Lymphoma & related disorders
Lymphoid proliferations and lymphomas associated with immune deficiency and dysregulation
Inborn error of immunity-associated lymphoid proliferations and lymphomas
Editorial Board Member: Elizabeth Courville, M.D.
Last author update: 9 June 2021
Last staff update: 17 October 2022
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search:
Lymphoproliferative disease[TI] free full text[sb] primary immune disorders
Cite this page: Cheng J. Inborn error of immunity-associated lymphoid proliferations and lymphomas. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomaprimaryimmuno.html. Accessed December 28th, 2024.
Definition / general
- Primary immunodeficiency disorders (PIDs) describe a group of diseases characterized by a decrease, absence or malfunction of at least 1 part of the immune system, caused by monogenic germline mutations, resulting in loss or gain of function of the encoded protein (Hematology Am Soc Hematol Educ Program 2019;2019:443)
- Can be dominant or recessive, autosomal or X linked and with complete or incomplete penetrance
- Generally manifest as increased susceptibility to a spectrum of infectious diseases, variable autoimmune or autoinflammatory conditions or malignancies
- The 2019 International Union of Immunological Societies (IUIS) classification reports 406 distinct disorders, with 430 different gene defects and a diversity of phenotypes (J Clin Immunol 2020;40:66)
- Due to compromised immunosurveillance and chronic infections, PID patients have a high risk of developing lymphoproliferative disorders, ranging from nonmalignant lymphoid hyperplasia to malignant lymphoma
Essential features
- Nonneoplastic or neoplastic lymphoid proliferation are common in PIDs
- Nonneoplastic lymphoid hyperplasia (such as follicular hyperplasia, marginal zone hyperplasia and paracortical hyperplasia) is associated with recurrent infections or autoimmune reactions
- Overall, B cell lymphoma is more frequent than T cell neoplasm; the lymphoma cells can be EBER positive or negative
- Diagnostic criteria of lymphoid neoplasm is nearly the same as immunocompetent counterparts
- Genetic mechanisms underlying transformation are largely unknown
Epidemiology
- Each individual primary immune disorder is generally rare but collectively primary immunodeficiency disorders are common
- Overall risk for malignancy in patients with PID is 4% (range: 0.7 - 15%), 10,000 times greater than the general population
- Risk for lymphoproliferative and related disorders varies among individual PID
Primary immune disorders associated with predominantly antibody deficiency or low immunoglobulins
Immunoglobulin A deficiency
- Prevalence 1:100 - 1,000
- Results from inherited genetic differences in chromosomes 18, 14 and 6
- Usually IgA < 7 mg/dL or absent; other immunoglobulin levels are normal or elevated
- Most patients are asymptomatic; some have infection, autoimmune and inflammatory disorders of gut; 10 - 20 times increased risk for celiac disease (Int J Surg Case Rep 2017;30:69)
- Microscopic (histologic) description: nodular lymphoid hyperplasia in the lamina propria of GI tract (see Microscopic (histologic) images)
- Overall the immunostaining pattern on tissue biopsies are nonspecific; occasionally, expansion of CD3+, CD8+, CD57+ LGLs can be identified in peripheral blood, lymph node or bone marrow by either flow cytometry or immunostains
- Molecular / cytogenetics description: usually IG and TCR gene rearrangements are poly or oligoclonal
- Rarely, B cell lymphoma and gastric carcinoma are reported (Dis Colon Rectum 1984;27:822)
Common variable immunodeficiency (CVID)
- CVID is the most prevalent symptomatic PID in adults (1 in 25,000 - 50,000)
- Results from variants in multiple genes of B cell development
- CVID represents a heterogeneous group with antibody deficiency of at least 2 serum Ig isotypes with normal or low B lymphocytes; T cell defects include increased memory T cells, loss of T cell proliferate and T cell associated cytokine defects; other PIDs excluded
- Good syndrome (GS) presents with thymoma and hypogammaglobulinemia in the fifth to sixth decade of life; patients can have low or absent B cells, variable defects in cell mediated immunity with a CD4 T lymphopenia, inverted CD4/CD8+ T cell ratio and reduced T cell mitogen proliferative responses (J Clin Immunol 2018;38:96)
- CVID patients present recurrent infections, GI villous atrophy and inflammatory bowel disease (IBD), liver and lung focal nodular hyperplasia and other symptoms (J Allergy Clin Immunol 2014;133:535)
- Microscopic (histologic) description: follicular hyperplasia with ill defined germinal centers lacking mantle cuffs or marginal zone hyperplasia; the differential diagnosis with B cell lymphoma, especially marginal zone B cell lymphoma is challenging (see Microscopic (histologic) images)
- Immunohistochemistry: nonspecific; similar to immunocompetent counterparts
- Aberrant T cell population, such as large granular lymphocytes (LGLs), can be identified in peripheral blood, lymph node or bone marrow by either flow cytometry or immunostains; TCR gene rearrangement can be monoclonal
- Molecular / cytogenetics description: IG and TCR gene rearrangements are usually poly or oligoclonal; the clonal IG does not warrant a lymphoma diagnosis in CVID patients
- 4.1% of CVID have malignant lymphoma, especially marginal zone lymphoma or diffuse large B cell lymphoma (DLBCL); usually EBV- (J Clin Immunol 2020;40:524)
- BCL6 gene rearrangement is reported in these B cell lymphomas
X linked agammaglobulinemia (XLA)
- Prevalence 1:250,000 - 379,000
- Results from mutations of Bruton tyrosine kinase or BTK on the X chromosome (Xq21.3-q22); recently, other genetic defects reported (Front Immunol 2020;11:2001)
- B cells often < 2% with maturation arrest, all immunoglobulins level decreased and absence of humoral responses
- Patients present recurrent infections since 6 - 9 months of age, with positive family history (33%); few patients have bronchiectasis and chronic lung disease, IBD or enteritis, neutropenia (Medicine (Baltimore) 2006;85:193, J Allergy Clin Immunol 2020;146:429)
- Microscopic (histologic) description: nonspecific microscopic features
- Overall the immunostaining pattern on tissue biopsies are nonspecific; occasionally, expansion of CD3+, CD8+, CD57+ LGLs can be identified in peripheral blood, lymph node or bone marrow by either flow cytometry or immunostains
- Molecular / cytogenetics description: TCR gene rearrangements are oligoclonal and rarely monoclonal
- Rare lymphoma is reported; 3.7% of XLA patients develop nonhematopoietic tumor in 1 study (Medicine (Baltimore) 2006;85:193, J Allergy Clin Immunol 2020;146:429)
Warts, hypogammaglobulinemia, infections and myelokathexis (WHIM)
- Approximately 0.23 cases per 1,000,000 live births
- Results from autosomal dominant mutation of CXCR4, encoding GPCR in many leukocytes and involving T / APC synapses; the mutations lead to prolonged receptor activation; the same mutation has been reported in Waldenström macroglobulinemia (Br J Haematol 2016;172:735)
- Severe congenital neutropenia and myelokathexis are characteristic; B or T lymphopenia and hypogammaglobulinemia were detected in 88% and 58% of patients, respectively (J Allergy Clin Immunol Pract 2019;7:1568)
- Patients present with severe bacterial infection (78% of patients), recurrent pneumonia (61%), skin warts (61%, at a mean age of 11 years), HPV related malignancies (16%) (N Engl J Med 2019;380:163, J Allergy Clin Immunol Pract 2019;7:1568)
- Microscopic (histologic) description: myelokathexis and right shifted granulocytosis in bone marrow biopsy
- EBV+ B cell lymphoma is reported (J Clin Immunol 2019;39:532)
Primary immune disorders associated with combined immunodeficiency
Severe combined immunodeficiency (SCID)
- 1 in 50,000 worldwide (J Allergy Clin Immunol 2014;133:335)
- Results from heterogeneous genetic defects involving VDJ (variable, diversity and joining regions) recombination process (RAG1, RAG2, DCLRE1C, PRKDC, NHEJ, LIG4), T cell maturation (such as ADA, AK2, PNP, CORO1A), T cell signaling (IL2RG, IL7R, JAK3) or T cell receptor (CD3D, CD3E, CD3Z) (Front Immunol 2021;11:623199, J Clin Immunol 2021;41:631, J Clin Immunol 2014;34:304, J Clin Immunol 2011;31:281)
- Absence or very low level of CD3+ T cells, proliferate response to mitogens < 10% of control, very low serum Ig and impaired specific antibody responses; B cell or natural killer (NK) cell deficiency is variable; T cell receptor excision circle (TREC) levels are absent or very low at birth
- Patients present with cytopenia of multiple lineages, autoimmunity (including vasculitis, enteropathy and endocrinopathy, at a median age of around 2 years) and granulomatous disease of multiple tissues (J Allergy Clin Immunol Pract 2019;7:1970)
- Microscopic (histologic) description: nonspecific microscopic features
- Overall the immunostaining pattern on tissue biopsies are nonspecific; occasionally, expansion of CD3+, CD8+, CD57+ LGLs can be identified in peripheral blood, lymph node or bone marrow by either flow cytometry or immunostains
- Risk for lymphoma is probably increased
Omenn syndrome
- Prevalence uncertain
- Results from hypomorphic mutations or leaky defects of gene involving VDJ recombination process (RAG1, RAG2, DCLRE1C, PRKDC, NHEJ, LIG4) (Blood 2005;105:4179, Clin Immunol 2005;116:246, J Clin Immunol 2016;36:46, Front Immunol 2020;11:900)
- Presents with severe erythroderma, lymphadenopathy and eosinophilia, associated with elevated IgE levels; other symptoms overlap with SCID; the symptoms are very similar to graft versus host disease (GVHD)
- Lymph node biopsy shows complete effacement of the architecture with increased dendritic cells, eosinophils and activated Th2 type CD3+ T cells expressing CD45RO, CD4, CD25 and CD30 (Virchows Arch A Pathol Anat Histopathol 1985;407:69, Am J Surg Pathol 1995;19:1082)
- Risk for lymphoma is probably increased
22q11.2 deletion syndrome (DiGeorge syndrome)
- 1 in 4,000 worldwide
- Results from a 1.5 - 3 Mb heterozygous deletion of chr22q11.2 including TBX1, a key gene for development; often diagnosed prenatally
- Presents with heart abnormalities, delayed growth, thymus aplasia, cleft palate and other features; usually no skin rash; T cell deficiency and autoimmune cytopenia, thrombocytopenia (may be due to deletion of GPIBB gene) (J Med Genet 1997;34:798, Front Immunol 2020;11:1837)
- Rarely, patients with congenital developmental defects, severe T cell immunodeficiency and rudimentary thymus have homozygous FOXN1 mutations (Front Immunol 2020;11:1837)
- CHARGE syndrome (caused by CHD7 mutations), 2p11.2 microdeletion (FOXI3 haploinsufficiency) can present with DGS features of thymic aplasia, resulting in combined immune deficiency (Eur J Med Genet 2007;50:338, J Allergy Clin Immunol 2020;145:358)
- Atypical teratoid rhabdoid tumor (associated with SMARCB1 deletion) is reported; uncertain risk for lymphoma (Cancer Genet 2014;207:415)
Primary immune disorders associated with immune dysregulation
Primary hemophagocytic lymphohistiocytosis (HLH)
- 1:50,000 live born children
- Results from autosomal recessive mutations of PRF1 gene and other genes, including UNC13D, STXBP2, STX11, LYST (overlaps with Chédiak-Higashi syndrome); RAB27A (Griscelli syndrome type 2); AP3B1 (Heřmanský-Pudlák syndrome type 2)
- Functionally impairs delivery of perforin and cytotoxic granules and prevents killing of antigen presenting or damaged cells and leads to NK / CTL overt activation (Blood Adv 2020;4:2578, Blood 2020;135:1332)
- Diagnostic criteria commonly used:
- Fever ≥ 38.5 °C
- Splenomegaly
- Cytopenias (affecting at least 2 of the below): hemoglobin < 9 g/dL (in infants < 4 week: hemoglobin < 10 g/dL) platelets < 100 x 109/L neutrophils < 1 x 109/L
- Hypertriglyceridemia or hypofibrinogenemia (< 150 mg/dL)
- Hemophagocytosis in bone marrow, spleen, lymph nodes, liver or other tissue
- Low or absent NK cell activity
- Ferritin > 500 ng/mL
- Elevated sIL2R: > 2,400 U/mL or elevated based on the laboratory defined normal range (Pediatr Blood Cancer 2007;48:124)
Clinically overlaps with macrophage activation syndrome (MAS) and HLH episodes during the clinical course of other PIDs (Open Access Rheumatol 2018;10:117)
Viral infections, especially EBV, trigger primary as well as secondary forms of HLH
See Hemophagocytic lymphohistiocytosis
X linked lymphoproliferative disease (XLP or Duncan disease)
- Approximately 1 - 3 in 1,000,000 males
- Firstly described in 1970s; XLP results from pathogenic mutation or deletion of SH2D1A gene, which causes its coding protein SLAM associated protein (SAP) unable to bind to signaling lymphocytic activation molecule (SLAM) and leads to aberrant IFNγ modulation, causing uncontrolled cell proliferation (Lancet 1978;1:554)
- Reduced memory B cells, defects of CD8+ / CD4- T and NK cells effector functions; hypogammaglobulinemia due to CD4- T cell defects (Immunol Rev 2005;203:180)
- Clinical presentation resembles that of infectious mononucleosis and includes hemophagocytic lymphohistiocytosis (HLH), lymphoma and dysgammaglobulinemia, often triggered by Epstein-Barr virus (Blood 2011;118:5060)
- Clinically overlaps with X linked inhibitor of apoptosis (XIAP) deficiency, caused by BIRC4 gene mutation, with increased risk of EBV- related hemophagocytic lymphohistiocytosis; recurrent splenomegaly, IBD with Crohn's disease-like features, especially in early infancy
- Microscopic (histologic) description: polyclonal infiltration by EBV infected B cells, as well as by reactive CD4+ and CD8+ T cells; macrophages with hemophagocytosis may be present
- Immunohistochemistry: similar to immunocompetent host counterparts
- Molecular / cytogenetics description: no specific genetic abnormalities reported in XLP associated LPD
- Hematopoietic malignancies include largely B cell lymphomas (Burkitt, DLBCL) and rarely Hodgkin disease or T cell lymphomas; mostly extranodal; EBV positive or negative (Immunol Rev 2005;203:180)
Autoimmune lymphoproliferative syndrome (ALPS or Canale-Smith syndrome)
- Prevalence unknown
- Results from pathogenic mutations in the FAS, FASL or CASP10 genes, which cause impairment of lymphocyte apoptosis
- Required diagnostic criteria: chronic (> 6 months), nonmalignant, noninfectious lymphadenopathy or splenomegaly; elevated CD3+ TCR alpha / beta positive CD4-, CD8- double negative T cells (≥ 1.5% of the total lymphocytes or 2.5% of the CD3 positive lymphocytes in the setting of normal or elevated lymphocyte counts) (Blood 2020;136:1933)
- Secondary diagnostic criteria: elevated biomarkers (s-FASL, IL10, vitamin B12, IL18), typical immunohistological findings, autoimmune cytopenia and elevated IgG and family history
- Patients present lymphadenopathy, hepatosplenomegaly and cytopenia
- Microscopic (histologic) description: prominent follicular hyperplasia, progressive transformation of germinal centers, expansion of CD4 / CD8 double negative T cells in peripheral blood, lymph nodes and spleen; or frank lymphoma similar to that seen in immunocompetent hosts (Haematologica 2017;102:364)
- Immunohistochemistry: CD3+, CD4-, CD8-, CD45RA+, CD45RO-, CD25-, variable CD57, may have an increase in CD5+ B cells
- Molecular / cytogenetics description: no specific genetic abnormalities reported in ALPS associated LPD
- Hematopoietic malignancies include Hodgkin and non-Hodgkin lymphomas; a subset are EBV positive; rare histiocytic sarcoma reported (Am J Surg Pathol 2010;34:589)
- Clinically overlaps with RAS associated lymphoproliferative disease (RALD), caused by somatic gain of function mutations in members of the RAS subfamily and lacked the defining increase in the CD3+ / TCRαβ+ CD4- / CD8- double negative T cell compartment, elevated biomarkers (i.e. vitamin B12) and the germline or somatic variants in FAS, FASL or CASP10 (Blood 2011;117:2883)
- Differential diagnosis of RALD from Rosai-Dorfman syndrome and occasionally systemic lupus erythematosus can be challenging; both of them have reported the same variant of somatic origin, KRAS p.G13C (Clin Immunol 2017;175:143)
- RALD and juvenile myelomonocytic leukemia (JMML) likely exist on a shared clinical continuum, where JMML undergoes additional genetic changes responsible for malignant transformation (Blood 2015;125:2753)
- Patients with Gaucher disease (GD) may have ALPS-like features and FAS mediated apoptosis defects (J Allergy Clin Immunol Pract 2020;8:3535)
PI3K related immunodeficiency
- Prevalence unknown
- Results from heterozygous gain of function mutations in PIK3CD gene or others genes in PI3K signaling pathway
- Usually normal B cells but memory B cells decrease while transitional / immature B cells increase; with variable immunoglobulin levels; progressive CD4+ T lymphopenia, reversed CD4:CD8 ratio; overlap with CVID and hyper-IgM syndrome (Front Immunol 2018;9:2172, Science 2013;342:866, J Clin Med 2020;9:3335, Clin Rev Allergy Immunol 2020;59:323)
- Patients present early in life with recurrent sinopulmonary infections, chronic virus infection (EBV, CMV), lymphadenopathy and extranodal lymphoid hyperplasia (J Allergy Clin Immunol 2016;138:210, J Allergy Clin Immunol 2017;139:597)
- Microscopic (histologic) description: atypical follicular hyperplasia with prominent naked germinal centers with ill defined outlines, monocytoid B cell proliferate; resembling CMV lymphadenitis
- Immunohistochemistry: IgM+ cells surrounding the naked germinal center and rare IgG+ cells
- Molecular / cytogenetics description: usually IG and TCR gene rearrangements are poly or oligoclonal
- Rare EBV associated lymphoma reported (Front Immunol 2018;9:1758)
Primary immune disorders associated with congenital syndromes or DNA repair defects
Ataxia telangiectasia (AT)
- 1 in 40,000 to 100,000 worldwide
- Results from autosomal recessive mutations in ataxia telangiectasia mutated (ATM) gene encoding a serine / threonine protein kinase and playing a key role in DNA double strand break repair (Am J Hum Genet 1995;57:112)
- PID in about 70% of AT patients; usually, B or T cell lymphopenia, decreased CD8 or CD4+ T cells, antibody production deficiency, Ig subclasses deficiency (mostly IgA and IgG2), hypogammoblobulinemia, occasionally hyper-IgM due to Ig class switch recombination defect (CSRD) and associated with high frequency of granulomas (J Pediatr 2004;144:505)
- Patients present with progressive cerebellar ataxia, mucocutaneous telangiectasia, sinopulmonary infections, variable immunodeficiency, radiosensitivity and metabolic disorders; patients often die from chronic pulmonary disease or malignancies (Pediatr Allergy Immunol 2019;30:277)
- High risk for leukemia and lymphoma (mostly T cell acute lymphoblastic leukemia and T cell prolymphocytic leukemia) in young age; EBV+ Hodgkin lymphoma and B cell non-Hodgkin lymphoma and other tumors are also common (J Clin Oncol 2015;33:202)
- TCR genes alterations on chromosomes 7 and 14 are seen in AT associated leukemia, including TCL1
Nijmegen breakage syndrome (NBS)
- 1 in 100,000 worldwide
- Results from biallelic mutations in NBN located at 8q21.3, encoding nibrin, 1 of 3 proteins that make up the MRN complex to activate and recruit ATM to double strand breaks (Acta Paediatr Scand 1981;70:557, Science 2005;308:551)
- Severe hypogammaglobulinemia in 20%, IgA deficiency in 50% and reduced B and T cells in > 80% of NBS patients, resulting in a spectrum from silent phenotype to recurrent, chronic respiratory tract infections (J Clin Immunol 2015;35:538)
- Patients present with microcephaly at birth with distinct, bird-like craniofacial features, growth retardation and intellectual disability
- High risk for diffuse large B cell lymphoma, T cell acute lymphoblastic leukemia and less frequently for Hodgkin lymphoma and acute myeloid leukemia (Front Pediatr 2020;8:570084)
- NBS overlaps with other PIDs; microcephaly and immunodeficiency are common in DNA ligase IV deficiency (LIG4 syndrome) and severe combined immunodeficiency with microcephaly, growth retardation and sensitivity to ionizing radiation due to NHEJ1 deficiency (NHEJ1 syndrome) (Orphanet J Rare Dis 2012;7:13)
Primary immune disorders associated with other deficiencies
Chronic granulomatous disease
- 1 in 200,000 worldwide
- Results from mutations in genes encoding any of the NADPH oxidase subunit proteins (gp91phox ~70% of cases; p47phox ~25%; p67phox ~2.5%; p22phox ~2.5%; and p40phox < 1%), leading to deficiency in production of antimicrobial reactive oxidative species (ROS) by granulocytes and monocytes (Blood Adv 2020;4:5976)
- Patients present with recurrent infections by catalase positive organisms; skin, gut, lungs, liver are commonly involved (Hematol Oncol Clin North Am 2013;27:89)
- Biopsy shows active chronic inflammation, pigmented macrophages, with or without necrotizing or non-necrotizing granuloma (Histopathology 2005;47:508)
- Granuloma prevalence in all PID patients is approximately 1 - 4% (J Clin Immunol 2018;38:717)
- Uncertain risk for lymphoma
Type I interferonopathies
- Unknown prevalence
- Genetically and phenotypically heterogeneous group of autoinflammatory and autoimmune disorders, characterized by constitutive activation of the antiviral type I interferon (IFN) axis
- Mutations in TREX1 and RNase H2 are reported in Aicardi-Goutieres syndrome (AGS) patients, retinal vasculopathy with cerebral leukodystrophy (RVCL), familial chilblain lupus (CHBL), systemic lupus erythematosus (SLE) (EMBO J 2018;37:e98506)
- Mutations in STING gene cause STING associated vasculopathy with onset in infancy (SAVI) (Front Pediatr 2020;8:577918, J Allergy Clin Immunol Pract 2021;9:803)
- Mutations in proteasome subunit genes cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) (Front Immunol 2017;8:927)
- Mutations or deficiency in adenosine deaminase 2 (ADA2) cause early onset vasculopathy resembling polyarteritis nodosa (PAN), early onset of recurrent strokes or cytopenia (Arthritis Rheumatol 2019;71:1747)
- Mutations in DNA polymerase POLA cause X linked reticulate pigmentary disorder (XLPRD), characterized by skin hyperpigmentation, sterile multiorgan inflammation, recurrent infections and distinct facial features; or O'Driscoll syndrome, characterized by growth retardation, microcephaly, hypogonadism and in some cases, overlapping immunological features to those seen in XLPDR (J Clin Immunol 2021;41:285)
- Biallelic mutations in ACP5, encoding tartrate resistant acid phosphatase (TRACP), cause the inherited immuno-osseous disorder, spondyloenchondrodysplasia (SPENCD)
- At least 1 autoimmune diagnosis is seen in most patients and some showing recurrent bacterial and viral infections and potential immunodeficiency (PLoS One 2020;15:e0230052, J Clin Immunol 2016;36:529)
Hyper-IgE syndrome
- Unknown prevalence
- Genetically and phenotypically heterogeneous group of PID, characterized by a triad of eczema, recurrent skin and pulmonary infections and elevated IgE levels
- Food and environmental allergies are common (J Allergy Clin Immunol Pract 2018;6:996)
- Most cases are sporadic but autosomal recessive (AR), autosomal dominant (AD) and X linked variants were described
- Heterozygous mutations in STAT3 gene with dominant negative effect cause autosomal dominant hyper-IgE syndrome (AD-HIES), showing eosinophilia, marked elevation in serum IgE (inversely correlated with patient age) and associated with abnormal B cell maturation (J Allergy Clin Immunol Pract 2018;6:996)
- Biallelic mutations in DOCK8 gene, regulating cytoskeletal rearrangement and immune synapse formation, cause autosomal recessive hyper-IgE syndrome (AR-HIES), showing eosinophilia, elevated IgE levels, T cell lymphopenia, variable B cell number and Ig levels (J Allergy Clin Immunol 2010;125:743)
- Other mutations (TYK2, PGM3, CARD11) have been reported in hyper-IgE patients (J Allergy Clin Immunol 2016;138:1222, J Pediatr 2004;144:93)
- Omenn syndrome, IPEX, Wiskott-Aldrich syndrome (WAS) and other PIDs with similar phenotypes are excluded
Treatment
- Treatment of PID is dependent on the underlying PID and clinical presentation
- Bone marrow transplant has been performed in some PIDs (J Allergy Clin Immunol Pract 2021;9:628)
- Treatment of lymphoproliferative disease or lymphomas is similar to immunocompetent counterparts
Microscopic (histologic) images
Contributed by Jinjun Cheng, M.D., Ph.D.
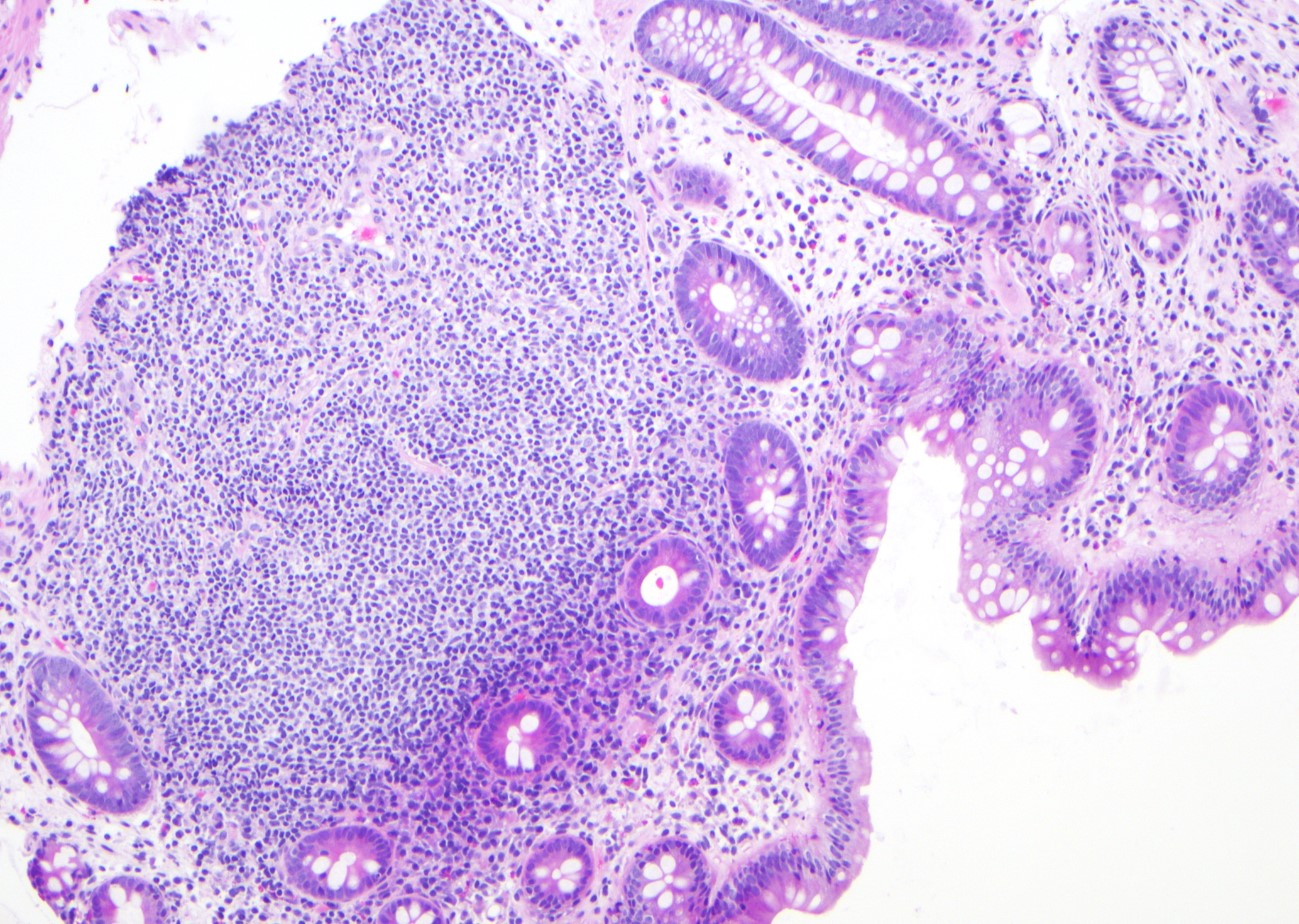
Lymphoid aggregate
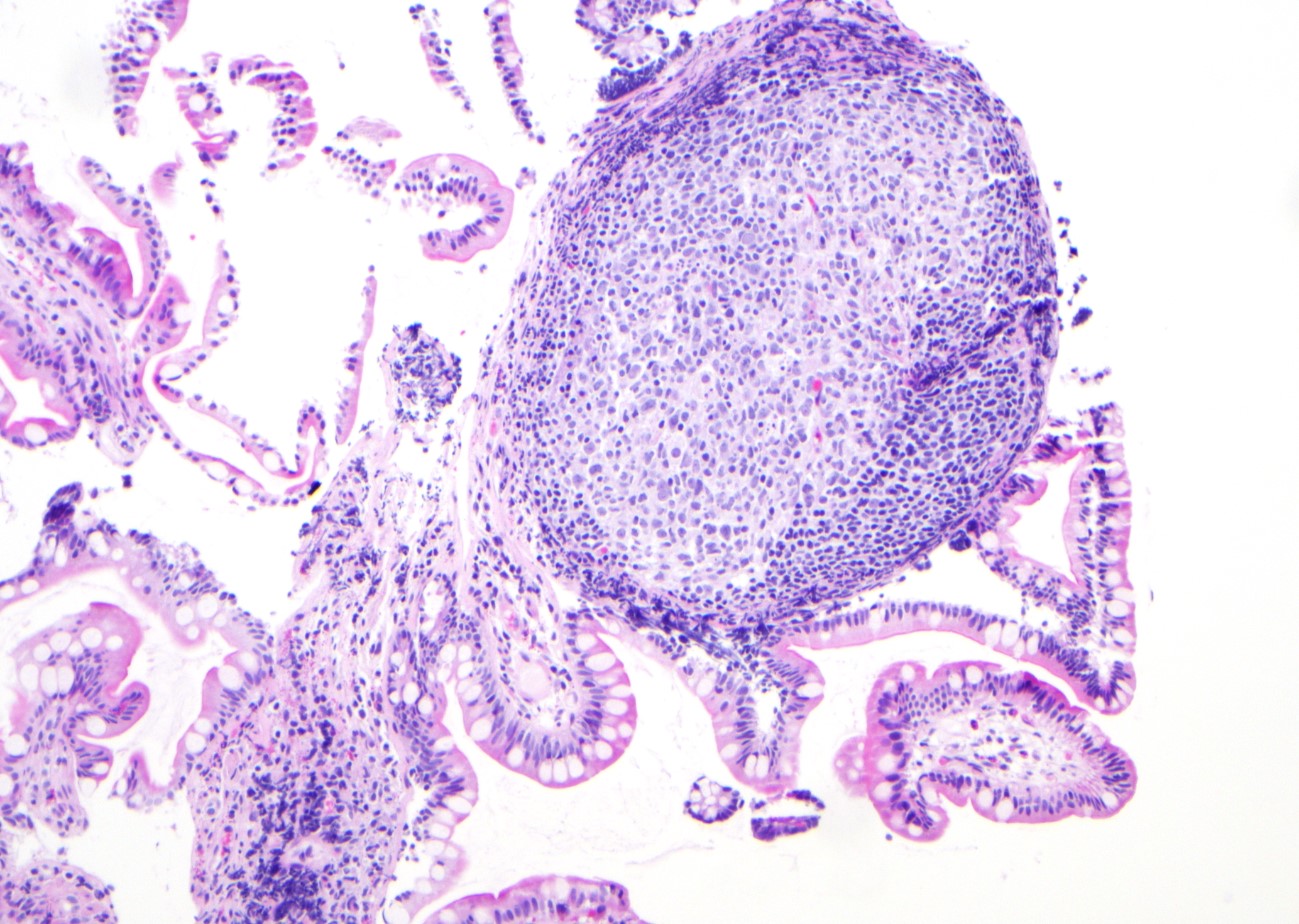
Lymphoid follicles
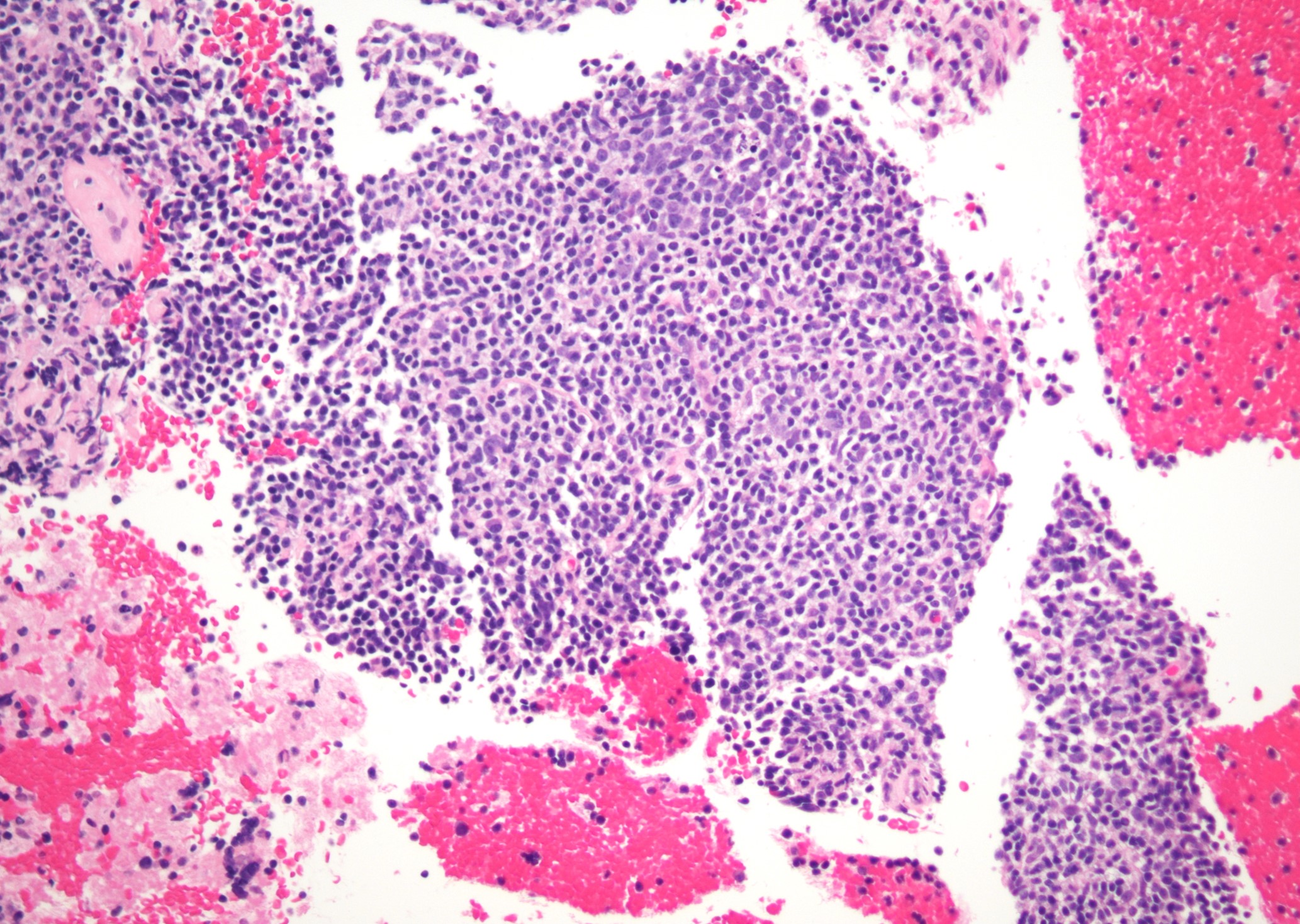
Marginal zone expansion
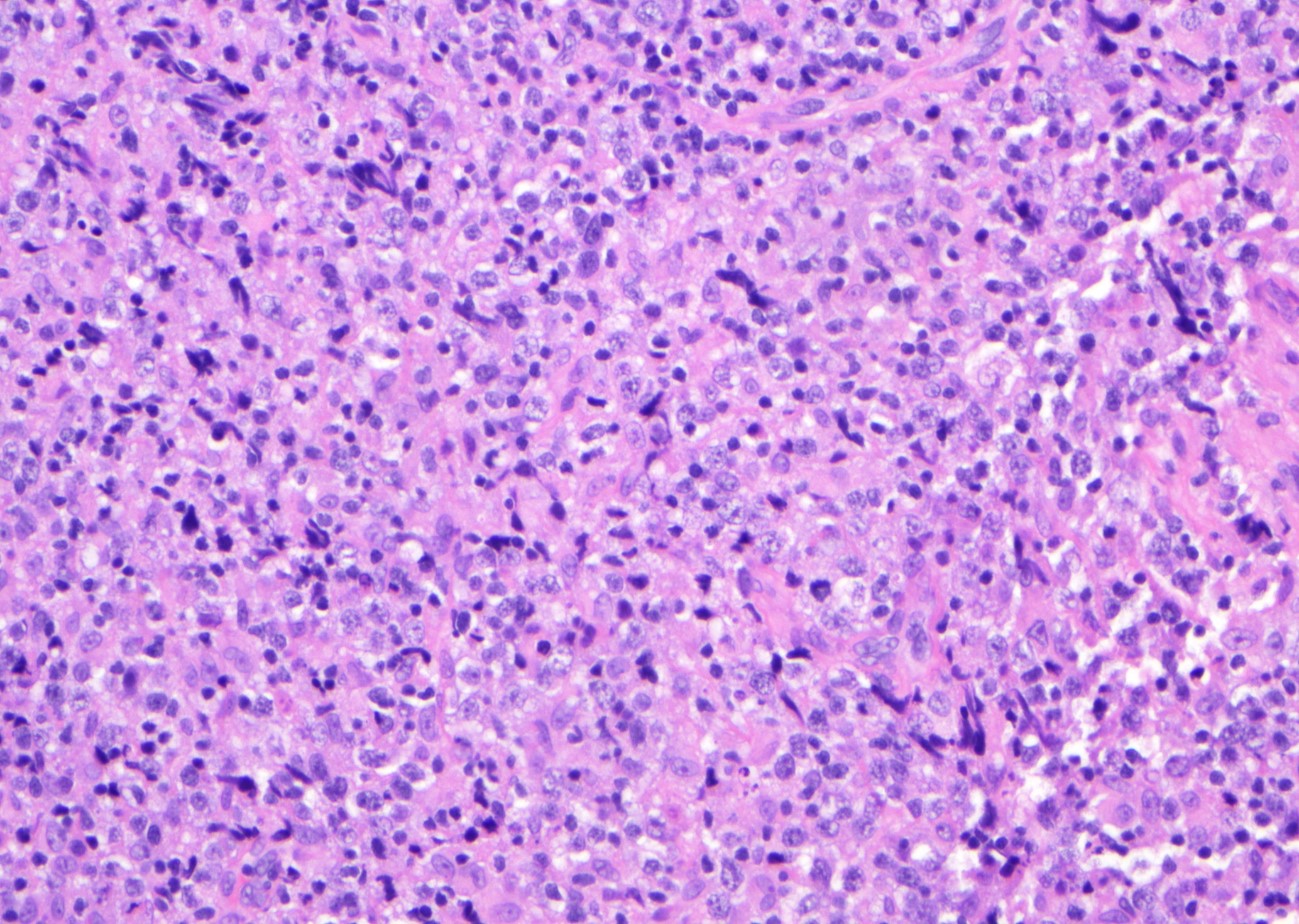
DLBCL
Flow cytometry images
Contributed by Jinjun Cheng, M.D., Ph.D.
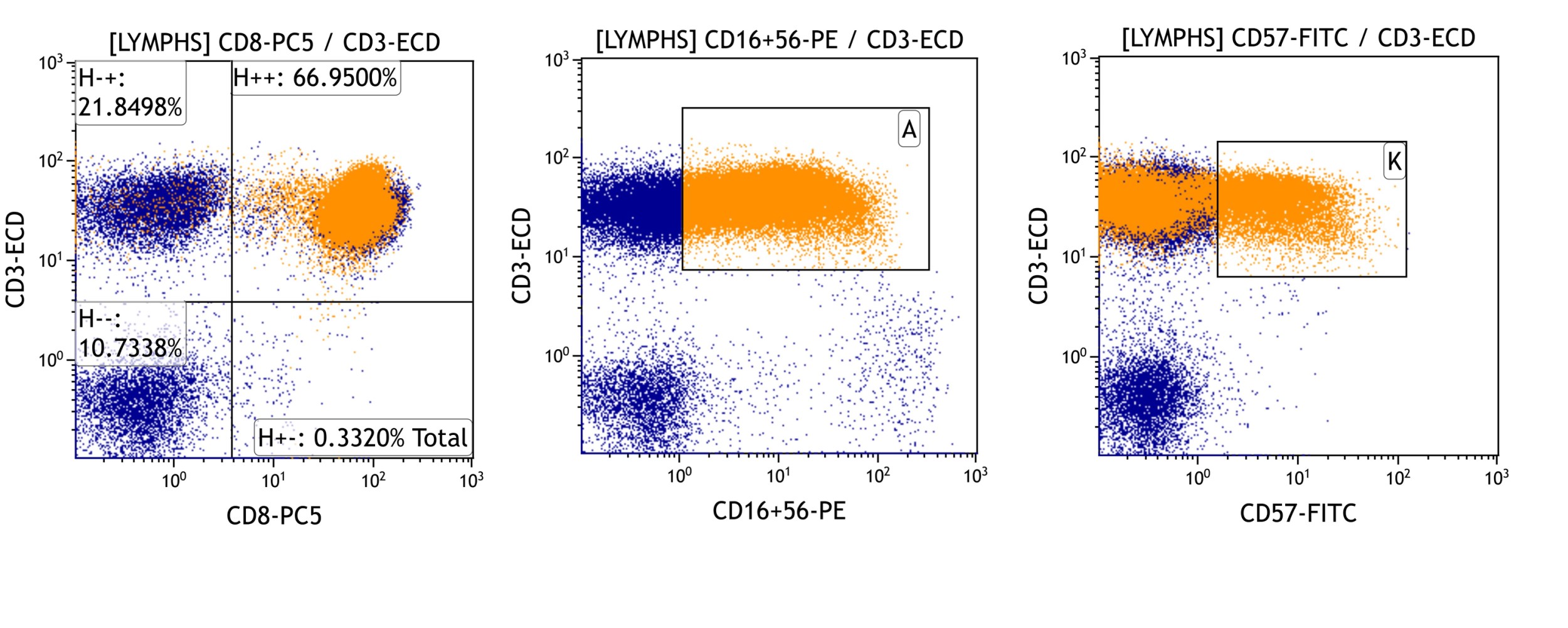
Large granular lymphocytosis
Board review style question #1
A 10 year old child was recently diagnosed with autoimmune lymphoproliferative syndrome (ALPS). The molecular studies identified a pathogenic mutation in the
FAS gene. The patient's peripheral blood was submitted for flow cytometry study. Which of the following aberrant T cell populations is expected to be elevated in this peripheral blood?
- CD4- CD8- T cells
- CD4+ CD8+ T cells
- CD4+ TFH cells
- CD4+ Treg cells
- Natural killer (NK) cells
Board review style answer #1
A. Elevated CD3+ TCR alpha / beta positive CD4- CD8- double negative T cells (≥ 1.5% of the total lymphocytes or 2.5% of the CD3+ lymphocytes in the setting of normal or elevated lymphocyte counts) is commonly seen in peripheral blood or lymph node of ALPS patients.
Comment Here
Reference:
Associated with primary immune disorders

Back to top




