
Heart & vascular pathology
Cardiomyopathy
Primary and secondary dilated cardiomyopathy
Author: R. Amita, M.D.

Last author update: 1 June 2015
Last staff update: 31 December 2020
Copyright: 2015-2025, PathologyOutlines.com, Inc.
PubMed Search: Dilated cardiomyopathy [title] primary secondary

Table of Contents
Definition / general | Epidemiology | Etiology | Clinical features | Laboratory | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Additional referencesCite this page: Amita R. Primary and secondary dilated cardiomyopathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/heartdilated.html. Accessed April 2nd, 2025.
Definition / general
- Dilated cardiomyopathy (DCM) is characterized by a poorly contracting dilated left ventricle with a normal or reduced left ventricular wall thickness
Epidemiology
- DCM is the most common cause of congestive cardiac failure (CCF), with an estimated prevalence of at least 36.5 per 100,000 persons in the USA
- It occurs more frequently in men than women and is most common between ages 20 and 60 years
Etiology
- In most cases, no definite cause is identifiable
- Most prevalent toxic cause is alcohol
- Single gene mutations involve:
- Structural proteins: dystrophin, metavinculin, lamin
- Mitochondrial DNA
- Coronary artery disease
- Infections: coxsackievirus, adenovirus, parvovirus, HIV, bacterial, fungal rickettsial, myobacterial and parasitic (Chagas disease)
- Nutritional deficiency: carnitine selenium deficiencies
- Cardiotoxins (anthracycline)
- Puerperium
- Metals (cobalt, lead, mercury, arsenic)
- Autoimmune and systemic disorders
- Pheochromocytoma
Clinical features
- Shortness of breath, syncope, angina
Laboratory
- Complete blood count
- Metabolic panel
- Thyroid function tests
- Cardiac biomarkers
- B type natriuretic peptide assay
- Chest radiography
- Echocardiography
- Cardiac magnetic resonance imaging (MRI)
- Electrocardiography (ECG)
Case reports
- 36 year old man with dilated cardiomyopathy secondary to hypothyroidism (J Cardiovasc Ultrasound 2014;22:32)
- 39 year old woman with alcoholic cardiomyopathy (The Internet Journal of Cardiology 2000;10(1))
Treatment
- Essentially the same as treatment of chronic heart failure (CHF)
Gross description
- The heart assumes a globular shape and there is pronounced ventricular chamber dilatation, diffuse endocardial thickening and atrial enlargement, often with thrombi in the appendages
Gross images
Images hosted on other servers:
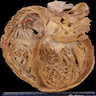
Ventricles dilated more than atria
Microscopic (histologic) description
- Increase in myocyte nuclear size
- Myofibrillary loss within myocyte
- Focal myocyte death
- Increase in interstitial T lymphocytes / macrophages
- Interstitial fibrosis
- The individual myocytes are increased in length rather than in width, lose the normal number of intracellular contractile myofibrils and appear empty and vacuolated
- The myocyte nuclei increase in size because of the synthesis of DNA and become polyploid
- Death of individual myocytes occurs both by apoptosis and necrosis
- Fibrosis characteristically is interstitial and begins to surround and isolate individual myocytes
- The number of macrophages and T lymphocytes in the interstitial spaces is often increased compared with normal hearts
Microscopic (histologic) images
Images hosted on other servers:
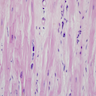
Cardiomyocyte multinucleation
Adenomatous hyperplasia
Molecular / cytogenetics description
- The pattern of inheritance of familial DCM (FDCM) is variable and includes autosomal dominant, X linked, autosomal recessive and mitochondrial
- Autosomal forms of FDCM are the most frequent
- Pure DCM phenotype: mutations of genes encoding cardiac actin, desmin, d-sarcoglycan, b-sarcoglycan, cardiac troponin T, tropomyosin
- DCM with cardiac conduction system disease: lamin A/C gene
- Autosomal dominant Emery-Dreifuss muscular dystrophy
- The X linked forms of DCM includes X linked dilated cardiomyopathy and Barth syndrome
- Caused by mutations in the dystrophin gene
- X linked dilated cardiomyopathy (XLCM) occurs in males during adolescence or early adulthood and has a rapidly progressive clinical course
- Female carriers develop a mild form of DCM with onset in middle age
- Creatine kinase levels are elevated but without clinical signs of skeletal myopathy
- The infantile form of X linked DCM or Barth syndrome typically presents
in male infants
- It is characterised by neutropenia, 3-methylglutaconic aciduria, growth retardation and mitochondrial dysfunction
- The cardiac manifestations include left ventricular dilatation, endocardial fibroelastosis or a dilated hypertrophied left ventricle
- Mutations in the gene G4.5, which encodes the protein tafazzin, cause Barth syndrome
Additional references
