
Hematology & immune disorders
Hemolytic anemia
Immune thrombotic thrombocytopenic purpura (TTP)
Authors: Jay S. Raval, M.D., Yara A. Park, M.D.
Board of reviewers: Frido Bruehl, M.D.
Editor-in-Chief: Patricia Tsang, M.D., M.B.A.


Last author update: 10 February 2025
Last staff update: 10 February 2025
Copyright: 2022-2025, PathologyOutlines.com, Inc.
PubMed Search: Immune thrombotic thrombocytopenic purpura

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology and ancillary testing | Prognostic factors | Case reports | Treatment | Clinical images | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Videos | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Raval JS, Park YA. Immune thrombotic thrombocytopenic purpura (TTP). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/hematologyTTP.html. Accessed April 1st, 2025.
Definition / general
- Rare, multiorgan disease characterized by the findings of thrombotic microangiopathy (TMA); i.e., a microangiopathic hemolytic anemia (MAHA) + thrombocytopenia, not attributable to another cause
- This is a different entity from congenital / hereditary TTP (i.e., Upshaw-Schulman syndrome), an inherited condition in which both copies of the ADAMTS13 gene have alterations that prevent the normal synthesis of the ADAMTS13 enzyme
Essential features
- MAHA
- Thrombocytopenia
- Caused by immune mediated inhibition or increased clearance of A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) enzyme
- Formation of platelet von Willebrand factor (VWF) microthrombi in small blood vessels carrying oxygen rich blood results in multiorgan ischemia, morbidity and ultimately mortality
Terminology
- Immune TTP
- Immune mediated TTP
- Autoimmune TTP
- Acquired TTP
- ADAMTS13 deficient TTP
- ADAMTS13 deficient TMA
- TTP due to anti-ADAMTS13 antibodies
- Adult onset TTP
ICD coding
- ICD-10: M31.19 - other thrombotic microangiopathy
Epidemiology
- Rare disease designation
- Annual incidence = 1 - 5/million, though this may vary among different populations
- While typically a diagnosis is made in early to middle adulthood, a de novo TTP presentation can occur at any age (from children to the elderly)
- More common in women and Black people (J Thromb Haemost 2005;3:1432)
Sites
- Blood
- Heart
- Brain
- Skin
- Gut
- Kidney
Pathophysiology
- Autoimmune formation of antibodies directed against ADAMTS13 enzyme
- ADAMTS13 enzyme is a protease responsible for cleaving ultra large von Willebrand factor (ULVWF) into small, physiologic VWF multimers that can participate in primary hemostasis (i.e., formation of platelet plugs at site of vascular injury)
- When ADAMTS13 is neutralized or cleared, ULVWF remains and hyperactively binds to platelets in peripheral blood
- Result is the formation of pathologic platelet VWF microthrombi in the higher shear stress microcirculation carrying oxygen rich red blood cells (RBC)
- Thrombocytopenia results and RBCs attempting to pass through microthrombi and damaged endothelium are sheared, causing mechanical hemolysis and associated schistocytes (fragmented RBCs)
- Multiple organs are deprived of oxygen and ischemia occurs throughout the body (Int J Hematol 2010;91:1)
Diagrams / tables

Clinical features
- MAHA
- Thrombocytopenia
- Petechiae or purpura
- Neurologic impairments
- Fever
- Myocardial infarction or arrhythmia
- Stroke or transient ischemic attack (TIA)
- Nausea / vomiting
- Abdominal pain
- Fatigue
- Mild kidney injury
- Reference: J Emerg Med 2021;61:674
Diagnosis
- TTP should be considered if the patient has an unexplainable combination of MAHA and thrombocytopenia (i.e., TMA) (J Thromb Haemost 2020;18:2503)
- Severe deficiency of ADAMTS13 activity (typically defined as < 10%) in addition to presence of an inhibitor or autoantibody directed against ADAMTS13 is present
- Clinical diagnosis is critical as ADAMTS13 activity, inhibitor and autoantibody testing can take a few hours to several days depending on the local availability of these assays
- Do not wait for the ADAMTS13 activity results to rule in or rule out the diagnosis of TTP; it is a clinical diagnosis that is supported by laboratory testing (J Thromb Haemost 2020;18:2503)
- Pentad of MAHA, thrombocytopenia, fever, neurologic impairment and kidney injury supports the diagnosis of TTP; it is now known that the risk of long term morbidity and mortality is significantly greater, even with prompt therapy, if the pentad is present before starting therapy
- Do not wait for the pentad to be present before initiating therapy (J Thromb Haemost 2020;18:2503)
- PLASMIC score consists of 7 components that may be used to aid in the diagnosis of TTP in patients with a new TMA, though this has not been found to be an accurate predictor of severe ADAMTS13 deficiency in all patients
- Per PLASMIC score validations, a score of 0 - 4 is low risk for TTP, a score of 5 is intermediate risk and a score of 6 - 7 is high risk for TTP (Lancet Haematol 2017;4:e148)
- PLASMIC score calculation: add 1 point for each of the following parameters that are true (Lancet Haematol 2017;4:e157, Transfusion 2020;60:2047)
- Platelet count that is < 30 x 109/L
- Hemolysis is present (reticulocyte count of < 2.5%, undetectable haptoglobin or indirect bilirubin of > 2.0 mg/dL)
- Mean corpuscular volume (MCV) of < 90 fL
- International normalized ratio (INR) of < 1.5
- Creatinine of < 2.0 mg/dL
- Patient does not have any active cancer
- Patient does not have a history of transplant (solid organ or stem cell)
Laboratory
- Anemia
- Thrombocytopenia
- Positive biomarkers of intravascular hemolysis (elevated lactate dehydrogenase [LDH], indirect bilirubin, reticulocyte count, plasma free hemoglobin; decreased or undetectable haptoglobin)
- Negative direct antiglobulin test (DAT) (i.e., Coombs test)
- Elevated cardiac troponins or lactate
- Schistocytes on peripheral blood film or complete blood count (CBC) differential (see Peripheral smear images)
- Etiologies of other causes of TMA must be simultaneously evaluated (such as human immunodeficiency virus [HIV] infection, pregnancy, drugs, typical or atypical hemolytic uremic syndrome [HUS] and mechanical heart valves)
- Renal pathology is consistent with hematuria and proteinuria with minimal impact on renal clearance function (Int J Hematol 2010;91:1)
Radiology and ancillary testing
- Brain imaging can demonstrate neurologic lesions due to ischemia or infarction
- Cardiac imaging can demonstrate wall motion abnormalities and other functional changes
- Electrocardiogram should be performed to identify the presence of any cardiac involvement (J Thromb Haemost 2020;18:2503)
Prognostic factors
- No reliable factors have been identified that correlate with outcomes
- However, as relapses of TTP occur, central nervous system (CNS) neurons and cardiac myocytes die and do not regrow (World J Cardiol 2018;10:254)
- Depression, cognitive impairments, heart failure and disability are some of the long term complications in survivors
- Underlying comorbidities may further complicate the patient's condition, magnify the impacts of ischemia and require more intensive supportive measures
Case reports
- 31 year old woman with acute ischemic stroke as initial finding in TTP (Cureus 2020;12:e7661)
- 56 year old woman with refractory TTP treated with caplacizumab (Cureus 2023;15:e42423)
- 76 year old woman with acute cardiac involvement in TTP (Proc (Bayl Univ Med Cent) 2022;35:832)
Treatment
- Emergent initiation of therapeutic plasma exchange (TPE); performed once daily through an apheresis compatible central venous catheter using plasma as replacement fluid until platelets have normalized for 2 consecutive days and LDH is normalizing (J Clin Apher 2019;34:171, J Thromb Haemost 2020;18:2486)
- High dose glucocorticoids (such as prednisone, methylprednisolone and dexamethasone) (J Thromb Haemost 2020;18:2496)
- Immunosuppressive medications (such as rituximab) (J Thromb Haemost 2020;18:2496)
- Anti-VWF therapies (such as caplacizumab and N-acetyl cysteine) (J Thromb Haemost 2020;18:2496)
- Splenectomy can be considered in refractory cases
- RBC transfusion
- Avoid platelet transfusion due to the risk of worsening disease (due to the thrombogenic effect of platelets) unless there is life threatening bleeding (such as in the airway or intracranial)
- Supportive care of the patient and any injured organs
- Place patient on telemetry whenever possible due to the high risk of early death from cardiac arrhythmias
- TTP is a lifelong autoimmune disease and is never cured; rather, it only goes into remission
- After treatment of de novo TTP, long term follow up with a provider familiar with TTP management is highly recommended to serially monitor CBC, ADAMTS13 activity and clinical status, as well as to adjust any treatments accordingly
- TTP recurrences (either early exacerbation or later relapse) can occur at any time
- See figures 1 and 2
Clinical images
Images hosted on other servers:

Purpura and petechiae in the skin
Microscopic (histologic) description
- Thrombi, which are rich in VWF are present in the arterioles and capillaries of multiple organs (e.g., brain, heart, kidney, pancreas, adrenals)
- Thrombi are not present in venules of these organs
- Endothelial cells are intact but contain VWF granules
- Inflammatory infiltrate is not often seen
- In the kidney, glomerulus architecture is preserved but shows multiple microthrombus foci
- In the liver, VWF is not seen in sinus endothelial cells and thrombi are not detected in sinusoids
Microscopic (histologic) images
Images hosted on other servers:

Immunohistopathology
of TTP
Peripheral smear description
- Thrombotic microangiopathy or thrombocytopenia, anemia and hemolysis with schistocytes (including helmet cells, triangulocytes and keratocytes) and microspherocytes on peripheral smear
- Evidence based reference values for schistocytes in normal healthy adults is ≤ 1%; any value greater than 1% is abnormal and warrants suspicion for thrombotic microangiopathy (Int J Lab Hematol 2021;43:1264)
- Degree of schistocytosis can vary at diagnosis and throughout the disease course, though this does not have impacts on outcomes (Ther Apher Dial 2018;22:662)
- As the TTP disease process evolves and worsens over time, schistocytes may not be appreciated early in the disease course and serial peripheral blood films may need to be reviewed
- Mechanical fragmentation of RBCs results in remnants with variable sizes and irregular shapes (i.e., schistocytes / smaller fragments, keratocytes / larger fragments); the International Council for Standardization in Haematology (ICSH) recommendations for the identification and quantification of schistocytes are available (Int J Lab Hematol 2021;43:1264)
- Reticulocytosis or polychromasia and anisocytosis are also seen (Clin Lab Sci 2020;33:48)
Peripheral smear images
Videos
Thrombotic thrombocytopenic purpura
Sample pathology report
- Peripheral blood film
- Normocytic normochromic anemia with polychromasia
- Schistocytosis >1%
- Marked thrombocytopenia with large platelets
- Leukocytosis with left shift
Differential diagnosis
- Congenital / hereditary TTP:
- TMA present
- Both copies of the ADAMTS13 gene have alterations that prevent the normal synthesis of the ADAMTS13 enzyme
- TMA due to other causes (do not typically demonstrate severe ADAMTS13 deficiency)
- Drug induced (e.g., chemotherapy, antibiotics, cardiovascular medication [i.e., clopidogrel])
- Trauma
- Atypical HUS and other complement defects
- Infections (e.g., Shiga-like toxin producing bacteria and S. pneumoniae)
- HIV
- B12 deficiency
- Cancer associated: mostly adenocarcinomas, particularly gastric, breast or prostate
- Disseminated intravascular coagulation (StatPearls: Thrombotic Thrombocytopenic Purpura [Accessed 22 October 2024]):
- Schistocytes are relatively uncommon
- Abnormal coagulation studies
- Fibrinolysis is more common than in TTP
- Endocarditis
- Glucose-6-phosphate dehydrogenase deficiency
- Malignant hypertension
- HELLP syndrome (hemolysis, elevated liver enzymes and low platelets) in pregnancy
- Systemic lupus erythematosus
- Autoimmune diseases
- Vasculitis / scleroderma
Additional references
Board review style question #1
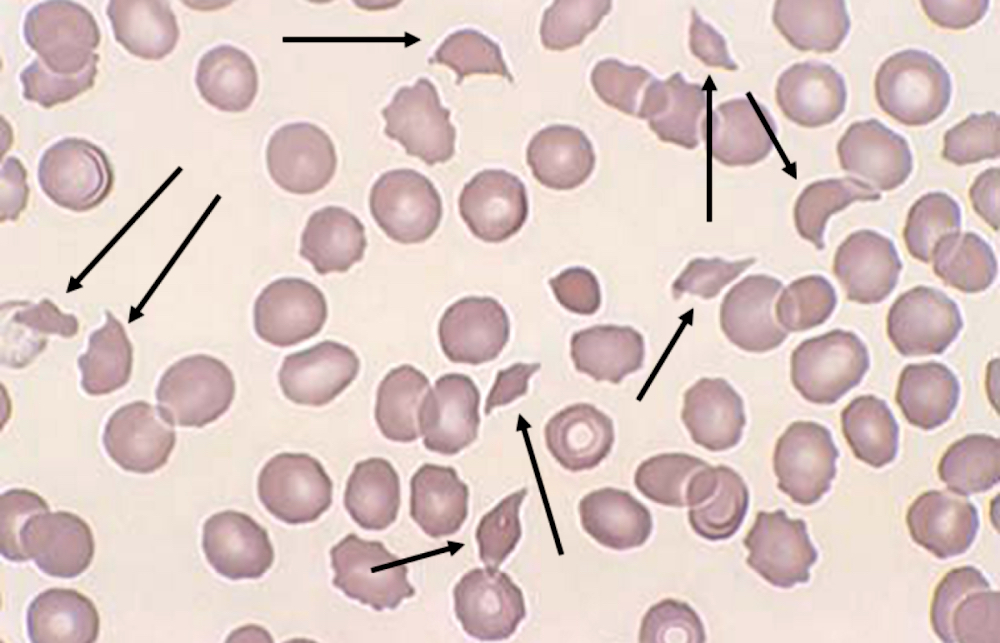
What are the characteristic microscopic peripheral blood findings in an acute episode of immune / acquired thrombotic thrombocytopenic purpura (TTP)?
- Anemia, schistocytosis and thrombocytopenia
- Anemia, schistocytosis and thrombocytosis
- Spherocytosis and thrombocytopenia
- Spherocytosis and thrombocytosis
Board review style answer #1
A. Anemia, schistocytosis and thrombocytopenia. TTP is a type of thrombotic microangiopathy, which consists of microangiopathic hemolytic anemia (i.e., anemia from intravascular hemolysis substantiated by peripheral blood findings of schistocytosis) and thrombocytopenia. Answers B, C and D are incorrect because thrombocytosis and spherocytosis are not characteristic peripheral blood findings of an acute episode of TTP.
Comment Here
Reference: Immune thrombotic thrombocytopenic purpura (TTP)
Comment Here
Reference: Immune thrombotic thrombocytopenic purpura (TTP)
Board review style question #2
What is the standard of care treatment for immune / acquired thrombotic thrombocytopenic purpura (TTP)?
- Aspirin therapy
- High volume plasma transfusion and immunosuppression
- Platelet and red blood cell (RBC) transfusion therapy
- Therapeutic plasma exchange and immunosuppression
Board review style answer #2
D. Therapeutic plasma exchange and immunosuppression. Treatment of TTP requires emergent initiation of therapeutic plasma exchange (TPE) and immunosuppressive medications (such as rituximab). Answer A is incorrect because although aspirin can be used for supportive care, it is not a mainstay of management (J Blood Med 2014;5:15). Answer B is incorrect because the high volume of plasma needed for TTP remission may lead to fluid overload (Medicine (Baltimore) 2003;82:27). Answer C is incorrect because platelet transfusion is contraindicated in TTP, except in the setting of severe bleeding.
Comment Here
Reference: Immune thrombotic thrombocytopenic purpura (TTP)
Comment Here
Reference: Immune thrombotic thrombocytopenic purpura (TTP)
