
Hematology & immune disorders
Hemolytic anemia
Paroxysmal nocturnal hemoglobinuria (PNH)
Editorial Board Member: Julie Feldstein, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 19 July 2021
Last staff update: 5 August 2022
Copyright: 2021-2025, PathologyOutlines.com, Inc.
PubMed Search: Paroxysmal nocturnal hemoglobinuria[title] pathology "last 5 years"[DP]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Peripheral smear description | Flow cytometry description | Flow cytometry images | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Erem AS, Asakrah S. Paroxysmal nocturnal hemoglobinuria (PNH). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/hematologyPNH.html. Accessed April 3rd, 2025.
Definition / general
- Acquired autosomal clonal mutation of hematopoietic stem cells (HSCs) characterized by altered surface protein expression of hematopoietic elements and increased hemolysis, risk of thrombosis and impaired bone marrow function
Essential features
- Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired somatic mutation in the X linked phosphatidylinositol glycan class A (PIGA) gene, which leaves hematopoietic cells unable to produce the glycosylphosphatidylinositol (GPI) anchor that links cell surface proteins to the plasma membrane (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Blood 2014;124:2804)
- PNH leads to episodic hemolytic anemia, recurrent thrombosis and bone marrow failure and cytopenias (EJIFCC 2019;30:355)
- Despite the name, PNH patients may experience hemoglobinuria at night, during the day or not at all (EJIFCC 2019;30:355)
- PNH clones frequently appear in patients with aplastic anemia and myelodysplastic syndromes (Int J Lab Hematol 2017;39:329)
Terminology
- Paroxysmal nocturnal hemoglobinuria (PNH)
- Marchiafava-Micheli (Q J Med 1949;18:105)
ICD coding
Epidemiology
- Global incidence: 1 - 2 per million (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Global prevalence: 10 - 20 per million (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Median age at diagnosis: early to mid-30s (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Globally, M = F (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- In a comparative analysis of 1,665 patients between Asia and Europe / America:
- Proportion of European / American women was significantly higher than in Asia
- Incidence of hemoglobinuria and thromboembolism: Europe / U.S. (F) > Asia (F) (Int J Hematol 2016;103:649)
Sites
- Vascular system: intravascular hemolysis, thrombosis (especially venous thrombosis) in atypical locations such as hepatic vein (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Bone marrow (J Hematopathol 2013;6:71):
- Classic PNH: 65% of patients have normal or hypercellular bone marrow; erythroid hyperplasia is common; myelopoiesis is decreased in 86% and megakaryopoiesis is decreased in 46%
- PNH patients with aplastic anemia: 95% have hypocellularity in bone marrow; myelopoiesis decreased in 85% and megakaryopoiesis decreased in 100%
- Kidneys: renal insufficiency or renal failure (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Esophagus: esophageal spasm, free hemoglobin in plasma depletes nitric oxide (NO), causing smooth muscle dystonia (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Blood 2014;124:2804)
- Penis: priapism or erectile dysfunction, also due to effect of nitric oxide scavenging by free hemoglobin (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Anemia 2011;2011:297364)
Pathophysiology
- PNH is caused by an acquired mutation in the phosphatidylinositol glycan class A (PIGA) gene; this gene produces the glycosylphosphatidylinositol (GPI) anchor proteins (GPI-APs), a posttranslational modification that links cell surface proteins to cell membranes (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Mechanism of mutation is unknown: proposed theory of immune attack on normal HSCs and sparing of PNH stem cell clones due to close association between PNH and acquired aplastic anemic (AA) (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Other factors leading to proliferation and expansion of PNH clones include:
- Mutation or aberrant expression of recombinant signal binding protein of immunoglobulin kJ region (RBPJ) (Exp Ther Med 2019;17:4536)
- A small subset of patients may alternately have a mutation in the phosphatidylinositol glycan anchor biosynthesis class T (PIGT) gene, responsible for transferring GPI to precursor proteins; patients with this mutation tend to have additional autoinflammatory symptoms (J Clin Invest 2019;129:5123)
- HSCs containing PIGA mutations lack GPI anchored cell surface markers, including complement inhibitors CD59, which interferes with C9 binding to the cell membrane and formation of C3 convertase; decay accelerating factor (DAF / CD55), homologous restriction factor (HRF), C8 binding protein, membrane inhibitor of reactive lysis (MIRL) (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383)
- Mutants HSCs undergo clonal expansion under bone marrow (BM) failure (J Clin Invest 2019;129:5123)
- Mature erythrocytes lacking GPI-APs are unprotected from the membrane attack complex (MAC or C5b9), leading to paroxysmal hemolysis (Indian J Hematol Blood Transfus 2016;32:383, Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- During hemolytic episodes, free hemoglobin (Hb) is released into the blood, which causes renal damage; free Hb can also contribute to hypercoagulability, thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Free hemoglobin scavenges nitric oxide in the bloodstream, which impairs its ability to regulate vascular tone and inhibit platelet activation, leading to smooth muscle dystonia and thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2017;33:453, Blood 2014;124:2804)
- Extravascular hemolysis: C3 fragments accumulate on GPI-AP negative red blood cells (RBCs) and act as an opsonin, which causes reticuloendothelial destruction in the liver and spleen (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
Etiology
- PNH clone found in:
- 40 - 70% of patients with aplastic anemia (AA); T cell mediated autoimmune attack may give PNH stem cells a survival advantage due to fewer surface markers (Indian J Hematol Blood Transfus 2017;33:453, Blood 2014;124:2804)
- 48% of patients with hemoglobinuria (Cytometry B Clin Cytom 2017;92:361)
- 33% of bone marrow failure patients have a PNH clone (Cytometry B Clin Cytom 2017;92:361)
- 10 - 19% of direct antiglobulin test (DAT) negative hemolytic anemia patients (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 5 - 10% of patients with myelodysplastic syndrome (MDS) (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 3 - 9% of patients with unexplained cytopenias (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 0.4 - 2% of patients with thrombosis (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
Diagrams / tables
Images hosted on other servers:

Suggested pathophysiology of PNH

Suggested pathophysiology of hemolysis in PNH
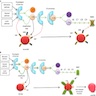
Complement action in healthy subjects and PNH patients
Clinical features
- Presentation varies, including intravascular hemolysis, thrombosis, AA and cytopenia, which precede or follow PNH (EJIFCC 2019;30:355)
- Possible symptoms include pulmonary hypertension, dyspnea, impaired renal function, erectile dysfunction, pain in the abdomen, fatigue and difficulty swallowing, although PNH may also be asymptomatic (EJIFCC 2019;30:355)
- Hemolytic episodes may be precipitated by infection, surgery or transfusions (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Hemolytic episodes may cause fever, headache, fatigue and back or abdominal pain (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Hemoglobinuria occurs in 90% of patients; it may occur at night, during the day or not at all (EJIFCC 2019;30:355)
- Thrombosis occurs in 50% of PNH patients, often in unusual locations, such as intraabdominal visceral veins or hepatic veins (Blood Cells Mol Dis 2020;80:102372, Pract Lab Med 2020;20:e00158)
- These complications are among the primary causes of morbidity and mortality for PNH patients (Sci Rep 2018;8:13458)
- Chronic kidney disease occurs in up to 65% of PNH patients, including stage III - V disease in 21% of PNH patients (Hematology 2018;23:558)
Diagnosis
- Indications for testing for PNH: cytopenia, diagnosis or suspicion of aplastic anemia, thrombosis, Coombs negative hemolysis or hemoglobinuria (Eur J Haematol 2019;102:36)
- Annual flow cytometry screening for PNH may be useful in patients with AA or MDS (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Other causes of anemia should be excluded before testing for PNH (Cytometry B Clin Cytom 2018;94:16)
- PNH is diagnosed by flow cytometry analysis of at least 2 GPI anchored markers on at least 2 cell types (Int J Lab Hematol 2016;38:5, Indian J Hematol Blood Transfus 2017;33:453)
- 3 classifications of PNH clones (Cytometry B Clin Cytom 2018;94:16, Pract Lab Med 2020;20:e00158):
- Classic or hemolytic PNH: patients have a PNH clone and symptoms related to intravascular hemolysis without evidence of a bone marrow failure disorder
- PNH in the context of other primary bone marrow disorders: patients have aplastic anemia or myelodysplastic syndrome with a PNH clone and symptoms primarily related to bone marrow failure
- Subclinical or asymptomatic PNH: patients have a PNH clone but lack clinical or laboratory signs of hemolysis or thrombosis
- Whole body magnetic resonance imaging can be used to detect and evaluate vascular complications (Sci Rep 2018;8:13458)
Laboratory
- Hemolysis with negative direct Coombs (DAT) test (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Ham test to RBC fragility when placed in a mild acid (replaced by flow cytometry) (Pract Lab Med 2020;20:e00158, Ann Clin Lab Sci 2003;33:401)
- Sucrose hemolysis test to RBC fragility when placed in hypotonic sucrose solution triggers complement activation in PNH patients (replaced by flow cytometry) (Pract Lab Med 2020;20:e00158)
- Normocytic anemia with inadequate reticulocytosis and variable neutropenia (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Thrombocytopenia is rare (Hematol Rep 2017;9:6862)
- BM appearance from erythroid hyperplasia to marked hypoplasia (J Hematopathol 2013;6:71)
- Increased levels of lactate dehydrogenase (LDH) and bilirubin and decreased levels of haptoglobin (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Flow cytometry (gold standard): absence of GPI anchored proteins (GPI-APs): CD14, CD16, CD24, CD48, CD52, CD55, CD58, CD59, CD66b, CD66c, CD73, CD87, CD157, CD177, with loss of at least 2 GPI-APs on RBCs and neutrophils (see below) (Pract Lab Med 2020;20:e00158, Cytometry B Clin Cytom 2007;72:167)
- Fluorescein labeled proaerolysin (FLAER) often used in conjunction, as it binds selectively to the GPI moiety on GPI-APs, resulting in increased sensitivity (Int J Lab Hematol 2016;38:5, Cytometry B Clin Cytom 2007;72:167)
- Organ damage resulting from hemolysis or thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
Prognostic factors
- 12 month remission rate: 79% (J Clin Lab Anal 2020;34:e23008)
- 5 year mortality rate: 35% with median survival time at 10 - 15 years (J Clin Lab Anal 2020;34:e23008)
- 10 year overall survival at 70 - 75% (J Clin Lab Anal 2020;34:e23008, Nihon Ketsueki Gakkai Zasshi 1989;52:1386, Blood 2008;112:3099)
- Other factors related to poor survival (J Korean Med Sci 2016;31:214):
- Initial clinical features at the time of diagnosis (Blood 2008;112:3099)
- Hemoglobin (Hb) < 10g/dL and neutropenia (Blood 2008;112:3099)
- Absence of specific treatment during the first year (hazard ratio [HR], 2.2) (Blood 2008;112:3099)
- Independent prognostic factors:
- Thromboembolism: odds ratio (OR) 7.11; 14 times higher risk of mortality (J Korean Med Sci 2016;31:214, J Clin Lab Anal 2020;34:e23008)
- Bone marrow failure (J Clin Lab Anal 2020;34:e23008)
- Elevated LDH > 1.5 times the upper limit of normal (ULN): OR 3.204; 4.8 times higher risk of mortality compared with general (non-PNH) population (J Korean Med Sci 2016;31:214, J Clin Lab Anal 2020;34:e23008)
- Renal impairment: OR 2.953; 8 times higher risk of mortality (J Korean Med Sci 2016;31:214)
- PNH cytopenia: OR 2.547; 6.2 times higher risk of mortality (J Korean Med Sci 2016;31:214)
- Patients with both hemolysis and 1 clinical symptom have a greater risk of mortality compared with patients with just hemolysis or just a clinical symptom (J Korean Med Sci 2016;31:214)
- Leading cause of death for PNH patients in Europe / North America is thromboembolism and for patients in Asia, serious infections (Int J Hematol 2016;103:649)
- Renal failure leads to death in 8 - 18% of PNH patients (Hematology 2018;23:558)
Case reports
- 17 year old Caucasian boy presented with several months of abdominal pain, fever and dark colored urine (Case Rep Pediatr 2019;2019:4930494)
- 23 year old woman was admitted to the emergency department due to bleedings of the jawbone after inflammation (Clin Case Rep 2018;7:175)
- 24 year old woman presented with fevers, epigastric pain, nausea and vomiting (Blood 2013;122:2795)
- 32 year old woman, G2P2A0, with no past medical history of any systemic illnesses (Bol Asoc Med P R 2015;107:9)
- 36 year old Hispanic man presented to the emergency department with 4 days of diffuse abdominal pain (Cureus 2020;12:e8941)
Treatment
- Pegcetacoplan is a pegylated peptide targeting complement protein C3
- New Food and Drug Administration (FDA) approved PNH therapy that showed its superiority to eculizumab in improving hemoglobin (Hb) and clinical and hematological outcomes (N Engl J Med 2021;384:1028)
- In a phase 3 open label controlled trial in adults with PNH and Hb levels lower than 10.5 g/dL despite eculizumab therapy, 85% of patients received pegcetacoplan as compared to 15% receiving eculizumab who no longer required transfusions (N Engl J Med 2021;384:1028)
- Eculizumab: anti-C5 antibody; interferes with activation of the complement system (Int J Lab Hematol 2016;38:5)
- Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved PNH therapy (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Not curative but symptomatic management (Indian J Hematol Blood Transfus 2016;32:383)
- In the initial randomized controlled trial of eculizumab, 49% of PNH patients had stabilized Hb levels following treatment; eculizumab also led to improved quality of life (N Engl J Med 2006;355:1233)
- A subsequent study found that eculizumab improved overall survival (92% survival at 6 years compared with 80% in nontreated historical controls), led to fewer thrombotic events (4% compared with 27%) and caused fewer transitions to MDS or leukemia (Am J Hematol 2016;91:366)
- Eculizumab improved renal function in some patients, especially in those with chronic kidney disease (CKD) stage I - II, while showing significant decrease in major clinical kidney event rates from 4.22 to 2.10 events per 100 patient years (Hematology 2018;23:558)
- Meningococcal vaccine is required before treatment with eculizumab due to increased risk of Neisseria meningitidis infections (Indian J Hematol Blood Transfus 2016;32:383)
- Ravulizumab is a new C5 inhibitor given every 8 weeks; 2 phase 3 trials showed it was noninferior to eculizumab and had a similar safety profile (Blood 2019;133:530, Blood 2019;133:540)
- Allogenic hematopoietic stem cell transplantation
- Only curative treatment (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Indicated for patients with very severe or life threatening disease or who have had eculizumab treatment failure (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383)
- Only curative treatment (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Supportive care (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383):
- Iron, folate and B12 supplementation
- Transfusion of RBCs
- Steroid therapy with glucocorticoids during hemolytic crises
- Immunosuppressive therapies in patients with bone marrow deficiency
- Anticoagulants to manage thrombosis
- Flow cytometry should be repeated every 3 - 6 months to monitor the size of the PNH clone (Indian J Hematol Blood Transfus 2017;33:453)
Microscopic (histologic) description
- Bone marrow:
- Bone marrow analysis is primarily done to determine if PNH arose in association with aplastic anemia (AA) or myelodysplastic syndrome (MDS) (Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758)
- There are no specific morphologic features for PNH (Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758, J Hematopathol 2013;6:71)
- Bone marrow cellularity may be increased, decreased or within a normal range for age (J Hematopathol 2013;6:71)
- Erythroid hyperplasia, decreased myelopoeisis and decreased megakaryocytes are features that can be seen in PNH bone marrow (J Hematopathol 2013;6:71, Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758, Semin Hematol 2019;56:65)
Peripheral smear description
- Peripheral smear may show spherocytosis or presence of sphere shaped erythrocytes; however, these findings are nonspecific (Clin Adv Hematol Oncol 2018;16:1)
Flow cytometry description
- At least 2 different hematopoietic cell types should be evaluated using antibodies against GPI anchored cell surface marker or using FLAER, which binds to all GPI anchored molecules (Int J Lab Hematol 2016;38:5)
- Because the majority of potential PNH cases will be negative, an initial screen with fewer antibodies can be performed before using a diagnostic confirmation assay using a full antibody panel (EJIFCC 2019;30:355)
- Screens may be accomplished with a simpler 2 color panel consisting of FLAER and CD15 labeling of neutrophils, although this is not a substitute for a full diagnostic assay (Int J Lab Hematol 2020;42:589)
- PNH clones will appear with these immunophenotypes:
- Granulocytes: absence of CD16, CD24, FLAER (Int J Lab Hematol 2016;38:5)
- Neutrophils or monocytes: absence of CD16, CD24, CD14, CD157, FLAER (Int J Lab Hematol 2016;38:5)
- Erythrocytes: absence of CD55, CD59, FLAER (Int J Lab Hematol 2016;38:5)
- Platelets: absence of CD55, CD59, FLAER (Blood Cells Mol Dis 2020;80:102372)
- Different types of RBC clones (Ann Hematol 2019;98:1083):
- Type I: presence of GPI-APs (CD55 and CD59; i.e. normal, non-PNH RBCs)
- Type II: partial absence of GPI-APs
- Type III: complete absence of GPI-APs
- 10% of patients present with both type II and type III clones (Ann Hematol 2019;98:1083)
- Analyze the size of PNH clone; if 1 - 5% of a given cell type is GPI deficient, treatment should be considered (Int J Lab Hematol 2016;38:5)
- Quantitation of the PNH clone is not as accurate when measuring RBCs if the patient has received blood transfusions containing normal RBCs or has had recent hemolytic crises (EJIFCC 2019;30:355)
- Quantification of RBCs is more accurate when adding anti-CD71 antibodies to the flow cytometry assay and using immature RBC populations, which are resistant to destruction by the complement system (Cytometry B Clin Cytom 2020;98:179)
- Because diagnosis relies on absence rather than the presence of specific markers, care should be taken to exclude apoptotic cells using cell viability dye (EJIFCC 2019;30:355)
- Using an 8 color panel can more accurately detect PNH clones compared with a 4 color panel; the sensitivity of the optimized 8 color panel is 0.01% when measuring granulocytes and 0.05% when measuring monocytes, after acquiring 100,000 events (Biomed Rep 2018;8:224)
Flow cytometry images
Contributed by Saja Asakrah, M.D.

High sensitivity PNH flow cytometry
Sample pathology report
- Peripheral blood sample
- Paroxysmal nocturnal hemoglobinuria (PNH) (see comment)
- Comment:
- Flow cytometric analysis of the submitted blood sample demonstrates the presence of population(s) of white blood cells (neutrophils or monocytes) or red blood cells (RBCs) with deficiency of tested glycosylphosphatidyl inositol (GPI) linked proteins.
- Percentages of the GPI deficient cell populations are:
- Total GPI deficient RBCs (type III plus type II): 13.8%
- Type III (GPI deficient) RBCs (CD235a+ CD59-): 10.2%
- Type II (partial GPIdeficient) RBCs (CD235a+ CD59- intermediate): 3.6%
- GPI deficient neutrophils (CD15+ CD24- FLAER-): 28.7%
- GPI deficient monocytes (CD64+ CD14- FLAER-): 59.4%
- The flow cytometric findings are consistent with a diagnosis of paroxysmal nocturnal hemoglobinuria (PNH). However, PNH cells may be seen in PNH, aplastic anemia, myelodysplastic syndromes and other conditions. Correlation with clinical and laboratory findings is suggested.
- Note: Discordance between the size of GPI deficient RBC and WBC populations may be due to hemolysis or transfusion.
- The lower limit of quantification (lower limit of enumeration) for the RBC assay is 0.01%, for the neutrophil assay is 0.05% and for the monocyte assay is 0.5%. Below these levels, PNH cells may be reported as detected but below the level of quantification. For leukopenic patients, the neutrophil and monocyte assay sensitivity may be decreased.
- References: Cytometry B Clin Cytom 2018;94:16, Cytometry B Clin Cytom 2018;94:23, Cytometry B Clin Cytom 2018;94:49, Cytometry B Clin Cytom 2018;94:67, CLSI: H52-A2 - Red Blood Cell Diagnostic Testing Using Flow Cytometry, 2nd Edition, 2014, EJIFCC 2019;30:355, Int J Lab Hematol 2019;41:73
Differential diagnosis
- Broad differential diagnosis includes other causes of hemolytic anemia and bone marrow failure
- Aplastic anemia (AA):
- Patients tend to have smaller clone sizes compared with patients with classic PNH (35% compared with 83%, although these clone sizes are variable) (Cytometry B Clin Cytom 2018;94:16)
- Have PNH clones that primarily affect granulocytes, rather than both granulocytes and erythrocytes (Exp Hematol 2019;77:41)
- Patients generally have leukopenia or thrombocytopenia (Pract Lab Med 2020;20:e00158)
- Patients tend to have smaller clone sizes compared with patients with classic PNH (35% compared with 83%, although these clone sizes are variable) (Cytometry B Clin Cytom 2018;94:16)
- Myelodysplastic syndrome (MDS):
- Patients also have GPI deficient cells but these arise in colony forming cells rather than hematopoietic stem cells and T cells are not affected (Cytometry B Clin Cytom 2018;94:16)
- Patients generally have leukopenia or thrombocytopenia (Pract Lab Med 2020;20:e00158)
- Autoimmune hemolytic anemia:
- Positive Coombs test, evidence of extravascular rather than intravascular hemolytic anemia (Oxf Med Case Reports 2020;2020:omz125)
- Various presentations of microangiopathic and macroangiopathic anemia
- Sickle cell disease:
- Vaso-occlusion and vasculopathy with early onset and chronic progression (Intern Emerg Med 2019;14:1051)
- Glucose-6-phosphate dehydrogenase (G6PD) deficiency:
- Acute hemolytic anemia occurs in response to infection, fava beans or drugs like primaquine (Hematol Oncol Clin North Am 2016;30:373)
- Pyruvate kinase deficiency:
- Congenital
- Family history
- Low enzyme activity detected during pyruvate kinase enzyme tests (Am J Hematol 2015;90:825)
Additional references
Board review style question #1
Which of the following tests is considered the gold standard test for paroxysmal nocturnal hemoglobinuria (PNH) detection?
- Bone marrow biopsy
- Coombs test
- Flow cytometry
- Serum haptoglobin level
Board review style answer #1
Board review style question #2
Which of the following is a glycosylphosphatidylinositol (GPI) anchored protein and is part of the paroxysmal nocturnal hemoglobinuria (PNH) flow cytometry panel?
- CD11b
- CD11c
- CD14
- CD64
Board review style answer #2
