
Bone marrow nonneoplastic
Alterations in cellularity
Aplastic anemia (AA)
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 30 March 2020
Last staff update: 18 September 2020
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Bone marrow[TI] aplastic anemia[TI] pathology full text[sb]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Immunostains | Flow cytometry description | Flow cytometry images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Sangiorgio V, Calaminici M. Aplastic anemia (AA). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrowaplasticanemia.html. Accessed March 29th, 2025.
Definition / general
- Bone marrow disorder characterized by pancytopenia due to bone marrow trilineage hypoplasia in the absence of any underlying neoplasia or reticulin fibrosis
- Mostly sporadic but can be constitutional (congenital) and occur as a consequence of different germ line genetic defects (Hematology 2015;20:433)
Essential features
- Acquired disease in the majority of cases characterized by pancytopenia due to ineffective hematopoiesis
- Bone marrow is hypocellular, with lacunar spaces replaced by fatty cells
- Residual nucleated cells include mostly lymphocytes, plasma cells and mast cells with isolated hematopoietic cells
- Sometimes mild atypia but no overt dysplasia
- No increase in blasts; normal CD34 positive count
- No overt reticulin fibrosis
- Careful distinction with other conditions characterized by bone marrow hypocellularity which can mimic aplastic anemia:
- Hypoplastic myelodysplastic syndrome / acute myeloid leukemia
- Subtle infiltration by hairy cell leukemia
- T cell large granular lymphocytic leukemia
- Other possible findings in aplastic anemia, whose detection does not affect diagnosis:
- Cytogenetic or molecular abnormalities (see Molecular / cytogenetics description)
- Small paroxysmal nocturnal hemoglobinuria (PNH) clone
Terminology
- Bone marrow failure: sometimes used as a synonym of acquired aplastic anemia
- This should be avoided as impaired hematopoiesis due to bone marrow failure can occur in different conditions (including reactive and neoplastic diseases)
- Idiopathic acquired aplastic anemia: should be used only in reference to secondary forms of unknown etiology
- Congenital cases named after the specific disease entity; most common are Fanconi anemia, dyskeratosis congenita and Schwachman-Diamond syndrome
ICD coding
- ICD-10: D61.9 - aplastic anemia, unspecified
Epidemiology
- Acquired aplastic anemia:
- Most common form of aplastic anemia
- Rare disease: estimated annual incidence of 2 - 5 cases per million (Eur J Haematol 2018;101:711)
- No sex predilection
- Bimodal age distribution: first peak at 10 - 25 years; second peak at > 60 years
- Geography: lower in Europe and North America; more common in Asia
- Constitutional:
- Rarely encountered in the clinical practice
- Except for X linked disorders, M = F
Pathophysiology
- Acquired aplastic anemia:
- Most commonly is the result of an autoimmune mediated process
- Likely antigen driven recruitment of cytotoxic and helper T lymphocytes
- Immune mediated destruction of hematopoietic stem cells
- Annihilation of hematopoietic stem cells also occurs as a consequence of both an inflammatory cytokine induced (TNFα and IFNγ) and Fas mediated apoptosis
- Most cases do not have a clear cause and are labeled idiopathic (Eur J Haematol 2018;101:711)
- Constitutional aplastic anemia:
- Results from specific loss of function germ line mutations
- Due to DNA repair defects (Fanconi anemia), telomere defects (dyskeratosis congenita), impairment of differentiation and self renewal pathways (GATA2 deficiency), ribosomopathies (Shwachman-Diamond syndrome and Diamond-Blackfan anemia) (N Engl J Med 2018;379:1643)
Etiology
- Acquired aplastic anemia (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017, Eur J Haematol 2018;101:711):
- Infectious agents:
- Hepatitis A, B, C viruses
- Other viruses: HIV, EBV, CMV, parvovirus
- Environmental exposures:
- Toxins: benzene, solvents, insecticides
- Drugs: allopurinol, carbamazepine, chloramphenicol, chloroquine, penicillamine, cotrimoxazole, diclofenac, naproxen, valproic acid; numerous other agents less commonly reported
- Others conditions:
- Pregnancy
- Thymoma (J Pediatr Hematol Oncol 2018;40:e464)
- Autoimmune diseases, most commonly systemic lupus erythematosus and rheumatoid arthritis
- Eosinophilic fasciitis
- Infectious agents:
- Constitutional aplastic anemia (see Pathophysiology)
Clinical features
- Signs and symptoms related to presence and severity of cytopenia(s) (i.e. anemia, thrombocytopenia, leukopenia); most common are fatigue, shortness of breath, bleeding, frequent or prolonged infections, bruising (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Constitutional forms clinically manifest at birth or early infancy (Hematology 2015;20:433)
Diagnosis
- The following clinical and pathologic findings required for diagnosis:
- Persistent (> 6 months) pancytopenia
- Characteristic features within bone marrow aspirate and biopsy
- Absence of abnormal bone marrow infiltrate and reticulin fibrosis
- Patients exhibit at least 2 of the following:
- Absolute neutrophilic count < 1.5 x 109/L
- Platelet count < 50 x 109/L
- Hemoglobin < 10 g/dL
- Further subclassification based on severity (Br J Haematol 2016;172:187)
- Severe aplastic anemia:
- Bone marrow cellularity < 25% or 25 - 50% with < 30% residual hematopoietic cells
- At least 2 of the following:
- Absolute neutrophilic count < 0.5 x 109/L
- Platelet count < 20 x 109/L
- Reticulocyte count < 0.6 x 109/L
- Very severe aplastic anemia:
- Meets the criteria for severe but absolute neutrophilic count < 0.2 x 109/L
- Nonsevere aplastic anemia:
- Cases not meeting criteria for above groups
- Severe aplastic anemia:
- Constitutional bone marrow failure panels in cases of suspected constitutional etiology (Blood Cells Mol Dis 2015;55:40)
Laboratory
- Main laboratory tests
- Complete blood count, showing pancytopenia (as defined above)
- Low reticulocyte count (< 30 x 109/L)
- Normal vitamin B12, folate and iron (to exclude vitamin deficiency anemias)
- Bone marrow aspirate and biopsy (see Microscopic (histologic) description)
- Additional studies (Br J Haematol 2016;172:187)
- Autoimmune screening (including antinuclear antibody, anti-dsDNA, rheumatoid factor, etc.) to exclude autoimmune etiologies
- Liver function tests and viral studies to rule out posthepatitis aplastic anemia
- Flow cytometry to screen for possible paroxysmal nocturnal hemoglobinuria clones
- Small paroxysmal nocturnal hemoglobinuria clones are detected in up to 60% of aplastic anemia
- They may fluctuate in size or even disappear on follow up studies
- Identification of a small paroxysmal nocturnal hemoglobinuria clone should not discourage a diagnosis of aplastic anemia
- In pediatric cases, additional tests needed (Hematology 2015;20:433, Blood Cells Mol Dis 2015;55:40):
- Chromosomal breakage analysis to exclude Fanconi anemia
- TERC and TERT mutation analysis with consideration of telomere length measurement (to investigate dyskeratosis congenita) (Blood 2009;113:309)
- Nowadays, comprehensive bone marrow failure panels to investigate simultaneously the most common inherited genetic defects
Prognostic factors
- Higher risk in patients > 45 years old
- Higher responses to immunosuppression in the presence of small paroxysmal nocturnal hemoglobinuria clones (Br J Haematol 2014;164:546)
- Aplastic anemia with somatic mutation(s) more likely to progress into myelodysplastic syndrome / acute leukemia compared to unmutated cases (Blood 2014;124:2698)
- Chromosome 7 abnormalities associated with poorer response to therapy and prognosis
- Cases carrying trisomy 8 or del(13q) show more favorable response to immunosuppressive therapy and better outcome (Blood 2002;99:3129)
Case reports
- 4 year old girl with quickly progressing peripheral retinal ischemia (Retin Cases Brief Rep 2017;11:108)
- 6 year old South Asian girl diagnosed with celiac disease, presenting with bruises and petechiae (J Med Case Rep 2018;12:16)
- 13 year old girl presenting with opportunistic fungal infection (J Infect Dev Ctries 2015;9:431)
- 14 year old boy with newly diagnosed thymoma (J Pediatr Hematol Oncol 2018;40:e464)
- 25 year old woman with aplastic anemia and delayed response to immunosuppressive therapy (Clin Case Rep 2018;6:1029)
- 32 year old man with acute promyelocytic leukemia in remission (Clin Adv Hematol Oncol 2009;7:670)
- 48 year old woman treated with ipilimumab and nivolumab for metastatic melanoma (Ann Oncol 2017;28:1672)
- 58 year old man with skin induration due to eosinophilic fasciitis (Arthritis Care Res (Hoboken) 2018;70:1095)
Treatment
- Different options varying by age, comorbidities, availability of a matched donor (Br J Haematol 2016;172:187)
- Bone marrow transplant is the only curative strategy; preferred option for immune aplastic anemia or young patients (Haematologica 2014;99:1784)
- Immunosuppressive agents (i.e. cyclosporine, antithymocyte globulin, corticosteroids) for patients who can't undergo a bone marrow transplant or for clearly immune mediated cases (Blood 2017;129:1428)
- Transfusion support only to relieve symptoms
- Bone marrow stimulating agents (as G-CSF) generally used combined with immunosuppressive agents
- Other agents less commonly used: alemtuzumab, androgens (regarded of poor clinical utility) (N Engl J Med 2018;379:1643, N Engl J Med 2016;374:1922)
Microscopic (histologic) description
- Bone marrow invariably hypocellular for age (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Lacunar spaces replaced by fatty cells
- Trilineage hematopoiesis virtually absent with no / few maturing cells
- Residual nucleated cells include mostly lymphocytes, plasma cells, macrophages, mast cells
- Megaloblastoid erythropoiesis sometimes seen but no overt dysplasia
- Normal blast count
- No evidence of fibrosis
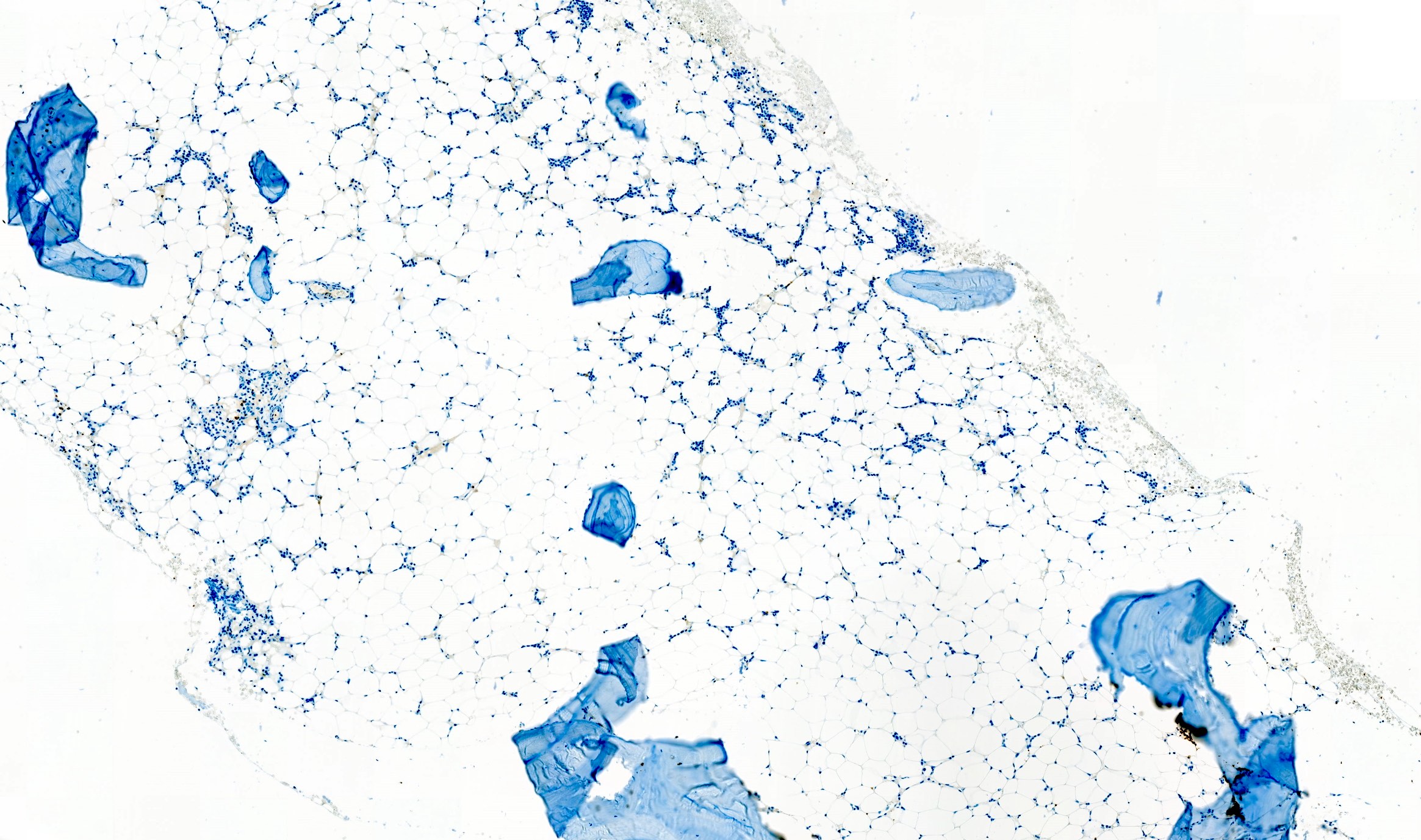
Microscopic (histologic) images
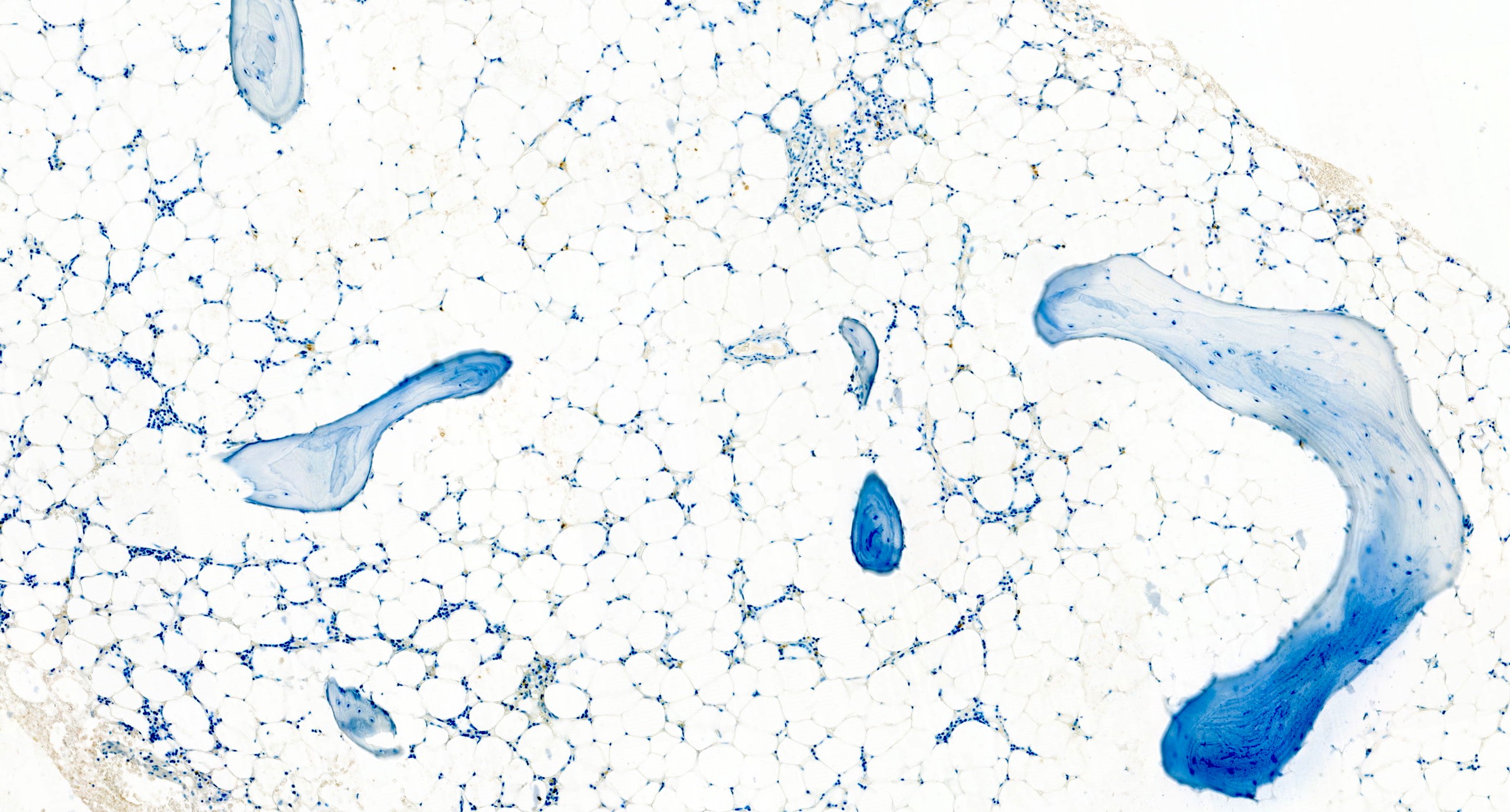
Contributed by Valentina Sangiorgio, M.D.
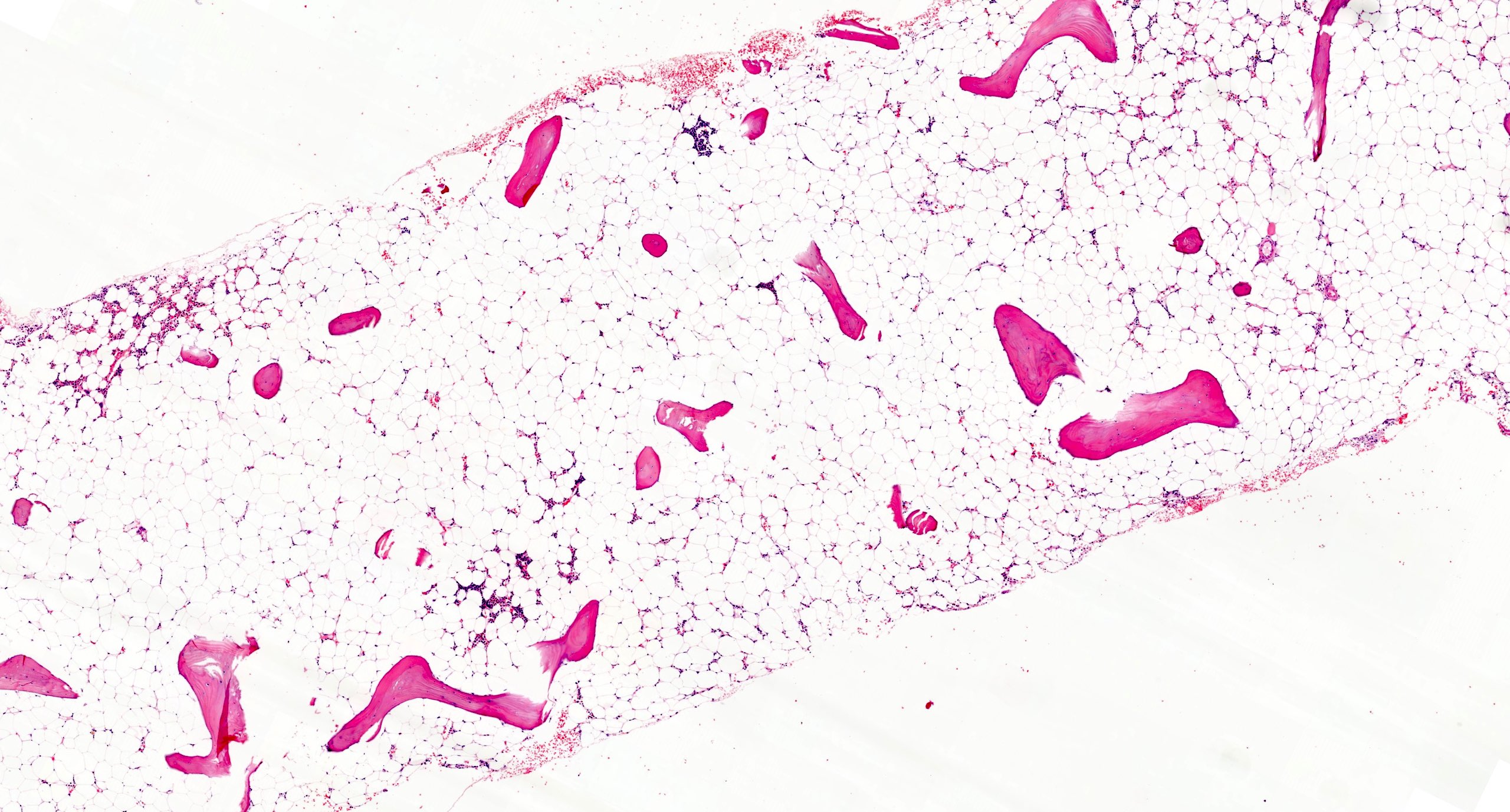
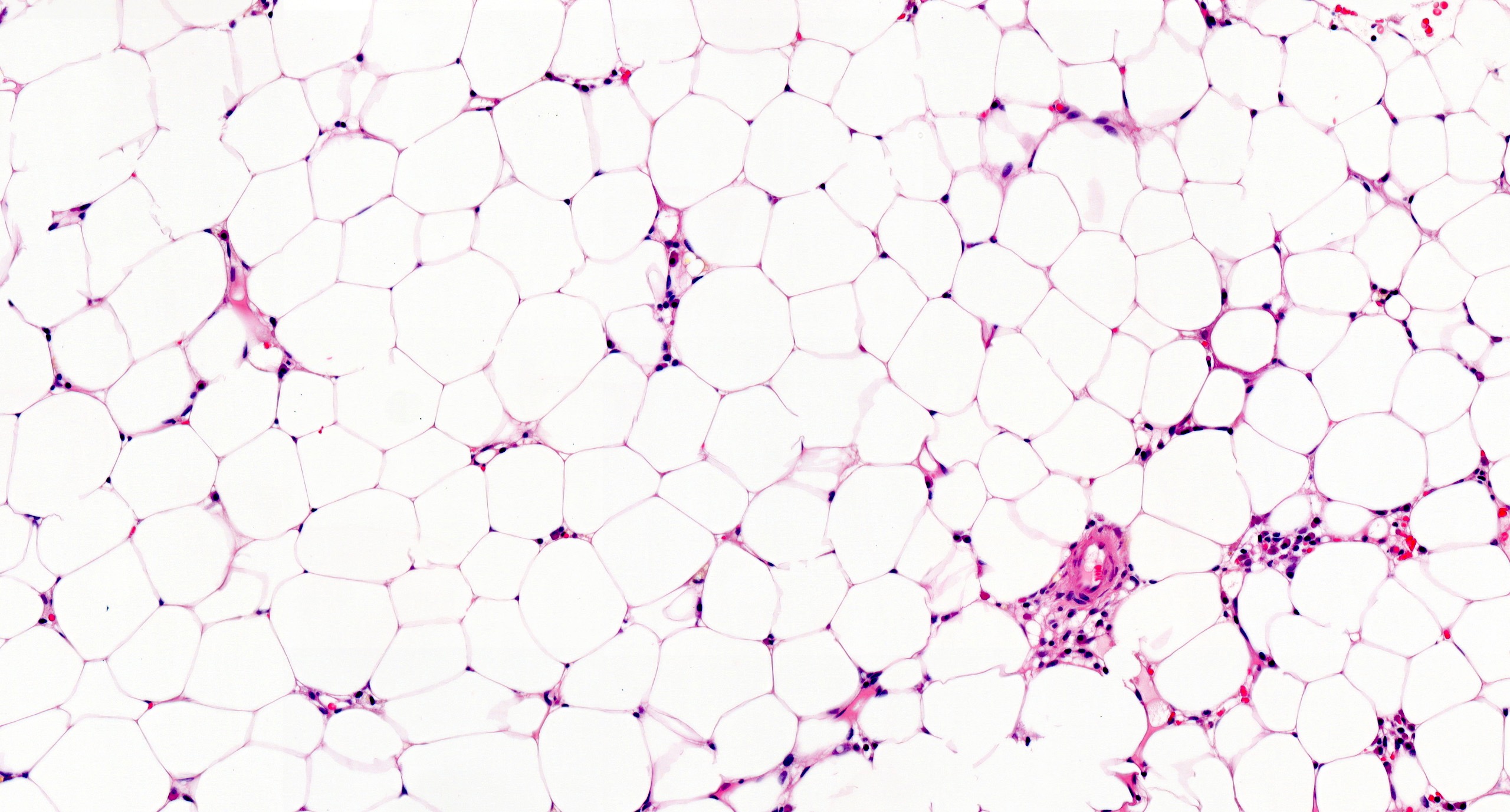
Hypocellular bone marrow
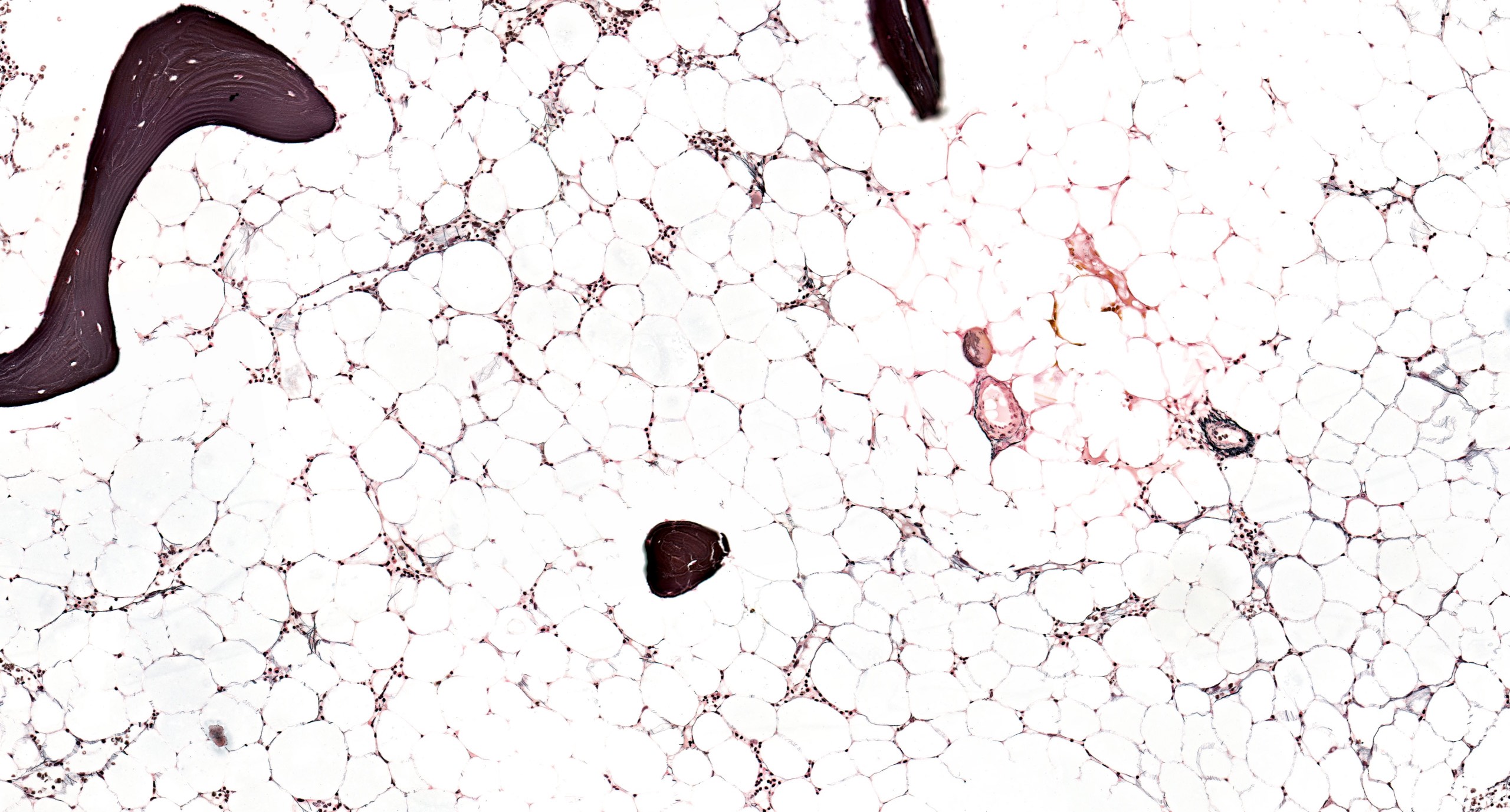
Absence of fibrosis
Virtual absence of myeloid cells
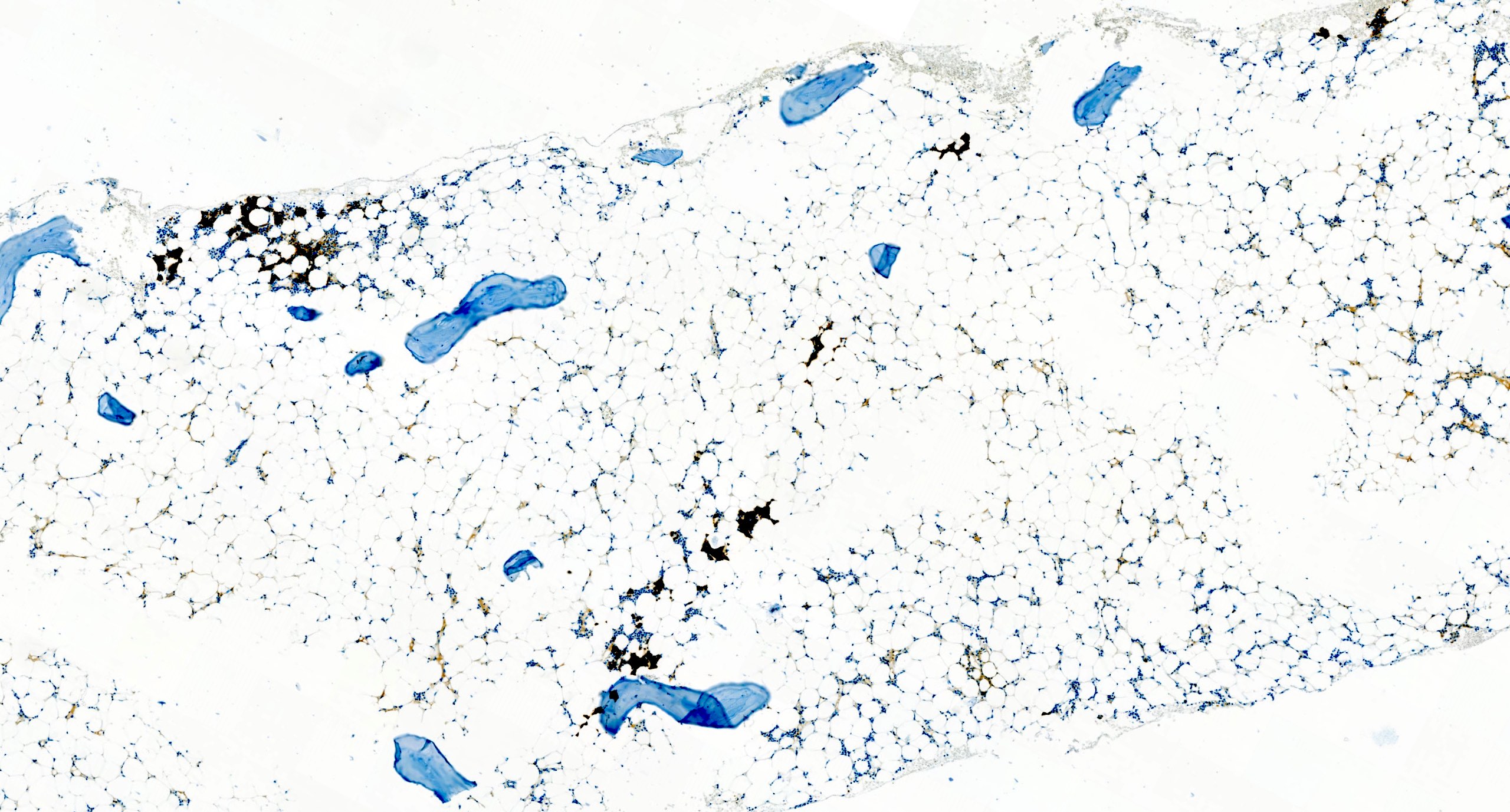
Isolated residual erythroid islands
Virtual absence of megakaryocytes

Normal CD34 count

Mast cell hyperplasia
Peripheral smear description
- Severe cytopenia but no overt dysplastic features (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Megaloblastoid changes sometimes seen
Immunostains
- CD34 to assess number and distribution of hematopoietic precursors; an increased CD34 positive count or the presence of abnormal localization of immature precursors (ALIP) suggest hypocellular myelodysplastic syndrome (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- CD20, CD3 to highlight any B and T lymphoid population, respectively; useful for differential diagnosis
Flow cytometry description
- Normal number and phenotype of hematopoietic cells (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- No monoclonal B cell proliferations or atypical T cell populations
- Paroxysmal nocturnal hemoglobinuria clones commonly detected (up to 60% of the cases); do not affect diagnosis if all other diagnostic criteria of aplastic anemia are met
Flow cytometry images
Contributed by Valentina Sangiorgio, M.D.
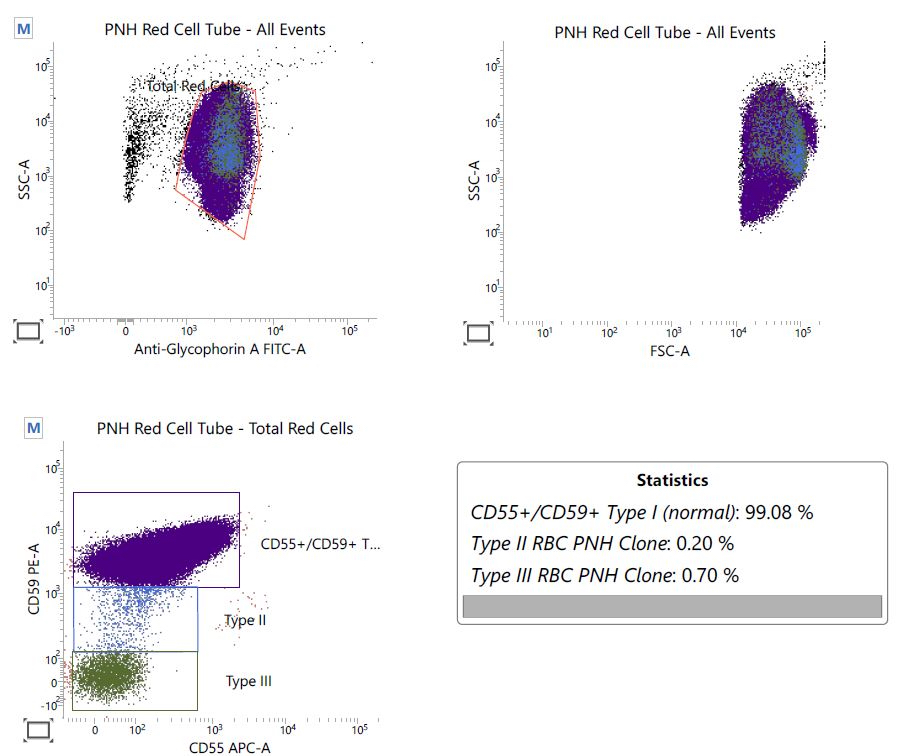
PNH red cell tube
Molecular / cytogenetics description
- Cytogenetic findings
- Clonal abnormalities are seen in up to 12% of AA
- Most common abnormalities involve chromosome 7 (around 13% of cases) and trisomy 8 (around 7% of cases) (Am J Hematol 2001;66:167)
- Detection of chromosome 7 abnormalities should prompt further investigations to exclude myelodysplastic syndromes
- Less common alterations: del(13q), trisomy 6, trisomy 15 (< 5% of cases) (N Engl J Med 2015;373:35, Blood 2002;99:3129)
- Unlike in primary myelodysplastic syndrome, aberrancies of chromosome 5 and 20 are infrequent in aplastic anemia
- Detection of any chromosomal abnormality does not suggest per se myelodysplastic syndrome over aplastic anemia
- Karyotyping sometimes problematic in hypocellular marrow; FISH for common myelodysplastic syndrome associated abnormalities should be considered if insufficient metaphases available
- Molecular findings
- Somatic mutations detected in around 20% of aplastic anemia (Blood 2014;124:2698)
- Most common mutations affect ASXL1 and DNMT3A
- Spliceosome gene mutations (SF3B1, SRSF2, U2AF1) and comutations patterns including TET2, DNMT3A and ASXL1 less common in aplastic anemia
- In general, no mutation is disease specific and molecular findings need clinical and pathologic correlation (Blood 2014;124:2698, Blood 2017;129:3371)
Sample pathology report
- Bone marrow, biopsy and aspirate:
- Hypocellular bone marrow for the patient's age, with features in keeping with aplastic anemia (see comment)
- Comment: The bone marrow is markedly hypocellular for the patient's age (cellularity < 5%). As such, the main histologic differential diagnoses include aplastic anemia and hypoplastic myelodysplastic syndrome. The residual nucleated cells include mostly lymphocytes, plasma cells, mast cells and macrophages. Moreover, overt dysplasia is not observed within any of the three hematopoietic lineages, blast count is within normal limits and there is no evidence of reticulin fibrosis. Given these findings, a diagnosis of aplastic anemia is favored. Correlation with molecular findings (including FISH, conventional cytogenetic and next generation sequencing, whether available) is advised.
- Bone marrow biopsy microscopic description:
- Hypocellular bone marrow for the patient's age (overall cellularity < 5%). Lacunar spaces are extensively replaced by fatty cells; the residual cellularity includes mostly lymphocytes, plasma cells, mast cells and macrophages with only few hematopoietic cells. There is no morphologic increase in blasts. The number of CD34 positive hematopoietic precursors is within normal limits. CD20 and CD3 highlight a population of small B and T lymphocytes, respectively, with a prevalence of the latter, which account for around 25% of the total nucleated cells. Plasma cells are polytypic for kappa and lambda light chains. Reticulin stain reveals a normal network of reticulin fibers.
- Bone marrow aspirate microscopic description:
- Marrow aspirate smear is spicular and shows marked hypocellularity for the patient's age. Myelopoiesis is reduced and left shifted, with only few segmented forms identified. Few normoblastic erythroid precursors are seen. There are scattered megakaryocytes (around 10); however, a reliable assessment of megakaryocytic dysplasia is not possible given the low number of megakaryocytes seen. The residual cellularity includes mostly lymphocytes, plasma cells, mast cells and macrophages. Prussian blue iron stain shows normal storage iron; no ring sideroblasts are noted.
- Bone marrow aspirate cell count (performed on 150 cells) reveals: 1% blasts, 25% promyelocytes / myelocytes, 11% maturing granulocyte forms, 9% erythroid precursors, 31% lymphocytes, 15% plasma cells, 2% eosinophils, 5% mast cells, 1% monocytes. Prussian blue iron stain shows normal storage iron; no ring sideroblasts are noted.
Differential diagnosis
- Paroxysmal nocturnal hemoglobinuria (PNH):
- In classical PNH, there is always evidence of intravascular hemolysis; reticulocyte count is generally high
- Bone marrow is normocellular to hypercellular with erythroid hyperplasia
- PNH clone generally relevant, above 50% and sometimes approaching representation of all circulating cells from the mutated clone (Br J Haematol 2018;182:730)
- Hypoplastic myelodysplastic syndrome:
- Dysplastic hematopoiesis, increased blasts or CD34 positive count and fibrosis suggest hypoplastic myelodysplastic syndrome (Haematologica 2009;94:264, Blood 2017;129:3371)
- Hypoplastic acute myeloid leukemia:
- Acute myeloid leukemia can present with hypocellular bone marrow; diagnosis relies on a proper blast count (≥ 20% defines leukemia) on peripheral blood or bone marrow samples (Haematologica 2009;94:264)
- T cell large granular lymphocytic leukemia:
- Generally presents with isolated anemia; pancytopenia observed only in a minority of the cases
- Lymphocyte count is usually 2 - 20 x 109/L
- Peripheral smears show a characteristic lymphocytosis of large granular lymphocytes
- Bone marrow biopsy shows a cytotoxic T cell infiltrate with a characteristic intrasinusoidal distribution (Blood 2017;129:1082)
- Constitutional bone marrow failure syndrome:
- With multilineage involvement: Fanconi anemia, dyskeratosis congenita
- With single lineage involvement: Diamond-Blackfan anemia, severe congenital neutropenia (Hematology 2015;20:433)
Board review style question #1
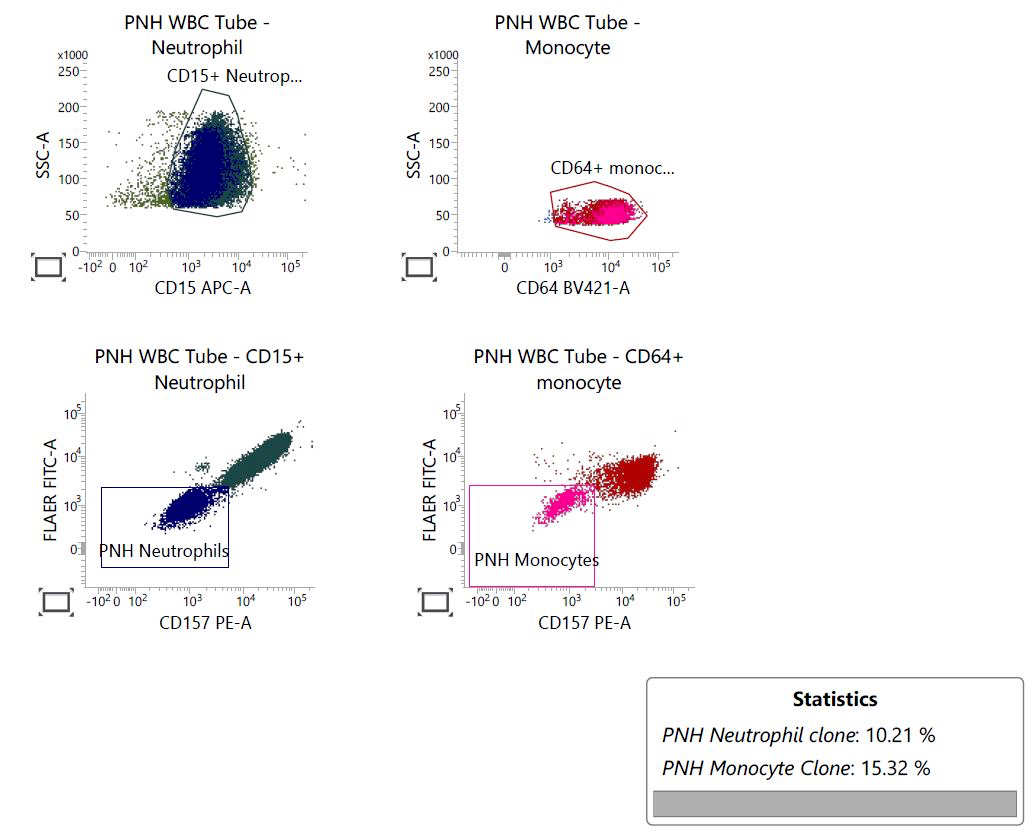
Are these flow cytometry findings compatible with aplastic anemia?
- No, they exclude completely aplastic anemia
- They are not relevant to a diagnostic workup of aplastic anemia
- They require clinical and pathologic correlation
- Yes, they are diagnostic of aplastic anemia
Board review style answer #1
C. They require clinical and pathologic correlation
Small paroxysmal nocturnal hemoglobinuria clones are common in aplastic anemia and their detection does not exclude such diagnosis. However, when more relevant clones are detected, as in this case, careful clinical and pathologic investigations are needed to rule out paroxysmal nocturnal hemoglobinuria. True paroxysmal nocturnal hemoglobinuria should not show severe cytopenia(s) and marrow hypocellularity, features most in keeping with aplastic anemia.
Comment Here
Reference: Aplastic anemia (AA)
Small paroxysmal nocturnal hemoglobinuria clones are common in aplastic anemia and their detection does not exclude such diagnosis. However, when more relevant clones are detected, as in this case, careful clinical and pathologic investigations are needed to rule out paroxysmal nocturnal hemoglobinuria. True paroxysmal nocturnal hemoglobinuria should not show severe cytopenia(s) and marrow hypocellularity, features most in keeping with aplastic anemia.
Comment Here
Reference: Aplastic anemia (AA)
Board review style question #2
Which of the following can be detected in aplastic anemia?
- 10% blast count
- Bone marrow collagen fibrosis
- Frank multilineage dysplasia
- Trisomy 8
Board review style answer #2
D. Trisomy 8
Trisomy 8 is one of the most common cytogenetic abnormalities detected in aplastic anemia. However, this finding is not specific, as it can occur also in other myeloid neoplasms and it has been described in nonneoplastic conditions.
Comment Here
Reference: Aplastic anemia (AA)
Trisomy 8 is one of the most common cytogenetic abnormalities detected in aplastic anemia. However, this finding is not specific, as it can occur also in other myeloid neoplasms and it has been described in nonneoplastic conditions.
Comment Here
Reference: Aplastic anemia (AA)
