
Kidney nontumor / medical renal
Glomerular disease
IgA related glomerulonephritis
IgA nephropathy
Editorial Board Member: Nicole K. Andeen, M.D.
Editor-in-Chief: Debra L. Zynger, M.D.

Last author update: 7 May 2020
Last staff update: 1 April 2025 (update in progress)
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: IgA nephropathy[TI] kidney review[PT] free full text[SB]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Virtual slides | Immunofluorescence description | Immunofluorescence images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Genetics | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Soares MF. IgA nephropathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyiga.html. Accessed April 2nd, 2025.
Definition / general
- Immunoglobulin A (IgA) nephropathy: dominant or codominant mesangial IgA deposits in the absence of systemic diseases
- Variable light microscopic and clinical features
Essential features
- Most common primary glomerulonephritis worldwide
- Macro / microscopic hematuria with or without proteinuria
- 80% renal survival in 10 years; 25 - 50% progression to end stage renal disease over 20 years; 30% recurrence in transplants
- Etiology: abnormally glycosylated IgA1
- Dominant or codominant IgA deposits in mesangium; variable light microscopic features; electron microscopy most often with mesangial electron dense deposits
- Histopathological grading: Oxford classification - mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), tubular atrophy / interstitial fibrosis (T) and crescents (C) are independent prognostic factors
Terminology
- Immunoglobulin A nephropathy
- Berger disease
ICD coding
- ICD-10: N02.8 - Recurrent and persistent hematuria with morphologic changes
- SNOMED CT: 68779003 - Primary immunoglobulin A nephropathy
- SNOMED II: D67300 - Mesangial IgA / IgG disease
- SNOMED II is no longer supported but is still in use in some histopathology databases (SNOMED International 2019: A Brief History of SNOMED Code Systems [Accessed 17 April 2020])
Epidemiology
- Most common primary glomerulonephritis worldwide (UptoDate: Clinical Presentation and Diagnosis of IgA Nephropathy [Accessed 6 April 2020])
- More common in Southern Europe, Asia and Native Americans; less common in individuals of African lineage
- First to seventh decade of life; peak 20 - 30 years
- M:F = 2:1 in Europe / U.S. and 1:1 in Asia
Sites
- Renal cortex
Pathophysiology
- Increased serum levels of poorly galactosylated IgA1 (galactose deficient IgA1; GD-IgA1)
- Overproduction of GD-IgA1 in response to immune challenges in mucosal sites (upper respiratory and GI tract infections)
- GD-IgA1 binds to CD71 on mesangial cells; it also triggers autoimmune response with autoantibody production against GD-IgA1 GalNAc epitopes (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
- Disease progression (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019):
- Mesangial cell proliferation
- IgA / anti-IgA complex deposition in mesangium
- Cytokine release by mesangial cells leading to podocyte injury
- 25 - 50% of patients will progress to renal failure at 20 years; recurs in 20 - 60% of allografts (average 30%)
Etiology
- Heightened IgA response to mucosal antigens
- Secretion of poorly galactosylated IgA1
- Autoantibodies to poorly galactosylated IgA1
Clinical features
- 40 - 50%: at least one episode of visible hematuria associated to upper respiratory tract infection (synpharyngitic hematuria) (UptoDate: Clinical Presentation and Diagnosis of IgA Nephropathy [Accessed 6 April 2020])
- 30 - 50%: microscopic hematuria
- < 10%: nephrotic syndrome or rapidly progressive glomerulonephritis
- Rare: acute kidney injury (due to either rapidly progressive glomerulonephritis or red cell cast nephropathy)
- Secondary IgA nephropathy: associated with chronic liver disease, celiac disease and HIV
Diagnosis
- Renal biopsy with immunofluorescence / immunohistochemistry is the cornerstone of the diagnosis (UptoDate: Clinical Presentation and Diagnosis of IgA Nephropathy [Accessed 6 April 2020])
Laboratory
- Serum polymeric IgA1: elevated in 30 - 50% of cases (UptoDate: Clinical Presentation and Diagnosis of IgA Nephropathy [Accessed 6 April 2020])
- Urine dip: hematuria with or without proteinuria
Prognostic factors
- Oxford classification MEST-C scores: mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), tubular atrophy / interstitial fibrosis (T) and crescents (C) are independent prognostic factors (Curr Opin Nephrol Hypertens 2017;26:165):
- MEST-C scores and eGFR can predict outcome as accurately as 2 year follow up eGFR and proteinuria (JAMA Intern Med 2019;179:942)
- Limitations: original cohort excluded patients with proteinuria < 0.5 g/day, those with eGFR < 30 ml/min and follow up < 1 year; angiotensin blockade and immunosuppressive regimens were uncontrolled, resulting in treatment bias to outcome data
- Online risk calculator (Calculate by QxMD: International IgAN Prediction Tool [Accessed 6 April 2020])
- In patients with proteinuria < 0.5 g/24 h: M1 and E1 predict proteinuria > 1 g/24 h over 5 year follow up; M1 independently predicts rapid loss of renal function
- E1: independently predicts rapid loss of renal function in patients not treated with steroids (J Nephrol 2016;29:367, Kidney Int 2017;91:235)
- S1 with podocyte hypertrophy and tip lesions (S1p): associated to higher proteinuria/24 h and worse renal survival in patients not treated with steroids (Kidney Int 2017;91:235)
- Endocapillary hypercellularity, crescents and necrosis seem to be responsive to immunosuppression (J Nephrol 2015;28:441)
- Extent of interstitial fibrosis / tubular atrophy: reflects chronic damage; associated with long term outcome
- Immunohistochemistry:
- Evidence of lectin pathway activation (deposition of mannose binding lectin, L ficolin, MASP1/3 and C4d): seen in 25 - 30% of cases and is associated with more severe histologic damage and proteinuria (Kidney Int Rep 2017;3:426)
- Mesangial C4d: predicts worse long term renal outcome (J Nephrol 2016;29:1)
- Glomerular deposition of complement factor H related protein 5 (CFHR5): associated to disease progression (Kidney Int Rep 2017;3:426)
- Clinical predictors (UptoDate: Treatment and Prognosis of IgA Nephropathy [Accessed 6 April 2020]):
- High serum creatinine / reduced eGFR
- Hypertension (> 140/90 mmHg)
- Proteinuria > 1 g/24 h for over 6 months
Case reports
- 24 year old man with atypical celiac disease presenting with nephrotic syndrome (Cent Eur J Immunol 2019;44:106)
- 36 year old man with recurrent crescentic disease in renal allograft (Clin Case Rep 2019;7:1773)
- 50 year old woman with concomitant primary membranous glomerulonephritis (Saudi J Kidney Dis Transpl 2019;30:531)
- 74 year old woman with systemic lupus erythematosus and acute kidney injury due to crescentic disease (Case Rep Nephrol 2019;2019:8354823)
- 81 year old man with differential diagnosis of IgA dominant infection associated glomerulonephritis (Am J Case Rep 2019;20:508)
Treatment
- Aims at slowing progression of renal damage (nonimmunosuppresive measures: control of blood pressure and proteinuria) and dampening inflammatory activity (immunosuppression: steroids) (UptoDate: Treatment and Prognosis of IgA Nephropathy [Accessed 6 April 2020])
- If hematuria, normal eGFR with or without proteinuria < 0.5 g/day: watchful monitoring at 6 to 12 month intervals
- If proteinuria > 500 mg/day over 6 months and or mildly reduced eGFR and mild to moderate histologic findings on renal biopsy: nonimmunosuppressive therapies (ACE-I / ARB)
- If rising creatinine, active lesions on renal biopsy (E1, C1/2) or nephrotic range proteinuria or proteinuria after 3 - 6 months of nonimmunosuppressive: consider association of immunosuppressive therapy in addition
- There is some evidence that patients with E1, podocytopathic S1 lesions and C1 and C2 have a markedly lower rate of decline in eGFR when immunosuppressed (J Am Soc Nephrol 2017;28:691)
- Fish oil: low quality evidence; tonsillectomy: evidence from randomized controlled trials missing
Microscopic (histologic) description
- Oxford classification: first evidence based glomerular disease classification (Semin Nephrol 2018;38:477)
- Variable light microscopic patterns; in the original classification 4 lesions were reproducible and predictive of outcome (Kidney Int 2017;91:1014):
- Mesangial hypercellularity (M0 ≤ 50% of glomeruli; M1 > 50% of glomeruli): > 3 mesangial cell nuclei / peripheral mesangial area, not adjacent to vascular pole, in 2 - 3 μm thick PAS stained section
- Endocapillary hypercellularity (E0 absent; E1 present): CD68 count > 6 in most affected glomerulus of the sample is associated with E1
- Segmental glomerulosclerosis / adhesions / synechiae (S0 absent; S1 present): due to hyperfiltration, healed necrotizing lesions or podocytopathy
- Tubular atrophy / interstitial fibrosis (T0 absent to involving ≤ 25% of the cortex; T1 26 - 50% of the cortex; T2 > 50% of the cortex)
- Crescents: added to the classification in 2016 (C0 absent; C1 1 - 24% of glomeruli with cellular / fibrocellular crescents; C2 ≥ 25% of glomeruli with cellular / fibrocellular crescents)
- Other lesions: fibrinoid necrosis (10% of biopsies), glomerular basement membrane duplication (< 10% of biopsies), thrombotic microangiopathy changes (5 - 50% of biopsies; usually associated with malignant hypertension) (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
Microscopic (histologic) images
Contributed by Maria Fernanda Soares, M.D., Ph.D.
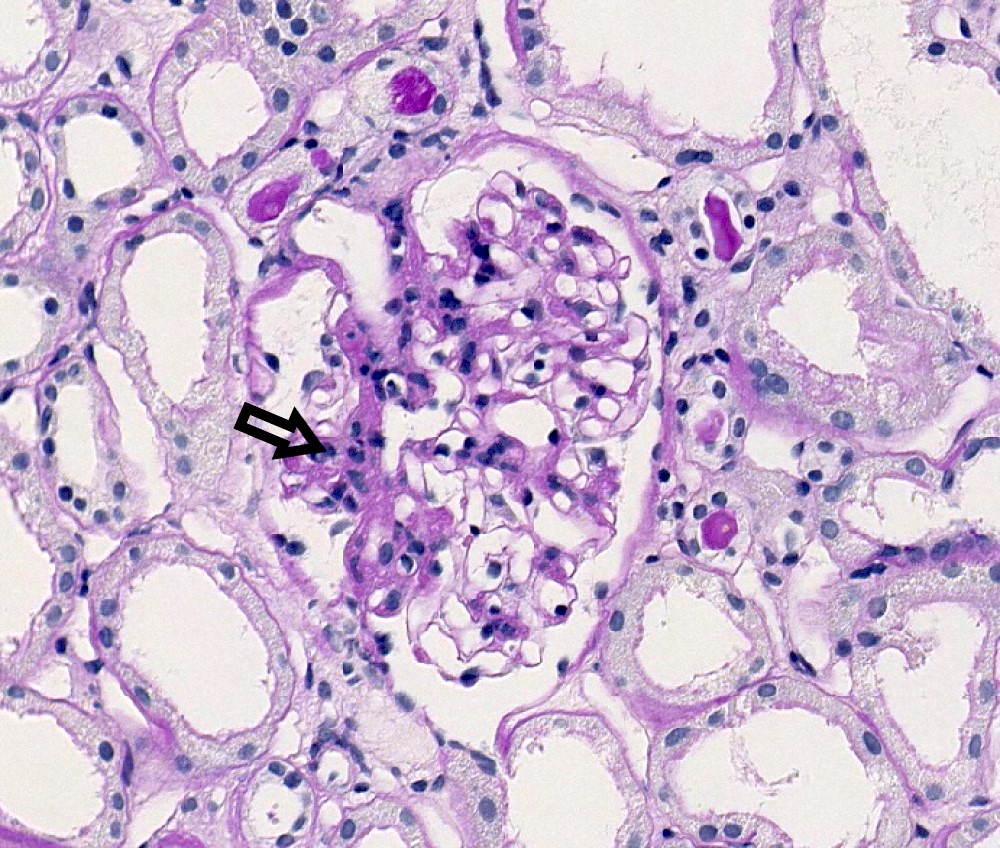
M1 lesion, PAS
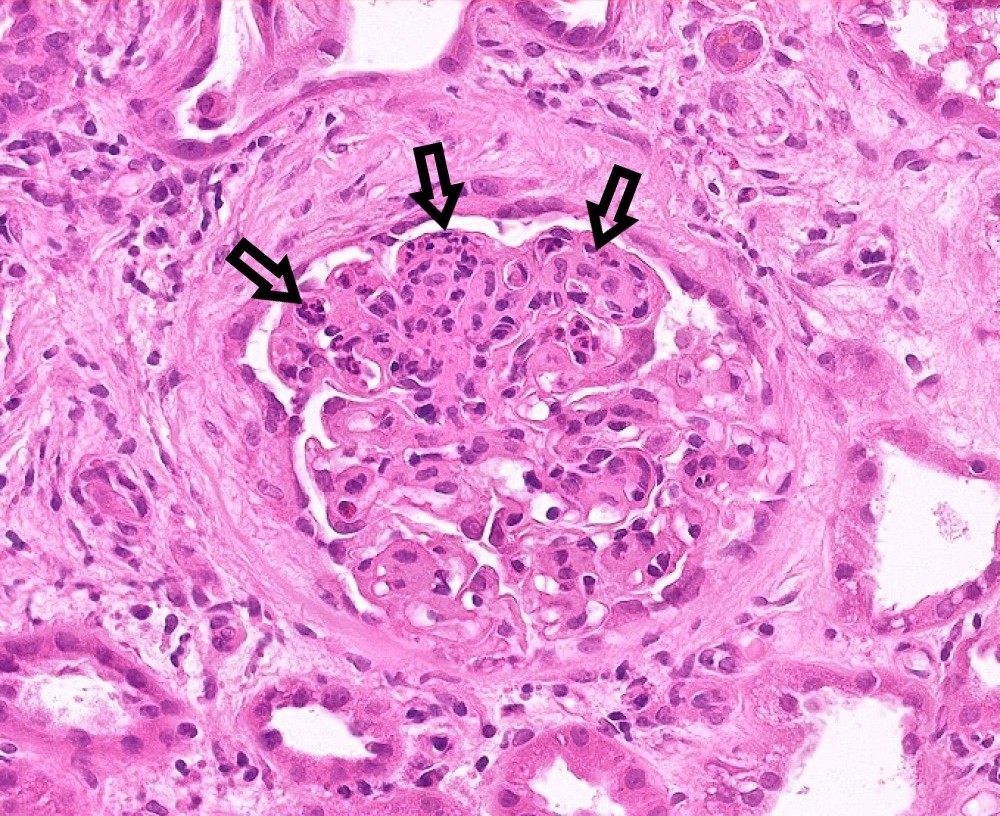
E1 lesion
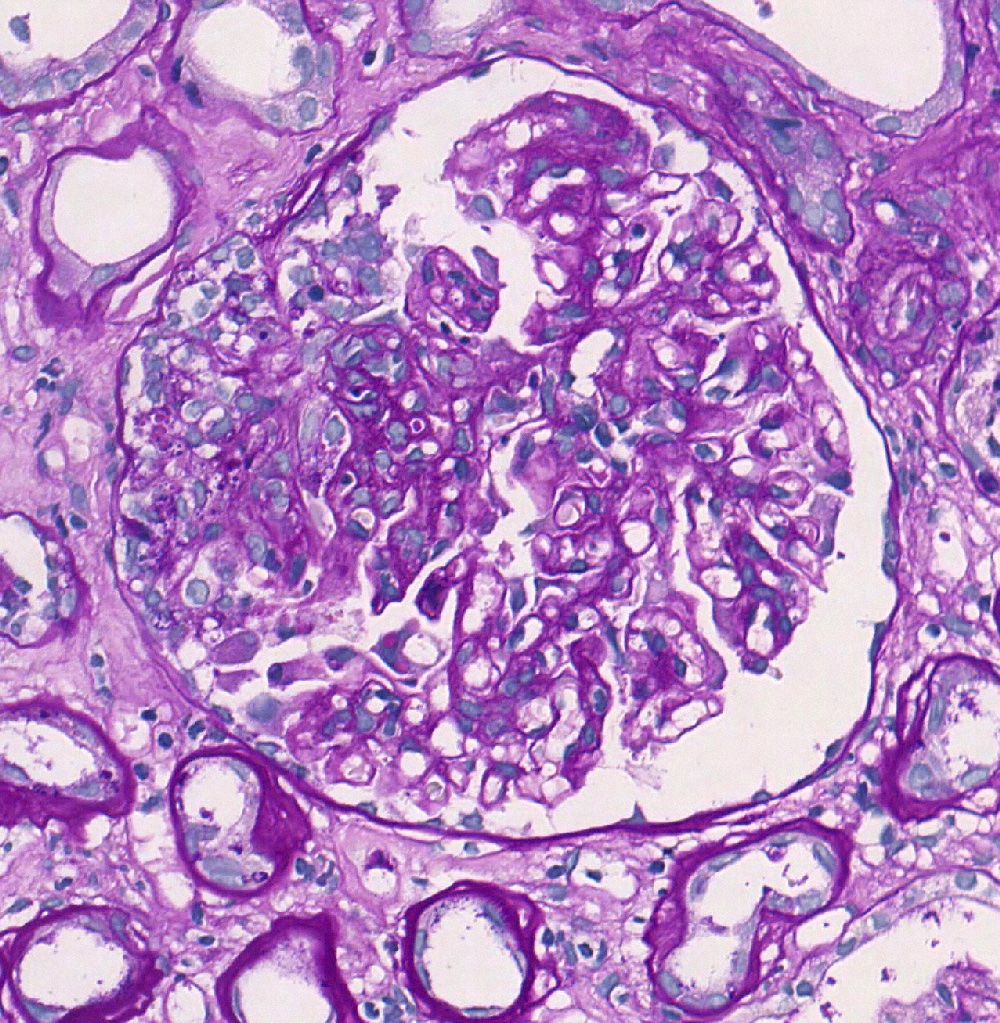
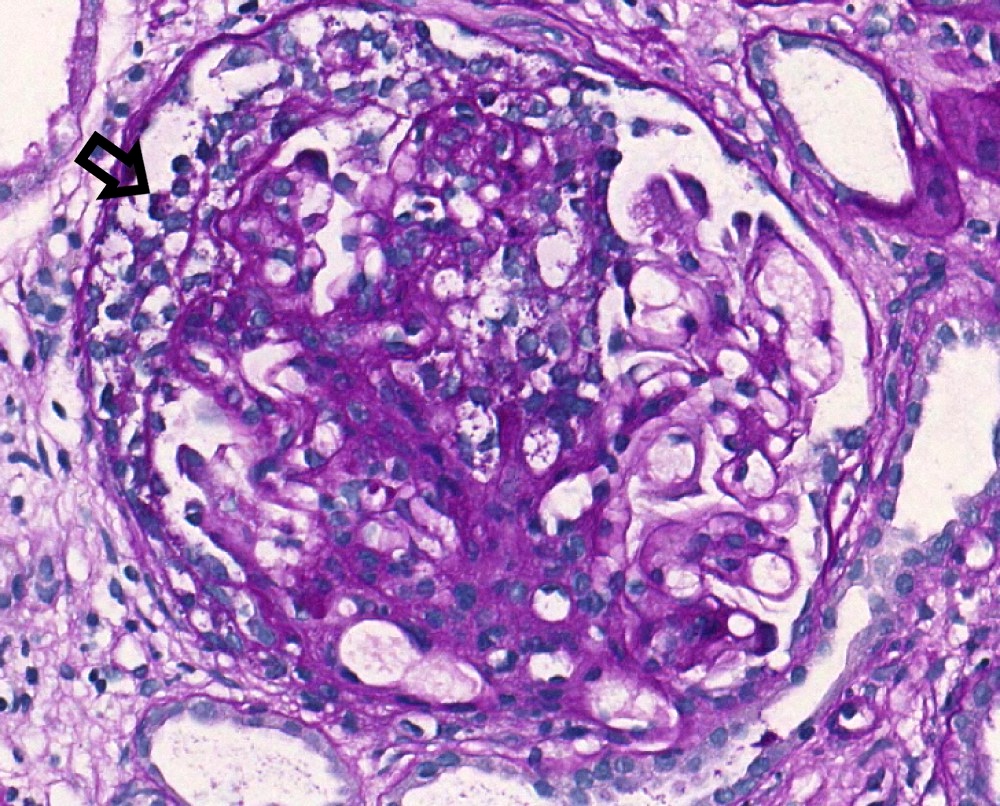
S1p lesion, PAS
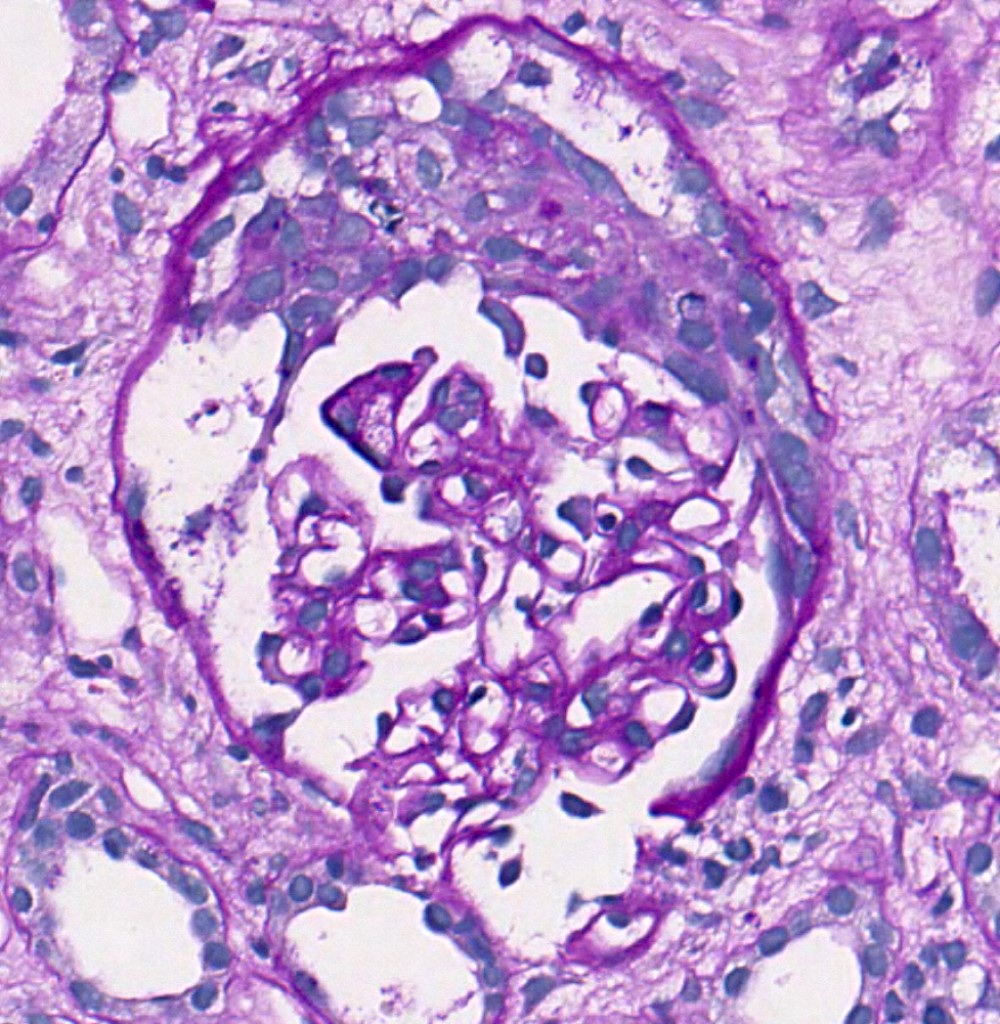
Crescent, PAS

Red cell casts

Acute arterial TMA
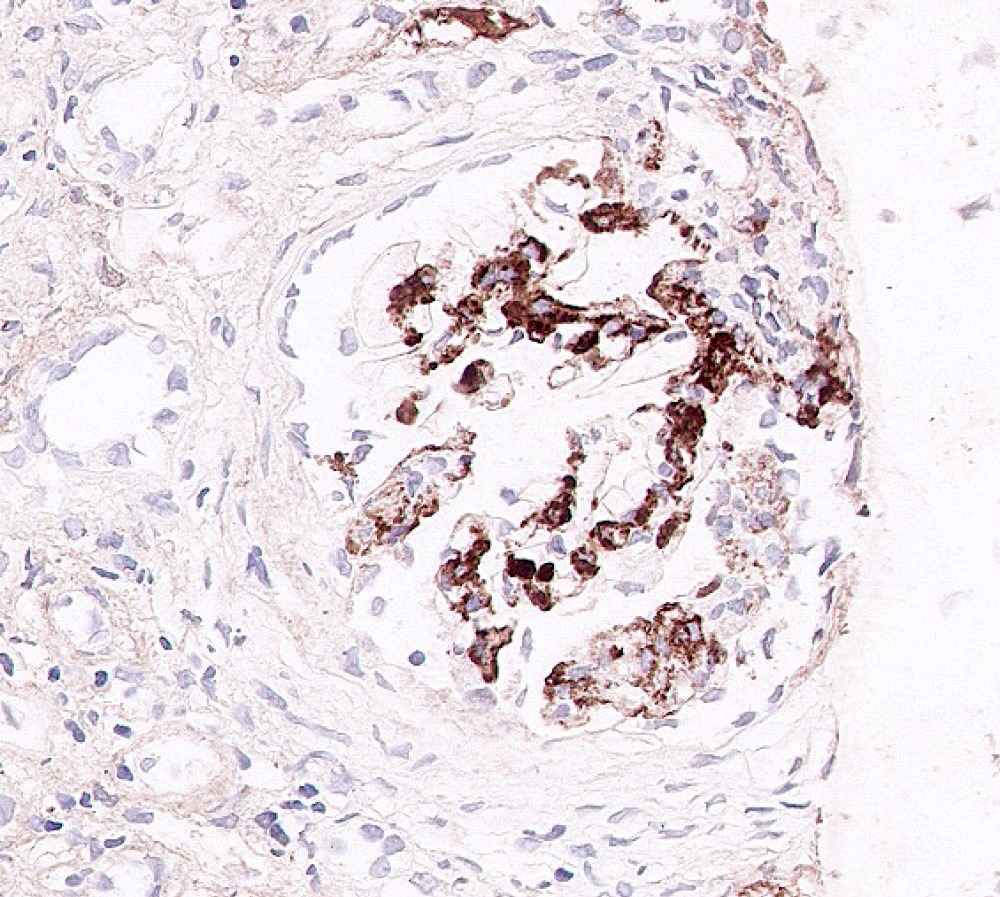
Mesangial IgA

CD68 and E1 lesion
Contributed by Arkana
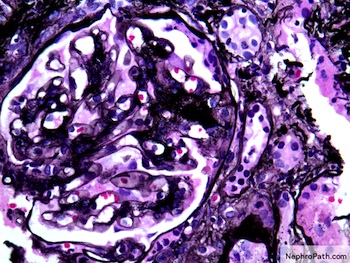
Mesangial expansion and loop adhesion, Jones silver
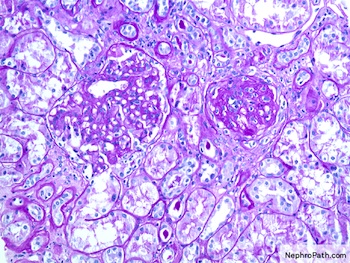
Mesangial expansion and global sclerosis, PAS

Mesangial expansion and loop adhesion, PAS
Virtual slides
Images hosted on other servers:

M0 E1 C1 S1 T1

M1 E0 C0 S1 T0

M0 E0 C0 S1 T1

M1 E1 C1 S1 T0
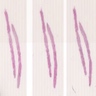
IgA nephropathy and TMA

IgA (same case as previous slide)
Immunofluorescence description
- Dominant IgA mesangial deposits (100%) - only defining and consistent feature of the disease
- Predominance of J chain containing IgA1 and lambda light chains
- Other mesangial deposits: C3 (90%), IgG (20 - 30%), IgM (50%), C1q (< 10% - almost always absent, indicating lack of activation of classical pathway) (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
- Occasional capillary loop subendothelial deposits
Immunofluorescence images
Contributed by Maria
Fernanda Soares, M.D., Ph.D.
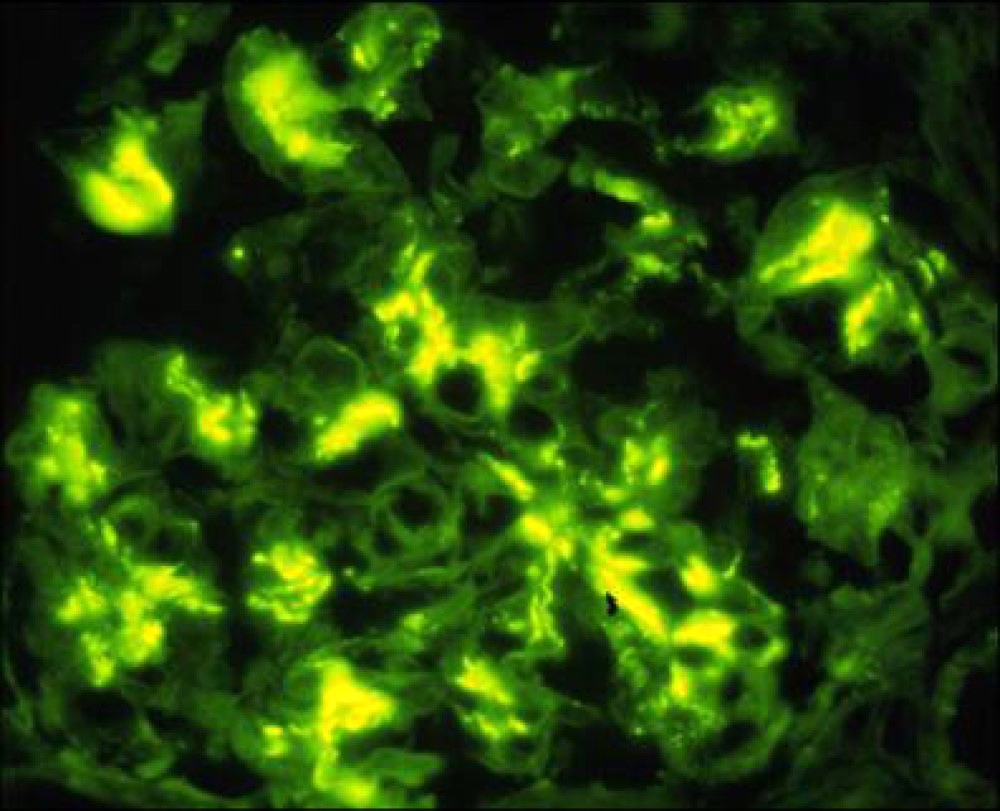
Mesangial IgA
Contributed by Arkana
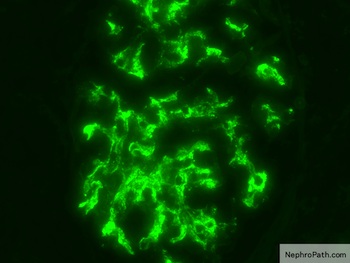
Mesangial IgA
Positive stains
- CD68: maximum count of glomerular CD68 positive shows relationship with E score (Histopathology 2019;74:629)
- C4d and mannan binding lectin (MBL): mesangial staining associated with worse prognosis (J Nephrol 2016;29:1)
Negative stains
Electron microscopy description
- Electron dense deposits without substructure (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
- Most commonly mesangial
- Subendothelial deposits associated to more active lesions on light microscopy
- Subepithelial deposits exceedingly rare
Electron microscopy images
Contributed by Maria Fernanda Soares, M.D., Ph.D.
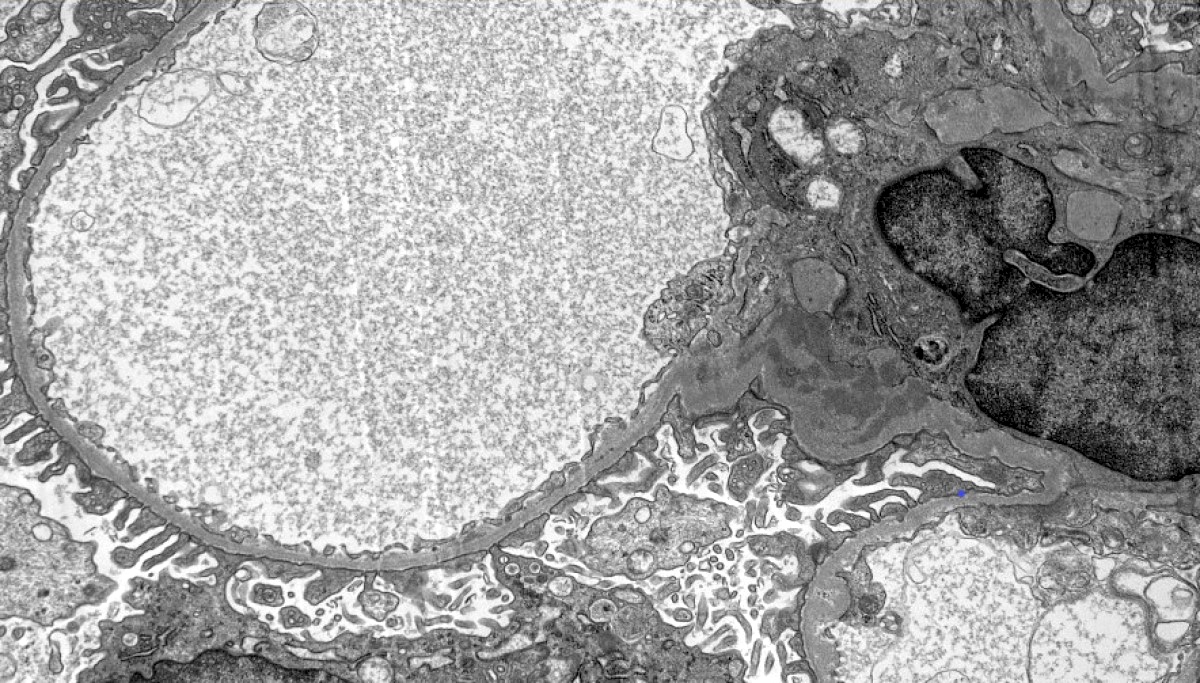
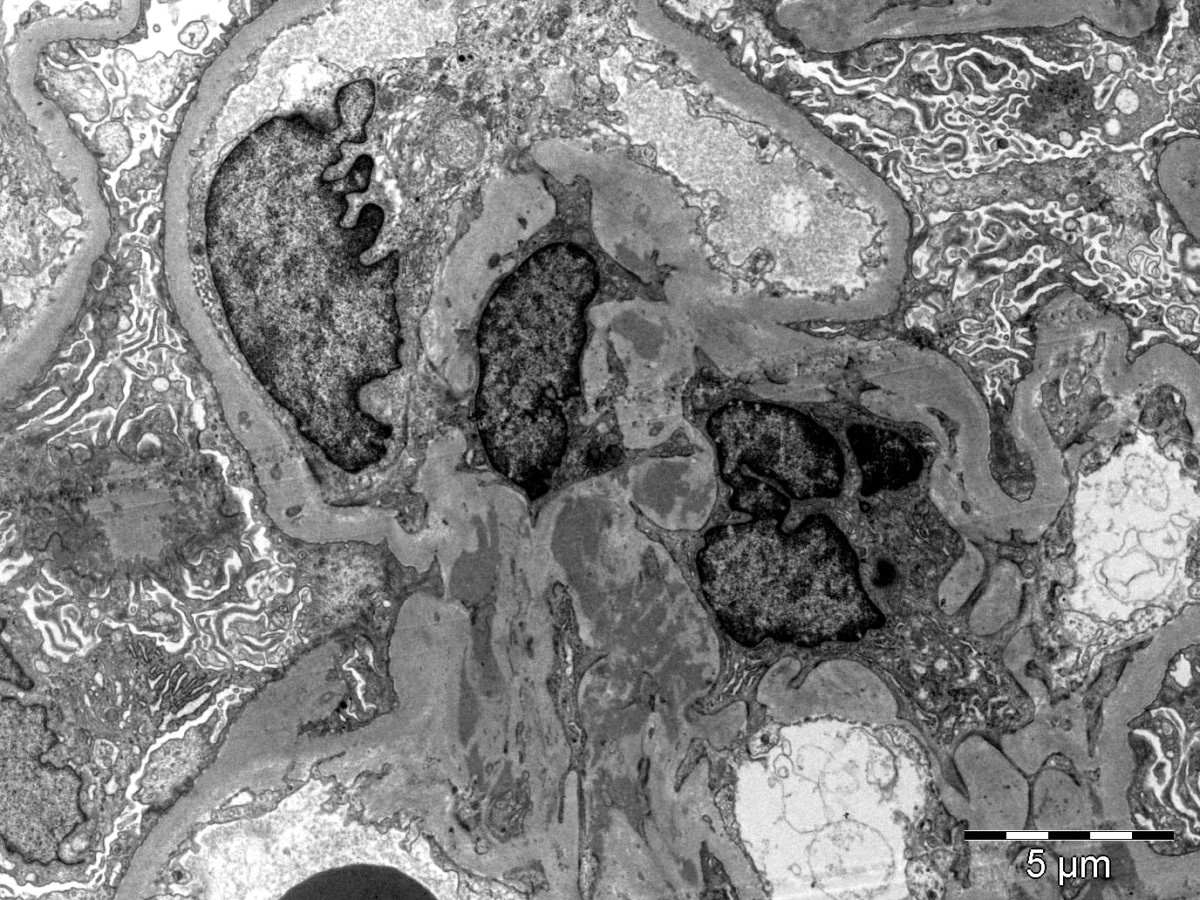
Mesangial deposits
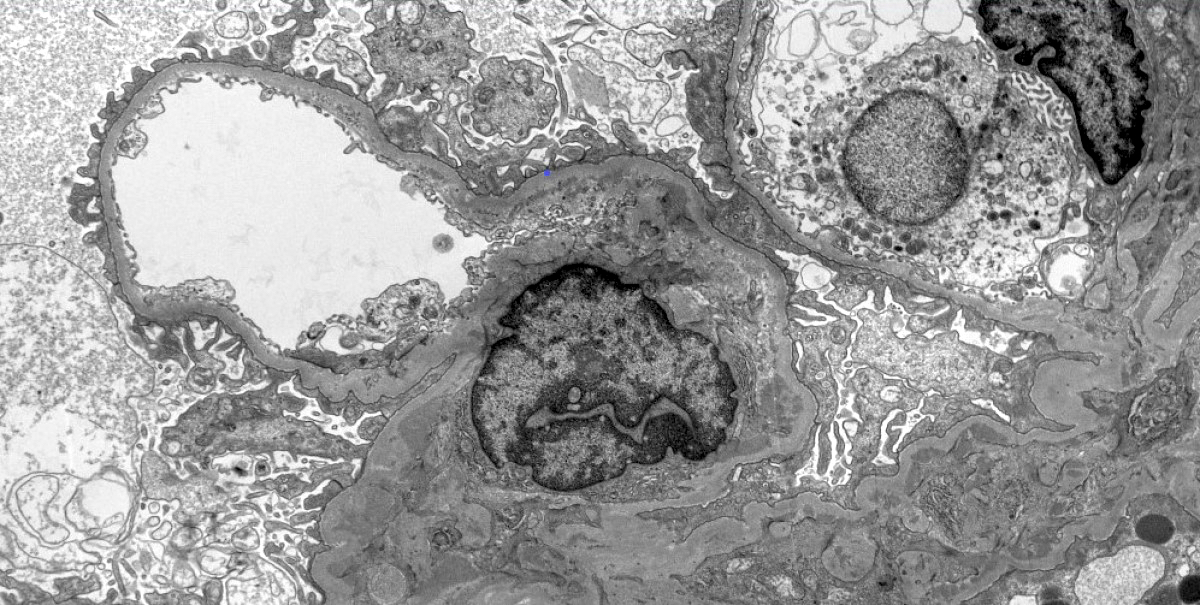
Mesangial and paramesangial deposits
Contributed by Arkana
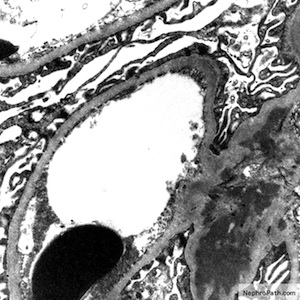

Mesangial deposits
Genetics
- Family history in 10 - 15% of cases (Colvin: Diagnostic Pathology - Kidney Diseases, 3rd Edition, 2019)
- Autosomal dominant IgA nephropathy identified in a Sicilian family with SPRY2 mutation that causes inhibition of inhibition of MAPK / ERK1/2
- Genome wide association study studies: susceptibility loci at 1q32 (CFH and CFHR1-5 genes), 16p11 (ITGAM / ITGAX), 17p13 (α defensin gene cluster), 8q23 (TNFSF13), 1p13 (VAV3) and 9q34 (CARD9)
Sample pathology report
- Left kidney, core biopsy:
- IgA nephropathy (see comment)
- Comment: There is an IgA nephropathy with diffuse mesangial hypercellularity and focal segmental glomerulosclerosis with podocytopathic features. There is severe chronic tubulointerstitial damage. Oxford classification: M1 E0 S1p T2 C0.
- Specimen: A core of renal cortex containing up to 28 glomeruli in the planes of section.
- Glomeruli: 15/28 are globally sclerosed. 6 show segmental sclerosis, 4 of which with podocytopathic features. There is diffuse mesangial hypercellularity but no endocapillary or extracapillary hypercellularity. Basement membranes appear normal on silver stain. Congo red stain is negative for amyloid.
- Tubules and interstitium: There is severe chronic damage with 60% of the cortex showing interstitial fibrosis and tubular atrophy. There is a mild mononuclear infiltrate, which is confined to the areas of fibrosis. There are uromodulin casts but no red cell casts.
- Vessels: Arteries show moderate fibroelastosis. There is focal arteriolar hyalinosis.
- Immunofluorescence microscopy: There is diffuse mesangial and focal capillary wall positivity for IgA (3+), C3 (1+), kappa (2+) and lambda (3+). IgG, IgM and C1q are negative.
Differential diagnosis
- Henoch-Schönlein purpura nephritis:
- Morphologically indistinguishable
- Clinical information is important for differential diagnosis (e.g. history of purpuric rash, abdominal pain, arthritis)
- IgA dominant infection associated glomerulonephritis (in general, Staphylococcus associated):
- Electron microscopy contains hump shaped deposits
- Mesangial IgA deposits in chronic liver disease:
- Present in 60% of autopsies of cirrhotic patients
- Rarely associated with active glomerular disease (UptoDate: Clinical Presentation and Diagnosis of IgA Nephropathy [Accessed 6 April 2020])
Additional references
Board review style question #1
A 32 year old man presents with mild reduction in eGFR, proteinuria and microscopic hematuria. Urine protein / creatinine ratio in nephrotic range. Immunofluorescence microscopy of a renal biopsy core showed mesangial deposition of IgA (strong - see image), C3, kappa and lambda (moderate) and IgG (weak and focal). IgM and C1q were negative. Based on these findings, what is the best diagnosis?
- Accurate diagnosis of a renal biopsy cannot be assigned on the basis of the immunofluorescence findings only
- Alport syndrome
- Immunoglobulin A nephropathy
- Lupus nephritis
- Membranous glomerulonephritis
Board review style answer #1
Board review style question #2
Indicate the correct association in the Oxford classification of IgA nephropathy
- Absence of necrosis / karyorrhexis - NO
- Crescents in 10% of glomeruli - C2
- Interstitial fibrosis / tubular atrophy in 25 - 50% of the cortex - T2
- Presence of endocapillary hypercellularity - E1
- Presence of segmental sclerosis in > 50% of glomeruli - S2
Board review style answer #2
