
Adrenal gland & paraganglia
Adrenal cortical carcinoma
Sarcomatoid
Author: Maria Tretiakova, M.D., Ph.D.
Editorial Board Members: Debra L. Zynger, M.D., Bonnie Choy, M.D.

Last author update: 10 January 2022
Last staff update: 2 December 2022
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Sarcomatoid variant adrenal


Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1Cite this page: Tretiakova M. Sarcomatoid. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/adrenocorticalcarcinomacarcinosarcoma.html. Accessed March 31st, 2025.
Definition / general
- Extremely rare malignant tumor of adrenal cortex with biphasic carcinomatous and sarcomatous morphology
Essential features
- Least common variant of adrenal cortical carcinoma (ACC), ~30 cases reported
- Sarcomatoid ACC is a particularly aggressive tumor with a dismal prognosis
- Composed of malignant epithelial adrenocortical cells and specialized mesenchymal elements resembling various sarcomas, often with heterologous features of rhabdomyoblastic, osteogenic, chondroid or PNET-like differentiation
- Compared to conventional ACC, sarcomatoid variant presents as:
- Predominately nonfunctioning tumors
- Predilection for the right adrenal
- Larger tumor size
- Higher rate of distant metastases and shorter median survival
- Older patient population
Terminology
- Adrenal cortical carcinoma (ACC), sarcomatoid variant
- Adrenocortical carcinoma, sarcomatoid type
- Adrenal carcinosarcoma
ICD coding
- ICD-O: 8370/3 - adrenal cortical carcinoma
Epidemiology
- Least common of the recognized histological variants (Virchows Arch 2012;460:9, Biomedicines 2021;9:175)
- Mean age: 56, range: 23 - 79 (Hum Pathol 2013;44:1947, Ulster Med J 2014;83:89, Biomedicines 2021;9:175)
- Slightly more common in females; M:F = 1:1.3 (Biomedicines 2021;9:175)
Sites
- Right adrenal more commonly affected; left to right ratio = 1:1.4 (Biomedicines 2021;9:175)
- 2 cases with bilateral disease (Biomedicines 2021;9:175)
Pathophysiology
- 4 theories of sarcomatoid ACC histogenesis (Virchows Arch 2012;460:9):
- Conversion tumor theory with neoplastic transformation of epithelial to mesenchymal cells
- Composition tumor theory (paradoxical reactive proliferation of nonepithelial component induced by the epithelial component via paracrine secretion)
- Collision or biclonal tumor theory (2 synchronous, histologically independent tumors of different clones)
- Combination or divergent tumor theory (deriving from a common monoclonal stem cell precursor); best supported by molecular studies (Hum Pathol 2016;58:113)
Etiology
- Sporadic tumors with no established risk factors
- Acquired genetic mutations of several driver genes (P53, CTNNB1, CDKN2A, TERT, ZNRF3, PRKAR1A) (Cancer Cell 2016;29:723, Turk Patoloji Derg 2015;31:98, Mod Pathol 2018;31:1257, Hum Pathol 2016;58:113)
Diagrams / tables
Images hosted on other servers:

Survival analysis:
sarcomatoid ACC
versus other
variants
Clinical features
- Vast majority presented with localized pain (abdominal pain and distension, loin pain, flank pain, back pain) related to the mass effect (64 - 90% of cases) (Biomedicines 2021;9:175)
- Hormone production in 11% of all reported sarcomatoid ACC (Biomedicines 2021;9:175)
- Hyperaldosteronism (Am J Surg Pathol 1993;17:941)
- Sex hormone production (virilization) (Am J Surg Pathol 1992;16:626)
- 75 - 80% with sarcomatoid ACC have metastases at presentation or within 4 months after surgery compared to 30% with conventional ACC (Biomedicines 2021;9:175)
- Metastatic sites include liver (60%), lung (30%), spleen, brain and heart (Biomedicines 2021;9:175, J Korean Med Sci 2017;32:764)
- Average survival of 6 - 7 months (Biomedicines 2021;9:175)
- Longest reported postoperative survival with no evidence of metastases was 17 months (Ulster Med J 2014;83:89)
Diagnosis
- Sarcomatoid ACC on imaging (CT, MRI) has large size, irregular contours, lipid poor characteristics and significant signal heterogeneity (Endocr J 2019;66:739, World J Surg Oncol 2015;13:117)
- Radiographically and even during gross evaluation, often difficult to confirm adrenal origin due to the advanced presentation
- By definition, tumor should be composed of adrenocortical epithelial malignant cells and sarcoma-like mesenchymal component on microscopy
- No minimum percentage of sarcomatoid cells required for the diagnosis (Biomedicines 2021;9:175)
- Weiss system does not account for sarcomatoid histology and may not be applicable to these tumors, especially the criteria of diffuse architecture (Virchows Arch 2012;460:9)
Laboratory
- Same as conventional ACC
Radiology description
- Same as conventional ACC
Radiology images
Images hosted on other servers:

CT scan: necrotic areas replacing adrenal gland
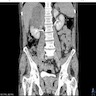
Coronal abdominal CT with large adrenal mass
Prognostic factors
- Sarcomatoid ACC has a dismal prognosis, worse than conventional ACC
- Despite aggressive treatment, overall median survival for sarcomatoid ACC is 7 months (Horm Cancer 2011;2:333, Biomedicines 2021;9:175, Ulster Med J 2014;83:89)
Case reports
- 29 year old woman with adrenal carcinosarcoma presenting with virilization (Am J Surg Pathol 1992;16:626)
- 45 year old man with oncocytic adrenal cortical carcinosarcoma with pleomorphic rhabdomyosarcomatous metastases (Am J Surg Pathol 2012;36:470)
- 48 year old woman with adrenocortical carcinosarcoma with oncocytic and primitive neuroectodermal-like features (Hum Pathol 2013;44:1947)
- 53 year old woman with sarcomatoid adrenal carcinoma with 2 liver metastases (Endocr Pathol 2017;28:139)
- 69 year old woman with bilateral sarcomatoid carcinoma of adrenal glands with adrenal insufficiency (Int J Surg Pathol 2016;24:743)
Treatment
- Surgical: radical resection is treatment mainstay (Surgery 2019;166:524)
- Systemic adjuvant chemotherapy, in addition to surgery, has no clear survival benefit compared with surgical resection alone (Hum Pathol 2013;44:1947, Ulster Med J 2014;83:89)
Gross description
- Slightly larger than conventional ACC (Biomedicines 2021;9:175)
- Median tumor size: 13 - 14.5 cm (range: 5 - 24 cm) (Hum Pathol 2013;44:1947, Biomedicines 2021;9:175)
- Median weight: 620 - 1,743 g (range: 199 - 6,500 g) (Hum Pathol 2013;44:1947, Biomedicines 2021;9:175)
- Partially encapsulated variegated yellow-gray or whitish fleshy mass
- Extensive hemorrhage and necrosis with cystic degeneration (Ulster Med J 2014;83:89)
- ~25% of sarcomatoid ACC had cystic appearance (Biomedicines 2021;9:175)
Gross images
Image hosted on other servers:

Whitish fleshy, variegated hemorrhagic and necrotic surface

Tumor adherent
to pancreas (A)
and compressing
kidney (B)
Microscopic (histologic) description
- 2 distinct morphologic presentations (Hum Pathol 2013;44:1947, Ulster Med J 2014;83:89):
- Both carcinomatous and sarcomatous components (83%)
- Pure sarcomatoid morphology (17%)
- Carcinomatous (epithelioid) component is similar to conventional ACC:
- Sheets and nests of loosely cohesive polygonal cells with clear or eosinophilic cytoplasm resembling adrenocortical cells
- Sarcomatous component:
- Mainly composed of spindle tumor cells arranged in haphazard fascicular pattern (Biomedicines 2021;9:175, J Korean Med Sci 2017;32:764, Hum Pathol 2016;58:113, Virchows Arch 2008;452:215)
- Heterologous elements resembling rhabdomyosarcoma, osteosarcoma, chondrosarcoma, angiosarcoma, liposarcoma or PNET-like (Pathol Res Pract 2010;206:59, Ulster Med J 2014;83:89, Cancer Cell 2016;29:723, Biomedicines 2021;9:175)
- Necrosis, brisk mitoses, numerous atypical mitoses and bizarre giant cells very common (Ulster Med J 2014;83:89, Virchows Arch 2012;460:9)
- Mitotic count is uniformly high; 3 - 4 times higher in sarcomatoid component compared to epithelioid (Virchows Arch 2012;460:9)
Microscopic (histologic) images
Contributed by Maria Tretiakova, M.D., Ph.D.
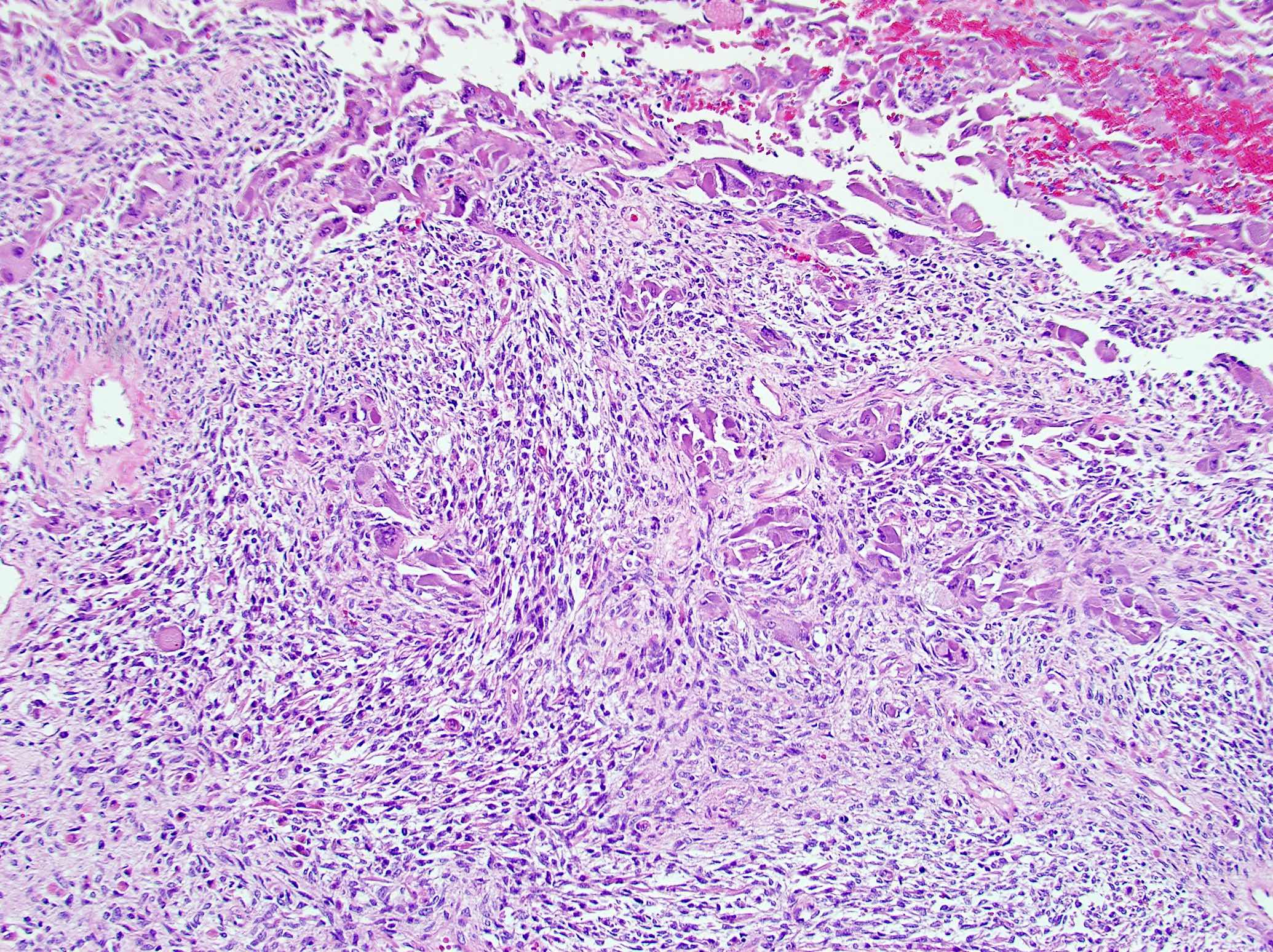
Sarcomatoid ACC biphasic
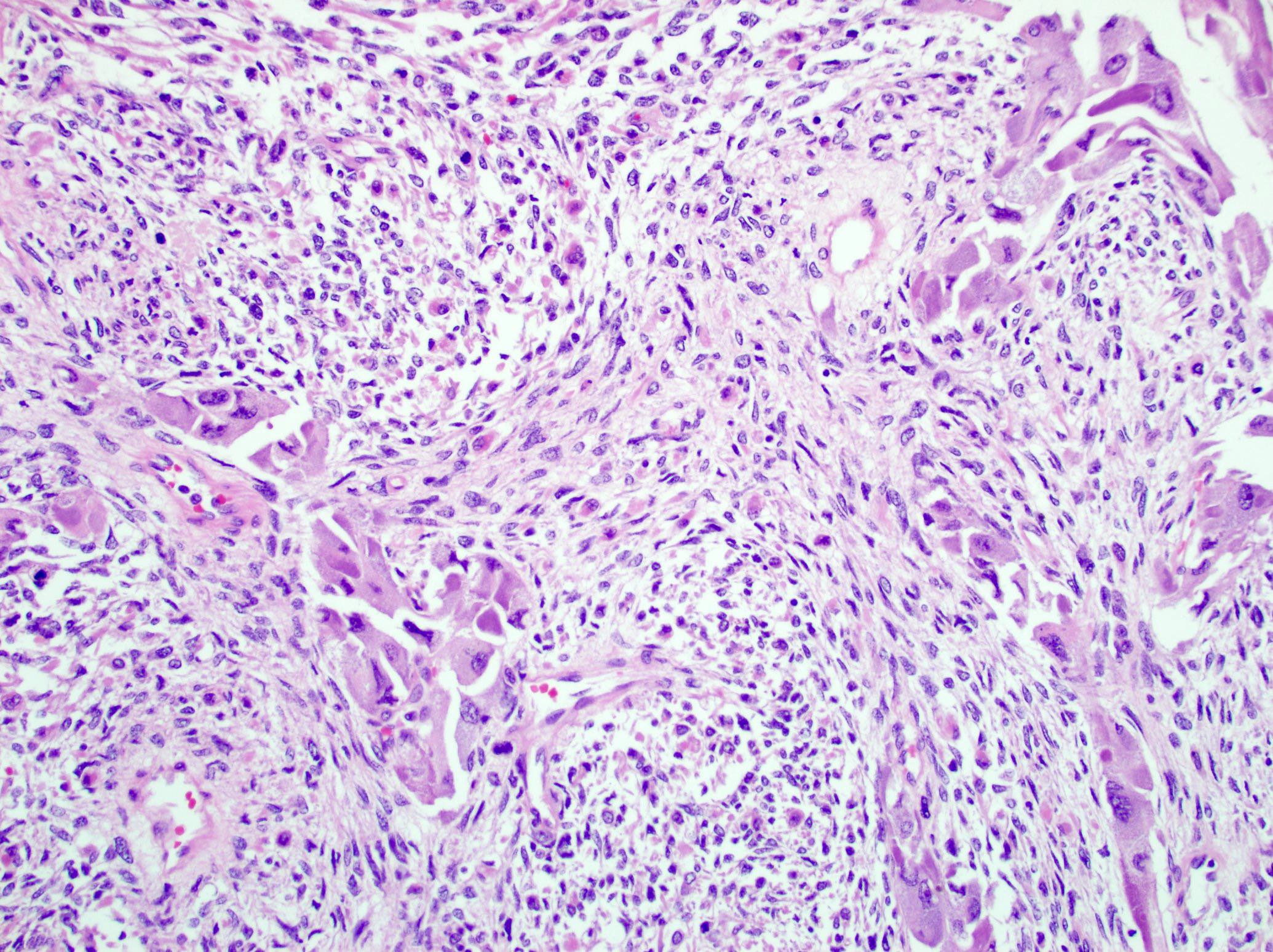
Epithelioid and spindle cells
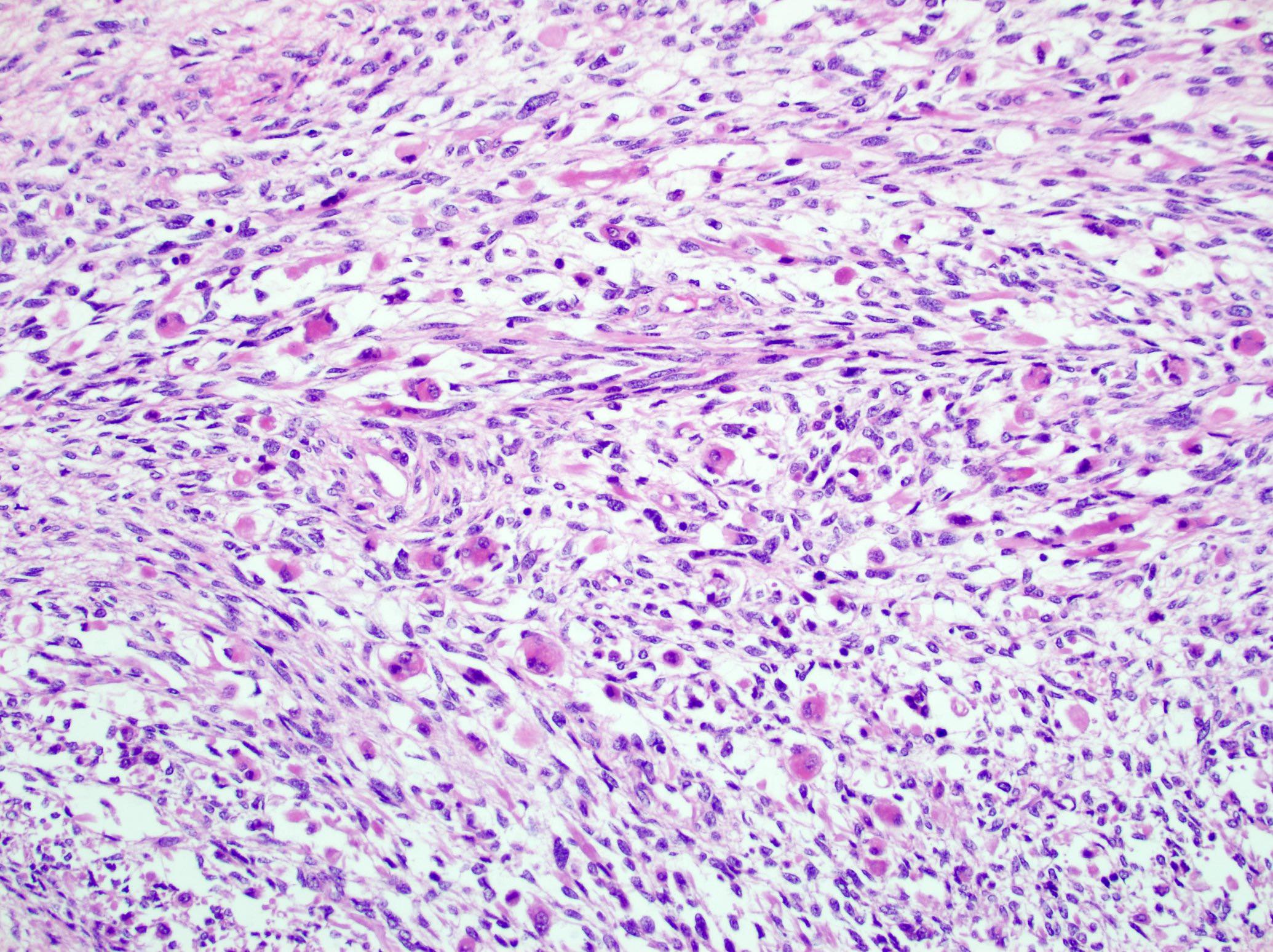
Heterologous differentiation
Positive stains
- Carcinomatous component positive for adrenocortical markers: SF1, inhibin, calretinin, MelanA, synaptophysin, NSE, S100, vimentin, AE1 / AE3, CAM 5.2
- Sarcomatoid component often has partial or complete loss of inhibin, MelanA and SF1 but is positive for cytokeratins, vimentin, SMA, myogenin, desmin, h-caldesmon, p53 and beta catenin (nuclear)
- Epithelial mesenchymal transition (EMT) related markers in sarcomatoid component: MMP2, MMP9, Slug, caveolin-1
- Stem cell markers in sarcomatoid component: nestin, SOX2, SOX17, LIN28
- Ki67 proliferation rates are very high (15 - 60%) (World J Surg Oncol 2015;13:117, J Korean Med Sci 2017;32:764, Ulster Med J 2014;83:89, World J Surg Oncol 2015;13:117)
Negative stains
Electron microscopy description
- Epithelioid component with tight junctions and abundant mitochondria, similar to conventional ACC
Electron microscopy images
Images hosted on other servers:

Epithelioid and spindle cell components
Molecular / cytogenetics description
- Sarcomatoid ACC had the lowest adrenocortical differentiation scores with very low expression of NR5A1 (SF1 regulator) and steroidogenic enzymes (Cancer Cell 2016;29:723)
- Wnt beta catenin signaling pathway dysregulation and mutational inactivation of TP53 are common genetic events in sarcomatoid ACC (Histopathology 2018;72:82)
- Enriched for EMT related markers and stem cell factors that may be associated with the poor prognosis of these tumors and may provide possible therapeutic targets (Histopathology 2018;72:82, Biomedicines 2021;9:175, Hum Pathol 2016;58:113)
- In differentiated epithelioid component, molecular alterations are similar to conventional ACC including high mutation burden, massive DNA loss followed by whole genome doubling and frequent somatic mutations of P53, CTNNB1, CDKN2A, TERT, ZNRF3, PRKAR1A (Histopathology 2018;72:82, Cancer Cell 2016;30:363)
- Concordant molecular alterations in phenotypically diverse components (i.e. presence of TP53 and CTNNB1 gene mutations) were detected in 50% of studies by NGS tumors and support a common clonal origin (Hum Pathol 2016;58:113)
Sample pathology report
- Adrenal gland, right, adrenalectomy:
- Adrenal cortical carcinoma with the following features:
- Tumor size: 19 cm x 18 cm x 12 cm
- Tumor (gland) weight: 600 g
- Tumor extent: invasion into inferior vena cava and inferior liver surface
- Histologic type: sarcomatoid variant
- Histologic grade: high grade
- Necrosis: present
- Lymphovascular invasion: present
- Margins: positive
- pTNM, AJCC 8th edition: pT4 N0
- Ancillary studies: Ki67 mitotic rate 40%
- Adrenal cortical carcinoma with the following features:
Differential diagnosis
- Adrenal cortical carcinoma, conventional:
- No sarcomatoid component
- Sarcomatoid renal cell carcinoma or urothelial carcinoma:
- Sarcomas (no epithelioid component):
Board review style question #1
A 68 year old woman presented with right sided back pain and weight loss. Abdominal ultrasonography and computed tomography (CT) showed the presence of a 12 cm heterogeneous adrenal mass. It was biopsied, showing biphasic morphology with large epithelioid polygonal cells and haphazardly arranged spindle cells. Which statement about this adrenal cortical tumor is accurate?
- Pure sarcomatoid morphology is more common than biphasic
- Hormonal production is typically abnormal
- Clinical presentation and prognosis are similar to conventional adrenocortical carcinoma (ACC)
- Adrenocortical immunomarkers are often lost in sarcomatoid areas
- Risk of metastatic disease and recurrence is low
Board review style answer #1
D. This is a case of sarcomatoid ACC, an extremely rare malignant tumor with biphasic epithelioid and mesenchymal differentiation. The carcinomatous component is strongly positive for inhibin, MelanA, calretinin and SF1, thus supporting an adrenocortical origin. However, the sarcomatoid component often has a partial or complete loss of adrenocortical markers. Fortunately, > 80% of sarcomatoid ACC has both carcinomatous and sarcomatoid components (A). Functional sarcomatoid ACCs with hormone overproduction are documented in only 11% of cases (B). Compared to conventional ACC, the sarcomatoid variant has a more advanced age at presentation, predominately nonfunctional tumors, predilection of the right side, larger tumor size, higher rate of distant metastases and shorter median survival (C). 75 - 80% of patients with sarcomatoid ACC develop metastases or recurrence within 4 months and have an overall dismal prognosis (E).
Comment Here
Reference: Adrenal cortical carcinoma, sarcomatoid variant
Comment Here
Reference: Adrenal cortical carcinoma, sarcomatoid variant
