
CNS nontumor
CNS myelin disorders
Leukodystrophies
Author: Jesse L. Kresak, M.D.
Editorial Board Member: Maria Martinez-Lage, M.D.

Last author update: 1 June 2015
Last staff update: 7 December 2020
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Leukodystrophies CNS

Table of Contents
Definition / general | Pathophysiology | Clinical features | Radiology description | Radiology images | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy images | Differential diagnosis | Additional referencesCite this page: Kresak J. Leukodystrophies. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnsleukodystrophies.html. Accessed March 31st, 2025.
Definition / general
- Leukodystrophy generally refers to a genetic disorder that affects white matter
- Most often present in children, can occur in adulthood
- Demyelination often progresses in occipital to frontal manner and there is often sparing of the U fibers (short association fibers, Wikipedia: Association Fiber)
- Can result from a wide range of genetic defects involving formation, maintenance and breakdown of myelin
- Most often classified based on etiology: peroxisomal, lysosomal and other
- Peroxisomal: adrenoleukodystrophy and neonatal adrenoleukodystrophy
- Lysosomal: Krabbe disease and metachromatic leukodystrophy
- Other: Alexander disease, Canavan disease, vanishing white matter disease, others
Pathophysiology
- Peroxisomal:
- Adrenoleukodystrophy: X linked, single enzyme defect in ATP binding transporter, leads to reduced capacity to form coenzyme A derivative of very long chain fatty acids
- Neonatal adrenoleukodystrophy: autosomal recessive, defective peroxisome assembly leads to decreased numbers of peroxisomes
- Lysosomal:
- Krabbe disease: autosomal recessive, galactocerebroside β-galactosidase deficiency
- Metachromatic leukodystrophy: autosomal recessive (rarely autosomal dominant), aryl sulfatase A deficiency, rarely SAP-1
- Other:
- Alexander disease: sporadic, gain of function mutation of GFAP leads to Rosenthal fiber accumulation
- Canavan disease: autosomal recessive, defective aspartoacylase activity
- Vanishing white matter disease: autosomal recessive, mutation of genes involved in translation of factor EIF-2B
Clinical features
- Varies depending on type and age at presentation
- In infancy: often includes motor disabilities, macrocephaly, seizures or spasticity and developmental failure
- In childhood: ataxia, vision changes, behavioral / educational issues
- In adulthood: behavioral changes, psychosis, spasticity
Radiology description
- MRI usually shows T2 hyperintense lesions in white matter, typically posterior greater than frontal (Mol Genet Metab 2015;114:501)
Radiology images
Images hosted on other servers:
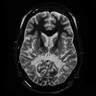
Marked loss of posterior white matter
Case reports
- 8 month old infant with multiple cranial nerve enhancement in early infantile Krabbe disease (J Neuroimaging 2010;20:195)
- 6 year old boy with cerebral X linked adrenoleukodystrophy (Acta Radiol Open 2015;4:2047981615573655)
- 38 year old woman with adult cerebral adrenoleukodystrophy and Addison disease (Gene 2014;544:248)
- Late onset Alexander disease presenting as cerebellar ataxia (J Neurol 2011;258:938)
Treatment
- Supportive
- Allogeneic haematopoietic cell transplantation (HCT) in early stage cerebral adrenoleukodystrophy may halt progression (Curr Neurol Neurosci Rep 2014;14:486)
Gross description
- Coronal autopsy specimens often show unaffected gray matter, spared subcortical U fibers and either firm (adrenoleukodystrophy), chalky white matter (Krabbe, metachromatic leukodystrophy) or markedly softened white matter (vanishing white matter disease, Canavan disease)
Gross images
Images hosted on other servers:
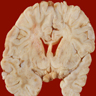
Krabbe disease
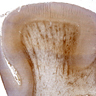
Vanishing white matter disease
Microscopic (histologic) description
- Peroxisomal:
- Adrenoleukodystrophy: severe demyelination in cerebral white matter, optic nerves and internal capsule, yet sparing of U fibers
- May see lesions of different ages: new lesions with large aggregates of macrophages with demyelination and perivascular lymphoid aggregates; older lesions with gliotic scar and no macrophage activity
- Adrenal glands show enlarged "ballooned" eosinophilic cells with striated cytoplasm
- Lysosomal:
- Krabbe disease: pathognomonic PAS+ "globoid macrophages" (may be multinucleated giant cells), extensive myelin and oligodendrocyte loss, reactive astrocytic gliosis
- Metachromatic leukodystrophy: accumulation of PAS and Luxol fast blue / LFB+ macrophages that show brown metachromasia with acidified cresyl violet, toluidine blue or thionine on frozen sections
- Also extensive myelin and axonal loss in white matter and corticospinal tracts
- Deposition of metachromatic sulfatides can also be seen in basal ganglia, numerous deep nuclei and peripheral nerves
- Other:
- Alexander disease: abundant Rosenthal fibers (especially perivascular, subpial and periventricular) with diffuse demyelination of white matter
- Rosenthal-like fibers can be seen in cell bodies of astrocytes
- Canavan disease: white matter vacuolation and demyelination predominantly at gray white junction; "spongiform leukodystrophy"
- Vanishing white matter disease: significant cavitation of white matter with excessive oligodendrocytes in pericavity residual white matter
- Alexander disease: abundant Rosenthal fibers (especially perivascular, subpial and periventricular) with diffuse demyelination of white matter
Microscopic (histologic) images
Images hosted on other servers:
Peroxisomal
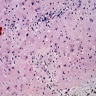
X linked adreno-leukodystrophy: gliosis and inflammation
Lysosomal:
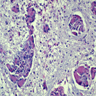
Krabbe disease: globoid cells

Metachromatic leukodystrophy: loss of myelin
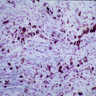
Metachromatic
leukodystrophy:
brown metachromasia
in peripheral nerve
Other:
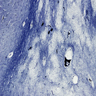
Pelizaeus-Merzbacher disease: loss
of myelin
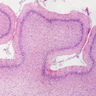
Pelizaeus-
Merzbacher
disease: cerebellar
degeneration
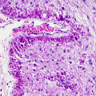
Alexander disease, Rosenthal
fibers
Canavan disease
Positive stains
- Demyelination is highlighted by loss of Luxol fast blue (LFB), often (but not always) with spared axons demonstrated by neurofilament protein and accompanied by macrophages
- Macrophages: CD68+, also positive for PAS in Krabbe and metachromatic leukodystrophy
- Reactive gliosis highlighted by GFAP
Electron microscopy images
Images hosted on other servers:
Peroxisomal:
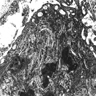
X linked adreno-leukodystrophy: trilamellar lipid products
Lysosomal:
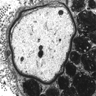
Metachromatic leukodystrophy: storage of sulfatides
Differential diagnosis
- Progressive multifocal leukoencephalopathy
- Toxic leukoencephalopathy
Additional references
