
Bone & joints
Other tumors
McCune-Albright syndrome
Deputy Editor-in-Chief: Borislav A. Alexiev, M.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 16 May 2022
Last staff update: 16 May 2022
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: McCune-Albright syndrome
See Also: Fibrous dysplasia, Mazabraud syndrome

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Alrohaibani A, Johnson TH, Davis JL. McCune-Albright syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonefibrousdysplasiamccunealbright.html. Accessed April 3rd, 2025.
Definition / general
- McCune-Albright syndrome (MAS) is a rare condition that results from postzygotic activating somatic mutations in the alpha subunit of the GNAS gene, leading to continued stimulation of endocrine glands
- Typically codon 201 and rarely codon 227 (Histopathology 2007;50:691)
- Exact presentation and severity depends on the extent of mosaicism and the involved tissues (Curr Osteoporos Rep 2015;13:146)
- Most patients are diagnosed in childhood or adolescence
Essential features
- Classically defined as the triad of
- Polyostotic fibrous dysplasia of bone
- Precocious puberty
- > 3 café au lait macules
- However, a more inclusive definition has been proposed, consisting of
- Monostotic fibrous dysplasia of bone or polyostotic fibrous dysplasia of bone
- Hyperfunctional endocrinopathy or café au lait macules (J Bone Miner Res 2006;21:P99)
Terminology
- McCune-Albright syndrome (MAS) has no synonyms
Epidemiology
- Very rare disorder affecting an estimated 1 in 100,000 to 1 in 1,000,000 people (Orphanet J Rare Dis 2008;3:12)
- F = M
Sites
- Fibrous dysplasia is most commonly found in the proximal femur and the skull base but can involve any bone (J Bone Miner Res 2007;22:1468)
- In contrast, nonsyndromic fibrous dysplasia is most commonly found in the ribs
- Any endocrine organ can be involved
- Café au lait macules are typically near, but do not cross, the midline (Orphanet J Rare Dis 2008;3:12)
Pathophysiology
- Constitutively active mutant GNAS results in increased cyclic adenosine monophosphate, which
- Promotes proliferation and inhibits differentiation of fibroblasts, which replace normal bone and produce fibrous dysplasia lesions
- Promotes proliferation and autonomous function of endocrine organs
- Fibrous dysplasia elaborates fibroblast growth factor 23, which
- Inhibits renal resorption of phosphate
- Causes hypophosphatemia
- Causes hyperphosphaturia
- Examples:
- Unregulated production of cortisol, leading to Cushing syndrome
- Unregulated production of gonadotropin releasing hormone, leading to precocious puberty (Curr Osteoporos Rep 2015;13:146)
Etiology
- Results from de novo mutations in GNAS (Curr Osteoporos Rep 2015;13:146)
- No inherited case has ever been reported
- Germline GNAS mutation is presumed embryonic lethal
- Has no risk factors
Clinical features
- 2 most common presentations are fibrous dysplasia related pain and precocious puberty
- Fibrous dysplasia
- May be monostotic or polyostotic
- When polyostotic, it may be unilateral, in keeping with postzygotic mutation
- Classical and most common endocrinopathy is precocious puberty; however, other endocrinopathies include
- Hypophosphatemia
- Hyperparathyroidism
- Cushing disease
- Acromegaly
- Macroorchidism
- Café au lait macules typically
- Are light brown in color but can vary depending on patient's skin color
- Are present at, or shortly after, birth
- Are recognized after another feature of the disease has prompted a skin survey
- Are near the midline
- Respect the lines of Blaschko
- Have jagged irregular borders, unlike the smooth borders that are characteristic of macules seen in neurofibromatosis
- Are ipsilateral with fibrous dysplasia (Orphanet J Rare Dis 2008;3:12)
Diagnosis
- Essentially a clinical diagnosis
- Genetic testing:
- Not routinely performed but available
- False negatives are common due to mosaicism and sampling
- Positive results merely confirm a clinical diagnosis
- No genotype / phenotype correlation, therefore no prognostic value (Orphanet J Rare Dis 2008;3:12)
- When there is clinical suspicion:
- Skeletal survey to identify lesions suspicious for fibrous dysplasia
- CT is more sensitive for craniofacial fibrous dysplasia
- Biopsy can confirm fibrous dysplasia
- Skin survey to identify café au lait macules
- If not apparent at presentation, endocrinopathies may become apparent with laboratory studies and focused examinations
Laboratory
- Some patients have multiple endocrinopathies; others have none
- The following laboratory abnormalities may be present in any combination (Orphanet J Rare Dis 2008;3:12):
- Depressed serum phosphate
- Elevated urine phosphate
- Elevated cortisol
- Elevated sex steroid hormones
- Elevated thyroid hormone
- Depressed thyroid stimulating hormone
Radiology images
Images hosted on other servers:

Ulnar fracture

Lucency in long bones
Prognostic factors
- Severity of hypophosphatemia and number / extent of fibrous dysplasia lesions correlates with the risk of bone pain and pathologic fracture (J Bone Miner Res 2016;31:2167)
- Sites of fibrous dysplasia are established early and burden of disease typically plateaus by age 15 (Expert Opin Investig Drugs 2007;16:761)
Case reports
- Preterm neonate girl with Cushing syndrome, hyperthyroidism and café au lait macules (J Med Case Rep 2015;9:189)
- Neonate girl with severe cholestasis requiring liver transplant (J Clin Res Pediatr Endocrinol 2019;11:100)
- 4 year old girl also diagnosed with Mazabraud syndrome (Acta Biomed 2017;88:198)
- 39 year old man whose autopsy revealed extensive mosaicism for GNAS R201C mutation and subclinical disease manifestations in multiple organs (J Clin Endocrinol Metab 2014;99:E2029)
Treatment
- There is no specific therapy
- Various endocrinopathies are treated just as their sporadic counterparts
- Fibrous dysplasia is treated just as its sporadic counterpart (StatPearls: McCune Albright Syndrome [Accessed 27 April 2022])
Clinical images
Images hosted on other servers:

5 year old girl: jagged "coast of Maine" borders
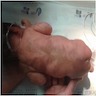
Café au lait spots
Gross images
Images hosted on other servers:
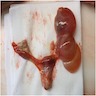
Ovarian abnormalities
Microscopic (histologic) description
- Fibrous dysplasia lesions are histologically indistinguishable from nonsyndromic fibrous dysplasia, characterized by (Arch Pathol Lab Med 2013;137:134)
- Woven bone with trabeculae that are thin, irregular and curvilinear
- Fibrous stroma
- Absence of osteoblastic rimming
Microscopic (histologic) images
Contributed by Jessica L. Davis, M.D.
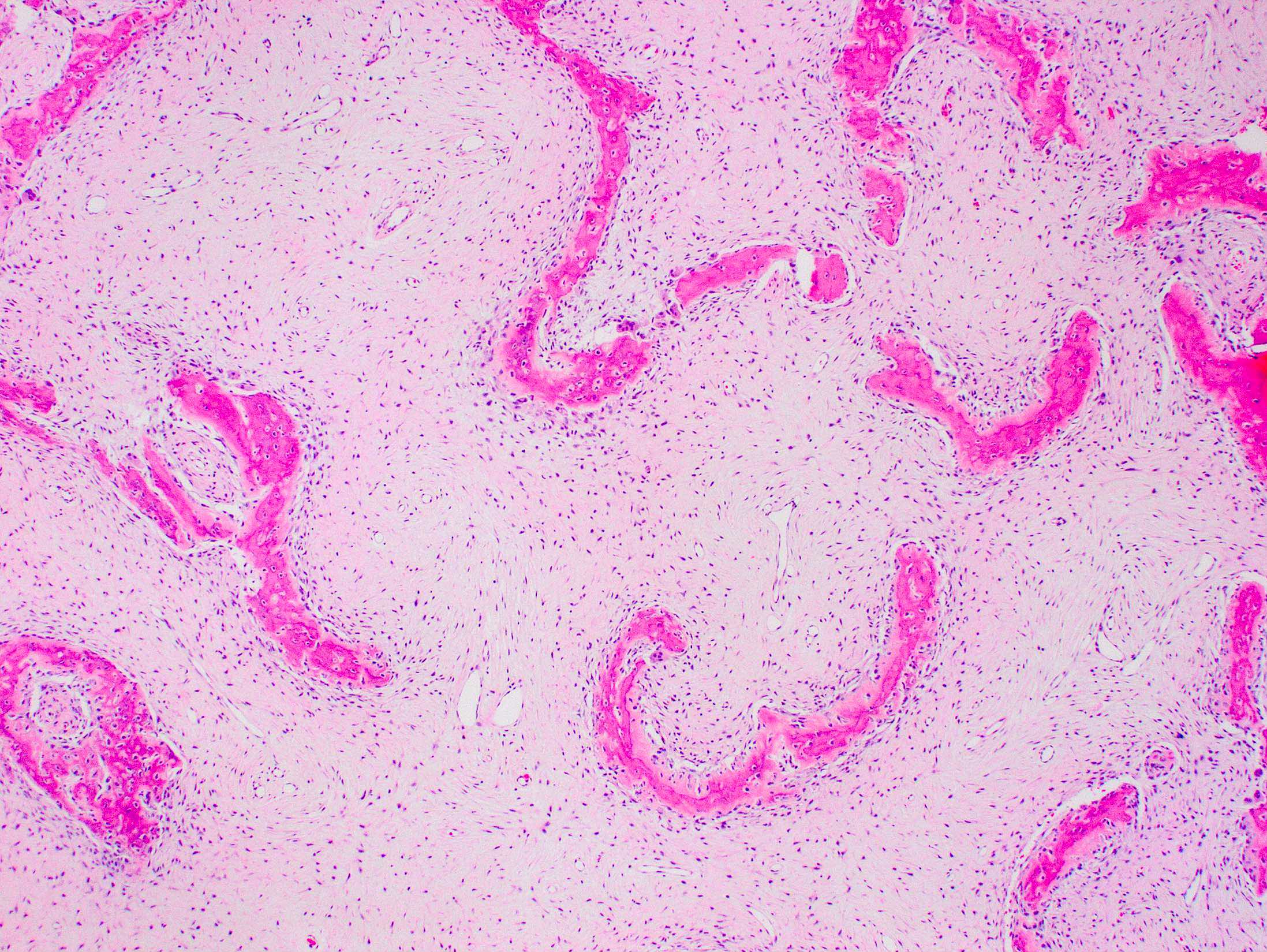
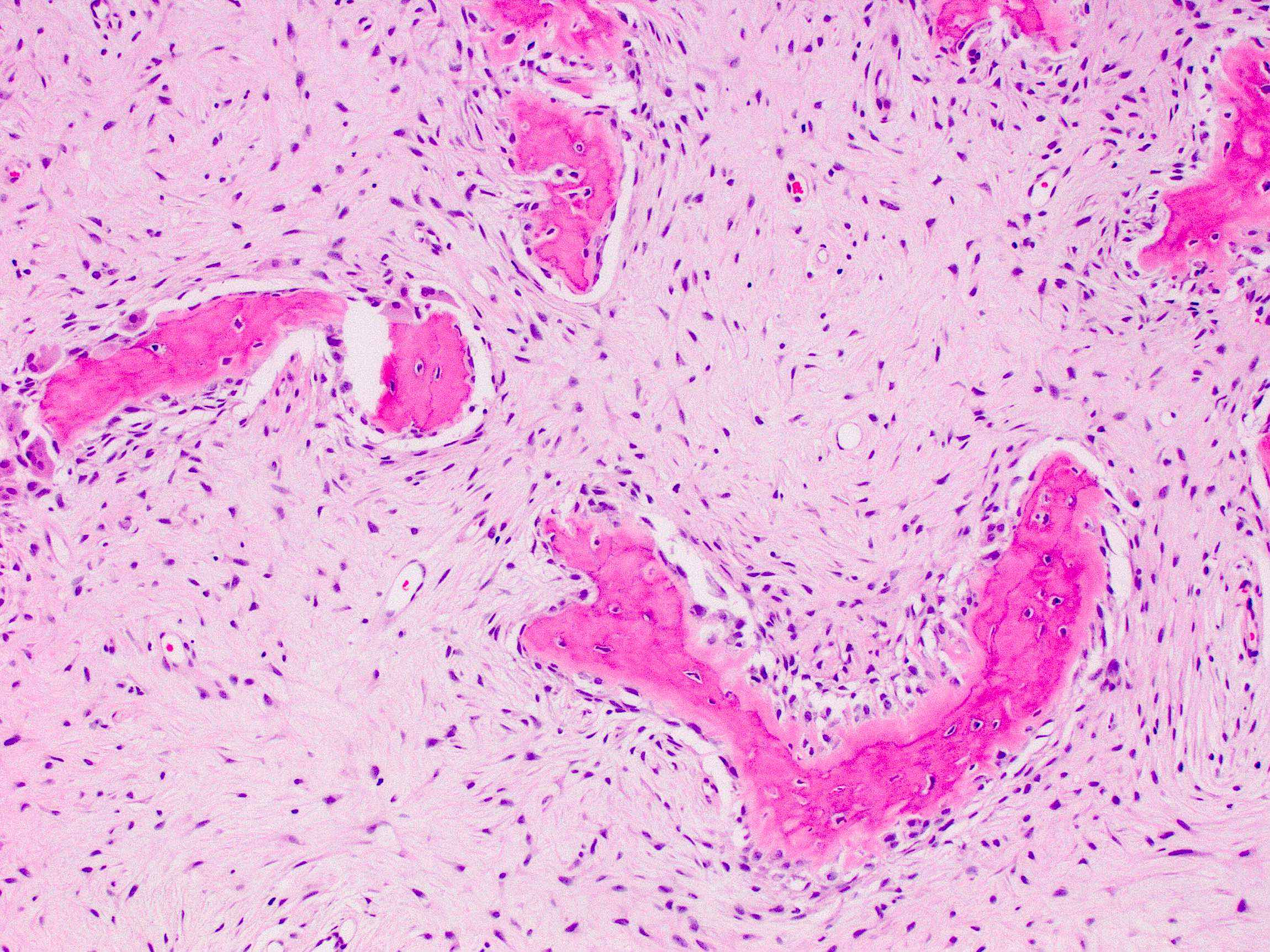
Fibrous dysplasia
Molecular / cytogenetics description
- Mutation specific restriction enzyme digest (MSRED) method may identify mutations in codon 201 or 227 of the GNAS1 gene (Histopathology 2007;50:691)
Sample pathology report
- Bone, biopsy:
- Fibrous dysplasia of bone (see comment)
- Comment: Fibrous dysplasia is most commonly sporadic but is also a typical presenting feature of McCune-Albright syndrome and Mazabraud syndrome. Clinical correlation is advised.
Differential diagnosis
- Nonsyndromic fibrous dysplasia of bone:
- Results from postzygotic GNAS mutations
- Other features of McCune-Albright syndrome are absent or as yet undeclared
- Endocrinopathies are absent
- Café au lait macules are absent
- Mazabraud syndrome:
- Results from postzygotic GNAS mutations
- Presents with polyostotic fibrous dysplasia and intramuscular myxomas
- Typically diagnosed in adulthood
- Patients may satisfy criteria for both McCune-Albright syndrome and Mazabraud syndrome
- Hyperparathyroidism jaw tumor syndrome:
- Severe hyperparathyroidism from parathyroid adenoma or carcinoma
- Ossifying fibromas of jaw may be confused with fibrous dysplasia:
- Randomly distributed mature (lamellar) bone spicules rimmed by osteoblasts admixed with a fibrous stroma
Additional references
Board review style question #1
A 23 year old man diagnosed at age 8 with McCune-Albright syndrome is considering starting a family and seeks genetic counseling. Which of the following best describes his likelihood of having an affected child?
- 0% chance of having an affected child
- 25% chance of having an affected female child
- 25% chance of having an affected male child
- 50% chance of having an affected child
- 100% chance of having an affected child
Board review style answer #1
A. 0% chance of having an affected child. McCune-Albright syndrome results from postzygotic mutations and an inherited case has never been reported. Germline mutation in GNAS is believed to be embryonic lethal. The likelihood that the patient will have an affected child is effectively 0% (Curr Osteoporos Rep 2015;13:146).
Comment Here
Reference: McCune-Albright syndrome
Comment Here
Reference: McCune-Albright syndrome
Board review style question #2
A 6 year old girl is brought to her pediatrician by her father who is concerned about her early breast development. On exam, the patient's breasts are Tanner stage 3 and the clinician also notes a large, irregularly contoured pigmented macule on her left flank. In addition to starting letrozole, which of the following is an appropriate next step?
- Diagnose the patient with McCune-Albright syndrome and order a skeletal survey
- Perform a complete skin survey in clinic and order a skeletal survey
- Perform a punch biopsy at the edge of the macule
- Send a blood sample for sequencing of GNAS
- Send a tissue sample for sequencing of GNAS
Board review style answer #2
B. Perform a complete skin survey in clinic and order a skeletal survey. A complete skin survey and skeletal survey would potentially reveal additional findings that would rule in or provisionally rule out McCune-Albright syndrome (MAS). As presented in the question, the patient has findings suggestive of but not diagnostic for MAS, making answer A wrong. A punch biopsy, as in answer C, would likely reveal melanocyte proliferation but would not aid in diagnosis or direct treatment. Sequencing studies, as in answers D and E, if negative, would not rule out MAS and if positive, would only confirm a clinical diagnosis; thus, neither is contributory (Curr Osteoporos Rep 2015;13:146).
Comment Here
Reference: McCune-Albright syndrome
Comment Here
Reference: McCune-Albright syndrome
