
Muscle & peripheral nerve nontumor
Muscle atrophy
Neurogenic atrophy
Author: Meggen Walsh, D.O., M.S., P.A.

Last author update: 1 July 2016
Last staff update: 14 February 2022
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: neurogenic atrophy[title] muscle

Table of Contents
Definition / general | Essential features | Terminology | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Treatment | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Differential diagnosis | Additional referencesCite this page: Walsh M. Neurogenic atrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/muscleneurogenicatrophy.html. Accessed March 29th, 2025.
Definition / general
- Neurogenic type atrophy is a descriptive diagnosis that has multiple different etiologies; underlying etiology generally cannot be further elucidated by the muscle biopsy itself and needs clinicopathologic or radiologic correlation
- Amyotrophic lateral sclerosis (ALS): sporadic and familial forms, progressive painless motor weakness with upper and lower motor symptoms; symptoms vary from fatigue, twitching, fasciculations, dropping items, tripping, difficulty with speech and swallowing; eye muscles and bowel and bladder are generally spared
- Primary lateral sclerosis (PLS): only upper motor neurons are affected
- Spinal muscular atrophy (SMA): lower limbs are affected more than upper, proximal muscles are affected more than distal
- 3 different types:
- I - infantile onset, has hypotonia with sparing of the diaphragm; patients usually succumb to respiratory infections around ages 1-2
- II - onset 6-12 months of age, able to sit but not stand / walk unaided, symmetrical weakness, hand tremor, fasciculation of tongue and joint laxity
- III - onset 2 years of age to adulthood, able to walk, difficulty with other activities (running, jumping), weakness can be progressive, static or regress
- Hereditary motor and sensory neuropathy (HMSN): CMT (Charcot Marie Tooth) is a broad category with mild deficits such as clumsiness, distal motor weakness, classic "stork leg deformity" and possible scoliosis; in more severe forms (CMT3) such as Dejerine-Sottas and congenital hypomyelination neuropathy (with hypotonia at birth) and congenital hypomyelination neuropathy, life expectancy may be only a few months
Essential features
- Atrophic myofibers with myofiber type grouping (groups of myofibers of the same histochemical type)
- No / minimal inflammation
Terminology
- No specific terminology
- Skeletal muscle with neurogenic atrophy
- Skeletal muscle with features of neurogenic atrophy
Epidemiology
- Specific to the underlying cause
- ALS incidence is 2 per 100,000
- Incidence for SMA type I is 1:10,000 and types II and III are 1:24,000
- HMSN incidence is approximately 1 in 2500 to 1 in 1214 in Scandinavian countries
Sites
- Muscle biopsy can be obtained from the more distal muscles depending on the disease process and the muscle that is affected
- Generally, distal is more affected than proximal in many of these disorders
Pathophysiology
- Has a variety of different causes
- Central theme is loss of innervation to the muscle
- Can be due to radiculopathy from tumor, cyst or herniated nucleus pulposus
- Other causes are loss of the anterior horn cell in SMA / ALS
- In CMT1, CMT3 and CMT4, there is a problem with myelination; in CMT2, there is axonal disease
Etiology
- Varies as to the cause of the neurogenic atrophy
- Main causes and significant chromosomal mutations are listed under Molecular Description
Clinical features
- Dependent on underlying etiology
Diagnosis
- Varies by etiology
- Most of the neurogenic muscle biopsies are descriptive
- Radiography, such as MRI, is ultimately required for diagnosis of radiculopathy
- Clinical correlation (upper motor neuron findings and lower motor neuron findings) and EMG / NCS findings are required for diagnosis of ALS
- Mutation studies will identify exact causes of HMSN, clinical and EMG findings may point to the type of mutation
- UpToDate has an excellent chart for clinicians to follow based on EMG / NCS and clinical findings on which mutation to test for first and then what to test if negative for mutation
- SMA is a clinicopathologic diagnosis, type is based on clinical presentation and genetic mutations can be assessed
Laboratory
- Occasional cases of SMA can have mild elevations in CK levels
Radiology description
- May reveal cause of nerve impingement
- Specifically, MRI can show herniated nucleus pulposus or spinal stenosis
Prognostic factors
- Prognosis rests on etiology
- ALS runs a fatal course; some mutations cause a fairly rapid progression (FUS), but other mutations (SOD-1 D90A) have survival of 10 years or greater
- Spinal muscular atrophy type I generally has a poor outcome and most succumb to respiratory compromise at age 1-2; other types have prognosis based on the type of mutations and underlying factors
- HMSN varies; form with Dejerine Sottas has a poor prognosis; CMT1 has minimal impact on survival
Treatment
- Therapy is aimed at the underlying etiology
- SMA III may require physical therapy
- ALS is generally progressive and requires additional support if bulbar symptoms; ie. PEG tube and ventilator dependency
Gross description
- No gross features of neurogenic atrophy are identifiable on the biopsy
Microscopic (histologic) description
- General / ALS / HMSN:
- Myofibers are smaller and angulated
- No increase in central nucleation
- In areas, myofiber may become atrophic to the point that there is only a "nuclear clump" or "nuclear bag" left
- In cases with deinnervation / reinnervation, there will be myofiber type grouping on ATPase staining or myofiber immunohistochemical stain
- Surrounding unaffected myofibers that are innervated differently may compensate and become hypertrophic
- Care should be also taken to ensure that both myofiber type groups are affected and not just a select type II atrophy for instance
- SMA:
- Large groups of atrophic myofibers
- Myofibers will be smaller and rounded
- There is associated hypertrophy of Type I myofibers (type I and type II)
- Type III may have no changes or show similar findings to type I and II
Microscopic (histologic) images
Contributed by Meggen Walsh, D.O., M.S., P.A.
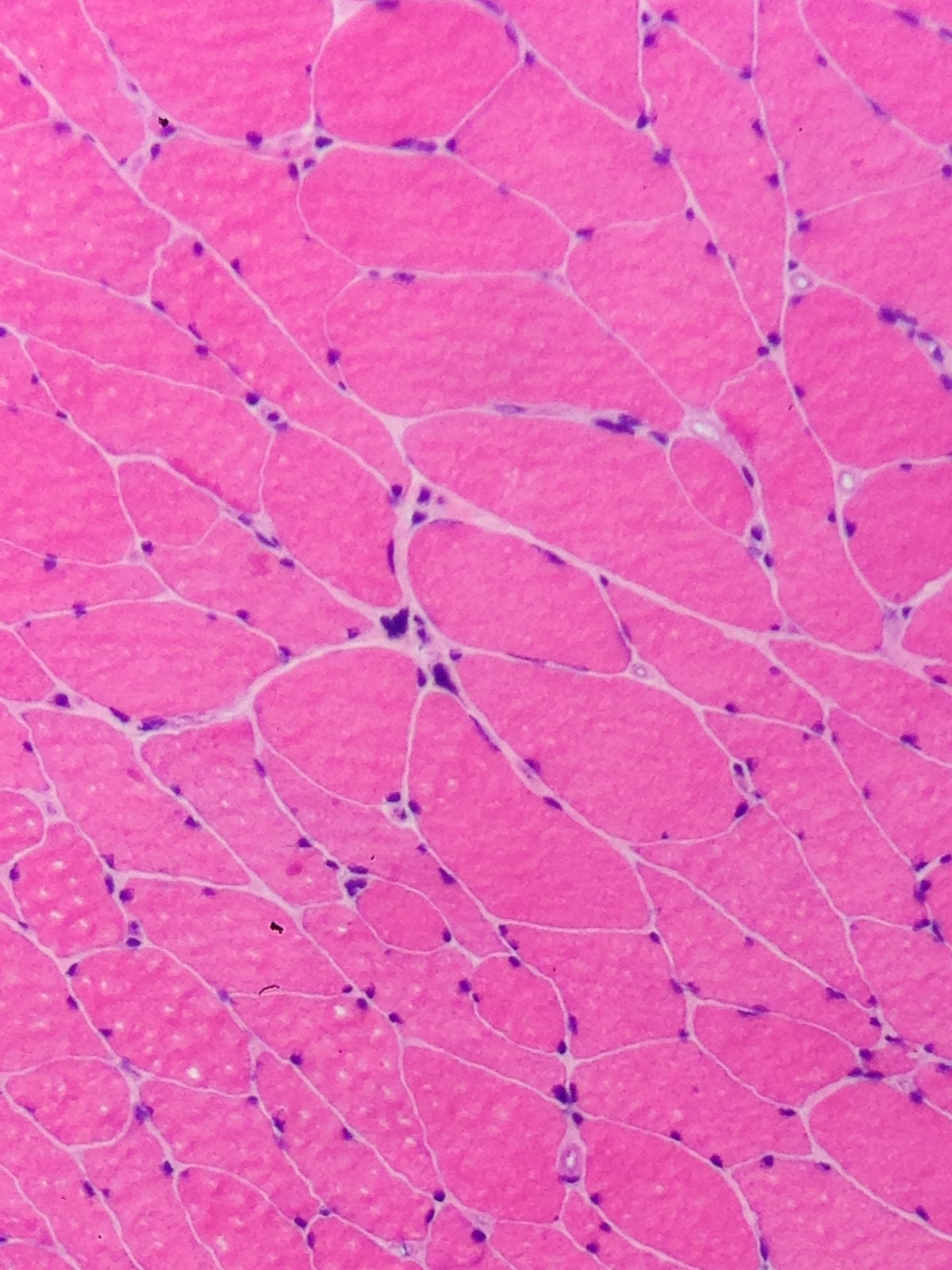
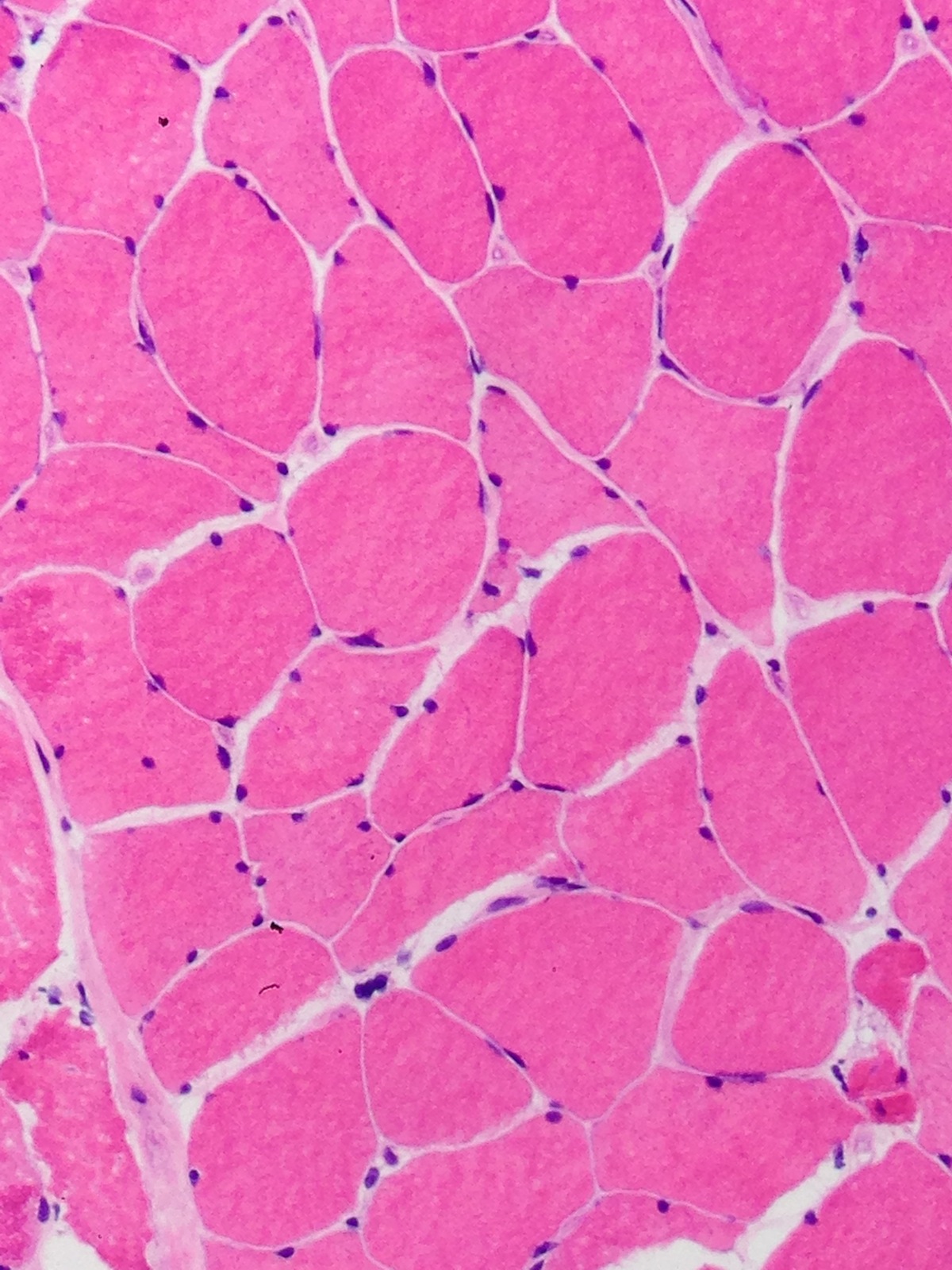
Skeletal muscle: multiple nuclear "clumps" or nuclear "bags"; also few atrophic angulated myofibers (H&E)
Positive stains
- In some angulated myofibers there can be expression of fetal myosin (Dubowitz: Muscle Biopsy: A Practical Approach, 4th ed, 2013)
- "Grouping of myofibers by type" is not a hard and fast criteria; grouping occurs if a myofiber of one type is completely surrounded by myofibers of the same type; normally, there are groups of both type I and type II myofibers
- Must rule out an "abundance" of one myofiber type, which resembles myofiber type grouping
- NADH-TR can show "target fibers", which have a dark staining rim around a pale central zone, similar to a central core
Negative stains
- Deinnervated myofibers may loose glycogen and not stain as strongly with PAS (Dubowitz: Muscle Biopsy: A Practical Approach, 4th ed, 2013)
Molecular / cytogenetics description
- ALS: molecular and neurodegenerative features
- C9ORF72: (familial 40%, sporadic 5-7%) 9q21-22; hexanucleotide repeat in the noncoding region of chromosome 9p (normal is 3 repeats and ALS is more than 30); ubiquitylated and TDP-43 positive neuronal cytoplasmic inclusions; bunina bodies can be seen; can see cytoplasmic inclusions in the granular cell layer of the cerebellum, dentate nucleus of the hippocampus and CA3/4
- SOD-1: (familial 15-20%,) 21q22; multiple forms, ubiquitylated only inclusions in some forms or hyalin inclusions that stain with non-phosphorylated neurofilament or phosphorylated neurofilament
- FUS: (familial 5%) 16q12.1-q12.2; glial and neuronal cytoplasmic inclusions that are positive with FUS-antisera and are negative for TDP-43 and few if any ubiquitylated inclusions
- Alsin2: (juvenile ALS form) 2q33; mutation can cause PLS, ALS and hereditary spastic paraplegia; therefore can have upper motor neuron only findings, upper and lower motor neuron findings
- Spinal muscular atrophy:
- SMA: most common is deletion of exon 7 or 8 on 5q
- Hereditary motor and sensory neuropathy:
- CMT1: pathology shows "onion bulb" formation on sural nerve biopsies; divided by mutations but all are clinically similar and have distal weakness, nerve hypertrophy but ambulation is maintained; CMT1A commonly is a mutation of PMP22 (17p11.2-p1); CMT1B is commonly MPZ, myelin protein zero (1q22); CMT1C generally LITAF, lipopolysaccharide-induced tumor necrosis factor alpha factor (16p13.1-p12.3); CMT1D EGR2, early growth response 2 gene (10q21.1-q22.1); CMT1E PMP22; CMT1F NEFL (8p21)
- CMT2: axonal loss is present but very little to no "onion bulb formation" as this is more an axonopathy than myelinating problem; can be wallerian degeneration of the nerves; sensation is lost more than motor skills and hypertrophic nerves are not clinically apparent; in early clinically presenting forms of the disease, ambulation can be severely affected; CMT2A is MFN2, mitochondrial fusion protein mitofusin 2 (1p35-36); CMT2B is RAB7 (6q21.3); CMT2C TRPV4 (12q24.1); CMT2D is GARS, glycyl tRNA synthetase (7p15); CMT2E is NEFL, neurofilament light polypeptide (8p); CMT2F is HSPB1, heat shock protein (7q11.23); CMT2G is 12q12-q13; CMT2I is MPZ; CMT2L HSPB8, heat shock protein (12q24.23); CMT2S IGHMBP2 (missense mutation)
- CMT3: these are the more severe forms; Dejerine-Sottas has "onion bulb formation" on biopsy and shows motor delays with distal weakness and contracture formation may occur; in congenital hypomyelinating neuropathy there is absent myelin, hence no onion bulb; congenital hypomyelination neuropathy presents with hypotonia and respiratory distress; death can occur in infancy; Dejerine Sottas has recessive and dominant forms mutations; mutations can be seen in PMP22, MPZ and EGR2 gene; congenital hypomyelinating neuropathy has mutations in the same genes and also MTMR2, myotubularin related 2 gene
- CMT4: CMT4 is more severe than 1 and 2; they are also rare; CMT4A shows mutations in GDAP1 (8q13-q21.1) and may be a possible mitochondrial disorder; CMT4B1 is MTMR2 (11q22-23); CMT4B2 is SBF2 (11p15); CMT4C SH3TC2 (5q32); CMT4D NDRG1 (8q24.3); CMT4E is EGR2 (10q21-22); CMT4F is PRX, periaxin (19q13.1-13.3); CMT4H is FGD4 (12p11.2); and CMT4J FIG4 gene (6q21)
- CMTX: CMT X linked is the most common form after CMT1A; weakness with occasional deafness is noted; cases with transient "stroke like episodes" have been noted; CMX1 is caused by GJB1, gap junction protein connexin (Xq13.1)
Differential diagnosis
- Myotonic dystrophies and limb girdle dystrophies can resemble SMA type III
