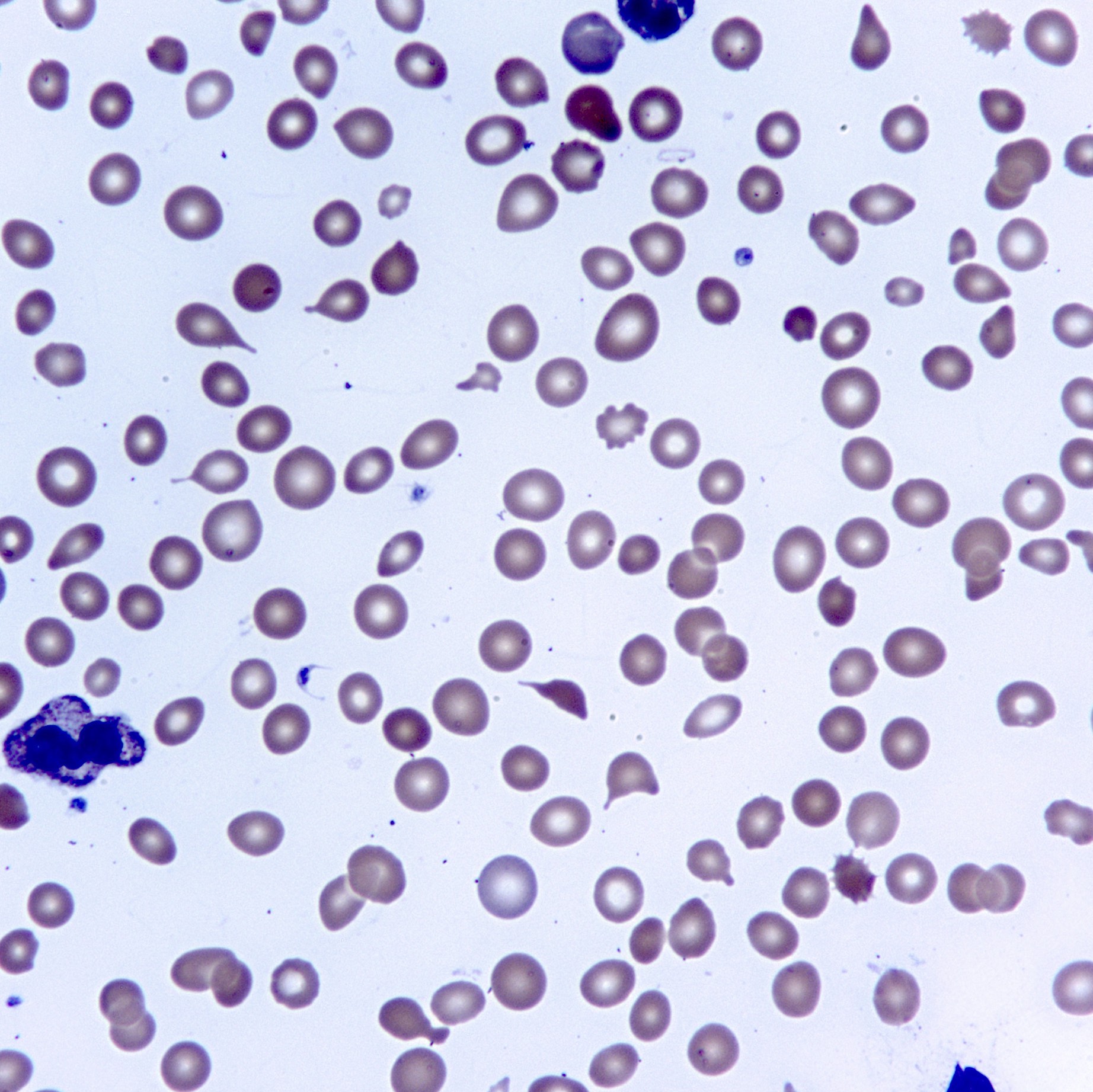
Dacrocytes
Positive stains
- VEGF (high expression) (Am J Clin Pathol 2007;128:966)
- CD105 (to assess microvascular density, may have prognostic value) (Mod Pathol 2004;17:1513)
- Chloroacetate esterase helps mark granulocytic proliferation in the prefibrotic phase
Flow cytometry description
- Some cases show low side scattering, aberrant CD56 expression in granulocytes and monocytes and an abnormal CD13 / CD16 maturation pattern (Am J Clin Pathol 2010;133:314)
Flow cytometry images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.

Normal myeloid maturation
Molecular / cytogenetics description
- No specific genetic marker
- No BCR::ABL fusion gene (or classify as chronic myeloid leukemia); presence of del(13)(q12-22) or der(6)t(1;6)(q21-23;p21.3) is strongly suggestive of primary myelofibrosis
- Shows karyotypic abnormalities in up to 50% of cases
- Frequent abnormalities include del(20q), del(13q), +8, +9 and abnormalities of 1q
- Unfavorable karyotype (complex karyotype; > 3 abnormalities) (Virchows Arch 2023;482:53)
- Isolated +8, isolated -7/7q-, i(17q), -5/5q-, 12p-, 11q23 rearrangement or inv(3) and an abnormal karyotype with abnormalities of chromosomes 5, 7, 17 or 12p-
- Mutations (Am J Hematol 2023;98:801)
- Driver mutations
- JAK2 V617F mutation (50 - 60%)
- CALR mutations (25 - 30%)
- MPL mutations (5 - 10%)
- Triple negative for mutations in JAK2, CALR and MPL (8 - 12%)
- Other mutations
- Include ASXL1, EZH2, TET2, IDH1 / IDH2, SRSF2 or SR3B1
- Driver mutations
- Driver mutational profile is associated with overall survival
- CALR mutated cases have better survival compared to others
- Triple negative cases have the shortest survival compared to others
Sample pathology report
- Bone marrow, biopsy and aspirate:
- Myeloproliferative neoplasm, favoring prefibrotic primary myelofibrosis (see comment)
- Comment: The marrow biopsy shows morphologic features of a myeloproliferative neoplasm, in conjunction with thrombocytosis. The major differential diagnosis includes essential thrombocythemia and prefibrotic stage of primary myelofibrosis. Overall, a diagnosis of the latter is favored due to hypercellular marrow, distribution of megakaryocytes (focal clusters) and markedly dysplastic cytology of megakaryocytes. Recommend correlation with clinical findings (presence of splenomegaly and disease progression) and molecular studies (JAK2, CALR and MPL mutational studies).
- Complete blood count (9/17/2018), by report: hemoglobin - 11.4 g/dL, mean corpuscular volume - 100 fL, white blood cells - 21.7 K/uL, platelets - 698 K/uL
- Bone marrow biopsy microscopic description:
- Hypercellular marrow for age (80% cellular). Megakaryocytes are increased in number, forming focal loose clusters and showing abnormal morphology including hypersegmented nuclei with hyperchromasia. The myeloid:erythroid ratio is markedly increased. Erythroid elements exhibit maturation. Myeloid elements exhibit maturation. Myeloblasts appear to be increased. No lymphoid aggregates are identified. Granulomas are absent. Reticulin stain reveals that reticulin is mildly increased (MF1/3). Trabecular bone is normal.
- Bone marrow aspirate microscopic description:
- The marrow aspirate smears are spicular and cellular. Megakaryocytes are increased and show prominent dysmegakaryopoiesis, including hyper and hyposegmented nuclei and increased N:C ratio. The myeloid:erythroid ratio is approximately 4:1. Erythroid maturation is identified. Myeloid maturation is slightly left shifted. Prussian blue iron stain shows # storage iron and no significant increase in ring sideroblasts.
- Aspirate cell count: A 207 cell count reveals 1% blasts, 15% promyelocytes / myelocytes, 57% maturing granulocyte forms, 20% erythroid forms, 3% lymphocytes, 2% eosinophils, 1% plasma cells, 1% basophils / mast cells and 2% monocytes.
Differential diagnosis
- Other causes of leukoerythroblastosis or dry taps:
- Granulomatous marrow disease, metastases to marrow (desmoplasia but no increased reticulin) or lymphoma
- Other myeloid disorders:
- Myeloproliferative neoplasms:
- Chronic myelogenous leukemia:
- Positive BCR::ABL translocation
- Polycythemia vera:
- Dispersed less atypical megakaryocytes and prominent erythroid element
- Essential thrombocythemia:
- Uniformly enlarged megakaryocytes with deeply lobulated (staghorn) nuclei, in loose clusters; normal myeloid:erythroid ratio
- Chronic myelogenous leukemia:
- Myelodysplastic / myeloproliferative neoplasms:
- Show hybrid myelodysplastic and myeloproliferative features manifested by at least 1 cytopenia and at least 1 cytosis in blood
- Myeloid maturation is intact and usually mature cells predominate
- Exact subtype should be diagnosed based on the specific criteria of the entity
- Myelodysplastic syndromes are
a heterogeneous group of entities that demonstrate:
- Variable degrees of ineffective hematopoiesis
- < 20% blasts in marrow
- Peripheral blood with unexplained and persistent cytopenia(s), < 20% blasts, < 1.0 x 109/L monocytes and dysplasia in 1 or more myeloid lineages
- Mastocytosis:
- Shows focal, compact aggregates of mast cells in bone marrow core biopsy
- Cells can show the following patterns of infiltration: paratrabecular, perivascular or parafollicular
- Presence of atypical mast cell supports the diagnosis
- Most cases will harbor a KIT D816V mutation
- Myeloproliferative neoplasms:
- Reactive thrombocythemia:
- Causes include exercise, allergic reaction, reaction to medications, inflammatory disorders, asplenism, infection, connective tissue disorders, metastatic cancer, lymphoproliferative disorders and iron deficiency
- Acute myelofibrosis:
- Subtype of acute myeloid leukemia that shows > 20% blasts in peripheral blood or bone marrow
- Patients also present with bone marrow fibrosis, fever and pancytopenia
- Autoimmune myelofibrosis:
- Generally shows diffuse reticulin fibrosis and an associated autoimmune disease such as rheumatoid arthritis
- Primary hyperparathyroidism:
- Rarely causes myelofibrosis and pancytopenia (Int J Lab Hematol 2007;29:464)
Additional references
Board review style question #1
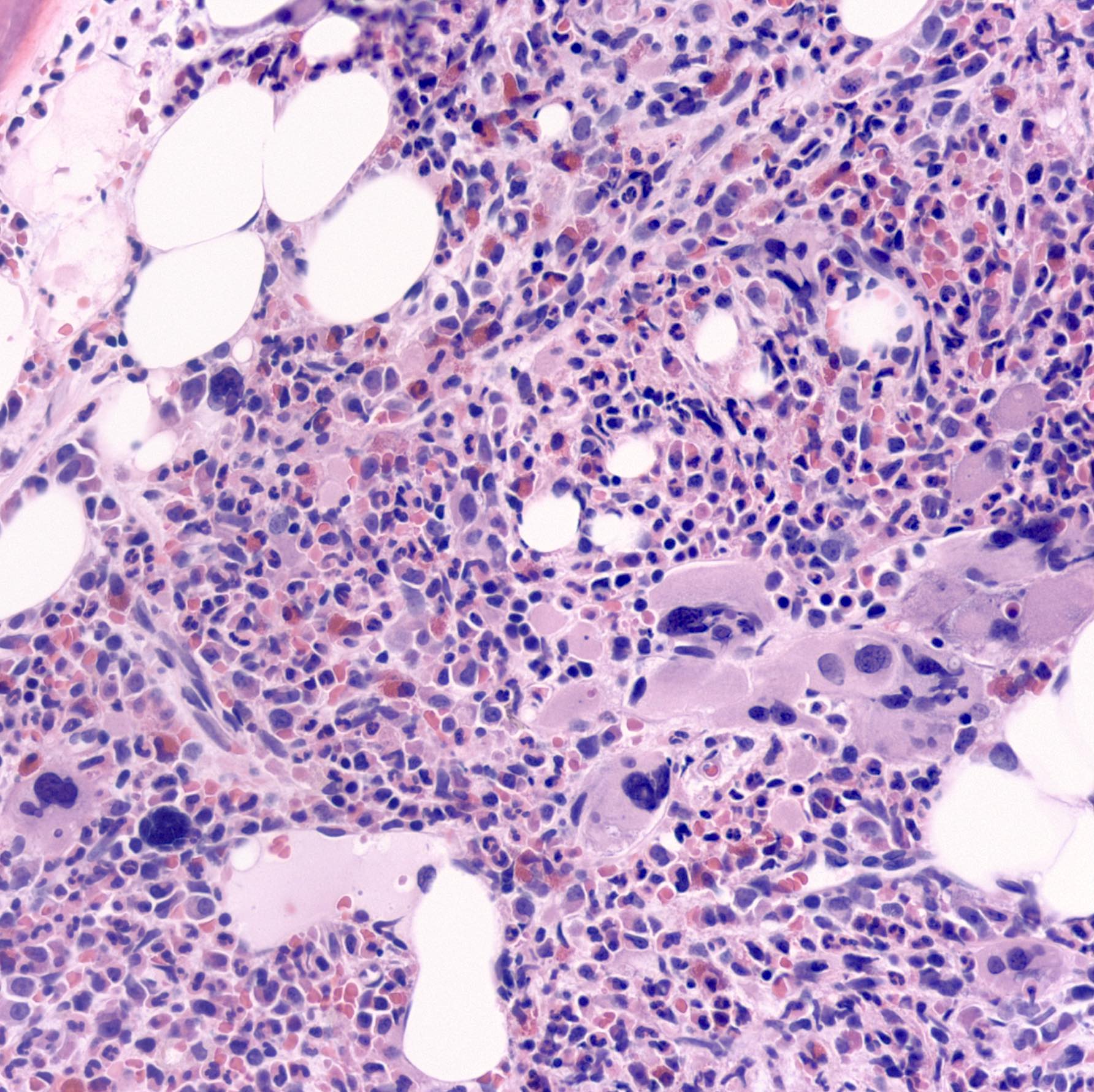
A 57 year old man presented with fatigue, splenomegaly and hepatomegaly. A complete blood count demonstrated anemia and thrombocytosis. A bone marrow aspirate and biopsy were ordered (shown above). What statement about this disease is correct?
- CALR mutation is the most common mutation encountered
- Complex karyotype is common
- MPL mutation is the most common mutation encountered
- Some cases can be triple negative for mutations in JAK2, CALR and MPL
Board review style answer #1
D. Some cases can be triple negative for mutations in JAK2, CALR and MPL. Primary myelofibrosis cases harbor a phenotypic driver mutation in JAK2 V617F mutation (50 - 60%), CALR mutations (25 - 30%) and MPL mutations (5 - 10%) and are triple negative for mutations in JAK2, CALR and MPL (8 - 12%). Answer B is incorrect because complex karyotype is uncommon. Answers A and C are incorrect because CALR and MPL are less common than JAK2 mutations.
Comment Here
Reference: Primary myelofibrosis
Comment Here
Reference: Primary myelofibrosis
Board review style question #2
What is the most common genetic mutation in primary myelofibrosis?
- ABL
- CALR
- JAK2
- MPL
Board review style answer #2
C. JAK2.
Primary myelofibrosis cases harbor a phenotypic driver mutation in JAK2 V617F mutation (50 - 60%), CALR mutations (25 - 30%) and MPL mutations (5 - 10%) and are triple negative for mutations in JAK2, CALR and MPL (8 - 12%). Answers B and D are incorrect because CALR and MPL are less common than JAK2 mutations. Answer A is incorrect because ABL mutations have not been described in primary myelofibrosis.
Comment Here
Reference: Primary myelofibrosis
Comment Here
Reference: Primary myelofibrosis
