
CNS nontumor
Movement disorders
Huntington disease
Editorial Board Member: Meaghan Morris, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 8 December 2023
Last staff update: 8 December 2023
Copyright: 2020-2025, PathologyOutlines.com, Inc.
PubMed Search: Huntington disease

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Kostelecky N, Ahrendsen JT. Huntington disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnshuntingtondisease.html. Accessed March 31st, 2025.
Definition / general
- Autosomal dominant neurodegenerative disorder characterized by abnormal movements, cognitive impairment and neuropsychiatric disturbances
- Secondary to expanded CAG trinucleotide repeat within the HTT gene encoding for the huntingtin protein
Essential features
- Hyperkinetic movement disorder causing choreiform movements, rigidity, cognitive decline, dementia and neuropsychiatric disturbances
- Autosomal dominant, polyglutamine disorder characterized by trinucleotide repeat expansion (CAG)n within the HTT gene on chromosome 4 (> 39 repeats resulting in disease)
- Characteristic atrophy of the striatum, particularly the caudate nucleus
- Variable atrophy of other brain regions, though cortex is often affected
- Loss of neurons and reactive gliosis, especially within affected areas of the basal ganglia
Terminology
- Huntington chorea
Epidemiology
- Disease of low prevalence: up to 10.6 - 13.7/100,000, with geographic and ethnic variation (Eur J Neurol 2018;25:24, PLoS Curr 2010;2:RRN1184, Lancet 2007;369:218)
- Lower prevalence in East Asia
- Higher prevalence among White people
- Age of onset is dependent on number of trinucleotide repeats (Eur J Neurol 2018;25:24)
- Juvenile (< 20 years old) cases infrequent (PLoS Curr 2010;2:RRN1184)
- Westphal variant associated with bradykinetic rigid phenotype and juvenile onset (Mov Disord 1998;13:920)
Sites
- Basal ganglia, specifically the striatum (J Neuropathol Exp Neurol 1998;57:369, Mov Disord 1999;14:398)
- Less obvious effect on the cerebral cortex, hippocampus, hypothalamus, substantia nigra and cerebellar cortex (Curr Drug Targets 2005;6:43)
Pathophysiology
- Mutant huntingtin protein (mHTT) has direct and indirect downstream effects leading to neuronal loss and dysfunction (Eur J Neurol 2018;25:24)
- Abnormal intranuclear aggregation of mHTT causes secondary dysregulation of transcription
- Abnormal cytoplasmic aggregation of mHTT causes impairment of proteostasis and impairment of synaptic function, mitochondrial function / energy metabolism and axonal transport
- Striatal neurons, specifically medium spiny neurons (MSNs), are selectively vulnerable to the effects mHTT (Curr Opin Neurobiol 2015;33:53)
- Selective early loss of striatal GABA / enkephalin neurons projecting to the globus pallidus externa (GPe) correlates with chorea, while nonselective loss is more associated with akinetic rigid phenotypes (Mov Disord 1999;14:398)
Etiology
- Autosomal dominant trinucleotide CAG repeat in the huntingtin gene (HTT) on chromosome 4 leading to a mutant protein product mHTT (Eur J Neurol 2018;25:24, Curr Opin Neurobiol 2015;33:53)
- Disease is notable for variable penetrance based on the number of CAG repeats and is notable for a process called anticipation (Eur J Neurol 2018;25:24)
- < 27 repeats: normal range
- 27 - 35 repeats: intermediate number
- 36 - 39 repeats: disease with reduced penetrance
- > 39 repeats: disease with full penetrance, with greater numbers resulting in earlier disease onset
- Anticipation phenomenon
- Paternal inheritance results in greater numbers of CAG repeats in offspring and subsequent earlier disease onset with more severe symptoms
Diagrams / tables
Images hosted on other servers:
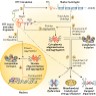
Pathogenic effects of mHTT

Clinical disease progression
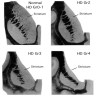
Vonsattel grading; gross pathology
Clinical features
- Disease progression can be divided into phases (Lancet 2007;369:218)
- Presymptomatic / prediagnostic phase
- Neuropsychiatric: irritability, disinhibition
- Diagnostic phase
- Hyperkinetic phenotype: prominent chorea (uncontrollable jerking / writhing movements) and dystonia (involuntary muscle contractions, often painful)
- Hypokinetic phenotypes: bradykinesia (slowness of movement), gait disturbance, imbalance
- Cognitive dysfunction: poor executive function and speech impairment
- Neuropsychiatric: depression and suicidal ideation
- Presymptomatic / prediagnostic phase
Diagnosis
- Utilization of the Unified Huntington Disease Rating Scale (UHDRS) (PLoS Curr 2010;2:RRN1184)
- Genetic testing (PLoS Curr 2010;2:RRN1184, Lancet 2007;369:218)
- Electrophysiology studies / electromyography (EMG) (Mov Disord 1998;13:920, Lancet 2007;369:218)
Laboratory
- Biomarkers to measure disease burden are under investigation and show promise, including measurement of mHTT in cerebrospinal fluid (CSF) (Eur J Neurol 2018;25:24)
Radiology description
- Magnetic resonance imaging (MRI): gray matter volume loss within the striatum, white matter volume loss around the striatum (Eur J Neurol 2018;25:24, Arch Neurol 1993;50:17, World J Radiol 2014;6:301, Lancet 2007;369:218)
- May be accompanied by cortical volume loss (World J Radiol 2014;6:301)
- Increased bicaudate diameter / intercaudate distance is a useful marker in the evaluation of Huntington disease (Arch Neurol 1993;50:17, AJNR Am J Neuroradiol 1995;16:1405)
- fMRI and PET imaging may provide functional and metabolic evidence of disease progression (World J Radiol 2014;6:301, J Cell Physiol 2017;232:1988)
Radiology images
Images hosted on other servers:
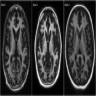
Caudate volume HDL2 and HD versus normal
Case reports
- 8 year old boy presented with progressive developmental regression (Radiol Case Rep 2020;16:113)
- 16 year old girl presented with excessive blinking and blepharoclonus (Mov Disord Clin Pract 2021;8:947)
- 59 year old woman with clinical presentation suggesting tauopathy, with genetic evidence of Huntington disease (Clin Case Rep 2021;9:e04547)
Treatment
- Key management strategies revolve around a multidisciplinary approach to target the main symptoms of Huntington disease; a curative treatment is not currently available (Eur J Neurol 2018;25:24)
- Motor symptoms: treatment with amine transport inhibitors (e.g., tetrabenazine, deutetrabenazine)
- Psychiatric symptoms: includes pharmacologic and nonpharmacologic interventions to treat depressive symptoms and irritability
- Cognitive symptoms: difficult to target with currently approved therapies
Gross description
- Atrophy of the striatum, particularly of the caudate nucleus (Eur J Neurol 2018;25:24)
- Variable gross cerebral atrophy affecting all 4 lobes (Brain Pathol 2016;26:726)
- Vonsattel grading scheme (see Microscopic description below for additional details) (J Neuropathol Exp Neurol 1998;57:369, J Neuropathol Exp Neurol 1985;44:559)
- Grade 0: macroscopically normal
- Grade 1: macroscopically normal
- Grade 2: macroscopically visible atrophy of the caudate but retains convex medial contour
- Grade 3: macroscopically severe atrophy of the caudate with flat medial contour
- Grade 4: macroscopically severe atrophy of the caudate with concave medial contour
Gross images
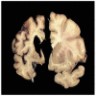
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
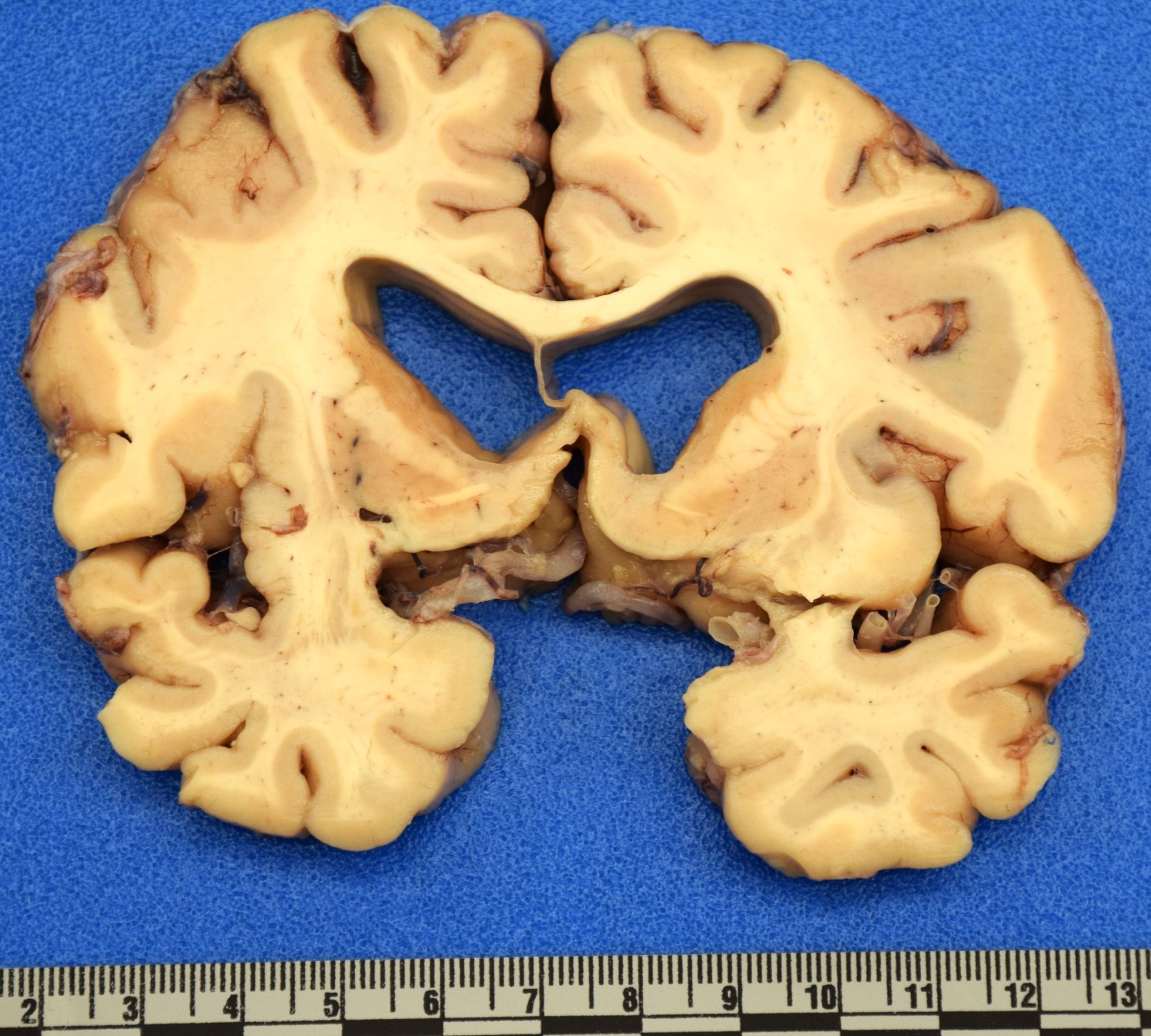
Vonsattel grade 3 atrophy
Images hosted on other servers:
Vonsattel grading
Microscopic (histologic) description
- Neostriatal atrophy with notable loss of neurons and astrogliosis (J Neuropathol Exp Neurol 1998;57:369)
- Vonsattel grading scheme (J Neuropathol Exp Neurol 1985;44:559, Ann Neurol 2001;49:29, Brain Pathol 2016;26:726)
- Grade 0: microscopy shows minimal to mild neuronal loss in the head of the caudate; ubiquitinated inclusions present
- Grade 1: microscopy shows mild neuronal loss and astrocytosis affecting the caudate head and putamen; ubiquitinated inclusions present
- Grade 2: microscopy shows neuronal loss and gliosis in the ventrolateral caudate; increased oligodendrocyte density appreciable
- Grade 3: microscopy shows neuronal loss and astrocytosis throughout striatum with sparing of nucleus accumbens
- Grade 4: microscopy shows severe neuronal loss and astrocytosis with involvement of nucleus accumbens
- Cortical neuronal loss and white matter thinning, with neuronal loss being most pronounced in cortical layers III, V and VI (Brain Pathol 2016;26:726)
Microscopic (histologic) images
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
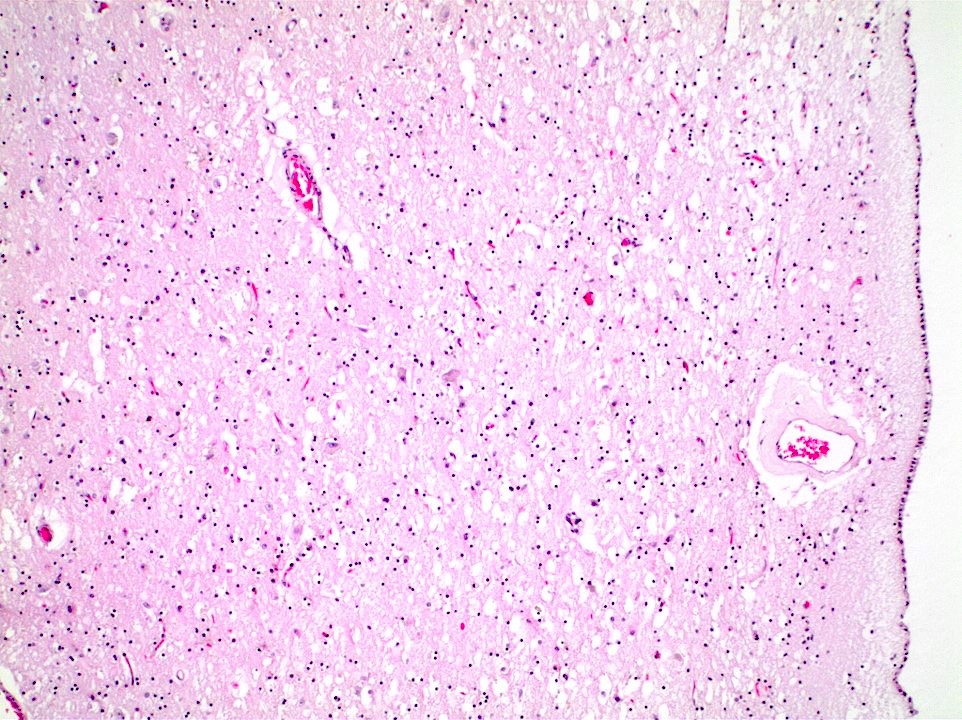
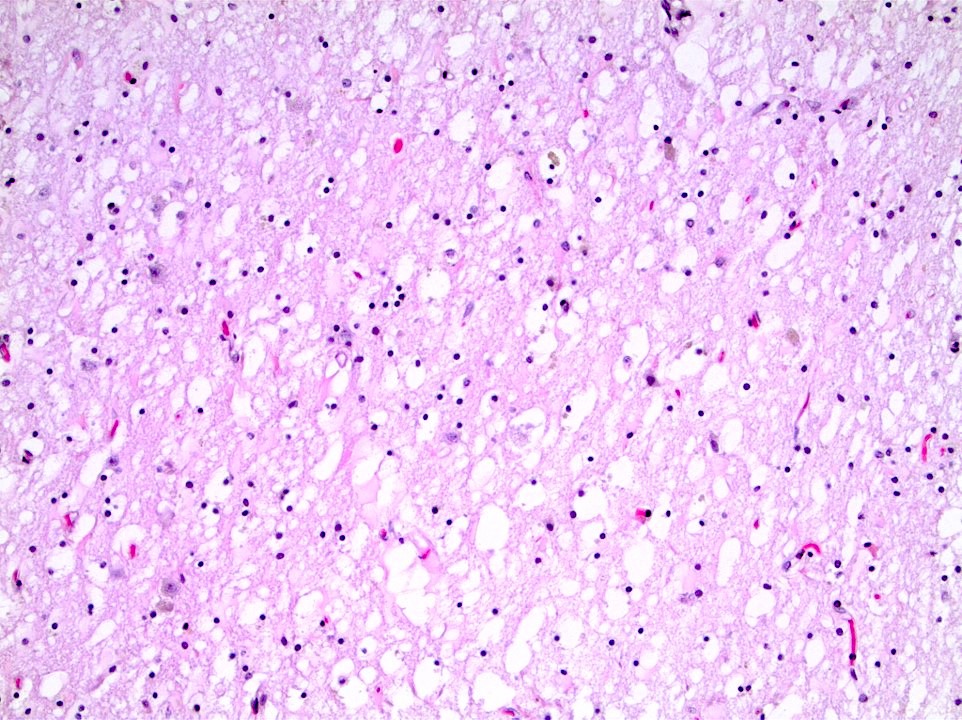
Caudate, neuronal loss
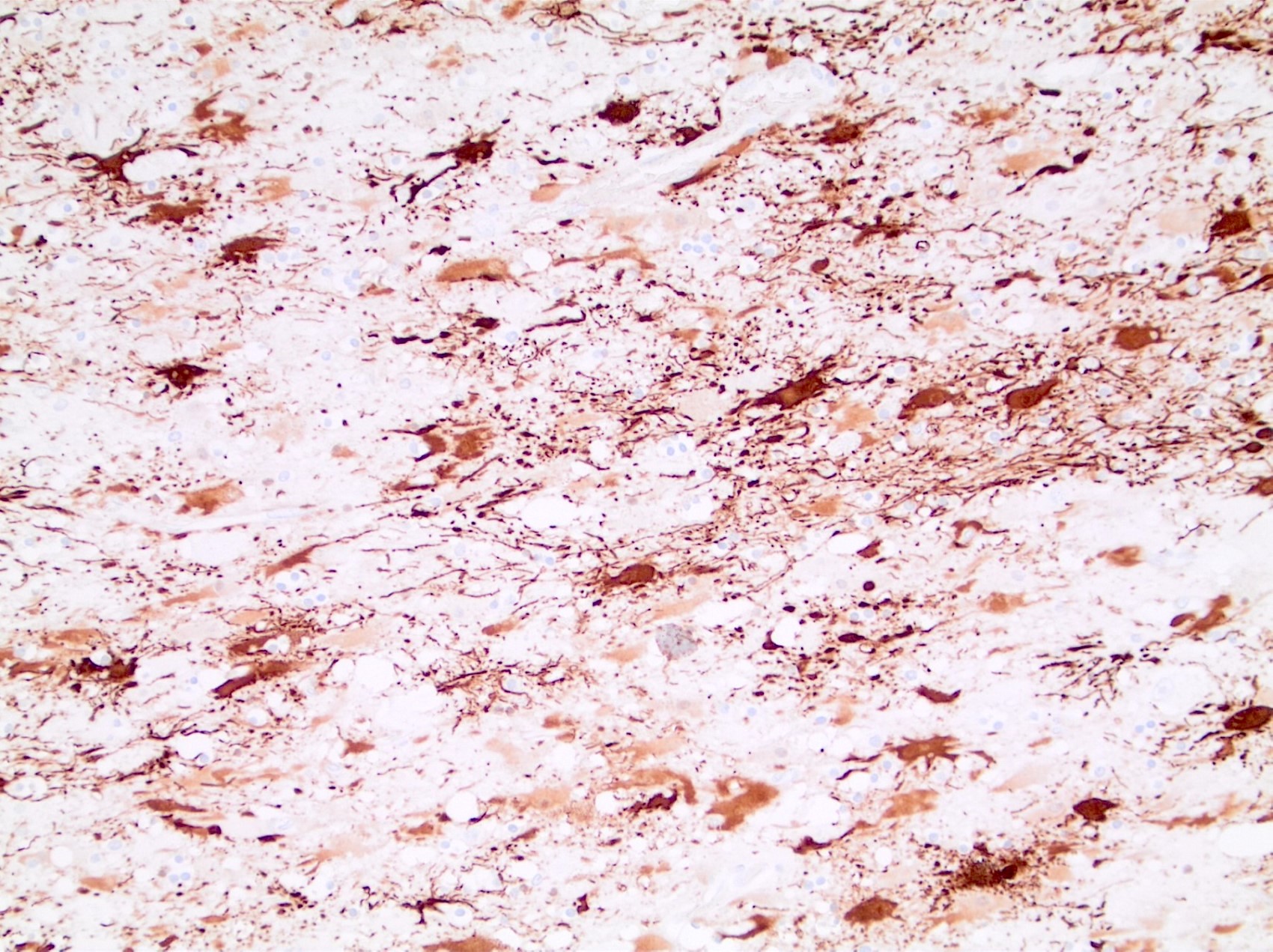
Caudate, reactive gliosis
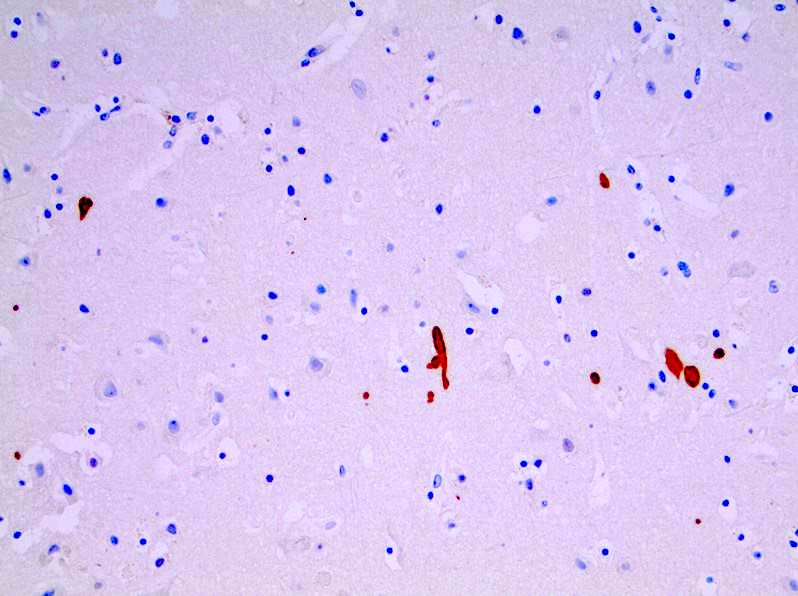
Ubiquitin positive inclusions
Positive stains
- Ubiquitin: highlights neuronal intranuclear inclusions (J Neuropathol Exp Neurol 1998;57:369)
- p62: highlights neuronal intranuclear inclusions and axonal neuronal inclusions (Brain Pathol 2016;26:726, Brain Pathol 2014;24:247)
Negative stains
- Tau: positive in neurodegenerative tauopathies such as progressive supranuclear palsy, corticobasilar degeneration and Pick disease, among others (Brain Sci 2020;10:972)
- Alpha-synuclein: positive in Parkinson disease and Lewy body disease
- TDP-43: positive in frontotemporal lobar degeneration with TDP-43 inclusions (Acta Neuropathol 2017;134:65)
- Of note, there is evidence suggesting co-occurrence of mtHTT and abnormal aggregates of TDP-43, tau and alpha-synuclein in advanced Huntington disease, though these phenomena are under active investigation (Acta Neuropathol 2018;135:249)
Molecular / cytogenetics description
- Autosomal dominant trinucleotide CAG repeat in the huntingtin gene (HTT) on chromosome 4 (Eur J Neurol 2018;25:24, Curr Opin Neurobiol 2015;33:53)
Sample pathology report
- Brain and spinal cord, autopsy examination:
- Neuropathologic findings in keeping with patient's known history of Huntington disease (see comment)
- Comment: The caudate nucleus shows marked gross atrophy with microscopic evidence of neuronal loss and prominent reactive astrogliosis. These findings are consistent with patient's known history of Huntington disease.
Differential diagnosis
- Huntington phenocopies:
- Disease states that include the triad of chorea, cognitive and neuropsychiatric decline in the absence of an mHTT mutation (Eur J Neurol 2018;25:24, Lancet 2007;369:218, Curr Opin Neurol 2013;26:420)
- Huntington disease-like (HDL) syndromes:
- HDL1:
- Autosomal dominant inheritance
- Octapeptide repeats in prion protein gene (PRNP)
- Prominent personality changes, chorea, rigidity and dysarthria
- Neuropathology shows basal ganglia atrophy and prion deposition without significant spongiosis
- HDL2:
- Autosomal dominant inheritance caused by CTG CAG triplet repeat expansion in JPH3 gene
- Anticipation phenomenon more common if maternally inherited
- More common among black Africans
- Similar neuropathology to HD
- HDL4:
- Also classified as spinocerebellar ataxia type 17 (SCA17)
- Caused by triplet repeat expansion in TATA box binding protein (TBP)
- Heterogenous clinical phenotype and neuropathology
- Dentatorubral pallidoluysian atrophy (Curr Opin Neurol 2013;26:420):
- Secondary to expanded CAG repeat in ATN1 gene
- Heterogenous clinical presentation
- MRI notable for cerebellar and brainstem atrophy
- More common in Japan
- Neuroacanthocytosis (Lancet 2007;369:218):
- Areflexia, raised creatine kinase and presence of acanthocytes
- Spinocerebellar ataxias
- Neuroferritinopathy (Curr Opin Neurol 2013;26:420):
- Caused by mutations in FTL (ferritin light chain), resulting in an autosomal dominant basal ganglia disease similar to Huntington disease
- MRI notable for cystic degeneration of the basal ganglia ad T2 hypointense lesions secondary to iron deposition
- More common in Cumbrian region of northern England
- HDL1:
- Isolated chorea may be secondary to other causes:
- Chorea of pregnancy
- Systemic lupus erythematosus
- Thyrotoxicosis
- Polycythemia vera
- Friedreich ataxia (Br Med Bull 2017;124:19):
- Autosomal recessive caused by homozygous GAA triplet repeat in frataxin (FXN)
- Gait and limb ataxia, dysarthria and loss of lower extremity reflexes
- Progressive disease may show scoliosis, pes cavus and talipes equinovarus
- Cardiomyopathy is common
- Symptom onset in adolescence
Additional references
Board review style question #1
A man of late middle age is found deceased by a passerby. An investigation reveals him to be a regular in the neighborhood and was known for abnormal, jerking movements and erratic obsessions. Examination of the heart demonstrated a transmural myocardial infarction. Examination of the brain revealed the gross findings shown in the image above. Which of following is the most likely diagnosis?
- Alzheimer disease
- Chronic infarct
- Huntington disease
- Parkinson disease
- Wernicke-Korsakoff syndrome
Board review style answer #1
C. Huntington disease. Although several of these answer choices can display global effects, the combination of motor and neuropsychiatric disturbance in a patient with striatal atrophy favors Huntington disease. Answer A is incorrect because Alzheimer disease presents with cognitive decline and memory deficits and is not associated with choreiform movements. Gross examination would reveal variable global atrophy. Answer D is incorrect because Parkinson disease is associated with cogwheel rigidity and a resting tremor but not choreiform movements. Gross examination would demonstrate pallor of the substantia nigra. Answer B is incorrect because chronic infarction would be associated with a neurologic deficit, particularly hemi or paraplegia but not choreiform movements. Gross examination would show cavitary tissue infarction within a vascular territory. Answer E is incorrect because Wernicke-Korsakoff syndrome may be associated with ataxic movements but not choreiform movements, with notable symptoms including vision difficulties and memory deficits. Gross examination would expect to find hemorrhage of the mamillary bodies or around the third ventricle.
Comment Here
Reference: Huntington disease
Comment Here
Reference: Huntington disease
Board review style question #2
Which of the following trinucleotide repeats is associated with Huntington disease?
- CAG
- CGG
- CTG
- CTG CAG
- GAA
Board review style answer #2
A. CAG. The CAG trinucleotide repeat in the HTT gene is associated with Huntington disease. Answer C is incorrect because the CTG trinucleotide repeat in the DMPK gene is associated with myotonic dystrophy. Answer D is incorrect because the CTG CAG repeat is associated with Huntington disease-like syndrome type 2. Answer E is incorrect because the GAA trinucleotide repeat in the FXN gene is associated with Friedrich ataxia. Answer B is incorrect because the CGG trinucleotide repeat in the FMR1 gene is associated with fragile X syndrome.
Comment Here
Reference: Huntington disease
Comment Here
Reference: Huntington disease
