
Bone marrow nonneoplastic
Alterations in cellularity
Fanconi anemia
Author: Dragos C. Luca, M.D.

Last author update: 1 March 2013
Last staff update: 26 September 2023
Copyright: 2002-2019, PathologyOutlines.com, Inc.
PubMed Search: Bone marrow Fanconi anemia

Table of Contents
Definition / general | Diagrams / tables | Clinical features and diagnosis | Laboratory | Case reports | Treatment and prognosis | Clinical images | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Molecular / cytogenetics images | Differential diagnosisCite this page: Luca DC. Fanconi anemia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrowfanconis.html. Accessed March 31st, 2025.
Definition / general
- Originally described in 1927 by Guido Fanconi, who reported 3 brothers with pancytopenia and multiple physical abnormalities (eMedicine: Fanconi Anemia)
- Most common inherited bone marrow failure syndrome; incidence at birth of 1 per 100 - 350K live births (Wikipedia: Fanconi Anemia)
- Typically autosomal recessive with intellectual disability and multiple bone, skin and renal abnormalities
- Associated with MDS / AML (15 - 20% of patients), hepatocellular adenoma
- Presence of significant birth defects correlates with early onset hematologic disease / anemia
- Note: Fanconi syndrome is a generalized dysfunction of proximal renal tubule transport with no primary glomerular involvement
Diagrams / tables
Images hosted on other servers:
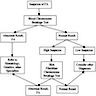
Diagnostic algorithm
Clinical features and diagnosis
- Usually diagnosed in infancy; 10% diagnosed at age 16 years or older; rarely diagnosed in 40's or 50's
- Prenatal diagnosis is possible by chromosome breakage analysis of chorionic villus, amniocentesis or cord blood lymphocyte samples
- Highly variable clinical features with occasional patients that are phenotypically normal (up to 30% according to some studies)
- Progressive lethal anemia associated with brown skin pigmentation, hypoplastic bone marrow, renal hypoplasia, absent or hypoplastic thumbs or radii, short stature and other anomalies; some patients have milder phenotype
- Thrombocytopenia: often the first evidence of impending hematopoietic failure
Laboratory
- Variable pancytopenia, macrocytosis, increased fetal hemoglobin and I antigen expression on erythrocytes (evidence of "stressed" erythropoiesis)
- Stimulated peripheral lymphocyte cultures: characteristic pattern of combined G2 phase prolongation and arrested G2 phase, detectable by flow cytometry
Case reports
- 4 year old boy diagnosed by genetic testing followed by prenatal diagnosis (Ann Lab Med 2012;32:380)
- 4 year old boy with Fanconi anemia and AML (Ann Clin Lab Sci 2011;41:66)
- 13 year old girl with Fanconi anemia and ALL (J Coll Physicians Surg Pak 2012;22:458)
Treatment and prognosis
- 50% improve with androgens; also G-CSF; stem cell transplantation
- Late development of myelodysplasia or AML in approximately 15 - 20% of patients
- Median survival is 7 years
Clinical images
Images hosted on other servers:
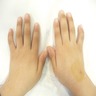
Clinodactyly with brachymesophalangia
Microscopic (histologic) description
- Early bone marrow specimens may be normocellular with megaloblastic features but gradual aplasia develops with severe pancytopenia by mid to late childhood
- Late stages: decreased / absent megakaryocytes, marked hypocellularity, rare small foci of erythropoiesis / myelopoiesis, fatty replacement
- Rare patients present directly with AML
Microscopic (histologic) images
Images hosted on other servers:
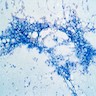
Hypocellular spicule with fatty infiltration
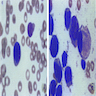
Aspirate smear findings
Molecular / cytogenetics description
- At the molecular level, 8 different genetic types of FA complementary groups have been described
- Several genes involved: FAC (9q22.3), FAA (16q24.3), FAG (19p13)
- Chromosomal instability due to increased number of chromosomal breaks, gaps, rearrangements and endoreduplication (OMIM: Fanconi Anemia, Complementation Group A; FANCA)
- 12 mutated genes identified to date; proteins are involved in repair of DNA damage
- Unique cytogenetic finding is increased sensitivity to apoptosis inducing effects of mitomycin C and diepoxybutane
Molecular / cytogenetics images
Images hosted on other servers:

Chromosome breakage test

Representative karyotypes
Differential diagnosis
