

CNS & pituitary tumors
Gliomas, glioneuronal tumors and neuronal tumors
Pediatric type diffuse low grade gliomas
Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
Author: Eman Abdelzaher, M.D., Ph.D.
Editorial Board Member: Jared T. Ahrendsen, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.

Last author update: 5 March 2024
Last staff update: 5 March 2024
Copyright: 2022-2025, PathologyOutlines.com, Inc.
PubMed Search: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Cytology images | Positive stains | Negative stains | Molecular / cytogenetics description | Molecular / cytogenetics images | Videos | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Abdelzaher E. Polymorphous low grade neuroepithelial tumor of the young (PLNTY). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnstumorplnty.html. Accessed January 18th, 2025.
Definition / general
- Indolent cerebral neoplasm characterized by a strong association with seizures in young individuals, diffuse growth patterns, frequent presence of oligodendroglioma-like components, calcifications, CD34 immunoreactivity and MAPK pathway activating genetic abnormalities
- CNS WHO grade 1
- Recently introduced in WHO 2021 classification of CNS tumors
Essential features
- Recently described, frequently epileptogenic, low grade neuroepithelial tumor (Acta Neuropathol 2017;133:417)
- Typically affects children and young adults
- Characterized by diffuse growth patterns, oligodendroglioma-like cellular components, CD34 immunopositivity and MAPK pathway activating genetic abnormalities
- Generally, exhibits a benign clinical course (Acta Neuropathol 2017;133:417)
Terminology
- Has been described under the generic designation of long term epilepsy associated tumors (Acta Neuropathol 2014;128:39)
- Not recommended by WHO
- Diffuse glioneuronal tumor
- Diffuse or nonspecific form of dysembryoplastic neuroepithelial tumor
- Massively calcified low grade glioma
ICD coding
Epidemiology
- Rare; population based incidence data are unavailable
- Age: mostly occurs in the second and third decades of life (mean age at diagnosis: 22.16 years; range: 4 - 57 years) (Acta Neurol Belg 2023;123:327)
- Slight male predominance (Acta Neurol Belg 2023;123:327)
Sites
- Cerebral tumors, usually with cortical and subcortical components (Neuroradiology 2022;64:1255)
- Mostly right sided (64.4%) (Acta Neurol Belg 2023;123:327)
- Majority involve temporal lobes (69%) with frequent involvement of medial / posteroinferior structures (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- Other sites include frontal, parietal and occipital lobes and rarely, third ventricular region (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
Pathophysiology
- Aberrant CD34 expression possibly reflects an origin from developmentally dysregulated neural precursors (Acta Neuropathol 1999;97:481)
- Somatic MAPK pathway activating genetic abnormalities (particularly BRAF mutations or FGFR fusions) definitely drive the pathogenesis of PLNTY; however, the specific mechanisms by which these genetic alterations contribute to the development of PLNTY are not clear (Acta Neuropathol 2017;133:417)
Etiology
- Unknown predisposing factors
- Germline ATM mutation was identified in a single case, which was associated with progressive disease (J Neuropathol Exp Neurol 2019;78:1100)
Diagrams / tables
Images hosted on other servers:
Prototypical PLNTY
Clinical features
- Typically presents with seizures (intractable epilepsy [47.6%] or a single seizure attack [42.9%]) (Acta Neurol Belg 2023;123:327)
- Less common: headache, dizziness, visual disturbances and mood disorders (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
Diagnosis
- Neuroimaging: MRI and CT (Acta Neurol Belg 2023;123:327)
- Biopsy
- WHO essential and desirable diagnostic criteria
- Essential diagnostic criteria (presence of the following 6 characteristics)
- Diffuse growth pattern (at least regionally)
- Oligodendroglioma-like components (may be minor)
- Few (if any) mitotic figures
- Regional CD34 expression by tumor cells and by ramified neural cells in associated cerebral cortex
- IDH wild type status
- Unequivocal expression of BRAF p.V600E on immunohistochemical assessment or molecular diagnostic evidence of BRAF V600E mutations, FGFR2 or FGFR3 fusions or potentially other MAPK pathway driving genetic abnormalities
- Desirable diagnostic criteria
- Conspicuous calcification (characteristic, although not constant)
- Absence of 1p / 19q codeletion
- Essential diagnostic criteria (presence of the following 6 characteristics)
Radiology description
- CT
- Often calcified (76.2%) (Neuroradiology 2019;61:1327, Acta Neurol Belg 2023;123:327)
- MRI
- Often superficially situated (Acta Neuropathol 2017;133:417, Neuroradiology 2019;61:1327)
- Circumscribed (76.7%) or blurred outlines (Acta Neurol Belg 2023;123:327)
- Usually cystic and solid (Acta Neurol Belg 2023;123:327, BMC Neurol 2020;20:123)
- Signal heterogeneity with T2 hyperintense or mixed signal, T1 hypointense or mixed signal (Acta Neurol Belg 2023;123:327)
- FLAIR hyperintense
- Majority shows no contrast enhancement (67.5%); patchy or nodular contrast enhancement in 32.5% of cases (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- No substantial edema or mass effect (Acta Neuropathol 2017;133:417)
Radiology images
Images hosted on other servers:
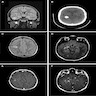
FLAIR, CT scan and T1 postcontrast
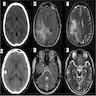
Radiological variability in CT and MRI
Prognostic factors
- Generally exhibits a benign clinical course (Acta Neuropathol 2017;133:417)
- Amenable to control by gross total resection with relief from seizures or reduced seizure frequency in most cases (Acta Neuropathol 2017;133:417)
- Rarely exhibits local recurrence (11%), tumor progression or malignant transformation (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
- Molecular events associated with less favorable prognosis include germline ATM mutation and somatic mutations involving RB1, TP53, ATRX, PTEN and TEK (J Neuropathol Exp Neurol 2021;80:821, BMC Neurol 2020;20:123, J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
- Radiological contrast enhancement seems to adversely impact prognosis, recurrence and seizure control (Acta Neurol Belg 2023;123:327)
Case reports
- 13 year old girl with PLNTY mimicking an intracranial granuloma (Indian J Radiol Imaging 2023;33:567)
- 15 year old girl with malignant transformation of PLNTY (Acta Neuropathol 2021;141:123)
- 37 year old man presented with progressive headache (Egyptian Journal of Neurosurgery 2023;38:7)
- 50 year old woman with longstanding epilepsy (Surg Neurol Int 2021;12:470)
Treatment
- Surgical resection
- Targeted therapies: the MAPK pathway activating genetic alterations are potentially targetable; this provides additional therapeutic options, particularly for tumors that cannot be completely resected or that are located in surgically challenging locations (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821, J Neuropathol Exp Neurol 2019;78:1100)
Clinical images
Images hosted on other servers:
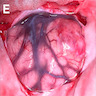
Operative view
Gross description
- Unencapsulated, indistinctly demarcated from normal brain
- Soft to friable, gray-white masses
Microscopic (histologic) description
- Exhibits both diffuse and compact growth patterns (Acta Neuropathol 2017;133:417)
- Typically shows an oligodendroglioma-like cellular component (Acta Neuropathol 2017;133:417)
- Usually dominant, may be minor
- Ranges from uniform cells with small and round nuclei with perinuclear haloes to cells exhibiting obvious nuclear pleomorphism with more oval or spindled nuclear contours, wrinkled or grooved nuclear membranes and intranuclear pseudoinclusions (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- Astrocytic or morphologically ambiguous elements may also be seen, including fibrillary, spindled and pleomorphic forms (Acta Neuropathol 2017;133:417)
- Subtle perivascular pseudorosettes may be seen
- Calcifications are present in the large majority, ranging from discrete calcospherules to confluent calcific masses with osseous metaplasia (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- Less common / rare: eosinophilic granular bodies, modestly dysmorphic neurons in small numbers and gemistocytic elements (7.7%) (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- Few (if any) mitotic figures; rarely, increased mitotic activity (up to 4 mitoses/10 high power fields) reported (15%) (J Neuropathol Exp Neurol 2021;80:821)
- Generally absent are foci of necrosis, microvascular proliferation, myxoid microcysts, neurocytic or ependymal rosettes, Rosenthal fibers and clearly neoplastic neuronal components (the nosological position of neoplasms otherwise qualifying as PLNTY but having nodular ganglion cell aggregates is unclear) (Acta Neuropathol 2017;133:417)
Microscopic (histologic) images
Contributed by Eman Abdelzaher, M.D., Ph.D. and Jared T. Ahrendsen, M.D., Ph.D.
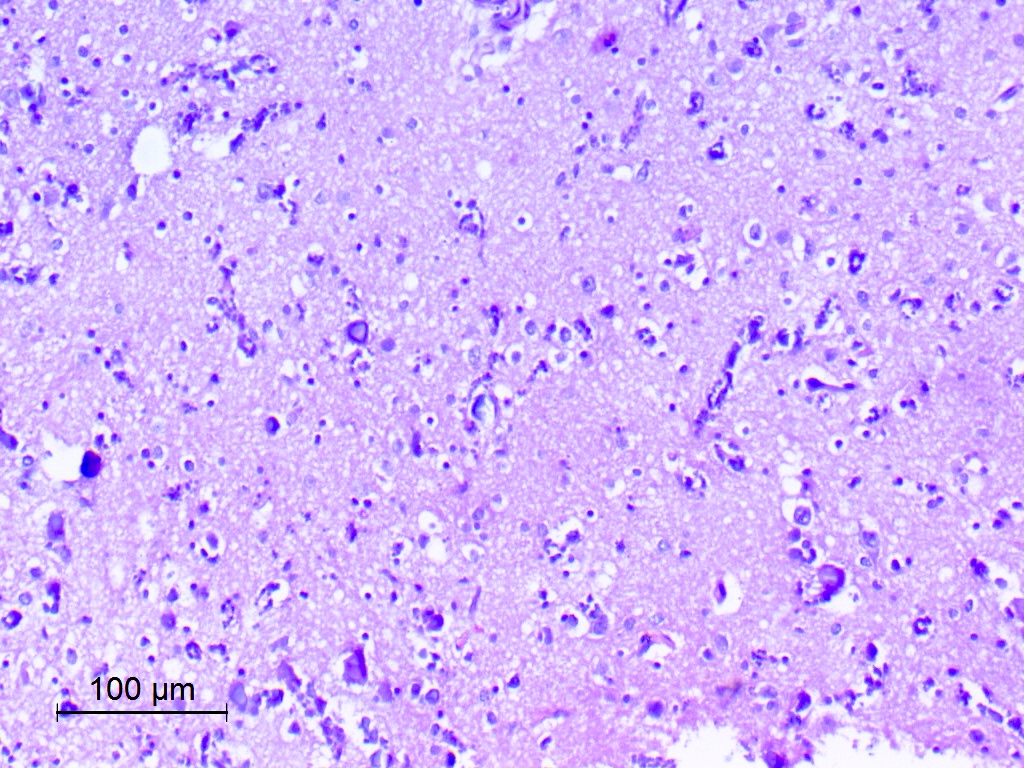
Diffuse growth pattern
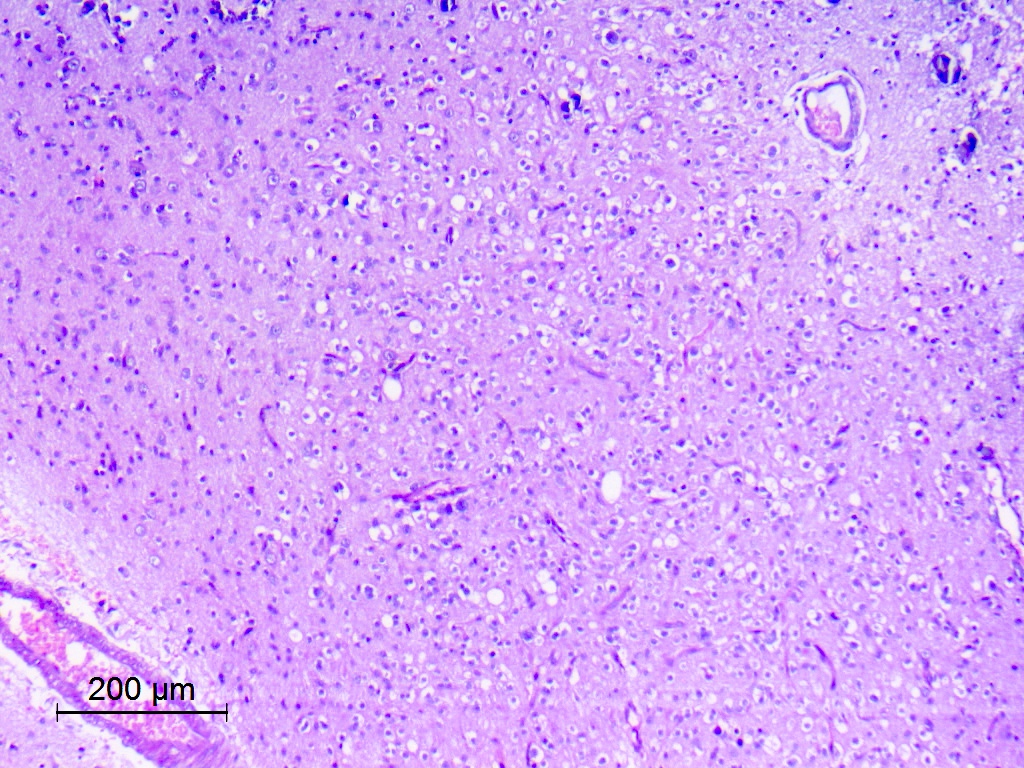
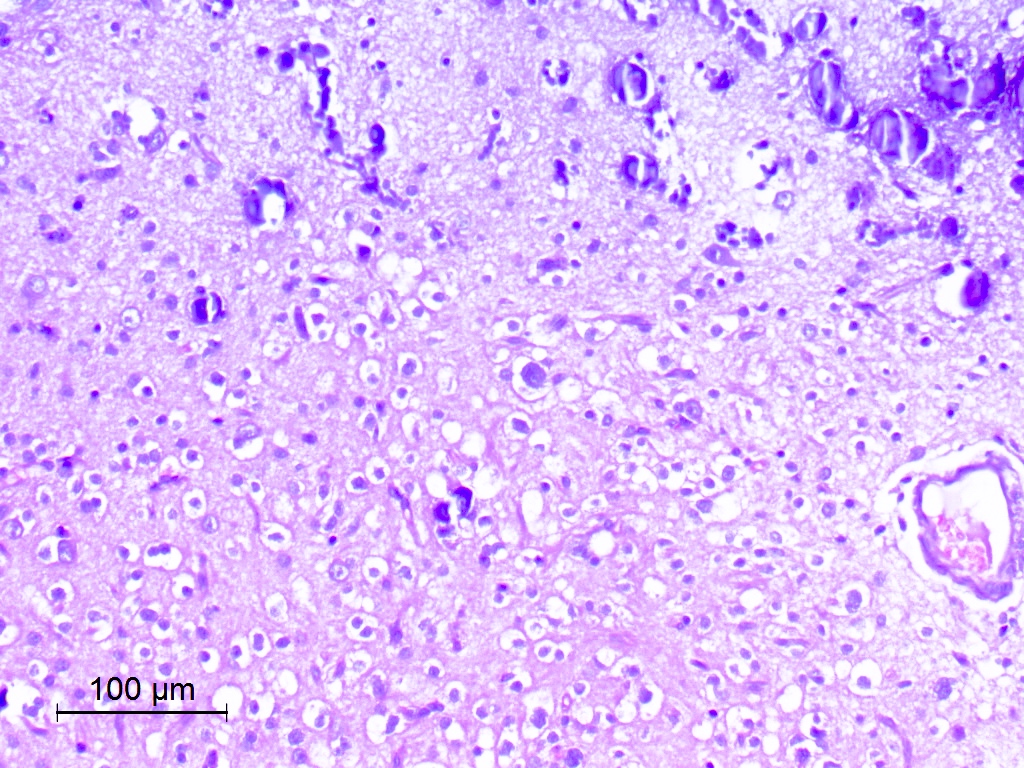
Diffuse growth pattern of oligodendroglioma-like cells
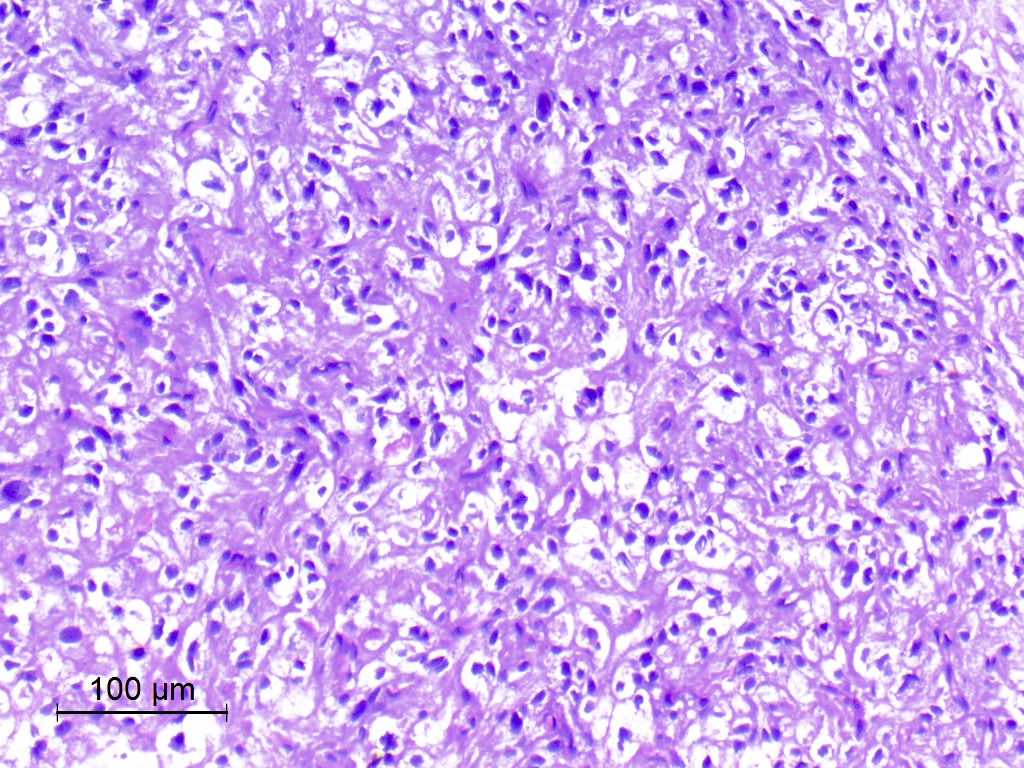
Oligodendroglioma-like cells with mild nuclear pleomorphism
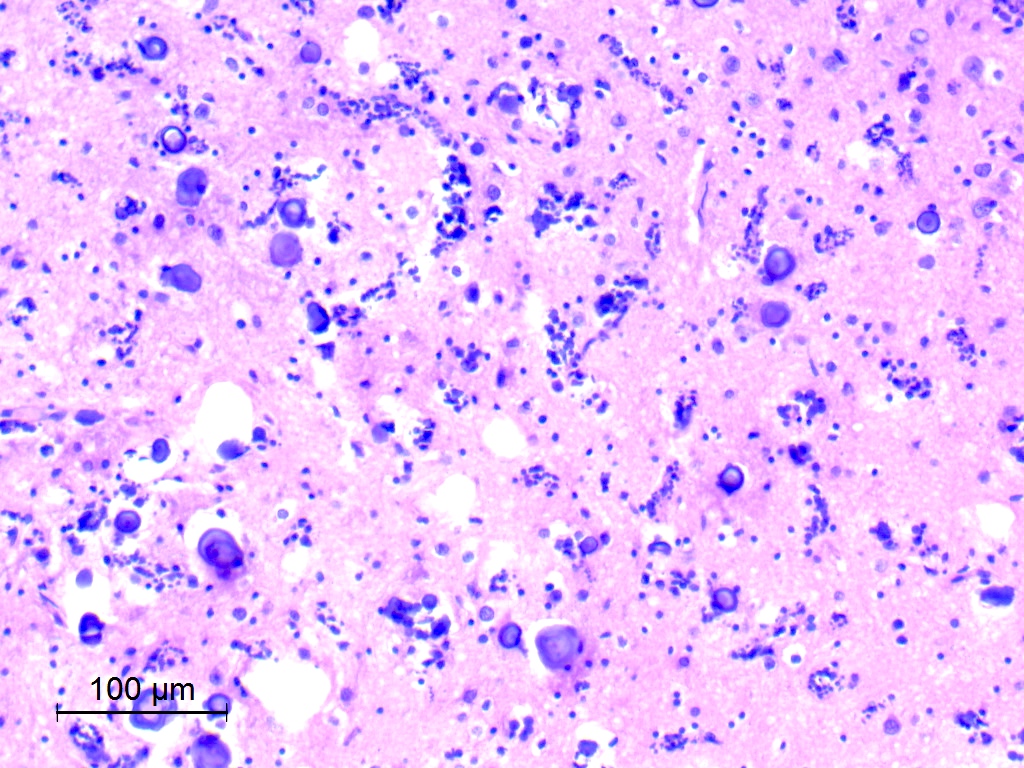
Calcification
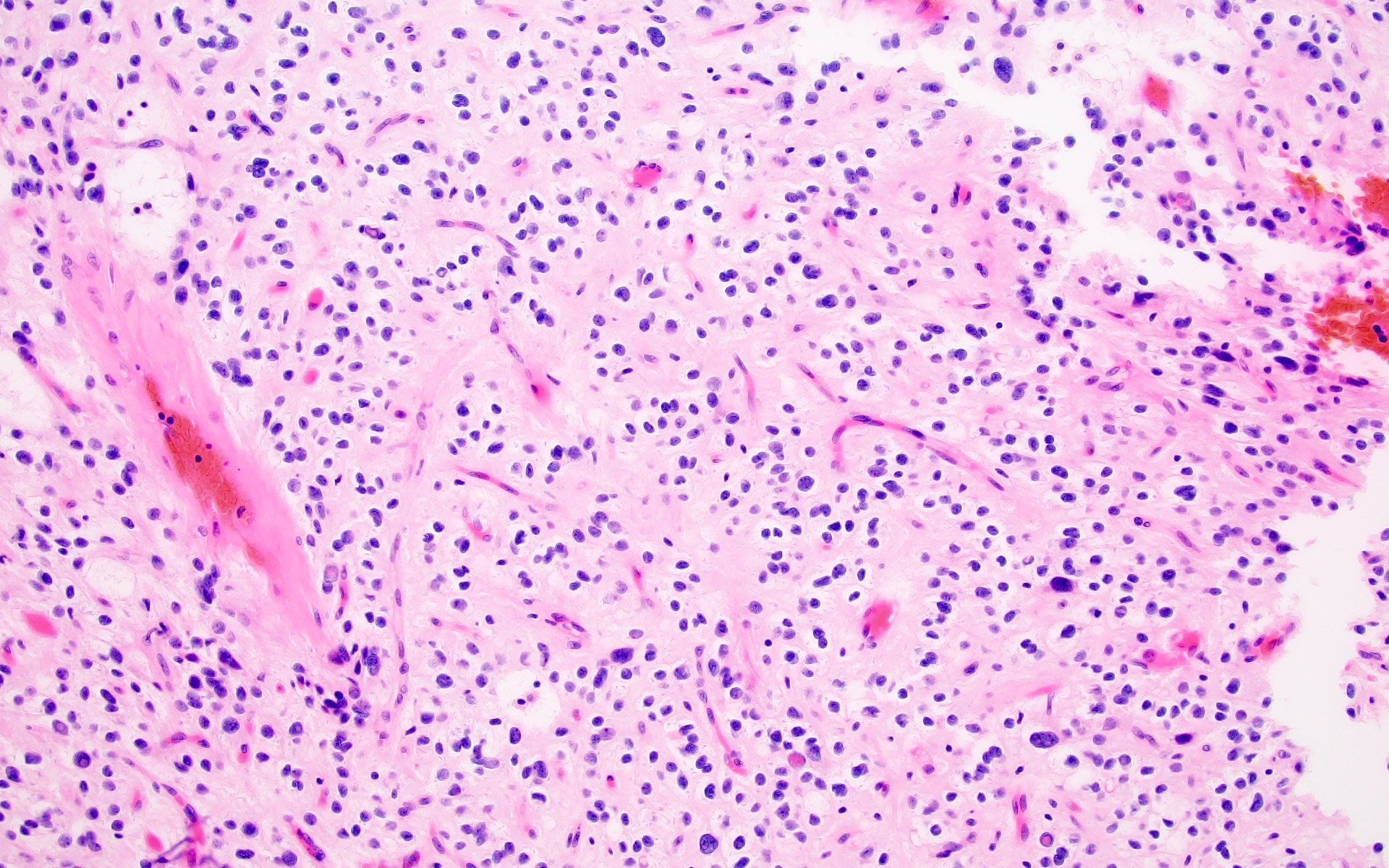
Oligo-like features
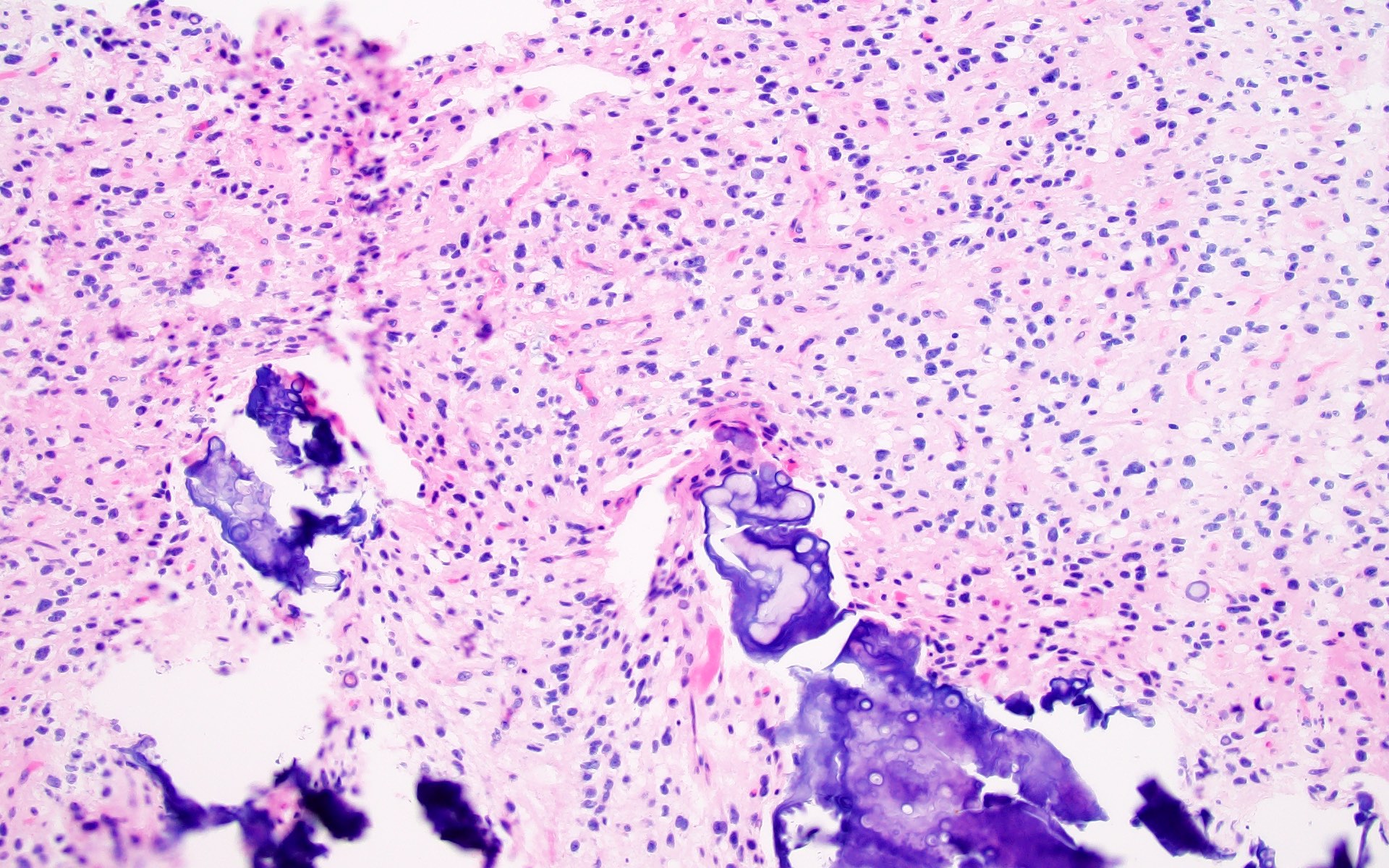
Dystrophic calcifications

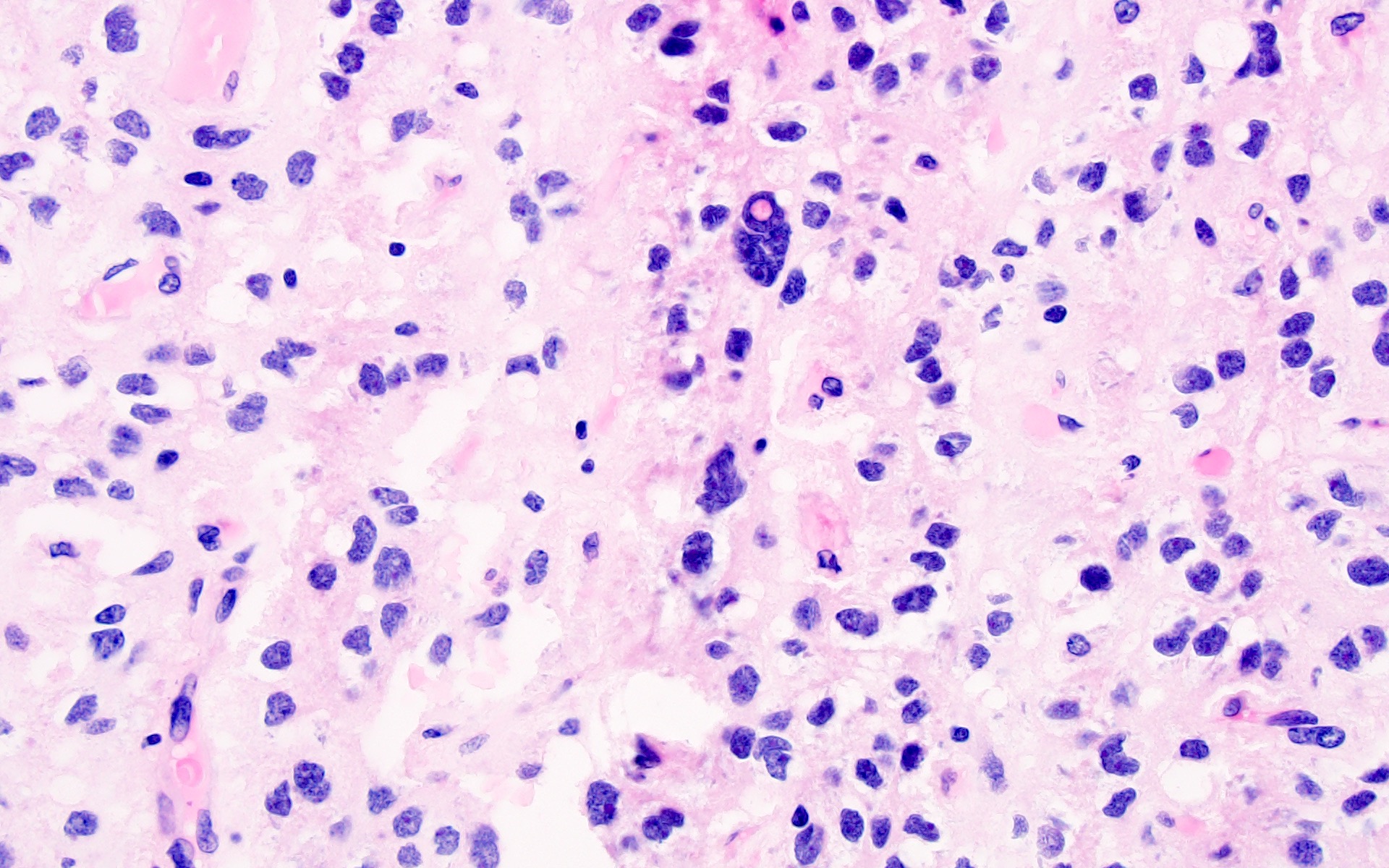
Polymorphic tumor cells
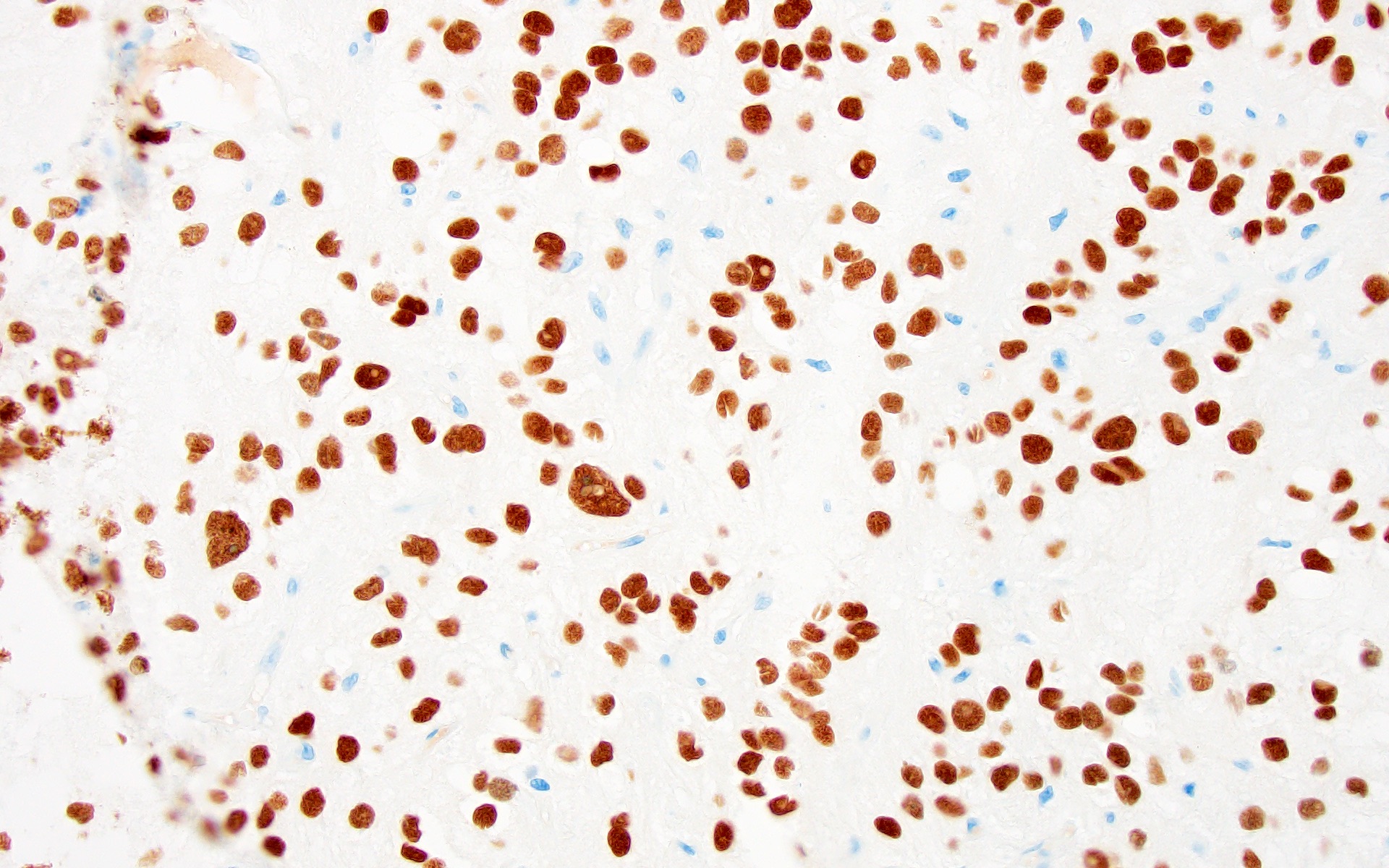
Olig2 diffusely positive
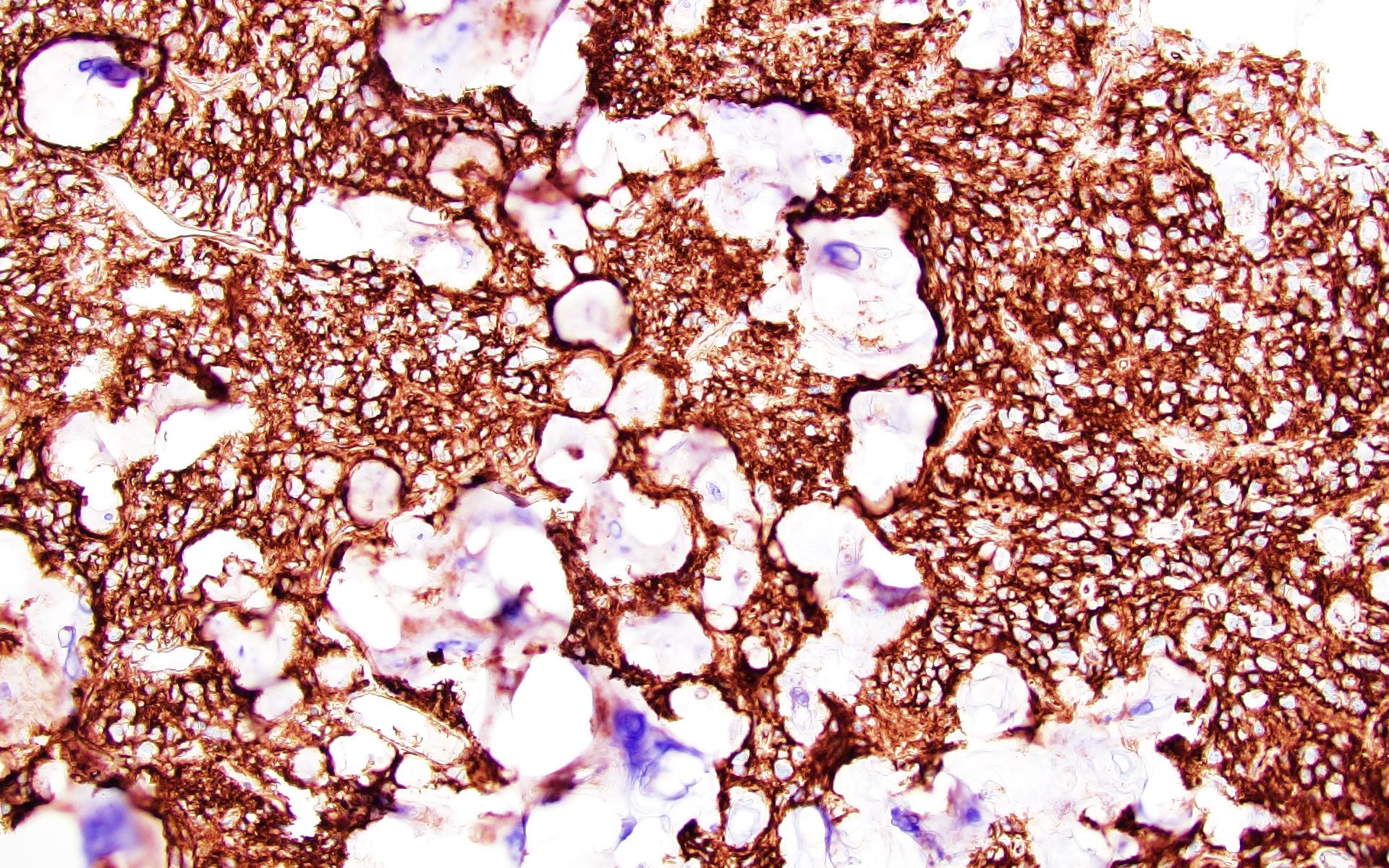
Strong, diffuse CD34 expression
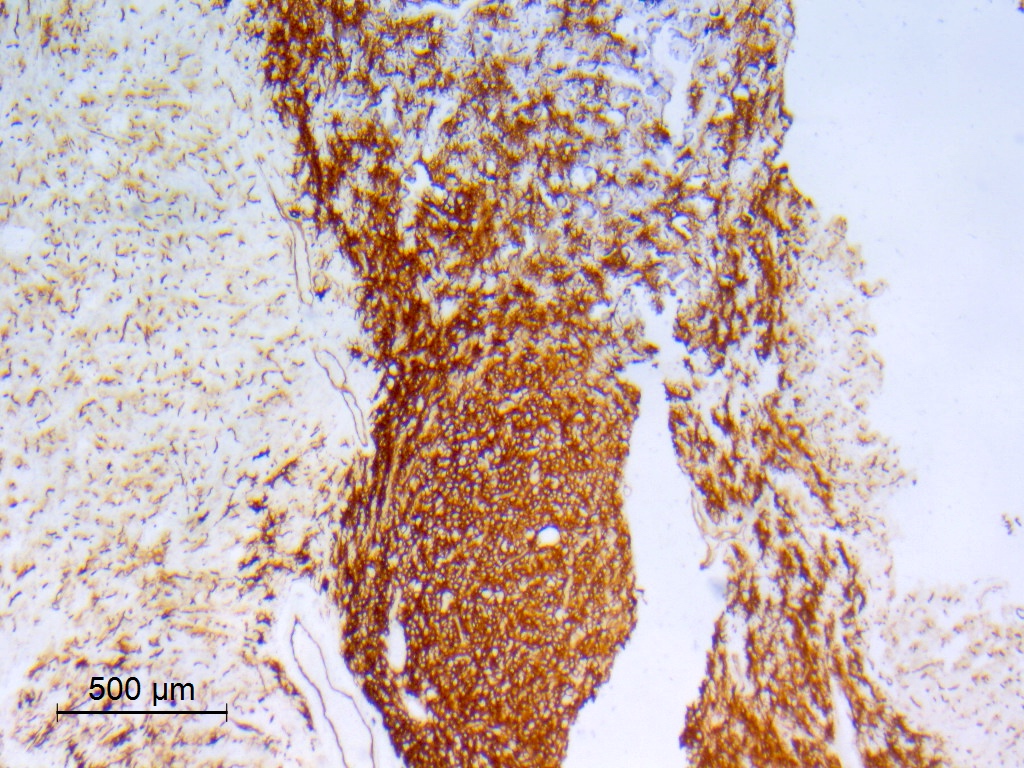
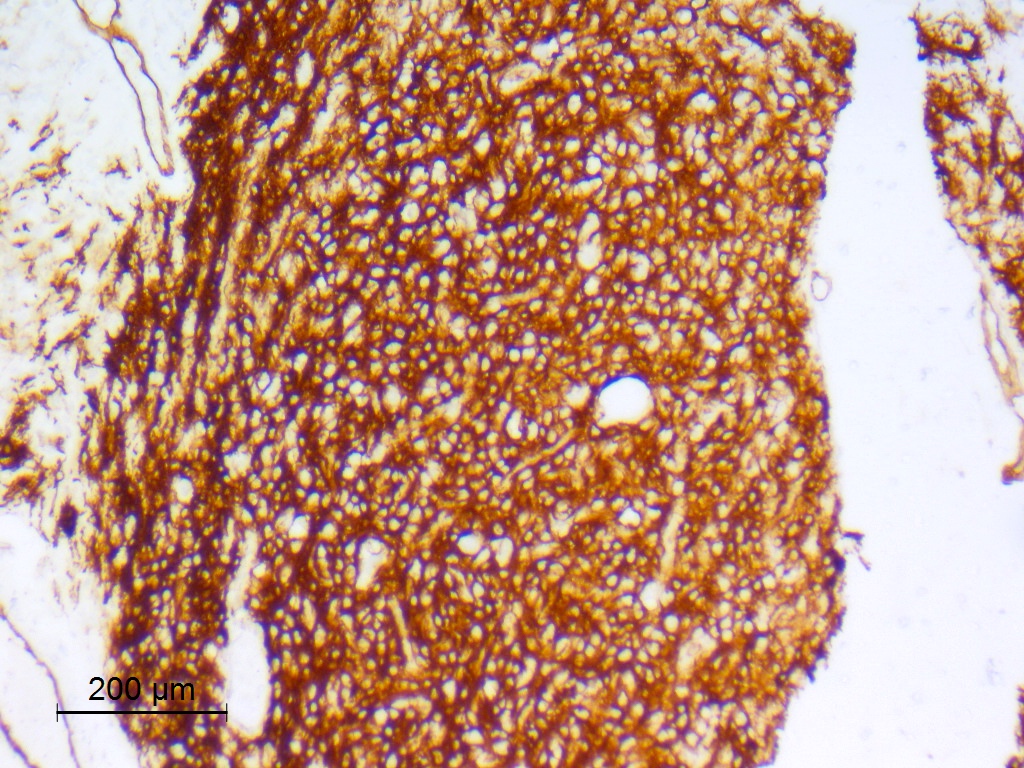
CD34
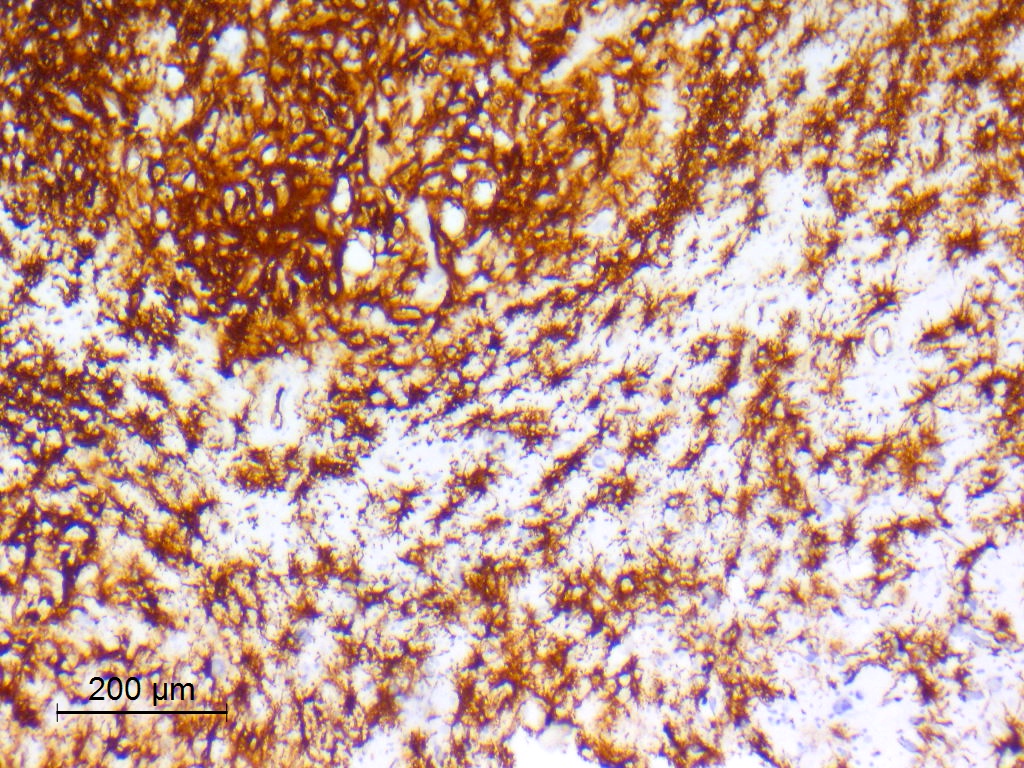
CD34 in tumor cells and ramified neural cells
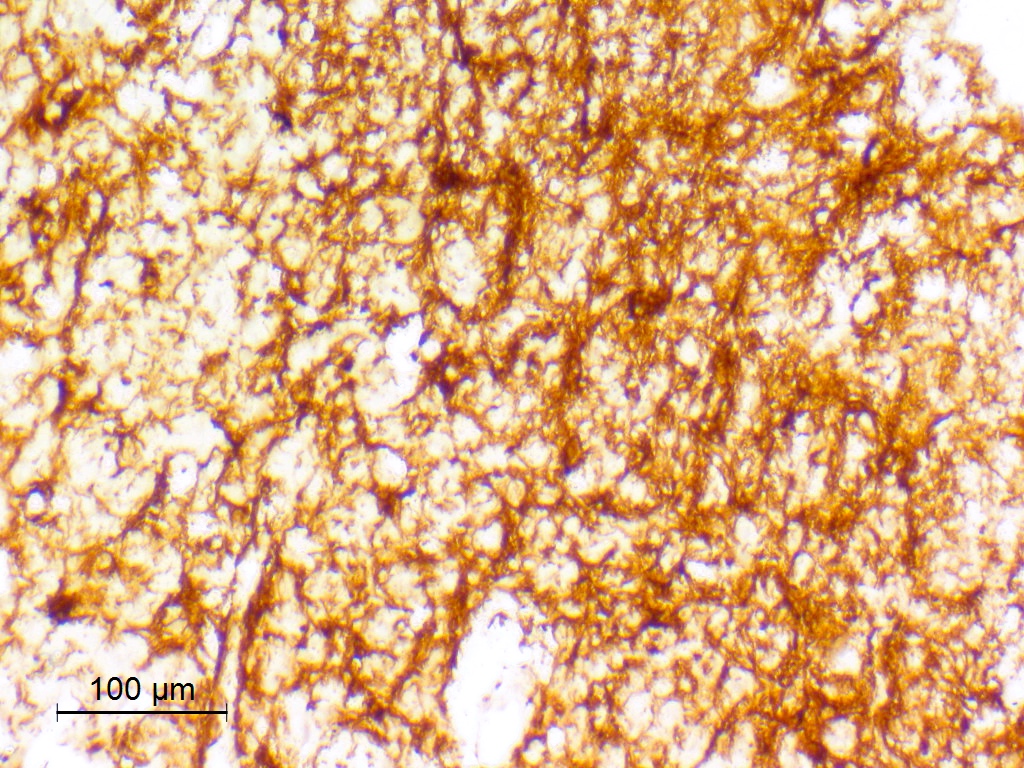
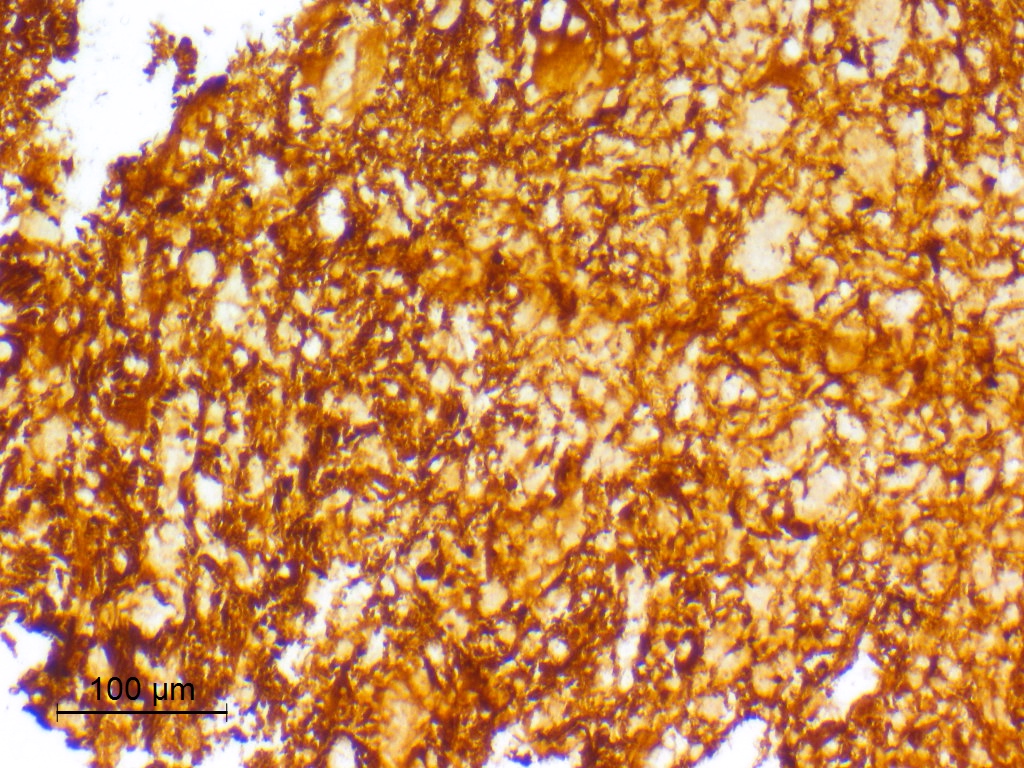
GFAP
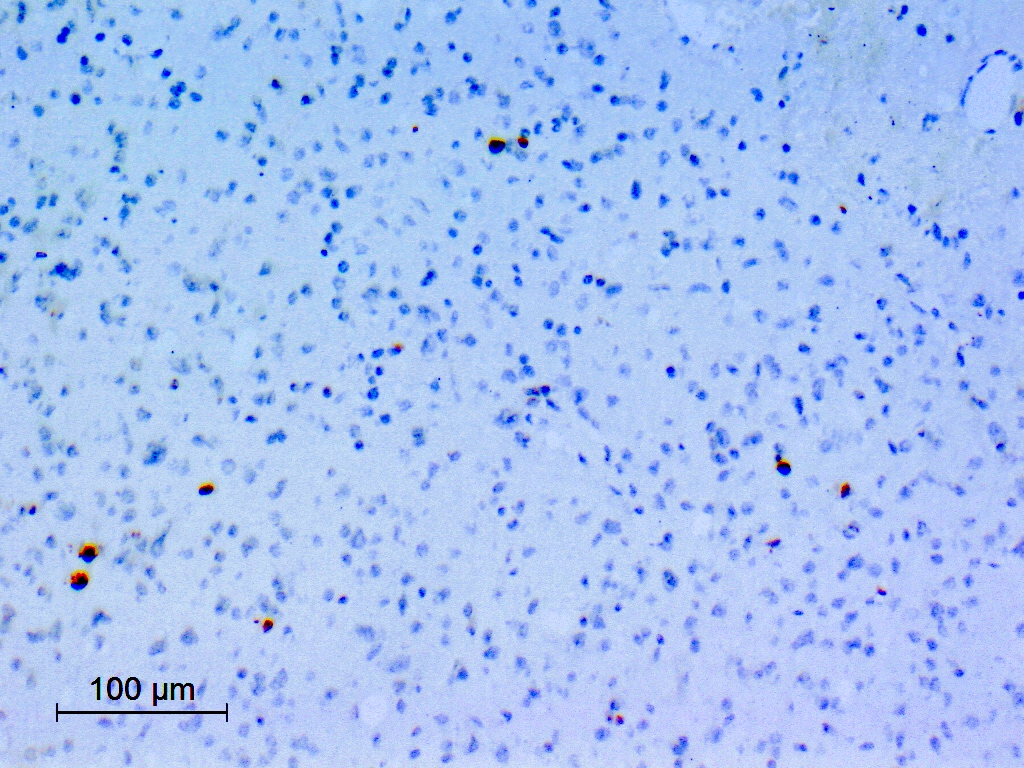
Ki67
Cytology description
- PLNTY cannot be identified on cytological grounds alone
- Intraoperative smear preparations may show
- Tumor cells with rounded nuclei and devoid of cytoplasm
- Tumor cells with irregular nuclear contours and cytoplasmic processes
- Tumor cells attached to blood vessels in a pseudorosette fashion (Acta Neuropathol 2017;133:417)
Cytology images
Images hosted on other servers:
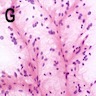
Vague perivascular pseudorosetting
Positive stains
- GFAP: may be regional or diffuse, may be weak (Acta Neuropathol 2017;133:417)
- OLIG2 (Acta Neuropathol 2017;133:417)
- CD34
- May be patchy, or diffuse and intense
- Expressed by both tumor cells and ramified neural elements in the associated cerebral cortex (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- ATRX: retained (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- BRAF p.V600E (30 - 54%) (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- Ki67: generally low (< 1 - 2%) but higher values (up to 5%) have been reported (Acta Neuropathol 2017;133:417)
Negative stains
Molecular / cytogenetics description
- Consistently associated with MAPK pathway activation by either BRAF V600E mutations or fusion events involving FGFR2 or FGFR3 in a mutually exclusive fashion; however, these abnormalities are hardly specific to PLNTY as a transforming molecular event (Acta Neuropathol 2017;133:417, Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2019;78:1100, J Neuropathol Exp Neurol 2021;80:821)
- FGFR fusions include FGFR2::CTNNA3, FGFR2:SHTN1 (KIAA1598), FGFR2::INA, FGFR3::TACC3 and FGFR2::MPRIP (J Neuropathol Exp Neurol 2021;80:821, Acta Neurol Belg 2023;123:327)
- Fusion positive tumors occur predominantly in pediatric patients and are more frequently associated with seizures / epilepsy, whereas BRAF V600E mutation positive tumors predominantly affect adult patients (J Neuropathol Exp Neurol 2021;80:821)
- Has a distinct DNA methylation profile most closely aligned to that of ganglioglioma (Acta Neuropathol 2017;133:417)
- Exhibits copy number changes predominantly involving whole chromosomes, suggesting that PLNTY is also characterized by chromosomal instability (J Neuropathol Exp Neurol 2021;80:821)
- PLNTY does not harbor IDH mutations and does not exhibit 1p / 19q codeletion
- Other reported molecular events include NTRK2 alterations, KIAA1549::BRAF fusion and low level FGFR3 amplification; molecular events associated with less favorable prognosis include germline ATM mutation and somatic mutations involving RB1, TP53, ATRX, PTEN and TEK (J Neuropathol Exp Neurol 2021;80:821, BMC Neurol 2020;20:123, J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
Molecular / cytogenetics images
Images hosted on other servers:

Molecular and clinical findings
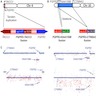
FGFR fusions

Distinct methylation signature

Genome wide copy number changes
Videos
Sample pathology report
- Brain mass lesion, temporal lobe, gross total resection:
- Polymorphous low grade neuroepithelial tumor of the young (PLNTY), CNS WHO grade 1
- Molecular genetics: evidence of BRAF V600E mutations, FGFR2 or FGFR3 fusions or potentially other MAPK pathway driving genetic abnormalities
Differential diagnosis
- Adult type diffuse glioma (oligodendroglioma, IDH mutant and 1p / 19q codeleted or low grade diffuse astrocytoma, IDH mutant):
- CD34- (Acta Neuropathol 2017;133:417, World Neurosurg 2019;127:47)
- IDH mutations
- 1p / 19q codeletion in oligodendroglioma
- Lacks MAPK pathway activating genetic abnormalities
- Ependymoma:
- Often OLIG2-
- Dot or ring-like EMA+ (Acta Neuropathol 2017;133:417)
- Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC):
- Characteristic feature is the presence of scattered multinucleated cells (pennies on a plate) and nuclear clusters composed of large pleomorphic nuclei
- Synaptophysin+, GFAP-, CD34- (Neuropathol Appl Neurobiol 2021;47:464)
- Diffuse leptomeningeal glioneuronal tumor:
- Leptomeningeal based
- Synaptophysin+, GFAP-
- Concurrent KIAA1549::BRAF fusion with isolated 1p deletion or 1p / 19q codeletion
- Ganglioglioma:
- Clearly neoplastic neuronal components
- Features that support a diagnosis of PLNTY over ganglioglioma include abundant calcifications, diffuse strong CD34 staining of tumor cells and FGFR2 or FGFR3 fusions (Acta Neuropathol 2017;133:417)
- Nosological position of neoplasms otherwise qualifying as PLNTY but having nodular ganglion cell aggregates is unclear (Acta Neuropathol 2017;133:417)
- Pilocytic astrocytoma:
- Biphasic pattern
- CD34- (Balkan Med J 2019;36:3)
- Diffuse astrocytoma, MYB or MYBL1 altered:
- Diffuse low grade glioma, MAPK pathway altered:
- CD34 is typically limited to a few cells, unlike the widespread expression seen in PLNTY
- PLNTYs likely represent a subset of a larger family of tumors reported as diffuse astrocytomas or oligodendrogliomas of pediatric type having MAPK pathway activating genetic abnormalities (Acta Neuropathol 2017;133:417)
Additional references
Board review style question #1
A 14 year old boy presented with refractory epilepsy. MRI showed a right temporal cystic and solid nonenhancing mass lesion. The mass was surgically resected and histopathological examination showed a low grade tumor composed of oligodendroglioma-like tumor cells arranged in a diffuse fashion as shown in the image above. Immunohistochemistry for CD34 showed regional positivity associated with positive ramified neurons in the adjacent cortex. Which of the following molecular events is characteristic of this tumor?
- 1p / 19q codeletion
- IDH mutation
- MAPK pathway activating abnormalities
- PRKCA gene fusion
Board review style answer #1
C. MAPK pathway activating abnormalities. PLNTY is characterized by a strong association with seizures in young individuals, diffuse growth patterns, frequent presence of oligodendroglioma-like components and CD34 immunoreactivity. It is molecularly characterized by MAPK pathway activating genetic abnormalities.
Answers A and B are incorrect because PLNTY does not harbor IDH mutations or 1p / 19q codeletion.
Answer D is incorrect because PRKCA gene fusion (mainly SLC44A1::PRKCA) is the hallmark of papillary glioneuronal tumor (PGNT).
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
Board review style question #2
Polymorphous low grade neuroepithelial tumor of the young (PLNTY) is characterized by which of the following features?
- Absence of calcification
- CD34 negative immunostaining
- Clinical presentation of headache
- Oligodendroglioma-like components
Board review style answer #2
D. Oligodendroglioma-like components. PLNTY is characterized by frequent presence of oligodendroglioma-like components.
Answer A is incorrect because calcification is a frequent finding in PLNTY.
Answer B is incorrect because CD34 immunoreactivity is a characteristic finding in PLNTY.
Answer C is incorrect because PLNTY is characterized by a strong association with seizures in young individuals.
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)

