
Muscle & peripheral nerve nontumor
Muscular dystrophies
Oculopharyngeal muscular dystrophy
Author: Chunyu Cai, M.D., Ph.D.
Editorial Board Members: Jared T. Ahrendsen, M.D., Ph.D., P.J. Cimino, M.D., Ph.D.

Last author update: 27 July 2023
Last staff update: 27 July 2023
Copyright: 2022-2025, PathologyOutlines.com, Inc.
PubMed search: Oculopharyngeal muscular dystrophy

Table of Contents
Definition / general | Essential features | ICD coding | Epidemiology | Sites | Pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Cai C. Oculopharyngeal muscular dystrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/muscleOCPmusculardystrophy.html. Accessed March 31st, 2025.
Definition / general
- Prototypical oculopharyngeal muscular dystrophy (OPMD) is a late adult onset myopathy clinically characterized by progressive ptosis, dysphasia and proximal weakness; also genetically characterized by GCN trinucleotide repeat expansion in exon 1 of the PABPN1 gene on chromosome 14q11
- Broader definition of OPMD includes a group of diseases that share the common features of ptosis, dysphagia and multinucleotide repeat expansion involving several different genes (see Table 1)
- This topic will focus on OPMD PABPN1
Essential features
- Prototypical OPMD is caused by GCN repeat expansion in exon 1 of the PABPN1 gene on chromosome 14q11 and is usually late adult onset; this is the most common type of OPMD and the only OPMD that can currently be confirmed by commercial genetic testing
- First described by E. W. Taylor in 1915 (J Nerv Ment Dis 1915;42:129)
- Oculopharyngeal muscular dystrophy was first termed in 1962 (N Engl J Med 1962;267:1267)
- Cause of OPMD was identified in 1998 as GCN trinucleotide repeat expansions in the PABPN1 gene on chromosome 14q11 (Nat Genet 1998;18:164)
- Muscle pathology was myopathy with rimmed vacuoles (Neuromuscul Disord 1997;7:S63)
- Diagnostic electron microscopy feature was intranuclear filamentous inclusions composed of short (0.1 μm), unbranched, 8.5 nm wide filaments, present in 3 - 6.5% of myofiber nuclei (Can J Neurol Sci 1989;16:446, Neurology 1996;46:1324)
ICD coding
- ICD-10: G71.0 - muscular dystrophy
Epidemiology
- Largest population of patients with OPMD are those with French Canadian ancestry who reside in Quebec (Surg Clin North Am 1983;63:825)
- Other clusters of this disease are found in Bukhara Jewish immigrants in Israel and individuals of Hispanic descent in the state of New Mexico (Neuromuscul Disord 1997;7:S38, JAMA 2001;286:2437)
- Mean age of presentation is 48; younger age at onset (< 30 years) can be observed in longer GCN expansion or in individuals who are compound heterozygous or homozygous for the GCN expansion (GeneReviews: Oculopharyngeal Muscular Dystrophy [Accessed 28 June 2023])
Sites
- Levator palpebrae superioris and pharyngeal muscles are most affected
- Proximal muscle groups can also be affected in late stage of disease
- Reference: N Engl J Med 1962;267:1267
Pathophysiology
- PABPN1 is an RNA binding nuclear protein that regulates the length of the mRNA poly(A) tail, the mRNA export from the nucleus and the processing of long noncoding RNA (lncRNA)
- GCN repeat expansion in the exon 1 of the PABPN1 gene leads to a short alanine expansion in the N terminal of PABPN1 protein, which results in nuclear accumulation of PABPN1 and other proteins and RNAs that are resistant to degradation
- Misregulation of PABPN1 dependent lncRNA affects downstream gene expression
- Reference: FEBS J 2013;280:4230
Diagrams / tables
Table 1: Genetic diseases with shared features of progressive ptosis and dysphagia
| Gene | Site | Heritance | Genetic defect | Clinical | Reference |
| PABPN1 | 14q11.2 | AD | GCN repeats | Late onset OPMD | Acta Neuropathol 2022;144:1157 |
| HNRNPA2B1 | 7p15.2 | AD | Frameshift | Early onset OPMD | Nat Commun 2022;13:2306 |
| LRP12 | 8q22.3 | AD | CGG repeats | Oculopharyngeal distal myopathy (OPDM) type 1 | JAMA Neurol 2021;78:853 |
| GIPC1 | 19p13.12 | AD | CGG repeats | OPDM type 2 | Am J Hum Genet 2020;106:793 |
| NOTCH2NLC | 1q21.2 | AD | CGG repeats | OPDM type 3 | Nat Genet 2019;51:1222 |
| RILPL1 | 12q27.31 | AD | CGG repeats | OPDM type 4 | Am J Hum Genet 2022;109:533 |
| NUTM2B::AS1 | 10q22.3 | AD | CGG repeats | Oculopharyngeal myopathy with leukodystrophy (OPML) | Nat Genet 2019;51:1222 |
Clinical features
- OPMD (PABPN1) usually presents insidiously in the 50s - 60s (Neurology 2017;88:359)
- Progressive, bilateral, myogenic ptosis
- Dysphagia, initially solid foods, progressing to difficulty swallowing liquids
- Aspiration pneumonia and malnutrition are the leading cause of death but usually do not shorten life expectancy because these tend to occur late in the disease
- Proximal limb weakness of varying severity, with some patients suffering from profound limb girdle weakness
- OPMD patients are often misdiagnosed as having myasthenia gravis
Diagnosis
- Definitive diagnosis is by genetic testing on blood showing expansion of the GCN trinucleotide repeat in the first exon of PABPN1 gene, usually by PCR (Neurology 2017;88:359)
- Normal individual has ≤ 10 GCN repeats
- 90% of OPMD patients had GCN trinucleotide repeat expansion of 11 to 18 repeats in the first exon of PABPN1
- 10% of OPMD patients had compound heterozygous or homozygous GCN trinucleotide repeat expansions
Laboratory
- Serum creatine kinase (CK) can be normal, mildly increased or > 1,000 IU/L, depending on number of GCN repeats (Neurology 2017;88:359)
Radiology description
- Muscle MRI: fatty involvement of the combination of tongue, soleus and adductor magnus muscles are highly suggestive of OPMD (J Neurol Neurosurg Psychiatry 2019;90:576)
Prognostic factors
- OPMD (PABPN1) patients with longer repeats present earlier and have higher CK (Neurology 2017;88:359)
- 11 - 12 GCN repeats: mean age of diagnosis is in the 70s, CK normal
- 13 - 15 GCN repeats: mean age is in the 60s, CK of 100 - 500
- 16 - 17 GCN repeats: mean age is in the 50s, CK of 500 - 1,000
- Patients with both alleles affected may present at even earlier ages (30s - 40s)
Case reports
- 53 year old woman with fatal choking (Medicine (Baltimore) 2018;97:e12935)
- 75 year old woman with OPMD and dementia (BMJ Case Rep 2019;12:e230521)
- 80 year old man with progressive dysphagia, frequent aspiration and change of voice (Ann Gastroenterol 2015;28:291)
Treatment
- Currently no cure; treatment is mostly supportive
- Ptosis repair / improve levator muscle function (Ann Plast Surg 2002;49:419, Ophthalmic Plast Reconstr Surg 2009;25:103)
- Cricopharyngeal muscle botulinum injection / dilatation / myotomy for dysphagia (J Clin Med 2021;10:1375)
- PABPN1 gene therapy is at early clinical trial stage (Nat Commun 2017;8:14848)
- First patient enrolled in a phase 1b / 2a clinical trial (BB-301, Benitec Biopharma) in January 2023 (Benitec Biopharma: Benitec Biopharma Receives FDA Clearance of the IND for BB-301 for the Treatment of Oculopharyngeal Muscular Dystrophy [Accessed 3 July 2023])
Microscopic (histologic) description
- All OPMD and OPDM subtypes share muscle pathology of myopathy with rare rimmed vacuoles (see references in Table 1)
- Significant fiber size variation
- May have fatty replacement and interstitial fibrosis
- Infrequent red rimmed vacuoles on Gomori trichrome and acid phosphatase
- Usually no inflammation or MHC1 myofiber upregulation
- Usually no significant increase in COX deficient fibers
- One of the most useful distinguishing feature is intranuclear filamentous inclusions identifiable by electron microscopy (see Electron microscopy description) or p62 stain (see Positive stains)
- References: Acta Neuropathol Commun 2022;10:176, Neurology 1996;46:1324
Microscopic (histologic) images
Contributed by Chunyu Cai, M.D., Ph.D.

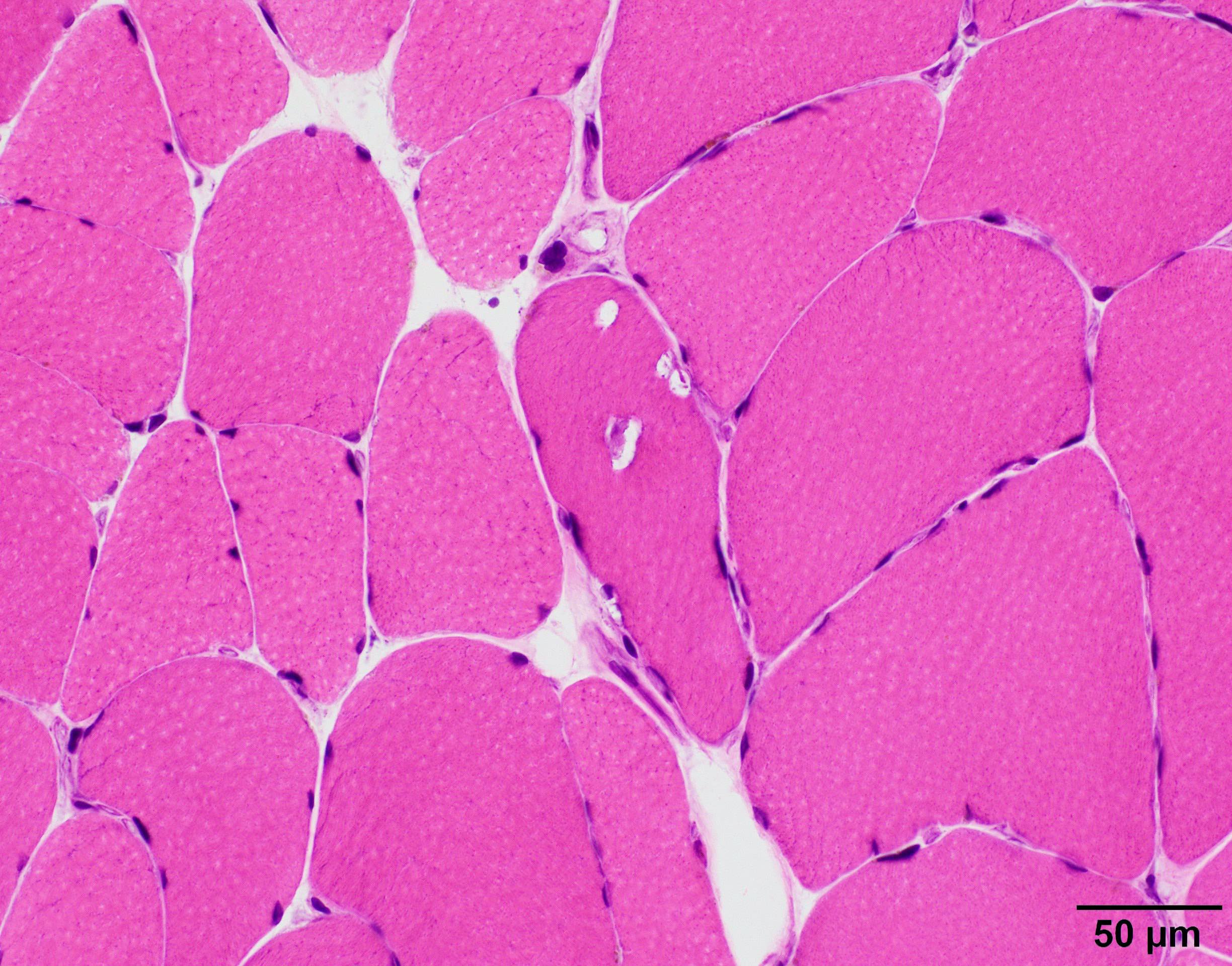
Myopathy with rimmed vacuoles

Gomori trichrome
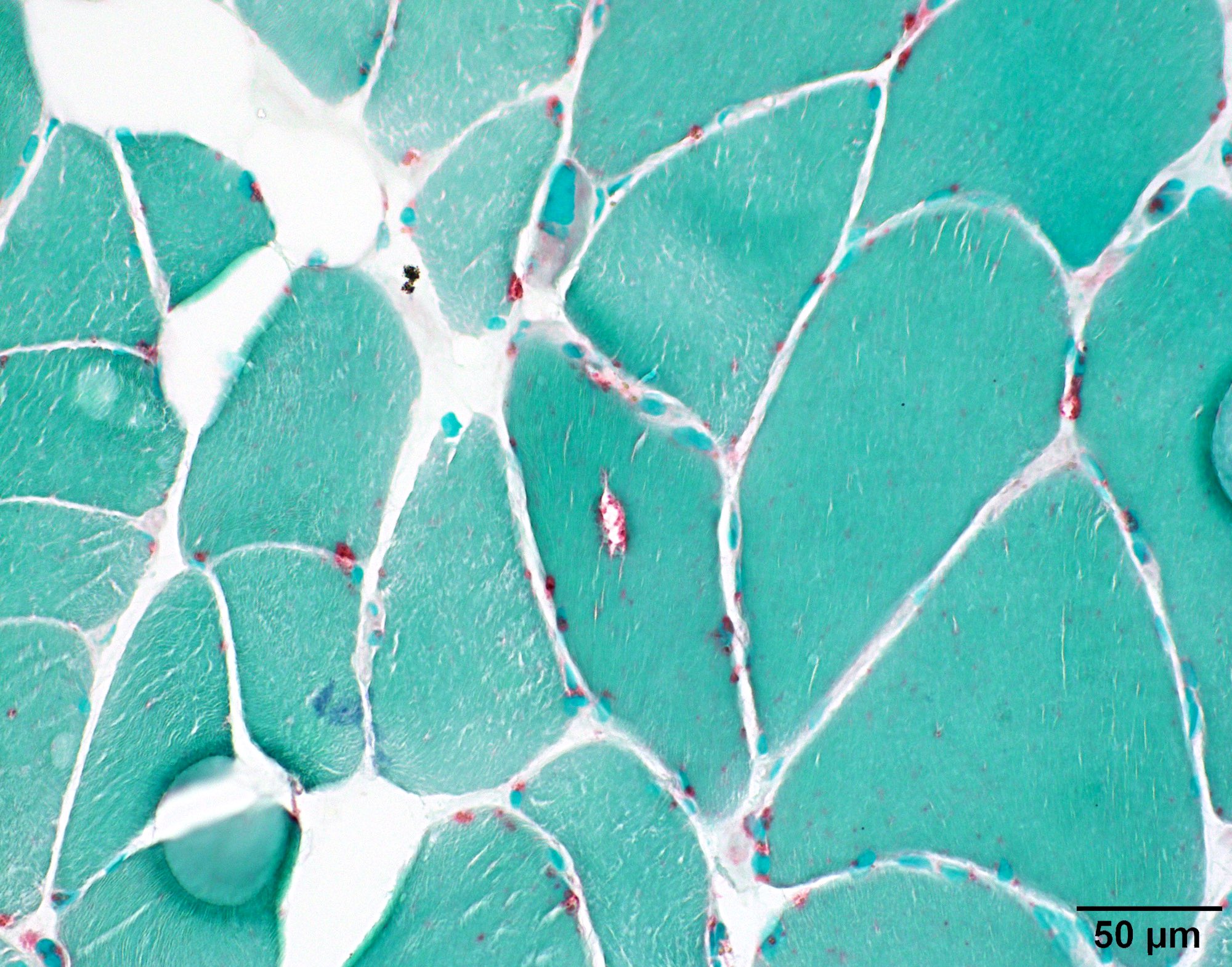
Acid phosphatase
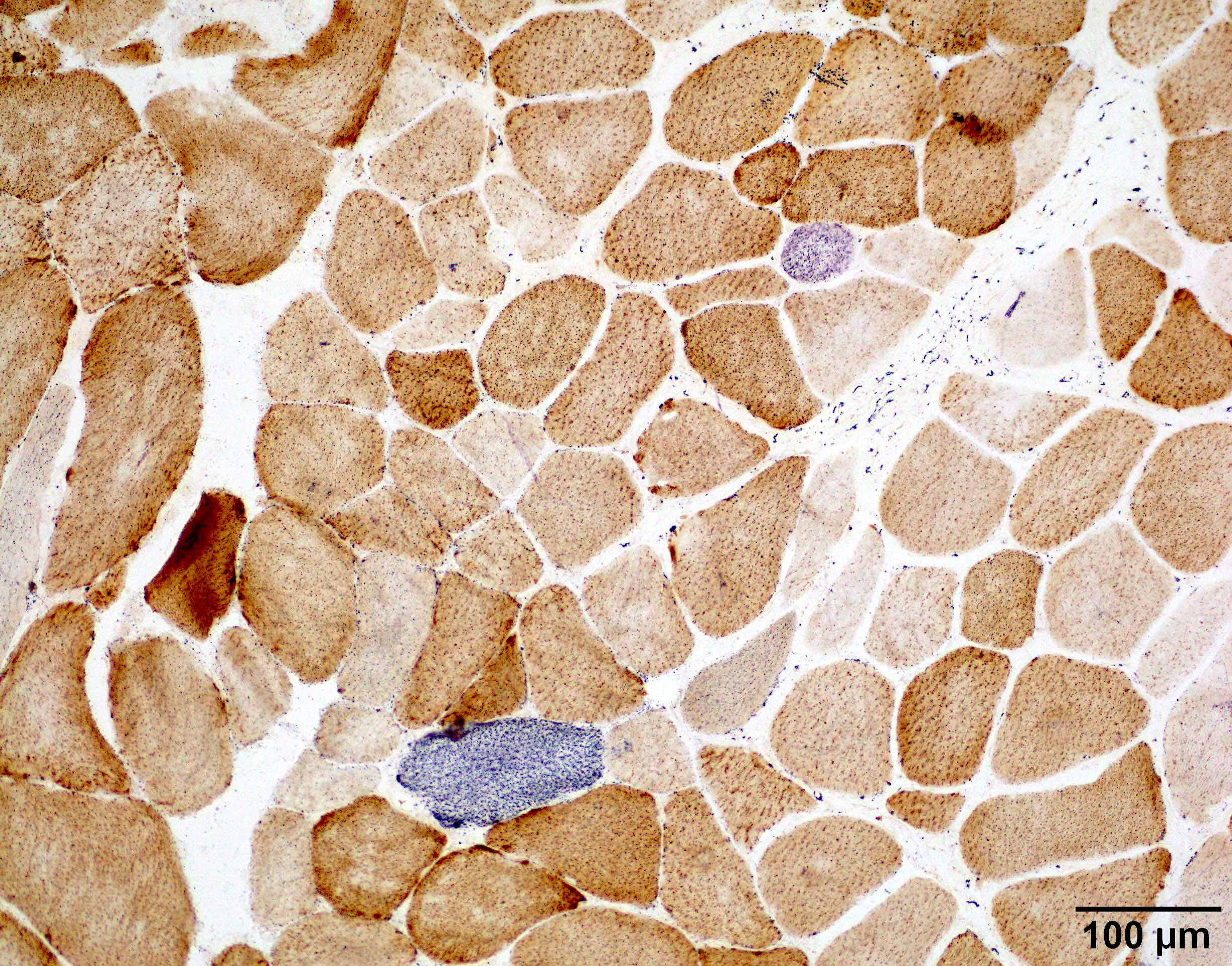
COX SDH double stain

MHC1
Positive stains
- Intranuclear inclusions in OPMD are positive for p62 and PABPN1 immunostains (Acta Neuropathol Commun 2022;10:176)
Negative stains
- Autophagic markers LC3, LAMP2 and p62 are negative in the sarcoplasmic vacuoles or intranuclear inclusions of OPMD (Acta Myol 2017;36:191)
- Ubiquitin and beta amyloid are negative in the sarcoplasmic vacuoles or intranuclear inclusions of OPMD (Neuromuscul Disord 1993;3:283)
Electron microscopy description
- Diagnostic electron microscopy feature was intranuclear filamentous inclusions composed of short (0.1 μm), unbranched, 8.5 nm wide filaments, present in 3 - 6.5% of myofiber nuclei (Can J Neurol Sci 1989;16:446, Neurology 1996;46:1324)
Electron microscopy images
Contributed by Chunyu Cai, M.D., Ph.D.
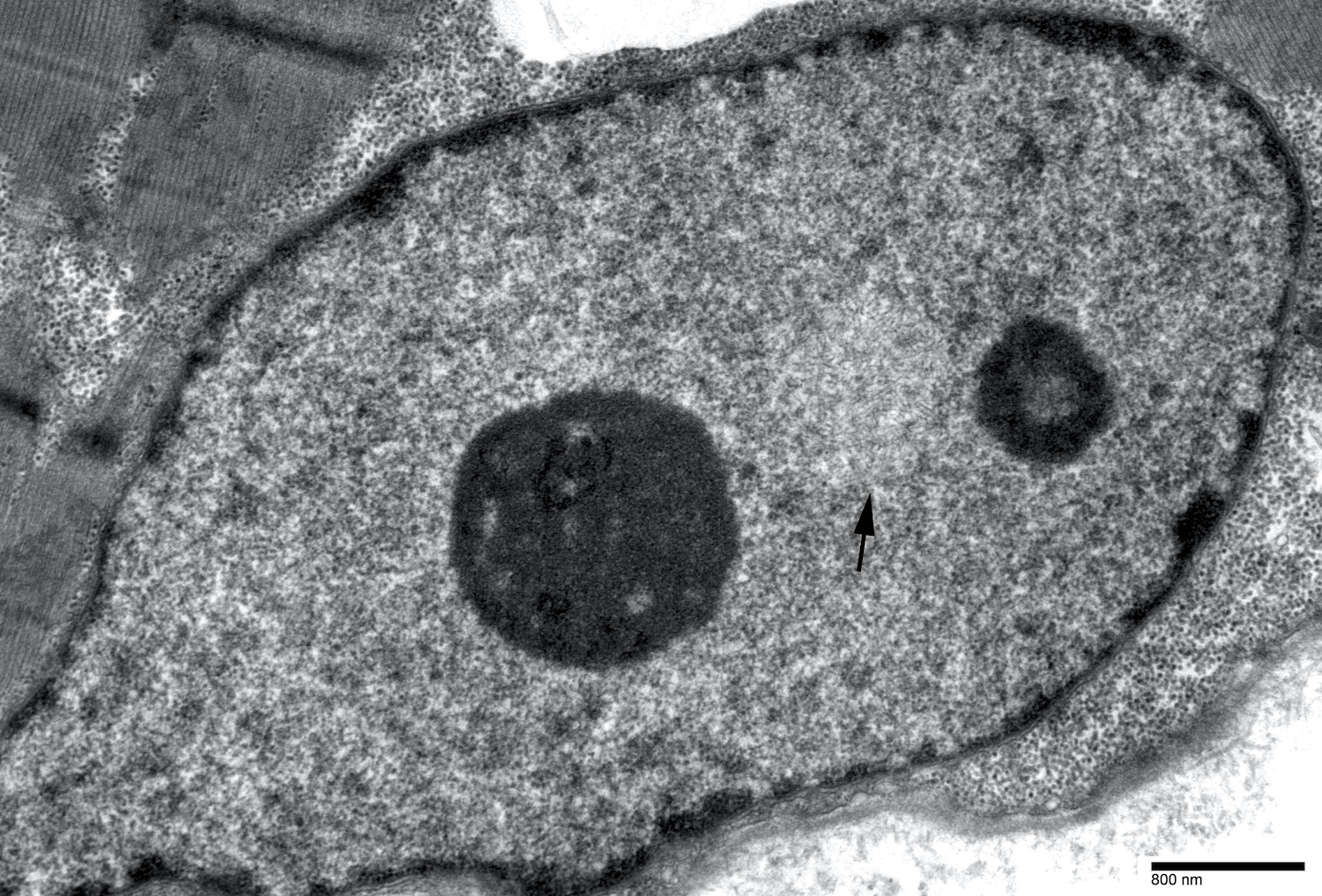
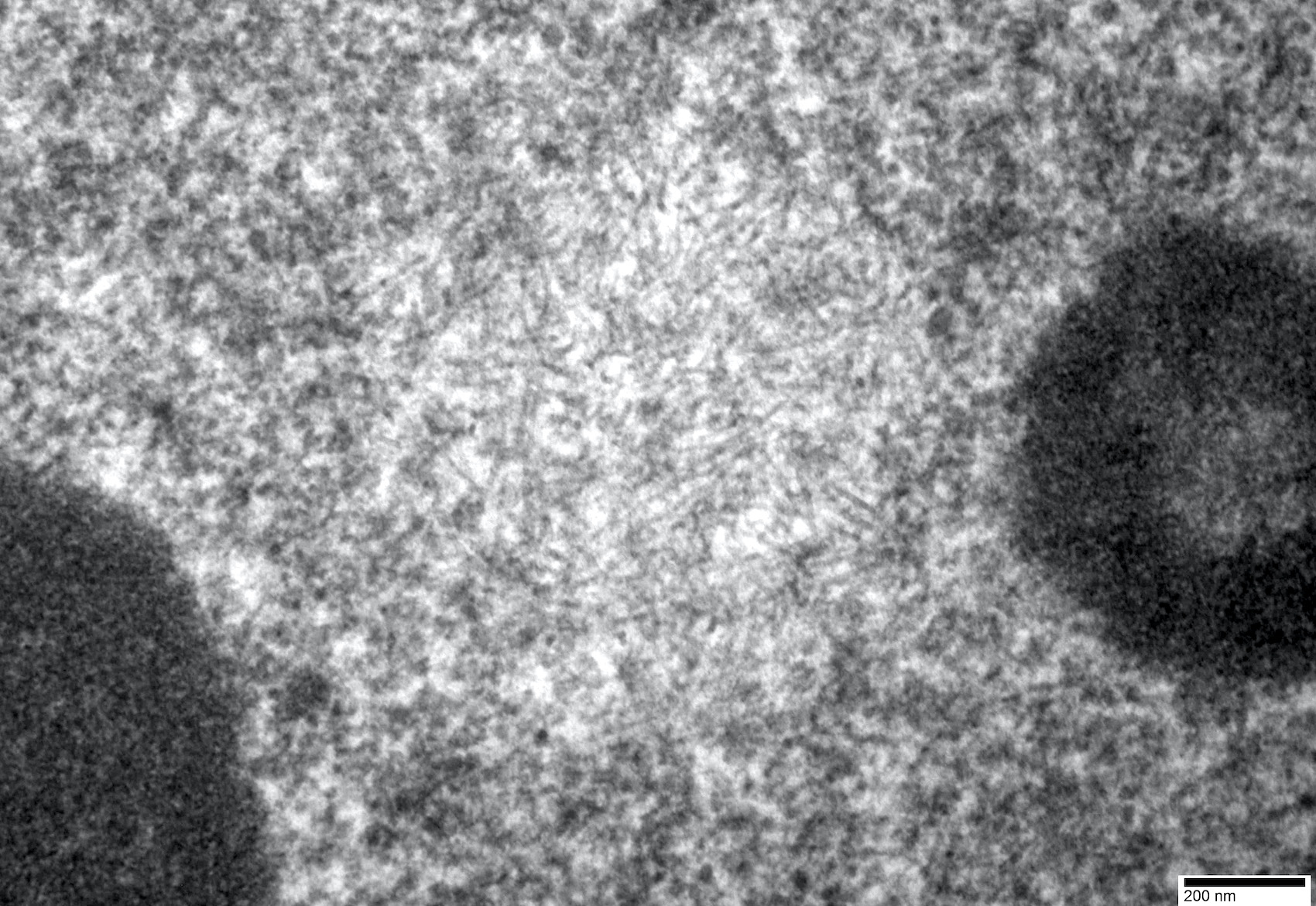
Intranuclear tubulofilamentous inclusion

Filaments reveal tubular nature
Molecular / cytogenetics description
- Definitive diagnosis is by genetic testing on blood showing expansion of the GCN trinucleotide repeat in the first exon of PABPN1 gene, usually by PCR (Neurology 2017;88:359)
- Normal individual: ≤ 10 GCN repeats
- OPMD patients: 11 - 18 GCN repeats
Sample pathology report
- Skeletal muscle, right vastus lateralis, biopsy:
- Chronic myopathy with rimmed vacuoles and intranuclear filamentous inclusions (see comment)
- Comment: The muscle biopsy shows mild myopathic changes with abnormal fiber size variation and rare red rimmed vacuoles but no significant inflammation or COX deficient fibers. Electron microscopy identified rare cytoplasmic bodies and rare intranuclear filamentous inclusions with diameter of 8.5 nm. These findings, in the correct clinical settings, are suggestive of oculopharyngeal muscular dystrophy (OPMD). Genetic analysis for GCN expansions in the PABPN1 gene may be of additional diagnostic value.
Differential diagnosis
- Sporadic inclusion body myositis (sIBM):
- Besides rimmed vacuoles, sIBM muscle usually contains lymphocytic inflammation and substantially increased COX deficient fibers; both are usually absent in OPMD
- Intranuclear inclusion of 8.5 nm filaments are nearly always present in OPMD, while sIBM only rarely has intranuclear tubulofilamentous inclusions, which are thicker (15 - 18 nm) in diameter (Semin Neurol 2012;32:237)
- Rimmed vacuoles and ubiquitin positive sarcoplasmic inclusions are less frequent in OPMD than sIBM (Neuromuscul Disord 1993;3:283)
- Oculopharyngeal distal myopathy (OPDM) and other rimmed vacuolar myopathies:
- p62 positive intranuclear inclusions are substantially more frequent in OPMD (~12% myofiber nuclei) than OPDM (~1% myofiber nuclei) and other rimmed vacuolar myopathies / hereditary inclusion body myositis (Acta Neuropathol Commun 2022;10:176)
- Otherwise, OPDM muscle pathology might be indistinguishable from OPMD; definitive diagnosis might require correlation with clinical weakness pattern and genetic test confirmation
Board review style question #1
A 63 year old man presented with bilateral ptosis and difficulty in swallowing. The patient had 5 years of progressive difficulty in swallowing, so now he can barely swallow solid food. The ptosis has been gradually progressive for 3 years. Tests for myasthenia gravis were negative. Aside from ptosis and dysphagia, a neurophysiological examination is also significant for proximal lower extremity weakness. A muscle biopsy shows pathology demonstrated in the image above. Electron microscopy of the muscle revealed rare intranuclear inclusions composed of aggregations of short filaments with diameter of 8.5 nm. Which of the following genetic defects are most likely to be present in this patient?
- CTG trinucleotide repeat of the DMPK gene
- GCN trinucleotide repeat of PABPN1 gene
- In frame deletion of the dystrophin (DYS) gene
- Missense mutation of the VCP gene
- Reduced D4Z4 microsatellite repeats upstream of the DUX4 gene
Board review style answer #1
B. GCN trinucleotide repeat of PABPN1 gene. The history and pathology are most consistent with OPMD, whose characteristic genetic change is GCN trinucleotide repeat of PABPN1 gene. Answer C is incorrect because in frame deletion of the dystrophin (DYS) gene is seen in Becker muscular dystrophy. Answer A is incorrect because CTG trinucleotide repeat of the DMPK gene is seen in myotonic dystrophy type 1. Answer D is incorrect because missense mutation of the VCP gene is seen in hereditary inclusion body myositis with Paget disease of the bone and frontotemporal dementia (IBMPFD). Answer E is incorrect because reduced D4Z4 microsatellite repeats upstream of the DUX4 gene is seen with facioscapulohumeral muscular dystrophy type 1 (FSHD1).
Comment Here
Reference: Oculopharyngeal muscular dystrophy
Comment Here
Reference: Oculopharyngeal muscular dystrophy
Board review style question #2
Which of the following additional stains on muscle biopsy is most helpful in reaching the diagnosis of oculopharyngeal muscular dystrophy?
- COX and SDH combination stains
- Dystrophin epitopes (DYS1 / DYS2 / DYS3)
- MHC class 1
- p62
- PAS with and without diastase
Board review style answer #2
D. p62 can highlight myofiber intranuclear inclusions, which are substantially more frequent in oculopharyngeal muscular dystrophy than other mimickers. Answer E is incorrect because PAS with and without diastase is used to detect glycogen or polyglucosan bodies. Answer C is incorrect because MHC class 1 is an inflammatory marker that is usually diffusely positive in myofibers in sporadic inclusion body myositis. Answer B is incorrect because dystrophin epitopes (DYS1 / DYS2 / DYS3) are used in Duchenne / Becker muscular dystrophy. Answer A is incorrect because COX and SDH combination stains are used in detecting mitochondria abnormality.
Comment Here
Reference: Oculopharyngeal muscular dystrophy
Comment Here
Reference: Oculopharyngeal muscular dystrophy
