
CNS nontumor
Movement disorders
Amyotrophic lateral sclerosis
Authors: Matthew McCord, M.D., Pouya Jamshidi, M.D., Rachel A. Multz, M.D., Margaret E. Flanagan, M.D.
Editorial Board Member: Meaghan Morris, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 17 April 2023
Last staff update: 17 April 2023
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: CNS amyotrophic lateral sclerosis

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: McCord M, Jamshidi P, Multz R, Flanagan ME. Amyotrophic lateral sclerosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnsALS.html. Accessed March 31st, 2025.
Definition / general
- Amyotrophic lateral sclerosis (ALS) is a chronic, progressive neurologic disease characterized by degeneration of upper and lower motor neurons in the central nervous system
- Motor neuron loss results in progressive and irreversible loss of motor function, muscle weakness and wasting and ultimately death, usually due to respiratory failure
Essential features
- Neurodegenerative disease that affects motor neurons; incurable and uniformly fatal
- Clinically, characterized by upper and lower motor neuron signs / symptoms
- Key pathologic findings: gross atrophy of anterior spinal nerve roots, neuronal loss and gliosis affecting anterior horn of spinal cord and primary motor cortex, pallor of corticospinal tracts, Bunina bodies (cystatin C positive) and skein-like inclusions (TDP-43 and ubiquitin positive) in motor neurons
- Familial ALS: C9orf72 expansion and SOD1 mutation are the 2 most common inherited genetic alterations
Terminology
- Motor neuron disease: in the U.S., motor neuron disease includes ALS and other related disorders; in the U.K., motor neuron disease is preferred term for ALS
- Upper motor neurons (UMN): eponymously known as Betz cells; originate in layer 5 of the primary motor cortex (located in the precentral gyrus) and project to lower motor neurons in brainstem and spinal cord
- Lower motor neurons (LMN): originate in brainstem motor nuclei and the anterior column of the spinal cord; project to skeletal muscle end plates
- Corticospinal tracts: white matter consisting of myelinated fibers of descending upper motor neurons
- Progressive bulbar palsy (PBP): syndrome characterized by progressive dysarthria and dysphagia
- Progressive muscular atrophy (PMA): syndrome characterized by lower motor neuron signs in the absence of upper motor neuron signs
- Primary lateral sclerosis (PLS): syndrome characterized by upper motor neuron signs in the absence of lower motor neuron signs, with pathologic changes restricted to the motor cortex and corticospinal tracts
- Some consider progressive bulbar palsy, progressive muscular atrophy and primary lateral sclerosis to be related variants of ALS; others consider ALS, progressive bulbar palsy and progressive muscular atrophy to be variants of the same clinicopathologic entity, while primary lateral sclerosis is considered distinct
- Frontotemporal dementia (FTD): clinically heterogeneous group of dementia syndromes with pathology characterized by preferential atrophy of the frontal and temporal lobes, typically mediated by pathologic accumulation of protein (e.g., TDP-43); may copresent with ALS
- ALS is popularly known as Lou Gehrig disease
ICD coding
Epidemiology
- Incidence of 2 per 100,000 annually (J Neurol Sci 2001;191:3)
- Peak incidence between ages 60 - 75 (Neuroepidemiology 2013;41:118)
- M:F = 1.3 - 1.5; slight male predominance (J Neurol Sci 2001;191:3)
Sites
- Primary motor cortex
- Brainstem motor nuclei
- Spinal cord: anterior horn motor neurons and corticospinal tracts
- Skeletal muscles are affected by motor neuron loss
Pathophysiology
- Loss of motor neurons results in skeletal muscle denervation and progressive loss of motor function
Etiology
- Sporadic:
- Accounts for 90 - 95% of cases (J Neurol Neurosurg Psychiatry 2011;82:623, Nature 2016;539:197)
- Etiology is not completely understood but mounting evidence suggests a role for altered RNA processing (Neurology 2011;77:1588, Muscle Nerve 2013;47:330, Nature 2013;495:467)
- Pathologic accumulation of TDP-43, an RNA binding protein that is central to disease development and progression (Arch Neurol 2009;66:180, Acta Neuropathol 2011;122:657)
- Familial:
- Accounts for 5 - 10% of cases
- Hexanucleotide expansion at C9orf72 and mutation in SOD1 are most frequent inherited genetic alterations (Neuron 2011;72:257, Science 2009;323:1208)
- Expansion of C9orf72 can cause overlapping syndromes of ALS and frontotemporal dementia
- Expansion of guanine rich sequences can form G quadruplexes, which are thought to interfere with RNA processing (Nature 2014;507:195)
- Free radical toxicity is thought to play a role in neuronal cell death in familial ALS with SOD1 mutation (Nature 1993;362:59)
Clinical features
- Progressively worsening muscle weakness, leading to loss of mobility and respiratory failure
- Upper motor neuron specific signs and symptoms
- Brisk tendon reflexes
- Spasticity
- Pseudobulbar signs (dysarthria, dysphagia, face and tongue weakness, emotional lability)
- Lower motor neuron specific signs and symptoms
- Skeletal muscle weakness and wasting
- Fasciculations
Diagnosis
- ALS diagnosis relies on history, clinical neurologic exam, electrophysiologic evaluation and neuropathologic evaluation (generally via autopsy examination of brain and spinal cord)
- El Escorial criteria for ALS diagnosis was originally published in 1994 and revised in 2000 to include electrophysiology; Awaji criteria further integrates clinical exam with electrophysiology (J Neurol Sci 1994;124:96, Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293, Amyotroph Lateral Scler 2009;10:53)
- Current Awaji criteria:
- ALS diagnosis requires the following:
- Evidence of lower motor neuron degeneration by clinical, electrophysiological or neuropathological examination
- Evidence of upper motor neuron degeneration by clinical examination
- Progressive spread of symptoms or signs within a region or to other regions, as determined by history, physical examination or electrophysiologic tests
- And the absence of:
- Electrophysiological or pathological evidence of other disease processes that might explain the signs of lower or upper motor neuron degeneration and
- Neuroimaging evidence of other disease processes that might explain the observed clinical and electrophysiological signs
- Diagnostic categories:
- Clinically definite ALS: clinical or electrophysiological evidence of lower as well as upper motor neuron signs in the bulbar region and at least 2 spinal regions or the presence of lower and upper motor neuron signs in at least 3 spinal regions
- Clinically probable ALS: clinical or electrophysiological evidence of lower and upper motor neuron signs in at least 2 regions with some upper motor neuron signs necessarily rostral to the lower motor neuron signs
- Clinically possible ALS: clinical or electrophysiological evidence of upper and lower motor neuron signs in only 1 region or upper motor neuron signs found alone in at least 2 regions or lower motor neuron signs found rostral to upper motor neuron signs
- El Escorial criteria previously included Clinically Probable Laboratory Supported ALS but this category was deemed redundant and eliminated from Awaji criteria
- ALS diagnosis requires the following:
Laboratory
- Routine laboratory studies may be helpful for ruling out metabolic or toxic etiology of symptoms
- Genetic testing can identify familial ALS
Radiology description
- Imaging may be more useful to rule out other etiologies than to provide direct evidence of ALS
- Notable features that have been described: increased FLAIR and T2 signal in corticospinal tracts and hypodensity on T2 of motor cortex (Radiology 1999;212:763, Radiology 1993;189:843)
Prognostic factors
- Median survival 20 - 48 months after symptoms develop (J Neurol Sci 1995;132:207, Neurology 1988;38:1604)
- Survival beyond 10 years: 5 - 10% (Amyotroph Lateral Scler 2009;10:310)
Case reports
- 48 year old man with rapidly progressive weakness and de novo SOD1 mutation (Exp Neurobiol 2016;25:347)
- 60 year old man with progressive weakness (Intern Med 2016;55:3511)
- 72 year old woman with slurred speech, family history of motor neuron disease and C9orf72 expansion by genetic testing (Innov Clin Neurosci 2016;13:37)
Treatment
- Disease is progressive, incurable and uniformly fatal
- 2 FDA approved disease modifying therapies:
- Riluzole: improves survival (N Engl J Med 1994;330:585, Lancet 1996;347:1425)
- Thought to reduce glutamate excitotoxicity by inhibition of glutamate release, NMDA receptor blockade and sodium channel modulation
- Edaravone: slows functional deterioration (Amyotroph Lateral Scler Frontotemporal Degener 2014;15:610)
- Thought to reduce oxidative stress by scavenging free radicals
- Riluzole: improves survival (N Engl J Med 1994;330:585, Lancet 1996;347:1425)
- Supportive care: management of symptoms (muscle spasms, fatigue, pain), mobility and functional assistance for activities of daily living, goals of care planning for patient and caregivers (wishes for nutritional or respiratory support)
Gross description
- Spinal cord (Nat Rev Neurosci 2013;14:248, Nat Rev Dis Primers 2017;3:17071)
- Atrophy of anterior spinal nerve roots
- Generalized thinning of spinal cord
- Brain (Nat Rev Neurosci 2013;14:248, Nat Rev Dis Primers 2017;3:17071)
- Primary motor cortex
- May show atrophy, particularly in patients with extended survival
- ALS with frontotemporal lobar disease (FTLD [TDP-43]), frontal and temporal lobes may show selective atrophy
- Primary motor cortex
Gross images
Contributed by Hannes Vogel, M.D.
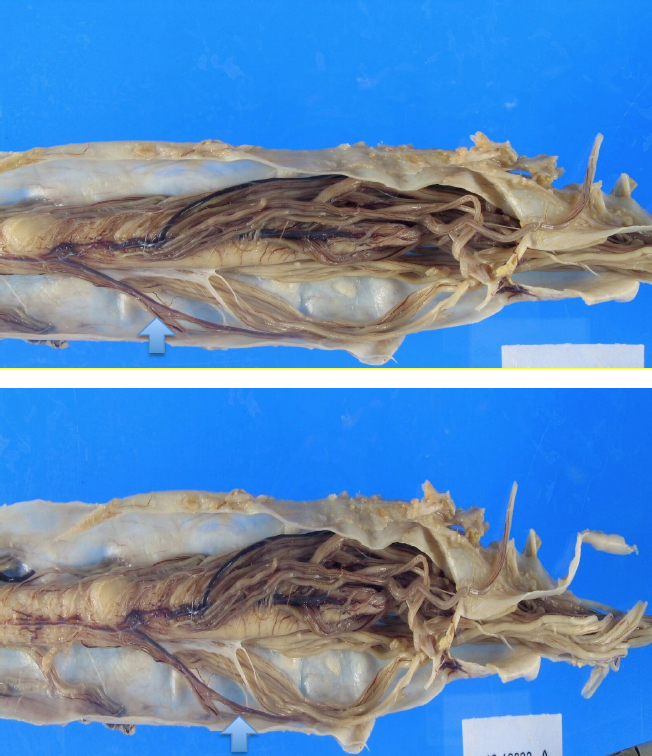
Spinal cord atrophy
Microscopic (histologic) description
- Loss of motor neurons and gliosis (Nat Rev Neurosci 2013;14:248, Nat Rev Dis Primers 2017;3:17071)
- Most evident in anterior horn of spinal cord
- Loss of Betz cells may be difficult to evaluate in the primary motor cortex; gliosis may be more obvious in this region
- Pallor of anterior and lateral corticospinal tracts (Nat Rev Neurosci 2013;14:248, Nat Rev Dis Primers 2017;3:17071)
- May be best visualized with myelin stain (Luxol fast blue)
- Inclusions in motor neurons:
- Bunina bodies: eosinophilic, granular intracytoplasmic inclusions composed of cystatin C and transferrin (Neuropathology 2008;28:109)
- Skein-like inclusions: intracytoplasmic protein aggregates appear like conglomerates of thread (yarn skeins); majority of cases positive for TDP-43 and ubiquitin (Nat Rev Neurosci 2013;14:248, Nat Rev Dis Primers 2017;3:17071)
- Lewy-like (spherical hyaline) inclusions: pale eosinophilic intracytoplasmic structures; positive for TDP-43 and ubiquitin (J Neurol Sci 1993;115:51, J Neuropathol Exp Neurol 2004;63:801)
- TDP-43 proteinopathy: different types of intracellular and extracellular pathologic accumulations in various parts of brain in frontotemporal dementia / ALS overlap syndrome (Acta Neuropathol 2011;122:111)
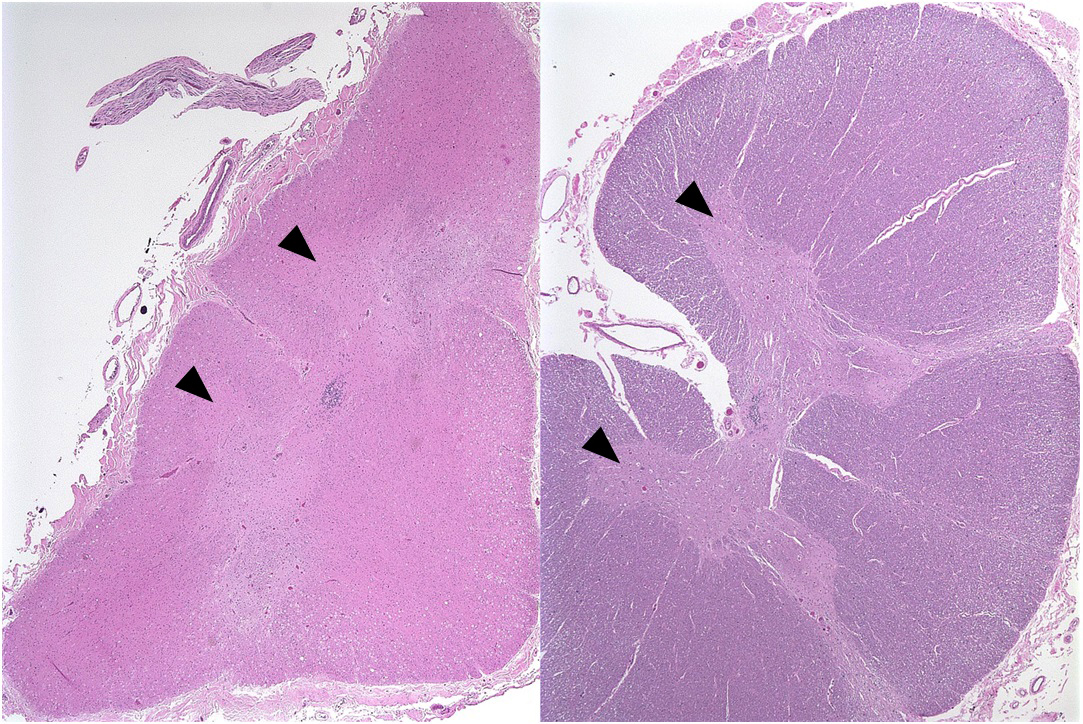
Microscopic (histologic) images
Contributed by Eileen Bigio M.D. and Qinwen Mao, M.D. Ph.D.
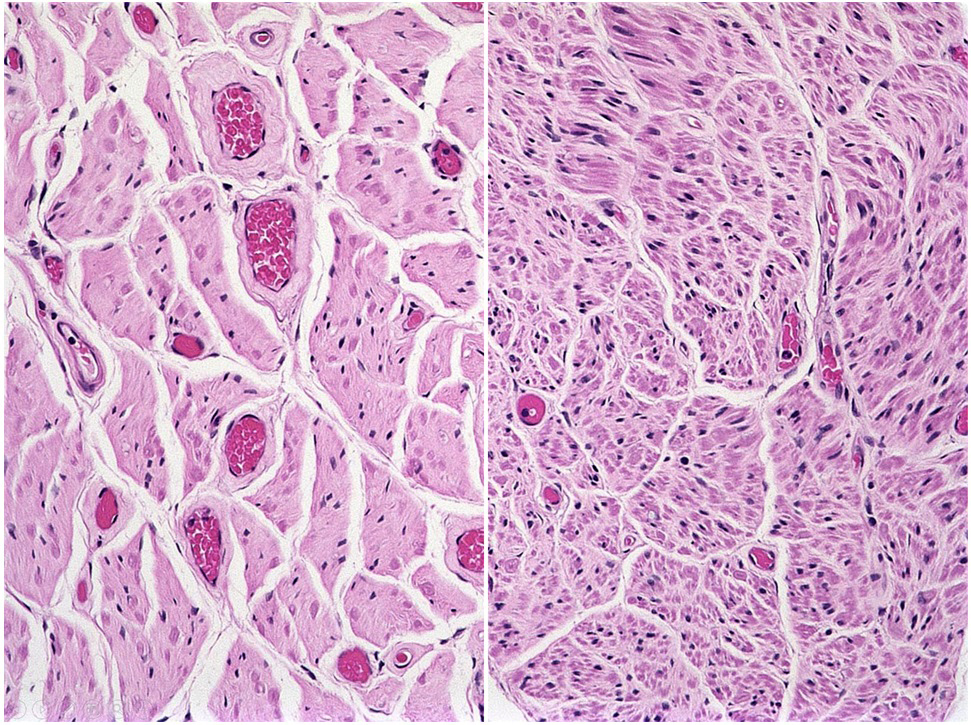
Spinal nerve roots
ALS versus normal spine
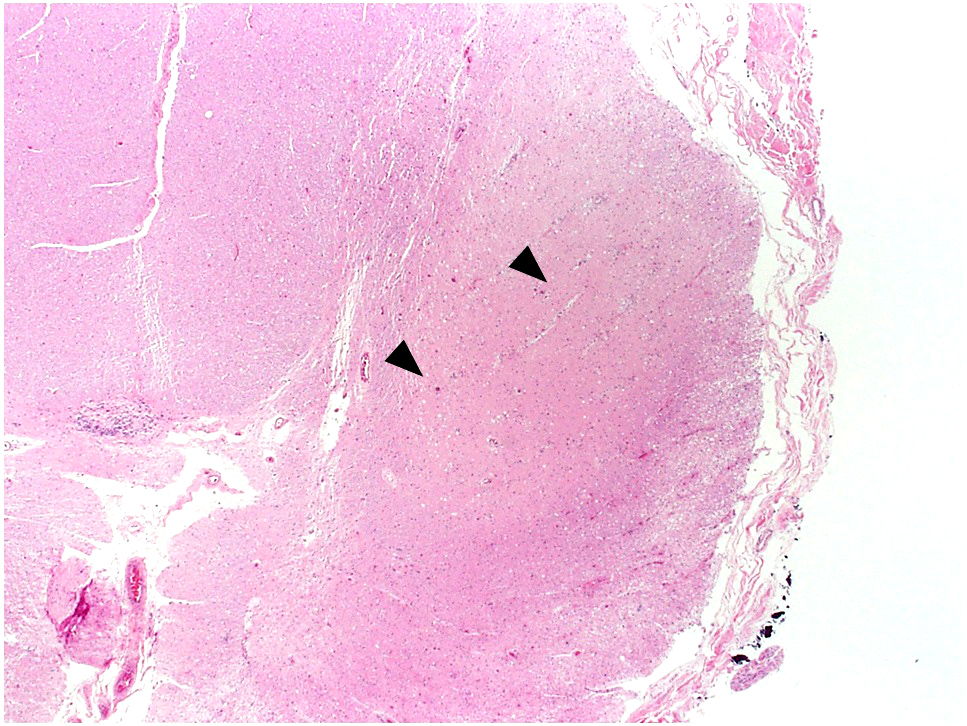
Corticospinal tract
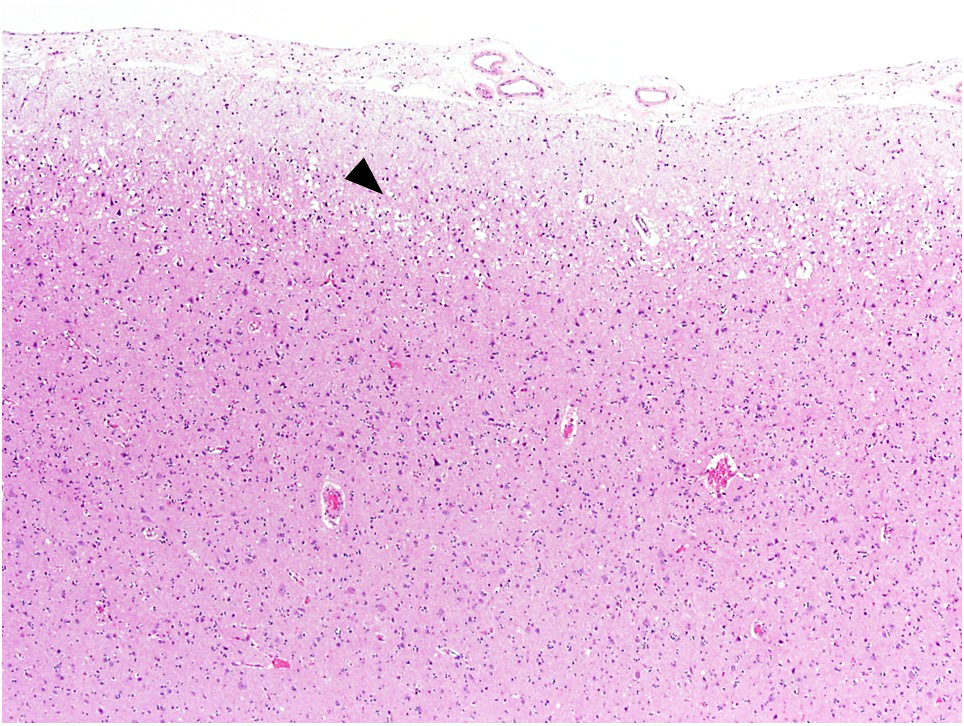
Motor cortex
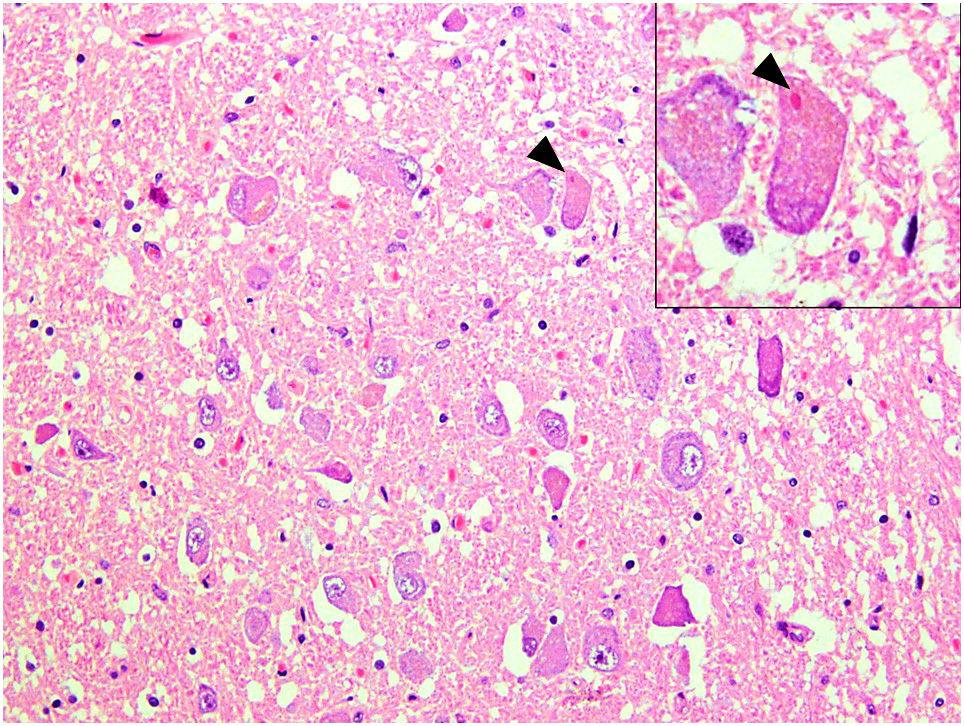
Bunina bodies
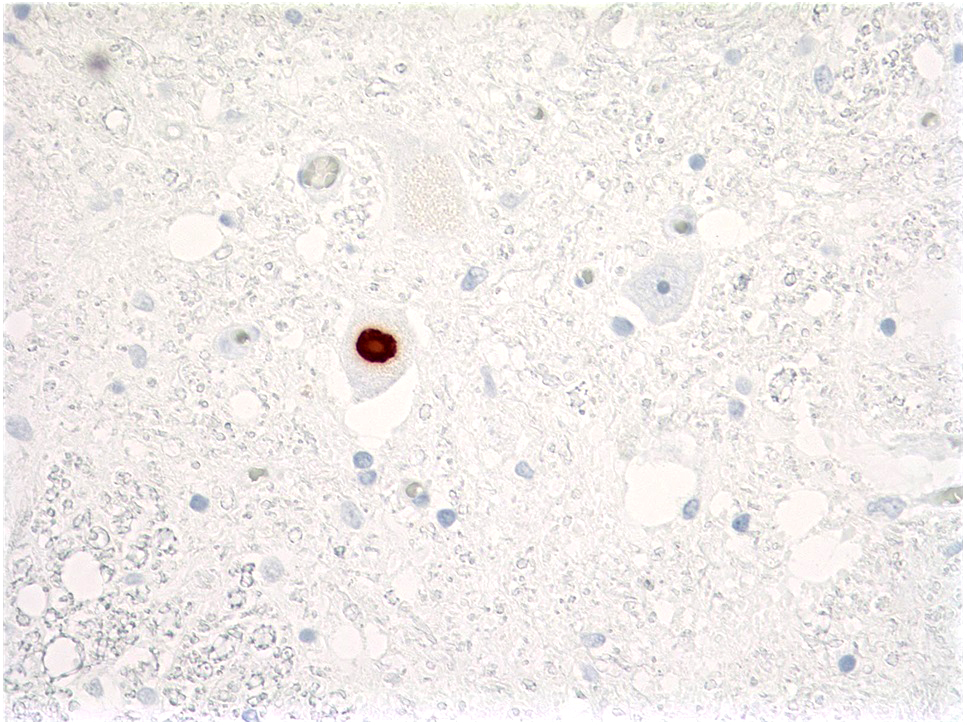
Lewy-like body
Contributed by Hannes Vogel, M.D., Matthew McCord, M.D., Christina Appin, M.D. and Missia Kohler, M.D.
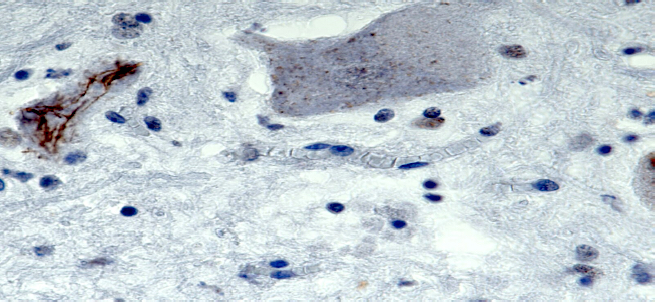
Inclusions immunoreactive for TDP-43
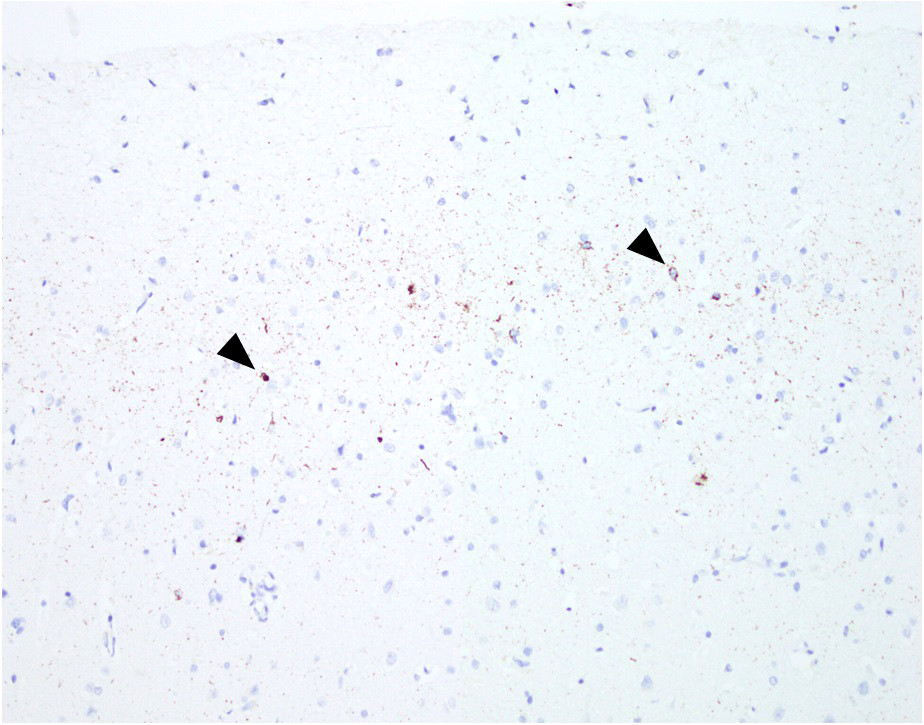
ALS / FTD overlap
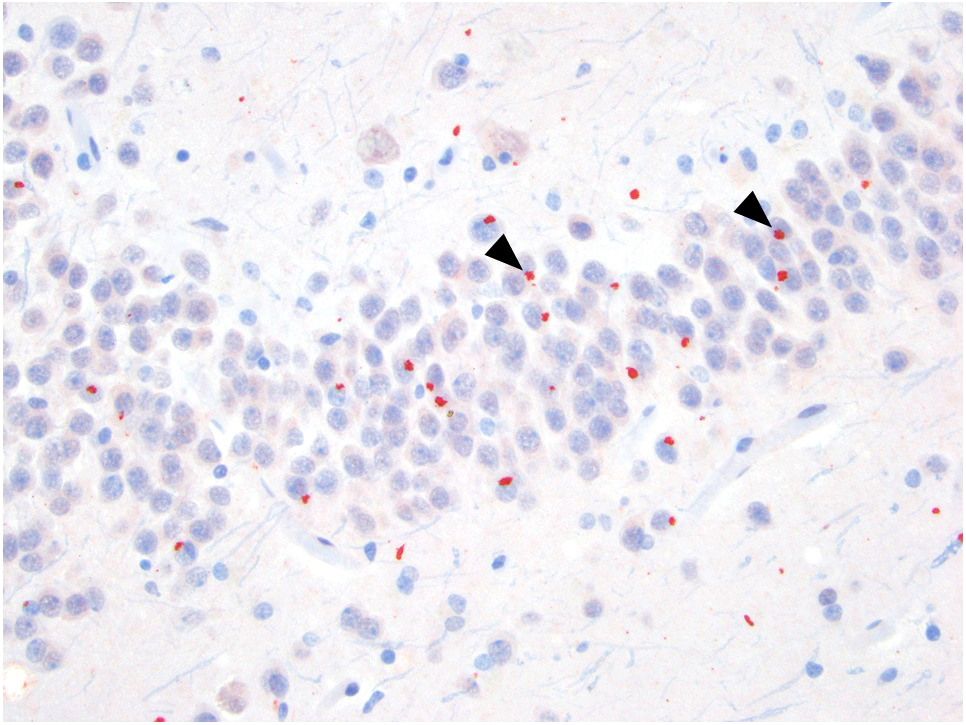
C9orf72 p62 positive inclusions
Positive stains
- TDP-43: positive in skein-like inclusions and Lewy-like inclusions
- Ubiquitin: positive in skein-like inclusions and Lewy-like inclusions
- Myelin stain (e.g., Luxol fast blue): loss of staining highlights pallor of corticospinal tracts
- p62: distinctive star shaped positive inclusions in cerebellar Purkinje cells and hippocampal dentate gyrus in C9orf72 linked ALS / frontotemporal dementia (Acta Neuropathol 2011;122:691)
Negative stains
- Tau: negative in all inclusions
- Alpha synuclein: negative in all inclusions
- Star shaped p62 positive inclusions of C9orf72 ALS / frontotemporal dementia are TDP-43 negative, notwithstanding the presence of TDP-43 positive inclusions elsewhere in the CNS (Acta Neuropathol 2011;122:691)
Electron microscopy description
- Bunina bodies: variable descriptions in the literature
- Generally described as electron dense, round to ovoid, containing ribosomal particles and cytoplasmic elements (Neuropathology 2008;28:109, Acta Neuropathol 1978;42:81)
Molecular / cytogenetics description
- 2 most common genetic defects in familial ALS (together, account for 50% of familial ALS):
- C9orf72
- Expansion of hexanucleotide repeat GGGGCC, typically 500 - 3,500 repeats
- Length of repeats does not show consistent relationship with survival (Neurology 2019;93:e1605, Neurobiol Aging 2016;38:217.e15, Hum Mol Genet 2014;23:749)
- SOD1 (ALS1) mutation, over 180 variants reported (JMIR Mhealth Uhealth 2013;1:e18)
- Most common: A4V missense mutation, associated with rapid progression (Amyotroph Lateral Scler Other Motor Neuron Disord 2003;4:62)
- I113T variant has variable penetrance (Amyotroph Lateral Scler Other Motor Neuron Disord 2001;2:S37)
- C9orf72
- Other genes associated with familial ALS:
- ALS3, 18q21 (Am J Hum Genet 2002;70:251)
- ALS4 (SETX), 9q34 (Am J Hum Genet 1998;62:633)
- ALS6 (FUS), chromosome 16 (Science 2009;323:1205)
- TDP-43 (Mol Cells 2018;41:818)
Sample pathology report
- Brain, autopsy:
- Histopathologic alterations compatible with amyotrophic lateral sclerosis (ALS):
- Upper motor neuron pathology, mild
- Lower motor neuron pathology, moderate
- Histopathologic alterations compatible with frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD TDP), type B
- Cerebrovascular disease, mild (nonocclusive):
- Mild atherosclerosis
- Mild arteriolosclerosis
- No cerebral amyloid angiopathy
- Up to moderate subcortical white matter pallor
- No Lewy body pathology
- Clinical information:
- The decedent is a 65 year old woman with an 8 year history of frontotemporal dementia and progressive motor neuron disease.
- Gross description:
- Brain weight: The unfixed brain weighs 1,376 g.
- Vascular: Examination of the circle of Willis shows mild cerebrovascular atherosclerosis. There are no large or lacunar infarcts and no hemorrhages.
- General: External examination of the brain reveals mild atrophy of the bilateral frontal and temporal cortices (symmetric) and no atrophy of the bilateral parietal or occipital cortices. Coronal sections of the cerebral hemispheres reveal moderate atrophy of the bilateral hippocampi (symmetric) and no atrophy of the bilateral caudate nuclei. There is mild dilatation of the ventricular system. External examination and transverse sections of the brainstem reveal no atrophy of the pons, mild pallor of the substantia nigra and moderate pallor of the locus coeruleus. External examination and sagittal and parasagittal sections of the cerebellum reveal no atrophy of the bilateral dentate nuclei. Spinal cord is not received for evaluation.
- General microscopic description:
- Microscopic sections are prepared from 23 regions, including bilateral dorsolateral prefrontal cortices (BA8-9), bilateral inferior frontal (Broca) gyri (BA44), bilateral superior temporal gyri (BA22), bilateral inferior parietal lobules (BA39), primary motor cortex (BA4), primary somatosensory cortex (BA3), primary visual cortex (BA17), anterior cingulate gyrus (BA24), posterior cingulate gyrus (BA23), striatum, basal ganglia with nucleus basalis of Meynert, thalamus with subthalamic nucleus, bilateral mammillary bodies, amygdala with entorhinal cortex, anterior hippocampus with entorhinal cortex, bilateral posterior hippocampi, cerebellum with dentate nucleus, cerebellar vermis, midbrain, pons and medulla. Sections are all from the left except as noted and are evaluated using H&E and fluorescent Thioflavin S and immunohistochemical preparations for tau (AT8), p62 (BD Biosciences), phosphorylated TDP-43 (CosmoBio), phosphorylated alpha synuclein (phospho S129) and beta amyloid (4G8).
- Microscopic examination reveals subcortical white matter pallor as summarized in the table below. There is mild hyaline arteriolosclerosis. Thioflavin S stains reveal no cerebral amyloid angiopathy. There is no Fahr vascular mineralization. There are no microinfarcts or lacunar infarcts.
- Microscopic examination reveals cortical layer II microvacuolation and gliosis and overall neuronal loss and gliosis as summarized in the table below. There is no pigment spheroid degeneration. No ballooned neurons are observed. There are no Pick bodies in the dentate gyri.
- Upper and lower motor neuron system:
- Microscopic examination reveals motor subcortical and descending corticospinal tract white matter pallor or sclerosis as summarized in the table below. There is mild neuronal loss and gliosis of the primary motor cortex, with no neuronophagia appreciated. In lower motor neurons, there is moderate neuronal loss and gliosis.
0 = absent, Min = minimal, + = mild, ++ = moderate, +++ = severe, L = left, R = right, B = bilateral,
General microscopic description summary
(Cortical and subcortical areas left sided unless otherwise indicated)White matter myelin pallor or sclerosis Motor cortex ++ Nonmotor cortex Up to ++ Internal capsule + Cerebral peduncle CSTs +, (left worse than right) Basis pontis CSTs Min Medullary pyramids + Cerebellum + Hyaline arteriolosclerosis + Fahr vascular mineralization 0 Microinfarcts 0 Lacunar infarcts 0 Superficial microvacuolation and gliosis Entorhinal cortex 0 Frontal cortex Up to ++ Temporal cortex 0 Motor cortex Min Neuronal loss & gliosis Hippocampus + (L) Entorhinal cortex L+, R++ Amygdala + Nonmotor cortex Up to + Motor cortex + Caudate + Putamen Min Globus pallidus 0 Nucleus basalis of Meynert + Thalamus 0 Subthalamic nucleus 0 Substantia nigra Min Locus coeruleus Min Cerebellar Purkinje cells 0 Cerebellar dentate nucleus + Hypoglossal nucleus ++ Pigment spheroid degeneration 0 Ballooned neurons 0 Pick bodies 0 Neuronophagia, motor cortex 0 Other significant histologic findings None
HGN = hypoglossal nucleus, CSTs = corticospinal tracts, C = cervical, T = thoracic, L = lumbar
- TDP-43 pathology:
- Phosphorylated TDP-43 immunostains of the neocortex highlight neuronal cytoplasmic inclusions (NCIs), both compact and granular, glial cytoplasmic inclusions (GCIs) and short, dystrophic neurites (DNs). Tau and alpha synuclein immunostains are negative in the inclusions and neurites. Threads and dots (ThDs) are also present in the background. TDP-43 pathology involves the full thickness of the cortex. No neuronal intranuclear inclusions (NIIs) are appreciated. DNs are of the short type, with long dystrophic neurites not appreciated. Semi quantitative estimates of NCIs / GCIs and DNs / ThDs are summarized in the table below. TDP-43 pathology in motor neurons is summarized separately, in the subsequent section.
TDP-43 pathology summary
(Cortical and subcortical areas left sided unless otherwise indicated)
CIs = cytoplasmic inclusions (including neuronal and glial cytoplasmic inclusions), NIIs = neuronal intranuclear inclusions,Region CIs NIIs DN / ThDs Cerebral cortex Middle frontal gyrus L++, R+ B 0 L+, R Min Superior temporal gyrus L+, R+ (left worse) B 0 L Min, R+ Inferior parietal lobule L+, R+ B 0 L+, R+ Sensory cortex Min 0 Min Entorhinal cortex L+++, R+++ B 0 L+, R++ Subcortical areas Amygdala +++ 0 ++ Dentate gyrus L+++, R+++ B 0 B 0 Hippocampus / subiculum L+, R+ B 0 L Min, R Min Caudate +++ 0 ++ Putamen ++ 0 Min Globus pallidus +++ 0 +++ Internal capsule 0 N/A 0 Nucleus basalis of Meynert + 0 Min Brainstem and cerebellum Inferior olivary nucleus +++ 0 + Cerebellar dentate nucleus 0 0 0 Cerebellar white matter 0 0 0
DN / ThDs = dystrophic neurites or threads and dots, 0 = absent, Min = minimal, + = sparse, ++ = moderate, +++ = frequent,
L = left, R = right, B = bilateral, N/A = not applicable, *focal
- ALS typical pathology and TDP-43 IHC:
- ALS pathology includes the above mentioned: neuronal loss and gliosis in the primary motor cortex; white matter rarefaction of motor subcortical white matter and descending corticospinal tracts; and neuronal loss and gliosis in the hypoglossal nuclei. The motor cortex and hypoglossal nuclei show chromatolysis, Bunina bodies and TDP-43 positive inclusions (or the lack thereof) as summarized in the table below.
Motor neuron disease pathology summary
0 = absent, + = sparse, ++ = moderate, +++ = frequent, N/A = not applicable, Chr = chromatolysis, BBs = Bunina bodies,Chr BBs TDP+ NCIs* TDP+ LLBs TDP+ GCIs Motor cortex N/A N/A + N/A + Hypoglossal nucleus 0 ++ ++ 0 +
NCIs = neuronal cytoplasmic inclusions, LLBs = Lewy-like bodies, GCIs = glial cytoplasmic inclusions,
*NCIs include granular and skein-like inclusions
- Lewy body pathology:
- H&E sections reveal no Lewy bodies in the brainstem or nucleus basalis of Meynert. Alpha synuclein immunostains of the left anterior and posterior cingulate gyrus, left amygdala with entorhinal cortex and right middle frontal gyrus reveal no Lewy bodies.
- p62 immunostains:
- p62 immunostains of the left middle frontal gyrus, left anterior hippocampus, bilateral posterior hippocampi and cerebellum highlight a subset of aging pathology only (FTLD TDP-43 and ADNC). There are no p62 positive / TDP-43 negative inclusions of the type seen in C9orf72 repeat expansion.
- Summary and comment:
- The histologic findings are compatible with the pathologic diagnosis of amyotrophic lateral sclerosis (ALS), also known as motor neuron disease. The upper motor neuron pathology is mild and the lower motor neuron pathology is moderate. Neocortical TDP-43 pathology is present, of the type seen in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD TDP), type B (Am J Pathol 2006;169:1343, Acta Neuropathol 2006;112:539, Acta Neuropathol 2011;122:111). TDP-43 proteinopathy may have different presentations, which vary across or even within specific pathology patterns. The pattern now recognized as FTLD TDP, type B may present with pure ALS, pure dementia or a combination of the 2 syndromes, as in this case (Acta Neuropathol 2006;112:539, Acta Neuropathol 2011;122:111). The patient tested negative for C9orf72 hexanucleotide expansion and there is no evidence of the distinctive p62 positive inclusions seen with this variant (Neuron 2011;72:245, Neuron 2011;72:257).
- This case was discussed at neurodegenerative disease clinicopathologic correlation conference and the above diagnoses were agreed upon.
- Microscopic sections (all from left side except as noted):
- A1 middle frontal gyrus (BA8-9)
- A2 inferior frontal gyrus (BA44)
- A3 superior temporal gyrus (BA22)
- A4 inferior parietal lobule (BA39)
- A5 anterior (BA24) and posterior (BA23) cingulate, posterior notched
- A6 motor (BA4) and somatosensory (BA3) cortex, sensory notched
- A7 amygdala and entorhinal cortex
- A8 anterior hippocampus and entorhinal cortex
- A9 bilateral posterior hippocampi, right notched
- A10 striatum
- A11 basal ganglia / nucleus basalis of Meynert
- A12 bilateral mammillary bodies
- A13 thalamus with subthalamic nucleus
- A14 occipital (visual) cortex (BA17)
- A15 cerebellum with dentate nucleus / vermis
- A16 left midbrain
- A17 pons with locus coeruleus
- A18 medulla with inferior olives
- A19 right middle frontal gyrus (BA8-9)
- A20 right inferior frontal gyrus (BA44)
- A21 right superior temporal gyrus (BA22)
- A22 right inferior parietal lobule (BA39)
- A23 right midbrain
- Histopathologic alterations compatible with amyotrophic lateral sclerosis (ALS):
Differential diagnosis
- Multifocal motor neuropathy:
- Immune mediated pathology of peripheral nerves rather than neurodegenerative disease
- Diagnosed clinically; nerve biopsy not often done
- Characterized by asymmetric weakness with subacute onset
- Lower motor neuron signs only
- Motor nerve conduction studies show conduction block, with normal sensory conduction
- Serology: may be associated with anti-GM1 antibodies
- Compressive radiculomyelopathy:
- Spondylolysis with spinal cord and nerve root compression
- Lower motor neuron signs corresponding to level of abnormality
- Radiology is key to diagnosis
- Inclusion body myositis:
- Disproportionate finger flexor weakness
- Electromyography shows myopathic signs
- Muscle biopsy classically shows variable inflammation, chronic myopathic features (e.g., regenerating fibers, endomysial fibrosis) and rimmed vacuoles (low sensitivity)
- Myasthenia gravis:
- Can present as bulbar syndrome with dysphagia and dysarthria, similar to ALS
- Clinical features favoring myasthenia gravis:
- Absence of either upper or lower motor neuron features
- Ocular findings
- Diurnal variation in symptom severity
- Serology: acetylcholine receptor binding antibodies, muscle specific tyrosine kinase antibodies
- Electromyography: cranial muscles show evidence if denervation / reinnervation in bulbar onset ALS but not in myasthenia gravis
- X linked bulbospinal neuronopathy:
- Usually progresses more slowly than ALS
- Genetic testing reveals CAG repeat expansion in androgen receptor gene
- Reference: Goldman: Cecil Textbook of Medicine, 21st Edition, 1999
Additional references
Board review style question #1
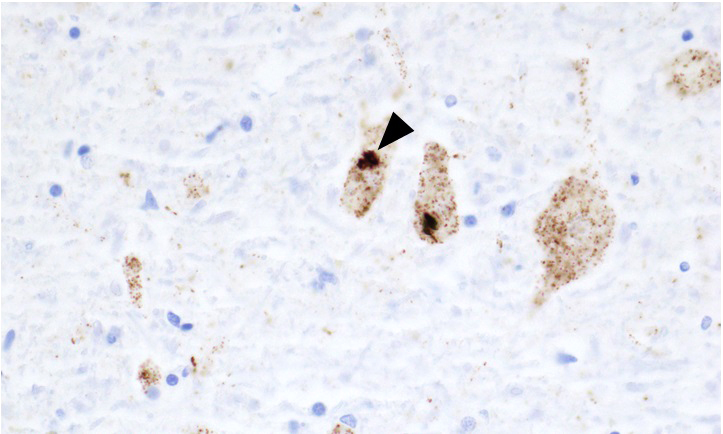
The image above shows inclusions in the hypoglossal nucleus in an autopsy brain from patient who succumbed to sporadic amyotrophic lateral sclerosis (ALS). Which of the following immunostains is represented in the image?
- 3 repeat tau
- 4 repeat tau
- Alpha synuclein
- Cystatin C
- TDP-43
Board review style answer #1
E. TDP-43. This protein is thought to be central to pathogenesis in sporadic ALS and skein-like inclusions found in motor neurons are positive. Neuronal inclusions positive for 3 repeat tau are found in Pick disease and 4 repeat tau positive inclusions are found in corticobasal degeneration and progressive supranuclear palsy. Lewy bodies and glial cytoplasmic inclusions of multiple system atrophy are positive for alpha synuclein. Cystatin C is a component of Bunina bodies.
Comment Here
Reference: Amyotrophic lateral sclerosis
Comment Here
Reference: Amyotrophic lateral sclerosis
Board review style question #2
In a case of amyotrophic lateral sclerosis (ALS), neuronal loss and gliosis would be most severe in which of the following CNS regions?
- Anterior horn of the spinal cord
- Basal ganglia
- Dentate nucleus of the cerebellum
- Posterior horn of the spinal cord
- Somatosensory cortex
Board review style answer #2
A. Anterior horn of the spinal cord. This region contains cell bodies of efferent lower motor neurons and is severely affected by neuronal loss and gliosis in ALS. The posterior horn of the spinal cord carries afferent sensory neurons and is usually preserved in ALS. The primary motor cortex (not the somatosensory cortex) also shows prominent neuronal loss and gliosis in ALS. The basal ganglia often show prominent neuronal loss and gliosis in multiple system atrophy, and sometimes in corticobasal degeneration. The dentate nucleus of the cerebellum shows severe neuronal loss and gliosis (grumose degeneration) in progressive supranuclear palsy.
Comment Here
Reference: Amyotrophic lateral sclerosis
Comment Here
Reference: Amyotrophic lateral sclerosis
