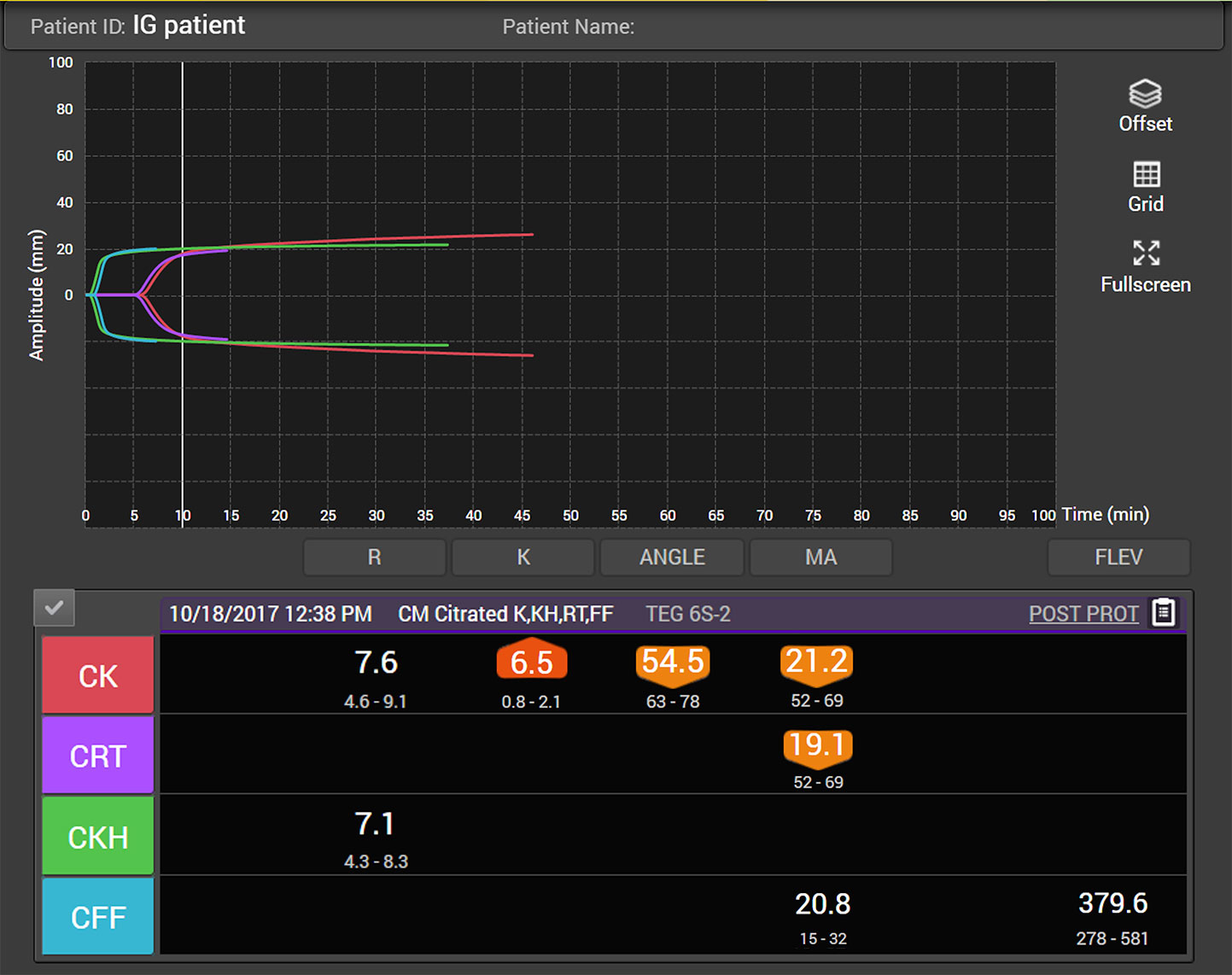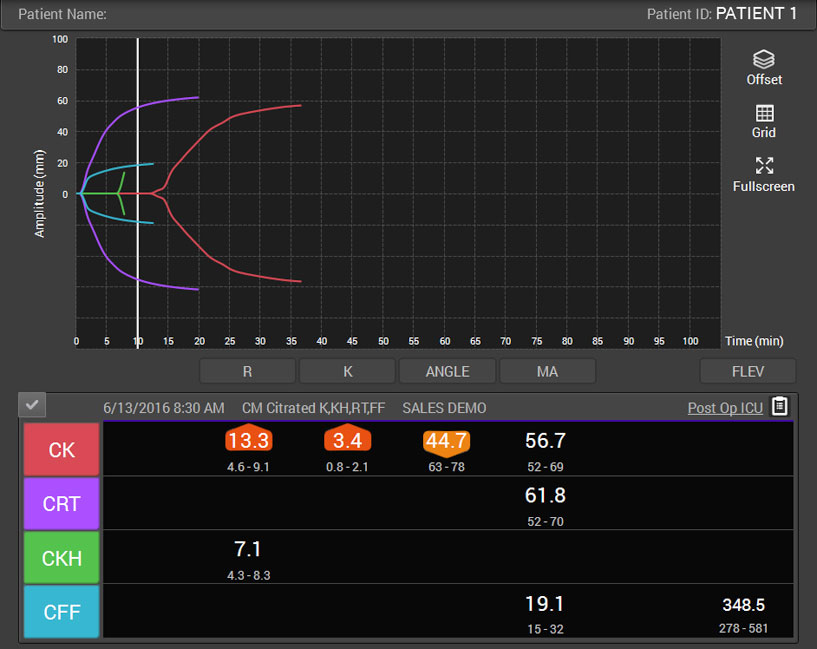









Primary hemostasis
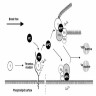
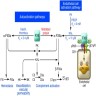
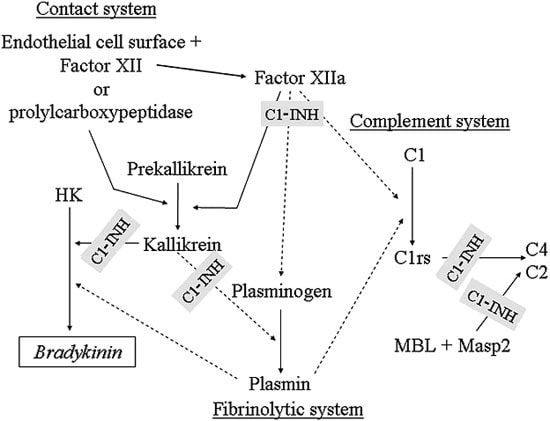
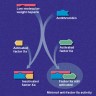
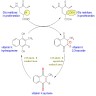
- During injury to vascular endothelium, the first response to prevent blood loss is vasoconstriction which activates endothelial cells (Ann Med 2012;44:405)
- Formation of the platelet plug in primary hemostasis is primarily orchestrated by interactions between the endothelium, von Willebrand factor (vWF) and platelets (Arterioscler Thromb Vasc Biol 2020;40:1441)
- Activated endothelial cells secrete vWF, which recruits platelets to the site of injury and results in a sequence of platelet adhesion, activation and aggregation (Int J Biochem Cell Biol 2021;131:105900, Hematol Oncol Clin North Am 2021;35:1069)
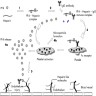
- Platelet adhesion occurs when vWF, bound to exposed subendothelial collagen, interacts with glycoprotein Ib / IX / V on platelets (Arterioscler Thromb Vasc Biol 2008;28:403)
- vWF is a plasma glycoprotein, stored in alpha granules secreted by platelets and Weibel-Palade bodies in the endothelium (Blood 2015;125:2019)
- Platelet activation results in changes in platelet and membrane morphology and secretion of microparticles from intracellular granules
- Upon activation, platelet cytoplasm and cytoskeleton rearrange to increase surface area and enable adherence to endothelium (Thromb Haemost 2002;88:186)
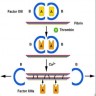
- Platelet aggregation is mediated by glycoprotein IIb / IIIa (GpIIb / IIIa) on the platelet surface
- During platelet activation, GpIIb / IIIa undergoes a conformational change into the active form; activated GpIIb / IIIa has high affinity for ligand binding and promotes platelet aggregation into the initial platelet plug (Blood 2007;109:5087)