

Bone marrow nonneoplastic
Noninfectious systemic disorders
Hemophagocytic lymphohistiocytosis
Editorial Board Member: Anamarija M. Perry, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 14 June 2023
Last staff update: 14 June 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Hemophagocytic lymphohistiocytosis

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Flow cytometry description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Evans MG, Balakrishna J, Rezk SA. Hemophagocytic lymphohistiocytosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrowhemophagocyticlymphohistiocytosis.html. Accessed December 21st, 2024.
Definition / general
- Pathologic immune activation syndrome
- Congenital or acquired defective natural killer (NK) / T cell function with uncontrolled hypercytokinemia and end organ damage
- Histiocytic infiltration of reticuloendothelial organs with hemophagocytosis of erythrocytes, leukocytes, platelets and their precursor cells
- Divided into genetic (primary) and acquired (secondary) forms
Essential features
- Sudden onset of a systemic inflammatory response syndrome (SIRS) with symptoms including high unremitting fevers, malaise, rash, jaundice and myalgia, along with accompanying cytopenias, hepatosplenomegaly and generalized lymphadenopathy
- Familial (primary) hemophagocytic lymphohistiocytosis (HLH) is a progressive disease characterized by early age of onset, autosomal recessive inheritance pattern, decreased NK cell function and frequent perforin mutations
- Acquired (secondary) hemophagocytic lymphohistiocytosis is often associated with immunodeficiency, normal or reduced NK cell numbers and normal perforin expression, and may be triggered by infectious agents, malignant neoplasms, graft versus host disease and (rarely) HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome
- Primary and secondary hemophagocytic lymphohistiocytosis are invariably fatal when left untreated but new therapy protocols (including HLH-2004) have reported 5 year survival rates up to 54%
Terminology
- Familial (primary) hemophagocytic lymphohistiocytosis (FHLH), FHL1 - FHL5 subtypes
- May occur in the setting of Chédiak-Higashi syndrome, Griscelli syndrome type 2, Hermansky-Pudlak syndrome type 2 and X linked lymphoproliferative disorder types 1 and 2 (Hematology Am Soc Hematol Educ Program 2011;2011:178)
- Acquired (secondary) hemophagocytic lymphohistiocytosis
ICD coding
- ICD-10: D76.1 - hemophagocytic lymphohistiocytosis
Epidemiology
- Familial hemophagocytic lymphohistiocytosis primarily affects young children with an incidence of approximately 1.5 per 100,000 live births (Pediatr Blood Cancer 2015;62:346)
- Acquired hemophagocytic lymphohistiocytosis is more common in older children and adults; its incidence is unknown but is likely underdiagnosed
- No race or sex predilection
Sites
- Involves all reticuloendothelial organs (bone marrow, spleen, liver and lymph nodes)
- Less commonly involves skin, lungs, meninges, cerebrospinal fluid and subcutaneous tissue
Pathophysiology
- Failure of NK cells to induce apoptosis of the target cell, remove the antigenic stimulus and terminate the inflammatory response
- Accentuated Th1 response leads to high levels of TNF alpha and TNF gamma, causing pancytopenia
- Upregulation of ferritin production secondary to increased heme oxygenase
- Hepatosplenomegaly due to organ infiltration by activated lymphocytes and histiocytes
- Fever induced by interleukin 1 and interleukin 6 (Am J Clin Pathol 2013;139:713)
Etiology
- Genetic or acquired impaired cytotoxic function of T lymphocytes and NK cells
- Familial hemophagocytic lymphohistiocytosis is an inherited autosomal recessive disease involving mutations in perforin and other genes
- Secondary hemophagocytic lymphohistiocytosis triggers include
- Infection (particularly viruses that include Epstein-Barr virus and cytomegalovirus)
- HLH has also been reported in severe COVID-19 and Mpox infections (Curr Med Sci 2021;41:39, N Engl J Med 2023;388:1246)
- Malignancy (most often NK / T cell neoplasms, such as panniculitis-like T cell lymphoma, nasal type extranodal NK / T cell lymphoma and cutaneous gamma / delta T cell lymphoma)
- Rheumatologic / autoimmune disease
- Immunosuppression
- Pregnancy / HELLP syndrome (rare) (Clin Case Rep 2018;6:2466)
- Infection (particularly viruses that include Epstein-Barr virus and cytomegalovirus)
- Similar triggers can initiate FHLH
- In children, a genetic defect characteristic of FHLH should be ruled out
- In adults, hemophagocytic lymphohistiocytosis should prompt an investigation for underlying malignancy
Clinical features
- Fever
- Malaise
- Myalgia
- Hepatosplenomegaly
- Jaundice
- Generalized lymphadenopathy
- Cytopenias
- Rash
- Neurologic symptoms
- Reference: Inflamm Intest Dis 2020;5:49
Diagnosis
- HLH-2004 criteria (updated 2007) include the molecular diagnosis of familial hemophagocytic lymphohistiocytosis or the presence of at least 5 of 8 criteria
- 1. Fever
- 2. Splenomegaly
- 3. Cytopenias (affecting at least 2 lineages in the peripheral blood)
- Hemoglobin levels < 90 g/L
- Platelets < 100 x 10⁹/L
- Neutrophils < 1.0 x 10⁹/L
- 4. Hypertriglyceridemia or hypofibrinogenemia
- Fasting triglycerides ≥ 265 mg/dL
- Fibrinogen ≤ 1.5 g/L
- 5. Documented hemophagocytosis in the bone marrow, spleen or lymph nodes
- 6. Low or absent NK cell activity
- 7. Ferritin ≥ 500 μg/L
- 8. Soluble CD25 ≥ 2,400 U/mL
- Familial hemophagocytic lymphohistiocytosis should have no evidence of malignancy
- Absence of hemophagocytosis during microscopic examination does not exclude the diagnosis of hemophagocytic lymphohistiocytosis (adapted from Pediatr Blood Cancer 2007;48:124)
Laboratory
- Hypofibrinogenemia
- Markedly elevated ferritin
- Hypertriglyceridemia
- Elevated transaminases
- Elevated lactate dehydrogenase
- Elevated serum soluble CD25 (interleukin 2 receptor)
- Reduced or absent NK cell activity by chromium 51 release assay
- Reference: J Blood Med 2014 Jun;5:69
Radiology description
- Hepatosplenomegaly
- Generalized lymphadenopathy
- Alveolar interstitial opacities with pleural effusions
- Gallbladder wall thickening
- Hyperechoic kidneys
- Ascites
- Nonspecific periventricular white matter abnormalities, brain volume loss and enlargement of extra-axial fluid spaces (Pediatr Radiol 2003;33:392)
Prognostic factors
- Almost invariably fatal if left untreated
- Familial hemophagocytic lymphohistiocytosis tends to recur within the first year of life in 70 - 80% of cases (Am J Clin Pathol 2013;139:713)
- Among the viral associated forms of hemophagocytic lymphohistiocytosis, Epstein-Barr virus carries the worst prognosis
- Delay in diagnosis and multiorgan involvement are associated with an inferior prognosis
- Hemophagocytic lymphohistiocytosis in the setting of HELLP syndrome as well as hemophagocytic lymphohistiocytosis development during the third trimester with associated Epstein-Barr viremia have a poor prognosis (Clin Case Rep 2018;6:2466)
Case reports
- Newborn twins with thrombocytopenia, coagulation defects and hepatosplenomegaly (N Engl J Med 2004;351:1120)
- 18 month old girl presenting with fluctuating fevers and leishmaniasis (Indian Pediatr 2004;41:605)
- 5 year old girl diagnosed with COVID-19 requiring hematopoietic stem cell transplantation (EJHaem 2022;3:1025)
- 6 year old boy with fever, headache, abdominal pain and typhoid fever (J Emerg Med 2003;25:321)
- 30 year old pregnant woman with HELLP syndrome and persistent fevers upon delivery (Clin Case Rep 2018;6:2466)
- 33 year old HIV positive man with disseminated Mpox infection (N Engl J Med 2023;388:1246)
Treatment
- Immediate treatment of the underlying disease in secondary hemophagocytic lymphohistiocytosis
- 8 week induction therapy with cyclosporine (HLH-2004 protocol), corticosteroids and etoposide (Am J Clin Pathol 2013;139:713)
- Cyclosporine inhibits T cell activation
- Corticosteroids suppress hypercytokinemia
- Etoposide blocks cell proliferation
- Has demonstrated a 5 year survival rate up to 54%
- Stem cell transplantation is utilized in cases of familial hemophagocytic lymphohistiocytosis
Microscopic (histologic) description
- Hemophagocytosis (histiocyte / macrophage ingestion of erythrocytes, leukocytes, platelets or their precursor cells), including erythrophagocytosis (ingestion of erythrocytes)
- Hemophagocytic cells are best visualized on bone marrow aspirate smear
- Liver sinusoids can demonstrate histiocytes with hemophagocytosis
- Splenic white pulp can be atrophic with extensive infiltration of histiocytes exhibiting hemophagocytosis
- Lymph node involvement in systemic HLH features
- Early disease shows an intense immunoblastic response
- Later stages demonstrate lymphoid depletion with massive sinusoidal infiltration by benign appearing histiocytes and hemophagocytosis
- Enlarged lymph nodes (especially of the axilla) displaying prominent erythrophagocytosis may not be indicative of systemic HLH but rather can be observed in the setting of
- Trauma to lymph drainage sites (i.e., following breast biopsy)
- Renal disease (malignant hypertension, glomerulonephritis, pyelonephritis)
- Infection
- Lymph node hemorrhage or congestion
- Sinus erythrocytosis
- Autoimmune or immunologic conditions
- Idiopathic
- Hemophagocytosis is not a specific finding and can be seen in up to 60% of patients with sepsis (Clin Case Rep 2018;6:2466)
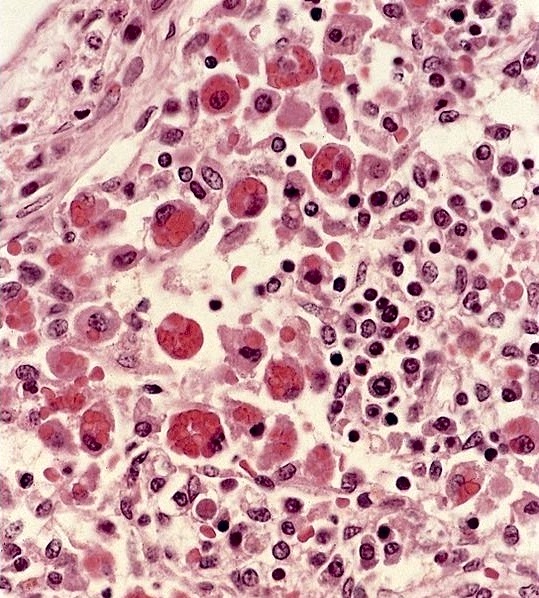
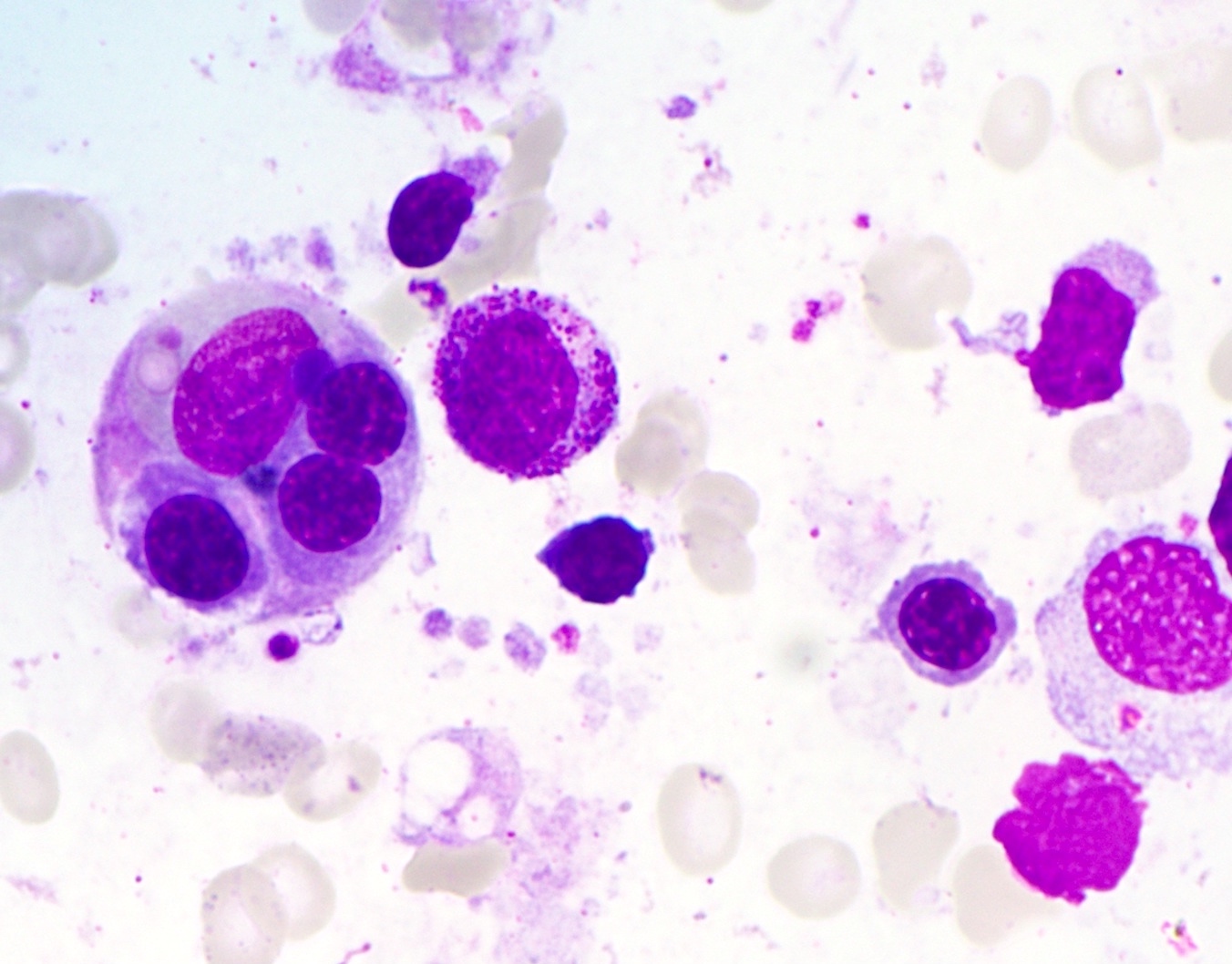
Microscopic (histologic) images
Contributed by Sherif A. Rezk, M.D. and AFIP

Bone marrow
hemophagocytosis
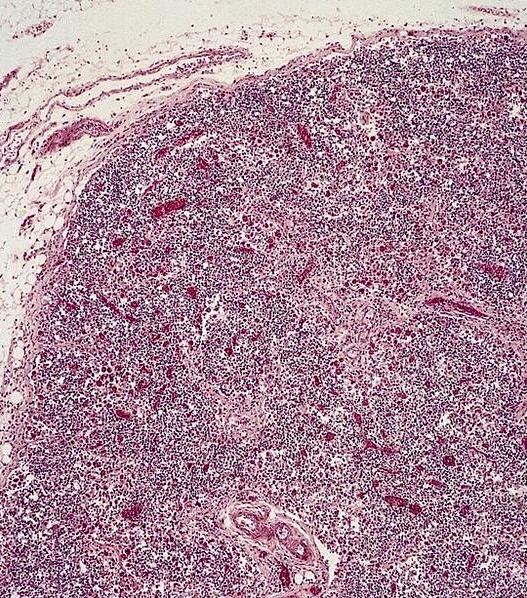
Reactive hemophagocytosis in lymph node
Erythrophagocytosis
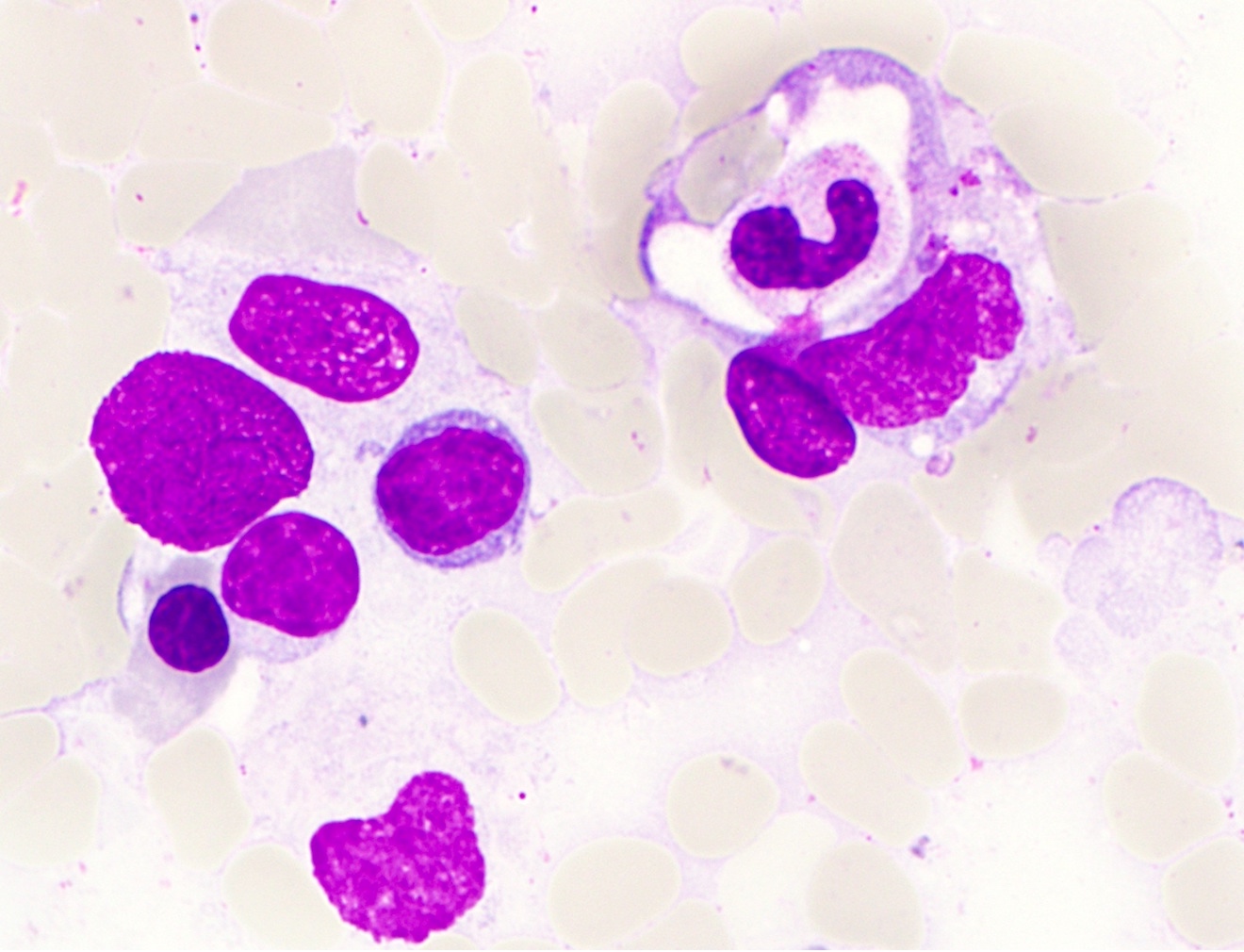
Neutrophil
hemophagocytosis
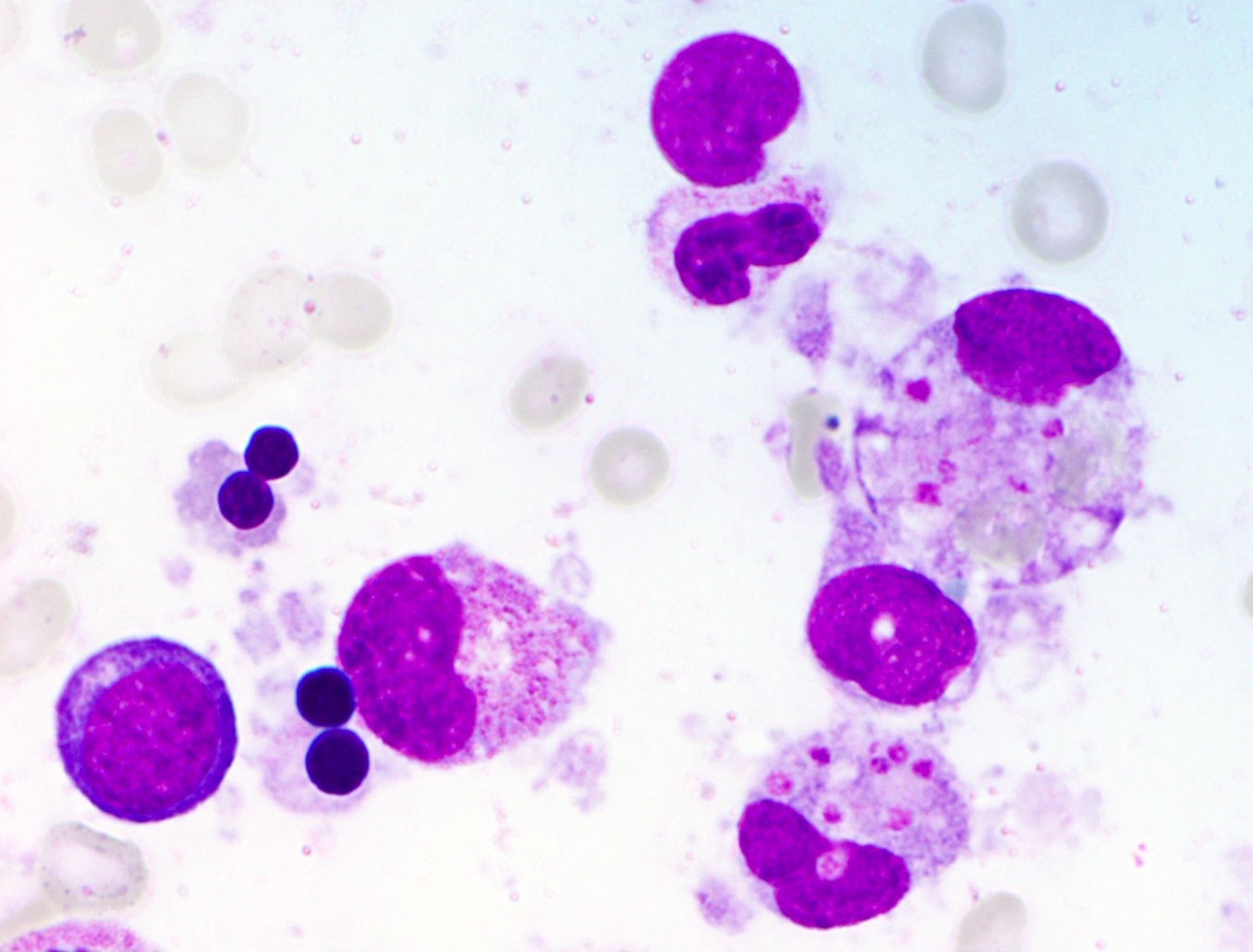
Platelet
hemophagocytosis
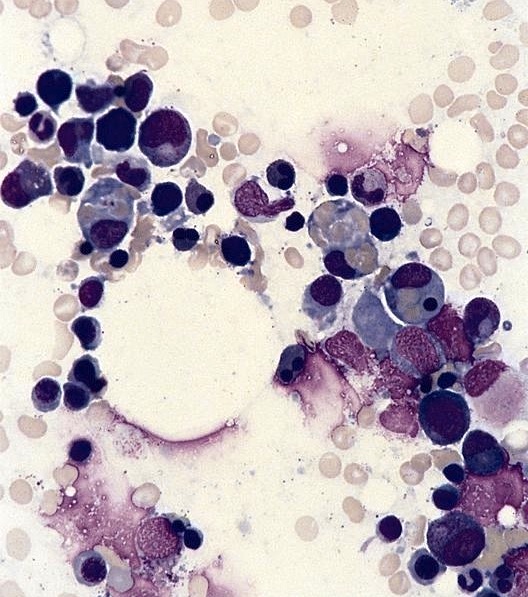
Bone marrow in
reactive
hemophagocytic
syndrome
Positive stains
Negative stains
- Absent or diminished intracellular staining for perforin can indicate mutations in the protein encoding gene
Flow cytometry description
- Flow cytometry may demonstrate decreased expression of CD107a (LAMP1) surface expression on cytotoxic NK / T cells (Am J Clin Pathol 2013;139:713)
- Reduced CD5 and CD7 expression has been observed in circulating and bone marrow CD8 positive T cells in some patients (Blood 2004;104:2007)
Molecular / cytogenetics description
- Familial hemophagocytic lymphohistiocytosis is divided into subtypes (FHL1 - FHL5) based on the specific genetic alteration
- Involved genes include PRF1 in 50% of cases (FHL2), UNC13D (FHL3), STX11 (FHL4) and STXBP2 (FHL5)
- Gene responsible for FHL1 has yet to be identified but is located at 9q21_3-22
- Some cases of FHLH can be seen in the setting of macrophage overactivation due to gain of function variants in NLRC4 and deficiency of XIAP (Blood 2020;135:1332)
- Secondary (acquired) hemophagocytic lymphohistiocytosis may demonstrate missense or splice site changes in PRF1, MUNC13-4 and STXBP2 (Blood 2011;118:5794)
Sample pathology report
- Bone marrow, right iliac crest, aspirate smears, touch imprints, clot and trephine biopsy section:
- Normocellular marrow with trilineage hematopoiesis, marked erythroid hyperplasia and evident hemophagocytosis
- By immunohistochemistry, increased phagocytic histiocytes identified
Differential diagnosis
- Sepsis due to various causes:
- Symptoms of hemophagocytic lymphohistiocytosis (including fever, malaise, myalgia, etc.) can often be seen in acute sepsis of various causes and the diagnosis of hemophagocytic lymphohistiocytosis requires fulfillment of the criteria defined previously
- Myeloproliferative neoplasm / myelodysplastic syndrome (MDS / MPN):
- Hemophagocytic lymphohistiocytosis can also present with cytopenias; careful examination of the bone marrow for dysplasia in various cell lineages will aid in diagnosing MDS / MPN
- Rosai-Dorfman disease:
- Prominent histiocytic infiltration may also be present in hemophagocytic lymphohistiocytosis; however, Rosai-Dorfman disease rarely presents with constitutional symptoms and generally features emperipolesis, as opposed to erythrophagocytosis
- Langerhans cell histiocytosis:
- In spite of the prominent histiocytes sometimes observed in hemophagocytic lymphohistiocytosis, these cells generally lack nuclear grooves and do not demonstrate erythrophagocytosis
- Leishmaniasis / histoplasmosis:
- Platelets phagocytosed by histiocytes in hemophagocytic lymphohistiocytosis can visually mimic intracellular Leishmania donovani or Histoplasma capsulatum organisms
- Clinical history and microbiology analyses, including indirect fluorescent antibody (IFA) testing and polymerase chain reaction (PCR), can confirm the infectious diagnosis
Additional references
Board review style question #1
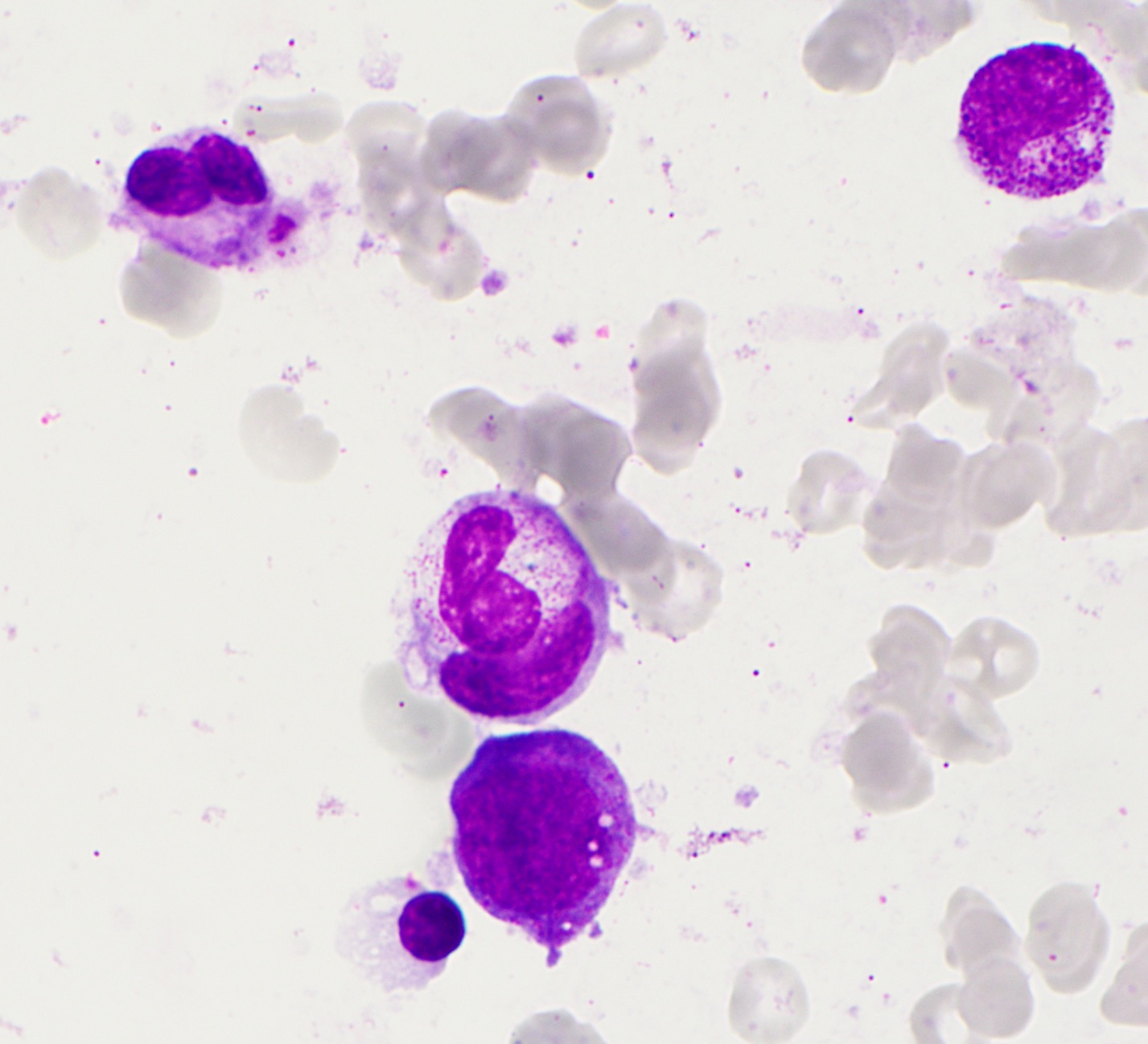
- A 45 year old Latina woman presents with mild normocytic anemia and thrombocytopenia. A representative photograph of her bone marrow aspirate smear is provided. Which of the following diagnoses is most likely in her medical history and could be contributing to the current findings in the bone marrow biopsy?
- Acute human immunodeficiency virus (HIV) infection
- Acute myeloid leukemia
- Panniculitis-like T cell lymphoma
- Triple negative breast cancer
Board review style answer #1
C. Panniculitis-like T cell lymphoma. The patient's aspirate smear demonstrates hemophagocytosis, consistent with hemophagocytic lymphohistiocytosis (HLH). Secondary (acquired) HLH is frequently associated with hematologic malignancies, in particular NK / T cell neoplasms like panniculitis-like T cell lymphoma. Answers B and D are incorrect because HLH has been only rarely reported in association with other cancers. Answer A is incorrect because HIV infection has been implicated in the development of HLH but typically in the setting of other acute infections or HIV associated lymphomas, especially EBV related lymphoproliferative processes.
Comment Here
Reference: Hemophagocytic lymphohistiocytosis
Comment Here
Reference: Hemophagocytic lymphohistiocytosis
Board review style question #2
- Among the infectious etiologies of hemophagocytic lymphohistiocytosis, which of the following has been most commonly implicated?
- Cytomegalovirus
- Epstein-Barr virus
- Influenza virus
- Parvovirus B19
Board review style answer #2
B. Epstein-Barr virus. While all of viruses listed have been associated with hemophagocytic lymphohistiocytosis, Epstein-Barr virus infection is most commonly observed and is associated with the worst prognosis. Answers A, C and D are incorrect because cytomegalovirus, influenza virus and parvovirus B19 are less commonly implicated than EBV.
Comment Here
Reference: Hemophagocytic lymphohistiocytosis
Comment Here
Reference: Hemophagocytic lymphohistiocytosis

