

Muscle & peripheral nerve nontumor
Other peripheral neuropathies
Amyloid neuropathy
Author: Chunyu Cai, M.D., Ph.D.
Editorial Board Members: Meaghan Morris, M.D., Ph.D., Jared T. Ahrendsen, M.D., Ph.D.

Last author update: 5 February 2024
Last staff update: 5 February 2024
Copyright: 2023-2025, PathologyOutlines.com, Inc.
PubMed Search: Amyloid neuropathy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Frozen section description | Frozen section images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Cai C. Amyloid neuropathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/muscleamyloid.html. Accessed January 1st, 2025.
Definition / general
- Peripheral neuropathy due to extracellular deposition of amyloid fibrils within the endoneurium, epineurium or the wall of endo or epineurial vessels
Essential features
- Most common cause of amyloid neuropathy is primary systemic immunoglobulin light chain (AL) amyloidosis in patients with plasma cell dyscrasia
- Most common hereditary amyloidosis is caused by mutations in transthyretin (hATTR)
- Other hereditary forms include mutations in apolipoprotein A1, gelsolin, lysozyme, fibrinogen, amyloid β and cystatin C
- Senile amyloidosis occurs in the elderly population (> 70 years old) and the amyloid deposits are composed of wild type transthyretin
- AA amyloidosis and β2 microglobulin dialysis associated amyloidosis rarely involve peripheral nerves
- Involved peripheral nerve pathology characterized by axonal degeneration preferentially affecting small myelinated and unmyelinated axons in early stages and extending to involve large myelinated axons in later stages
Terminology
- Familial amyloid polyneuropathy (FAP)
- Neuromuscular amyloidosis
ICD coding
Epidemiology
- AL amyloidosis is the most common form of systemic amyloidosis with a prevalence of 2.5 per 100,000 in the U.S.
- 17 - 35% of patients with AL amyloidosis have peripheral neuropathy (Semin Neurol 2019;39:578)
- Hereditary transthyretin amyloidosis polyneuropathy overall incidence rate is 0.87/100,000 but is endemic in some regions such as Portugal, Japan and Sweden (Neuroepidemiology 2018;51:177, Orphanet J Rare Dis 2019;14:34)
Sites
- Systemic disease with multiorgan involvement
- Peripheral nerves, cardiovascular system, kidneys and musculoskeletal system are commonly involved
- In the peripheral nerve system, small myelinated and unmyelinated fiber damage predominates in amyloidosis
- References: J Neurol Sci 1983;59:237, Arch Neurol 1969;20:490
Pathophysiology
- Amyloidosis is caused by extracellular deposition of insoluble aggregates of amyloid fibrils in tissue
- Different types of amyloidosis are classified by the amyloid precursor proteins
- Amyloid deposition leads to direct damage in blood vessels, disruption of the blood nerve barrier, Schwann cell damage, local compression and potentially toxic effects on peripheral nerve axons
- Reference: Neurology 2016;87:2220
Etiology
- Acquired forms arise from excessive or misfolded monoclonal κ or λ light chains in primary systemic amyloid (AL), serum amyloid A protein in secondary amyloidosis (AA) and β2 microglobulin (β2M) in dialysis associated amyloidosis
- Hereditary forms include transthyretin (TTR), apolipoprotein A1, gelsolin, lysozyme, fibrinogen, amyloid β and cystatin C; TTR is by far the most common hereditary form of amyloidosis
- References: Semin Neurol 2019;39:578, J Neurol Sci 1983;59:237
Diagrams / tables
Images hosted on other servers:
hATTR clinical epidemiologic characteristic
Clinical features
- Length dependent sensory neuropathy is the most common clinical presentation of amyloid neuropathy; other presentations include predominant upper limb neuropathy, pure small fiber neuropathy or carpal tunnel syndrome
- Early in the disease course there is selective involvement of distal thermal and pain sensation and later involvement of touch, vibration, joint sensations and motor function; this is due to selective small fiber involvement and later larger fiber involvement
- Autonomic dysfunction is common due to small fiber involvement, including orthostatic hypotension, alternating postprandial diarrhea and constipation, erectile dysfunction and neurogenic bladder dysfunctions
- Besides neurologic symptoms; patients may also have cardiac conduction defects or cardiomyopathy and other organ involvement, such as kidneys, gastrointestinal tract and brain
- References: Semin Neurol 2019;39:578, Arch Neurol 1969;20:490
Diagnosis
- Hereditary forms of amyloid neuropathy may be diagnosed on the grounds of family history, clinical examination / genetic analysis or demonstration of amyloid in tissue and genetic analysis
- Serum free light chain assay (SFLC) is more sensitive than serum and urine protein electrophoresis (SPEP or UPEP) in detection of abnormal immuno light chains and plasma cell dyscrasia
- Liquid chromatography with tandem mass spectrometry (LC MS / MS) is the current gold standard for amyloidosis diagnosis and subtyping on pathology tissue
- Reference: Lancet 2016;387:2641
Laboratory
- AL amyloidosis: SPEP identifies monoclonal gammopathies
- SFLC is more sensitive than SPEP in detecting abnormal immuno light chains and plasma cell dyscrasia
- Reference: Semin Neurol 2019;39:578
Radiology description
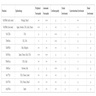
- 99mTc-3,3-diphosphono-1,2-pyrophosphate (99mTc-DPD) whole body scintigraphy with single photon emission computed tomography (SPECT) / CT may help detect AL and ATTR type amyloid in vivo (Medicine (Baltimore) 2020;99:e18905, Amyloid 2023 Aug 2 [Epub ahead of print])
Radiology images
Images hosted on other servers:
99mTc-DPD scan of cardiac ATTR
Prognostic factors
- Prognosis is influenced by the extent of organ damage, especially by cardiac involvement
- Median survival of AL amyloidosis patients presenting with neuropathy is 25 - 35 months
- Usual cause of death is from congestive heart failure or kidney failure
- Reference: Semin Neurol 2019;39:578
Case reports
- 45 year old woman with painful neuropathy, deafness and cardiac pacemaker (Ann Afr Med 2022;21:296)
- 64 year old man with diarrhea, anemia and peripheral neuropathy (J Med Case Rep 2022;16:248)
- 70 year old woman being treated for refractory painful neuropathy (J Palliat Med 2021;24:1579)
Treatment
- Principle of treatment is to reduce the supply of amyloid precursor protein
- Mainstay treatment for AL type amyloidosis is to eliminate abnormal plasma cells or lymphocytes by chemotherapy and autologous peripheral blood stem cell transplantation (Ann Intern Med 2004;140:85)
- Treatment for hATTR disease
- Liver transplant: eliminate the source of mutant TTR
- Tafamidis, diflunisal: TTR tetramer stabilizers
- TTR gene silencers: patisiran (siRNA) and inotersen (antisense oligonucleotide) decrease abnormal TTR production in liver
- Reference: Curr Neuropharmacol 2023;21:471
Frozen section description
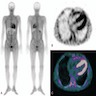
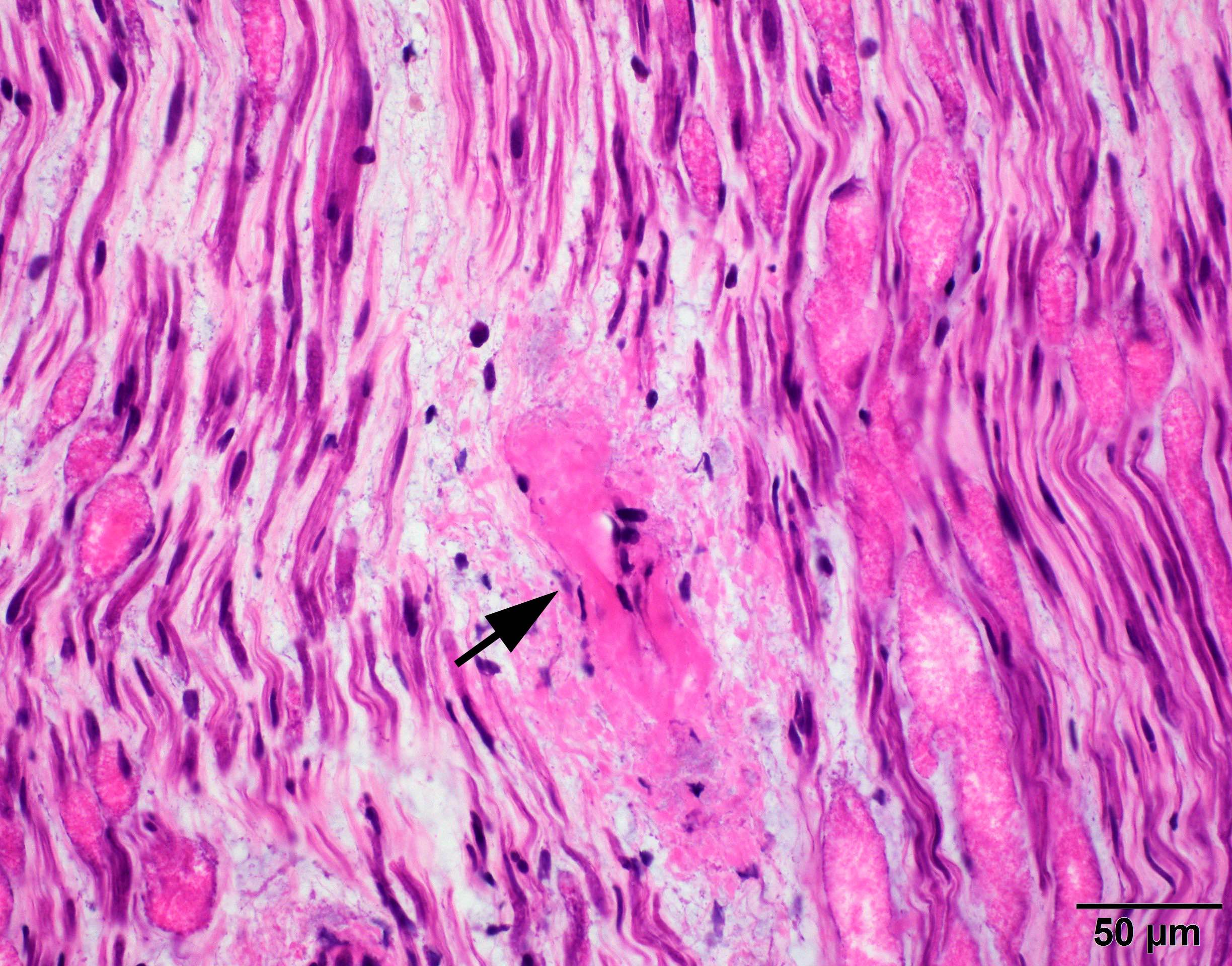
- Amorphous pink material, usually in association with irregular expansion of blood vessel wall
Frozen section images
Contributed by Chunyu Cai, M.D., Ph.D.
Amyloid in peripheral nerve
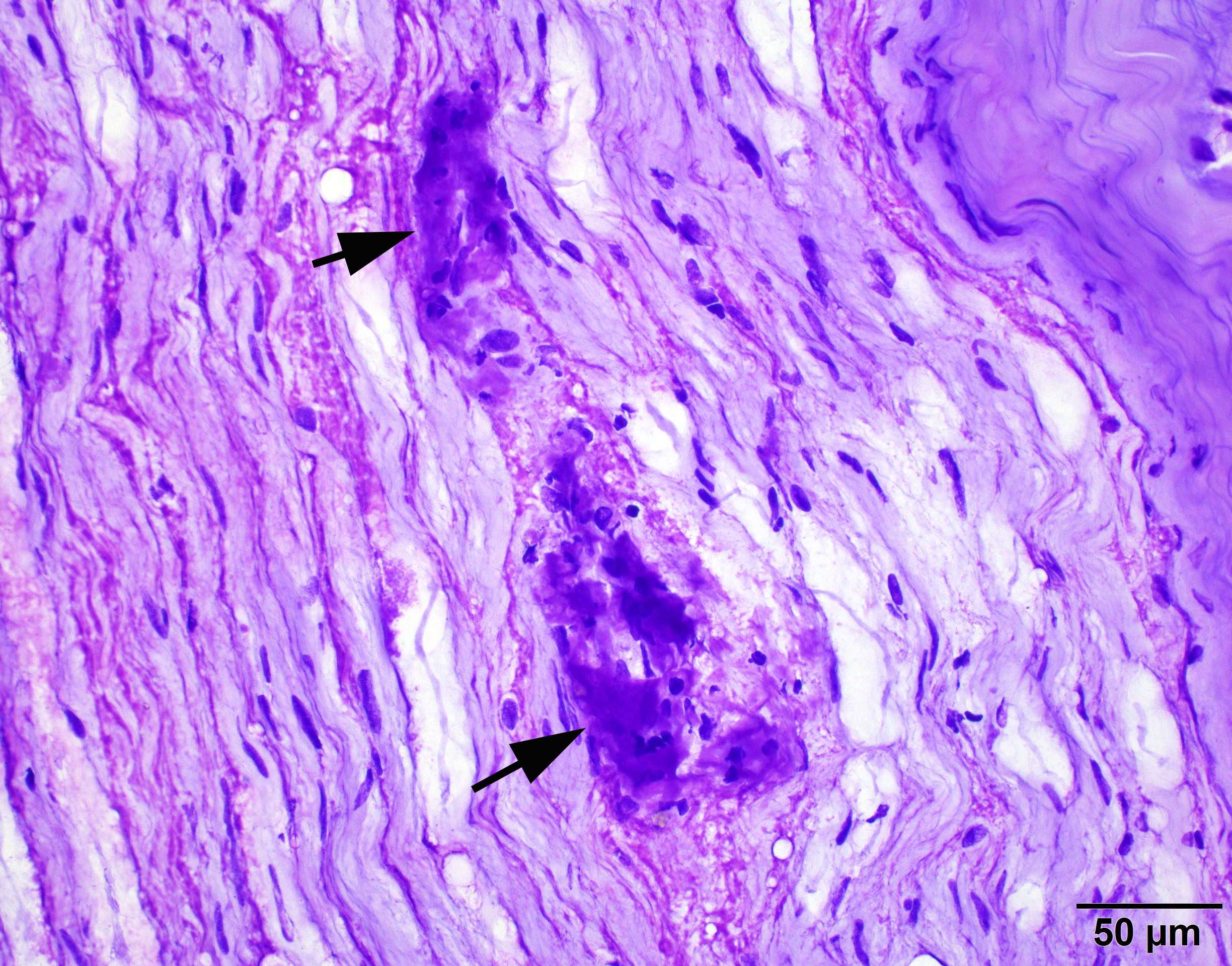
Crystal violet stain amyloid
Microscopic (histologic) description
- Axonal degeneration preferentially involving small myelinated and unmyelinated axons
- Amyloid deposits can be found in vessel walls or connective tissue in the epineurium, perineurium and endoneurium
- Transthyretin IHC stain highlights both mutant and wild type forms of TTR amyloid deposits
- References: Blood 2012;119:488, Arch Neurol 1969;20:490
Microscopic (histologic) images
Contributed by Chunyu Cai, M.D., Ph.D.
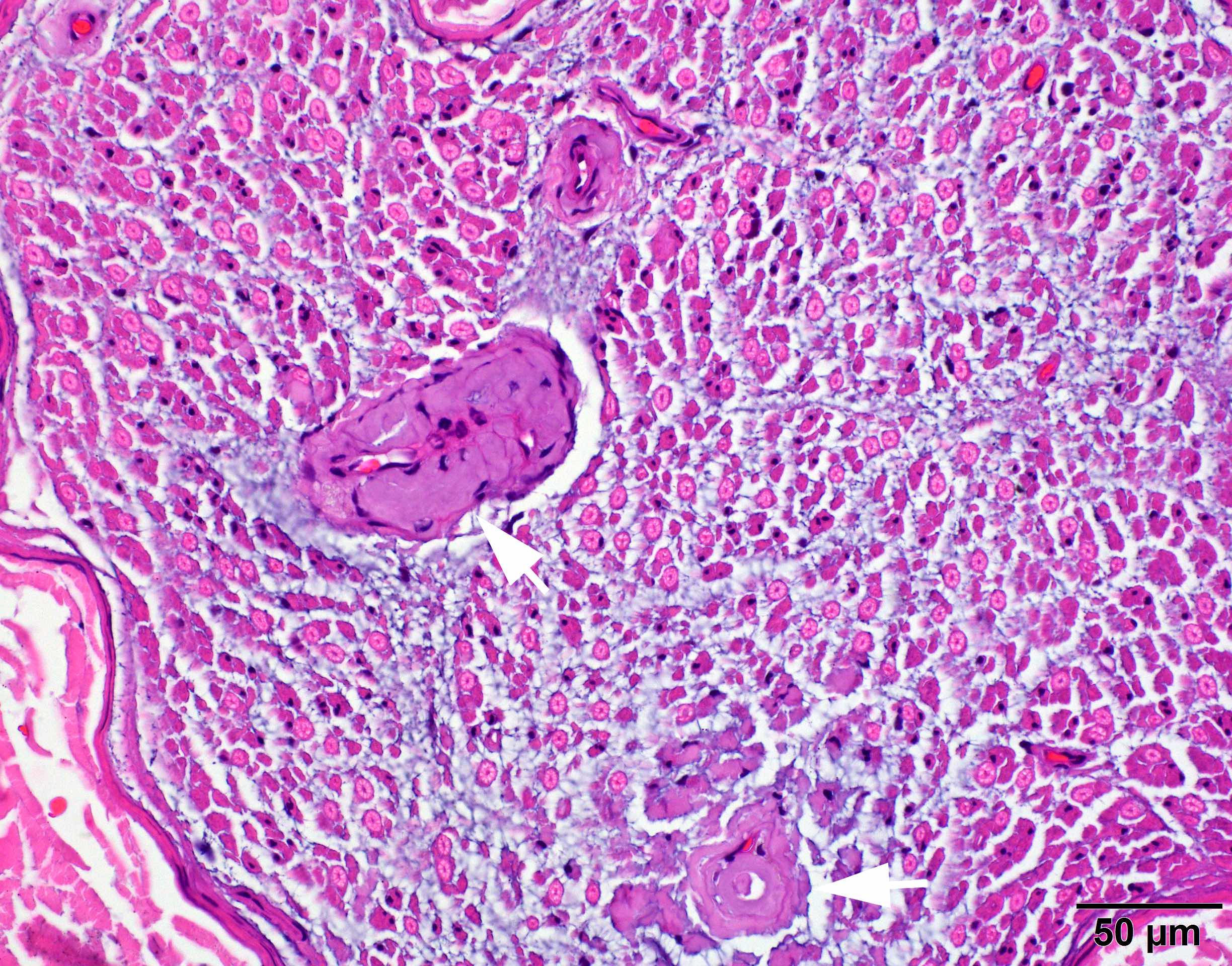
Early amyloid neuropathy

Nerve edema
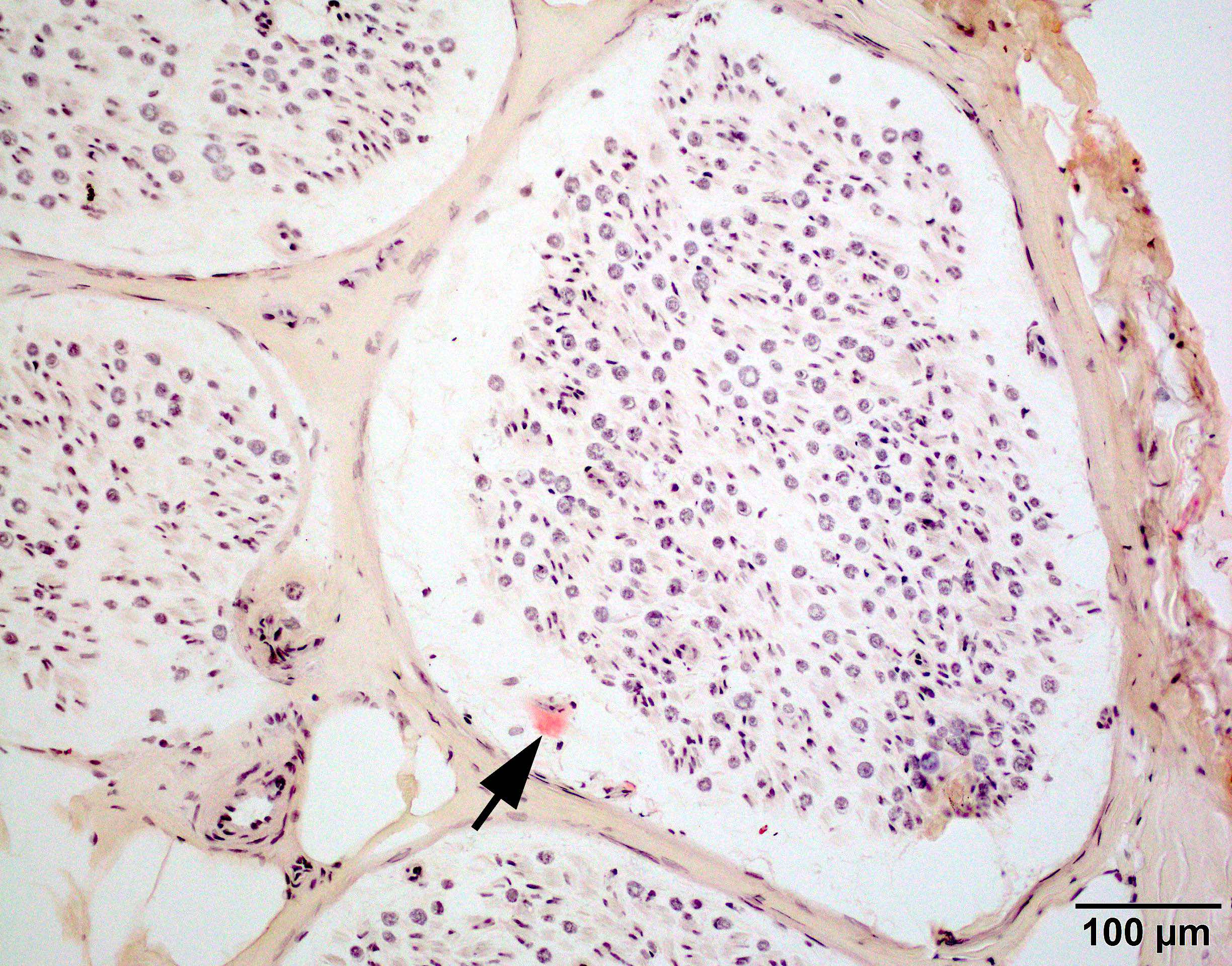
Congo red amyloid deposit
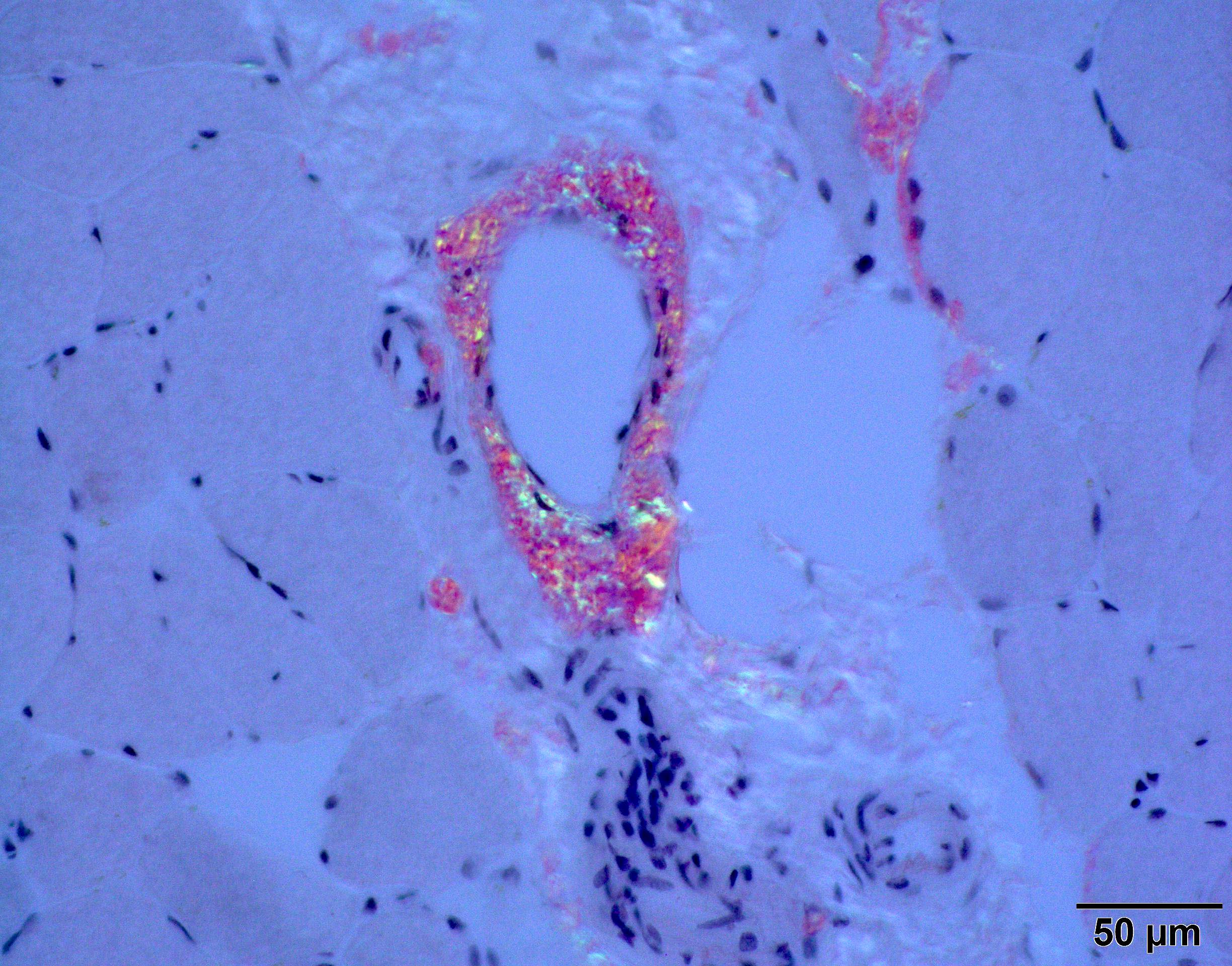
Congo red polarized
Loss of small myelinated axons
Late amyloid neuropathy
Nerve edema
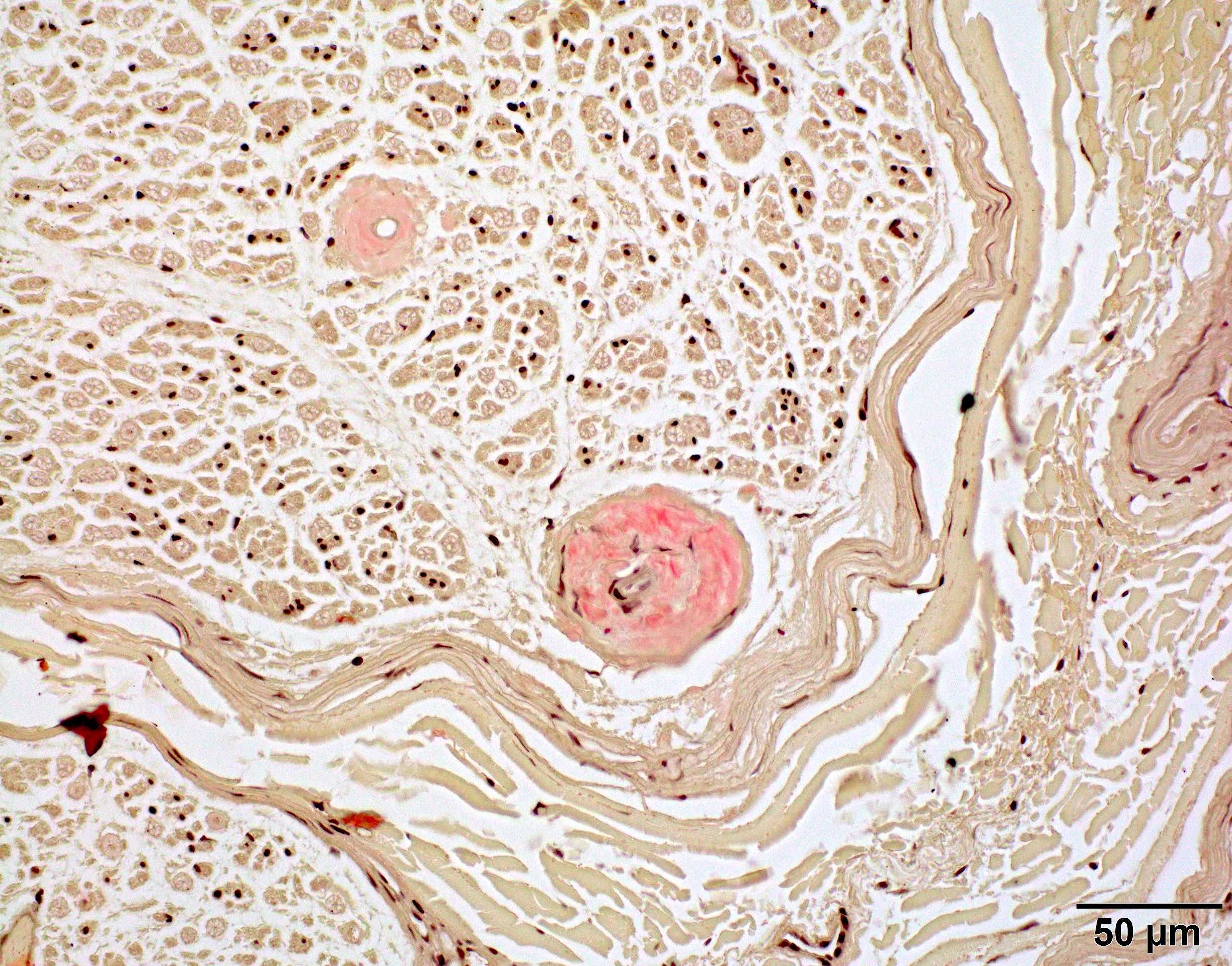
Congo red amyloid deposit
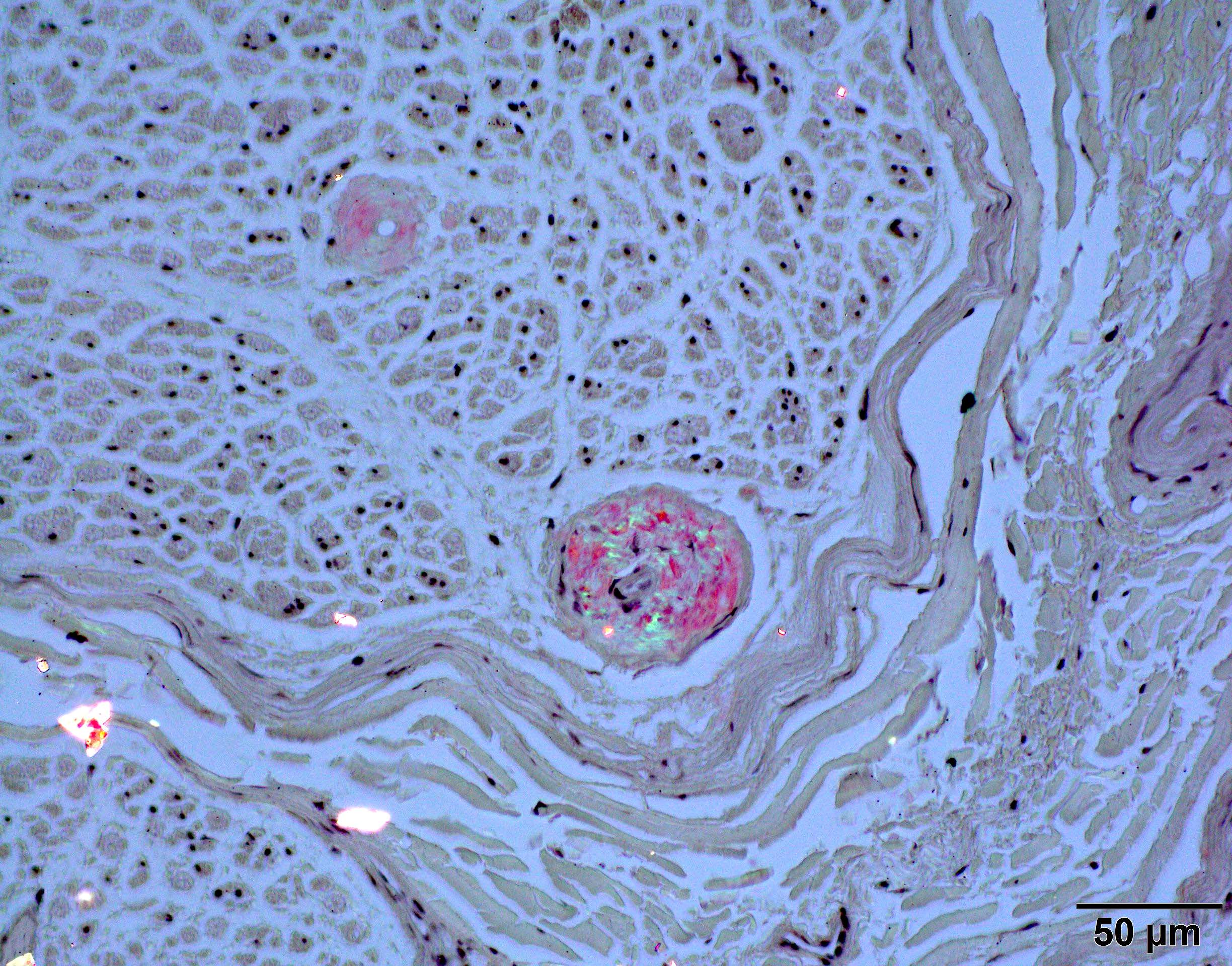
Congo red polarized
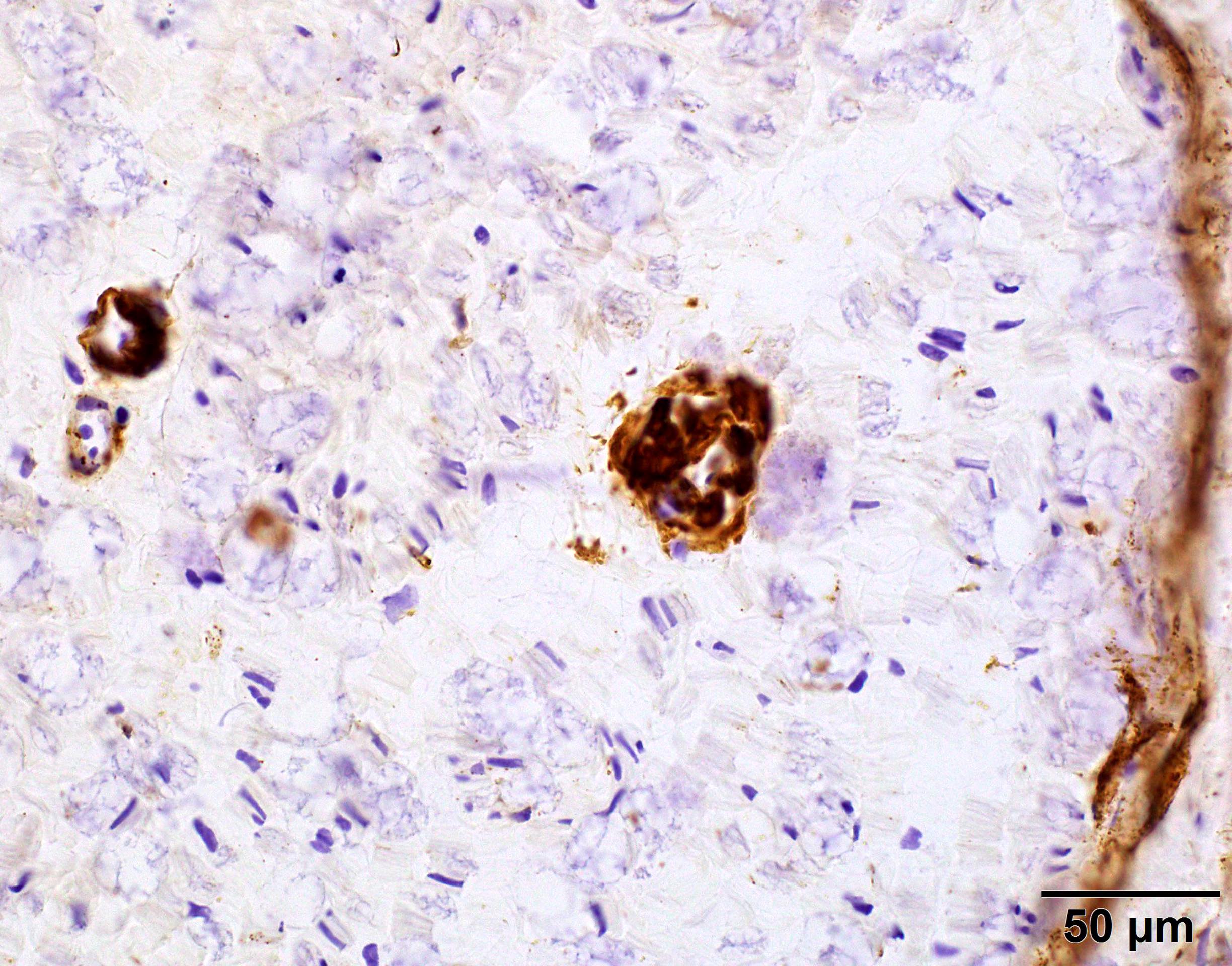
C5b9 amyloid deposit
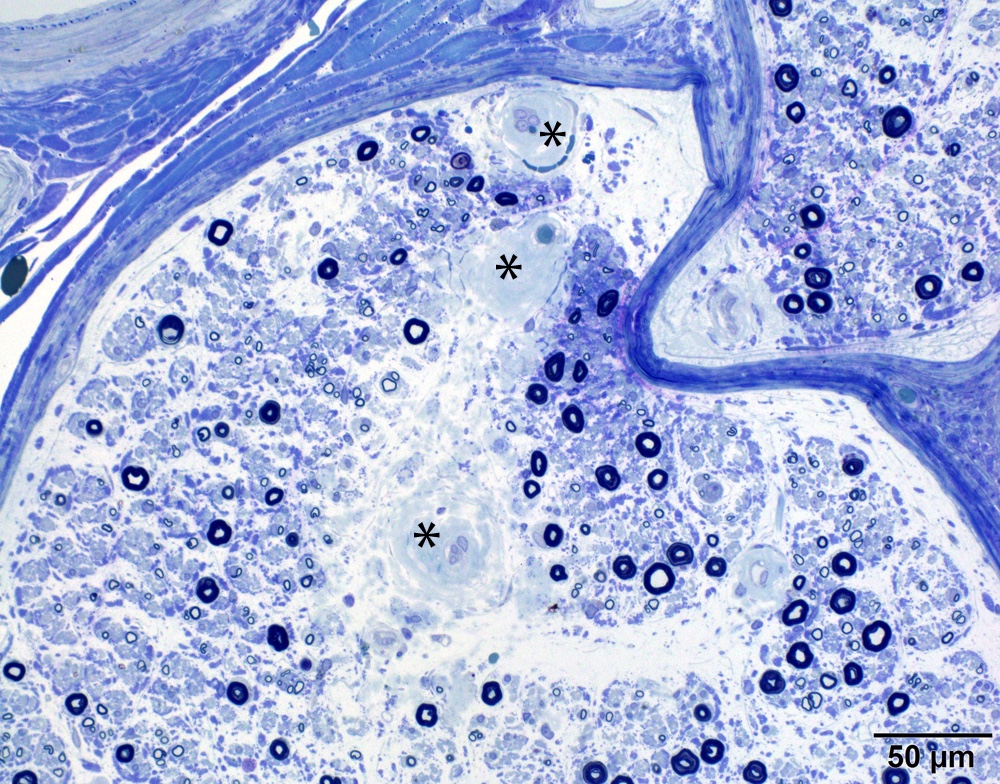
Small and large axon loss
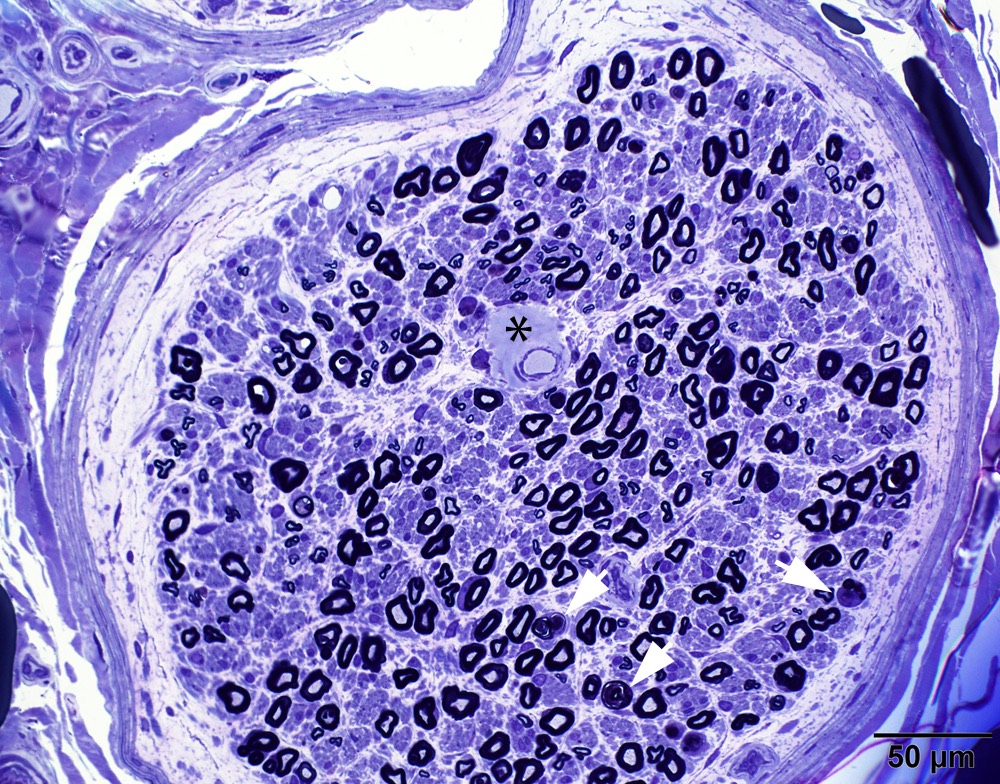
Hereditary transthyretin amyloid neuropathy
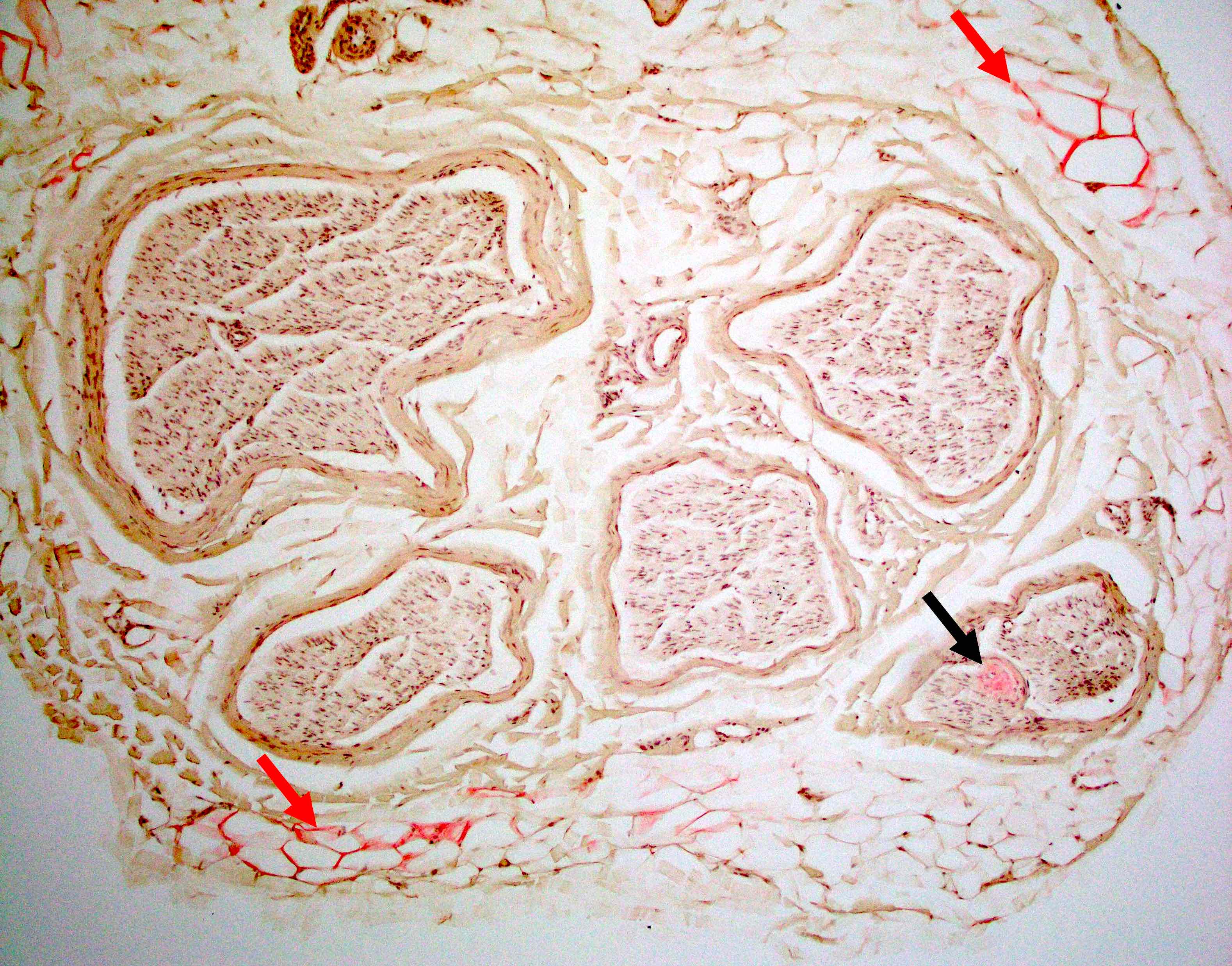
Nerve Congo red
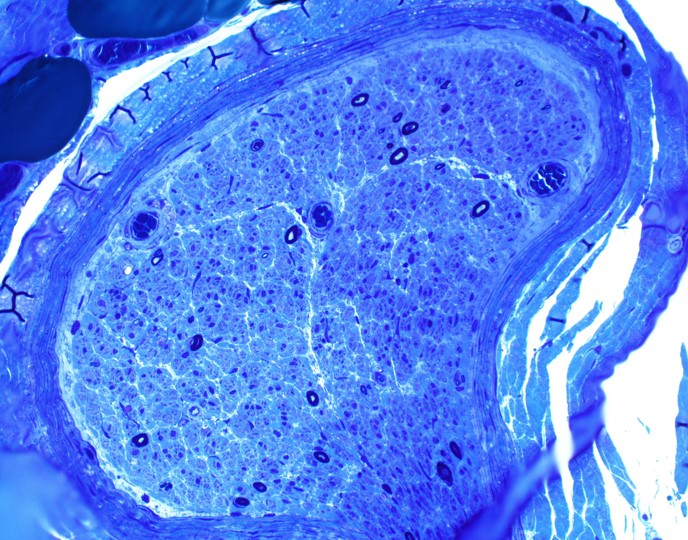
Nerve plastic section
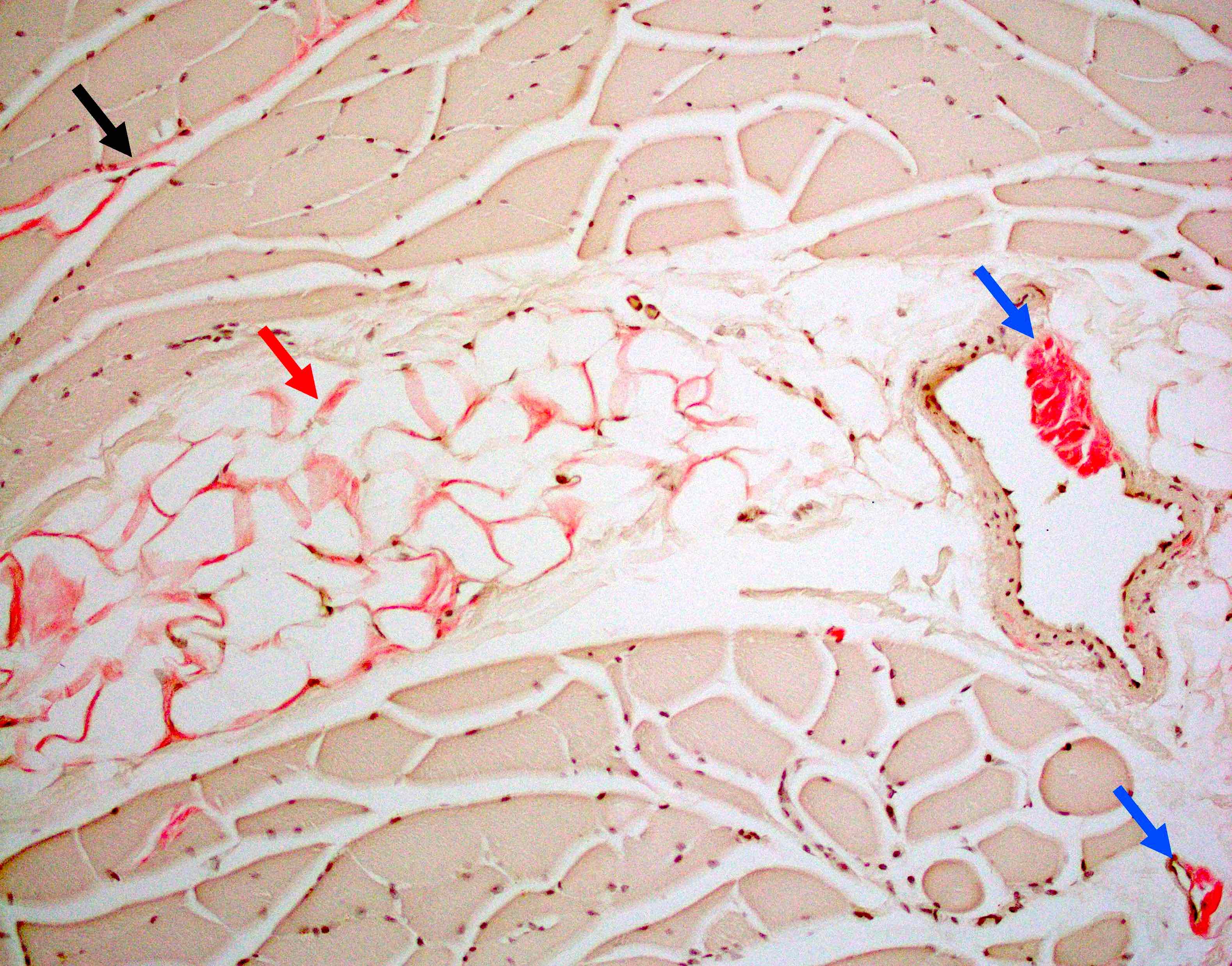
Muscle Congo red
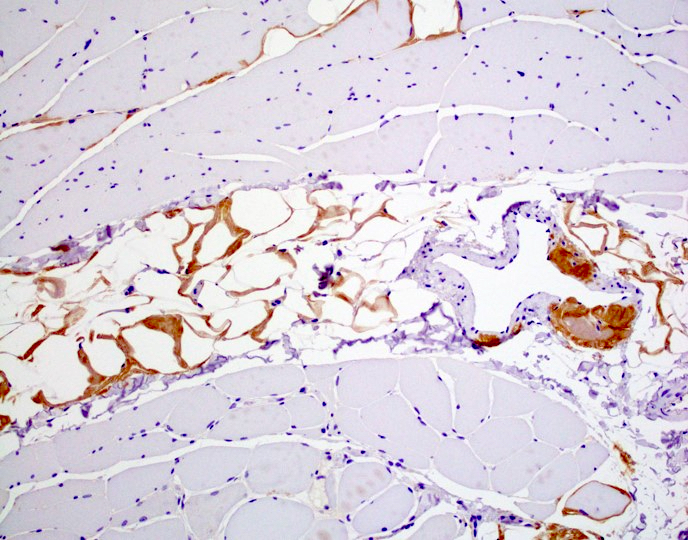
Muscle TTR
AL amyloid neuropathy
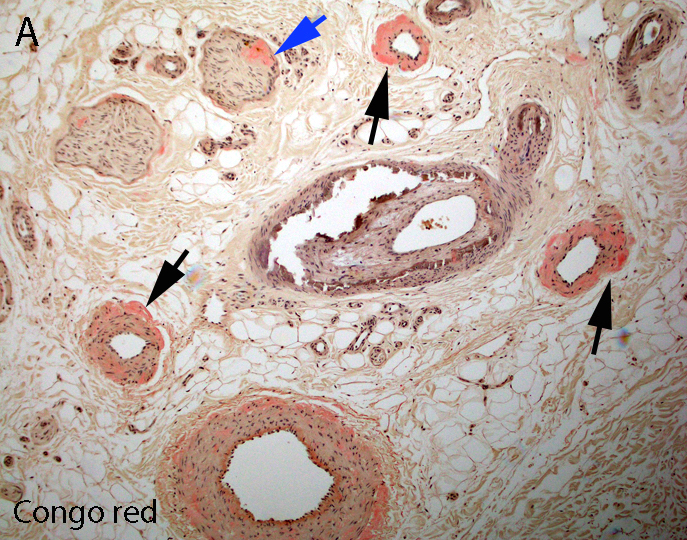
Congo red
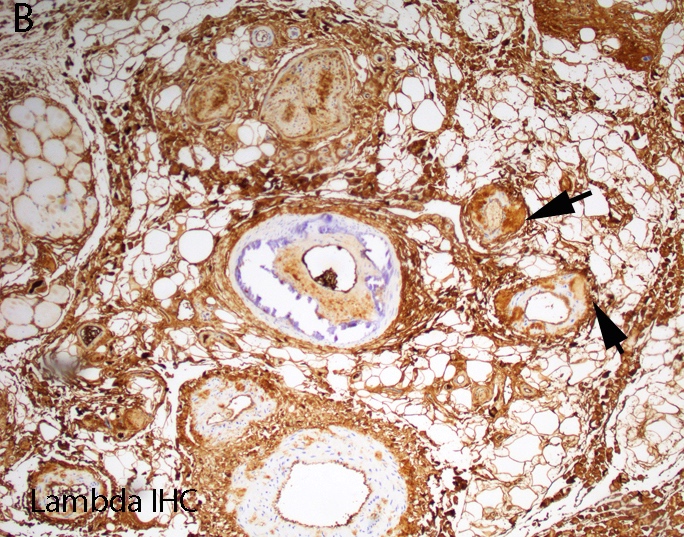
Lambda IHC
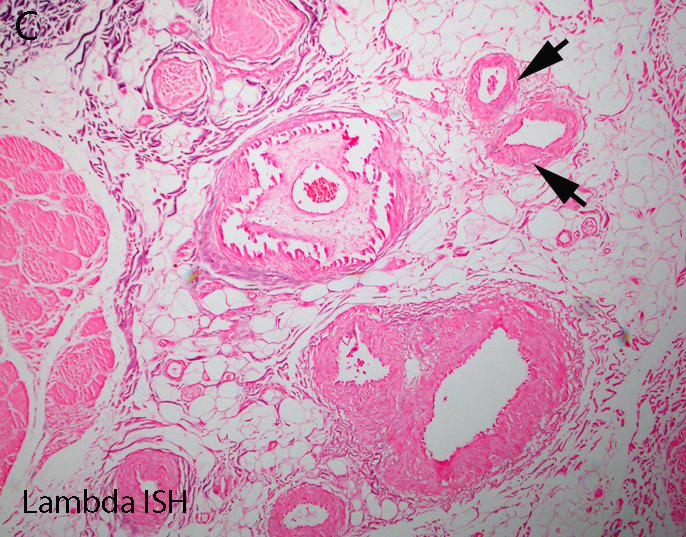
Lambda in situ hybridization
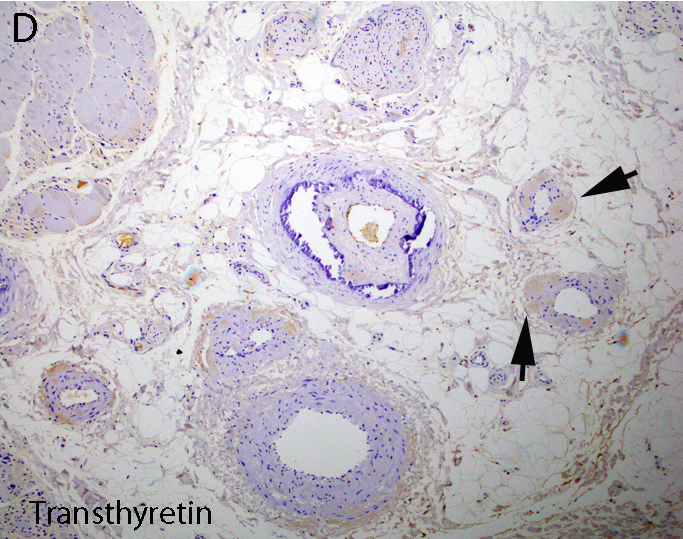
Transthyretin IHC
Positive stains
- General amyloid markers
- Congo red
- Thioflavin T or S
- Terminal complement complex (C5b9)
- Crystal violet
- Subtype specific amyloid markers
- Transthyretin (Blood 2012;119:488)
Electron microscopy description
- Nerve damage (J Neurol Sci 2021:421:117305, Neurology 2016;87:2220)
- Prominent loss of small myelinated and unmyelinated axons; relatively preserved large myelinated axons
- Atrophy of Schwann cells
- Damage of endothelial cells and disrupted blood nerve barrier
- Amyloid fibrils
- Amyloid deposits are composed of haphazardly arranged, nonbranching, nonperiodic fibrils with a diameter of 7 - 15 nm (Ultrastruct Pathol 2020;44:325)
Electron microscopy images
Contributed by Chunyu Cai, M.D., Ph.D.
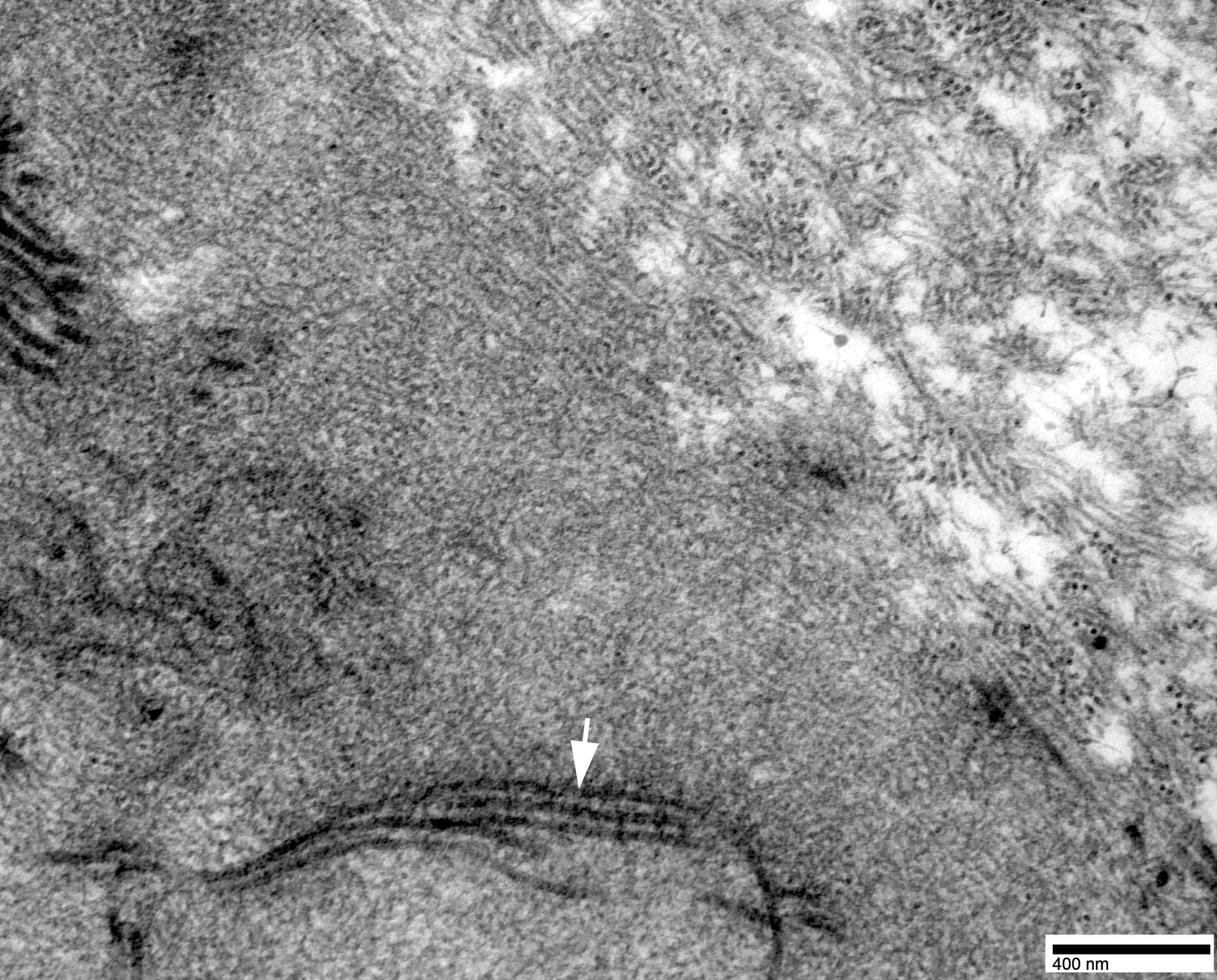
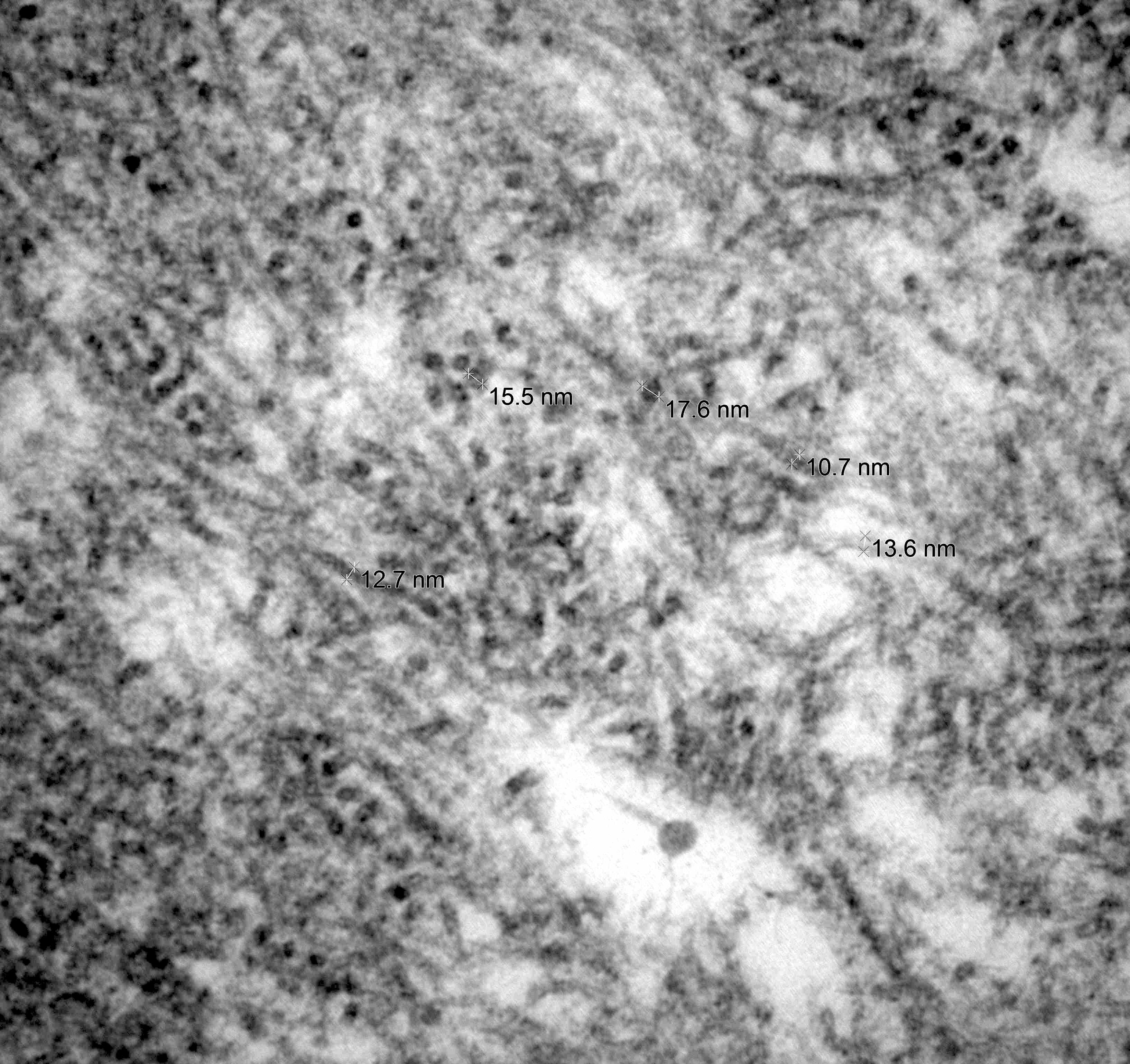
Amyloid deposit
Molecular / cytogenetics description
- TTR amyloidosis is by far the most common cause for hereditary amyloidosis
- Of the > 140 mutations reported in the TTR gene, the p.Val50Met mutation is by far the most common (Curr Neuropharmacol 2023;21:471)
Videos
The principles for pathology confirmation of amyloidosis and significance of subtyping
Sample pathology report
- Peripheral nerve, right sural, biopsy:
- Amyloid neuropathy (see comment)
- Comment: The nerve shows amyloid deposition in multiple endoneurial blood vessels. There is severe loss of small myelinated and unmyelinated axons but relatively preserved large myelinated axons, compatible with amyloid neuropathy. Transthyretin immunostain is negative in the amyloid deposits. Given the patient’s history of Waldenström lymphoma, immune light chain associated amyloidosis (AL) is suspected. Liquid chromatography mass spectrometry (LC MS) is recommended for definitive subtyping of amyloid.
Differential diagnosis
- Positive Congo red stain with green-yellow birefringence under polarized light is considered pathognomonic for amyloidosis
- Main amyloid subtypes in peripheral nerve include AL, hereditary and senile types
- Precise amyloid subtyping relies on LC MS / MS
- Absence of amyloid on nerve biopsy:
- Since amyloid deposition is patchy, absence of amyloid on nerve biopsy does not completely exclude the possibility of amyloidosis
- Paraproteinemic neuropathy:
- Diseases with circulating paraprotein such as Waldenström macroglobulinemia and POEMS syndrome may cause peripheral neuropathy without amyloid deposition
- Other small fiber neuropathies:
- Other peripheral neuropathies with predominant involvement of small myelinated or unmyelinated fibers and relatively preserved large myelinated axons include diabetic neuropathy, alcoholic neuropathy, Fabry disease, Tangier disease, hereditary sensory and autonomic neuropathies (Bilbao: Biopsy Diagnosis of Peripheral Neuropathy, 2nd Edition, 2015)
Board review style question #1
Congo red
Transthyretin IHC
A 70 year old man with history of progressive weakness and sensory loss for 4 years has reported several falls. No bulbar or respiratory symptoms are reported. The symptoms progressed despite receiving intravenous immunoglobulin treatment. Electrophysiologic studies demonstrated severe sensory motor polyneuropathy with demyelinating features and evidence of active denervation in proximal and distal limb muscles. A right sural nerve and right quadriceps muscle biopsy demonstrated the findings above. Which of the following is the best choice to confirm the diagnosis?
- Cardiac ultrasound
- Electron microscopy of the nerve
- Liquid chromatography with tandem mass spectrometry (LC MS / MS)
- Serum protein electrophoresis (SPEP)
- Whole body scintigraphy with single photon emission computed tomography (SPECT) / CT
Board review style answer #1
C. Liquid chromatography with tandem mass spectrometry (LC MS / MS). The pathology images demonstrate amyloid deposition in muscle and nerve. The amyloid deposits are strongly positive for transthyretin, which suggests hereditary transthyretin amyloidosis (hATTR) or senile amyloidosis. The next step is to determine the amyloid subtype and treat accordingly. LC MS / MS is the current gold standard for subtyping of amyloid on pathology specimens.
Answer D is incorrect because SPEP will be useful in identifying serum paraproteins for AL type amyloidosis but not in cases of hATTR or senile amyloidosis. Answer E is incorrect because whole body scintigraphy with SPECT / CT is useful in assessing the extent of amyloid deposition in the whole body but has a limited role in differentiating amyloid subtypes. Answer B is incorrect because electron microscopy of the nerve can help assess the extent of nerve damage and visualize the amyloid fibrils but cannot determine the type of amyloid by ultrastructural morphology. Answer A is necessary since the heart is often involved in amyloidosis and the extent of cardiac involvement is an important prognostic factor; however, it is not the correct answer since it cannot determine the amyloid subtype.
Comment Here
Reference: Amyloid neuropathy
Answer D is incorrect because SPEP will be useful in identifying serum paraproteins for AL type amyloidosis but not in cases of hATTR or senile amyloidosis. Answer E is incorrect because whole body scintigraphy with SPECT / CT is useful in assessing the extent of amyloid deposition in the whole body but has a limited role in differentiating amyloid subtypes. Answer B is incorrect because electron microscopy of the nerve can help assess the extent of nerve damage and visualize the amyloid fibrils but cannot determine the type of amyloid by ultrastructural morphology. Answer A is necessary since the heart is often involved in amyloidosis and the extent of cardiac involvement is an important prognostic factor; however, it is not the correct answer since it cannot determine the amyloid subtype.
Comment Here
Reference: Amyloid neuropathy
Board review style question #2
What type of amyloid is the most common cause of amyloid neuropathy in the U.S.?
- AA
- Aβ
- Aβ2 microglobuli
- AL
- ATTR
Board review style answer #2
D. AL is the most common cause of amyloid neuropathy (> 50% of patients with amyloidosis) (Lancet 2016;387:2641). Answer E is incorrect because ATTR is the most common hereditary form of amyloidosis but only makes up < 10% of all patients with amyloidosis (Lancet 2016;387:2641). Answer A is incorrect as AA amyloidosis usually does not involve peripheral nerves. Answer B is incorrect as Aβ is usually restricted in the central nervous system and does not involve peripheral nerves. Answer C is incorrect as Aβ2 microglobuli is usually associated with dialysis and usually doesn't involve peripheral nerves.
Comment Here
Reference: Amyloid neuropathy
Comment Here
Reference: Amyloid neuropathy

