
Kidney nontumor / medical renal
Tubulointerstitial disease
Genetic
Nephrocalcinosis
Editorial Board Members: Nicole K. Andeen, M.D., Jonathan E. Zuckerman, M.D., Ph.D.


Last author update: 10 March 2025
Last staff update: 10 March 2025
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Nephrocalcinosis

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Positive stains | Negative stains | Electron microscopy description | Genetics | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2 | Board review style question #3 | Board review style answer #3Cite this page: Jankowski K, Mikhailov A. Nephrocalcinosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneynephrocalcinosis.html. Accessed April 1st, 2025.
Definition / general
- Nephrocalcinosis is the generalized deposition of calcium phosphate within renal tubules and interstitium
Essential features
- Nephrocalcinosis is a tubulointerstitial nephropathy characterized by tubular and interstitial calcium phosphate deposition and slowly progressive renal insufficiency (Hum Pathol 2004;35:675)
Terminology
- Nephrocalcinosis falls into 3 categories
- Molecular or chemical: an increase in intracellular calcium concentration that is quantifiable but not microscopically or radiologically visible
- Microscopic: deposits visible by light microscopic evaluation of kidney biopsy specimen but not radiologically visible
- Macroscopic: calcifications visible radiologically (StatPearls: Nephrocalcinosis [Accessed 17 February 2025])
- Strictly, the term nephrocalcinosis in pathology refers to the generalized deposition of calcium phosphate or calcium oxalate (calcium salts) in the kidney; it is an example of metastatic calcification (Urology 2012;79:277)
- Nephrocalcinosis belongs to the group of crystalline nephropathies, patterns of renal injury that share the distinctive finding of abundant crystals, most frequently involving the tubules and interstitium (Arch Pathol Lab Med 2012;136:713)
- Many pathologists limit the definition of nephrocalcinosis to the deposition of calcium phosphate by histopathologic detection of metastatic renal calcium deposits without associated tissue necrosis (StatPearls: Nephrocalcinosis [Accessed 17 February 2025], Hum Pathol 2004;35:675, Nephrol Dial Transplant 2012;27:1122)
- Deposition of calcium oxalate carries the name oxalosis
- Acute phosphate nephropathy is a clinical pathological entity characterized by acute and subsequent chronic renal failure following exposure to oral sodium phosphate bowel purgatives (Kidney Int 2009;76:1027)
ICD coding
- ICD-10: E83.59 - other disorders of calcium metabolism, including nephrocalcinosis
Epidemiology
- Nephrocalcinosis
- Is the main diagnosis in 0.4% of native kidney biopsies (Nephrol Dial Transplant 2012;27:1122)
- Tends to manifest equally between males and females (Kidney Int 2015;87:623)
- Has been reported in up to 22% of cases of primary hyperparathyroidism (Medicine (Baltimore) 1968;47:53)
- Is seen in up to 50% of cases of sarcoidosis with renal involvement (StatPearls: Nephrocalcinosis [Accessed 17 February 2025])
- Is seen in up to 29% of patients with renal tubular acidosis (N Engl J Med 1982;307:217)
- Hypocitraturia is a risk factor for nephrocalcinosis and renal stone formation (Rev Urol 2009;11:134)
- Nephrocalcinosis prevalence in kidney donors and patients with chronic kidney disease stage 1 - 2, 3 - 4 and 5 - 5D is 4.6%, 14.3%, 20.2% and 54%, respectively (Nephrol Dial Transplant 2015;30:843)
- Children with nephrotic syndrome (Nephrol Dial Transplant 2012;27:1122)
- In a study of medullary sponge kidney, nephrocalcinosis was observed in up to 50% of cases (N Engl J Med 1982;306:1106)
- Various genetic and hereditary disorders are associated with nephrocalcinosis, including X linked hypophosphatemic rickets, Bartter syndrome, Dent disease and many others (Kidney Int 1998;53:3, Am J Physiol Renal Physiol 2016;311:F1243)
- In kidney transplants, the prevalence of calcium phosphate crystals within the first 6 months posttransplantation is 26 - 32% (Transplantation 2009;87:618)
- Acute phosphate nephropathy: risk factors are female gender, age, hypertension and chronic kidney disease; also angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), diuretics and a history of chronic kidney disease (Kidney Int 2009;76:1027, Jennette: Heptinstall's Pathology of the Kidney, 8th Edition, 2023)
Sites
- Kidney tubules, tubular basement membranes and interstitium
- Usually involves the renal medulla (in 97% of patients) or less often, the cortex (Kidney Int 2015;88:35)
Pathophysiology
- Renal calcium deposits occur in disorders associated with
- Increased renal excretion of potassium or phosphate
- Altered solubility
- Factors facilitating crystal adhesion to the tubular epithelium
- Increased renal excretion of calcium or phosphate can be seen with
- Hypercalcemia or hyperphosphaturia
- Normal or low serum calcium or phosphate levels but impaired reabsorption in the tubules
- High calcium concentration, by activating the calcium sensing receptor in the collecting duct, is able to reduce its vasopressin stimulated water permeability, leading to an increased urinary volume, diluting the calcium ions (Am J Physiol Renal Physiol 2010;298:F485)
- On the opposite, decreased urine volume and urinary supersaturation are factors in intratubal calcium crystal formation
- In the renal tubules, a complex of citrate with calcium increases calcium solubility, limiting aggregation of calcium salts in association with Tamm-Horsfall protein (Am J Physiol 1993;265:F784)
- Acidosis (systemic, tubular, intracellular) decreases renal citrate excretion and accelerates citrate metabolism; hypokalemia has a similar effect (Rev Urol 2009;11:134)
- Alkalosis decreases calcium solubility (NDT Plus 2009;2:314)
- Crystals do not adhere to normally differentiated tubular epithelium but epithelial damage plays a role (Kidney Int 1999;55:1426)
- Injured / regenerating tubular epithelial cells express multiple crystal binding molecules on the apical surface: sialic acid containing proteins, phospholipids, phosphatidyl serine, nucleolin related protein, annexin II hyaluronan, osteopontin and CD44 (Kidney Int 2015;88:35, J Am Soc Nephrol 2003;14:107)
- After the failure of the mechanisms preventing crystal formation / adhesion, calcium phosphate crystals are observed adherent to the apical plasma membrane of the damaged tubular epithelial cells or the denuded tubular basement membrane (Kidney Int 2009;75:41)
- Then, epithelial cells neighboring the adhesion site proliferate over the adhered crystals (Kidney Int 2009;75:41)
- This newly formed tubular epithelial cell layer differentiates into mature epithelium, preventing further growth of the crystal and new crystal formation
- Crystals are directed towards the interstitium and eventually disintegrate
- This process may be assisted by monocytes / macrophages and other inflammatory cells (Kidney Int 2009;75:41)
- This clearing mechanism has been proposed based on observation of different cortical and medullary crystals of different types and animal experiments (Kidney Int 2009;75:41)
- Thus, nephrocalcinosis may be due to the failure of mechanisms preventing the formation of calcium phosphate crystals and the crystal clearing mechanism
- In acute phosphate nephropathy, massive phosphate intake inhibits proximal tubule phosphate reabsorption and the consequent diarrhea induced hypovolemia leads to a marked increase in calcium phosphate concentration in the distal tubule and collecting ducts; hypovolemia associated tubular injury also plays a role (Kidney Int 2009;76:1027)
Etiology
- Nephrocalcinosis is caused by diseases that result in hypercalcemia, hyperphosphatemia, hypercalciuria and hyperphosphaturia (StatPearls: Nephrocalcinosis [Accessed 17 February 2025])
- Primary and secondary hyperparathyroidism
- Malignancy
- Milk alkali syndrome: the third most common cause of hypercalcemia, with high incidence likely due to the consumption of calcium carbonate for prevention or treatment of osteoporosis (Kidney Int 2009;75:856)
- Hypervitaminosis D increases the intestinal absorption of calcium, which results in hypercalcemia and hypercalciuria (StatPearls: Nephrocalcinosis [Accessed 17 February 2025]) (see Vitamin D)
- Hypocitraturia due to hypokalemia, bowel dysfunction, a high protein, low alkali diet, medications
- Loop diuretics inhibit the Na-K-2Cl transporter and increase calcium excretion (Semin Nephrol 2011;31:535)
- Carbonic anhydrase inhibitors decrease bicarbonate absorption and the resultant metabolic acidosis, while increasing calcium excretion (Semin Nephrol 2011;31:535)
- Sarcoidosis
- Medullary sponge kidney
- Chronic kidney disease due to acid base and mineral metabolism disturbances and secondary hyperparathyroidism
- In renal allografts, nephrocalcinosis likely occurs due to persistent hyperparathyroidism, calcineurin inhibitors (inhibitor associated low citrate excretion) and hyperphosphaturia due to denervation (Pediatr Nephrol 2005;20:652, Nephrol Dial Transplant 2012;27:1122)
- Hereditary disorders
- Distal renal tubular acidosis: primary (hereditary) forms, usually type 1, lead to early onset of nephrocalcinosis (N Engl J Med 1982;307:217, Kidney Dis (Basel) 2023;9:371) (see Genetics)
- Dent disease (X linked recessive nephrolithiasis, X linked hypercalciuric hypophosphataemic rickets) (Orphanet J Rare Dis 2010;5:28) (see Genetics)
- Lowe syndrome (the oculocerebral syndrome of Lowe) (J Urol 1995;153:1244) (see Genetics)
- In X linked hypophosphatemia, nephrocalcinosis detected by renal ultrasound was diagnosed in 22% of children and in 38% of adults (J Bone Miner Res 2024;39:1493) (see Genetics)
- Nephrocalcinosis is associated with therapy consisting of active vitamin D combined with oral phosphate salts
- Idiopathic infantile hypercalcemia (see Genetics)
- Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (see Genetics)
- Idiopathic hypocitraturia (see Genetics)
- Bartter syndrome (BS): 5 different forms of salt losing nephropathies (see Genetics)
- Hypercalciuria and subsequent nephrocalcinosis are seen in BS1 and BS2 (Kidney Int 2021;99:324) (see Hyperaldosteronism)
- Enamel renal syndrome: amelogenesis imperfecta observed concomitantly with nephrocalcinosis (see Genetics)
- Acute phosphate nephropathy
- Ingestion of oral sodium phosphate bowel purgatives
- Also observed in renal allografts and may be associated with hyperphosphatemia in end stage renal disease (Transplantation 2009;87:618)
Clinical features
- Nephrocalcinosis is typically asymptomatic
- Hypercalcemia patients may present with hypercalcemia related symptoms such as polyuria and polydipsia
- History of high calcium / phosphate consumption
- Hypocitraturia (J Nephrol 2017;30:411)
- Hyperphosphatemia
- Specific clinical features depend on the etiology of nephrocalcinosis
Diagnosis
- Microscopic nephrocalcinosis is diagnosed on kidney biopsy
- Finding of nephrocalcinosis should prompt investigation into hypercalcemia etiology
- After the radiologic determination of nephrocalcinosis, serum electrolytes, calcium and phosphate should be measured; urine pH will help to determine the presence of distal renal tubular acidosis (StatPearls: Nephrocalcinosis [Accessed 17 February 2025])
Laboratory
- Laboratory test results depend on the underlying cause of nephrocalcinosis
- Serum electrolytes, calcium and phosphate, as well as urine pH, should be measured
- 24 hour urine is collected for measuring the excretion of calcium, phosphate and oxalate
- Chronic kidney disease patients with nephrocalcinosis show lower kidney function, lower serum bicarbonate levels and higher serum parathyroid hormone (PTH) and calcium levels (Nephrol Dial Transplant 2015;30:843)
Radiology description
- The most sensitive nephrocalcinosis imaging tests, ultrasound and computed tomography (CT), can detect macroscopic (but not microscopic) nephrocalcinosis
Prognostic factors
- Prognosis depends on the etiology of the underlying condition and the treatment efficacy
- Nephrocalcinosis may be reversed when a causative agent is stopped
- Extensive nephrocalcinosis may lead to renal damage and kidney failure (J Am Soc Nephrol 2005;16:3389)
- Posttransplant nephrocalcinosis is associated with poor renal allograft function (Ochsner J 2015;15:25)
Case reports
- 26 year old muscular man with nephrocalcinosis from suspected Dent disease (Am J Kidney Dis 2018;71:A12)
- 41 year old Japanese man with nephrocalcinosis in the distal tubules following a kidney transplant caused by hereditary renal hypouricemia (Nephrology (Carlton) 2016;21:67)
- 42 year old Caucasian man with nephrocalcinosis due to milk alkali syndrome (Kidney Int 2009;75:856)
- 47 year old woman with severe hypercalcemia from primary hyperparathyroidism (Am J Med 1987;83:355)
- 50 year old man with multiple medical problems resulting in calcium deposits along the glomerular basement membranes and tubular basement membranes (Am J Kidney Dis 2006;47:e23)
Treatment
- Treatment is directed at treating the underlying cause of nephrocalcinosis
- Goal of treatment is to reduce symptoms, prevent more calcium from building up in the kidney and reduce kidney damage
- Treatment plans may involve dietary restrictions of protein and sodium while increasing potassium intake for patients with hypercalciuria (StatPearls: Nephrocalcinosis [Accessed 17 February 2025])
Microscopic (histologic) description
- Calcium phosphate deposits within tubular lumina, interstitium and tubular basement membranes (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
- Calcium phosphate crystals arise more often in the medulla, especially near the loop of Henle but can arise in the cortex (Jennette: Heptinstall's Pathology of the Kidney, 8th Edition, 2023)
- Deposits are purple on H&E stained sections and blue on PAS staining
- 5 different patterns of nephrocalcinosis can be distinguished by H&E stain: clumpy, finely granular, globular, shell-like and onion skin-like (Nephrol Dial Transplant 2012;27:1122)
- In diseases and conditions associated with hypercalcemia or hypercalciuria, the clumpy and the finely granular deposits predominate (Nephrol Dial Transplant 2012;27:1122)
- In diseases and conditions associated with hyperphosphatemia or hyperphosphaturia, the most frequent patterns are globular and shell-like deposits (Nephrol Dial Transplant 2012;27:1122).
- The 2 different patterns likely reflect 2 distinct mechanisms of crystal formation; the calcium type could be composed predominantly of calcium hydroxyl apatite crystals, whereas phosphate could precipitate due to a self organizing phenomenon forming concentric lamellated structures (Liesegang rings) (Nephrol Dial Transplant 2012;27:1122)
- Concurrent hydroxyapatite and calcium oxalate crystal deposition may occur in the same patient with nephrocalcinosis (Kidney Int 2015;88:35)
- In the acute phase, acute tubular injury with possible foci of tubular regeneration is seen; in the chronic phase, there are fewer calcium phosphate crystals and less acute tubular injury but the interstitial fibrosis and tubular atrophy are predominant (Am J Kidney Dis 2017;69:e17)
- Deposits may be surrounded by mild lymphocytic infiltration
- In very severe hypercalcemia, calcifications may also be seen in glomeruli involving the Bowman capsule, the juxtaglomerular apparatus, glomerular basement membranes, mesangium and the Bowman capsule space (Arch Pathol Lab Med 2003;127:E80)
- Progressive acute tubular injury, interstitial fibrosis and tubular atrophy
- X linked hypophosphatemia: calcium phosphate / oxalate deposits within the renal medulla or less often, the cortex (J Bone Miner Res 2024;39:1493)
- Medullary sponge kidney: calcifications composed of calcium oxalate, calcium phosphate and uric acid predominantly in the dilated collecting ducts in the medulla (Urology 2012;79:277)
- Acute phosphate nephropathy: abundant distal tubular collecting duct and less prominent interstitial calcium phosphate deposits more prominent in the renal cortex; in biopsies with ≥ 10 glomeruli, > 30 calcifications are typically encountered (Kidney Int 2009;76:1027)
Microscopic (histologic) images
Contributed by Alexei Mikhailov, M.D., Ph.D.

Multiple phosphate crystals

Large calcium phosphate crystal

Clumpy and globular calcifications

Interstitial calcifications

2,8-DHA tubular crystals

2,8-DHA birefringence

Oxalate crystal birefringence
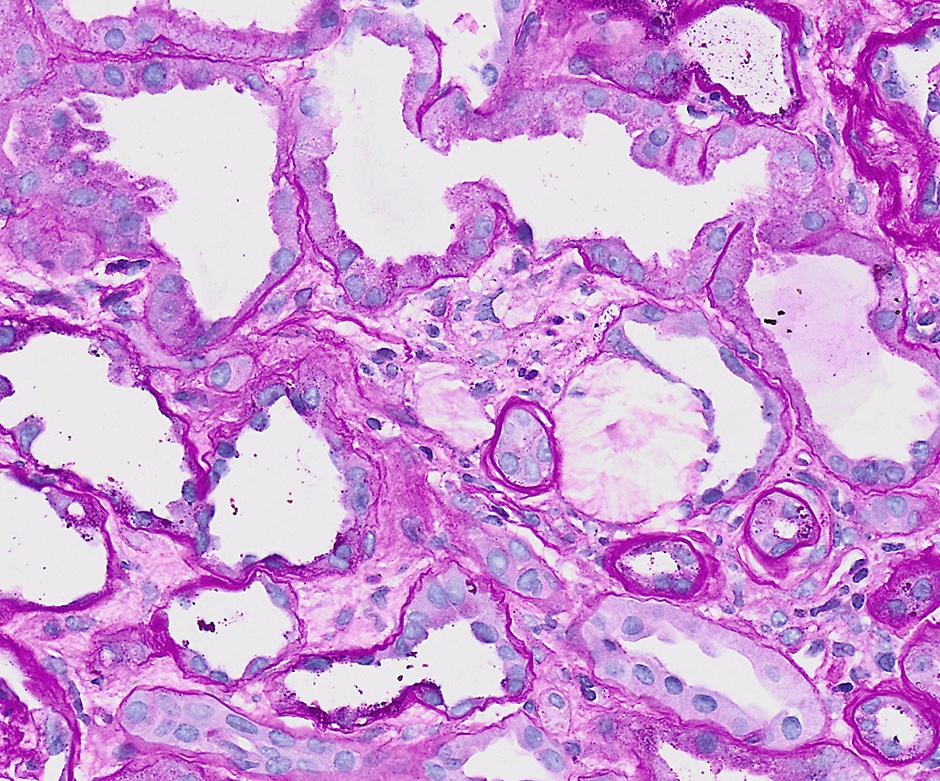
Oxalosis
Immunofluorescence description
- Immunofluorescence study is noncontributory in nephrocalcinosis
Positive stains
- Calcium phosphate is black with stains detecting phosphate (von Kossa)
- Alizarin red S stains calcium phosphate at pH of 7.0 and 4.2 but calcium oxalate crystals stain only at pH of 7.0 (Arch Pathol Lab Med 1985;109:186)
Negative stains
- Calcium phosphate deposits in H&E stained specimens are not birefringent under polarized light, as opposed to calcium oxalate (see Oxalosis)
Electron microscopy description
- Electron microscopy is not used for the diagnosis of nephrocalcinosis
- In the experimental setting, intracellular calcification is detected before intraluminal crystals appear (Am J Pathol 1964;44:365)
- Needle shaped deposits are found on many membrane surfaces throughout the cells and later, large masses involve the apical portion of the tubular epithelial cell and protrude into the tubular lumen (Am J Pathol 1964;44:365)
- Calcium aggregates in the glomerulus appear as dark, electron dense deposits (Arch Pathol Lab Med 2003;127:E80)
Genetics
- Determined by the underlying etiology
- Autosomal dominant or recessive
- Primary (inherited) distal renal tubular acidosis variants in the SLC4A1 gene encoding the basolateral Cl- HCO3- exchanger on type A distal tubule intercalated cells and on human erythrocytes (anion exchanger 1 [AE1]) (Kidney Dis (Basel) 2023;9:371)
- Idiopathic hypocitraturia: nucleotide polymorphisms in vitamin D receptor (VDR gene) and sodium citrate cotransporter-1 (also called solute carrier family 13 member 2 [SLC13A2]) (J Nephrol 2017;30:411)
- Autosomal recessive
- Idiopathic infantile hypercalcemia: mutations in CYP24A1, which encodes 25-hydroxyvitamin D 24-hydroxylase, the key enzyme of 1,25-dihydroxyvitamin D3 degradation (N Engl J Med 2011;365:410)
- Bartter syndrome forms with hypercalciuria: BS1 (SLC12A1 gene) and BS2 (KCNJ1 gene) (Kidney Int 2021;99:324)
- Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: an autosomal recessive disorder caused by mutations in the CLDN16 (paracellin-1) or CLDN19 genes encoding tight junction proteins claudin16 and claudin19 and characterized by renal loss of magnesium and calcium (Clin Kidney J 2015;8:656)
- Enamel renal syndrome: associated with recessive mutations in the FAM20A gene (family with sequence similarities 20 member A) (Orphanet J Rare Dis 2014;9:84)
- X linked recessive
- Dent disease: mutations affecting the CLCN5 (inactivation of the CLC-5 voltage gated chloride channels, Dent disease 1) or OCRL1 gene (phosphatidylinositol bisphosphate [PIP2] 5-phosphatase, Dent disease 2) on chromosome X (Orphanet J Rare Dis 2010;5:28)
- Lowe syndrome: mutation in the OCRL1 gene (phosphatidylinositol bisphosphate [PIP2] 5-phosphatase, Dent disease 2) on chromosome X (Orphanet J Rare Dis 2006;1:16)
- X linked dominant or recessive
- X linked hypophosphatemia: a rare disorder due to pathogenic variants in the phosphate regulating endopeptidase homolog (PHEX) gene leading to elevated circulating levels of fibroblast growth factor that impairs renal phosphate reabsorption of 1,25-dihydroxy vitamin D leading to chronic hypophosphatemia (J Bone Miner Res 2024;39:1493)
Sample pathology report
- Kidney, biopsy:
- Acute and chronic tubulointerstitial nephropathy with numerous tubular and interstitial calcium phosphate deposits, consistent with nephrocalcinosis (see comment)
- 5% global glomerulosclerosis (1/20)
- Mild arteriosclerosis
- Mild to moderate interstitial fibrosis and tubular atrophy
- Comment:
- Clinical information: The patient is a 50 year old Caucasian man admitted with generalized weakness, poor oral intake, nausea and vomiting for 4 weeks. To relieve acid reflux, the patient was frequently taking antacids and milk. The patient reports not having seen a physician for many years.
- Laboratory data: serum creatinine 5.2 mg/dL, hypercalcemia, severe nonanion gap metabolic alkalosis and low parathyroid hormone
- Light microscopy:
- The biopsy material consists of tissue from the renal cortex and medulla. The tissue is sampled at 10 levels of the section with H&E, PAS, trichrome and PAMS stains. Per section level up to 20 glomeruli are available, of which 1 is globally sclerosed. The nonsclerosed glomeruli show patent capillary lumens, single contoured capillary walls and no segments of sclerosis, adhesion or crescents. Approximately 30 tubules per biopsy core contain intraluminal calcifications of the calcium phosphate type (blue by H&E, nonbirefringent under polarized light). Tubules display markedly flattened epithelium with absent brush borders and prominent nucleoli, irregular luminal contours and foci of the denuded tubular basement membrane, associated with prominent and numerous intraluminal, intracellular and interstitial basophilic calcifications, nonbirefringent under polarizing light.
- A few lymphocytes are seen in the peritubular interstitium adjacent to the calcifications. There is mild to moderate interstitial fibrosis and tubular atrophy. Interlobular arteries of the medium caliber are present and show mild intimal fibroelastosis; there is mild arteriolar hyalinosis.
- The von Kossa special stain is positive in the distribution of the tubular calcifications, confirming the calcium phosphate content.
- Immunofluorescence:
- Up to 5 nonsclerosed glomeruli are available for evaluation by immunofluorescence microscopy.
- No diagnostic immunofluorescence staining is identified with antibodies to IgG-, IgA-, IgM-, C3-, C1q-, kappa light chain-, lambda light chain-.
- Electron microscopy:
- 2 nonsclerosed glomeruli are available for evaluation on semithin sections. The glomeruli do not show significant diagnostic abnormalities. There is mild interstitial fibrosis and tubular atrophy.
- Ultrastructural examination of 1 glomerulus shows glomerular basement membranes of normal thickness. There is no significant foot process effacement. Capillary lumens are patent. The mesangial matrix does not demonstrate any substantial expansion or hypercellularity. There are no electron dense (immune complex) deposits in any location. The tubular basement membranes are unremarkable.
Differential diagnosis
- Aging kidneys, acute tubular injury, chronic kidney disease of any etiology:
- Incidental finding of sparse tubular or interstitial calcifications
- Dystrophic calcinosis:
- Result of dystrophic calcification after parenchymal tissue destruction rather than urinary calcium crystallization
- Typically focal and seen in infarction, infection or malignancy
- Oxalosis:
- Abundant fan shaped crystals nonstaining with H&E or PAS stains, birefringent under polarized light
- Light chain proximal tubulopathy:
- History of monoclonal light chains, crystals with different tinctorial properties in the proximal tubules that react with either kappa or lambda light chain antibody (J Am Soc Nephrol 2016;27:1555)
- Light chain (myeloma) cast nephropathy:
- History of monoclonal light chains, hard wavy intratubular casts in the tubular lumens that react with either kappa or lambda light chain antibody
- Crystal storing histiocytosis:
- Accumulation of monoclonal light chain crystals in tubular epithelial histiocytes (Arch Pathol Lab Med 2012;136:713)
- Drug induced crystalline nephropathy associated with sulfadiazine:
- History of sulfadiazine use, crystalluria, hourglass shape crystals (Arch Pathol Lab Med 2012;136:713)
- Drug induced crystalline nephropathy associated with acyclovir:
- History of acyclovir antiviral treatment, crystals are needle shaped and polarizable (Arch Pathol Lab Med 2012;136:713)
- Drug induced crystalline nephropathy associated with indinavir:
- History of protease inhibitor treatment of HIV infection, needle shaped crystals that may form obstructive casts (Arch Pathol Lab Med 2012;136:713)
- Uric acid nephropathy or chronic urate nephropathy:
- Urate crystals dissolved during processing in formalin fixed tissue, staining blue with hematoxylin in frozen or alcohol fixed specimens (Arch Pathol Lab Med 2012;136:713)
- Cystinosis:
- Crystals are dissolved during processing with aqueous solutions but may be seen in the podocytes, mesangial cells, macrophages and tubular epithelium in frozen sections or unfixed tissues (Arch Pathol Lab Med 2012;136:713)
- Adenine phosphoribosyltransferase (APRT) deficiency (2,8-dihydroxyadeninuria):
- Tubular crystals resemble oxalate crystals but are tinted brownish green (Arch Pathol Lab Med 2012;136:713)
Board review style question #1
Kidney biopsy shows intratubal globular and clumpy crystals, staining intense blue to purple with the H&E stain, located in the lumens and adjacent to the denuded tubular basement membranes and associated with acute tubular injury. These deposits are expected to display which of the following properties?
- Association with intense lymphoplasmacytic infiltration
- Birefringence under the polarized light
- Black color with the von Kossa stain
- Positive alizarin red staining at pH 7.0 only
- Staining with either kappa or lambda light chains
Board review style answer #1
C. Black color with the von Kossa stain. The histological description points to calcium phosphate crystals staining blue to purple with the H&E stain. Von Kossa stain primarily detects calcium phosphate.
Answers B and D are incorrect because these are the properties of calcium oxalate crystals.
Answer E is incorrect because crystals of proximal light chain tubulopathy or casts of light chain cast nephropathy do not share the tinctorial properties of calcium phosphate crystals.
Answer A is incorrect because calcium phosphate crystals are sometimes associated with only mild lymphocytic infiltration.
Comment Here
Reference: Nephrocalcinosis
Comment Here
Reference: Nephrocalcinosis
Board review style question #2
A 60 year old patient is admitted with a history of uncontrolled hypertension and markedly elevated serum creatinine. A kidney biopsy is performed that shows a well circumscribed subcapsular focus of parenchymal atrophy, mononuclear infiltration and fibrosis with obliteration of the kidney structure, associated with large basophilic crystal aggregates consistent with calcium phosphate. What is the likely etiology of these crystals?
- Dystrophic calcification
- Incidental finding of interstitial calcification
- Nephrocalcinosis
- Oxalosis
- Uric acid nephropathy
Board review style answer #2
A. Dystrophic calcification. The patient history, the location, focal nature and the histology of the lesion strongly suggest a site of previous ischemic damage, possibly a cortical infarct.
Answer C is incorrect because the usual diffuse distribution of calcium phosphate in the tubules and the interstitium is not seen.
Answer E is incorrect because uric acid crystals are dissolved during formalin fixation.
Answer D is incorrect because the tinctorial properties of the calcification do not point to calcium oxalate and the distribution is not characteristic.
Answer B is incorrect because incidental calcifications are typically small, scattered and are not associated with the area of parenchymal damage.
Comment Here
Reference: Nephrocalcinosis
Comment Here
Reference: Nephrocalcinosis
Board review style question #3
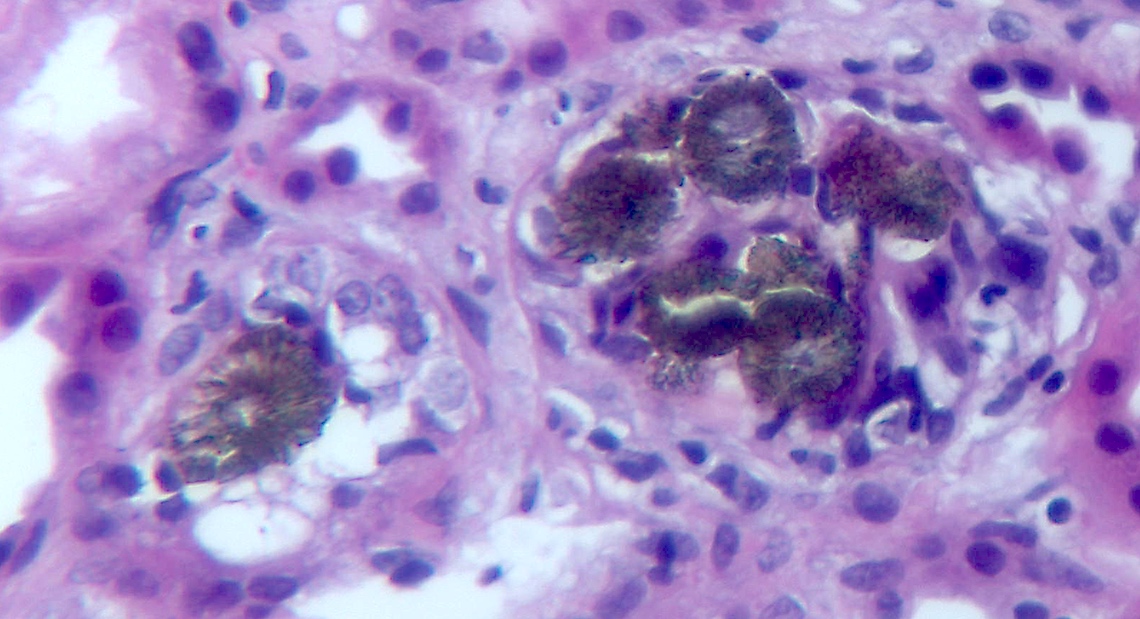

A 40 year old man is admitted to the hospital with frequent vomiting, diarrhea and general weakness. His family history is positive for urinary tract stones. Severe acute kidney injury was noted on admission. Computed tomography showed a single small stone in the left kidney. Aggressive rehydration failed to improve the kidney function and a kidney biopsy was performed. A representative field of view is shown in the images above. What is the likely diagnosis?
- Acute phosphate nephropathy
- Adenine phosphoribosyltransferase (APRT) deficiency
- Congenital nephrocalcinosis
- Nephrocalcinosis
- Oxalosis
Board review style answer #3
B. Adenine phosphoribosyltransferase (APRT) deficiency. The family history shows this is an inherited disease, the personal history shows nephrolithiasis and the kidney biopsy shows intratubular brown-green polarizing crystals characteristic of 2,8-dihydroxyadenine, consistent with the diagnosis of APRT deficiency.
Answers C and D are incorrect because the crystals do not show features of calcium phosphate aggregates (basophilic nonpolarizing crystals).
Answer E is incorrect because the biopsy does not show features of calcium oxalate (translucent fan shaped crystals).
Answer A is incorrect because there is no history of phosphate ingestion and the tinctorial properties of the crystals are consistent with 2,8-dihydroxyadenine.
Comment Here
Reference: Nephrocalcinosis
Comment Here
Reference: Nephrocalcinosis
