
Lung
Interstitial lung diseases
Interstitial pneumonia with autoimmune features
Last author update: 1 May 2017
Last staff update: 19 August 2022
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: Lung IPAF



Table of Contents
Definition / general | Essential features | Terminology | Epidemiology | Sites | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Bychkov, A. Interstitial pneumonia with autoimmune features. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lungnontumorIPAF.html. Accessed December 4th, 2024.
Definition / general
- Interstitial pneumonia is one of the common clinical manifestation of connective tissue disease (CTD); this entity is called CTD associated interstitial lung disease (CTD-ILD)
- Some patients initially diagnosed with idiopathic interstitial pneumonias (IIPs) eventually present with systemic autoimmune symptoms and develop a complete form of CTD; thus, it is often difficult to distinguish CTD-ILD from IIPs before the symptoms appear (Respiration 1995;62:248)
- IIP cases with certain features suggesting an underlying autoimmune process but insufficient to fulfill the diagnostic requirements of any specific CTD were previously categorized as an idiopathic or unclassifiable interstitial pneumonia (Am J Respir Crit Care Med 2013;188:733)
- In 2015, ATS / ERS working group proposed the term "interstitial pneumonia with autoimmune features (IPAF)" as a possible spectrum of CTD-ILD along with its provisional criteria (Eur Respir J 2015;46:976)
Essential features
- An interstitial lung disease with clinical, serologic or histologic features suggestive of CTD which does not conclusively meet current rheumatologic criteria
- Histologically, nonspecific interstitial pneumonia (NSIP) and organizing pneumonia (OP) patterns are prevalent; lymphocytic inflammation is suggestive of IPAF
Terminology
- Before the proposal from ATS / ERS, studies used nomenclature of:
- Lung dominant connective tissue disease (lung dominant CTD)
- Undifferentiated connective tissue disease associated interstitial lung disease (UCTD-ILD)
- Autoimmune featured interstitial lung disease (autoimmune featured ILD)
Epidemiology
- Mean age 50 - 60 years old
- Female predominant
- No correlation with smoking status
Sites
- Unilateral or bilateral lower lobes of the lungs
Etiology
- Although interstitial pneumonia had not been accepted as the sole manifestation of CTD in the current rheumatologic criteria, several studies suggested that interstitial pneumonia may appear as one of the preliminary manifestation of subclinical or undifferentiated CTD (Autoimmun Rev 2016;15:61, Respir Med 2005;99:234, Am J Respir Crit Care Med 2007;176:691)
Clinical features
- Mild to moderate chronic respiratory failure
- Shortness of breath
- Dyspnea on exertion
- Cough
- Fatigue
- Weight loss
- Abnormal chest auscultation
- End inspiratory fine crackles in lower lobes of the lungs
- Restrictive pattern in pulmonary function tests:
- Decreased forced vital capacity (FVC)
- Decreased diffusing capacity of the lung for carbon monoxide (DLCO)
- Extrathoracic autoimmune manifestation or serologic abnormality accompanythe respiratory symptoms (See Diagnosis)
Diagnosis
- The proposed criteria consists of a priori requirements and three domains: clinical, serologic and morphologic, as below (Eur Respir J 2015;46:976)
- Findings of "multi compartment involvement" are detected by various modalities such as simple chest X ray, high resolution computed tomography (HRCT), surgical lung biopsy, pulmonary function tests and right heart catheterization
|
|
|
|
Laboratory
- Not included in the IPAF criteria because of their low specificity for CTD: low titer ANA, low titer RF, erythrocyte sedimentation rate, C reactive protein, creatine phosphokinase (Eur Respir J 2015;46:976)
Radiology description
- NSIP pattern
- Diffuse ground glass opacity
- Reticular opacity
- Traction bronchiectasis
- Often difficult to make a definite diagnosis without histopathological examination
- OP pattern
- Patchy and migratory consolidation in a subpleural and peribronchial area
- Band-like pattern
- LIP pattern
- Reticular opacity
- Air space opacity
- Often difficult to make a definite diagnosis without histopathological examination
- UIP and other radiological patterns can also be seen
Radiology images
Images hosted on other servers:
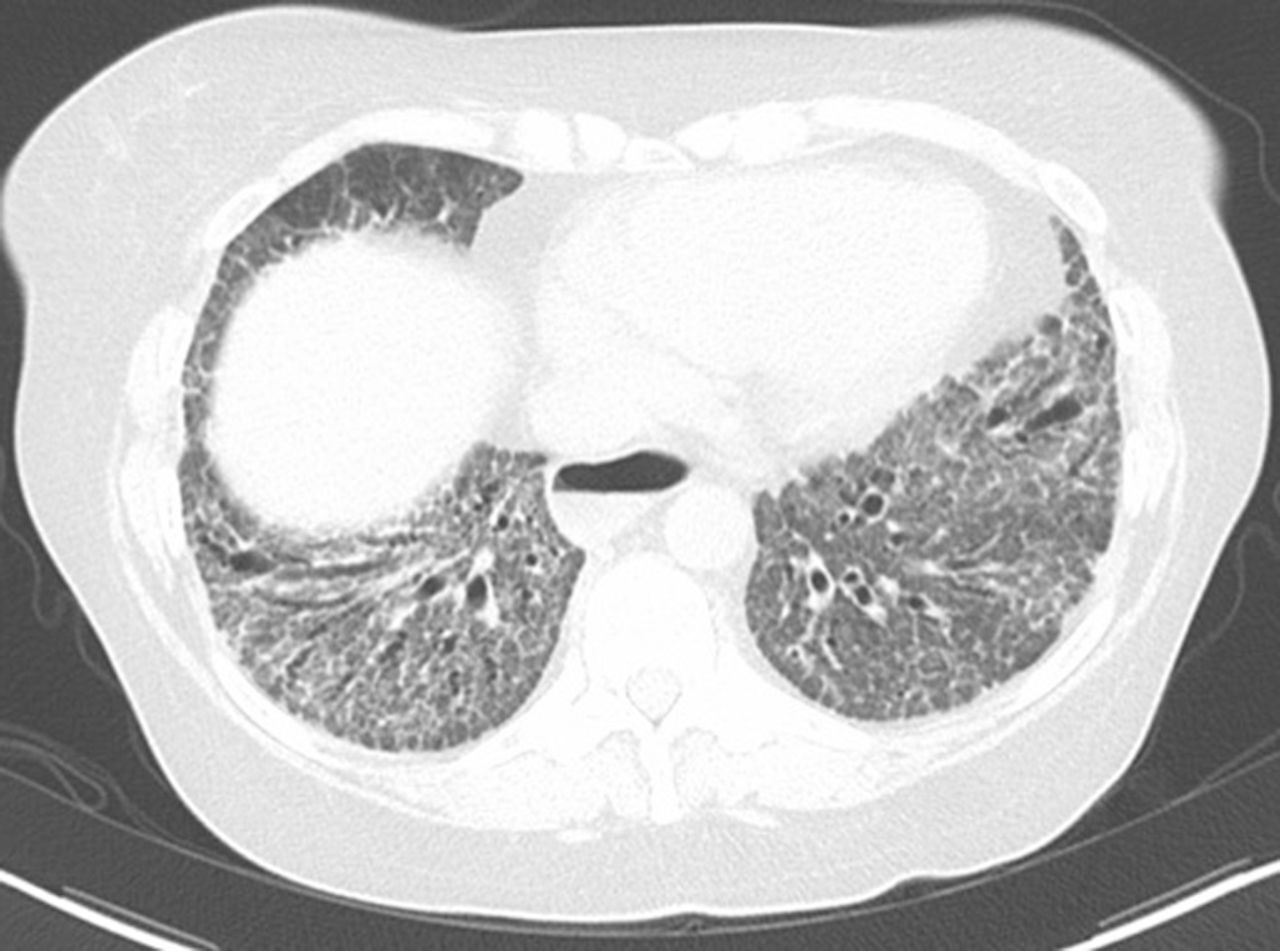

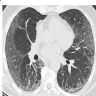
NSIP pattern on CT
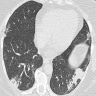
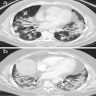
OP pattern on CT
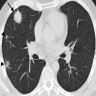
LIP pattern on CT
Prognostic factors
- Limited data are currently available
- Retrospective studies observed different prognosis of IPAF, either better (Eur Respir J 2016;47:1767, Respir Med 2015;109:1326, Respir Med 2016;119:150) or similar (Respir Med 2017;123:56) than that of IIPs (IPF)
Treatment
- Corticosteroids or immunosuppression drugs may improve the prognosis and stabilize pulmonary function deterioration (Respir Med 2016;119:150, Eur Respir J 2016;47:1767, Chest 2017 Mar 11 [Epub ahead of print], Respir Med 2015;109:1326), but evidence is limited to date
Clinical images
Images hosted on other servers:
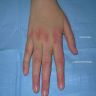
Clinical features in systemic sclerosis
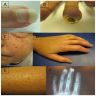

Digital ulceration
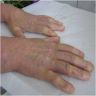
Telangiectasia
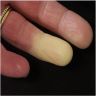
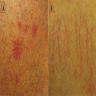
Raynaud's phenomenon

Raynaud's phenomenon
and telangiectasia
Clinical features in dermatomyositis


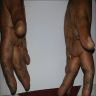
Mechanic hand
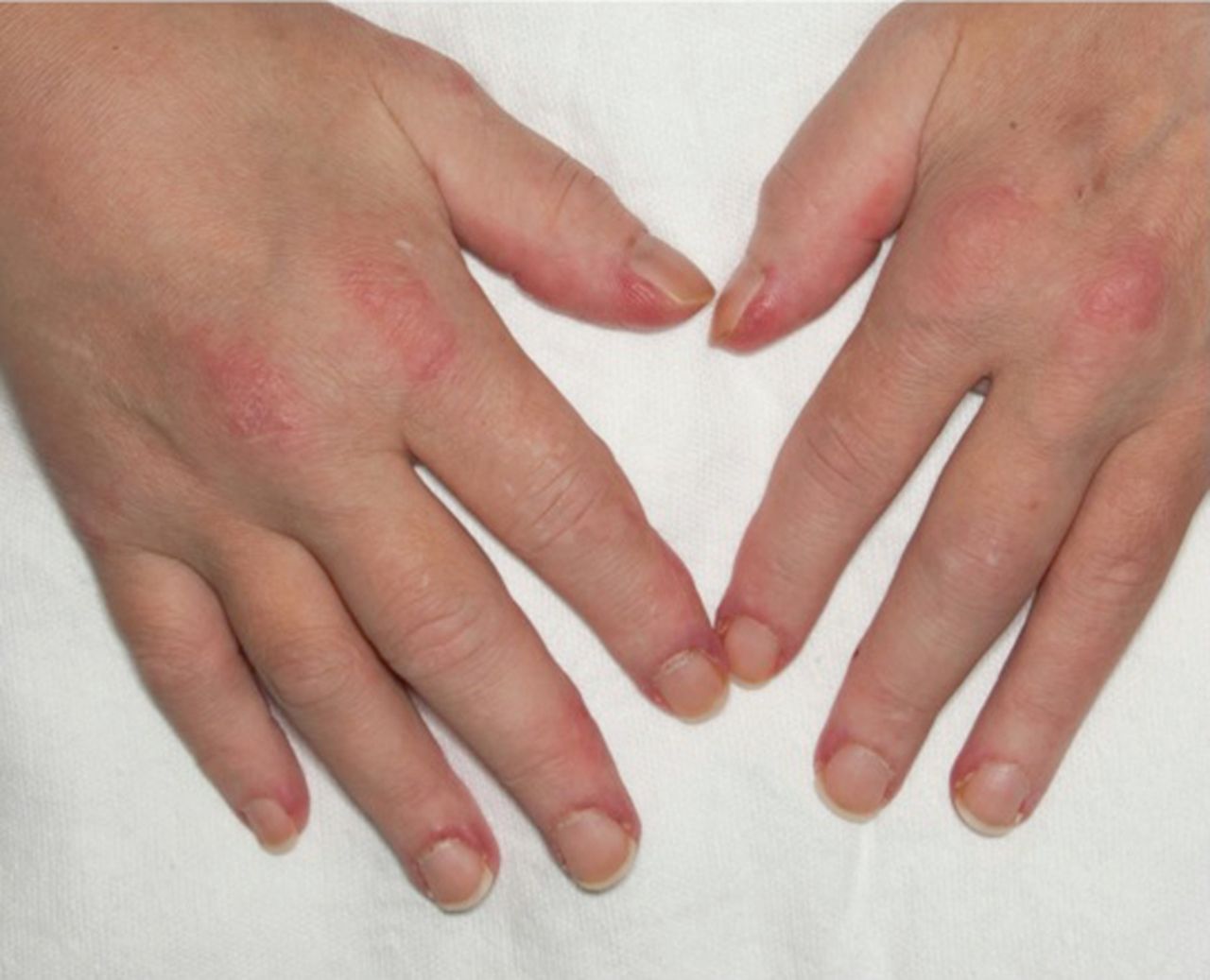
Gottron's sign
Gross description
- Diffuse involvement with mild to moderate increase in weight
- Shrunken lung due to fibrotic changes in lower lobes
- Pleural thickening
Microscopic (histologic) description
- Histological NSIP pattern is the most common (about 50%) in IPAF cases, followed by NSIP with OP overlap pattern (Respir Med 2016;119:150, Eur Respir J 2016;47:1767)
- NSIP pattern
- Diffuse and uniform inflammation in alveolar walls
- Cellular or fibrotic changes
- OP pattern
- Polyps of fibroblastic organization obstructing alveolar ducts
- LIP pattern
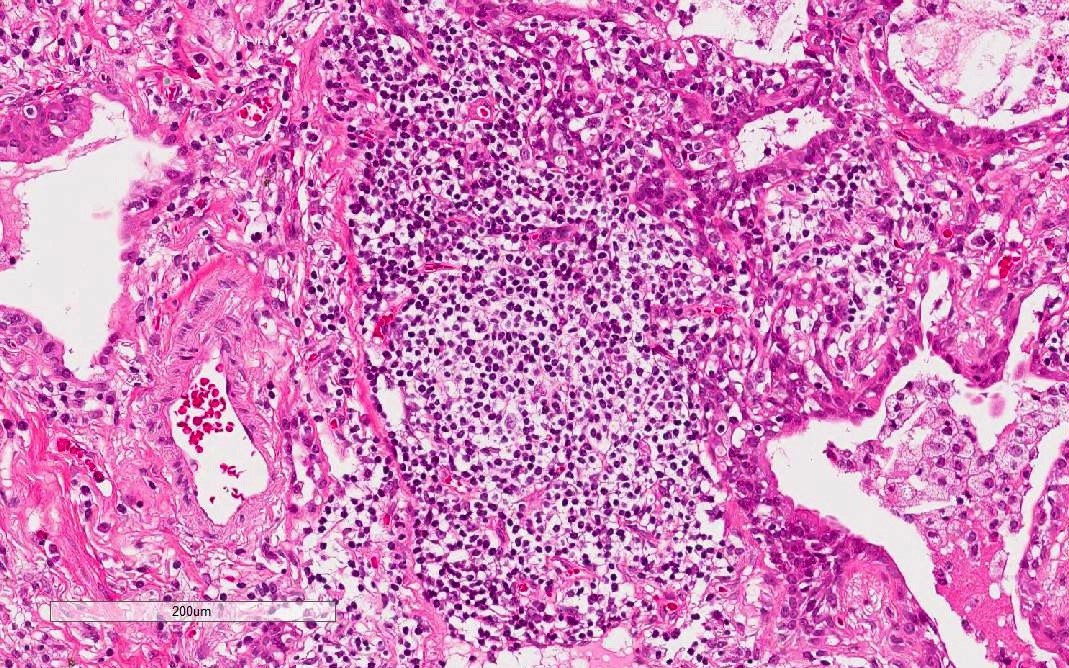
- Marked and extensive infiltration of lymphocytes in alveolar wall
- Other findings suggestive of IPAF
- Lymphoid aggregates with germinal centers
- Diffuse lymphoplasmacytic infiltration
- Additional supportive findings (Chest 2010;138:251)
- Extensive pleuritis
- Dense perivascular collagen
- Other histological patterns, such as UIP pattern and respiratory bronchiolitis interstitial lung disease (RB-ILD) pattern, can be seen with lesser frequency
- UIP pattern may be related to poorer prognosis than NSIP pattern (Chest 2015;147:165)
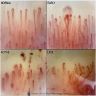
Microscopic (histologic) images
Contributed by Akira Yoshikawa, M.D.
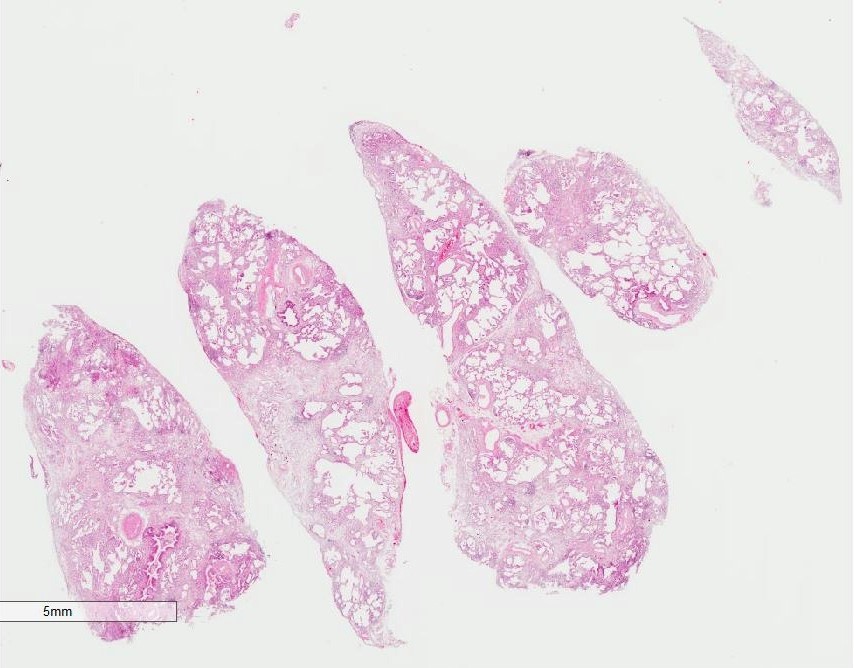
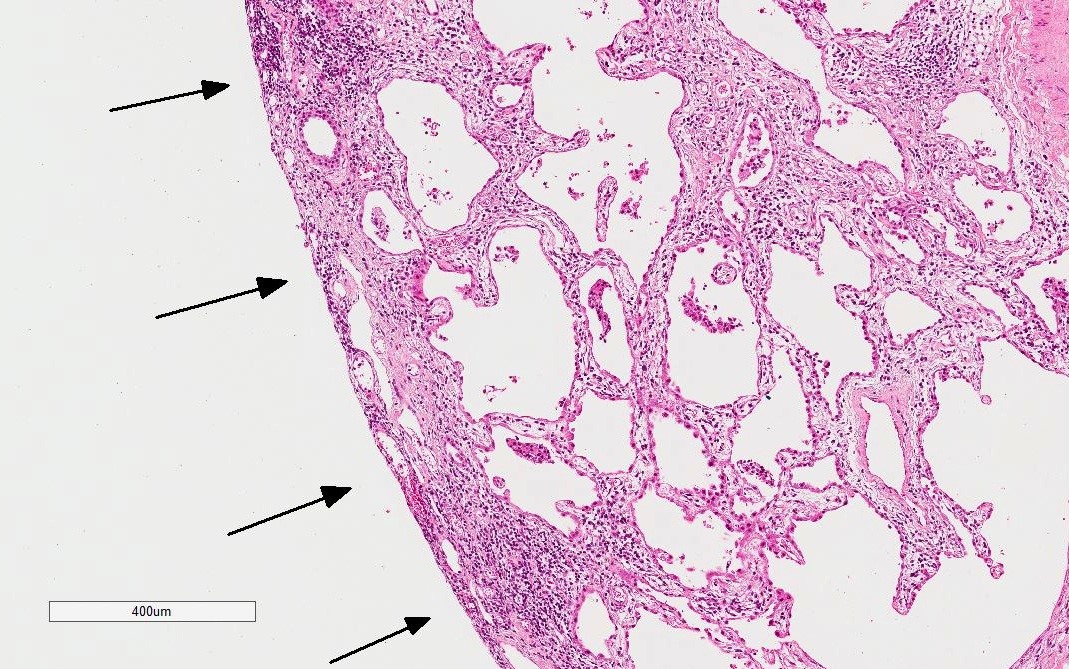
Low power
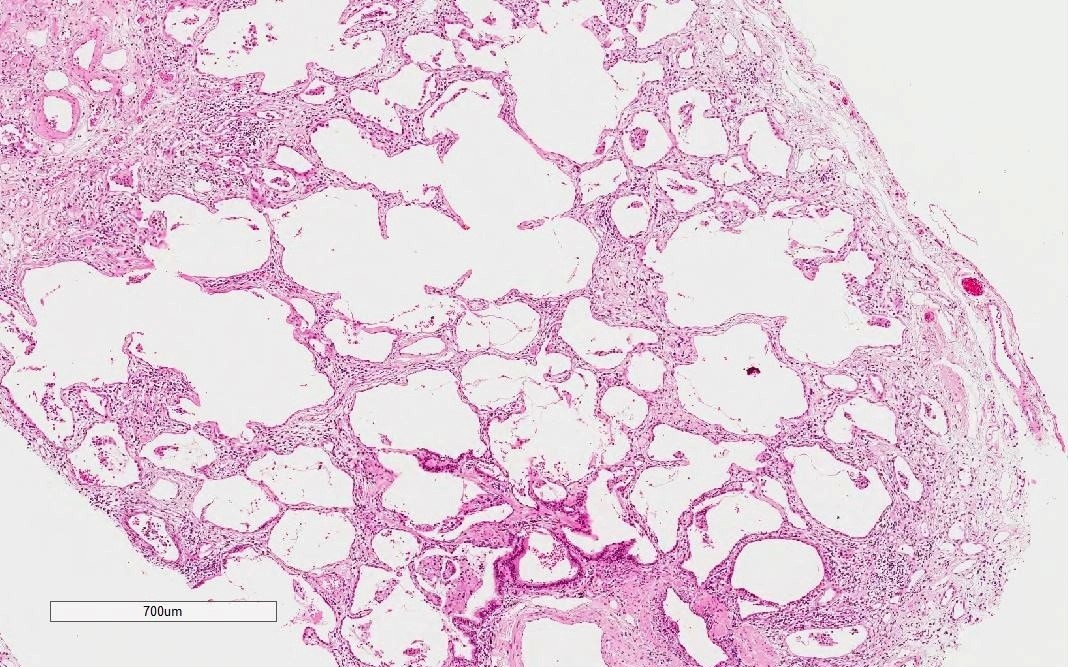
NSIP pattern
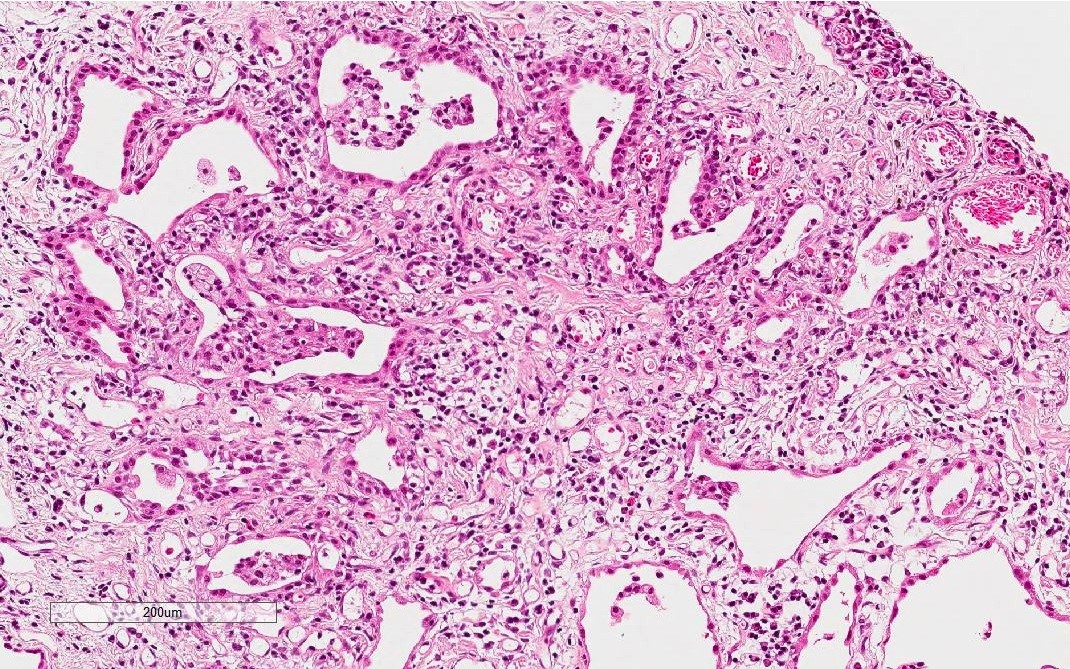
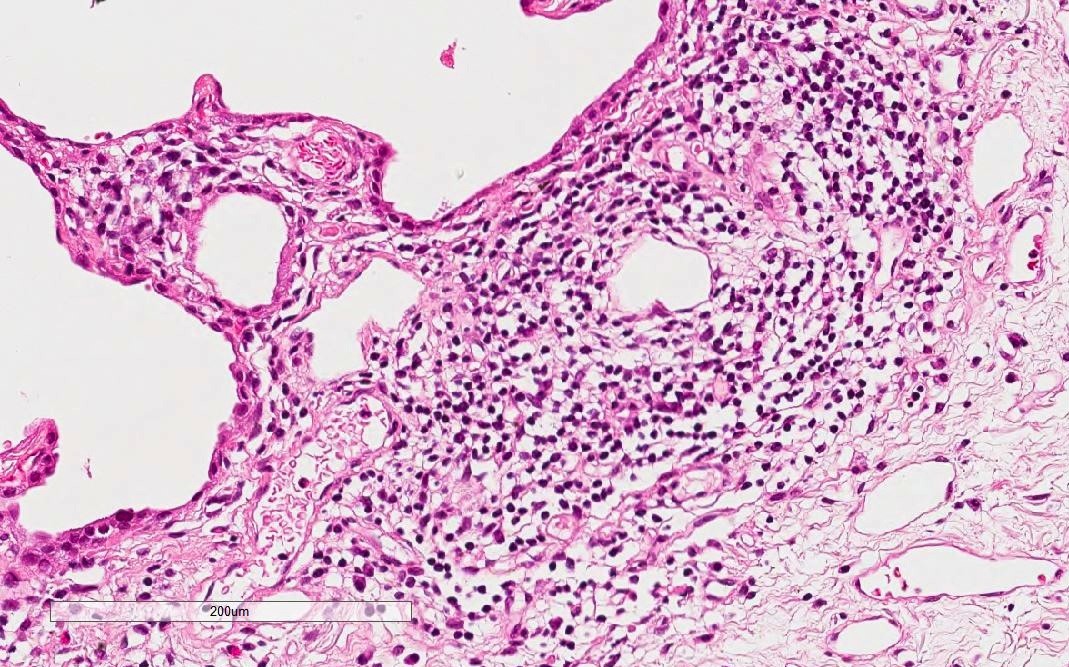
Lymphoplasmacytic infiltration without lymphoid follicles
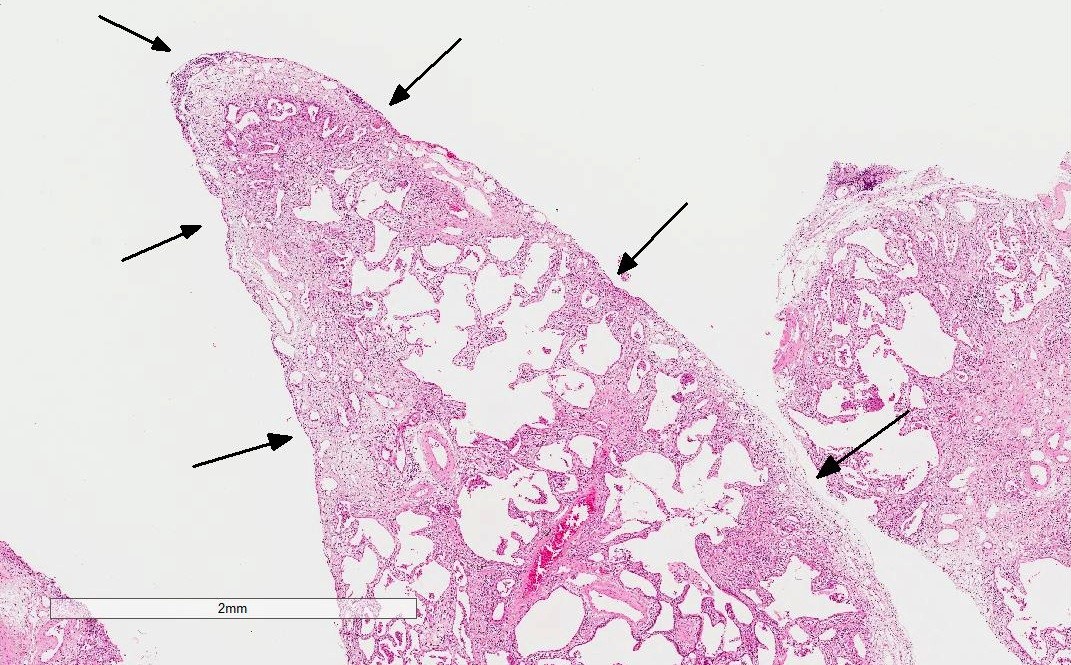
Pleural thickening and pleuritis
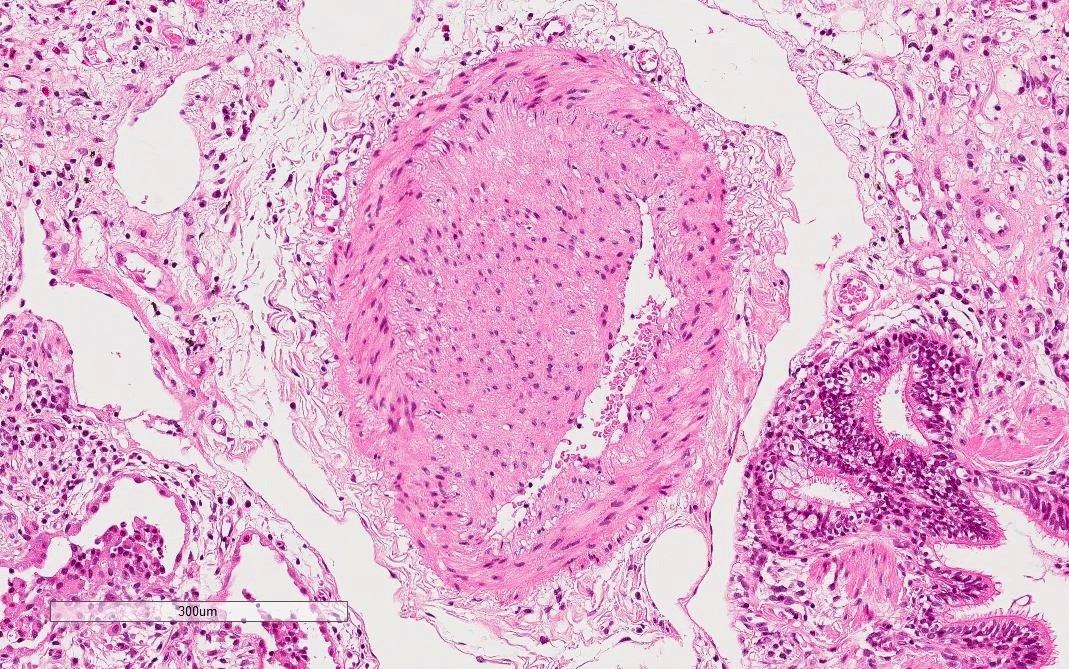
Vascular wall thickening
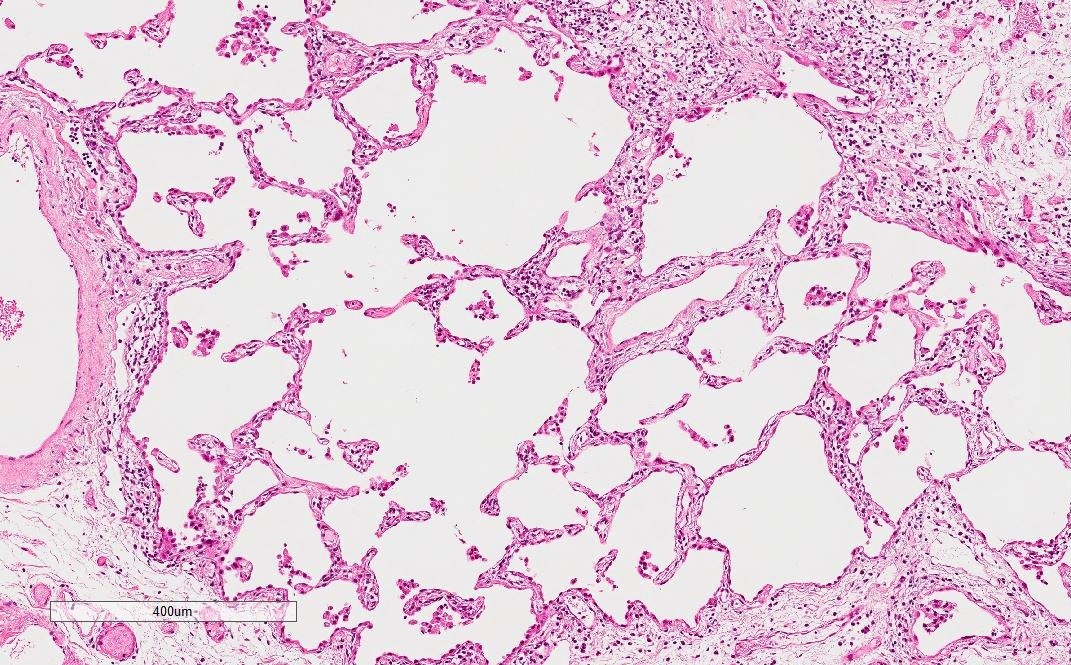
Emphysematous change
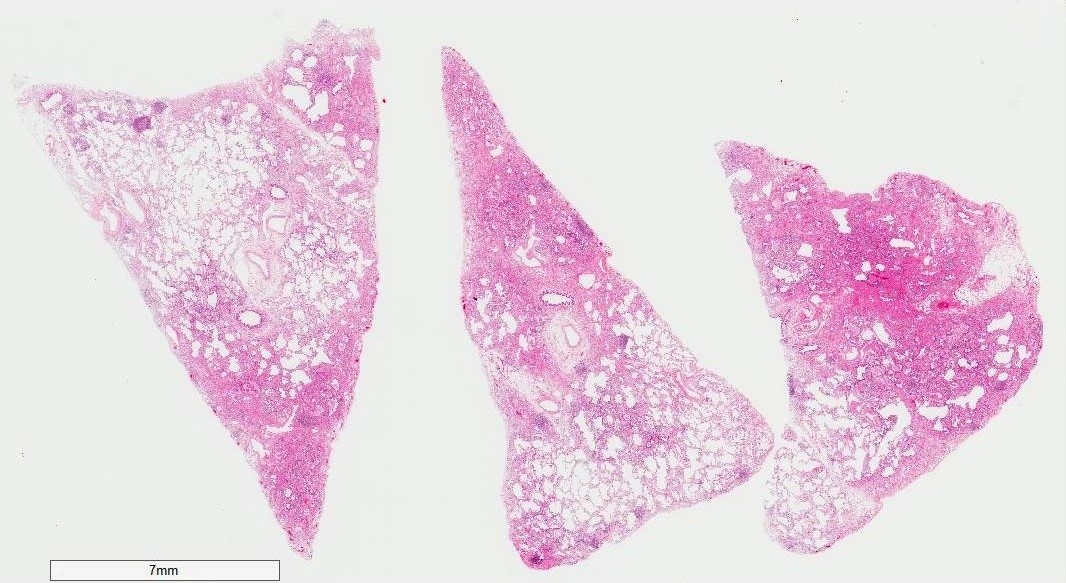
Low power
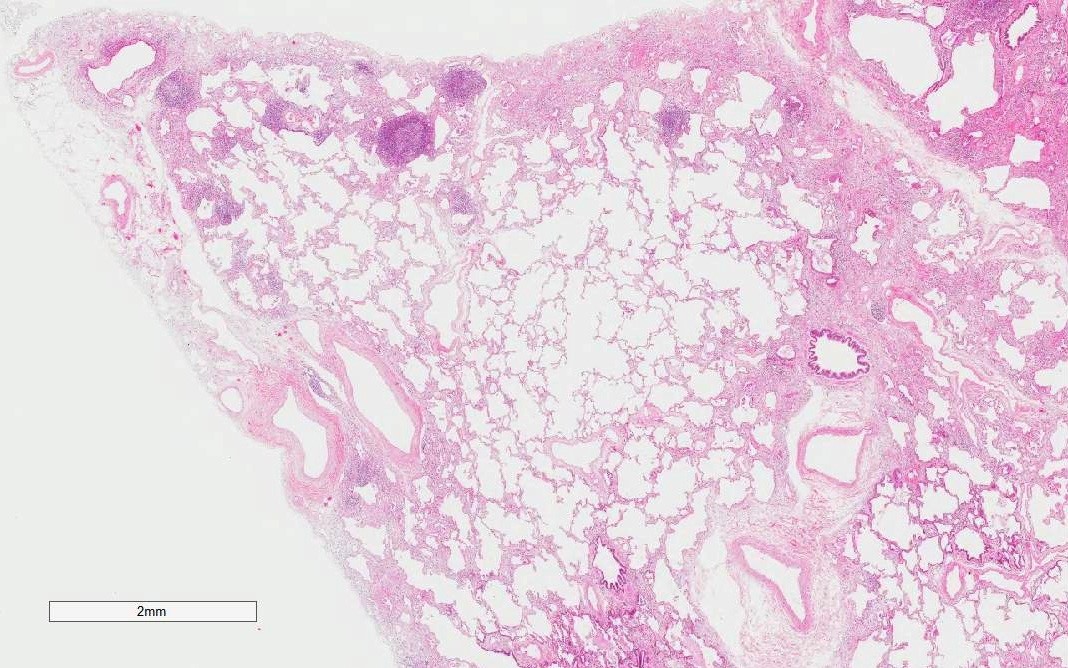
UIP pattern
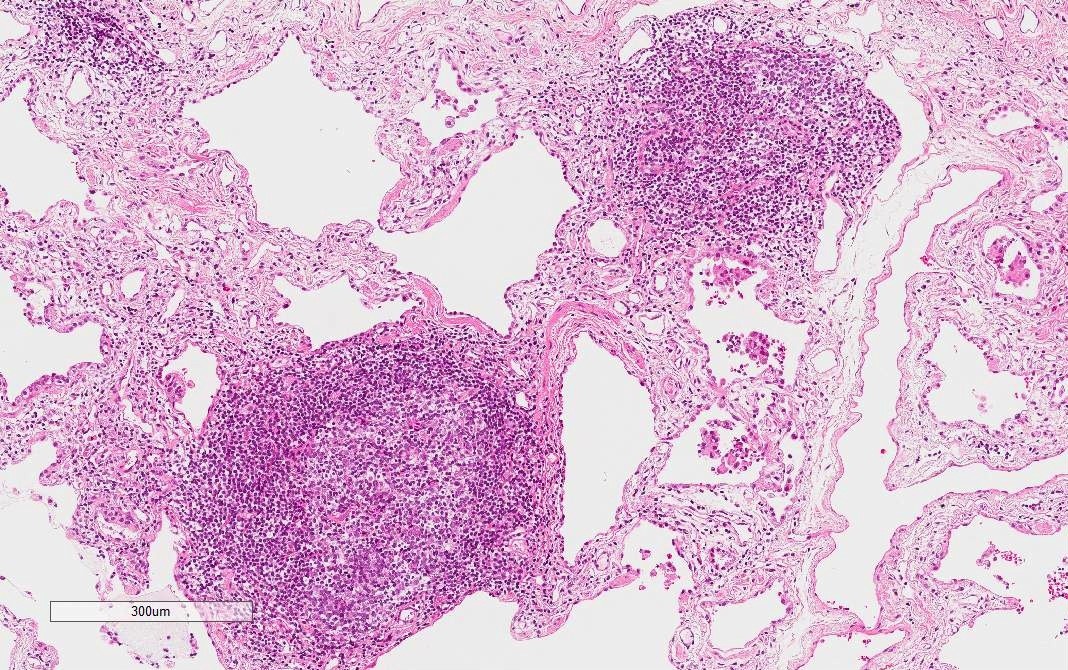
Lymphoid follicles with germinal centers
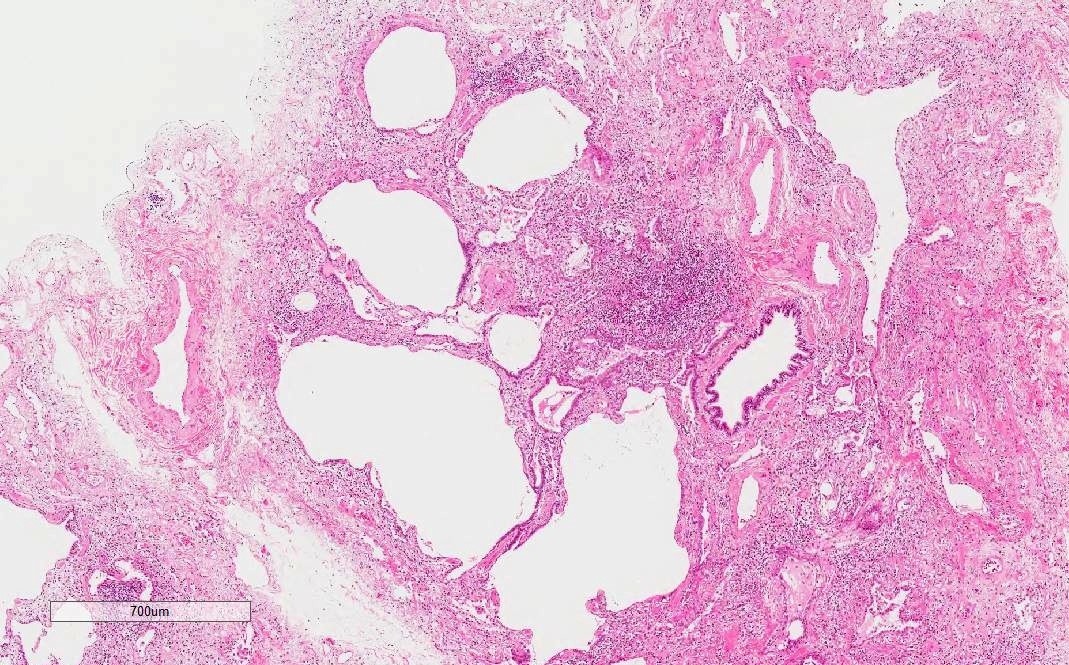
Honeycomb change
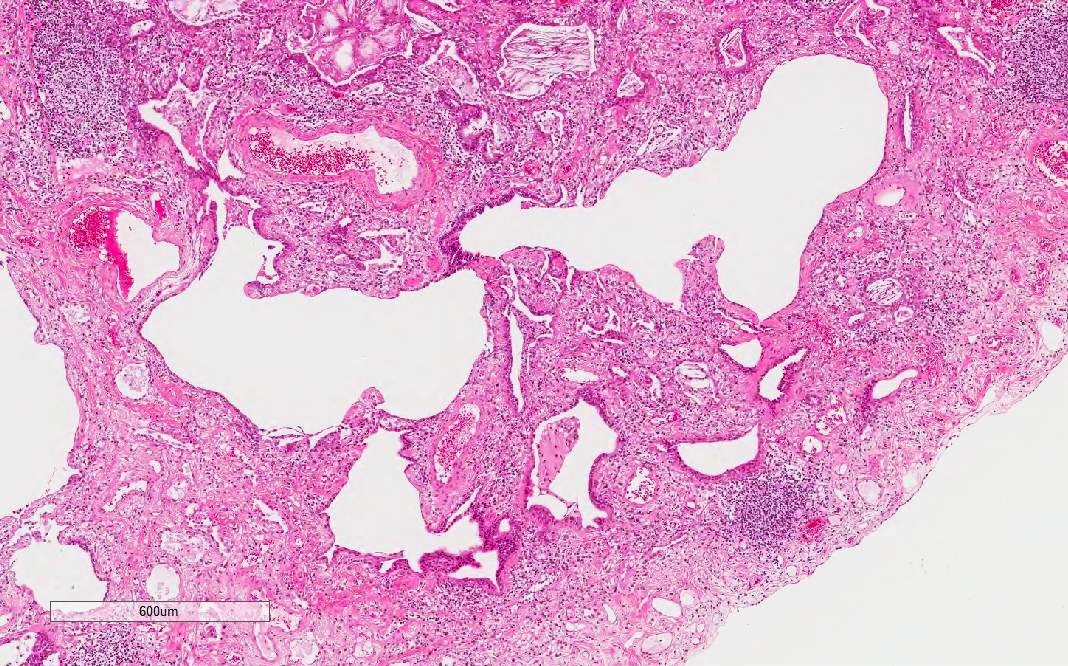
Honeycomb change
Lymphoplasmacytic infiltration
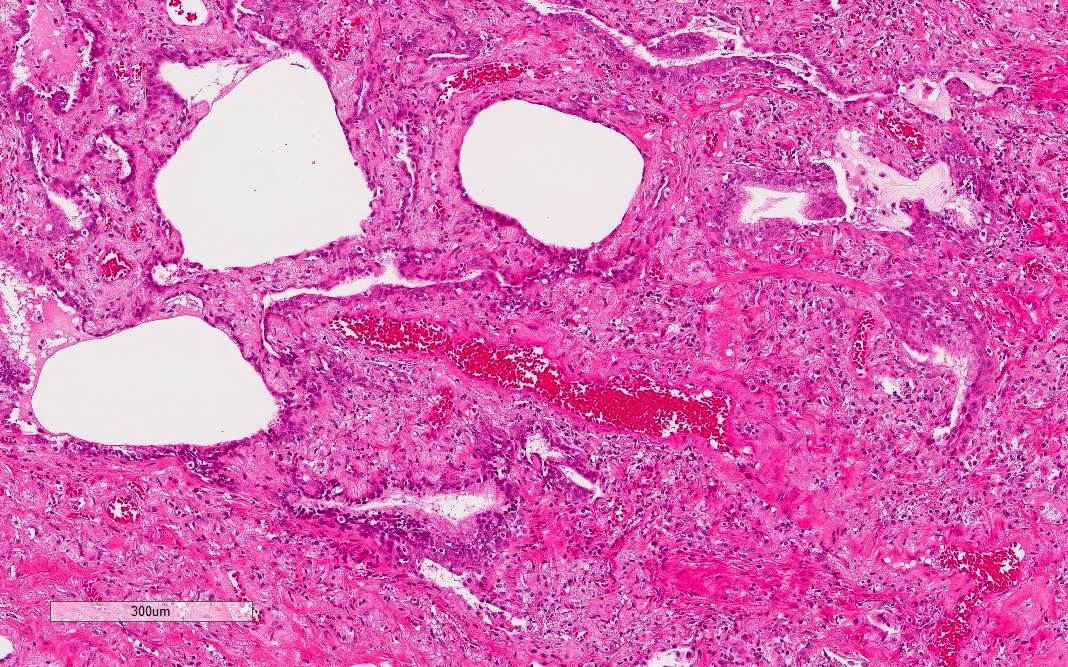
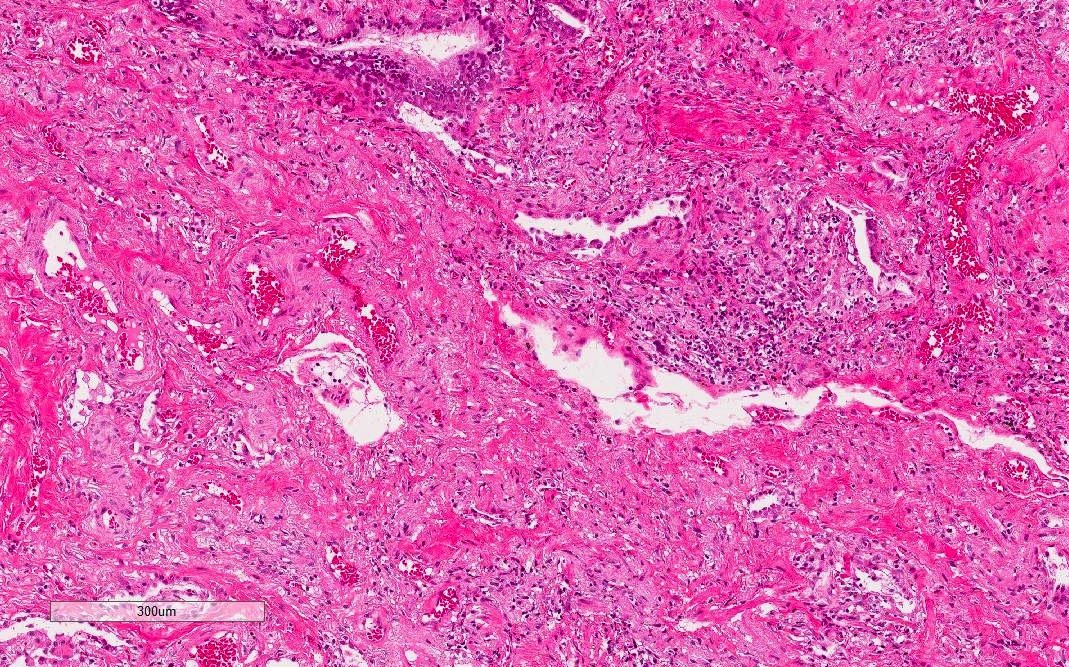
Dense fibrosis with lymphocytic infiltration
Also see: NSIP
Images hosted on other servers:

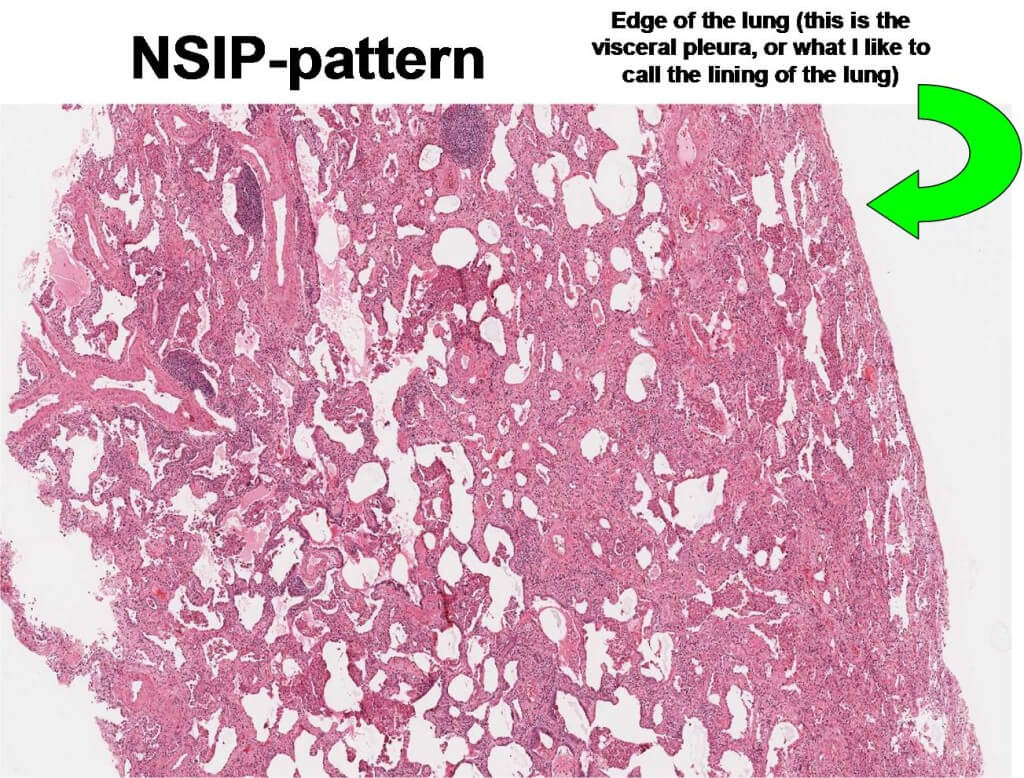
NSIP pattern

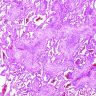

OP pattern
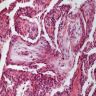
OP pattern


_2.jpg)
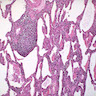
LIP pattern
Differential diagnosis
- Connective tissue disease associated interstitial lung disease (CTD-ILD): clinical and serologic manifestations conclusive to meet rheumatologic criteria
- Cryptogenic organizing pneumonia (COP): no clinical or serologic autoimmune features
- Hypersensitivity pneumonitis: history of exposure to causative antigens, airway centered change, granuloma or interstitial giant cells
- Idiopathic NSIP: no clinical or serologic autoimmune features
- LIP: no clinical or serologic autoimmune features
- UIP / IPF: no or only one autoimmune features
Additional references
Board review style question #1
Which 2 cases best meet the criteria for IPAF?
- A current smoker with Raynaud’s phenomenon and a histological OP pattern. No serological abnormality is detected.
- Patient hospitalized for shortness of breath during the past 3 weeks, now improving over the past week. High titer ANA and radiological NSIP patterns are detected.
- Patient with swelling in both wrists for the past year, high titer anti-CCP antibody, high titer CRP, NSIP pattern on CT.
- Patient with digital tip ulceration for 6 months, high titer Scl-70 histological UIP pattern with little cellular inflammation.
- Patient with 2 year history of pulmonary hypertension who died of acute exacerbation. Histology shows NSIP with OP overlap pattern, diffuse lymphocytic infiltration and pleural thickening.
Board review style answer #1
A & D.
Comment Here
Reference: Interstitial pneumonia with autoimmune features
- Comments:
- IPAF. This case meets clinical and morphologic criteria of IPAF and meets no rheumatologic criteria.
- Acute hypersensitivity pneumonitis. This case shows the IPAF findings of serologic and morphologic domain. However, the clinical course is more suggestive for hypersensitivity pneumonitis. NSIP pattern is also common in hypersensitivity pneumonitis.
- CTD-ILD. This case meets the classification criteria for rheumatoid arthritis (Ann Rheum Dis 2010;62:2569).
- IPAF. This case is suspicious for systemic sclerosis, but does not fulfill the rheumatologic criteria (Ann Rheum Dis 2013;72:1747).
- Idiopathic NSIP (with OP). This case shows the findings of morphologic domain only.
Comment Here
Reference: Interstitial pneumonia with autoimmune features

