

Lymphoma & related disorders
Mature T/NK cell disorders
T/NK cell disorders with a leukemic component
T prolymphocytic leukemia
Editorial Board Member: Roberto N. Miranda, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 6 January 2023
Last staff update: 9 January 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: T cell prolymphocytic leukemia

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Positive stains | Negative stains | Flow cytometry description | Flow cytometry images | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Shi M, Jevremovic D. T prolymphocytic leukemia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomanonBTcellpro.html. Accessed December 23rd, 2024.
Definition / general
- T prolymphocytic leukemia (T PLL) is an aggressive, mature T cell leukemia, composed of small to medium sized mature T cells, usually with high white blood cell (WBC) count and widespread organ involvement
- No significant changes in WHO 5 and ICC classifications (Leukemia 2022;36:1720, Blood 2022;140:1229)
Essential features
- Aggressive leukemia of mature T cells
- High white blood cell count (lymphocytosis)
- Usually CD4+
- Usually TCL1 positive by immunophenotyping and TCL1 rearranged by FISH
- Difficult to treat, poor prognosis
Terminology
- T cell chronic lymphocytic leukemia (small cell variant of T cell prolymphocytic leukemia)
ICD coding
Epidemiology
- Rare (2% of mature lymphocytic leukemias)
- Adults and elderly (> 30 years, median age: 65 years)
Sites
- Widespread: peripheral blood, bone marrow, lymph nodes, spleen, liver, skin
Pathophysiology
- Combination of overexpression of TCL1 family of proteins (which stimulate AKT / protein kinase B driven proliferation) and functional deficit of ATM protein (Nat Commun 2018;9:697)
Etiology
- Unknown at this time
- Higher risk in patients with ataxia-telangiectasia (germline ATM mutations)
Clinical features
- High tumor burden with high white blood cell count (median 50 - 60 x 109/L), bone marrow involvement (in 100% of patients) with cytopenias, hepatosplenomegaly, lymphadenopathy, skin and mucosal lesions, other organ involvement / dysfunction (Am J Hematol 2017;92:441)
- Prominent constitutional symptoms
- 20 - 30% present with inactive disease and progress to active disease within 1 - 2 years (criteria for progression: lymphocyte doubling time (LDT) of less than 6 months or lymphocyte count increase by > 50% in 2 months) (Blood 2019;134:1132)
Diagnosis
- Peripheral blood: morphology + flow cytometry with or without fluorescent in situ hybridization (FISH)
- Bone marrow or solid tissue biopsy (lymph node, spleen, liver, skin, other): morphology + phenotyping (immunohistochemistry or flow cytometry) with or without FISH
- Note: FISH not necessary if TCL1 overexpression in neoplastic T cells can be shown by IHC or flow (J Clin Pathol 2018;71:309)
- Consensus criteria for diagnosis by the T PLL International Study Group (Blood 2019;134:1132)
- All 3 major criteria or the first 2 major and 1 minor criteria are required for diagnosis
- Major criteria:
- 5 x 109/L cells of T PLL phenotype in peripheral blood or bone marrow
- T cell clonality by molecular or flow cytometry methods
- Abnormalities of 14q32 or Xq28 or expression of TCL1A/B or MTCP1
- Minor criteria:
- Abnormalities involving chromosome 11 (11q22.3; ATM)
- Abnormalities in chromosome 8: idic(8)(p11), t(8;8), trisomy 8q
- Abnormalities in chromosome 5, 12, 13, 22 or complex karyotype
- Involvement of specific sites (spleen, effusions)
Laboratory
- Increased peripheral blood lymphocytes, often > 100 x 109/L, increased lactate dehydrogenase (LDH) and beta2 microglobulin (Ann Oncol 2017;28:1554)
- Negative serology for HTLV1
Radiology description
- Prominent hepatosplenomegaly and widespread lymphadenopathy; moderate to high FEV on PET scan
Prognostic factors
- Overall poor prognosis; median survival with active disease is 1 - 2 years
- Worse prognosis: pleural effusion, high LDH (> 1668 IU/l), low hemoglobin (< 9.3 g/dl), complex karyotype (Ann Oncol 2017;28:1554, Am J Hematol 2017;92:441)
Case reports
- 59 year old man with the history of renal transplant (J Med Case Rep 2019;13:223)
- 60 year old woman with a history of chronic myeloid leukemia (CML) (J Clin Pathol 2019;72:511)
- 64 year old man with TCL1 positive hematogones after T cell prolymphocytic leukemia therapy (Hum Pathol 2017;65:175)
- 68 year old man with heart failure (Blood 2017;130:691)
- 75 year old man with unusual phenotype of T cell prolymphocytic leukemia (Blood 2018;132:111)
Treatment
- Only for active disease
- Standard treatment: alemtuzumab (anti-CD52) variable allogeneic bone marrow transplant
- Experimental therapies with BCL2, JAK3 or HDAC inhibitors (Blood 2019;134:1132)
Microscopic (histologic) description
- Perivascular and diffuse tissue infiltrates of uniform small to medium sized lymphocytes
- Red pulp involvement in the spleen
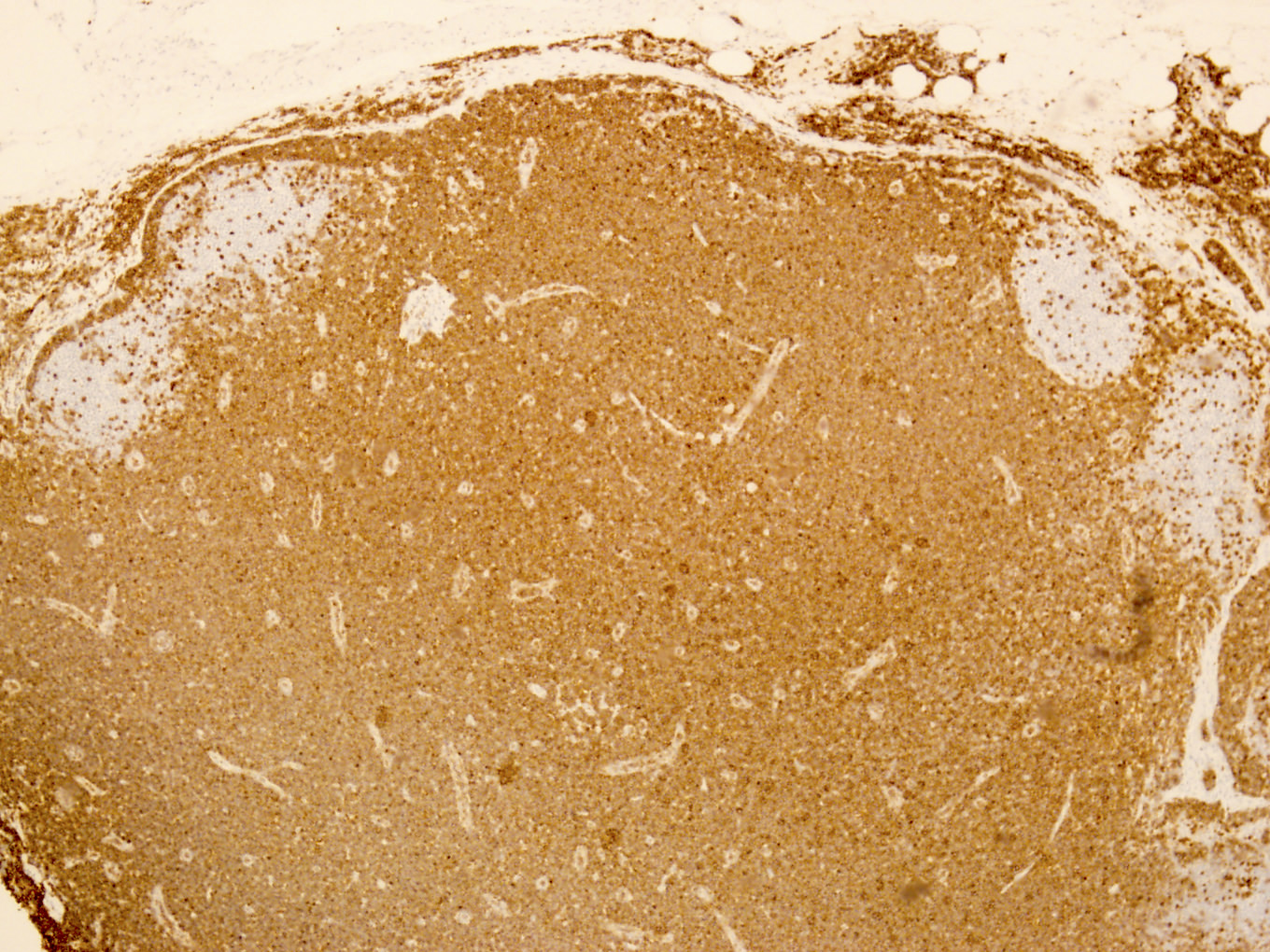
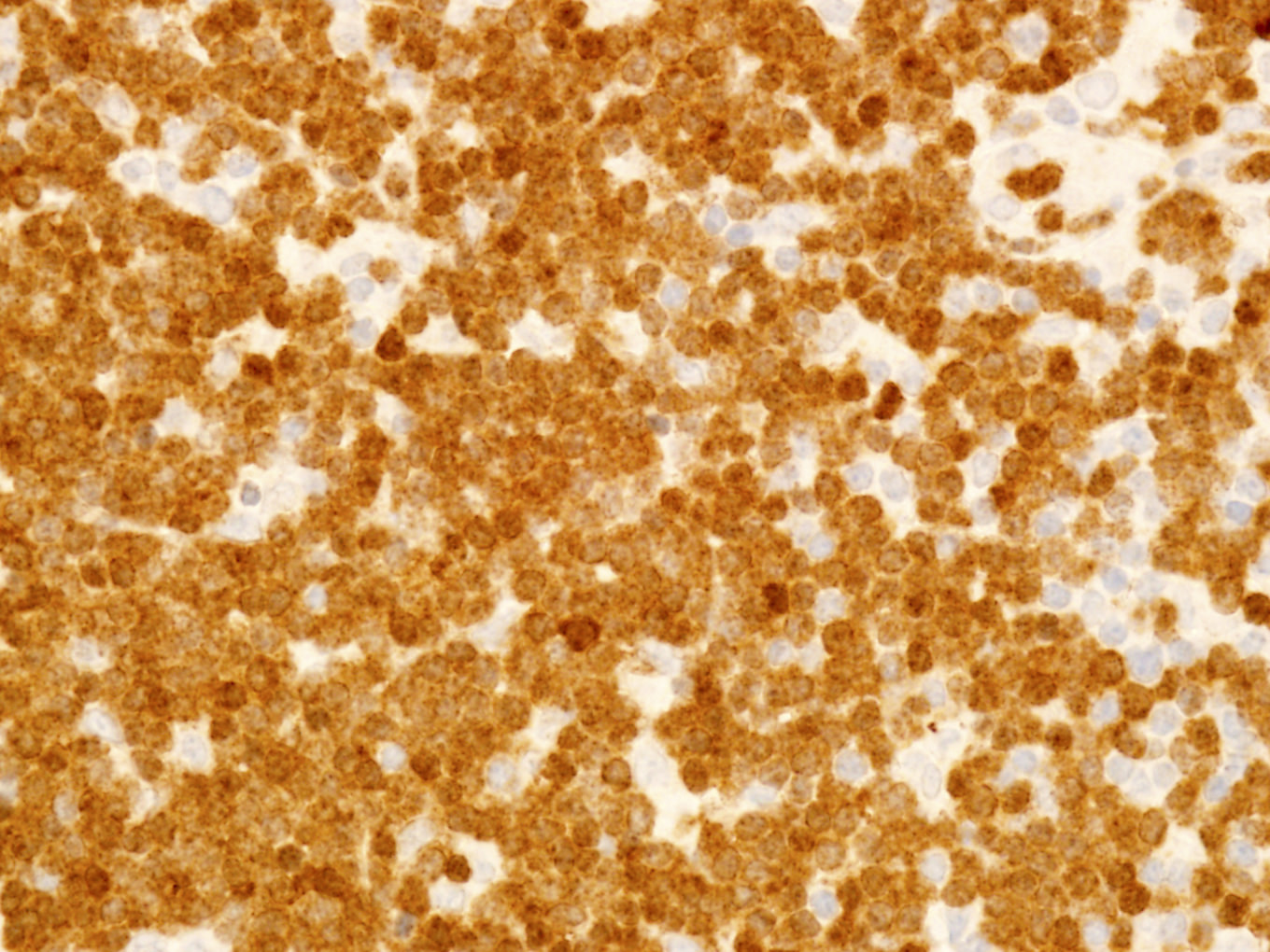
Microscopic (histologic) images
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
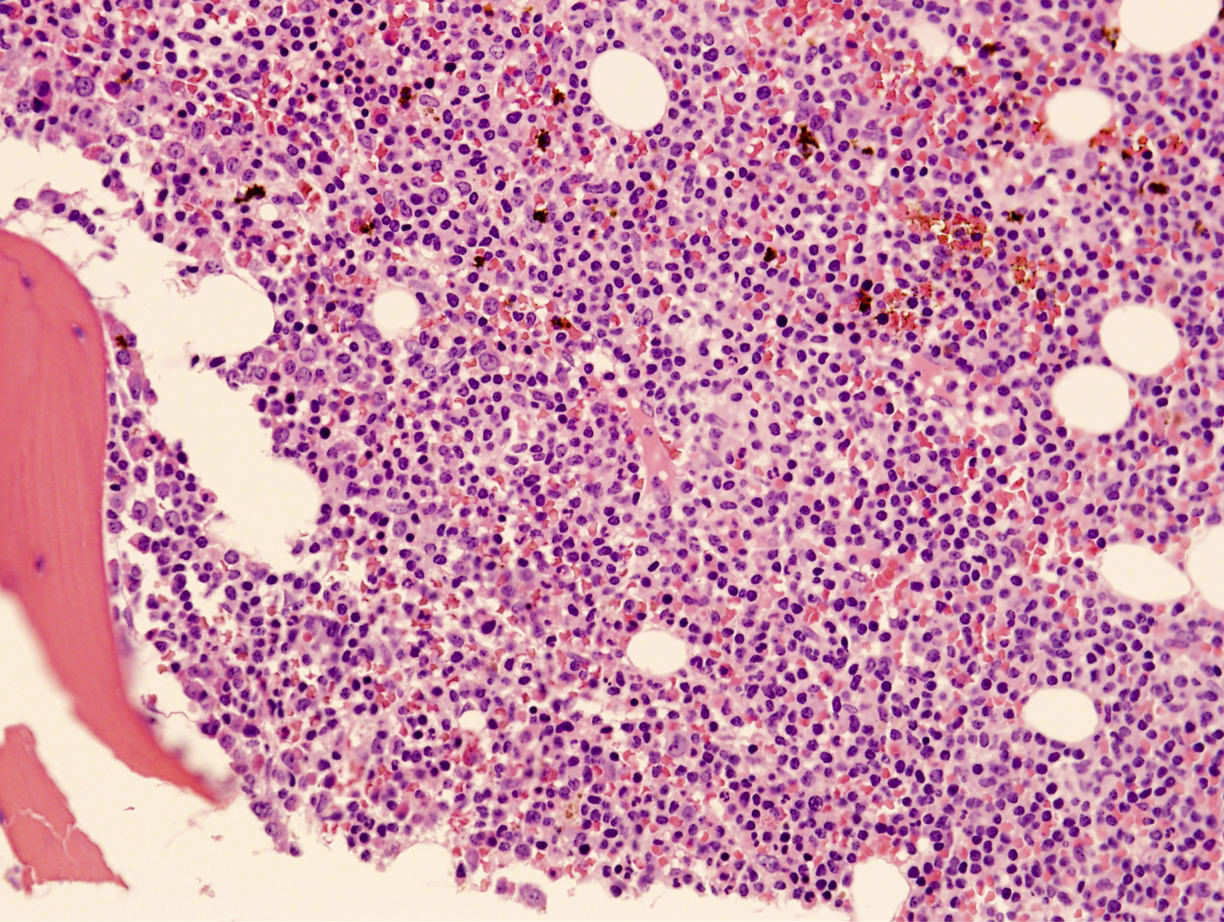
Diffuse bone marrow involvement
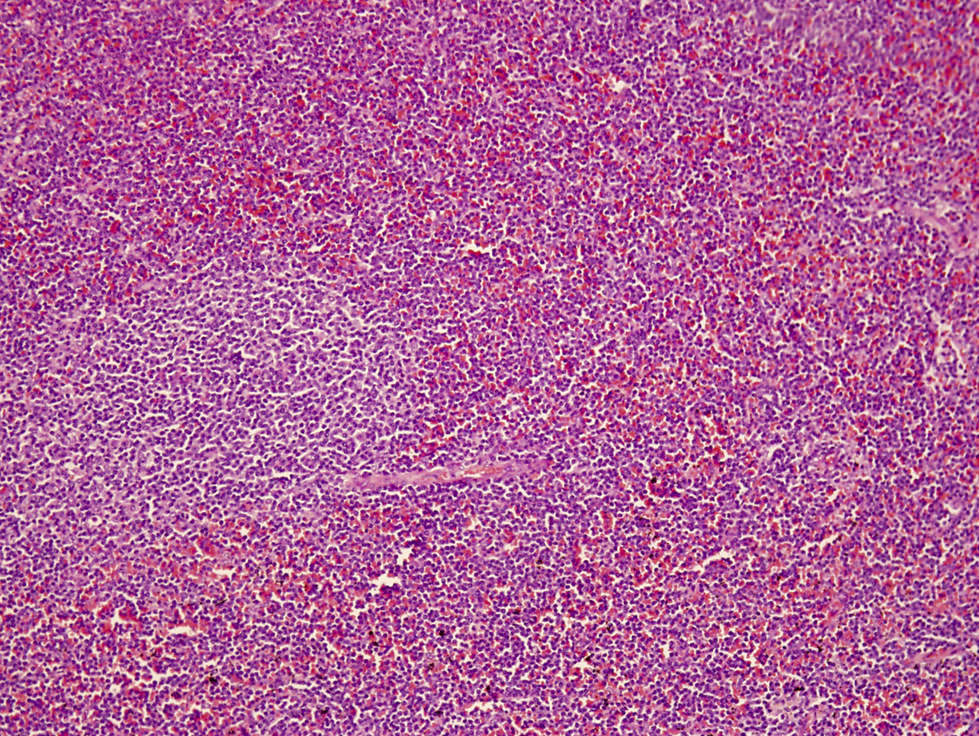
Splenic red pulp infiltrate
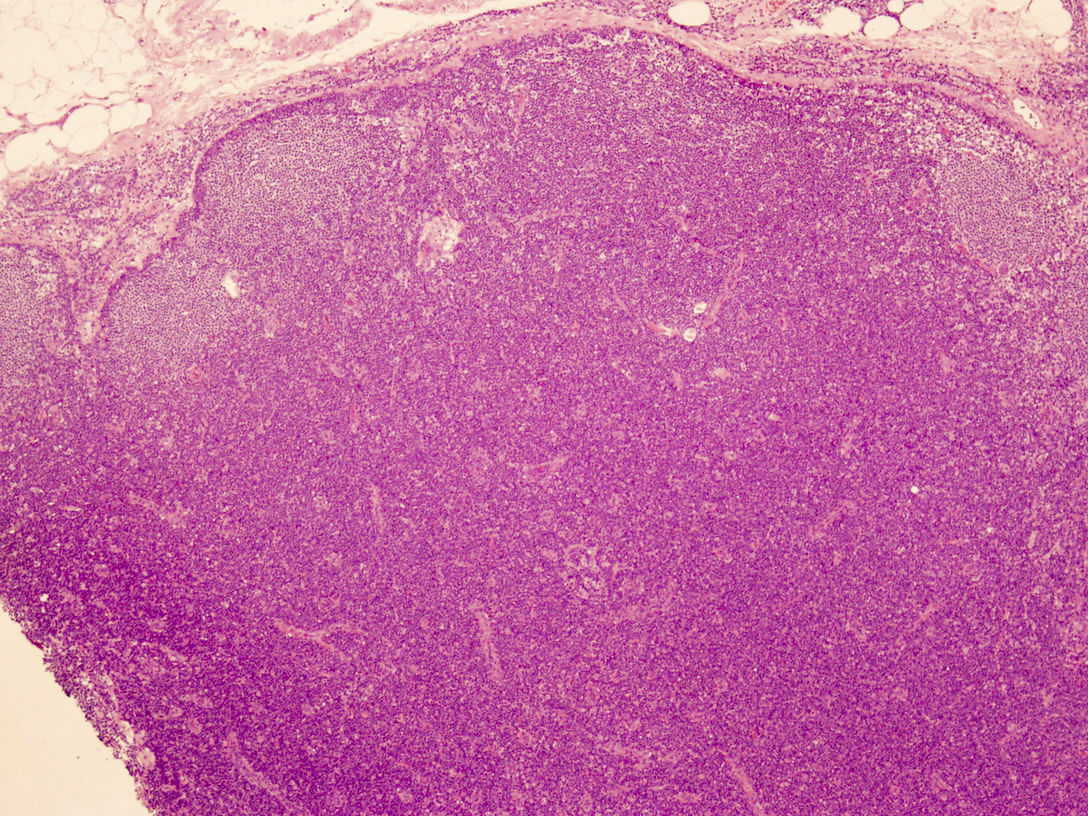
Monotonous, paracortical lymphoid infiltrate
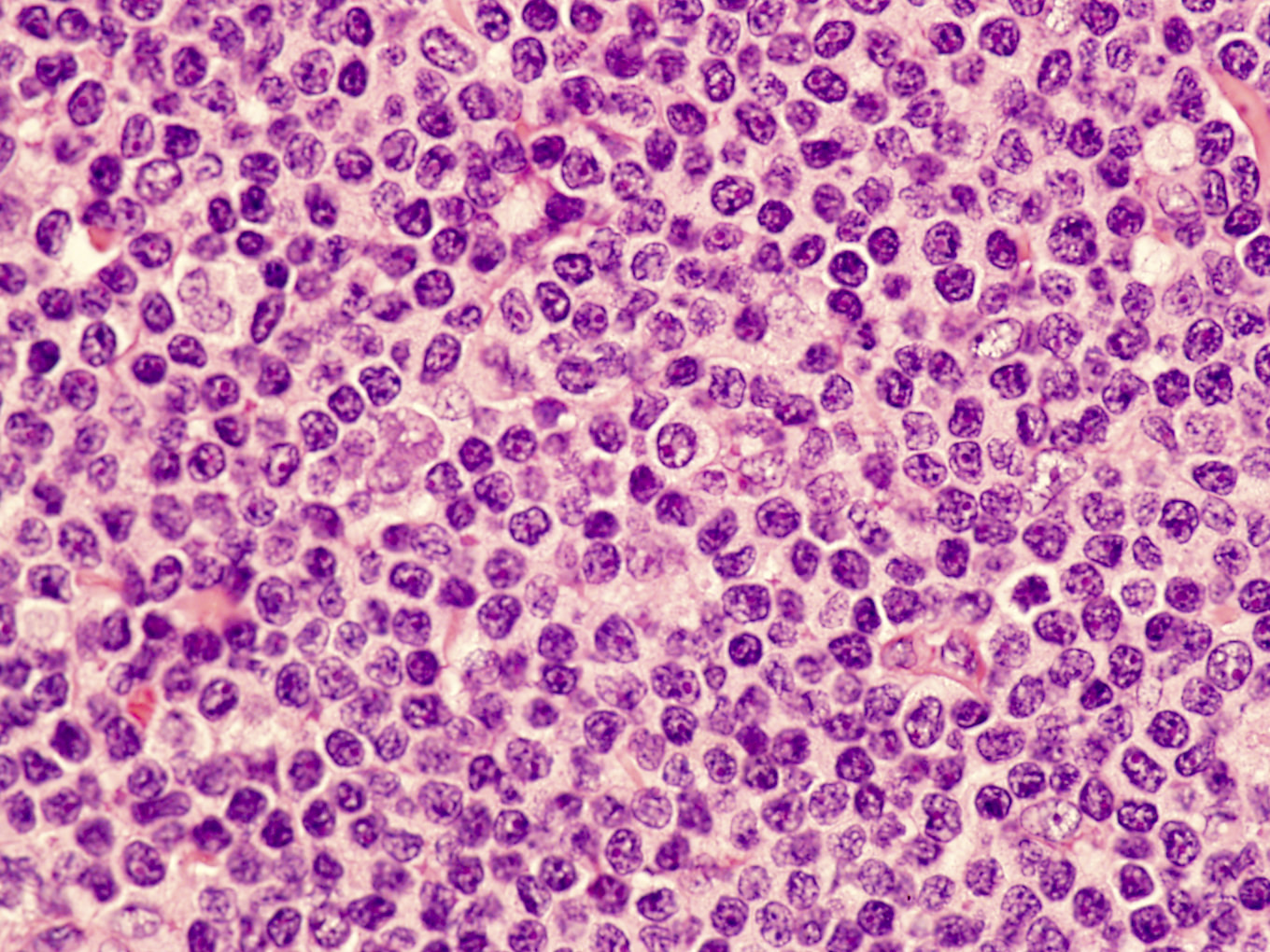
Intermediate size, atypical lymphocyte
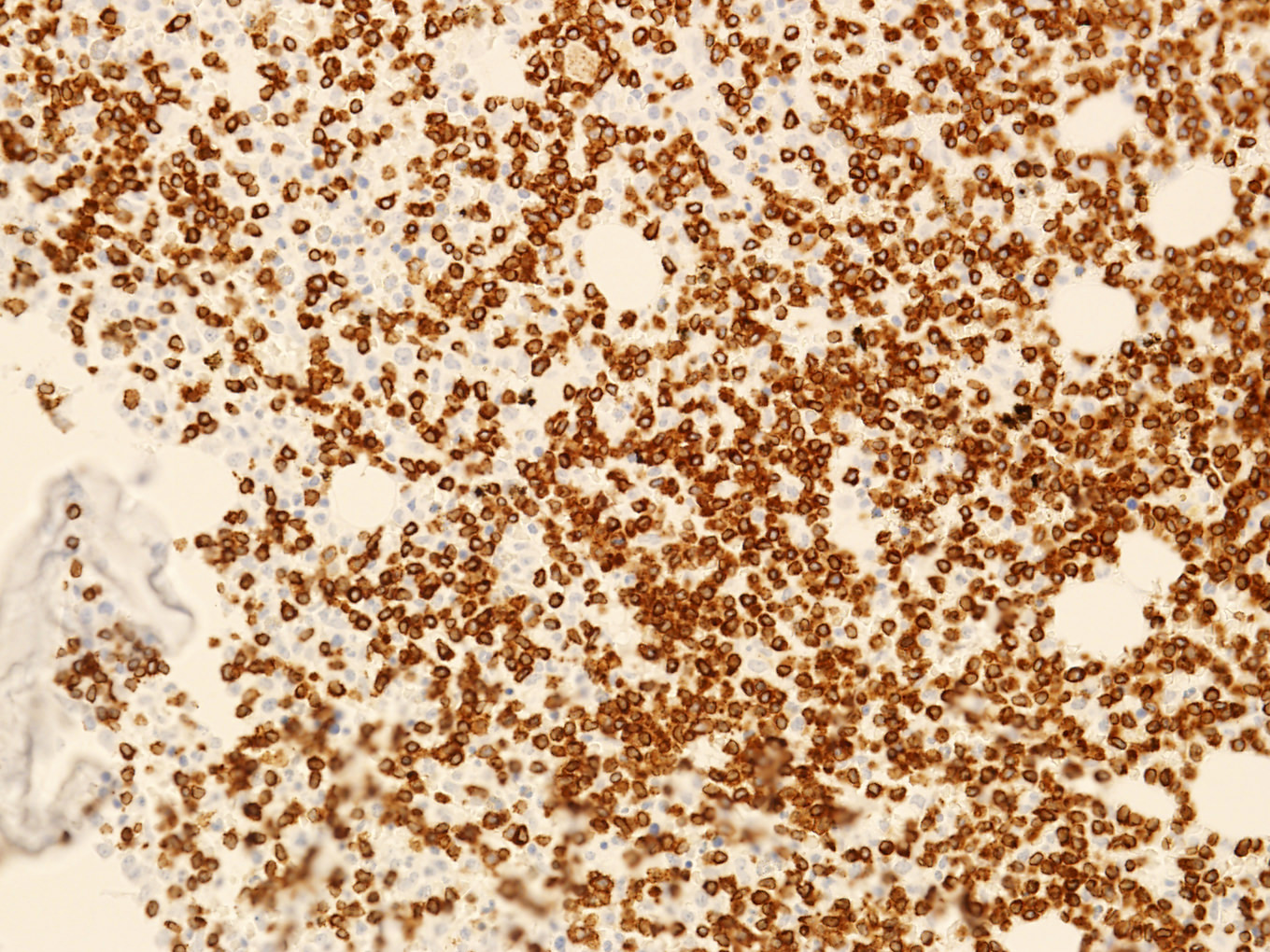
CD3 positivity in bone marrow
Monotonous T cell infiltrate
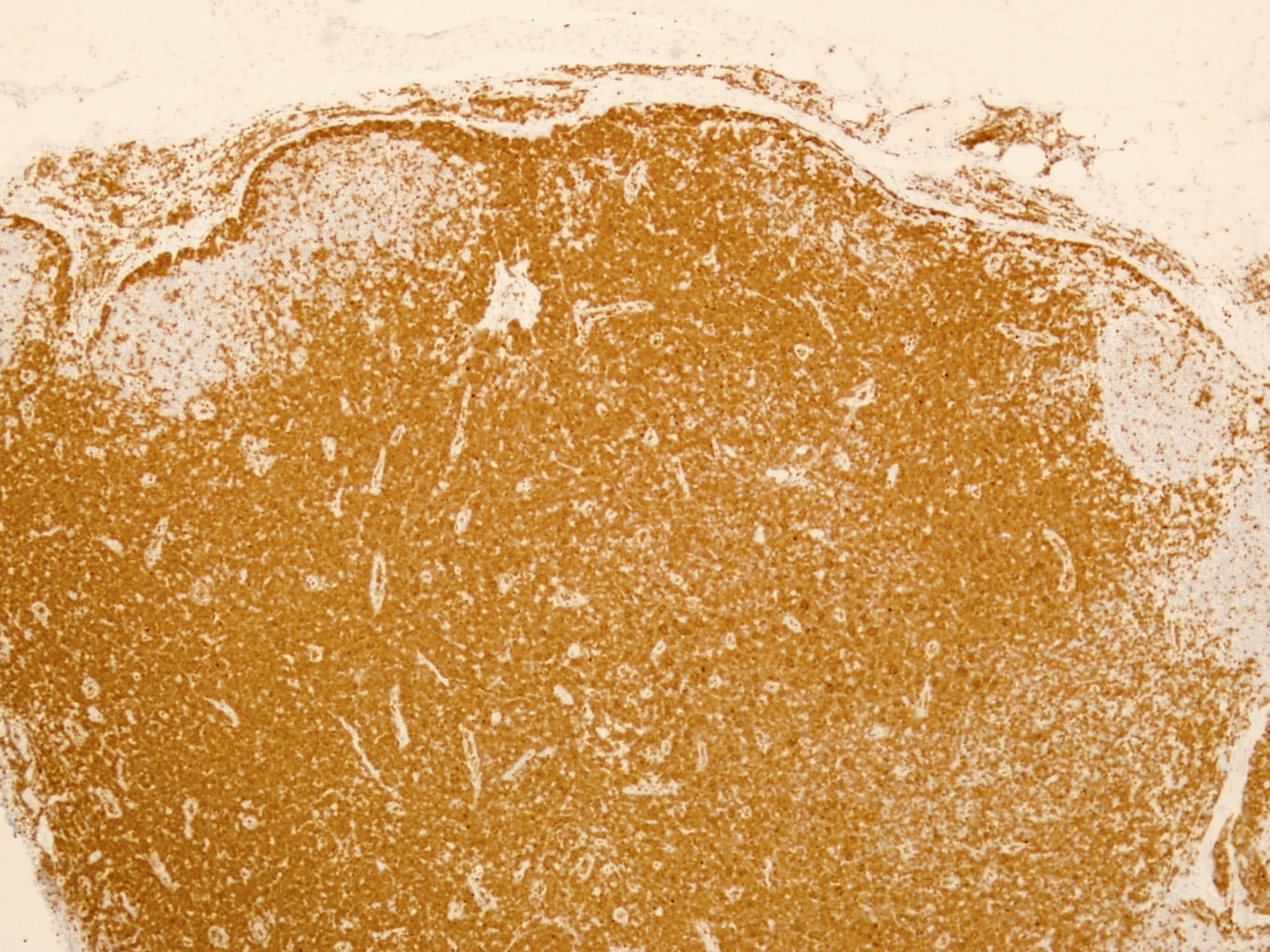
TCL1A positivity
Uniform strong TCL1A staining
Peripheral smear description
- Lymphocytosis with small to medium sized lymphocytes with cytoplasmic blebs, clumped chromatin and variably prominent central nucleolus
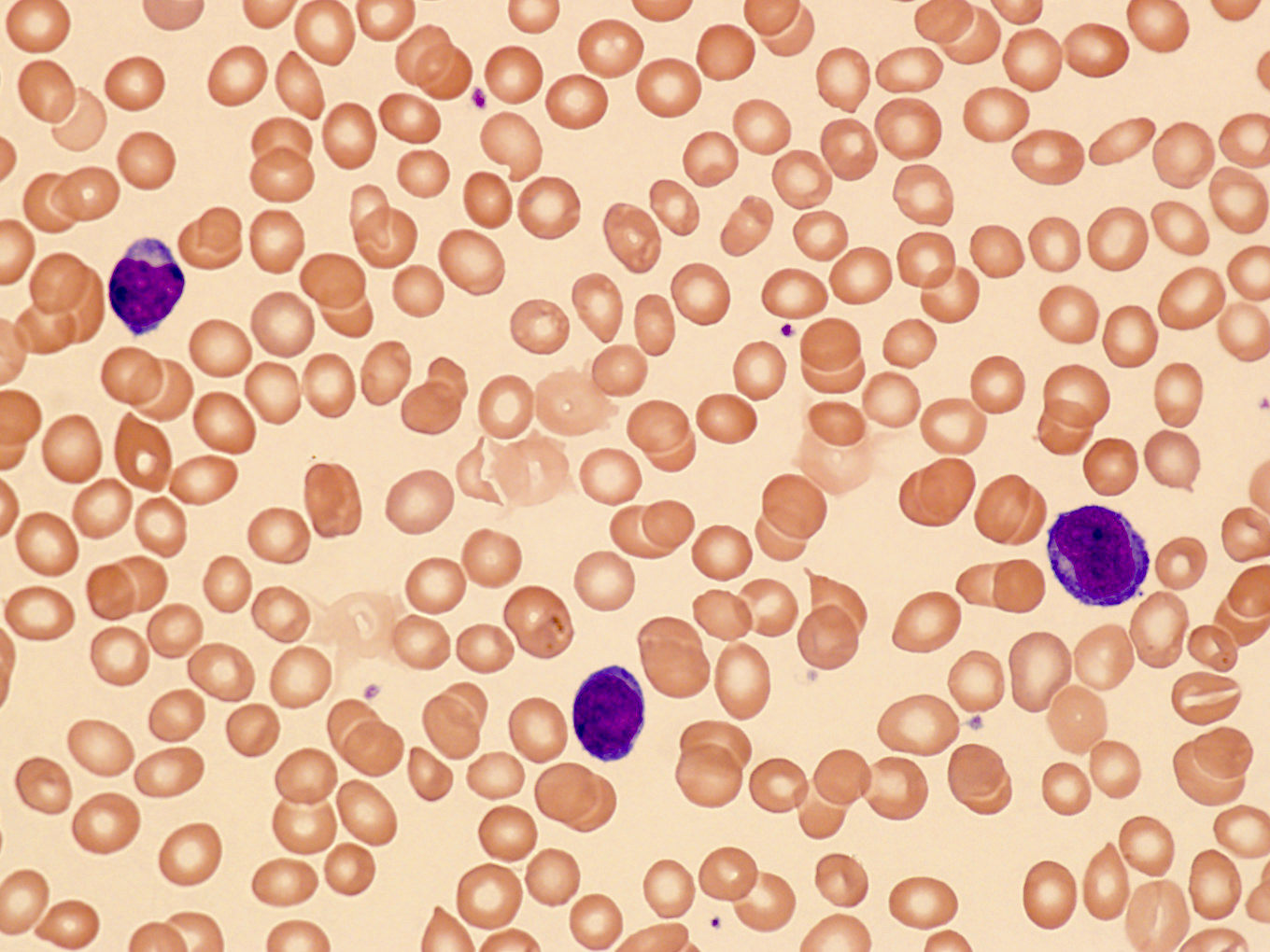
- Small cell variant in 25% of cases: smaller cells without obvious nucleolus
- Cerebriform variant in 5% of cases
- Reference: Blood 2019;134:1132
Peripheral smear images
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
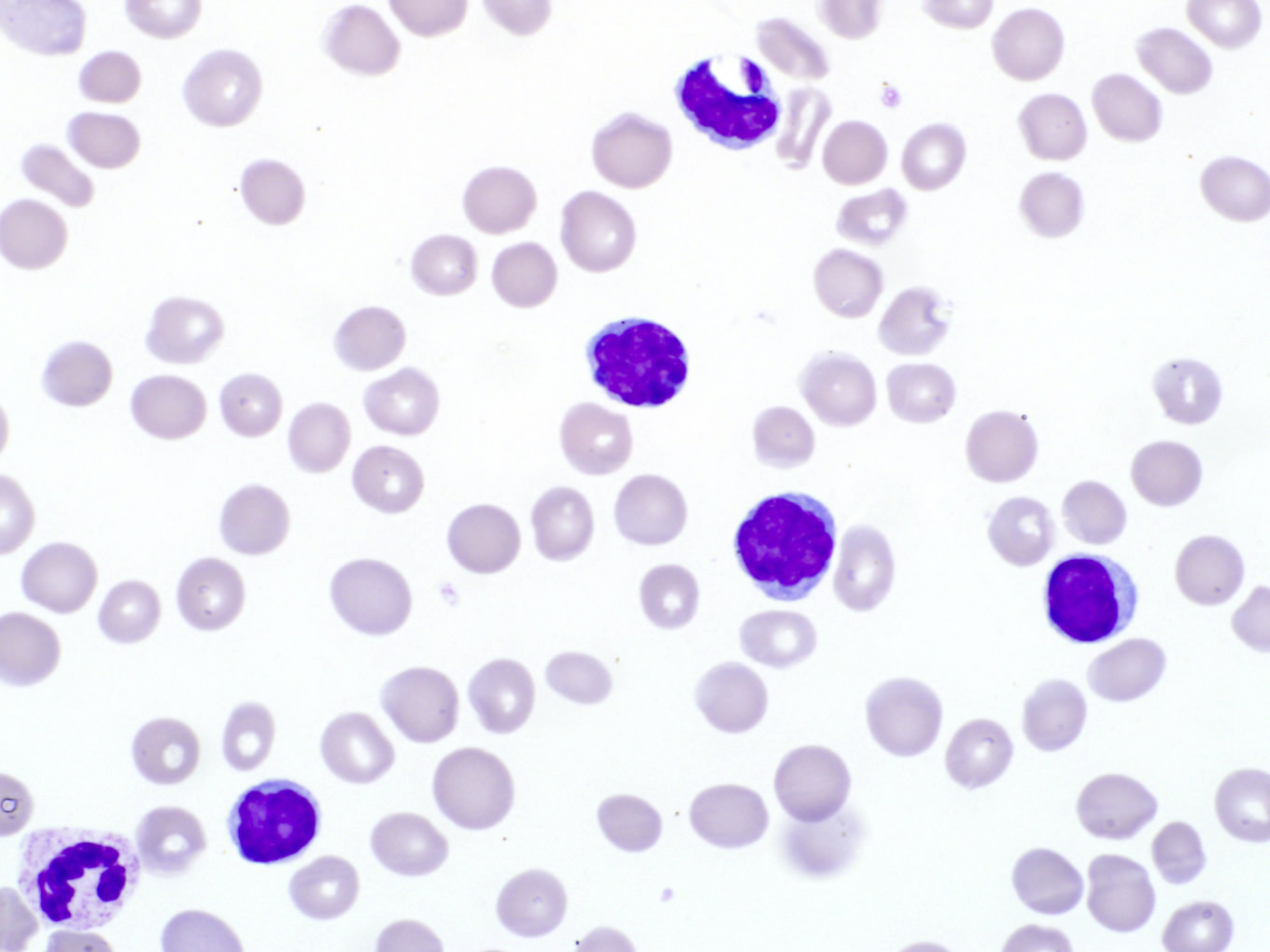
Cerebriform variant
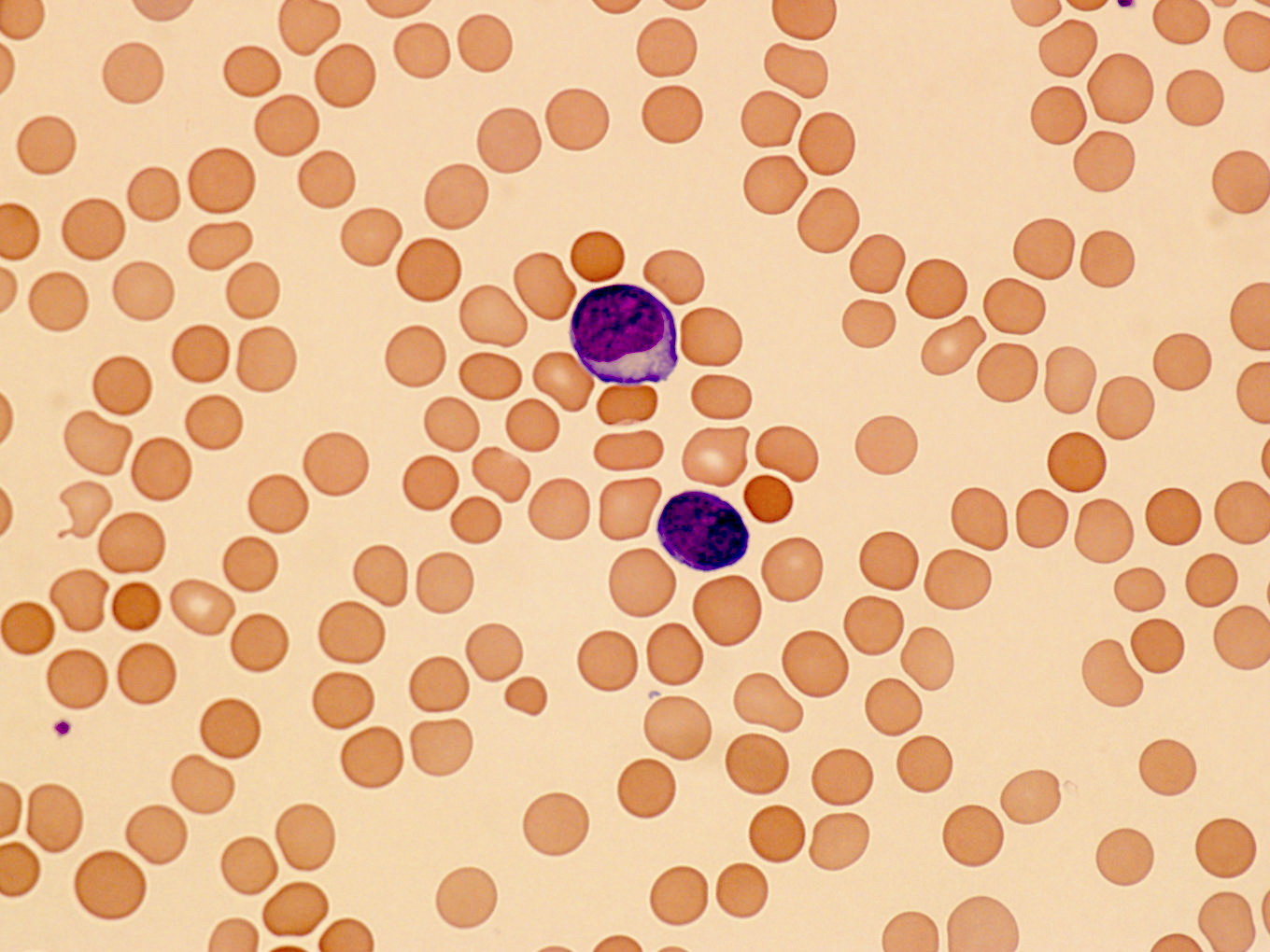
Prolymphocytes in peripheral blood
Small cell variant
Positive stains
Flow cytometry description
Flow cytometry images
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.

T PLL flow cytometry
Molecular / cytogenetics description
- Clonal T cell receptor gene rearrangements (TRB@ and TRG@)
- FISH is commonly used for diagnosis; T cell receptor locus rearranged with the TCL1 family of genes:
- TCL1A / TCL1B rearrangement: inv(14)(q11;q32) in 80%, t(14;14)(q11;q32) in 10%
- Rarely MTCP1 rearrangement t(X;14)(q28;q11)
- Rarely negative for TCL1A / TCL1B or MTCP1 rearrangements (Blood 2019;134:1132)
- Complex karyotype in 70 - 80%; abnormalities of chromosomes 6, 8, 12p, 17p
- Mutations in ATM gene on 11q23 in 80 - 90%
- Other mutations / alterations: TP53, JAK / STAT pathway genes IL2RG, JAK1, JAK3, STAT5B (Blood 2014;124:1460)
Molecular / cytogenetics images
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
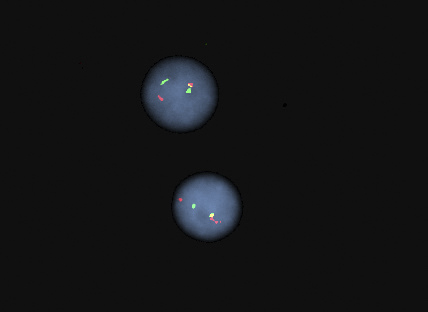
FISH for TCL1A separation
Sample pathology report
- Peripheral blood, bone marrow aspirate and biopsy, iliac crest:
- T cell prolymphocytic leukemia, extensively involving peripheral blood and bone marrow, with decreased trilineage hematopoiesis.
- Peripheral blood:
- Complete blood count: hemoglobin 11.2 g/dL; red blood cell 3.55 x 1012/L; mean corpuscular volume 97.2 fL; red blood cell distribution width 15.2%; white blood cell 470.0 x 109/L; PLT 111 x 109/L
- Cell percentage of total cells: neutrophils (4%), lymphocytes (95%), monocytes (1%)
- Peripheral smear: lymphocytosis; small to intermediate lymphocytes with mature chromatin, prominent nucleoli, eccentric nuclei and moderate amounts of basophilic cytoplasm
- Bone marrow aspirate and biopsy:
- Aspirate quality: cellular
- Biopsy quality: adequate
- Myeloid to erythroid (M:E) ratio: normal, 3:1
- Cellularity: hypercellular, 90%
- Erythroid precursors: markedly decreased quantity, normal morphology
- Myeloid precursors: markedly decreased quantity, normal morphology, blasts not increased
- Megakaryocytes: markedly decreased quantity, normal morphology and distribution
- Lymphocytes: abnormal (diffuse) infiltrates of small to intermediate sized cells present (90% of the total marrow cellularity)
- Plasma cells: not increased
- Ancillary studies:
- Flow cytometry, bone marrow:
- Blasts: not increased by CD45 / side scatter and CD34
- B cells: no monotypic; normal expression pattern of CD19, CD10, surface kappa and lambda
- T cells / NK cells: distinct T cell population
- Express: CD4, CD2, CD3, CD5, CD7
- Do not express: CD8, CD16, TCR gamma / delta, CD1a, nTdT, cMPO, cCD79a, cCD22
- Estimated size: 95% gated lymphoid events; 87% total analyzed events
- T cell lymphoma FISH, bone marrow: the result is abnormal and indicates 88.5% of nuclei had a rearrangement involving TCL1A; this observation may indicate a clone with inv(14) or t(14;14), which are common rearrangements in T cell prolymphocytic leukemia
- Flow cytometry, bone marrow:
Differential diagnosis
Additional references
Board review style question #1
What is the most common phenotype of T cell prolymphocytic leukemia (T PLL)?
- CD3-, CD19+, CD20+, CD5+, CD23+
- CD3+, CD4+, CD5+, CD7-, CD8-, CD26-
- CD3-, CD4+, CD8+, TdT+
- CD3+, CD4+, CD5+, CD7+, CD8-
- CD3+, CD4-, CD7-, CD8+, CD16+
Board review style answer #1
D. CD3+, CD4+, CD5+, CD7+, CD8-. The most common phenotype of T PLL is that of mature CD4+ T cells expressing pan T cell antigens, including CD7. Answer A is a common B CLL / SLL phenotype. Answer B is a common Sézary syndrome phenotype. Answer C is a common T lymphoblastic leukemia / lymphoma phenotype. Answer E is a common T cell large granulocytic lymphocyte leukemia phenotype.
Comment Here
Reference: T cell prolymphocytic leukemia
Comment Here
Reference: T cell prolymphocytic leukemia
Board review style question #2
Which genetic abnormality is used to define T cell prolymphocytic leukemia (T PLL)?
- 17p (TP53) deletion
- CCND1 rearrangement
- MYC rearrangement
- TCL1 rearrangement
- TP53 mutations
Board review style answer #2
D. TCL1 rearrangement on chromosome 14 is the most common genomic alteration, which results in the overexpression of TCL1 protein and drives proliferation of neoplastic cells in T cell prolymphocytic leukemia.
Comment Here
Reference: T cell prolymphocytic leukemia
Comment Here
Reference: T cell prolymphocytic leukemia

