
Muscle & peripheral nerve nontumor
Congenital myopathies
Multiminicore myopathy
Author: Chunyu Cai, M.D., Ph.D.
Editorial Board Members: Jared T. Ahrendsen, M.D., Ph.D., Meaghan Morris, M.D., Ph.D.

Last author update: 5 March 2025
Last staff update: 5 March 2025
Copyright: 2020-2025, PathologyOutlines.com, Inc.
PubMed Search: Multiminicore myopathy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Cai C. Multiminicore myopathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/musclemultiminicore.html. Accessed April 2nd, 2025.
Definition / general
- Type of congenital myopathy histologically characterized by multifocal small pale areas within myofiber on oxidative stains (NADH-TR, SDH and COX) and ultrastructurally by multifocal small areas of sarcomeric disarray devoid of mitochondria
Essential features
- Multicore disease was first described by A. G. Engel et al. in 1971, as a group of early onset, autosomal recessive congenital myopathies, ultrastructurally characterized as small areas of sarcomere disruption, spanning only a few sarcomeres and often multifocal in 1 fiber (Mayo Clin Proc 1971;46:666)
- Heterogeneous group of congenital myopathy with mutations identified in SELENON, RYR1, TTN, MYH7, ACTN2, MEGF10
- Given the significant overlap with other core myopathies and widespread applications of paneled genetic analysis for congenital myopathies, the morphologic distinction between central core, dusty core or multiminicore becomes less important and can be collectively referred to as core myopathy (Neuromuscul Disord 2021;31:968)
- Pathologically characterized by multiminicores involving both type I and type II fibers visible on oxidative stains, typically in a background of type I predominance, type I hypotrophy (smallness) and increased centralized nuclei
Terminology
- Multicore disease or multicore myopathy
- Minicore disease or minicore myopathy
- Multiminicore disease
ICD coding
Epidemiology
- Overall prevalence of congenital myopathy in the U.S. is estimated at 1 in 26,000; core myopathies represent the most common subgroup (Ann Neurol 2011;70:662)
- Most have infant or early childhood onset, although some variants can have adult onset
Sites
- Mostly involving skeletal and cardiac muscles
- See Radiology description and Clinical features for gene specific features
Pathophysiology
- Most proteins implicated in congenital myopathies are related to primary or secondary defects of muscle excitation - contraction coupling, intracellular calcium homeostasis and abnormal sarcomeric assembly and function (Nat Rev Neurol 2018;14:151)
- SELENON: 1p36.13 encodes selenoprotein N, a calcium sensor in the endoplasmic reticulum that monitors calcium levels and activates the sarcoplasmic reticulum calcium pump (SERCA) when calcium is low (J Mol Med (Berl) 2012;90:1095)
- RYR1: 19q13.1 encodes ryanodine receptor 1, a calcium channel located in the sarcoplasmic reticulum membrane, which upon activation releases calcium ions from the SR into the sarcoplasm and stimulates muscle contraction (Acta Neuropathol Commun 2016;4:121)
- MYH7: 14q12, encodes beta (β) myosin heavy chain protein, a key component of the thick filament assembly in both cardiac and skeletal muscles (Neuromuscul Disord 2012;22:1096)
- TTN: 2q31, encodes titin, a large, filamentous protein that spans the length of half sarcomere that regulates sarcomere length and provides passive contraction force like a spring (Biophys Rev 2018;10:1187)
- ACTN2: 1q42, encodes alpha actinin 2, a component of the Z disk; functions as a link between the actin filaments and titin that is essential for sarcomere contractility and structural stability (Ann Clin Transl Neurol 2024;11:629)
- MEGF10: 5q23.2, encodes multiple epidermal growth factor-like domains protein 10, a regulator of satellite cell myogenesis (Nat Genet 2011;43:1189)
Etiology
- Driven by familial inherited or de novo mutations
Clinical features
- Common features of multiminicore myopathy are similar to other congenital myopathies, include neonatal hypotonia, delayed motor development, generalized muscle weakness and atrophy and normal intelligence; weakness usually remains stable (nonprogressive)
- Gene specific clinical features
- SELENON: autosomal recessive; classical severe clinical phenotype of axial myopathy, scoliosis and respiratory failure
- Significant subset shows rigid spine syndrome (Am J Hum Genet 2002;71:739)
- RYR1: autosomal recessive (dominant RYR1 mutations usually cause central core myopathy); weakness, malignant hyperthermia, external ophthalmoplegia and ligament laxity in distal upper extremity (Neurology 2005;65:1930)
- MYH7: autosomal dominant and recessive; hypertrophic or dilated cardiomyopathy, distal weakness and respiratory impairment (Neuromuscul Disord 2012;22:1096)
- TTN: autosomal recessive: cardiomyopathies (dilated cardiomyopathy, septal defects, noncompaction), arthrogryposis (Hum Mol Genet 2014;23:980)
- ACTN2: autosomal recessive; adult onset, progressive proximal and distal low extremity weakness, with facial, respiratory and ocular muscle involvement in a subset (Ann Clin Transl Neurol 2024;11:629, Acta Neuropathol 2019;137:501)
- MEGF10: autosomal recessive; pediatric or adult onset respiratory insufficiency, scoliosis and distal joint hyperlaxity (Nat Genet 2011;43:1189, Muscle Nerve 2016;53:984)
- SELENON: autosomal recessive; classical severe clinical phenotype of axial myopathy, scoliosis and respiratory failure
Diagnosis
- Diagnosis is typically made by muscle biopsy and genetic testing
Laboratory
- Serum creatine kinase (CK) is usually normal
Radiology description
- SELENON mutant patients show severe wasting of sternocleidomastoid muscle and atrophy of semimembranosus, paraspinal, gluteus maximus and thigh muscle (Muscle Nerve 2015;52:728)
- In RYR1 mutant patients, quadriceps are usually diffusely involved with relative sparing of rectus femoris; sartorius is more severely affected than gracilis (Neurology 2005;65:1930)
- In MYH7 mutant patients, there is more prominent involvement of distal compared to proximal muscle groups and more prominent involvement of the posterior compared to the anterior compartment in the lower extremities (Neuromuscul Disord 2012;22:1096)
- ACTN2 mutant patients demonstrate selective involvement of the hamstrings and adductors in the thigh and anterior tibial group and soleus in the lower leg (Ann Clin Transl Neurol 2024;11:629)
Prognostic factors
- Muscle weakness is usually stable or slowly progressive over time
- Prognosis depends on multidisciplinary care and monitoring of life risk factors such as cardiac involvement, respiratory impairment or malignant hyperthermia
Case reports
- 10 year old girl and 18 year old man (siblings) with congenital multicore myopathies harboring TTN mutations (Ann Clin Transl Neurol 2020;7:846)
- 40 year old woman and 45 year old man (unrelated patients) with progressive early onset muscle weakness and respiratory involvement and ACTN2 mutations (Acta Neuropathol 2019;137:501)
- 41 and 44 year old sisters with multiminicore disease due to novel MEGF10 mutations and adult onset respiratory insufficiency, scoliosis and distal joint hyperlaxity (Muscle Nerve 2016;53:984)
- 57 year old man with lifelong neck muscle weakness, multicore myopathy and MYH7 mutation (BMC Med Genet 2017;18:105)
Treatment
- Currently no cure; treatment is mostly supportive: specialized neuromuscular clinic with a multidisciplinary team that includes neurologist, lung specialist, orthopedic surgeon, cardiologist, physiotherapist, occupational therapist, speech and language therapist, dietician, psychologist
- Patients with RYR1 mutation are at high risk for malignant hyperthermia and should avoid triggering anesthetics (succinylcholine, volatile anesthetics); dantrolene is used for emergency treatment of malignant hyperthermia
- Patients with SELENON mutation are at higher risk of respiratory impairment, respiratory support is often needed
- Patients with TTN, MYH7 and ACTN2 mutations are at higher risk for cardiomyopathy; cardiac evaluation and regular cardiac monitoring are indicated
- Orthopedic surgeries for scoliosis, joint contractures or other deformities
- Reference: Dev Med Child Neurol 2020;62:297
Microscopic (histologic) description
- By definition, multiminicore myopathy is defined by the presence of multifocal small pale areas within myofibers on oxidative stains
- Background muscle usually shows type I predominance with or without type I hypotrophy
- Centralized nuclei are usually increased, particularly prominent in TTN mutant cases (Hum Mol Genet 2014;23:980)
Microscopic (histologic) images
Contributed by Chunyu Cai, M.D., Ph.D.
Case 1: Muscle biopsy from a 7 year old boy with clinical phenotype of congenital myopathy
and pathology finding of multiminicore disease; genetic analysis was denied

Classic multiminicore low
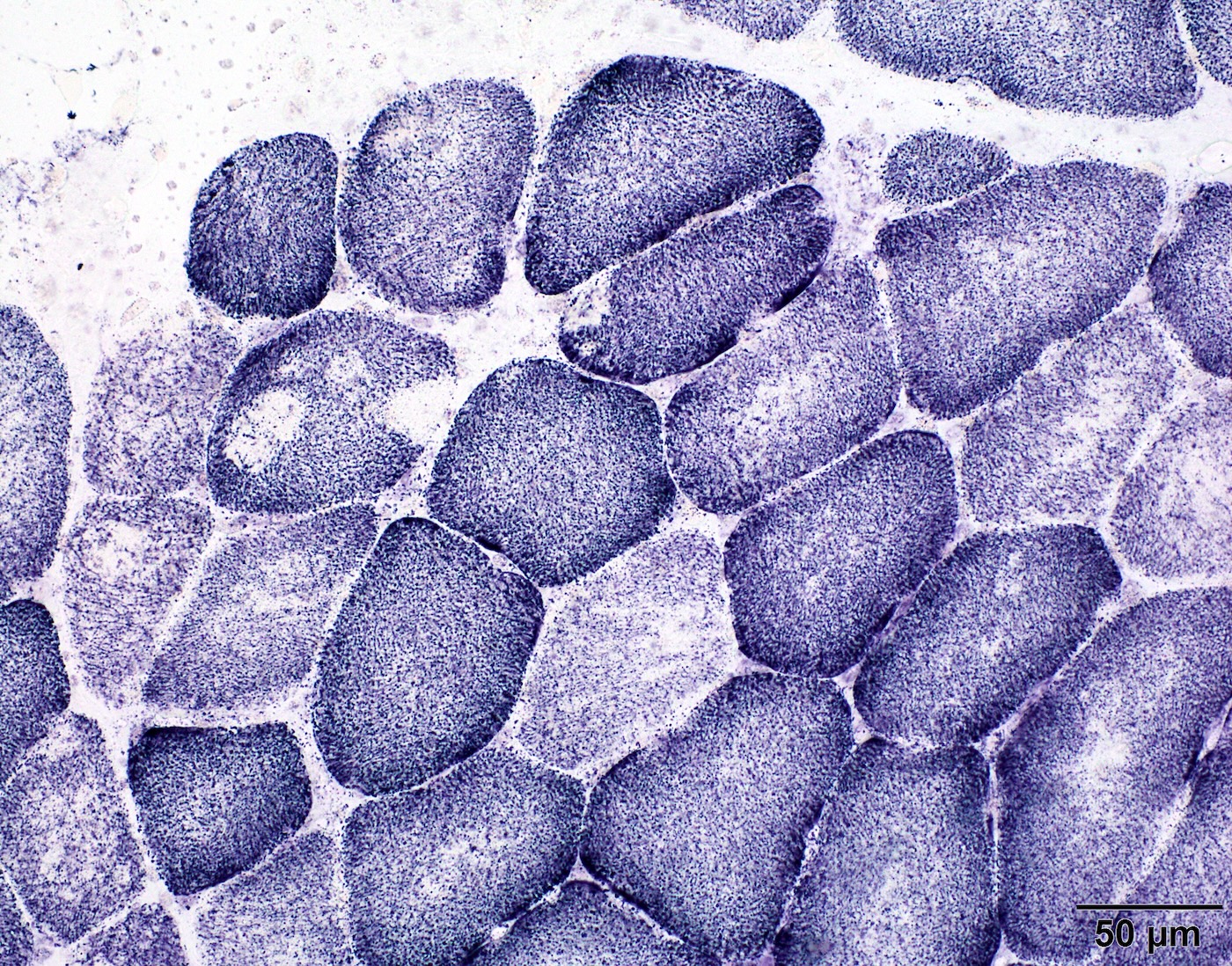
Classic multiminicore high
Case 2: Muscle biopsy from a 12 year old boy with multiple heterozygous RYR1 mutations

Minicore RYR1 FSV and internal nuclei

Minicore RYR1 type I smallness

Multiminicores RYR1 on NADH

Multiminicore RYR1 on COX

Multiminicore RYR1 on toluidine blue
Case 3: Muscle biopsy from a 2 year old girl with 2 heterozygous TTN mutations

Multiminicore TTN low

Multiminicore TTN high

NADH type I predominance

NADH minicores
Positive stains
Negative stains
- Cores are not visualized on ATPase stains
Electron microscopy description
- Small areas of sarcomere disruption spanning only a few sarcomeres and often multifocal in 1 fiber (Mayo Clin Proc 1971;46:666)
Electron microscopy images
Contributed by Chunyu Cai, M.D., Ph.D.
Case 2: Muscle biopsy from a 12 year old boy with multiple heterozygous RYR1 mutations

Multiminicore RYR1

Variable sized cores RYR1

Central core RYR1
Case 3: Muscle biopsy from a 2 year old girl with 2 heterozygous TTN mutations

Small multiminicore TTN low

Small multiminicore TTN high
Molecular / cytogenetics description
- Classical multiminicore disease is described in children with SELENON (selenoprotein N1 / SEPN1) autosomal recessive mutations; the gene is located on chr 1p36 (RSMD1 locus) and is associated with rigid spine syndrome (Am J Hum Genet 2002;71:739)
- Autosomal dominant RYR1 mutations are associated with central core disease and near normal RYR1 protein expression, while recessive RYR1 mutations are associated with multiminicore myopathy and severely depleted RYR1 protein (Brain 2007;130:2024)
- Multiminicore myopathy has also been reported in association with mutations in TTN, MYH7, ACTN2 (Hum Mol Genet 2014;23:980, Neuromuscul Disord 2012;22:1096, Acta Neuropathol 2019;137:501, Ann Clin Transl Neurol 2024;11:629)
Sample pathology report
- Skeletal muscle, left thigh, biopsy:
- Congenital myopathy with multiminicore formation (see comment)
- Comment: The morphologic changes of this biopsy include type I myofiber predominance and multiminicores on oxidative stains. Electron microscopy examination identified multiple small areas of Z disk streaming involving 1 to 3 sarcomeres; rare fibers demonstrate areas of myofibrillar disarray oriented along the longitudinal axis of the fiber. In this case, the biopsy features may best be categorized as multiminicore myopathy. A congenital myopathy genetic panel analysis is recommended to confirm the diagnosis. Precautions should be taken if this patient undergoes general anesthesia because patients with central core disease and some with multiminicore disease are at increased risk for malignant hyperthermia.
Differential diagnosis
- The finding of cores (center core, multiminicore or dusty core) on muscle biopsy is a nonspecific morphological finding and needs to be differentiated from a wide range of myopathies or neurogenic changes; pediatric onset, a background of diffuse type I predominance or diffuse selective type I atrophy and absence of active myofiber necrosis / regeneration are several key features that help distinguish congenital myopathy from other diseases
- Target fibers:
- Classic target fibers contain a single and central area with 3 concentric zones on both mitochondrial stains and electron microscopy (EM), a center pale zone (EM: Z band streaming and sarcomeric disarray), an intermediate dark zone (EM: mild Z band irregularity) and an outer normalizing zone (EM: normal sarcomere pattern); however, less well defined targetoid fibers without concentric zones are also common and difficult to distinguish from cores by morphology
- Differential point is that target / targetoid fibers are typically seen in adult muscle biopsy with a background of denervation atrophy

- Moth eaten fibers:
- Refer to irregular pale areas on mitochondria stains due to any causes that resulted in disruptions of the myofibrillar network
- This term is often used when there are obvious chronic or active myopathic changes in the muscle, such as necrotic fiber, phagocytosis and regenerating myofibers
- As a differential point, the vast majority of congenital myopathies do not lead to frank myofiber necrosis; therefore, acute / chronic myopathic changes are typically lacking on muscle biopsy
- Normal muscle:
- Multiminicores can be very small and sparse and the changes on NADH or other mitochondria stains can be very subtle or easily obscured by staining artifacts on light microscopy; therefore, EM is recommended for all pediatric muscle biopsies even if the muscle appears normal under light microscopy
- Vacuoles:
- Sarcoplasmic vacuoles and cores both appear pale on mitochondrial stains; however, they can be easily distinguished on ATPase stains
- Vacuoles will also be visible on ATPase stains as pale areas, while cores retain their ATPase reactivity and are thus not visible on ATPase stains
Board review style question #1
A 7 year old boy presented with generalized weakness. Per his mother, he had an onset of symptoms at birth but has since been stable. Serum creatine kinase (CK) was normal. A left quadriceps muscle biopsy was performed and appeared unremarkable on H&E stained cryostat sections. However, on NADH stain, the muscle showed changes represented by the image above. What is the most likely diagnosis?
- Congenital muscular dystrophy
- Congenital myasthenic syndrome
- Juvenile dermatomyositis
- Multiminicore myopathy
- Nemaline rod myopathy
Board review style answer #1
D. Multiminicore myopathy. The history is most consistent with a congenital myopathy. The NADH shows multiple irregular pale areas involving both type I (darker fibers) and type II (lighter fibers) muscle fibers, characteristic of multiminicore myopathy. Answer A is incorrect because congenital muscular dystrophy is usually progressive and has high CK. Answer B is incorrect because congenital myasthenic syndrome is not associated with core-like changes in muscle fibers. Answer C is incorrect because juvenile dermatomyositis is characterized by perifascicular myofiber damage and atrophy, which is not present in the image. Answer E is incorrect because nemaline rod myopathy is another type of congenital myopathy characterized by the presence of nemaline rods, best visualized in Gomori trichrome stain.
Comment Here
Reference: Multiminicore myopathy
Comment Here
Reference: Multiminicore myopathy
Board review style question #2
A 7 year old boy presented with generalized weakness. Per his mother, he had an onset of symptoms at birth but has since been stable. Serum creatine kinase (CK) was normal. A left quadriceps muscle biopsy was performed and appeared unremarkable on H&E stained cryostat sections. However, on NADH stain, the muscle showed changes represented by the image above. Subsequent genetic analysis identified a homozygous RYR1 mutation in this patient. Which of the following is indicated in this patient?
- Avoid succinylcholine or inhaled anesthetics
- Cardiac evaluation and monitoring
- Chest CT and respiratory support
- CNS evaluation
Board review style answer #2
A. Avoid succinylcholine or inhaled anesthetics. The history is most consistent with a congenital myopathy. The NADH shows multiple irregular pale areas involving both type I (darker fibers) and type II (lighter fibers) muscle fibers, characteristic of multiminicore myopathy. Genetic analysis identified RYR1 mutation, which is commonly associated with central core myopathy or multiminicore myopathy. Patients with RYR1 mutations are at high risk for malignant hyperthermia and thus need to avoid trigging anesthetics including succinylcholine or inhaled anesthetics. Answers B and D are incorrect because RYR1 mutations typically do not cause cardiomyopathy or CNS abnormality. Answer C is incorrect because respiratory muscle weakness is common in multiminicore myopathy caused by SELENON but not common in RYR1 mutant patients. Therefore, chest CT and respiratory support are not indicated.
Comment Here
Reference: Multiminicore myopathy
Comment Here
Reference: Multiminicore myopathy
