

Kidney nontumor / medical renal
Developmental & cystic diseases
Autosomal dominant polycystic kidney disease
Author: Mandolin S. Ziadie, M.D.

Last author update: 1 December 2011
Last staff update: 11 January 2024 (update in progress)
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: autosomal dominant (adult) polycystic kidney disease
Table of Contents
Definition / general | Epidemiology | Pathophysiology | Clinical features | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Additional referencesCite this page: Ziadie MS. Autosomal dominant polycystic kidney disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneytumoradultpkd.html. Accessed April 25th, 2024.
Definition / general
- Autosomal dominant renal cystic disorder due to mutations in genes coding for polycystin 1 (PKD1, chromosome 16p, most common) and polycystin 2 (PDK2, chromosome 4q)
- Also associated with TSC2 / PKD1 contiguous gene syndrome (Am J Surg Pathol 2002;26:198)
- Usually inherited; new mutations without a family history occur in approximately 10%
Epidemiology
- 1 - 2 / 1,000 births
- M = F
Pathophysiology
- Mutated proteins are involved in cell differentiation, polarization, proliferation and membrane transport
- The exact mechanism of cyst formation is not yet understood
- Cysts form in all regions of the nephron, enlarging and expanding throughout life
- Normal renal function is maintained until midadulthood in most patients
Clinical features
- Third most common cause of endstage renal disease
- Patients present with hematuria, abdominal pain, hypertension, urinary tract infection or urolithiasis
- Associated with von Meyenburg complexes in liver (97%, Mod Pathol 1996;9:233); hepatic cysts (40% - 88%); berry aneurysms (10% - 30%, cause death in 4% - 10%); mitral valve prolapse (20%); cysts in pancreas, lung, spleen, pineal gland and seminal vesicles; aortic aneurysms; hepatic fibrosis; intestinal diverticula
- 25% die from infection, 40% from hypertension and heart disease and 15% from berry aneurysms or stroke
Prognostic factors
- Poor prognostic factors: sickle cell trait, male sex, early disease onset, early hypertension onset and proteinuria
Case reports
- 15 year old girl with no family history but classic findings and oral-facial-digital syndrome type I (Arch Pathol Lab Med 1991;115:519)
- 38 year old man with littoral cell angioma of spleen (Arch Pathol Lab Med 2001;125:1505)
- 43 year old woman with polycystic liver disease and hemorrhagic hereditary telangiectasia (Am J Surg Pathol 1998;22:368)
Treatment
- Laparoscopic nephrectomy (Can J Urol 2006;13:3340), transplant
Clinical images
Images hosted on other servers:
Angiogram
Gross description
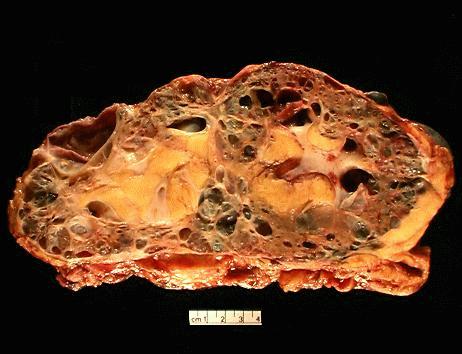
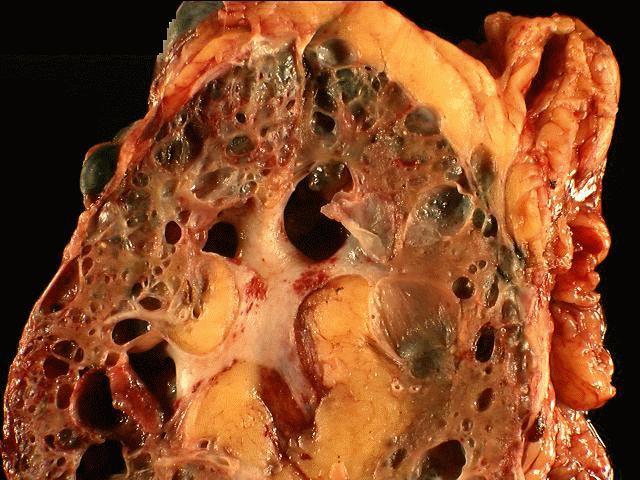
- Markedly enlarged kidneys with bosselated surface (up to 8 kg) composed of subcapsular cysts up to 4 cm
- Cysts contain clear to brown fluid
Gross images
Contributed by Debra L. Zynger, M.D.
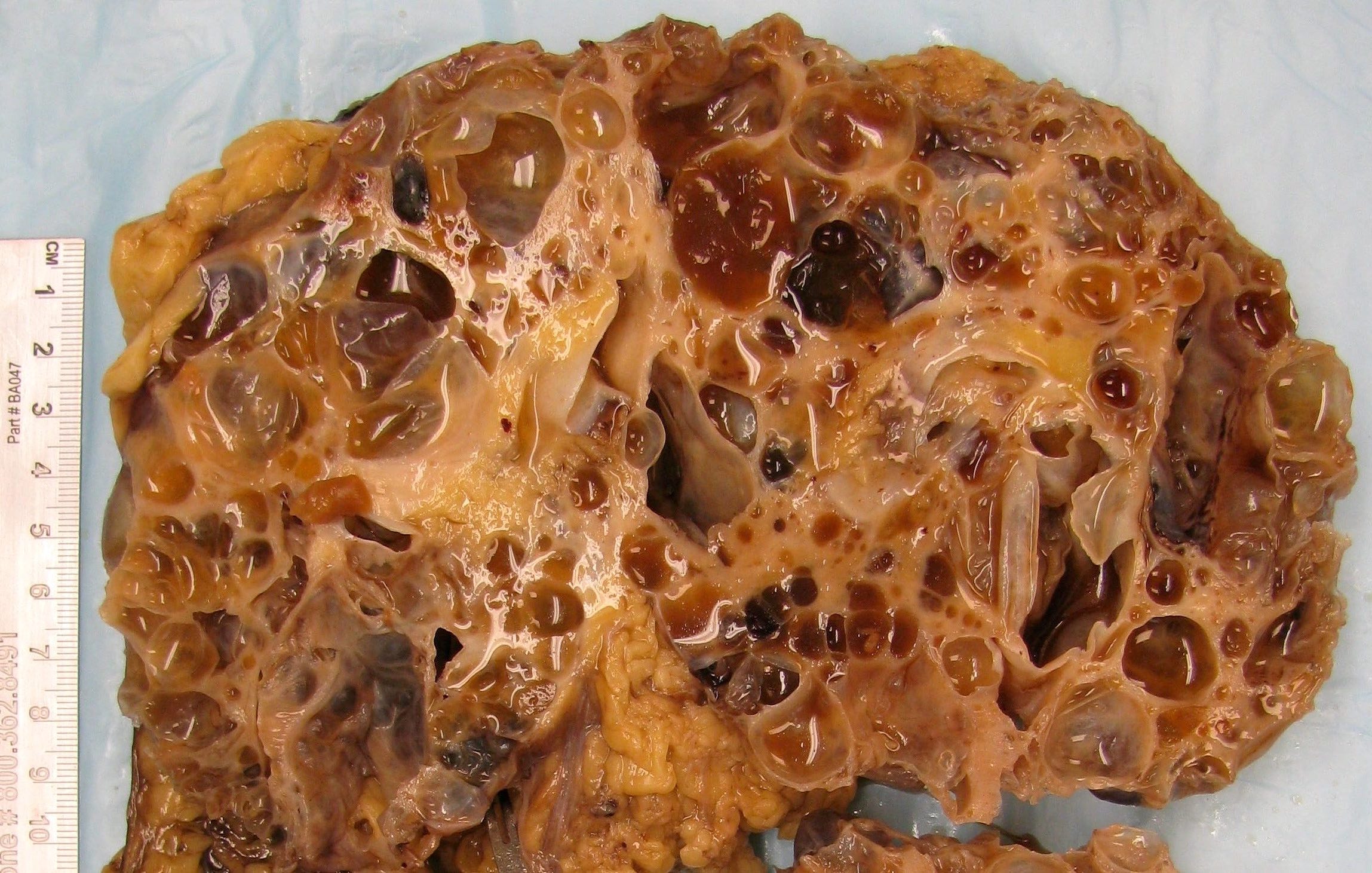
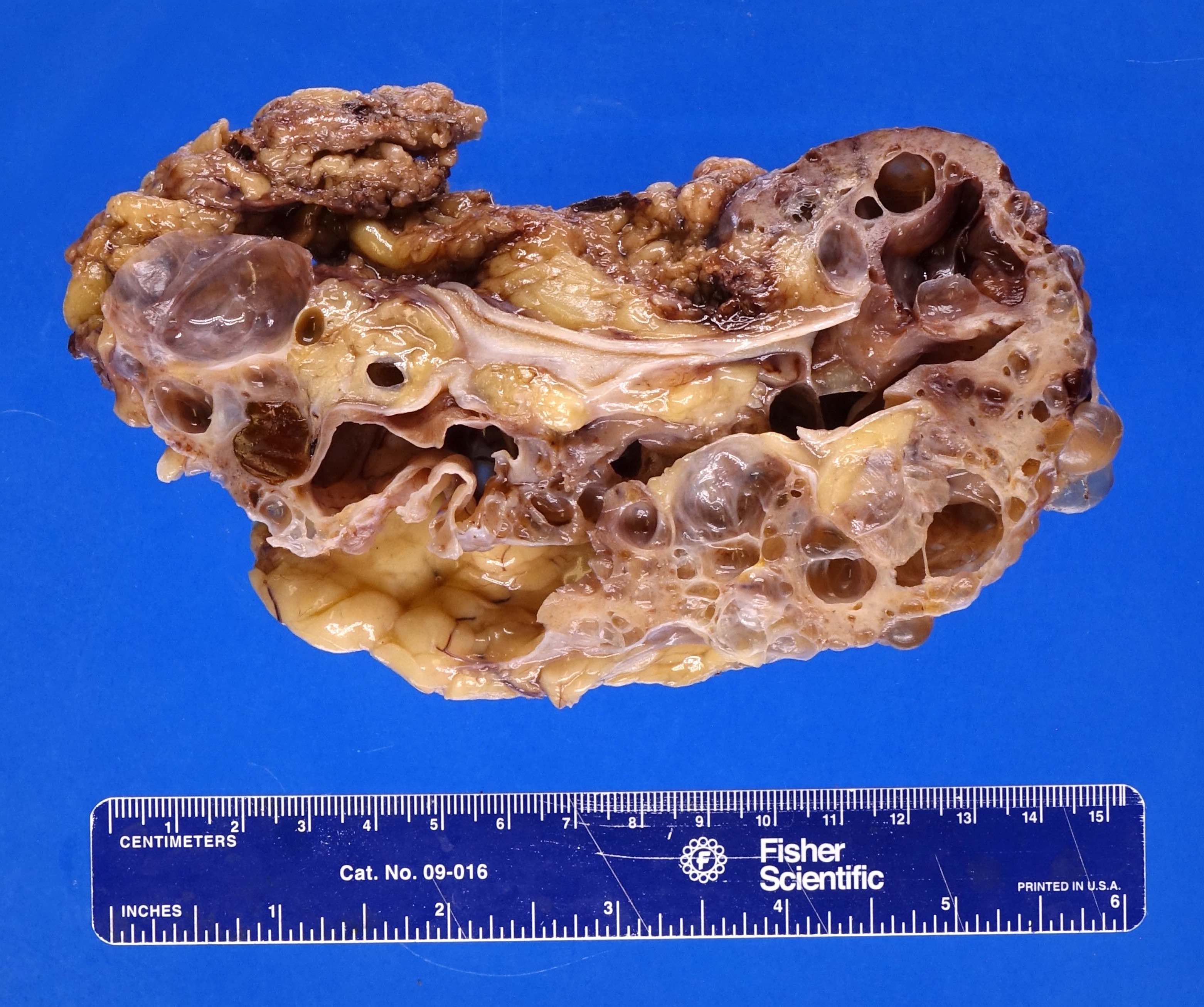
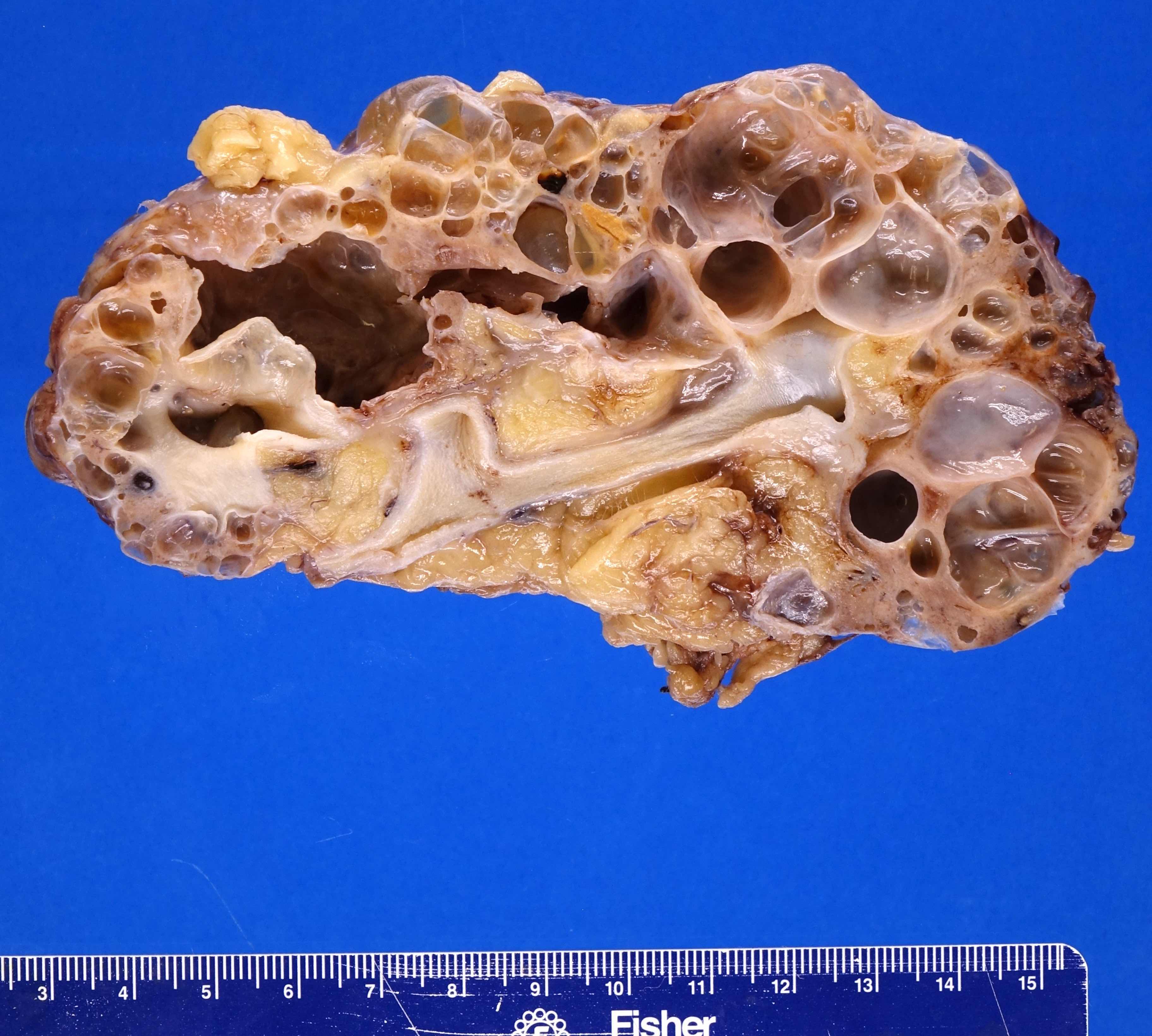
Numerous cysts of varying sizes
Images hosted on other servers:


Enlarged kidney with variably sized cysts
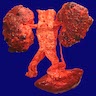
With transplanted kidney

Compared to normal kidney
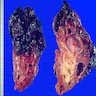
Hemorrhagic infarct with rupture
Microscopic (histologic) description
- Saccular expansions or diverticula of all portions of renal tubule and glomerular capsule that later become disconnected and filled with fluid
- Cysts are lined by cuboidal or flattened epithelium, may have papillary projections or polyps
- Functional nephrons exist between cysts with areas of global sclerosis, tubular atrophy, interstitial fibrosis and chronic inflammation
- Infants may show primarily cystic dilatation of Bowman's space
- 20% have renal adenomas
Microscopic (histologic) images
Images hosted on other servers:


Cysts of varying sizes; glomerular cyst present at birth (rare)
Molecular / cytogenetics description
- PKD1 gene on 16p13.3 (altered in 85% - 90% of cases) produces polycystin 1; function unknown (OMIM 601313)
- PKD2 gene on 4q13-23 (altered in 10% of cases) produces polycystin 2; later onset and development of chronic renal failure than PKD1 (OMIM 173910)
- PKD3 gene: minority of cases, gene unmapped (OMIM 600666)
- 10% lack a family history and are considered new mutations
Additional references
