
Bone marrow neoplastic
Bone marrow - neoplastic myeloid
Myeloproliferative neoplasms (MPN)
Essential thrombocythemia
Editorial Board Members: Alexa J. Siddon, M.D., Anamarija M. Perry, M.D.


Last author update: 24 May 2023
Last staff update: 22 September 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Essential thrombocythemia
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Pathophysiology | Etiology | Clinical features | Diagnosis | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Flow cytometry description | Flow cytometry images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Kaseb H, Hudnall SD. Essential thrombocythemia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/myeloproliferativeET.html. Accessed April 19th, 2024.
Definition / general
- Essential thrombocythemia is a chronic myeloproliferative neoplasm that primarily involves the megakaryocytic lineage
Essential features
- Characterized by sustained thrombocytosis (platelet count > 450 x 109/L) in the peripheral blood and increased numbers of large, atypical megakaryocytes in the bone marrow; clinically by the occurrence of thrombosis or hemorrhage
Terminology
- Idiopathic thrombocythemia / thrombocytosis; essential hemorrhagic thrombocythemia; idiopathic hemorrhagic thrombocythemia; idiopathic thrombocythemia
- Has also been called essential thrombocytosis and primary thrombocytosis
ICD coding
- ICD-10: D47.3 - essential (hemorrhagic) thrombocythemia
Epidemiology
- In the U.S. and Europe the incidence is 0.2 - 2.3 cases/100,000
- Median age of 60 years at diagnosis but may occur at any age
- Higher incidence among African Americans
- More common in women (F:M = 2:1) (Br J Haematol 2016;174:382)
- Rare in children (must rule out hereditary thrombocytosis)
Pathophysiology
- Clonal stem cell disorder characterized by increased platelet production
- Serum thrombopoietin levels can be normal or elevated
- Harbor a phenotypic driver mutation in JAK2 (present in 50 - 60% of cases), CALR (30%) or MPL (3%); ~12% of cases are triple negative for these mutations (Leuk Lymphoma 2017;58:2786)
- JAK2, CALR or MPL mutations result in the upregulation of JAK-STAT pathway
- JAK2 is a nonreceptor tyrosine kinase and has been found to play a crucial role in hematopoiesis
- JAK2 mutations leads to a gain of function leading to the activation of intracellular signaling pathways associated with the receptors of hematopoietic cytokines
- CALR is involved in cellular proliferation, differentiation and apoptosis
- JAK2, CALR or MPL mutations result in the upregulation of JAK-STAT pathway
Etiology
- Etiology unknown
- Most cases are sporadic
- Some mutations of JAK2 and GSN genes have been found in families with hereditary thrombocytosis
Clinical features
- Patient can either be asymptomatic or symptomatic
- Asymptomatic
- More than half of cases
- Detected on routine peripheral blood count
- Less than half present with symptoms related to thrombosis or hemorrhage
- Long asymptomatic periods alternating with thrombotic or hemorrhagic crises
- May rarely progress to myelofibrosis
- Symptoms are due to qualitative and quantitative abnormalities in platelets and include
- Bleeding tendency
- Microvascular occlusion
- Thrombosis of major arteries leading to symptoms such as stroke and transient ischemic attacks
- Vasomotor manifestations such as headache, transient visual disturbances and syncope
- Mild splenomegaly
- Hepatomegaly and lymphadenopathy are uncommon
- Unexplained abortion (Blood 2007;110:485)
- Closely related to polycythemia vera but no increase in red blood cell mass
- Increased proliferation is usually confined to megakaryocytes with platelet count > 450 x 109/L
Diagnosis
- Diagnostic criteria for early / prefibrotic essential thrombocythemia: requires either all major criteria or the first 3 major criteria plus the minor criterion (Blood 2022;140:1200, Leukemia 2022;36:1703) (WHO 2022 and ICC 2022)
- Major
- Criterion #1: sustained platelet count ≥ 450 x 109/L
- Criterion #2: megakaryocyte proliferation, large and mature with hyperlobulated nuclei (atypical), little to no granulocyte or erythroid proliferation; very rarely a minor (grade P) increase in reticulin fibers
- Criterion #3: does not meet WHO criteria for chronic myeloid leukemia, polycythemia vera, primary myelofibrosis, myelodysplastic syndrome or other myeloid neoplasm
- Criterion #4: demonstration of JAK2 or CALR or MPL mutation
- Minor
- Presence of a clonal marker
- Absence of evidence of reactive thrombocytosis
- Major
- Diagnostic criteria for post essential thrombocythemia myelofibrosis (Blood 2022;140:1200, Leukemia 2022;36:1703)
- 2 major criteria and at least 2 minor criteria (WHO 2022 and ICC 2022)
- Required criteria
- Established diagnosis of essential thrombocythemia
- Bone marrow fibrosis grade 2 - 3 (on 0 - 3 scale)
- Additional criteria (2 required)
- Anemia or decrease in hemoglobin by 2 gm/dL from the baseline level
- Leukoerythroblastic peripheral blood
- Splenomegaly
- Rise in lactate dehydrogenase (LDH) levels
- Constitutional symptoms (at least 2 or all 3): unexplained fever (> 37.5 °C), night sweats, > 10% weight loss in 6 months
- Required criteria
- Bone marrow examination is necessary to exclude other myeloproliferative neoplasms, myelodysplastic syndromes associated with isolated del(5q) and myelodysplastic / myeloproliferative neoplasm
- 2 major criteria and at least 2 minor criteria (WHO 2022 and ICC 2022)
Prognostic factors
- Essential thrombocythemia is generally an indolent disease with a good prognosis
- Initial management is largely dictated by the risk of thrombotic complication based on age, JAK2 mutation status and the history of thrombosis, and is stratified as follows (Blood Cancer J 2015;5:e369)
- High risk: history of thrombosis at any age or age > 60 with a JAK2 V617F mutation
- Intermediate risk: age > 60, no JAK2 mutation detected and no history of thrombosis
- Low risk: age ≤ 60 with JAK2 mutation and no history of thrombosis
- Very low risk: age ≤ 60, no JAK2 mutation detected and no history of thrombosis
- Variable rate of transformation to acute myeloid leukemia or myelodysplastic syndrome; generally these patients have a poor prognosis
- Can undergo delayed disease progression into a fibrotic state called post essential thrombocythemia myelofibrosis; < 5% conversion rate after 10 - 20 years
- Median survival is 10 - 15 years, hence life expectancy is almost normal (Am J Med 2004;117:755)
- CALR mutations are associated with a favorable prognosis in essential thrombocythemia compared to other genetic changes (Semin Hematol 2018;55:215)
Case reports
- 35 year old woman with calreticulin mutation presenting with postpartum hemorrhage (Blood Coagul Fibrinolysis 2016;27:727)
- 37 year old woman presenting with acute renal infarction (Kidney Int 2017;92:1292)
- 50 year old man presenting with coronary artery dissection (WMJ 2015;114:26)
- 61 year old woman with abdominal surgery (Medicine (Baltimore) 2017;96:e8856)
- 3 patients with essential thrombocythemia presenting as acute coronary syndrome (J Thromb Thrombolysis 2017;44:57)
Treatment
- Goal of management is to control symptoms arising from thrombotic and hemorrhagic complications
- Aspirin is given to the majority of patients, to reduce the odds of vascular events and vasomotor symptoms
- Low risk and very low risk patients (Br J Haematol 2000;110:577)
- Should undergo an annual or semi annual visits with aspirin treatment
- No randomized trials comparing low risk and very low risk treatment options
- Intermediate risk and high risk patients (Br J Haematol 2000;110:577)
- Should be treated with cytoreductive therapy (preferably hydroxyurea followed by anagrelide) or systemic anticoagulation (such as low dose aspirin)
Microscopic (histologic) description
- Bone marrow
- Normal / mildly hypercellular
- Atypical megakaryocytes (Blood 2016;127:2391)
- Increased number and large atypical forms with deeply lobulated or hyperlobulated nuclei (staghorn nuclear appearance)
- Dispersed throughout the marrow with occasional loose clusters; dense clusters are uncommon
- No highly bizarre / dysplastic forms, no predominance of small megakaryocytes with monolobulated nuclei
- Delicate reticulin fibers (myelofibrosis absent / minimal) but no overt fibrosis (usually grade 1)
- Usually normal erythroid and granulocytic proliferation
- Usually no dyserythropoiesis, dysgranulopoiesis, macrocytosis or monocytosis
- Features in bone marrow aspirate / biopsy suggesting an alternative diagnosis (Blood 2016;127:2391)
- Megakaryocytes with highly atypical morphology
- Increased myeloblasts
- Myelodysplastic features
- Significant (> grade 1) reticulin fibrosis or collagen fibrosis
Microscopic (histologic) images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
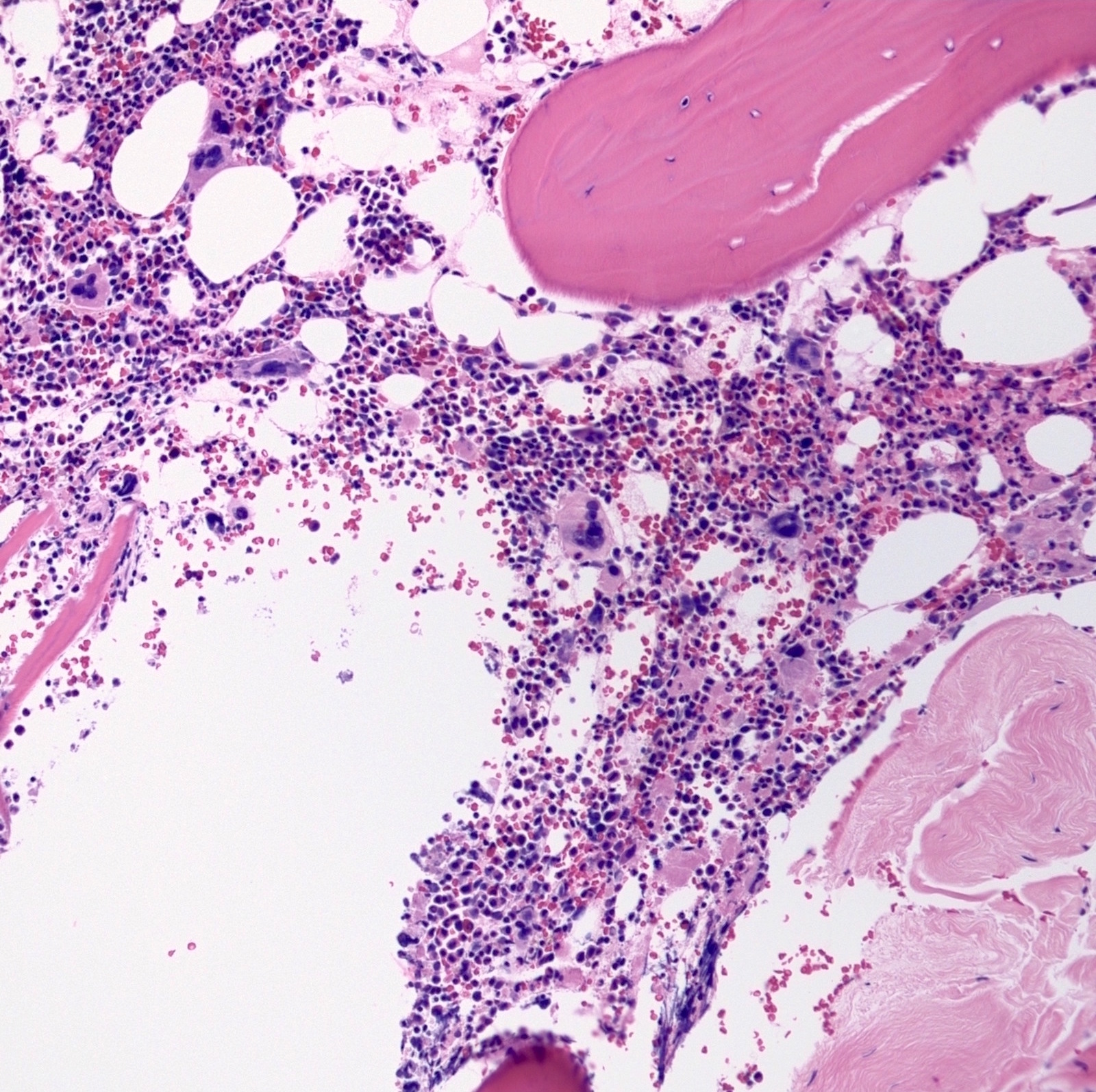
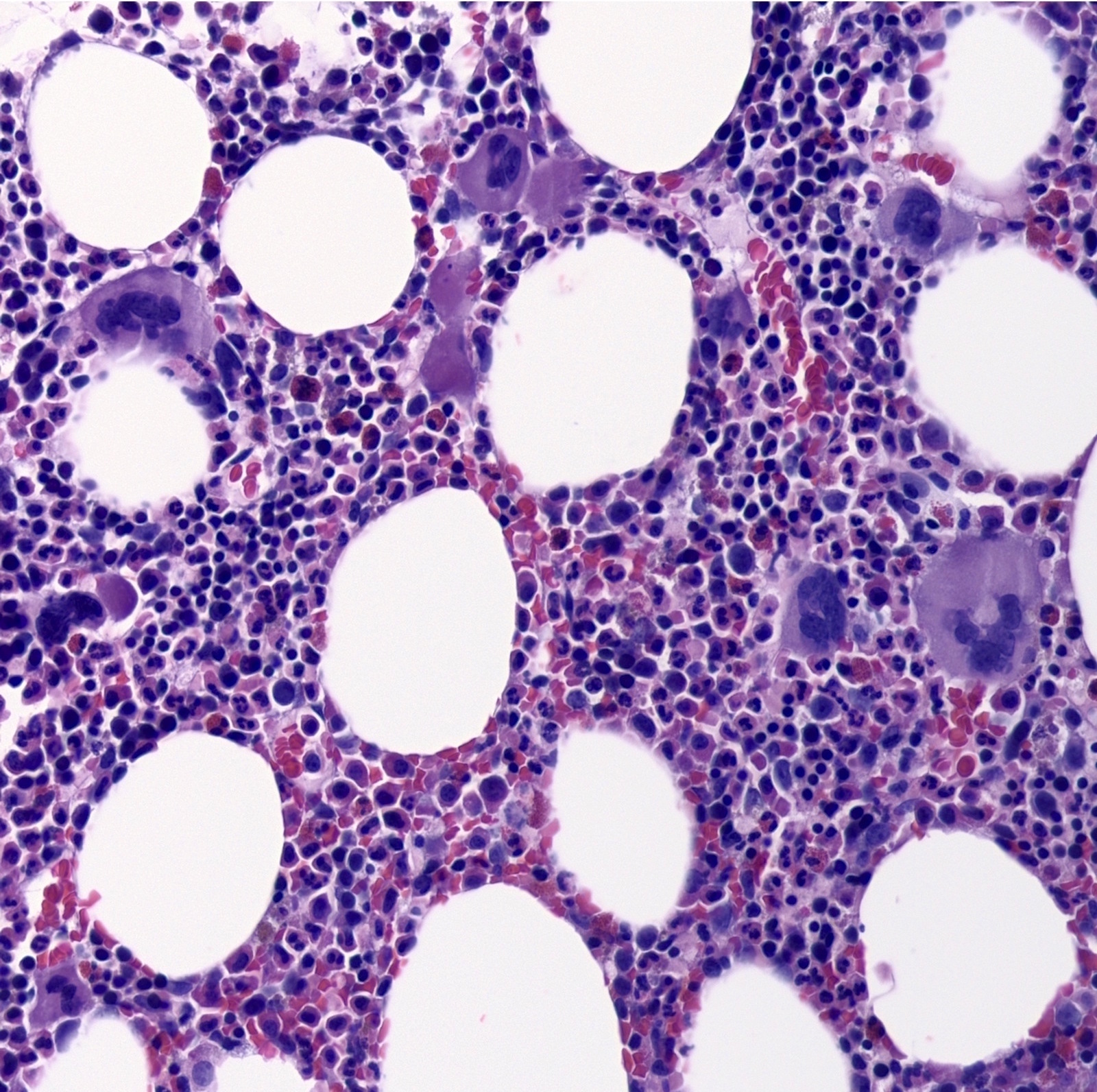
Bone marrow biopsy with atypical megakaryocytes
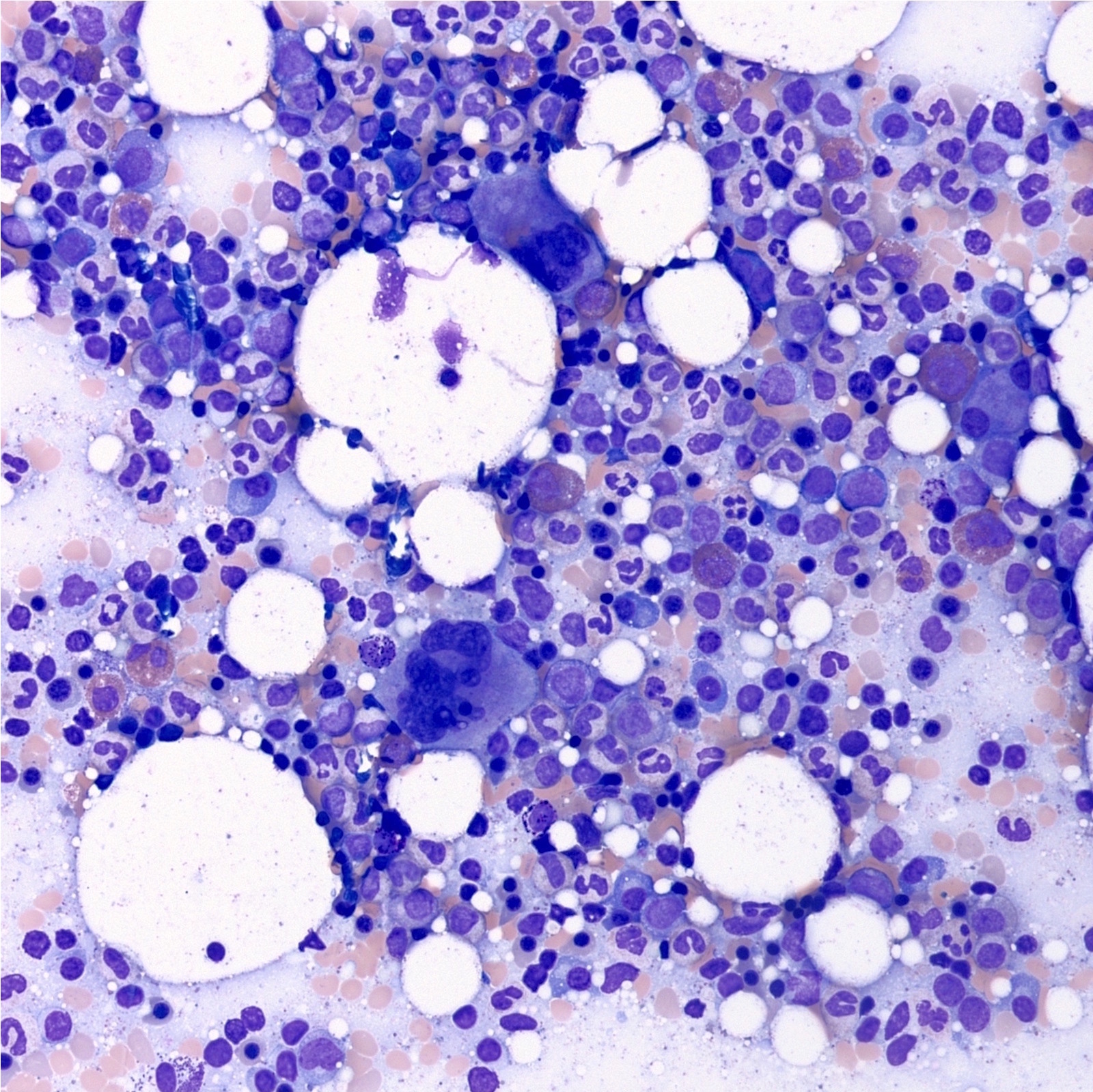
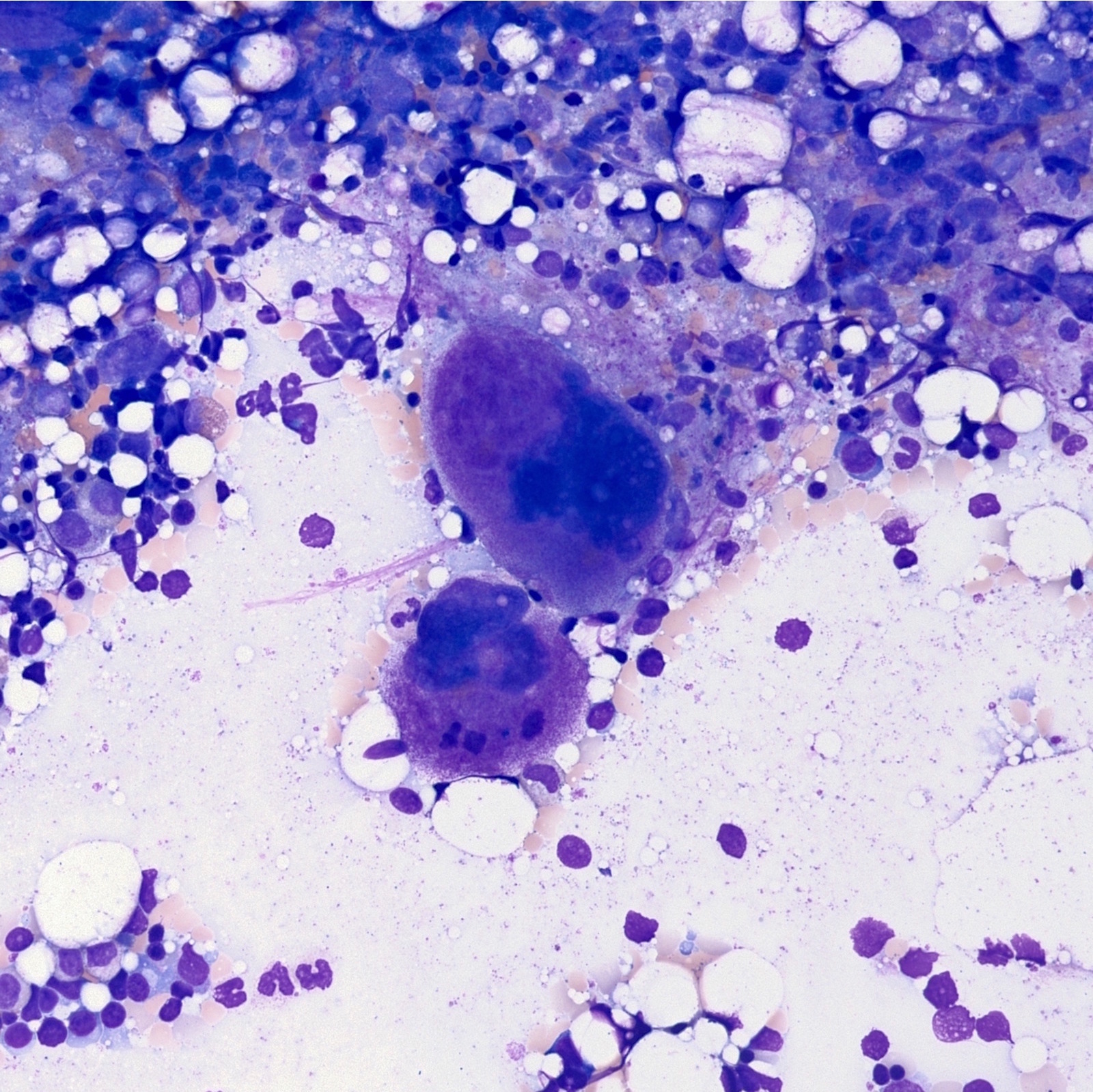
Bone marrow aspirate with atypical megakaryocytes
Peripheral smear description
- Normal / mild increase in the white blood cell count
- Moderate to marked thrombocytosis
- Platelet anisocytosis, abnormally large platelets and increased platelets; bizarre shapes, pseudopods and agranular forms
Peripheral smear images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
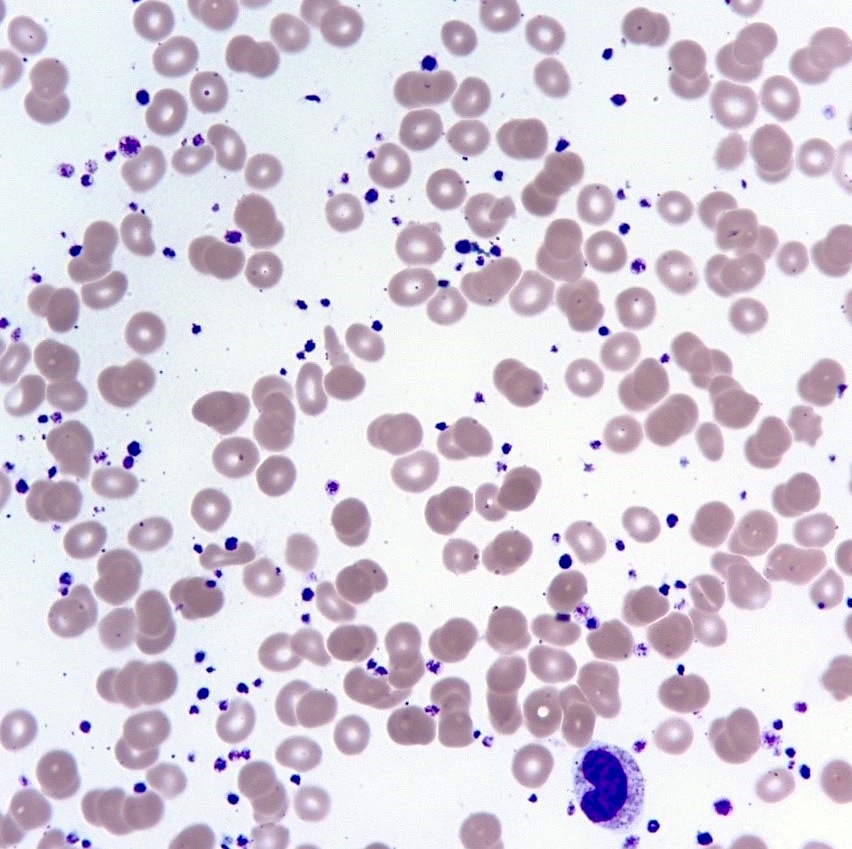
Anisocytosis and bizarre forms
Flow cytometry description
- Normal number and phenotype myeloblasts with normal myeloid scatter by CD45 / SSC
- Normal CD10 / CD13 / CD16 / CD11b myeloid maturation pattern and all other myeloid markers are normally expressed; hence, there is no immunophenotypic evidence of myelodysplasia
- No evidence for monoclonal B cell lymphoproliferative disease
- Although no unusual phenotype T cells are identified, surface markers will not routinely detect monoclonal T lymphocytes
Flow cytometry images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
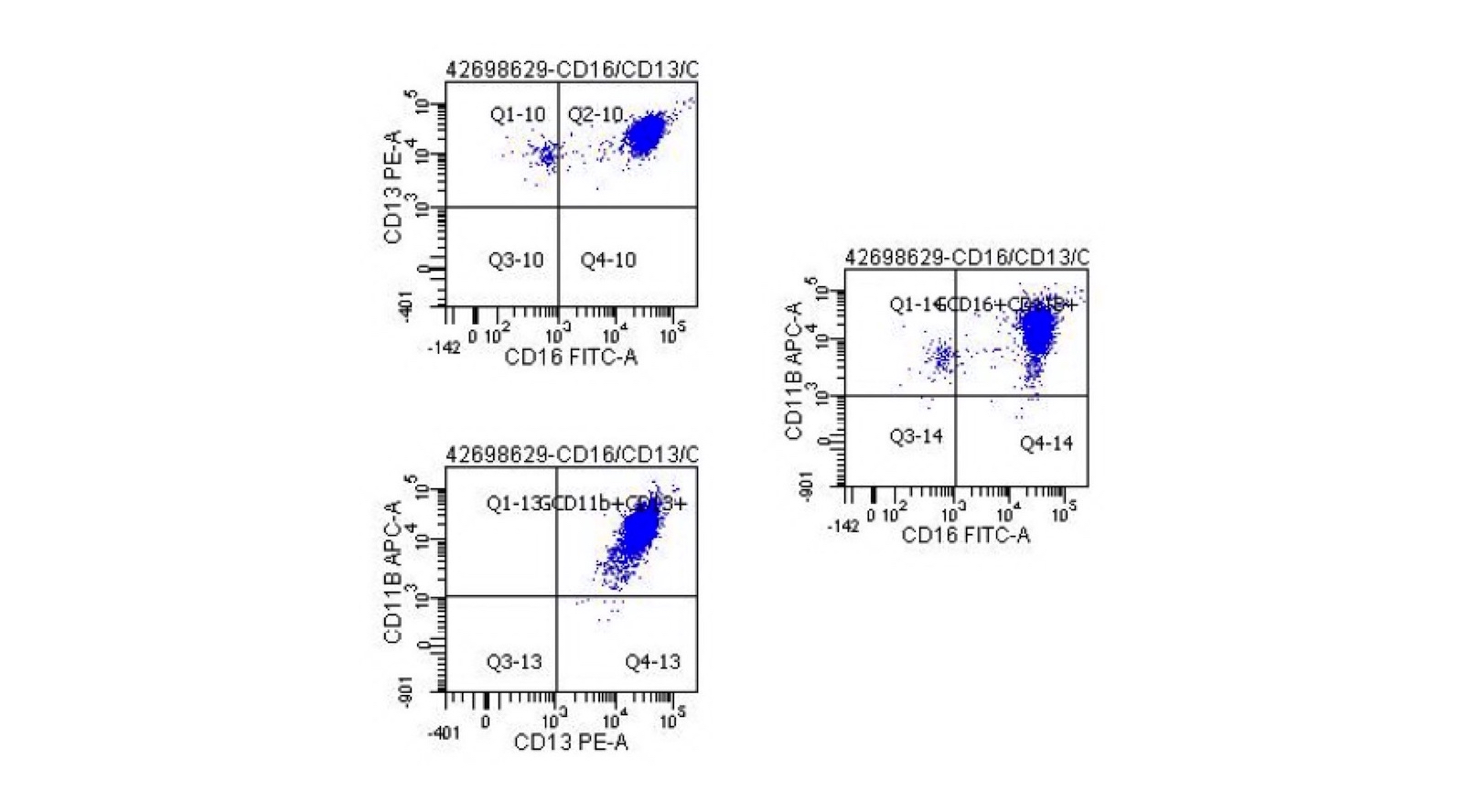
No myelodysplasia
Molecular / cytogenetics description
- Phenotypic driver mutation
- JAK2 (present in 50 - 60% of cases), CALR (30%) or MPL (3%), ~12% of cases are triple negative for these mutations
- Mutations are specific for essential thrombocythemia but their presence excludes reactive thrombocytosis
- Abnormal karyotype is found in 5 - 10% of cases
- No consistent abnormality
- Reported abnormalities include gain of chromosome 8, abnormalities in 9q and del(20q) and unbalanced translocations between 1q and 7p
- JAK2 mutant allele burden contributes to clinical phenotype (Haematologica 2009;94:144)
- Subset of triple negative cases have been found to have
- Gain of function mutations (e.g., MPL S204P and MPL Y591N) through whole exome sequencing or other molecular techniques
- W515L and W515K mutations in thrombopoietin receptor c-Mpl (J Transl Med 2006;4:41)
- Must rule out BCR::ABL1 fusion gene status
- Children usually do not have JAK2 mutation
Sample pathology report
- Bone marrow, biopsy and aspirate:
- Myeloproliferative neoplasm (see comment)
- Comment: The patient's positive JAK2 mutation is noted. The morphologic findings are compatible with a myeloproliferative neoplasm and in conjunction with the thrombocytosis, essential thrombocythemia is favored. Correlation with clinical, cytogenetic and laboratory findings is recommended.
- CBC (4/29/2019): HGB: 12.7 g/dL; MCV: 89.7 fL; WBC: 9.7 K/uL; PLT: 735 K/uL
- Bone marrow biopsy microscopic description:
- Slightly hypercellular marrow for age (30% cellular). Megakaryocytes are increased in number, occasionally in clusters and show frequent atypical forms, including large and hyperlobated forms. The myeloid:erythroid (M:E) ratio is normal. Erythroid elements exhibit maturation. Myeloid elements exhibit maturation. Prominent lymphoid aggregates are not seen. Granulomas are not seen. Reticulin stain reveals mild and patchy reticulin fibrosis. Trabecular bone is unremarkable. The clot section is mainly blood and shows small fragments with findings similar to the core biopsy. CD34 highlights blasts comprising < 5% of the marrow cellularity.
- Bone marrow aspirate microscopic description:
- The marrow aspirate smears are spicular and cellular. Megakaryocytes are increased. The M:E ratio is approximately 1:1. Erythroid maturation is seen. Myeloid maturation is seen. Prussian blue iron stain shows decreased iron storage and no significant increase in ring sideroblasts.
- Aspirate cell count: A 226 cell count reveals 2% blasts, 9% promyelocytes / myelocytes, 34% maturing granulocyte forms, 38% erythroid forms, 8% lymphocytes, 5% eosinophils, 2% plasma cells, 0% basophils / mast cells and 0% monocytes.
Differential diagnosis
- Other myeloproliferative neoplasms:
- Chronic myeloid leukemia:
- BCR::ABL translocation is diagnostic
- Polycythemia vera:
- Most cases harbor a clonal JAK2 mutation
- Increase in erythroid / granulocytic population should raise suspicion of polycythemia vera; polycythemia vera patients also present with decreased erythropoietin (EPO)
- Polycythemia vera patients usually present with some symptoms related to hyperviscosity such as headaches or visual disturbance; some patients may present with pruritus exacerbated by hot water
- Primary myelofibrosis and prefibrotic myelofibrosis:
- Increased bone marrow reticulin fibrosis, markedly bizarre megakaryocytes and a leukoerythroblastic peripheral blood smear favor myelofibrosis
- Prefibrotic myelofibrosis:
- Will show features of primary myelofibrosis
- The main feature that can help differentiate this entity from essential thrombocythemia is the presence of reticulin fibrosis in pre-PMF
- Myelodysplastic syndrome:
- Dysplastic features and genetics such as 5q loss
- Familial essential thrombocythemia:
- Autosomal dominant disease
- Genetic analysis will show mutations in thrombopoietin, TPO receptor (c-MPL) or others related to thrombopoiesis
- Reactive thrombocythemia:
- Causes include exercise, allergic reaction, reaction to medications, inflammatory disorders, asplenism, infection, connective tissue disorders, metastatic cancer, lymphoproliferative disorders and iron deficiency
- Spurious thrombocytosis:
- Due to the miscount of structures that are not platelets (e.g., cryoglobulin crystals, cytoplasmic fragments of circulating leukemic cells and bacteria) as platelets leading to thrombocytosis (Thrombosis 2011;2011:536062)
- Chronic myeloid leukemia:
Additional references
Board review style question #1
What is the prevalence of genetic mutation in essential thrombocythemia?
- CAL > JAK2 > MPL > triple negative
- JAK2 > CAL > MPL > triple negative (JAK2, CAL and RET)
- JAK2 > CAL > triple negative > MPL
- JAK2 > MPL > CAL > triple negative
Board review style answer #1
C. JAK2 > CAL > triple negative > MPL. Essential thrombocythemia cases harbor a phenotypic driver mutation in JAK2 (50 - 60% of cases), CALR (30%) or MPL (3%) and ~12% of cases are triple negative for these mutations.
Comment Here
Reference: Essential thrombocythemia
Comment Here
Reference: Essential thrombocythemia
Board review style question #2
Which of the following is a characteristic of essential thrombocythemia?
- Abnormal karyotype
- JAK2 mutation
- Leukoerythroblastic reaction
- Positive BCR::ABL translocation
Board review style answer #2
B. JAK2 mutation. Most cases of essential thrombocythemia show JAK2 mutation is present in up to 60% of cases and normal karyotype. Negative BCR::ABL excludes chronic myeloid leukemia and is one of the diagnostic criteria of essential thrombocythemia. Leukoerythroblastic reaction favors primary myelofibrosis.
Comment Here
Reference: Essential thrombocythemia
Comment Here
Reference: Essential thrombocythemia
