

Lymphoma & related disorders
Lymphoid proliferations and lymphomas associated with immune deficiency and dysregulation
Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation
Editorial Board Members: Roberto N. Miranda, M.D., Julie Feldstein, M.D.


Last author update: 27 April 2023
Last staff update: 27 August 2024
Copyright: 2022-2024, PathologyOutlines.com, Inc.
PubMed Search: Polymorphic lymphoproliferative disorders in immune deficiency

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Flow cytometry description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Gonzalez-Mancera M, Kitahara S. Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomapolymorphicimmunedeficiency.html. Accessed November 28th, 2024.
Definition / general
- Lymphoproliferative disorders (LPDs) arising in patients with immune deficiency or dysregulation are usually Epstein-Barr virus (EBV) driven and composed of a heterogeneous lymphoid cell infiltrate with a variable number of B cells that efface the architecture of involved tissues
- New standardized nomenclature for reporting this histological diagnosis is based on an integrated approach and consists of 3 parts
- Histological diagnosis (polymorphic lymphoproliferative disorder)
- Viral association
- Immune deficiency / dysregulation setting
Essential features
- Polymorphous lymphoproliferative disorder arising in any immune deficiency / dysregulation setting
- Typically associated with EBV infection
- Histologically, there is effacement of the architecture of the involved tissue by a mixed inflammatory process and absence of sheets of monotonous cells
Terminology
- Encompasses the previous polymorphic posttransplant lymphoproliferative disorders, other iatrogenic immunodeficiency associated lymphoproliferative disorders, among others such as lymphomatoid granulomatosis (LYG) grade 1 and 2 in IDD settings (except immunosenescence) and lymphoid proliferations in the HIV setting (although usage of these terms is not recommended)
- Not recommended: polymorphic lymphoma
ICD coding
Epidemiology
- Polymorphic LPDs can arise in any immunodeficiency setting
- Account for 20 - 80% of LPDs in the posttransplant setting (Cancer 1998;82:1978, Hematol Oncol 2005;23:61, Am J Transplant 2006;6:2735)
- Account for 15 - 30% of LPDs in the autoimmune / therapy related immunodeficiency setting (Arthritis Rheum 2007;56:1433, Clin Rheumatol 2012;31:631)
- In the HIV setting, they account for < 5% (Am J Surg Pathol 2003;27:293, Virchows Arch 2012;461:93)
- No gender predilection has been recognized
Sites
- Primarily involves lymph nodes or other lymphoid organs; extranodal sites are involved less frequently, including the lung, gastrointestinal tract, liver and central nervous system (Am J Surg Pathol 2019;43:869, Blood 2011;117:4726)
Pathophysiology
- First step in the development of polymorphic LPDs is a disruption in T cell immune surveillance that results from an immunodeficient status
- In the setting of EBV positive LPDs, associated proteins promote cellular proliferation and immune evasion
- EBV is transmitted by oral transfer; EBV shedding can also be detected in cervical secretions of the gynecologic tract
- EBV latent membrane proteins LMP2A (that mimics the B cell receptor signaling) and LMP1 (that mimics CD40 signaling) activate NFκB, inducing cell proliferation (N Engl J Med 1998;338:1413, N Engl J Med 2004;350:1328)
- EBV nuclear antigen (EBNA1) plays a role in DNA regulation, transcription and immune evasion, while EBNA2 regulates viral and cellular genes
- EBV can present as multiple latency patterns depending on which proteins are expressed
- Latency patterns II (LMP1+ / EBNA2-) and III (LMP1+ / EBNA2+) are the most common patterns associated with immune deficiency / dysregulation associated LPDs
- In the setting of EBV negative LPDs, multiple theories have been proposed
- Some cases may be related to EBV infections that are no longer detectable
- Unidentified viruses or other causes of chronic antigenic stimulation (Br J Haematol 2001;113:425, Am J Surg Pathol 1999;23:1393)
- Studies have shown that EBV positive and EBV negative LPDs in the posttransplant setting have distinct genomic and transcriptomic landscape (Front Oncol 2020;10:506, Diagn Mol Pathol 2007;16:158)
- These authors have suggested that EBV negative LPDs develop coincidentally in transplant recipients
Etiology
- Any immunodeficiency setting, typically associated with EBV infection
- In therapy related (e.g., methotrexate) polymorphic LPD, EBV has been found in 75% of cases (Eur J Haematol 2013;91:20)
- Within all settings, EBV was found in 97% of cases (Am J Clin Pathol 2017;147:129)
Diagrams / tables
Images hosted on other servers:

Immunodeficiency
associated LPDs
in WHO-HAEM4R
and WHO-HAEM5

3 part nomenclature
for immunodeficiency
associated LPDs
Clinical features
- Associated with a wide spectrum of clinical features
- Isolated lymphadenopathy
- Widespread lymphadenopathy with or without splenomegaly
- Involvement of extranodal sites
- B symptoms have been reported in 35% of cases (Blood 2011;117:4726)
- Can be associated with hemophagocytic lymphohistiocytosis (Am J Clin Pathol 2017;147:129, Int J Hematol 2023;117:769)
- Disease severity, duration of disease and length of the treatment in autoimmune diseases have been associated with the development of EBV driven LPDs (Am J Clin Pathol 2017;147:129, J Rheumatol 2015;42:564)
- In general, in the posttransplant setting, lymphoproliferative disorders can present from months up to several years after transplant (Curr Oncol Rep 2010;12:383, Leuk Lymphoma 2013;54:2433)
- 15 - 25% of cases occur > 10 years after transplant
Diagnosis
- Known history of immunodeficiency or immune dysregulation
- CT imaging
- Biopsy of the lesional tissue
- References: Br J Haematol 2010;149:675, Am J Clin Pathol 2017;147:129
Laboratory
- Elevated lactate dehydrogenase (LDH) levels have been associated with a poor response to treatment and overall survival (Blood 2006;107:3053, Am J Transplant 2006;6:569)
- EBV serology (Am J Transplant 2009;9:S87)
- Its most important role in the posttransplant setting is the determination of pretransplant donor and recipient EBV serostatus for posttransplant lymphoproliferative disorder risk assessment
- In immunocompromised patients, serology is an unreliable diagnostic tool due to delayed or absent humoral response
- If these patients are receiving blood products, the passive transfer of antibodies is another important drawback to consider
- EBV viral load can be used to monitor therapeutic response (Am J Transplant 2008;8:1016, Transplantation 1998;66:1641, Transplantation 2002;74:656)
Radiology description
- Depending on the involved site, imaging studies may show localized or diffuse lymphadenopathy, organomegaly or a mass lesion
Prognostic factors
- There are no well defined histopathologic, immunophenotypic or molecular prognostic / predictive factors within the group of polymorphic LPDs
- Staging with PET / CT imaging should be done following the Lugano staging system (J Clin Oncol 2014;32:3059)
- Lymphadenopathy resolves in up to 20% of cases with no therapy
- Clinical progression has been reported in 38% of patients, with 10% progressing to DLBCL (Blood 2011;117:4726)
- Improvement may be seen upon discontinuation of immunosuppression in the posttransplant setting or in therapy related immunodeficiency (Med Oncol 2009;26:1)
- In the posttransplant setting, factors that have been associated with lack of response to decreased immunosuppression include elevated LDH levels, organ dysfunction, multiorgan involvement, advanced age, bulky disease and older age of presentation (Am J Transplant 2011;11:336, Transplantation 2001;71:1076)
Case reports
- 26 year old woman presented with pharyngeal foreign body sensation after peripheral blood stem cell transplant (PBSCT) (Int J Clin Exp Med 2014;7:1904)
- 36 year old HIV infected man presented with acute abdominal pain (EJHaem 2021;2:562)
- 67 year old man status post-renal transplant presented with multiple allograft parenchymal cystic lesions (Am J Transplant 2012;12:245)
Treatment
- Discontinuation of the immunosuppression generally leads to regression
- Chemotherapy alone has been used in 56% of cases (Blood 2011;117:4726)
- 6% required the addition of radiotherapy
- Systemic chemotherapy
- 3 step approach has been used in the posttransplant setting (Ther Adv Hematol 2011;2:393, Br J Haematol 2010;149:693)
- Reduction of immunosuppression by 50%
- Single agent rituximab
- Rituximab and cytotoxic chemotherapy with CHOP
- 3 step approach has been used in the posttransplant setting (Ther Adv Hematol 2011;2:393, Br J Haematol 2010;149:693)
- Initiation of antiretroviral therapy in HIV patients has shown response (Am J Surg Pathol 2003;27:293, Virchows Arch 2012;461:93)
Gross description
- Lesions can present as a mass with ulceration or necrosis
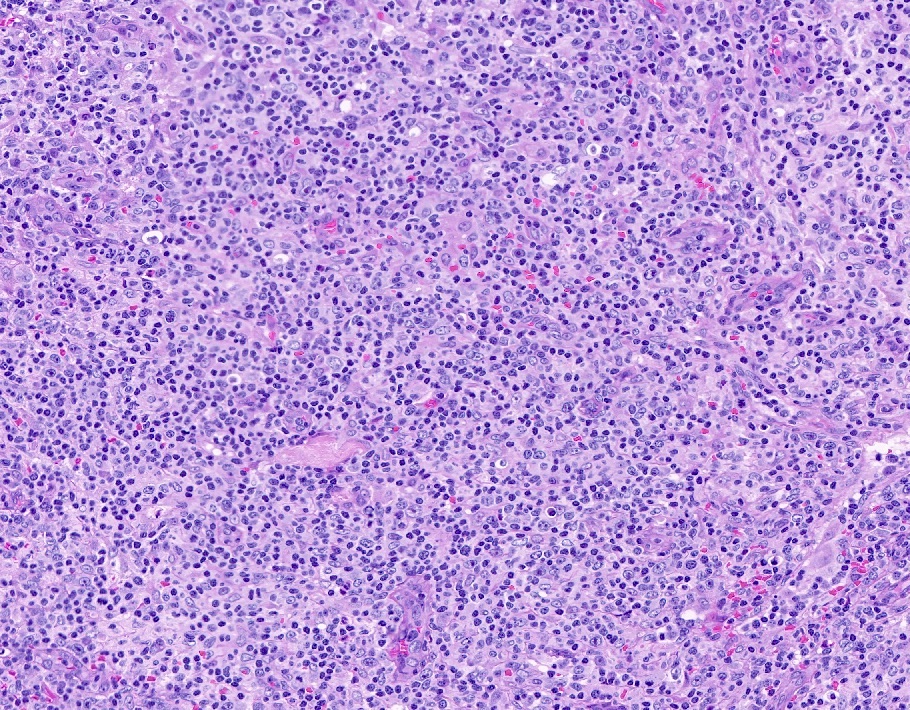
Microscopic (histologic) description
- Histologic features include architectural destruction and a mixed infiltrate of cells, including B cells at different stages of maturation such as immunoblasts, small lymphoid cells, plasmacytoid cells, plasma cells (Am J Clin Pathol 2017;147:129, Blood 2011;117:4726)
- Intermixed T cells and histiocytes are commonly seen
- Immunoblasts may show Hodgkin / Reed-Sternberg (HRS)-like morphology
- Sheets of monotonous cells are not present
- Necrosis may be seen and angiocentric / angiodestructive polymorphous infiltrate may be seen in lymphomatoid granulomatosis type lesions
Microscopic (histologic) images
Contributed by Miguel Gonzalez-Mancera, M.D.
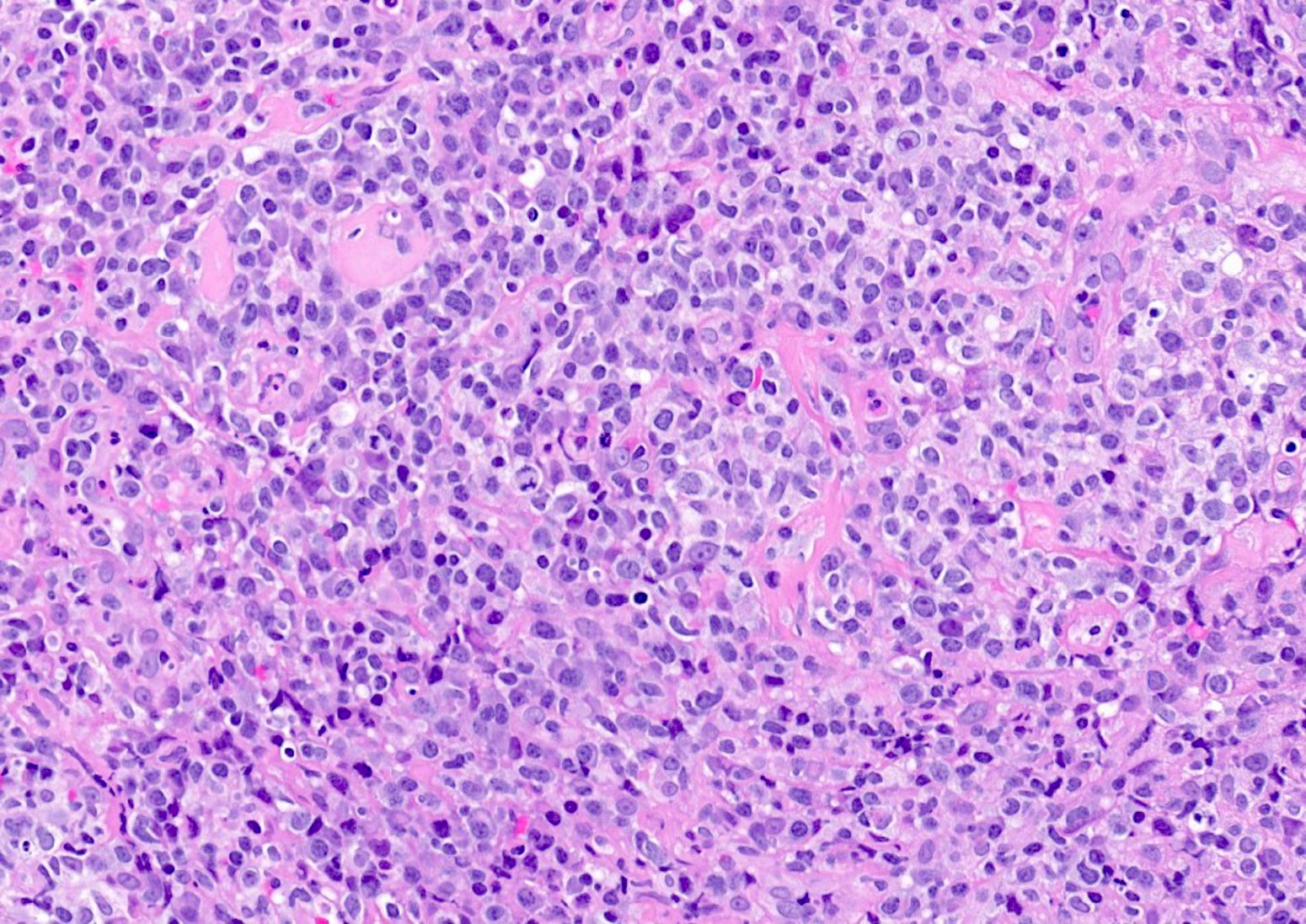
Heterogeneous infiltrate
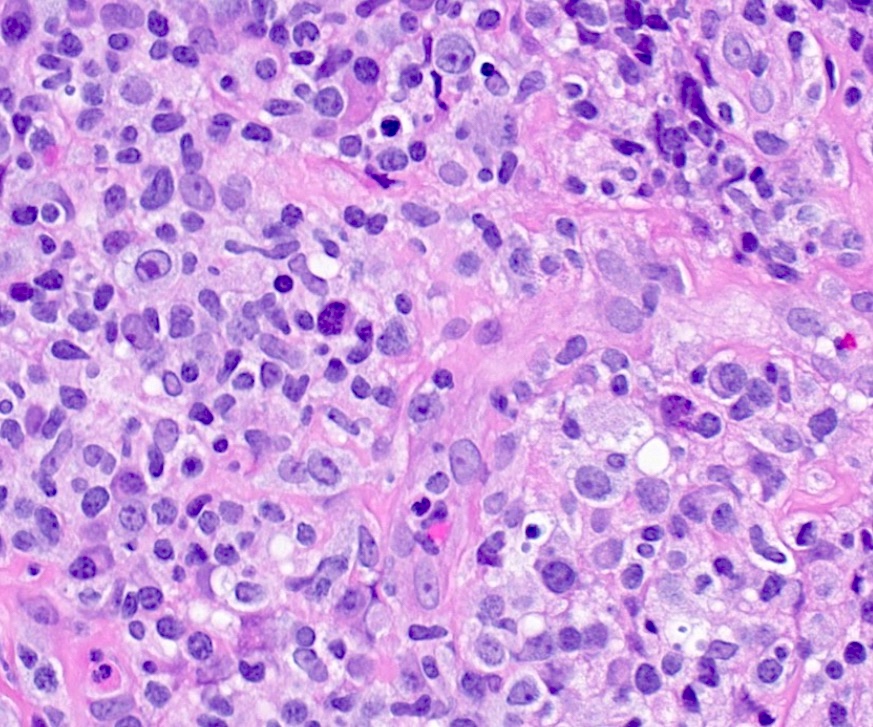

Heterogeneous infiltrate

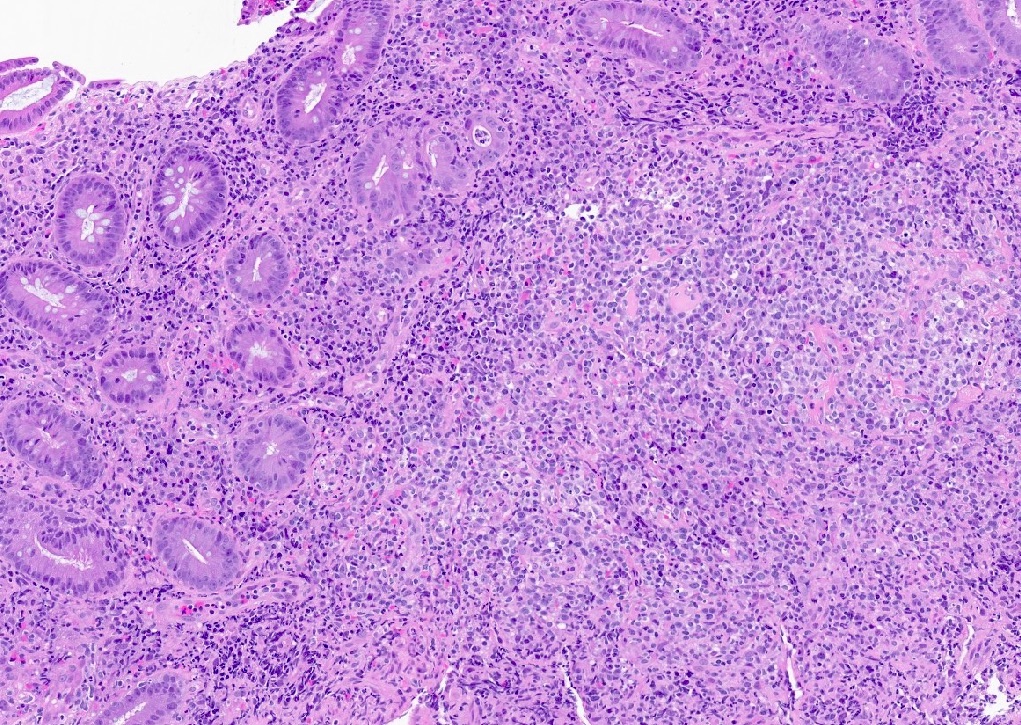
Gastrointestinal biopsy

CD3
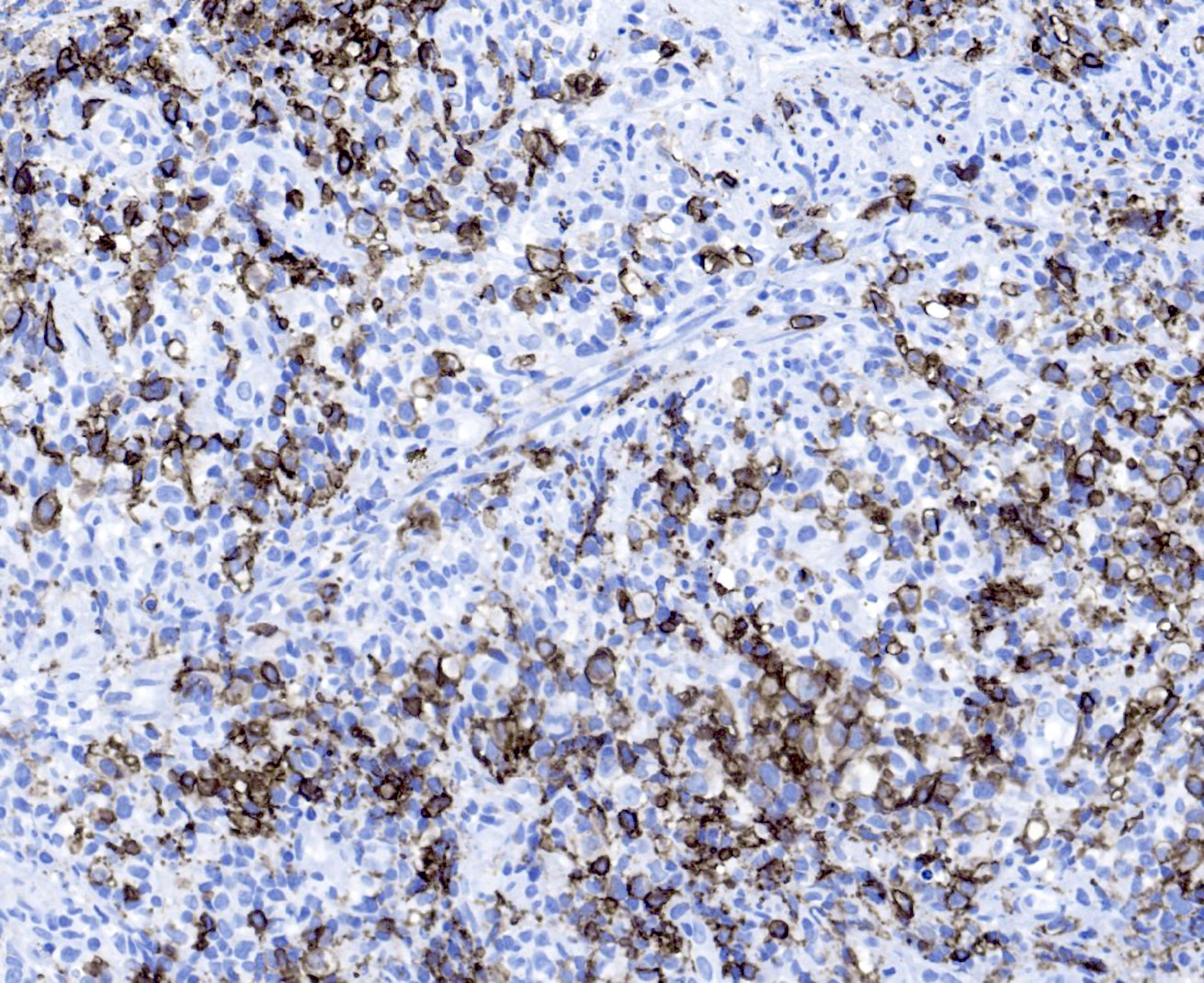
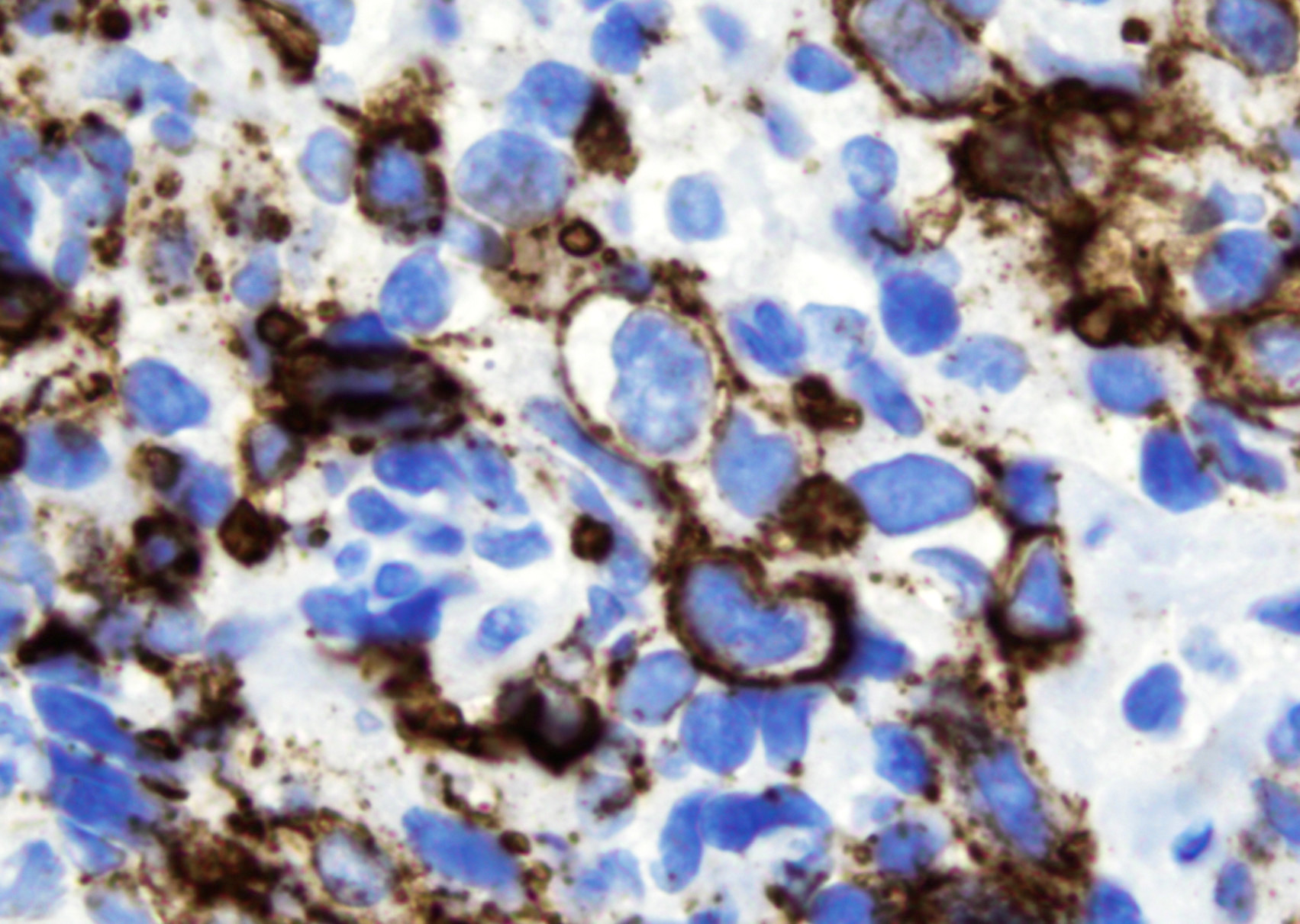
CD20

CD68
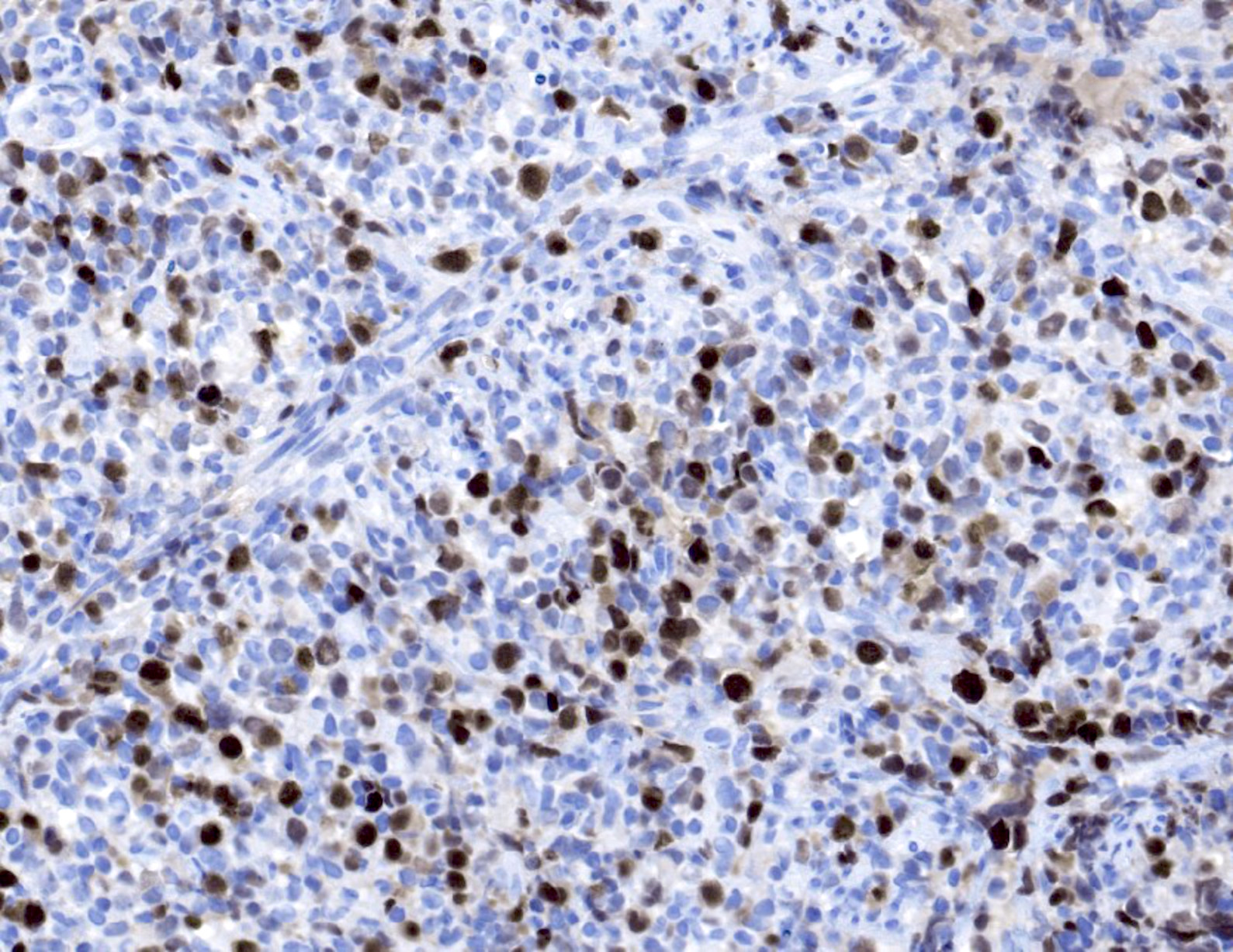
MUM1
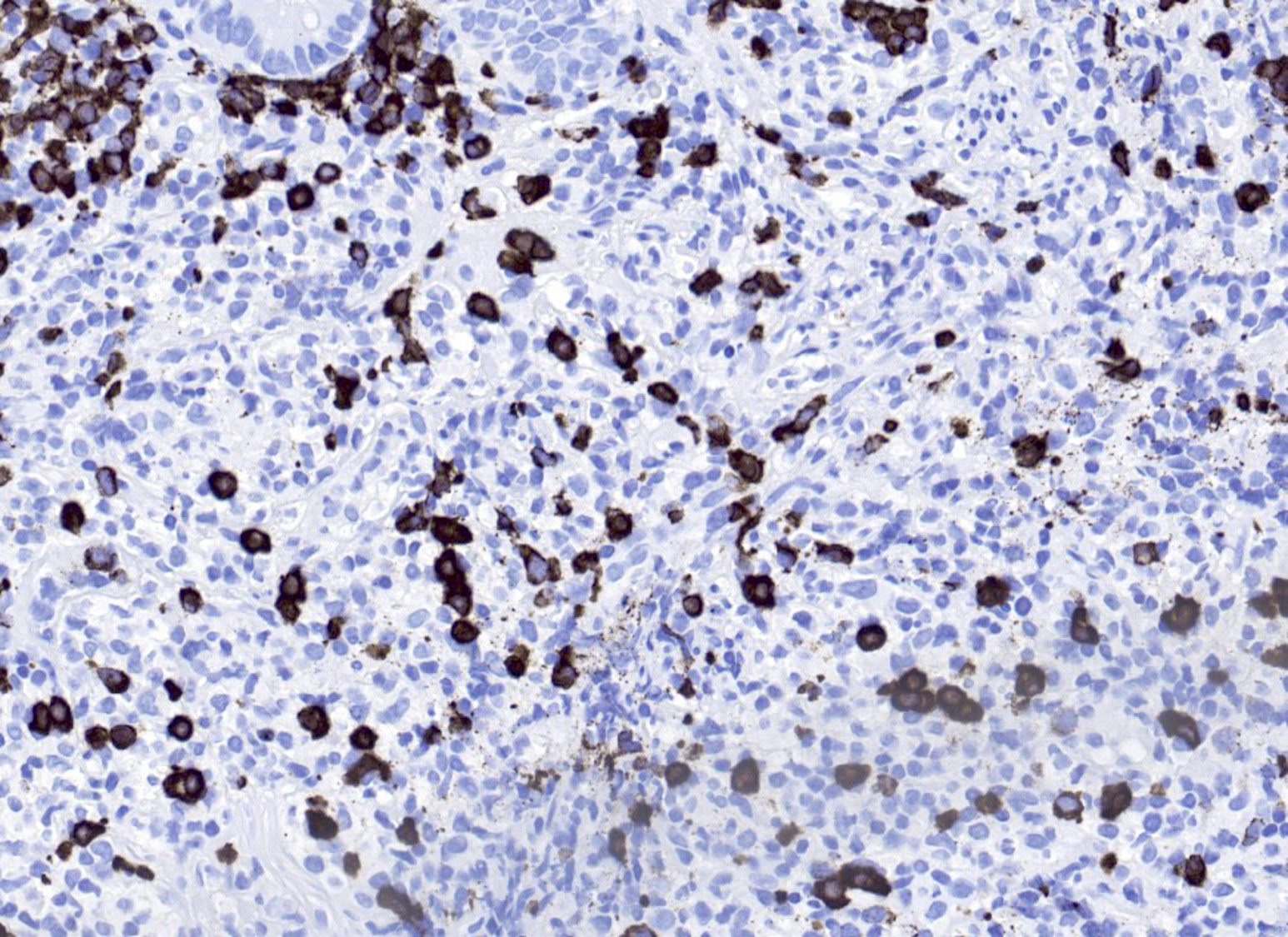
Kappa light chain

Lambda light chain
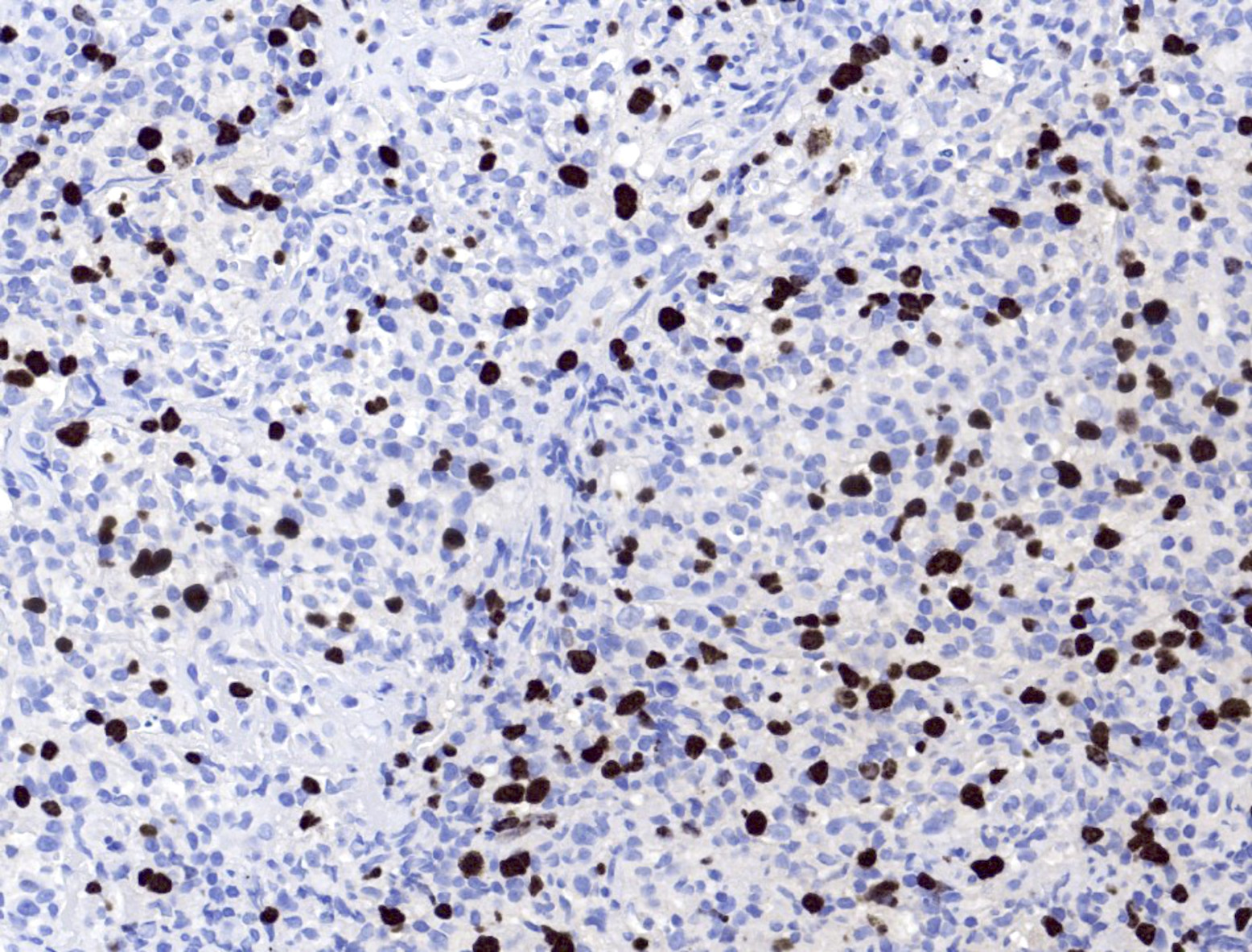
EBV EBER
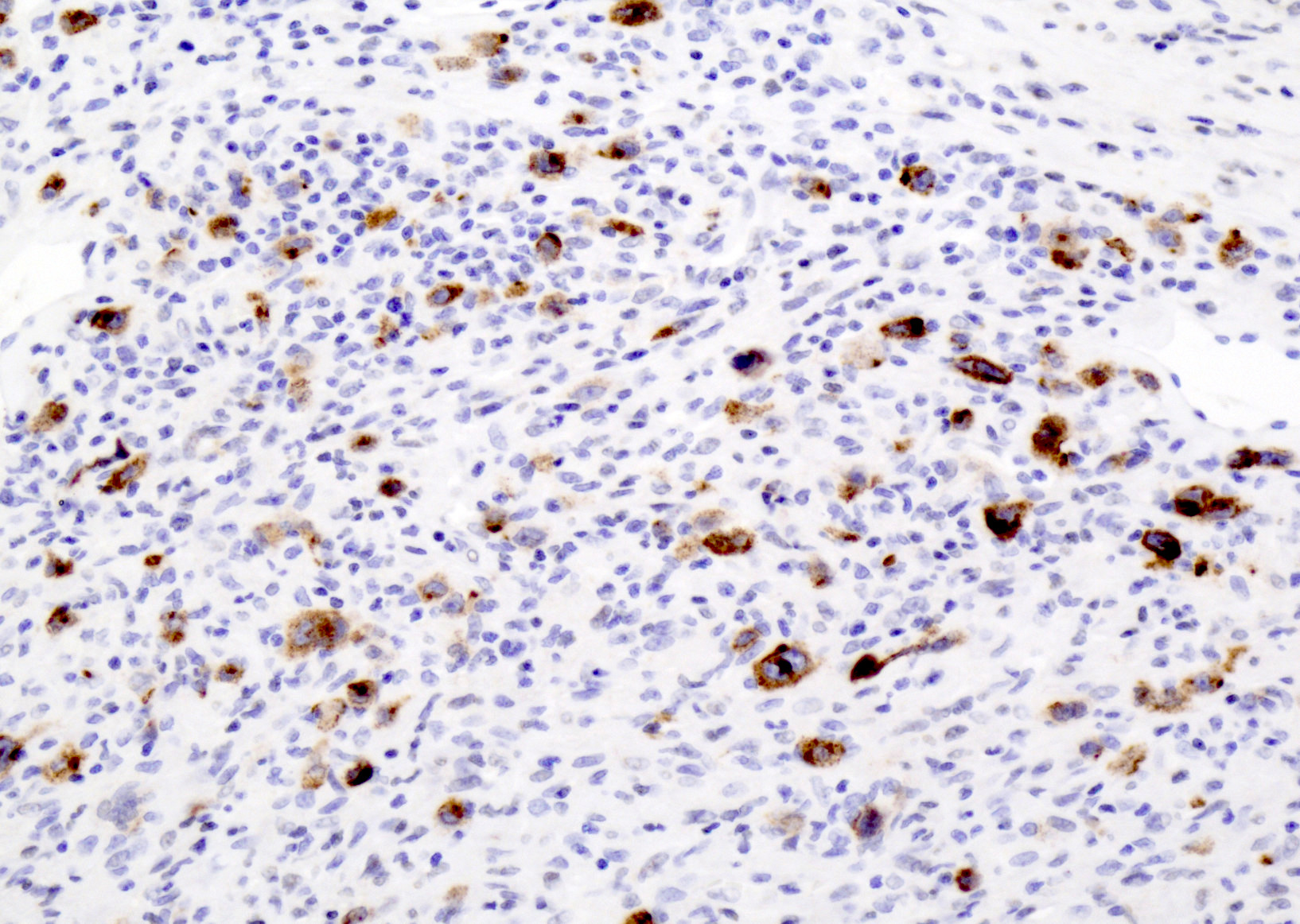
LMP1
Positive stains
- Intensity and proportions of all markers listed below are variable depending on the predominating cell type
- B cell markers: CD20, CD79a, PAX5, OCT2, BOB1
- T cell markers: CD3, CD5, CD7, CD4, CD8
- CD30: immunoblasts, HRS-like cells (variable staining on cells and between cells)
- CD15: less frequently positive in HRS-like cells
- EBV EBER (variably positive in cells of varying size)
- EBV LMP
- Kappa / lambda light chains showing polytypic staining in plasma cells
- Reference: Am J Clin Pathol 2017;147:129
Negative stains
- Not relevant to the diagnosis
Flow cytometry description
- Not essential for diagnosis; mixed lymphoid population expected to be seen
Molecular / cytogenetics description
- These cases typically lack genetic alterations, which explains its regression upon reduction in immunosuppression (N Engl J Med 1998;338:1413, N Engl J Med 2004;350:1328)
- Gene rearrangement studies show monoclonal or oligoclonal background in most cases
- Although uncommon, few karyotype abnormalities have been reported (Genes Chromosomes Cancer 2006;45:313, Hum Pathol 2007;38:315)
- Rearrangement of 1q21.3
- Gain of X
Sample pathology report
- Rectum, biopsy:
- Polymorphic lymphoproliferative disorder, EBV+, in the setting of HIV infection (see comment)
- Comment: The patient's history of HIV / AIDS with proctitis and lymphadenopathy is noted. The current specimen shows a polymorphic lymphoid infiltrate composed of small cells, plasma cells, immunoblasts and large, atypical cells. Occasional Reed-Sternberg-like cells are seen. EBV EBER is positive in many cells and plasma cells are polytypic. Immunoglobulin heavy chain and kappa light chain gene rearrangement studies are negative.
Differential diagnosis
- Chronic inflammation (e.g., gastritis):
- No history of immune deficiency / dysregulation
- Histologic features of inflammatory processes characteristic of the site of involvement
- Architecture is generally preserved
- Negative for EBV
- Infectious mononucleosis (IM)-like hyperplasia (Am J Clin Pathol 2017;147:129):
- Lacks architectural destruction
- Varying degrees of follicular hyperplasia
- Hyperplasias arising in immune deficiency / dysregulation (IDD):
- Heterogeneous proliferations with preserved underlying architecture involved by varying histologic features:
- Follicular hyperplasia (FH)
- Infectious mononucleosis-like hyperplasia (IMH)
- Plasmacytic hyperplasia
- Usually driven by EBV of KSHV / HHV8
- Heterogeneous proliferations with preserved underlying architecture involved by varying histologic features:
- EBV positive mucocutaneous ulcer (EBVMCU) (Int J Mol Sci 2021;22:1053):
- Confined to the oral mucosa, skin or gastrointestinal tract
- Sharply circumscribed ulcerative lesion
- No adenopathy is seen
- Lymphomas arising in immune deficiency / dysregulation (IDD):
- Meets diagnostic criteria for B cell or T / NK cell neoplasms in immunocompetent hosts:
- B cell neoplasms (Ann Oncol 1991;2:289):
- Diffuse large B cell lymphoma
- Burkitt lymphoma
- Classic Hodgkin lymphoma (JCO Glob Oncol 2021;7:277)
- Multiple myeloma
- Plasmacytoma
- Other
- Indolent small B cell lymphomas arising in IDD setting are not included in LPD-IDD, with the exception of EBV positive marginal zone lymphoma
- T cell neoplasms (rare) (Am J Surg Pathol 1994;18:1092, Am J Surg Pathol 1999;23:1208, Cardiovasc Pathol 2000;9:49, J Clin Exp Hematop 2019;59:56, Mod Pathol 2019;32:1135)
- B cell neoplasms (Ann Oncol 1991;2:289):
- Meets diagnostic criteria for B cell or T / NK cell neoplasms in immunocompetent hosts:
- Lymphomatoid granulomatosis (LYG):
- Angiocentric and angiodestructive polymorphous lymphoid infiltrate
- Pulmonary involvement seen in > 90% of cases
- Classic HRS cells are generally not present
- Skin lesions may show prominent granulomatous reaction
- Prominent vascular changes such as lymphocytic vasculitis and fibrinoid necrosis
- Not associated with IDD setting
- Cases associated with IDD should be classified as polymorphic LPD, LYG type or monomorphic LPD, diffuse large B cell lymphoma, LYG type
- Diffuse large B cell lymphoma, EBV positive / negative:
- Does not show the spectrum of B cell differentiation stages
- Histologically can present as sheets of large, atypical B cells
- Clonal
- Classic Hodgkin lymphoma:
- Nodal T follicular helper (TFH) lymphoma, angioimmunoblastic type:
- Proliferation of variably sized lymphoid cells with clear cytoplasm (Am J Surg Pathol 2007;31:1077)
- Shows T follicular helper (TFH) phenotype (Leuk Lymphoma 2016;57:2804)
- TCR rearrangements seen in 75 - 90% of cases (Sci Rep 2017;7:11301)
- RHOA and IDH2 mutations are seen in up to 70% of cases (Nat Genet 2014;46:171)
- Plasmablastic lymphoma:
- Diffuse proliferation of immunoblastic or plasmablastic cells
- MYC rearrangements are common
- Clonal
- Low grade B cell lymphomas:
- Monotonous proliferation of small lymphoid cells with typical diagnostic features of each entity
- Lacks history of immune deficiency / dysregulation
- EBV negative
Additional references
Board review style question #1
An excisional lymph node biopsy from a patient with rheumatoid arthritis on methotrexate shows the histologic findings seen in the image above. EBV EBER is positive in many cells of varying sizes. What is the most appropriate diagnostic interpretation?
- Diffuse large B cell lymphoma, EBV+, iatrogenic methotrexate related
- Infectious mononucleosis
- Polymorphic lymphoproliferative disorder, EBV+, iatrogenic methotrexate related
- Reactive follicular hyperplasia
Board review style answer #1
C. Polymorphic lymphoproliferative disorder, EBV+, iatrogenic methotrexate related. These findings are consistent with polymorphic lymphoproliferative disorder (LPD) arising in immune deficiency / dysregulation. Polymorphic LPDs in the posttransplant setting are among the most commonly seen.
Comment Here
Reference: Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation
Comment Here
Reference: Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation
Board review style question #2
A 40 year old woman post-renal transplant presents with iliac lymphadenopathy. Excisional biopsy is consistent with polymorphous posttransplant lymphoproliferative disorder. Which of the following histologic features is characteristic of this entity?
- Florid reactive follicles with prominent germinal centers
- Mixed population of plasma cells, histiocytes, variably sized lymphocytes and immunoblasts effacing normal lymphoid architecture
- Necrotizing granulomatous lymphadenitis with palisading histiocytes
- Sheets of immunoblasts diffusely replacing normal architecture
Board review style answer #2
B. Mixed population of plasma cells, histiocytes, variably sized lymphocytes and immunoblasts effacing normal lymphoid architecture. In polymorphic LPDs, these are the usual histologic findings.
Comment Here
Reference: Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation
Comment Here
Reference: Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation

