

Liver & intrahepatic bile ducts
Developmental anomalies / cysts
Polycystic liver disease
Author: Alexander Kikuchi, M.D., Ph.D.
Editorial Board Member: Kimberley J. Evason, M.D., Ph.D.
Deputy Editor-in-Chief: Aaron R. Huber, D.O.

Last author update: 2 October 2024
Last staff update: 2 October 2024
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Polycystic liver disease

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Frozen section description | Frozen section images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Kikuchi A. Polycystic liver disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverpolycysticliverdisease.html. Accessed December 22nd, 2024.
Definition / general
- Form of ductal plate malformation that leads to benign cystic dilation of intrahepatic bile ducts
- Most frequently associated with autosomal dominant polycystic kidney disease (ADPKD)
- May develop in absence of renal cystic disease in association with a heterogeneous group of genetic disorders
Essential features
- Numerous, simple biliary cysts of varying size and shape with associated biliary hamartomas (von Meyenburg complexes)
- Form of abnormal development of intrahepatic biliary system (ductal plate malformation)
- Predominantly associated with autosomal dominant polycystic kidney disease (ADPKD)
- Rarely isolated polycystic liver disease can occur (isolated polycystic liver disease, PCLD)
- Symptomatic management (medical versus surgical) predominantly to alleviate mechanical effects of liver enlargement
- This entry addresses adult polycystic liver disease; please see separate related entries for congenital hepatic fibrosis and Caroli disease
Terminology
- Fibropolycystic liver disease: more generic term to describe heterogeneous group of genetic diseases including adult polycystic liver disease (frequently associated with ADPKD) as well as congenital hepatic fibrosis and Caroli disease
Epidemiology
- ADPKD prevalence: 1/400 - 1/1,000 (Lancet 2019;393:919)
- Liver cysts present in > 79 - 94% of ADPKD patients ≥ 35 years (Clin J Am Soc Nephrol 2006;1:64, Clin Gastroenterol Hepatol 2015;13:155)
- Most cases of polycystic liver disease arise in ADPKD (see Etiology subsection)
- ADPLD (or PCLD) with prevalence of 1/100,000 to 1/1,000,000 (Orphanet J Rare Dis 2014;9:69, Liver Int 2011;31:92)
Sites
- Liver (by definition)
- Predominantly associated with renal cyst development (ADPKD)
- Asymptomatic cyst formation in other organs in ADPKD (Kidney Int 1997;51:2022, Int J Mol Sci 2024;25:2554, Liver Int 2008;28:264)
- Pancreas, spleen, lungs, seminal vesicles, central nervous system (CNS) (arachnoid cysts, spinal meningeal diverticula) (Medicine (Baltimore) 2020;99:e19511)
- Valvular heart disease (e.g., mitral valve prolapse) (Liver Int 2008;28:264)
- Intracranial aneurysms
Pathophysiology
- Ductal plate is a layer of hepatoblasts (multipotent liver progenitor cells) that envelops portal vein branches and accompanies them as they spread through liver (beginning at hilum) during embryonic development (Anat Rec (Hoboken) 2008;291:628)
- From ~12 weeks of gestation onward, ductal plate remodels and forms intrahepatic biliary tree
- Ductal plate malformation
- Aberrant remodeling or lack of remodeling of ductal plate
- Defects in biliary precursor cell differentiation, maturation of primitive bile ducts and abnormal bile duct enlargement (Nat Rev Gastroenterol Hepatol 2014;11:750)
- Can lead to persistence of primitive duct structures
- Abnormal dilation of intrahepatic ducts leading to biliary cystic formation in polycystic liver disease (Orphanet J Rare Dis 2014;9:69)
- Abnormalities in signaling function of cholangiocyte cilia (Annu Rev Pathol 2022;17:251)
- Enhanced cholangiocyte proliferation and fluid secretion (Nat Rev Gastroenterol Hepatol 2014;11:750)
- Cysts become detached from biliary tree as disease progresses
- May arise from cystic dilation of biliary ducts in biliary hamartomas (von Meyenburg complexes)
Etiology
- Cystic dilation of intrahepatic biliary tree related to ductal plate malformation (see Pathophysiology section)
- In adults, most frequently associated with autosomal dominant polycystic kidney disease (ADPKD)
- ~80 - 85% due to mutations in PKD1 gene (encoding polycystin 1, PC1) (Adv Anat Pathol 2005;12:126, Adv Chronic Kidney Dis 2010;17:118)
- ~15% due to mutations in PKD2 gene (encodes polycystin 2, PC2) (Annu Rev Pathol 2022;17:251)
- Remaining small percentage of cases due to mutations in GANAB, LRP5, DNAJB11, ALG9 (Eur J Med Genet 2021;64:104160)
- PC1 and PC2 form a heterodimeric plasma membrane channel with roles in regulation of tubular morphology, cell proliferation, migration and differentiation (Nat Cell Biol 2005;7:1202, J Med Genet 2003;40:311)
- Isolated polycystic liver disease (PCLD) / autosomal dominant polycystic liver disease (ADPLD)
- Occurs independently from renal cystic disease (Clin Genet 1986;30:29)
- Mutations in PRKCSH gene encoding hepatocystin (Hepatology 2004;39:924, Nat Genet 2003;33:345, Am J Hum Genet 2003;72:691)
- Mutations in SEC63 gene encoding Sec63p (Nat Genet 2004;36:575)
- Mutation in LRP5 gene encoding LRP5 transmembrane protein (Proc Natl Acad Sci U S A 2014;111:5343)
- Other rare syndromes can also be associated with polycystic liver disease
- Nephronophthisis (Am J Med Genet C Semin Med Genet 2009;151C:296)
- Congenital disorders of glycosylation (J Pediatr Gastroenterol Nutr 2021;73:444)
Clinical features
- Usual manifestation is asymptomatic liver enlargement
- Number and size of liver cysts increases with age (Hepatology 1990;11:1033)
- Often clinically detected in fourth decade of life
- Symptomatic in a minority of individuals
- Abdominal pain / distension, orthopnea, dyspnea, early satiety
- Symptoms considered secondary to mechanical disruption / compromise of thoracoabdominal visceral function
- Compression of inferior vena cava
- Can lead to lower extremity edema, cyst infection / hemorrhage / rupture
- Rarely, venous compression and ascites can lead to fatal hepatic failure (Br J Surg 1992;79:562)
Diagnosis
- Hepatic and renal cysts can be detected by ultrasound, computed tomography (CT) or magnetic resonance imaging (MRI) (Abdom Radiol (NY) 2022;47:2356, Semin Ultrasound CT MR 2009;30:368)
- Numerous cysts (typically > 20) with varying size and distribution
- Presence of numerous hepatic cysts without renal cystic disease may be consistent with ADPLD (isolated polycystic liver disease)
- Positive family history of renal / hepatic cystic disease in autosomal dominant inheritance pattern
- Molecular diagnostics can confirm the diagnosis
- PKD1 and PKD2 mutations in ADPKD
- PRKCSH and SEC63 mutations in ADPLD
Laboratory
- Liver function enzymes are normal in a majority of cases
- Serum gamma glutamyl transferase (GGT) and alkaline phosphatase (ALP) can be elevated in severe disease (Liver Int 2011;31:92, Nat Rev Gastroenterol Hepatol 2013;10:101)
- CA19-9 raised in a subset of patients with polycystic liver disease, correlating with liver volume (Liver Int 2009;29:1389)
Radiology description
- Cysts typically homogeneously anechoic on ultrasound, T2 hyperintense on MRI without associated enhancement (Abdom Radiol (NY) 2022;47:2356, Semin Ultrasound CT MR 2009;30:368)
- Detection of complications arising from cyst formation (Abdom Radiol (NY) 2022;47:2356)
- Hemorrhage (T1 hyperintense dependent fluid with absence of enhancement on subtraction)
- Infected cysts (thick wall enhancement, occasionally restricted diffusion on diffusion weighted imaging)
Radiology images
Images hosted on other servers:


CT and MRI
Prognostic factors
- Autopsy series evidence of positive correlation between number of biliary microhamartomas (von Meyenburg complexes) and clinical severity of disease (Arch Pathol Lab Med 1990;114:180)
- Risk factors for high number and increased size of liver cysts (Hepatology 1990;11:1033)
- Older age
- Female sex
- Previous pregnancy, number of pregnancies
- Hormone replacement therapy
Case reports
- 23 month old girl with isolated polycystic liver disease (Int J Surg Case Rep 2023;112:108950)
- 51 year old man with polycystic liver disease and aneurysms (Ann Transl Med 2016;4:167)
- 57 year old man with polycystic liver disease who underwent liver transplantation (J Surg Case Rep 2023;2023:rjad507)
- 72 year old man with polycystic liver disease associated with autosomal dominant polycystic kidney disease with obstructive jaundice and polycystic lung (Medicine (Baltimore) 2020;99:e19511)
Treatment
- Somatostatin analogues
- Surgical interventions (Orphanet J Rare Dis 2014;9:69, Hepat Med 2022;14:135)
- Laparoscopic cyst deroofing or fenestration
- Aspiration with sclerotherapy
- Partial hepatectomy
- Liver transplantation
Gross description
- Enlarged, diffusely cystic liver
- Numerous smooth walled cysts of varying size (ranging from < 1 mm to 12 cm or greater in diameter each) (Burt: MacSween's Pathology of the Liver, 8th Edition, 2023)
- Contain clear or yellow fluid
- May have little or no intervening normal liver tissue
Gross images
Images hosted on other servers:

ADPKD patient

Enlarged polycystic liver
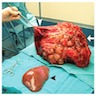
Massive hepatomegaly and numerous cysts
Frozen section description
- Frozen section generally not indicated for benign appearing cysts
- May be performed if there is macroscopic suspicion of underlying malignancy
- Solid or thickened cyst wall
- Associated nodules
- Typical findings are fibrous cyst wall fragments with simple, bland epithelial lining
- Areas of cyst denudation / epithelial sloughing common
- With or without von Meyenburg complexes
- Increased stromal cellularity underlying cyst lining (ovarian type stroma) raises consideration of mucinous cystic neoplasm
- Formal hepatic resection may be indicated for malignancy (e.g., invasive carcinoma associated with mucinous cystic neoplasm)
Frozen section images
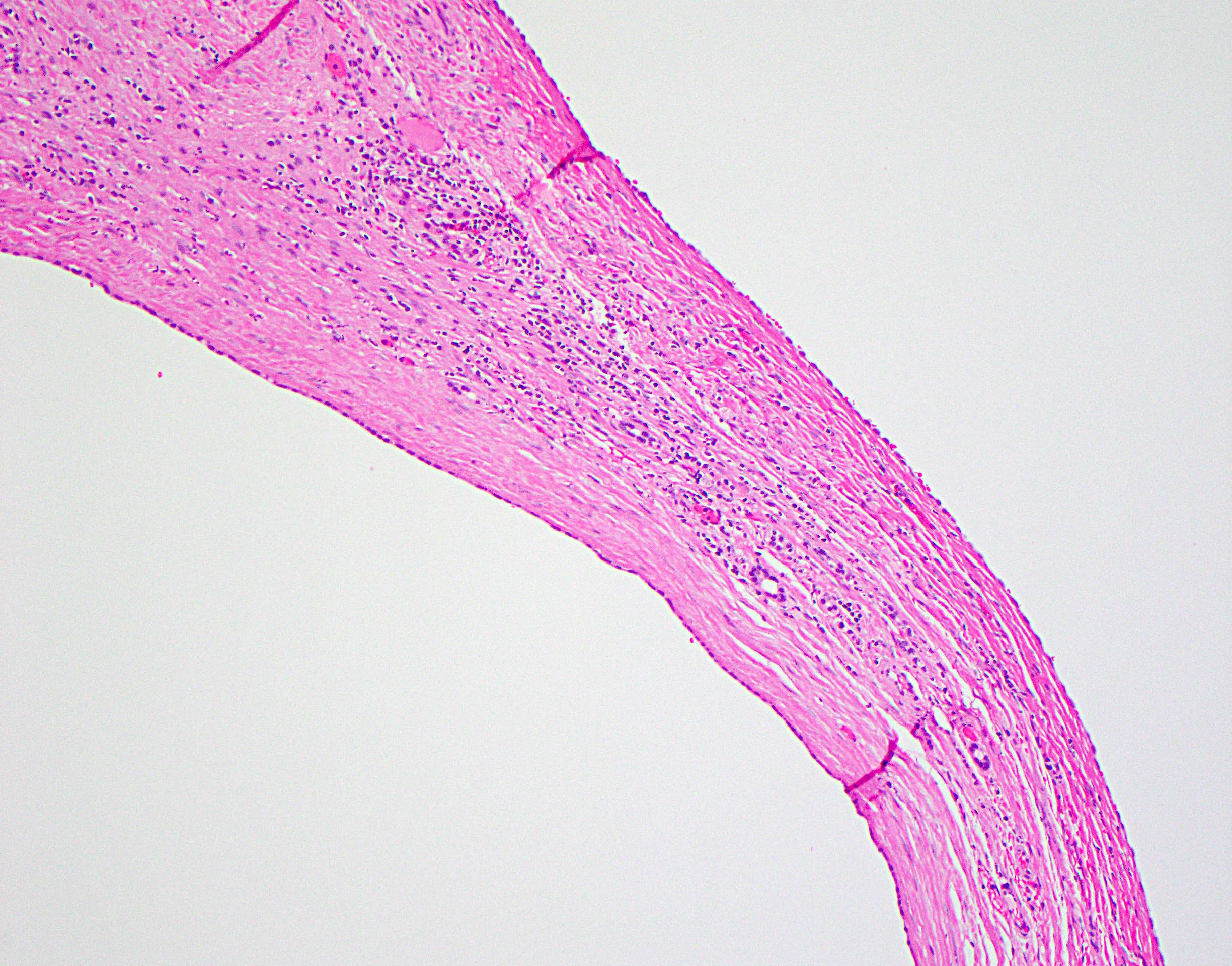
Contributed by Alexander Kikuchi, M.D., Ph.D.
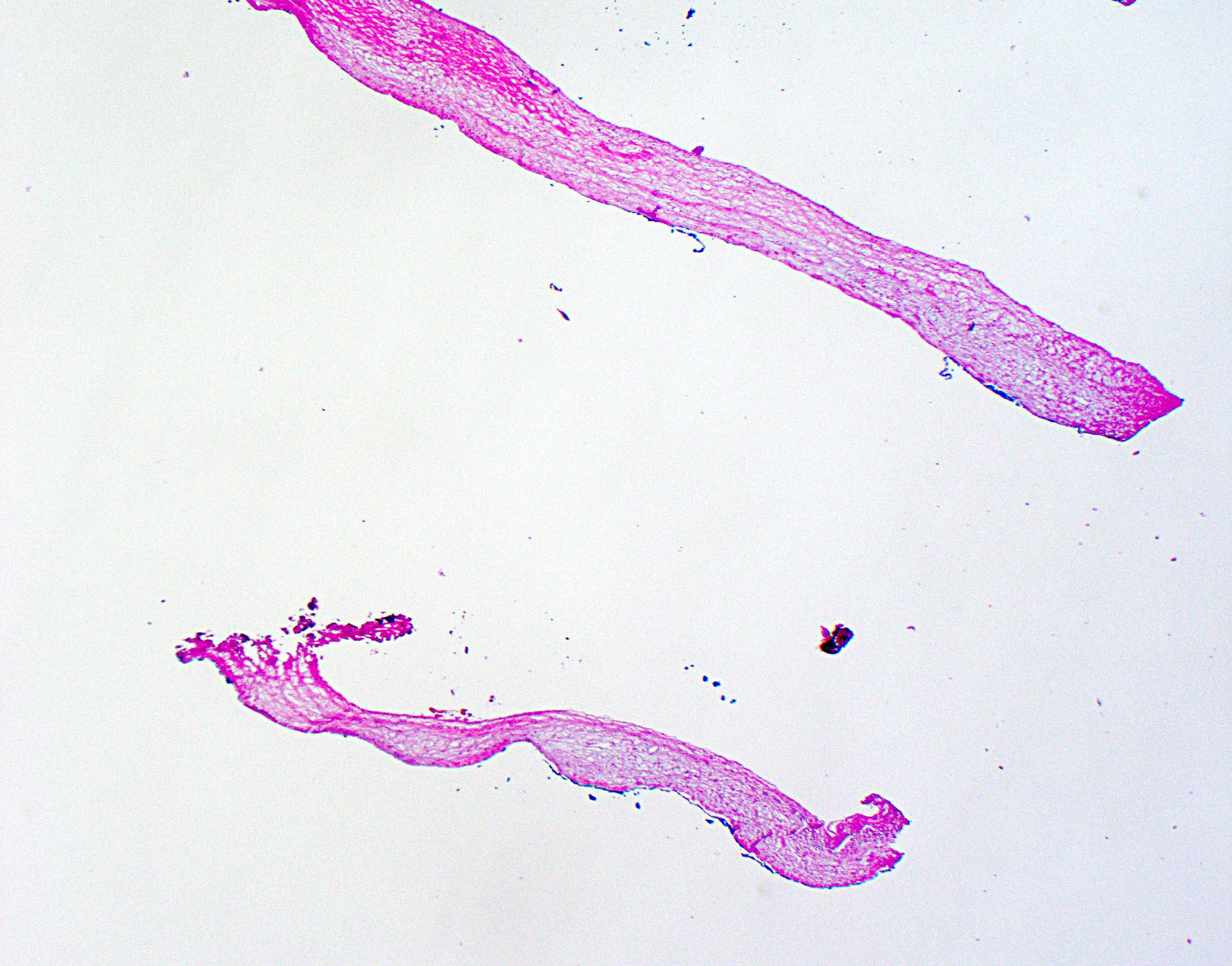
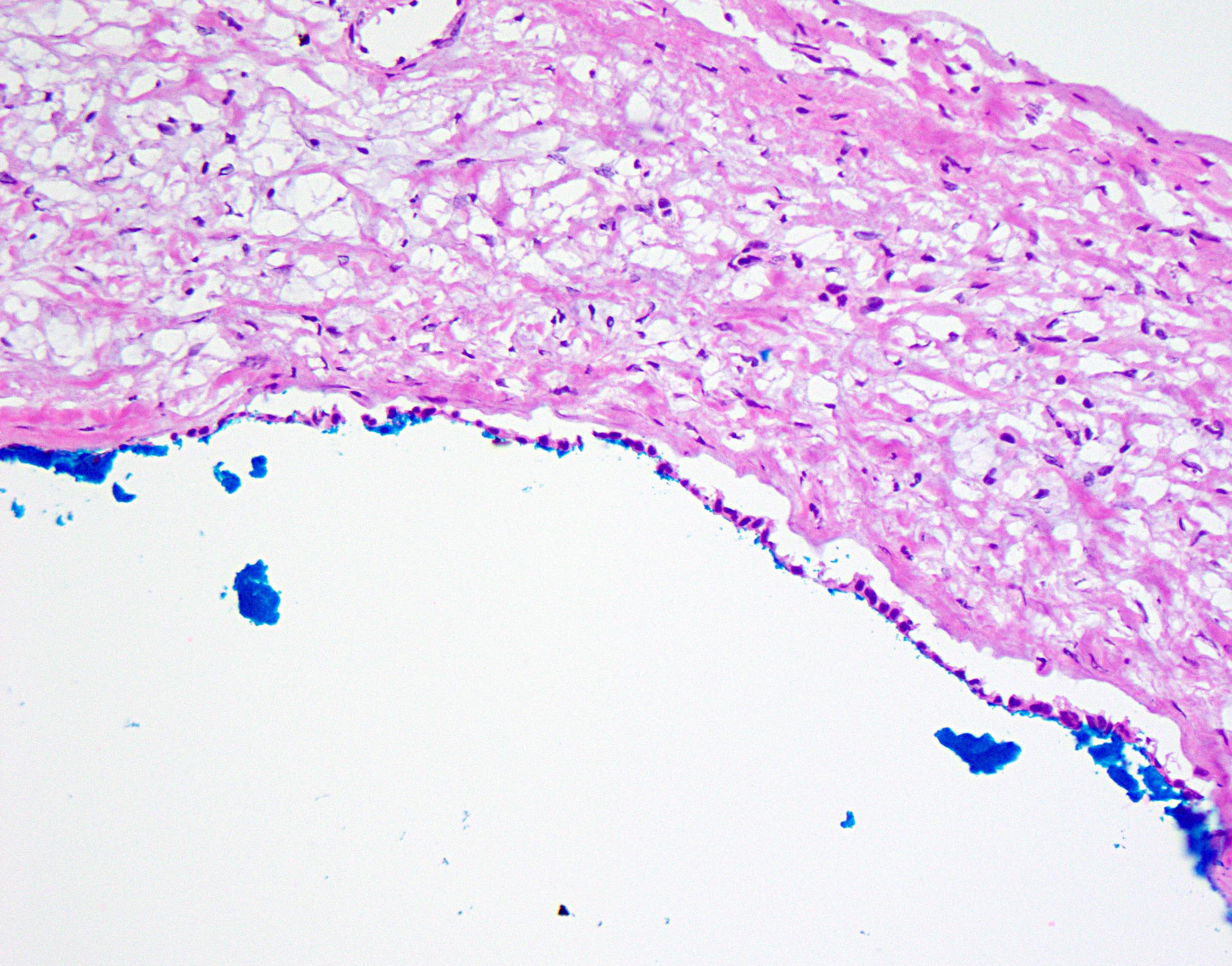
Cyst wall
Microscopic (histologic) description
- Variably sized, irregular and branching cysts lined by single layer of biliary epithelium
- Columnar or cuboidal epithelium
- Larger cysts with flattened epithelium
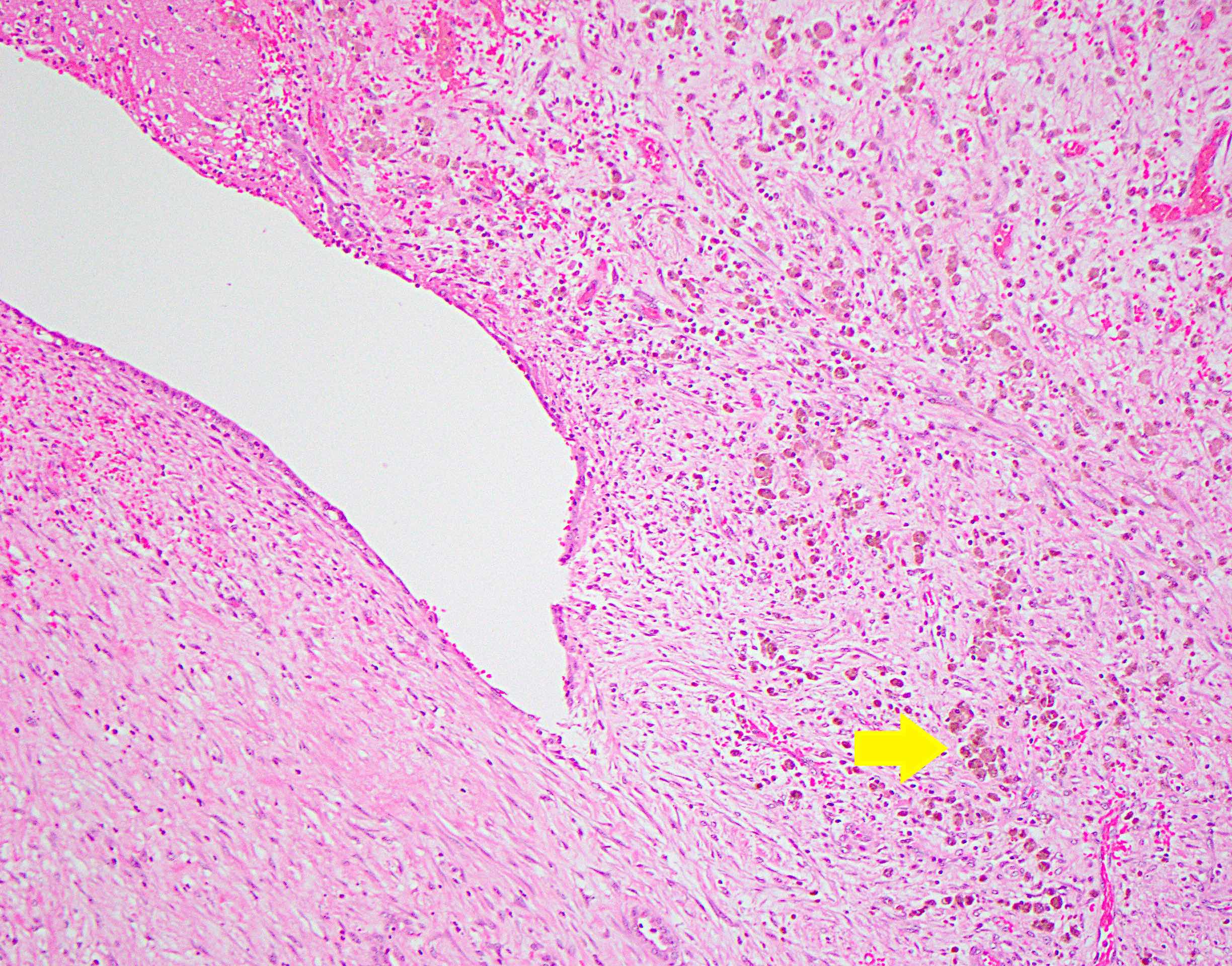
- Often associated with von Meyenburg complexes
- Discrete, round or irregular benign bile duct proliferations
- Constituent bile ducts with dilated lumina, inspissated bile
- Connective tissue between cysts generally scant
- May be dense or hyalinized
- May have associated hemorrhage, hemosiderin filled macrophages, granulation tissue
- Infected cysts may become filled with pus (neutrophilic abscesses) and rupture
- Calcification of cyst wall can be seen
- Collapsed cysts with fibrosis and hyalinization
- References: Odze: Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas, 4th Edition, 2022, Burt: MacSween's Pathology of the Liver, 8th Edition, 2023
Microscopic (histologic) images
Contributed by Alexander Kikuchi, M.D., Ph.D. and Debra L. Zynger, M.D.

Multiple simple hepatic cysts
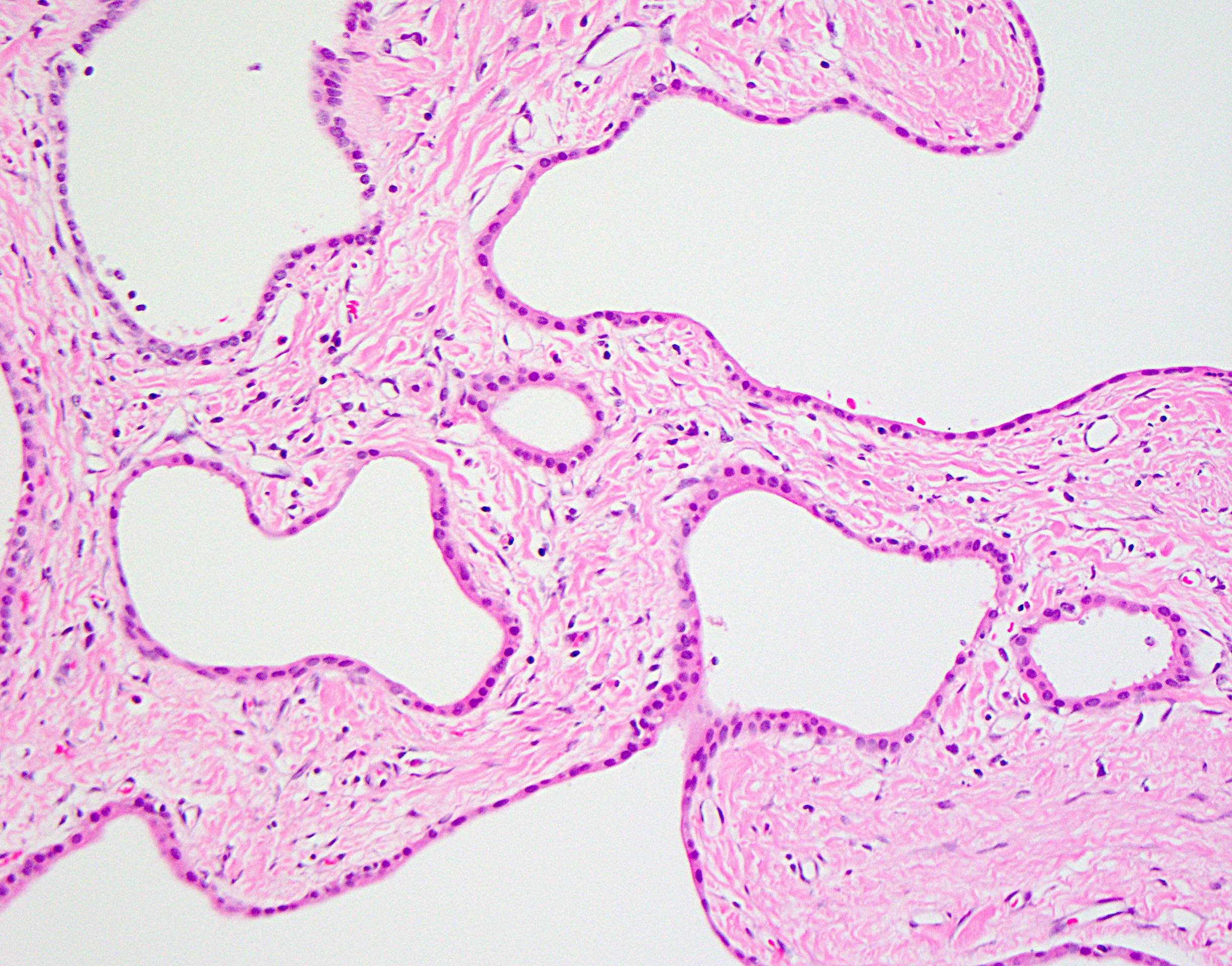
Biliary epithelial cyst lining
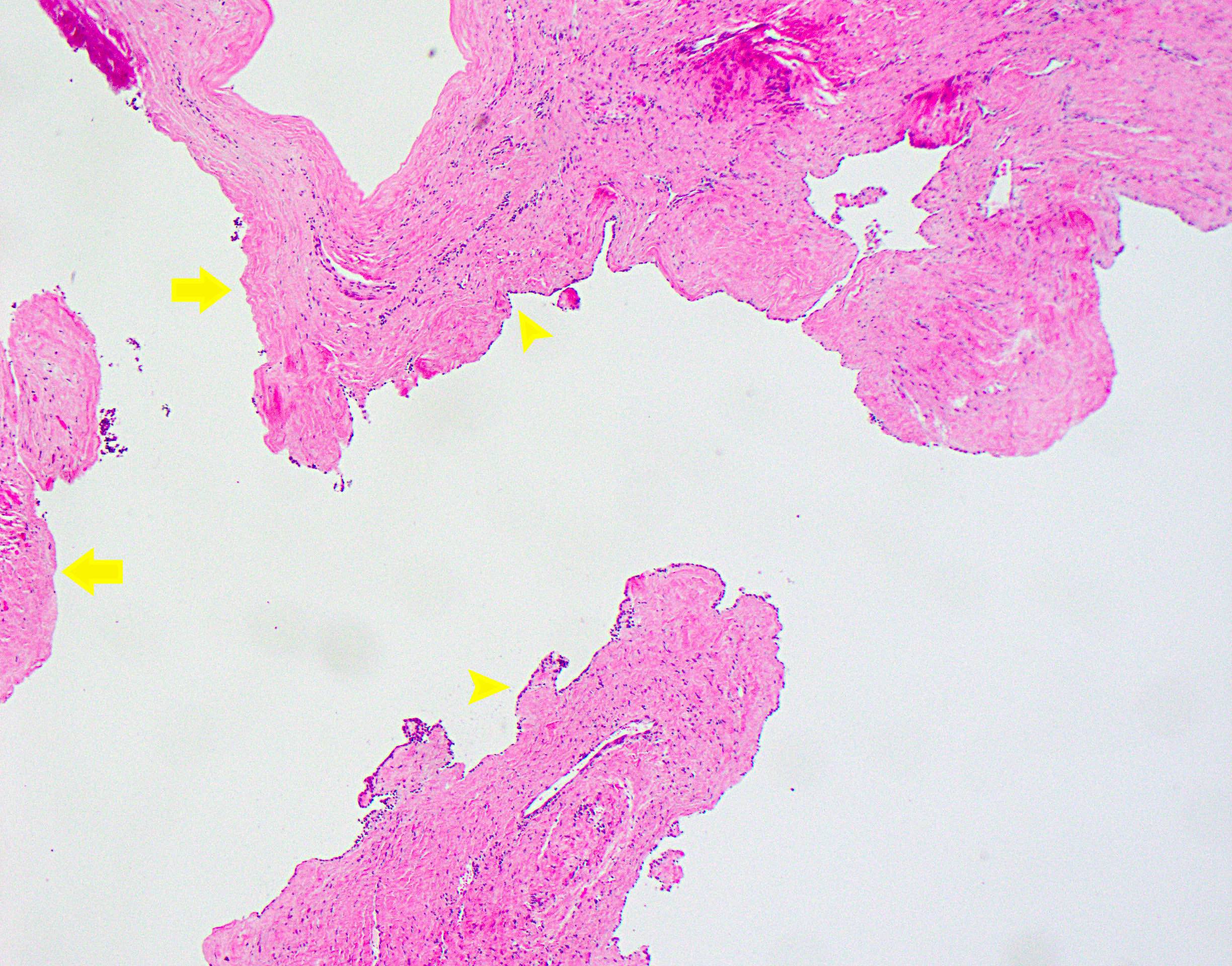
Fragmented hepatic cysts
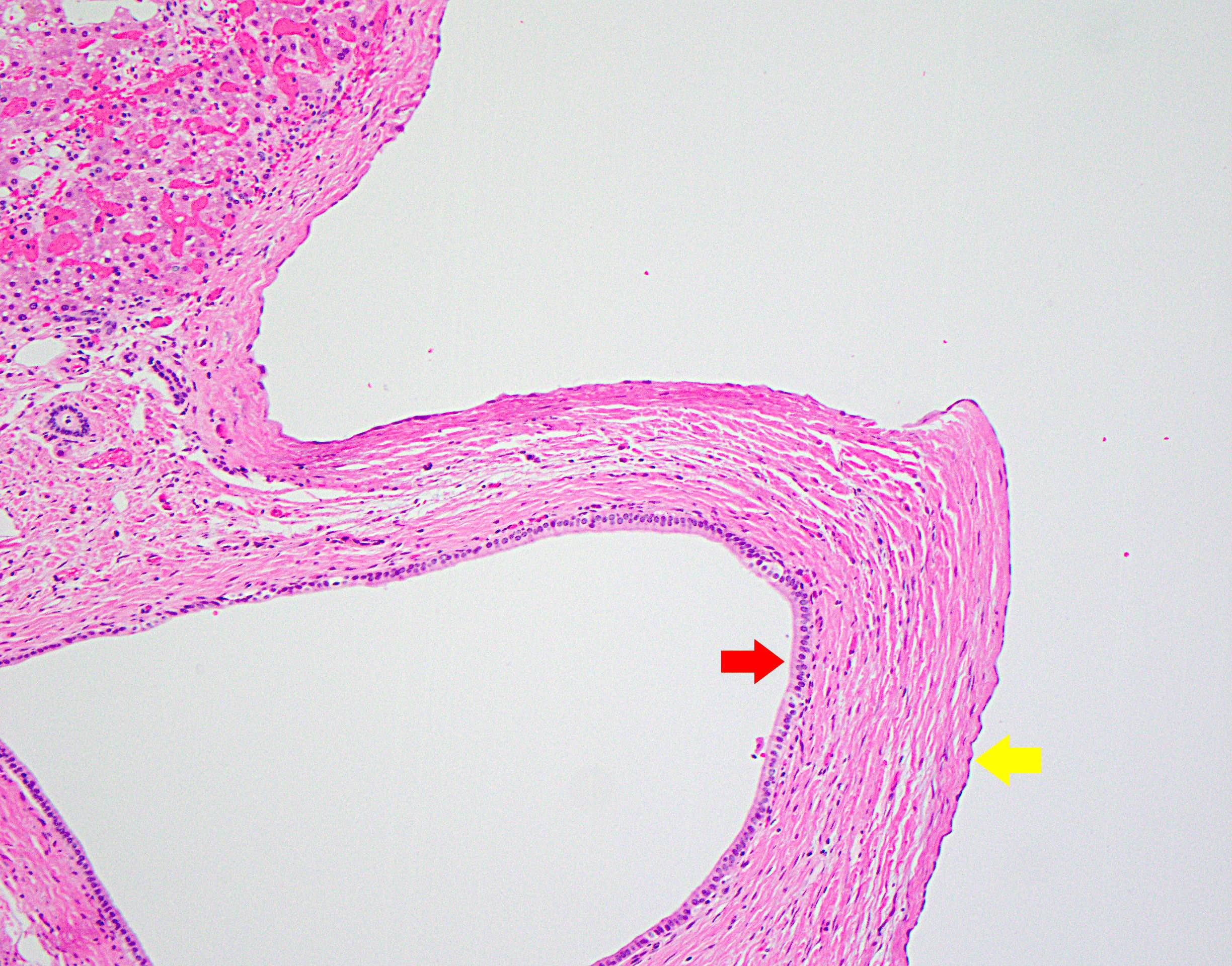
Variable cyst lining morphology
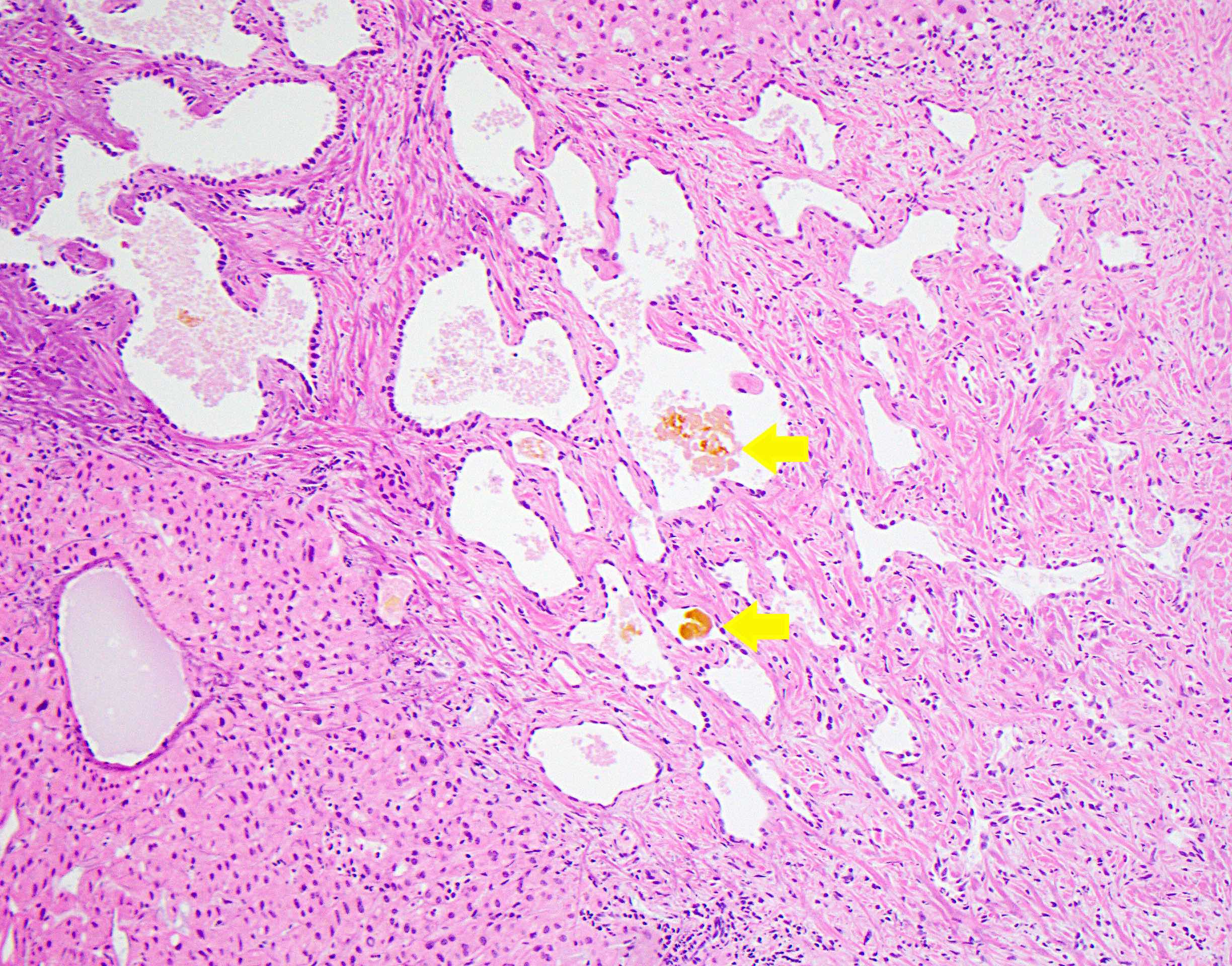
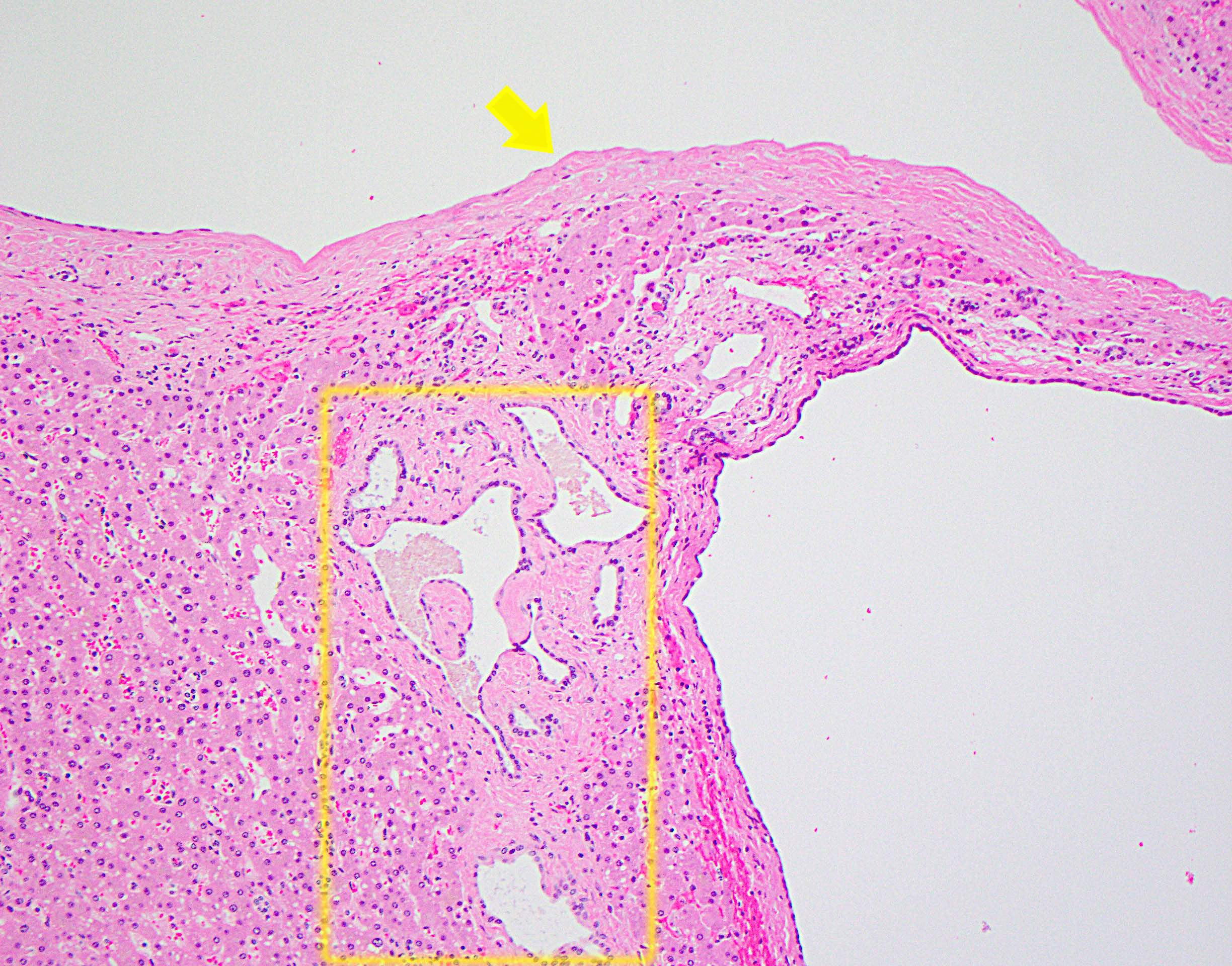
Von Meyenburg complex
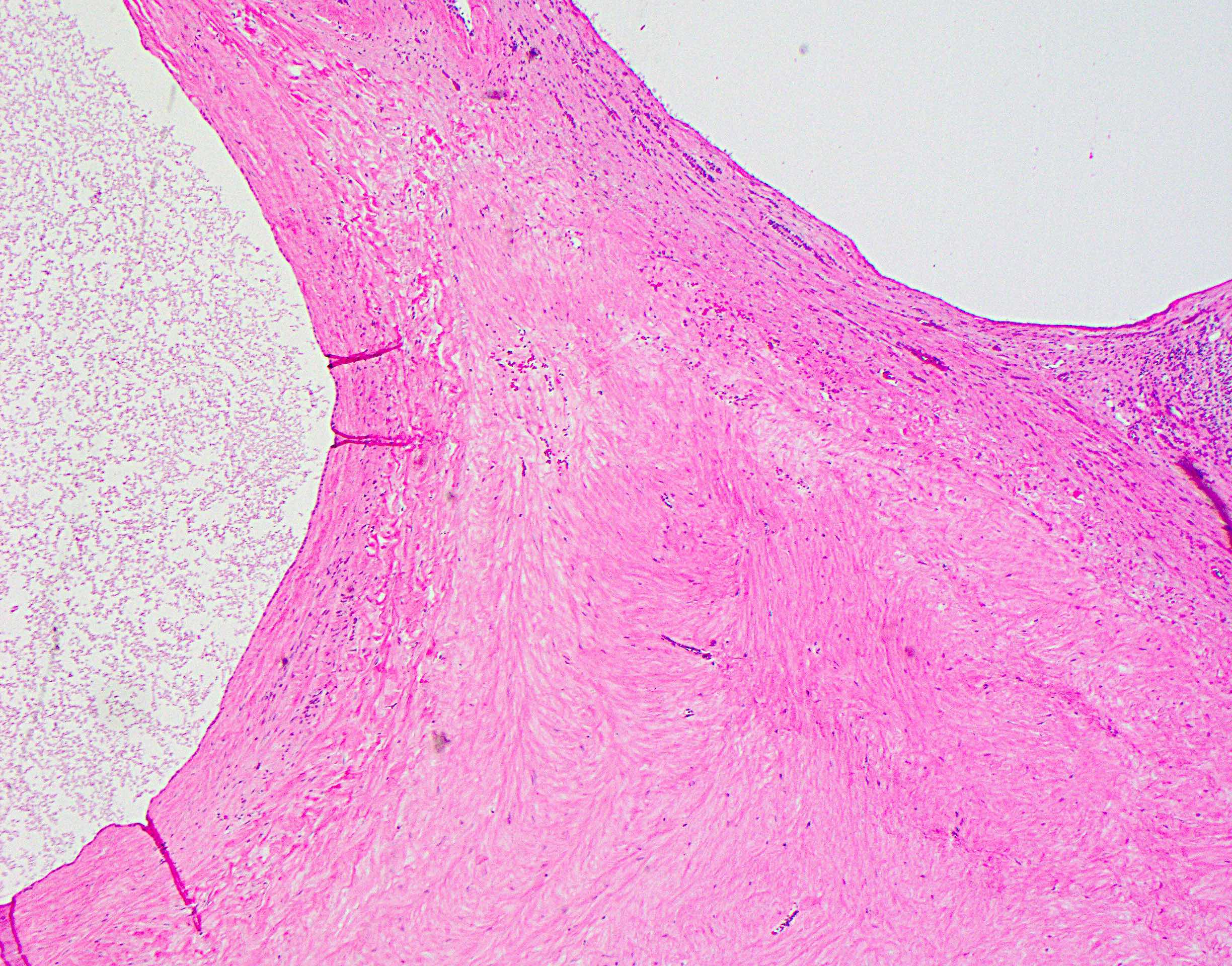
Hyalinized stroma between cysts
Hyalinized stroma, intervening parenchyma
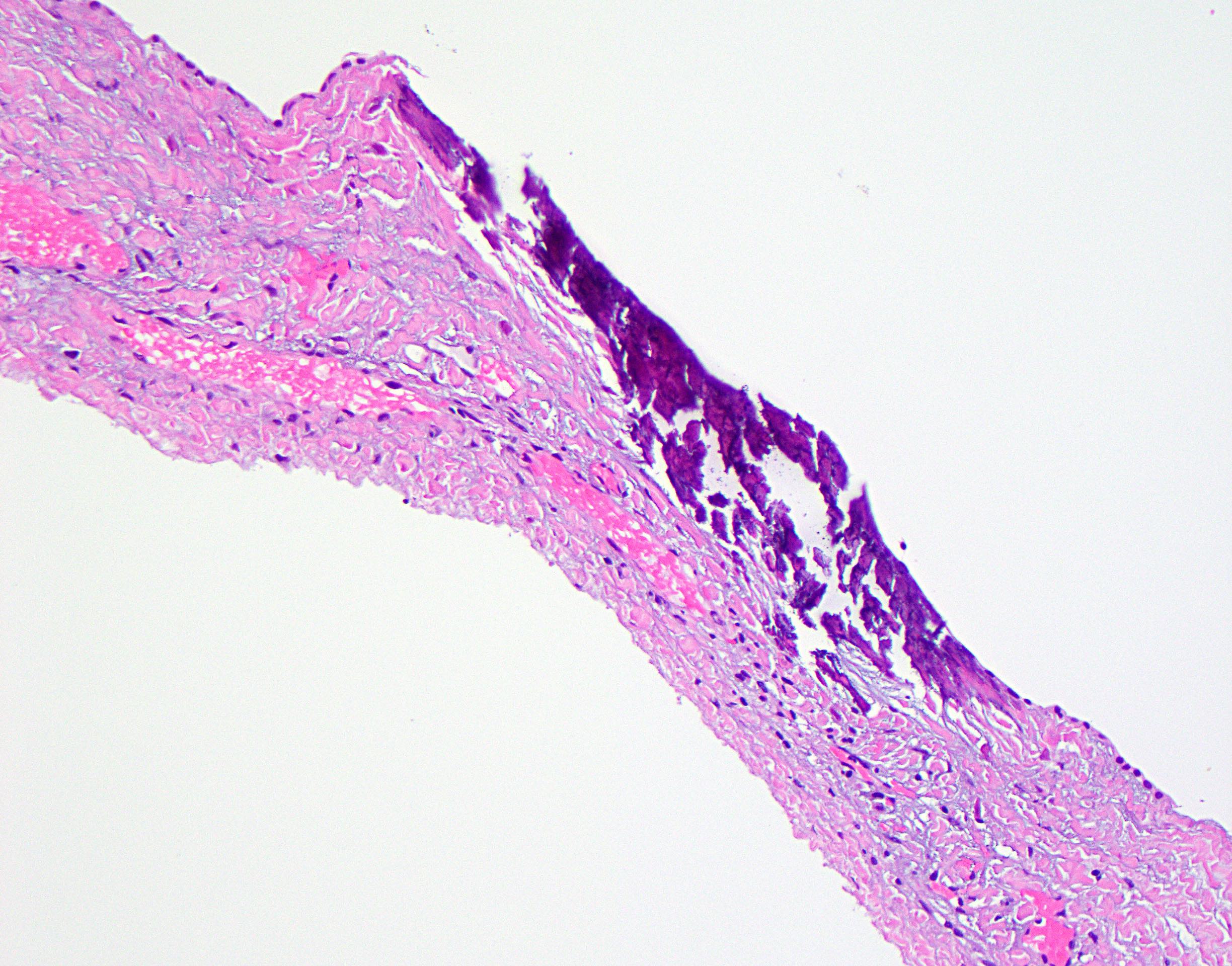
Cyst wall calcification
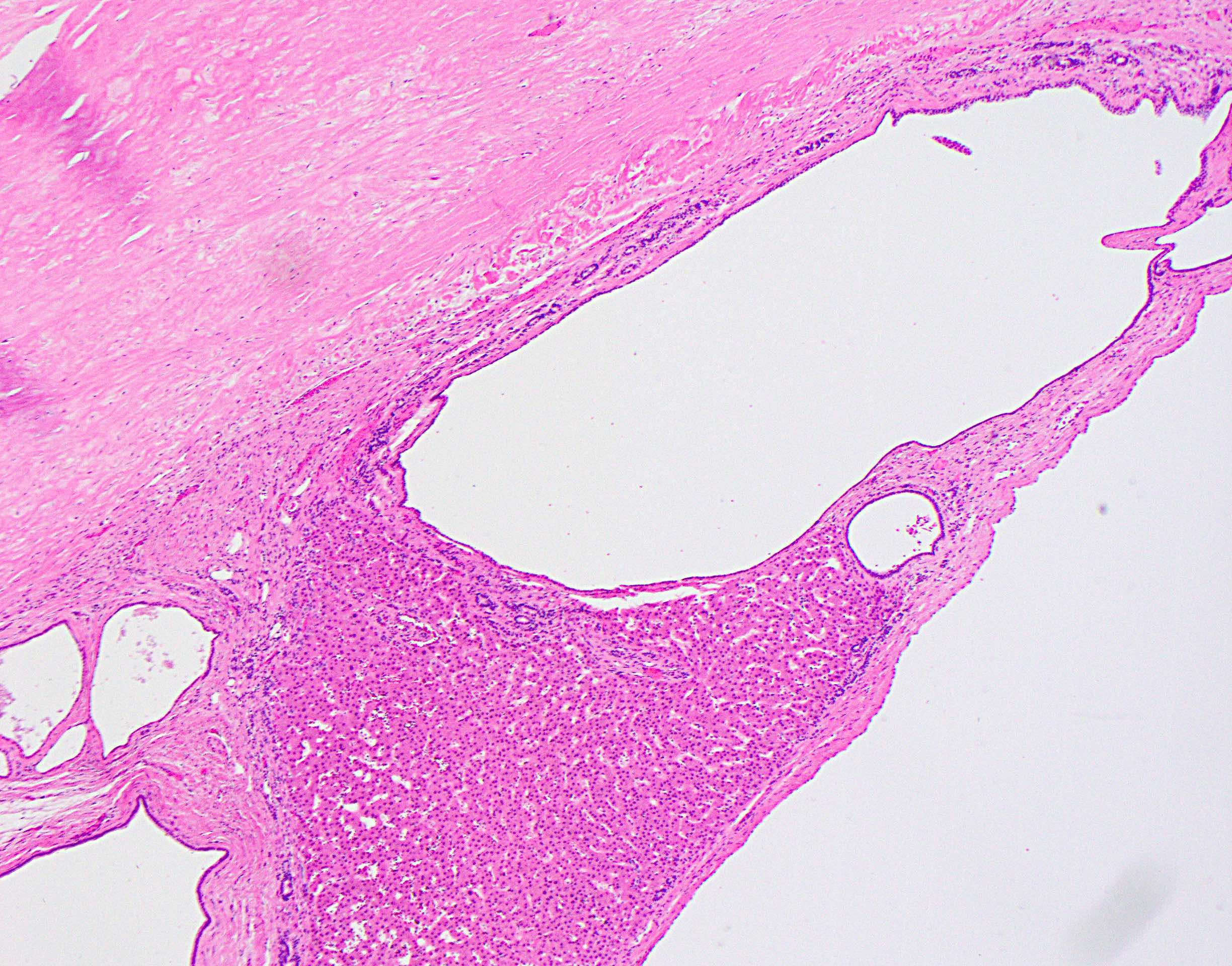
Cyst with adjacent hemorrhage


Collapsed cyst
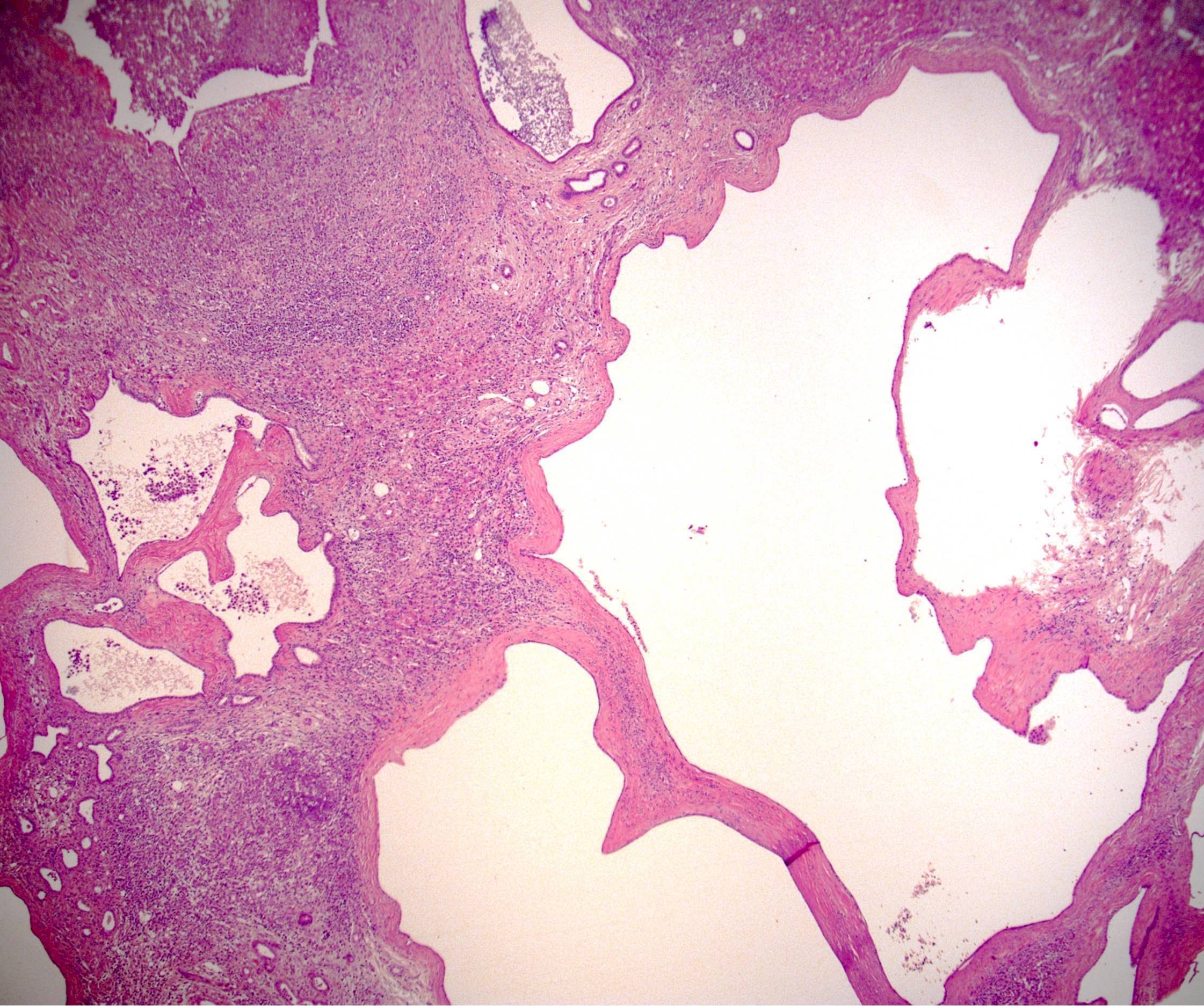
Cysts of varying sizes

Cysts

Von Meyenburg complex
Biliary epithelium versus mesothelium

HNF-1B IHC

WT1 IHC
Positive stains
Negative stains
Molecular / cytogenetics description
- Relevant genetic mutations described under Etiology subsection
Videos
Liver cysts and polycystic liver disease
Sample pathology report
- Liver, cyst wall, fenestration:
- Fragments of benign biliary cyst with associated bile duct hamartomas (see comment)
- Comment: Sections of the specimen show fragments of bland biliary type epithelium with underlying fibrous tissue and chronic inflammation consistent with cyst wall fragments. A few bile ductular proliferations as well as bile duct hamartomas (von Meyenburg complexes) are seen adjacent to the cyst lining. Rare calcification is present and patchy hemosiderin filled macrophages are seen, suggestive of prior hemorrhage. No dysplasia or evidence of malignancy is identified. These findings correlate with the imaging impression of numerous simple hepatic cysts and are consistent with polycystic liver disease.
- Native liver, explant:
- Numerous benign biliary cysts, consistent with hepatic involvement by polycystic liver disease (see comment)
- Comment: Sections of this explant demonstrate hepatic parenchyma with numerous, variably sized cysts lined by simple biliary type epithelium and separated by dense fibrous tissue with hyalinization and mild inflammatory cell infiltrates (predominantly lymphocytic). Frequent von Meyenburg complexes (bile duct hamartomas) are also identified. The background hepatic parenchyma is otherwise unremarkable, with portal areas showing mild ductular reaction with minimal inflammation and no significant duct loss or injury. The lobular parenchyma shows mild, patchy sinusoidal dilatation and congestion. No significant lobular inflammation, steatosis or cholestasis is identified. Trichrome staining shows no well established fibrosis. Iron stain is negative. PASD staining shows no intracytoplasmic globules. These findings are consistent with hepatic involvement (polycystic liver disease) by the patient's known autosomal dominant polycystic kidney disease.
Differential diagnosis
- Caroli disease:
- Associated with autosomal recessive polycystic kidney disease (ARPKD)
- May be associated with congenital hepatic fibrosis (Caroli syndrome)
- Dilated ducts with periductal fibrosis and mixed inflammation
- Epithelial lining of dilated ducts may be ulcerated, hyperplastic or dysplastic
- Transluminal fibrovascular bridges or polypoid intraluminal protrusions
- Cysts communicate with intrahepatic biliary system
- Congenital hepatic fibrosis:
- Associated with ARPKD
- Liver enlarged but does not typically contain macroscopically visible cysts
- Diffuse periportal fibrosis
- Irregularly shaped, branching duct structures arranged circumferentially around portal vein or at interface of portal tracts / fibrous septae with hepatocytes
- Hydatid cyst:
- Infectious cyst caused by Echinococcus granulosus or Echinococcus multilocularis
- Characteristic trilayered cyst wall histology: inner nucleated germinal layer, middle hyalinized acellular layer and outer layer with granulation tissue and fibrosis
- Small organism hooklets may be seen
- Other infectious hepatic cysts:
- Etiologies include pyogenic bacteria, amoebae (e.g., Entamoeba histolytica)
- Amoebic liver abscesses characteristically hemorrhagic, with liquefactive necrosis
- Cysts demonstrate eroded lining with adherent fibrous material
- Pyogenic cyst / abscess formation consists of purulent exudate (neutrophils and fibrin), which becomes walled off by host fibrotic response
- Mucinous cystic neoplasm:
- Solitary biliary cyst:
- Incidental, solitary unilocular cyst
- No associated hereditary predisposition
Additional references
Board review style question #1
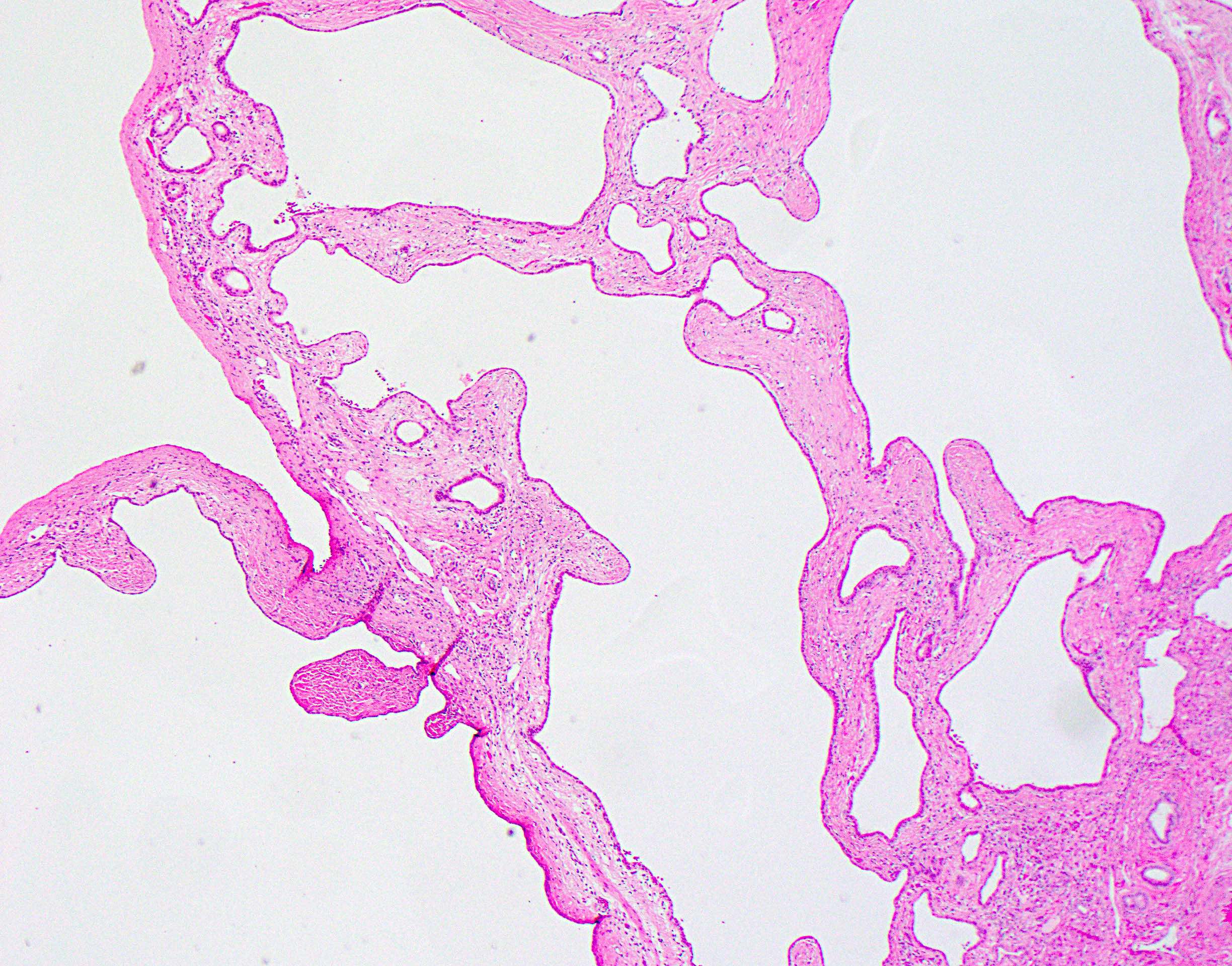
A 35 year old woman is found to have numerous liver cysts on imaging and the above is seen on partial hepatectomy. This finding is most likely seen in which disease process?
- Autosomal dominant polycystic kidney disease
- Cholangiocarcinoma
- Congenital hepatic fibrosis
- Endometriosis
- Mucinous cystic neoplasm
Board review style answer #1
A. Autosomal dominant polycystic kidney disease. The finding in the image shows multiple benign simple cysts of varying sizes, each with bland biliary epithelial lining. The finding is consistent with polycystic liver disease. Polycystic liver disease in adults can rarely occur in isolation but is most frequently associated with autosomal dominant polycystic kidney disease, driven by mutations in PKD1 and PKD2.
Answer B is incorrect because cholangiocarcinoma is typically composed of small, irregular, infiltrative biliary glands with associated desmoplastic stroma. Large duct cholangiocarcinomas can be composed of some larger glands histologically resembling cysts but these are typically associated with mucin production and demonstrate greater degree of cytomorphologic atypia and architectural complexity than the enlarged simple cysts in this image.
Answer C is incorrect because congenital hepatic fibrosis is a form of ductal plate malformation affecting predominantly children and adolescents and is characterized by irregular ductal structures and prominent periportal fibrosis rather than frank biliary cyst formation.
Answer D is incorrect because endometriosis is characterized by glands lined by pseudostratified, columnar proliferative type endometrium and underlying spindled, ovarian type stroma.
Answer E is incorrect because mucinous cystic neoplasm (MCN) is characterized by areas of ovarian type stroma in the cyst wall underlying the cystic epithelial lining. Additionally, the radiologic description of numerous cysts is uncharacteristic of MCN, which typically manifests as a solitary cyst (although this can be multiloculated).
Comment Here
Reference: Polycystic liver disease
Comment Here
Reference: Polycystic liver disease

