
Kidney nontumor / medical renal
Renal allograft
Recurrent and de novo diseases
Author: Arzu Sağlam, M.D.
Editorial Board Members: Jonathan E. Zuckerman, M.D., Ph.D., Nicole K. Andeen, M.D.

Last author update: 21 September 2022
Last staff update: 21 September 2022
Copyright: 2019-2025, PathologyOutlines.com, Inc.
PubMed Search: Kidney recurrent de novo

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Etiology / pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) and immunofluorescence images | Immunofluorescence description | Positive stains | Electron microscopy description | Genetics | Videos | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Sağlam A. Recurrent and de novo diseases. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyrecurrentdenovo.html. Accessed March 31st, 2025.
Definition / general
- Diseases that recur or develop de novo in the renal allograft
Essential features
- Recurrent or de novo diseases can injure the renal allograft
- Most native kidney diseases can recur or develop de novo in the allograft, including cardiovascular diseases / hypertension, diabetes, metabolic diseases, infectious diseases, drug toxicities, perfusion problems and cancer, in addition to de novo / recurrent primary glomerular diseases
- Cause of end stage renal disease has to be known in order to differentiate recurrence from de novo disease
- Morphology is similar to those in the native kidney but frequently has additional features related to allograft rejection or other complications of transplantation
- Clinical course of the disease usually differs from those of the native kidney due to presence of iatrogenic immunosuppression
Terminology
- Recurrent "name of disease entity" of the renal allograft: original disease that damaged the native kidney recurring in the allograft
- De novo "name of disease entity" of the renal allograft: development of a new disease in the allograft, different than that which led to end stage renal disease
- Unknown: when native kidney disease is not known
ICD coding
- ICD-10: T86.19 - other complication of kidney transplant
Epidemiology
- With improvement in treatment of rejection related complications, recurrent and de novo diseases have become a significant contributing factor to graft dysfunction (Transplantation 1999;68:635)
- Today recurrent / de novo cardiovascular diseases and cancer are the most common cause of morbidity and mortality
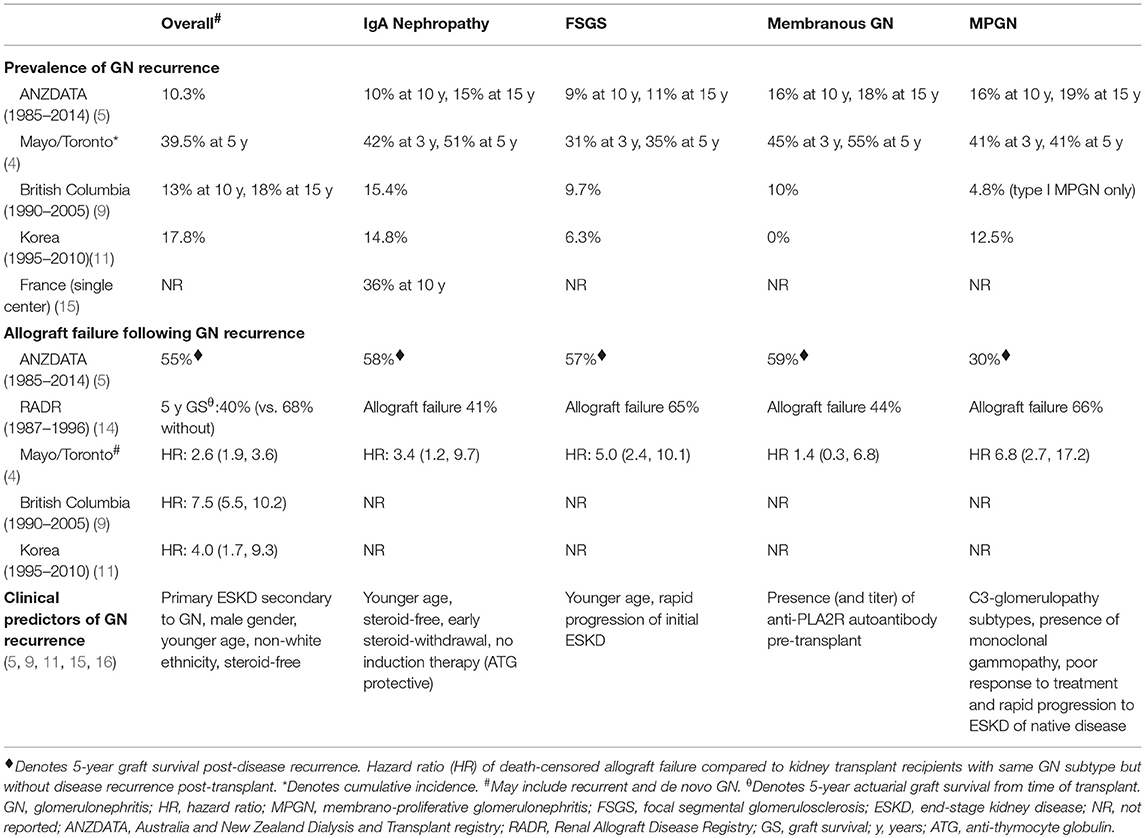
- Focal segmental glomerulosclerosis, IgA nephropathy, membranous glomerulonephritis and membranoproliferative glomerulonephritis are among the most common recurrent glomerular diseases that lead to graft loss (Nat Clin Pract Nephrol 2008;4:446)
- True incidence of recurrent or de novo diseases has not been clearly established, predominantly due to complexity in initial classification of the primary disease leading to end stage kidney
- Figures vary widely between studies and with regard to disease subtype; exact figures are hard to determine due to
- Anonymity of primary cause of end stage renal disease
- Possibility of subclinical disease going underdiagnosed where protocol biopsies are not performed (Nephrology (Carlton) 2014;19:6)
- Possibility of newly developed disease being donor derived
- Coexistence or morphological overlap of certain primary renal diseases and forms of rejection (World J Transplant 2017;7:285)
- Differences in morphology of early recurrent / de novo disease (especially those observed in protocol biopsies of asymptomatic patients) from those of clinically symptomatic diseases in the native kidney (Kidney Int 2017;91:304)
- Lower utilization of immunofluorescence and electron microscopy in transplant biopsies
- Risk of recurrence of glomerular disease is higher in patients with living related transplantation (BMC Nephrol 2017;18:25)
- Better HLA compatibility is also associated with increased risk of recurrence of glomerular disease but longterm outcome is better despite this drawback (Kidney Int 2018;93:482)
- Estimated rates given, in a comprehensive review article, in relation to glomerular diseases can be seen in table 1 below (Nephrology (Carlton) 2014;19:6)
- Individual disease entities
- Membranous nephropathy
- Recurrence rates vary (10 - 50%); recurrence usually takes place within the first year
- Most cases of de novo membranous nephropathy develop after the first year
- Detectable serum phospholipase A2 receptor before or at transplantation or reemergence after transplantation increases risk of recurrence
- Focal segmental glomerulosclerosis
- Recurrence rates vary: 30 - 80%, higher in pediatric patients and in patients with primary / idiopathic focal segmental glomerulosclerosis; lower in those with secondary or genetic focal segmental glomerulosclerosis in the native kidney (Pediatr Transplant 2010;14:314, Front Immunol 2019;10:1944)
- Usually develops within the first 2 years posttransplant
- Risk factors for recurrence are (Nephrol Dial Transplant 2006;21:1053, Transplant Proc 2007;39:737)
- Childhood onset disease
- Age < 15 years
- Rapid progression from diagnosis to end stage renal disease
- Presence of mesangial hypercellularity
- Recurrence of focal segmental glomerulosclerosis in a previous allograft (risk of recurrence increases to 80 - 100%)
- Presence or absence of nephrotic syndrome in presentation of native focal segmental glomerulosclerosis (Clin J Am Soc Nephrol 2021;16:1730)
- Subclass of focal segmental glomerulosclerosis: primary / idiopathic, genetic and secondary; risk of recurrence being highest in primary / idiopathic focal segmental glomerulosclerosis
- Focal segmental glomerulosclerosis may also develop / progress over time, in which case separation of recurrent versus de novo disease can be problematic
- IgA nephropathy
- IgA deposition is frequently encountered in the renal allograft and recurrence rates vary between 10 - 53% depending on whether protocol or indication biopsies are involved and the follow up period (Nephrology (Carlton) 2018;23:4, Front Immunol 2019;10:1944)
- Risk of recurrence increases with time after transplant
- Latent IgA deposition should be differentiated from IgA nephropathy
- Latent IgA deposition: mesangial IgA deposition in an asymptomatic patient (no hematuria or proteinuria) established by protocol biopsies
- IgA nephropathy: IgA deposition in a patient with urinary abnormalities
- Membranoproliferative glomerulonephritis - immune complex mediated (BMC Nephrol 2016;17:7)
- Polyclonal immunoglobulins
- Recurrence risk is lower (30 - 35%)
- Presents later, during the first 5 years
- Monoclonal immunoglobulins
- Recurrent disease is more common, 66%
- Usually occurs within the first year
- Risk of recurrence is correlated with
- Living related donation: possible genetic background
- Preemptive transplants
- Low complement levels
- Presence of monoclonal gammopathy
- Polyclonal immunoglobulins
- C3 glomerulopathy (World J Transplant 2016;6:632)
- Genetic C3 glomerulopathy has high rates of recurrence (up to 66.7%) (J Am Soc Nephrol 2014;25:1110)
- Thrombotic microangiopathy (Curr Opin Organ Transplant 2014;19:283, Transplant Rev (Orlando) 2018;32:58)
- Most cases are due to development of de novo thrombotic microangiopathy
- Risk of recurrence depends on etiology
- Typical hemolytic uremic syndrome due to infections rarely recurs (Pediatr Nephrol 2002;17:809)
- Recurrence is most common in atypical hemolytic uremic syndrome; overall recurrence is 60% and usually occurs during the first year posttransplant (Clin J Am Soc Nephrol 2006;1:88)
- Risk of atypical hemolytic uremic syndrome changes in relation to genetic abnormality; i.e., risk of recurrence is 4 times higher in patients with mutations in complement genes (Am J Transplant 2013;13:663)
- ADAMTS13 deficiency: if deficiency is genetic, disease can recur (BMC Nephrol 2013;14:156)
- De novo immune complex glomerulonephritis, unclassified (Hum Pathol 2015;46:1521, Hum Pathol 2018;71:109)
- Various immune complexes and complement components can be identified in the renal allograft with immunofluorescence
- Although the majority of immune complex glomerulonephritis in the allograft have a correlate in the native kidney, some do not and are not readily classifiable
- Cardiovascular diseases and posttransplant diabetes mellitus: a common cause of mortality and morbidity in kidney transplant recipients
- Infectious diseases: especially opportunistic microorganisms
- Cancer (Nat Rev Nephrol 2018;14:508)
- Is the second most common cause of mortality and morbidity in kidney transplant recipients
- Kidney transplant recipients have at least a twofold higher risk of developing or dying from cancer than the general population
- Types with greatest risk increase are: Kaposi sarcoma, nonmelanoma skin cancers, lip cancer (> 10 times) and cancers with viral oncogenesis (posttransplant lymphoproliferative disorder and anogenital cancers)
- Membranous nephropathy
Sites
- Renal disease
Etiology / pathophysiology
- Persistence of original inciting agents related to glomerular diseases increases the chances of recurrence of glomerular disease; examples are the presence of
- Putative circulating factors that are toxic to the podocyte in primary / idiopathic focal segmental glomerulosclerosis (Am J Transplant 2013;13:266)
- Resulting podocyte injury is thought to be reversible early in the disease course
- Antiphospholipase A2 receptor antibody in membranous glomerulonephritis (Transplantation 2015;99:1709, Nephrol Dial Transplant 2014;29:2334, Clin Transplant 2016;30:1394)
- High titer of antiglomerular basement membrane antibody in antiglomerular basement membrane antibody glomerulonephritis (J Am Soc Nephrol 2001;12:394)
- Paraproteins / free light chains and abnormal kappa/lambda ratio in AL amyloidosis and other glomerulopathies due to paraproteins (Nephrology (Carlton) 2014;19:6)
- Persistence of high level acute phase reactants in AA amyloidosis (Transplantation 2020;104:1703)
- Low ADAMTS13 activity in atypical hemolytic uremia syndrome (Nephrology (Carlton) 2014;19:6)
- Mutations of or autoantibodies to complement regulatory factors in atypical hemolytic uremia syndrome and C3 nephropathies (Clin J Am Soc Nephrol 2006;1:88)
- Genetic deficiency of liver peroxisomal enzyme alanine:glyoxylate aminotransferase in primary hyperoxaluria (World J Nephrol 2015;4:235)
- Autoantibodies directed against phospholipid binding proteins in antiphospholipid syndrome (Nat Rev Nephrol 2014;10:279)
- Poor diabetic control in diabetic nephropathy (Diabetes 1983;32:948)
- Poor blood pressure control in hypertensive nephropathy
- Putative circulating factors that are toxic to the podocyte in primary / idiopathic focal segmental glomerulosclerosis (Am J Transplant 2013;13:266)
- Certain factors are associated with risk of de novo disease
- Hyperfiltration (chronic allograft injury with graft loss, transplantation of pediatric kidney into adult), vascular diseases (allografts from diseased donors) and medications (calcineurin inhibitors, mTOR inhibitors) for focal segmental glomerulosclerosis (World J Transplant 2017;7:285, Clin Transplant 2011;25:6)
- Antiglomerular basement membrane antibody disease in transplants for Alport disease due to development of antibodies against α3, α4 or α5 chains of collagen IV of the allograft (Clin J Am Soc Nephrol 2017;12:1162)
- Risk is higher in patients with large COL4A5 gene deletions and genetic testing is recommended to detect risk pretransplant (J Am Soc Nephrol 2013;24:364)
- Steroid / immunosuppression use: diabetic nephropathy (Diabetes Metab Syndr Obes 2011;4:175)
- Thrombotic microangiopathy due to use of calcineurin inhibitors and antibody mediated transplant rejection (Transplant Rev (Orlando) 2018;32:58, Exp Clin Transplant 2018;16:131)
- Individual disease entities
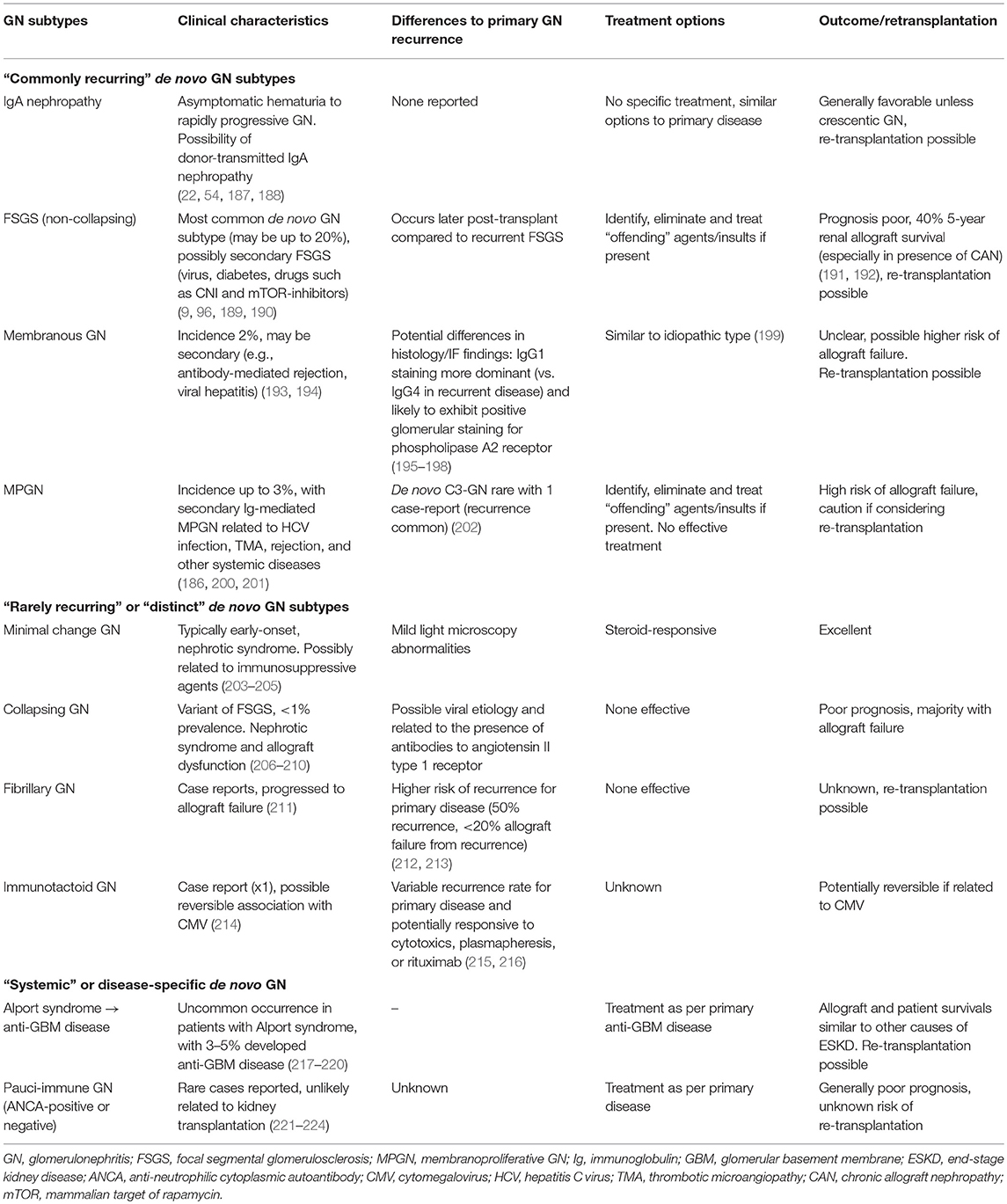
- Membranous nephropathy (Clin Transplant 2016;30:1394)
- IgG4 is dominant in recurrent disease (similar to primary membranous nephropathy), whereas IgG1, IgG2 and IgG3 are dominant in de novo disease (similar to secondary membranous nephropathy) (Transplant Proc 2011;43:3743)
- There is lack of phospholipase A2 receptor in de novo disease (Transplantation 2013;95:1259)
- De novo disease can be associated with Alport syndrome, ureteral obstruction, newly acquired hepatitis B virus, hepatitis C virus and rejection
- It is hypothesized that renal injury (including alloimmune injury due to transplant reactions) may result in exposure of hidden epitopes and autoantibody production (Transpl Int 2012;25:1205)
- Focal segmental glomerulosclerosis
- Recurrent primary / idiopathic focal segmental glomerulosclerosis is thought to be associated with continued presence of circulating factors and can take place within hours after transplantation, presenting as heavy proteinuria (Am J Transplant 2013;13:266, Clin J Am Soc Nephrol 2010;5:2115)
- De novo focal segmental glomerulosclerosis is similar to secondary focal segmental glomerulosclerosis and is associated with multiple pathogenetic factors that can be seen in the allograft much like those in the native kidney
- These factors include decreased nephron mass and hyperfiltration injury, obesity, hypertension, diabetes, urinary reflux, infections and medications (World J Transplant 2017;7:285, Clin Transplant 2011;25:6, Indian J Nephrol 2015;25:82)
- Membranoproliferative glomerulonephritis (BMC Nephrol 2016;17:7)
- Classical or lectin complement system is involved in the pathogenesis of immune complex mediated type whereas the alternative complement system is involved in the pathogenesis of C3 glomerulopathy
- Type of membranoproliferative glomerulonephritis (immune complex mediated versus alternative complement mediated / C3 glomerulopathy) may change after transplant
- Thrombotic microangiopathy (Curr Opin Organ Transplant 2014;19:283, Transplant Rev (Orlando) 2018;32:58)
- De novo thrombotic microangiopathy development is associated with
- Medications
- Antibody mediated rejection
- Infections
- Acquired deficiencies in complement regulatory proteins or ADAMTS13 (rare) (Am J Transplant 2008;8:1694)
- De novo thrombotic microangiopathy development is associated with
- C3 glomerulopathy and atypical hemolytic uremic syndrome / thrombotic microangiopathy (World J Transplant 2016;6:632)
- Associated with genetic or acquired abnormalities of the complement system
- Posttransplant diabetes mellitus and diabetic nephropathy
- De novo disease development is frequently associated with the use of immunosupression (Diabetes Metab Syndr Obes 2011;4:175)
- De novo immune complex glomerulonephritis, unclassified (Hum Pathol 2015;46:1521, Hum Pathol 2018;71:109)
- These cases are sometimes associated with infections and alloimmune injury
- Cancer: risk of developing de novo or recurrent cancer is multifactorial and associated with (Nat Rev Nephrol 2018;14:508)
- Oncogenic viruses
- Immunosuppression and altered T cell immunity
- Membranous nephropathy (Clin Transplant 2016;30:1394)
- References: Kidney Int 2017;91:304, Transplant Proc 2013;45:3
Diagrams / tables

Clinical features
- Diseases may recur / present subclinically and be diagnosed on protocol biopsies - histological and immunophenotypic recurrence / presentation
- Clinical picture may be complicated due to accompanying rejection
- Individual disease entities
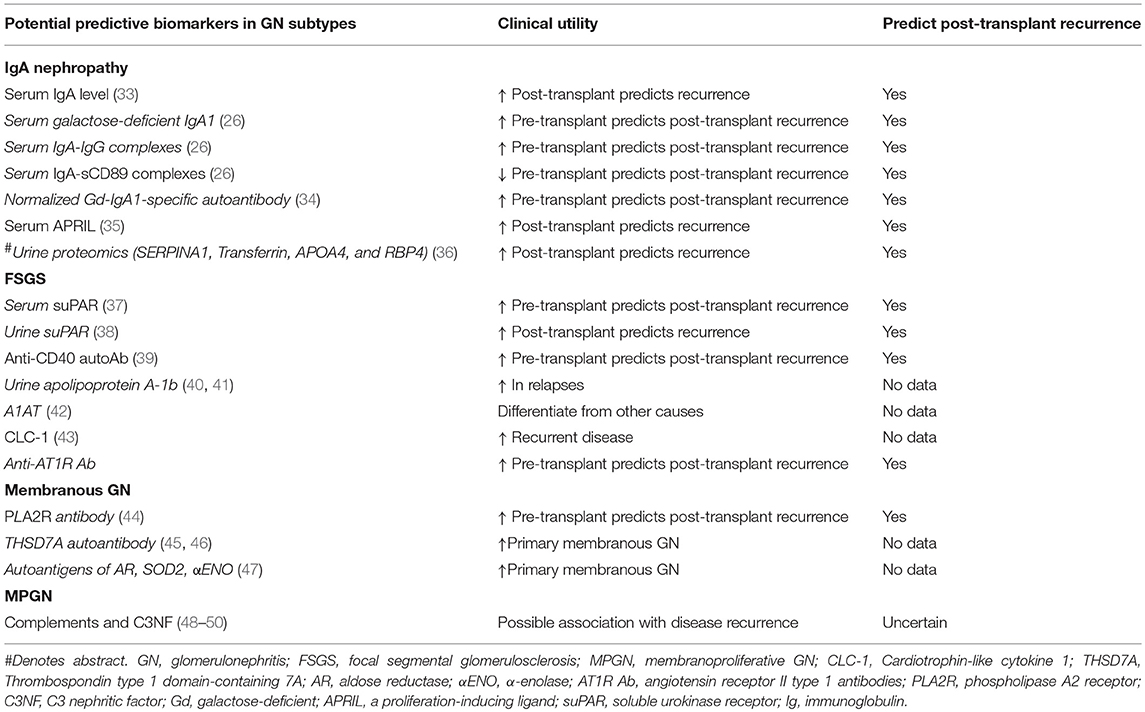
- Membranous nephropathy: proteinuria and antiphospholipase A2 receptor (PLA2R) can be used to monitor recurrence (Clin Transplant 2016;30:1394)
- Thrombotic microangiopathy: clinical triad of findings (hemolytic anemia, thrombocytopenia and acute kidney injury) may be incomplete or absent and require biopsy for diagnosis (Curr Opin Organ Transplant 2014;19:283, Transplant Rev (Orlando) 2018;32:58)
- De novo immune complex glomerulonephritis, unclassified: these patients usually have nonnephrotic proteinuria and are biopsied due to allograft dysfunction (Hum Pathol 2015;46:1521, Hum Pathol 2018;71:109)
Diagnosis
- Similar to that of individual native kidney diseases; however, take into consideration indication of biopsy (protocol biopsy versus biopsy due to clinical symptoms) and accompanying alterations due to the effects of
- Immunosuppressive agents (most notably calcineurin inhibitors)
- Immunological injury
- Posttransplant infections (especially viral)
- Ischemia reperfusion injury
- Hyperfiltration injury
Laboratory
- Abnormalities in urine analysis: proteinuria, hematuria, leukocyturia
- Abnormalities in serum creatinine, albumin and protein levels
- No abnormal lab findings in early disease recurrence / de novo development (established via protocol biopsies)
Prognostic factors
- Recurrent glomerulonephritis is associated with increased risk of graft failure
- Allograft glomerulopathy in general, both de novo and recurrent is responsible for about 25% of death censored kidney allograft failures and is among the top 3 leading causes of graft failure
- Early age at transplantation is a major risk factor for recurrence / de novo disease development
- Persistence of original inciting agents related to glomerular diseases adversely effects prognosis
- Lupus has a very good prognosis (Biomed Res Int 2014;2014:746192)
- Membranous nephropathy has a good prognosis, possibly due to maintenance immunosuppression (Clin Transplant 2016;30:1394)
- Focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis and atypical hemolytic uremia syndrome have the worst prognosis (Transplantation 1999;68:635)
- Development of posttransplant antiglomerular basement membrane disease in patients with Alport disease has a high risk of graft loss (Pediatr Transplant 2006;10:651)
- IgA nephropathy
- Latent IgA deposition (asymptomatic, absence of hematuria and proteinuria) can be observed in the donated kidney and is not associated with worse prognosis even if mesangial expansion is present; most of these depositions disappear in about a year, which may be due to use of immunosuppression (Kidney Int 2003;63:2286, Clin Transplant 2013;27:14, Nephrology (Carlton) 2018;23:4)
- Presence of crescents is associated with worse graft outcome (Am J Transplant 2019;19:145)
- Membranoproliferative glomerulonephritis - immune complex mediated
- Polyclonal immunoglobulins: progresses slowly, risk of graft loss low (10%)
- Monoclonal immunoglobulins: aggressive course, graft loss due to disease is common (50%) (Kidney Int 2017;91:304)
- Alternative complement mediated membranoproliferative disease / C3 glomerulopathy and atypical hemolytic uremic syndrome / thrombotic microangiopathy (World J Transplant 2016;6:632)
- Genetic C3 glomerulonephritis that cannot be effectively controlled by immunosuppression, has a high rate of graft loss (50%) (J Am Soc Nephrol 2014;25:1110)
- Graft loss is more common in atypical hemolytic uremic syndrome in patients with mutations in the CFH gene or carriers of the hybrid gene CFH / CFHR1 (Am J Transplant 2013;13:663)
- Also see table 3 above (Front Immunol 2019;10:1944)
Case reports
- 12 year old boy with de novo lupus nephritis (Transplant Proc 2014;46:648)
- 17 year old boy with de novo amyloidosis (Pediatr Transplant 2014;18:E259)
- 22 year old man with granulomatous interstitial nephritis (Transplant Proc 2016;48:946)
- 22 year old man with de novo C3 glomerulonephritis in a renal allograft with morphologic transformation to dense deposit disease (Ultrastruct Pathol 2016;40:112)
- 40 year old man, 42 year old woman and 53 year old man with successful renal transplantation for antiphospholipid syndrome (Medicine (Baltimore) 2016;95:e5419)
- 41 year old woman who had a late diagnosis of 2,8-dihydroxyadenine nephropathy induced end stage renal disease, made on the native nephrectomy that accompanied the renal transplant (Exp Clin Transplant 2017;15:574)
- 43 year old man with nephropathy of unknown etiology developed a nephrotic syndrome with kidney failure at 2 years followup (J Med Case Rep 2014;8:205)
- 47 and 53 year old women, transplant recipients treated with long term plasmapheresis therapy for management of recurrent focal segmental glomerulosclerosis (Transfus Apher Sci 2021;60:103046)
- 52 year old woman receiving retransplant of an allograft due to focal segmental glomerulosclerosis recurrence in the initial recipient (Am J Transplant 2018;18:2818)
- 55 year old man presented with rapid deterioration of renal function (Saudi J Kidney Dis Transpl 2016;27:381)
- 6 cases of kidney transplantation in patients with monoclonal immunoglobulin deposition disease (Am J Kidney Dis 2021;78:755)
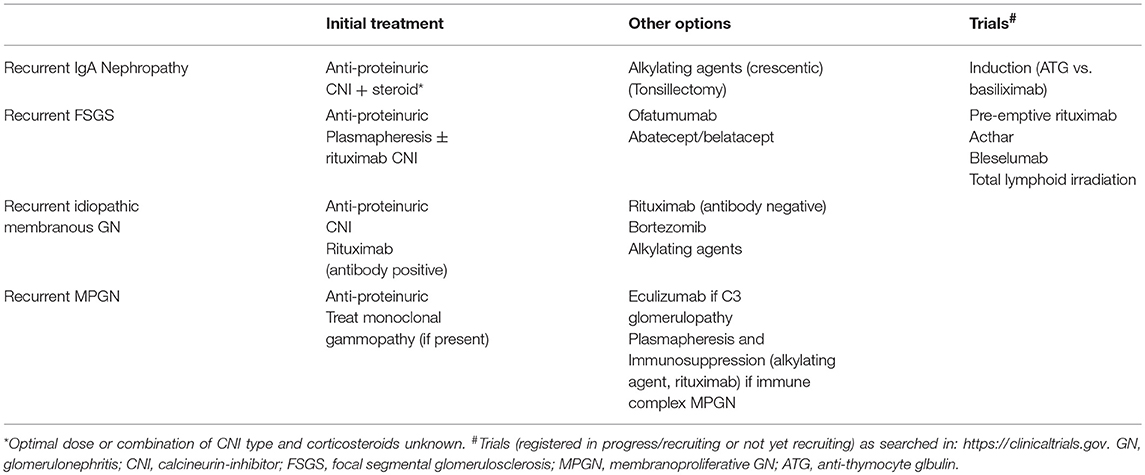
Treatment
- Maintenance immunosuppression to prevent rejection generally alters disease course in immune mediated glomerulonephritis in comparison to native kidneys
- Due to reduction of antibody levels, recurrence of some diseases, particularly systemic lupus erythematosus, antineutrophil cytoplasmic antibody associated vasculitis, IgA and membranous nephropathy, is lower and disease progression slower
- Concomitant or subsequent pancreas transplantation may be considered in diabetic nephropathy (N Engl J Med 1998;339:69)
- Concomitant or subsequent liver transplantation may be considered in oxalosis (Curr Med Res Opin 2014;30:2109)
- Membranous nephropathy (Clin Transplant 2016;30:1394, Clin J Am Soc Nephrol 2021;16:1730)
- Treatment at early stages is best to prevent progression (unlike observation due to possibility of spontaneous remission in primary membranous nephropathy); with effective treatment, recurrence does not portend worse graft survival
- Renin-angiotensin-aldosterone blockade recommended for all patients
- Rituximab is treatment of choice given allograft related immunosuppression (Clin J Am Soc Nephrol 2009;4:1083, Clin J Am Soc Nephrol 2021;16:1730)
- Recurrent focal segmental glomerulosclerosis
- Treatment involves plasmapheresis (especially for early aggressive disease) / immunoabsorption and anti-CD20 monoclonal antibodies (rituximab, ofatumumab); however, these therapies do not seem to be effective in prevention of disease in high risk patients (Artif Organs 2011;35:420, Transplantation 2018;102:e115, World J Transplant 2021;11:303)
- Thrombotic microangiopathy
- Address underlying etiology, including management of calcineurin or mTOR inhibitors; plasma exchange, intravenous immunoglobulin, complement inhibition (Curr Opin Organ Transplant 2014;19:283, Transplant Rev (Orlando) 2018;32:58)
- C3 glomerulopathy
- Plasma exchange and rituximab
- Eculizumab for patients with worsening or high grade proteinuria and decline in kidney function
- De novo immune complex glomerulonephritis, unclassified (Hum Pathol 2015;46:1521, Hum Pathol 2018;71:109)
- These cases are usually self limited and do not require additional therapy
- Also see table 4 above (Front Immunol 2019;10:1944)
- References: Transplant Proc 2013;45:3, Semin Dial 2000;13:195, Clin J Am Soc Nephrol 2021;16:1730
Microscopic (histologic) description
- Histology of early disease may be very subtle and different from that of typical findings one is used to seeing in native biopsies; strict adherence to diagnostic criteria can lead to delays in diagnosis
- Membranous nephropathy (Clin Transplant 2016;30:1394)
- Light microscopy: normal initially; development of basement membrane thickening, vacuolization and spikes later on
- Immunofluorescence: peripheral granular IgG, C4d, kappa and lambda staining
- Lack of phospholipase A2 receptor staining in de novo disease (Transplantation 2013;95:1259)
- Electron microscopy: only small subepithelial deposits initially, may even be absent; formation of well-formed subepithelial deposits later during the disease course
- De novo disease is more likely to have segmental distribution of immune deposits and accompanying mesangial hyperplasia / expansion
- Changes in relation to accompanying allograft related complications may also be present
- Focal segmental glomerulosclerosis (Adv Chronic Kidney Dis 2014;21:448, Clin Transplant 2011;25:6)
- Light microscopy: may resemble minimal change disease initially, with development of segmental sclerosis later on
- Recurrence is usually in the same morphological variant as that of the disease in the native kidney (J Am Soc Nephrol 2008;19:2219)
- Perihilar and not otherwise specified focal segmental glomerulosclerosis variants are more common in de novo disease (Clin Transplant 2011;25:6)
- Electron microscopy: ultrastructural findings of podocyte effacement can be seen very early (Transplantation 2012;93:1238)
- In general, proteinuria precedes the histologic findings of focal segmental glomerulosclerosis seen by light microscopy
- Changes in relation to accompanying allograft related complications may also be present (J Clin Diagn Res 2017;11:EC39)
- IgA nephropathy
- Light microscopy: near normal glomeruli to mesangial and endocapillary hypercellularity
- Immunofluorescence: dominant IgA in the mesangium
- Changes in relation to accompanying allograft related complications may also be present
- Membranoproliferative glomerulonephritis - immune complex mediated
- Early findings show sole mesangial proliferation without characteristic double contours of the glomerular capillary basement membrane
- Typical findings can evolve rapidly as disease progresses
- Changes in relation to accompanying allograft related complications may also be present
- Alternative complement mediated membranoproliferative disease / C3 glomerulopathy (World J Transplant 2016;6:632)
- Light microscopy: membranoproliferative pattern is common in C3 glomerulonephritis and chronic thrombotic microangiopathy
- Immunofluorescence: isolated C3 deposition in C3 glomerulonephritis
- Changes in relation to accompanying allograft related complications may also be present
- Thrombotic microangiopathy (Curr Opin Organ Transplant 2014;19:283, Transplant Rev (Orlando) 2018;32:58)
- Biopsy usually cannot identify the underlying cause (Curr Opin Organ Transplant 2014;19:283)
- Light microscopy: thrombi within glomerular capillaries, arteries / arterioles
- Bloodless glomeruli: glomerular capillary lumina occluded by endothelial swelling and amorphous material
- Fragmented red blood cells within arterial / arteriolar walls or glomeruli
- Severe mucoid intimal thickening of small arteries and arterioles
- Concentric thickening of the small arteries / arterioles
- Focal and segmental glomerular capillary thickening due to endothelial swelling (causing double contours)
- Immunofluorescence: negative
- Electron microscopy: subendothelial space expansion due to electron lucent material, new matrix formation and duplication of glomerular basement membranes
- Chronic thrombotic microangiopathy has a membranoproliferative pattern of glomerular injury and resembles transplant glomerulopathy
- Presence of donor specific antibodies, C4d staining in peritubular capillaries or microvascular inflammation represents chronic and active antibody mediated rejection, not chronic thrombotic microangiopathy
- Diabetic nephropathy
- Light microscopy: vascular changes predominate in recurrence; fewer mesangial nodules than in de novo disease (Transplantation 1996;62:632)
- Immunofluorescence: negative
- Changes in relation to accompanying allograft related complications may also be present
- De novo immune complex glomerulonephritis, unclassified (Hum Pathol 2015;46:1521, Hum Pathol 2018;71:109)
- Light microscopy: minimal mesangial expansion
- Immunofluorescence: mesangial deposits of IgM or IgG
Microscopic (histologic) and immunofluorescence images
Contributed by Arzu Sağlam, M.D.
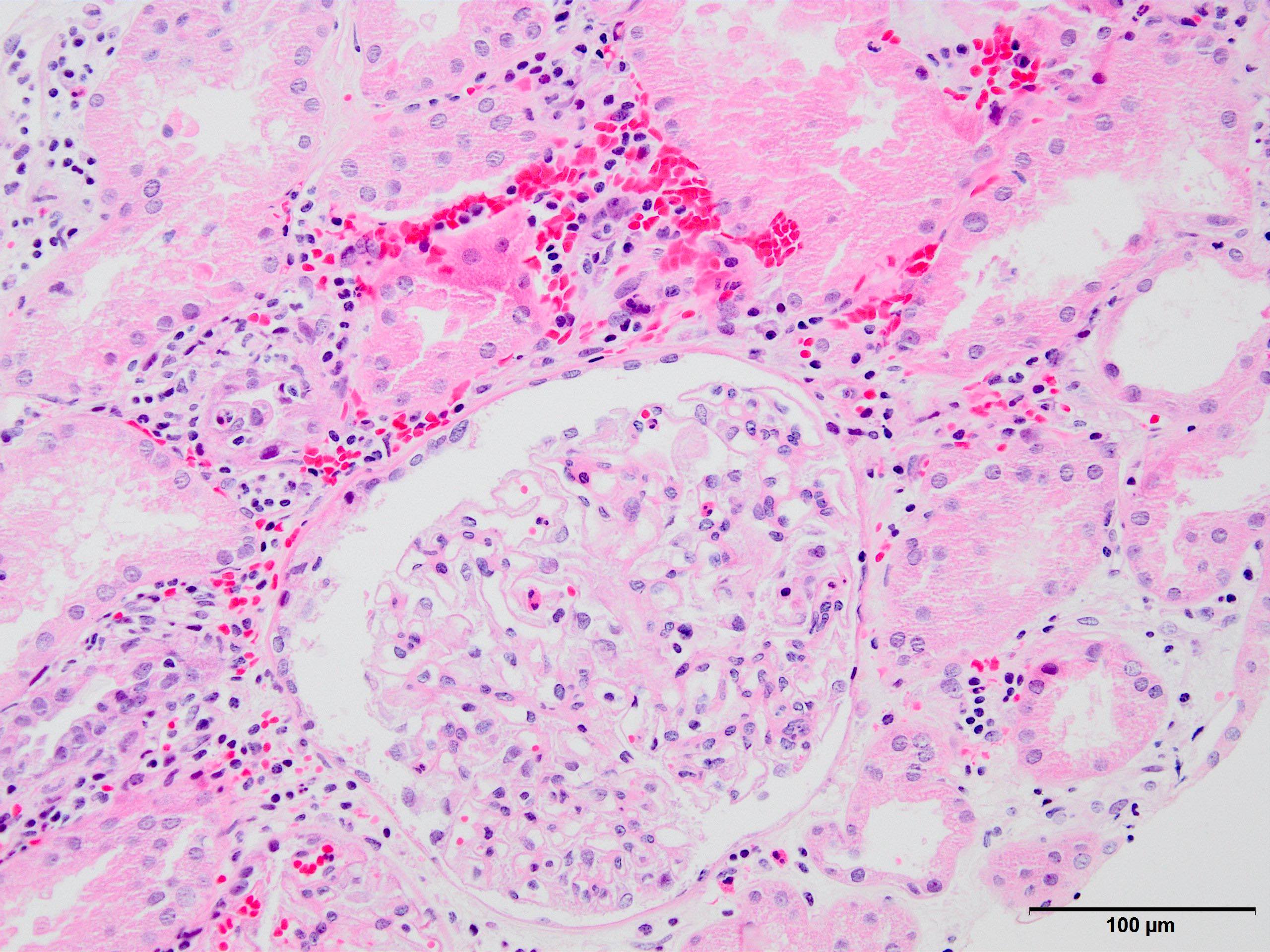
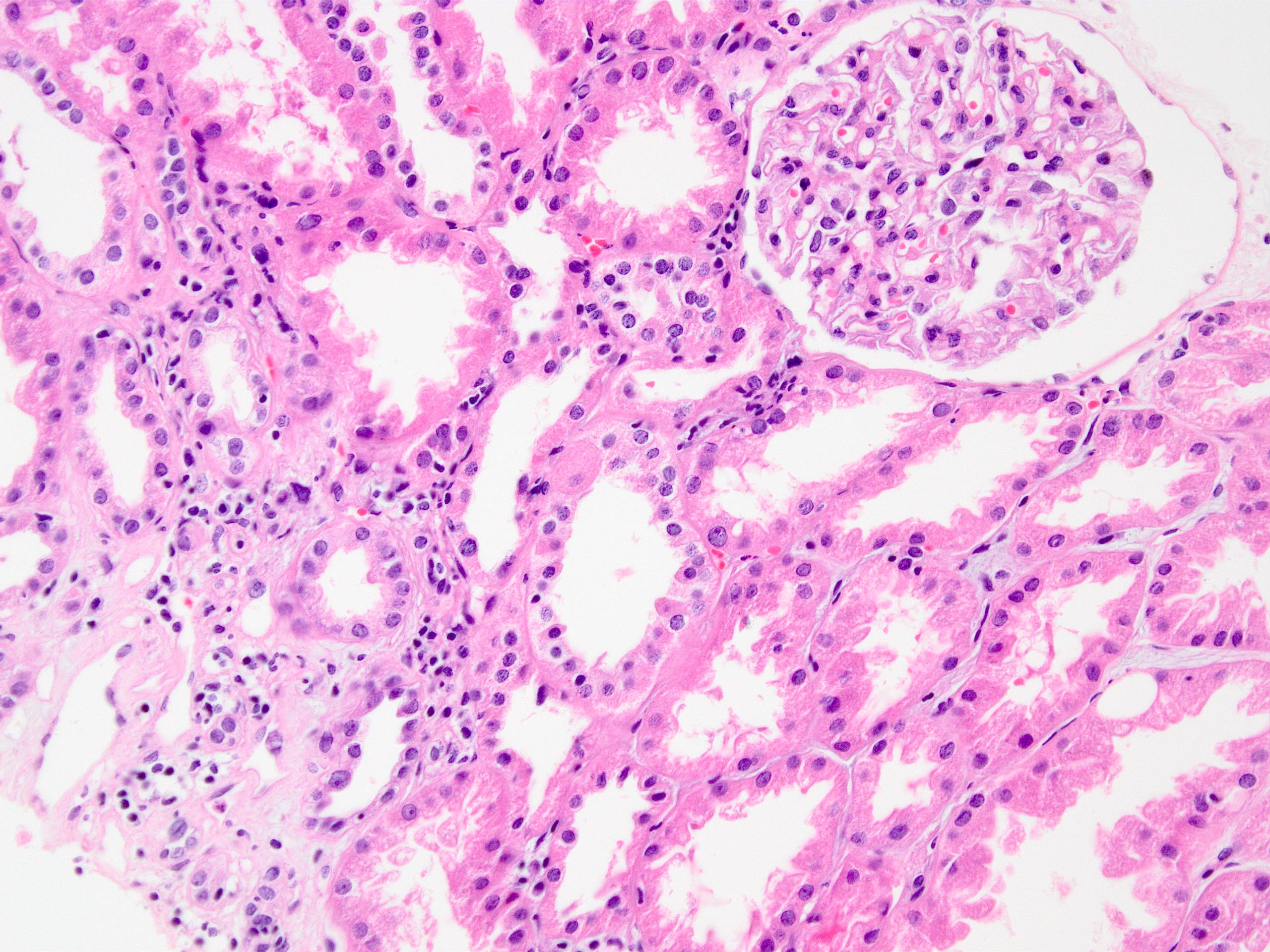
Membranous nephropathy in allograft
IgG deposition,
immunofluorescence
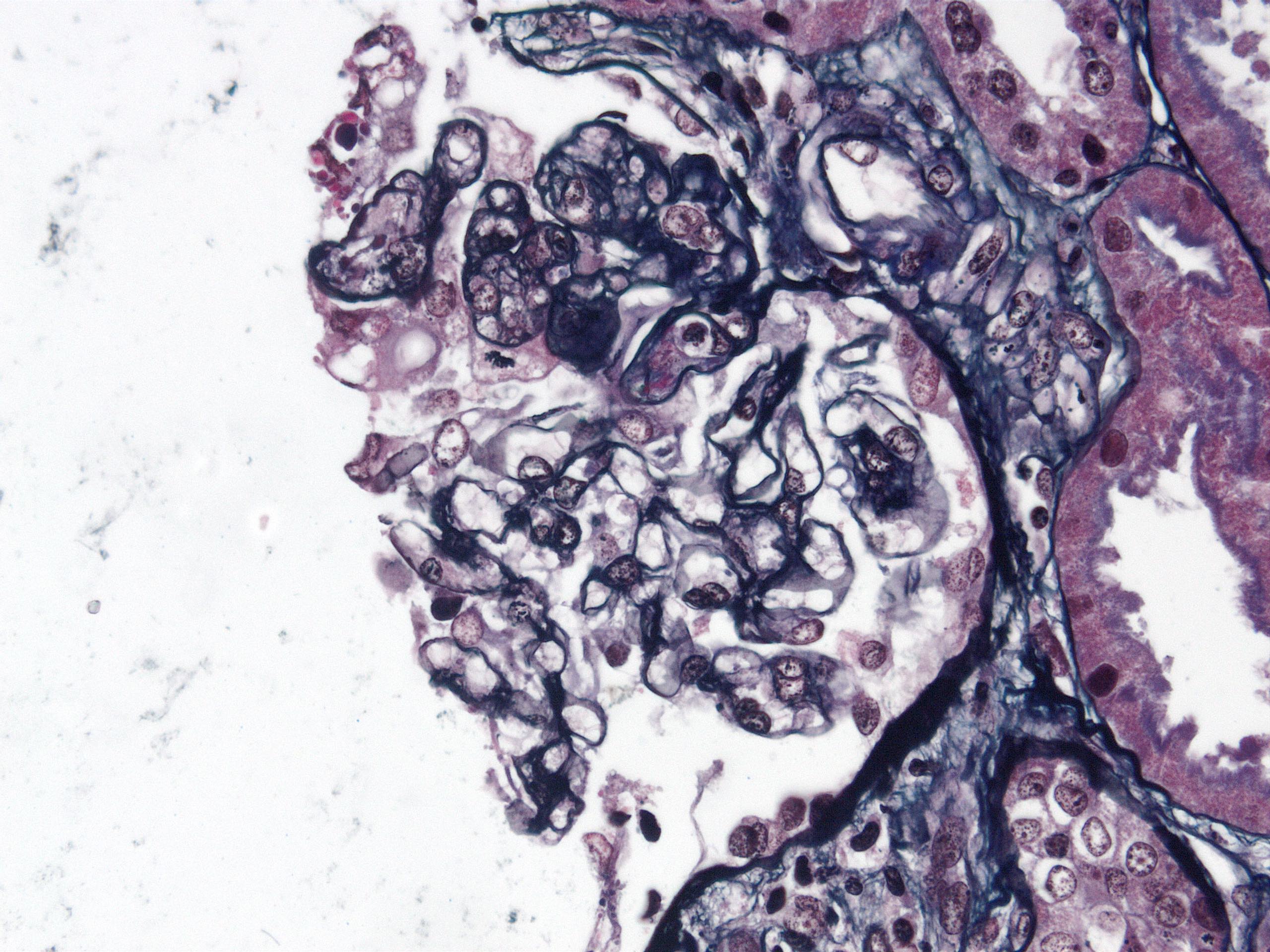
Recurrent focal
segmental
glomerulosclerosis
in allograft
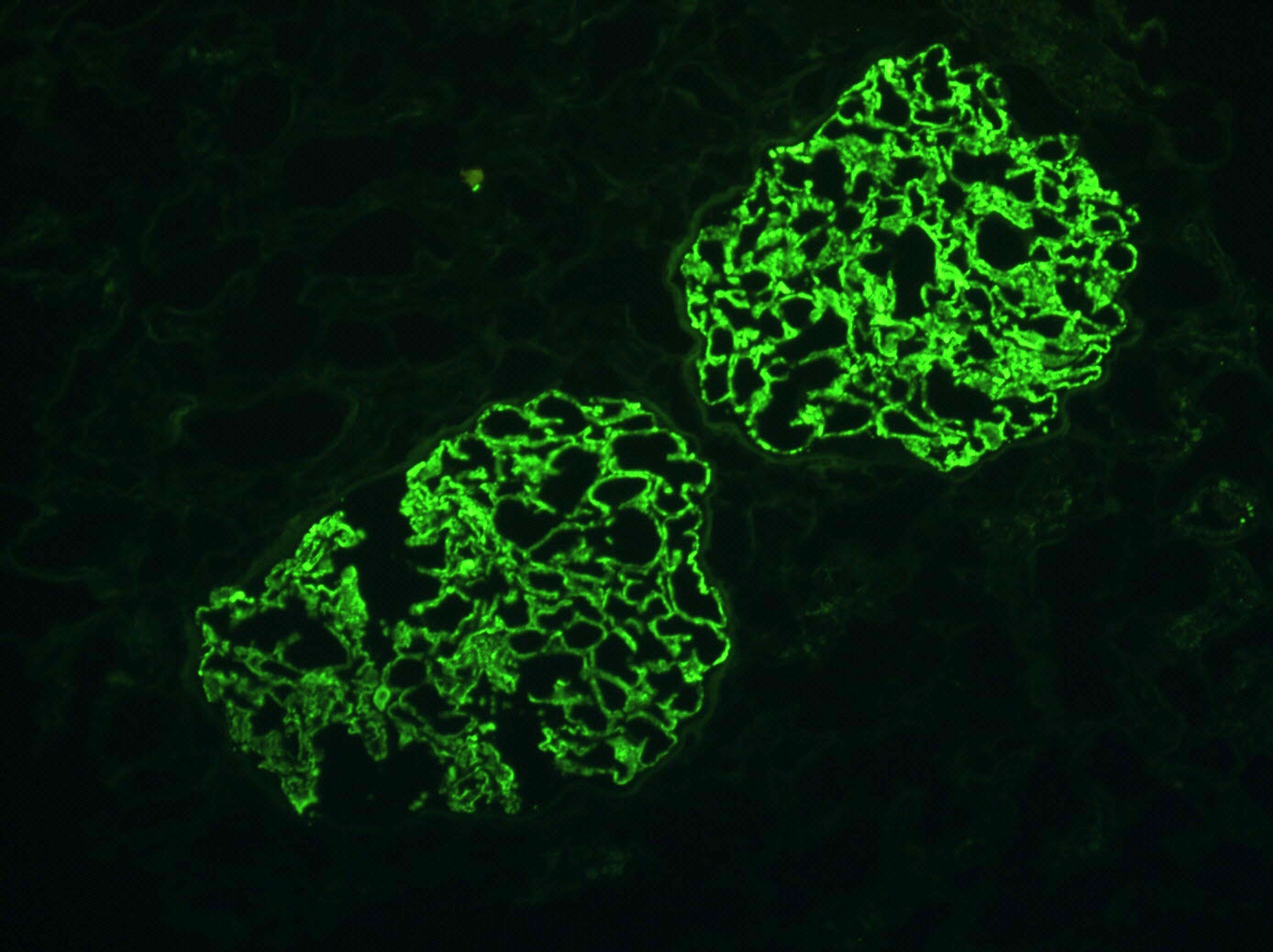
IgA nephropathy in allograft
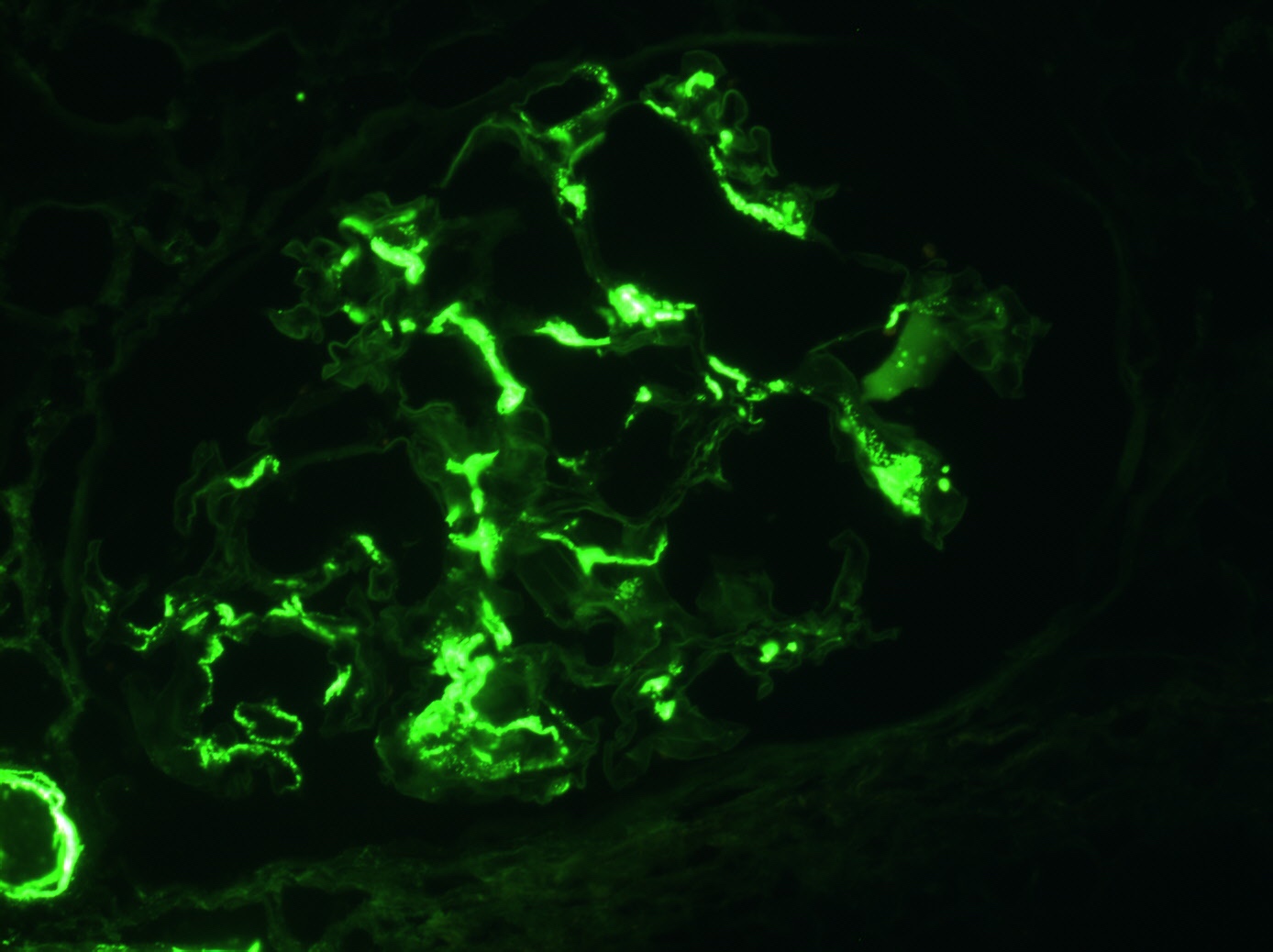
Mesangial IgA
deposition,
immunofluorescence
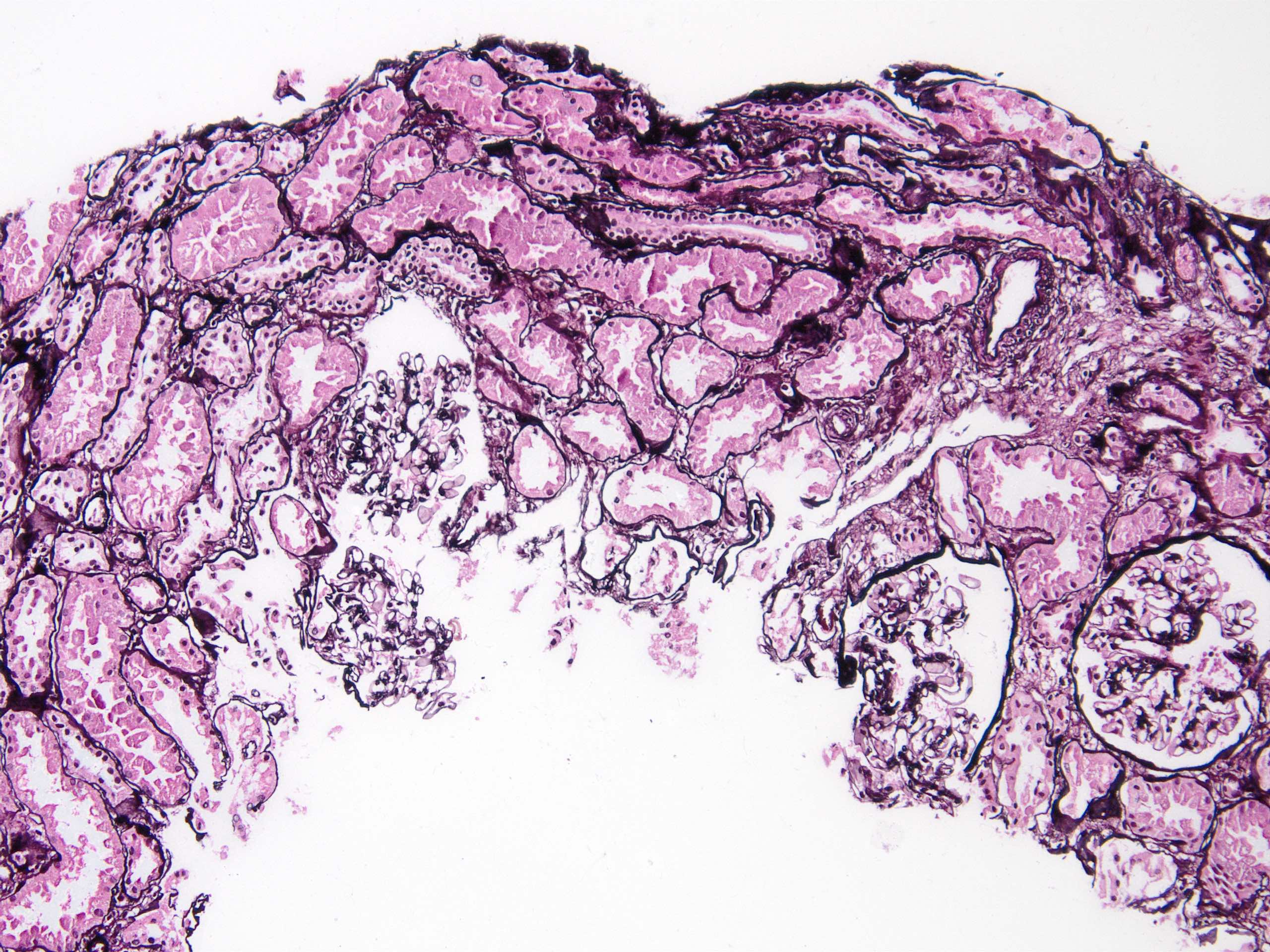
IgA nephropathy in allograft
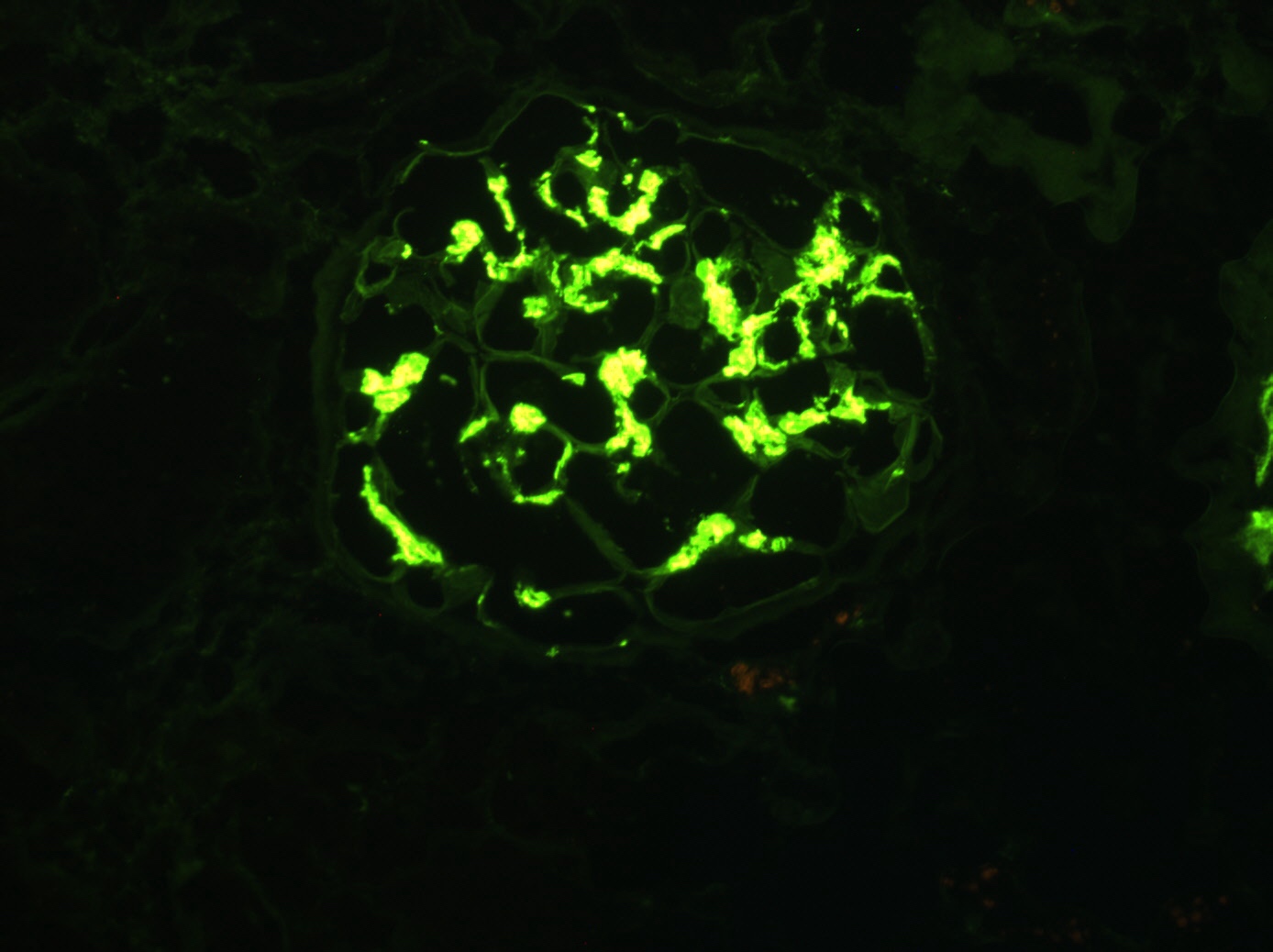
Mesangial IgA
deposition,
immunofluorescence

Recurrent amyloidosis in allograft
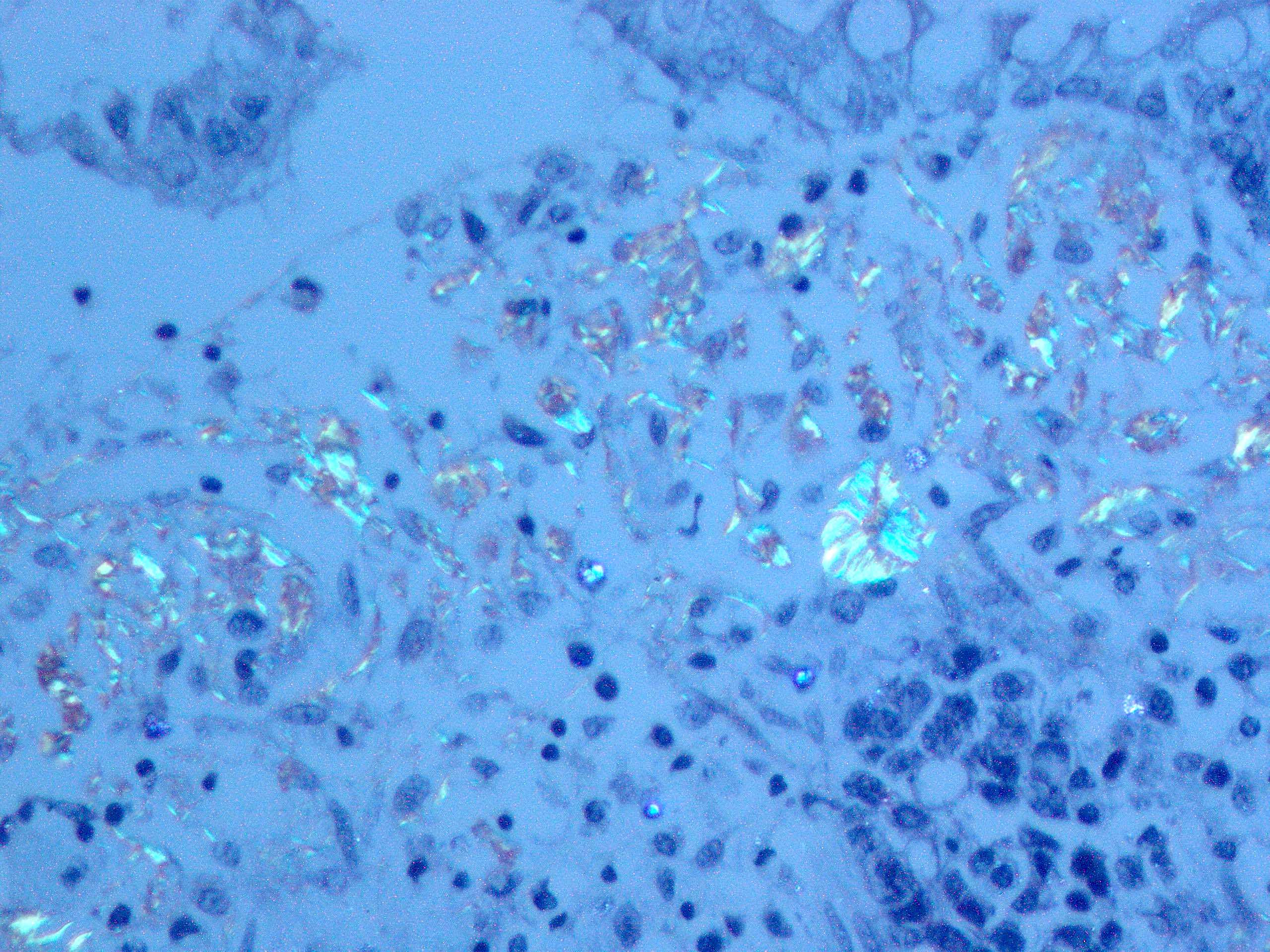
Recurrent amyloidosis
in allograft, Congo
stain polarized
microscopy
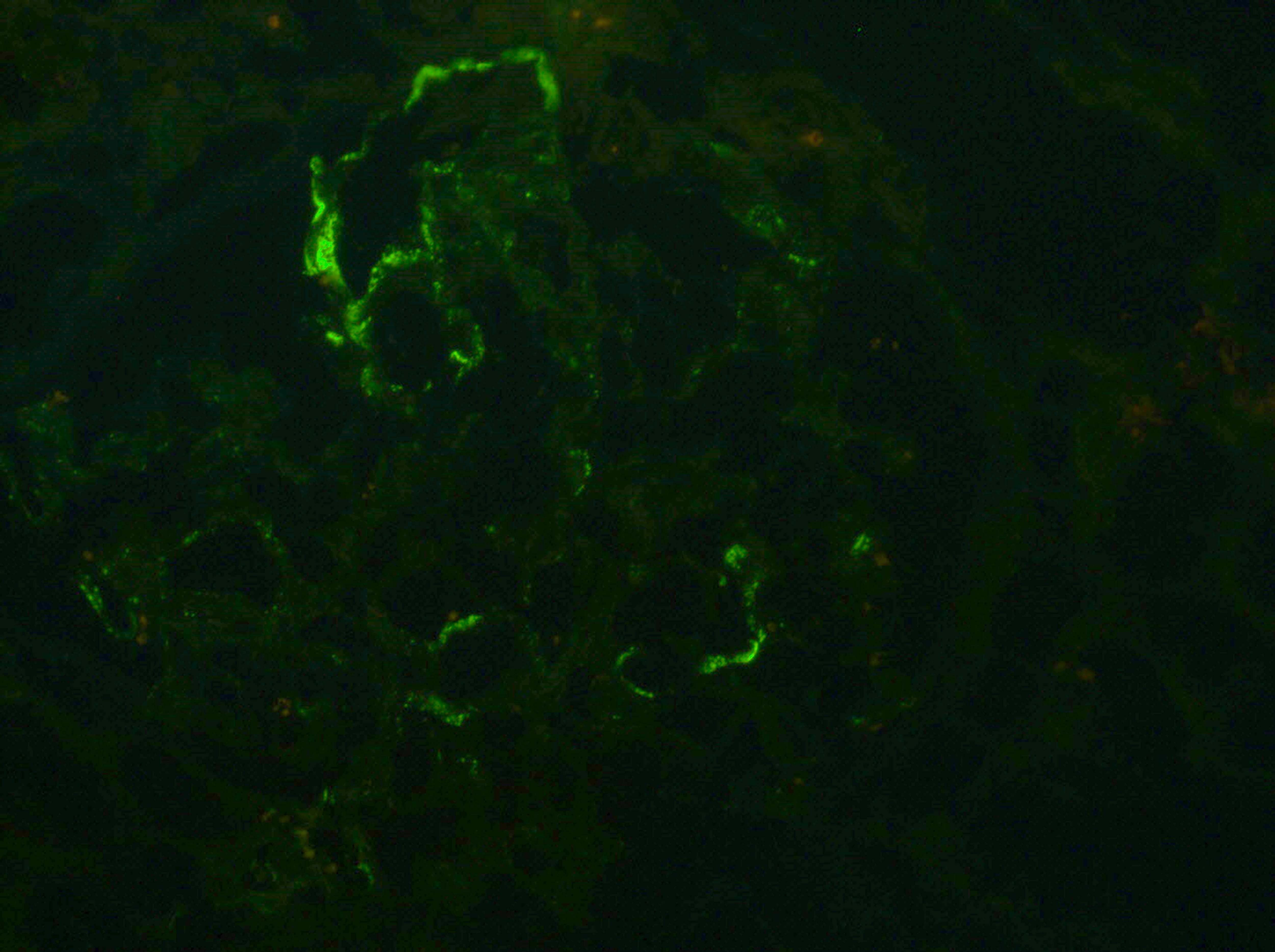
Nonspecific IgM
deposition,
immunofluorescence
Immunofluorescence description
- See disease specific descriptions above
Positive stains
- See disease specific descriptions above
Electron microscopy description
- See disease specific descriptions above
Genetics
- Genetic forms of focal segmental glomerulosclerosis have very low risk of recurrence, therefore, genetic testing may be considered in children and those adult onset focal segmental glomerulosclerosis patients where cause of focal segmental glomerulosclerosis is uncertain, especially if there is a family history
- Screening of the donors for patients undergoing live related donor transplantation may also be considered in these circumstances (Transplant Proc 2019;51:3077)
- Some studies suggest a genetic link between HLA antigens and risk of membranous nephropathy recurrence (N Engl J Med 2011;364:616, Kidney Int 2021;99:671, Kidney Int 2021;100:243)
- Increased risk of recurrence of atypical hemolytic uremic syndrome (HUS) in patients with mutations in complement factor genes and complement regulatory factor genes, therefore genetic complement testing is recommended for all atypical HUS patients undergoing transplant (Am J Transplant 2013;13:663)
Videos
Case #34 - HacettepePathology
Sample pathology report
- Kidney, allograft biopsy:
- Recurrent focal segmental glomerulosclerosis (see comment)
- No significant deposition on immunofluorescence microscopy. No evidence of T cell or antibody mediated rejection. Interstitial fibrosis / tubular atrophy absent. Arterial intimal fibrosis (mild).
- Microscopic description: Serial sectioning shows 26 glomeruli, one of which is globally sclerotic. 2 glomeruli with segmental sclerosis are identified; these segments display obliteration of the capillary tuft and adhesion to the Bowman membrane, accompanied by hyalinosis, lipid vacuoles and hypertrophy of the parietal epithelial cells. Other glomeruli appear close to normal by light microscopy. Endocapillary or extracapillary proliferation, necrosis, intracapillary inflammatory cells (g0), intracapillary thrombi or double contouring of the capillary walls are absent (cg0). Interstitium shows multifocal edema and minimal inflammatory infiltrate comprising < 10% of the nonscarred cortical parenchyma (i0). No significant tubulitis or peritubullary capillaritis is seen (t0, ptc0). Narrow foci of IFTA can be identified, comprising less than 10% of the cortical parenchyma with scattered mononuclear cells (i-IFTA0, ci0, ct0). Immunohistochemical staining for C4d and SV40 is negative. Arteries display mild intimal fibrosis (cv1). Intimal arteritis is not identified (v0).
- Immunofluorescence microscopy: 3 glomeruli observed
- Anti IgG Ab: No deposits
- Anti IgA Ab: No deposits
- Anti IgM Ab: Segmental peripheral 1+ granular staining (interpreted as nonspecific)
- Anti C3 Ab: No glomerular deposits, blush segmental reactivity on walls of arterioles
- Anti C1q Ab: No deposits
- Anti kappa Ab: No glomerular deposits, 2+ staining of tubular casts
- Anti lambda Ab: No glomerular deposits, 2+ staining of tubular casts
- Comment: Biopsy findings are consistent with recurrent focal glomerulosclerosis in this patient biopsied due to proteinuria during the early posttransplant period. There is no evidence of accompanying T cell or antibody mediated rejection or significant chronic tubulointerstitial damage. Vascular sclerosis is most probably donor related.
Differential diagnosis
- De novo versus recurrence:
- Differentiation of late recurrence versus development of de novo disease may be challenging
- In glomerular diseases early onset (< 1 - 2 years after transplant) favors recurrent disease
- Antibody mediated rejection (ABMR) may have similar morphology with:
- Immune complex mediated glomerular disease with membranoproliferative pattern of injury:
- Can resemble chronic active ABMR
- Immunofluorescence findings of immunoglobulins or complement deposition provides evidence for immune complex or complement mediated disease
- Acute and chronic thrombotic microangiopathy:
- Can resemble active and chronic active ABMR, respectively
- Negative immunofluorescence
- Absence of microvascular inflammation, C4d staining in peritubular capillaries and donor specific antibodies
- Presence of microvascular inflammation (transplant glomerulitis, peritubular capillaritis and transplant glomerulopathy), C4d staining in peritubular capillaries, presence of donor specific antibodies in the serum and lack of glomerular immune complex deposition by immunofluorescence are characteristics of antibody mediated rejection
- Immune complex mediated glomerular disease with membranoproliferative pattern of injury:
- Donor transmitted glomerular diseases:
- Can be diagnosed with time zero biopsies
- IgA nephropathy:
- IgA deposits usually disappear following the first 3 months of transplantation (Kidney Int 2003;63:2286)
- Membranous nephropathy:
- Deposits may be asymptomatic and resolve over time (Nephrol Dial Transplant 2014;29:2343)
Additional references
Board review style question #1
Which patient group below is at an increased risk of developing antiglomerular basement membrane glomerulonephritis following transplant?
- Patients with Alport syndrome
- Patients with diabetic nephropathy
- Patients with IgA nephropathy
- Patients with membranous glomerulonephritis
- Patients with primary oxalosis
Board review style answer #1
A. Patients with Alport syndrome. Patients with Alport syndrome lack α3, α4 or α5 chains of collagen IV after transplantation the α3, α4 or α5 chains of collagen IV of the donor kidney are recognized as foreign by the host immune system, which results in development of antibodies against these components and antiglomerular basement membrane glomerulonephritis.
Comment Here
Reference: Recurrent and de novo diseases
Comment Here
Reference: Recurrent and de novo diseases
Board review style question #2
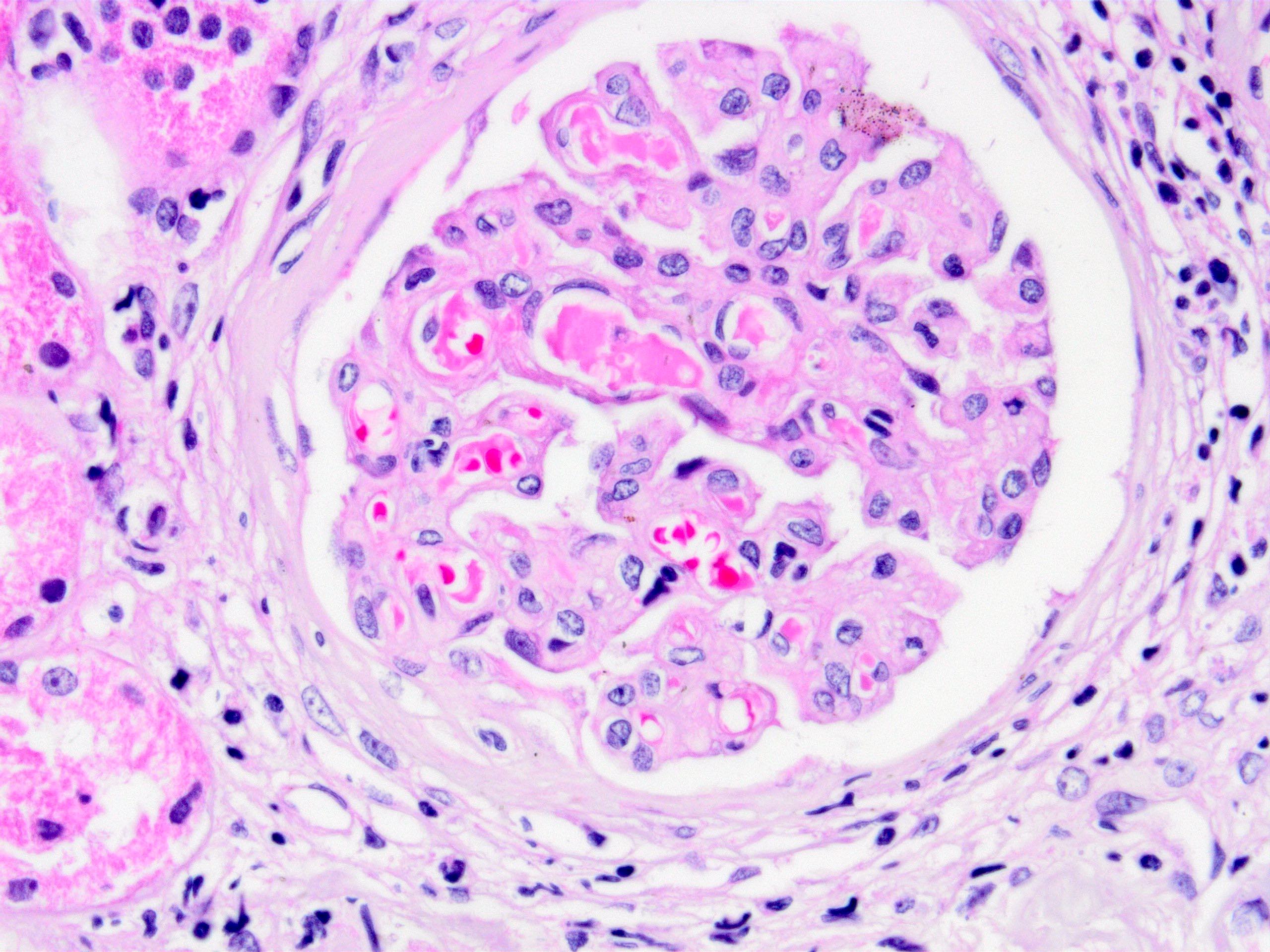
Above is a photomicrograph of the renal allograph biopsy of a patient who has been transplanted 3 weeks ago. The patient has a clinical history of hemolytic uremic syndrome due to infection. Immunohistochemistry reveals presence of peritubular and glomerular C4d staining. Which of the following differential diagnostic entities is the most likely diagnosis?
- Active antibody mediated rejection
- Acute calcineurin inhibitor toxicity
- Recurrent C3 glomerulopathy
- Recurrent hemolytic uremic syndrome
- Recurrent membranoproliferative glomerulonephritis
Board review style answer #2
A. Active antibody mediated rejection. The photomicrograph shows thrombotic microangiopathy; although almost all the mentioned entities enter the differential diagnosis, the presence of C4d staining initially points to active antibody mediated rejection; furthermore, hemolytic uremic syndrome due to infections typically does not recur.
Comment Here
Reference: Recurrent and de novo diseases
Comment Here
Reference: Recurrent and de novo diseases
