
Lymphoma & related disorders
Posttransplant lymphoproliferative disorders (PTLD)
PTLD-polymorphic
Authors: Daniel Cassidy, M.D., Jennifer Chapman, M.D.
Editorial Board Member: Patricia Tsang, M.D., M.B.A.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 12 April 2021
Last staff update: 17 July 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Polymorphic[TIAB] (PTLD[TI] OR posttransplant lymphoproliferative[TI])
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Etiology | Clinical features | Prognostic factors | Case reports | Treatment | Gross description | Frozen section description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Immunohistochemistry & special stains | Flow cytometry description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Cassidy D, Chapman J. PTLD-polymorphic. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomanonBpolymorphicB.html. Accessed April 19th, 2024.
Definition / general
- Polymorphic posttransplant lymphoproliferative disorders (P-PTLDs) are a heterogenous group of hematolymphoid proliferations composed of a polymorphic population of immunoblasts, plasma cells and small to intermediate sized lymphoid cells that efface normal tissue architecture and do not meet criteria for the diagnosis of any well established form of lymphoma recognized in immunocompetent hosts (Blood 2016;127:2375)
Essential features
- Occurs in the posttransplant setting
- Effacement of normal tissue architecture
- Presence of a polymorphic lymphoid infiltrate with a complete spectrum of maturation and variable number of transformed cells / immunoblasts
- Variable resolution following the cessation of immunosuppressive therapy
- Frequently EBV related (Lancet 2006;367:233, Jaffe: Hematopathology, 2nd Edition, 2017)
Terminology
- Polymorphic posttransplant lymphoproliferative disorder (P-PTLD)
ICD coding
- ICD-O: 9971/1 - posttransplant lymphoproliferative disorder, NOS
Epidemiology
- PTLDs develop in approximately 2% of all transplant patients and are more common in liver, lung, intestinal and multivisceral transplant recipients
- P-PTLD accounts for the minority of PTLDs in older individuals
- P-PTLD is a much more common form of PTLD in children, related to primary EBV infection
- Occurs across a wide age range without sex predilection (Lancet 2006;367:233, Jaffe: Hematopathology, 2nd Edition, 2017)
Sites
- Can occur nearly anywhere throughout the body, including both nodal and extranodal sites and within the graft or in native tissue
- Most common extranodal sites are the gastrointestinal tract, lung and liver
Etiology
- Majority of PTLDs following solid organ transplantation are of host origin, while those following bone marrow transplant are more often derived from the donor
- P-PTLDs are caused by expansion of clonal lymphoid or plasma cells, which are frequently infected by EBV and inadequately controlled by the immune system of an immunosuppressed host
- Most P-PTLDs demonstrate a type III latency pattern, with fewer demonstrating a type II latency pattern and still fewer demonstrating a type I latency pattern
- Genetic predisposition likely also plays a role in the risk for development of PTLD, as evidenced by increased and early onset disease in patients with certain interferon gamma and human leukocyte antigen (HLA) polymorphisms
- PTLDs are thought to exist on a spectrum beginning with an expanded EBV infected polyclonal population that develops increasingly pure clonal populations, resulting in the development of P-PTLD and monomorphic PTLD (World J Transplant 2016;6:505, Cancers (Basel) 2020;12:328)
Clinical features
- Variable frequency but generally considered to comprise the minority of PTLDs in adults
- More common in children, often following primary EBV infection
- Historically, 80% of PTLDs were reported to occur within the first year; however, recent studies suggest an interval of several years on average
- May present as multiple mass-like lesions with symptomatology related to site of involvement (i.e. obstructive symptoms with an intestinal mass)
- Reduction in immunosuppression leads to regression in a variable number of cases; some cases progress and warrant treatment as overt lymphoma (Cancer Treat Res 2015;165:305)
Prognostic factors
- Good response to reduced immunosuppression is associated with superior outcome
- PTLD development following solid organ transplant generally fares better than after bone marrow transplant
- PTLD limited to the allograft is associated with better outcome
- CNS involvement, multiorgan involvement and organ dysfunction predict poor response to reduced immunosuppression
- Other supposed prognostic factors in P-PTLD are a subject of debate:
- Cases with demonstrable clonality are purported by some to be more resistant to reduction of immunosuppression
- Absence of EBV appears to be associated with a more aggressive clinical course according to some studies (Exp Hematol Oncol 2017;6:26)
Case reports
- 1 year old boy with diffuse lymphadenopathy and 25 year old man with a lung mass (Am J Clin Pathol 2017;147:129)
- 8 year old girl with extensive lymphadenopathy (Pediatr Dev Pathol 2003;6:449)
Treatment
- Reduction in immunosuppression is often used as first line therapy, if possible
- Additional treatment modalities such as chemotherapy (including rituximab), surgical excision and local radiation have also been used, depending on the clinical scenario (Cancer Treat Res 2015;165:305, Jaffe: Hematopathology, 2nd Edition, 2017)
Gross description
- These lesions can have a nearly normal gross appearance or appear as a mass forming lesion with variable cut surface, depending on the histologic composition of the proliferation
Frozen section description
- Outright diagnosis of P-PTLD should not be made on frozen section examination
- Histologic appearance will be similar to that described for permanent sections below, although lymphocyte atypia can often be more striking in frozen sections
- Better to describe an atypical lymphoid infiltrate and defer to permanent sections for diagnosis
- In this setting, tissue can be triaged for flow cytometric studies (Jaffe: Hematopathology, 2nd Edition, 2017)
Microscopic (histologic) description
- Distinction of P-PTLD from nondestructive lesions hinges on the presence of effacement of normal tissue architecture
- Unlike most lymphomas, there is characteristically a full range of B cell maturation, including immunoblasts, plasma cells and small to intermediate sized lymphoid cells
- Necrosis, cells resembling Hodgkin or Reed-Sternberg cells, and numerous mitoses may be present
- Relatively more monomorphic areas may be present, suggesting a continuum between P-PTLD and monomorphic PTLD (Jaffe: Hematopathology, 2nd Edition, 2017)
Microscopic (histologic) images
Contributed by Daniel Cassidy, M.D. and Jennifer Chapman, M.D.
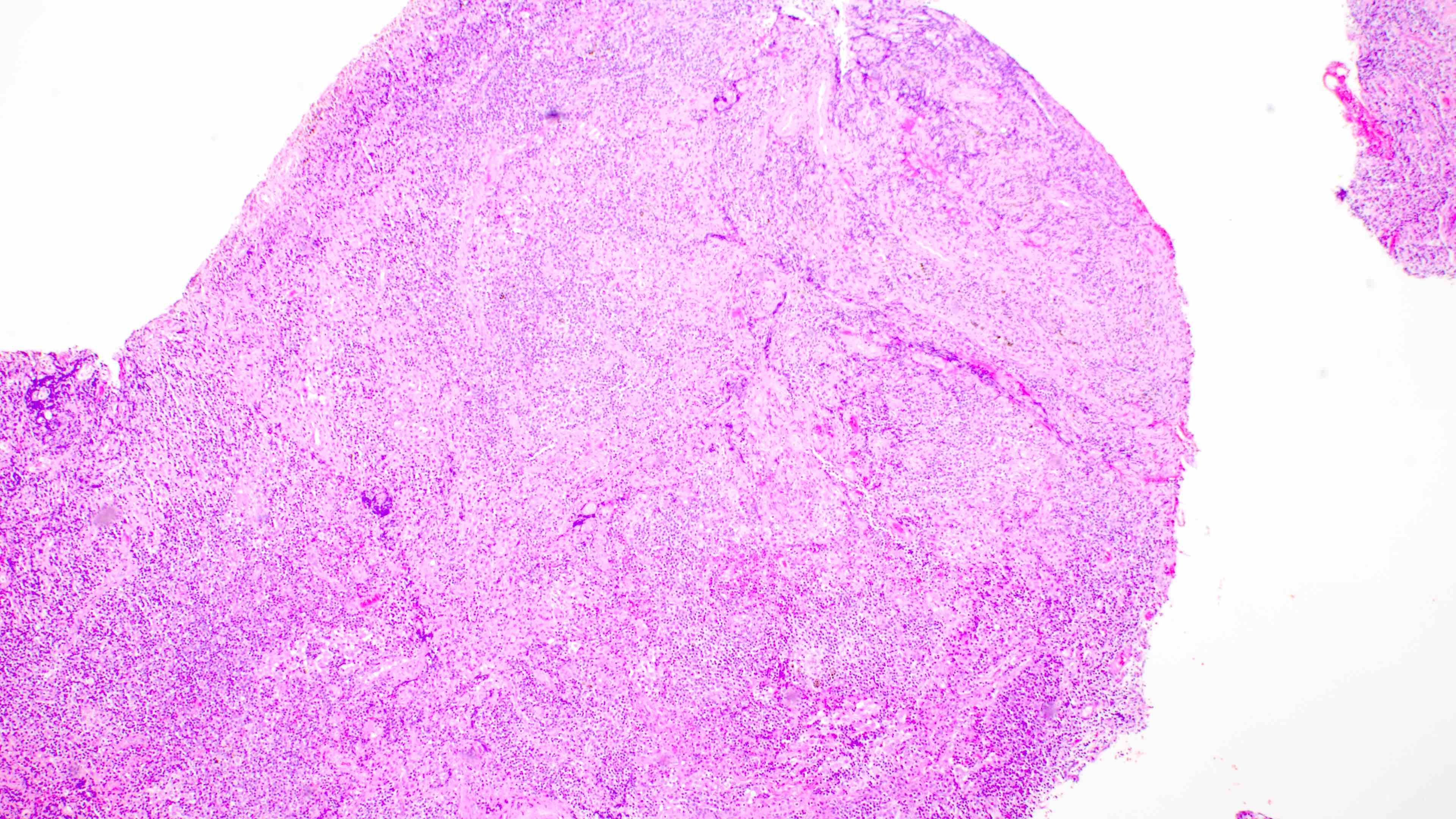
Architectural distortion
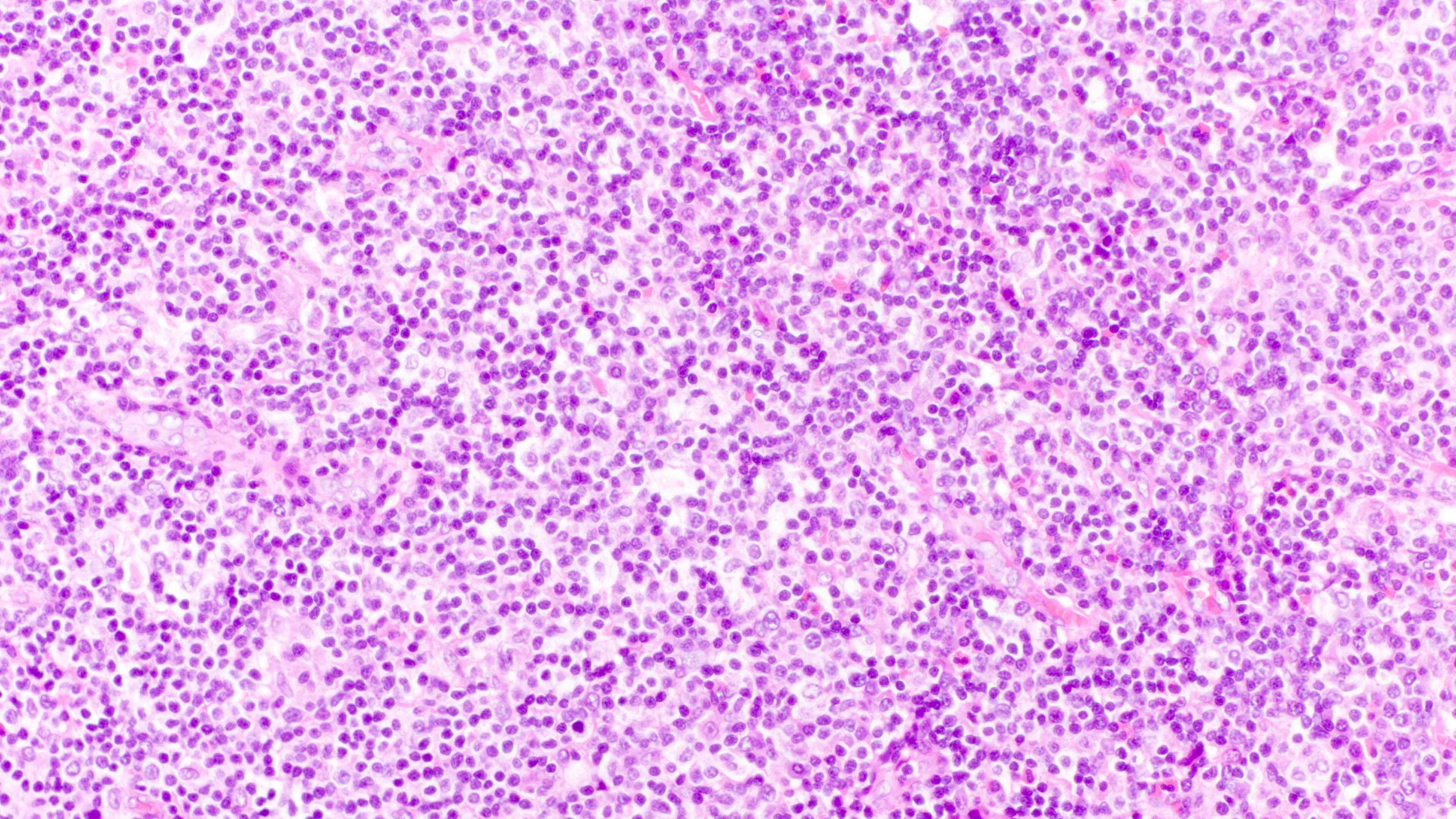
Polymorphic lymphoid infiltrate
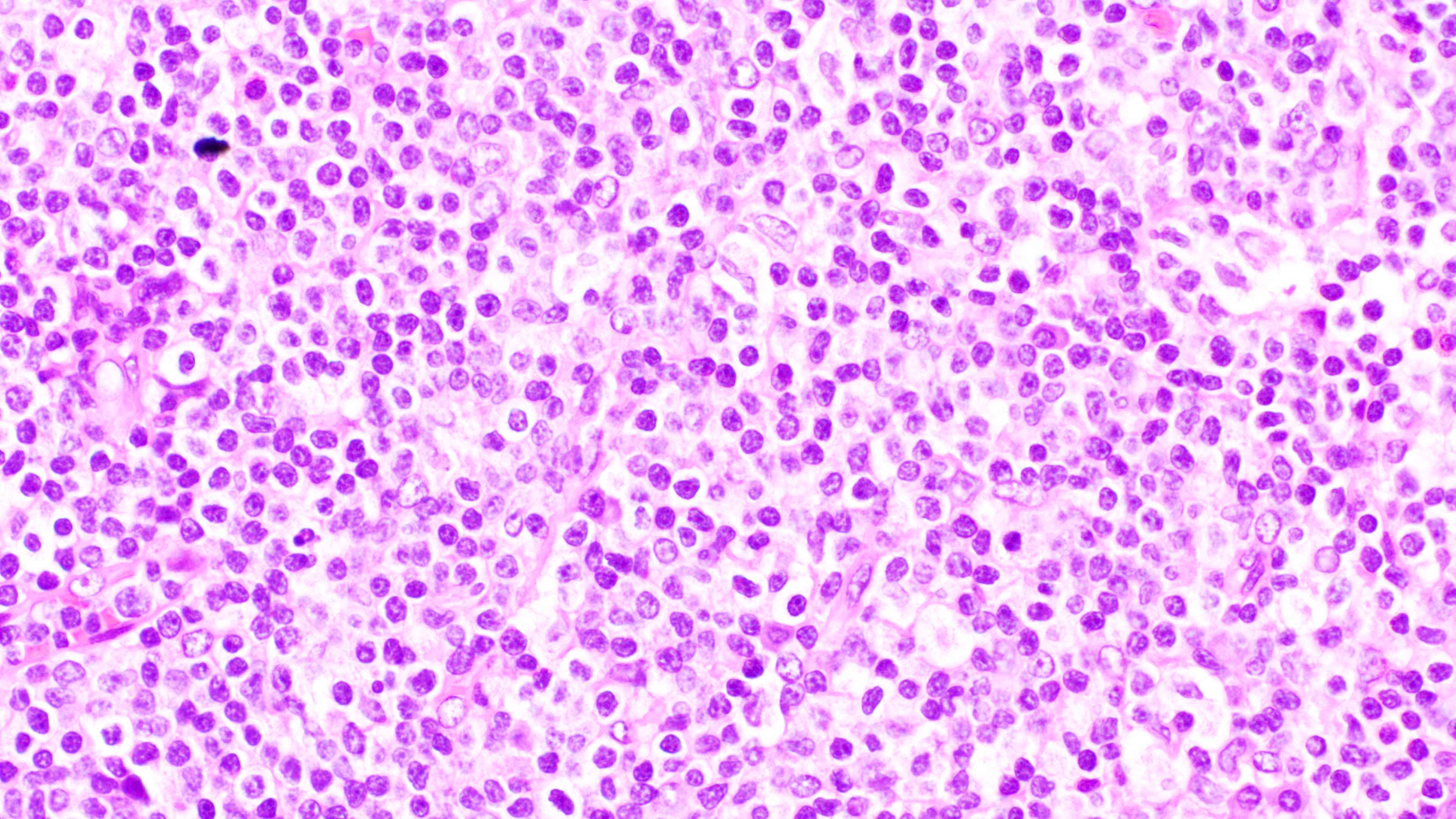
Full spectrum of maturation
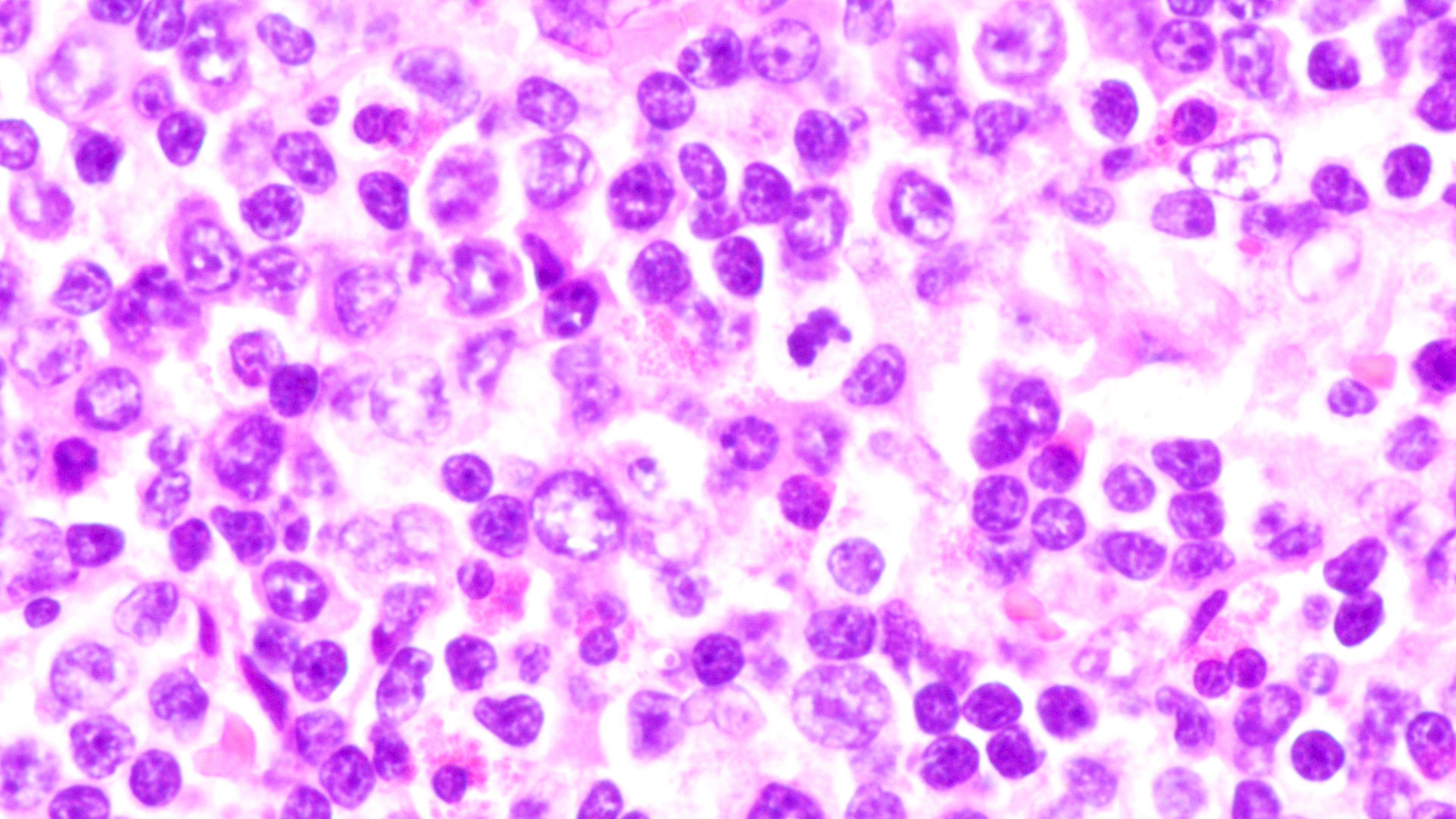
Composition of lymphoid infiltrate
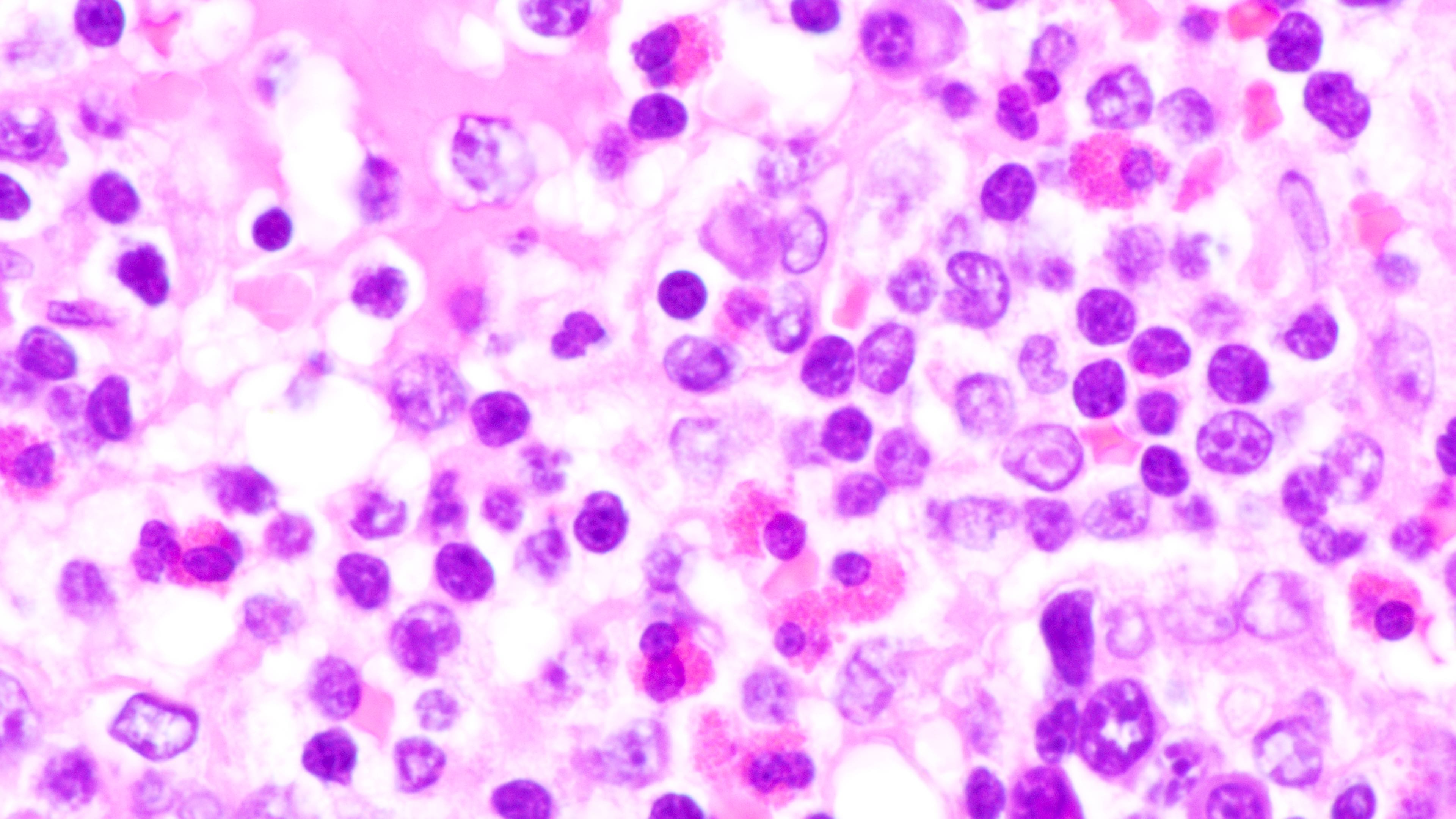
Polymorphic infiltrate
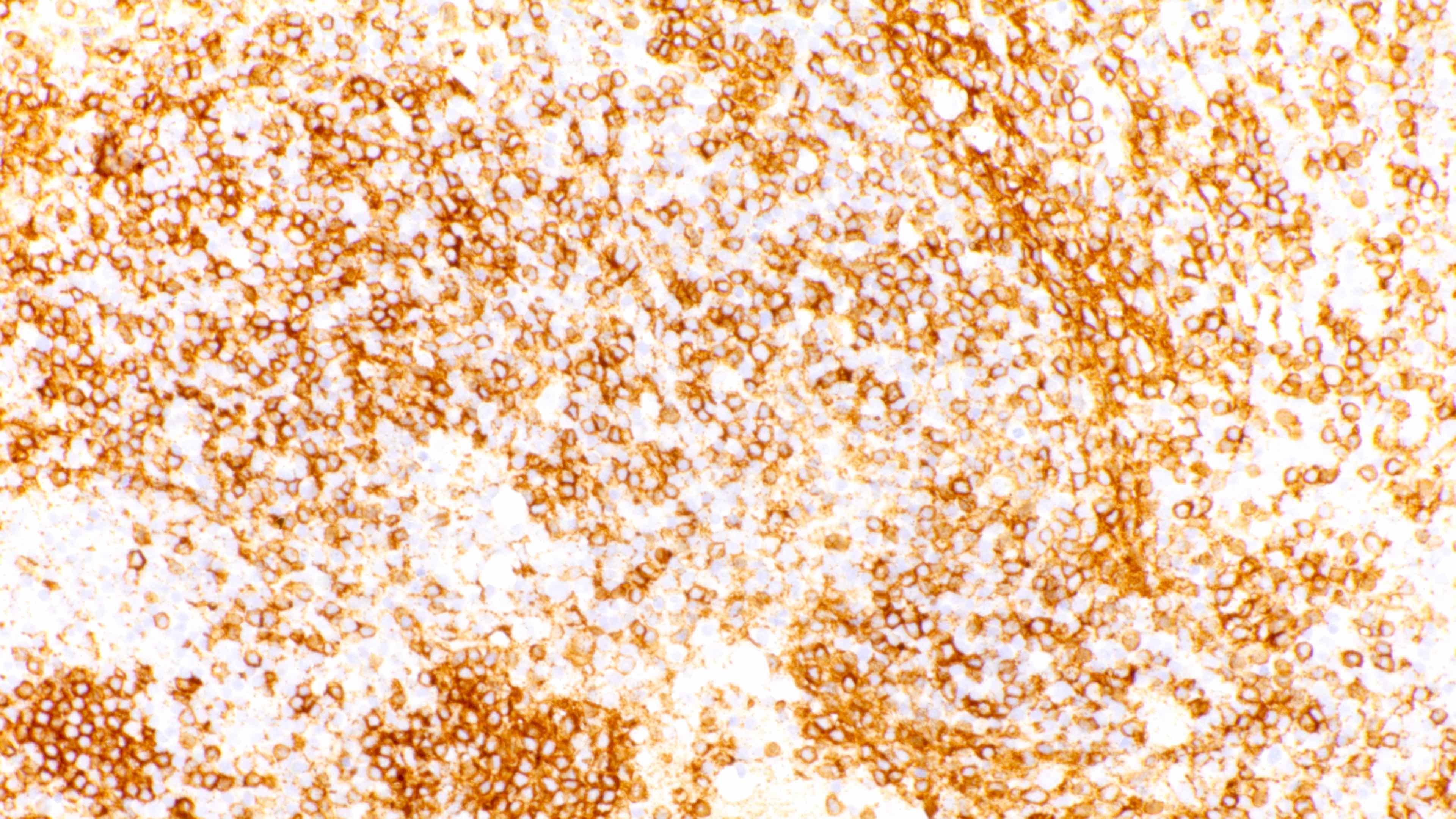
Variably sized B cells
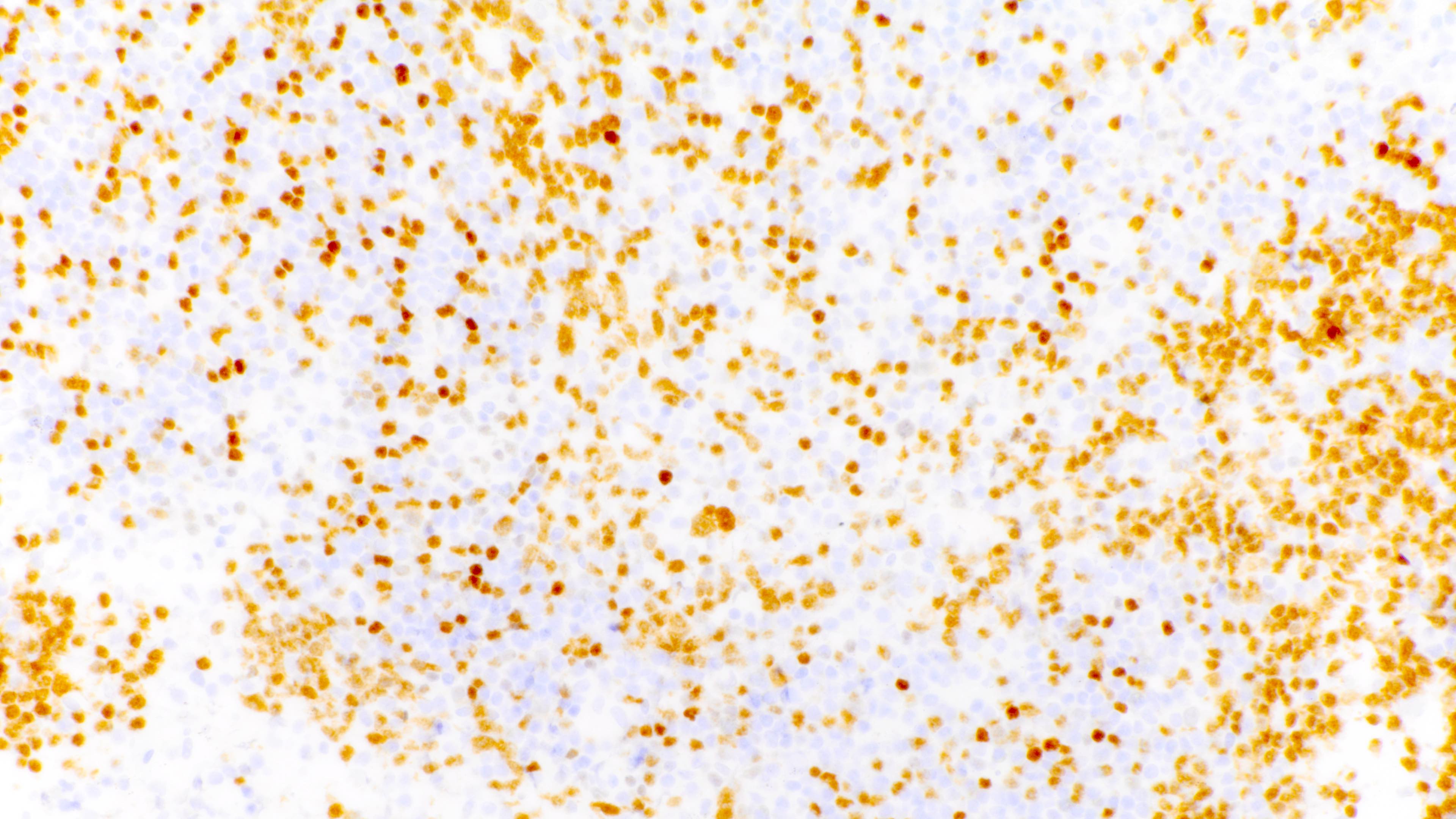
PAX5 variable cell size
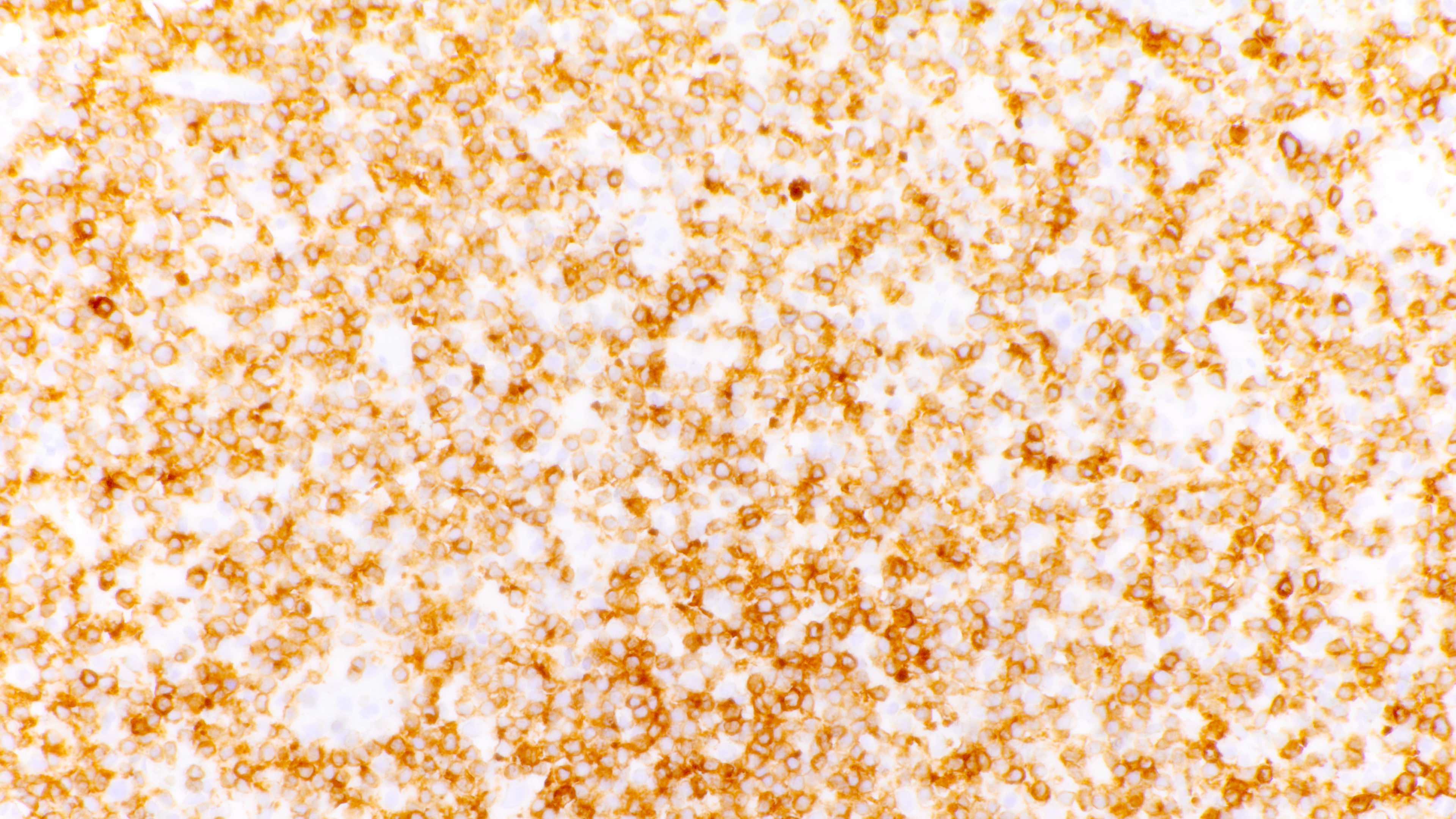
Variably sized T cells
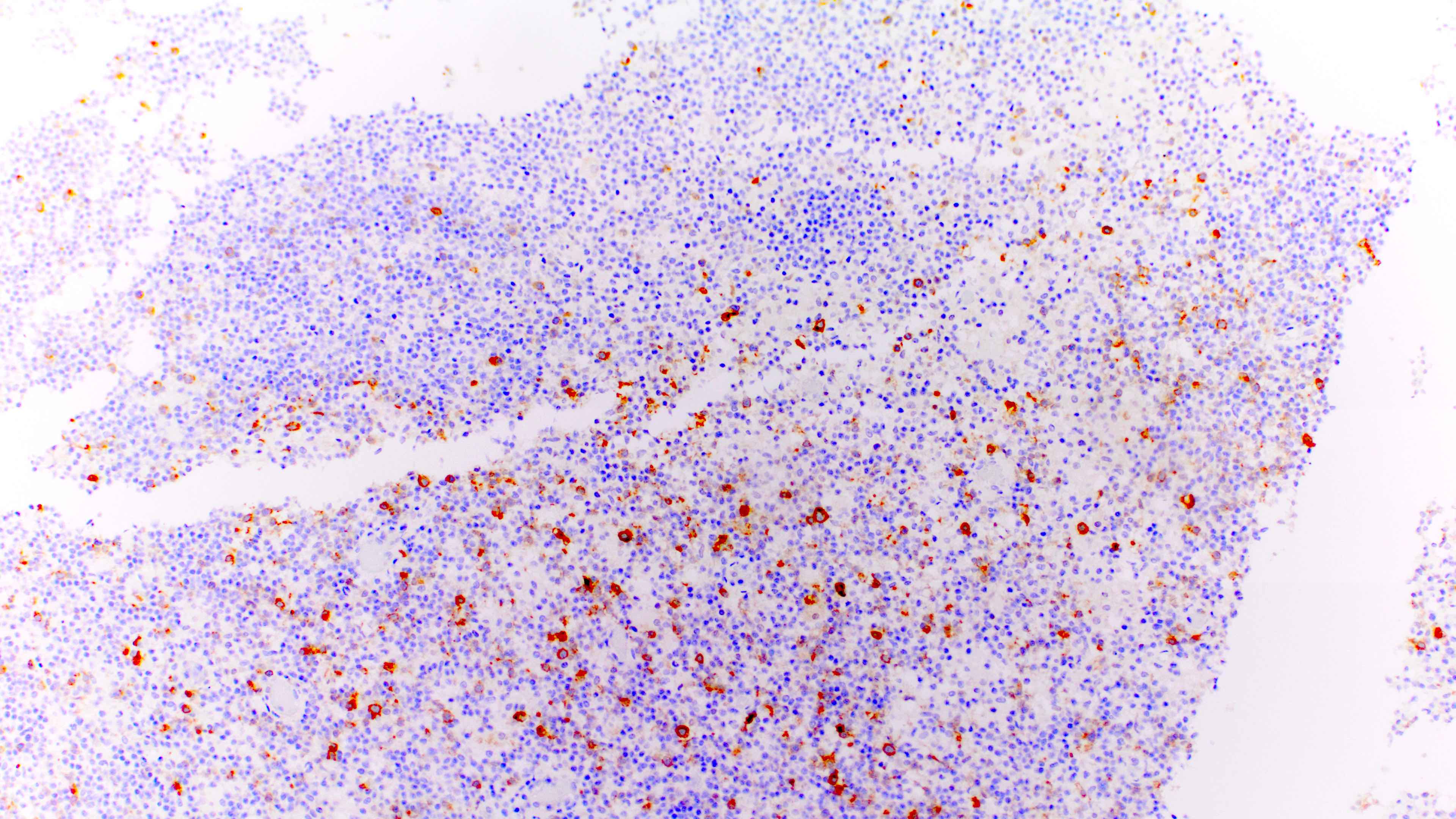
Scattered immunoblasts
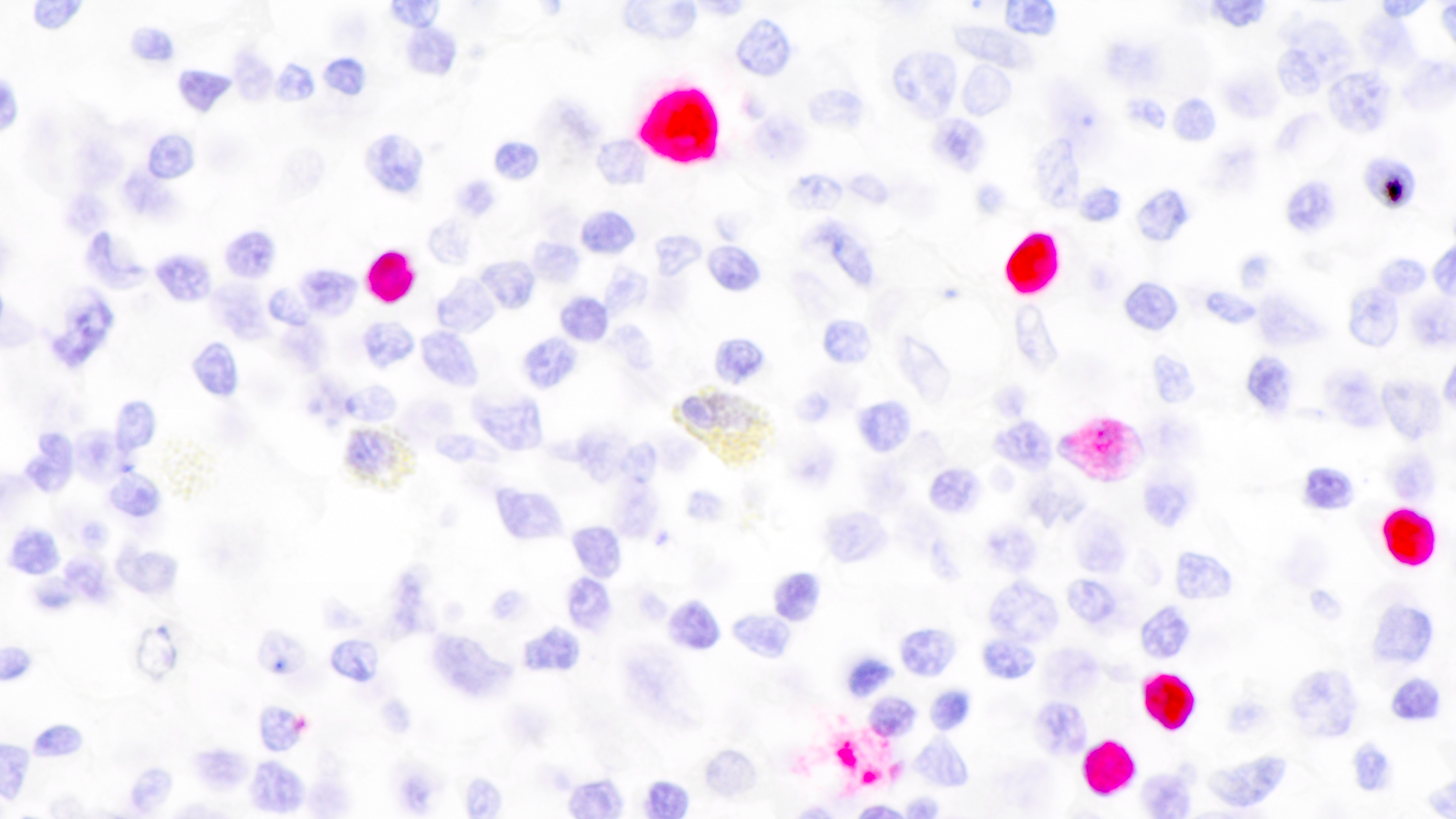
EBER positivity
Cytology description
- Heterogeneous population of cells including small to intermediate sized lymphoid cells, which may have irregular nuclear contours, plasma cells and a variable number of immunoblasts with more open chromatin and prominent, centrally located nucleoli
- Variable amount of necrotic debris and large cells with features of Hodgkin or Reed-Sternberg cells can be seen
- Reference: Diagn Cytopathol 1997;16:489
Immunohistochemistry & special stains
- Admixture of B cells (expressing B cell markers including CD19, CD20, PAX5, etc.) and T cells (expressing T cell markers including CD2, CD3, CD5, CD7, etc.)
- Light chain restriction may be present and does not exclude this diagnosis, although it should be noted if present
- Variable or even strong CD30 expression can be seen; however, these CD30+ large cells are CD20 positive and CD15 negative, unlike classic Hodgkin cells
- Moreover, the large CD30 positive cells are more heterogeneous in morphologic appearance and degree of CD30 expression compared to classic Hodgkin lymphoma
- Most cases are positive for EBER by in situ hybridization
- Ki67 nuclear proliferation index may be brisk (Jaffe: Hematopathology, 2nd Edition, 2017)
Flow cytometry description
- In many cases, a demonstrable monotypic B cell or plasma cell population is present
Molecular / cytogenetics description
- Clonally rearranged IGH / IGK / IGL genes frequent, albeit less prominent than in monomorphic PTLD cases
- EBV terminal repeat analysis - most sensitive method for clonal populations in EBV+ cases
- Tumors at different sites may be either clonally distinct or related
- No significant T cell clone should be present
- Cytogenetic studies may demonstrate abnormalities, such as trisomy 9 or trisomy 11, in some cases
- BCL6 mutations occur in up to half of P-PTLD
- P-PTLD does not segregate from nongerminal center monomorphic PTLD by gene expression profiling (Am J Clin Pathol 1994;101:590, Genes Chromosomes Cancer 2006;45:313, Hematol Oncol 2008;26:199)
Sample pathology report
- Left colon, biopsy:
- EBV positive posttransplant lymphoproliferative disorder, favor polymorphic type (see comment)
- Comment: This patient is a 6 year old female status post orthotopic liver transplant, now with nodularities identified in the colon. Biopsies show colonic mucosa within which the lamina propria is abnormally expanded by a heterogeneous population of hematolymphoid cells, including small and large, transformed lymphoid cells, plasma cells, neutrophils, immunoblasts and eosinophils. Histologic features of graft versus host disease are not seen.
- By immunohistochemistry, CD20 identifies scattered and loosely aggregated B cells, which are heterogeneous in appearance, ranging from small to large. Large (transformed) CD20 positive B cells are few and are not present in sheets. CD3 immunostain confirms the presence of an associated population of small T cells. BCL6 is positive in scattered large B cells. Plasma cells are few and appear polytypic based on kappa and lambda in situ hybridization. EBER is detected in the scattered large and transformed lymphoid cells.
- The above histologic and immunophenotypic features support the presence of a posttransplant lymphoproliferative disorder. Based on the architectural distortion observed in the biopsy, we would classify this PTLD as polymorphic type. We have also considered the possibility of an infectious mononucleosis type PTLD; however, we do not favor this interpretation due to the presence of architectural distortion within the colonic mucosa and lack of diffuse EBER expression, the latter being seen more frequently in infectious mononucleosis type PTLD.
Differential diagnosis
- Reactive inflammatory infiltrate:
- Nondestructive posttransplant lymphoproliferative disorder - infectious mononucleosis-like type:
- Differential diagnosis, particularly at tonsillar site
- IM-like PTLD usually should not have overt architectural effacement
- At tonsillar site, florid IM-like PTLD may be indistinguishable from P-PTLD
- At this site, as long as changes are in keeping with diagnosis of IM in immunocompetent host, a diagnosis of IM-PTLD is favored
- Classic Hodgkin lymphoma posttransplant lymphoproliferative disorder:
- Differential diagnosis given the presence of large, sometimes Hodgkin-like cells in P-PTLD
- Most effective way to make distinction is an abnormal immunophenotype of HL cells, including CD15+, CD30+ and CD45-
- A variable number of cases may be CD15-, however, further complicating differential diagnosis with P-PTLD
- Of note, CHL-PTLD Hodgkin-like cells more commonly express B cell markers than CHL
- Background of CHL-PTLD is more sparse in B cells and rich in T cells than P-PTLD tends to be
- In CHL, Hodgkin cells are histologically and immunophenotypically distinct from background lymphoid cells
- In P-PTLD, there is a spectrum of B cells, ranging from small to immunoblastic and Hodgkin-like, which may show EBV expression, in contrast to EBV being largely restricted to the Hodgkin Reed-Sternberg cells in CHL or CHL-PTLD
- Monomorphic posttransplant lymphoproliferative disorder:
- By definition, M-PTLD demonstrates features that would be diagnostic of a well established lymphoma entity in an immunocompetent host, while P-PTLD does not (Jaffe: Hematopathology, 2nd Edition, 2017)
Additional references
Board review style question #1
Which of the following features would be most helpful in favoring a diagnosis of polymorphic posttransplant lymphoproliferative disorder over infectious mononucleosis-like posttransplant lymphoproliferative disorder?
- Architectural effacement
- CD30 positive large B cells
- EBER ISH positivity
- Hodgkin-like cells
Board review style answer #1
A. Architectural effacement. Architectural effacement is one of the key distinguishing histologic features between P-PTLD and IM-PTLD, as other morphologic features tend to overlap significantly. Of note, the distinction between P-PTLD and IM-PTLD at tonsillar sites can be exceptionally difficult, if not impossible, on morphologic grounds alone, as florid IM at this site can cause architectural distortion. While a prominent clonal B cell population favor a diagnosis of P-PTLD in this setting, small clonal peaks have been described in IM-PTLD of unknown significance.
Comment Here
Reference: PTLD-polymorphic
Comment Here
Reference: PTLD-polymorphic
Board review style question #2
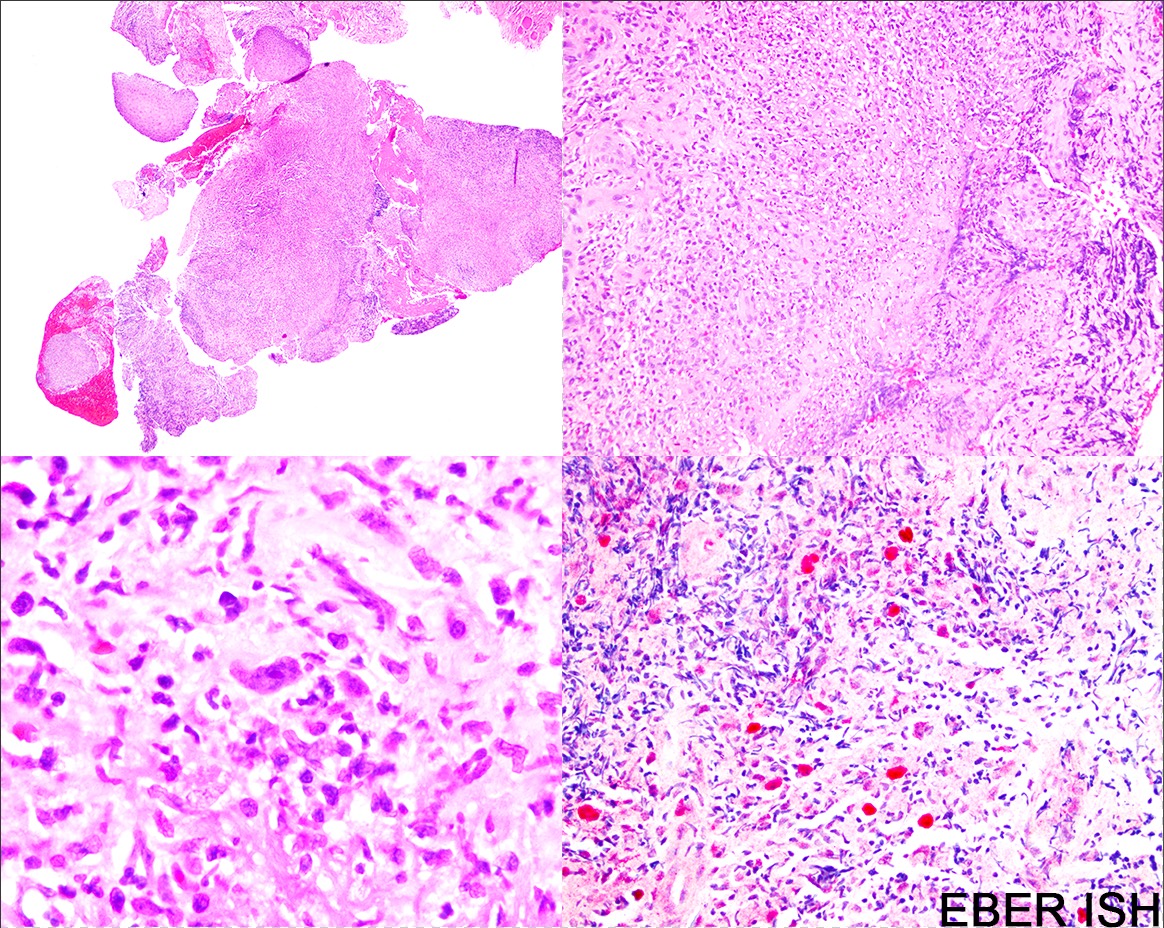
A 73 year old man status post renal transplant 3 years prior for end stage renal disease secondary to chronic, uncontrolled hypertension presents with an ulcerated lesion involving the oral mucosa. Histologic sections of the lesion are shown above. The large cells are positive for CD30, CD20, CD45 and EBER ISH and negative for CD15. Which of the following is the best diagnosis according to the current WHO classification schema?
- Classic Hodgkin lymphoma posttransplant lymphoproliferative disorder
- EBV positive mucocutaneous ulcer
- Monomorphic posttransplant lymphoproliferative disorder
- Polymorphic posttransplant lymphoproliferative disorder
- Reactive immunoblastic proliferation
Board review style answer #2
B. EBV positive mucocutaneous ulcer. Based on the current WHO classification, PTLDs with clinical and morphologic features consistent with EBV MCU should be classified as such.
Comment Here
Reference: PTLD-polymorphic
Comment Here
Reference: PTLD-polymorphic
