

Liver & intrahepatic bile ducts
Developmental anomalies / cysts
Navajo hepatopathy
Authors: Ryan Rebbe, M.D., Joshua A. Hanson, M.D.
Deputy Editor-in-Chief: Raul S. Gonzalez, M.D.


Last author update: 12 August 2020
Last staff update: 16 June 2021
Copyright: 2020-2024, PathologyOutlines.com, Inc.
PubMed Search: Navajo hepatopathy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Rebbe R, Hanson JA. Navajo hepatopathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/livernavajohepatopathy.html. Accessed December 23rd, 2024.
Definition / general
- A rare hepatocerebral mitochondrial DNA (mtDNA) depletion syndrome (MDS) with nonspecific clinical and pathologic features aside from Navajo ancestry (Acta Gastroenterol Belg 2016;79:463)
Essential features
- mtDNA depletion syndrome caused by MPV17 missense mutation (Am J Hum Genet 2006;79:544)
- Nonspecific cirrhotic findings in a patient with Navajo ancestry
- Megamitochondria within hepatocytes
- Mixed macrovesicular and microvesicular steatosis
Terminology
- Navajo neurohepatopathy
- MPV17 related hepatocerebral mitochondrial DNA depletion syndrome
ICD coding
- ICD-10: K74.60 - liver cirrhosis NOS
Epidemiology
- 1 in 1,600 live births of Navajo persons; Southwestern United States predominance (Am J Hum Genet 2006;79:544)
- Presents in infancy / early childhood (Am J Hum Genet 2006;79:544)
- M:F ratio of 1.1:1 (Hum Mutat 2018;39:461)
- There are other MPV17 missense mutations reported, causing similar mtDNA depletion syndrome (Am J Hum Genet 2006;79:544); see Molecular / cytogenetics description section for more details
Sites
- Affects sites that need abundant source of adenosine triphosphate (ATP):
- Liver (Acta Gastroenterol Belg 2016;79:463)
- Cardiac and skeletal muscle (Acta Gastroenterol Belg 2016;79:463)
- Nerves
- ATP is needed for maintenance of myelin
- Lack of ATP leads to degradation of myelin by reactive oxygen species and eventual axonal injury (Acta Neuropathol Commun 2019;7:86)
Pathophysiology
- Autosomal recessive inheritance pattern (Am J Hum Genet 2006;79:544)
- MPV17 encodes channel forming protein of inner mitochondrial membrane and is thought to be involved in deoxynucleotide homeostasis (Am J Hum Genet 2006;79:544)
- Exact pathophysiology is still unclear (Am J Hum Genet 2006;79:544)
- The mutation results in:
- Depletion of mtDNA
- Quantitative decreased synthesis of respiratory chain complexes I, III and IV (Hepatology 2001;34:776)
- Decreased ATP synthesis from electron transport chain (Hepatology 2001;34:776)
- Deformation of mitochondrial cristae (folds within inner membrane to increase surface area for electron transport chain to occur) (Hepatology 2001;34:776)
- Creating megamitochondria
- Decreased ATP causes cascade of effects:
- Increased reliance on glycolysis for ATP production, leading to hypoglycemia and increased lactate production
- Compensatory decrease in CO2
- Affects sites most reliant on ATP; see Sites for more details
- Depletion of mtDNA
Etiology
- Autosomal recessive germline MPV17 missense mutation (Am J Hum Genet 2006;79:544)
- See Molecular / cytogenetics description section for more details
Diagrams / tables
Images hosted on other servers:
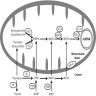
Proteins involved in mtDNA replication
Clinical features
- General features:
- Failure to thrive, with hypoglycemic incidents (J Pediatr 1999;135:482)
- Non anion gap metabolic acidosis (Acta Gastroenterol Belg 2016;79:463)
- Nonspecific elevated liver enzymes, though aspartate transaminase (AST) > alanine aminotransferase (ALT), as in other forms of hepatocyte mitochondrial injury (Acta Gastroenterol Belg 2016;79:463)
- Jaundice and ascites with eventual progression to cirrhosis and liver failure (J Pediatr 1999;135:482)
- Neurologic degradation of myelin and eventually axonal injury (J Pediatr 1999;135:482)
- Muscle weakness due to neuronal degradation and lack of adenosine triphosphate production
- Hepatocerebral presentation is associated with R50Q mutation; see Molecular / cytogenetics description section for more details (Neurotherapeutics 2013;10:186)
- 3 different presentation phenotypes:
- Infantile: presents before 6 months of age with jaundice, failure to thrive and death by 2 years of age (Am J Hum Genet 2006;79:544)
- Childhood: presents between 1 - 5 years old with liver failure and death within 6 months (Am J Hum Genet 2006;79:544)
- Classical: variable onset age of liver disease but with progressive neurological deterioration (Am J Hum Genet 2006;79:544)
- All 3 have peripheral and CNS demyelination
Diagnosis
- Screening: flash frozen tissue sent for quantitative mtDNA analysis
- Liver or muscle biopsy can be used; liver is preferred (Acta Gastroenterol Belg 2016;79:463)
- Flash frozen to -80°C with liquid nitrogen (Acta Gastroenterol Belg 2016;79:463)
- Decreased quantitative mtDNA is a positive screen (< 20% of age matched controls) (Acta Gastroenterol Belg 2016;79:463)
- Diagnosis: whole blood genetic sequencing to show MPV17 homozygous R50Q missense mutation (Am J Hum Genet 2006;79:544)
Laboratory
- Hypoglycemia (Acta Gastroenterol Belg 2016;79:463)
- Elevated lactate (Acta Gastroenterol Belg 2016;79:463)
- Non anion gap metabolic acidosis
- Decreased carbon dioxide (Acta Gastroenterol Belg 2016;79:463)
- Nonspecific findings:
- Elevated liver enzymes: alkaline phosphatase, AST, ALT and gamma glutamyl transferase (GGT)
- AST elevated more than ALT
- AST is a cytoplasmic and mitochondrial enzyme while ALT is cytoplasmic only (Acta Gastroenterol Belg 2016;79:463)
- Prolonged PT / INR and partial thromboplastin time (PTT) (Acta Gastroenterol Belg 2016;79:463)
- Hypofibrinogenemia (Acta Gastroenterol Belg 2016;79:463)
- Hypoalbuminemia (Acta Gastroenterol Belg 2016;79:463)
Radiology description
- Liver ultrasound: nodular and echogenic liver consistent with cirrhosis (Acta Gastroenterol Belg 2016;79:463)
Prognostic factors
- Overall poor prognosis (Acta Gastroenterol Belg 2016;79:463)
- Mortality rates (Hum Mutat 2018;39:461):
- 66% die during infancy
- 13% die in early childhood (1 - 5 years old)
- 2% die in adolescence
- 1% die in early adulthood
- 18% reported alive in 2018 across all age groups
- Oldest reported case at 25 years old
Case reports
- 3 year old boy with nonpainful abdominal swelling (Acta Gastroenterol Belg 2016;79:463)
- 3 children with hepatocerebral mtDNA depletion syndrome (BMC Med Genet 2019;20:167)
- 4 Navajo children with mutilating neuropathy with severe motor involvement (Arch Neurol 1976;33:733)
- 5 families with MPV17 mutations (Am J Hum Genet 2006;79:544)
- 6 children with liver dysfunction (J Pediatr 1999;135:482)
Treatment
- No curative treatment
- Liver transplant
- Roughly 20% of affected children have a liver transplant; this is controversial and considered palliative (Acta Gastroenterol Belg 2016;79:463)
- Transplant can alleviate hepatic dysfunction in the short term; patient will still progress to neurologic dysfunction (Acta Gastroenterol Belg 2016;79:463)
- Roughly 60% of transplanted individuals die during posttransplant period secondary to sepsis, respiratory failure or multi organ failure (Hum Mutat 2018;39:461)
- Nutritional modulation and cofactor supplementation may be beneficial (Neurotherapeutics 2013;10:186)
- Selenium may be beneficial (eNeurologicalSci 2016;2:8)
- Formulas enriched with medium chain triglycerides, uncooked corn starch and frequent feedings are required to prevent hypoglycemia (Hum Mutat 2018;39:461)
- Nucleoside supplementation has been a suggested potential therapy (Hum Mutat 2018;39:461)
Microscopic (histologic) description
- The following features are characteristic but not pathognomonic:
- Mixed macrovesicular and microvesicular steatosis with hepatocanalicular cholestasis (J Pediatr 1999;135:482)
- Ballooning hepatocellular degeneration indistinguishable from typical steatohepatitis
- Red granular hepatocytes containing abnormal megamitochondria (Acta Gastroenterol Belg 2016;79:463)
- Scattered acidophil bodies (Acta Gastroenterol Belg 2016;79:463)
- Bridging fibrosis and cirrhosis with nonspecific portal / septal ductular reaction (J Pediatr 1999;135:482)
- Juxtaposed cirrhotic nodules appear fatty and oncocytic due to variable accumulations of fat and megamitochondria
Microscopic (histologic) images
Contributed by Joshua A. Hanson, M.D.
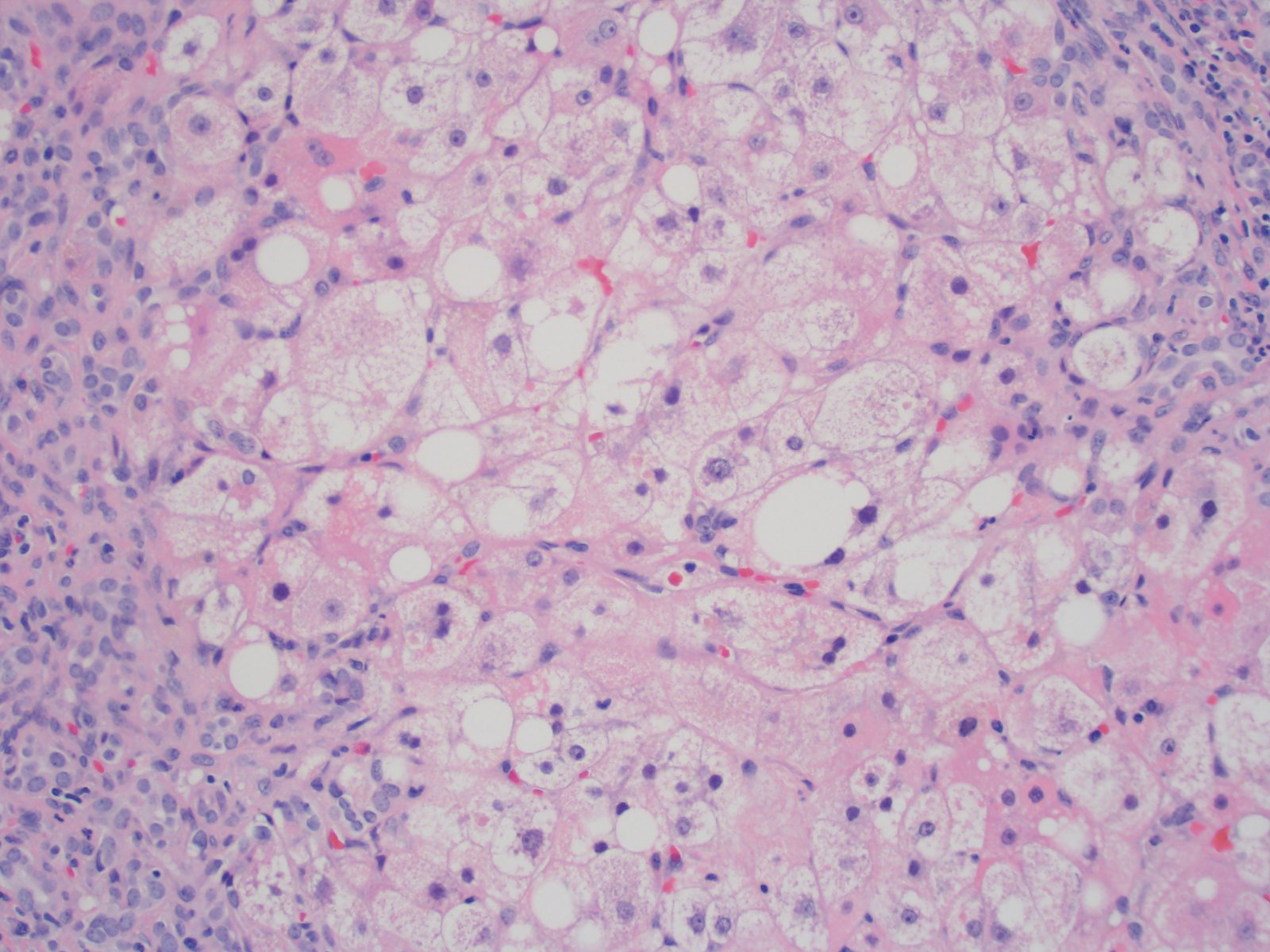
Steatohepatitis
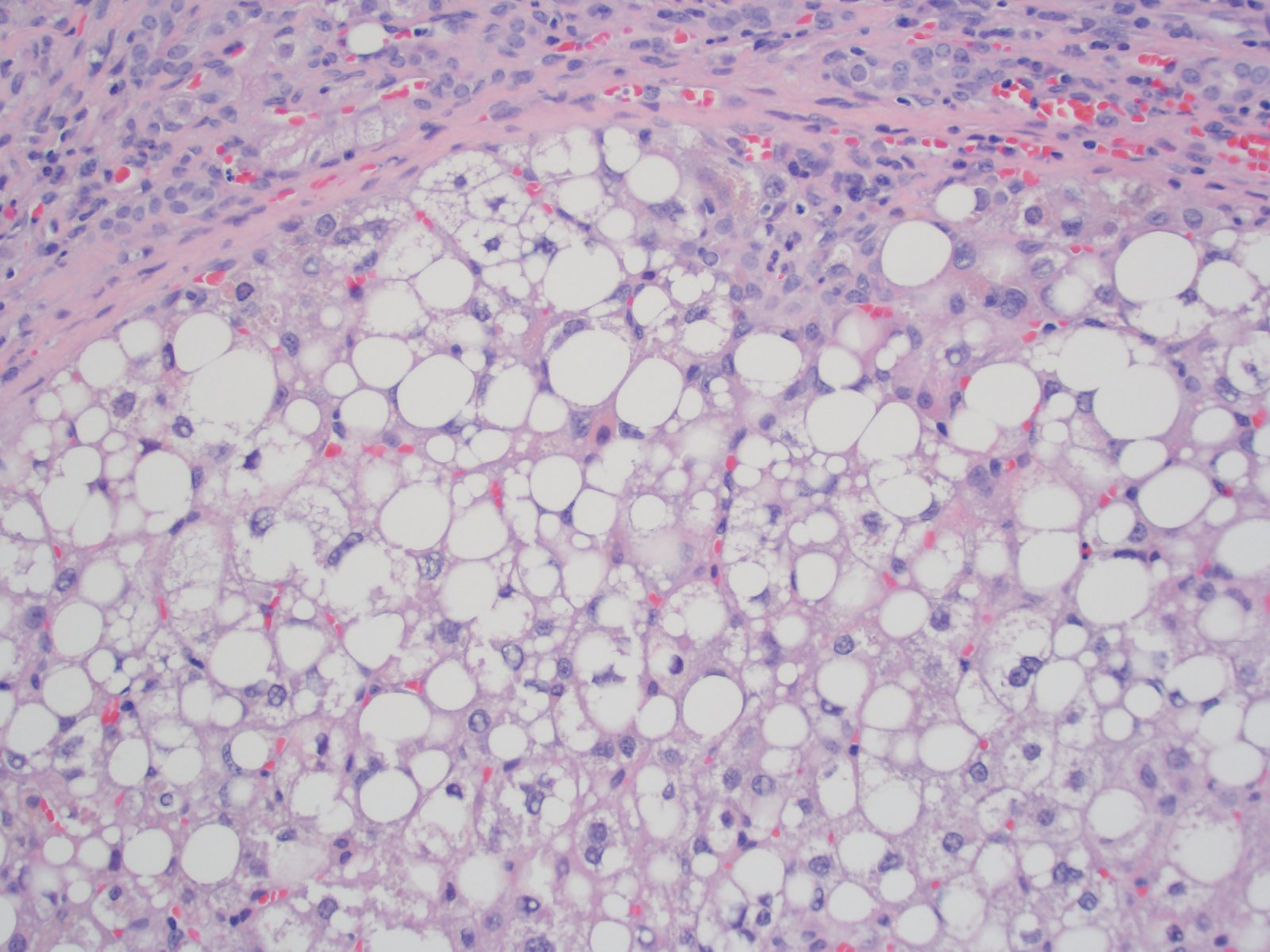
Macrovesicular steatosis

Megamitochondria
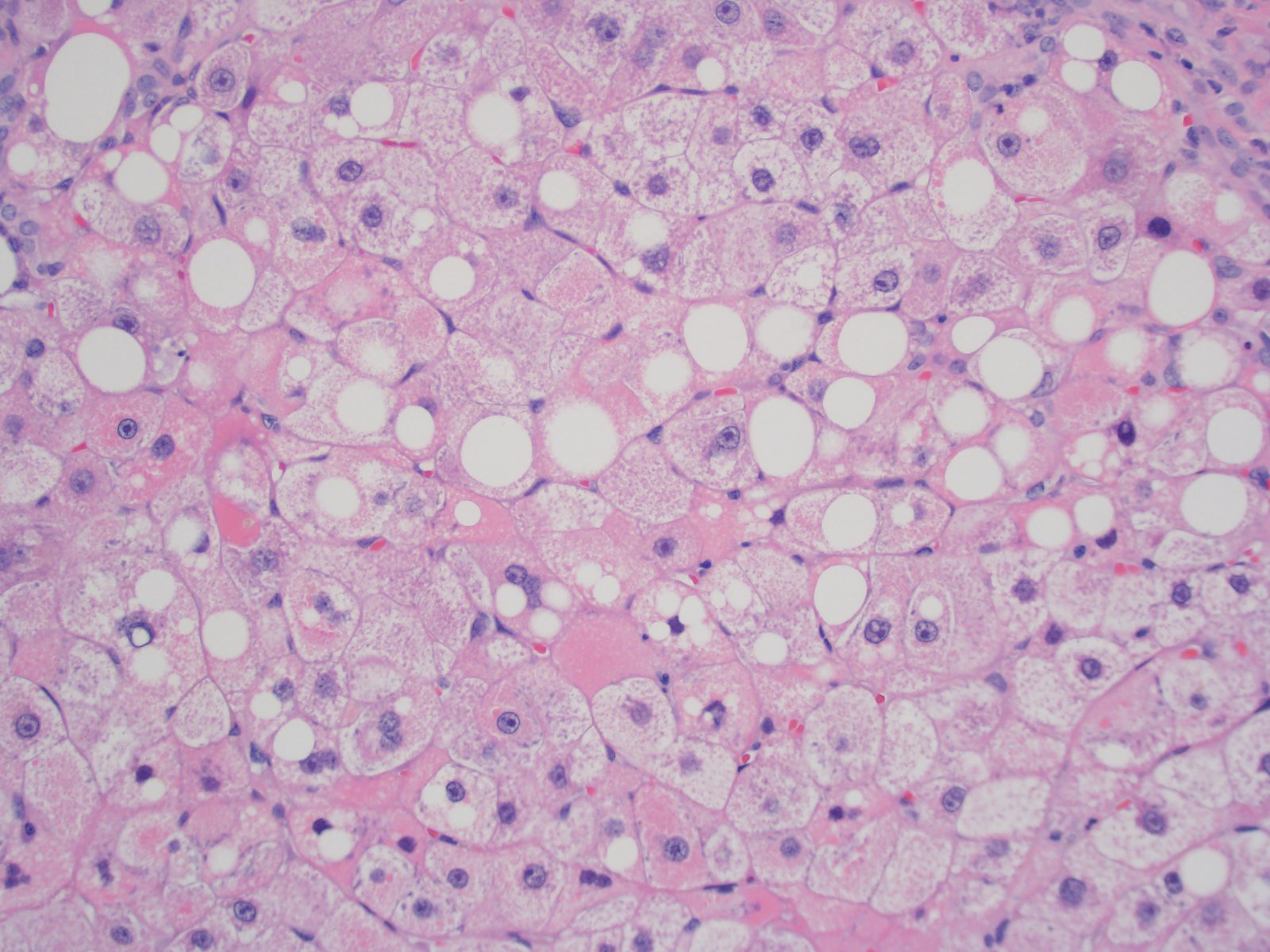
Mixed steatosis and megamitochondria
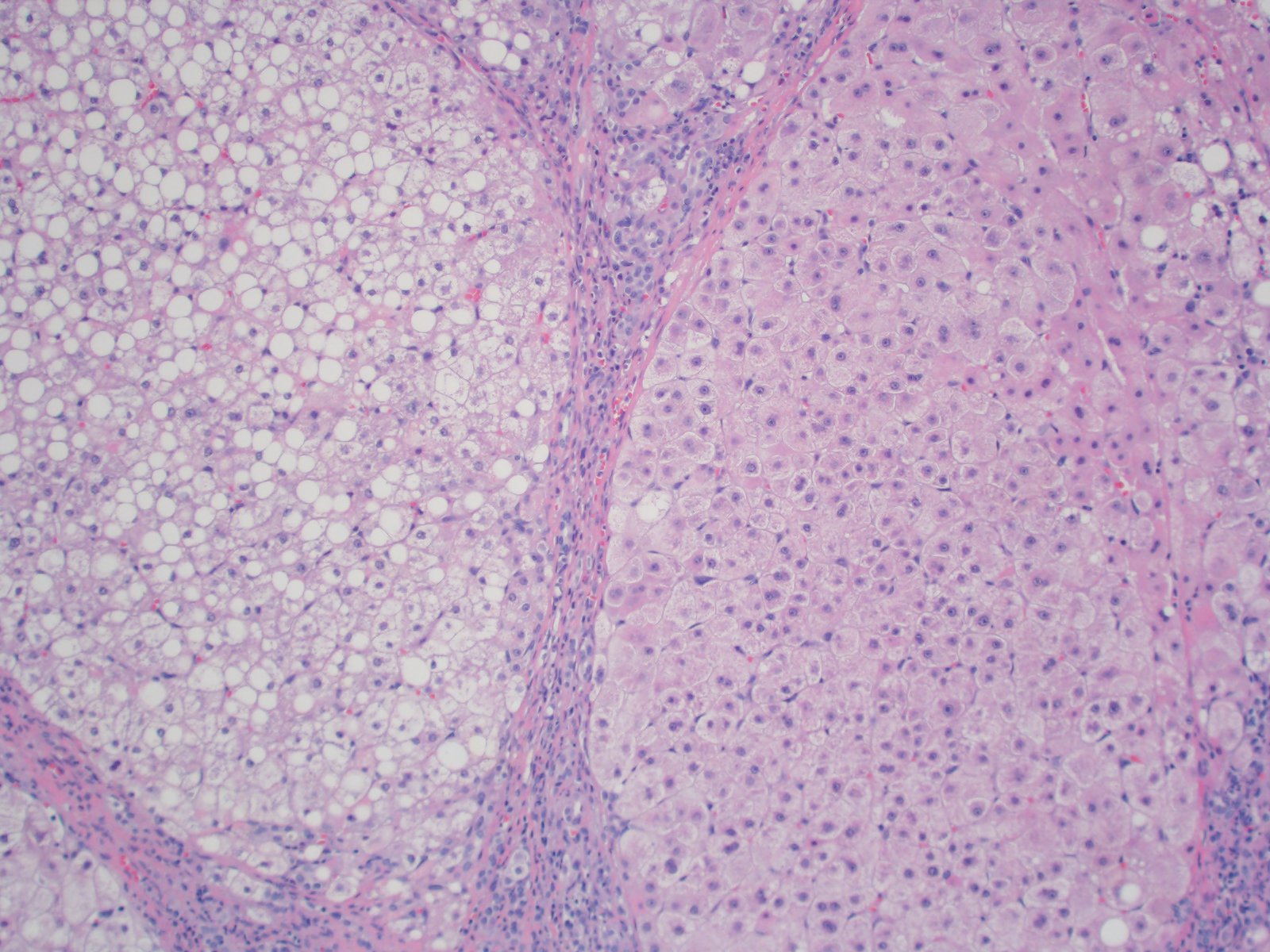
Juxtaposed cirrhotic nodules
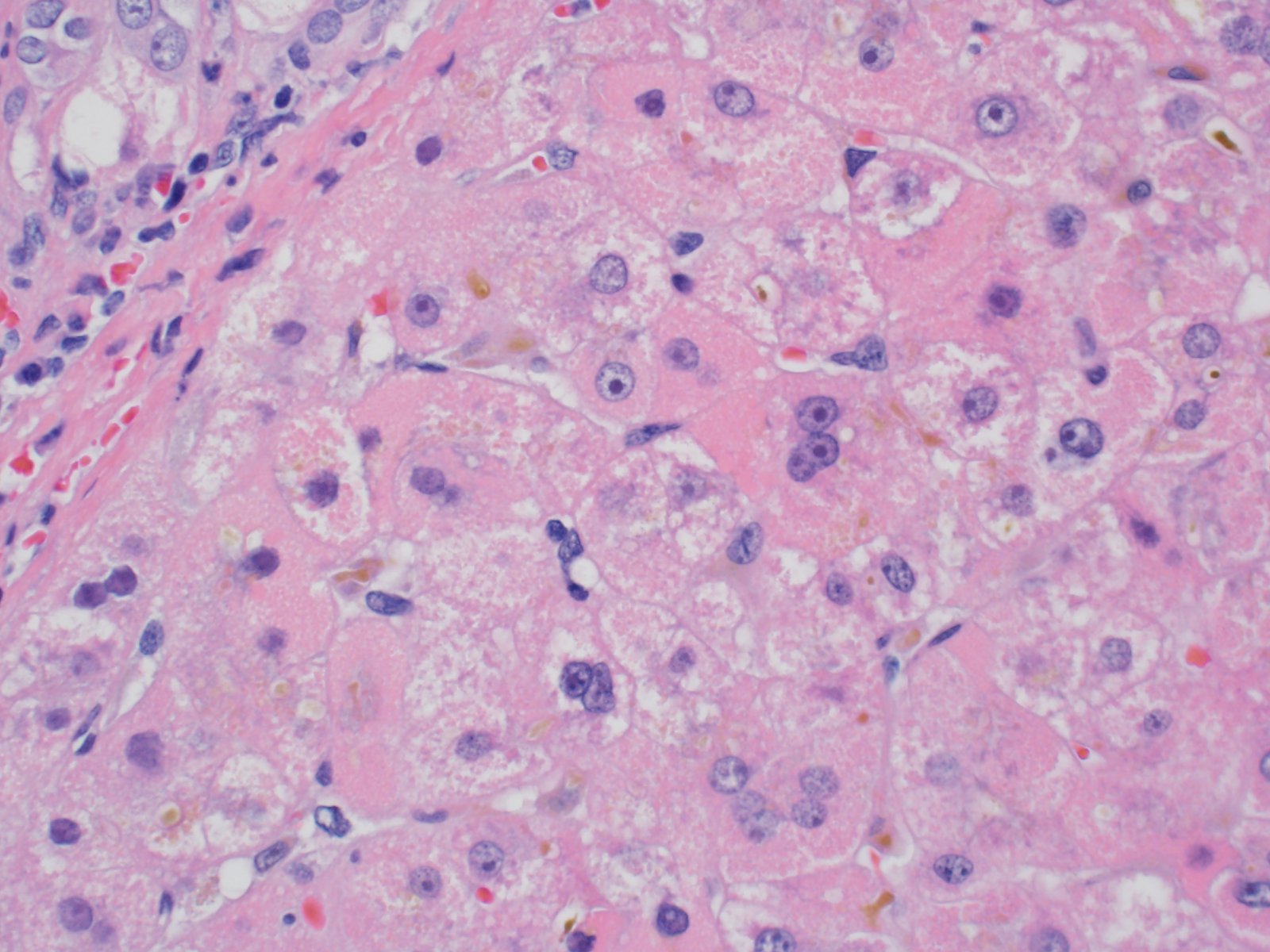
Canalicular cholestasis
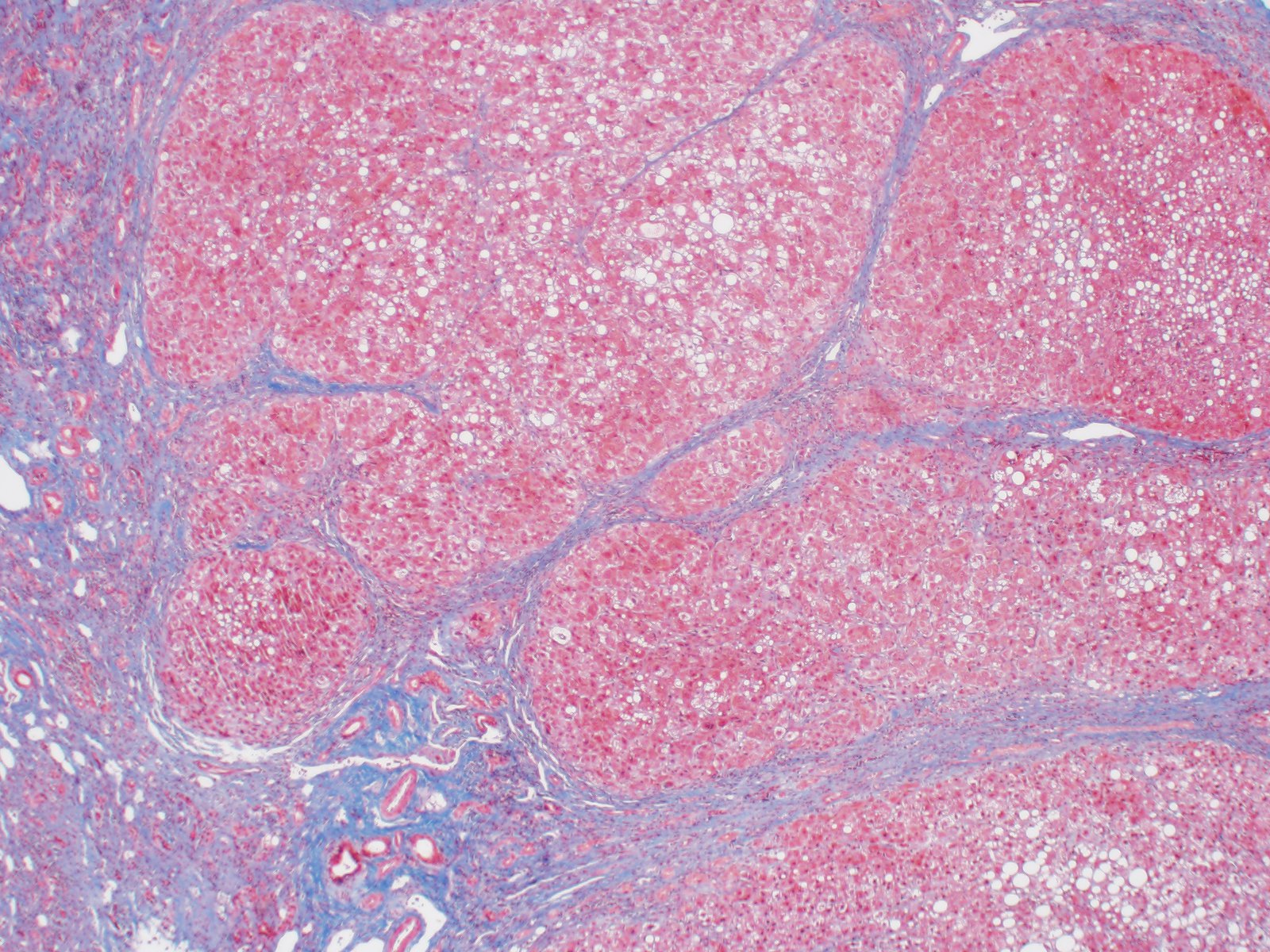
Cirrhosis
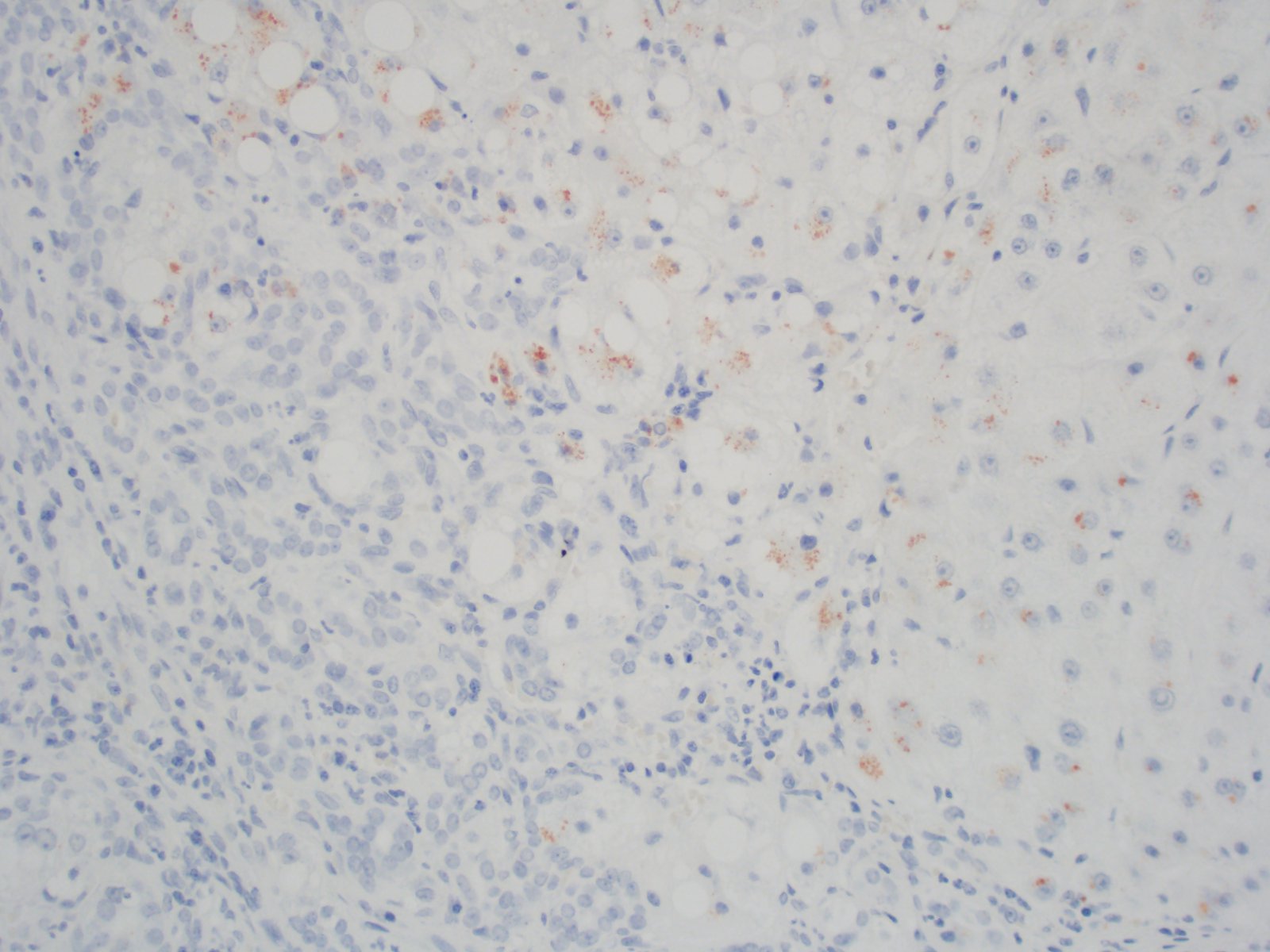
Copper stain
Positive stains
- Trichrome: effaced nodular architecture with dense bands of bridging fibrosis (Acta Gastroenterol Belg 2016;79:463)
- Copper IHC: positive staining in cirrhotic nodules (nonspecific finding in cirrhosis), so do not use as a positive finding to suggest Wilson disease (Acta Gastroenterol Belg 2016;79:463)
Negative stains
- Iron stain (Acta Gastroenterol Belg 2016;79:463)
- PASD may stain few scattered small intracytoplasmic granules but not the large globules seen in alpha-1 antitrypsin deficiency (Acta Gastroenterol Belg 2016;79:463)
Electron microscopy description
- Enlarged pleomorphic mitochondria with dilated cristae
- Lack of dense mitochondrial matrix granules
- Numerous minute lipid droplets within hepatocytes (Hepatology 2007;46:1218)
Molecular / cytogenetics description
- Autosomal recessive germline MPV17 missense mutation; gene is located on short arm of chromosome 2 (2p23.3)
- Homozygous R50Q: glutamine (Q) replaces arginine (R) at position 50 on both alleles (Am J Hum Genet 2006;79:544)
- Diagnosis is done by sequencing
- There are 22 other reported missense mutations of MPV17 protein that cause similar pathology, i.e. R50W, P64R, etc. (Hum Mutat 2018;39:461)
- However, Navajo ancestry has exclusively been reported with the R50Q mutation (Am J Hum Genet 2006;79:544)
- R50Q mutation has also been reported in patients of Italian, Moroccan and other Arabic ancestries (Am J Hum Genet 2006;79:544)
- Other mutations that cause mtDNA depletion syndrome:
- Mutations in mitochondrial nucleotide synthesis genes includingTK2, SUCLA2, SUCLG1, RRM2B, DGUOK and TYMP (Neurotherapeutics 2013;10:186)
- Mutations in mtDNA replication genes including POLG and C10orf2 (Neurotherapeutics 2013;10:186)
Sample pathology report
- Liver, biopsy:
- Cirrhotic liver with steatohepatitis, hepatocanalicular cholestasis and intracytoplasmic megamitochondria (see comment)
- Comment: A trichrome stain demonstrates an effaced nodular architecture with dense bands of bridging fibrosis (cirrhosis). There is mild portal / septal chronic inflammation consisting predominantly of lymphocytes with rare plasma cells. The inflammation is confined to the fibrous areas and the native bile ducts are not preferentially involved. There is no evidence of ductopenia. There is mixed macrovesicular and microvesicular steatosis with areas of ballooning hepatocellular degeneration indicative of steatohepatitis. There is canalicular and hepatocellular cholestasis. Acidophil bodies are seen and many hepatocytes contain course pink granules, consistent with megamitochondria. These findings in a pediatric patient with cirrhosis and Navajo ancestry would be compatible with Navajo neurohepatopathy in the proper clinical setting. This diagnosis would, however, need to be supported by additional testing (quantitative mtDNA analysis on fresh frozen tissue or genetic testing).
Differential diagnosis
- Other metabolic liver disease such as tyrosinemia, glycogen storage disease, fatty acid oxidation defects and urea cycle disorders:
- Often identified on newborn screen
- Wilson disease:
- Usually presents at an older age
- Hepatic quantitative copper in Wilson disease is much higher compared to the mildly elevated levels seen in Navajo neurohepatopathy
- Drug / toxin mediated injury, especially Reye syndrome and valproate toxicity:
- Both cause mitochondrial dysfunction and subsequent microvesicular steatosis
- Clinical evaluation for offending drugs is indicated
Board review style question #1
- The finding highlighted by the image is discovered in a liver biopsy of a 2 year old male of Navajo ancestry with liver failure. What is the mutation associated with this disease?
- ATP7B
- C282Y
- MPV17
- TP53
Board review style answer #1
Board review style question #2
- Which of the following ancillary studies supports a diagnosis of Wilson disease as opposed to Navajo hepatopathy?
- Markedly elevated liver copper concentration
- Positive copper immunohistochemistry
- Positive MPV17 germline testing
- Reduced hepatic mtDNA content
Board review style answer #2

