
CNS nontumor
Movement disorders
Multiple system atrophy
Editorial Board Member: P.J. Cimino, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 3 August 2023
Last staff update: 3 August 2023
Copyright: 2022-2025, PathologyOutlines.com, Inc.
PubMed Search: Multiple system atrophy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Pinarbasi E, Lieberman A. Multiple system atrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnsmultiplesystematrophy.html. Accessed March 31st, 2025.
Definition / general
- Multiple system atrophy (MSA) is a sporadic, neurodegenerative disease with core clinical features of parkinsonism, autonomic dysfunction and cerebellar ataxia
- Pathologically, MSA is characterized by glial cytoplasmic inclusions (GCIs) that are predominantly within oligodendroglia and composed of alpha-synuclein
Essential features
- Clinically characterized by a triad of autonomic dysfunction, levodopa resistant parkinsonism and cerebellar ataxia
- Pathologically defined by widespread frequent alpha-synuclein glial cytoplasmic inclusions (GCIs) and striatonigral or olivopontocerebellar degeneration
Terminology
- Shy-Drager syndrome (autonomic dysfunction)
- Olivopontocerebellar atrophy (OPCA)
- Striatonigral degeneration
ICD coding
Epidemiology
- Incidence of 3 - 4 per 100,000 person years (Lancet 1999;354:1771, Neurology 1997;49:1284)
- Usually presents between fourth and sixth decades (Neurology 1997;49:1284)
- In Europe, the clinical subtype parkinsonism predominant (MSA-P) is more common (Mov Disord 2010;25:2604)
- In Japan, the clinical subtype cerebellar ataxia predominant (MSA-C) is more common (Brain 2002;125:1070)
Sites
- Central and peripheral nervous system
- Cerebellum
- Putamen
- Pons
- Substantia nigra
- Spinal cord (intermediolateral column neurons)
- Musculoskeletal
- Atrophy of the posterior cricoarytenoid muscles
Pathophysiology
- Unknown
- Hypotheses of alpha-synuclein accumulation in oligodendroglia include SNCA gene reactivation in oligodendroglia and neuron oligodendroglia protein transfer (Acta Neuropathol Commun 2019;7:113)
Etiology
- Unclear
- Predominantly sporadic, although rare familial cases reported (Arch Neurol 2007;64:545, N Engl J Med 2013;369:233)
- Some gene variants, including FBXO47, ELOVL7, EDN1 and MAPT, are associated with increased risk for MSA (Neurology 2016;87:1591)
Clinical features
- 2 clinical subtypes: parkinsonism predominant (MSA-P) and cerebellar ataxia predominant (MSA-C)
Diagnosis
- There are 4 diagnostic categories of MSA: neuropathologically established, clinically established, clinically probable and possible prodromal (Mov Disord 2022;37:1131)
- Neuropathologic diagnosis can only be made by postmortem examination
- Widespread alpha-synuclein positive GCIs in association with neurodegenerative changes in nigrostriatal or olivopontocerebellar structures
- Clinically established diagnosis requires a sporadic, progressive, adult onset disease with
- Autonomic dysfunction
- Either cerebellar ataxia syndrome or levodopa unresponsive parkinsonism
- 2 supportive clinical features (features include rapid disease progression, postural instability, levodopa induced craniocervical dystonia, severe dysphagia, Babinski sign, postural deformities, stridor and pathologic laughter / crying)
- 1 MRI marker (markers include atrophy of cerebellum, middle cerebellar peduncle, pons or putamen; hot cross bun sign; increased diffusivity of putamen or middle cerebellar peduncle)
- Clinically probable diagnosis requires a sporadic, progressive, adult onset disease with
- At least 2 of the following: parkinsonism, cerebellar ataxia or autonomic dysfunction
- 1 supportive clinical feature (same as listed above)
- Possible prodromal diagnosis requires a sporadic, progressive, adult onset disease with
- At least 1 clinical nonmotor entry criteria (REM sleep behavior disorder, urogenital failure or neurogenic orthostatic hypotension)
- At least 1 clinical motor entry criteria (subtle parkinsonian signs, subtle cerebellar signs)
Radiology description
- Atrophy of putamen and signal decrease on iron sensitive sequences
- Atrophy of cerebellum, pons and middle cerebellar peduncle
- Hot cross bun sign (T2 hyperintense signal in axial MRI of pons; thought to be due to gliosis of pontocerebellar fibers)
- Increased diffusivity of putamen and middle cerebral peduncle (J Mov Disord 2018;11:107)
Radiology images
Images hosted on other servers:
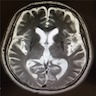
Bilateral putaminal atrophy
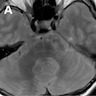
Hot cross bun sign
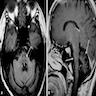
Hot cross bun sign and cerebellopontine atrophy
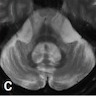
MRI findings
Prognostic factors
- Average survival time is 7 - 9 years from diagnosis (J Neurol Neurosurg Psychiatry 2007;78:327, Mov Disord 2008;23:294)
- Patients with disease onset before age 40 may have longer survival (Mov Disord 2018;33:1099)
- Risk factors for shorter survival include MSA-P variant, female sex and early or severe autonomic dysfunction (Arch Neurol 2007;64:256, Med Welt 1975;26:1957)
Case reports
- 31 year old man carrying LRRK2 G2019S mutation and presenting with REM behavior disorder who was diagnosed with neuropathologically confirmed multiple system atrophy at autopsy (Mov Disord 2019;34:1080)
- 66 year old woman with 4 year history of MSA and sudden death (Intern Med 2019;58:1643)
- 70 year old man with parkinsonism and autonomic dysfunction (Acta Neuropathol 2019;137:167)
- 6 patients (37 - 66 years old) presented with severe urinary retention, bowel and sexual dysfunction and abnormal anal sphincter electromyelography (EMG) (J Neurol 2020;267:659)
Treatment
- No disease modifying or neuroprotective therapies are available
- Treatment is aimed at symptomatic relief, with physical therapy and occupational therapy as mainstay
- Autonomic dysfunction is treated similar to other neurogenic autonomic dysfunction
- Parkinsonism symptoms are generally poorly responsive to levodopa but ~33% of patients will have transient clinical benefit (Mov Disord 2007;22:2141)
Clinical images
Images hosted on other servers:
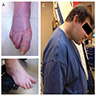
Clinical signs of MSA-C
Gross description
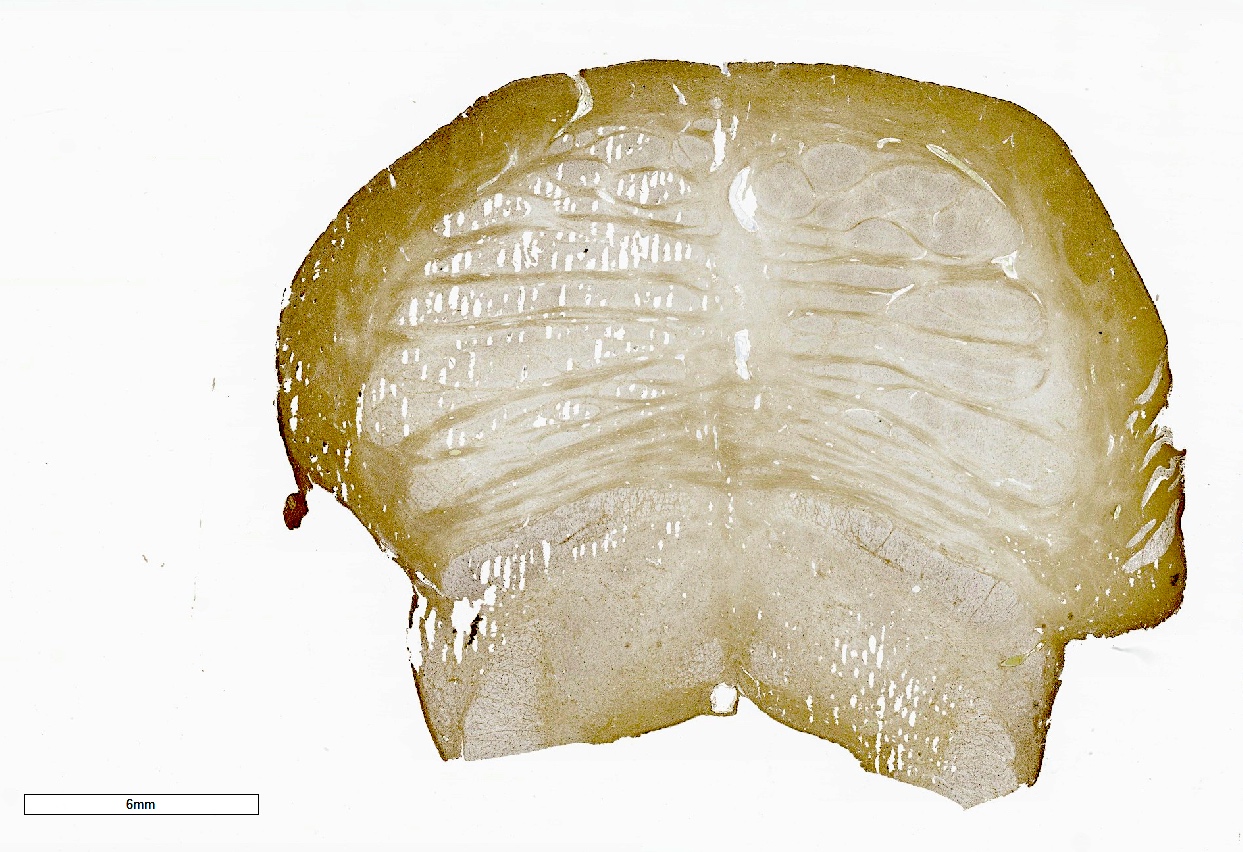
- Atrophy of the cerebellum, middle cerebellar peduncles and pons
- Pallor of the substantia nigra and locus coeruleus
- Atrophy and gray-brown discoloration of the putamen (Brain Pathol 1999;9:721)
Gross images
Contributed by Emile Pinarbasi, M.D., Ph.D.

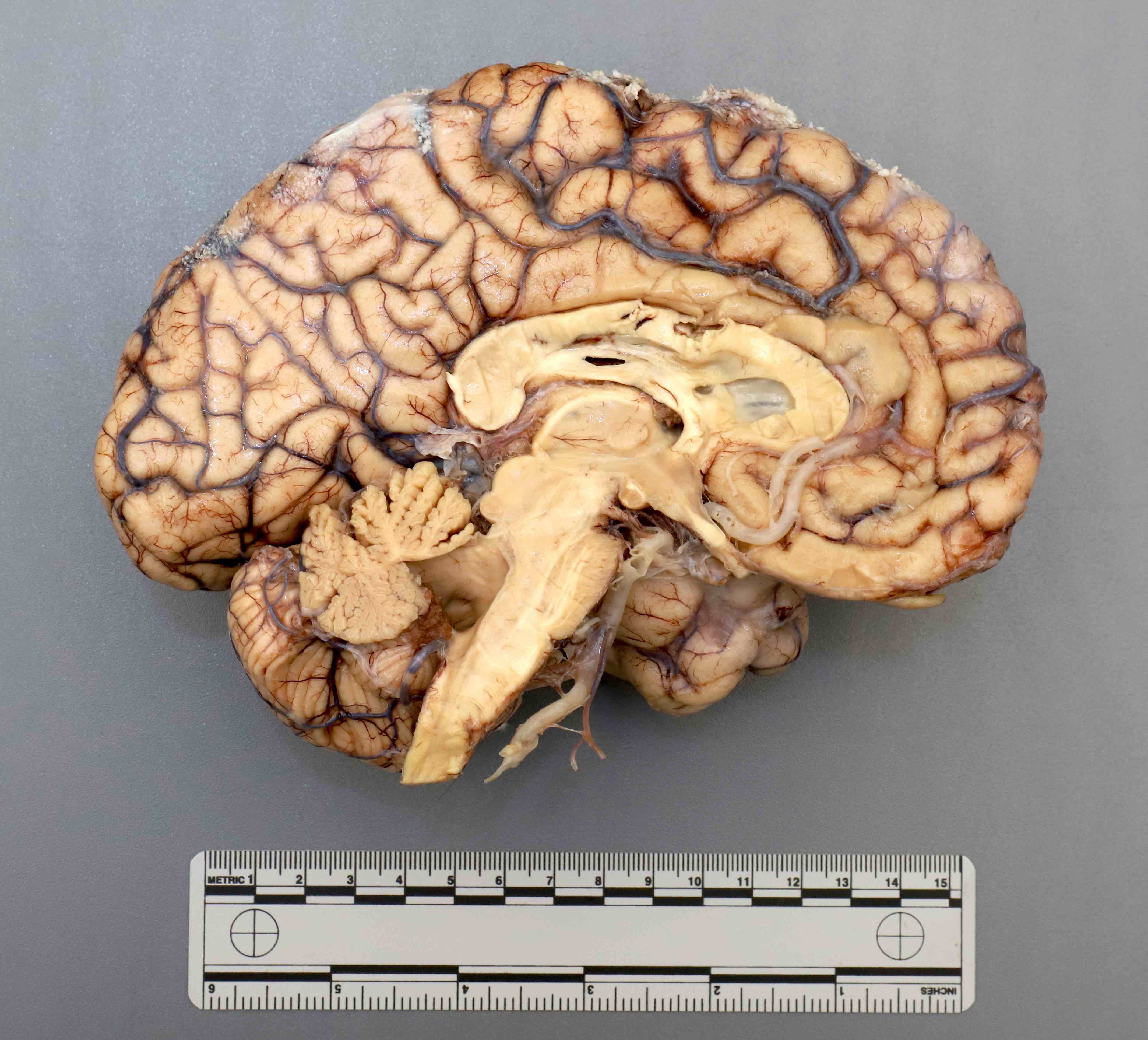
Cerebellar atrophy
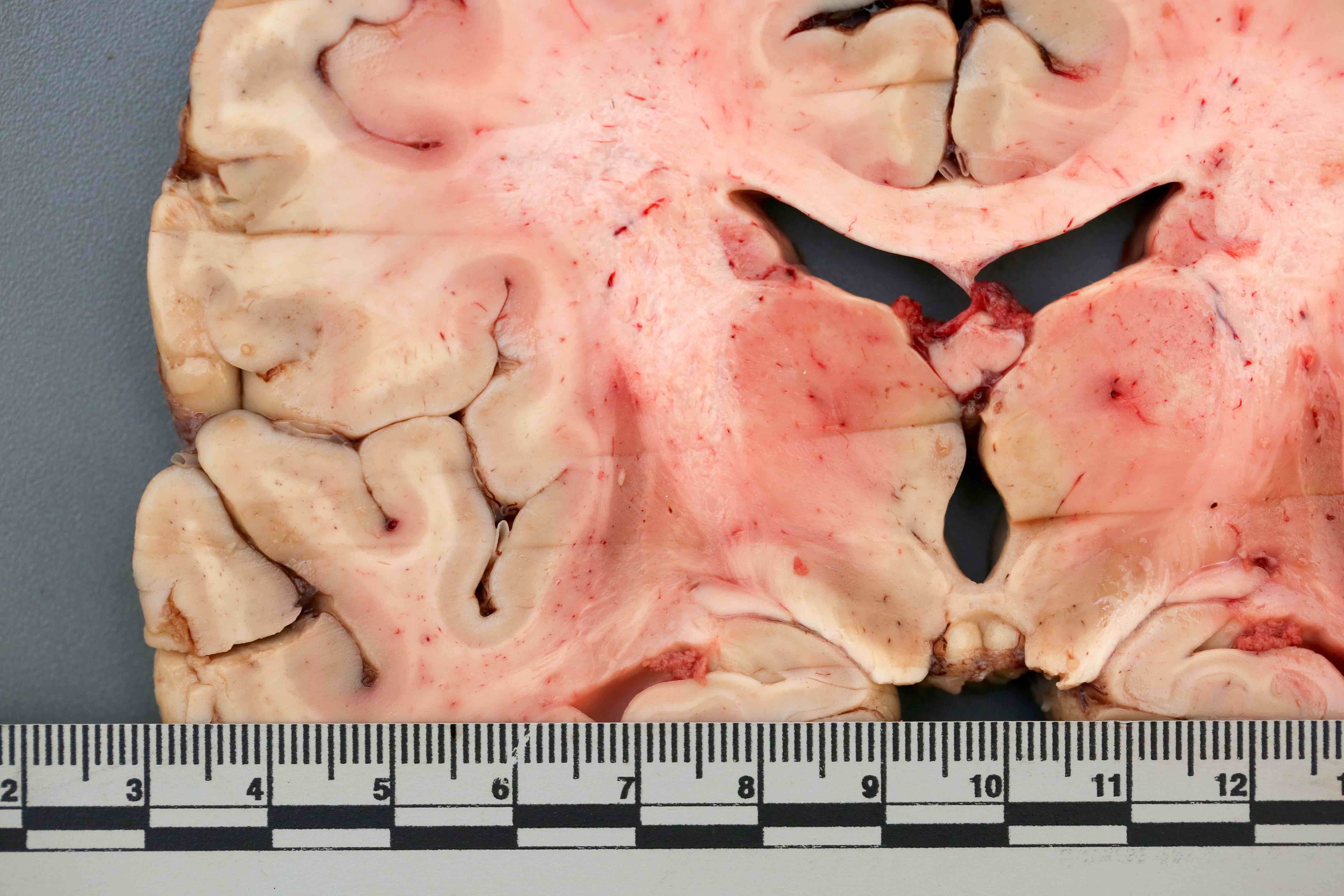
Discoloration of the putamen

Depigmentation of substantia nigra
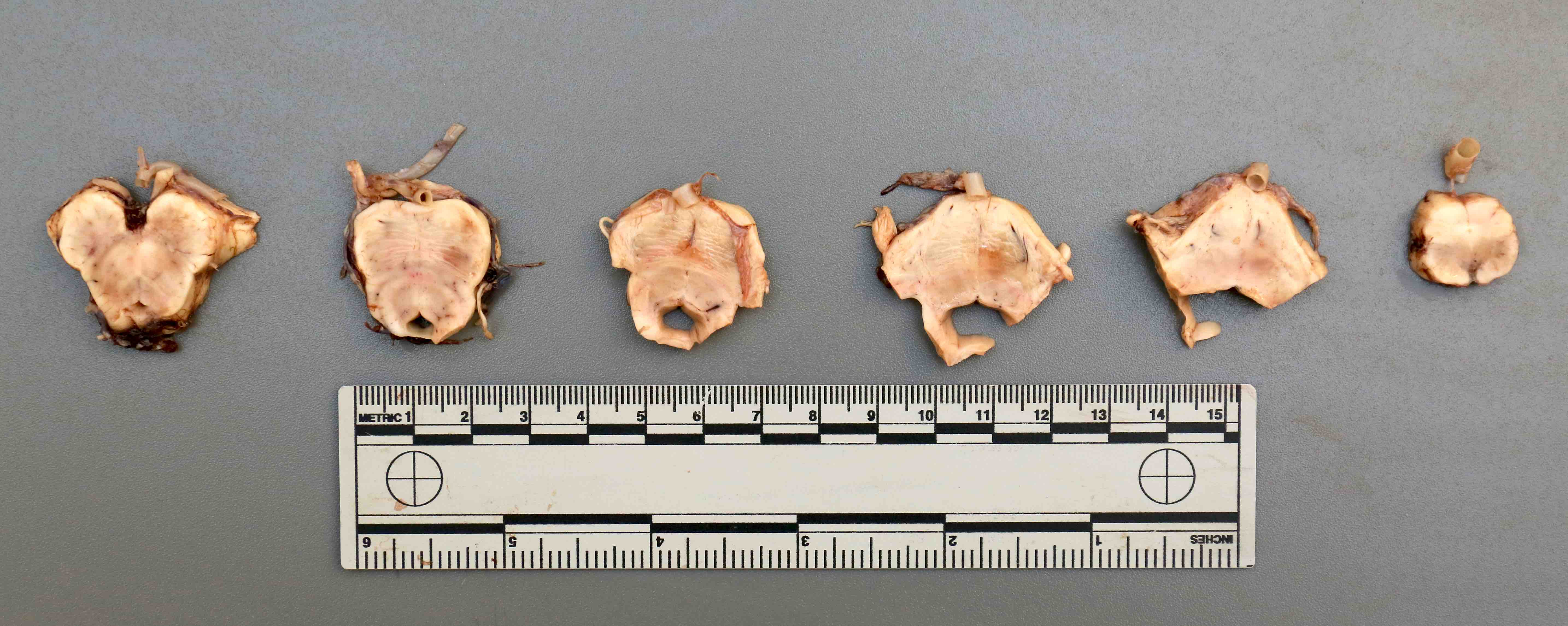
Pontine atrophy
Microscopic (histologic) description
- Diagnostic criteria (Neuropathol Appl Neurobiol 2007;33:615)
- Widespread abundant CNS alpha-synuclein positive GCIs
- Neurodegenerative changes (atrophy, gliosis) in striatonigral or olivopontocerebellar structures
- GCIs are in oligodendrocytes (in gray and white matter) and can be flame-like, triangular or sickle shaped
- GCIs are composed of alpha-synuclein
- GCIs can also be seen with Gallyas and other silver stains and are immunoreactive for ubiquitin and alpha B crystallin
- Other alpha-synuclein inclusions, including intraneuronal inclusions (nuclear and cytoplasmic) and neuropil threads can also be seen, particularly in the pons and putamen but are not required for diagnosis
- Tau positive granular glial inclusions can be seen, primarily in the cerebellum and putamen, particularly in cases of long disease duration (J Neurol Sci 2020;416:117010)
Microscopic (histologic) images
Contributed by Emile Pinarbasi, M.D., Ph.D.
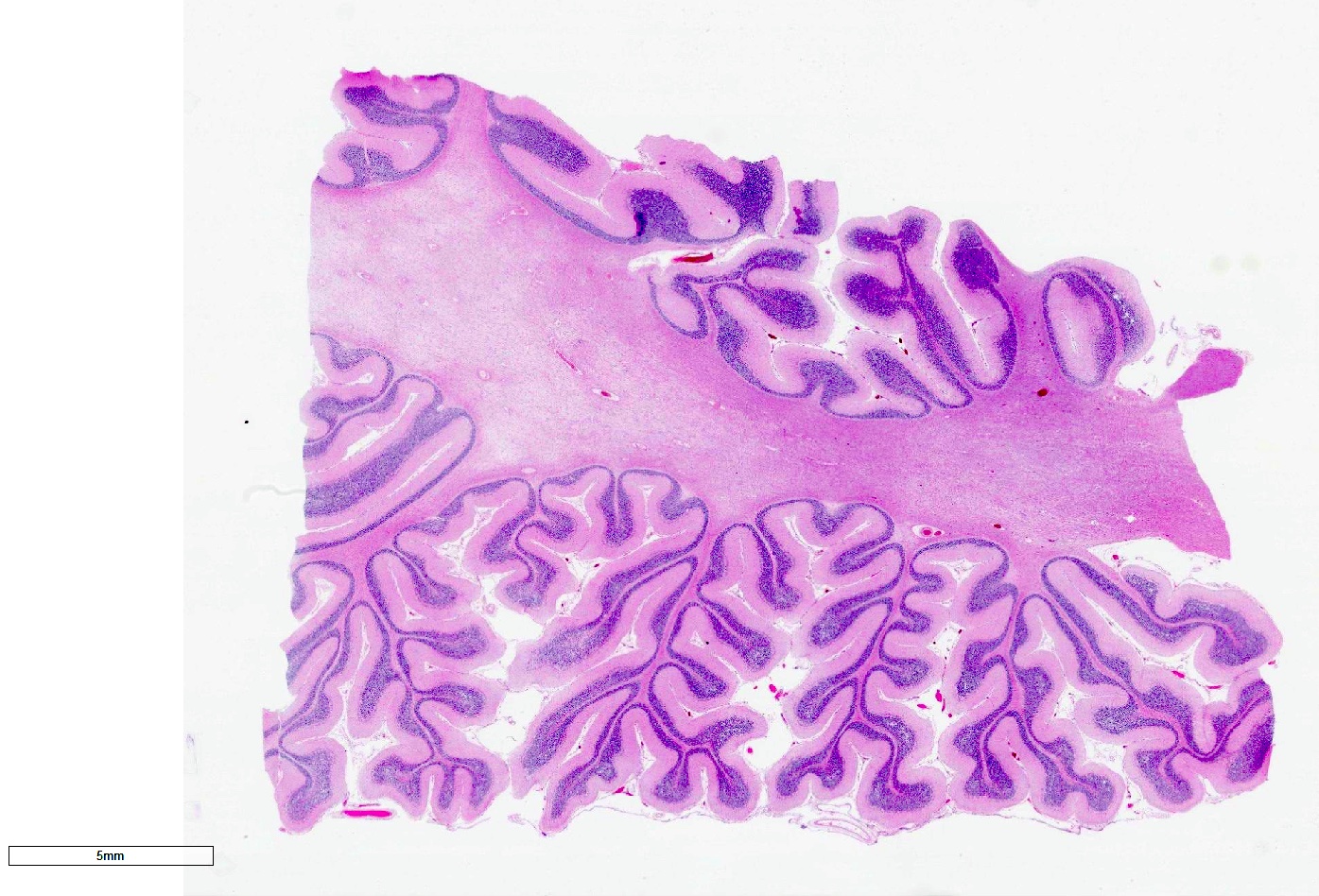
Cerebellar atrophy with prominent white matter loss
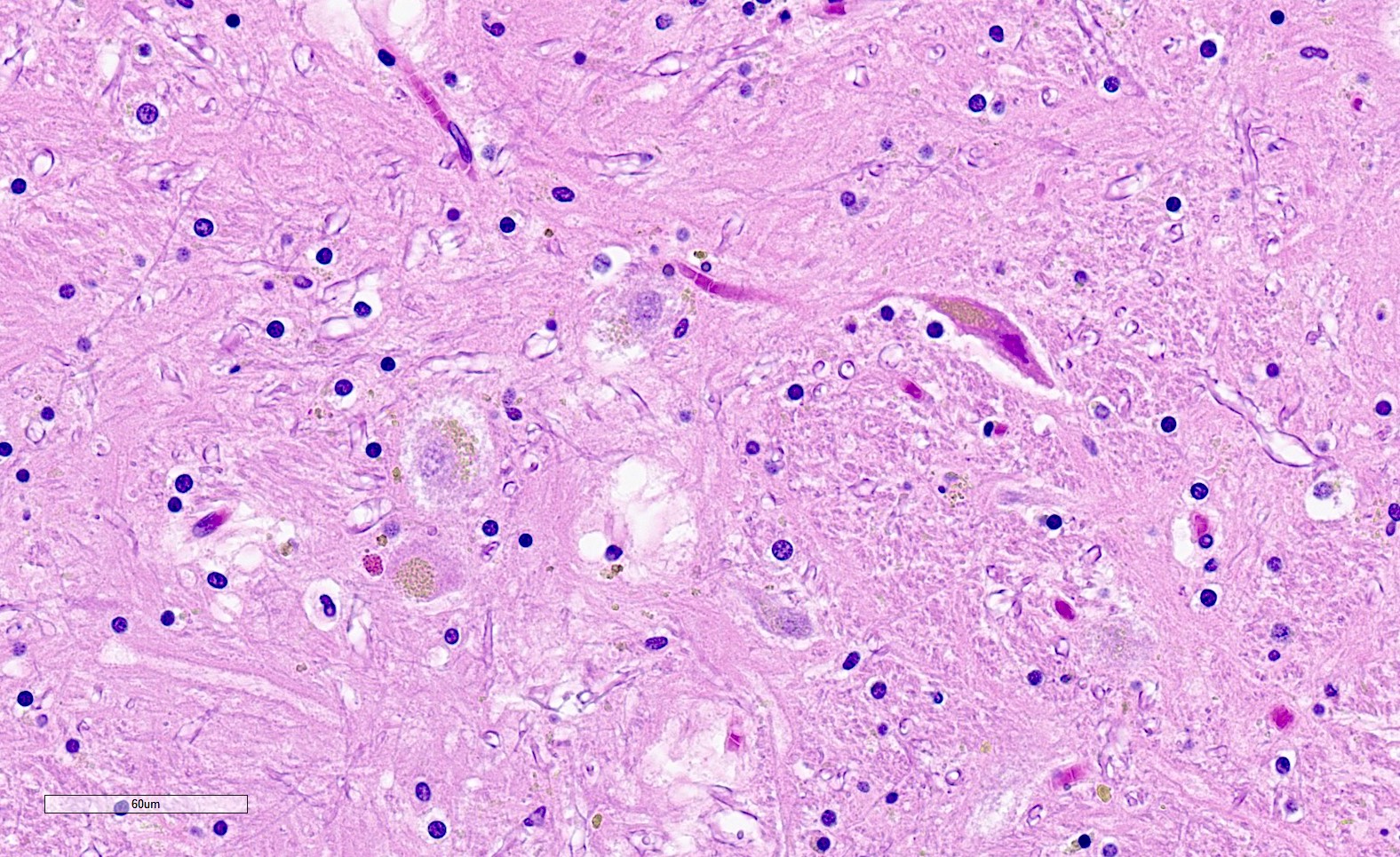
Lipofuscin accumulation in putamen
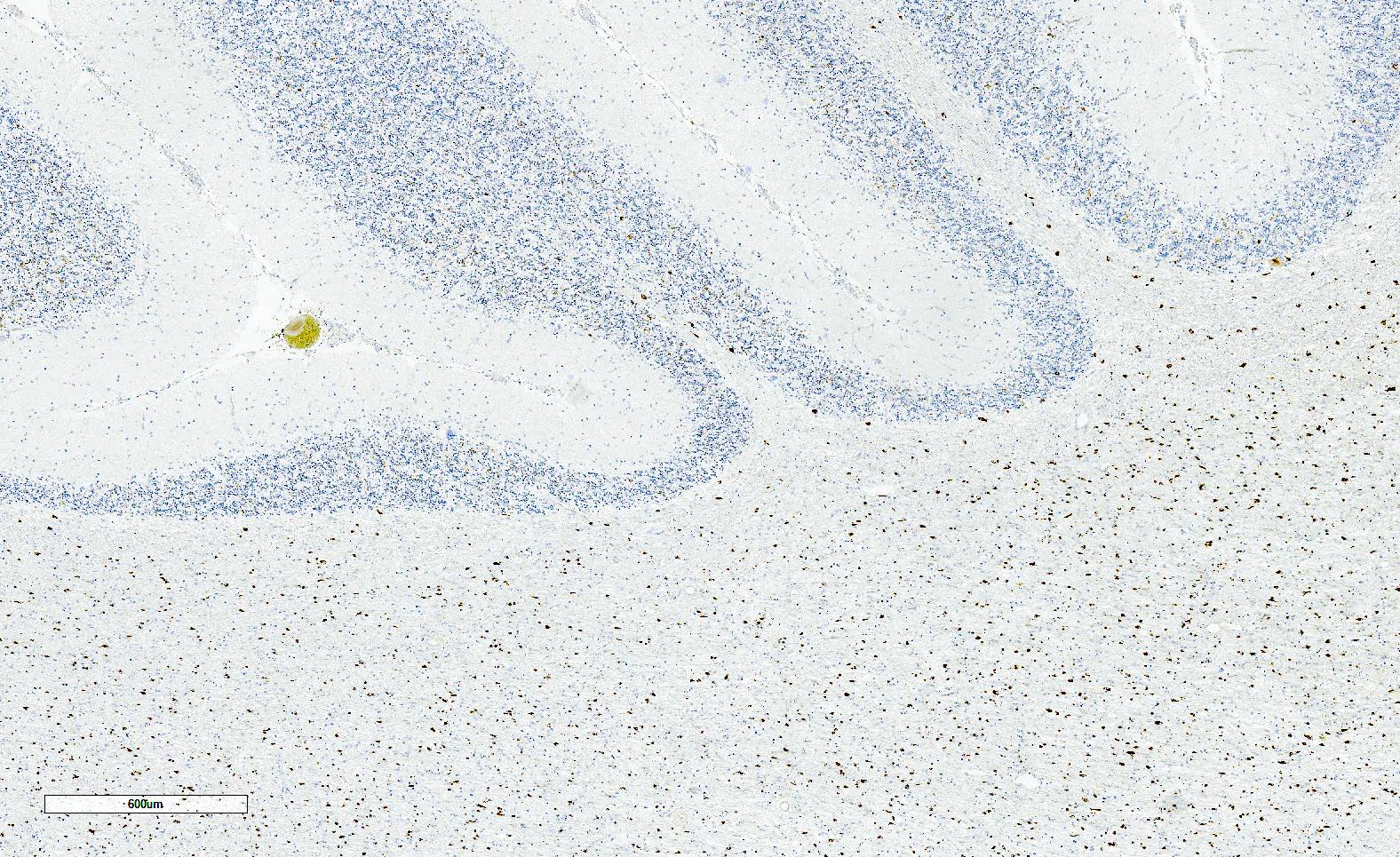
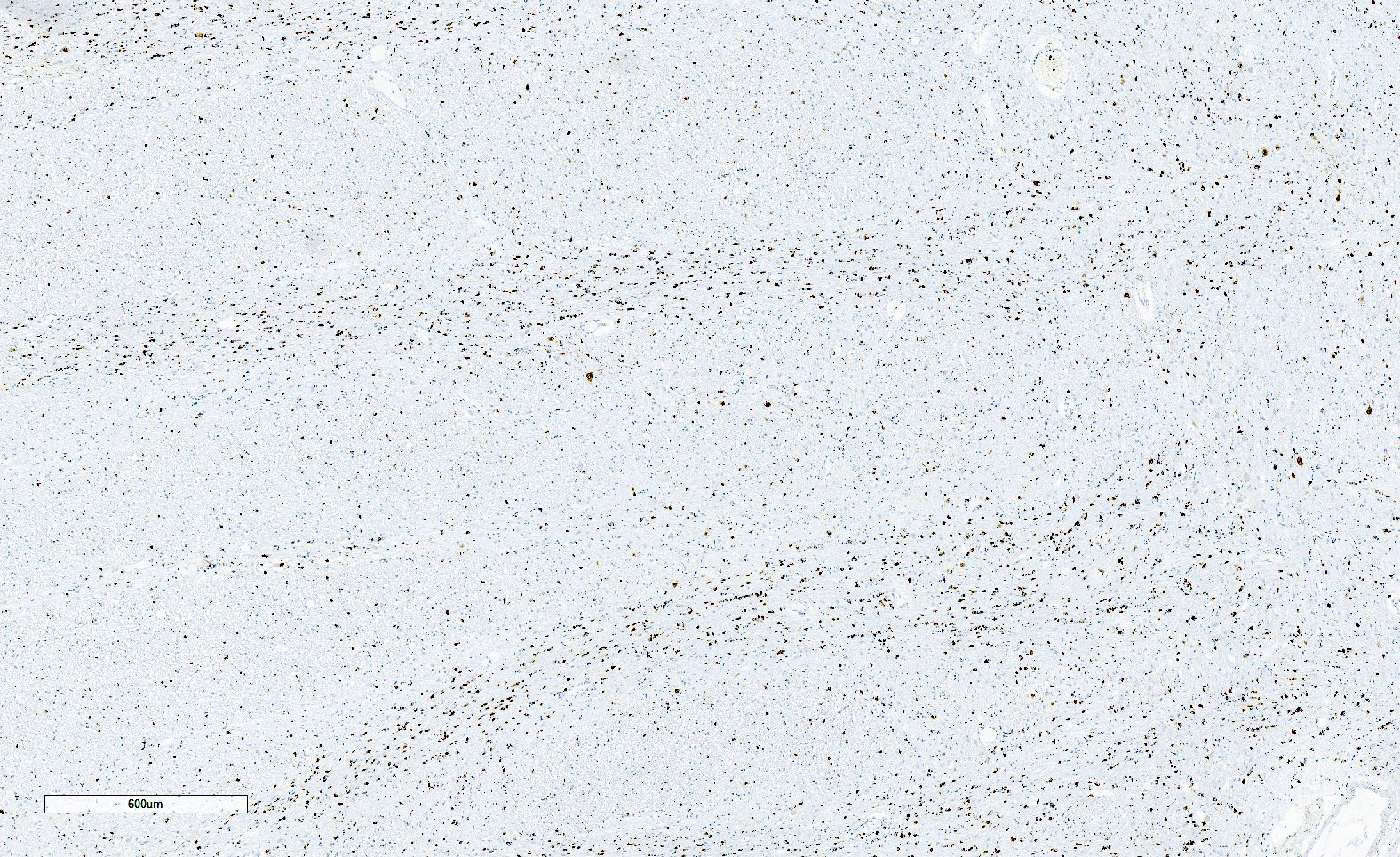
Alpha-synuclein positive GCIs
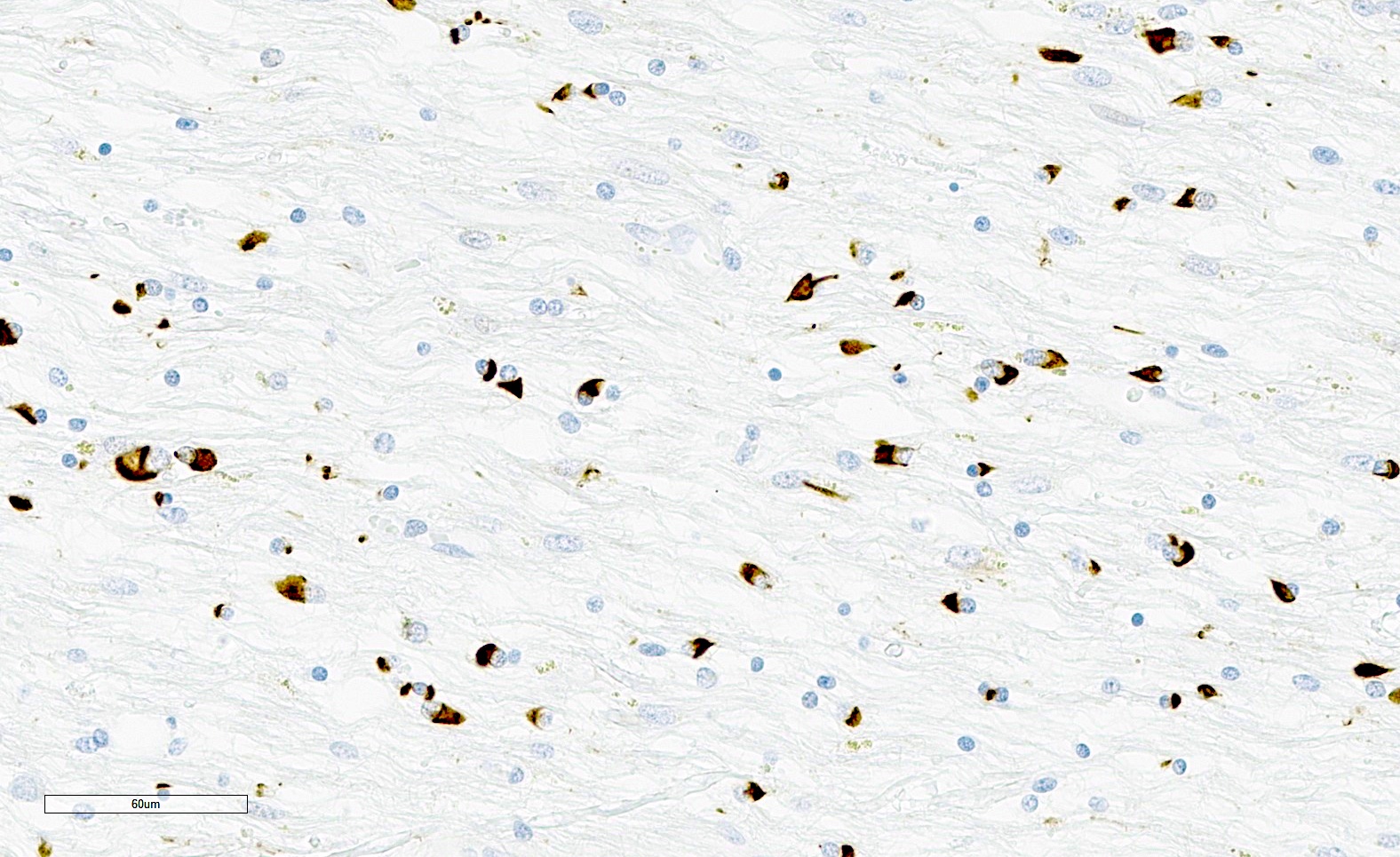
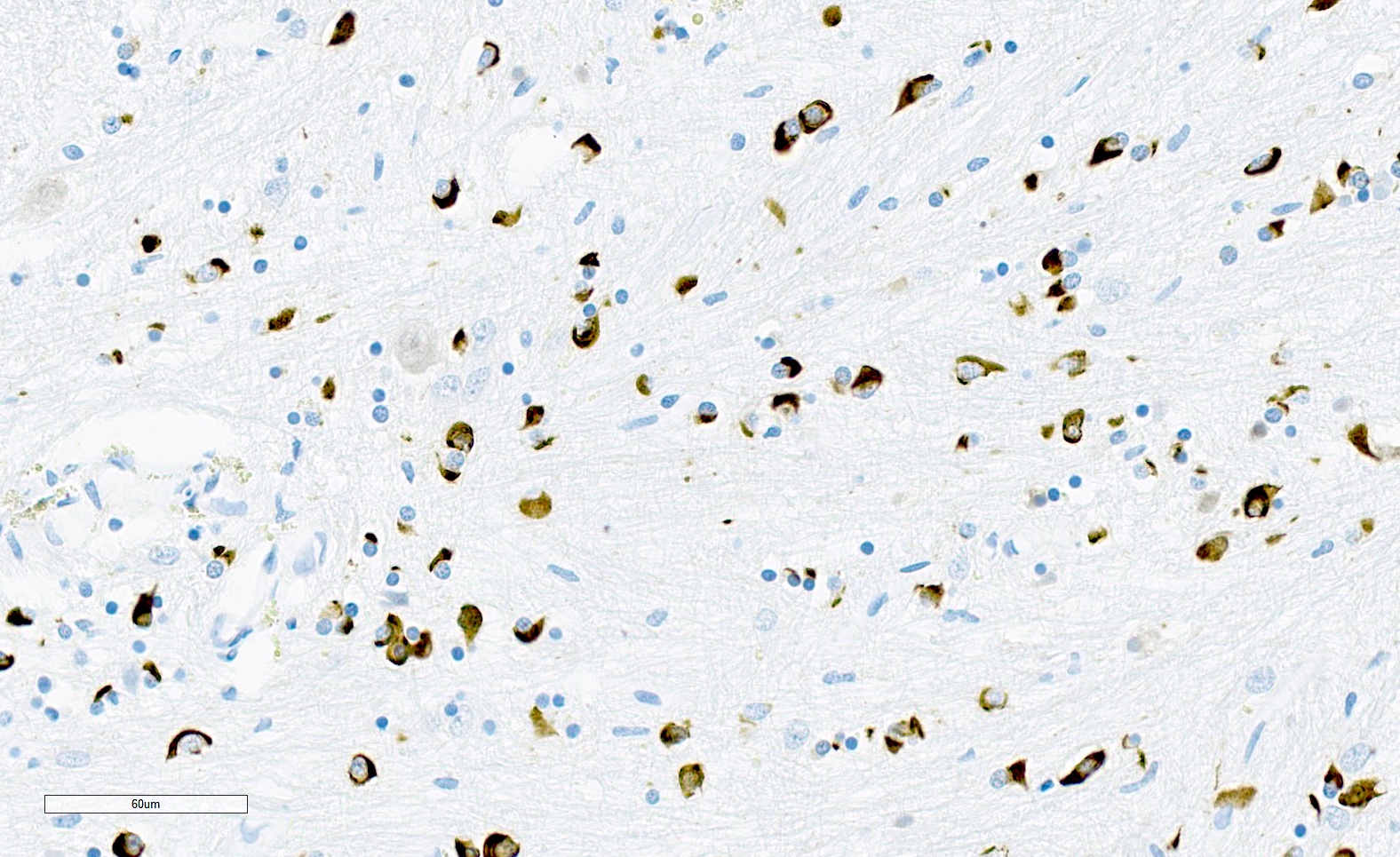
Flame and sickle shaped GCIs
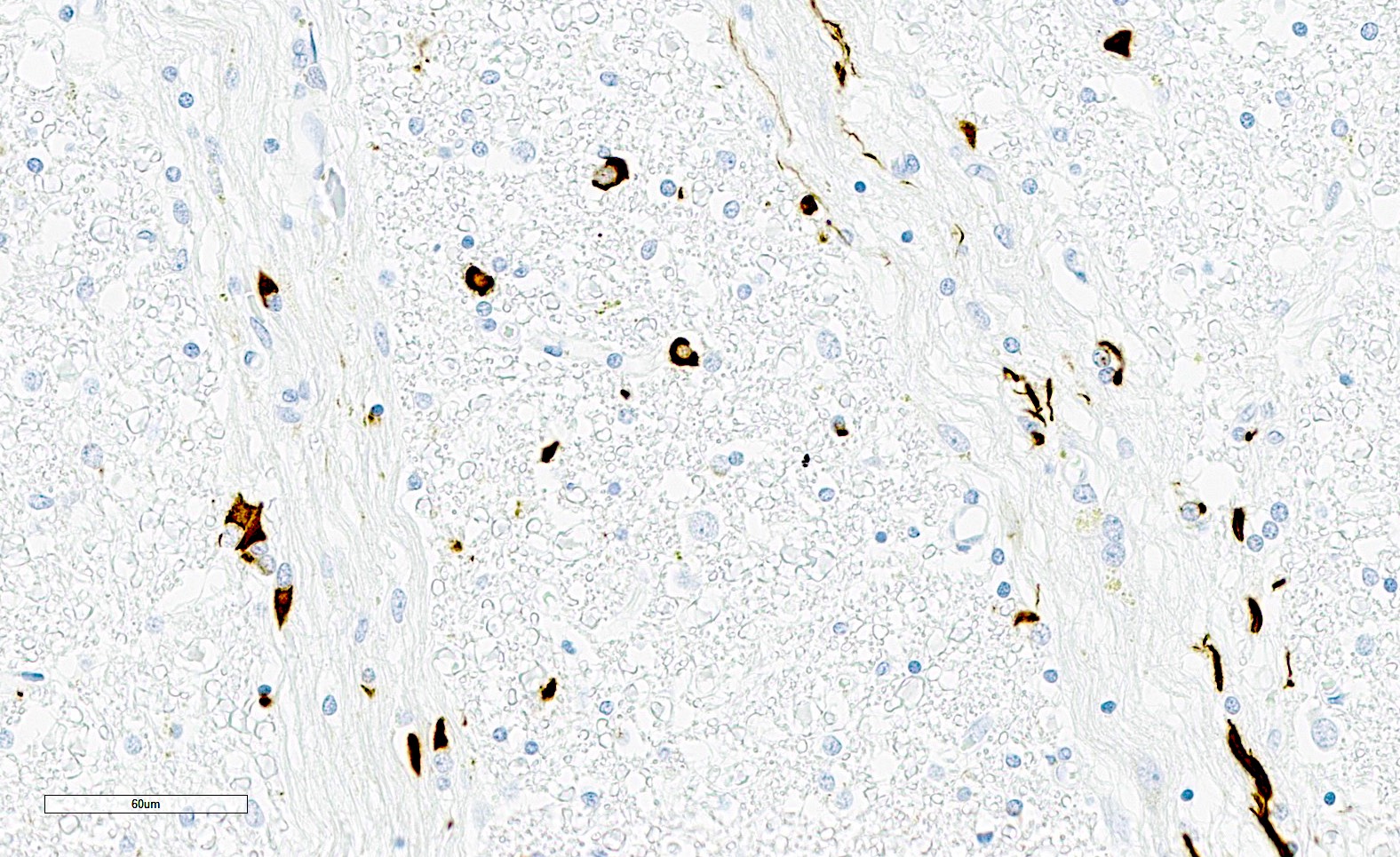
Alpha-synuclein GCIs and neuropil threads
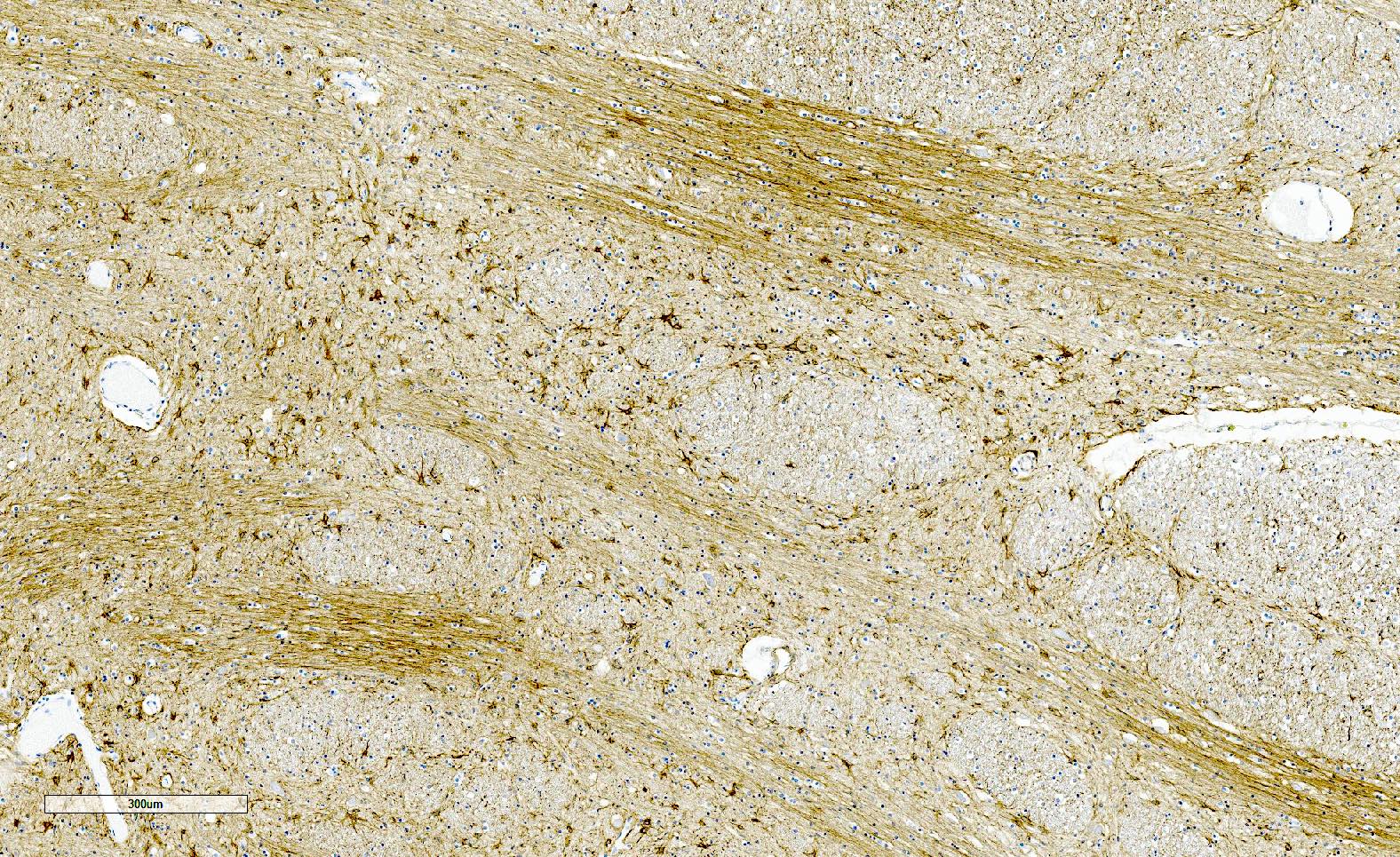
Gliosis in pontine crossing fibers
GCIs in pontine crossing fibers
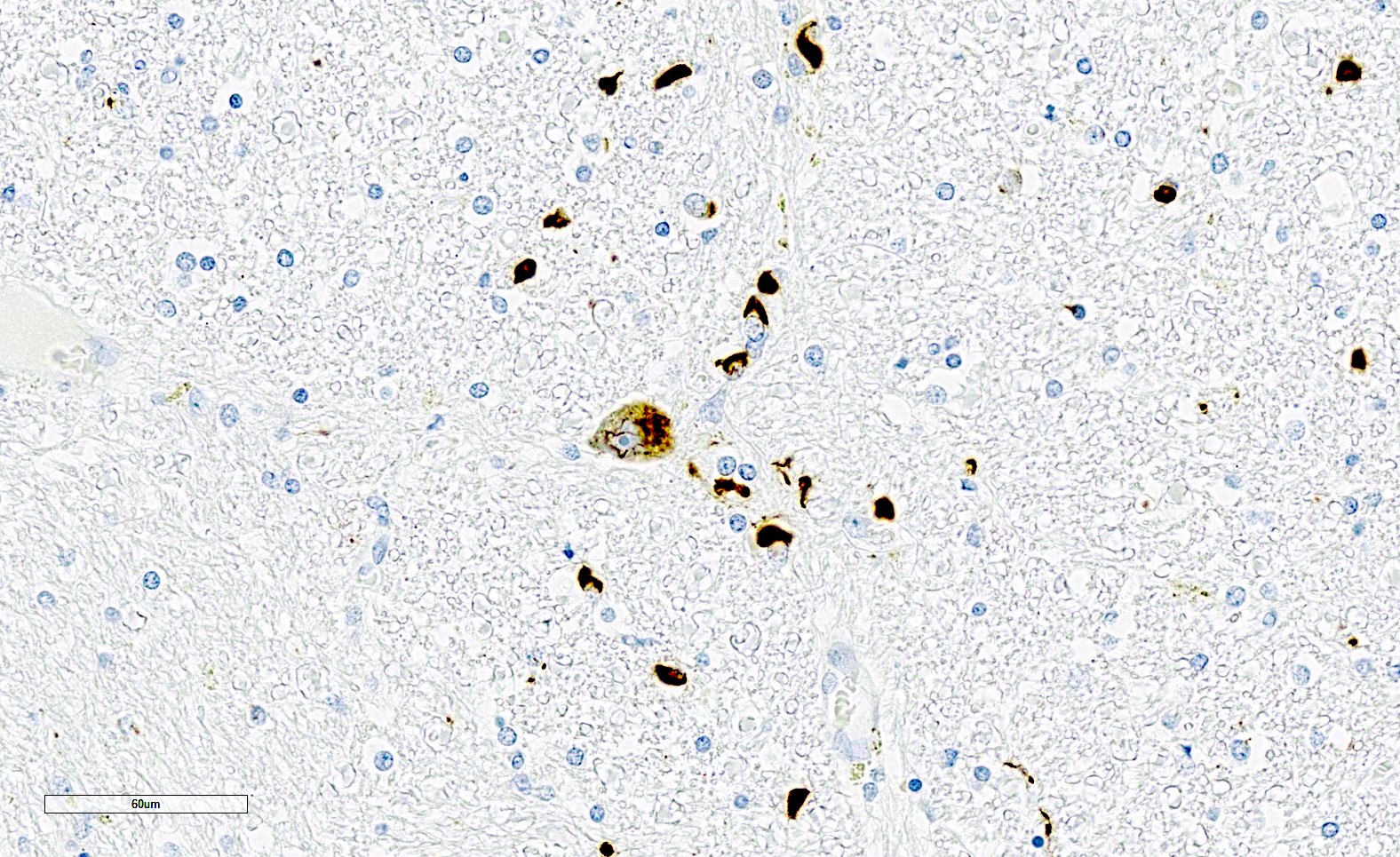
Filamentous intraneuronal inclusions
Positive stains
- Alpha-synuclein is positive in GCIs (required for diagnosis)
- GFAP highlights gliosis
- Thioflavin S and Gallyas silver stain are positive in GCIs
- References: Acta Neuropathol 2005;110:255, Neuropathol Appl Neurobiol 2007;33:615
Negative stains
- GCIs are negative for phospho-tau, amyloid beta, TDP-43 and phospho-TDP-43
Electron microscopy description
- GCIs are composed of a loose meshwork of randomly arranged, loosely packed filaments with cross sectional diameters of 15 - 30 nm
Electron microscopy images
Images hosted on other servers:
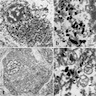
Glial cytoplasmic inclusions
Sample pathology report
- Brain, 1,410 grams, autopsy:
- Multiple system atrophy (see comment)
- Comment: This case demonstrates classic gross and microscopic findings of multiple system atrophy (MSA) with pronounced atrophy of the pons and cerebellum, depigmentation of the substantia nigra and locus coeruleus and widespread alpha-synuclein glial cytoplasmic inclusions. MSA is a rare progressive neurodegenerative disease of unknown etiology and there are no effective disease modifying or neuroprotective therapies.
Differential diagnosis
- Lewy body pathology (Parkinson disease and Lewy body dementia):
- No alpha-synuclein GCIs
- No cerebellar or pontine atrophy
- Depigmentation of substantia nigra and locus coeruleus
- Round to amorphous intraneuronal alpha-synuclein inclusions (Lewy bodies)
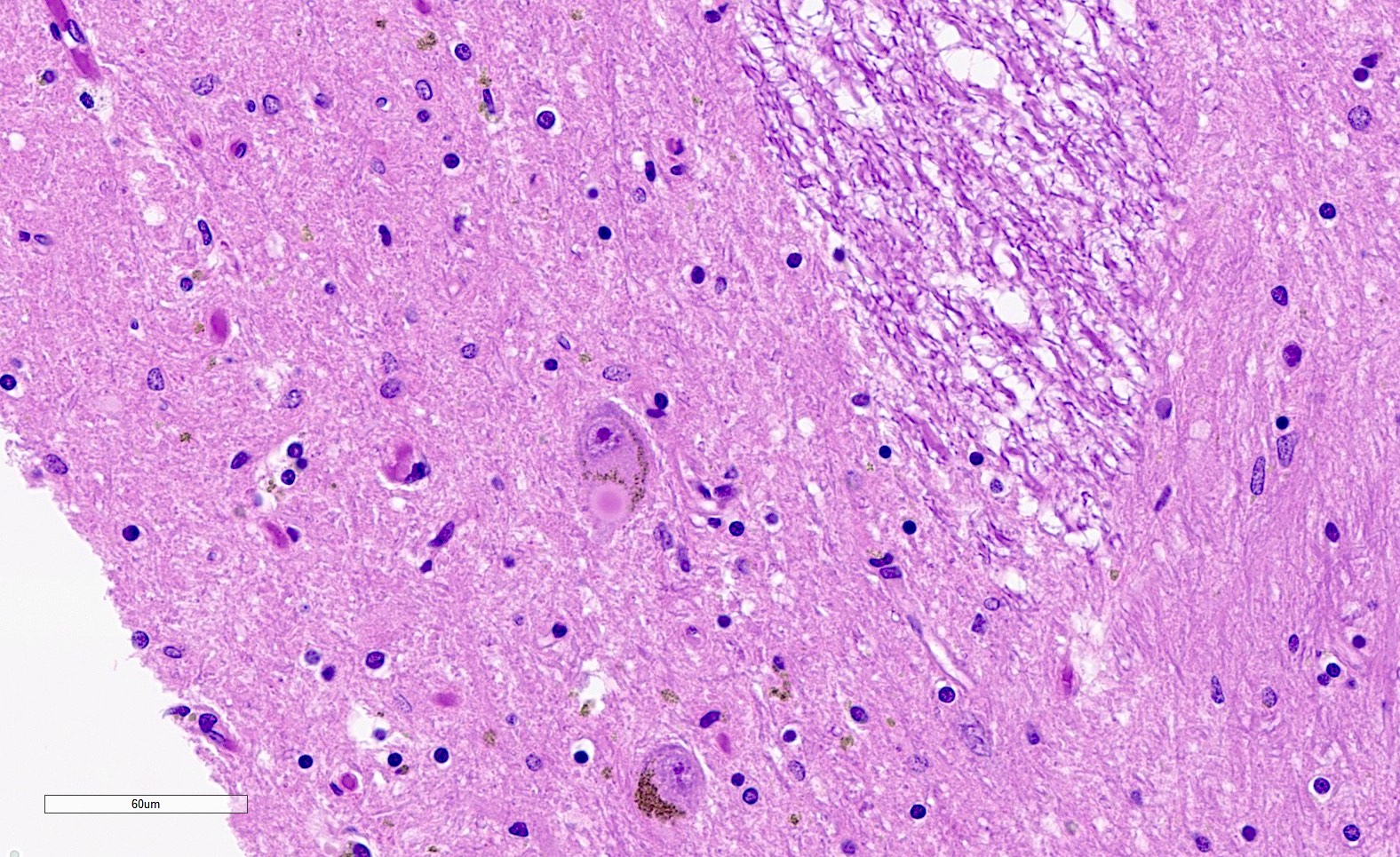
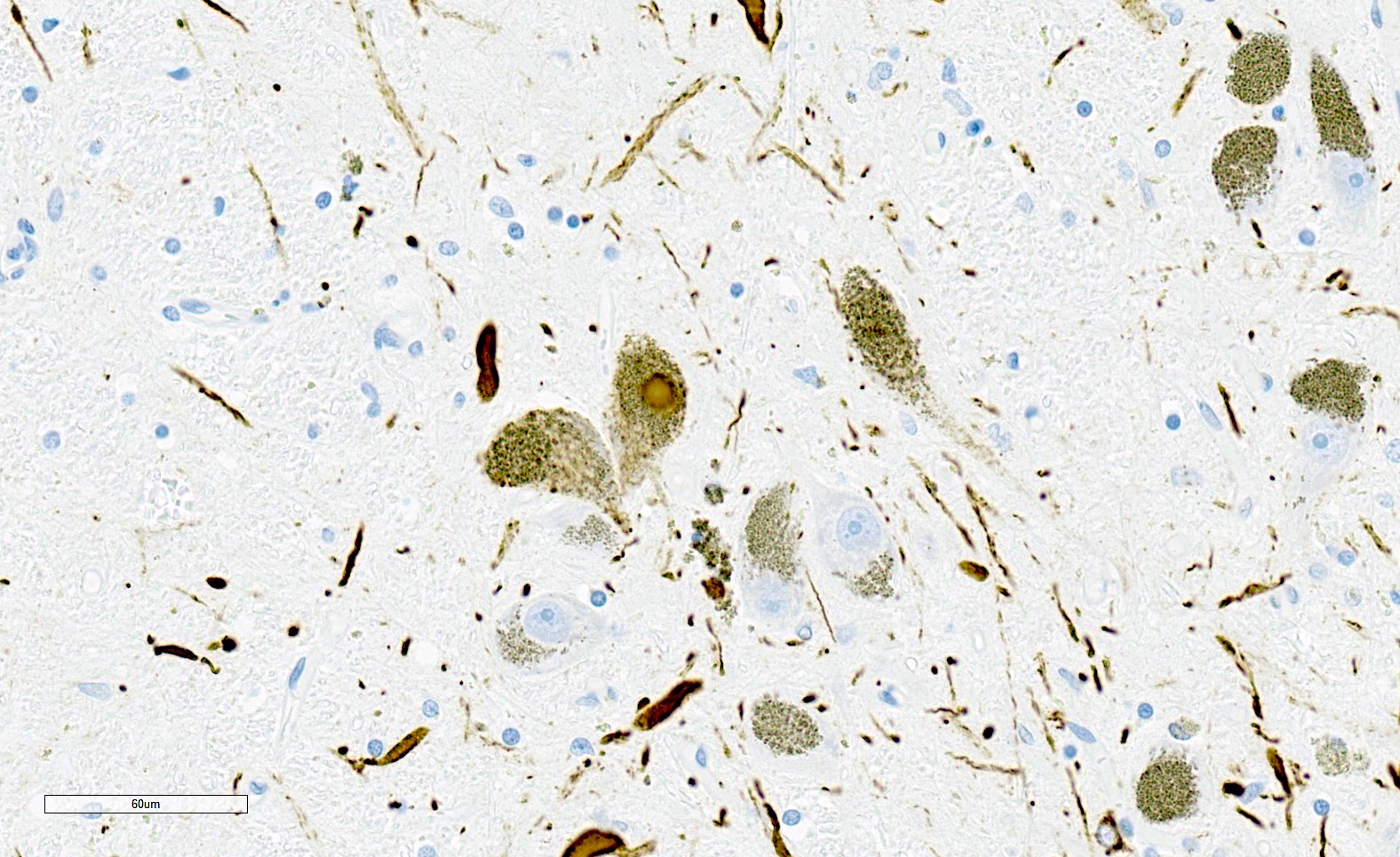
- Inherited sporadic spinocerebellar ataxias:
- No alpha-synuclein GCIs
- No depigmentation of substantia nigra and locus coeruleus
- Cerebellar atrophy
- Corticobasal degeneration:
- No alpha-synuclein GCIs
- No cerebellar pontine atrophy
- Prominent cortical and thalamic atrophy
- Depigmentation of substantia nigra and locus coeruleus
- Tau positive inclusions: astrocytic plaques and coiled bodies
- Progressive supranuclear palsy:
- No alpha-synuclein GCIs
- No cerebellar atrophy
- Prominent atrophy of frontal cortex, midbrain and pons
- Tau positive inclusions: tufted astrocytes and coiled bodies
Board review style question #1
The images above are taken from the postmortem brain examination of a 72 year old man with 6 years of progressive ataxia, prominent autonomic dysfunction, bradykinesia and pill rolling tremor. Microscopic examination shows widespread oligodendroglial inclusions. These inclusions are positive for which of the following?
- Alpha-synuclein
- Amyloid beta
- Phospho-tau
- Phospho-TDP-43
Board review style answer #1
A. Alpha-synuclein
The gross image shows pronounced cerebellar and pontine atrophy and the microscopic image shows frequent, widespread oligodendroglial inclusions. These features are diagnostic for multiple system atrophy (MSA). This correlates with the clinical presentation of parkinsonism, ataxia and autonomic dysfunction. The oligodendroglial inclusions in this sporadic, fatal neurodegenerative disease are composed of alpha-synuclein.
Answer B is incorrect because amyloid beta forms extracellular plaques in Alzheimer disease neuropathologic change (ADNC) and deposits in vessel walls in cerebral amyloid angiopathy (CAA). Amyloid beta does not form oligodendroglial inclusions and B amyloid inclusions are not a feature of MSA. Answer C is incorrect because phospho-tau white matter inclusions can be seen in primary tauopathies, most notably corticobasal degeneration (CBD), characterized by teeming white matter disease; however, gross CBD will not have atrophy of cerebellum and pons. Additionally, the inclusions fill processes of astrocytes, giving a characteristic bushy / spiked rather than a flame shaped appearance.
Answer D is incorrect because phospho-TDP-43 inclusions are found in frontotemporal lobar disease and amyotrophic lateral sclerosis. Frontotemporal lobar disease presents with dementia and is characterized by pronounced frontal and temporal atrophy. Amyotrophic lateral sclerosis classically presents with upper and lower motor symptoms and grossly shows atrophy of ventral nerve roots. Neither of these would be expected to show gross atrophy of the cerebellum and pons. Additionally, while phospho-TDP-43 inclusions are infrequently seen in oligodendroglia, they are far more frequent in neurons and astrocytes.
Comment Here
Reference: Multiple system atrophy
The gross image shows pronounced cerebellar and pontine atrophy and the microscopic image shows frequent, widespread oligodendroglial inclusions. These features are diagnostic for multiple system atrophy (MSA). This correlates with the clinical presentation of parkinsonism, ataxia and autonomic dysfunction. The oligodendroglial inclusions in this sporadic, fatal neurodegenerative disease are composed of alpha-synuclein.
Answer B is incorrect because amyloid beta forms extracellular plaques in Alzheimer disease neuropathologic change (ADNC) and deposits in vessel walls in cerebral amyloid angiopathy (CAA). Amyloid beta does not form oligodendroglial inclusions and B amyloid inclusions are not a feature of MSA. Answer C is incorrect because phospho-tau white matter inclusions can be seen in primary tauopathies, most notably corticobasal degeneration (CBD), characterized by teeming white matter disease; however, gross CBD will not have atrophy of cerebellum and pons. Additionally, the inclusions fill processes of astrocytes, giving a characteristic bushy / spiked rather than a flame shaped appearance.
Answer D is incorrect because phospho-TDP-43 inclusions are found in frontotemporal lobar disease and amyotrophic lateral sclerosis. Frontotemporal lobar disease presents with dementia and is characterized by pronounced frontal and temporal atrophy. Amyotrophic lateral sclerosis classically presents with upper and lower motor symptoms and grossly shows atrophy of ventral nerve roots. Neither of these would be expected to show gross atrophy of the cerebellum and pons. Additionally, while phospho-TDP-43 inclusions are infrequently seen in oligodendroglia, they are far more frequent in neurons and astrocytes.
Comment Here
Reference: Multiple system atrophy
Board review style question #2
A 62 year old woman with a 5 year history of parkinsonism, ataxia and orthostatic hypotension dies of respiratory failure. Postmortem brain examination reveals pronounced cerebellar atrophy and widespread alpha-synuclein positive oligodendroglial inclusions. Which of the following regarding this entity is true?
- Amyloid plaques are a characteristic microscopic finding
- Gross features include depigmentation of the substantia nigra
- Prominent cortical atrophy is a common feature
- The diagnostic alpha-synuclein inclusions shown above are also known as Lewy bodies
Board review style answer #2
B. Gross features include depigmentation of the substantia nigra
The combination of cerebellar atrophy and frequent widespread oligodendroglial alpha-synuclein inclusions is diagnostic of multiple system atrophy. This correlates with the clinical presentation of parkinsonism, ataxia and autonomic dysfunction. One of the common gross findings in multiple system atrophy is depigmentation of the substantia nigra.
The diagnostic alpha-synuclein inclusions in multiple system atrophy are known as glial cytoplasmic inclusions (GCIs). Answer D is incorrect because Lewy bodies are round alpha-synuclein positive intraneuronal inclusions seen in Lewy body pathology (Parkinson disease and Lewy body dementia). Answer C is incorrect because cortical atrophy is not a gross feature of multiple system atrophy. Answer A is incorrect because amyloid plaques are a histologic finding in Alzheimer disease and are not seen in multiple system atrophy.
Comment Here
Reference: Multiple system atrophy
The combination of cerebellar atrophy and frequent widespread oligodendroglial alpha-synuclein inclusions is diagnostic of multiple system atrophy. This correlates with the clinical presentation of parkinsonism, ataxia and autonomic dysfunction. One of the common gross findings in multiple system atrophy is depigmentation of the substantia nigra.
The diagnostic alpha-synuclein inclusions in multiple system atrophy are known as glial cytoplasmic inclusions (GCIs). Answer D is incorrect because Lewy bodies are round alpha-synuclein positive intraneuronal inclusions seen in Lewy body pathology (Parkinson disease and Lewy body dementia). Answer C is incorrect because cortical atrophy is not a gross feature of multiple system atrophy. Answer A is incorrect because amyloid plaques are a histologic finding in Alzheimer disease and are not seen in multiple system atrophy.
Comment Here
Reference: Multiple system atrophy
