
Small intestine & ampulla
Other malignancies
Neuroendocrine tumor
Authors: Megan Kinn, D.O., M.S., Yue Xue, M.D., Ph.D.
Editorial Board Member: Aaron R. Huber, D.O.
Deputy Editor-in-Chief: Catherine E. Hagen, M.D.


Last author update: 26 July 2021
Last staff update: 12 July 2023
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Neuroendocrine tumor[title] small intestine


Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Kinn M, Xue Y. Neuroendocrine tumor. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/smallbowelcarcinoidtumor.html. Accessed March 28th, 2025.
Definition / general
- Well differentiated duodenal, jejunal and ileal epithelial neoplasms with neuroendocrine differentiation
- Subtypes:
- Neuroendocrine tumor, grades 1 - 3
- Gastrinoma, NOS
- Somatostatinoma, NOS
- Enterochromaffin cell carcinoid
Essential features
- Neuroendocrine tumors (NETs) of the small intestine and ampulla are rare, although the incidence is increasing
- Majority of tumors are nonfunctioning; functional (hormone secreting) neuroendocrine tumors are rare
- Histologic features include monotonous tumor cells with finely stippled chromatin arranged in nests, trabeculae or tubuloglandular architecture
- Grading is based on mitotic rate and Ki67 index
Terminology
- Intestinal NET: carcinoid (not preferred), well differentiated neuroendocrine tumor
ICD coding
Epidemiology
- Prevalence increased from 0.006% in 1993 to 0.048% in 2012
- NETs are more prevalent in women than in men (2.5:1)
- Incidence of GI-NET 3.56 per 100,000 population
- Incidence of small intestinal NET 1.05 per 100,000 persons
- References: World J Gastrointest Oncol 2020;12:791, JAMA Oncol 2017;3:1335
Sites
- > 95% duodenal NETs located in part 1 or 2 (those in part 2 are predominantly in the ampullary region)
- Gastrin expressing NETs: duodenum
- Somatostatin expressing NETs: ampulla
- Majority of jejunoileal NETs are located in the distal ileum, with only 11% of jejunal origin:
- Serotonin expressing enterochromaffin cell NETs: Meckel diverticula
Pathophysiology
- Poorly understood at this time
Etiology
- Etiology of sporadic intestinal NETs is unknown
- Minority of NETs arise in setting of hereditary cancer predisposition syndromes
- Multiple endocrine neoplasia (MEN) type 1: develop multiple duodenal gastrinomas (Neuroendocrinology 2017;104:112)
- 10% of ampullary somatostatinomas arise in the setting of neurofibromatosis type 1 (NF1) (J Gastrointest Surg 2010;14:1052)
Clinical features
- Nonfunctioning NETs: patients may present with symptoms of mass effect (small bowel obstruction, jaundice)
- Functioning NETs: can cause symptoms related to hormone secretion
- Gastrinomas: cause Zollinger-Ellison syndrome (hypergastrinemia, gastric hypersecretion, refractory peptic ulcer disease, diarrhea) (Int J Endocr Oncol 2017;4:167)
- Patients with Zollinger-Ellison syndrome have worse prognosis and higher rate of metastasis
- Somatostatinomas: rarely cause somatostatinoma syndrome (diabetes mellitus, diarrhea, steatorrhea, hypohydrea or achlorhydia, anemia, gallstones) (Endocr Relat Cancer 2008;15:229)
- Carcinoid syndrome (diarrhea, bronchospasms, flushing, tricuspid valve fibrosis) occurs in < 10% of cases and only if liver metastasis is present (Endocr Relat Cancer 2008;15:229)
- Gastrinomas: cause Zollinger-Ellison syndrome (hypergastrinemia, gastric hypersecretion, refractory peptic ulcer disease, diarrhea) (Int J Endocr Oncol 2017;4:167)
Diagnosis
- Most small intestine and ampullary NETs are detected on endoscopy and diagnosed with biopsy
Laboratory
- Well differentiated NETs are associated with elevated serum chromogranin A
- Increases with larger tumor burden
- Not recommended as a screening test due to low specificity
- May be used to assess disease progression, response to therapy or recurrence in patients with established advanced NET
- Clinical importance decreasing due to day to day variation in chromogranin levels and lack of a standard assay
- 24 hour urinary excretion of 5-HIAA is the preferred initial diagnostic test for carcinoid syndrome (sensitivity > 90%, specificity 90%)
- 5-HIAA is the end product of serotonin metabolism
- Most useful for jejunoileal, appendiceal and ascending colon NETs because they secrete the highest levels of serotonin
Radiology description
- CT or MRI considered mandatory for radiologic staging of NETs
- CT and MRI have poor sensitivity for detection of primary small bowel NETs
- Somatostatin receptor scintigraphy has been used for decades to image and survey small intestinal NETs
- Currently, PET and concomitant CT with 68Ga-DOTA somatostatin analogs (SSAs) are preferred, given the better detection of distant metastases
Radiology images
Images hosted on other servers:

PET / CT
Prognostic factors
- Ampullary NETs:
- 5 year survival rate of 82%; 10 year survival rate 71% (Arch Pathol Lab Med 2010;134:1692)
- Tumors < 2 cm or limited to ampulla have better prognosis
- Lymph node metastases are frequent but do not influence overall and disease free survival rates (Langenbecks Arch Surg 2012;397:933)
- Liver is the most common site of metastasis
- Small intestine NETs:
- Often present with metastatic disease (locoregional lymph nodes and liver most common)
- Most have prolonged survival due to low proliferative rate (World J Surg 2012;36:1419)
- Localized disease 5 year overall survival rate: 70 - 100%
- Distant metastasis 5 year overall survival rate: 35 - 60%
- Long term recurrence rates are ~50% (HPB (Oxford) 2010;12:427)
- Nodal metastasis, mesenteric involvement, lymphovascular invasion and perineual invasion increase risk of recurrence
Case reports
- 38 year old woman with occult duodenal microgastrinoma with lymph node metastases (Am J Surg Pathol 2008;32:1101)
- 53 year old woman undergoing papillectomy for ampullary adenoma found to have accompanying ampullary NET (World J Gastroenterol 2016;22:3687)
- 58 year old man diagnosed with ampullary NET, pheochromocytoma and multiple gastrointestinal stromal tumors as first manifestation of NF1 (Diagn Pathol 2019;14:77)
- 60 year old man with periampullary neuroendocrine tumor presenting with acute pancreatitis (Am J Case Rep 2018;19:1063)
Treatment
- Surgical excision of tumor and regional lymph nodes
Clinical images
Images hosted on other servers:


Ampullary NET on endoscopy
Gross description
- Duodenal and periampullary NETs are small polypoid lesions within the submucosa (usually < 2 cm); rarely can be large and infiltrating
- Jejunoileal NETs can be multifocal; deep infiltration of muscularis propria and subserosa is common
Gross images
Contributed by Megan Kinn, D.O., M.S.
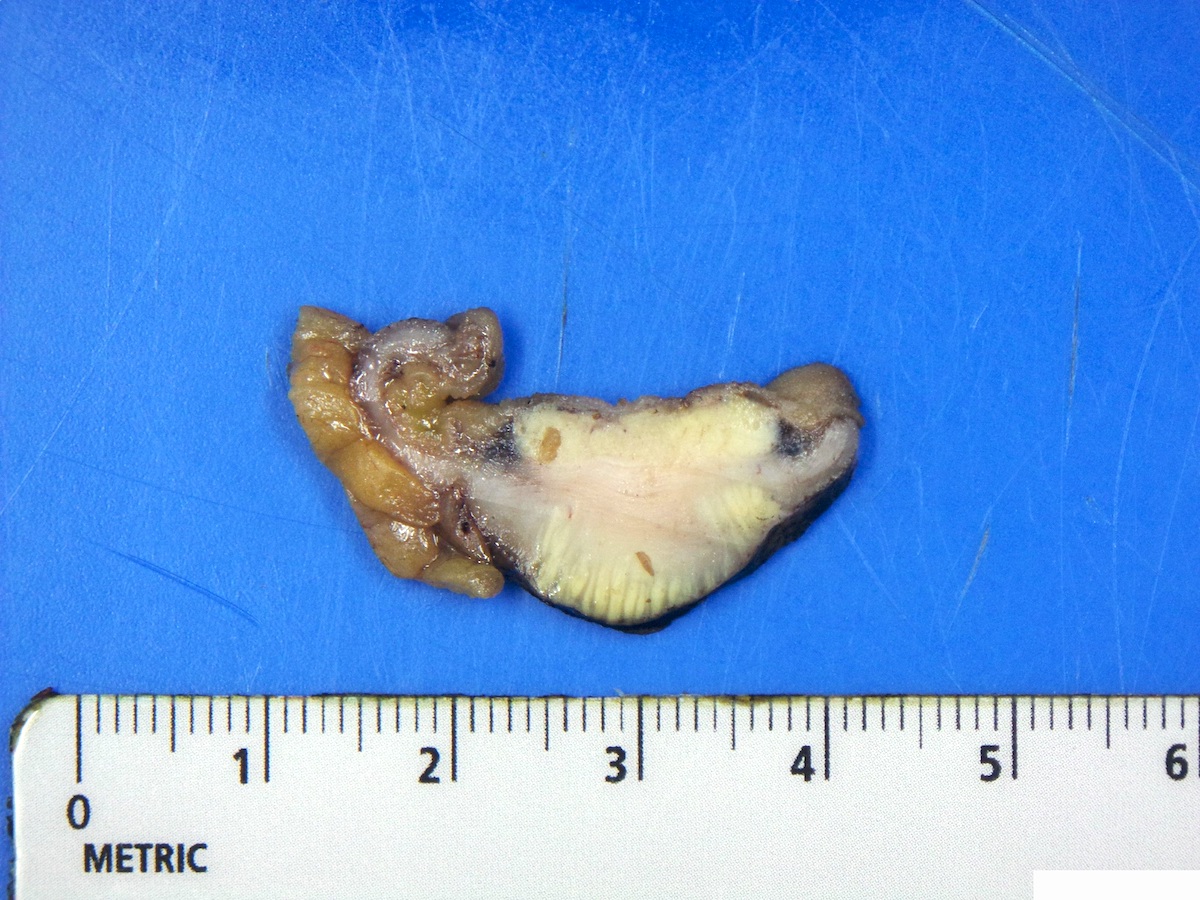
Ileal neuroendocrine tumor
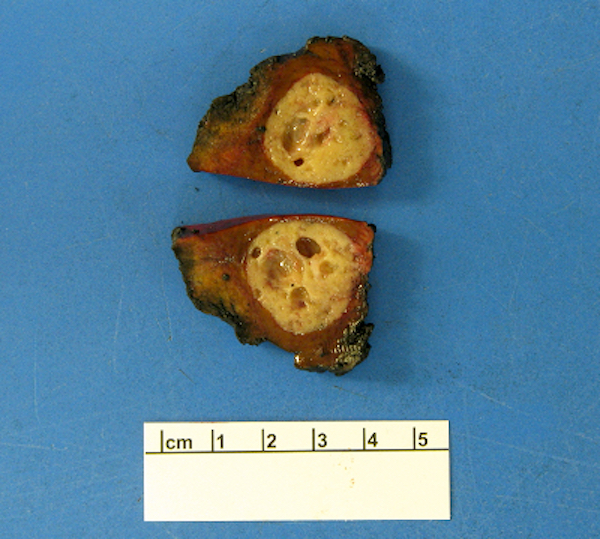
Liver metastasis
Microscopic (histologic) description
- Composed of uniform tumor cells showing round to oval nuclei with salt and pepper chromatin
- Varying architectural patterns
- Gastrin expressing G cell NETs: trabecular growth pattern
- Somatostatin expressing D cell NETs: tubuloglandular pattern; may have psammoma bodies
- Serotonin expressing EC cell NETs: nests of tumor cells with peripheral palisading, often minor pseudoglandular component
- Grading:
- Well differentiated neuroendocrine neoplasms:
- Neuroendocrine tumor, grade 1: low grade, < 2 mitoses/2 mm2, Ki67 index: < 3%
- Neuroendocrine tumor, grade 2: intermediate grade, 2 - 20 mitoses/2 mm2, Ki67 index: 3 - 20%
- Neuroendocrine tumor, grade 3: high grade, > 20 mitoses/2 mm2, Ki67 index: > 20%
- Well differentiated neuroendocrine neoplasms:
Microscopic (histologic) images
Contributed by Megan Kinn, D.O., M.S.
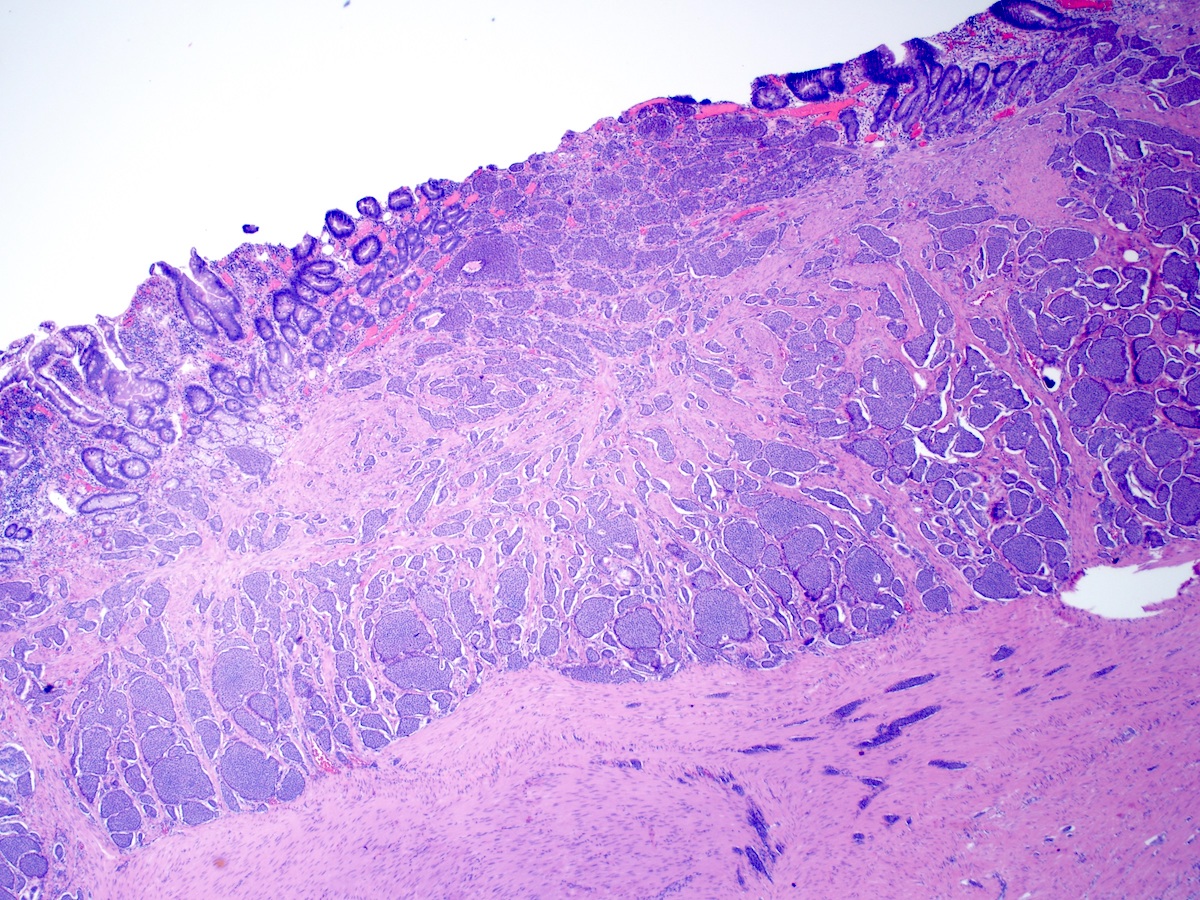
Ileal NET extending to mucosa
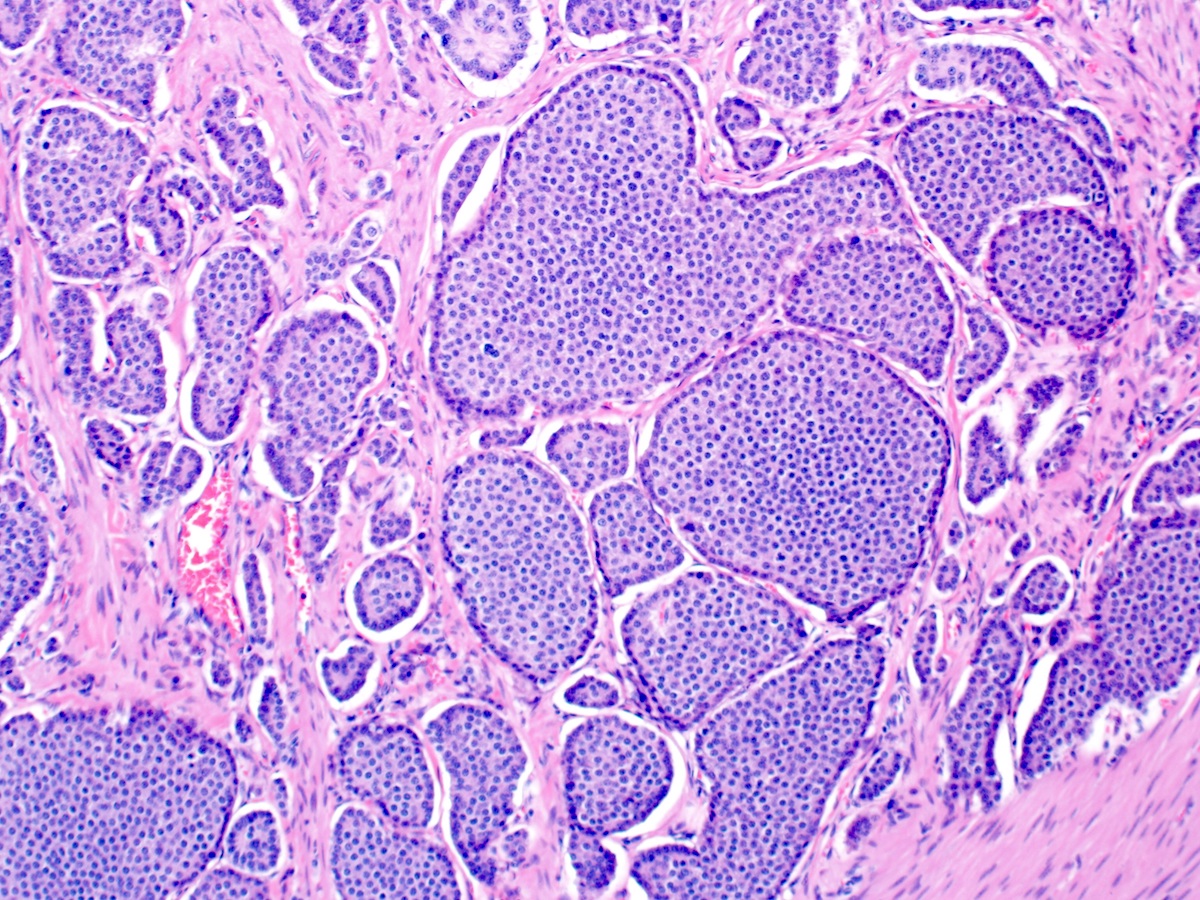
Nests of tumor cells
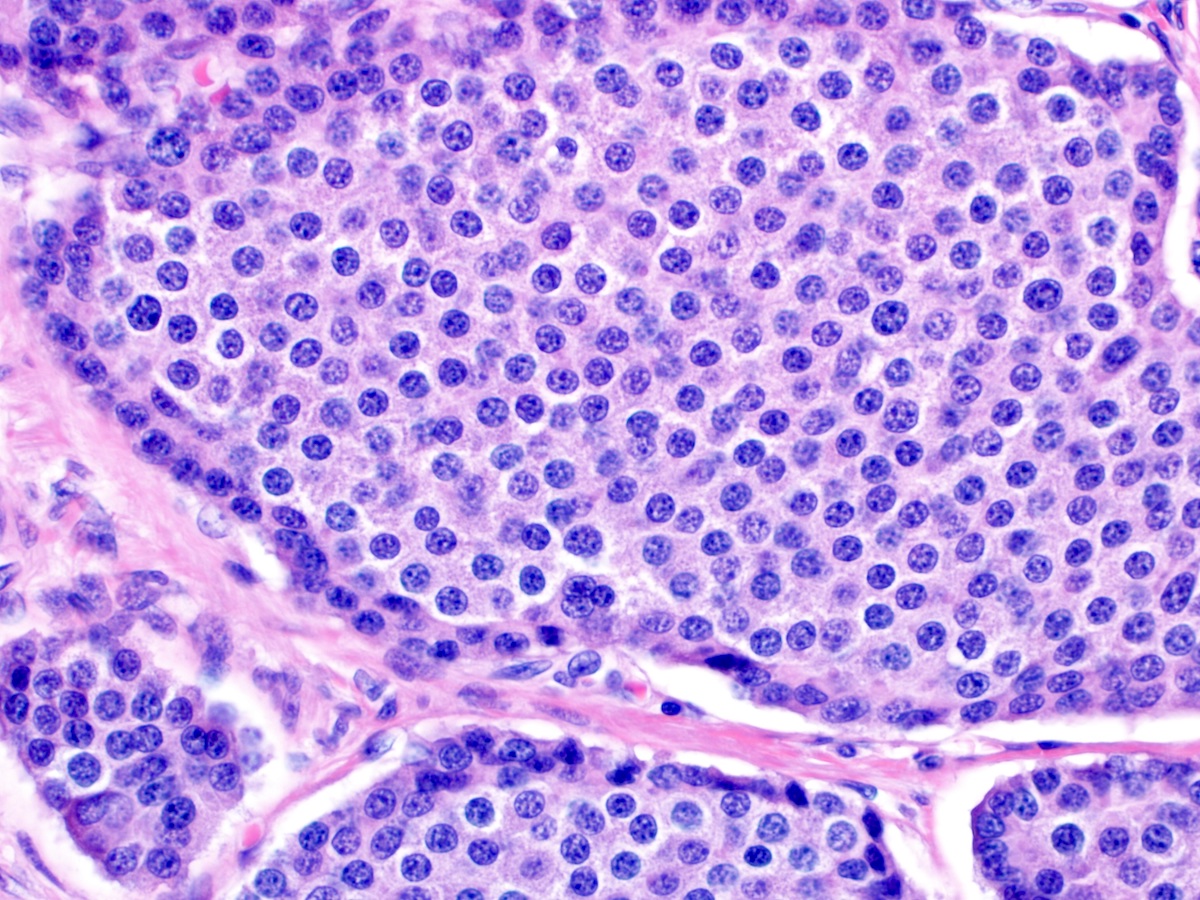
Salt and pepper chromatin
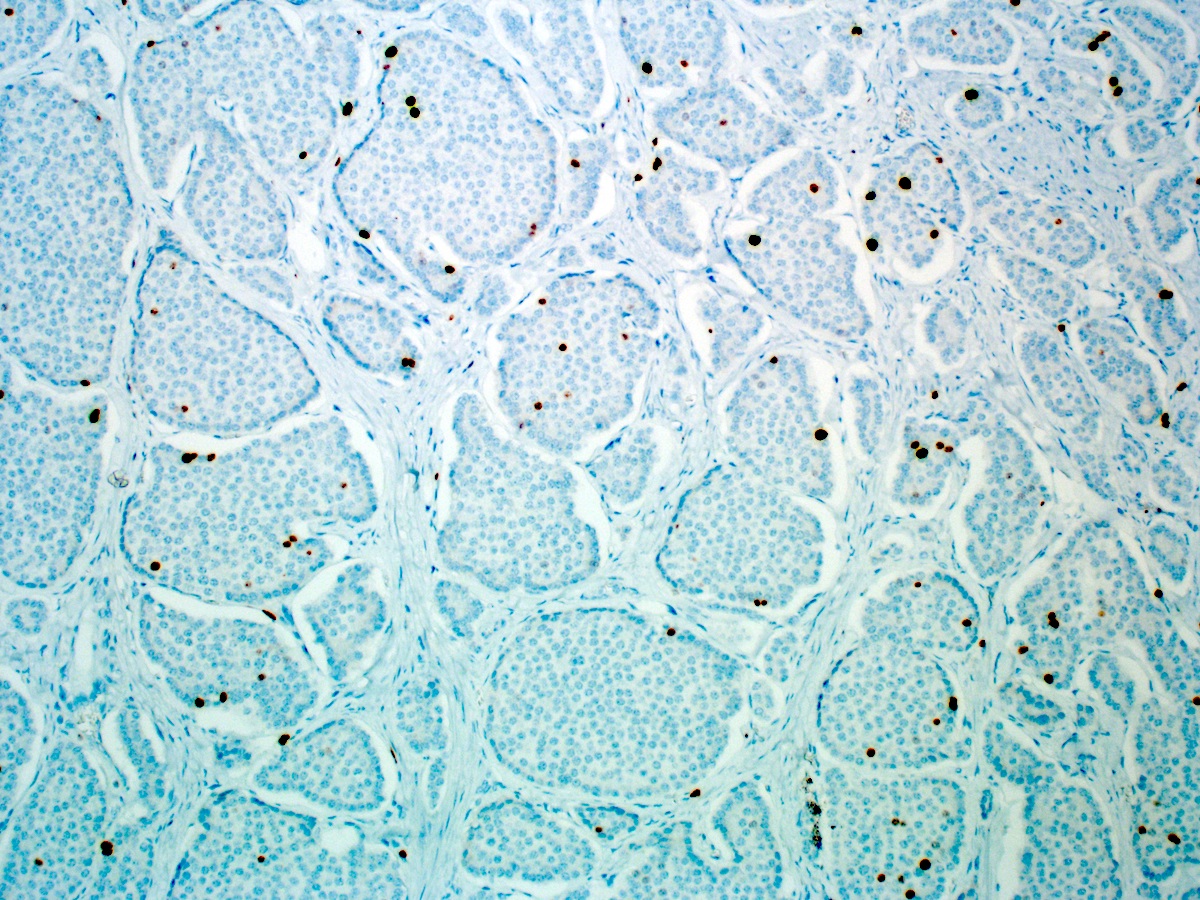
Ki67
Contributed by Wei Zheng, M.D., Ph.D.
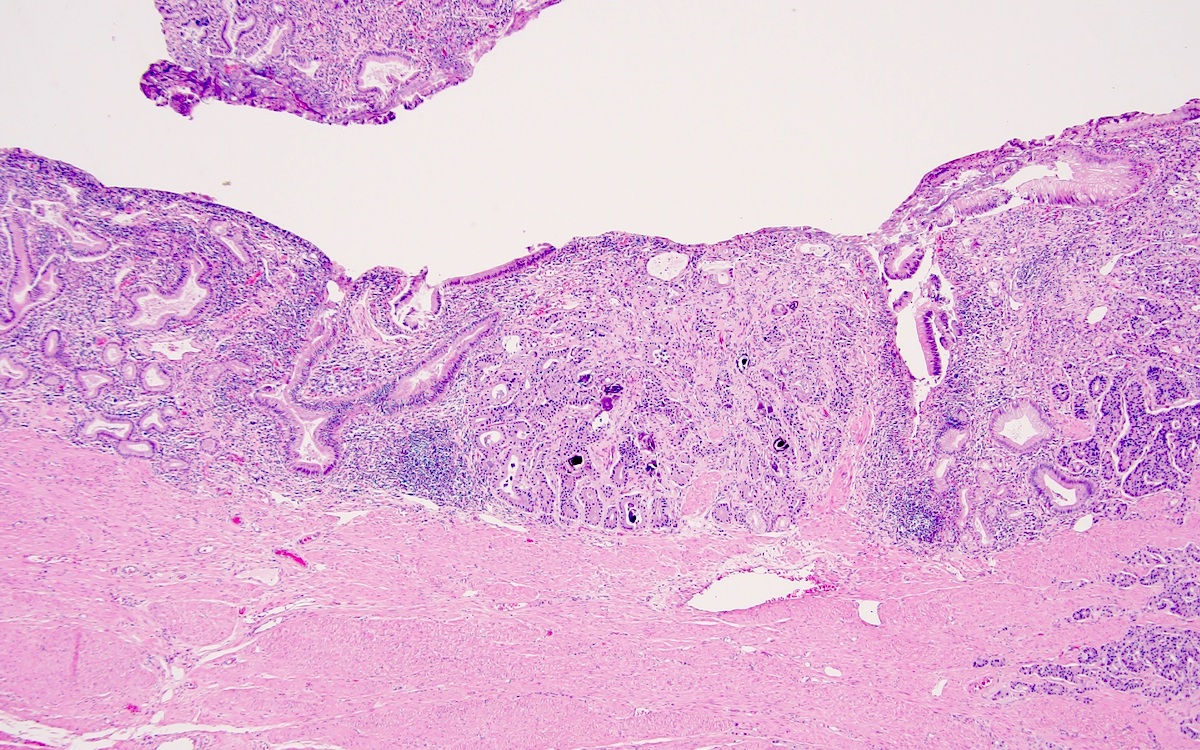
Ampullary NET
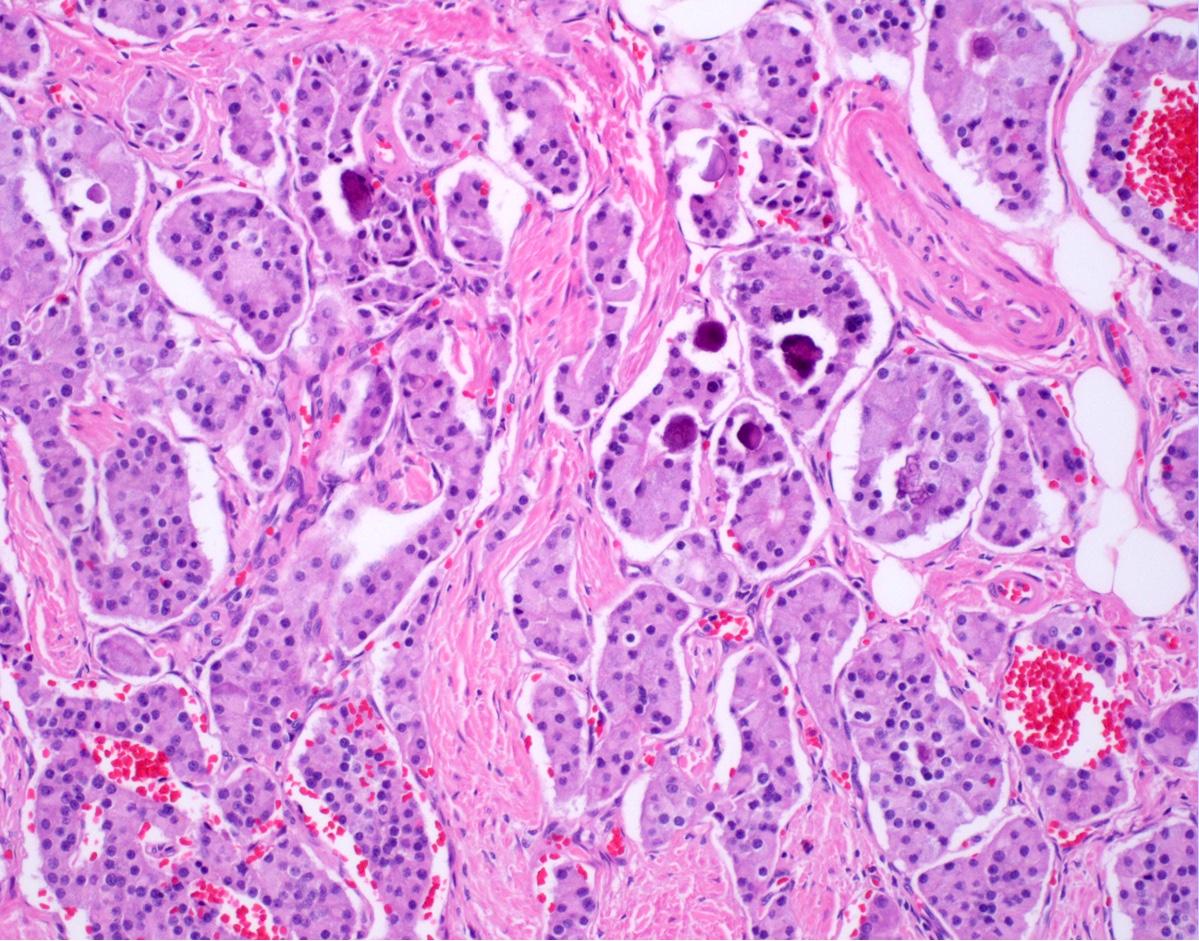
Trabecular and glandular architecture
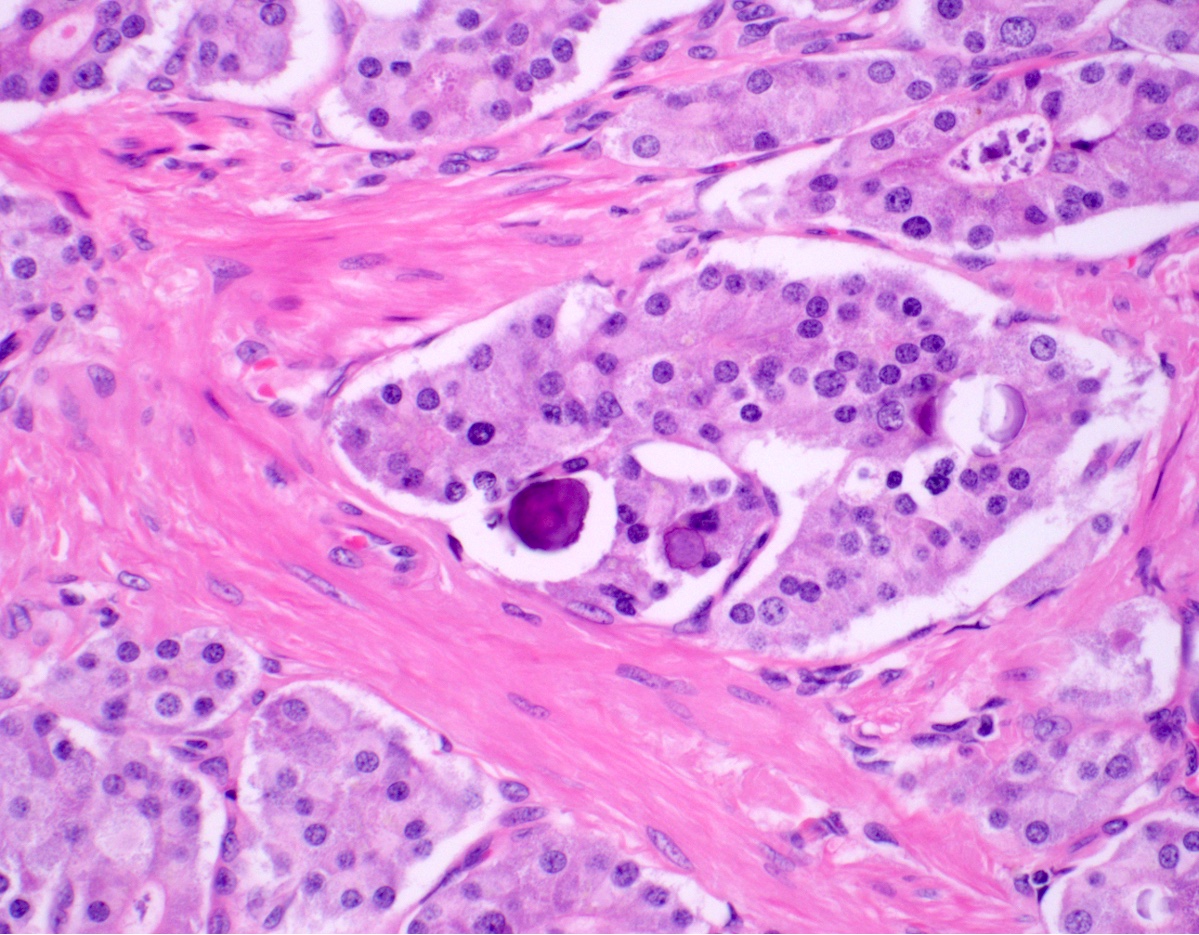
Psammoma bodies
Positive stains
- AE1 / AE3, CAM5.2
- Chromogranin A, synaptophysin
- Somatostatin expressing NETs less likely to express chromogranin A
- CDX2 and SSTR2A are positive in serotonin producing EC cell NETs
Electron microscopy description
- Well formed, membrane bound, dense core secretory granules
Molecular / cytogenetics description
- Sporadic duodenal and ampullary neuroendocrine tumor: no molecular data available
- Small intestinal NETs have a complex genomic landscape:
- 60 - 90% of jejunoileal NETs show chromosome 18 deletion (Mod Pathol 2005;18:1079)
- Chromosome 14 gain in advanced lesions and metastases (Endocr Relat Cancer 2009;16:953)
- CDKN1B mutations in ≤ 10% of ileal NETs (Ann Surg Oncol 2015;22:S1428)
- Epigenetic changes are common (Epigenomics 2015;7:1245)
- > 50% of ileal NETs are CpG island methylator phenotype
- Substantial epigenetic dysregulation found in 70 - 80% of tumors
- 3 molecular subgroups with potential prognostic impact (Clin Cancer Res 2016;22:250):
- Favorable prognosis: small intestinal NET with chromosome 18 loss (55%)
- Correlates with CpG island methylator phenotype negativity and CDKN1B mutation
- Intermediate prognosis: small intestinal NET with no whole arm copy number variation (19%)
- Correlates with CpG island methylator phenotype positivity
- Poor prognosis: small intestinal NET with multiple whole arm copy number variations (26%)
- Favorable prognosis: small intestinal NET with chromosome 18 loss (55%)
Sample pathology report
- Ileum, resection:
- Well differentiated neuroendocrine tumor (2.4 cm), WHO grade 2 of 3 (see synoptic report)
- Ki67 proliferation index: 6%
- Tumor involves mucosa up to subserosa
- Lymphovascular invasion present
- Resection margins are negative for tumor
- 10 lymph nodes, negative for tumor
Differential diagnosis
- Neuroendocrine carcinoma:
- Poorly differentiated neoplasm with sheets of tumor cells or poorly formed trabeculae or nests that is almost exclusive to the ampullary region
- Pleomorphic cells with large cell or small cell patterns
- Chromogranin A and synaptophysin may be negative
- Loss of RB1 and aberrant p53 expression support the diagnosis of neuroendocrine carcinoma
- Poorly differentiated neoplasm with sheets of tumor cells or poorly formed trabeculae or nests that is almost exclusive to the ampullary region
- Mixed neuroendocrine - nonneuroendocrine neoplasms (MiNENs):
- Exocrine (usually adenocarcinoma) and neuroendocrine components, each ≥ 30%
- Adenocarcinoma:
- Negative or focally positive for neuroendocrine markers
- More prominent nuclear atypia
- Gangliocytic paraganglioma:
- Triphasic (neuroendocrine, Schwannian and ganglion cell-like components)
- Predominantly in ampullary region
Additional references
Board review style question #1
A 49 year old woman presented with epigastric abdominal pain, diarrhea and significant weight loss in the last 2 years. Clinical examination revealed the presence of a few café au lait spots located on the upper limbs, large, poorly circumscribed areas of hyperpigmentation on the trunk, bilateral axillary freckling and multiple asymptomatic, soft, flesh colored, sessile or dome shaped cutaneous tumors 1 - 2 cm in diameter, nonadherent to the underlying structures, distributed on the trunk and limbs. An abdominal MRI revealed an infiltrative duodenal mass. Microscopy and immunohistochemistry uncovered a well differentiated neuroendocrine tumor (Ki67 = 1%) with psammoma bodies. A mutation in which of the following genes is most likely responsible for the patient’s disease?
- MEN1
- RET
- MLH1
- APC
- NF1
Board review style answer #1
E. NF1. The patient’s clinical examination (the presence of a few café au lait spots located on the upper limbs, large, poorly circumscribed areas of hyperpigmentation on the trunk, bilateral axillary freckling and multiple asymptomatic, soft, flesh colored, sessile or dome shaped cutaneous tumors, 1 - 2 cm in diameter, nonadherent to the underlying structures, distributed on the trunk and limbs) and well differentiated neuroendocrine tumor with psammoma bodies (somatostatinomas) in the duodenum are highly suggestive of neurofibromatosis type 1 (NF1).
MEN1 (answer A) is incorrect because mutations in the MEN1 gene cause multiple endocrine neoplasia type 1 (MEN 1). MEN1 is a hereditary syndrome characterized by hyperplasia or sometimes adenomas of the parathyroid glands, pancreatic neuroendocrine tumors or pituitary gland tumors. RET (answer B) is incorrect because the RET proto-oncogene mutations are responsible for multiple endocrine neoplasia (MEN) type 2. MEN2 syndrome is divided into three subtypes (MEN2A, MEN2B and FMTC). People with all subtypes of MEN2 syndrome have an increased risk of medullary thyroid cancer, pheochromocytoma and parathyroid gland cancer. MLH1 (answer C) is incorrect because MLH1 is one of the mismatch repair gene mutations that have been implicated in the development of hereditary nonpolyposis colon cancer (Lynch) syndrome. This patient does not have colon cancer. APC (answer D) is incorrect because the autosomal dominant APC gene mutation is responsible for familial adenomatous polyposis (FAP). This condition causes multiple adenomatous polyps in the colon of affected individuals.
Comment Here
Reference: Neuroendocrine tumor
MEN1 (answer A) is incorrect because mutations in the MEN1 gene cause multiple endocrine neoplasia type 1 (MEN 1). MEN1 is a hereditary syndrome characterized by hyperplasia or sometimes adenomas of the parathyroid glands, pancreatic neuroendocrine tumors or pituitary gland tumors. RET (answer B) is incorrect because the RET proto-oncogene mutations are responsible for multiple endocrine neoplasia (MEN) type 2. MEN2 syndrome is divided into three subtypes (MEN2A, MEN2B and FMTC). People with all subtypes of MEN2 syndrome have an increased risk of medullary thyroid cancer, pheochromocytoma and parathyroid gland cancer. MLH1 (answer C) is incorrect because MLH1 is one of the mismatch repair gene mutations that have been implicated in the development of hereditary nonpolyposis colon cancer (Lynch) syndrome. This patient does not have colon cancer. APC (answer D) is incorrect because the autosomal dominant APC gene mutation is responsible for familial adenomatous polyposis (FAP). This condition causes multiple adenomatous polyps in the colon of affected individuals.
Comment Here
Reference: Neuroendocrine tumor
