

Kidney nontumor / medical renal
Glomerular disease
Inherited glomerular disease
Alport syndrome and thin basement membrane lesion
Author: Alexei Mikhailov, M.D., Ph.D.
Editorial Board Members: Jonathan E. Zuckerman, M.D., Ph.D., Nicole K. Andeen, M.D.

Last author update: 17 January 2024
Last staff update: 17 January 2024
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Alport syndrome
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Immunofluorescence images | Electron microscopy description | Electron microscopy images | Genetics | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Mikhailov A. Alport syndrome and thin basement membrane lesion. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyalport.html. Accessed April 19th, 2024.
Definition / general
- Alport syndrome (AS) is a progressive renal disease associated with mutations in the genes COL4A3, COL4A4 and COL4A5, affecting the synthesis, assembly, deposition or function of the collagen IV α345 network of the basement membranes; this syndrome has a range of clinical findings from microscopic hematuria to end stage renal disease (ESRD), sensorineural hearing loss and ocular abnormalities
- Thin basement membrane disease is a diffuse and global thinning of the glomerular basement membrane (GBM) not associated with extrarenal features
Essential features
- Alport syndrome and thin basement membrane disease are genetically heterogeneous conditions associated with abnormalities in glomerular basement membrane collagen
- Alport syndrome and thin basement membrane disease present as structural abnormalities in the glomerular basement membrane
- Alport syndrome and thin basement membrane disease present initially as hematuria
- Alport syndrome often progresses to deterioration of the organ function
- Thin basement membrane disease is largely considered to be a nonprogressive condition
- Some patients with thin basement membrane disease display proteinuria and renal impairment; they may in fact have Alport syndrome but this requires further study (Kidney Int 2004;66:909)
Terminology
- Alport syndrome, historical term: hereditary nephritis
- Thin basement membrane disease, alternative terms: benign familial hematuria, familial benign essential hematuria, thin membrane nephropathy, thin basement membrane disease, thin basement membrane syndrome, hereditary hematuria
ICD coding
Epidemiology
- Exact prevalence of Alport syndrome is unknown, although it is frequently reported as ranging from 1 in 5,000 in the United States to 1 in 50,000 in Europe; predicted pathogenic COL4A3 - COL4A5 variants show a much higher frequency, suggesting that other genetic and environmental factors mitigate the clinical manifestation of the disease (Kidney Med 2023;5:100631, J Am Soc Nephrol 2021;32:2273)
- X linked Alport syndrome accounts for up to 80% of Alport syndrome cases
- Autosomal recessive Alport syndrome is estimated at 15% of cases
- Autosomal dominant Alport syndrome accounts for 20% of cases (Clin Kidney J 2020;13:1025)
- Digenic inheritance is rare
- Thin basement membrane disease is the most common cause of persistent hematuria in children and affects at least 1% of the population
Sites
- Kidney (glomerulus), eye (cornea, lens capsule and retina), ear (cochlea)
Pathophysiology
- Glomerular basement membrane contains 5 isoforms of collagen IV (α1, α2, α3, α4, α5) encoded by genes COL4A1, COL4A2, COL4A3, COL4A4 and COL4A5, providing tensile strength to the glomerular basement membrane and serving as scaffold for assembly of other proteins
- Immature glomerular basement membrane contains isoforms α1 and α2 only, produced by both podocytes and endothelial cells
- At the capillary loop stage of development, the α1α1α2 network is replaced by the α3α4α5 network
- The latter provides greater strength to resist the intracapillary hemodynamic pressure increasing with age
- Collagen IV α3, α4 and α5 isoforms are produced by the podocytes only and form the thicker subepithelial network, whereas collagen IV α1 and α2 forms the subendothelial network (Elife 2013;2:e01149)
- Absence or functional compromise of one of the components of the α3α4α5 network results in reduced ability of the glomerular basement membrane to counter the hemodynamic intracapillary pressure, stressing the filtration barrier and leading to an adaptive reaction of the podocytes, endothelium and mesangial cells; compensatory expansion of the subendothelial α1α2 network provides temporary support (Matrix Biol 2017;57:45)
- An early pathogenetic event in Alport syndrome glomeruli is elevated expression of endothelin 1 by the endothelial cells, leading to subsequent activation of the mesangial cells, deposition of the mesangial matrix proteins in the GBM causing a podocyte reaction with production of proinflammatory cytokines and matrix metalloproteinases (Front Med (Lausanne) 2022;9:846152)
- Slowly progressing structural and biochemical changes in the glomerular basement membrane lead to deterioration of the podocyte architecture and eventually focal and segmental glomerulosclerosis (FSGS)
- Collagen IV is also a major component of the cochlea, cornea, lens capsule and retinal basement membranes, accounting for the sensorineural hearing loss and ocular abnormalities
Etiology
Clinical features
- A classification system for Alport syndrome and related disorders has been proposed by the Alport Syndrome Working Group and is based on the mode of inheritance and genetic state (see Genetics) (Kidney Int 2018;93:1045)
- X linked Alport syndrome: in addition to the kidney symptoms, there is bilateral sensorineural hearing loss which may be detected in boys during the first decade
- Anterior lenticonus develops progressively in male patients and is very rare in female patients
- Also specific to Alport syndrome is the progressive appearance of asymptomatic, perimacular, yellowish flecks
- Median age to reach end stage renal disease in men is 25 years with large deletion / protein truncating variants, 28 years with splice site variants and 37 years with missense variants (J Am Soc Nephrol 2010;21:876)
- Women with X linked Alport syndrome have a variable phenotype due to X linked inactivation
- 12% of heterozygous X linked women with Alport syndrome reached end stage renal disease by age 40 and 30 - 40% by age 60 (J Am Soc Nephrol 2003;14:2603)
- Hypertension is usually seen with end stage renal disease
- Autosomal recessive Alport syndrome: clinical symptoms are similar to those seen in X linked Alport syndrome; progression to end stage renal disease usually occurs by the age of 30 years in both men and women
- Autosomal dominant Alport syndrome: less severe phenotype than in X linked Alport syndrome, similar presentation in men and women, variable progression to end stage renal disease after the age of 50 years; hearing loss and ocular involvement are rare
- It has been proposed that thin basement membrane disease be classified as a form of collagen IV related renal disease along with Alport syndrome; this approach has not been generally adopted (Kidney Int 2018;93:1045)
- Thin basement membrane disease is suspected clinically if there is persistent microhematuria in the absence of proteinuria, impaired renal function and extrarenal symptoms of Alport syndrome
Diagnosis
- Genetic testing is the most sensitive method for detection of COL4A3 - COL4A5 variants; not only should individuals with the typical features of Alport syndrome be tested but also those with proteinuria, FSGS, familial IgA glomerulonephritis or with end stage renal disease without an obvious cause (Eur J Hum Genet 2021;29:1186)
- Given the high prevalence of collagen IV variants in the population (see Epidemiology), it is important that clinical diagnostic features of Alport syndrome are documented as well as histologic features by light, immunofluorescence and electron microscopy
- Since the differential diagnosis for characteristic Alport syndrome ultrastructural lesions is wide (see Differential diagnosis), genetic testing is absolutely necessary for the correct diagnosis
- Anterior lenticonus is diagnostic of Alport syndrome
- Ultrastructural finding of thin glomerular basement membrane does not exclude Alport syndrome nor does the normal renal expression of collagen IV or detection of the COL4A3 / COL4A4 variants in the heterozygous state, as the same may be observed in Alport syndrome; regular follow up examination and reevaluation of patients with thin basement membrane disease is recommended
Laboratory
- In X linked Alport syndrome, microscopic or macroscopic hematuria is often the first sign and can occur as early as infancy (Kidney Med 2023;5:100631)
- Hematuria is the cardinal symptom; it is observed in all male patients and nearly all female patients
- Albuminuria occurs later, followed by decrease in glomerular filtration rate
- Autosomal recessive Alport syndrome: laboratory symptoms similar to those seen in X linked disease (Clin Exp Nephrol 2019;23:158)
- Autosomal dominant Alport syndrome: less severe symptoms (Am J Kidney Dis 2021;78:560)
- Thin basement membrane disease: isolated microhematuria and infrequent episodes of macroscopic hematuria, with no proteinuria and normal kidney function (Nephron 2023;147:383)
Prognostic factors
- X linked hemizygous Alport syndrome has 100% progression to end stage renal disease; timing of progression depends on genotype
- X linked heterozygous Alport syndrome has up to 25% progression to end stage renal disease; risk factors are gross hematuria, sensorineural hearing loss, proteinuria, glomerular basement membrane thickening or lamellation
- Autosomal recessive Alport syndrome has 100% progression to end stage renal disease; timing depends on genotype
- Autosomal dominant Alport syndrome has 20% progression to end stage renal disease among those with risk factors for progression and < 1% in the absence of risk factors; risk factors are proteinuria, FSGS, glomerular basement membrane thickening and lamellation, sensorineural hearing loss or evidence of progression in patient or family
- Digenic Alport syndrome exhibits COL4A3 and COL4A4 mutations in trans with up to 100% risk of end stage renal disease (Kidney Int 2018;93:1045)
- Digenic Alport syndrome exhibits COL4A3 and COL4A4 mutations in cis with up to 20% risk of end stage renal disease (Kidney Int 2018;93:1045)
- Prognosis for thin basement membrane disease has been considered excellent for many years but development of end stage renal disease at an advanced age is now considered possible (Nephron 2023;147:383)
Case reports
- 16 year old boy with proteinuria for 15 years and a 31 year old man with hearing loss and end stage renal disease, both with positive family history of renal disease (Biomed Res Int 2021;2021:6664973)
- 20 year old man with Alport leiomyomatosis syndrome (diffuse esophageal leiomyomatosis) (Gen Thorac Cardiovasc Surg 2020;68:199)
- 20 year old man with both antiglomerular basement membrane disease and thin basement membrane disease (Medicine (Baltimore) 2021;100:e26095)
- 50 year old woman presenting with microhematuria and proteinuria (BMC Nephrol 2023;24:97)
Treatment
- Current clinical practice recommendations (Kidney Int 2018;93:1045)
- Observation for hematuria with normal urine albumin / creatinine ratio
- Consider ACE inhibitors for hematuria and microalbuminuria
- ACE inhibitors for hematuria and overt proteinuria
- Angiotensin receptor blockers, aldosterone antagonists, calcium channel blockers or diuretics for progressive proteinuria despite ACE inhibitors
- Potential therapies currently undergoing clinical trials: sodium glucose cotransporter 2 (SGLT2) inhibitors, aminoglycoside analogs (prevent premature peptide termination), endothelin type A antagonists, lipid modifying drugs, gene replacement therapy
Microscopic (histologic) description
- Early in the course of Alport syndrome
- Glomeruli appear normal (Glomerular Dis 2021;1:135)
- In young children, features of glomerular immaturity may be seen (small glomeruli with indistinct capillaries, circumference of the capillary segments lined with cuboidal epithelial cells); such features have no association with Alport syndrome
- Tubulointerstitial compartment appears normal (Glomerular Dis 2021;1:135)
- Glomeruli appear normal (Glomerular Dis 2021;1:135)
- With progression of the disease
- Segmental thickening of the glomerular capillary walls (more appreciable on the silver stain) and mesangial expansion become visible
- Red blood cell casts may be seen in the tubules
- Foci of interstitial fibrosis and tubular atrophy develop (Glomerular Dis 2021;1:135)
- Lipid laden interstitial foam cells are considered a marker of Alport syndrome; they appear as rows or clusters in the cortex or at the corticomedullary junction (Glomerular Dis 2021;1:135)
- Eventually, with marked thickening of the glomerular basement membranes and expansion of the mesangium, segmental tuft scarring develops in an increasing number of glomeruli; the end point is global glomerulosclerosis
- In patients with thin basement membrane disease, the renal tissue is usually normal by light microscopy (Kidney Int 2003;64:1169)
- Mesangial expansion and mild mesangial proliferation may occur
- Red blood cells may be seen within tubules
Microscopic (histologic) images
Contributed by Alexei Mikhailov, M.D., Ph.D.
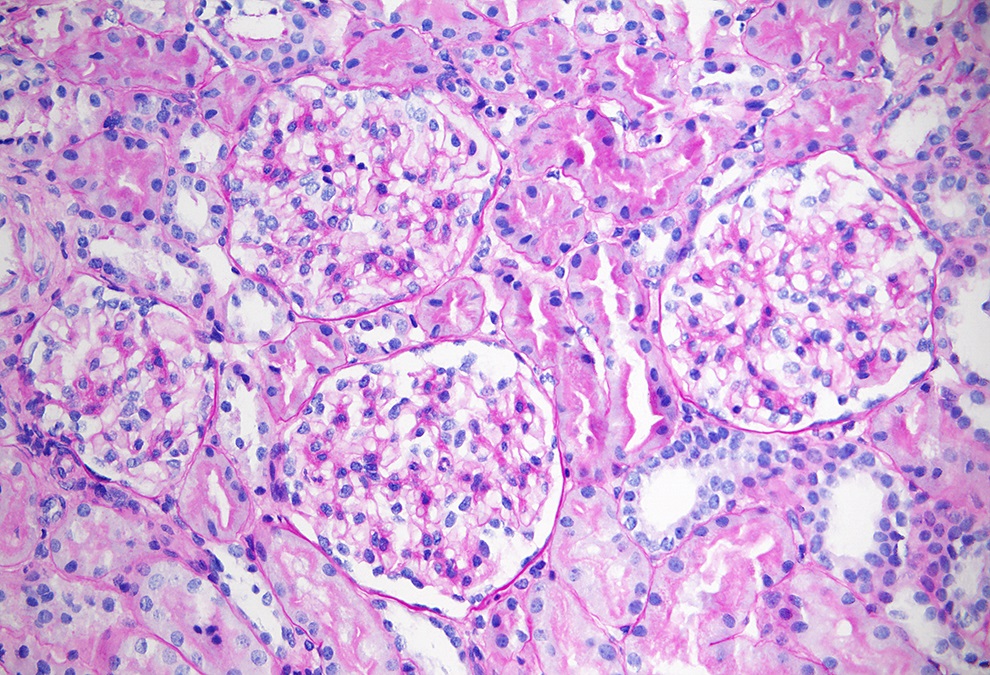
Early Alport syndrome
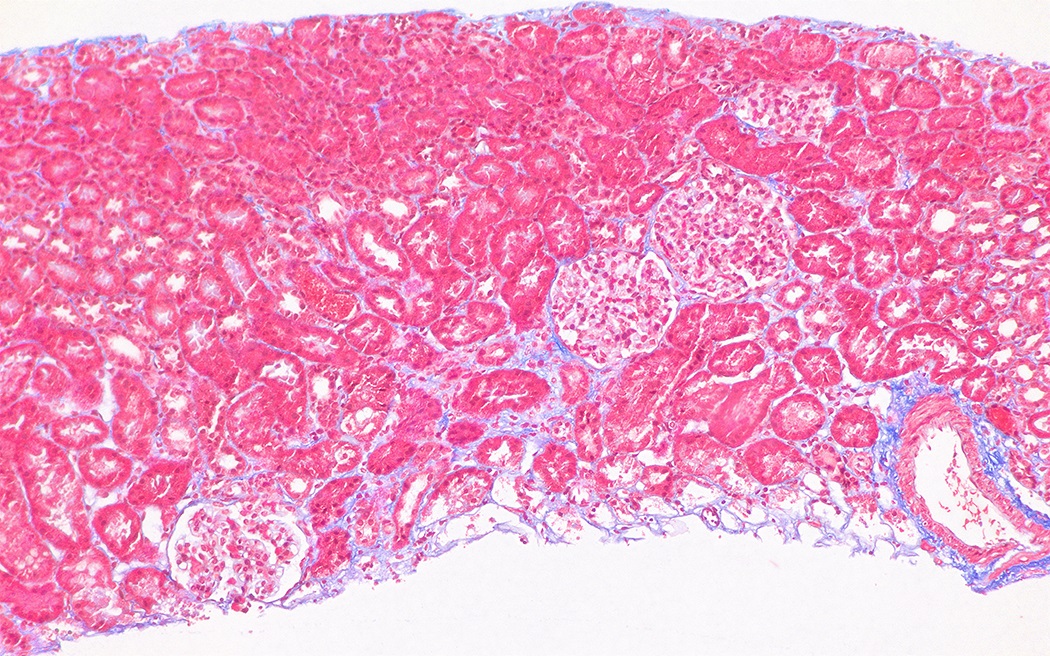
Early chronic changes
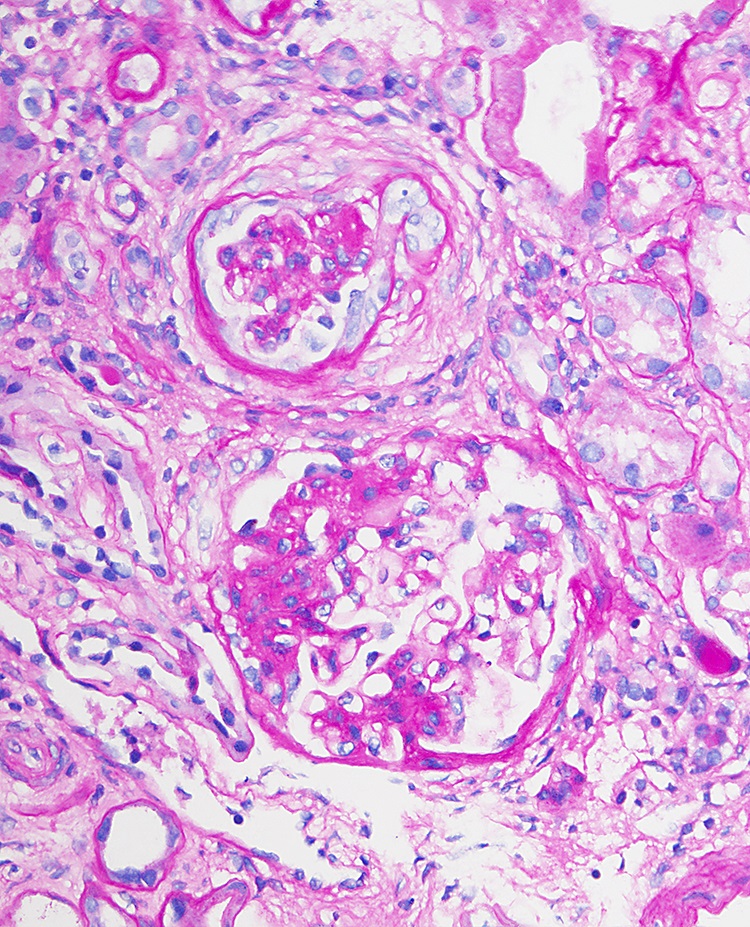
Focal segmental glomerular scarring
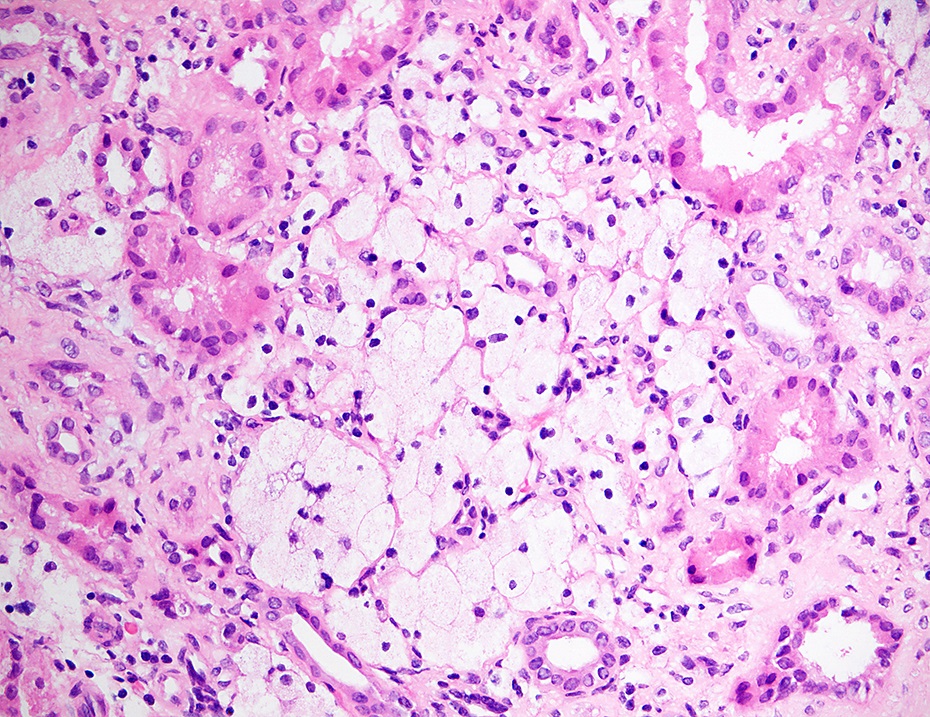
Foam cells, Alport syndrome
Immunofluorescence description
- Immunofluorescence for the antibodies and complement is usually negative, although with progression of Alport syndrome, faint deposits of IgM, C3 or C1q may be seen
- Biopsies from most male patients with X linked Alport syndrome are completely negative for collagen IV α5 isoform in the glomerular capillary walls and the Bowman capsule (Glomerular Dis 2021;1:135)
- Female patients with X linked Alport syndrome show a mosaic pattern of collagen IV α5 expression in the glomerular capillary segments (Glomerular Dis 2021;1:135)
- In autosomal recessive Alport syndrome, the glomerular capillary walls are negative for α3, α4 and α5 staining, whereas the Bowman capsule is positive for α5 (Glomerular Dis 2021;1:135)
- Biopsies from patients with thin basement membrane disease show normal distribution of the collagen IV isoforms in the glomeruli (Arch Pathol Lab Med 2001;125:631)
Immunofluorescence images
Contributed by Alexei Mikhailov, M.D., Ph.D., Volker Nickeleit, M.D. and Sharan Singh, M.D.
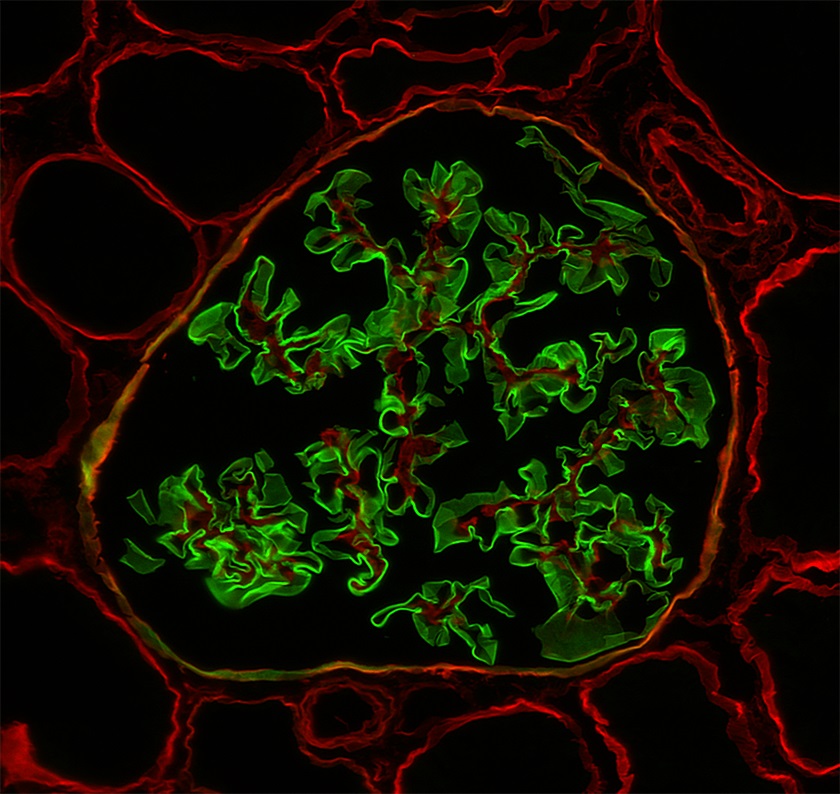
Normal collagen 1 and 5
Electron microscopy description
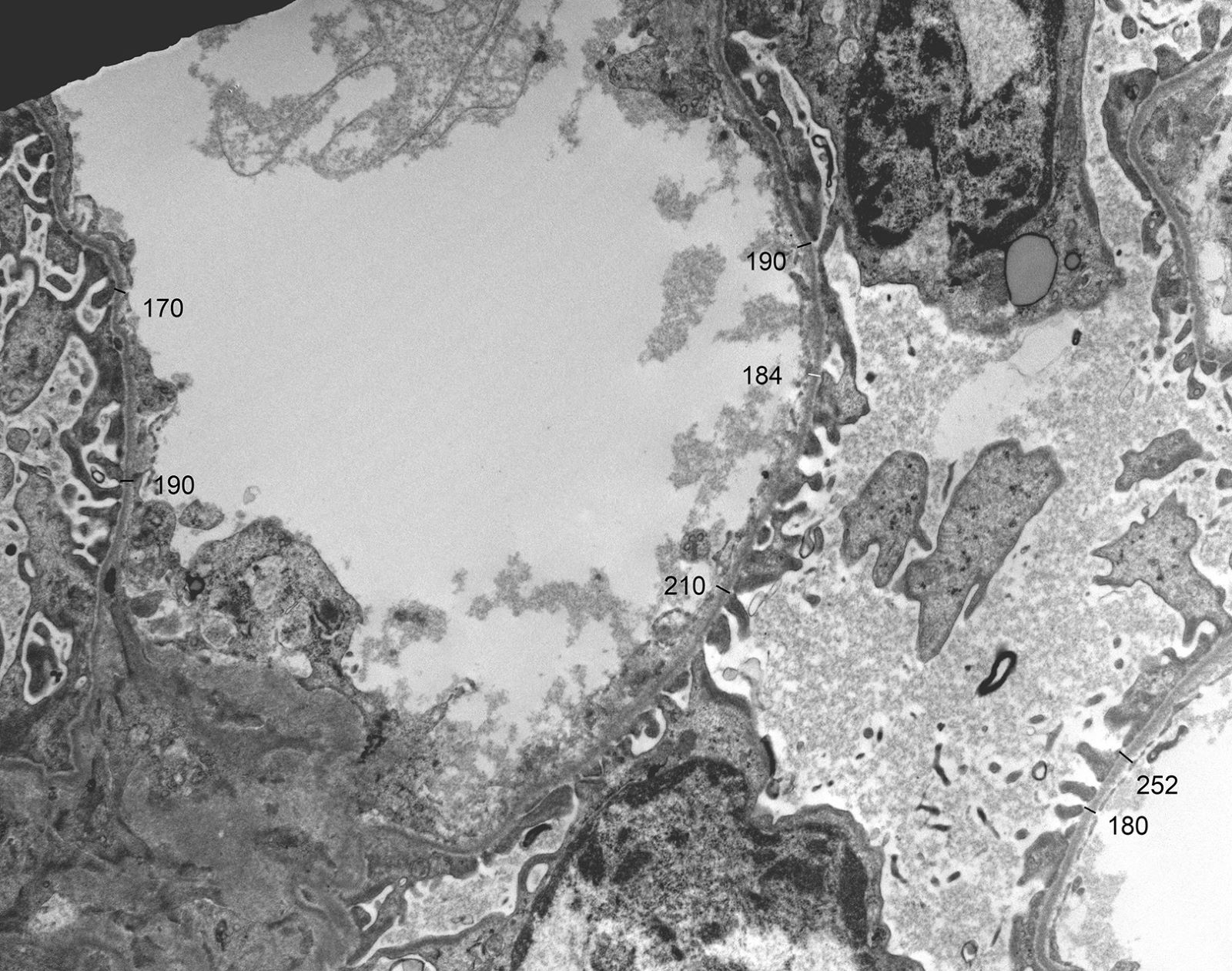
- In younger Alport syndrome patients, a thin glomerular basement membrane may be the only finding (Glomerular Dis 2021;1:135)
- Typical feature in older children is irregular glomerular basement membrane appearance with close alternation of extremely thin and thick split segments
- Breaking of the thin glomerular basement membrane segments is the histological basis of hematuria in such patients; an image of a red blood cell traversing the glomerular basement membrane is provided in N Engl J Med 2007;357:2687
- Characteristic lesion of Alport syndrome seen in many but not all patients consists of marked irregular thickening of the glomerular basement membrane with splitting of the lamina densa into a basket weave pattern, lucent zones containing small electron dense granules (bread crumbs) and sometimes small electron dense deposits
- Fragmentation of the lamina densa may give a laminated appearance to the glomerular basement membrane
- Inner and outer contours are corrugated and the overlying podocytes are hypertrophied and often show foot process effacement (Glomerular Dis 2021;1:135)
- Severity and extent of the Alport syndrome lesions vary very widely depending on the type of mutation and other factors; marked variability in the glomerular basement membrane width within the glomerulus of a patient with persistent hematuria should raise suspicion of Alport syndrome (GeneReviews: Alport Syndrome [Accessed 25 August 2023])
- Mesangium is initially normal but with time, an increase in the mesangial matrix is observed, with focal mesangial hypercellularity and mesangial expansion; the mesangial material appears heterogeneous and contains inclusions (Ren Fail 2000;22:751)
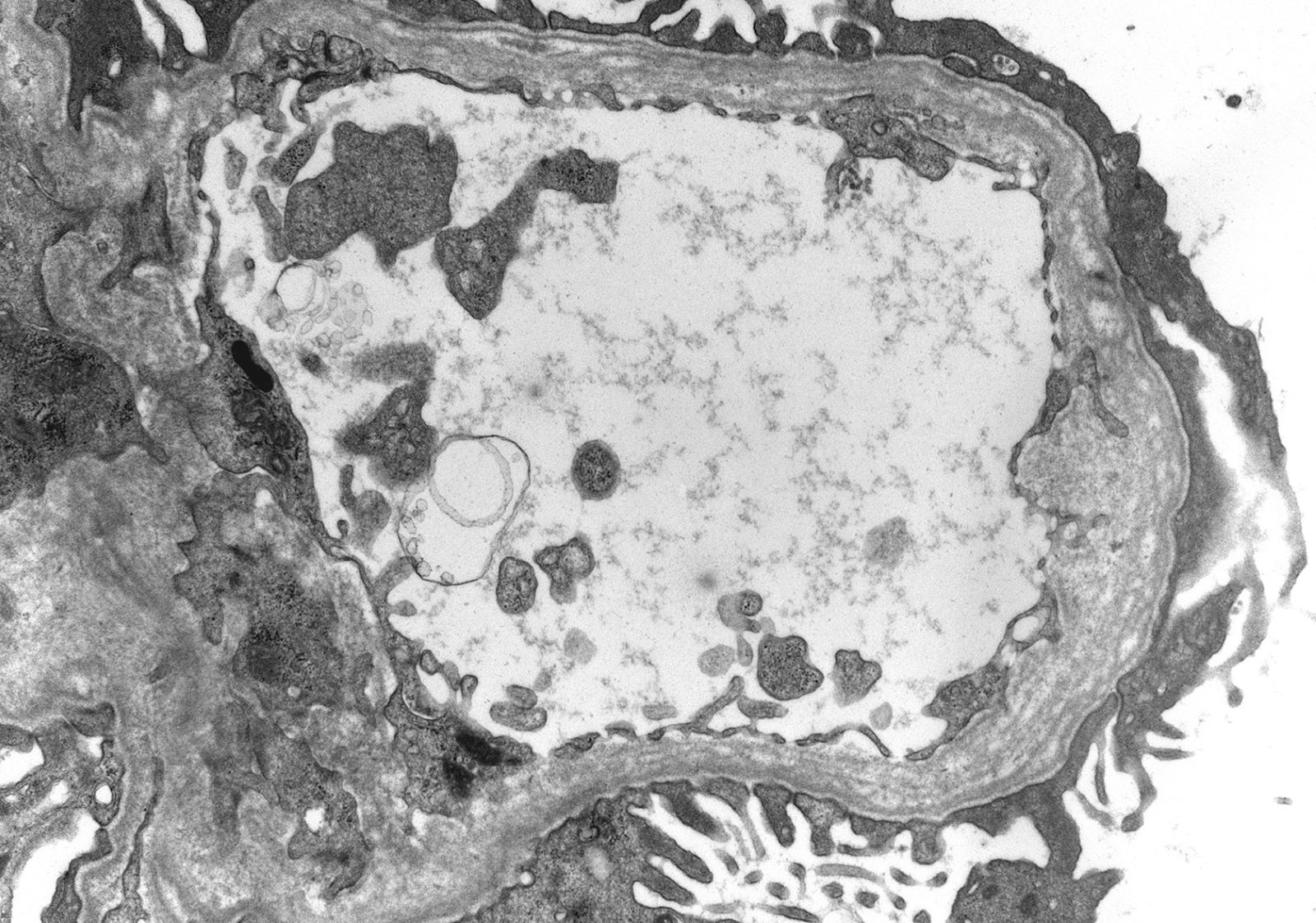
- Thin basement membrane disease manifests ultrastructurally as global and diffuse thinning of the glomerular basement membrane (Kidney Int 1996;50:1445)
- Method of glomerular basement membrane measurement and estimated normal ranges of glomerular basement membrane thickness in children are indicated in Arch Pathol Lab Med 2009;133:224 and Glomerular Dis 2021;1:135
- Total of 30 measurements are made on 10 randomly selected capillaries; the average and at least 50% of the measurements should be below the lower limit of the normal range for the patient's age and sex
- Alport syndrome changes involving > 5% of the glomerular basement membrane length examined as well as evidence of Alport syndrome on immunofluorescence excludes thin basement membrane disease
- Glomerular basement membrane thickness in children increases linearly between 1 and 9 years of age and then plateaus (Arch Pathol Lab Med 2009;133:224)
- Thin basement membrane disease cannot be reliably diagnosed in deparaffinized formalin fixed tissue (Matrix Biol 2018;71:250)
- Measurements of glomerular basement membranes can be reliably interpreted from reclaimed frozen tissue if routine processed EM tissue is unavailable (Kidney Med 2022;4:100562)
Electron microscopy images
Contributed by Alexei Mikhailov, M.D., Ph.D.
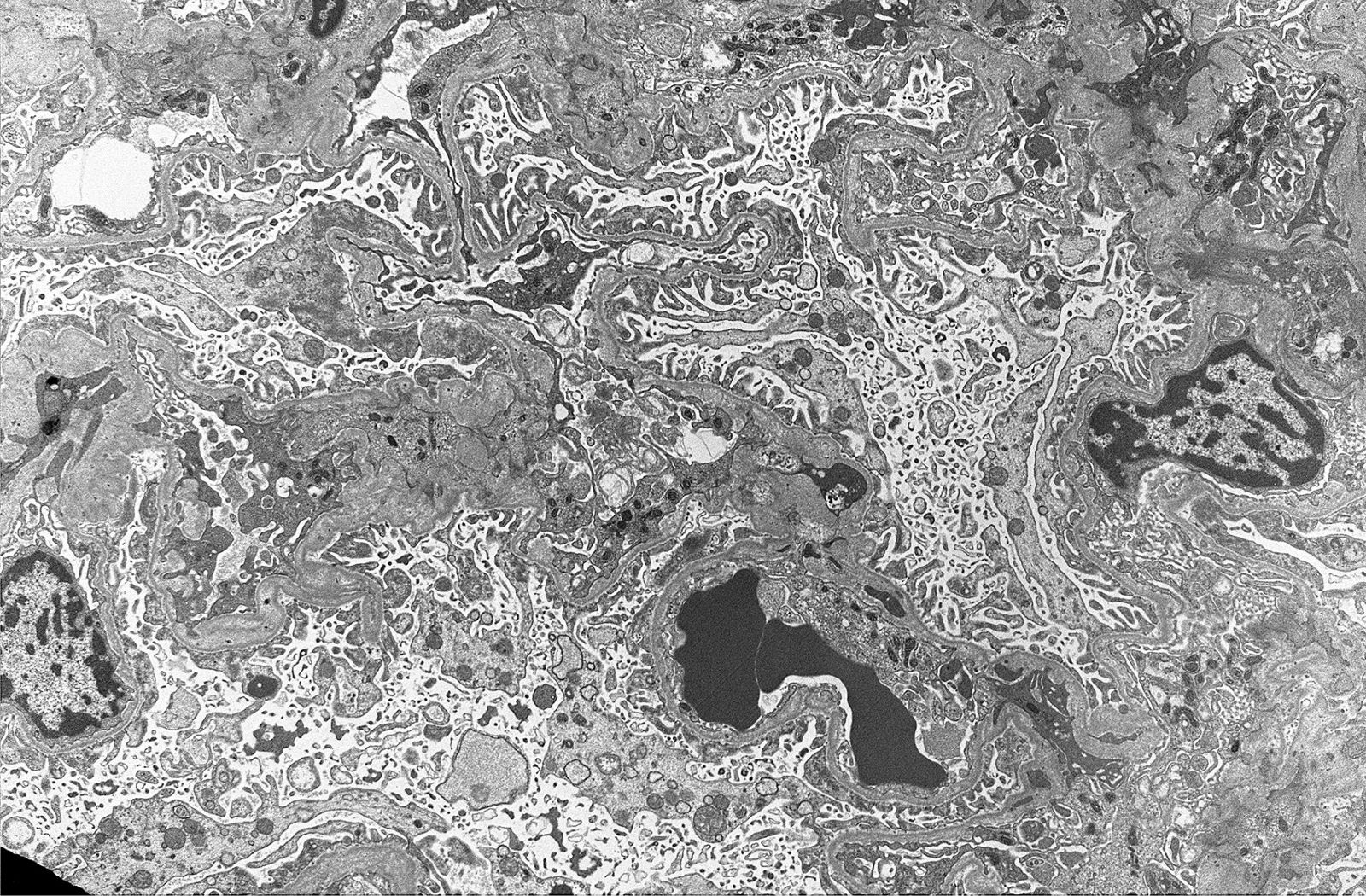
Microvillus transformation
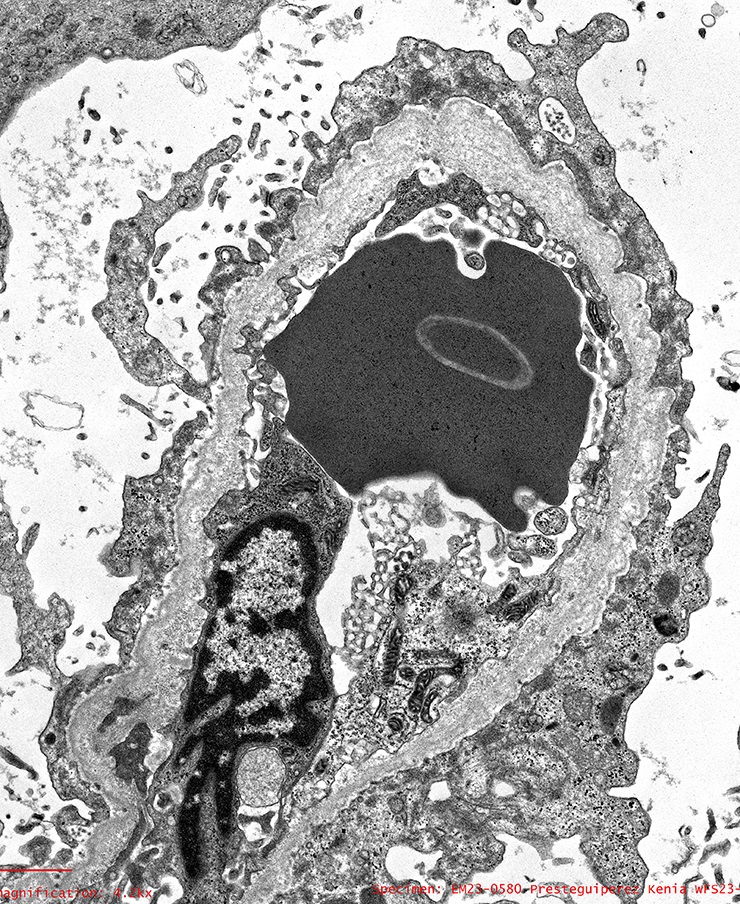
Characteristic changes
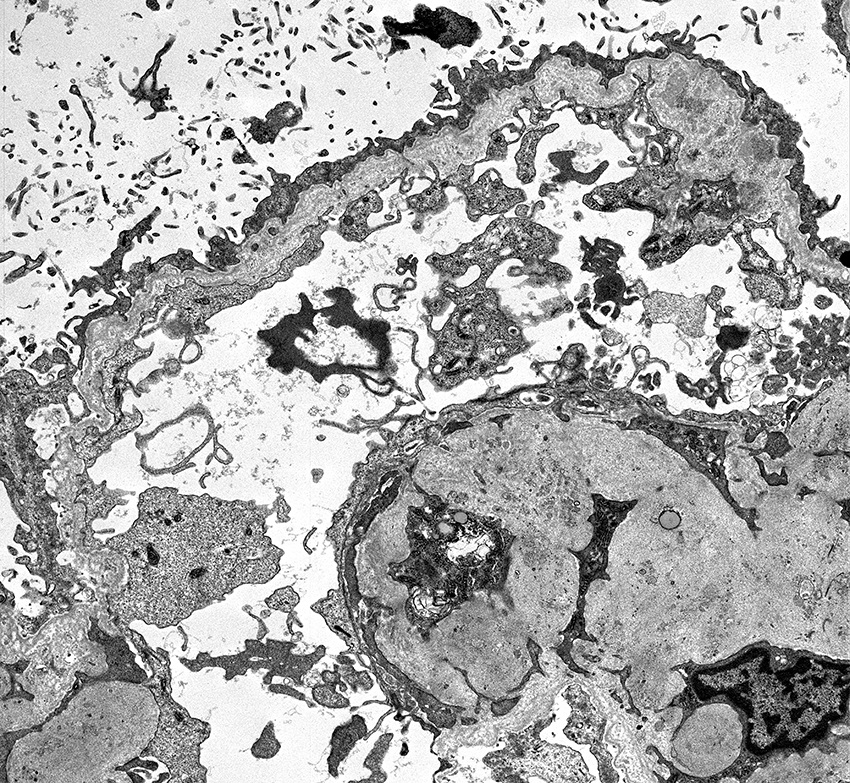
More advanced changes
Thin basement membrane disease
Alport-like changes
Genetics
- COL4A3, COL4A4 and COL5A5 are the most frequent causative genes of monogenic chronic kidney disease after the polycystic kidney disease genes PKD1 and PKD2 (N Engl J Med 2019;380:142)
- COL4A3 and COL4A4 genes reside on chromosome 2, while COL4A5 gene I is located on the X chromosome
- There are 4 modes of inheritance of Alport syndrome
- X linked Alport syndrome: pathogenic variants in the COL4A5 gene on the X chromosome, genetic state - hemizygous (male subjects) and heterozygous (female subjects)
- Autosomal recessive: pathogenic variants in COL4A3 / COL4A4 genes
- Autosomal dominant: pathogenic variants in COL4A3 / COL4A4 genes due to heterozygous COL4A3 and COL4A4 mutations
- Digenic: combination of 2 mutations in different genes, typically a pathogenic variant in COL4A3 and COL4A4 (Clin Genet 2017;92:34)
- ClinVar database currently documents 680 pathogenic variants of COL4A5, 239 pathogenic variants of COL4A4 and 236 pathogenic variants of COL4A3 (NIH: ClinVar - COL4A5[gene] [Accessed 25 August 2023], NIH: ClinVar - COL4A4[gene] [Accessed 25 August 2023], NIH: ClinVar - COl4A3[gene] [Accessed 25 August 2023])
- Thin basement membrane disease shows autosomal dominant inheritance in 50% of cases with heterozygous COL4A3 or COL4A3 mutations (carrier state for autosomal recessive Alport syndrome) (Pediatr Nephrol 2009;24:877)
- Other genetic causes such as COL4A1 variants have also been proposed (Nephrol Dial Transplant 2016;31:1758)
- Mutations are usually different in each family and there is no hotspot; some sources report that the mutation detection rate in thin basement membrane disease is low (Kidney Int 2003;64:1169)
Sample pathology report
- Kidney, biopsy:
- Alport syndrome (see comment)
- Focal global glomerulosclerosis (1/12)
- Mild focal interstitial fibrosis and tubular atrophy
- Comment: Genetic study is recommended.
- Clinical history: The patient is an 8 year old boy with a history of microscopic hematuria and proteinuria. Family history is positive for mother developing end stage renal disease at the age of 40 years. Laboratory data: serum creatinine 0.6 mg/dL, BUN 15 mg/dL, serum albumin 4.4 mg/dL, UPC 900 mg/g creatinine, C3 124 mg/dL, C4 25 mg/dL, ANA negative, anti-DNA antibody and ANCA screen negative.
- Light microscopy: The biopsy material consists of tissue from the renal cortex. The tissue is sampled at 10 levels of section with H&E, PAS, trichrome and PAMS stains. Per level of section up to 12 glomeruli are available, of which 1 is globally sclerosed. The nonsclerosed glomeruli show patent capillary lumens, single contoured capillary walls and no segments of sclerosis, adhesion or crescents. A few perihilar capillaries contain hyaline. Tubules show mostly intact epithelium with preserved brush borders. There is mild focal interstitial fibrosis and tubular atrophy; scattered lymphocytes are present in the interstitium. Some distal tubules contain proteinaceous casts. Interlobular arteries of the medium caliber are present and do not show diagnostic arteriosclerotic changes; there is no significant arteriolar hyalinosis.
- Immunofluorescence microscopy: 9 nonsclerosed glomeruli are available for evaluation by immunofluorescence microscopy. No diagnostic immunofluorescence signal is detected with antibodies to IgG-, IgA-, IgM-, C3-, C1q-, kappa light chain-, lambda light chain-. Immunofluorescence staining for the collagen type IV α2 and α5 isoforms was performed at our request and showed normal staining for the α2 isoform with enhanced staining at the glomerular capillary walls and loss of staining for the α5 isoform.
- Electron microscopy: 2 nonsclerosed glomeruli are available for evaluation on semithin sections and do not show significant diagnostic abnormalities. There is no significant interstitial fibrosis and tubular atrophy. Ultrastructural examination of 2 glomeruli shows alternating segments of thick and thin glomerular basement membrane segments with irregular internal and external contours. Thicker glomerular basement membrane segments display lamina densa splitting (basket weave), microgranular inclusions (bread crumbs), large electron lucent areas and contain a few faded electron dense deposits. There is segmental variable mild to marked effacement of the podocyte foot processes and pronounced podocyte microvillus transformation. The mesangial matrix demonstrates mild segmental expansion. Tubular basement membranes are unremarkable.
- Alport syndrome (see comment)
Differential diagnosis
- Alport syndrome associated conditions
- AMME syndrome (Alport syndrome, midface hypoplasia, intellectual deficit and elliptocytosis) (J Med Genet 2017;54:269)
- Associated with contiguous gene deletions in the chromosomal region Xq22.3-Xq23 including the entire COL4A5 gene and its adjacent genes
- Abovementioned dysmorphic, developmental and hematologic abnormalities form the basis for the differential diagnosis
- Diffuse and global Alport-like changes are seen in
- Focal and segmental glomerulosclerosis (FSGS) associated with variants in nonmuscle myosin myosin1e (Pediatr Nephrol 2023;38:439):
- These patients may become proteinuric early in life, with early onset of FSGS and early progression to renal failure
- Fechtner and Epstein syndrome associated with mutations in myosin heavy chain 9 gene (MYH9) (Hum Genet 2002;110:182):
- Platelet macrocytosis, thrombocytopenia, sensorineural hearing loss, cataracts, nephritis and characteristic MYH9 aggregates in the granulocyte cytoplasm (Pediatr Nephrol 2023;38:439)
- Steroid resistant nephrotic syndrome:
- Caused by activation of a Rac1 GTPase as a result of mutation in a regulatory protein ARHGAP24 manifesting ultrastructurally as subepithelial extensions of the glomerular basement membrane and localized areas of glomerular basement membrane lamellation (The Open Urology & Nephrology Journal 2016;9:88)
- Renal coloboma syndrome due to mutated podocyte PAX2 (Pediatr Nephrol 2012;27(:1189)
- Presents as optic disk coloboma and renal insufficiency
- Collagen IV α5 immunostaining is normal
- Frasier syndrome with male pseudohermaphroditism and slowly progressing nephropathy associated with mutation in Wilms tumor suppressor gene WT1 (Am J Kidney Dis 2003;41:1110)
- Focal and segmental glomerulosclerosis (FSGS) associated with variants in nonmuscle myosin myosin1e (Pediatr Nephrol 2023;38:439):
- Conditions that may resemble Alport syndrome
- Nail patella syndrome (hereditary osteo-onychodysplasia) associated with mutations in LIM homeodomain transcription factor LMX1B and showing collagen fibrils in the GBM (Pflugers Arch 2017;469:927)
- Collagen type III glomerulopathy (collagenofibrotic glomerulopathy):
- Seen mostly in Asian patients and manifested by accumulation of collagen type III in the glomerular extracellular matrix (Pflugers Arch 2017;469:927)
- Pierson syndrome:
- Congenital nephrotic syndrome with massive proteinuria, ocular abnormalities and rapid progression to end stage renal disease due to variants in the β2 laminin chain (LAMB2 gene) (Matrix Biol 2018;71:250)
- HANAC syndrome:
- COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms and muscle cramps
- Renal manifestations include hematuria and bilateral large cysts (N Engl J Med 2007;357:2687)
- Renal perfusion mismatched transplants (Am J Surg Pathol 1999;23:437, Clin Nephrol 2017;88:364):
- Alport-like GBM changes may be seen
- Membranous glomerulopathy and sometimes glomerulonephritis of various etiologies:
- GBM rearrangements resembling Alport syndrome may be seen
- Thin basement membrane disease:
- Alport syndrome
- IgA nephropathy
- Segments of thin glomerular basement membrane may be seen
- AMME syndrome (Alport syndrome, midface hypoplasia, intellectual deficit and elliptocytosis) (J Med Genet 2017;54:269)
Board review style question #1
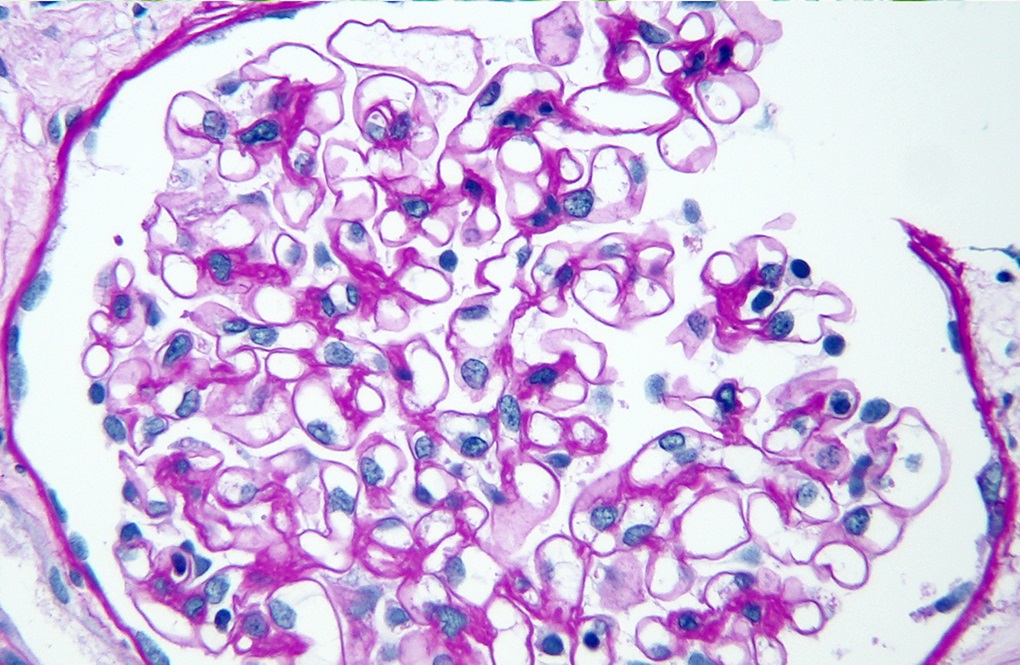
A 7 year old girl presents with hematuria and very mild proteinuria; she has not undergone laboratory testing previously. Her mother states that the men in the family develop renal failure. A biopsy is done and a representative glomerulus is displayed in the image shown above. Immunofluorescence shows segmental loss of Collagen IV α5 isoform in the glomerular capillary walls. What is the most likely diagnosis?
- Alport syndrome, autosomal recessive
- Alport syndrome, X linked heterozygous
- Minimal change disease
- Normal kidney
- Thin basement membrane disease
Board review style answer #1
B. Alport syndrome, X linked heterozygous. Family history suggests an X linked disease. Segmental loss of Collagen IV α5 or mosaic pattern of expression is due to differential X inactivation. Answer C is incorrect because minimal change disease does not show a family history of renal failure and loss of Collagen IV α5 expression. Answer E is incorrect because thin basement membrane disease does not show loss of Collagen IV α5 expression. Answer A is incorrect because autosomal recessive Alport syndrome affects male and female family members equally and shows complete loss of the Collagen IV α345 glomerular expression. Answer D is incorrect because the patient presents with nephrological symptoms and family history and displays an abnormal distribution of glomerular Collagen IV α5.
Comment Here
Reference: Alport syndrome and thin basement membrane lesion
Comment Here
Reference: Alport syndrome and thin basement membrane lesion
Board review style question #2
A man with end stage renal disease due to X linked Alport syndrome and associated focal and segmental glomerulosclerosis (FSGS) receives a renal transplant at the age of 16 years. 6 months after the procedure, his serum creatinine increases from 0.9 g/dL to 4 mg/dL in a matter of days. The allograft kidney biopsy shows 90% global cellular crescents and segmental fibrinoid glomerular tuft necrosis with no evidence of mesangial expansion or endocapillary hypercellularity. What is the most likely diagnosis?
- Acute cell mediated rejection
- Antiglomerular basement membrane disease
- De novo immune complex glomerulonephritis
- FSGS relapse
Board review style answer #2
B. Antiglomerular basement membrane disease. The patient is a man with X linked Alport syndrome who developed end stage renal disease early; this strongly suggests that the mutation completely prevented the synthesis of collagen IV α5 isoform. Thus, the patient's glomerular basement membranes do not contain the collagen IV α345 network. The transplanted kidney glomeruli contain the normal collagen IV α345 network, which may induce the generation of antibodies. This usually occurs during the first year after transplantation. De novo anti-glomerular basement membrane nephritis develops in approximately 3% of transplanted men (UpToDate: Clinical Manifestations, Diagnosis and Treatment of Alport Syndrome (Hereditary Nephritis) [Accessed 25 August 2023]). Answer A is incorrect because glomerular crescents are not seen in rejection. Answer D is incorrect because the patient's FSGS was secondary to the Alport syndrome and thus cannot relapse after transplantation, as primary FSGS may. Answer C is incorrect because the histological presentation (lack of capillary tuft hypercellularity and mesangial reaction) is not consistent with immune complex glomerulonephritis.
Comment Here
Reference: Alport syndrome and thin basement membrane lesion
Comment Here
Reference: Alport syndrome and thin basement membrane lesion
