

Kidney tumor
Childhood tumors
Nephroblastoma
Editorial Board Member: Maria Tretiakova, M.D., Ph.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 16 September 2020
Last staff update: 6 October 2021
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed search: Nephroblastoma / Wilms tumor[title]
Related topics: Cystic partially differentiated nephroblastoma

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: D'Hooghe E, Vujanic GM. Nephroblastoma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneytumorwilmkids.html. Accessed December 21st, 2024.
Definition / general
- Nephroblastoma (or Wilms tumor) is a malignant embryonal tumor originating from nephrogenic blastema, which imitates the histology of developing kidney
- Primarily occurs in children
- Named after the German surgeon Max Wilms (who is often wrongly attributed to be the first one describing this entity)
Essential features
- Tumor consists of blastemal, epithelial and stromal elements (triphasic) which can be present in variable amounts
- Biphasic and monophasic tumors are not infrequent
- Epithelial and stromal elements may show different lines and degrees of differentiation (from poor to well differentiated)
- There are 2 main treatment approaches:
- SIOP (International Society of Paediatric Oncology) which uses preoperative chemotherapy as the first line of treatment
- COG (Children’s Oncology Group) which advocates for primary surgery
- Both followed by postoperative chemotherapy and radiotherapy, if necessary
- Both approaches have their own histologic classification and risk assessment (based on prognostic factors)
- Overall survival rate is approximately 90%
Terminology
- Wilms tumor
- Nephroblastoma
ICD coding
Epidemiology
- Most common: 3 - 4 years of age (Pritchard-Jones: Renal Tumors of Childhood - Biology and Therapy, 2014)
- European and North American rates are about the same (8 per million)
- More common in Africans; least common in East Asian population
- Congenital Wilms tumors very rare (J Pediatr Surg 1995;30:856)
- Rare adult cases (Expert Rev Anticancer Ther 2011;11:1105)
- Slight female preponderance
- Majority of patients with Wilms tumor are nonsyndromic
- 10 - 15% associated with syndromes and congenital anomalies (J Med Genet 2006;43:705)
- 1 - 2% familial (Am J Med Genet C Semin Med Genet 2004;129C:29)
- Relapses occur in ~15% of children, the majority within 2 years of diagnosis
Sites
- Kidney
- 5 - 10% are bilateral (Expert Rev Mol Med 2017;19:e8)
- Very rarely extrarenal (J Pediatr Surg 2013;48:E33)
Pathophysiology
- Nephrogenic rests are precursor lesions (Am J Med Genet 1998;79:268)
- Nephrogenic rests are found in 40% of unilateral and > 90% of bilateral Wilms tumors (Pediatr Blood Cancer 2017;64)
- Not clear how nephrogenic rests progress to tumor
- 2 main types of nephrogenic rests:
- Perilobar nephrogenic rests
- Intralobar nephrogenic rests
- Perilobar nephrogenic rests in children under 1 year of age are associated with a markedly increased risk of developing a contralateral Wilms tumor
Etiology
- Thought to develop from persistent metanephric tissue or nephrogenic rests
- Believed to be due genetic alterations of embryological development of the genitourinary tract
- WT1 gene at chromosome 11p13 implicated in tumorigenesis
- WT2 gene at chromosome 11p15 (still not isolated) is a second locus
- Other genes, still to be identified, involved (see below)
Clinical features
- Usually presents as abdominal mass with no associated symptoms
- Other signs or symptoms (present in 20 - 30% of cases):
- Abdominal pain
- Hematuria
- Hypertension
- Anemia
- Most common predisposition syndromes and conditions with different risk of Wilms tumor include:
- High risk (> 20%)
- WAGR (Wilms tumor - aniridia - genitourinary anomalies - intellectual disability)
- Denys-Drash syndrome
- Moderate risk (5 - 20%)
- Beckwith Wiedemann syndrome
- Simpson-Golabi-Behmel syndrome
- Frasier syndrome
- Low risk (< 5%)
- Bloom syndrome
- DICER1 syndrome
- Li-Fraumeni syndrome
- Isolated hemihypertrophy
- High risk (> 20%)
Diagnosis
- Imaging techniques can reliably diagnose 80 - 85% of Wilms tumors (Lancet Child Adolesc Health 2020;4:232)
Laboratory
- No specific / diagnostic laboratory findings
- Catecholamine levels to exclude neuroblastoma
- Complete blood count
- Biochemistry profile
- Renal functions
- Coagulation studies
Radiology description
- Abdominal ultrasound
- Chest Xray (to look for metastases)
- Abdominal and chest CT
- Abdominal MRI
- Recently, MRI diffusion studies showed that Wilms tumor can be differentiated from neuroblastoma
Radiology images
Images hosted on other servers:

Large renal mass

Renal mass
Prognostic factors
- Differ between 2 main approaches; COG (followed mainly in the U.S. and Canada) and SIOP (followed in the rest of the world) (Am Soc Clin Oncol Educ Book 2014;215)
- In both SIOP and COG, tumor histology and stage are important prognostic factors (see Microscopic description)
- In SIOP, which advocates preoperative chemotherapy, followed by surgery and further chemo or radiotherapy; if necessary, prognostic factors also include:
- Risk stratification into three treatment groups: low, intermediate and high risk group
- Tumor volume before and after preoperative chemotherapy in defined cases
- Responsiveness of lung metastases to initial chemotherapy in some groups
- In COG, which advocates primary surgery, followed by chemo or radiotherapy, if necessary, prognostic factors also include:
- Age
- Tumor weight
- Rapidity of lung nodule response
- Molecular markers (LOH at chromosomes 1p, 16q)
- In SIOP, which advocates preoperative chemotherapy, followed by surgery and further chemo or radiotherapy; if necessary, prognostic factors also include:
- 1q gain associated with poor outcome; it will be used in the coming COG trial as a prognostic factor, whereas SIOP will be testing its importance in the current SIOP-RTSG UMBRELLA 2016 study
- Overall survival for Wilms tumor is approximately 90%
Case reports
- Newborn girl with congenital Wilms tumor (BMJ Case Rep 2019;12:e228651)
- 28 month old girl and 41 month old boy with bilateral Wilms tumor with TP53 related anaplasia (Pediatr Dev Pathol 2013;16:217)
- 2 year old girl with hemihypertrophy underwent partial nephrectomy for multifocal, unilateral Wilms tumor (Urology 2019 Nov;133:243)
- 2 year old boy with extrarenal Wilms tumor (J Pediatr Surg 2013;48:E33)
Treatment
- Multimodal treatment which includes a combination of surgery, chemotherapy and, in higher stages, radiotherapy, depending on which approach is followed (COG or SIOP) (Am Soc Clin Oncol Educ Book 2014;215)
- In SIOP, preoperative chemotherapy is given to all patients > 6 months of age, followed by surgery and:
- No further treatment is given for low risk Wilms tumors (completely necrotic) stage 1
- Further chemo and radiotherapy, if necessary, for all other Wilms tumors
- No molecular or other markers are used for risk stratification at present
- Focal anaplasia is regarded as an intermediate risk tumor
- Diffuse anaplasia and blastemal type are regarded as a high risk tumors
- In COG:
- Very low risk Wilms tumors (nonanaplastic, stage 1, negative lymph nodes, < 24 months of age, no LOH for 1p and 16q, specimen weight < 550 g) are treated with surgery only
- All other Wilms tumors are treated with surgery, chemo and radiotherapy if necessary, depending on histology, stage and molecular markers
Clinical images
Images hosted on other servers:

Intraoperative appearance
Gross description
- Usually large, solitary, spherical mass, sharply demarcated from the renal parenchyma, distorting kidneys contours
- In ~10% multinodular
- In primarily operated cases, tumor is gray-white or pink-gray in color, lobulated, soft and friable
- Some tumors may have a prominent cystic appearance
- In pretreated cases, hemorrhage and necrosis are usually extensive
- Gross sampling notes:
- Very important to adequately sample tumor, at least 1 slice of the whole tumor, for assessment of histologic features
- Renal sinus, its vessels and ureter should be included
- Origin of all tissue blocks should recorded, preferably at a gross picture (block identification key) (see video below) (J Clin Pathol 2010;63:102)
Gross images
Contributed by Ellen D’Hooghe, M.D. and Gordan M. Vujanic, M.D., Ph.D.
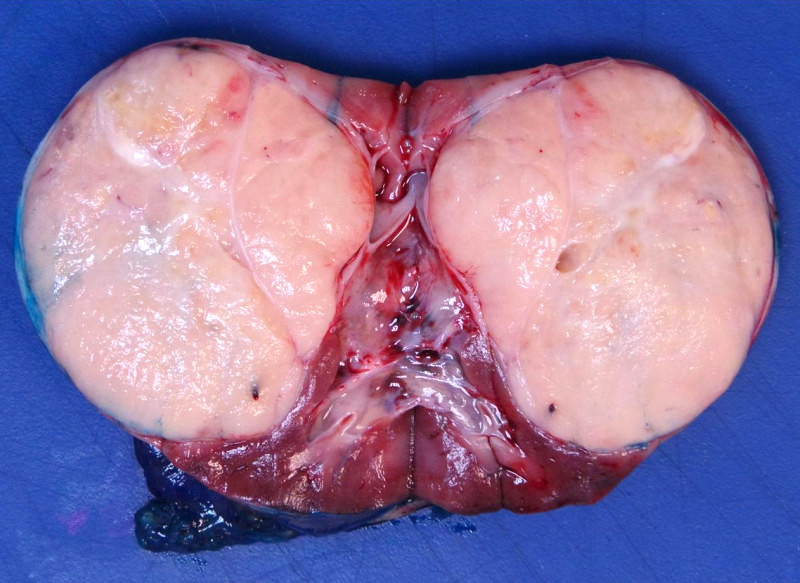
Encapsulated tumor
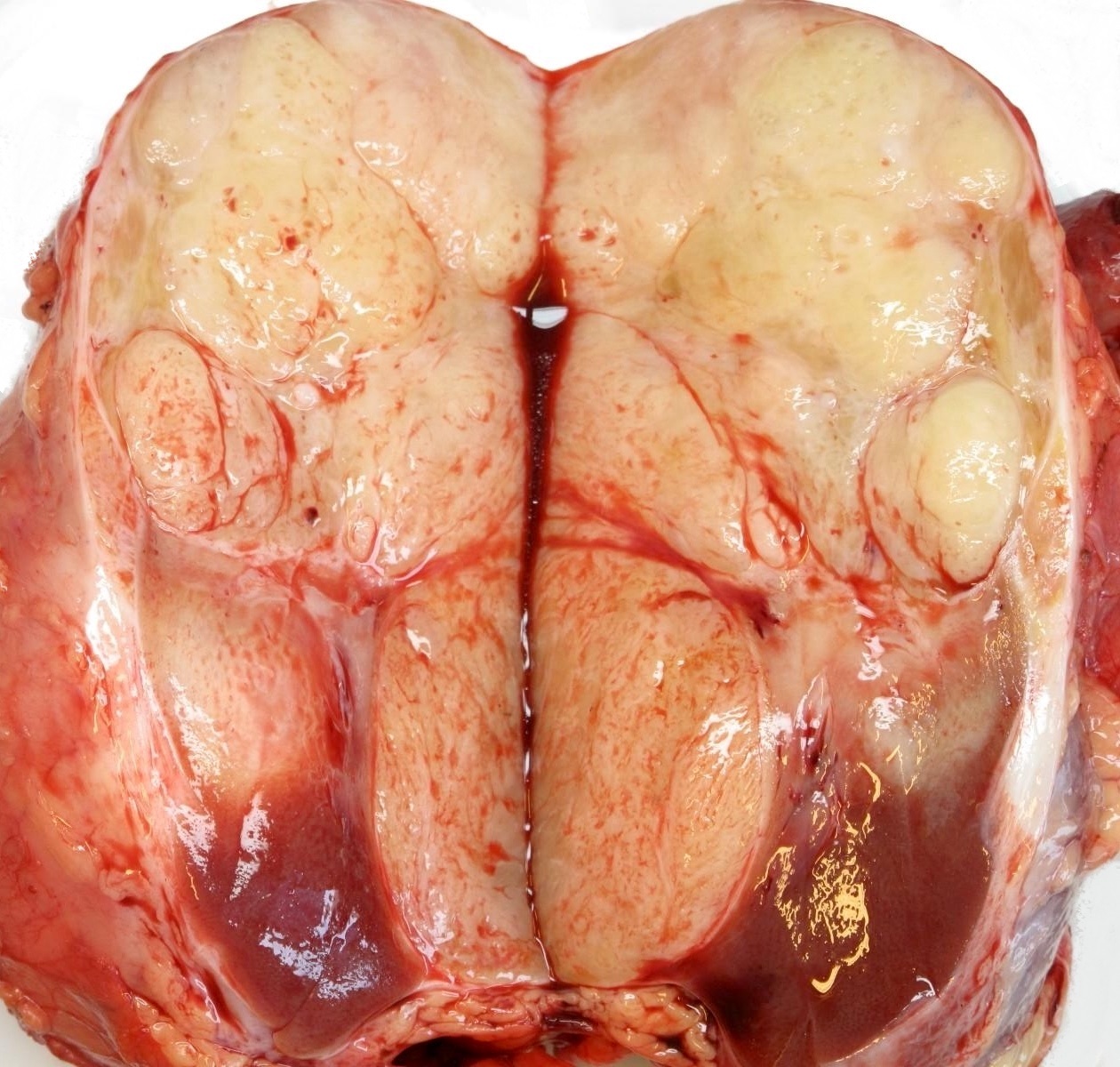
Lobulated tumor

Necrotic tumor
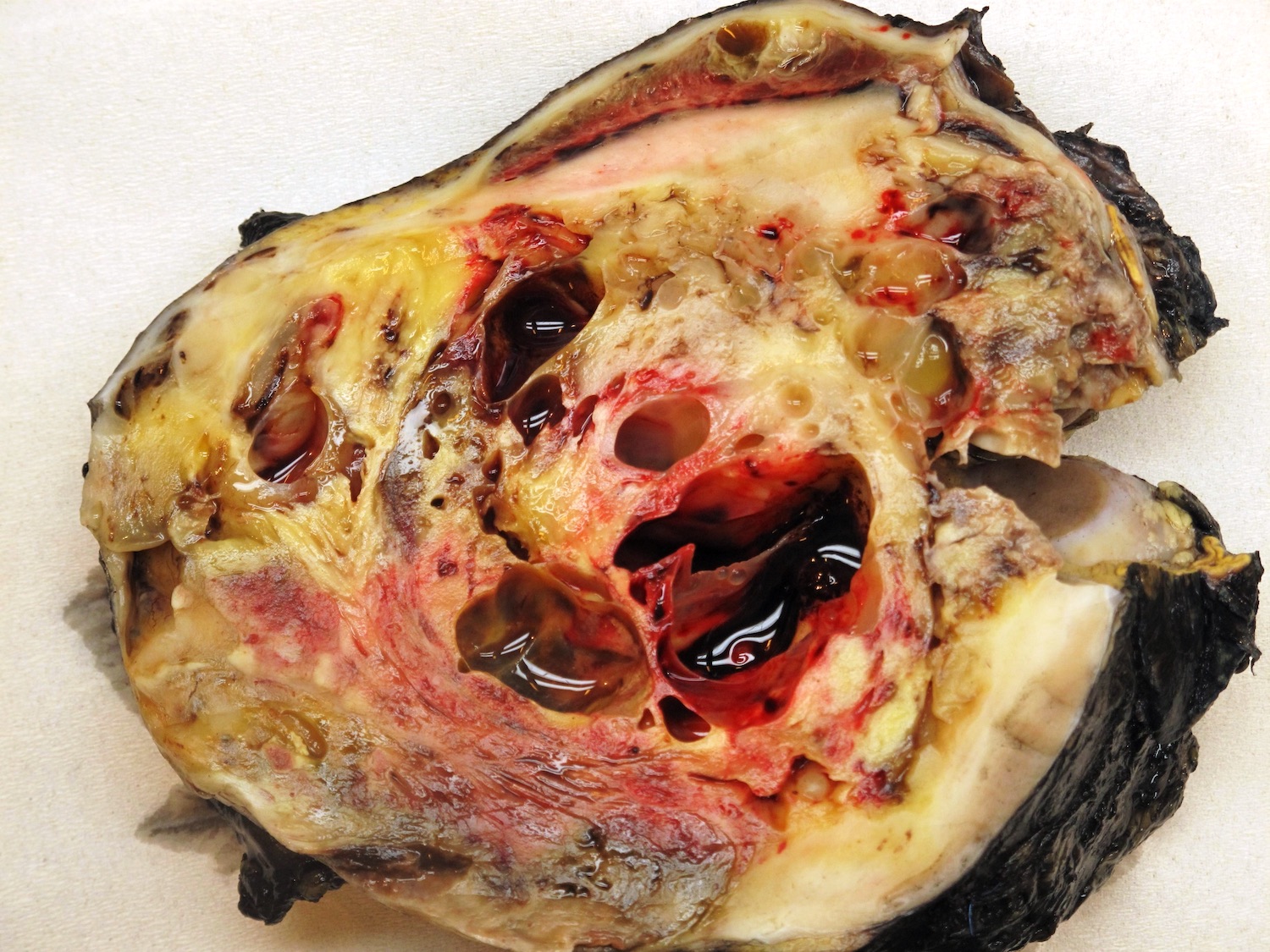
Postchemotherapy tumor
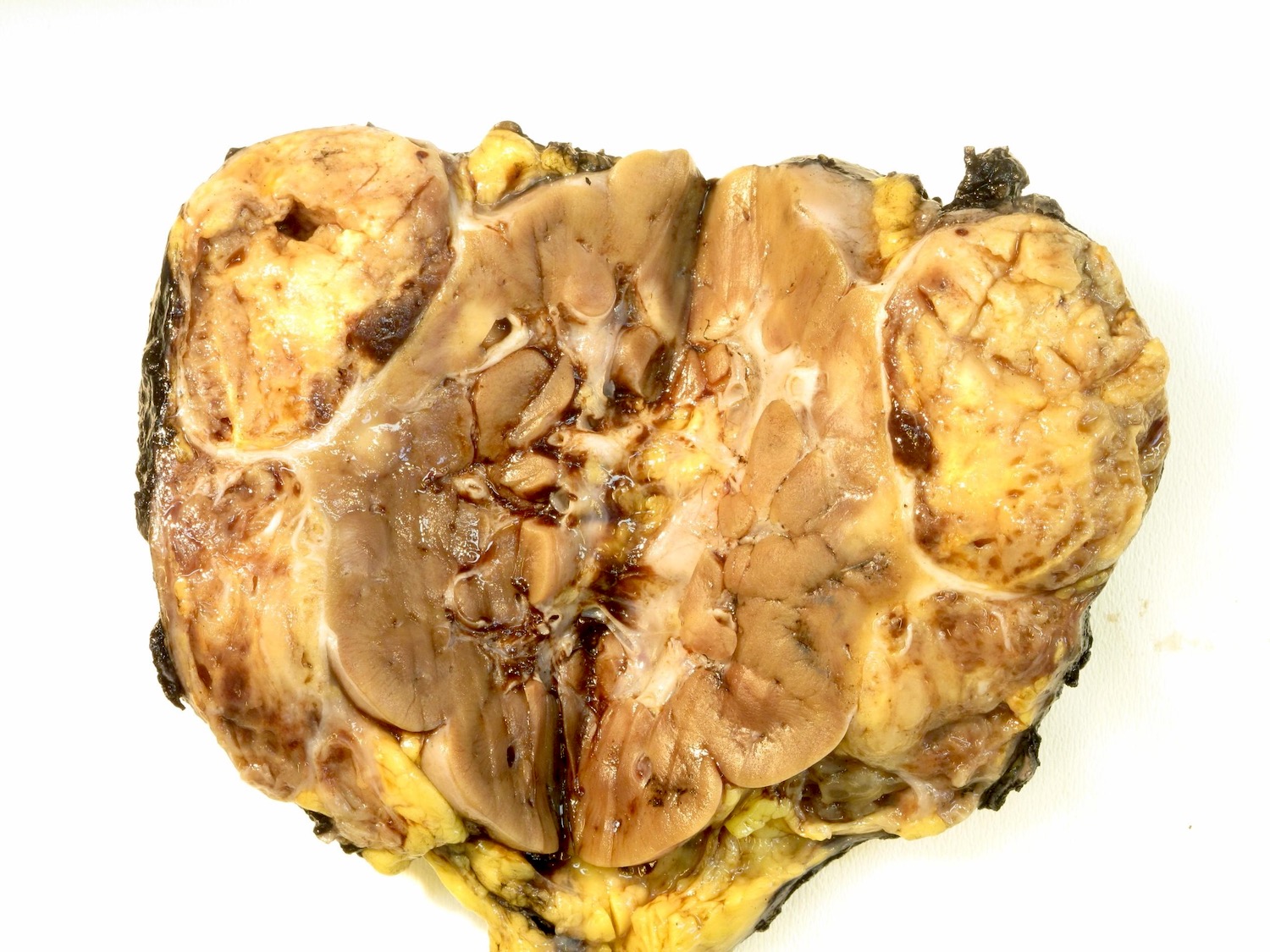
Extrarenal growth of tumor
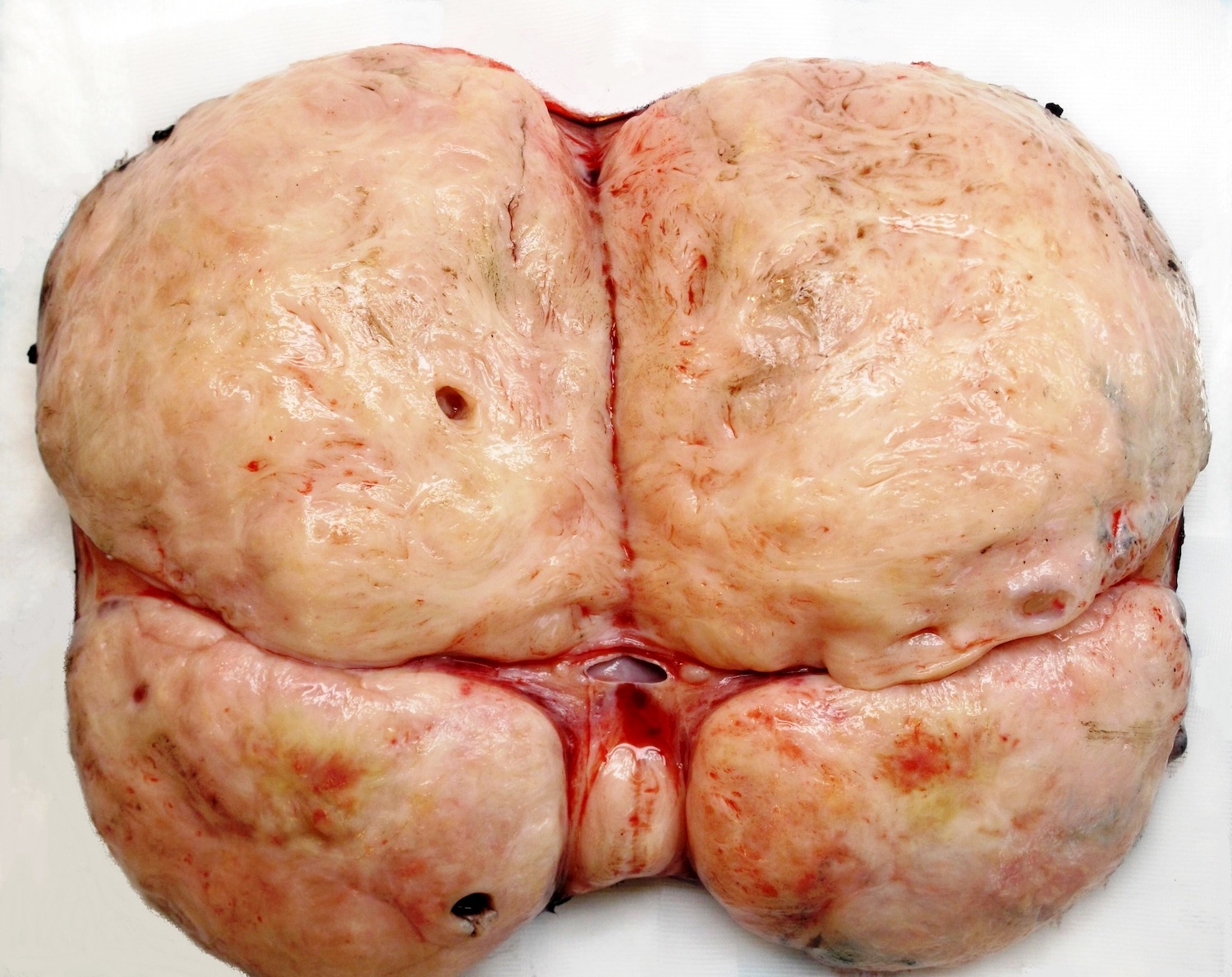
Multifocal tumor
Microscopic (histologic) description
- Typically consists of 3 components: blastemal, epithelial and stromal
- Tumors showing 2 or only 1 component not rare
- Blastema
- Least differentiated component
- Consists of small to medium sized undifferentiated cells with relatively small regular nuclei and small nucleoli
- May show different patterns (diffuse, serpentine, nodular and basaloid patterns) which are of no prognostic significance
- Mitotic figures frequent
- Epithelial component includes:
- Poorly differentiated (rosette-like structures)
- Moderately differentiated (tubules and papillary structures)
- Well differentiated (glomerular-like structures and small, mature tubules) elements
- May contain heterologous elements (squamous and mucinous epithelium, glial tissue)
- Stromal component
- From hypo to hypercellular undifferentiated areas to well differentiated areas
- Cells showing no clear cell borders, oval to spindle shaped nuclei with bland nucleoli
- Often shows heterologous elements (rhabdomyoblasts, adipose tissue, cartilage) (Am J Surg Pathol 2019;43:1583)
- The following descriptive terms should not be used as they are not separate entities, do not exist in any classification and have no prognostic significance
- Fetal rhabdomyomatous Wilms tumor
- Botryoid Wilms tumor
- Teratoid Wilms tumor
- Anaplasia
- Present in 5 - 10% of tumors
- Defined as the presence of enlarged, atypical, tripolar or multipolar mitotic figures, marked nuclear enlargement and hyperchromasia
- All 3 features have to be present
- May occur in any of the components
- Focal anaplasia is defined as a clearly defined focus within a primary intrarenal tumor
- In SIOP: up to 2 foci of up to 15 mm in size
- In COG: up to 4 small foci, size not specified
- Diffuse anaplasia is defined as nonlocalized anaplasia; focal anaplasia with marked nuclear unrest in the remaining tumor; anaplasia beyond the tumor capsule; anaplastic cells in intrarenal or extrarenal vessels, renal sinus, extracapsular sites, metastatic deposits or anaplasia in a biopsy
- Histologic classification
- SIOP histologic classification is based on the assessment of percentage of chemotherapy induced changes and viable tumor components (Tables 1 and 2) (Nat Rev Urol 2018;15:693)
- COG histologic classification includes anaplastic and nonanaplastic Wilms tumors (Table 1) (Am Soc Clin Oncol Educ Book 2014;215)
- Staging criteria in the COG and SIOP are similar but there are some differences (for example, any biopsy in COG but only wedge / open biopsy in SIOP is regarded as a criterion for stage III) (Table 3)
Table 1: histologic risk classifications for Wilms tumor
| International Society of Paediatric Oncology (SIOP) | Children's Oncology Group (COG) | |
| Low risk | Cystic partially differentiated nephroblastoma* Completely necrotic Wilms tumor | Cystic partially differentiated nephroblastoma* |
| Intermediate risk | Epithelial, stromal, mixed, regressive types Focal anaplasia | Favorable histology Wilms tumor No evidence of anaplasia |
| High risk | Diffuse anaplasia Blastemal type | Diffuse anaplasia Focal anaplasia |
Table 2: Histological criteria for Wilms tumor types in SIOP for pretreated cases
| Tumor type | |||||
| Blastema | Epithelium | Stroma | |||
| Completely necrotic | 100% | 0% | 0% | 0% | |
| Regressive | > 66% | 0 - 100% | 0 - 100% | 0 - 100% | |
| Mixed | < 66% | 0 - 65% | 0 - 65% | 0 - 65% | |
| < 66% | 11 - 65% | 0 - 89% | 0 - 89% | ||
| Epithelial | < 66% | 0 - 10% | 66 - 100% | 0 - 33% | |
| Stromal | < 66% | 0 - 10% | 0 - 33% | 66 - 100% | |
| Blastemal | < 66% | 66 - 100% | 0 - 33% | 0 - 33% | |
**The percentage of the components of the viable tumor should add up to 100%
***The presence of diffuse anaplasia in any of the above types supersedes the underlying types. Focal anaplasia also needs to be specifically mentioned in the diagnosis (for example, "focal anaplasia in mixed type")
Table 3: staging criteria for Wilms tumor in SIOP and COG
| Stage | SIOP (pre-operative chemotherapy) | COG (primary surgery) |
| I | Tumor is limited to the kidney and completely resected | Tumor is limited to the kidney and is completely resected |
| Tumor is present in the perirenal fat but is surrounded by a fibrous (pseudo)capsule; the (pseudo)capsule might be infiltrated by viable tumor, which does not reach the outer surface | Renal capsule intact; not penetrated by tumor | |
| Tumor might show protruding ('botryoid') growth into the renal pelvis or ureter but does not infiltrate their walls | Tumor not ruptured or biopsied prior to removal | |
| The vessels or the soft tissues of the renal sinus and not involved by tumor; intrarenal vessel involvement might be present | No tumor invasion of veins or lymphatics of renal sinus | |
| * Fine needle or tru-cut biopsy do not upstage tumor | No nodal or hematogenous metastases | |
| No rupture or biopsy prior to removal | ||
| II | Viable tumor is present in the perirenal fat and is not covered by a (pseudo)capsule but is completely resected (resection margins are clear) | Tumor in the perirenal fat but completely resected |
| Viable tumor infiltrates the soft tissues of the renal sinus | Tumor infiltrates the renal sinus or blood and lymphatic vessels outside the renal parenchyma but is completely resected | |
| Viable tumor infiltrates blood or lymphatic vessels of the renal sinus or of the perirenal tissue | Tumor infiltrates adjacent organs or vena cava but is completely resected | |
| Viable tumor infiltrates the wall of the renal pelvis or the ureter | ||
| Viable tumor infiltrates the vena cava or adjacent organs (except the adrenal gland) but is completely resected | ||
| III | Viable tumor present at resection margin(s) | Residual tumor or nonhematogenous metastases confined to abdomen |
| Abdominal lymph nodes contain viable or nonviable tumor | Involved abdominal lymph nodes | |
| Viable or nonviable thrombus present at resection margins of ureter, renal vein or inferior vena cava | Peritoneal tumor implants | |
| Viable or nonviable tumor thrombus in the inferior vena cava removed piecemeal by a surgeon | Tumor spillage before or during surgery | |
| Preoperative or intraoperative tumor rupture, if confirmed by microscopic examination (viable tumor at the surface of the specimen at the area of rupture) | Gross residual tumor in abdomen | |
| Wedge or open biopsy before preoperative chemotherapy or surgery | Biopsy of tumor (including fine needle biopsy) prior to removal of kidney | |
| Tumor implants (viable or nonviable) in the abdomen | Resection margins involved by tumor or transection of tumor during resection (i.e. piecemeal excision of tumor) | |
| Tumor (viable or nonviable) penetrated through the peritoneal surface | ||
| IV | Hematogenous metastases (lung, liver, bone, brain) or lymph node metastases outside the abdominopelvic region | Hematogenous metastases or spreads beyond abdomen |
| V | Bilateral tumors at diagnosis; each side should be substaged according to the above criteria | Bilateral tumors at diagnosis; each side should be substaged according to the above criteria |
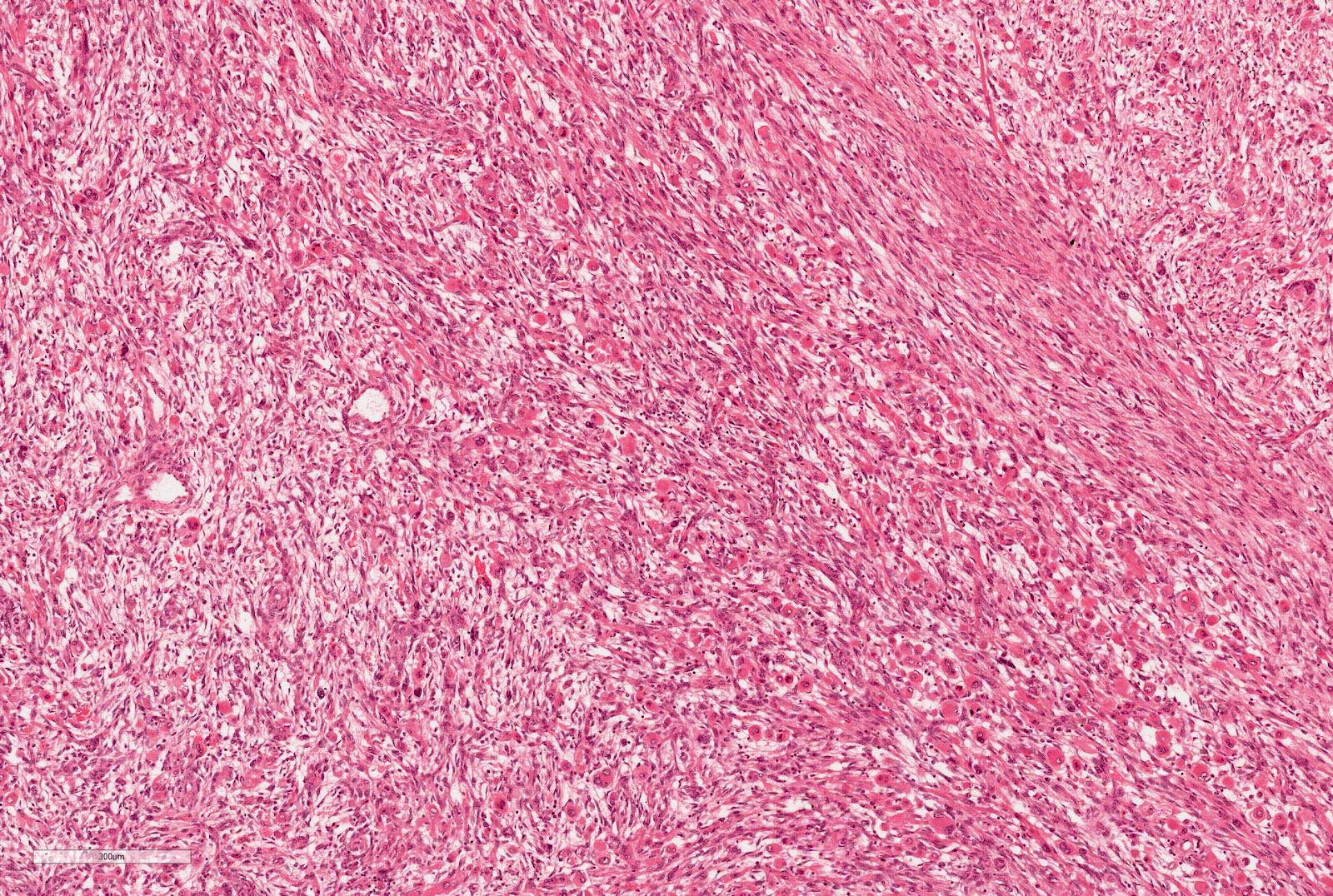
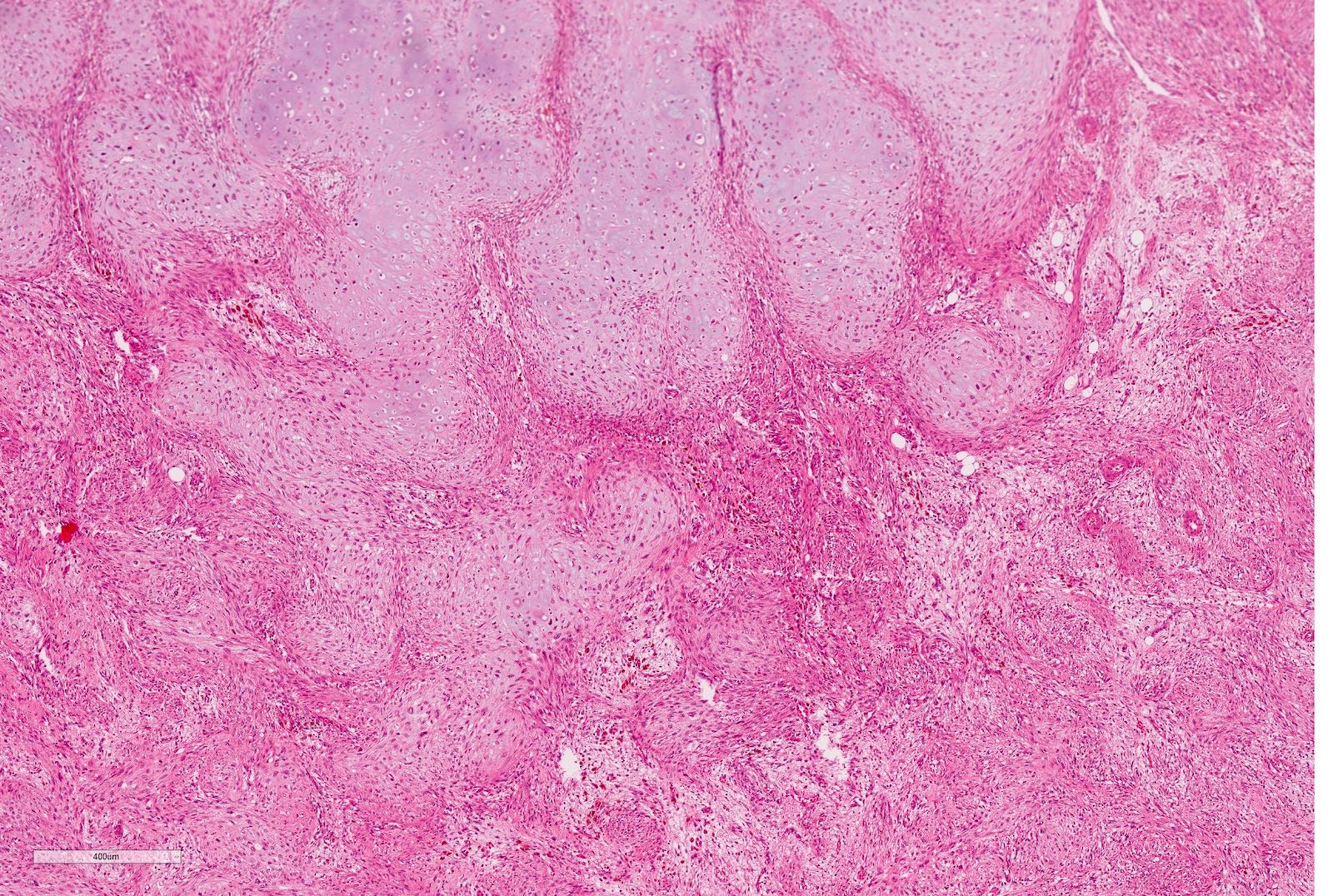
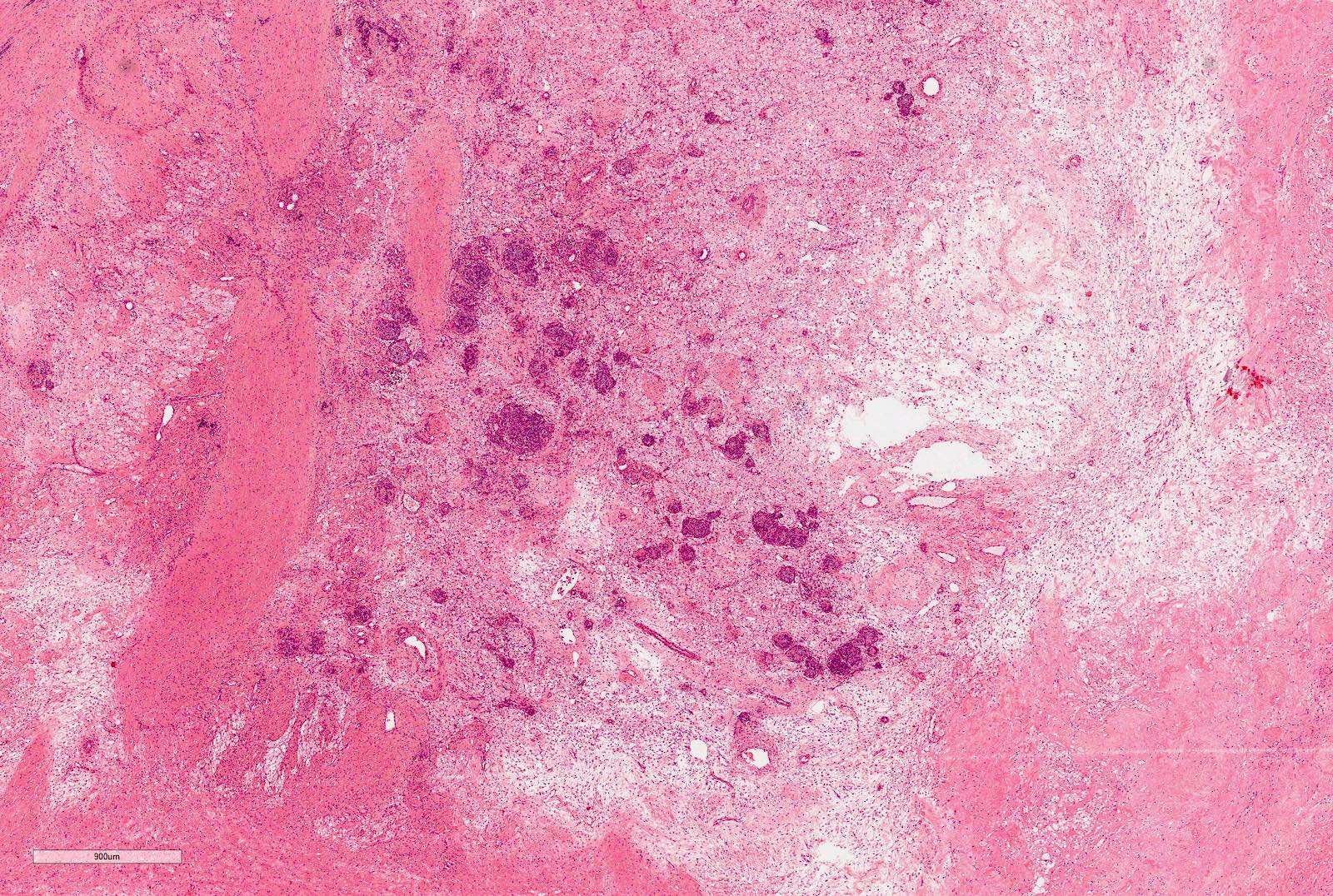
Microscopic (histologic) images
Contributed by Ellen D’Hooghe, M.D. and Gordan M. Vujanic, M.D., Ph.D.
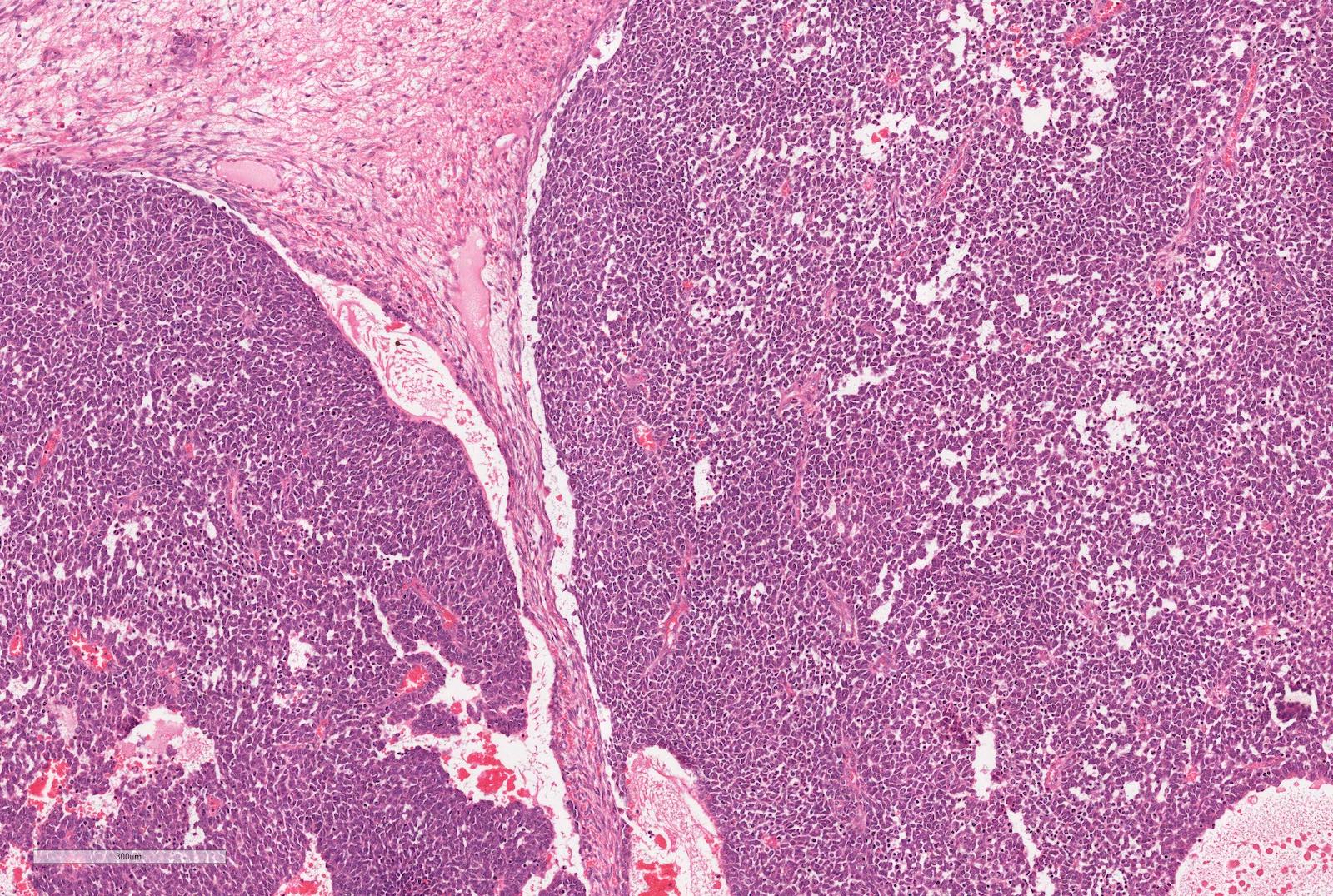

Blastemal component

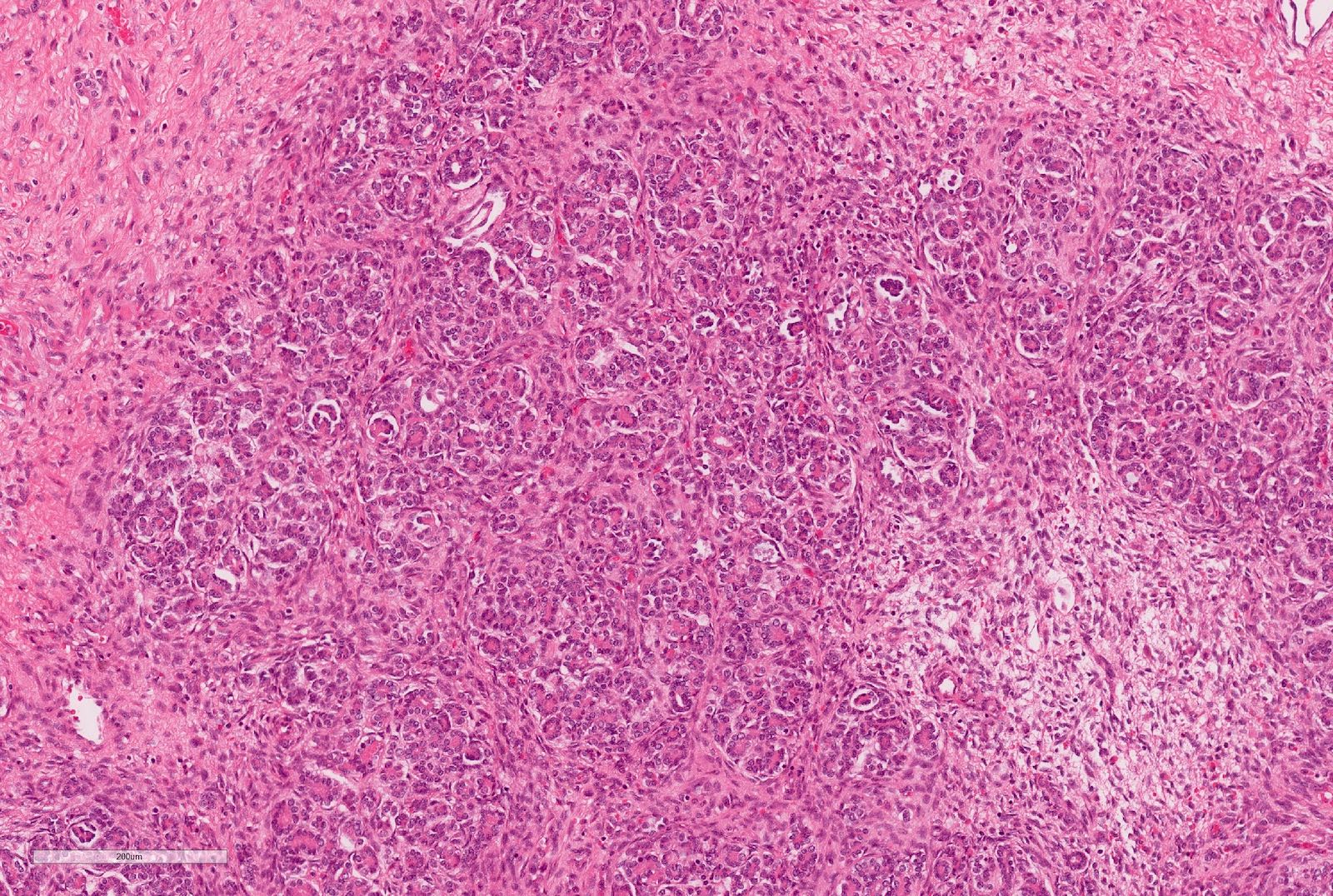
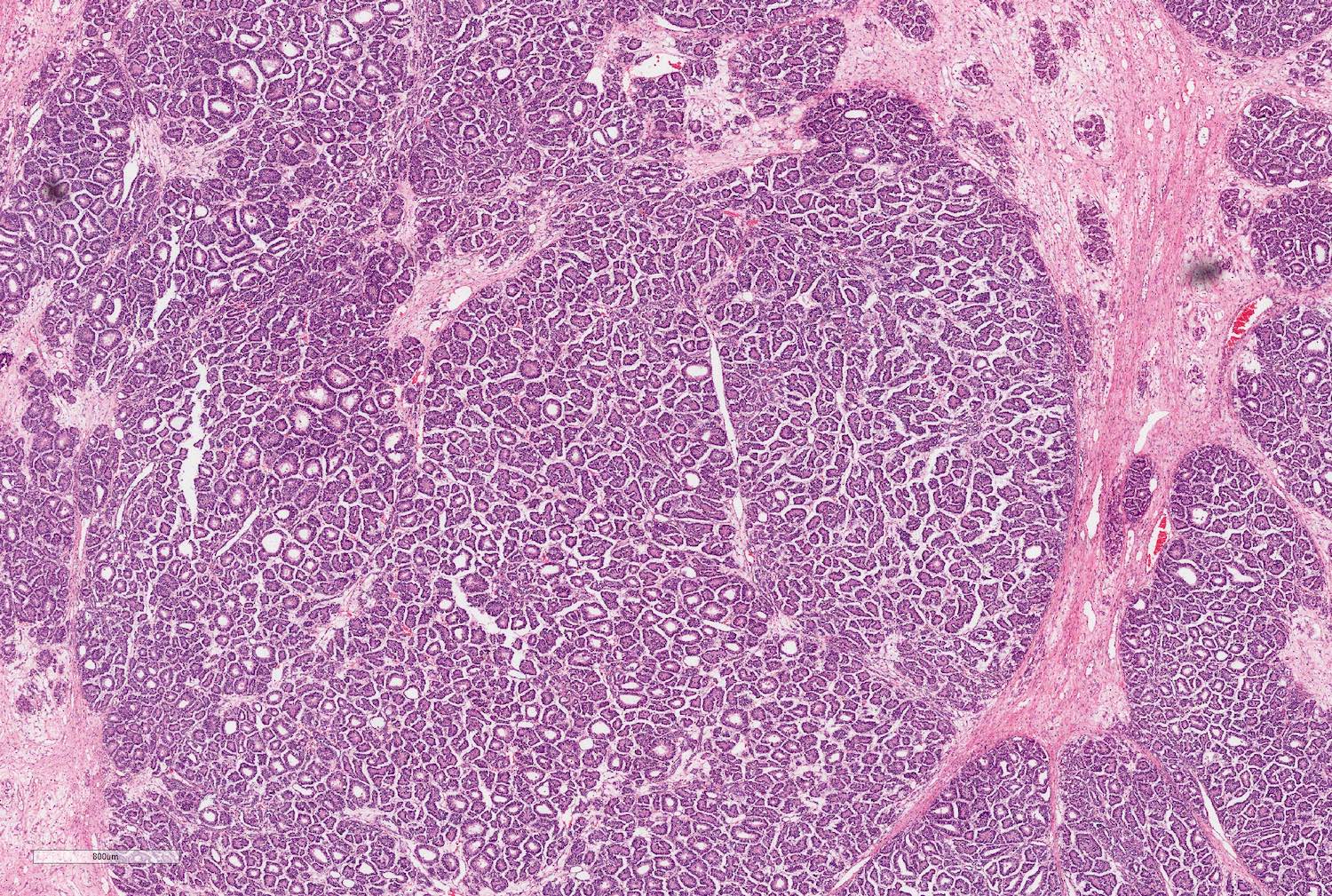
Epithelial component
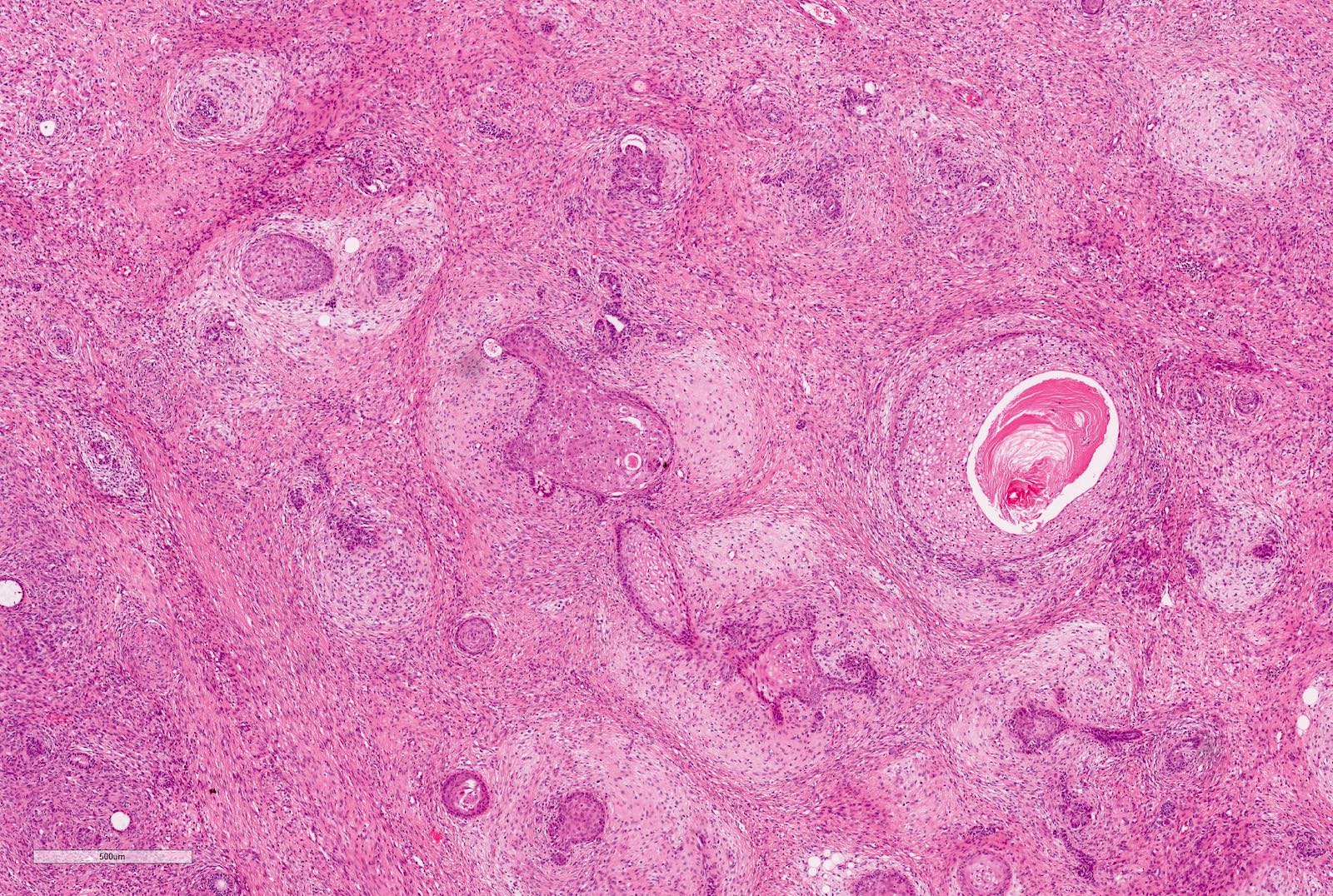
Squamous epithelium

Undifferentiated stroma
Differentiated stroma

Adipose tissue
Mature cartilage
Subtotally necrotic tumor
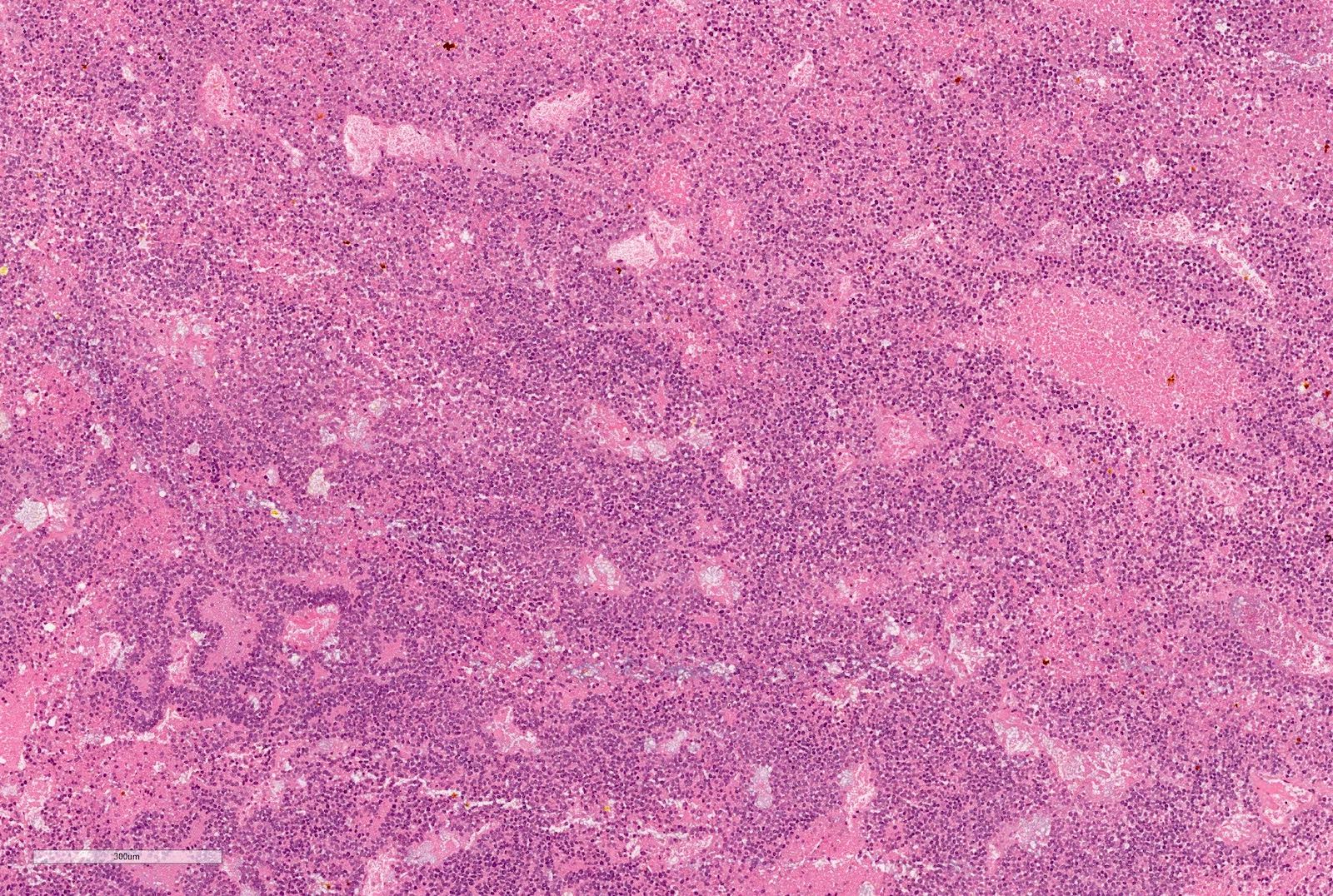
"Dying" blastema
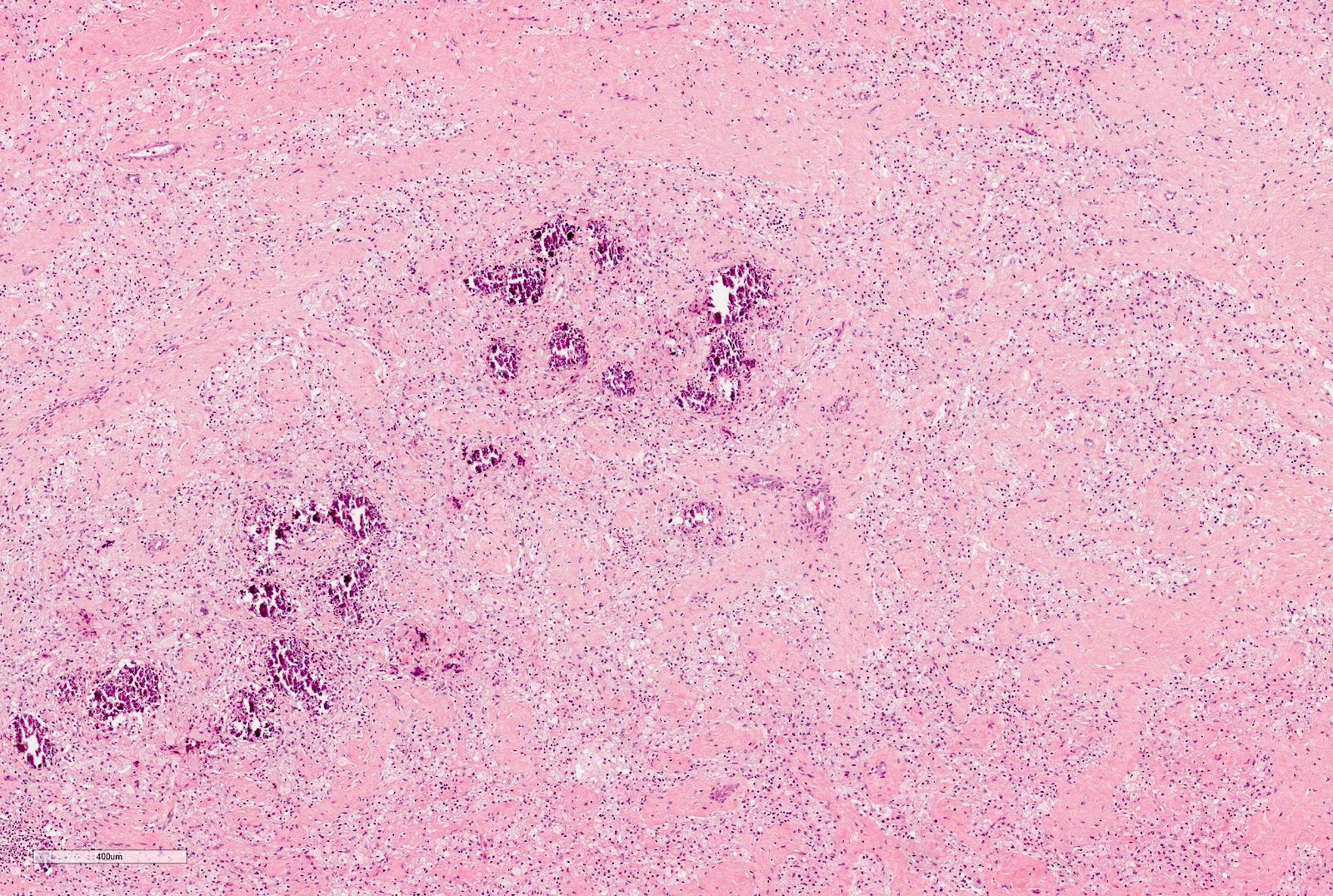
Chemotherapy induced changes
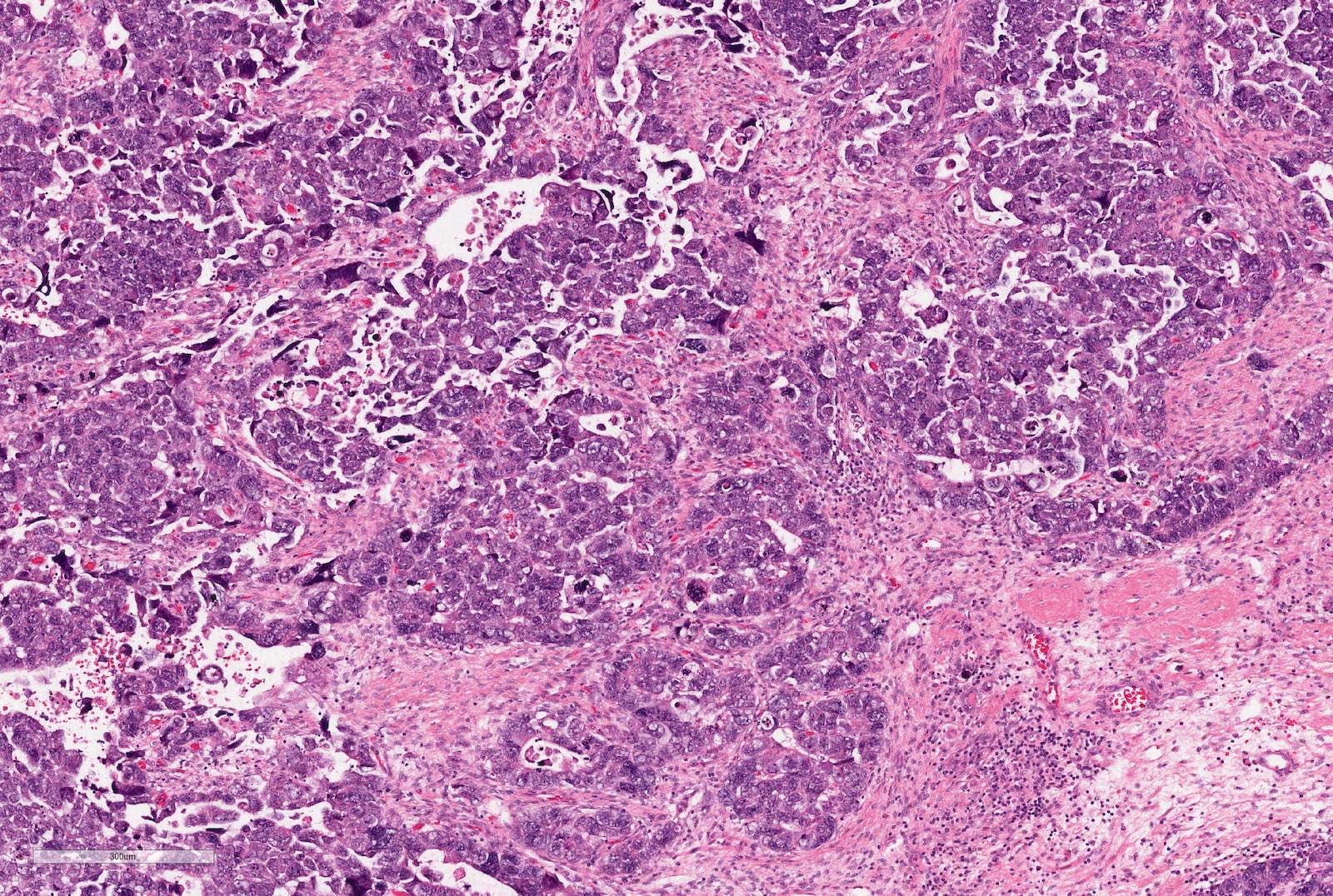
Anaplasia
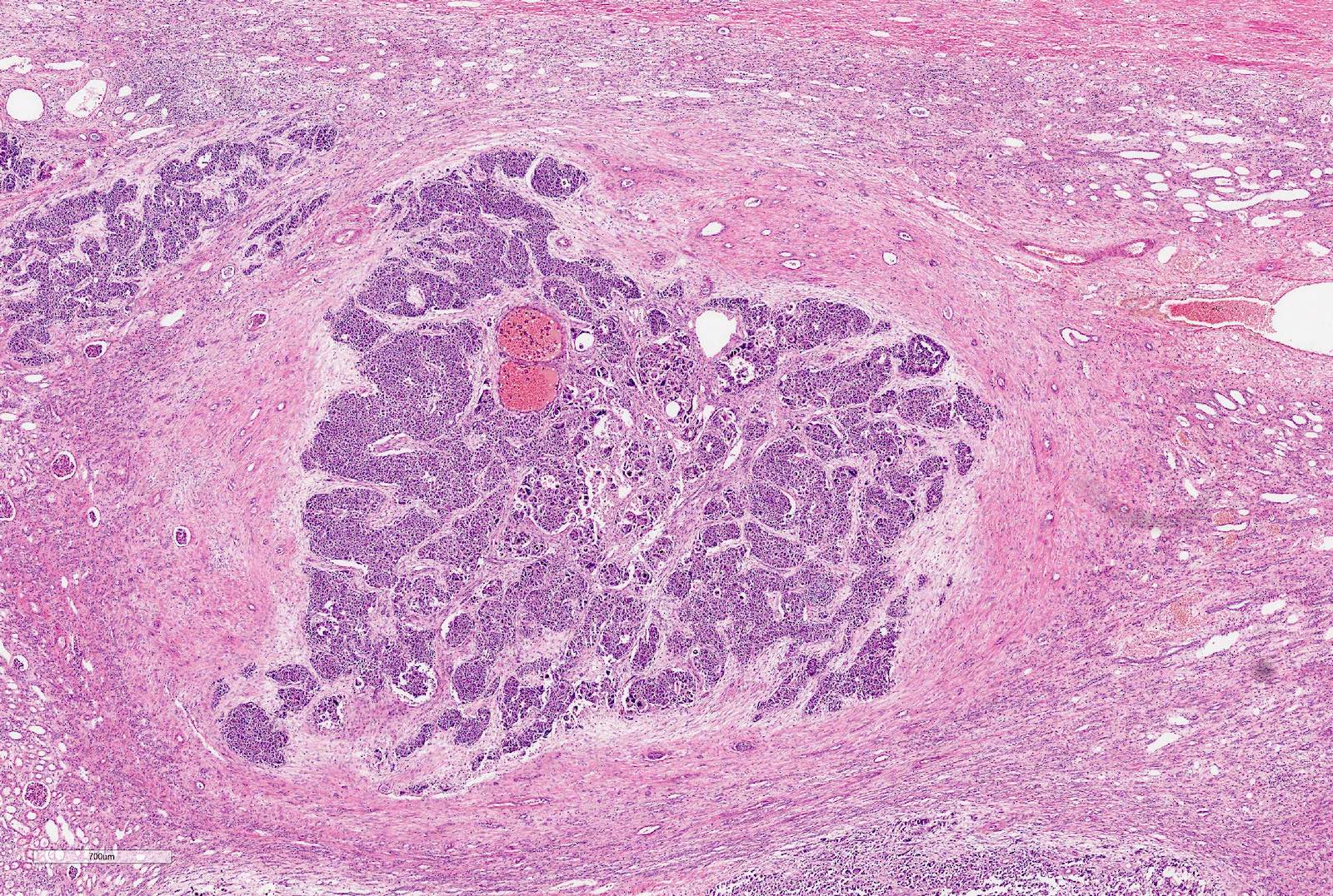
Focal anaplasia
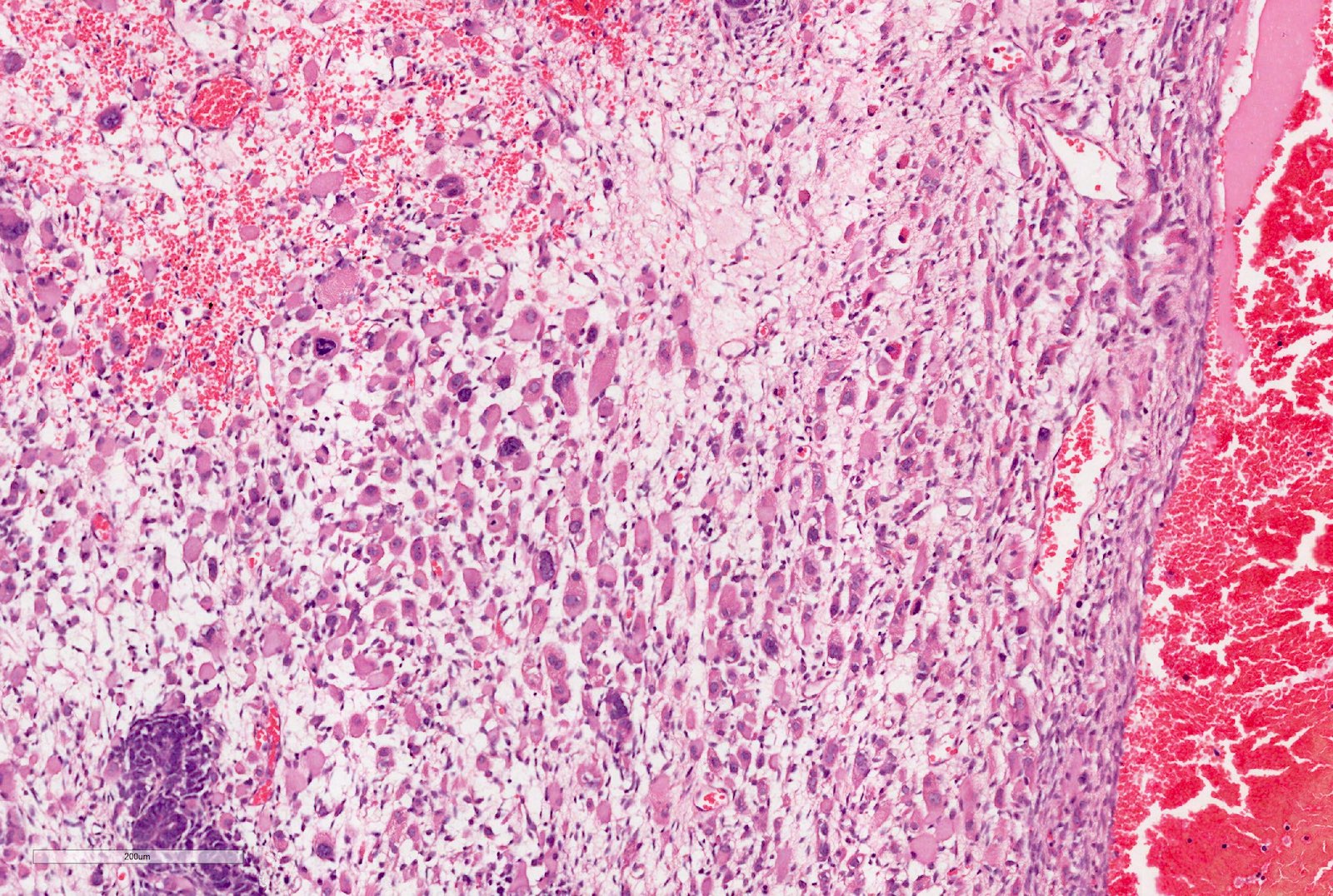
Pseudoanaplasia

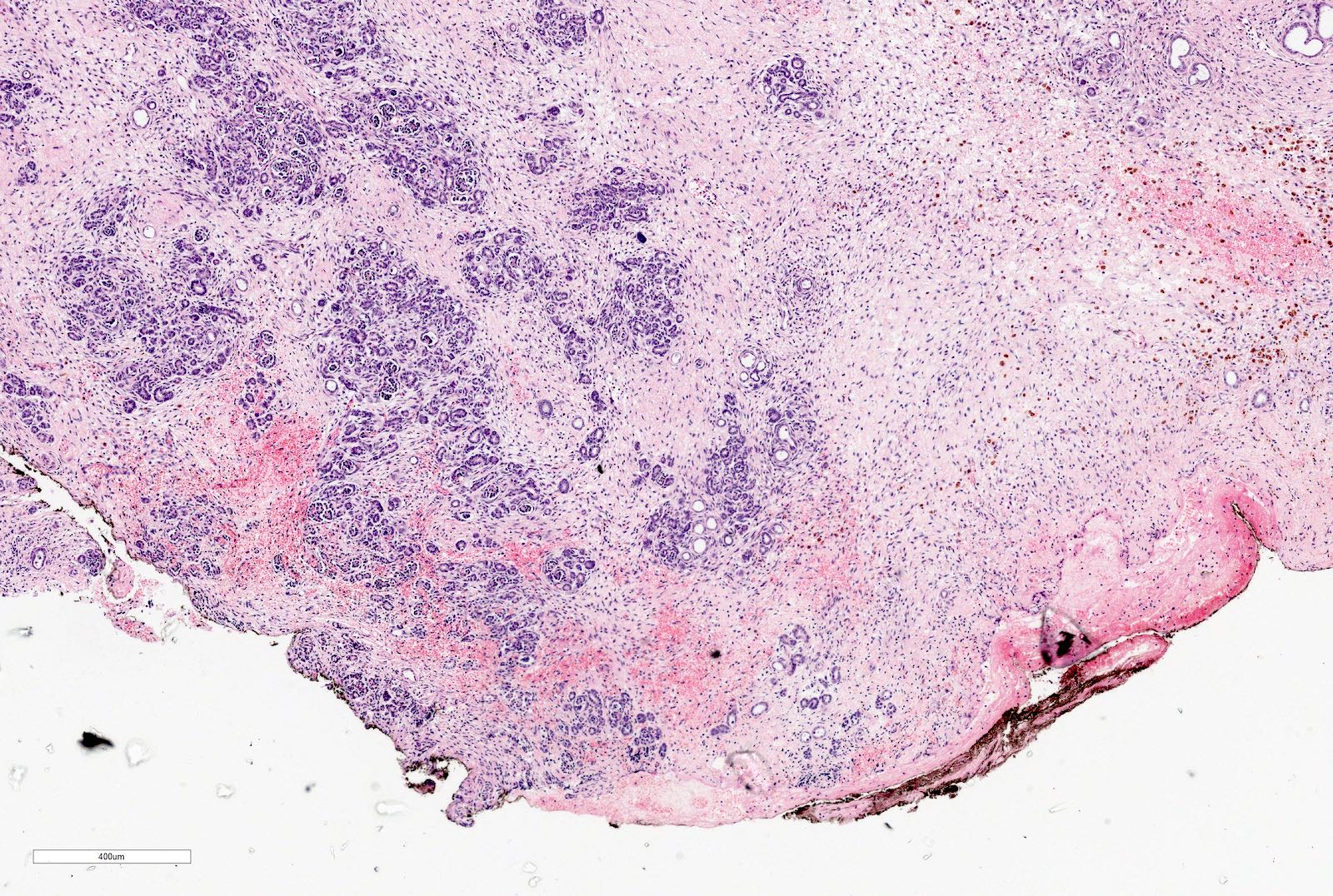
Renal sinus involvement
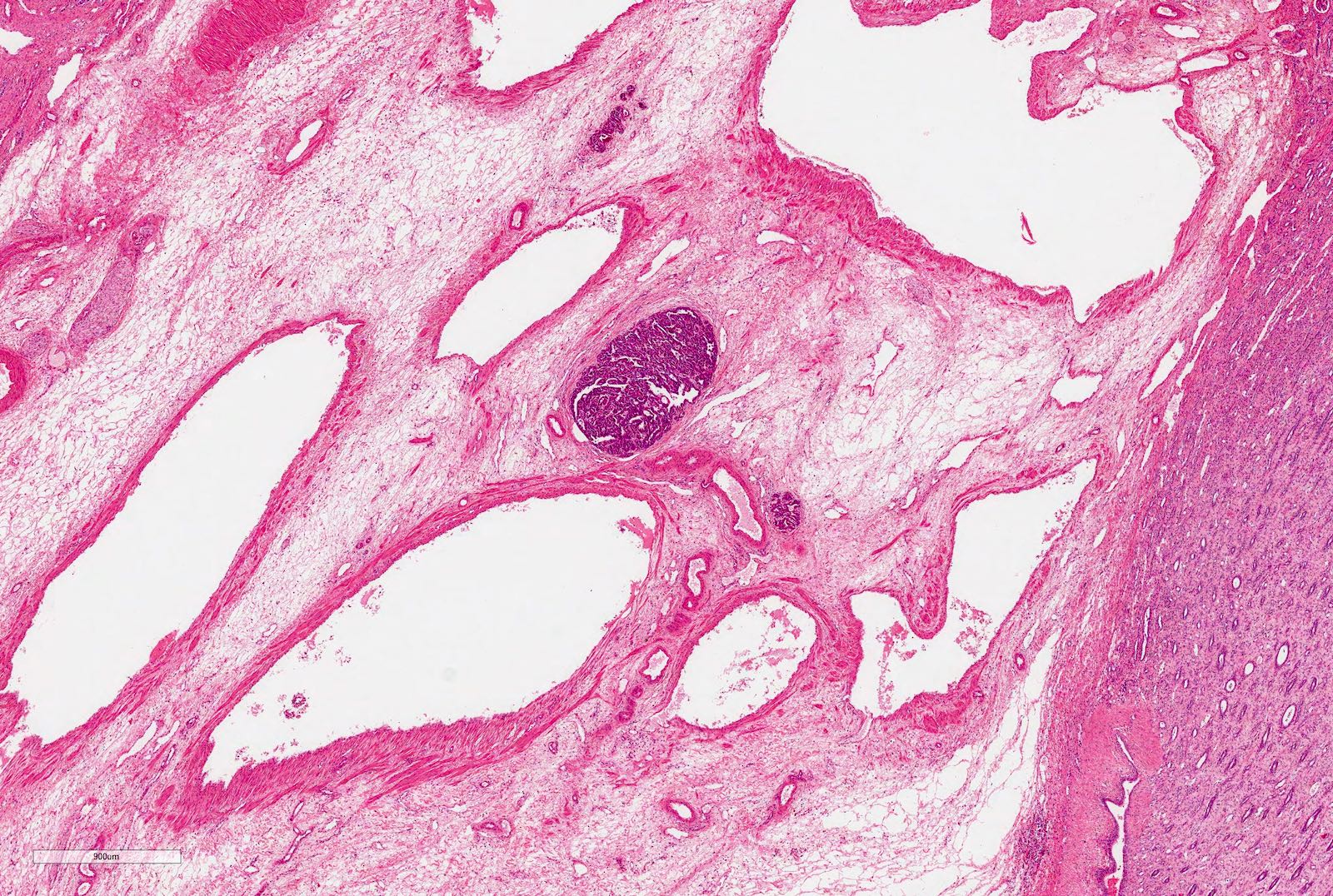
Tumor thrombus
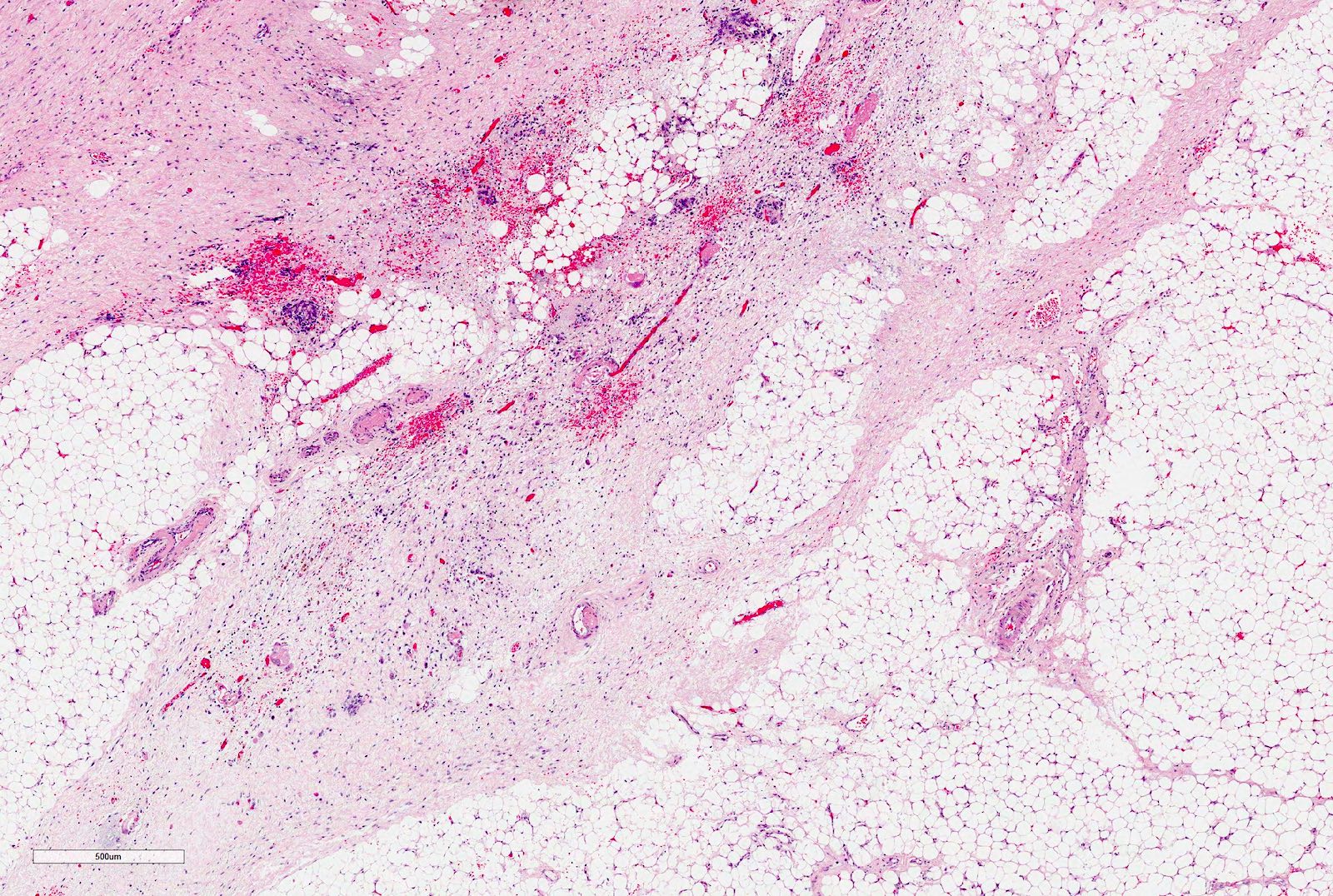

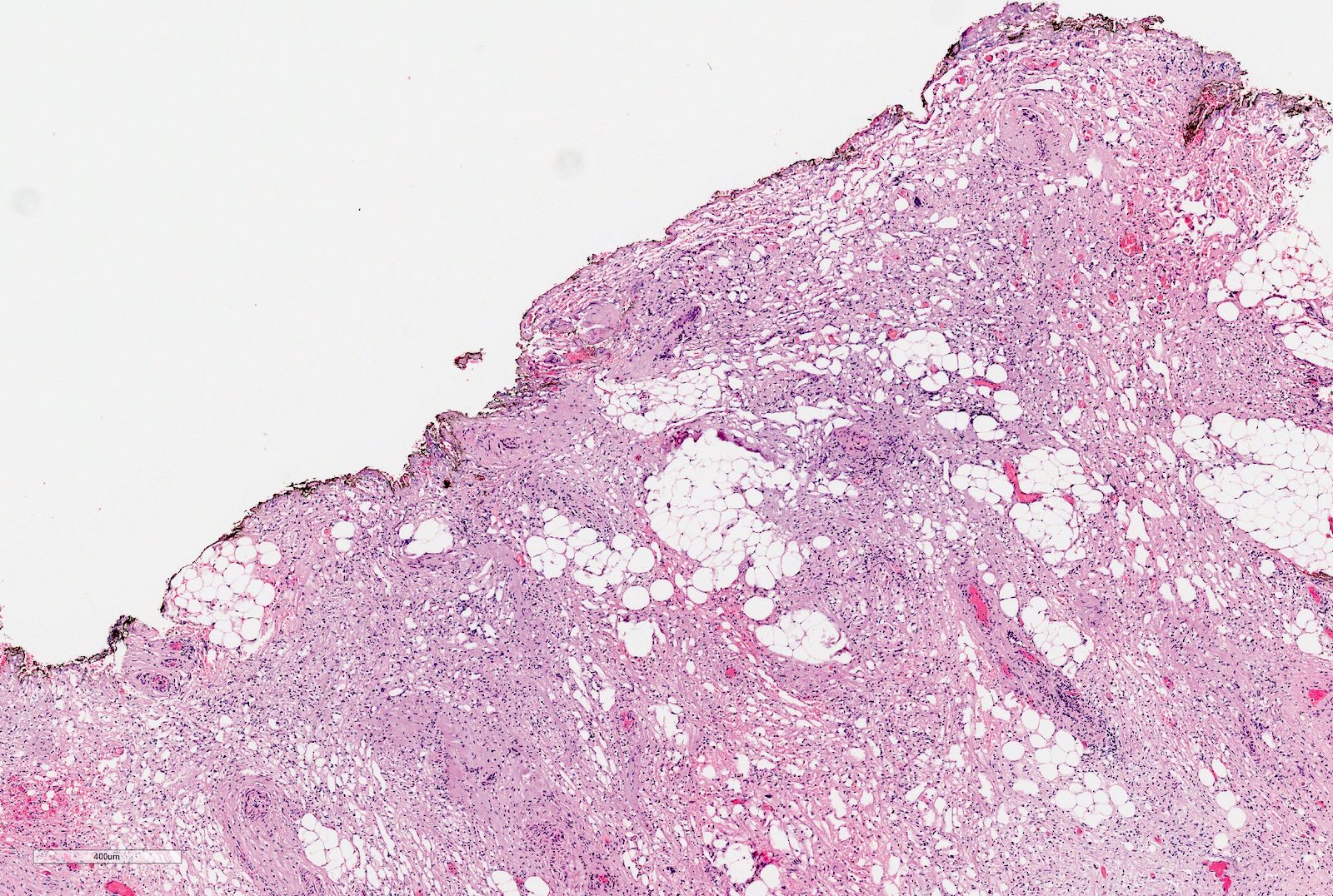
Perirenal fat invasion
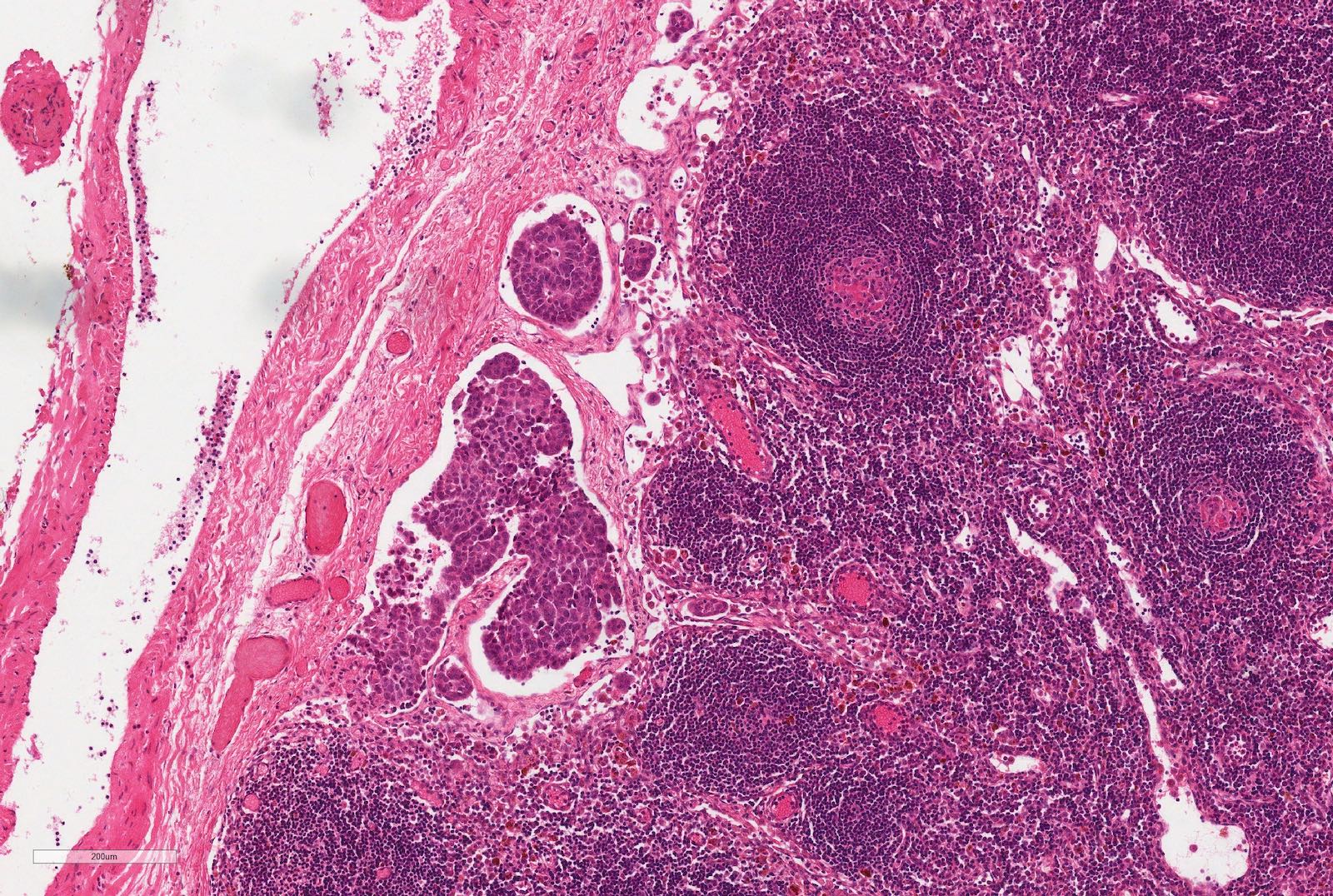
Lymph node metastasis - viable
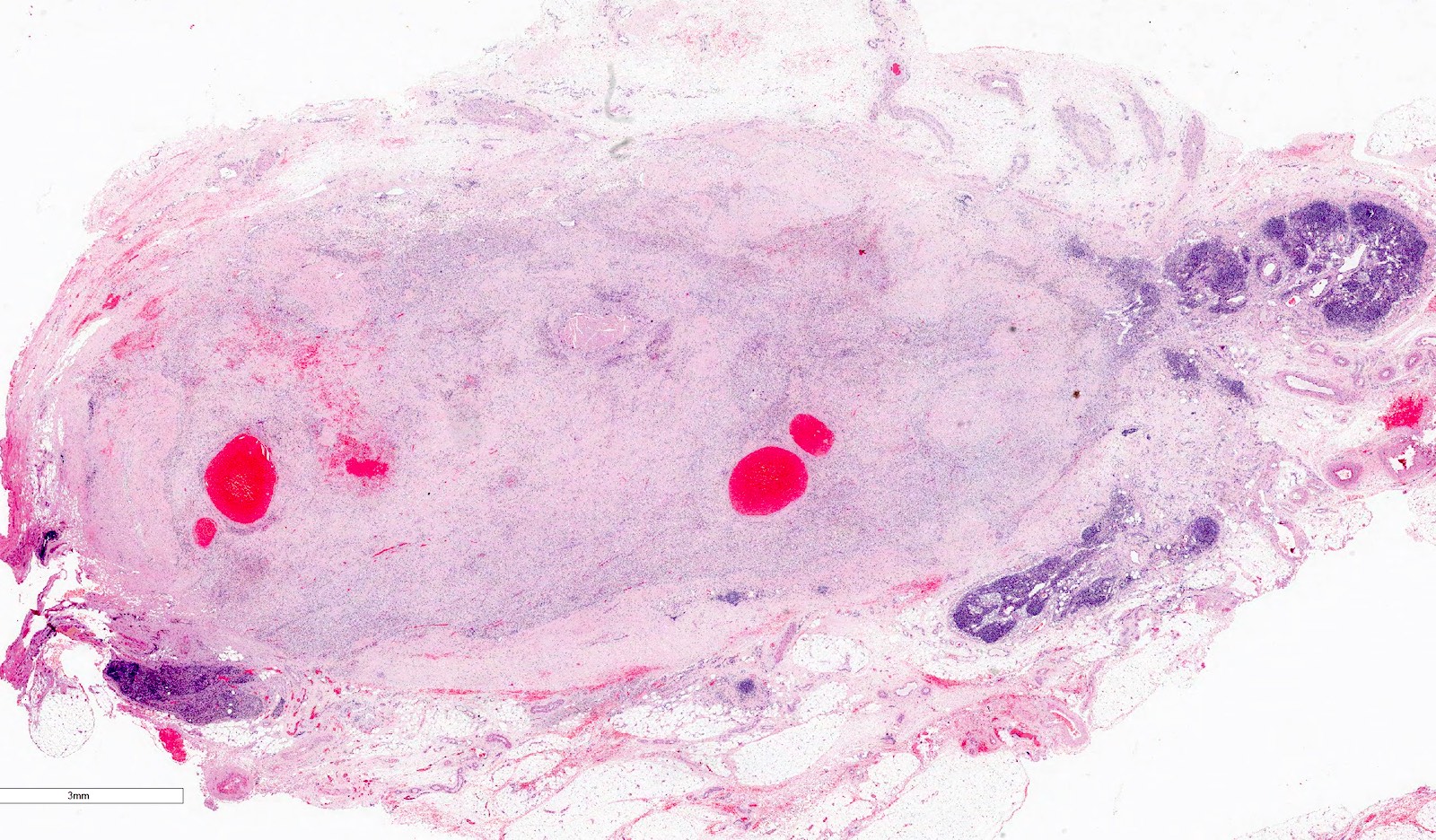
Lymph node metastasis - nonviable
Resection margin - nonviable tumor
Incompletely resected tumor
Cytology description
- Cytology (FNA) not recommended or routinely done
Positive stains
- WT1
- The most sensitive and specific marker (positive in ~90% of cases) (Cancers (Basel) 2020;12:729)
- Only nuclear staining should be evaluated, seen with antibodies directed against C terminal and N terminal
- Cytoplasmic expression can be seen, especially using antibodies to the N terminal
- Blastema
- Epithelium
- Stroma
- INI1 retained
- Glypican 3: expressed in the majority of Wilms tumors (blastema and epithelium) (Virchows Arch 2015;466:67)
Electron microscopy description
- Electron microscopy not routinely used
Molecular / cytogenetics description
- A number of genetic changes with variable prevalence have been described, including (Nat Rev Nephrol 2019;15:240):
- WT1 (11p13), prevalence 10 - 20%, early event, associated with stromal histology
- CTNNB1 (3p22), prevalence ~15%, late event, not associated with nephrogenic rests
- IGF2 (11p15), prevalence ~70%, early event, associated with perilobar nephrogenic rests and with epithelial and blastemal histology
- TP53 (17p13), prevalence ~70%, associated with anaplasia and with reduced event free and overall survival
- MYCN (2p24), prevalence ~15%, associated with anaplasia and with reduced event free and overall survival
- 1q gain, prevalence ~30 - 40%, associated with reduced event free and overall survival
Sample pathology report
- Left kidney, total nephrectomy:
- If primarily operated:
- Wilms tumor, nonanaplastic type, stage II (due to renal sinus invasion)
- Comment: Tumor comprises blastemal, epithelial and stromal components, present in equal proportion. No evidence of anaplasia is seen. Tumor is infiltrating the renal sinus but is not reaching the resection margins. Lymph nodes are free of tumor.
- If treated with preoperative chemotherapy:
- Wilms tumor, mixed type, intermediate risk tumor, stage III (due to viable and nonviable lymph node metastases)
- Comment: Tumor shows chemotherapy induced changes occupying 40% of the mass. The viable tumor consists of blastemal (20%), epithelial (50%) and stromal (30%) elements, with no evidence of anaplasia. The renal sinus and perirenal fat are free of tumor. Lymph nodes contain viable and nonviable metastases.
- If primarily operated:
Differential diagnosis
- Triphasic Wilms tumors relatively easy to diagnose on H&E slides
- Pure blastemal, epithelial and stromal tumors may pose a diagnostic challenge
- Extensive sampling may be helpful in identifying other diagnostic features
- Immunohistochemistry and molecular biology investigations help in the majority of cases (Cancers (Basel) 2020;12:729)
- Pure blastemal Wilms tumor needs to be distinguished from:
- Rhabdoid tumor:
- Neuroblastoma:
- Synaptophysin+, WT1-
- Ewing sarcoma:
- Desmoplastic small round cell tumor:
- WT1+, diagnosis established by EWS-WT1 gene fusion
- Pure epithelial Wilms tumor may be difficult to distinguish from:
- Hyperplastic perilobar nephrogenic rests:
- No reliable histologic, immunohistochemical or molecular findings to distinguish from Wilms tumor
- Papillary renal cell carcinoma:
- Metanephric adenoma:
- BRAF V600E+, CD57+, WT1+
- Hyperplastic perilobar nephrogenic rests:
- Pure stromal Wilms tumor may be difficult to distinguish from:
- Clear cell sarcoma of the kidney:
- Mesoblastic nephroma:
- WT1-, 70% of cellular mesoblastic nephromas show ETV6-NTRK3 gene fusion
- Metanephric stromal tumor:
- CD34+, BRAF V600E+, WT1 variable
- Other rare tumors (synovial sarcoma, inflammatory myofibroblastic tumors and renal cell carcinoma with sarcomatous differentiation)
Board review style question #1
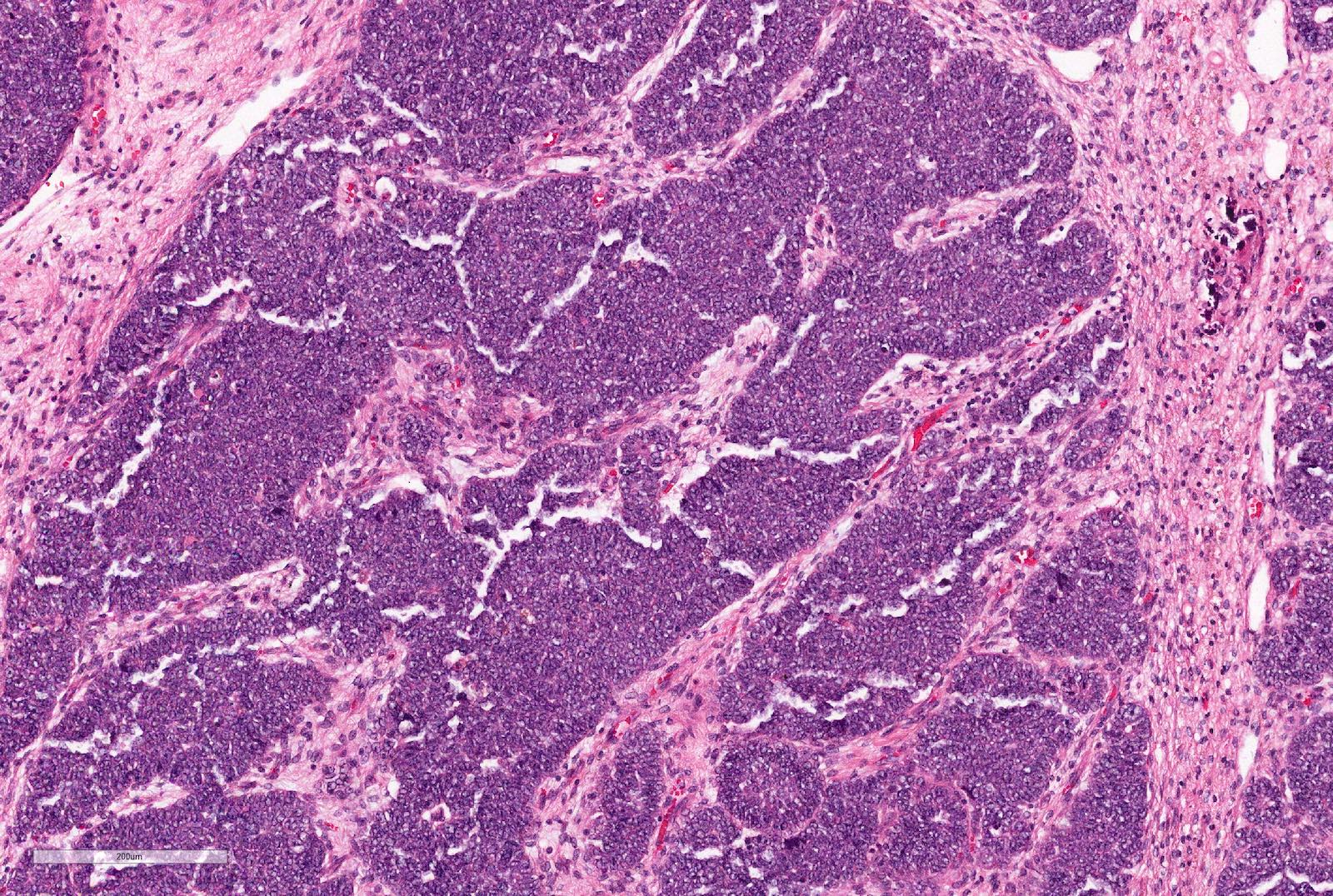
A 2 year old girl presents with an abdominal mass noticed by parents. Imagining studies show a left sided renal mass. No obvious lung metastases. Preoperative chemotherapy given as per the SIOP (International Society of Paediatric Oncology) protocol. Total left nephrectomy performed. The tumor is composed of undifferentiated cells with no particular pattern, shown above. The tumor cells are positive for WT1, CD56 and PAX8, negative for synaptophysin and cyclin D1 and INI1 is retained.
If the whole tumor shows the same pattern, the most likely diagnosis is
- Wilms tumor (nephroblastoma), blastemal type
- Desmoplastic small round cell tumor
- Hyperplastic nephrogenic rest
- Neuroblastoma
- PNET / extraosseous Ewing sarcoma
Board review style answer #1
Board review style question #2
Which of the following histologic features is important for risk stratification in nephroblastoma (Wilms tumor)?
- Anaplasia
- Botryoid growth
- Fetal rhabdomyomatous pattern
- Teratoid elements
Board review style answer #2

