

Bone marrow neoplastic
Bone marrow - neoplastic myeloid
Hematologic neoplasms with germline predisposition and platelet disorders
Myeloid or lymphoid neoplasms with germline RUNX1 mutation
Author: Madina Sukhanova, Ph.D.
Editorial Board Member: Alexa J. Siddon, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.

Last author update: 17 June 2024
Last staff update: 2 January 2025
Copyright: 2024-2025, PathologyOutlines.com, Inc.
PubMed Search: Myeloid or lymphoid neoplasms with germline RUNX1 mutation

Table of Contents
Definition / general | Essential features | Terminology | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Sukhanova M. Myeloid or lymphoid neoplasms with germline RUNX1 mutation. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrowneoplasticrunx1mutation.html. Accessed January 10th, 2025.
Definition / general
- Myeloid or lymphoid neoplasms with germline RUNX1 variant
Essential features
- Confirmation of germline origin of RUNX1 variant in skin fibroblasts or persistent presence of RUNX1 variant at ~50% allelic frequency in all complete remission samples
Terminology
- World Health Organization (WHO)-5 (Leukemia 2022;36:1703)
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorders
- Germline RUNX1 P/LP variant (familial platelet disorder with associated myeloid malignancy)
- International Consensus Classification (ICC) (Blood 2022;140:1200)
- Hematologic neoplasms with germline predisposition associated with a constitutional platelet disorder
- Myeloid or lymphoid neoplasms with germline RUNX1 mutation
Epidemiology
- Up to 10% of acute myeloid leukemia (AML) cases with mutated RUNX1 at diagnosis have the RUNX1 mutation confirmed as germline change (Blood 2021;137:1428, Blood Adv 2018;2:146)
- Carriers of familial RUNX1 variants are at a ~30 - 40% lifetime risk of having a myeloid neoplasm (Leukemia 2020;34:2519)
- Incidence of development of T cell or B cell lymphoid neoplasm is unknown but recognized (see Case reports) (Pediatr Blood Cancer 2023;70:e30184, Blood Adv 2021;5:3199)
Sites
- Variable tissues, including skin, peripheral blood and bone marrow
- Frequency of tissue and germline mosaicism is unknown
Pathophysiology
- RUNX1 encodes a transcription factor, a binding partner for core binding factor beta (CBFB) and plays a crucial role in thrombocyte differentiation
- Germline pathogenic or likely pathogenic (P / LP) RUNX1 variants are common in familial platelet disorder with lifelong thrombocytopenia and a risk of progression to myeloid neoplasms and less frequently, T cell lymphoblastic leukemia / lymphoma and B cell neoplasms (OMIM: Runt related transcription factor 1; RUNX1 [Accessed 11 January 2024], OMIM: Platelet disorder, familial, with associated myeloid malignancy; FPDMM [Accessed 11 January 2024], Blood Adv 2019;3:2962, Haematologica 2020;105:870)
- Germline P / LP RUNX1 variants in patients with familial platelet disorder demonstrate a wide spectrum of RUNX1 mutations: missense, stop codon, frameshift, splice site and intragenic or whole gene deletions, which result in loss of function of mutated RUNX1 allele (Blood Adv 2019;3:2962, Blood Adv 2020;4:1131, Leukemia 2016;30:999)
- Majority of germline variants in the RUNX1 cluster are in the Runt domain and result in loss of binding activity
- Germline RUNX1 P / LP variants in the transactivation domain have also been noted
- Stop codon, splice site and frameshift mutations, as well as gene deletions, result in loss of function of 1 allele and haploinsufficiency
- Patients with germline RUNX1 variants experiencing malignant transformation acquire additional mutation(s), affecting either the second RUNX1 allele or other gene (DNMT3A, FLT3, GATA2, PHF6, BCOR, WT1, TET2, ASXL1) (Blood Adv 2020;4:1131, Leukemia 2016;30:999)
Etiology
- History of thrombocytopenia and progression to hematologic neoplasm in individuals with germline RUNX1 P / LP variant can occur at any age from early infancy to late adulthood (Blood Adv 2020;4:1131, Pediatr Blood Cancer 2023;70:e30184, Blood Adv 2021;5:3199, Leukemia 2020;34:2519)
Clinical features
- Easy bruising and prolonged bleeding following trauma or dental procedures (Blood Adv 2020;4:1131)
- Thrombocytopenia (Blood Adv 2020;4:1131)
- Eczema, psoriasis, arthritis (Blood Adv 2020;4:1131)
Diagnosis
- Detection of heterozygous germline P / LP variant in RUNX1 by molecular genetics test
Laboratory
- Mild to moderate thrombocytopenia (50 - 150 x 109/L)
- Qualitative platelet defect with impaired platelet aggregation and dense granule deficiency
Case reports
- 10 year old boy with history of thrombocytopenia, germline RUNX1 variant and new diagnosis of T cell lymphoblastic lymphoma (Pediatr Blood Cancer 2023;70:e30184)
- 12 year old girl with revertant mosaicism for the familial RUNX1 mutation (Haematologica 2020;105:e535)
- 16 year old girl and 16 year old boy (unrelated patients) with history of mild thrombocytopenia, germline RUNX1 variants and new diagnosis of B cell acute lymphoblastic leukemia (Blood Adv 2021;5:3199)
Treatment
- Best practice consensus guidelines from the UK Cancer Genetics Group, CanGene-CanVar and the NHS England Hematological Oncology Working Group recommend complete blood count (CBC) every 3 - 4 months and, if abnormal, thorough clinical and bone marrow examination for all carriers of familial RUNX1 P / LP variants (Br J Haematol 2023;201:25)
- For bleeding: recommendation to use clotting promoters during procedures with bleeding risk
- For early onset of hematologic malignancy: consideration for allogeneic stem cell transplantation from related donor who does not carry RUNX1 germline variant or matched unrelated donor
- For skin manifestations: topical steroids
- Genetic counseling to identify unaffected relatives at risk of being a carrier of germline RUNX1 variant for further surveillance
Microscopic (histologic) description
- Hypocellular or normocellular bone marrow for patient's age (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661)
- Atypical but not dysplastic small megakaryocytes with hypolobated nuclei (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661)
- Eosinophilia (Haematologica 2017;102:1661)
Microscopic (histologic) images
Contributed by Barina Aqil, M.D.

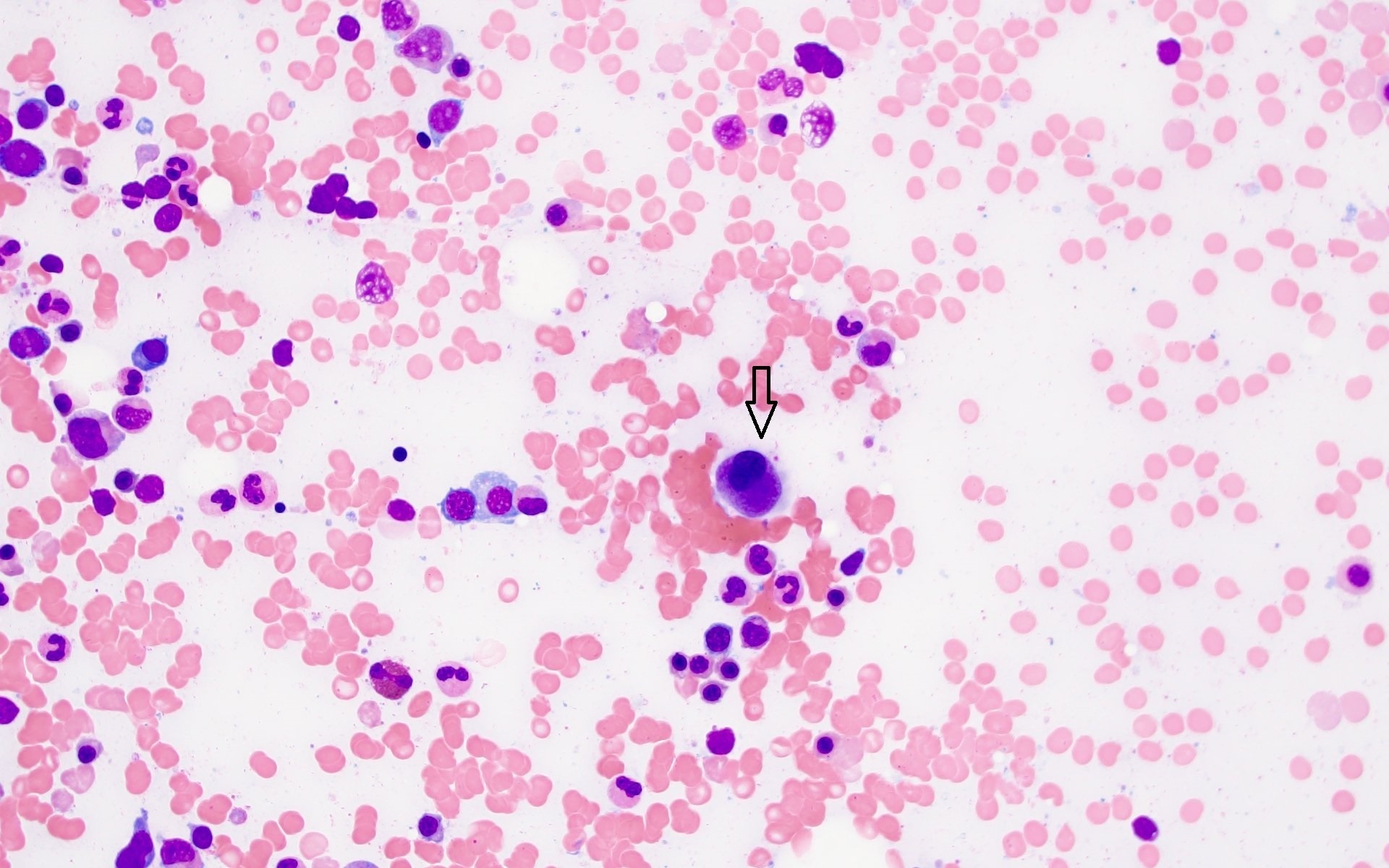

Small hypolobated megakaryocyte

Erythroid dysplasia

Scattered blasts

CD61 highlights small megakaryocytes
Positive stains
- CD34 highlights blasts
- MPO highlights myeloid precursors
- CD61 highlights megakaryocytes including small hypolobated / monolobated forms (Haematologica 2017;102:1661)
- CD71 highlights erythroid precursors
Negative stains
- Reticulin does not show increased fibrosis
- Prussian blue shows decreased iron storage with no ring sideroblasts identified
Molecular / cytogenetics description
- Sequence analysis (single gene sequencing or next generation sequencing [NGS] of gene panel) detects heterozygous P / LP RUNX1 variant in ~80% of cases (Blood Adv 2020;4:1131, Blood Adv 2019;3:2962, Eur J Hum Genet 2008;16:1014)
- Copy number analysis (chromosomal microarray, multiplex ligation dependent probe amplification [MLPA], quantitative PCR, fluorescence in situ hybridization) detects a whole RUNX1 gene deletion or intragenic deletion in ~20% of cases (Cancers (Basel) 2022;14:3431, Am J Med Genet A 2016;170:2580, Blood Adv 2019;3:2962, Eur J Hum Genet 2008;16:1014)
- In some patients, analysis of hematopoietic tissue can be negative for mutation or deletion of RUNX1 due to somatic loss of heterozygosity event with a loss of chromosome 21 with P / LP RUNX1 variant; thus, genetic testing of cultured skin fibroblasts is a preferred method of confirmation of the presence of P / LP germline RUNX1 variant, if possible (see Case reports) (Haematologica 2020;105:e535)
Molecular / cytogenetics images
Contributed by Madina Sukhanova, Ph.D.
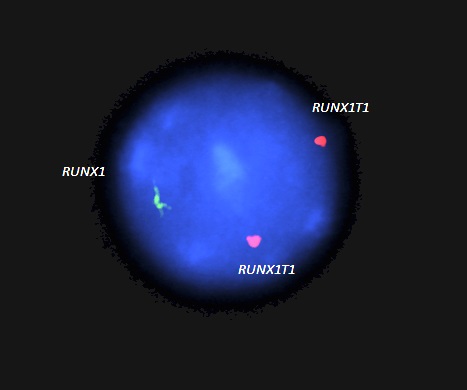
RUNX1 whole gene deletion
Images hosted on other servers:

RUNX1 germline mutations

Reverse mosaicism due to LOH
Sample pathology report
- Peripheral blood, bone marrow aspirate and bone marrow core biopsy:
- Myelodysplastic syndrome involving a variably cellular bone marrow (see comment)
- Comment: There is dysplasia in the granulocyte, erythroid and megakaryocyte lineages. Immunochemical stain for CD34 highlights 3 - 4% blasts. No ring sideroblasts are identified. Myeloid NGS detected DNMT3A (Q656*, VAF 47%) and TET2 (Q1828*, VAF 46%) mutations. The overall findings are consistent with myelodysplastic syndrome with low blasts (MDS LB) per the WHO, 5th edition, 2022 and myelodysplastic syndrome, not otherwise specified (MDS, NOS), with multilineage dysplasia per ICC (Blood 2023;141:437).
- Peripheral blood smear
- Peripheral blood smear shows mild normocytic anemia
- Red blood cells show anisopoikilocytosis including occasional teardrop forms, ovalocytes and acanthocytes
- Absolute neutropenia with shift to immaturity
- Thrombocytopenia with unremarkable morphology
- Bone marrow aspirate smear / touch preparation
- Bone marrow aspirate smear slides show several cellular spicules for evaluation
- Myeloid precursors show shift to immaturity with many hypolobated / monolobated neutrophils
- Erythroid precursors show progressive maturations with megaloblastic changes and occasional nuclear budding and irregular nuclear contour
- Megakaryocytes are decreased but occasional small monolobated megakaryocytes are present
- Touch preparations are cellular with similar findings to the bone marrow aspirates
- Small hypolobated / monolobated megakaryocytes are present
- Bone marrow core biopsy / particle clot
- Bone marrow core biopsy is fragmented and has variable cellularity, overall normocellular
- Myeloid precursors show progressive maturation with unremarkable morphology
- Small megakaryocytes are present
- Particle clot sections show many bone marrow particles for evaluation
- Marrow is hypercellular for age (~80% cellular)
- Myeloid precursors show progressive maturation with many hypolobated / monolobated neutrophils
- Erythroid precursors are relatively decreased and show progressive maturation with unremarkable morphology
- Megakaryocytes are present with small hypolobated / monolobated forms
- Bone marrow core biopsy is fragmented and has variable cellularity, overall normocellular
- Addendum: This addendum is being issued to report the results of additional germline genetic testing. Molecular testing of patient’s skin fibroblasts identified germline pathogenic intragenic deletion of RUNX1 (exons 5-6). This finding in conjunction with lifelong history of thrombocytopenia is consistent with myelodysplastic syndrome with germline predisposition (familial platelet disorder).
Differential diagnosis
- Presentation with pre-existing platelet disorder and risk of hematologic neoplasms
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorder due to germline ETV6 P / LP variant (familial platelet disorder with associated myeloid malignancy):
- Presentation with red cell macrocytosis and neutropenia
- Higher likelihood of progression to lymphoid neoplasms
- Thrombocytopenia 2 due to germline ANKRD26 P / LP variant:
- Presentation with erythrocytosis and leukocytosis
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorder due to germline ETV6 P / LP variant (familial platelet disorder with associated myeloid malignancy):
- Presentation without pre-existing platelet disorder and risk of hematologic neoplasms
- Acute myeloid leukemia with germline CEBPA P / LP variant (also known as CEBPA associated familial AML):
- Presentation with anemia and leukopenia
- Myeloid neoplasms with germline DDX41 P / LP variant:
- Bone marrow examination reveals megakaryocytic and erythroid dysplasia in some cases
- Myeloid neoplasms with germline TP53 P / LP variant (Li-Fraumeni syndrome):
- Patients with Li-Fraumeni syndromes are at high risk of not only hematologic neoplasms but also soft tissue sarcoma, osteosarcoma and breast cancer
- Myeloid neoplasms with germline GATA2 P / LP variant (GATA2 deficiency):
- Presentation with immunodeficiency
- Acute myeloid leukemia with germline CEBPA P / LP variant (also known as CEBPA associated familial AML):
Additional references
Board review style question #1
Which of the following is a characteristic phenotype of megakaryocytes in bone marrow biopsy of an asymptomatic individual with familial RUNX1 mutation (shown in the image above)?
- Atypical megakaryocytes with separated nuclear lobes
- Dysplastic megakaryocytes with nonlobated or bilobed nuclei
- Numerous micromegakaryocytes
- Small hypolobated / monolobated megakaryocytes
Board review style answer #1
D. Small hypolobated / monolobated megakaryocytes is a characteristic finding during bone marrow examination of individuals with germline RUNX1 pathogenic or likely pathogenic (P / LP) variant. It should be noted that megakaryocytes are atypical but not dysplastic (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661); thus, answer B is incorrect because nonlobated or bilobed megakaryocytes are common in acute myeloid leukemia (AML) with MECOM rearrangements. Answer A is incorrect because megakaryocytes with separated nuclear lobes are typical for GATA2 deficiency syndrome. Answer C is incorrect because the presence of numerous micromegakaryocytes in the bone marrow is frequently observed in myelodysplastic syndrome (MDS) with isolated del(5q) syndrome.
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation
Board review style question #2
A 48 year old patient with a history of lifelong thrombocytopenia and family history of thrombocytopenia and myeloid malignancies in their mother and maternal uncle was found to have pancytopenia during routine monitoring. Bone marrow examination revealed 3 - 4% blasts, erythroid precursors with unremarkable morphology and megakaryocytes with small monolobated forms. Conventional cytogenetic analysis did not reveal any chromosomal aberrations. Single nucleotide polymorphism (SNP) array analysis detected intragenic deletion of RUNX1 and next generation sequencing (NGS) revealed pathogenic mutations in TET2, DNMT3A and ASXL1. Which of the following detected genetic abnormalities is most likely a germline one?
- ASXL1 mutation
- DNMT3A mutation
- RUNX1 intragenic deletion
- TET2 mutation
Board review style answer #2
C. RUNX1 intragenic deletion. The patient's personal history and family history as well as small monolobated megakaryocytes are concerning for RUNX1 pathogenic or likely pathogenic (P / LP) variant, in this case intragenic deletion, which should be confirmed in cultured skin fibroblasts (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661). Answers A, B and D are incorrect because mutations in TET2, DNMT3A and ASXL1 are common during oncogenic progression and are somatic events.
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation

