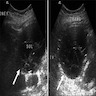
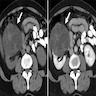
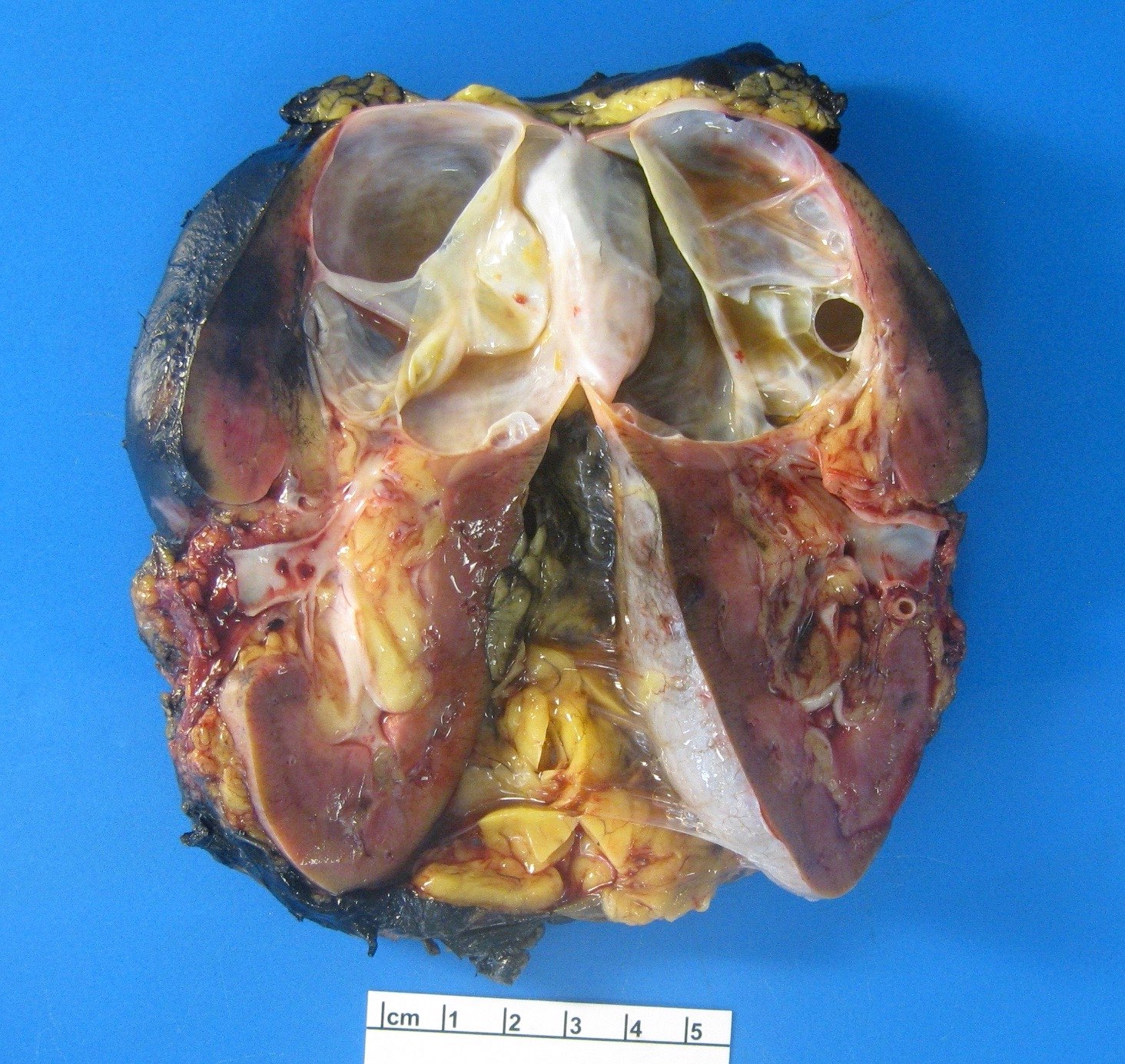
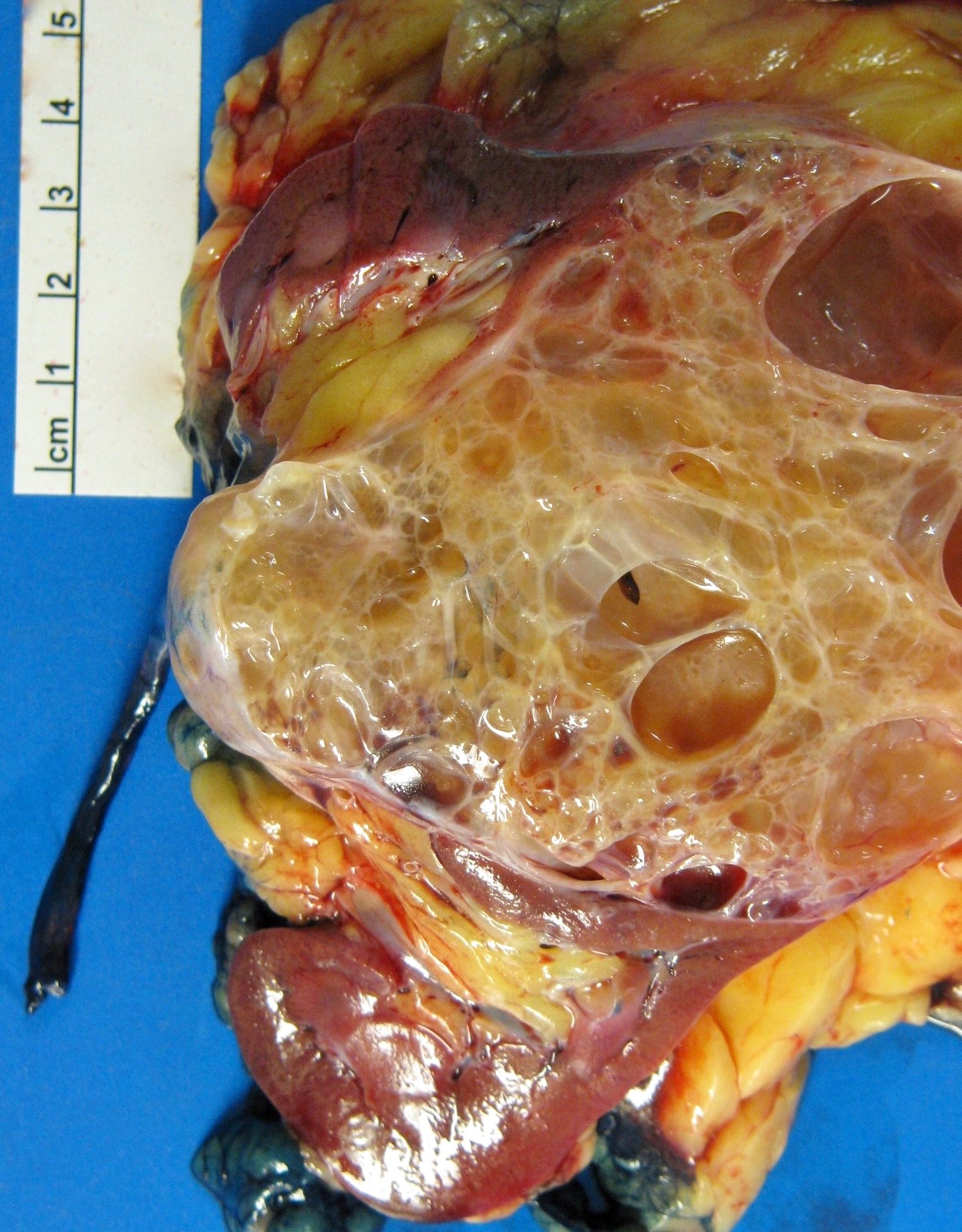
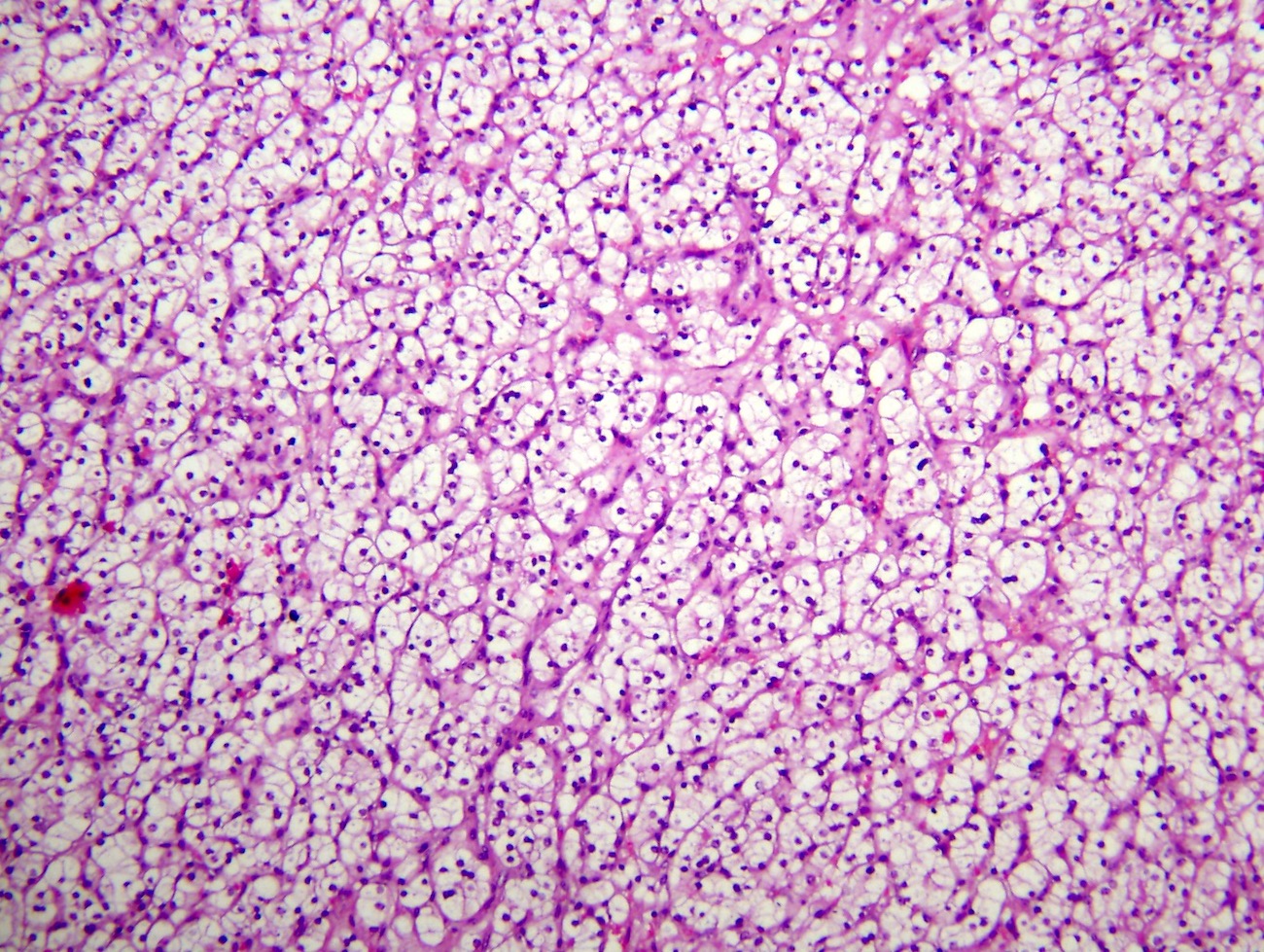
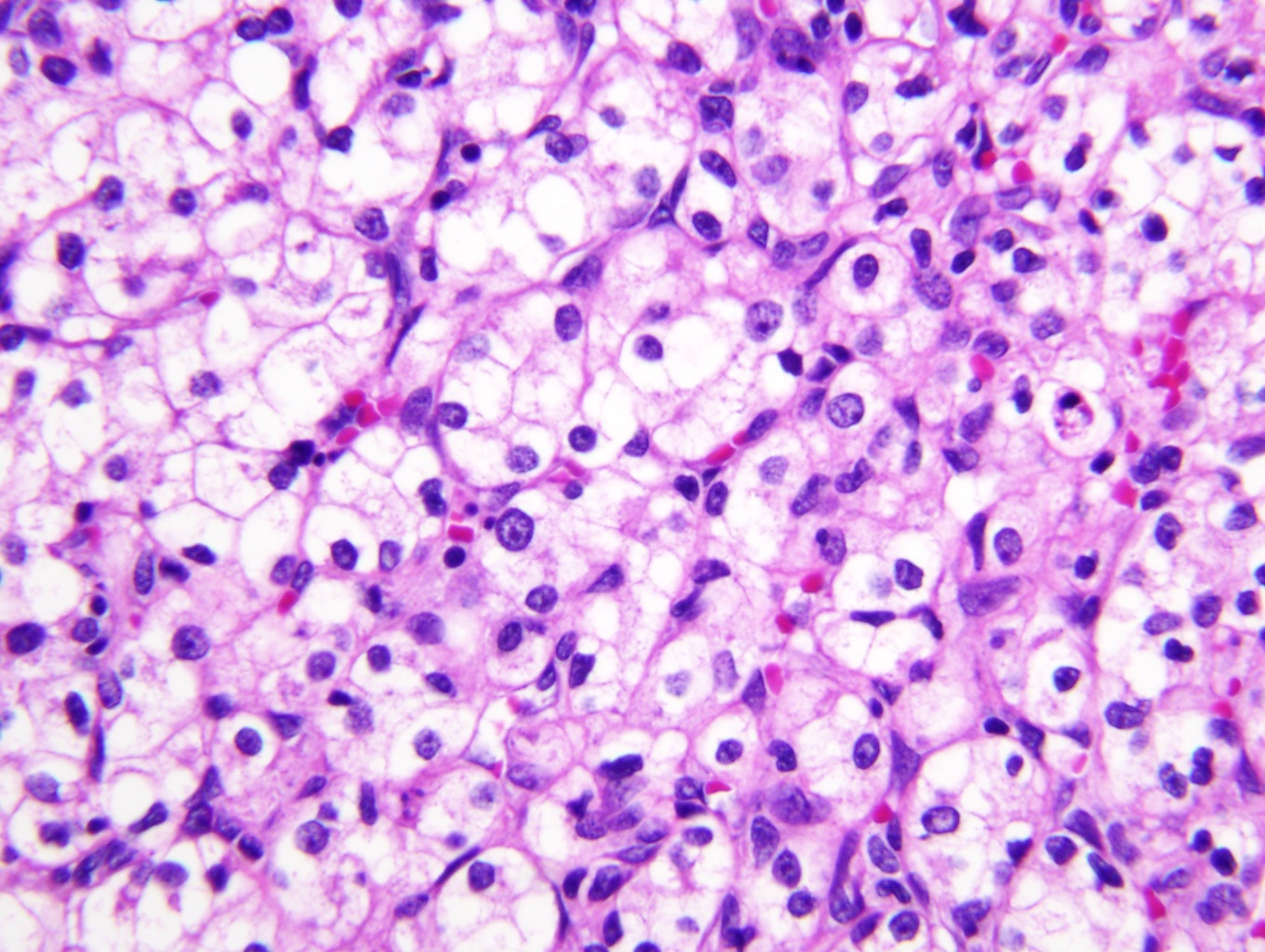
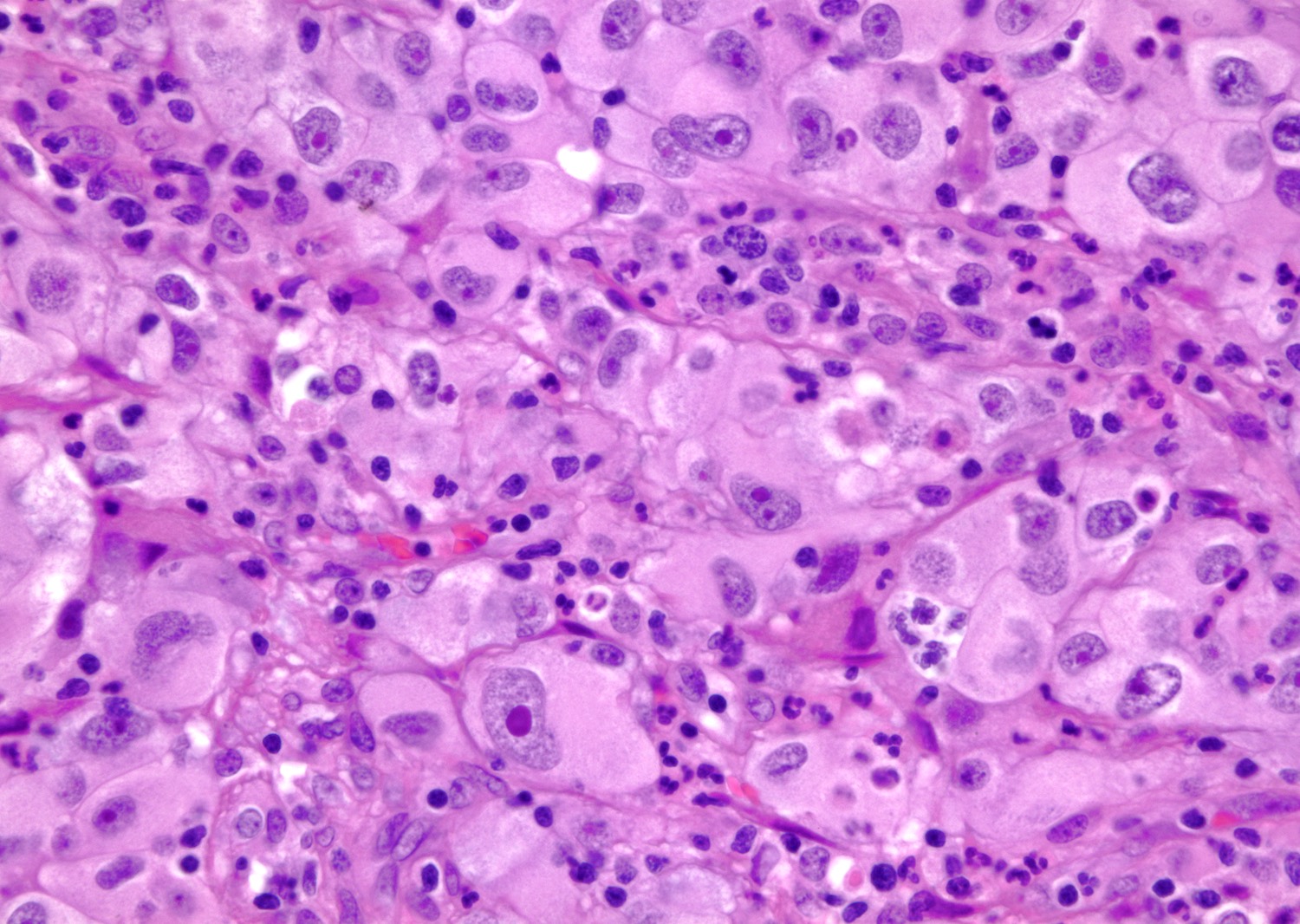
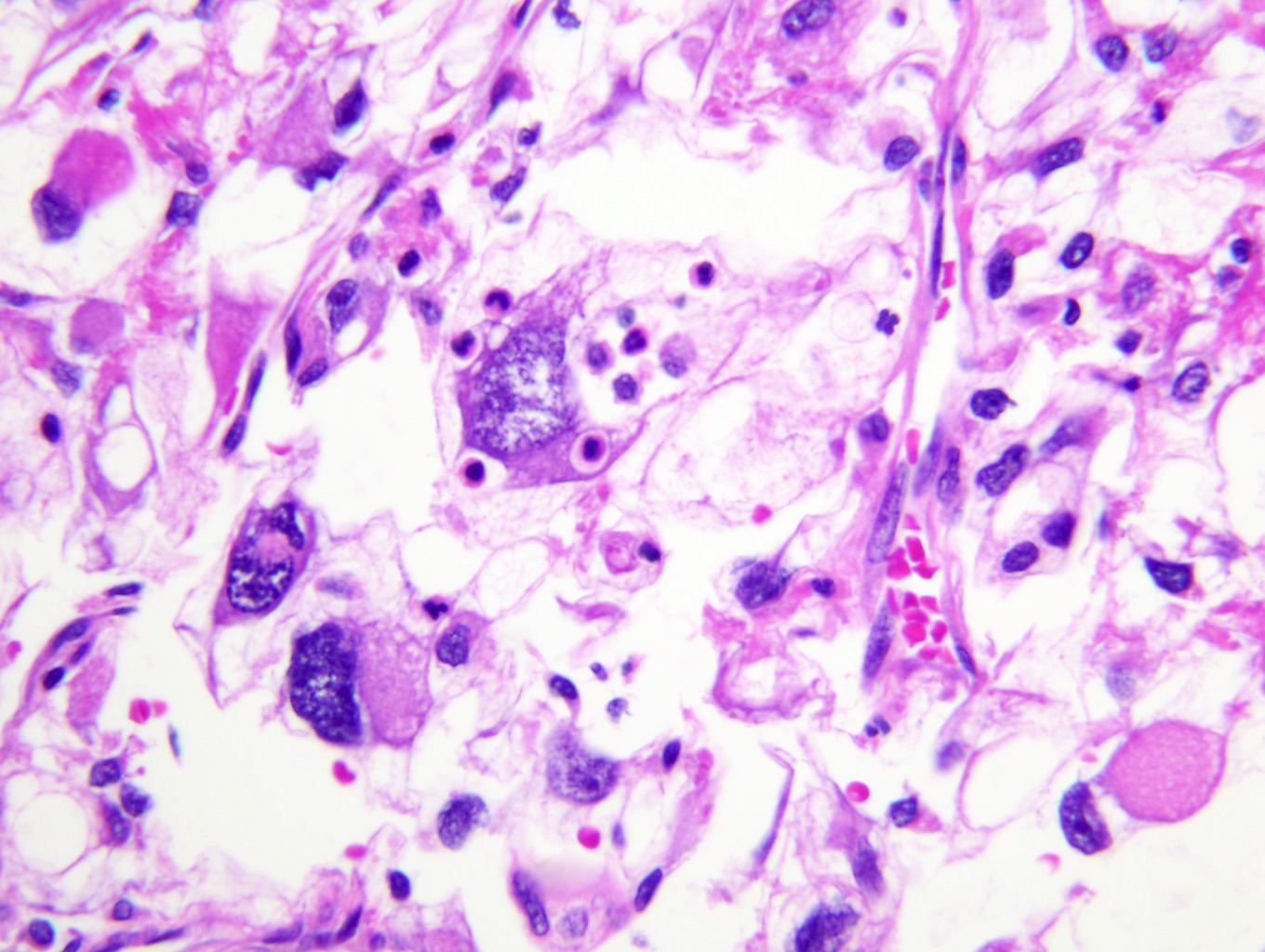
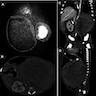
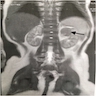
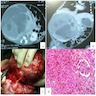
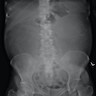
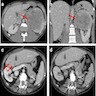
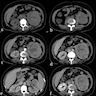
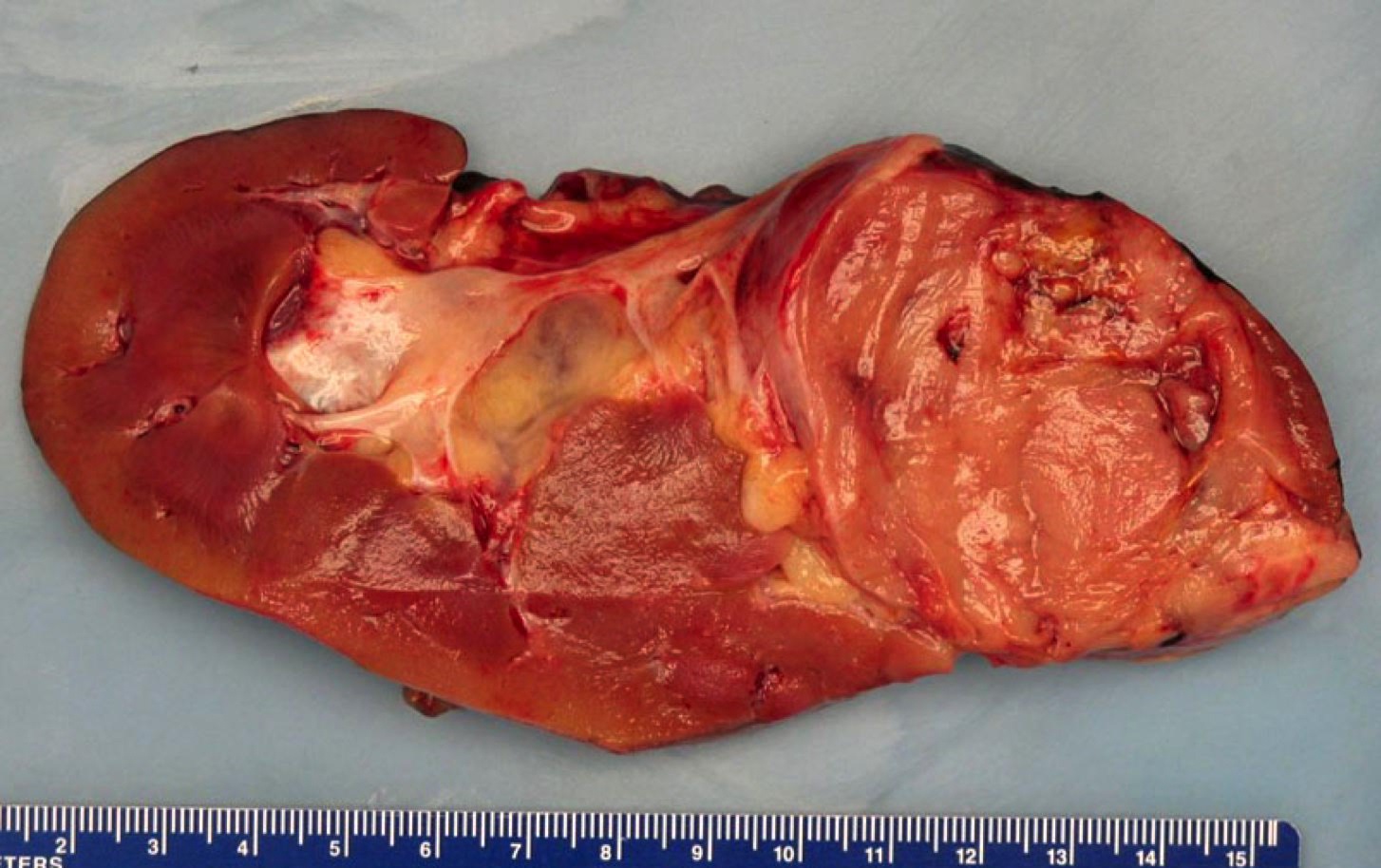
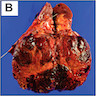
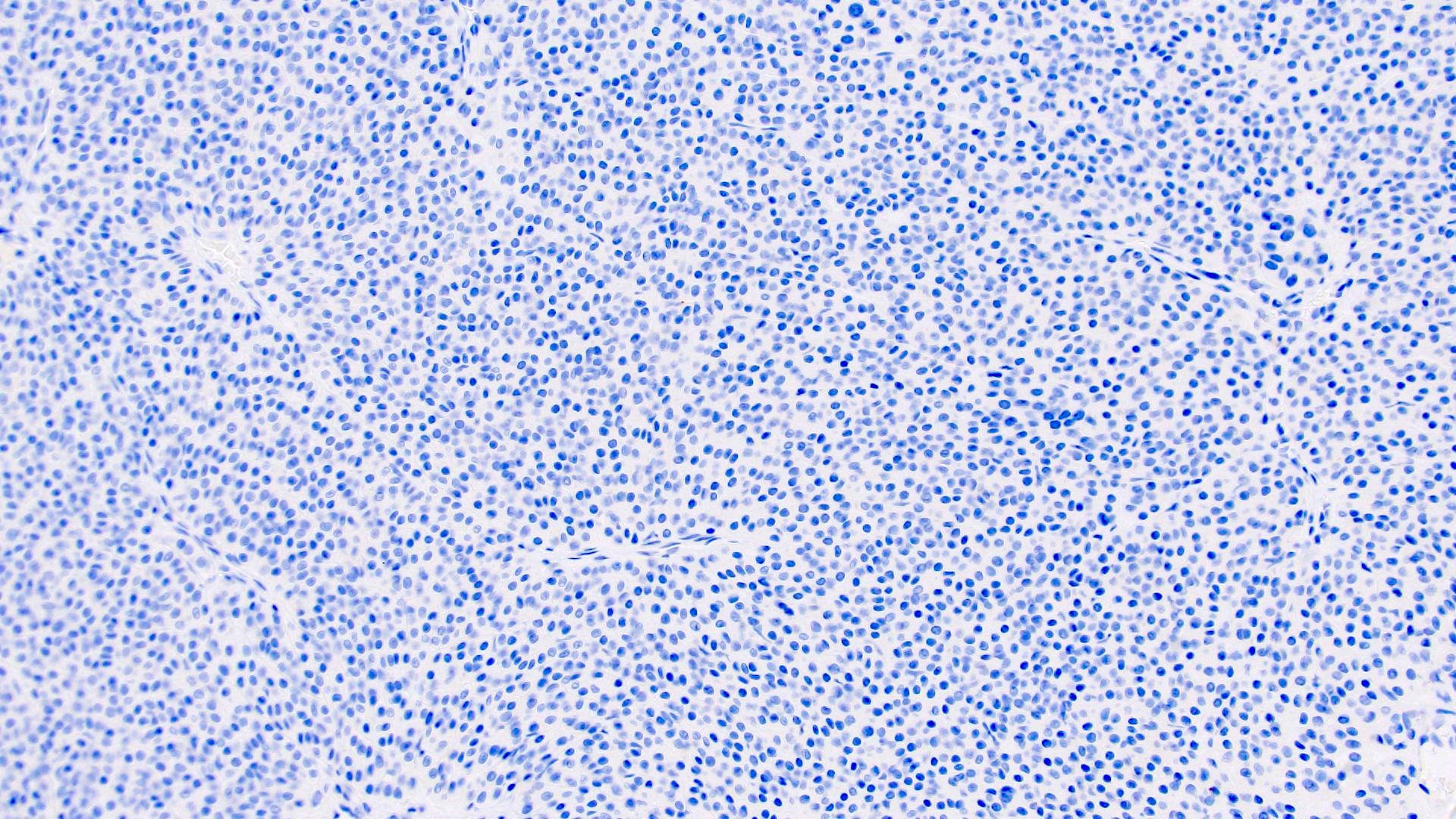
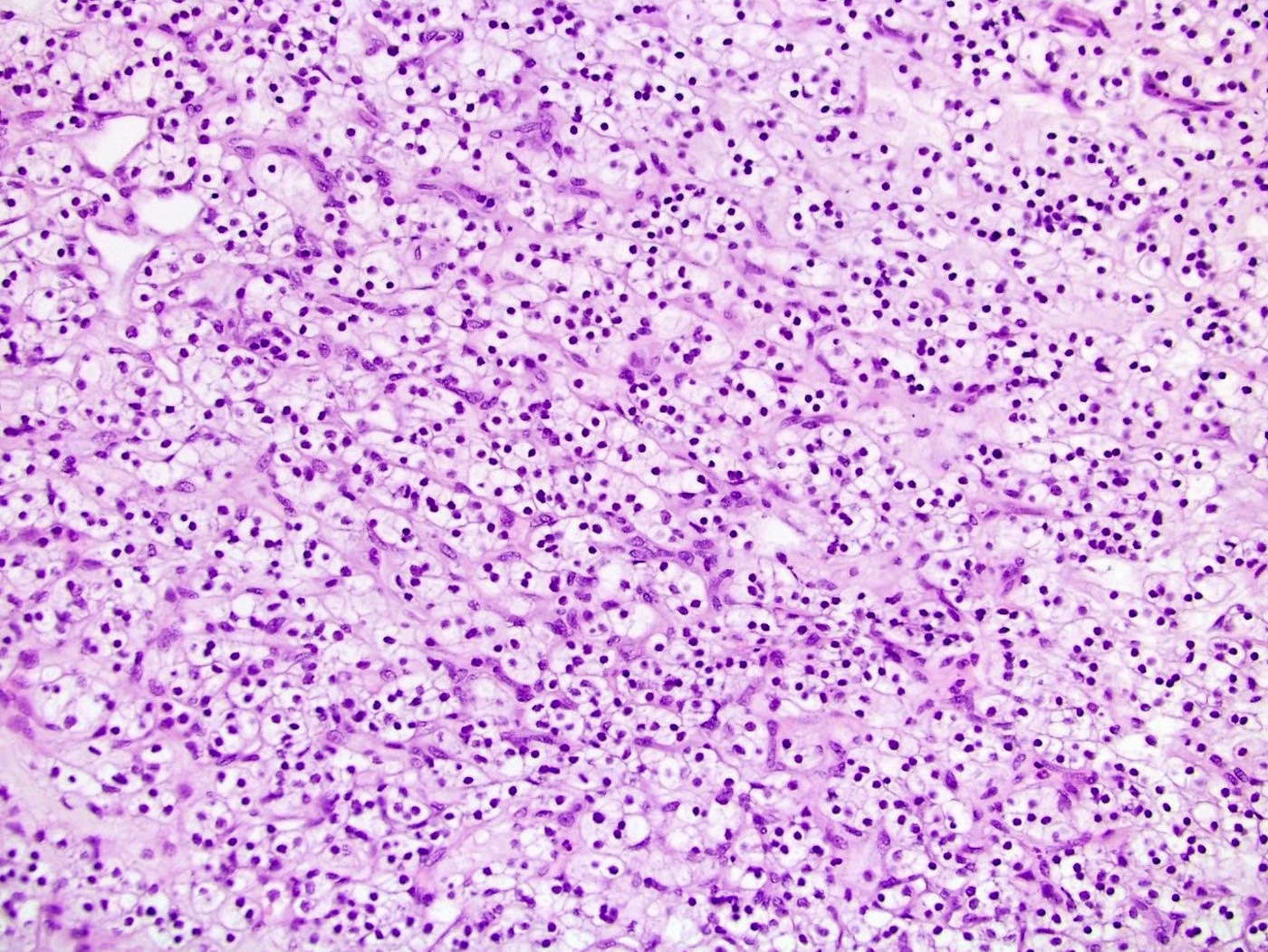
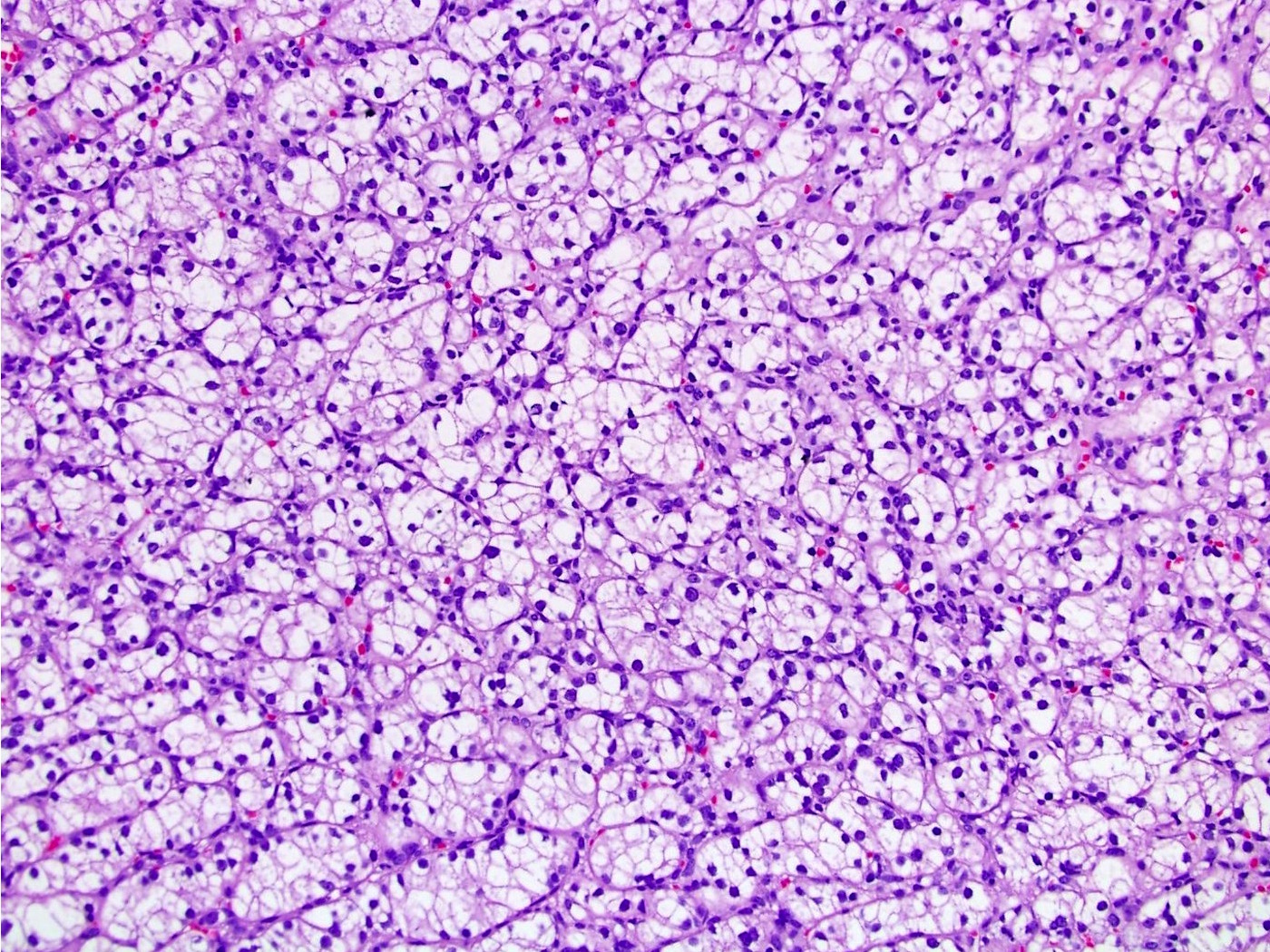
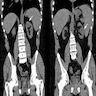
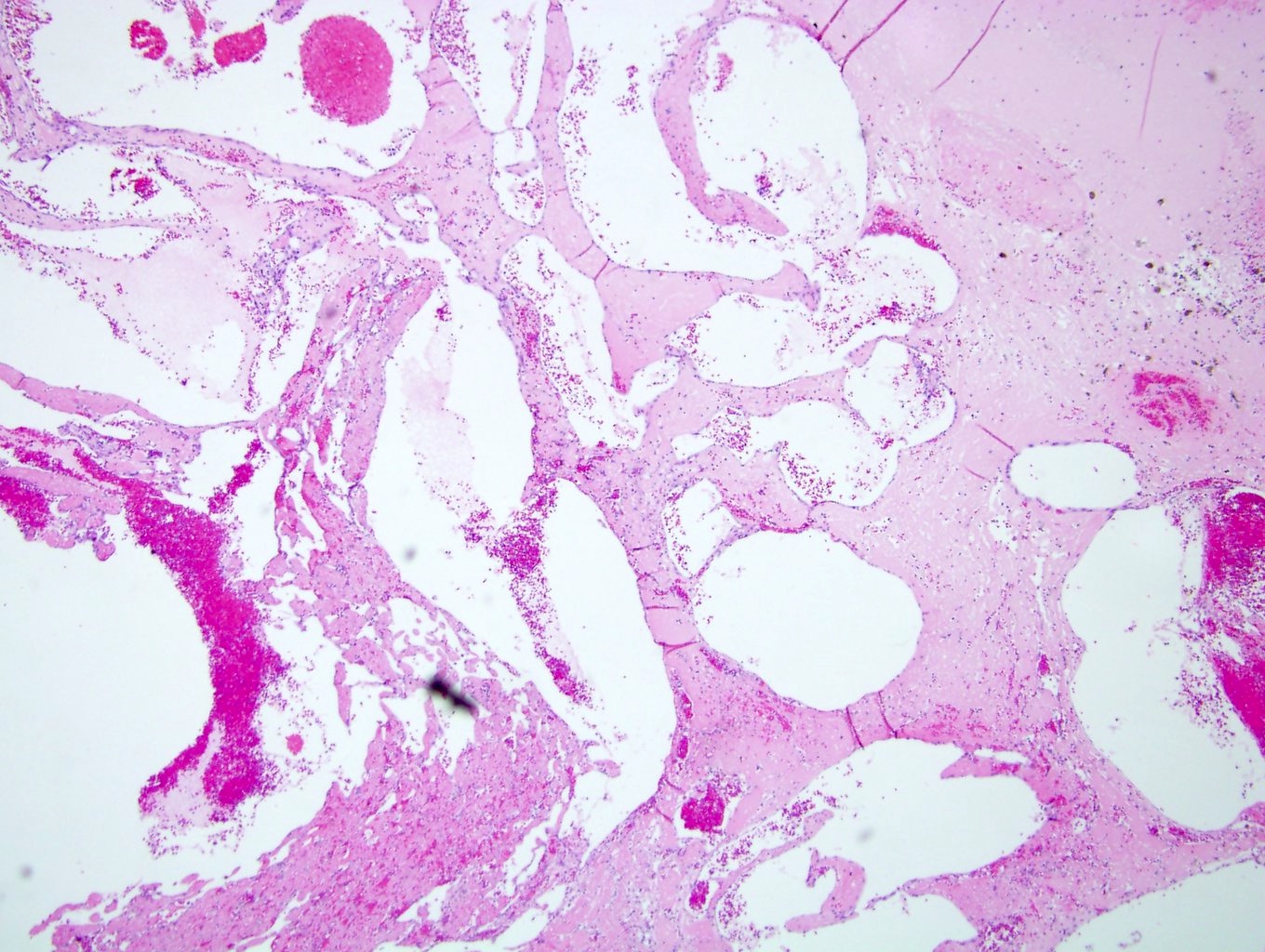
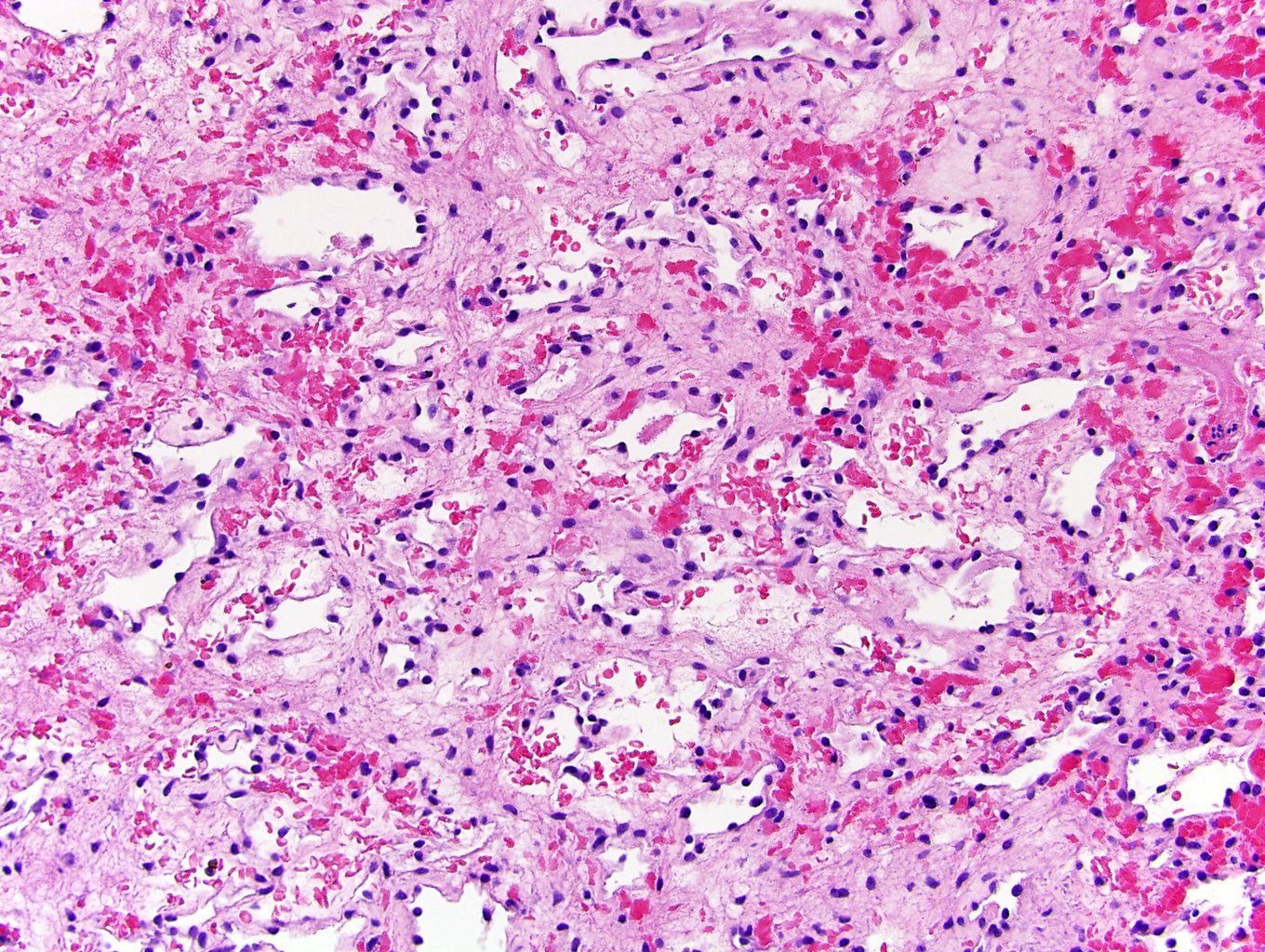
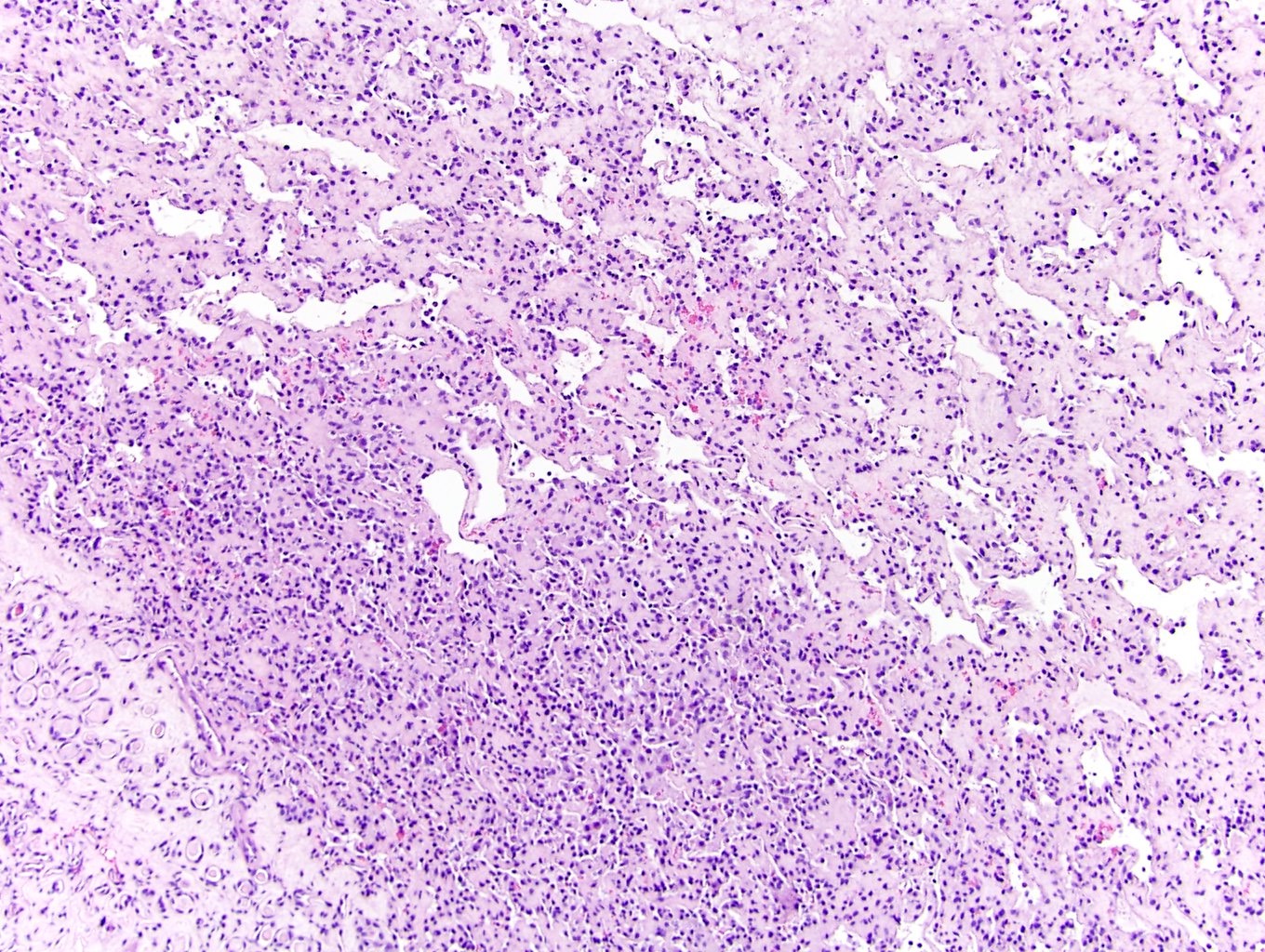
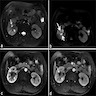
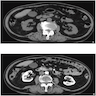
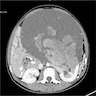
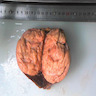
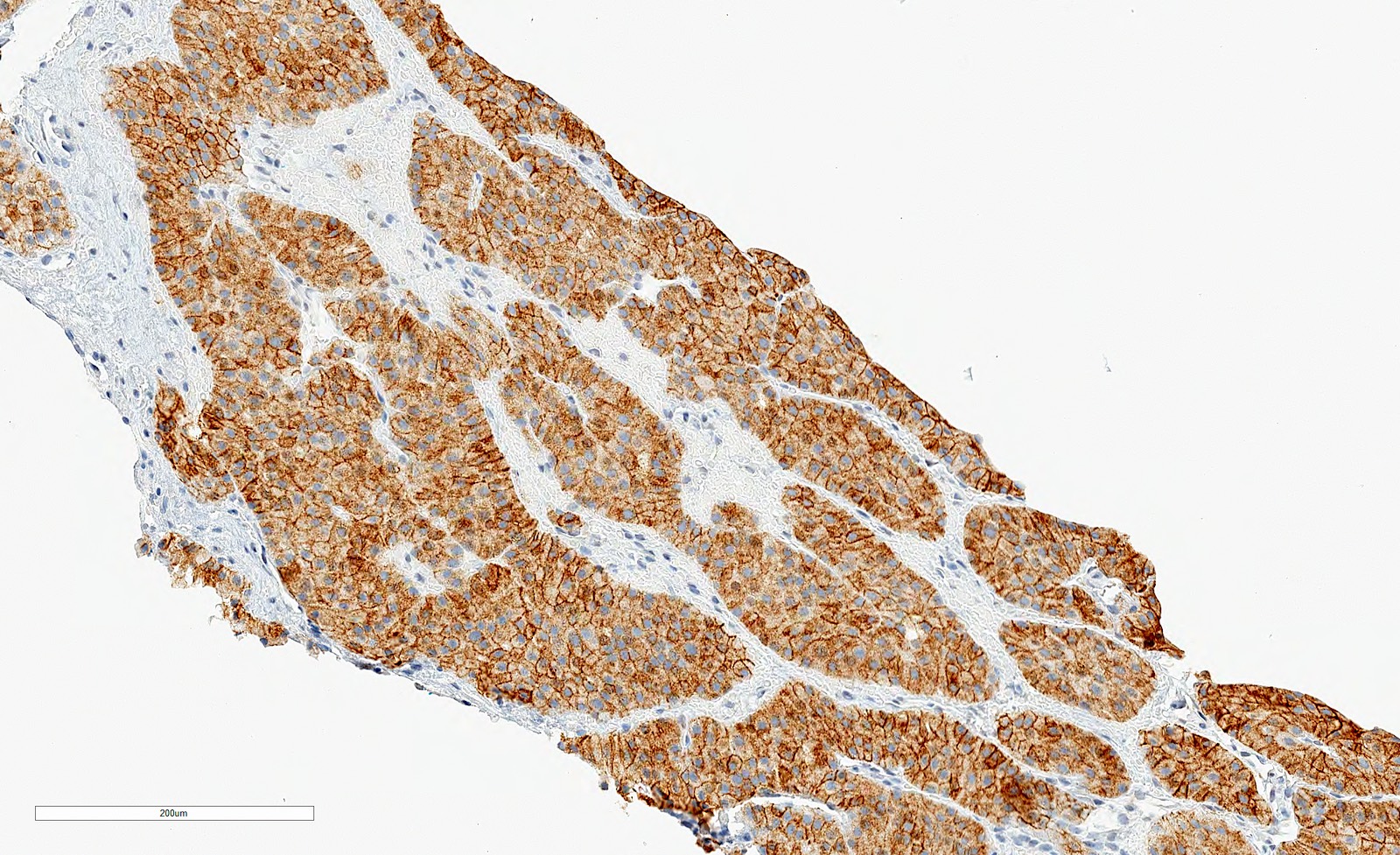
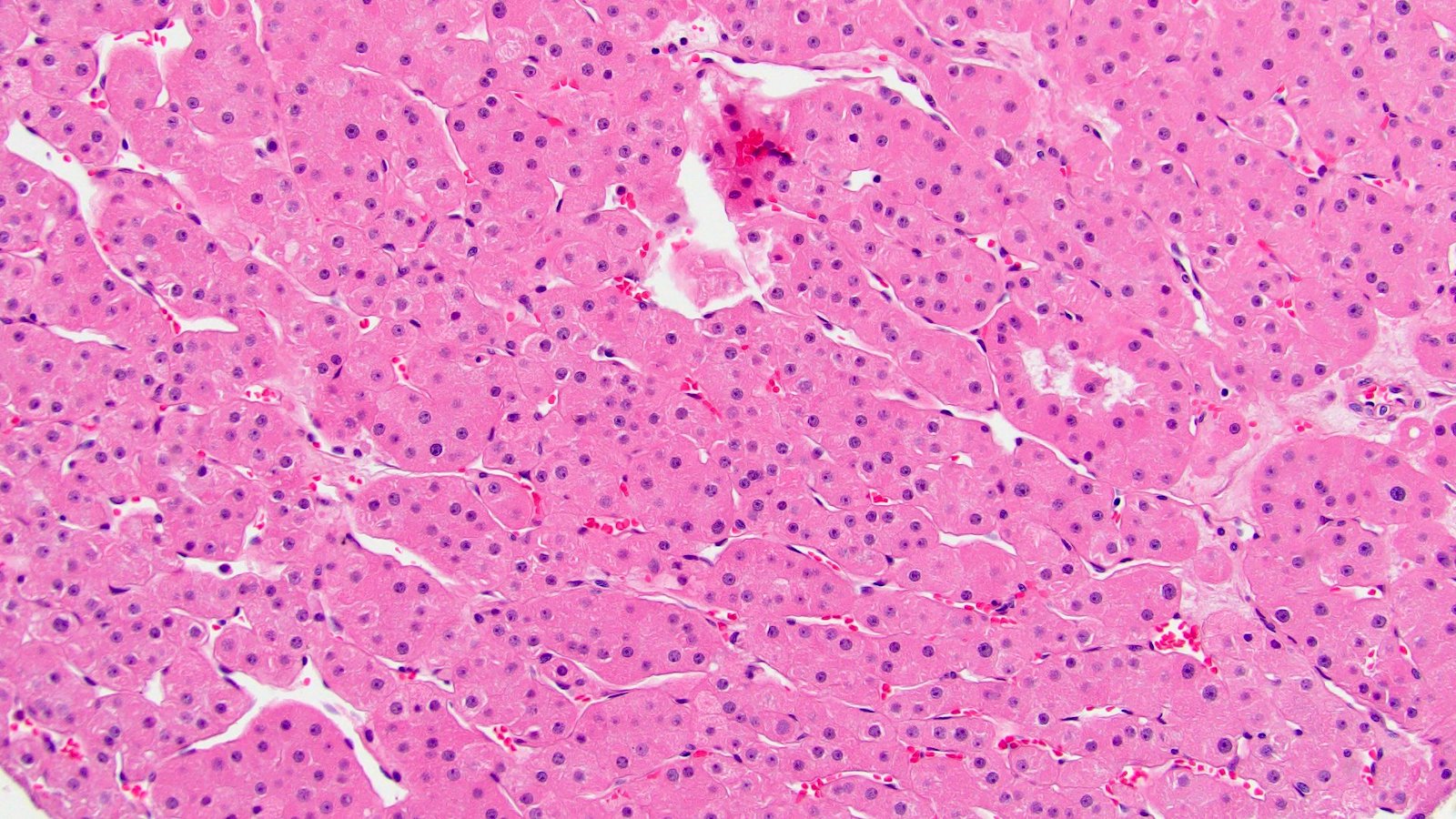
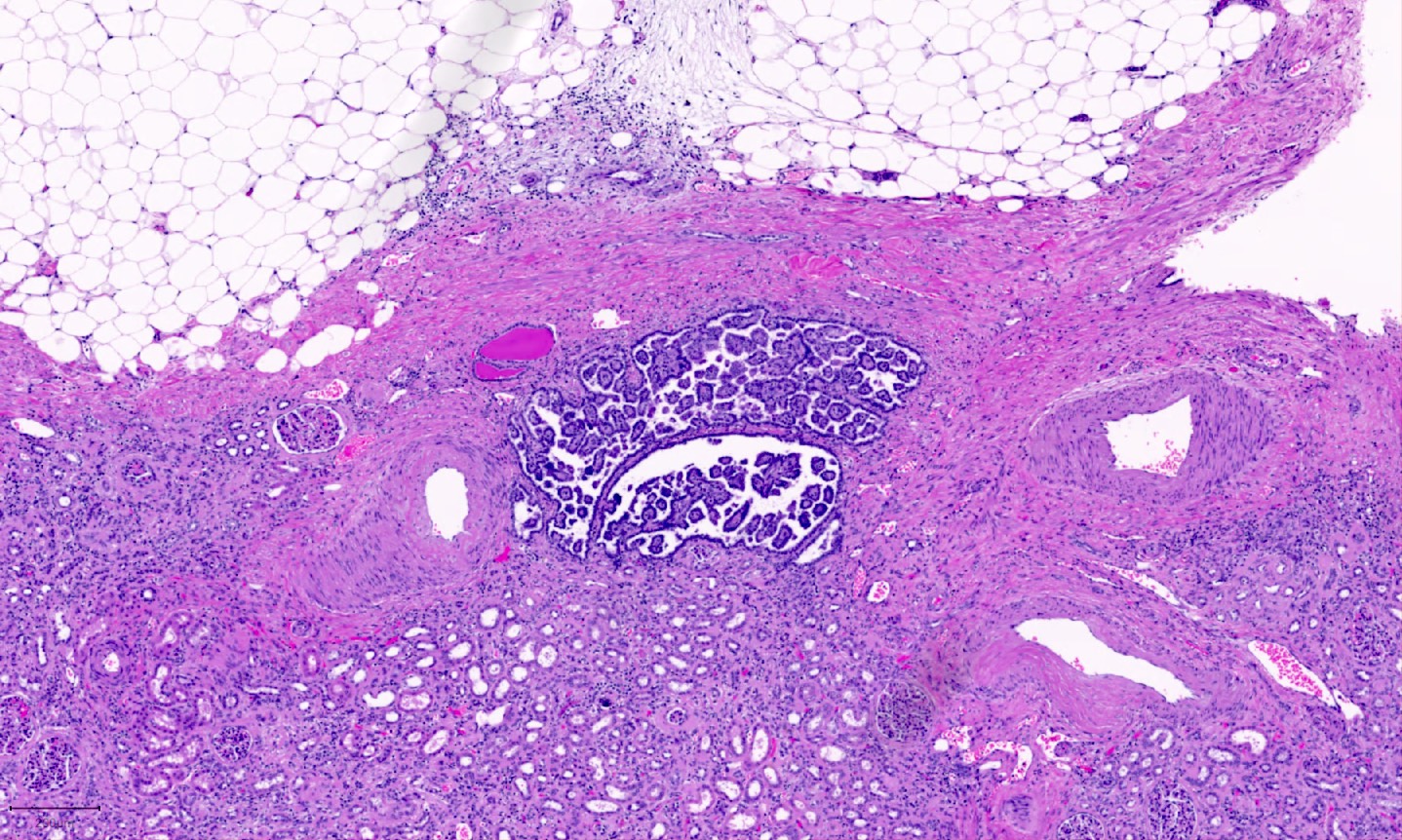
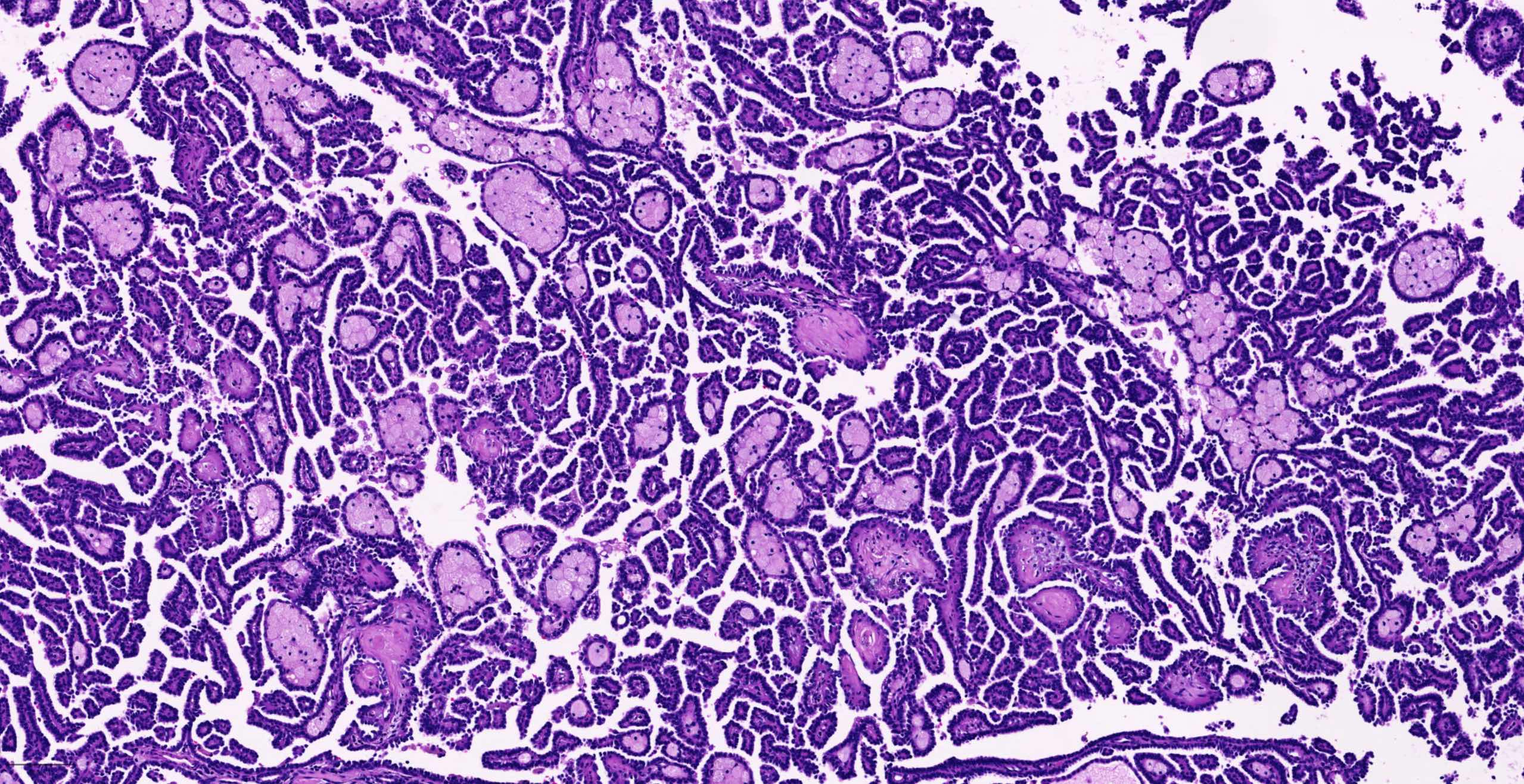
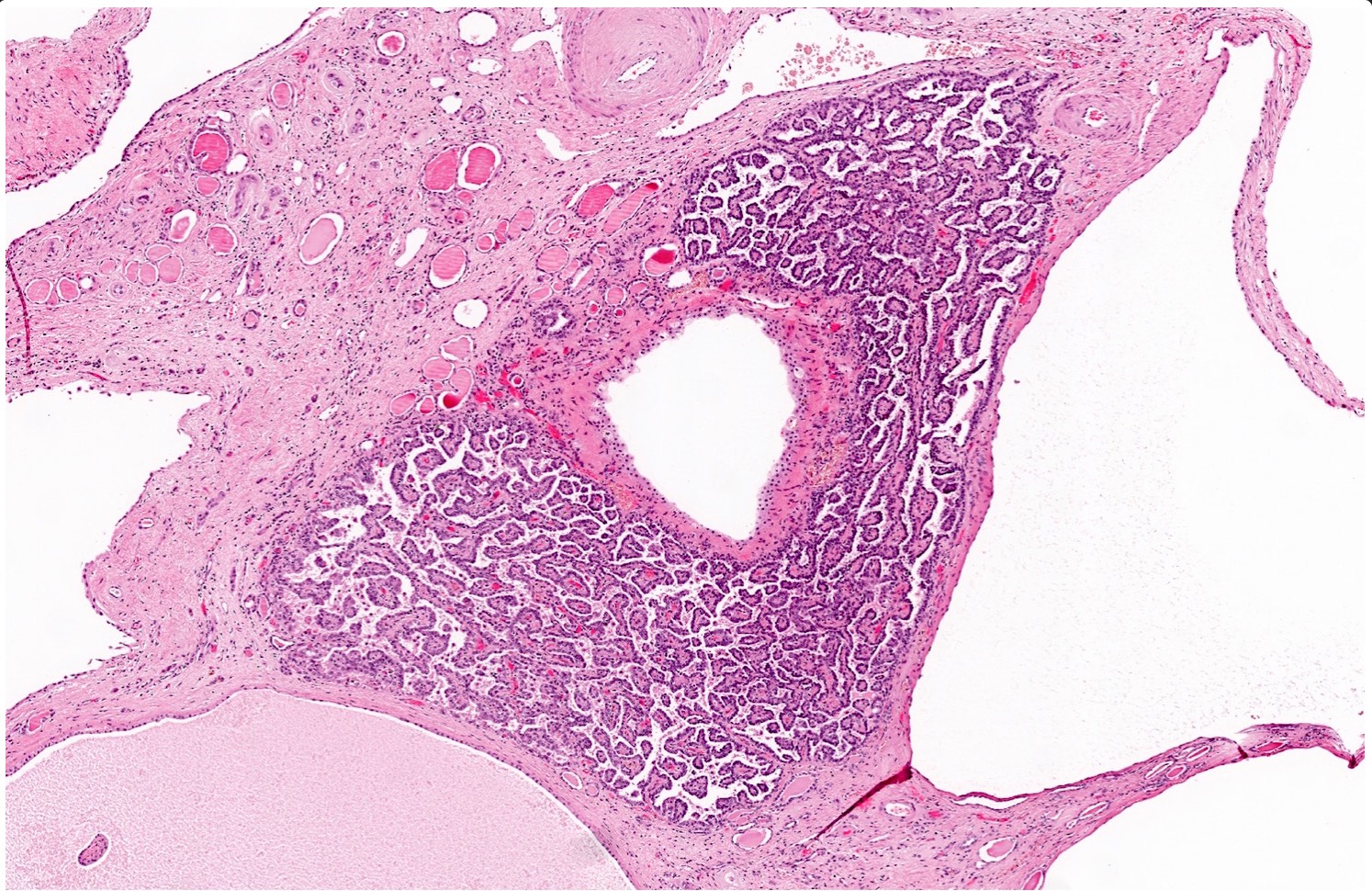
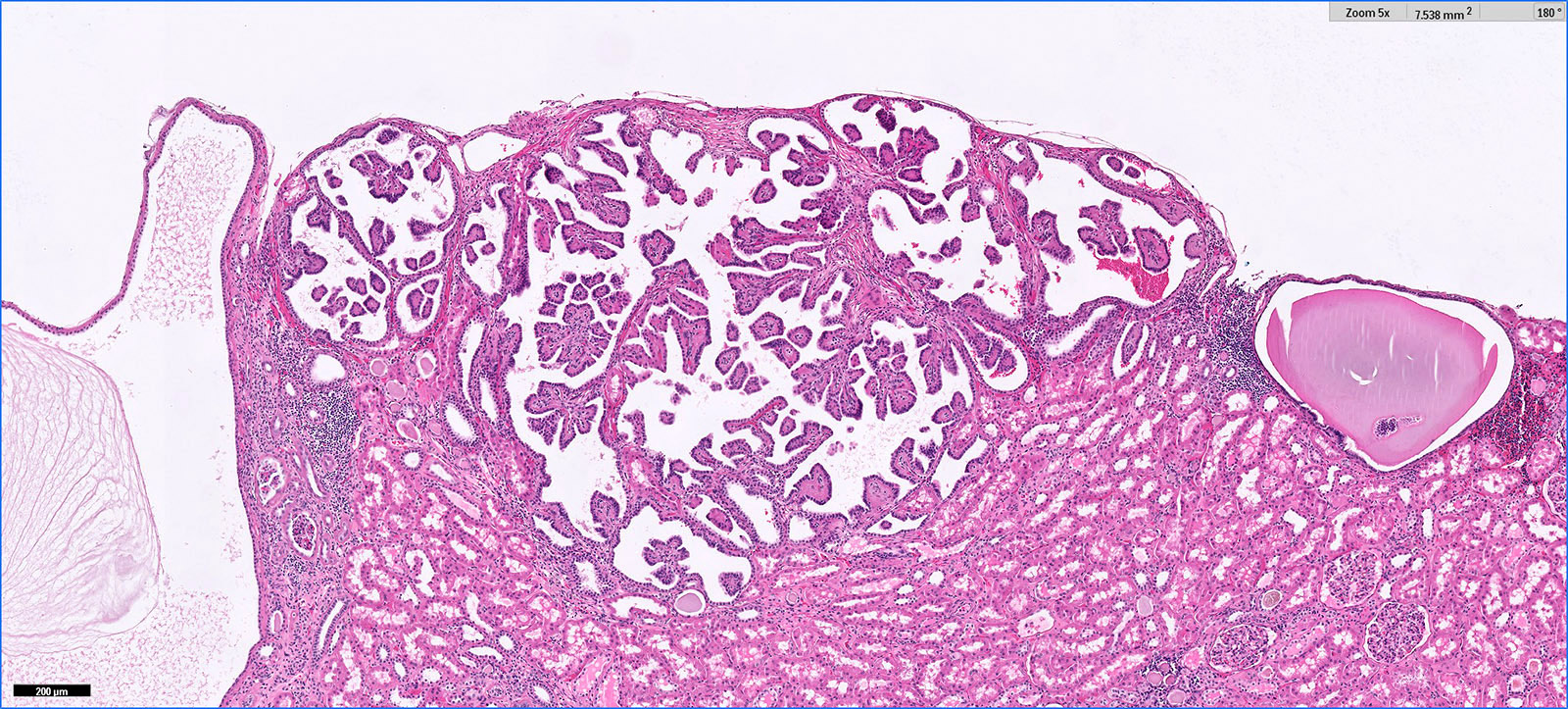
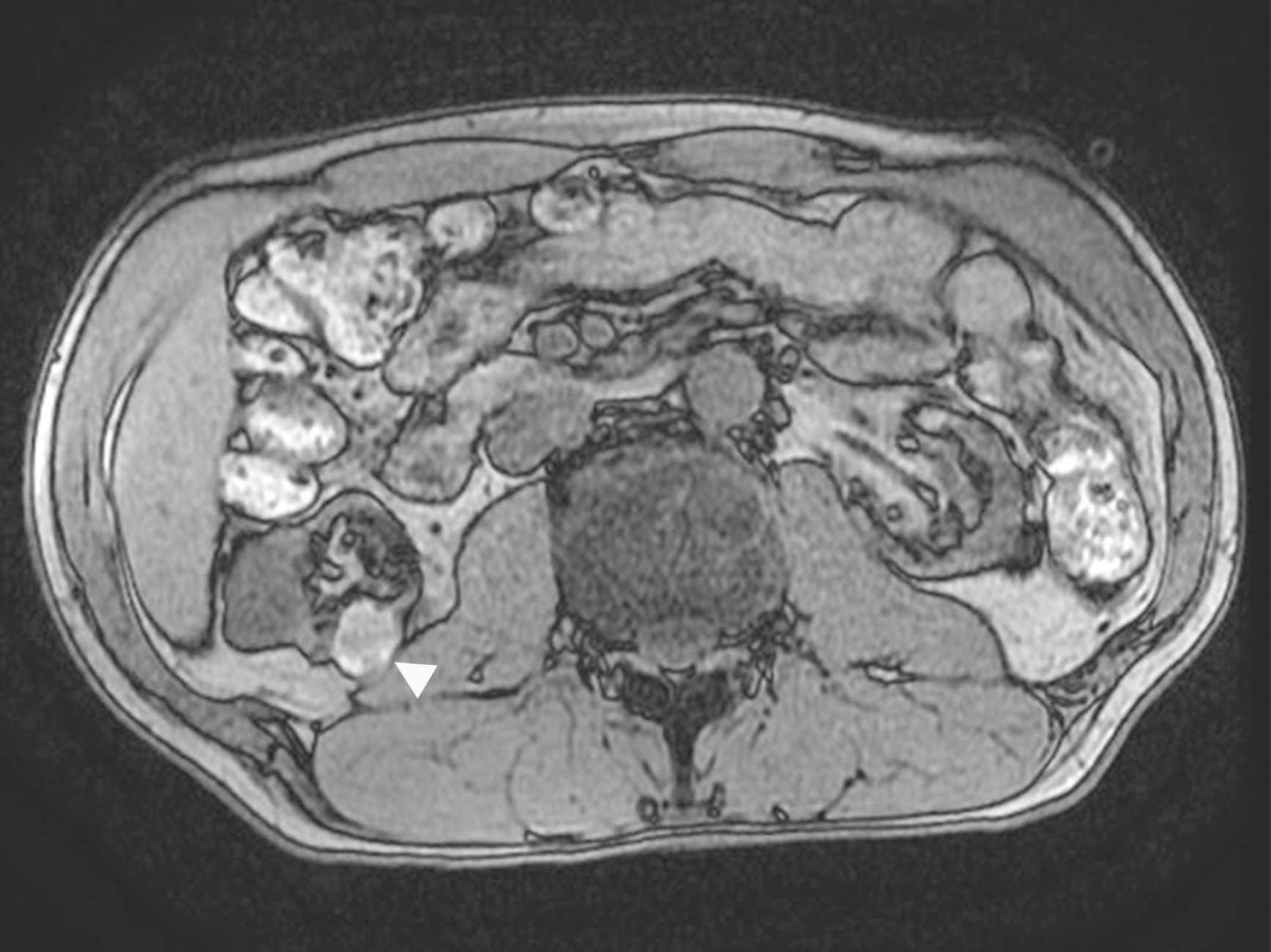
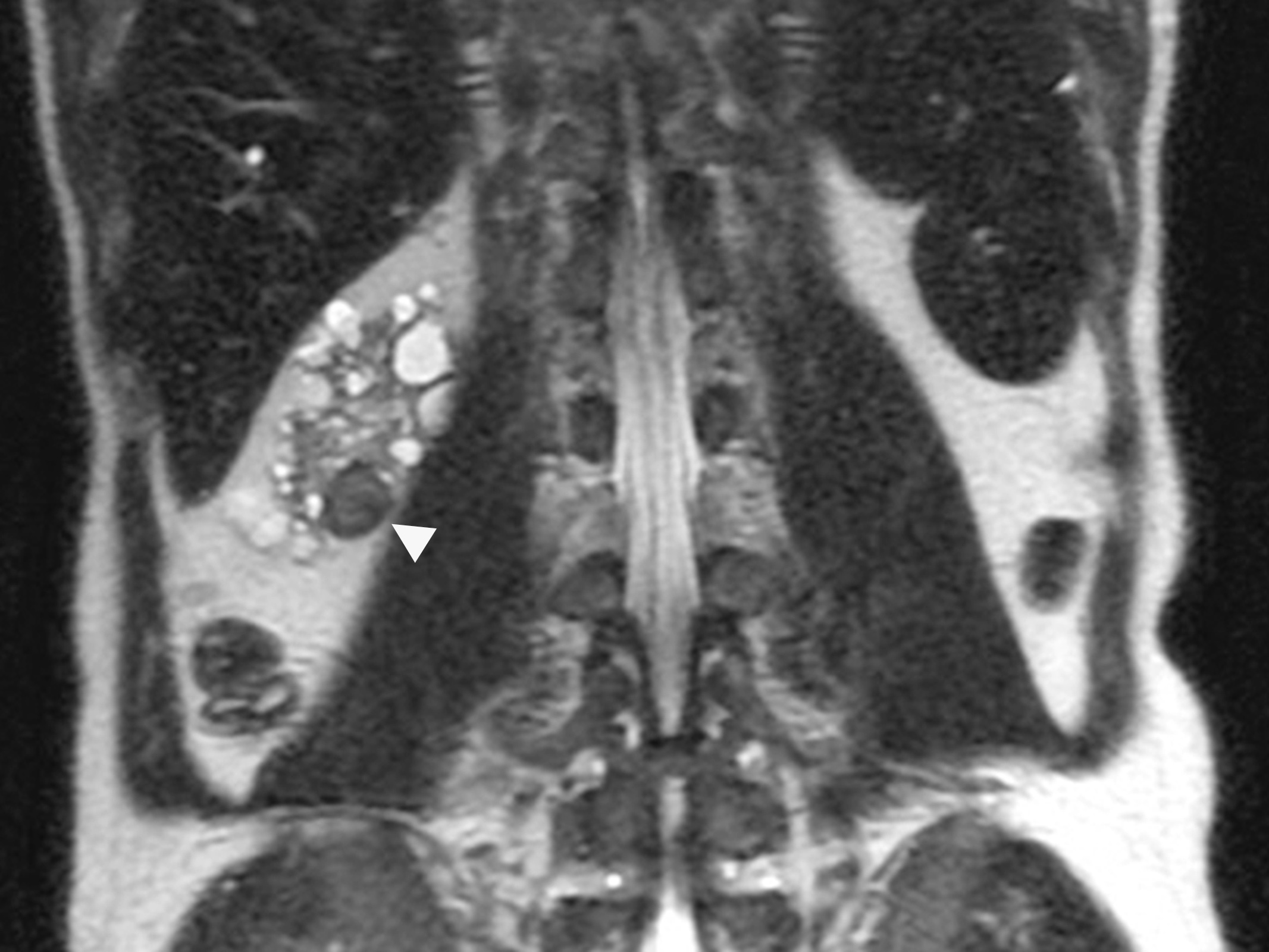
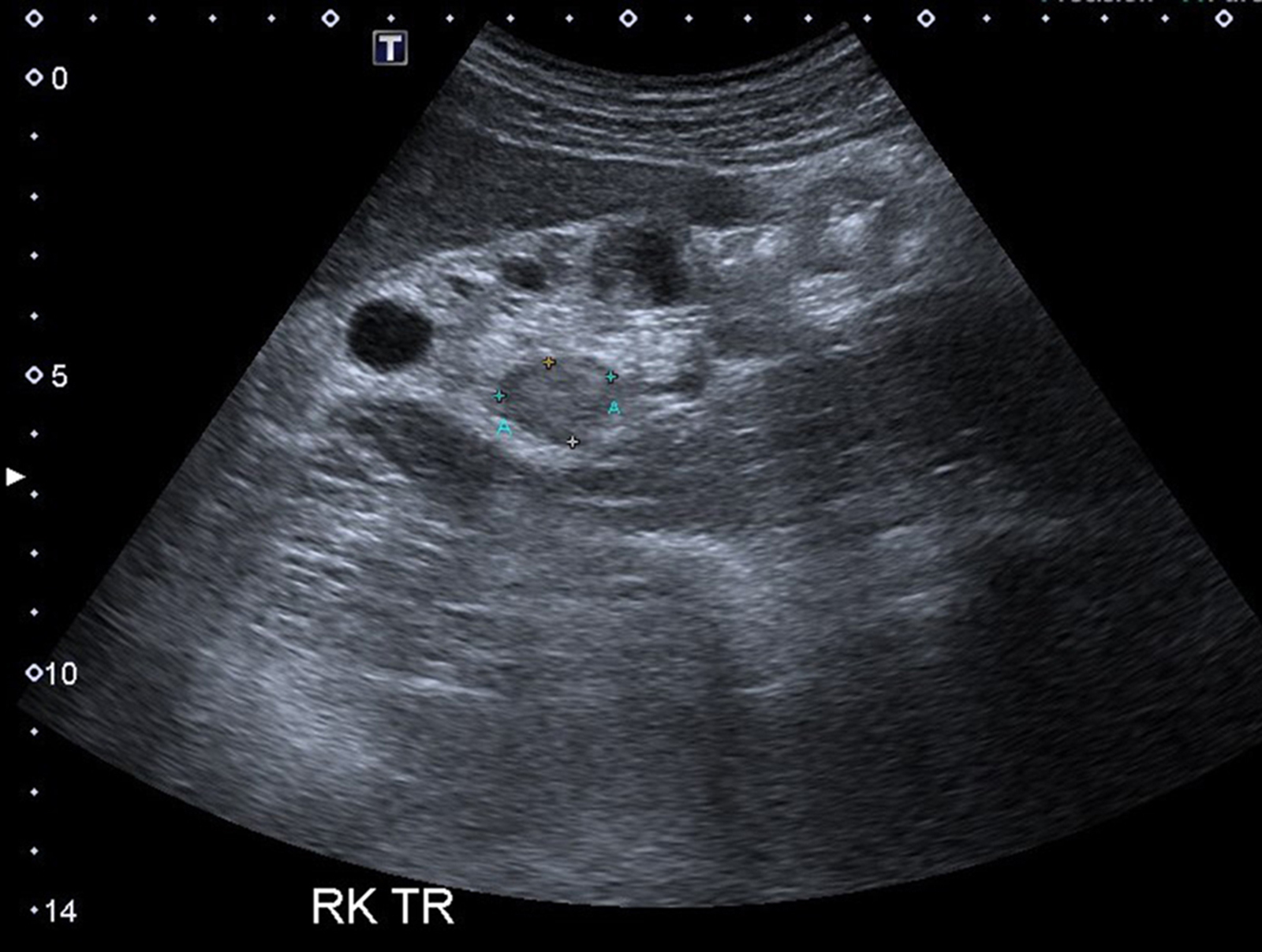
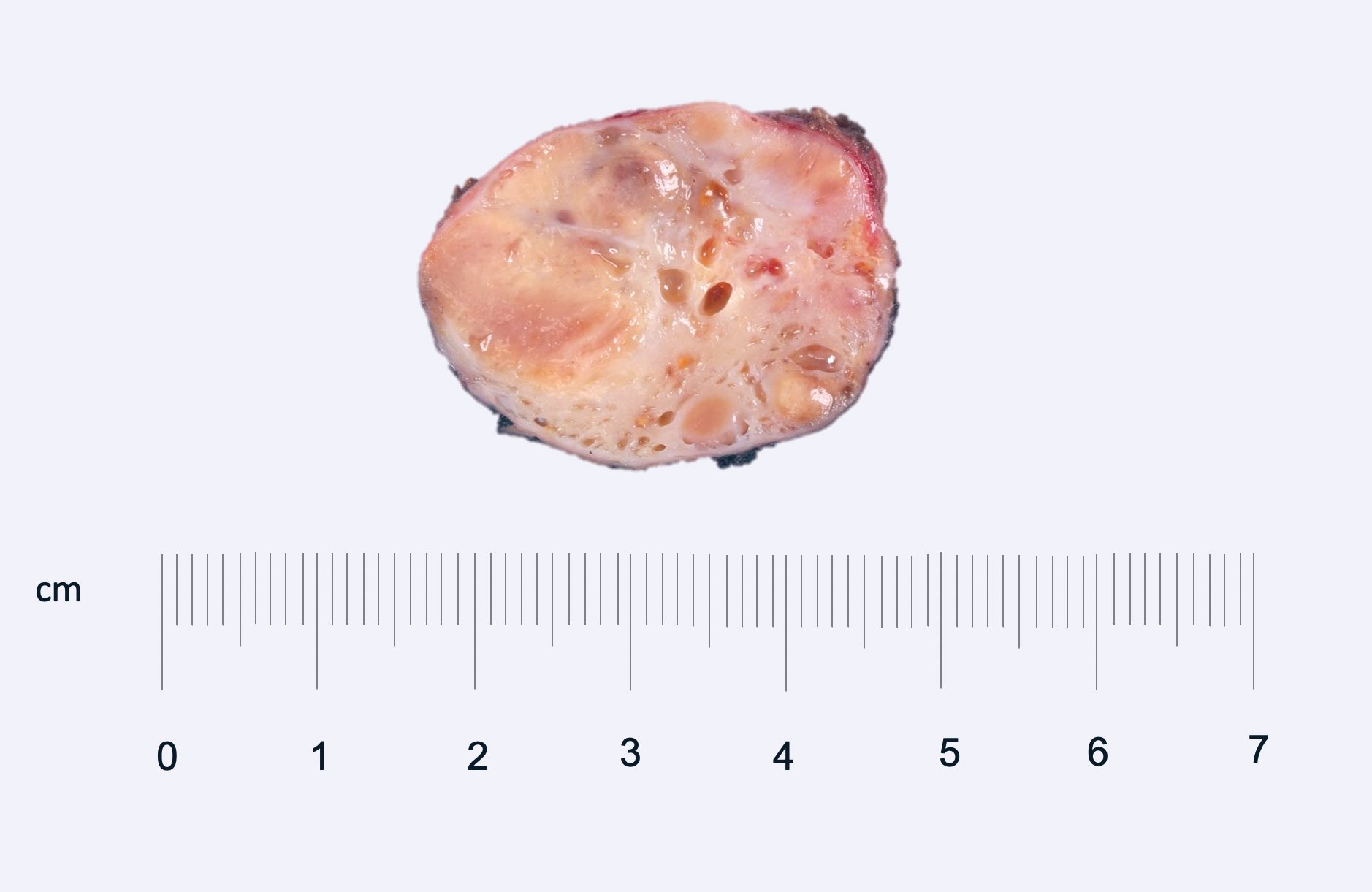
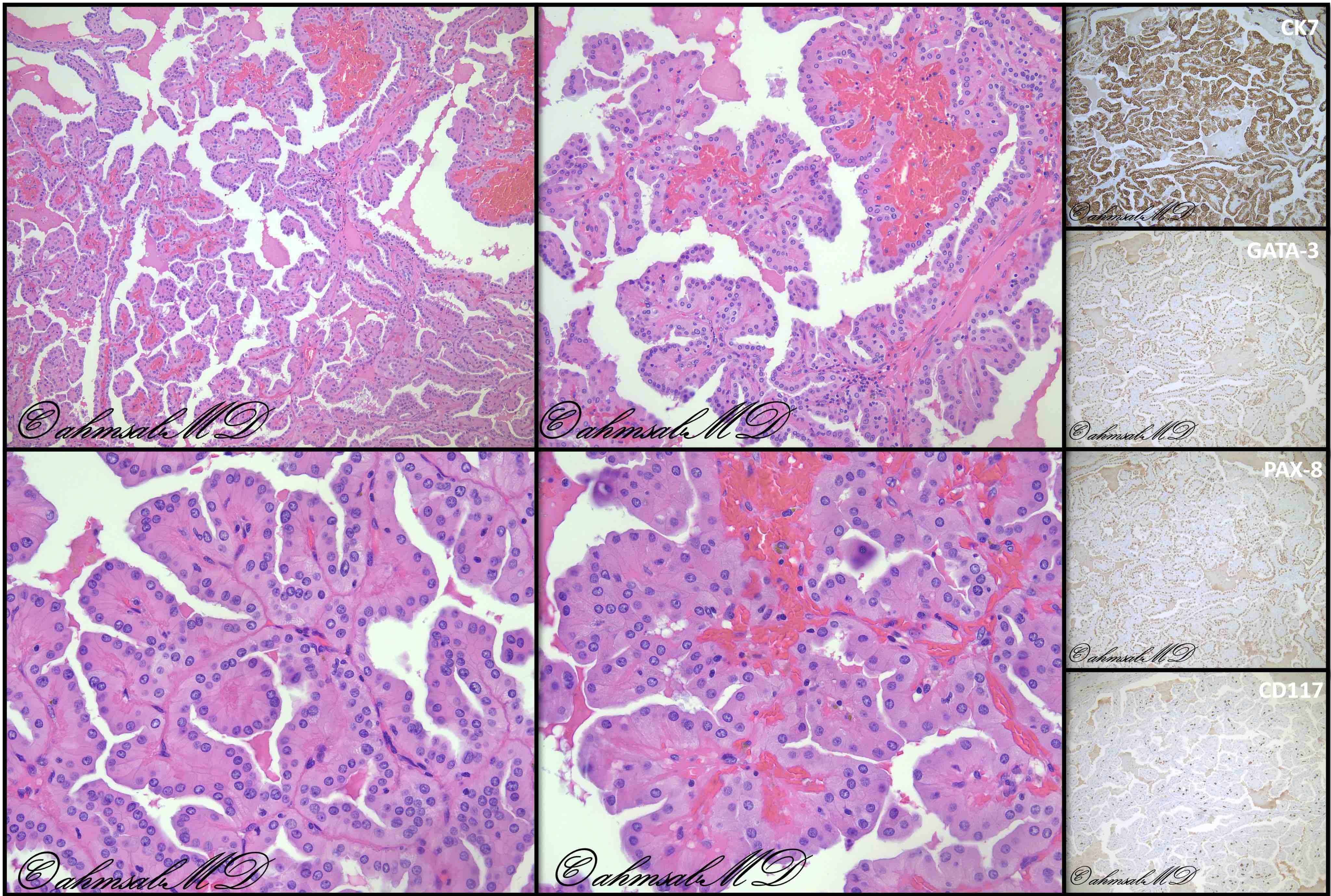
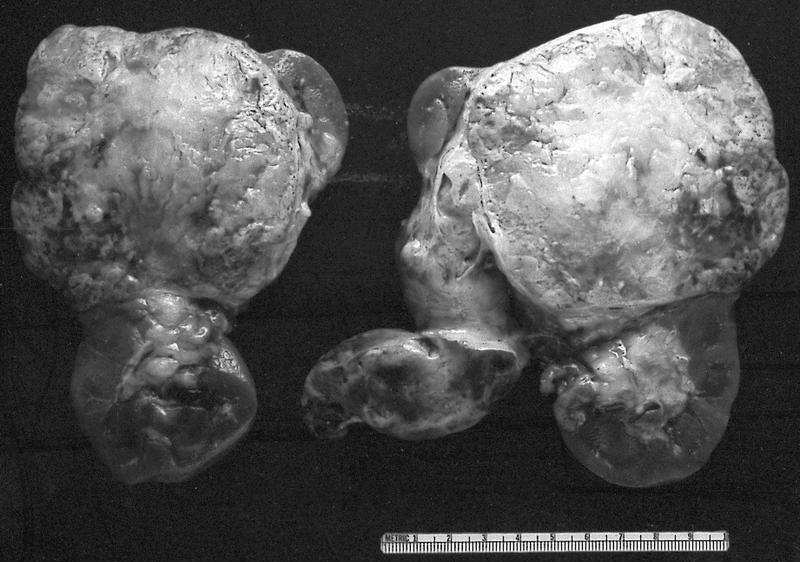
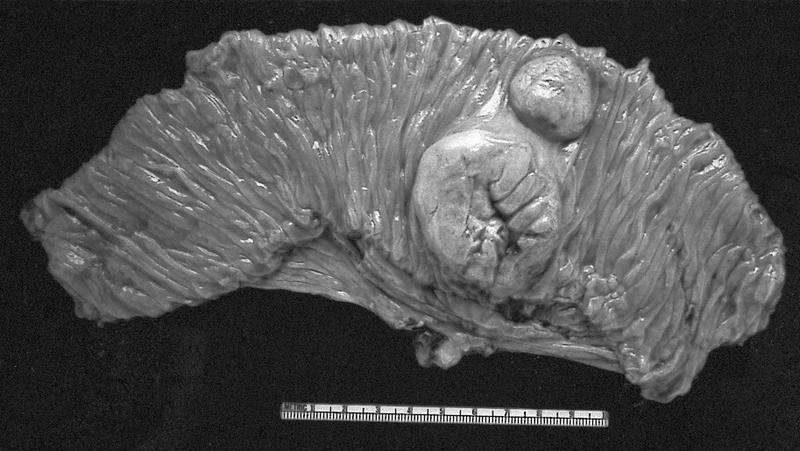
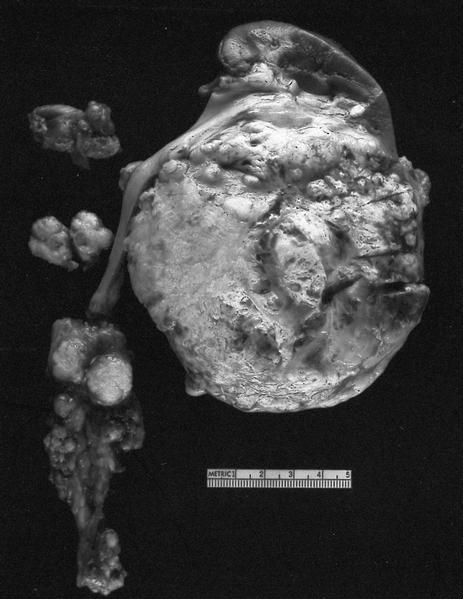
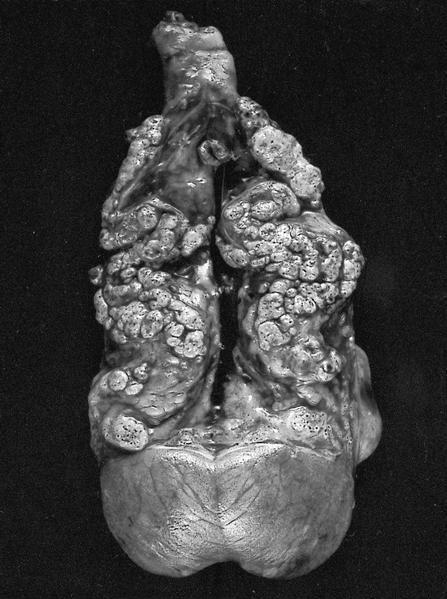
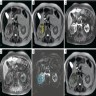
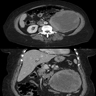
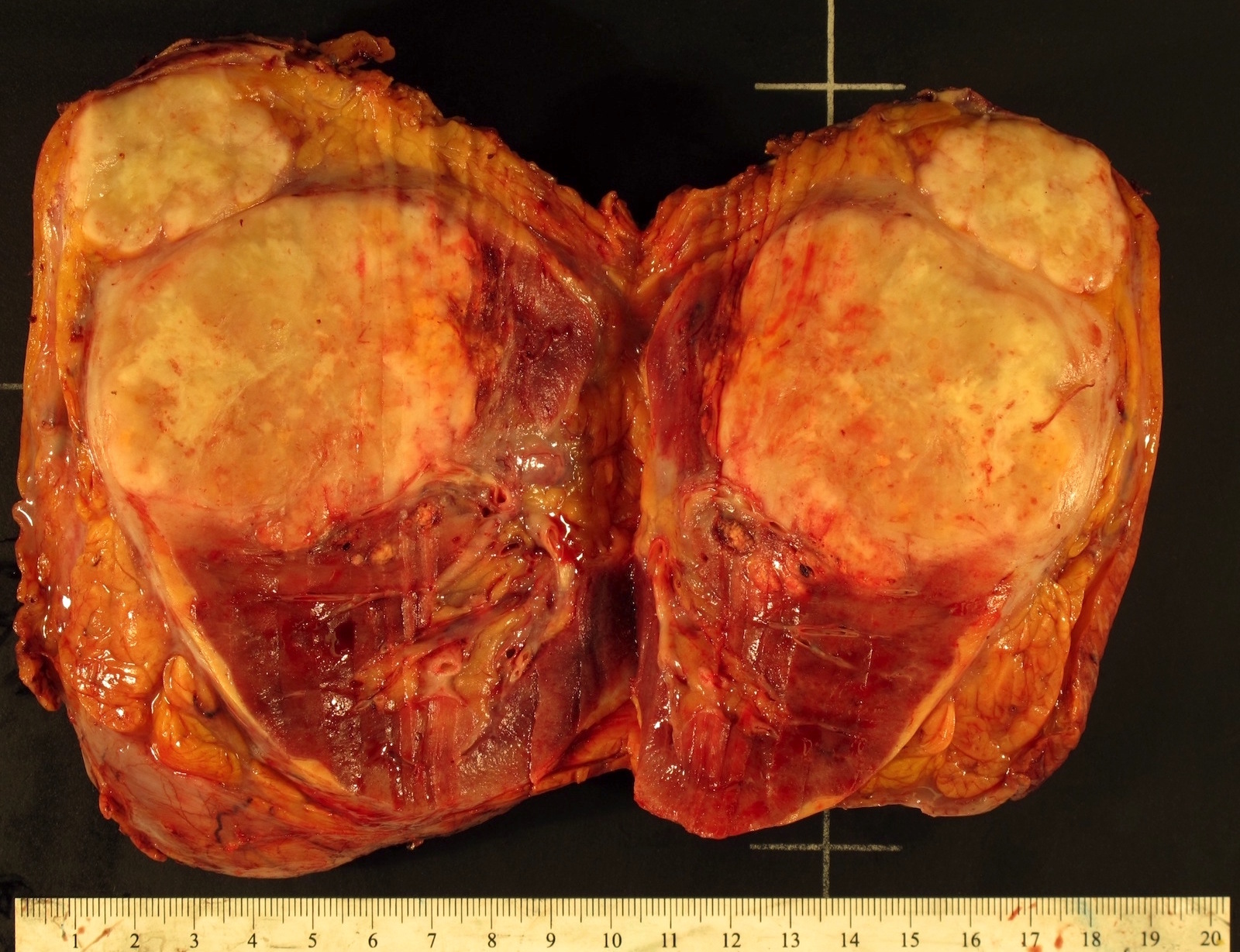
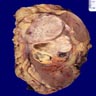
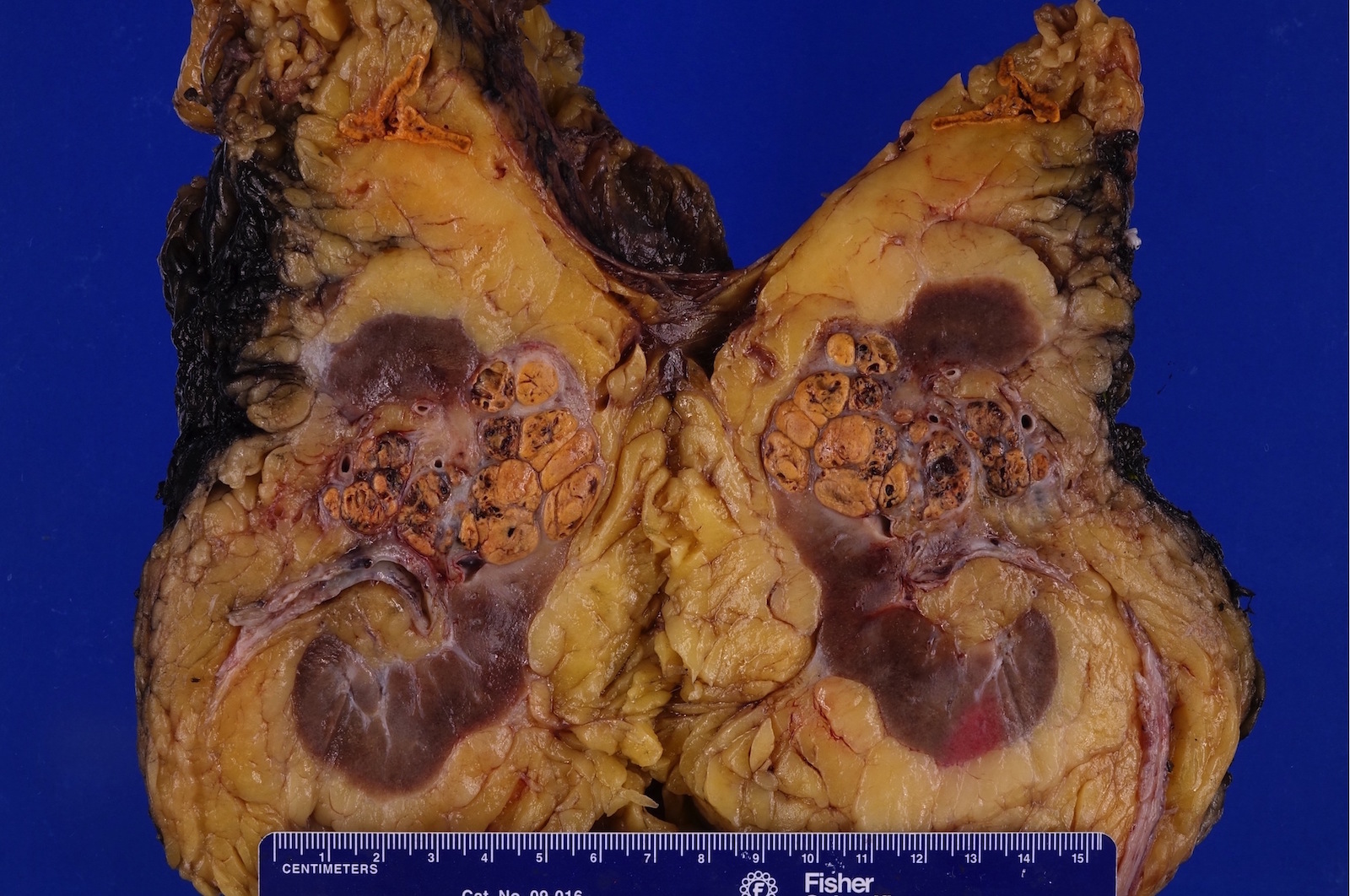
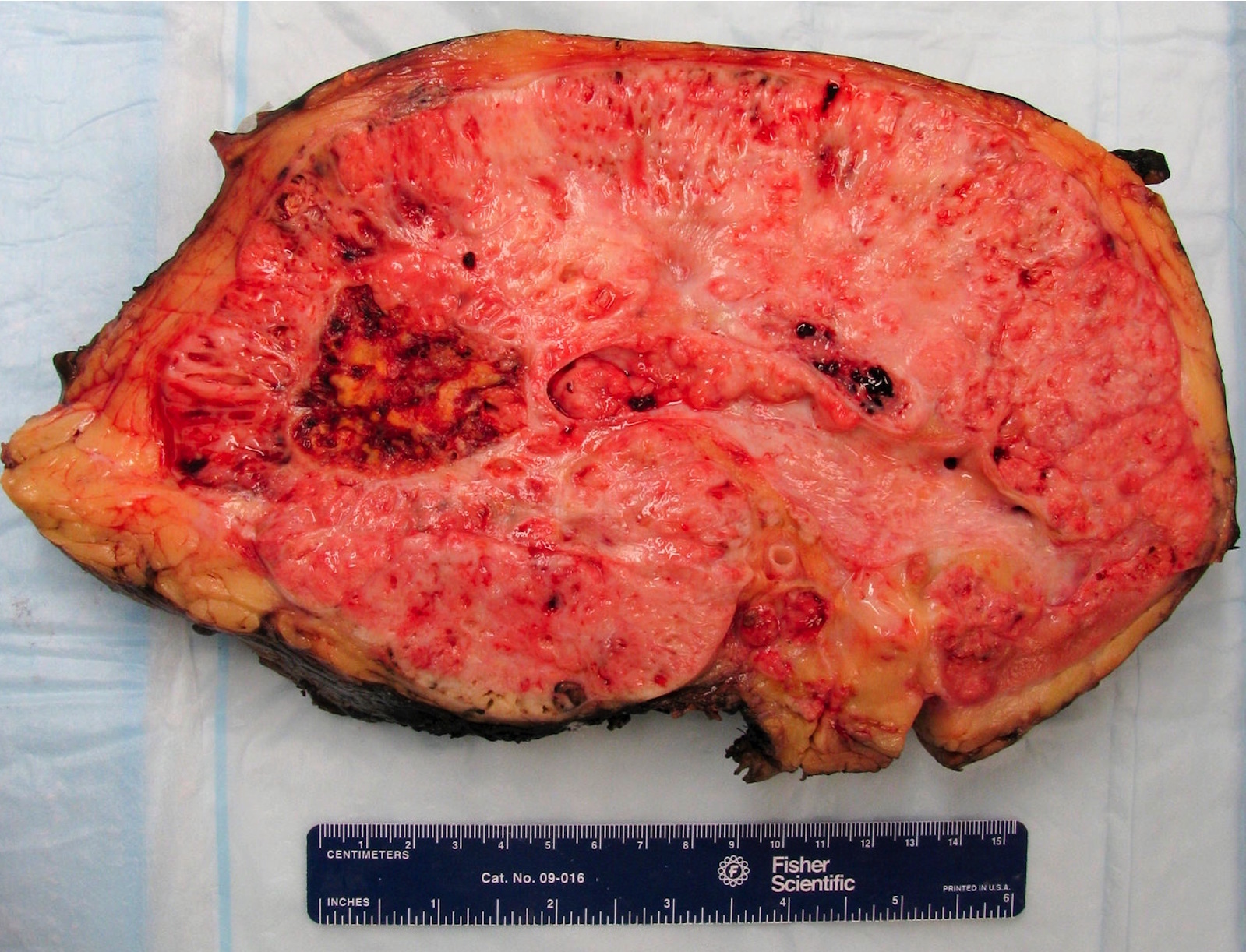
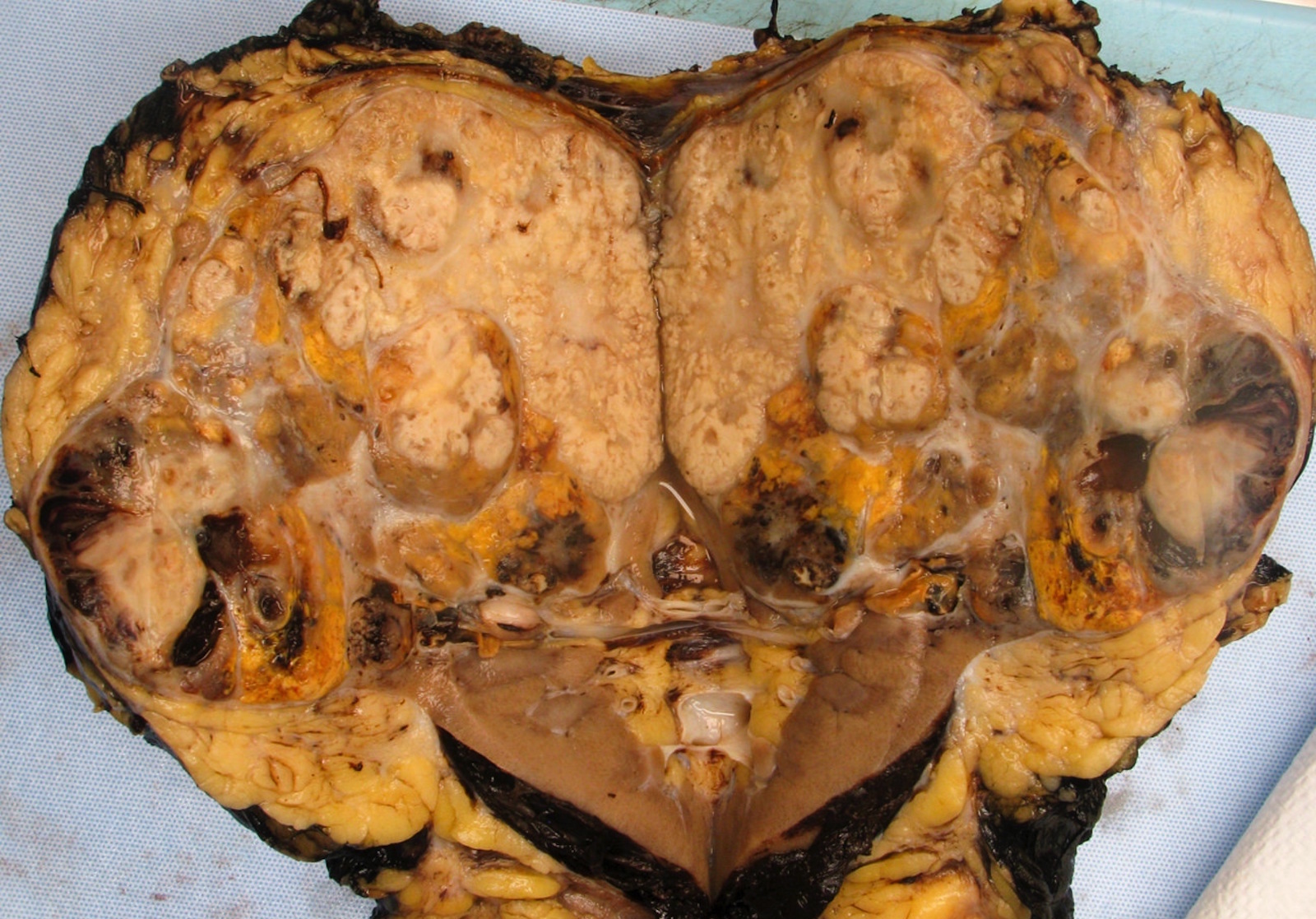
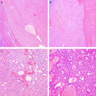
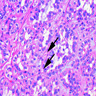
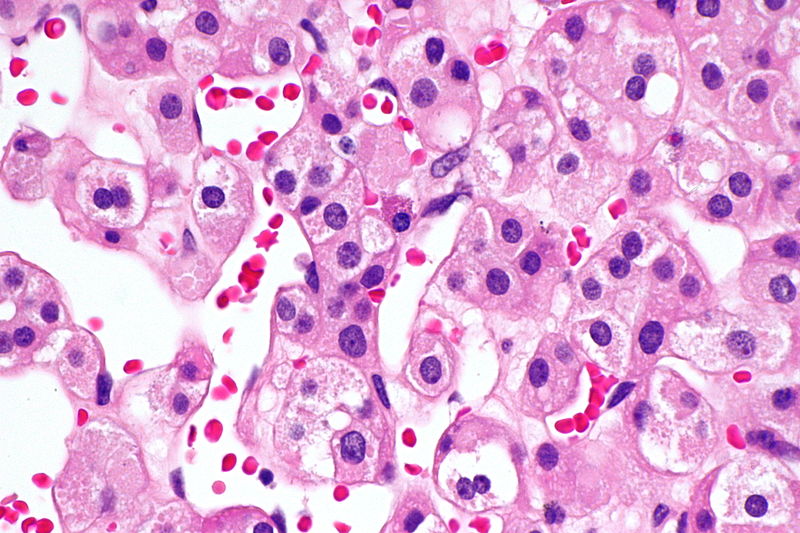
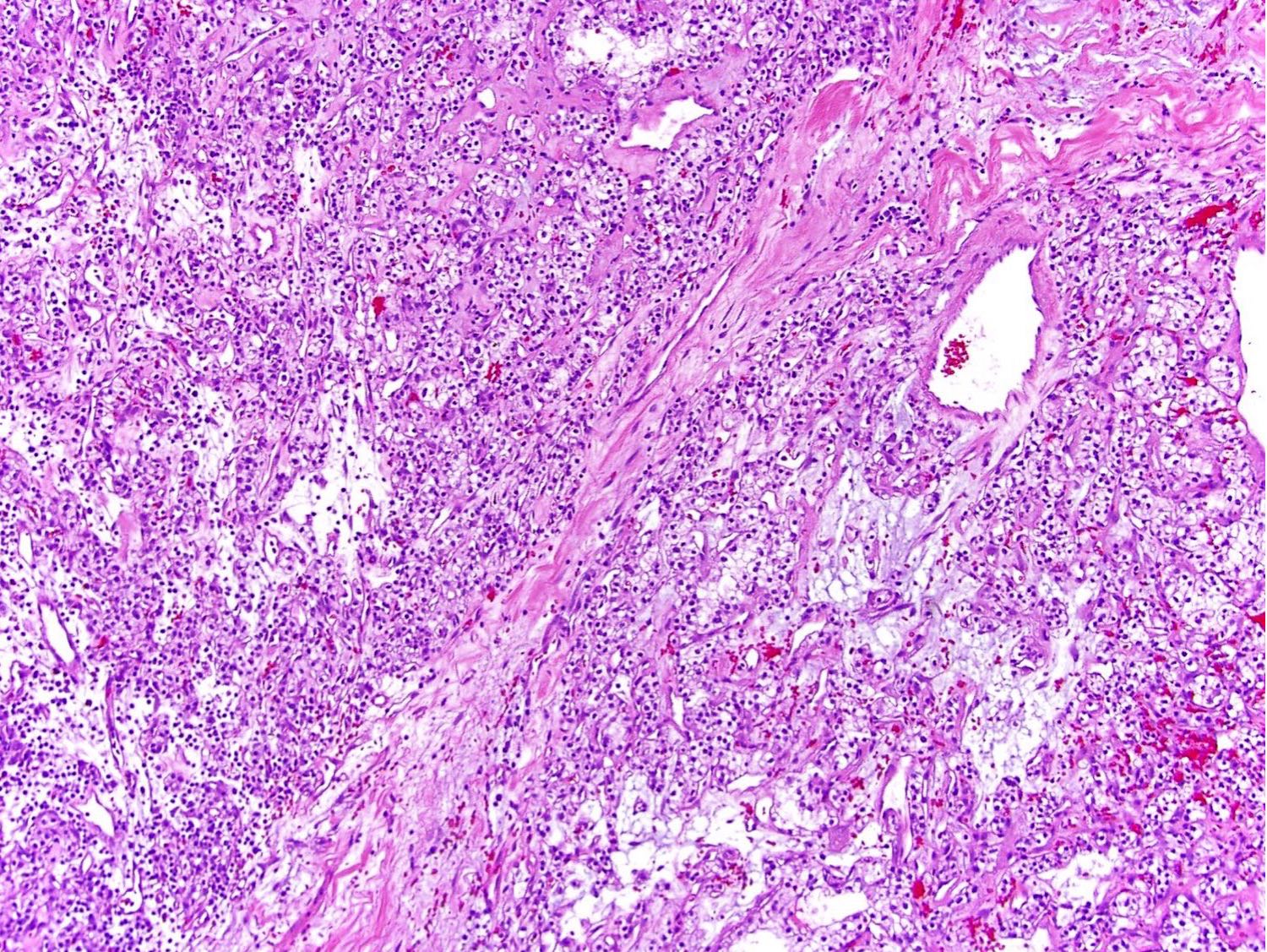
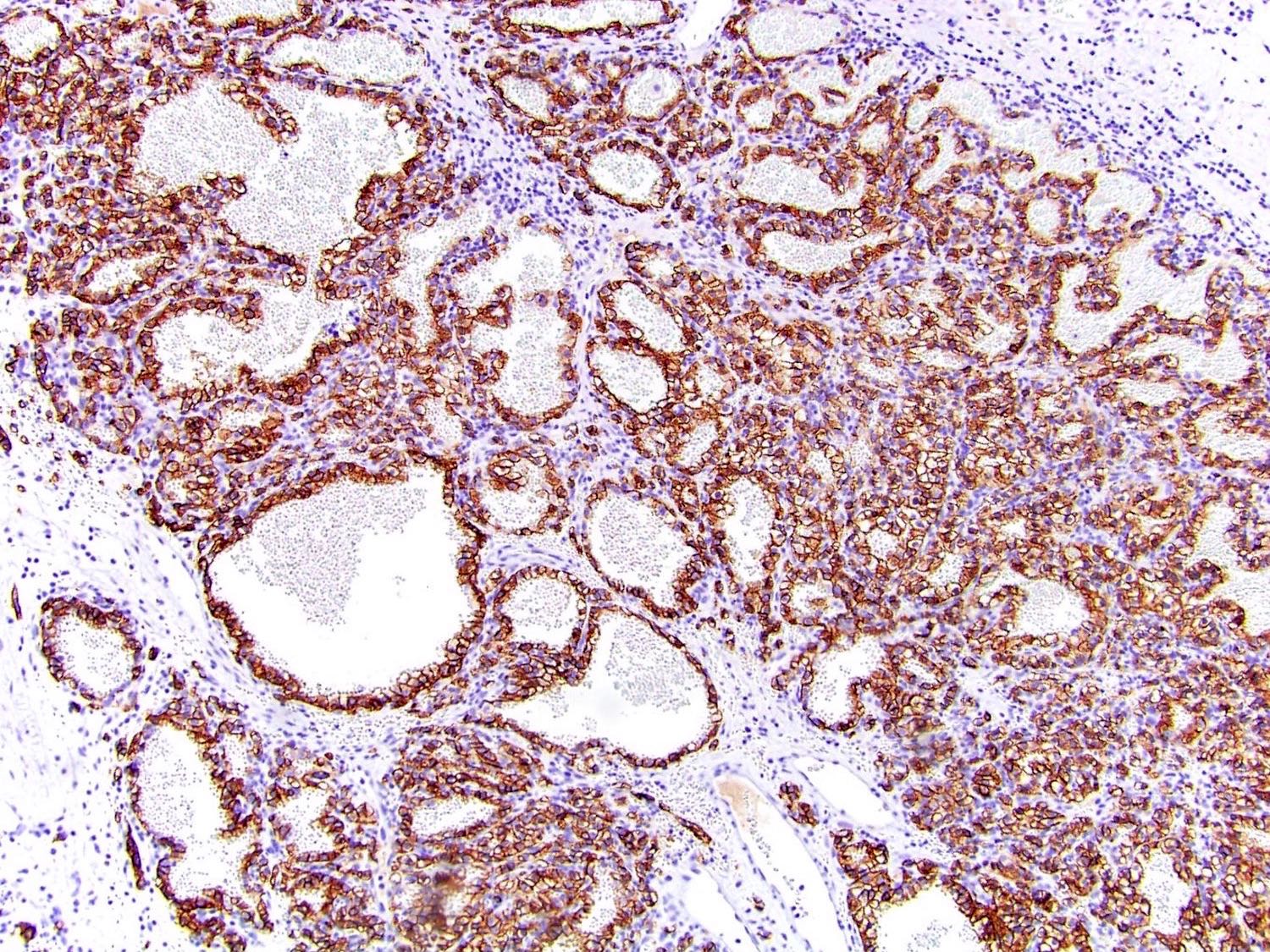
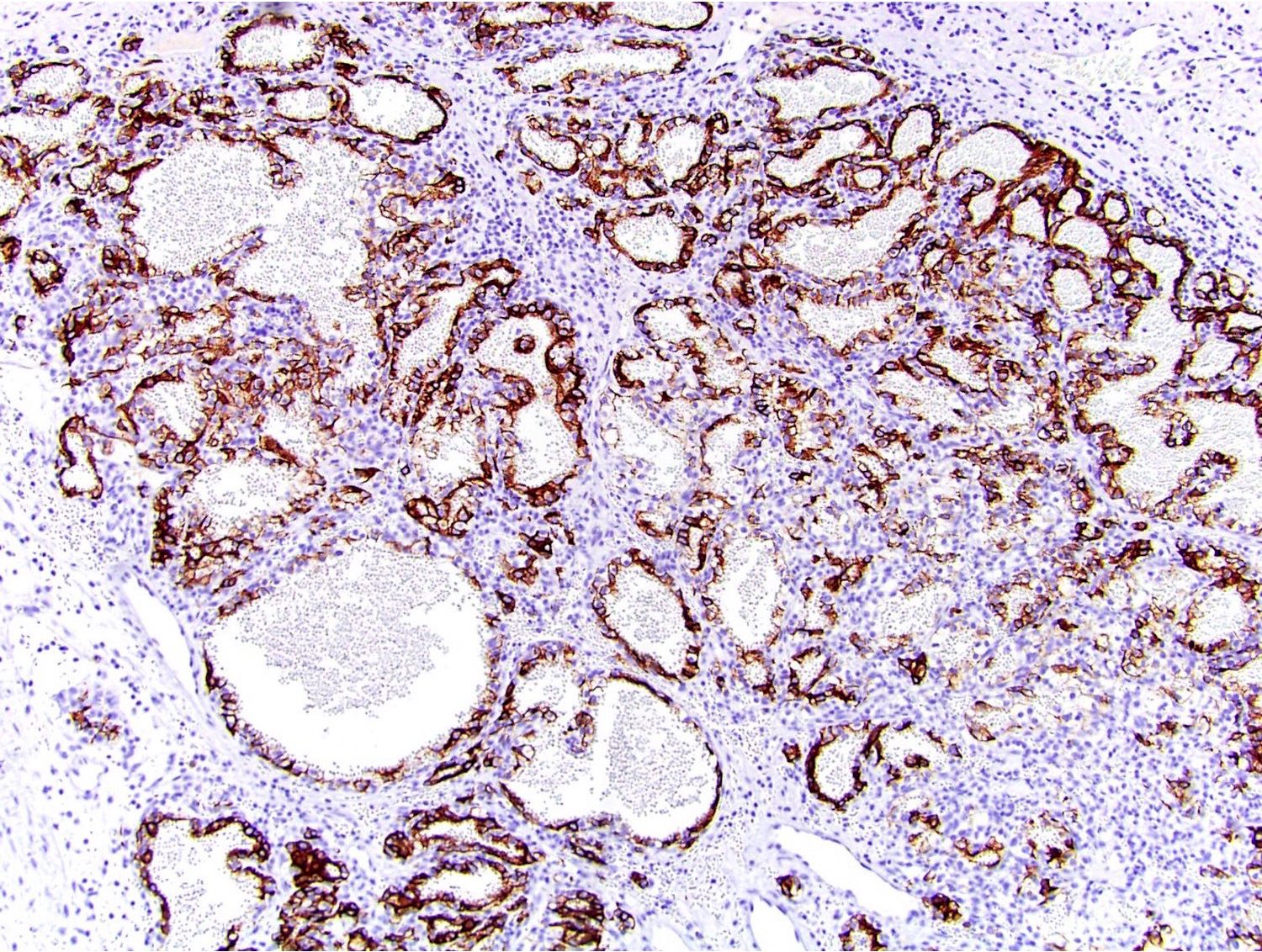
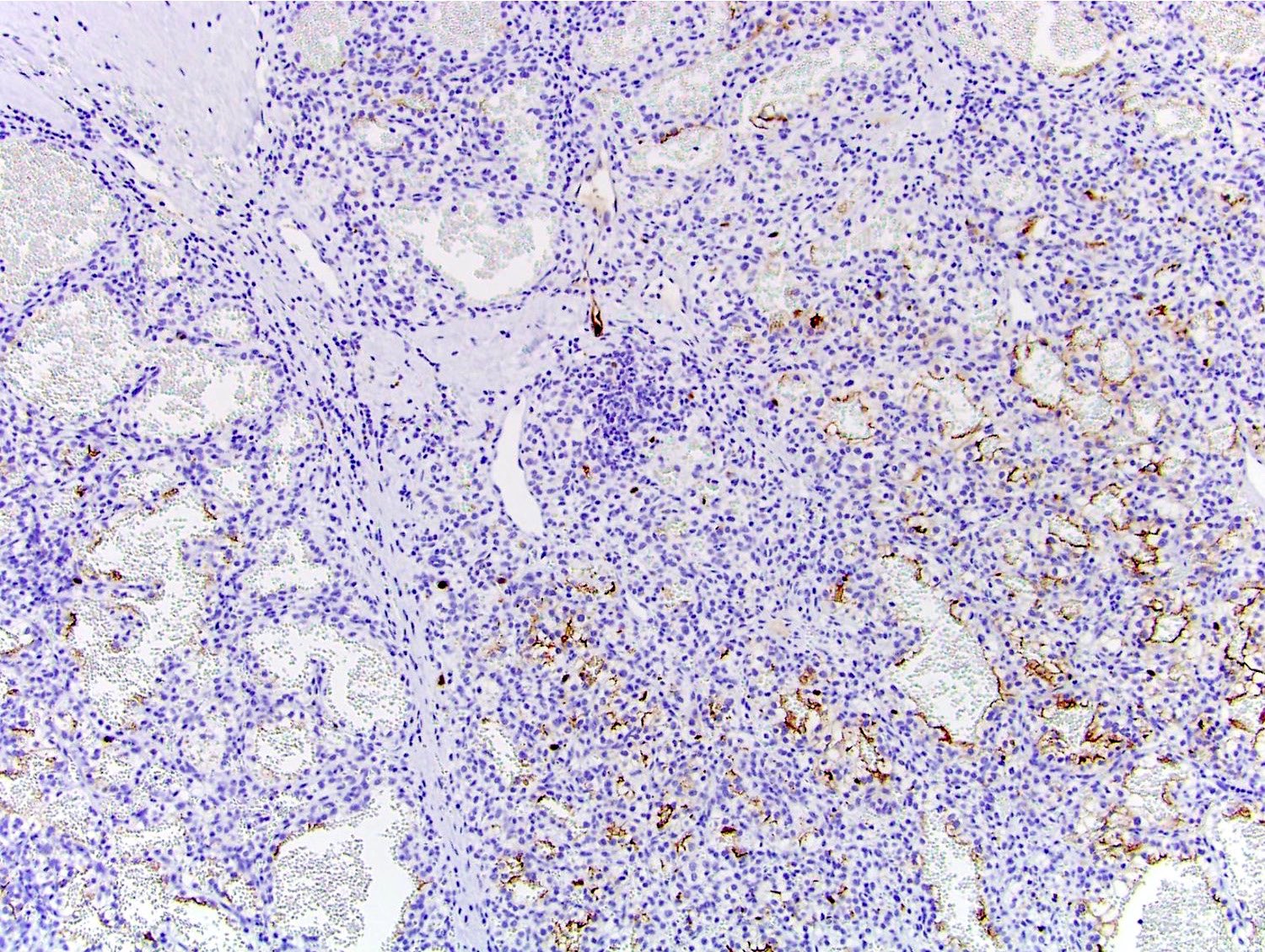
B. Detection of
ALK rearrangement is critical for diagnosis.
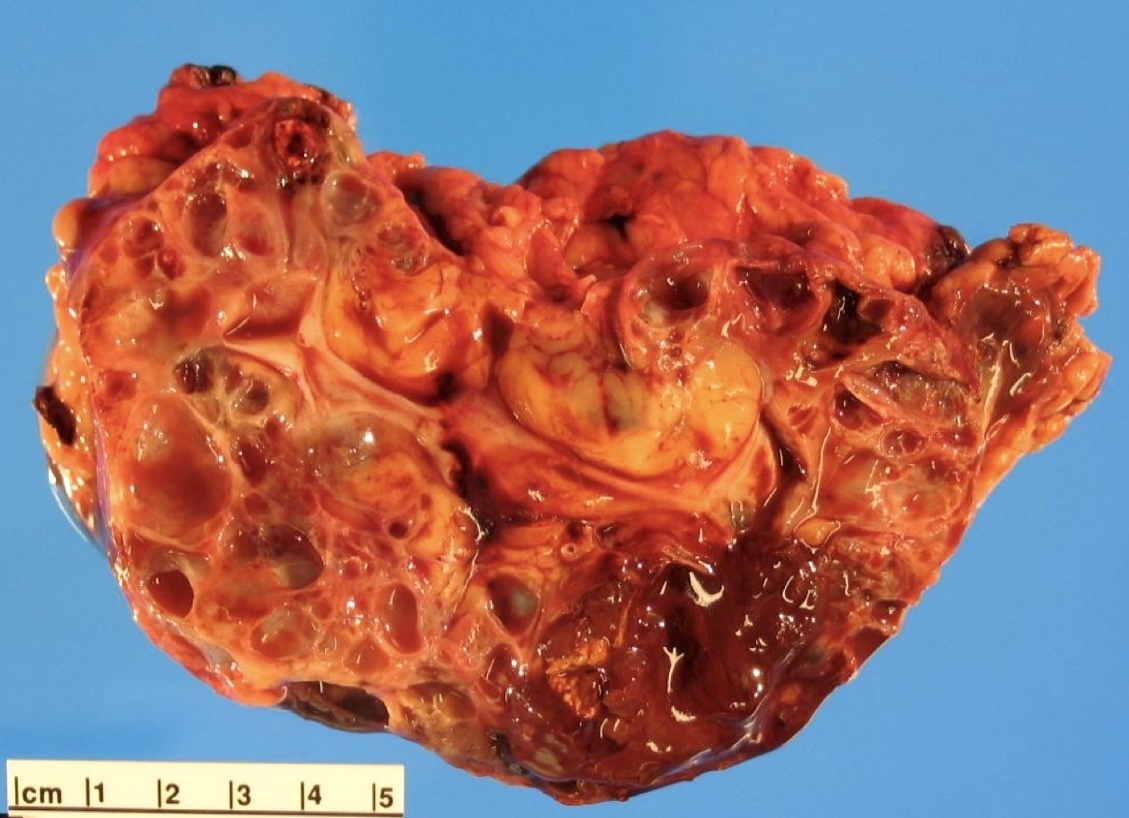
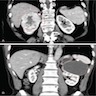
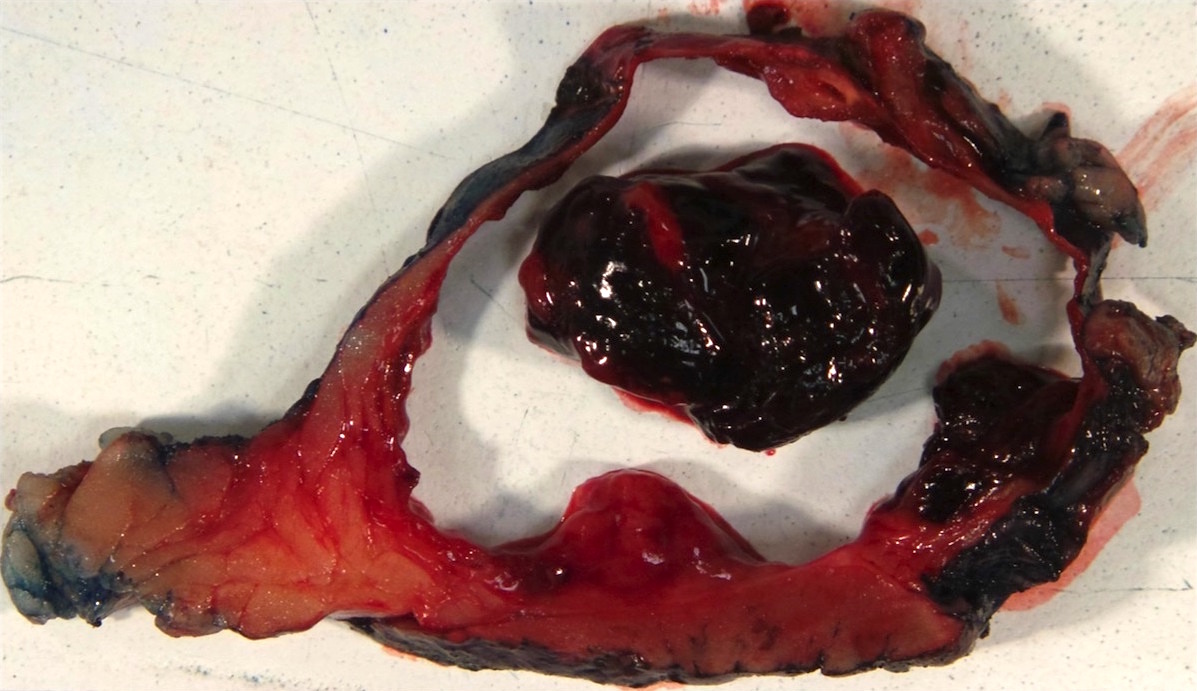
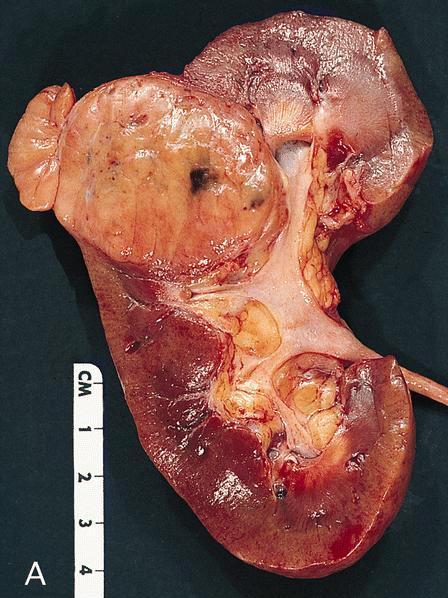
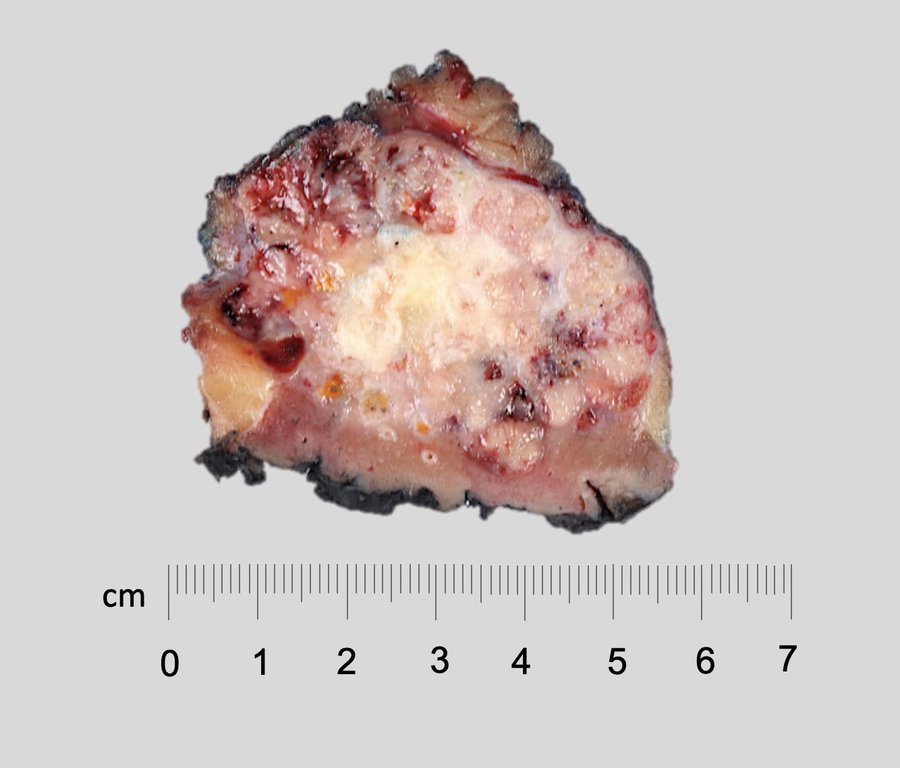
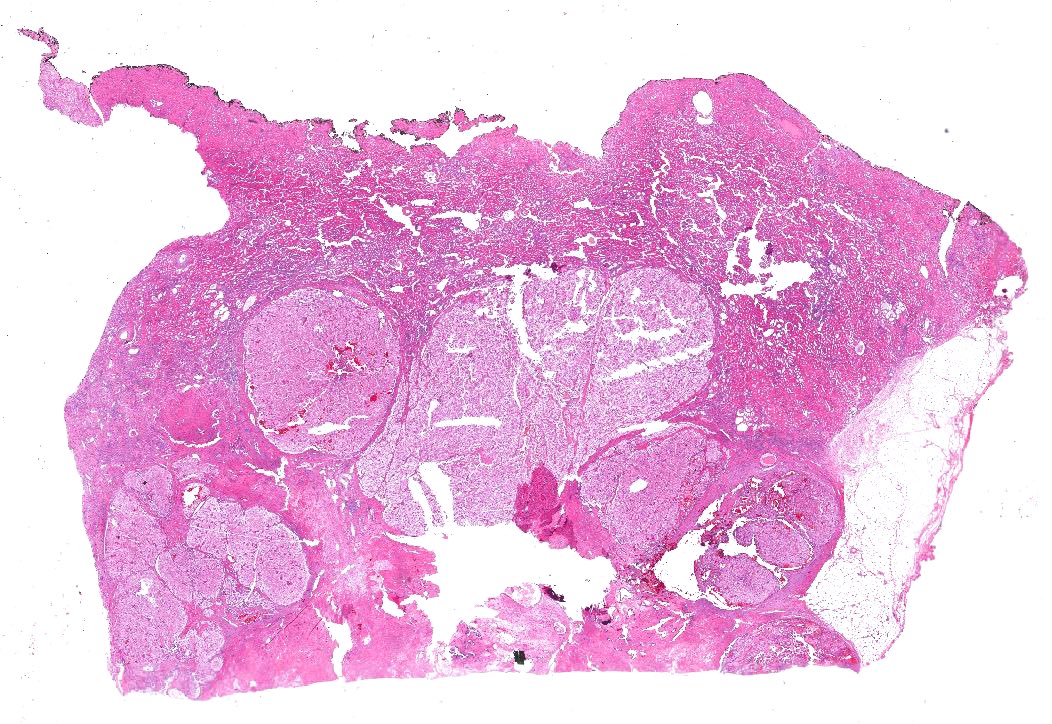
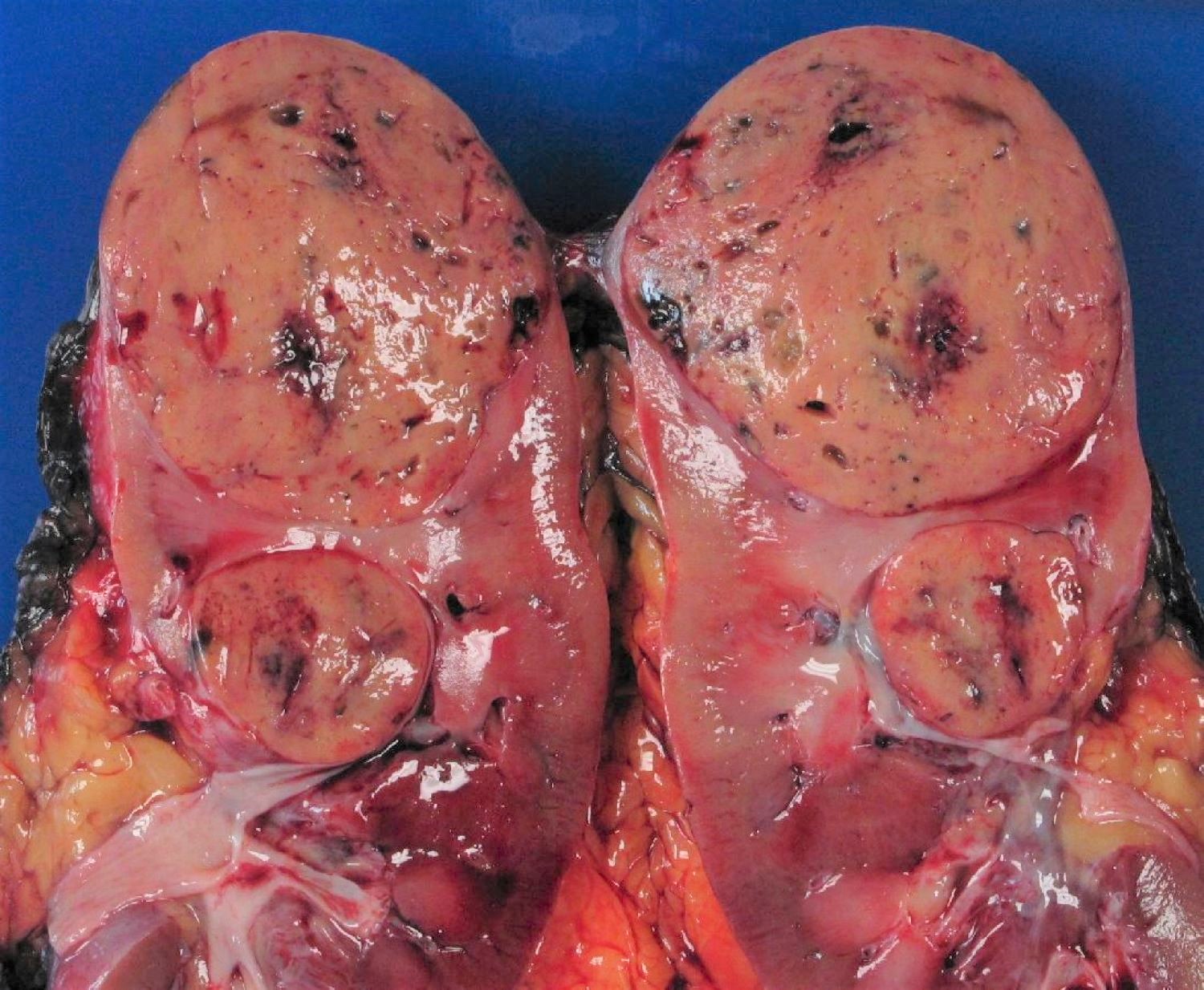
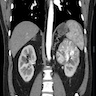
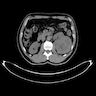
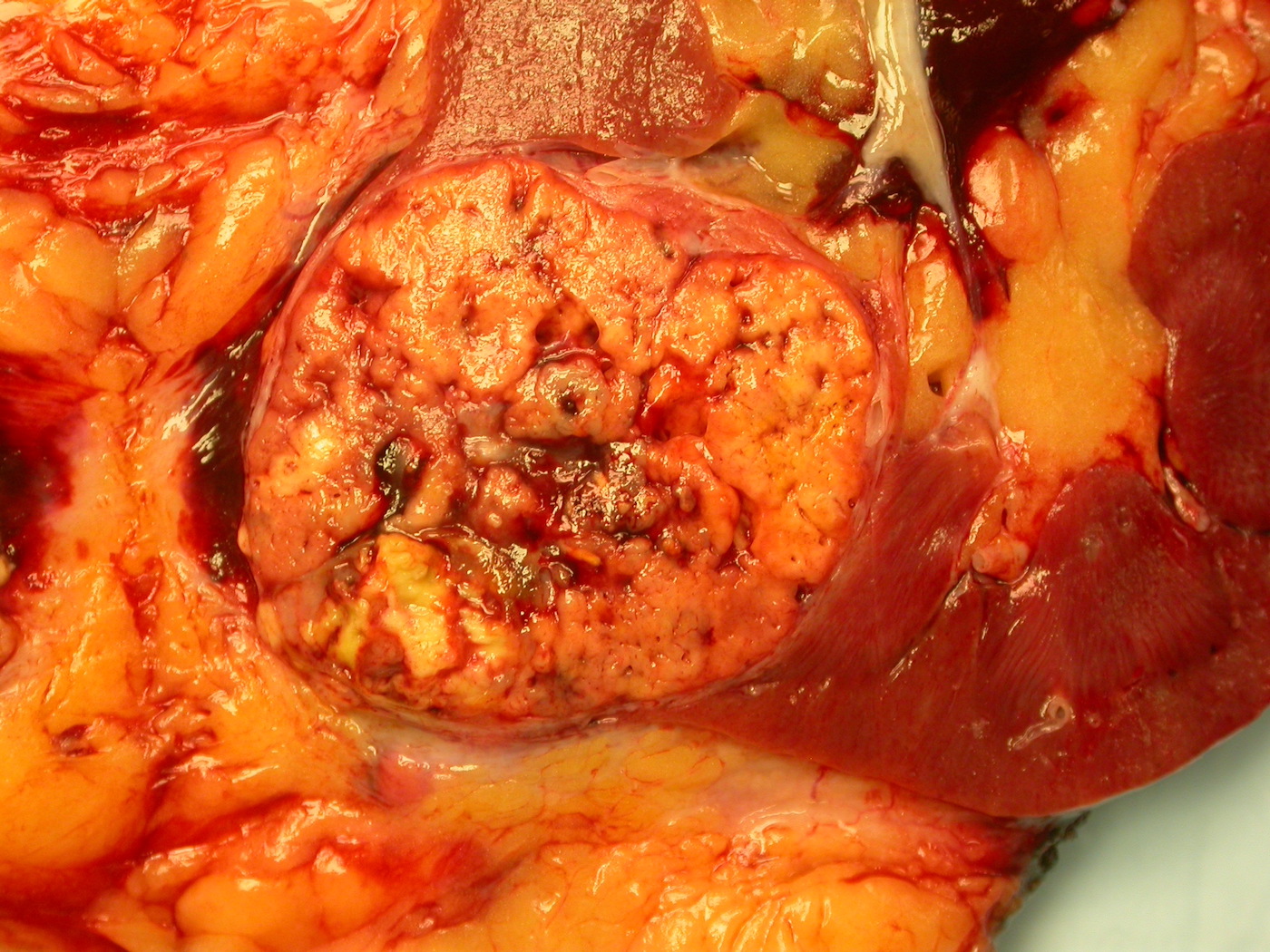
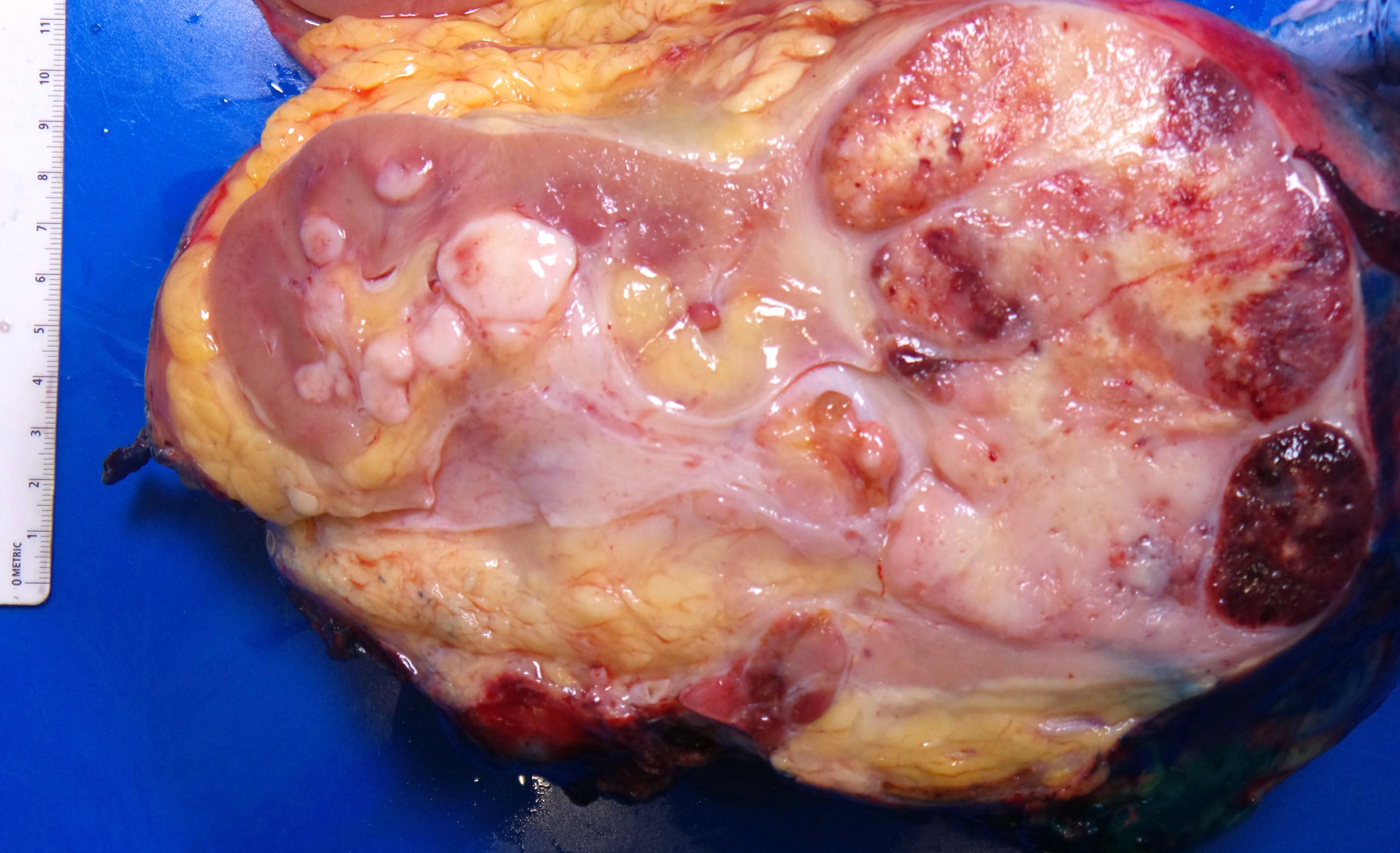
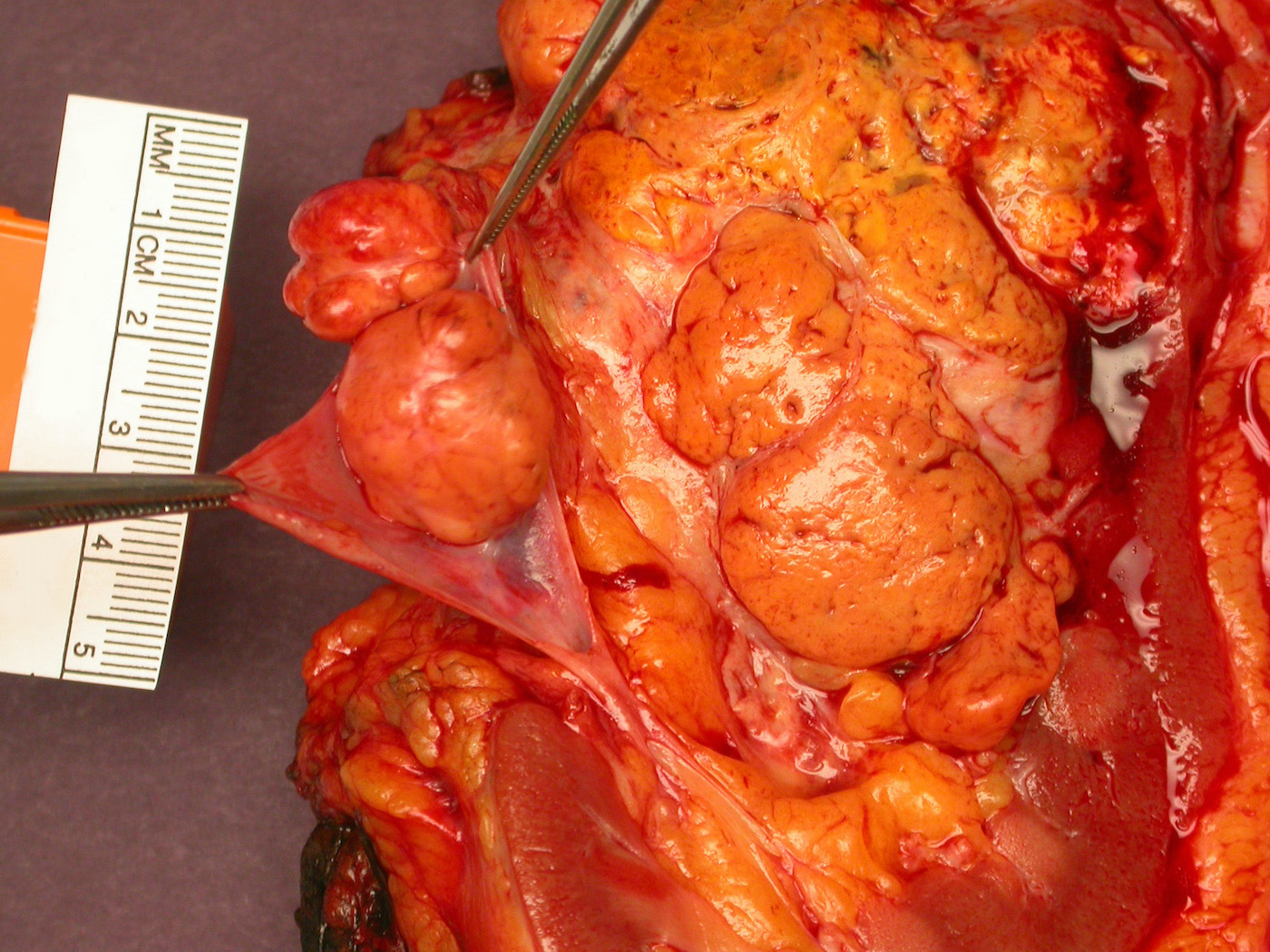
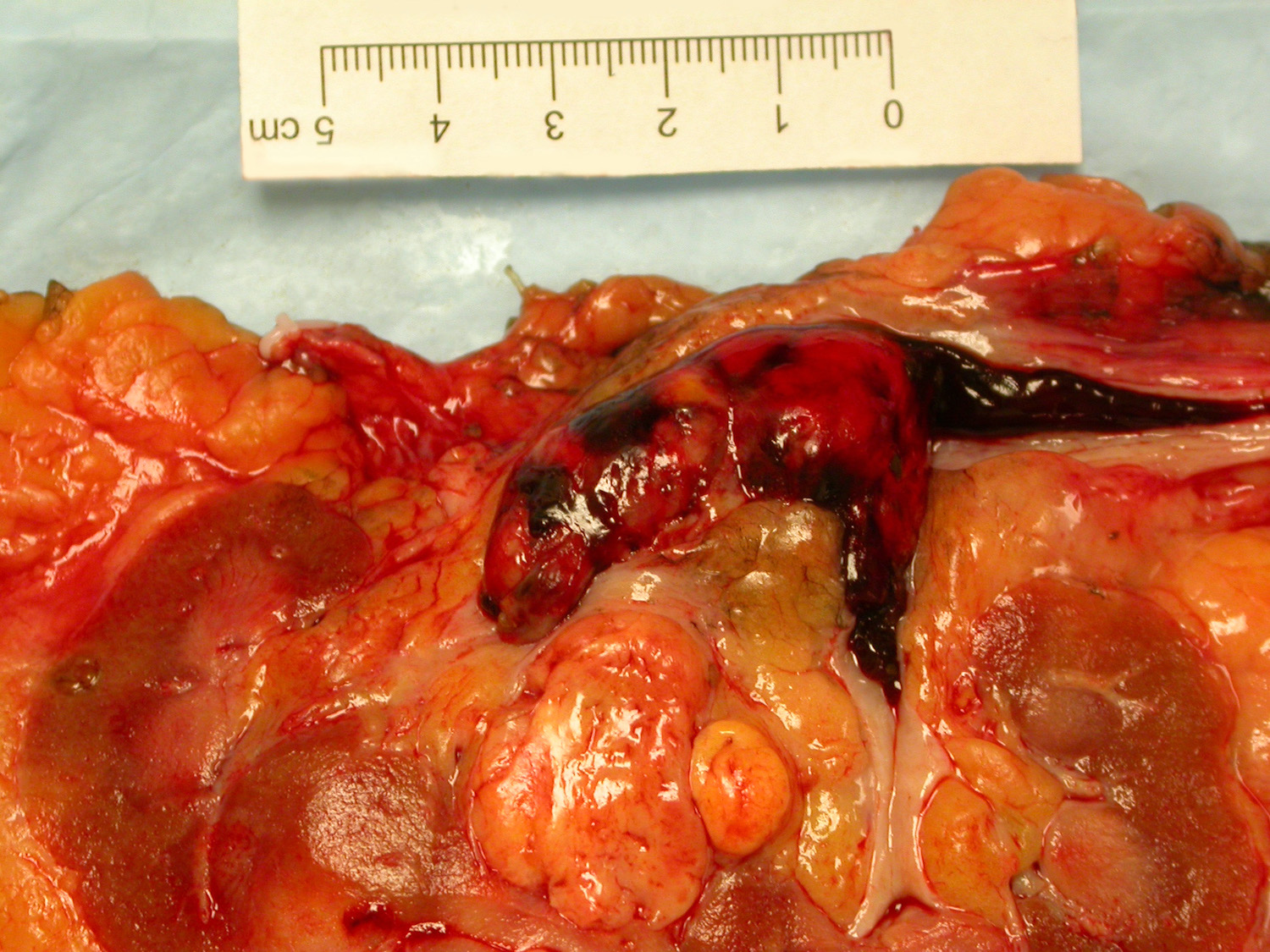
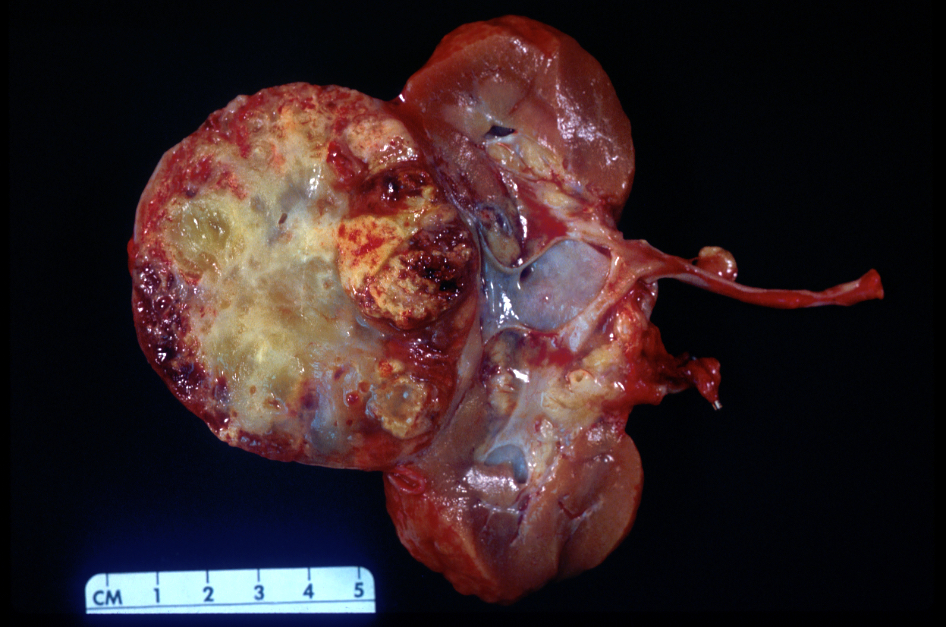
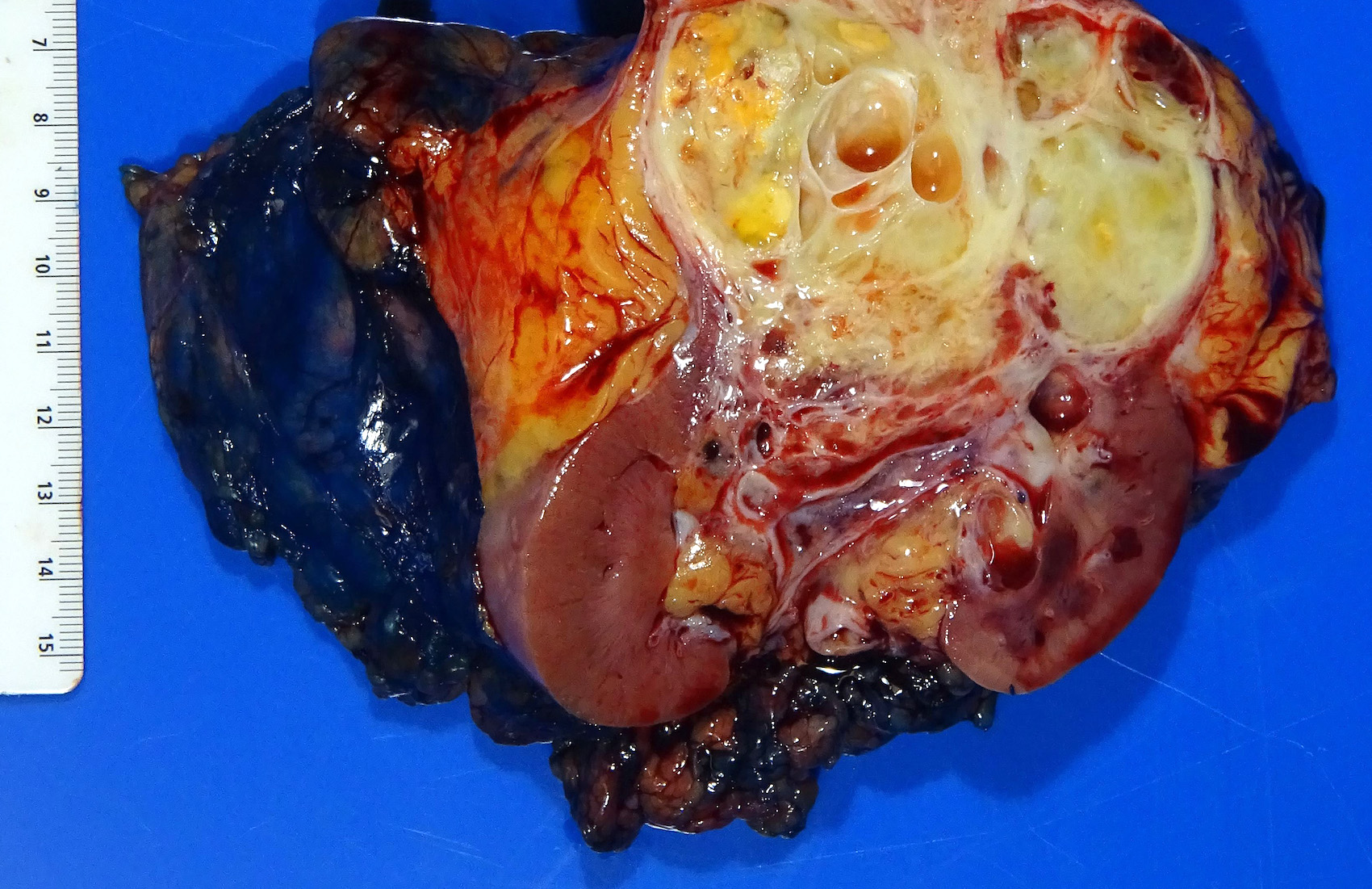
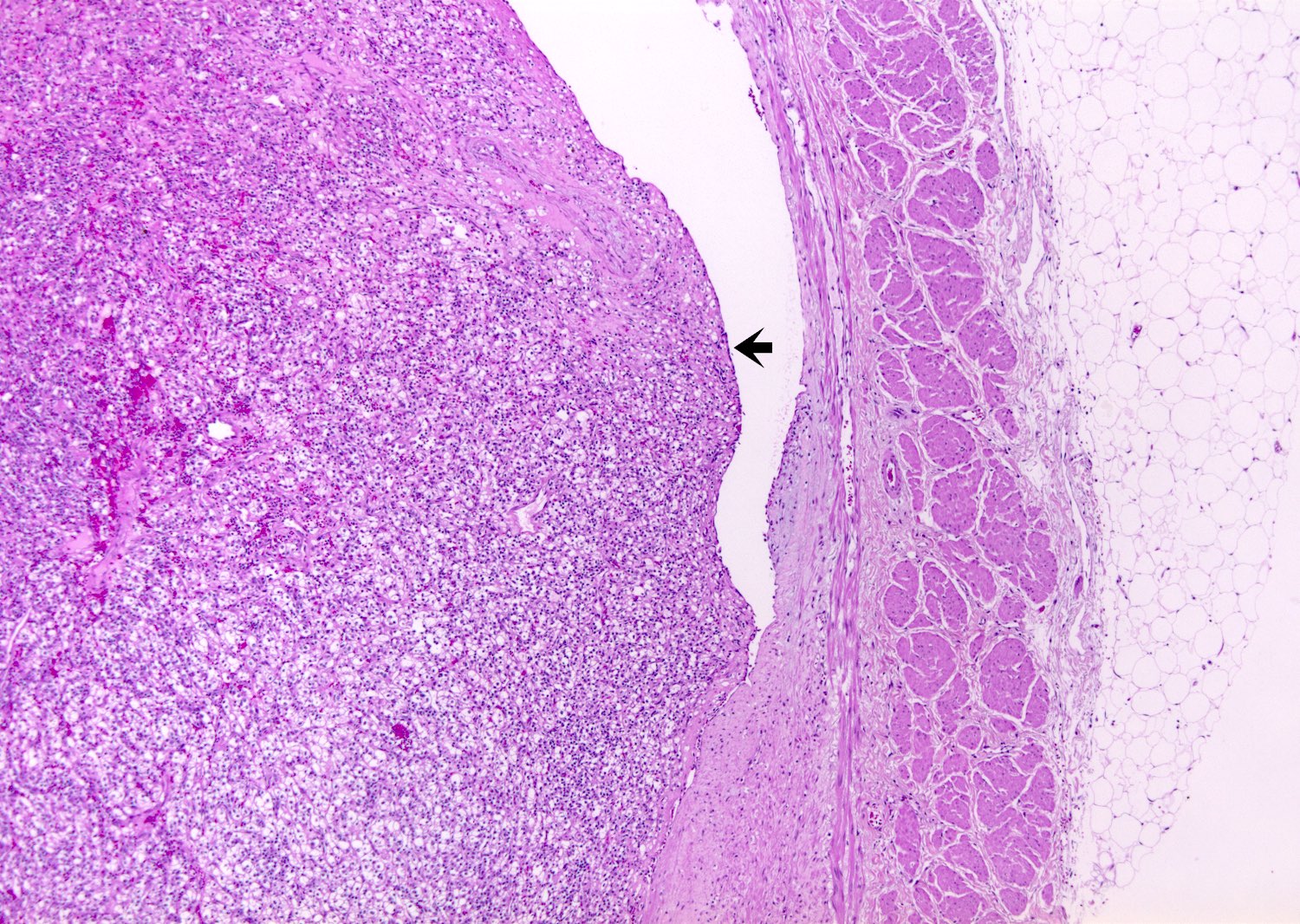
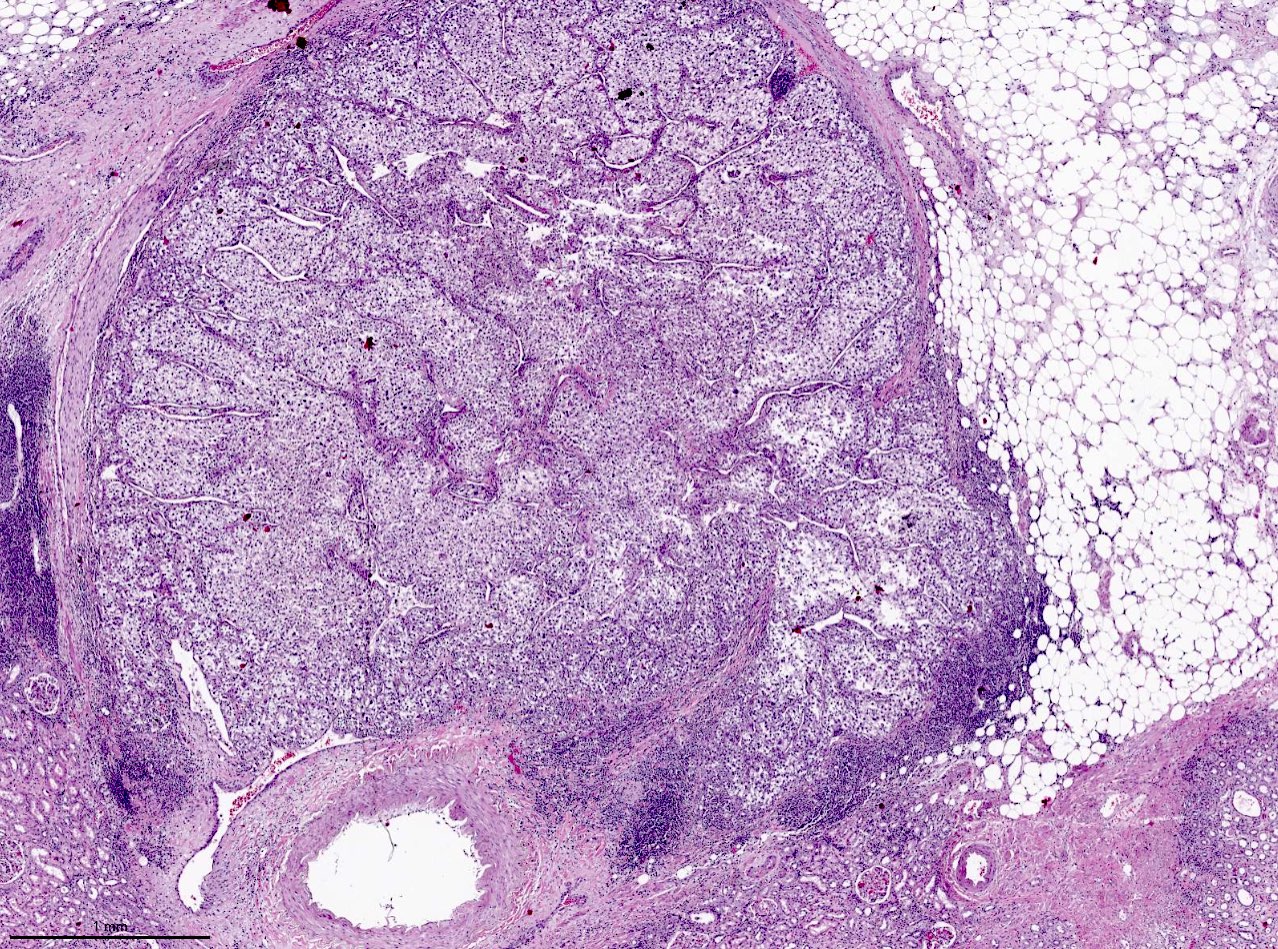
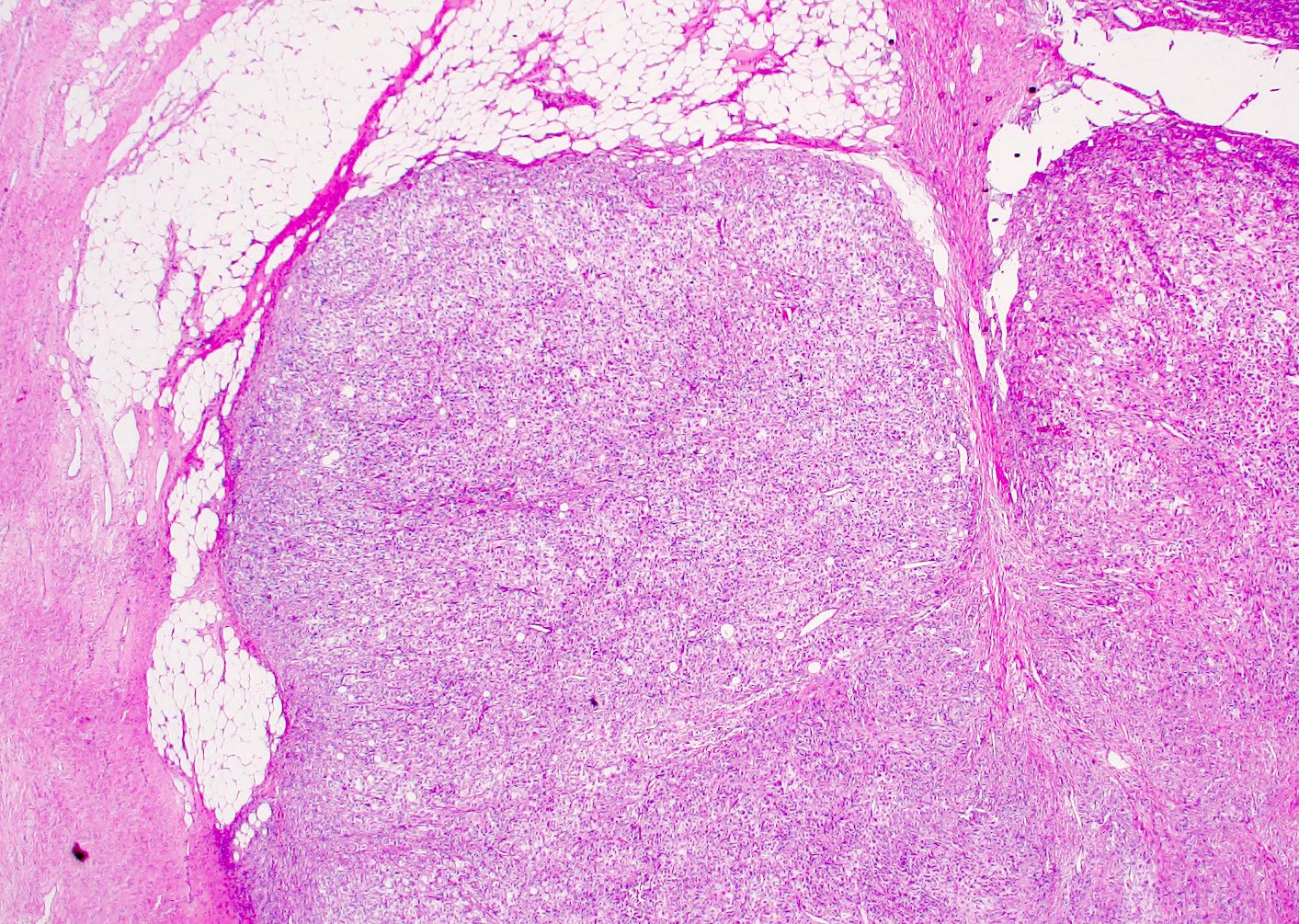
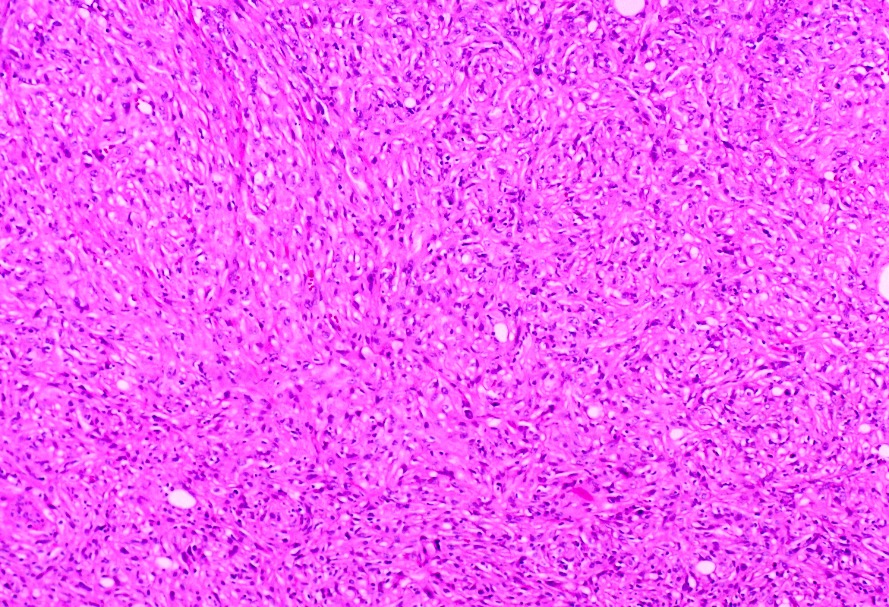
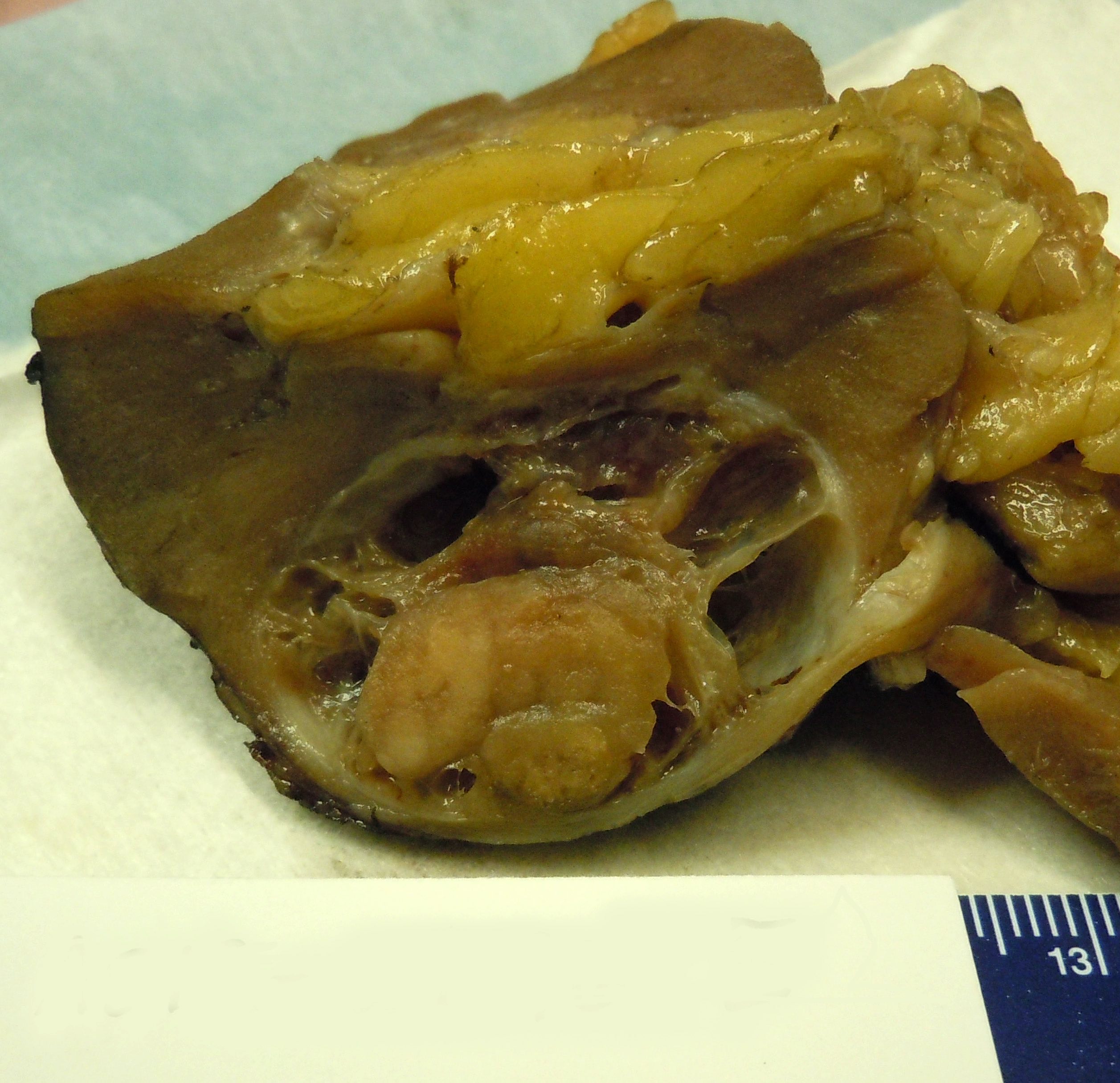
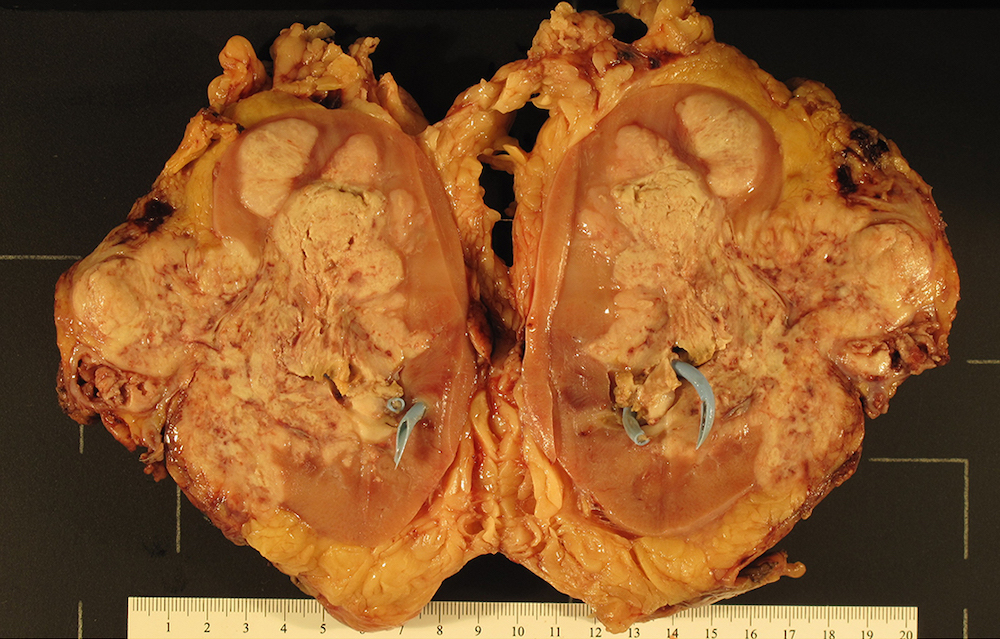
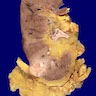
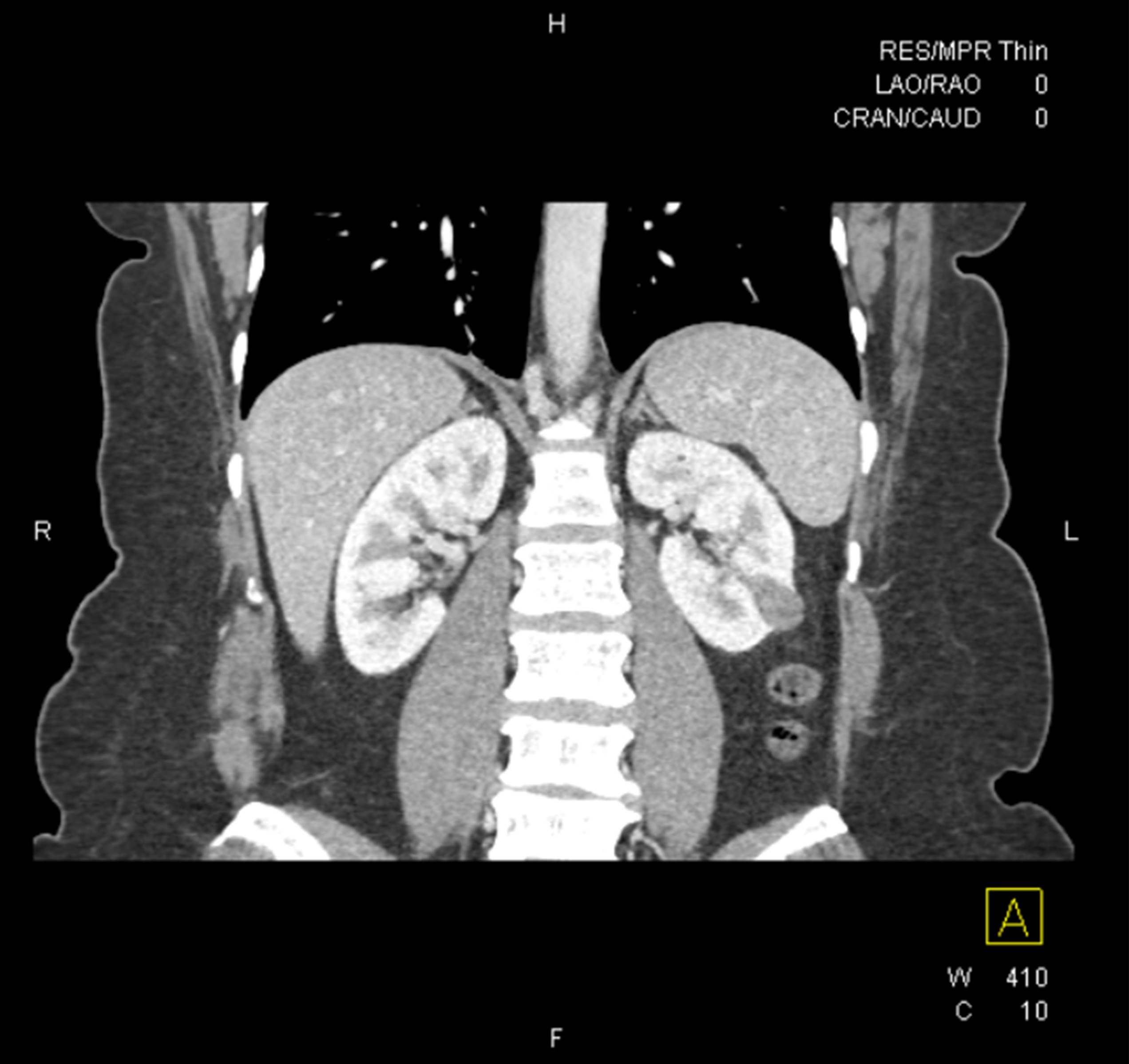
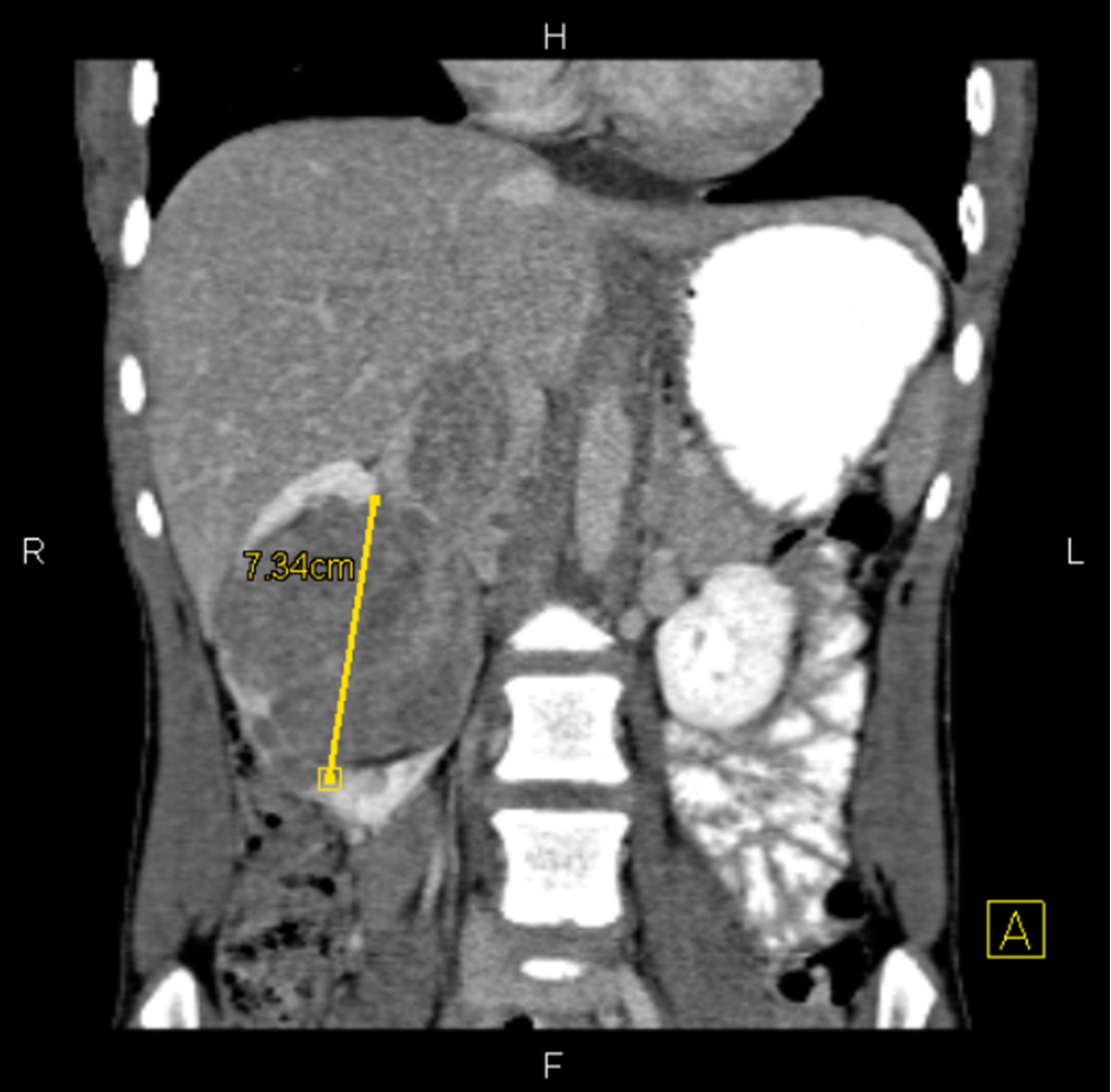
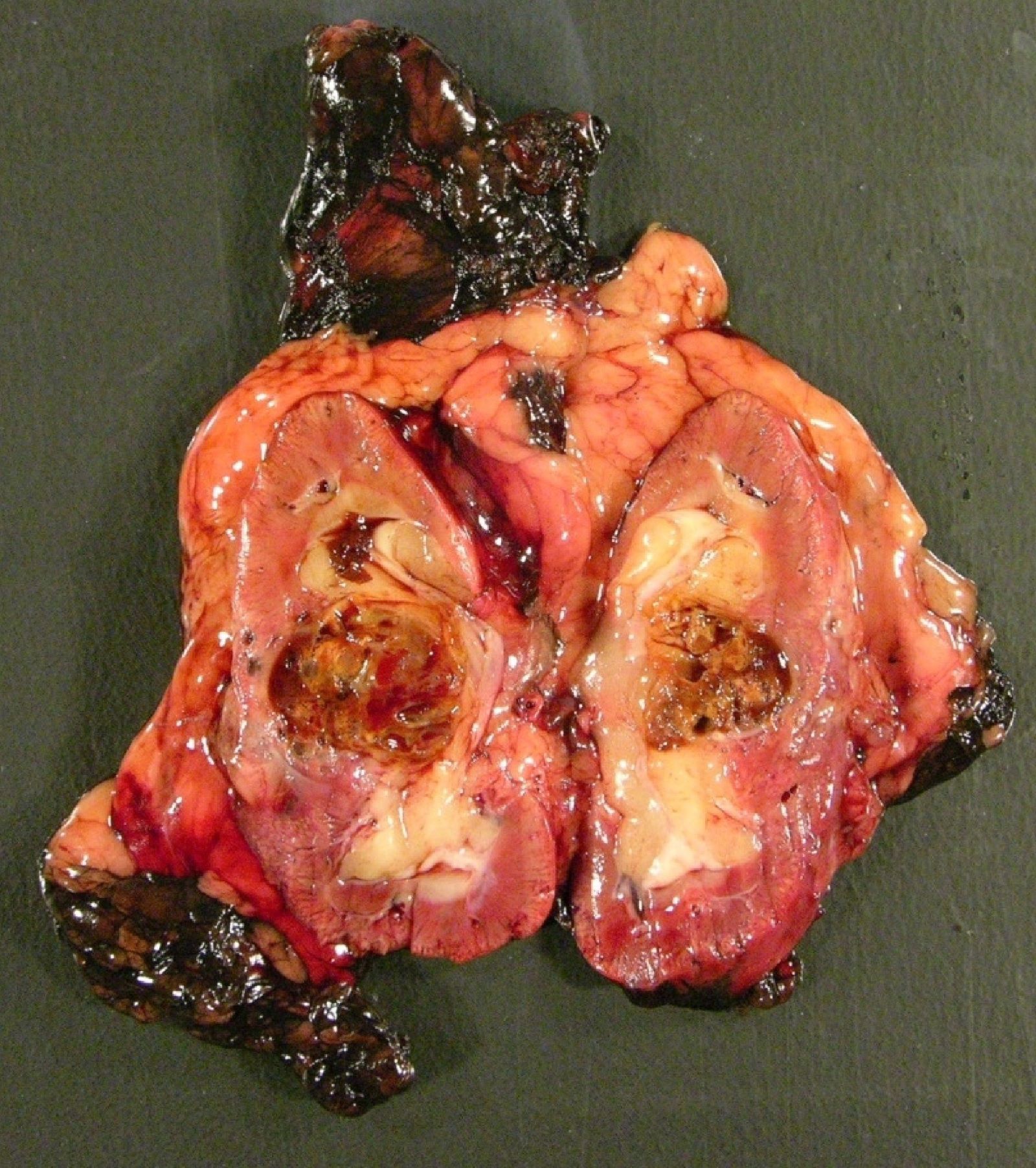
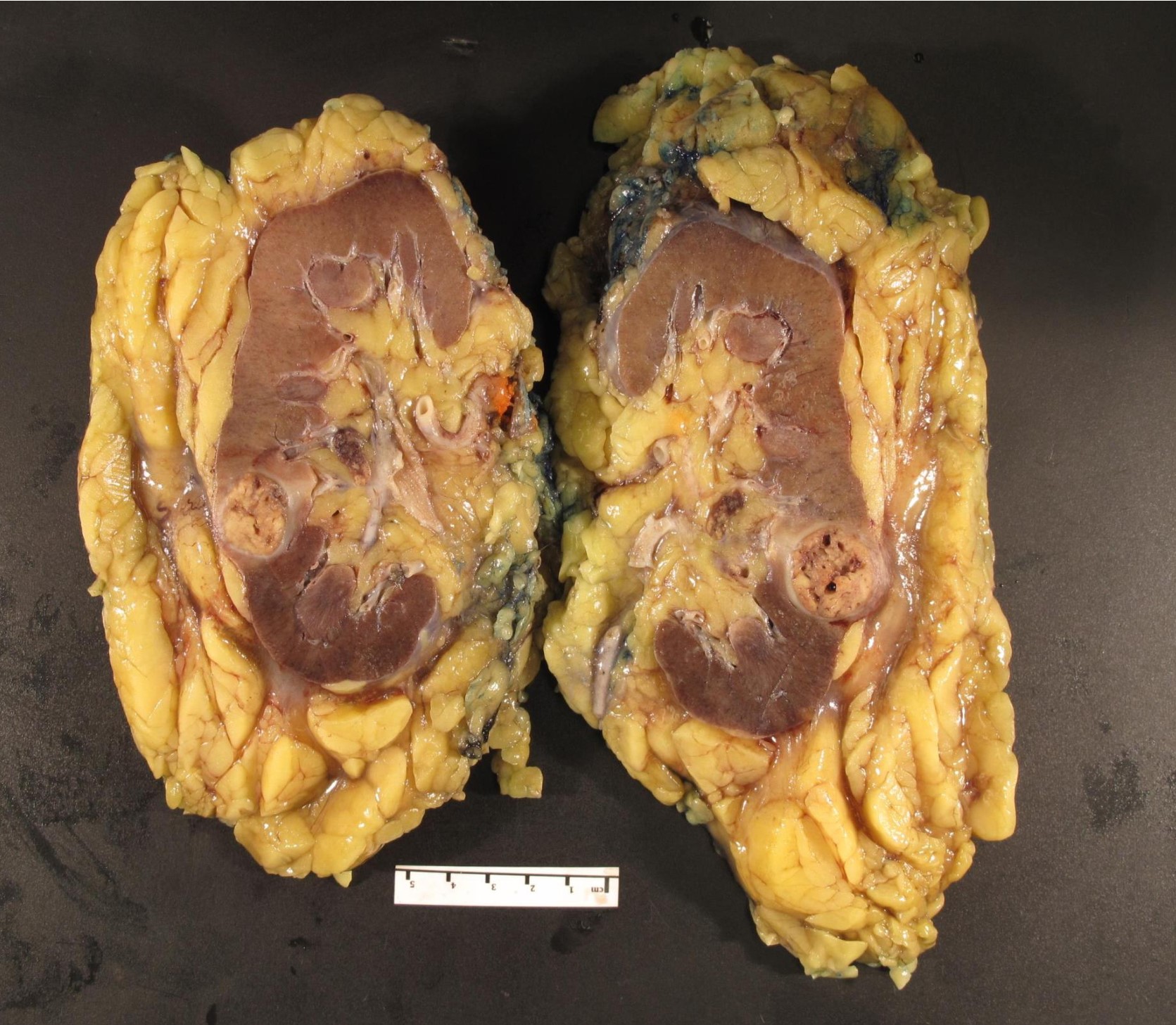
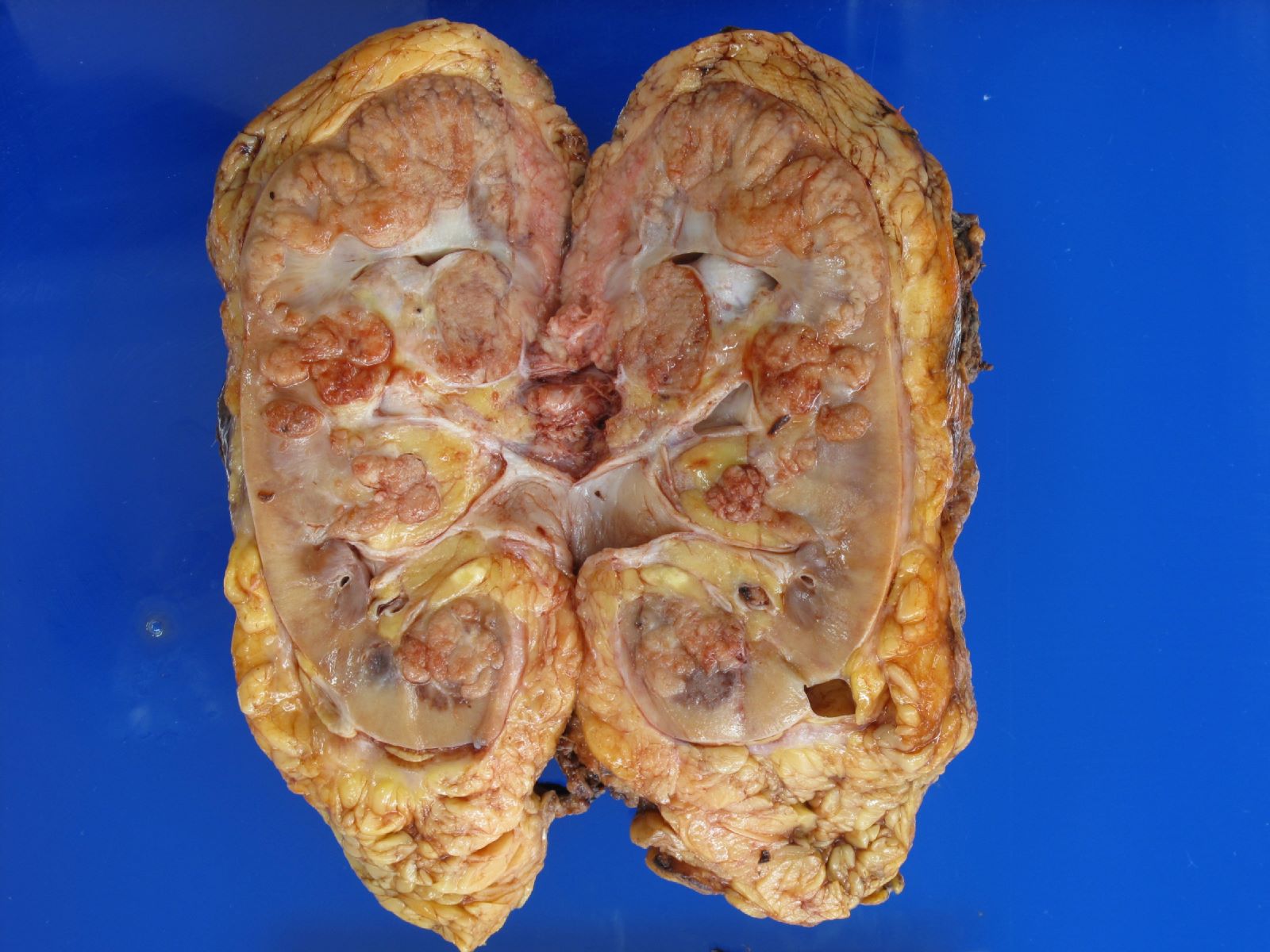
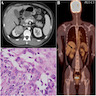
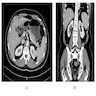
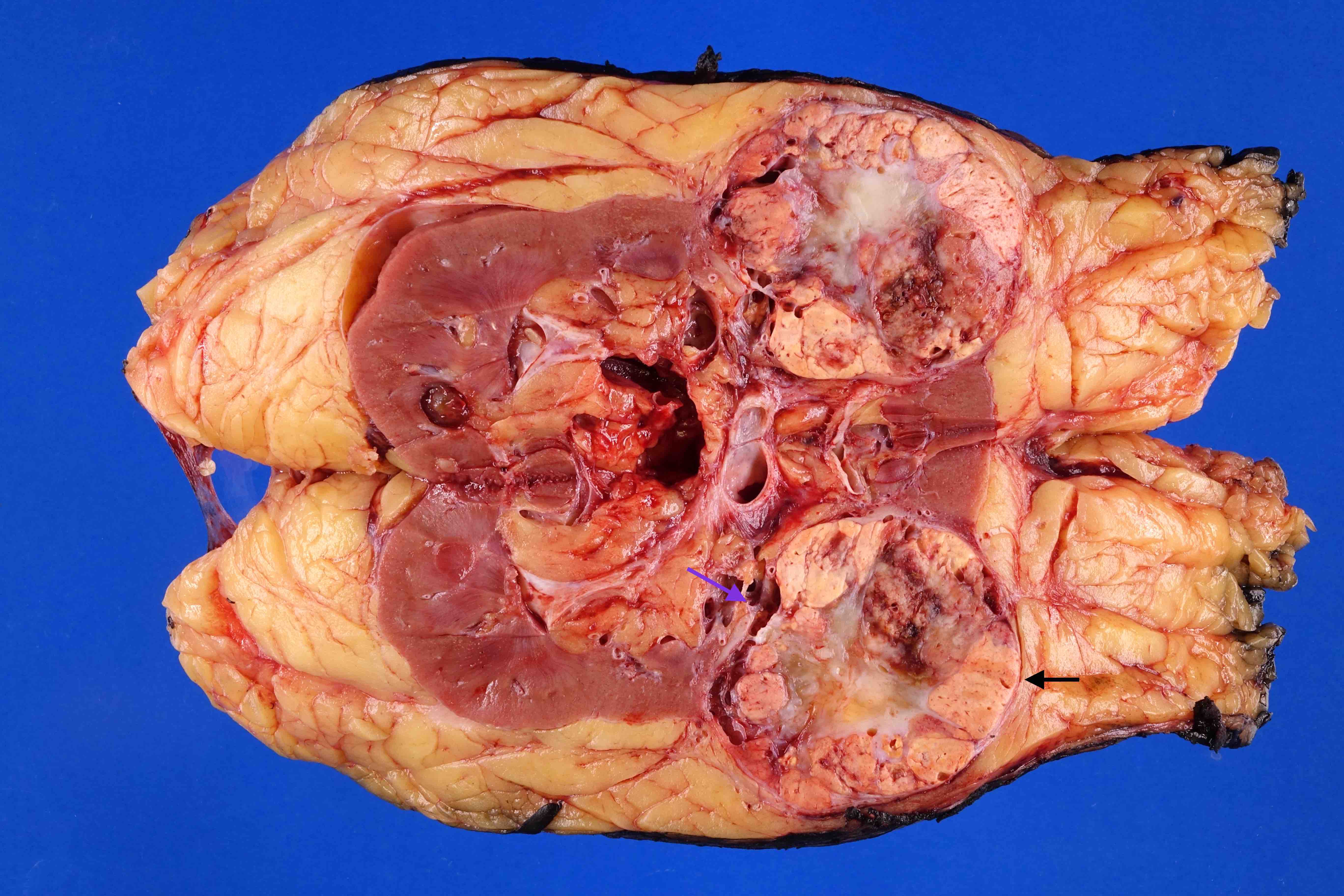
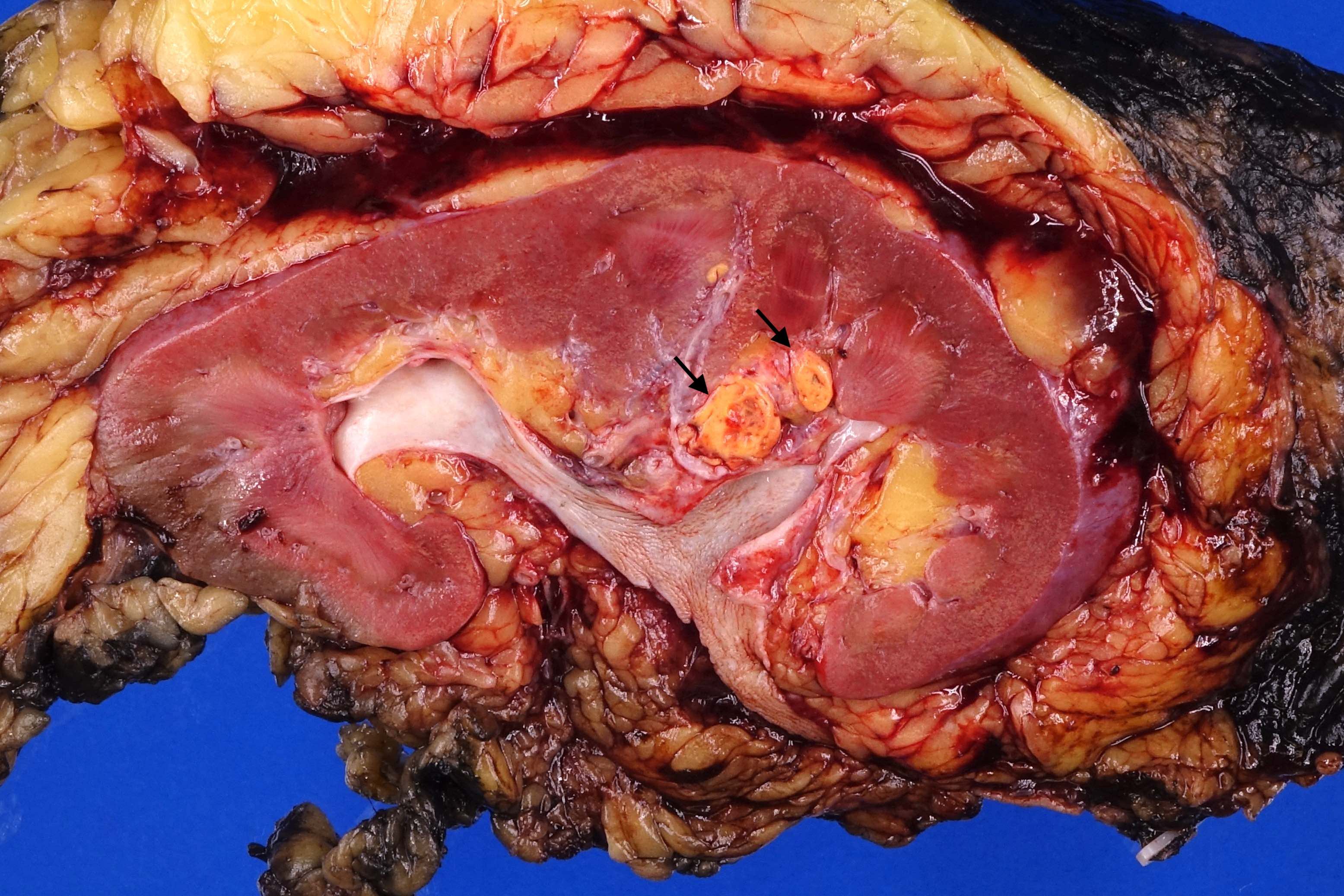
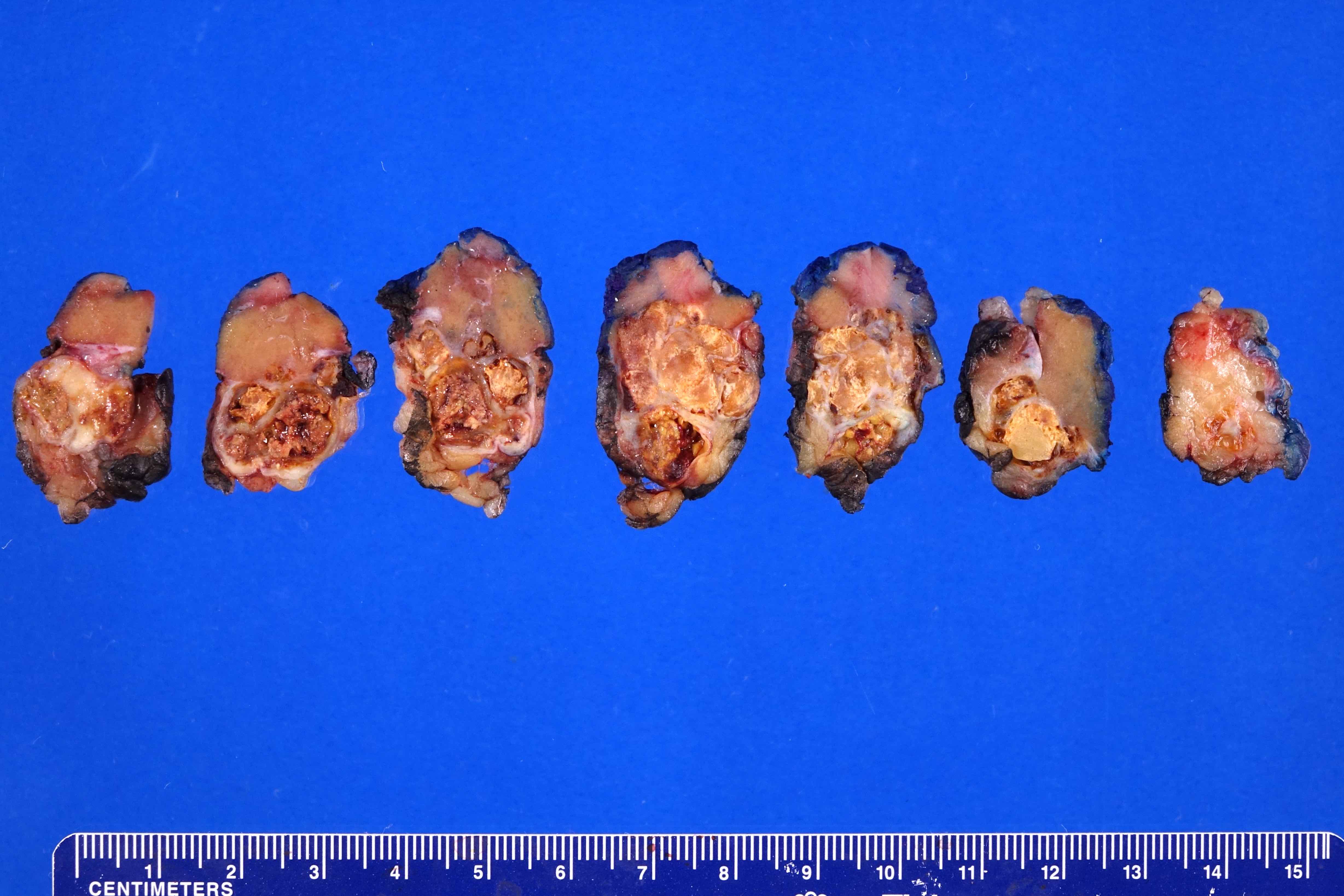
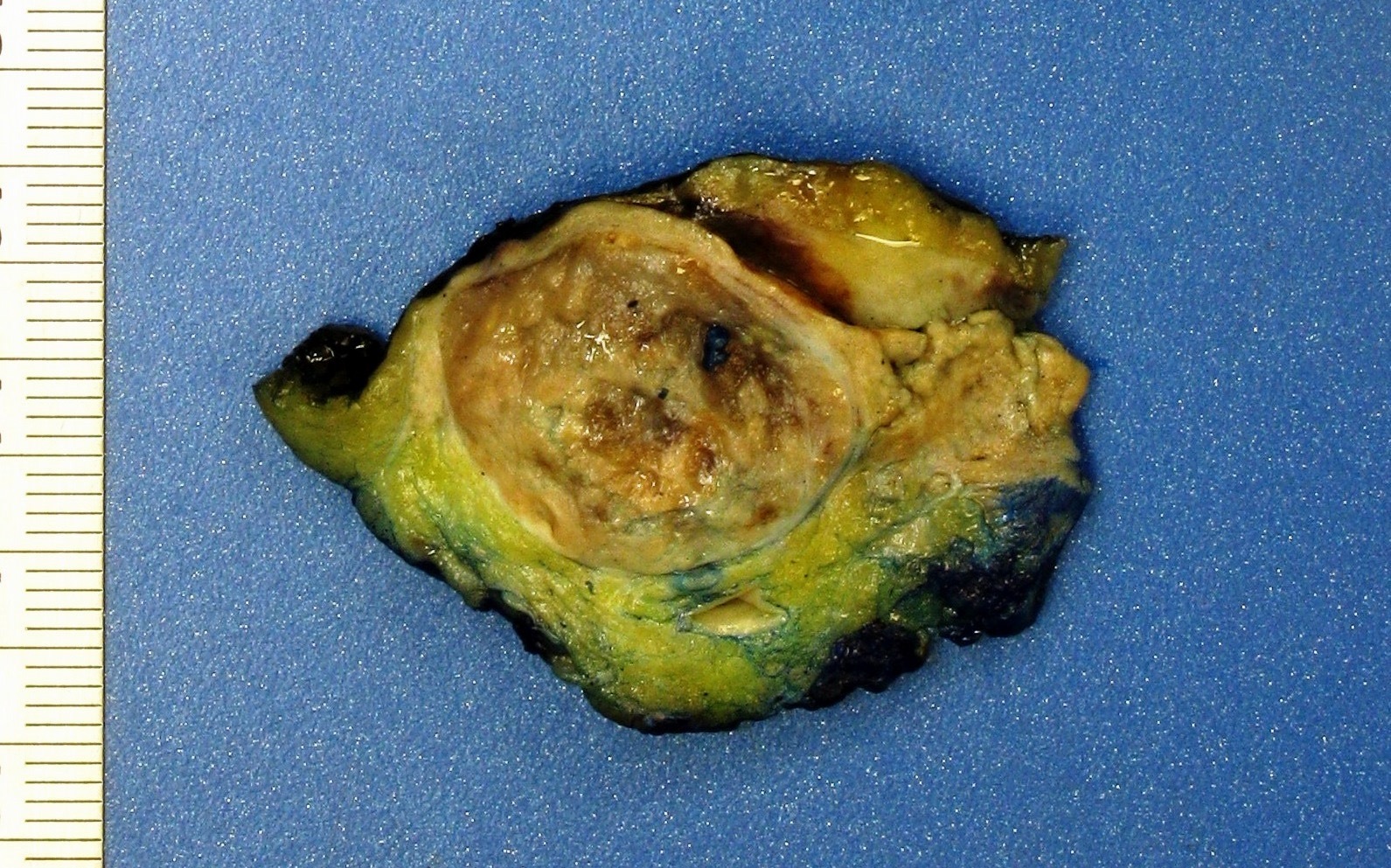
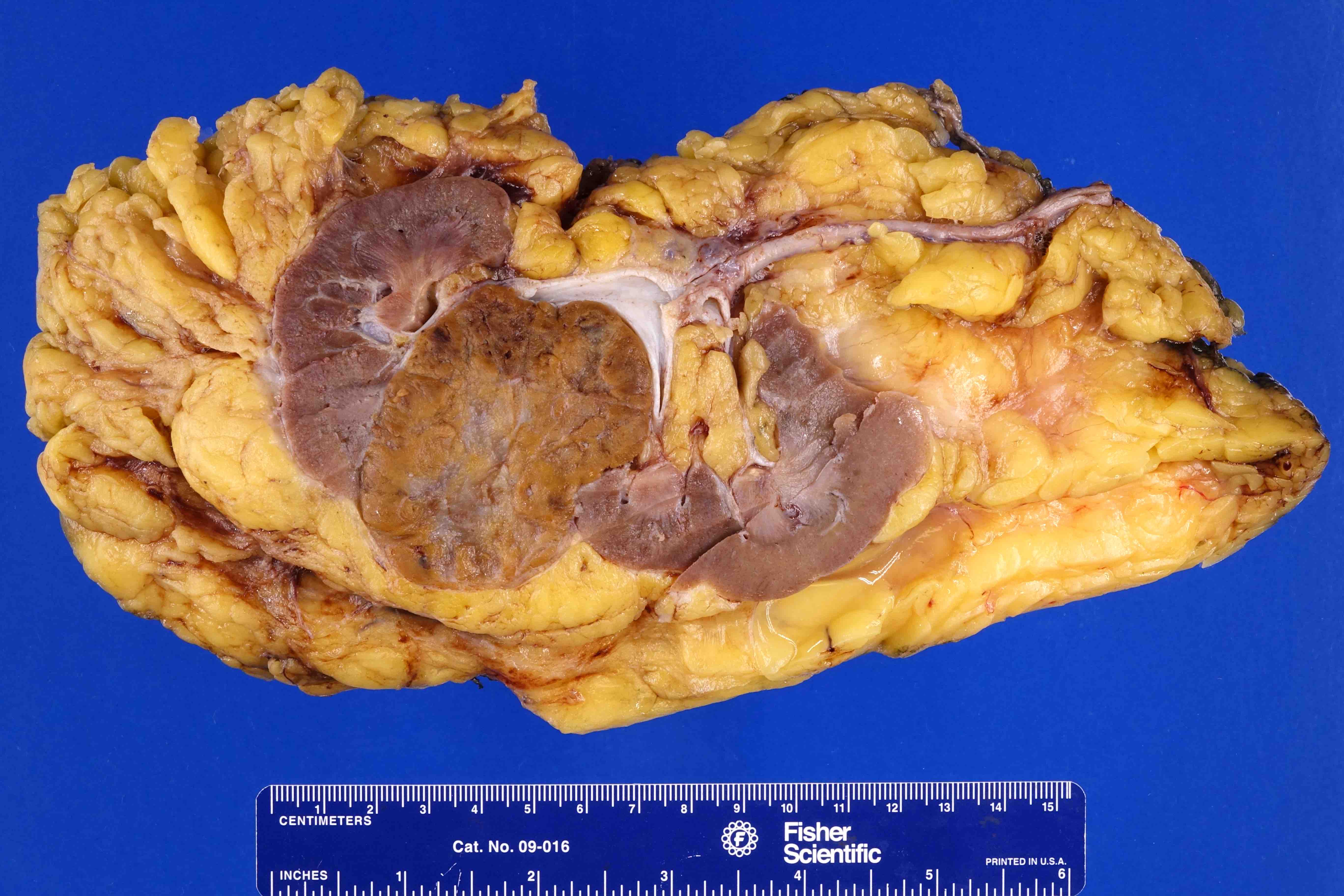
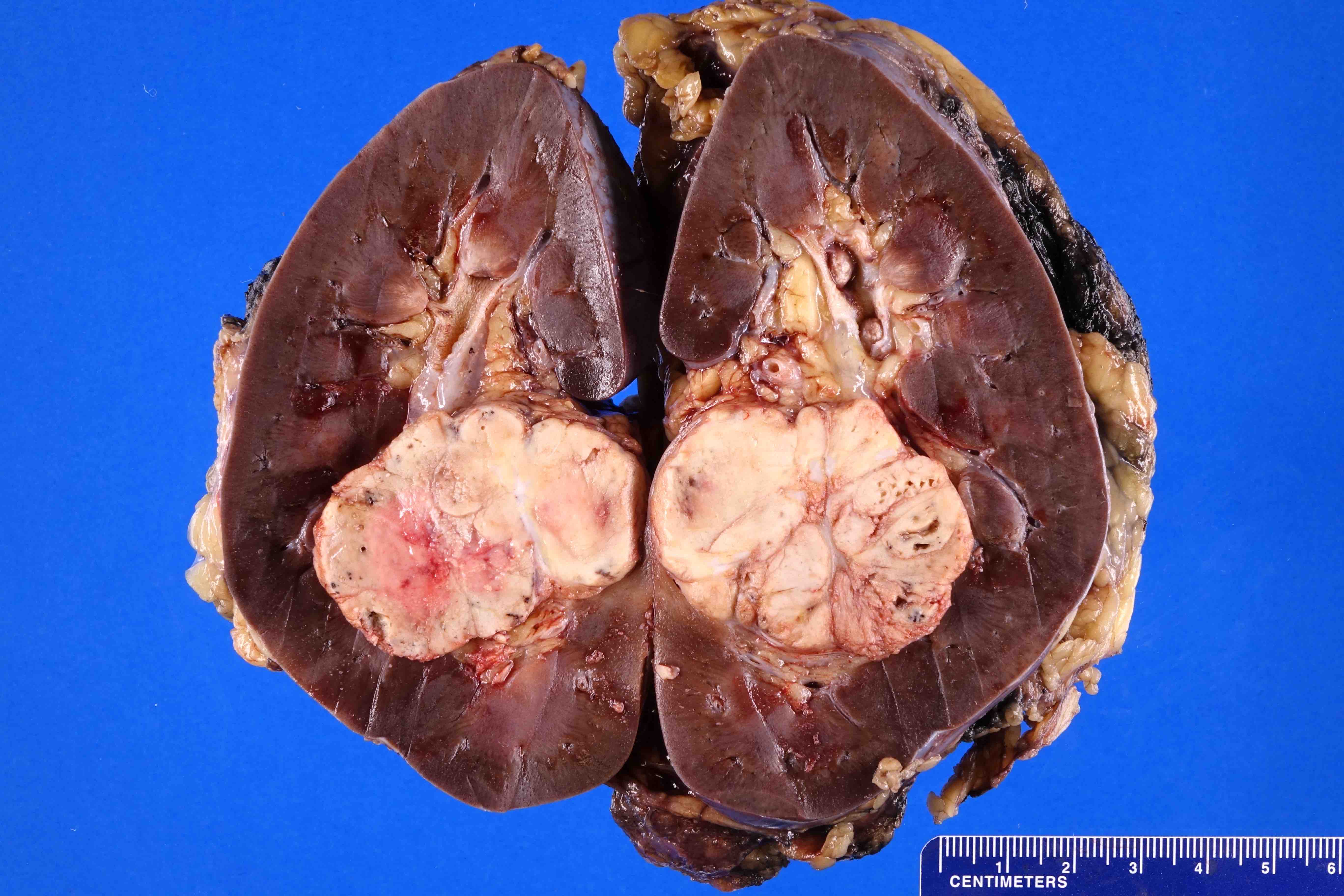
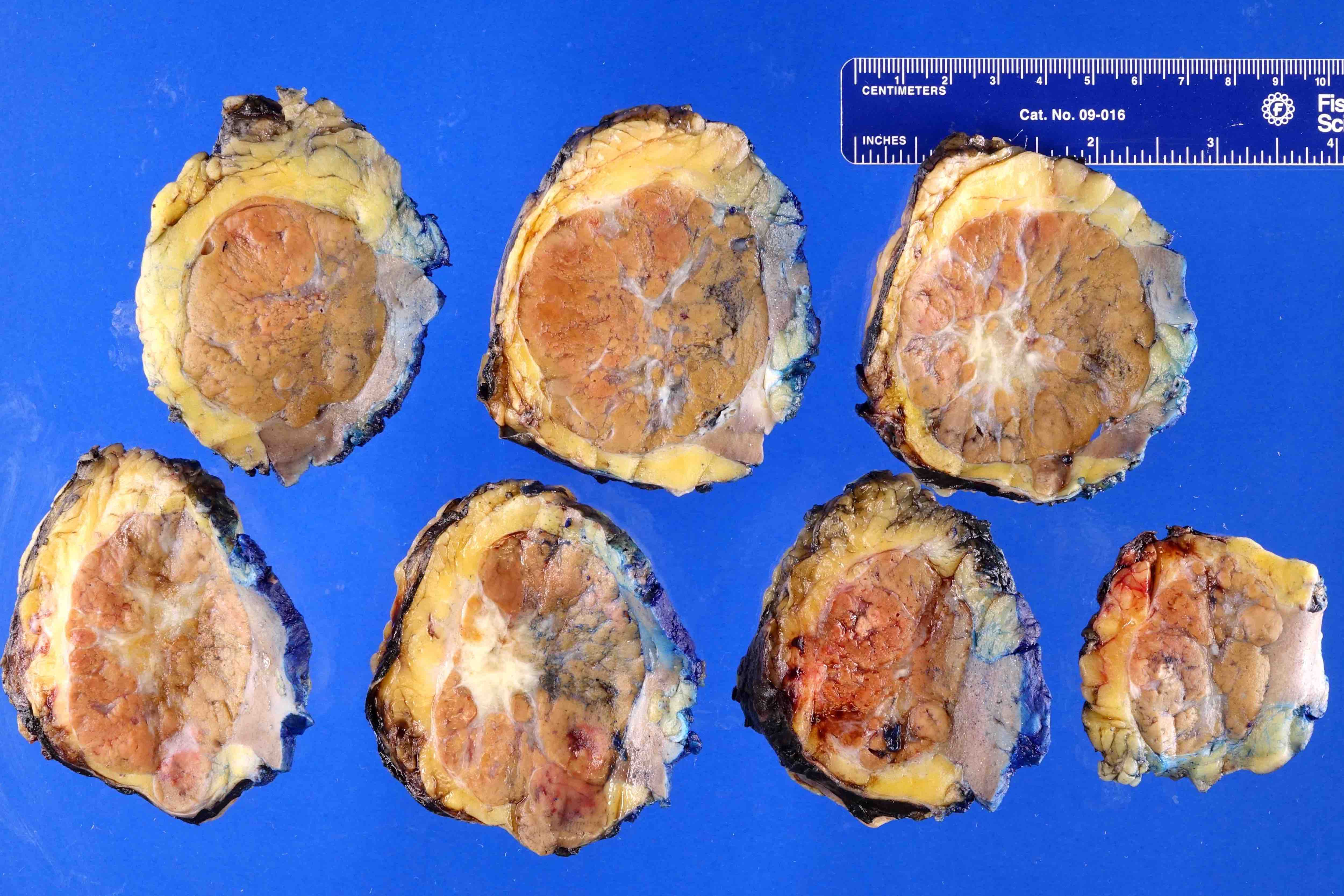
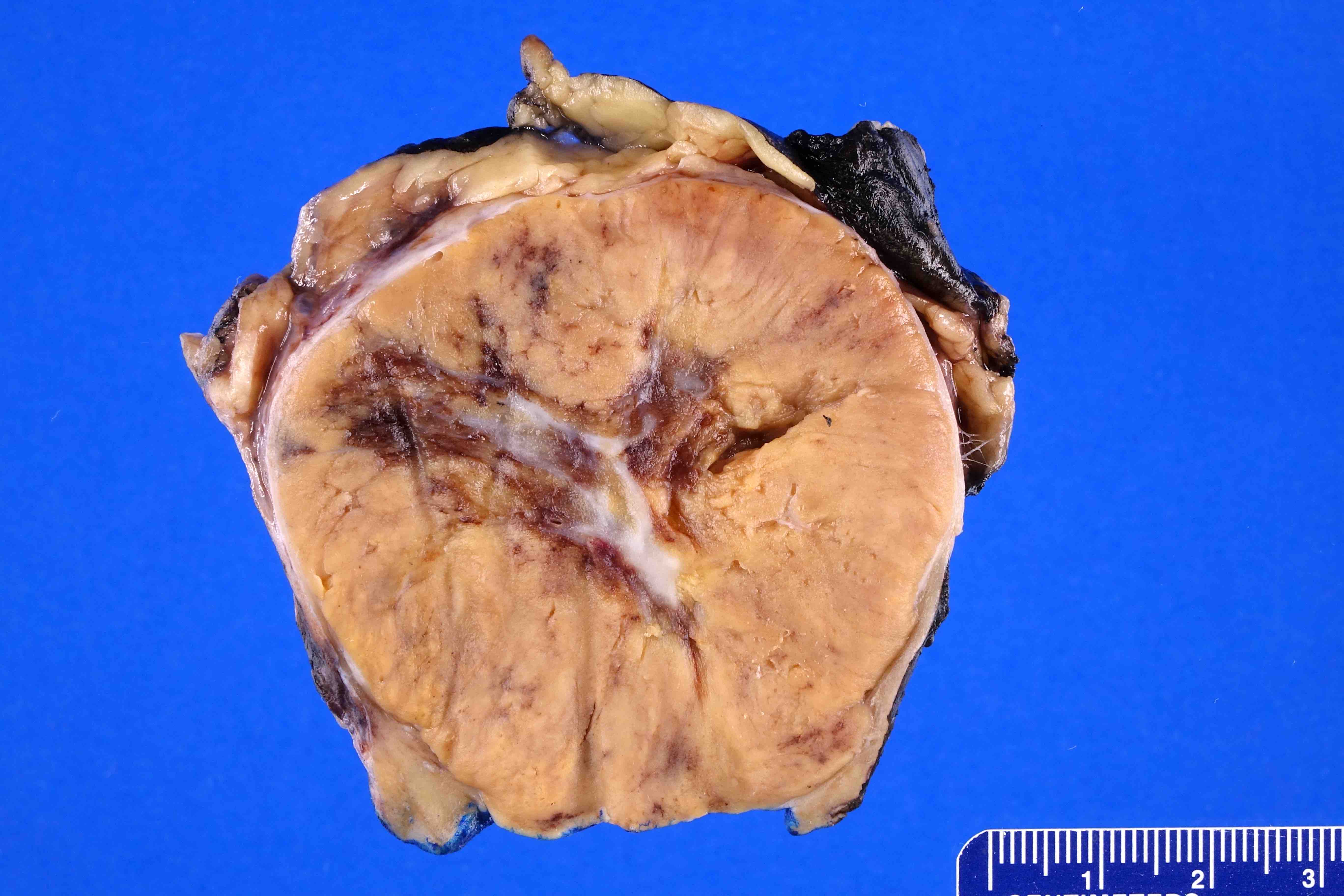
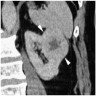
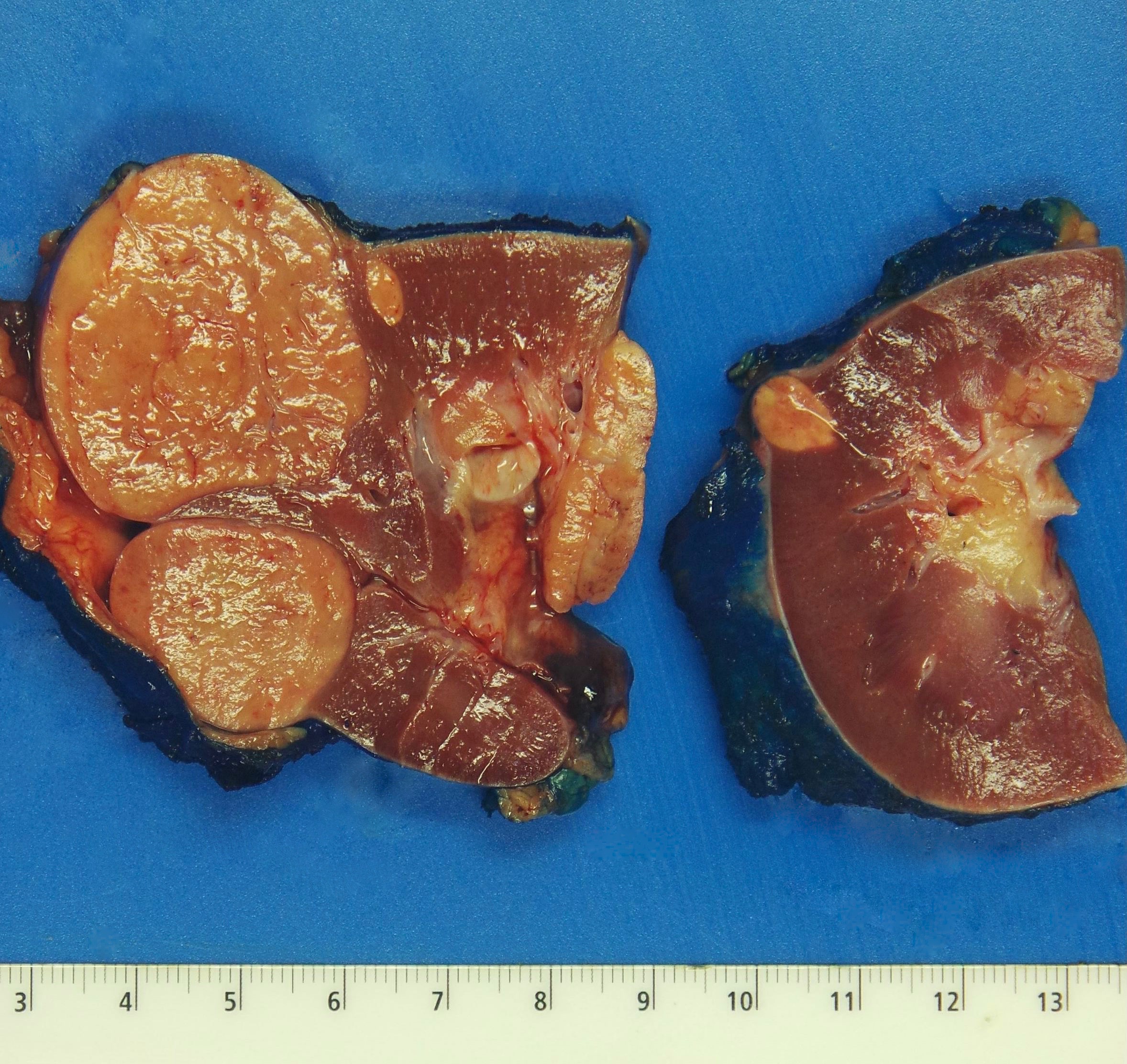
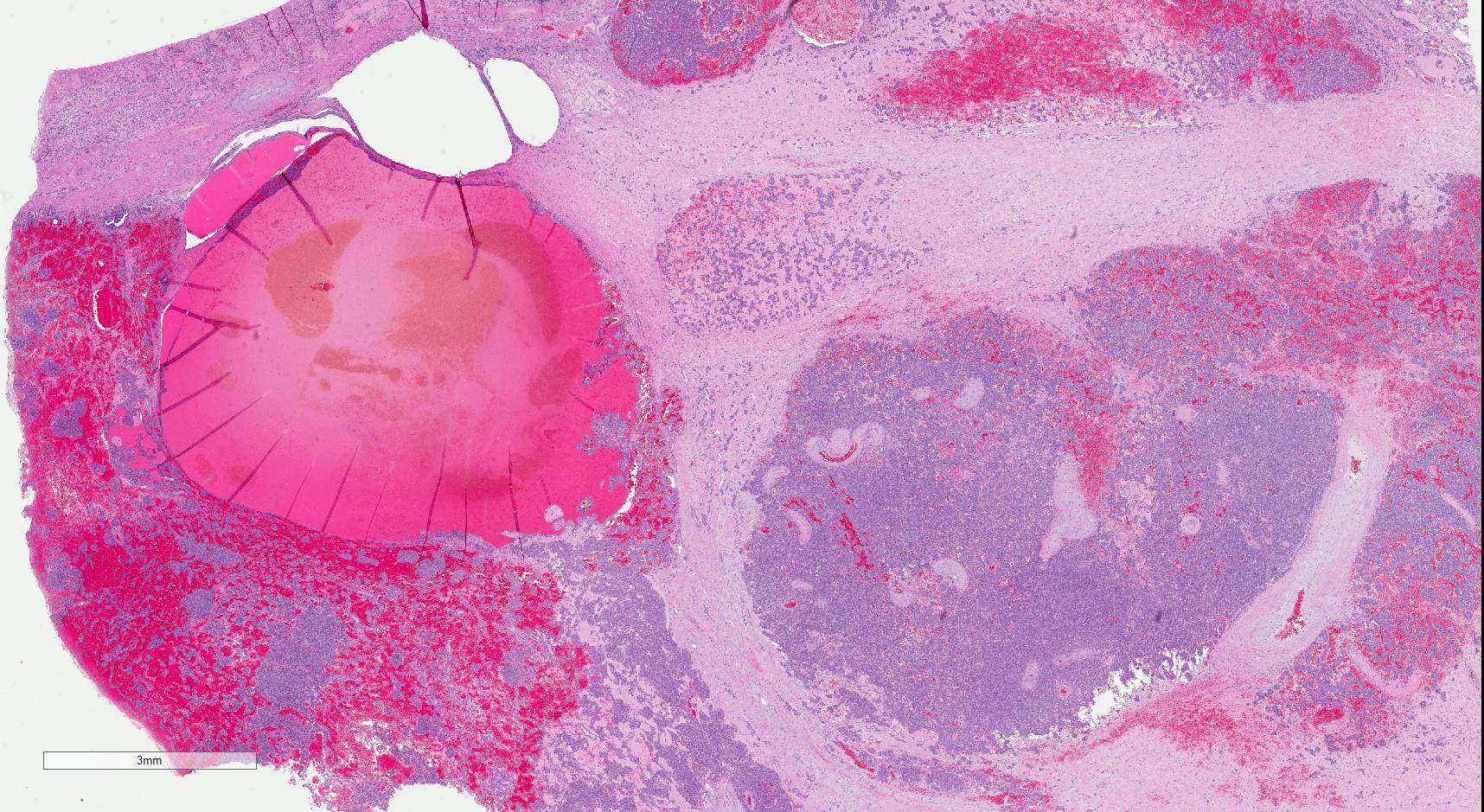
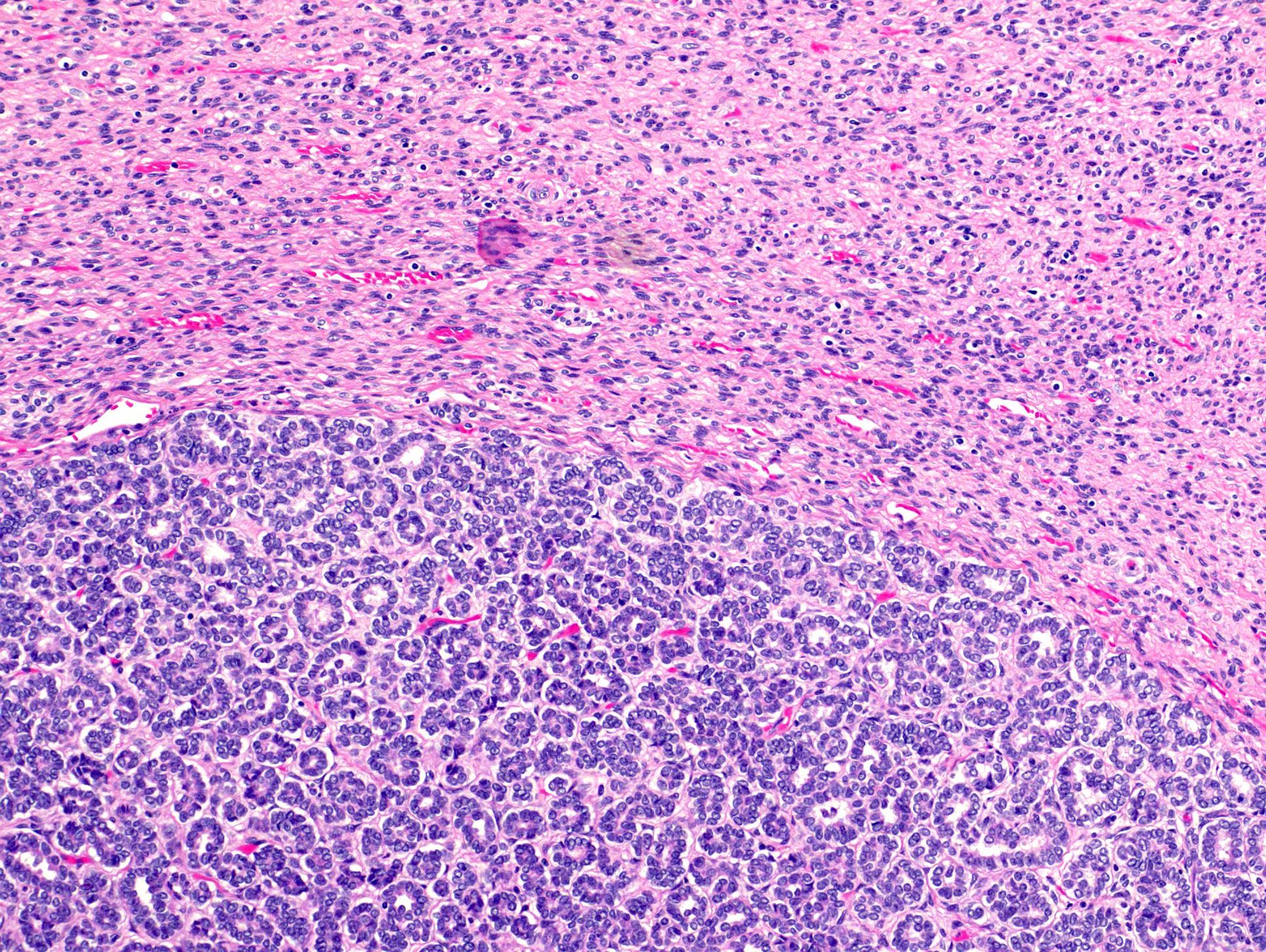
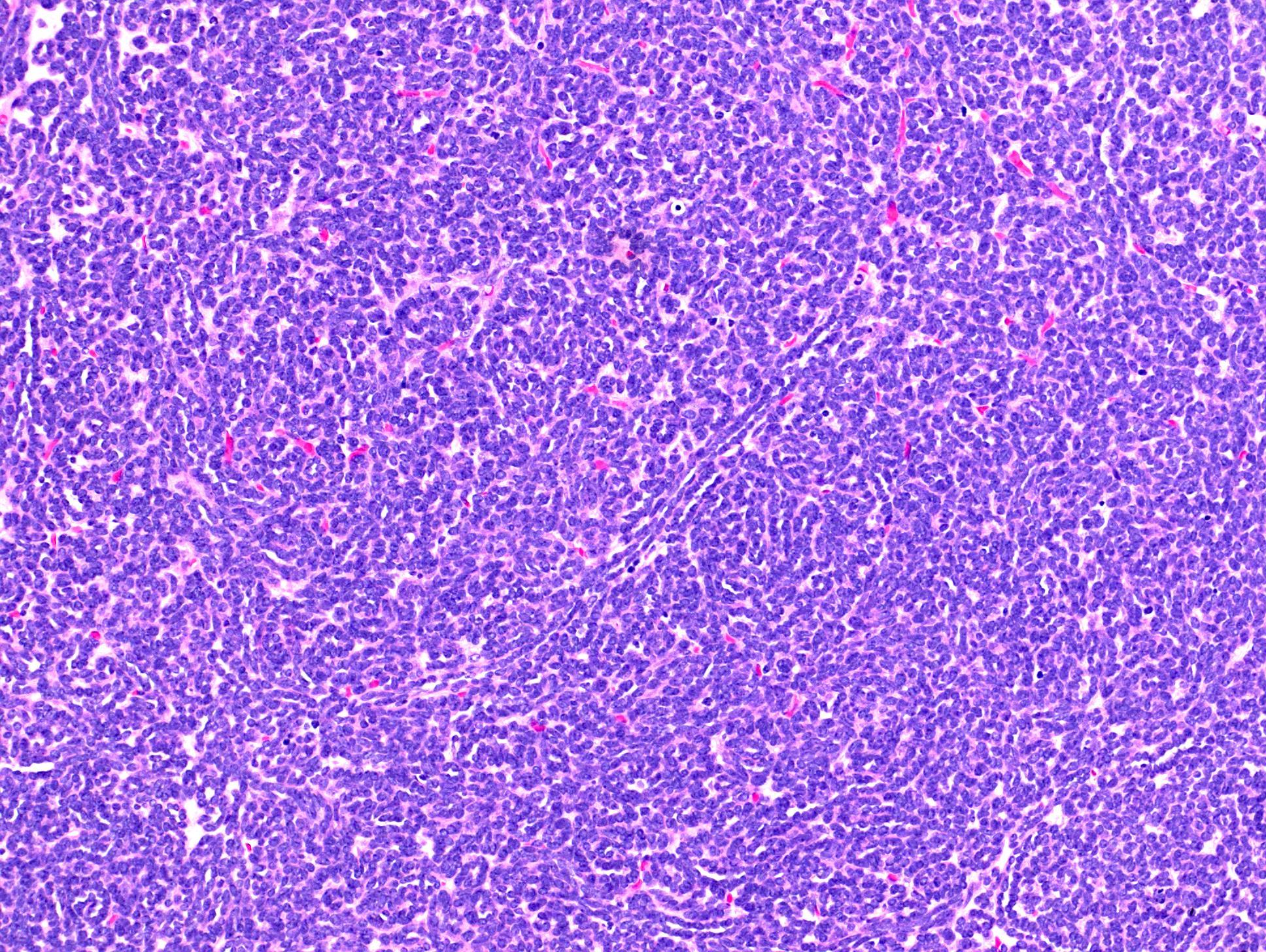
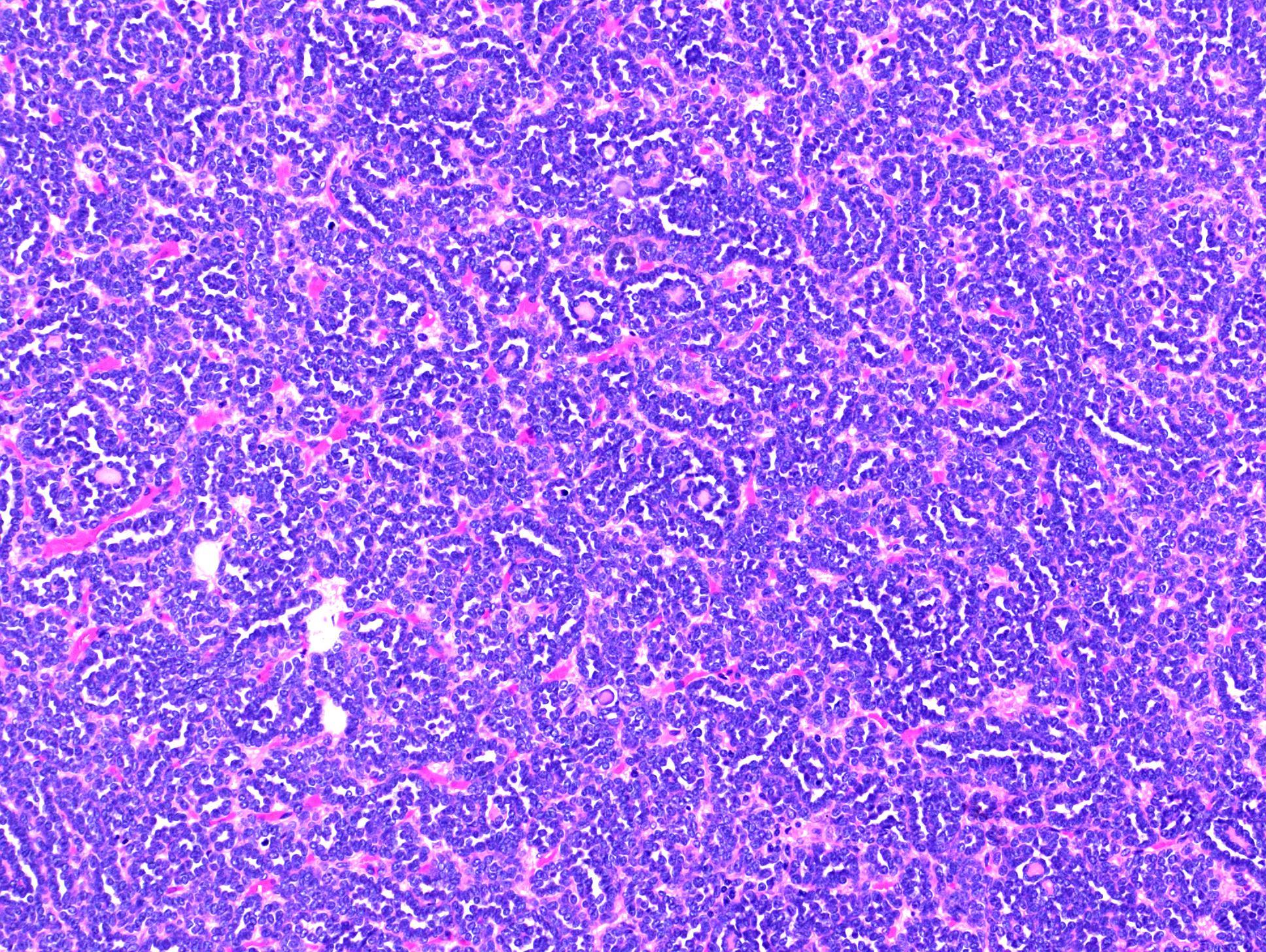
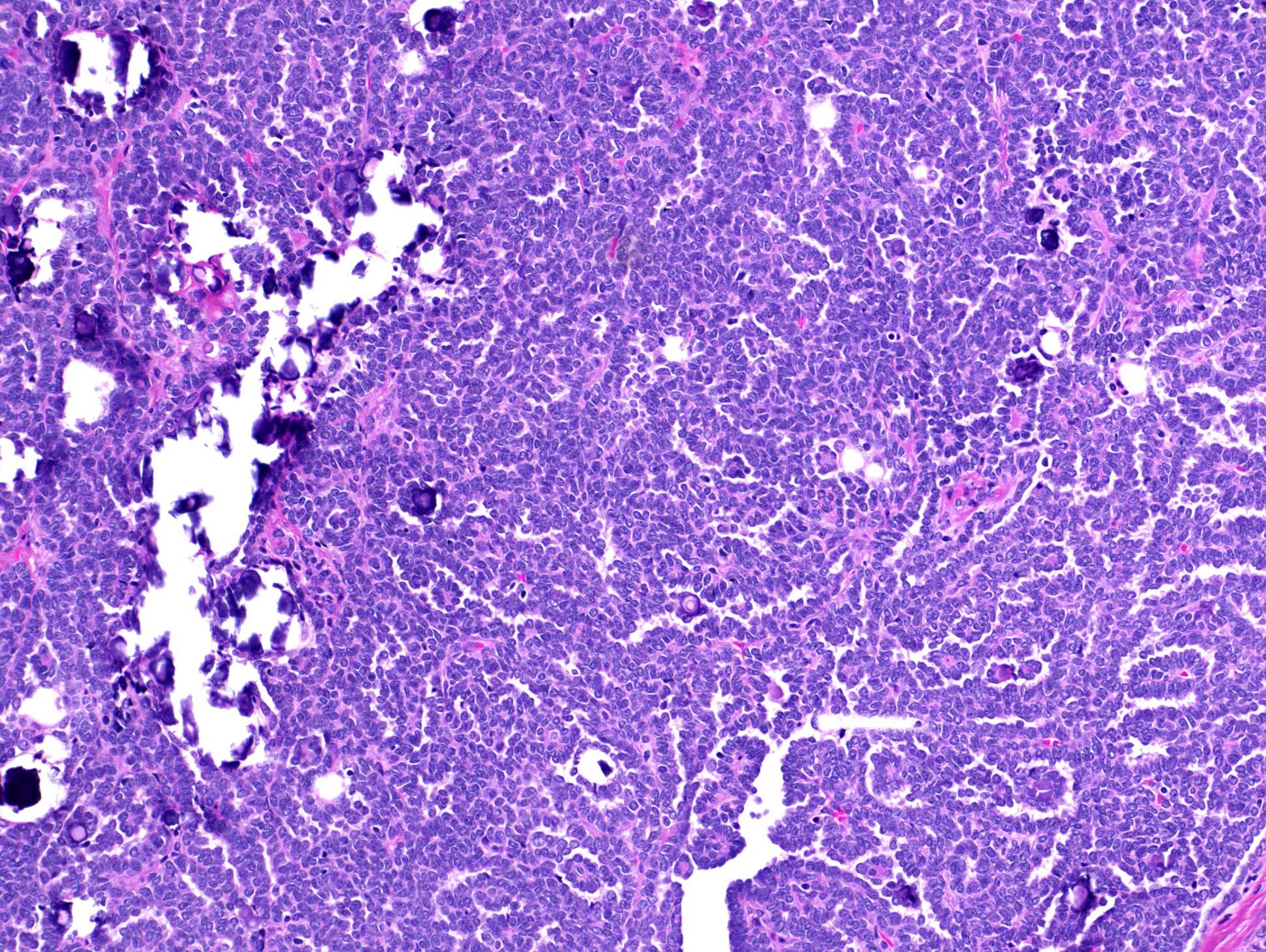
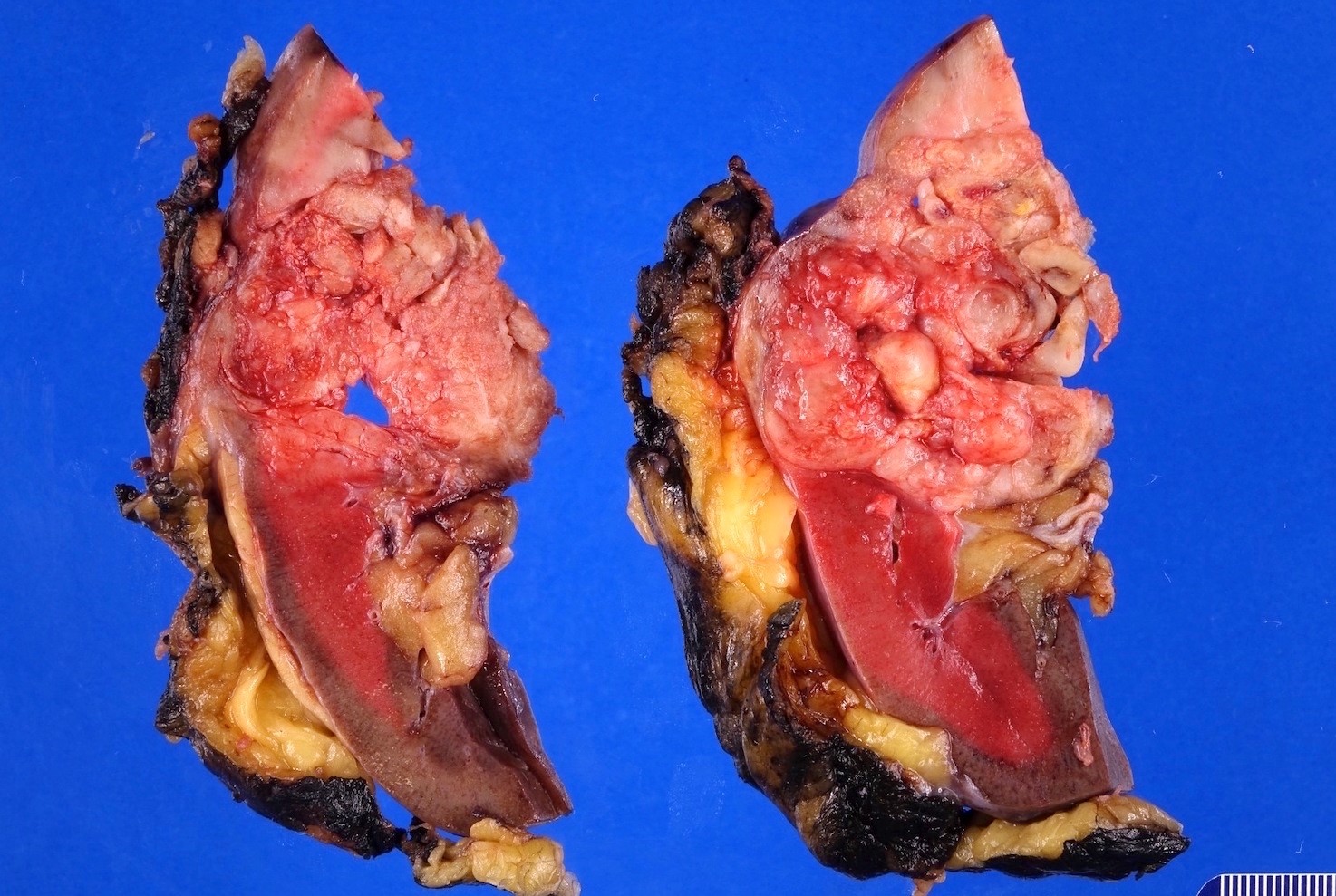
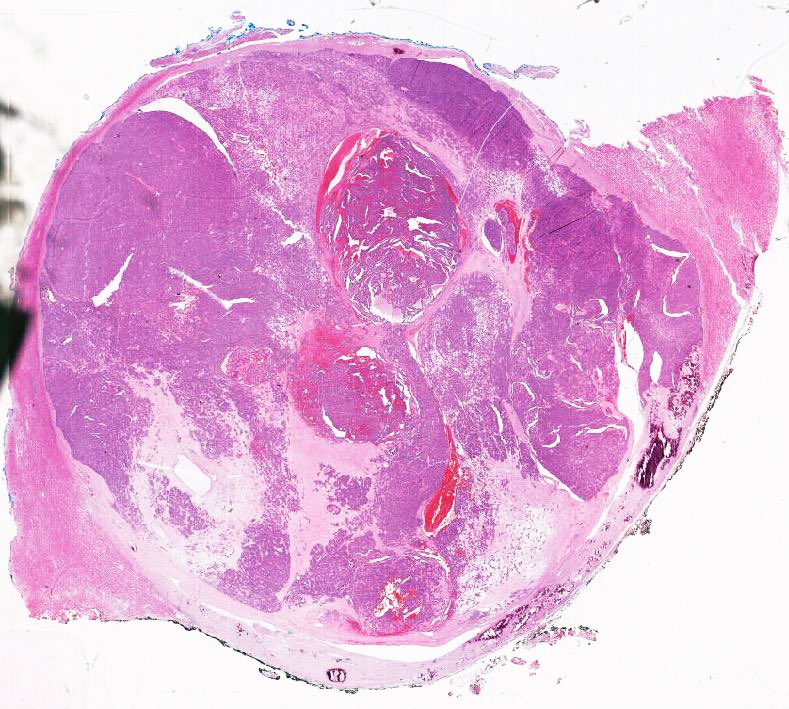
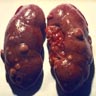
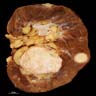
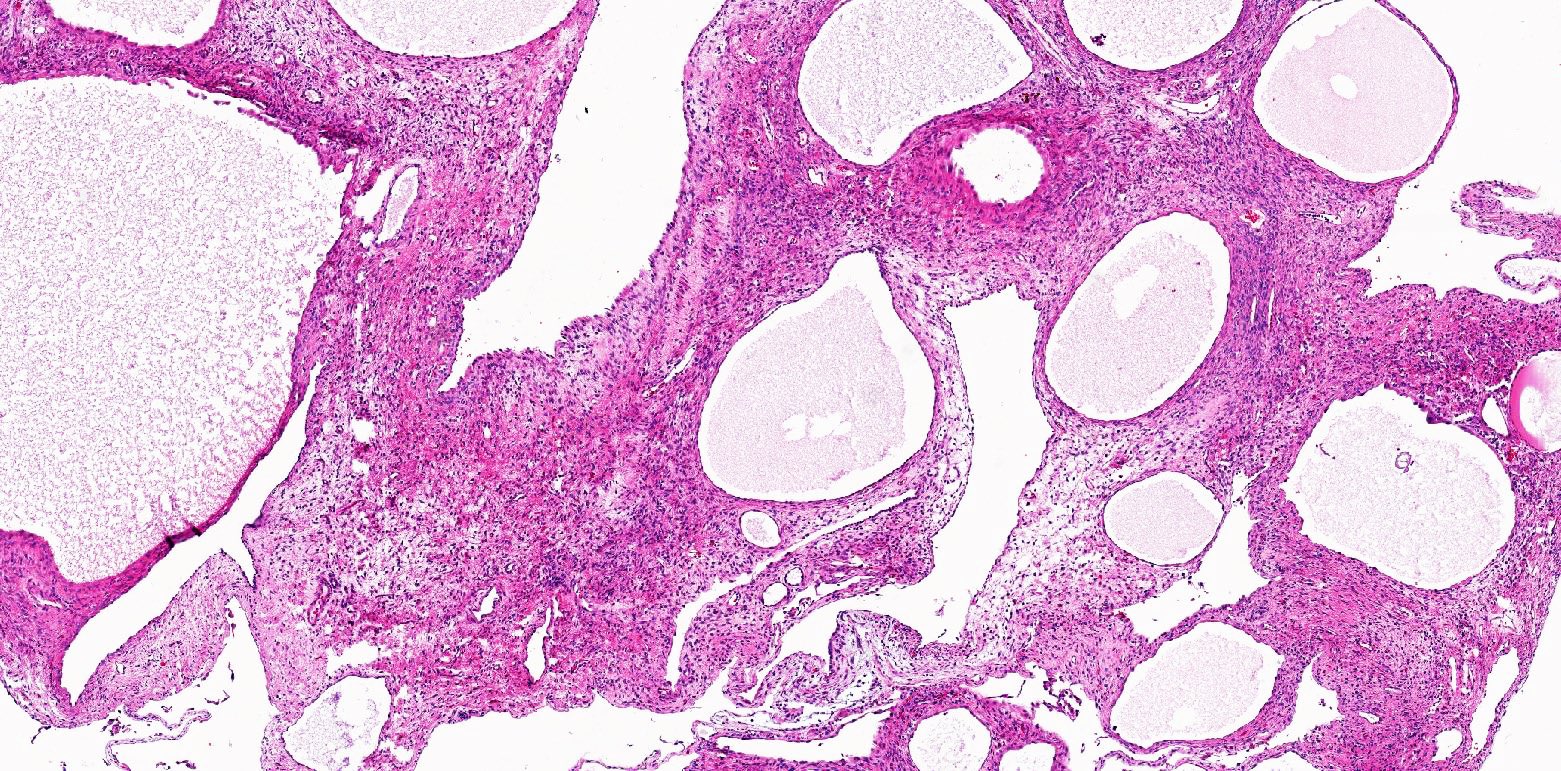
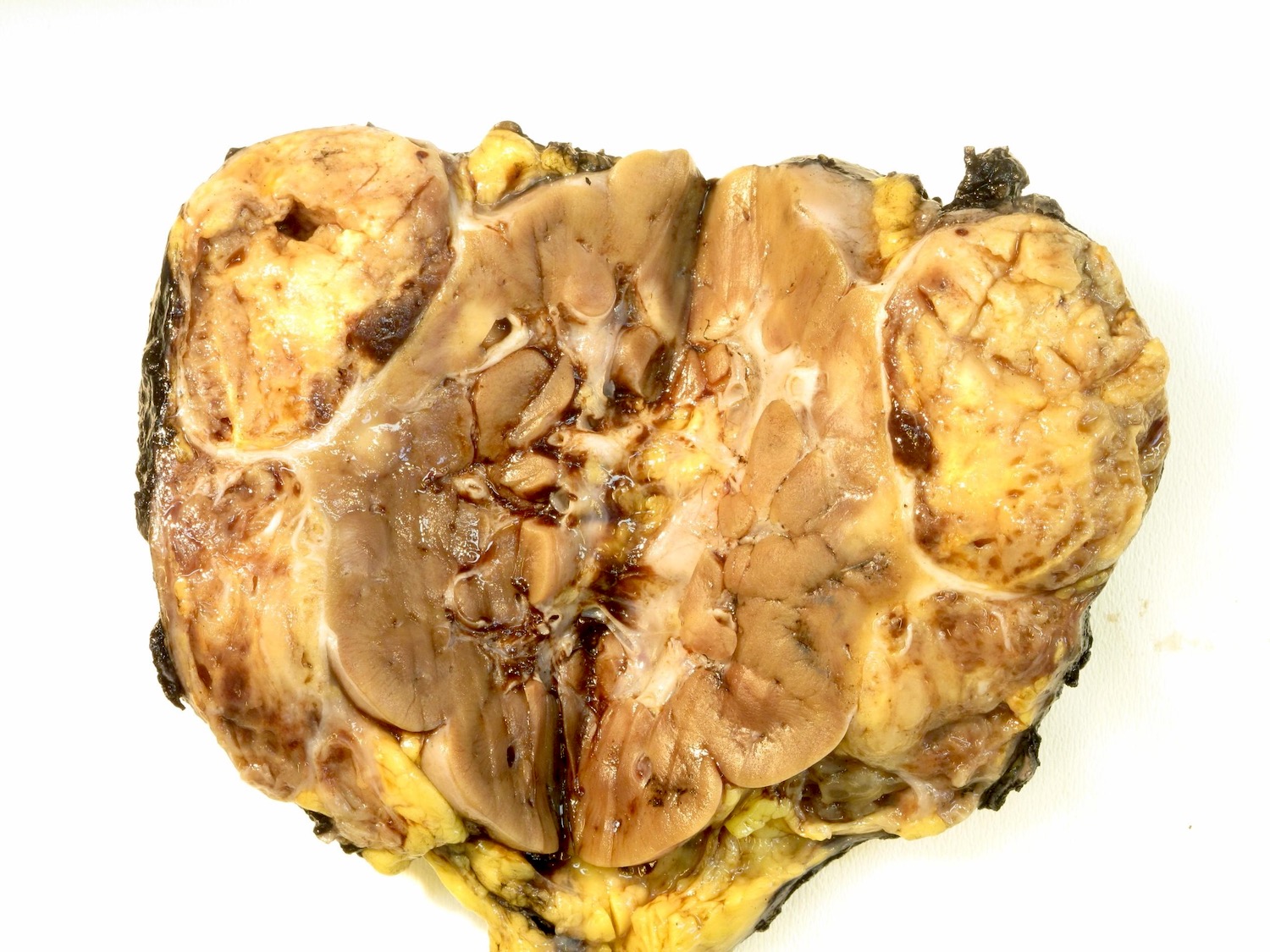
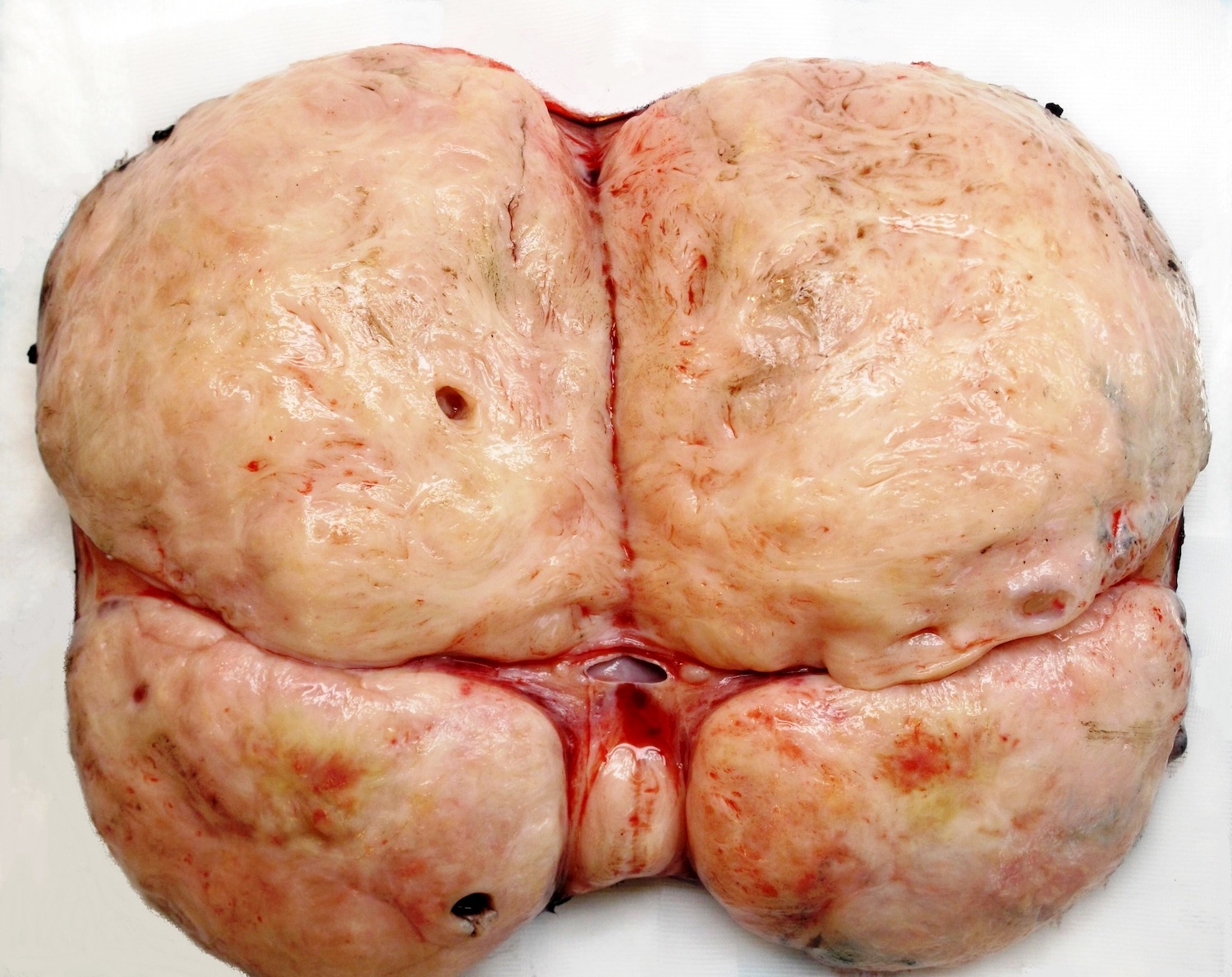
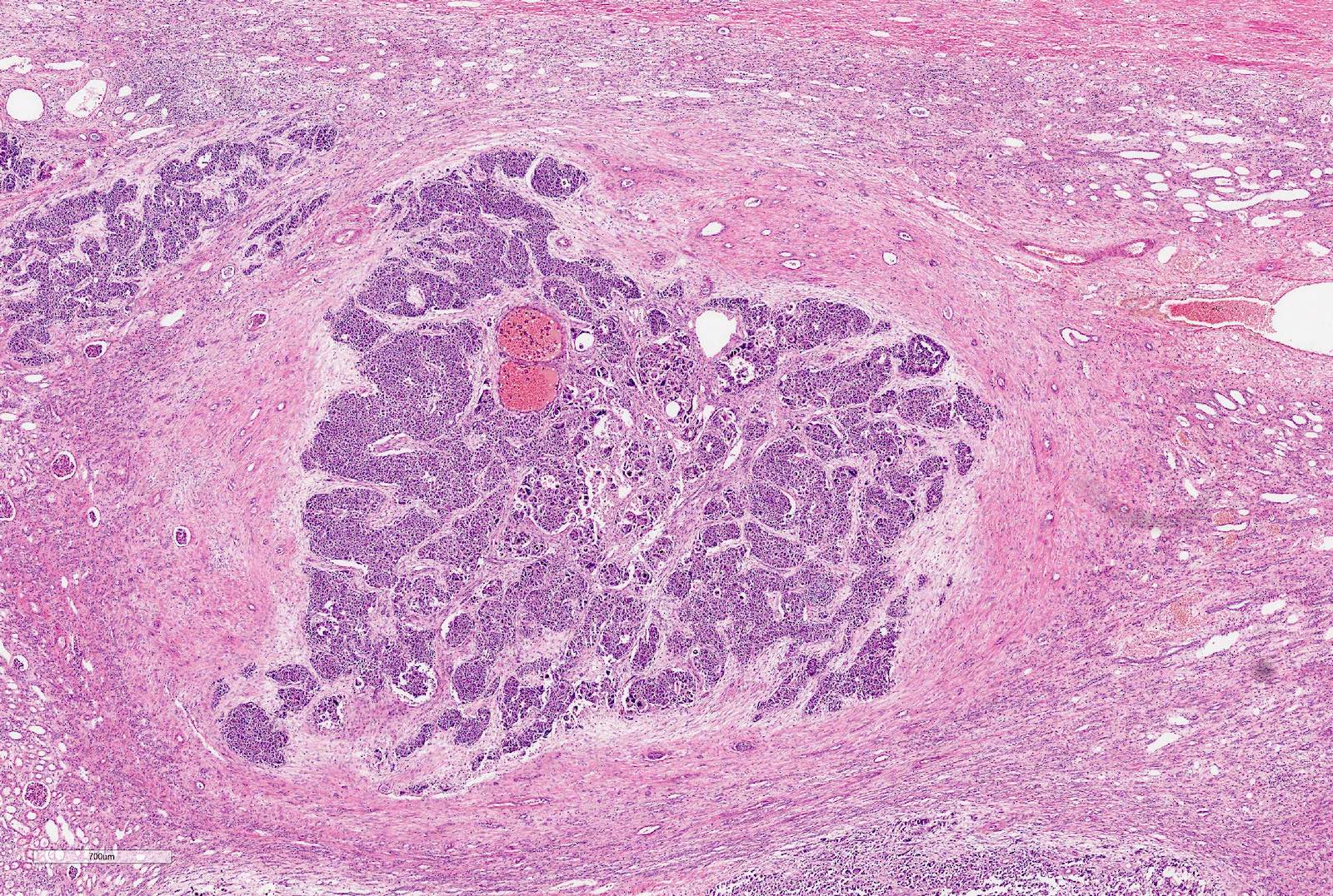
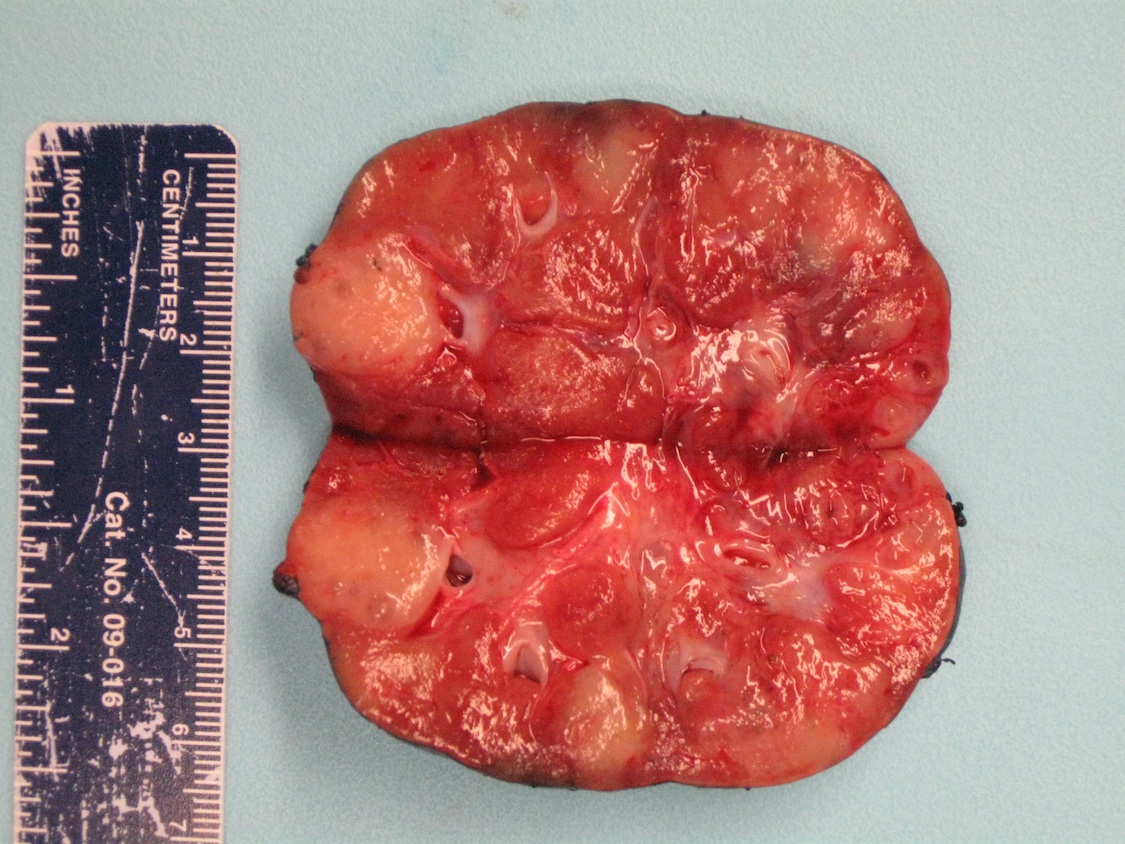
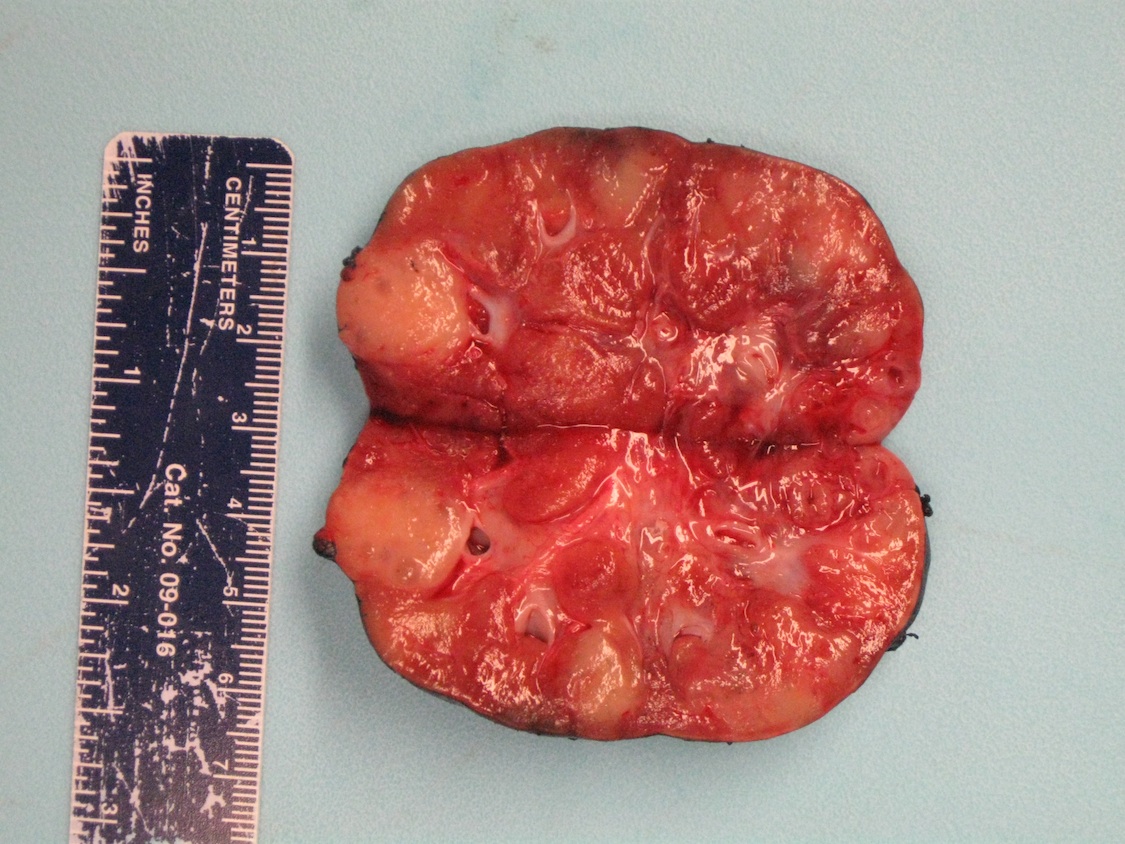
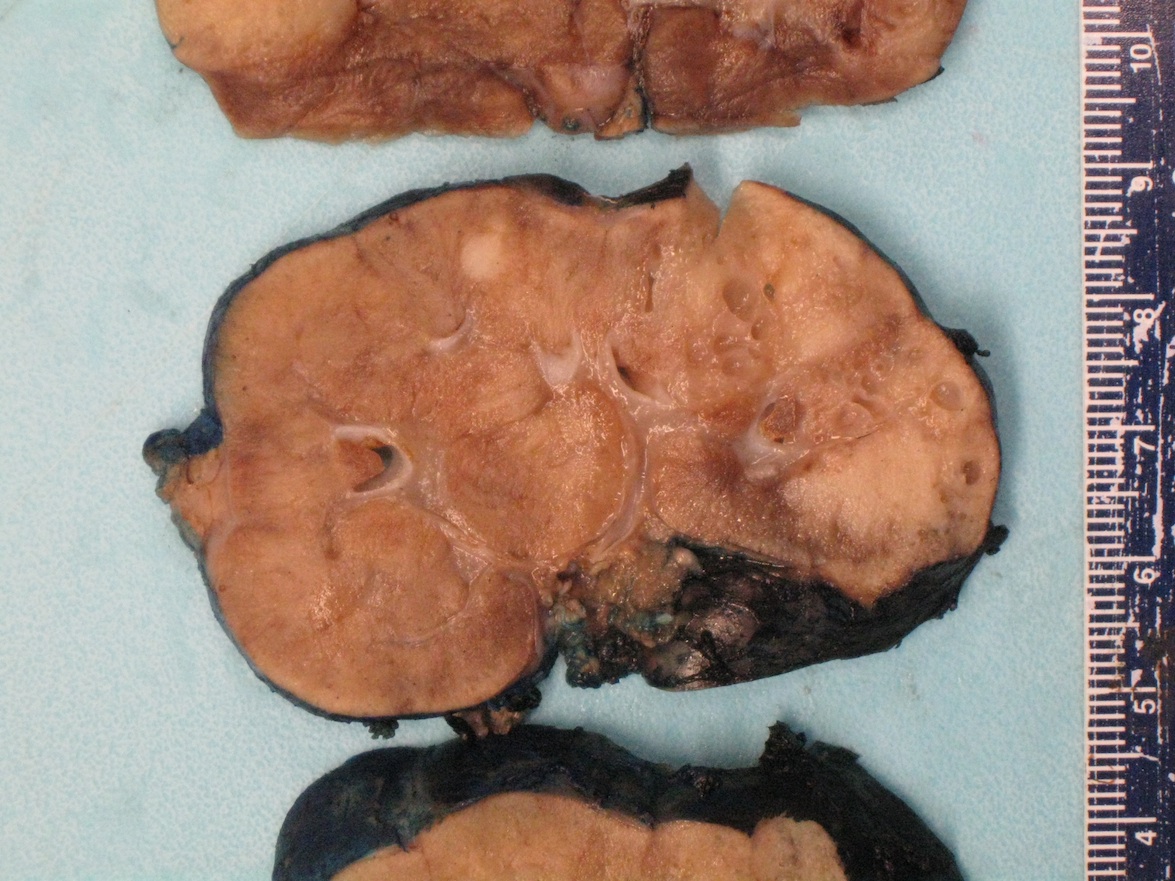
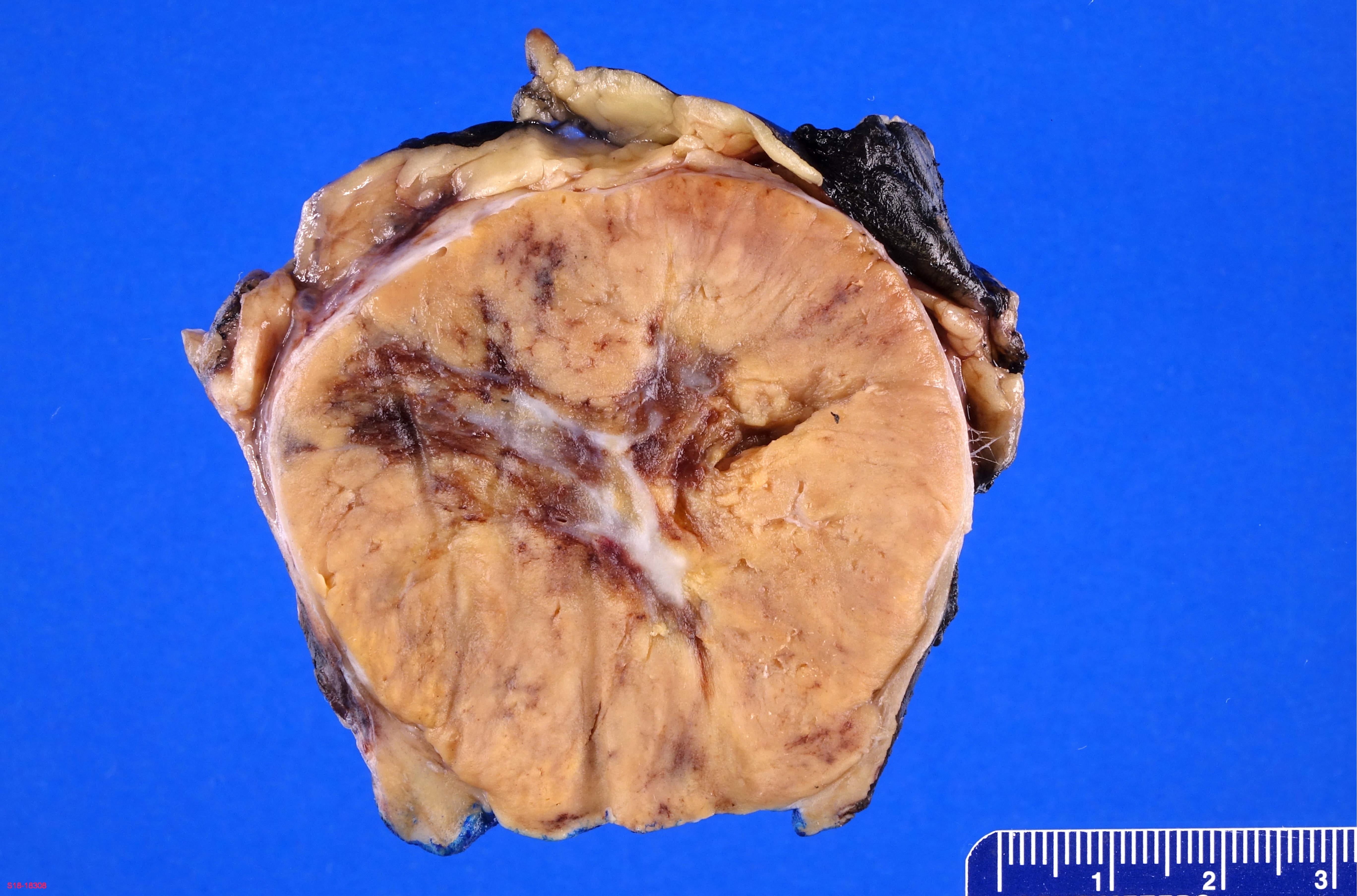
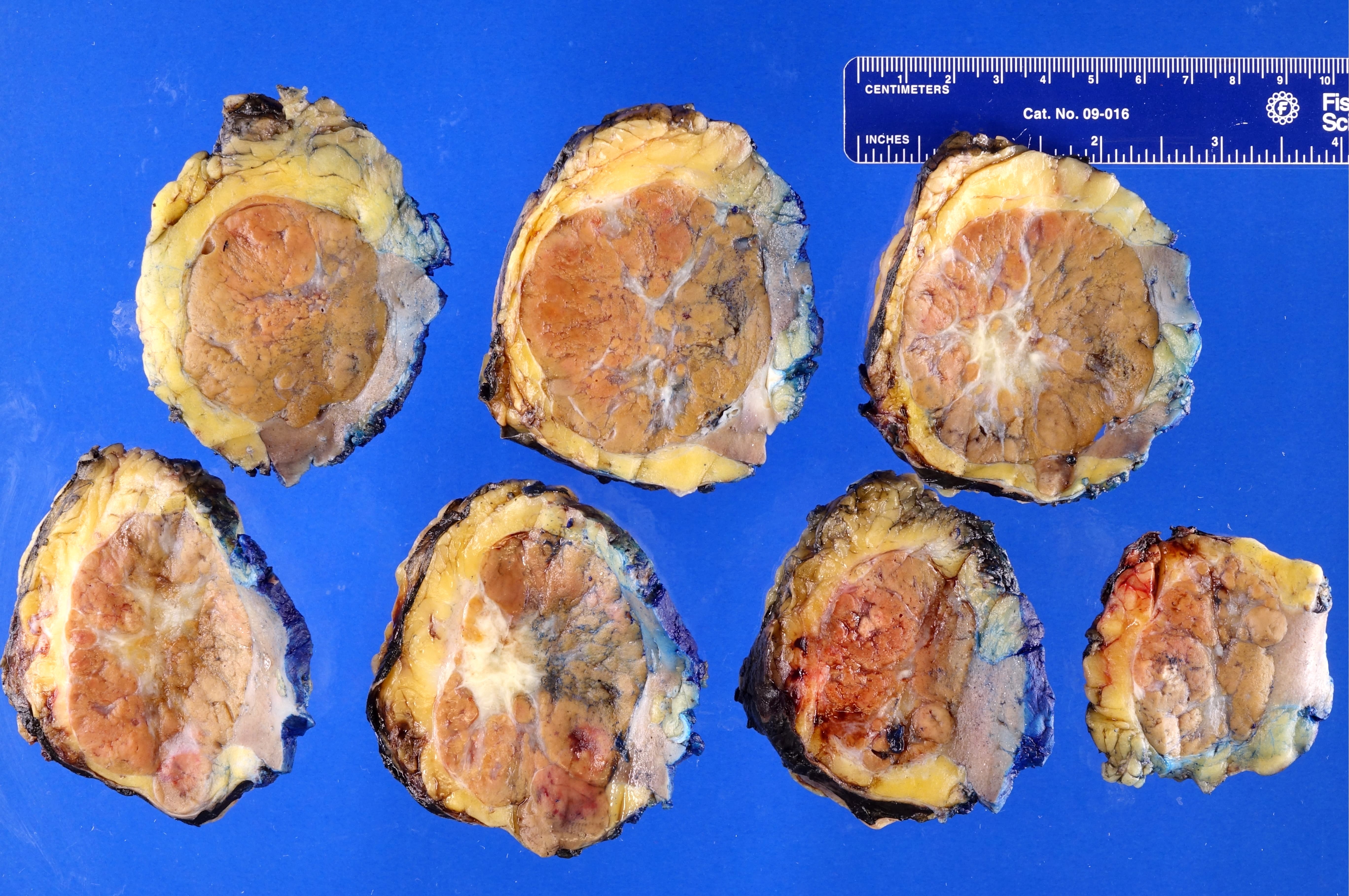
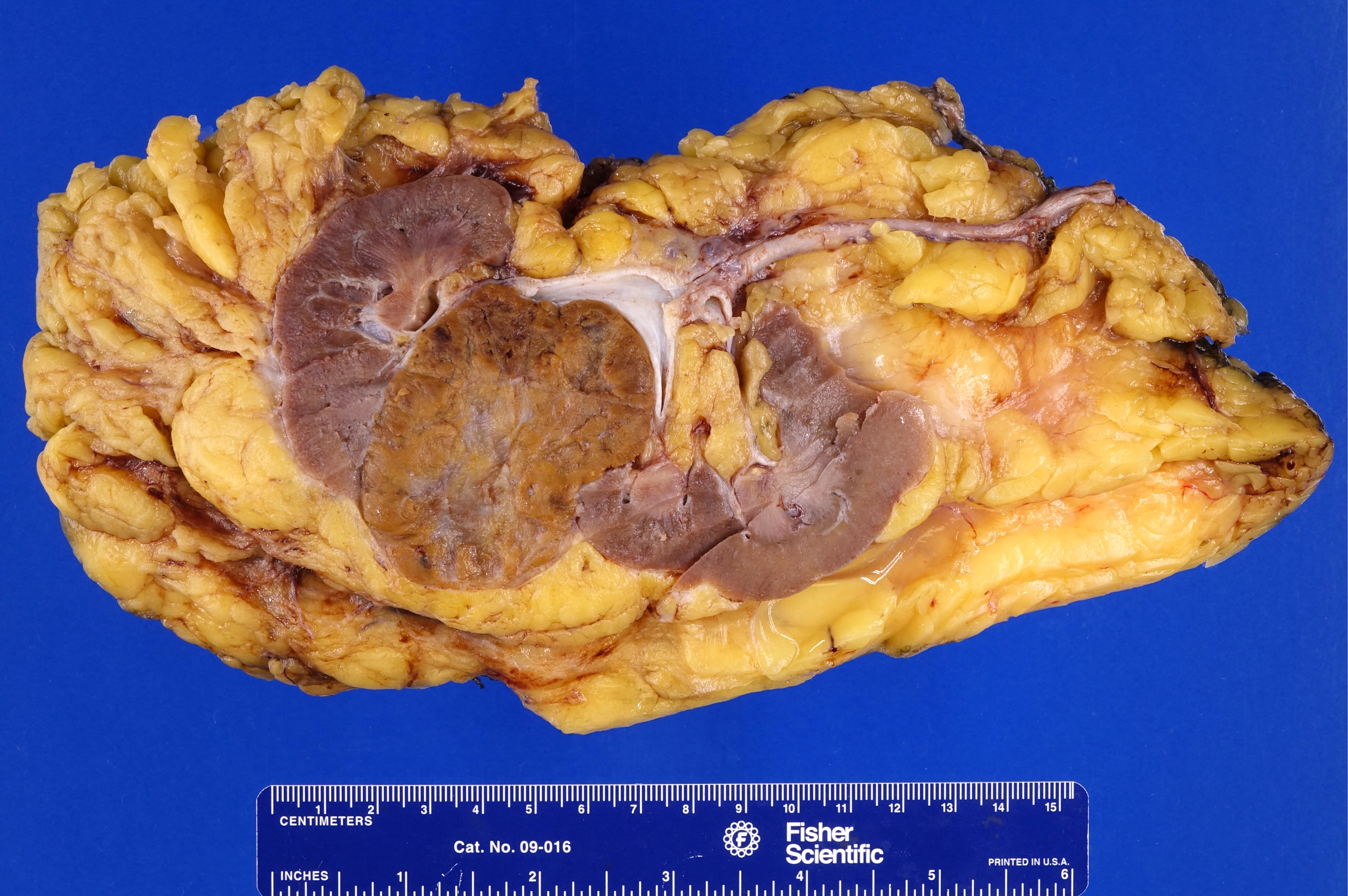
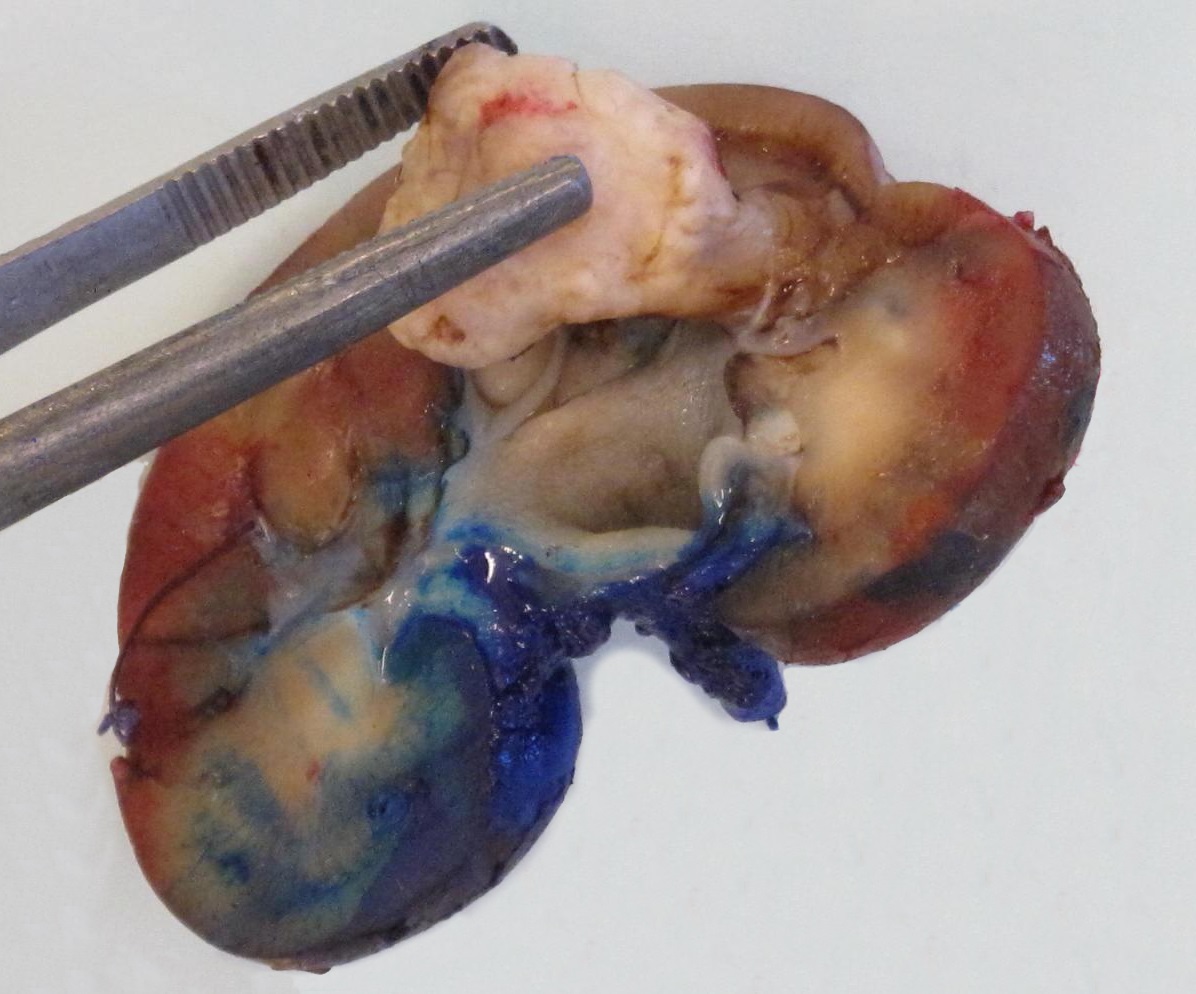
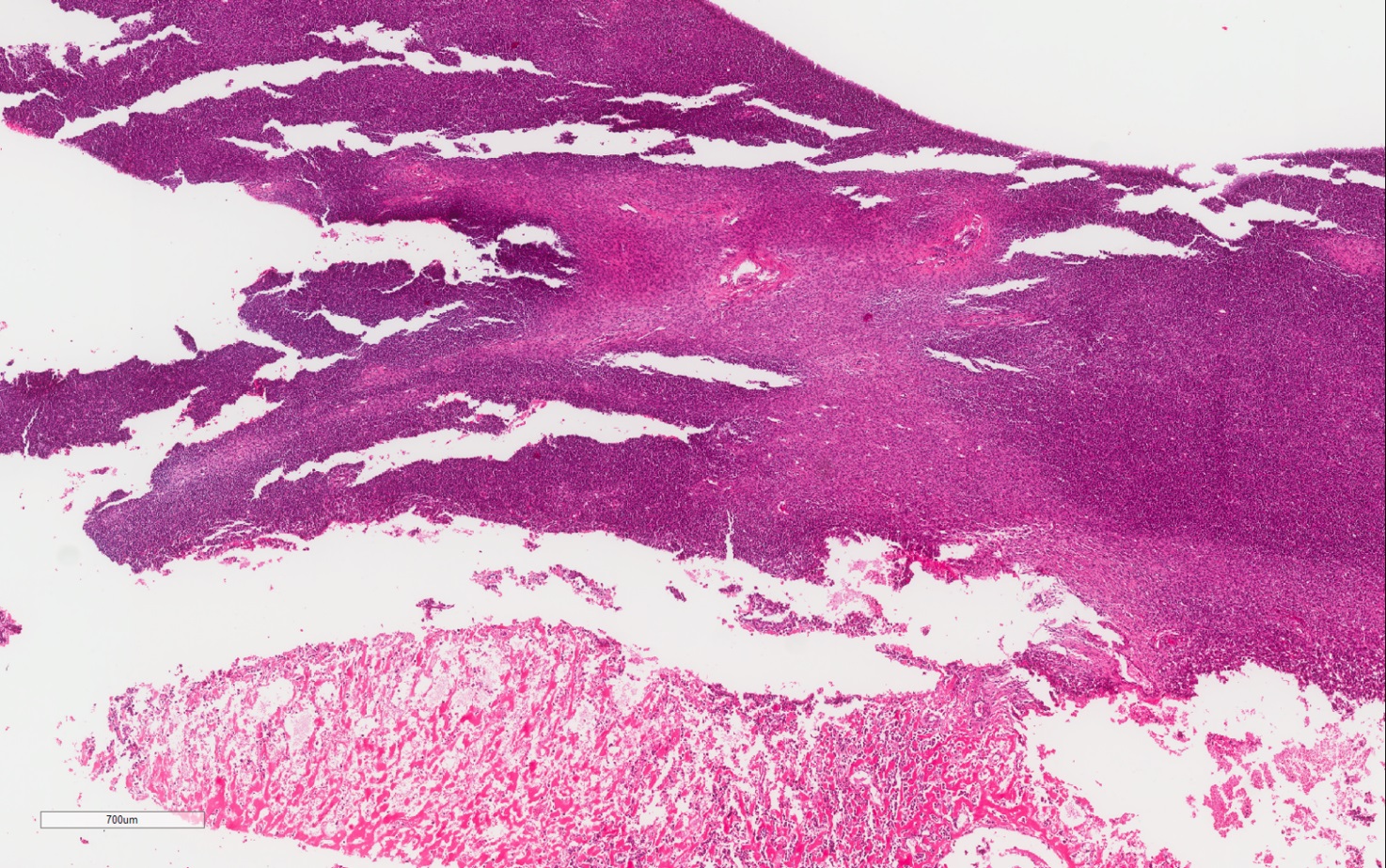
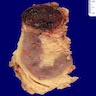
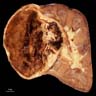
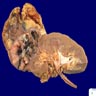
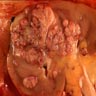
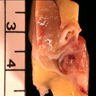
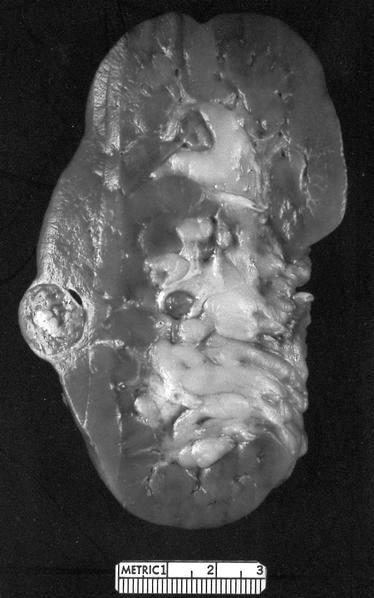
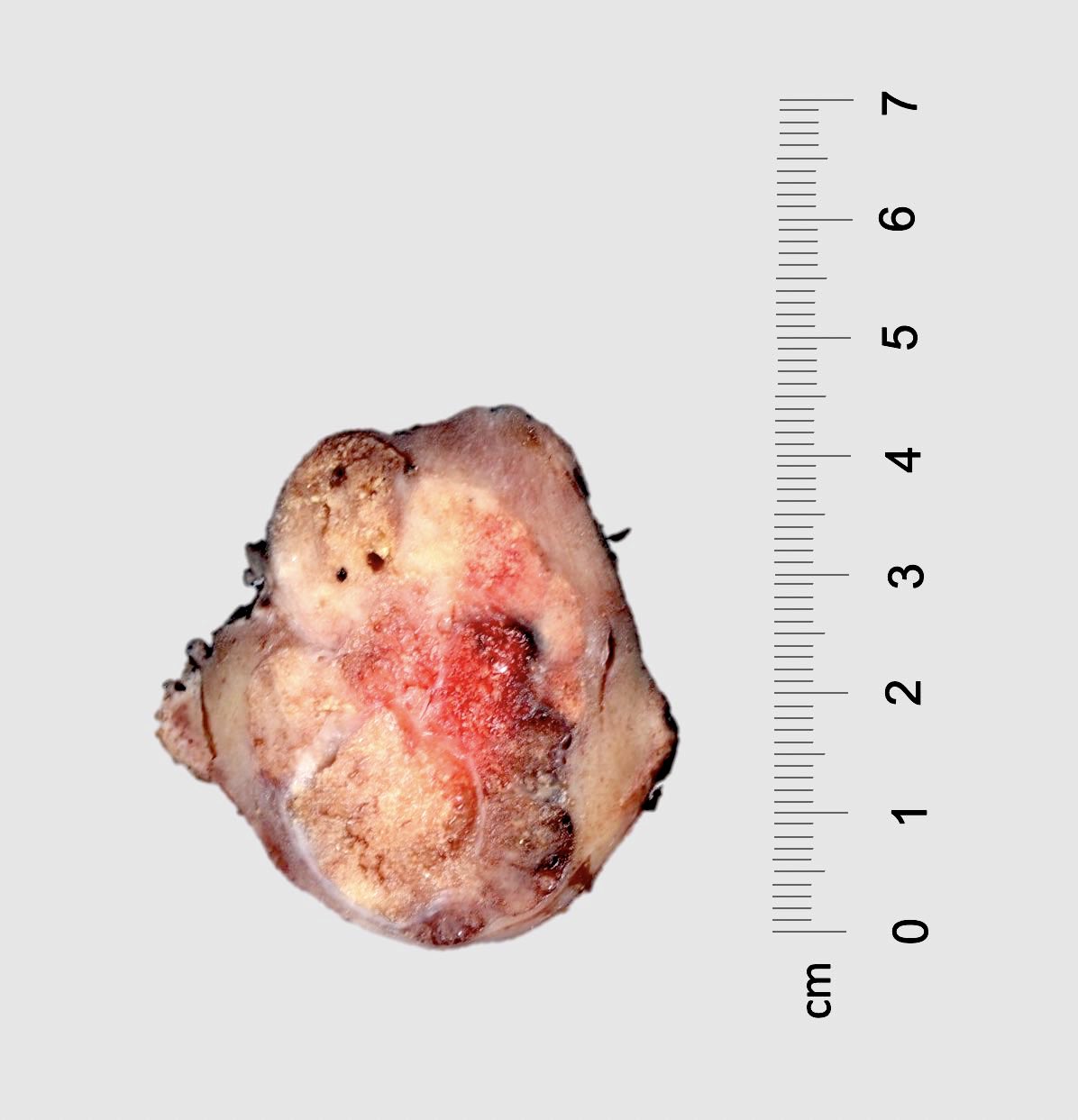
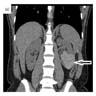
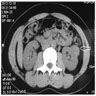
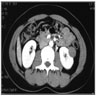
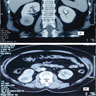
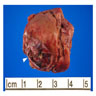
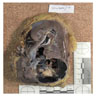
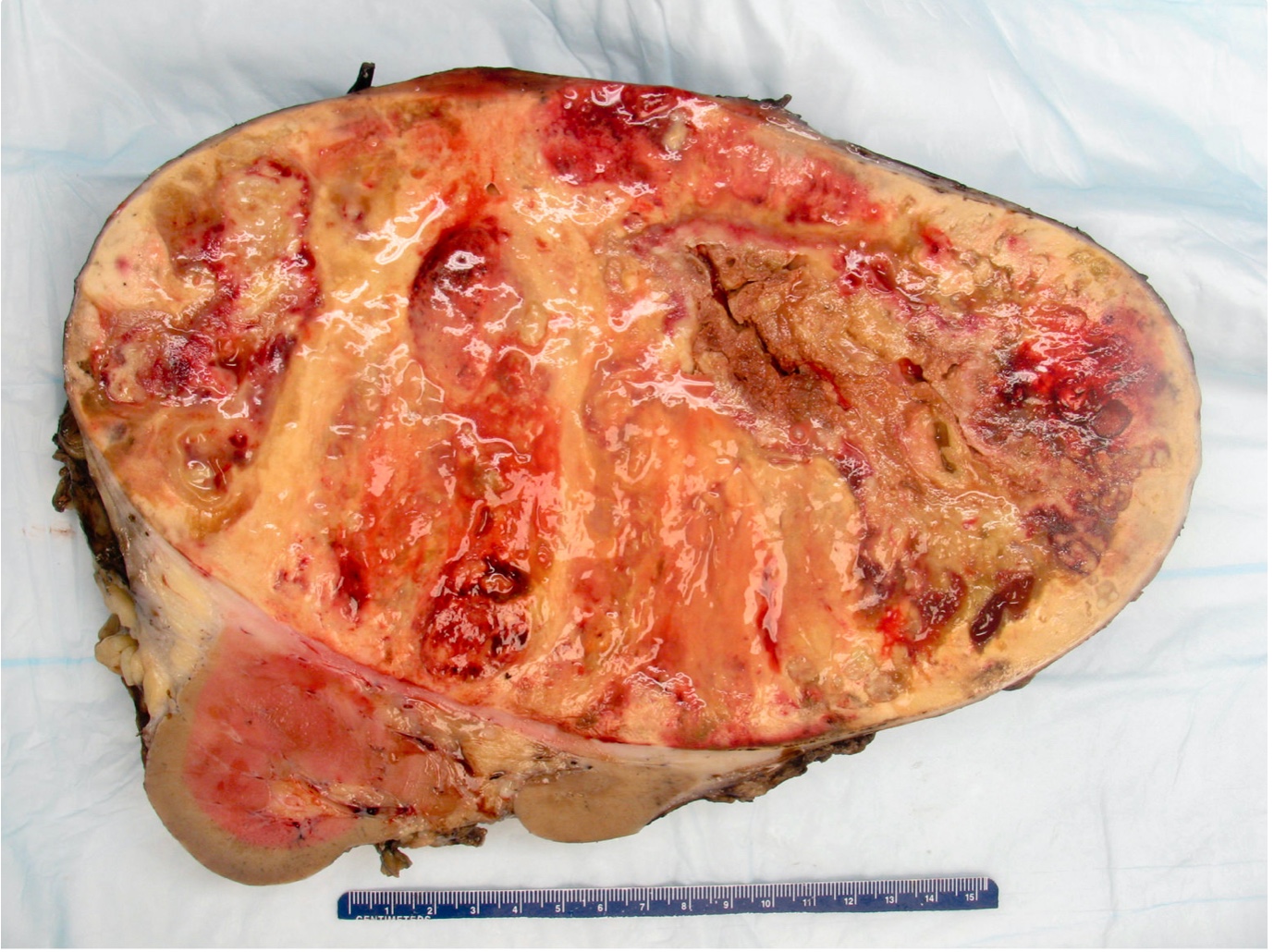
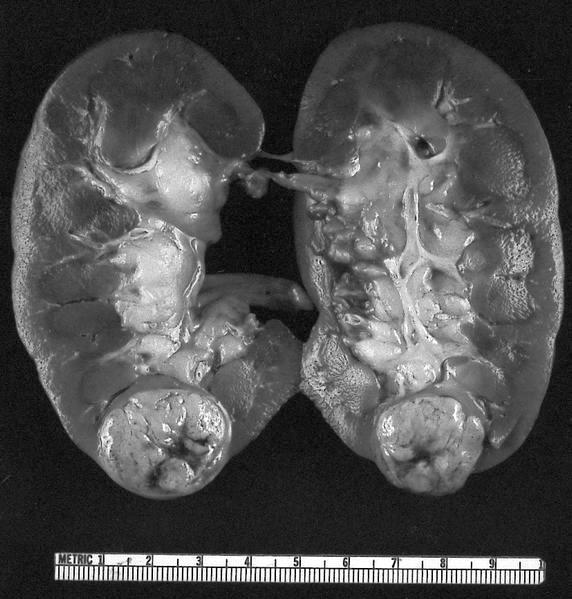
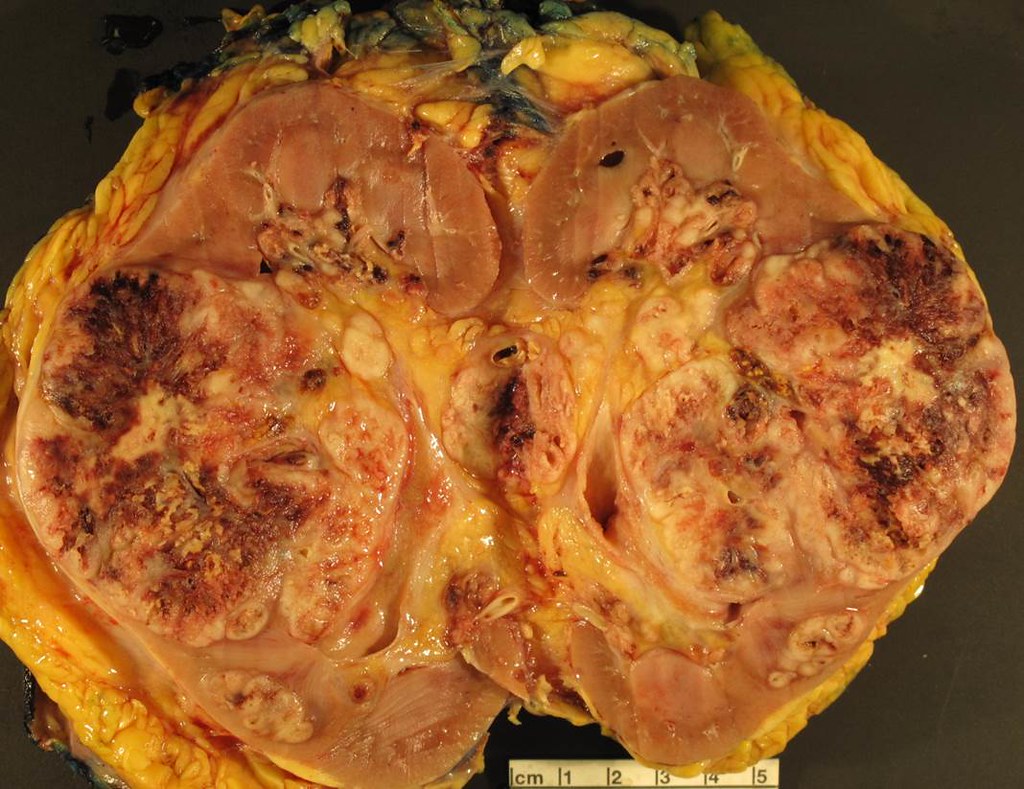
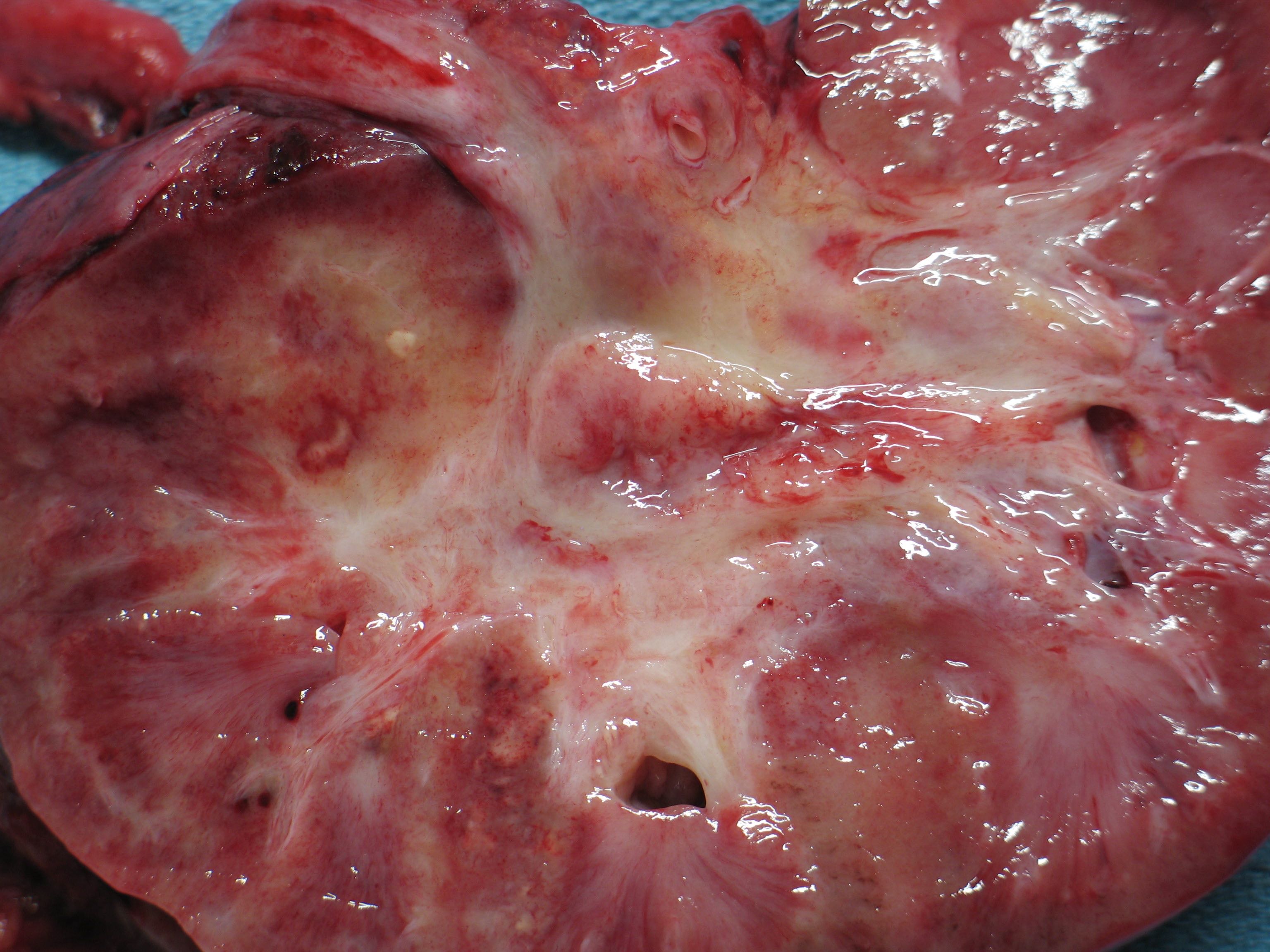
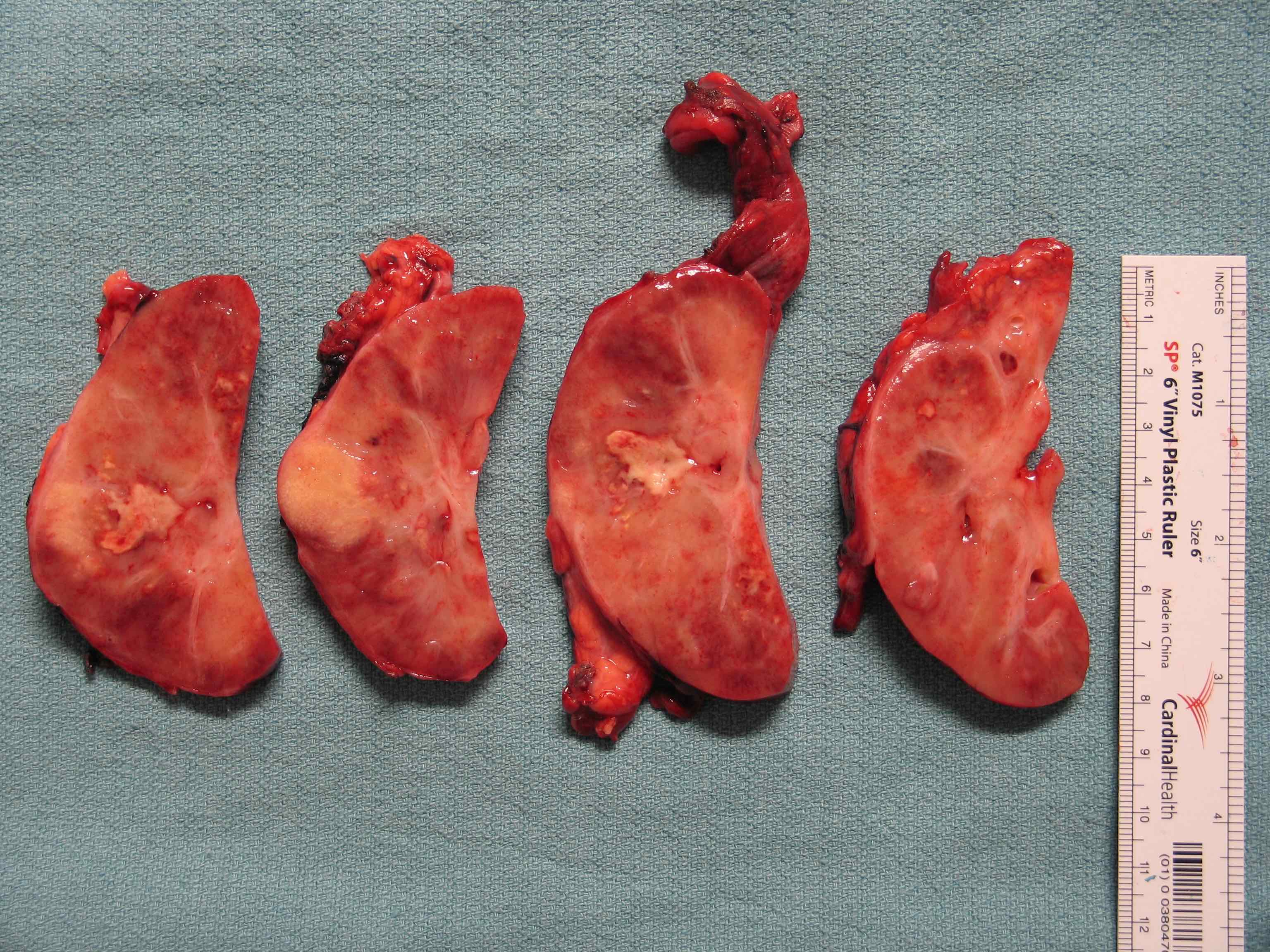
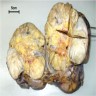
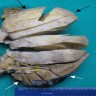
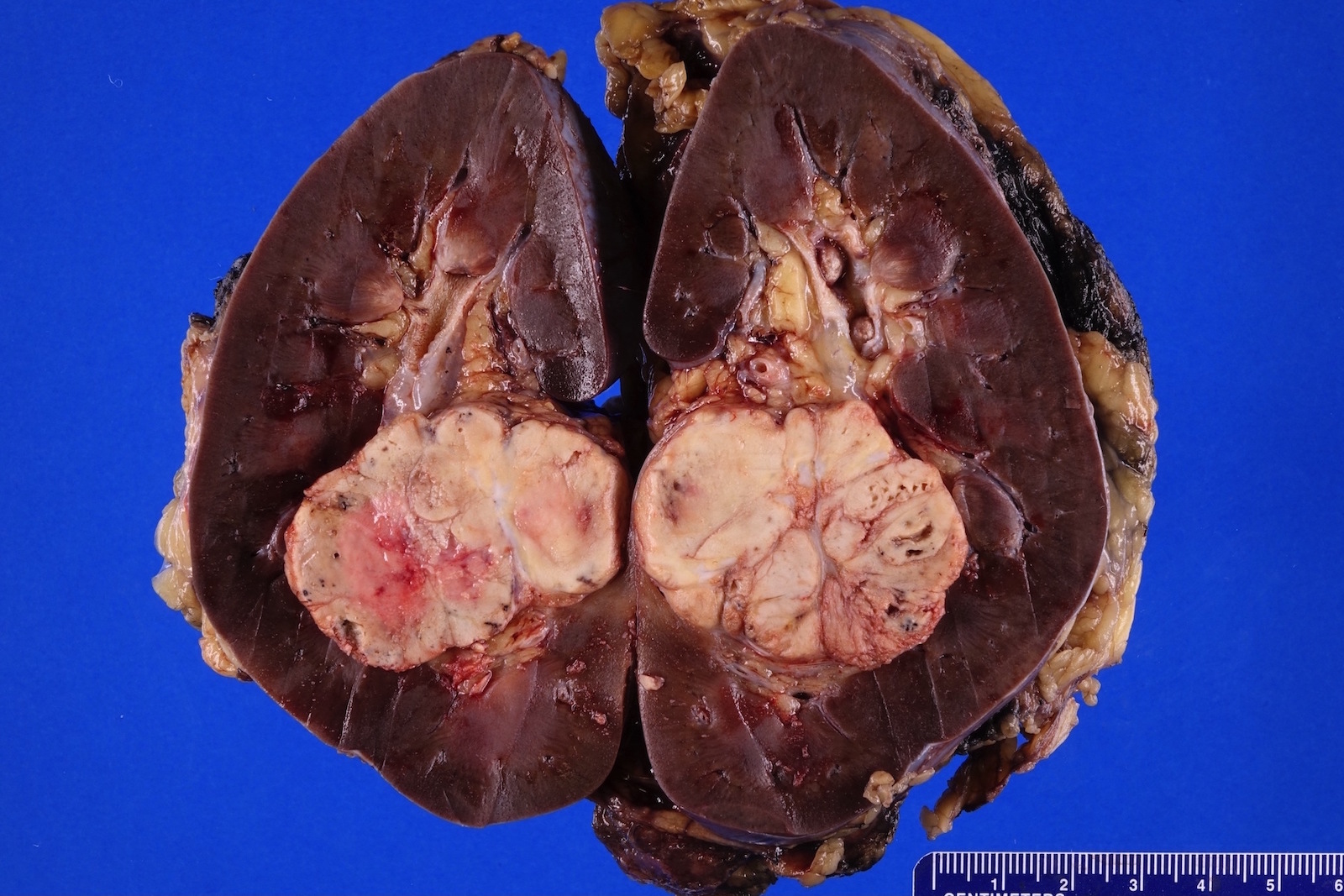
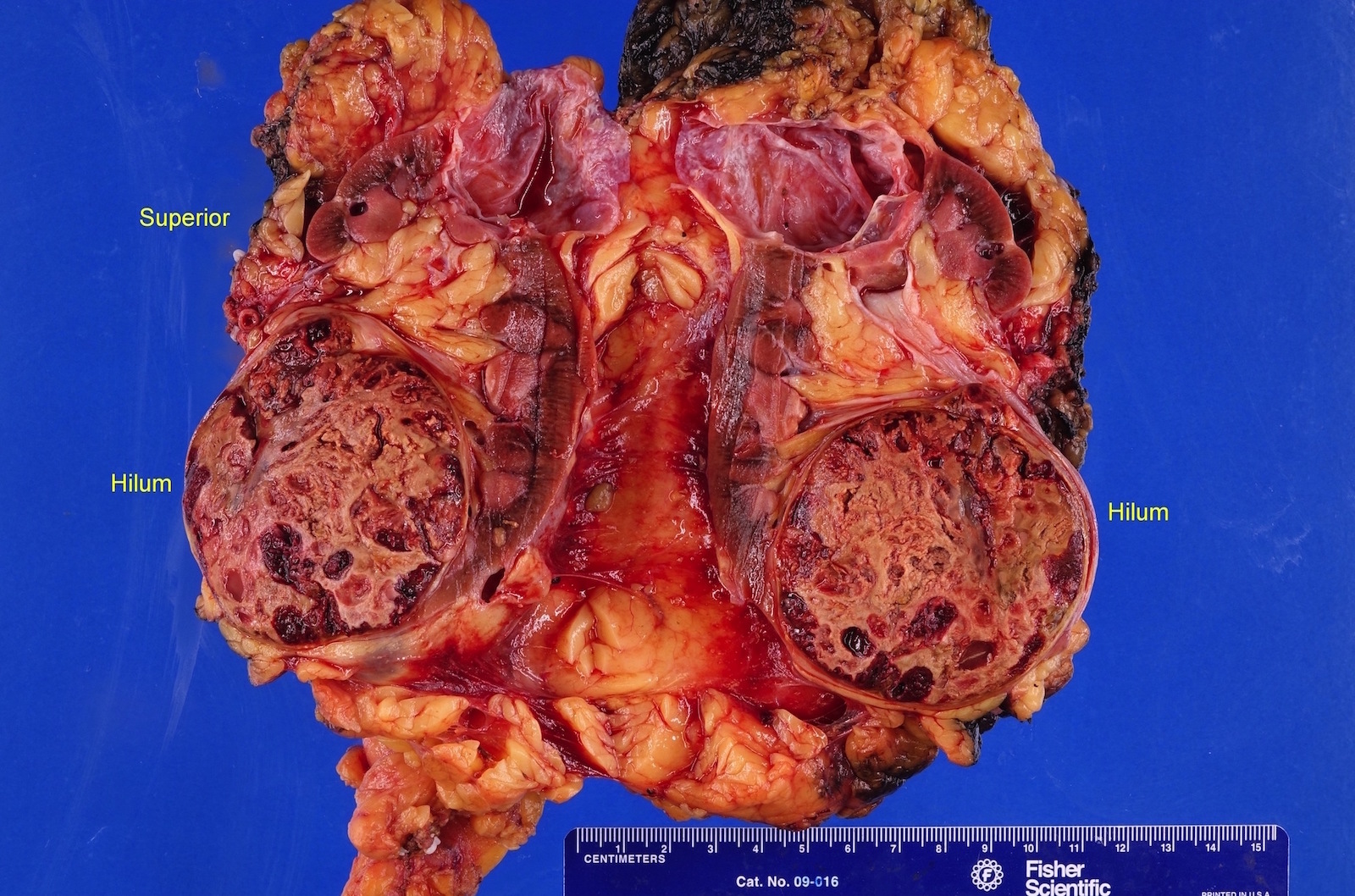
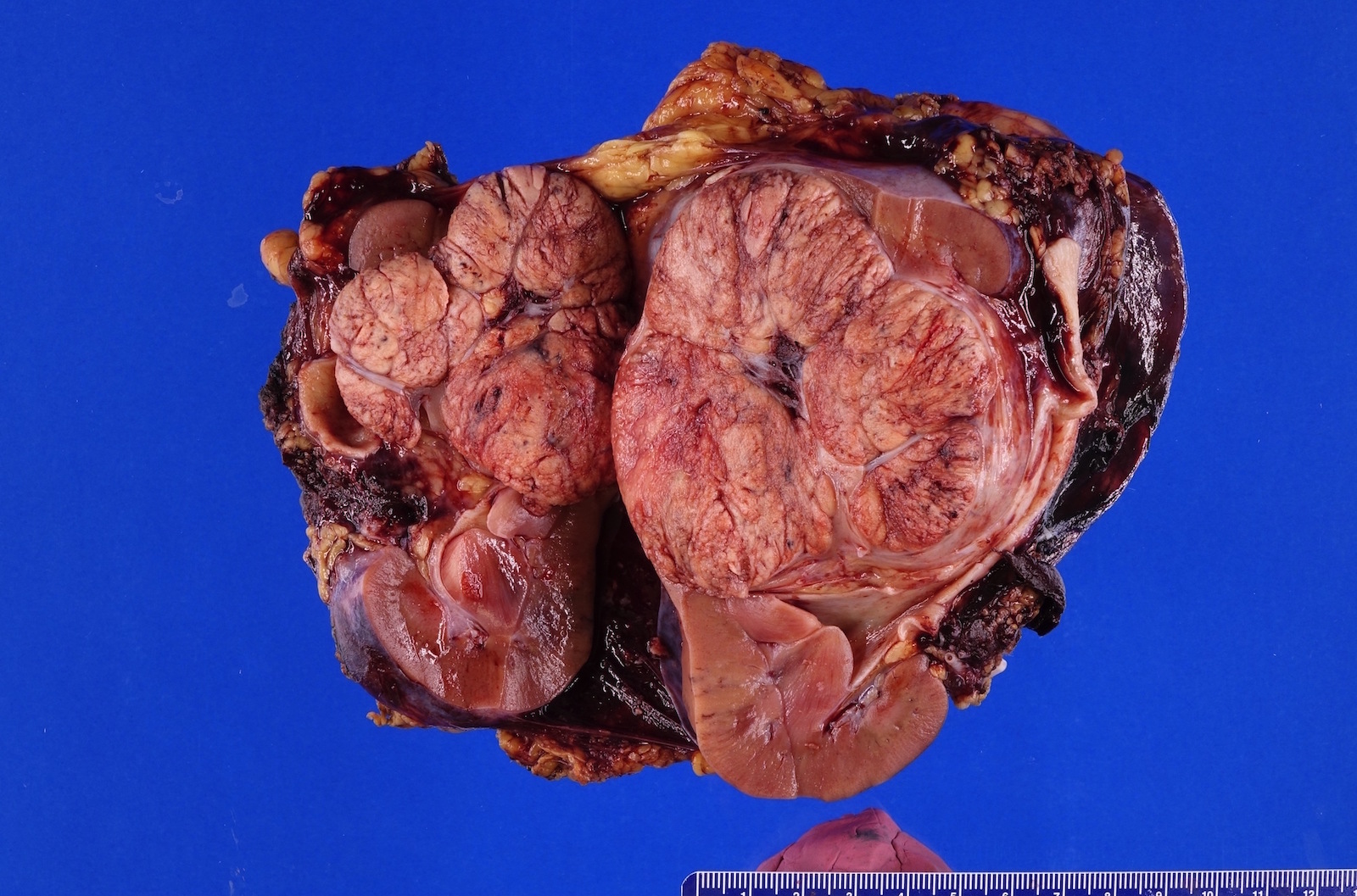
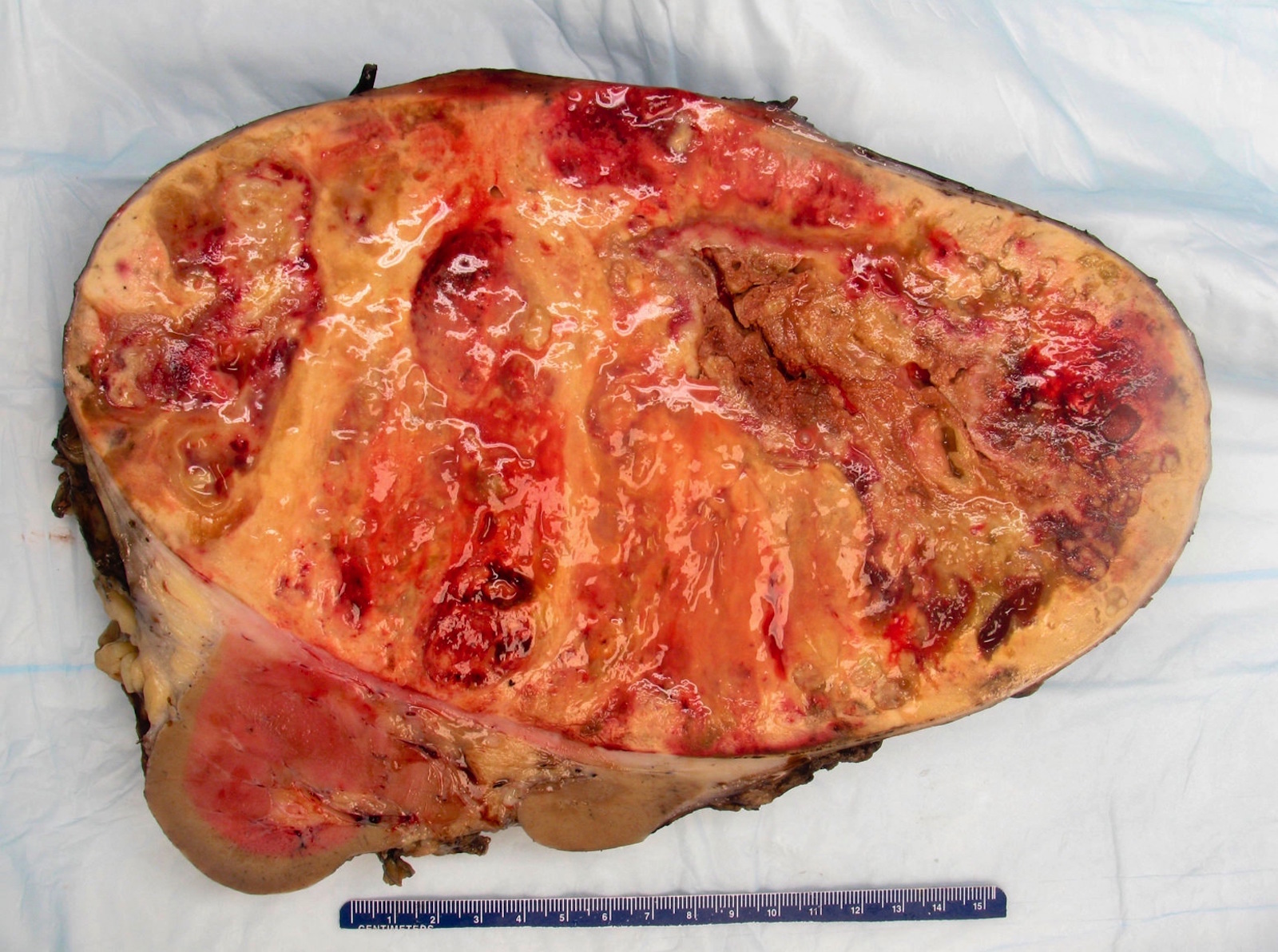
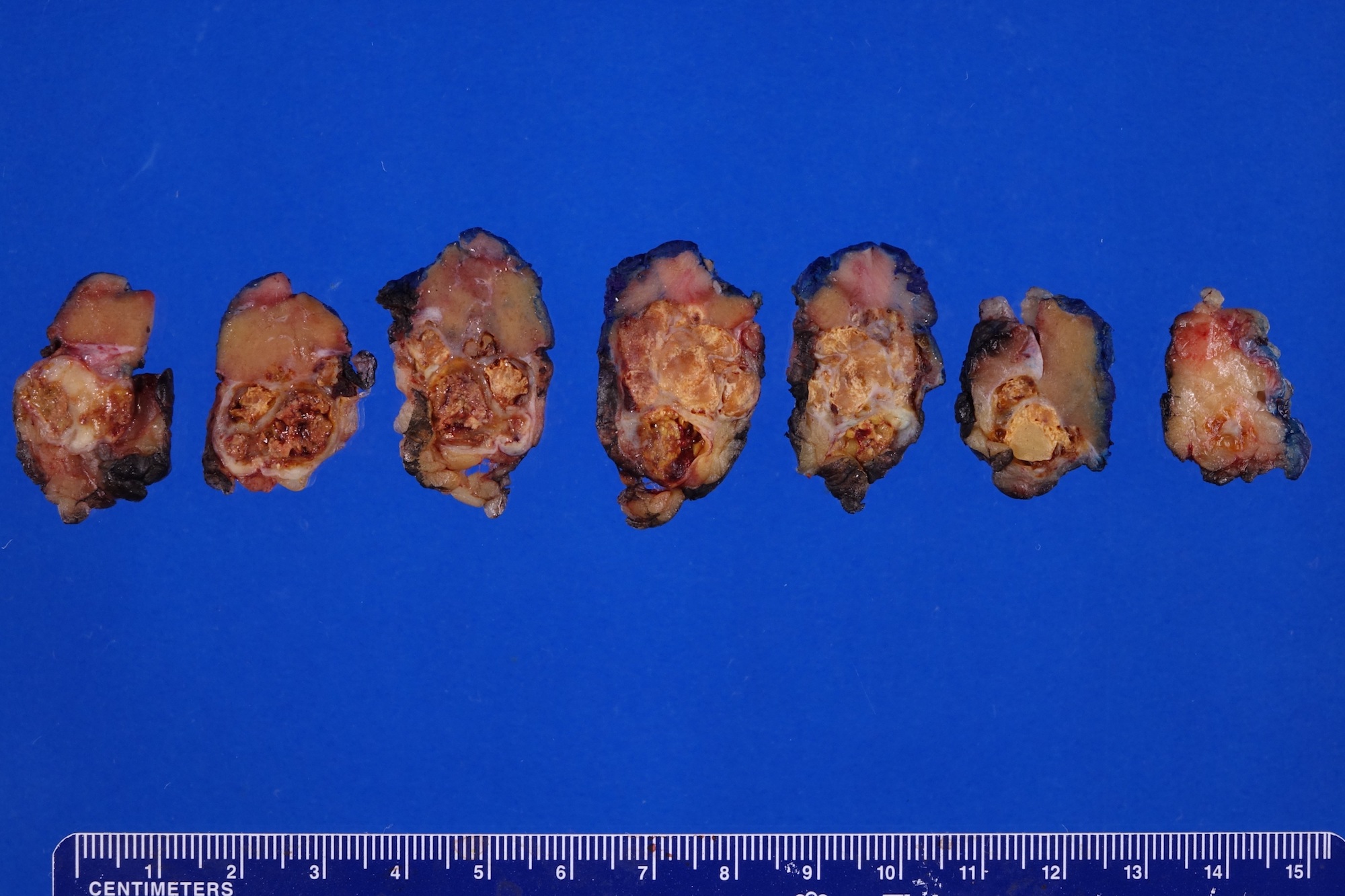
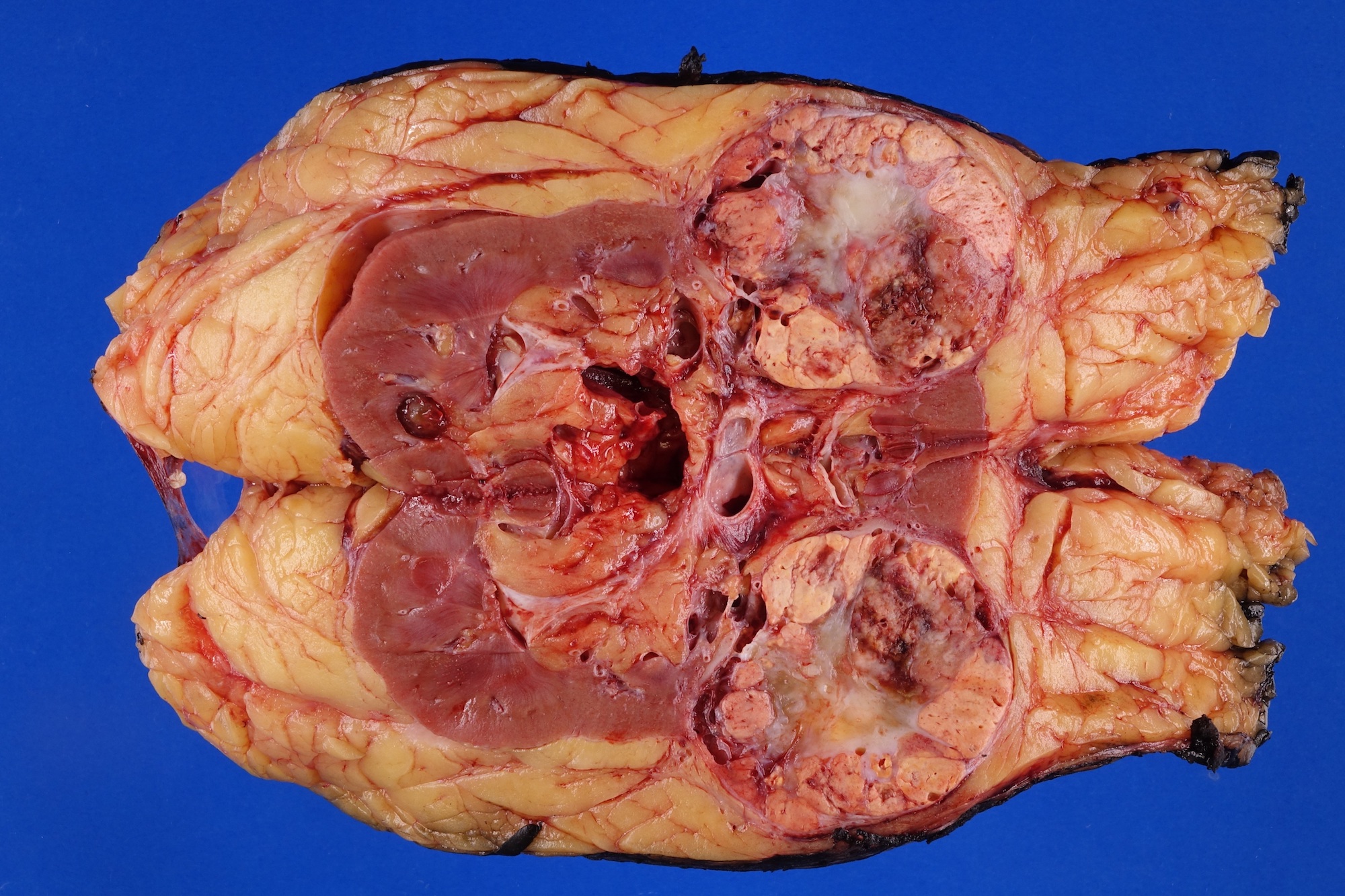
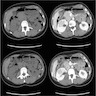
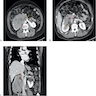
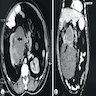
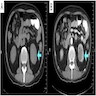
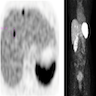
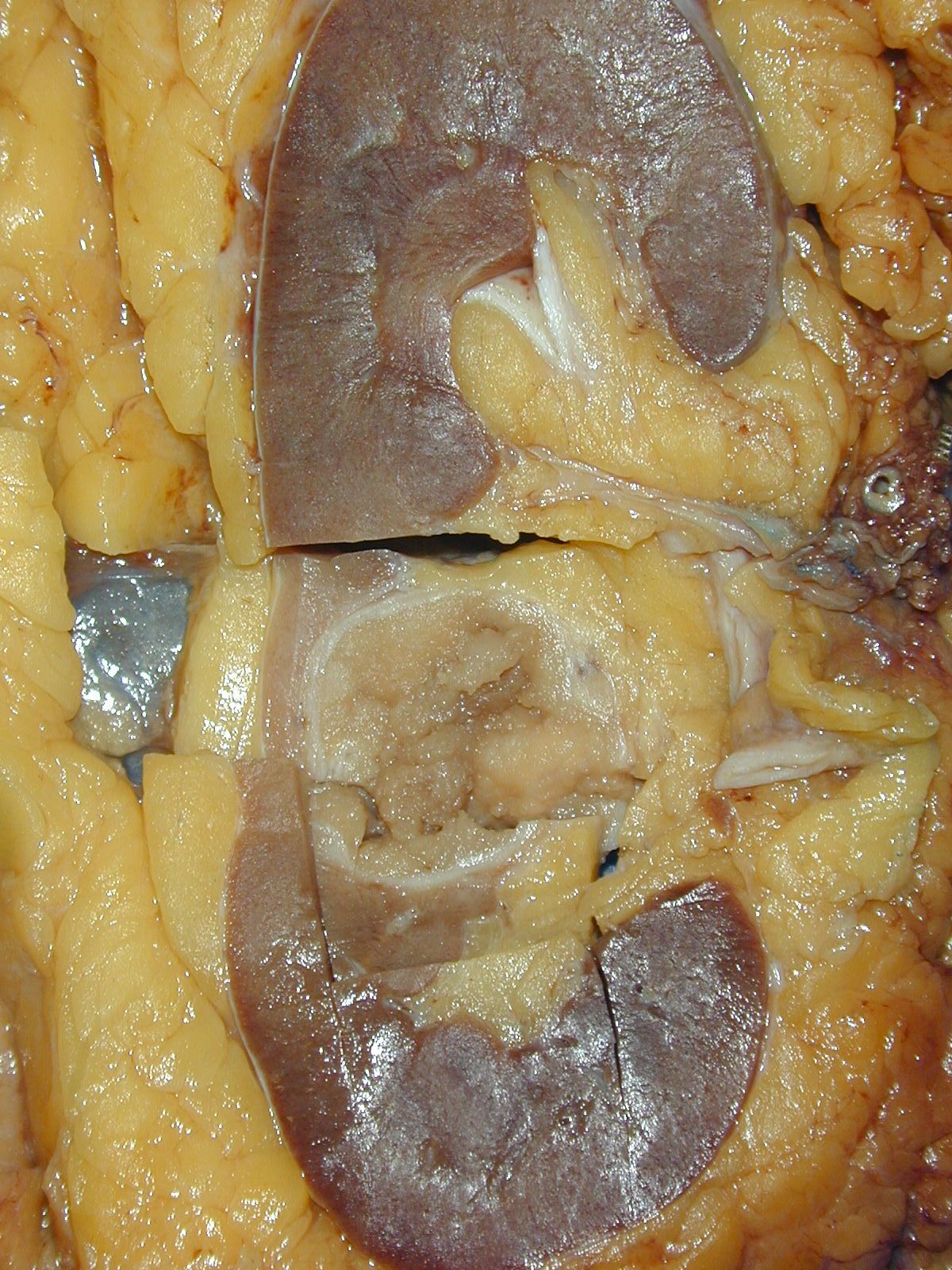
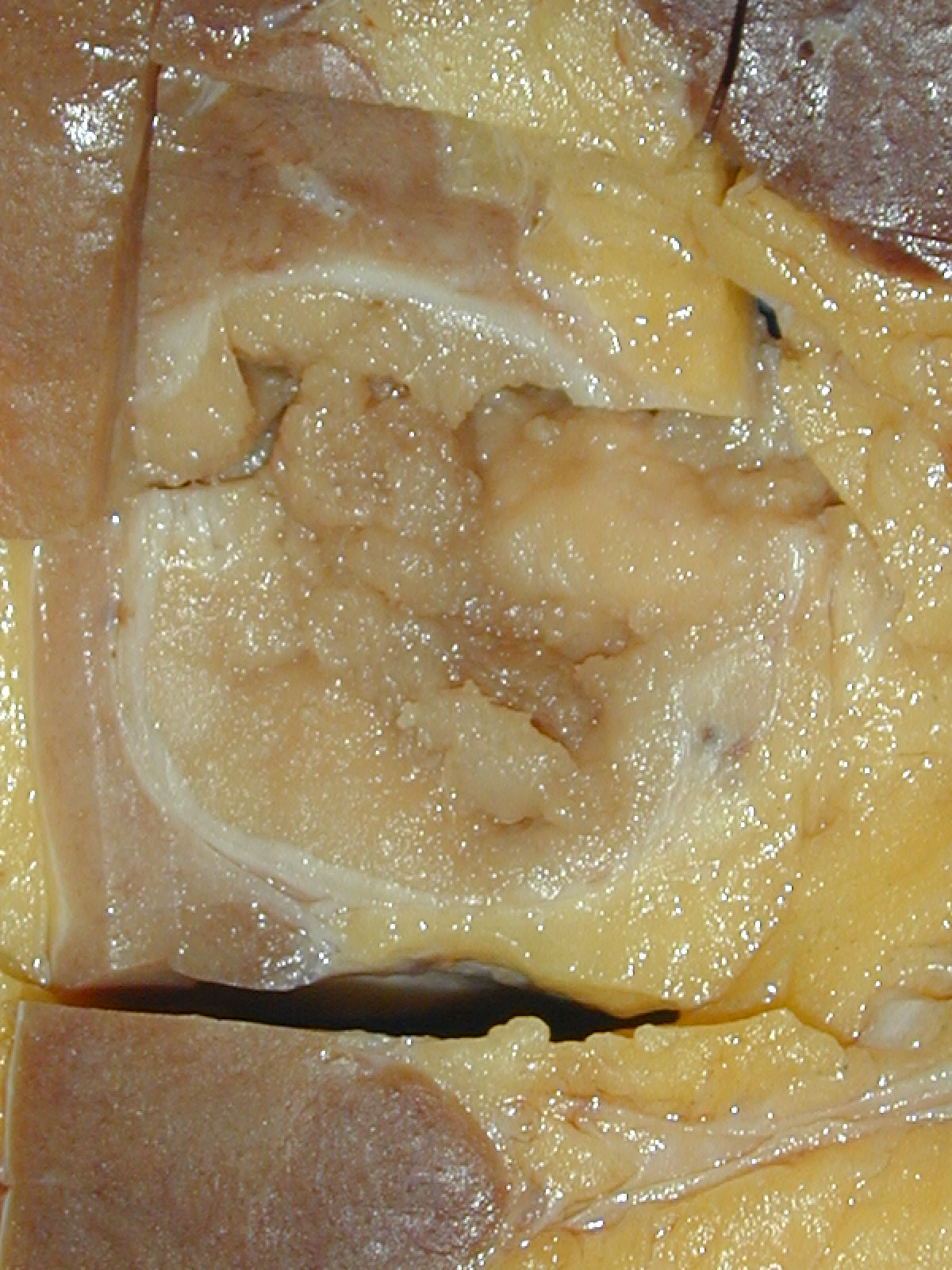
ALK rearranged renal cell carcinoma (
ALK-RCC) has been recently added as a provisional entity into the 2016 World Health Organization classification.
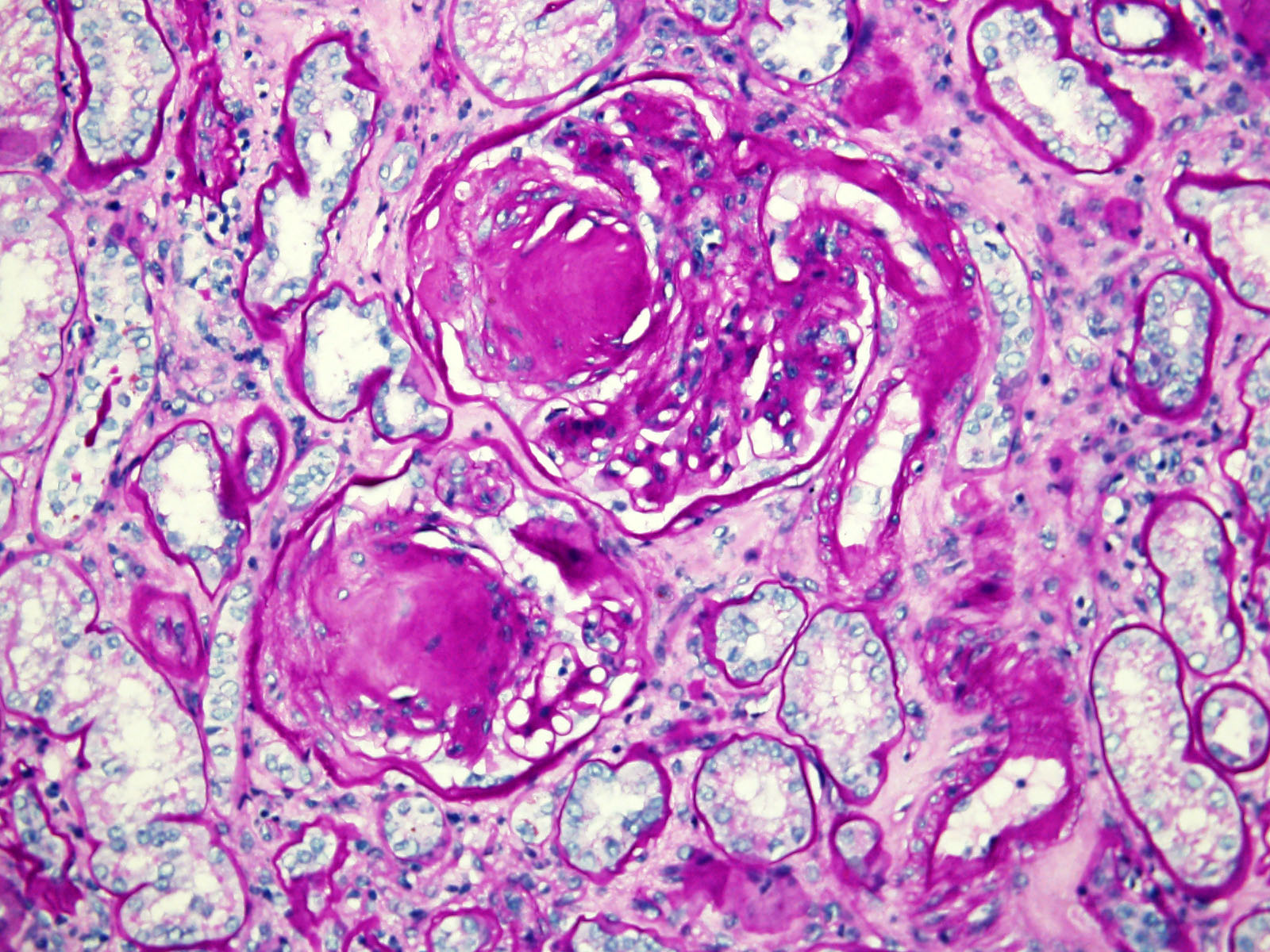
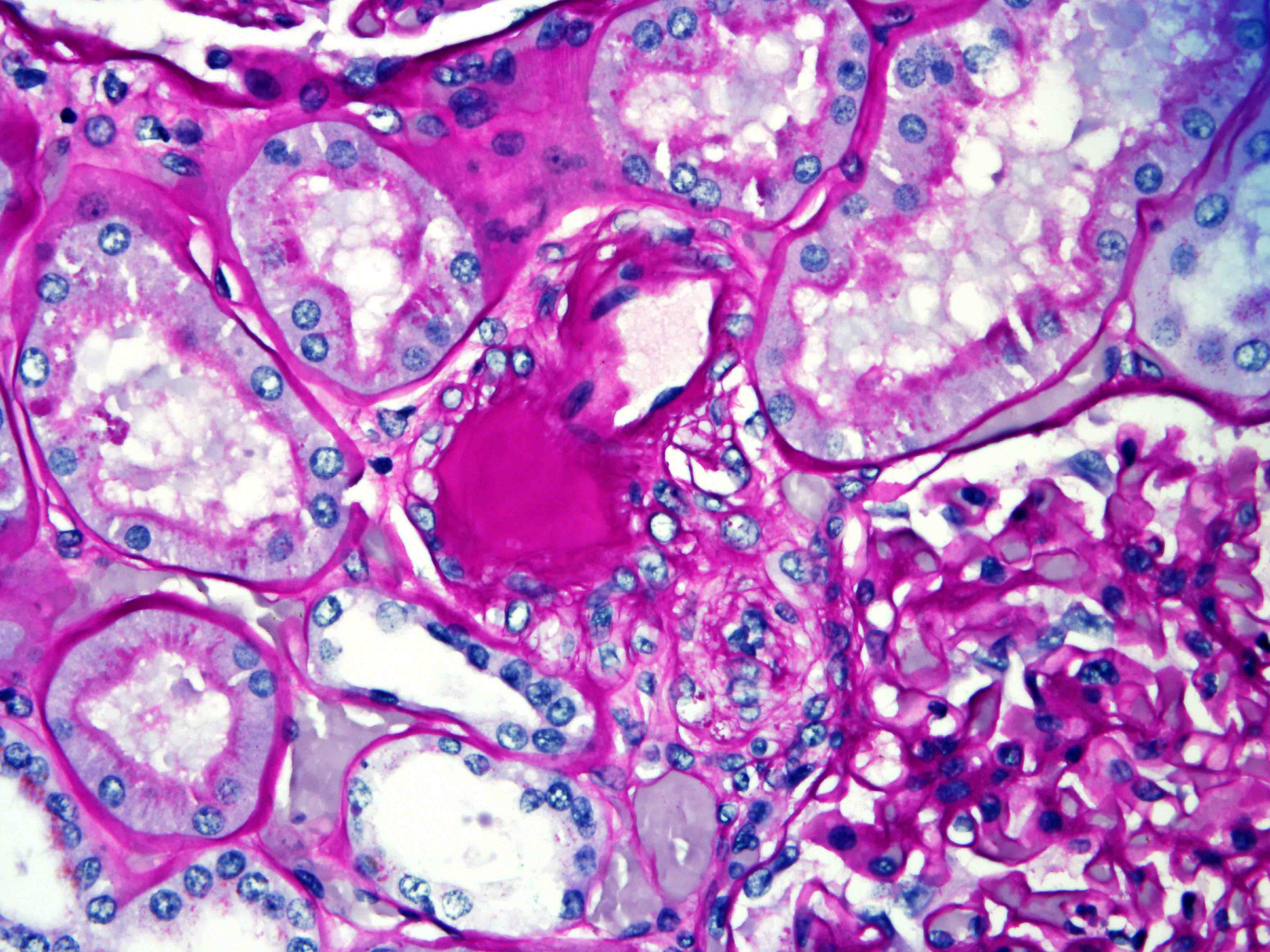
ALK-RCC is characterized by fusion of a variety of genes with the anaplastic lymphoma kinase (
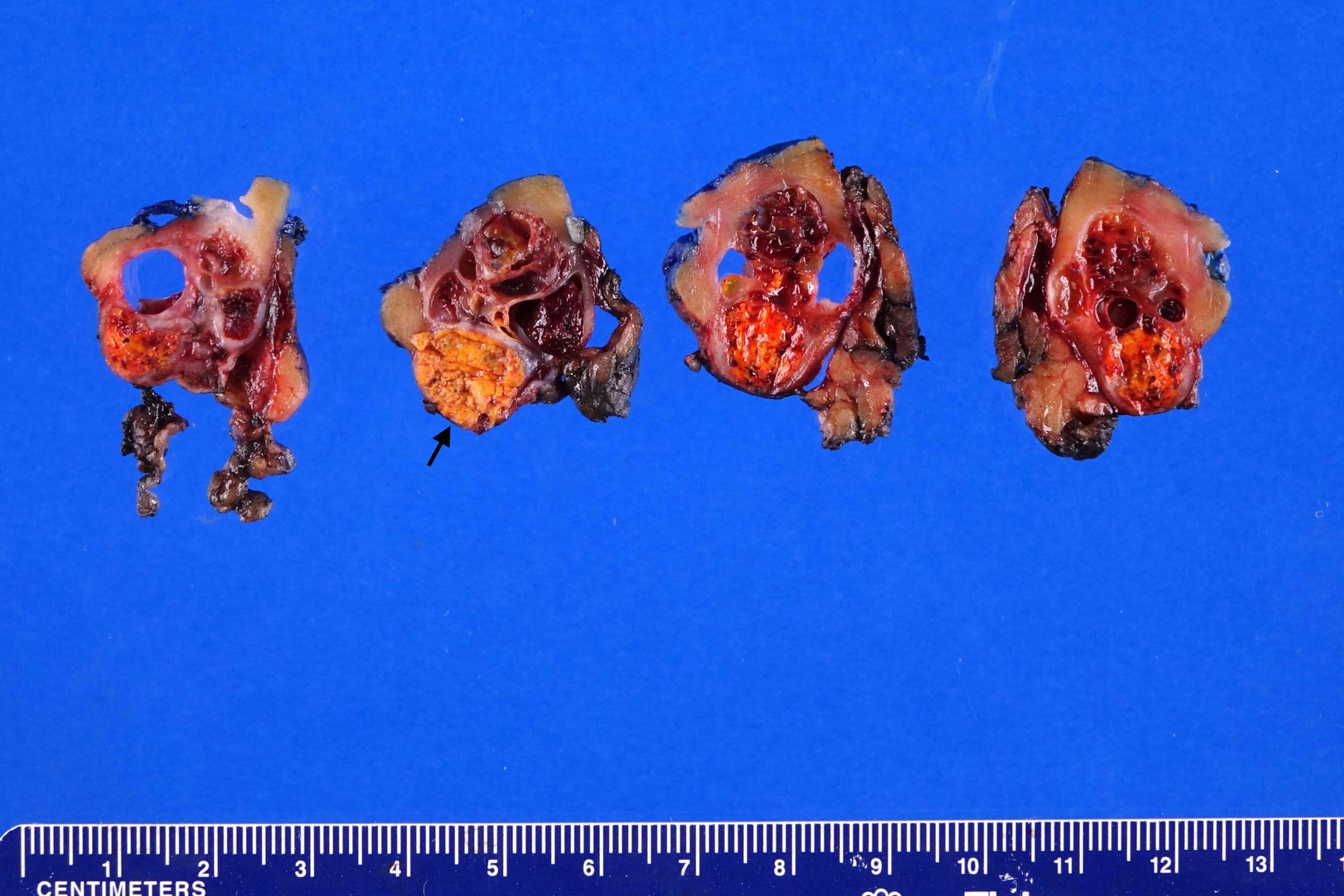
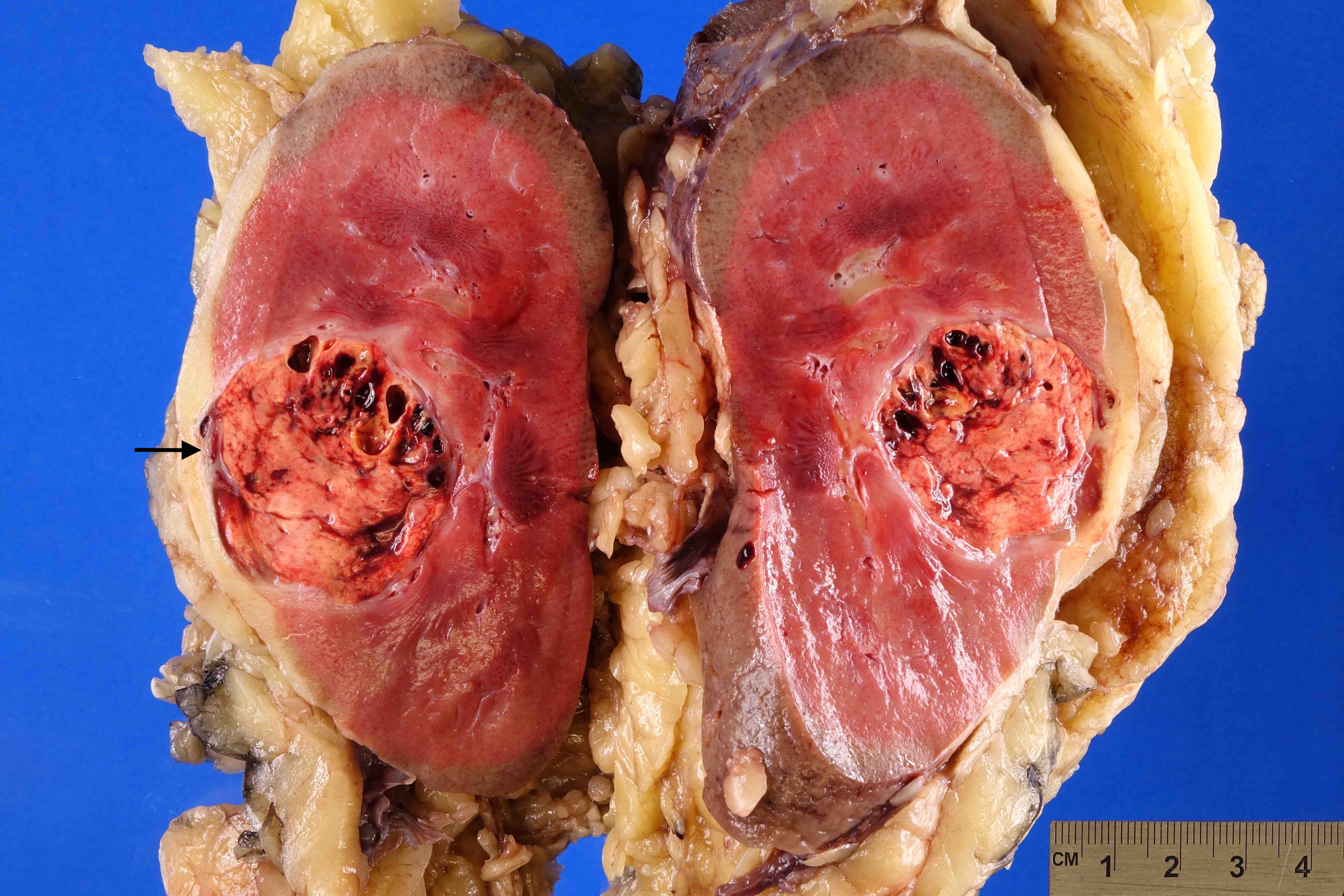
ALK) gene occurring in children with sickle cell trait and adults without sickle cell trait. It is a rare tumor affecting patients from 6 to 61 years old (mean 30 years). In children,
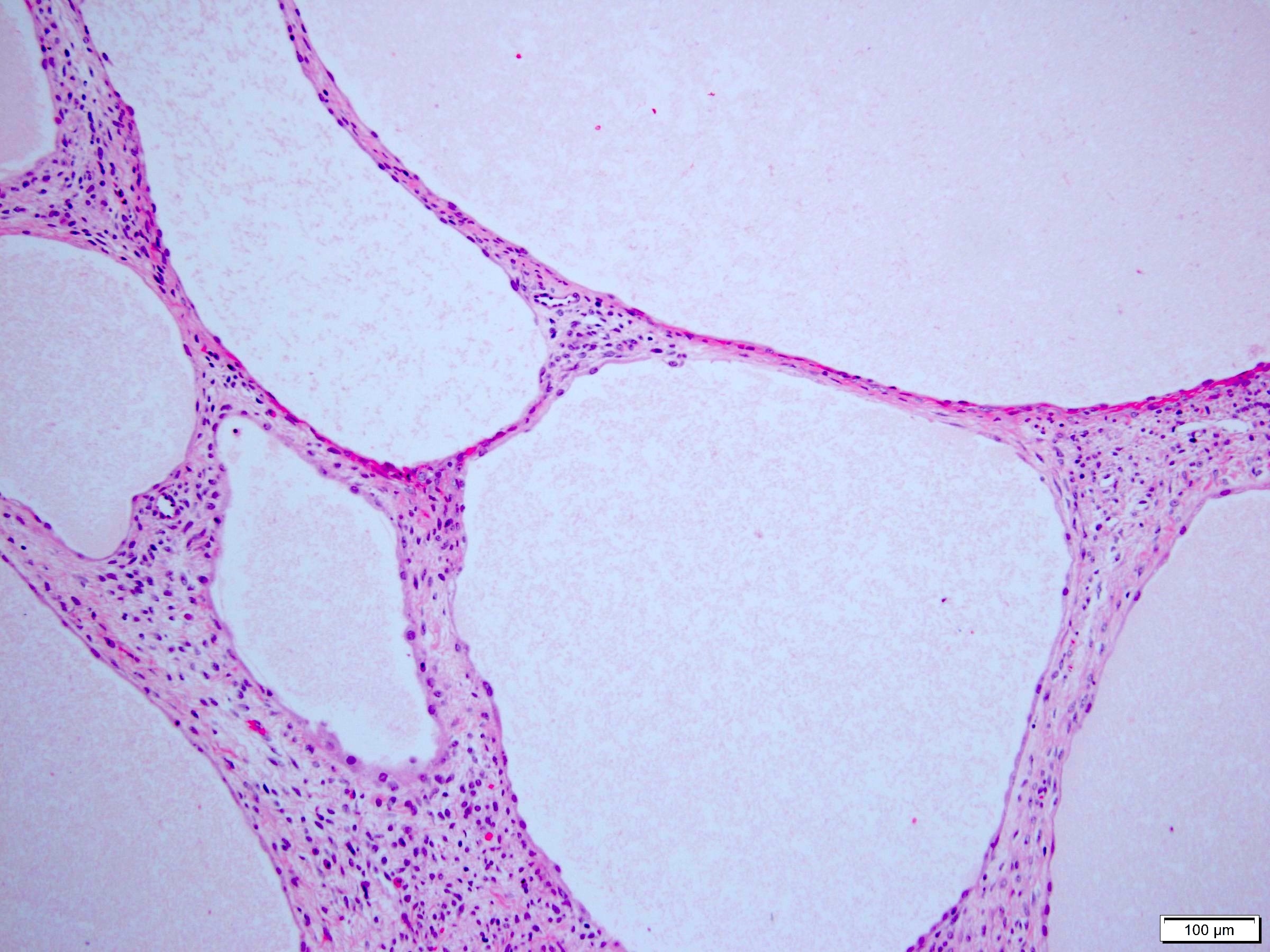
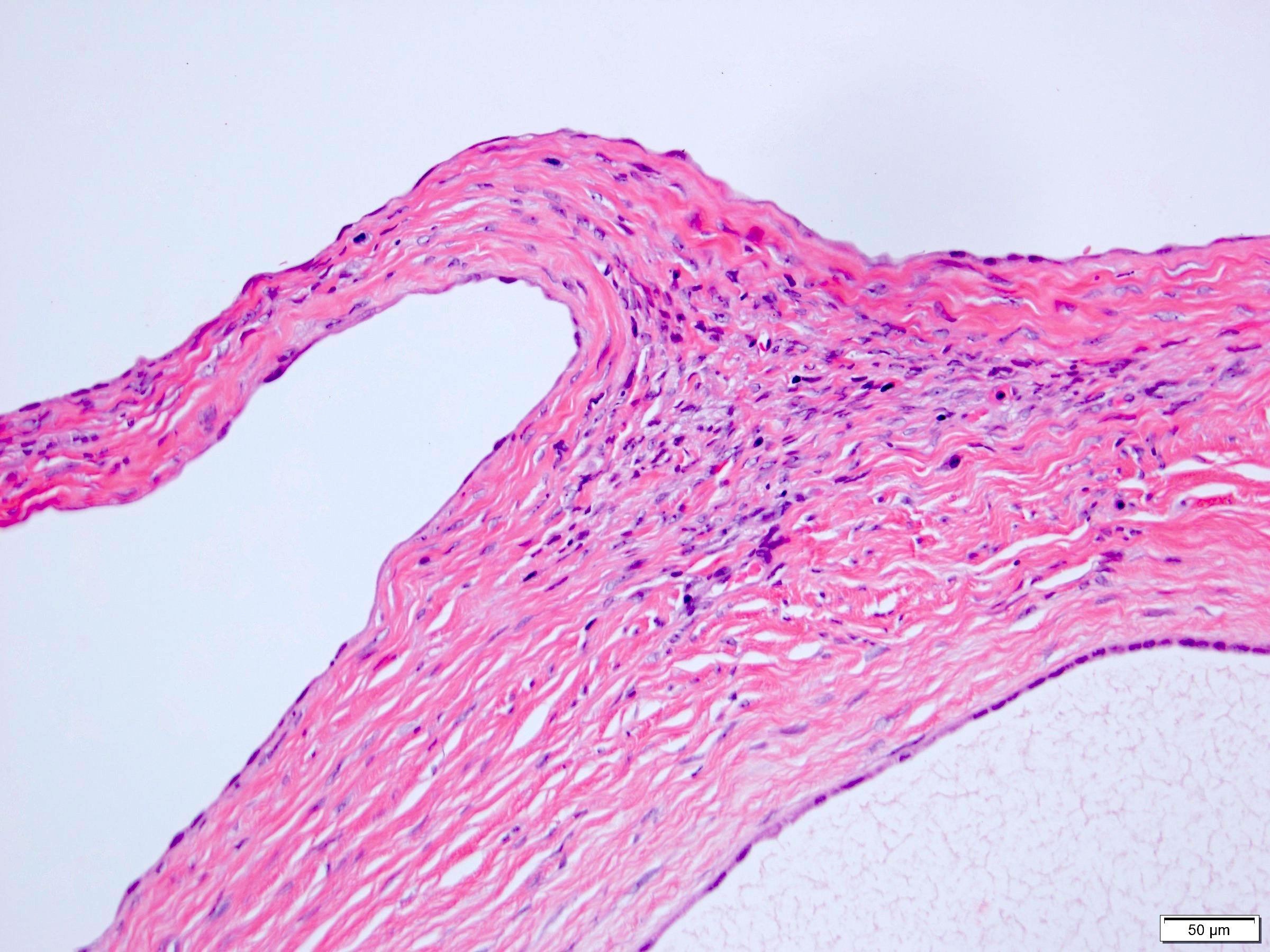
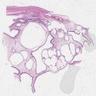
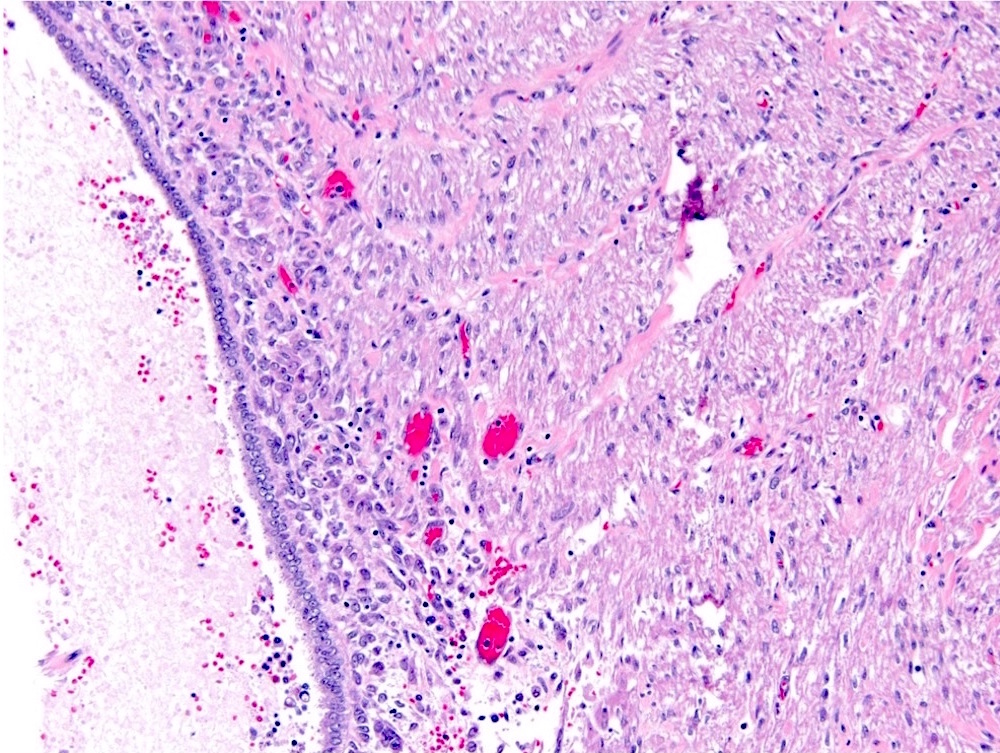
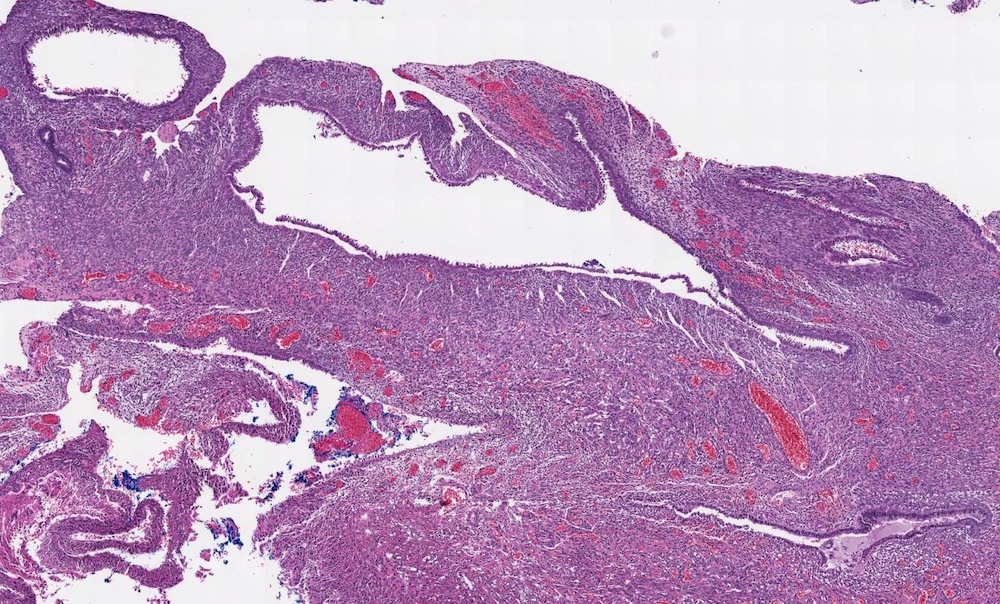
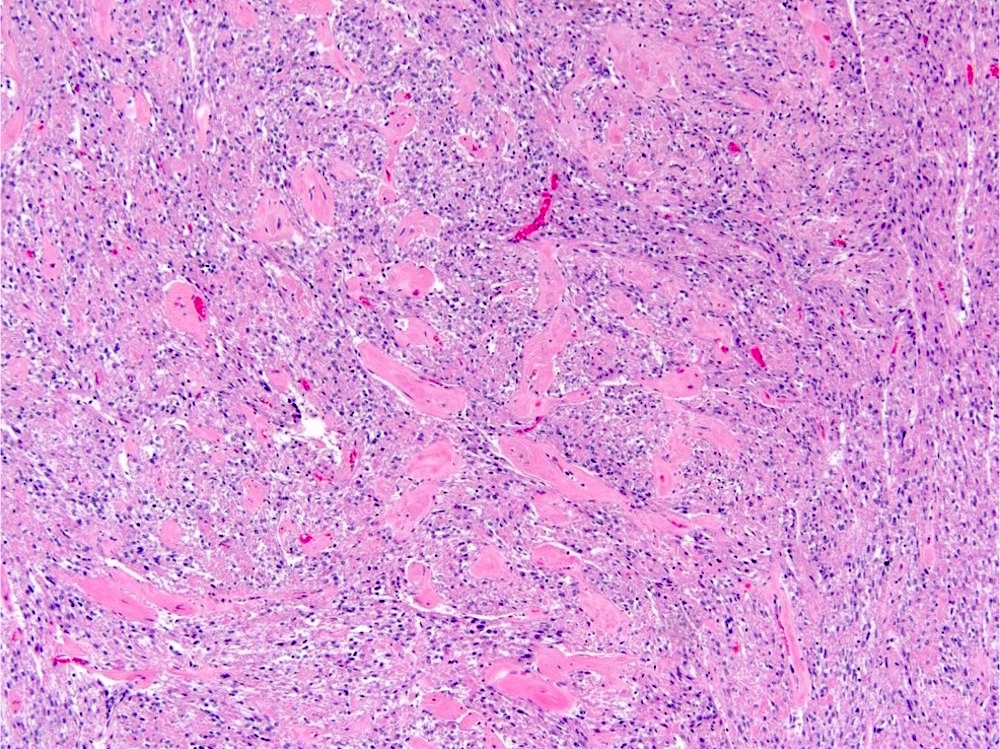
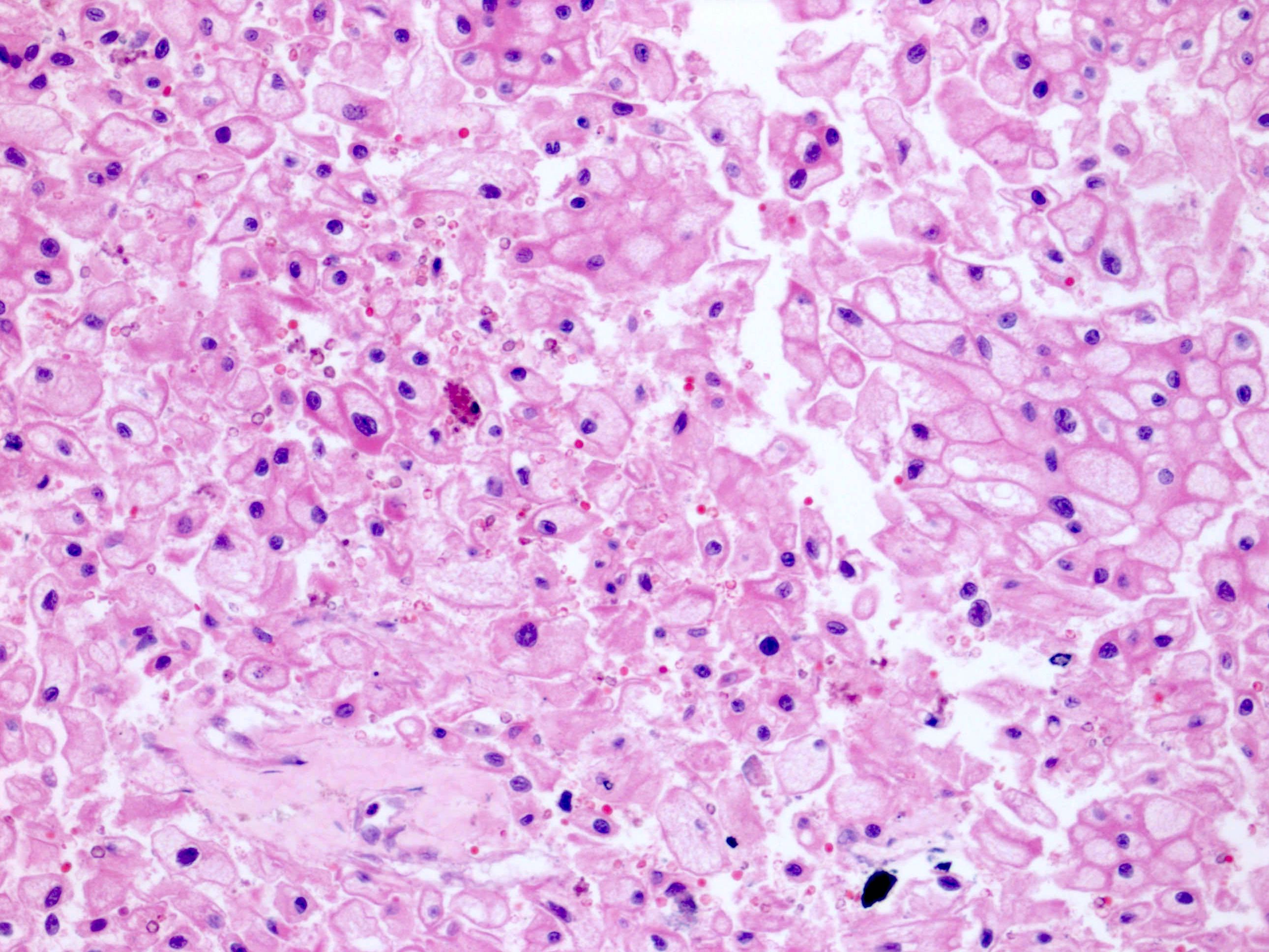
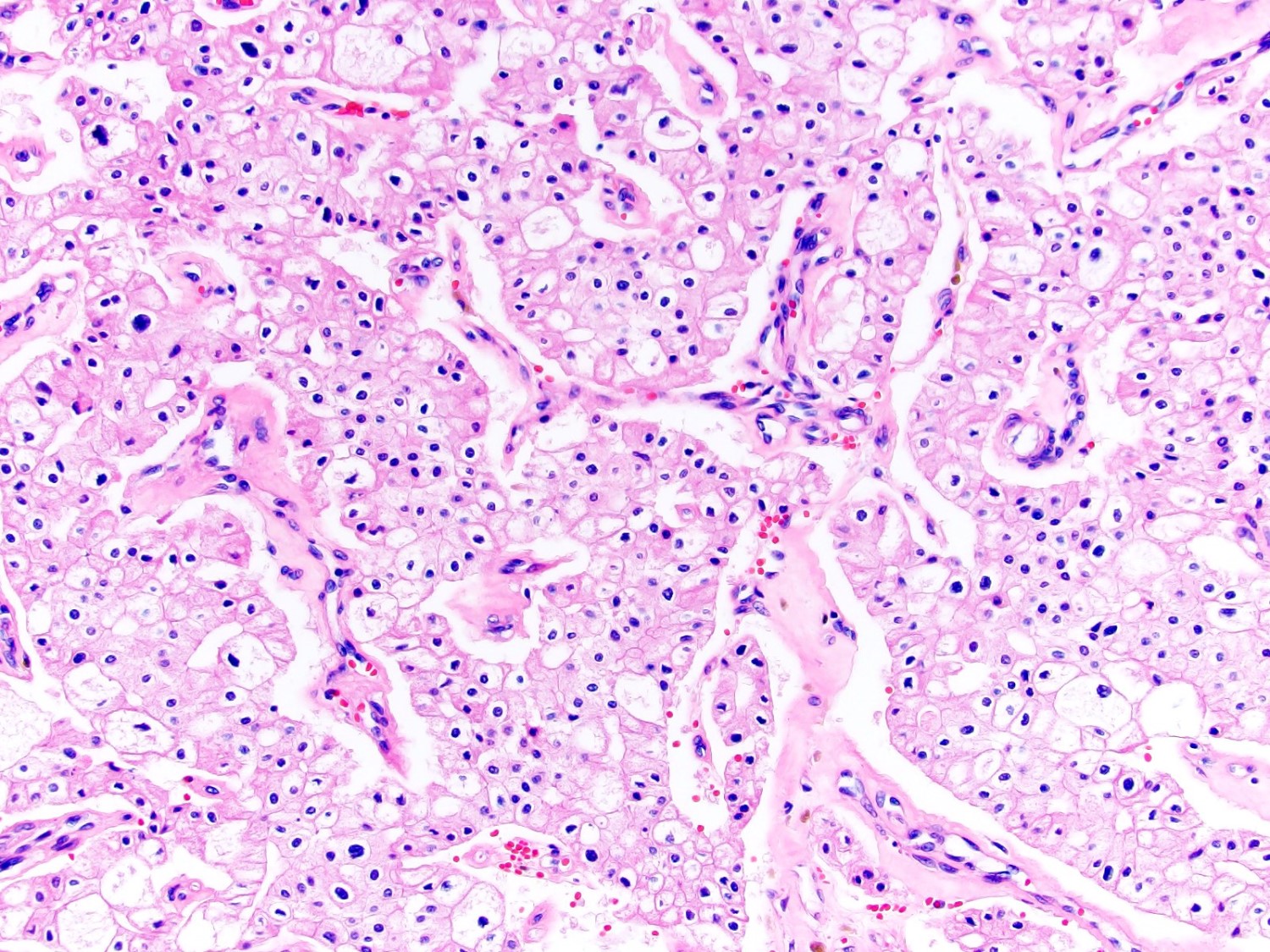
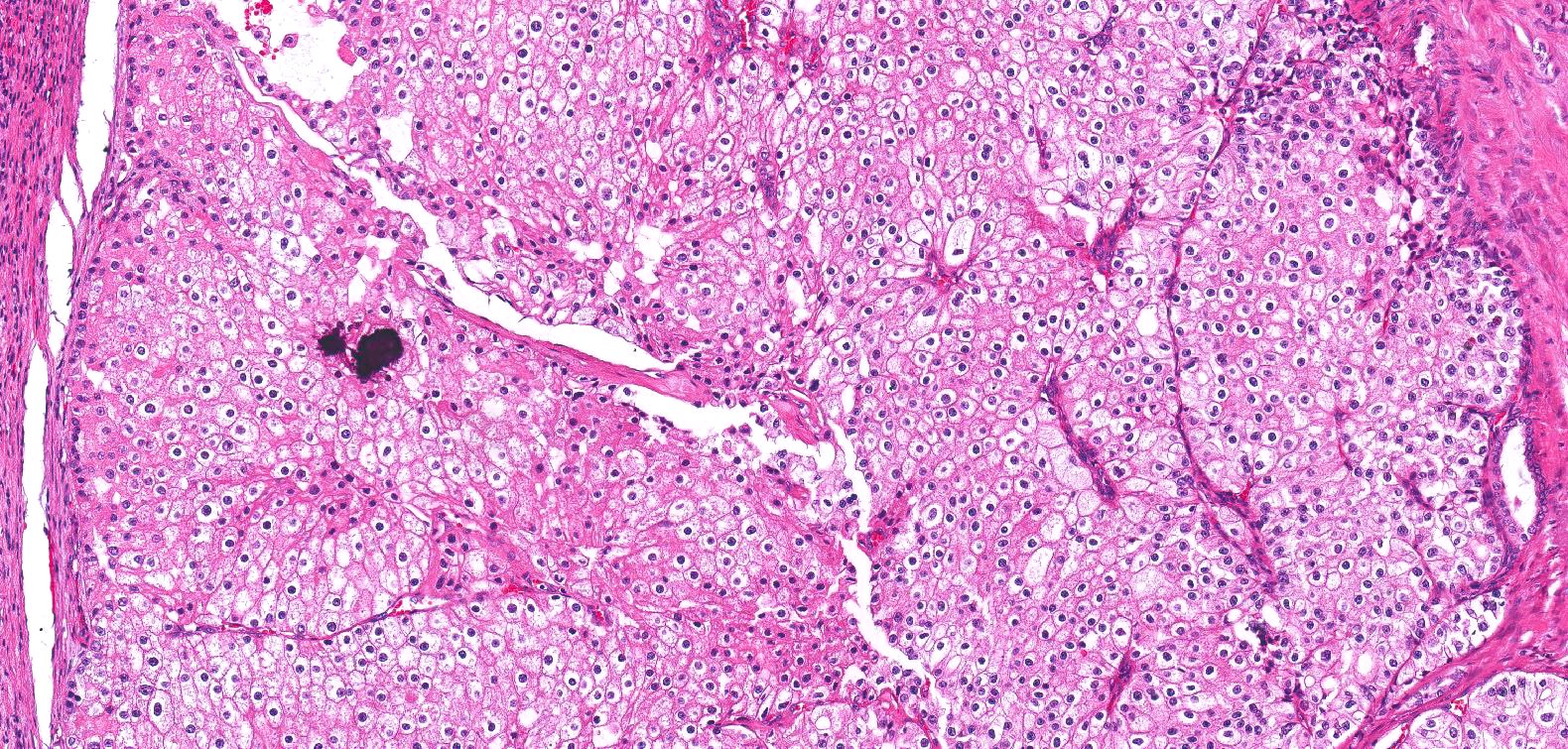
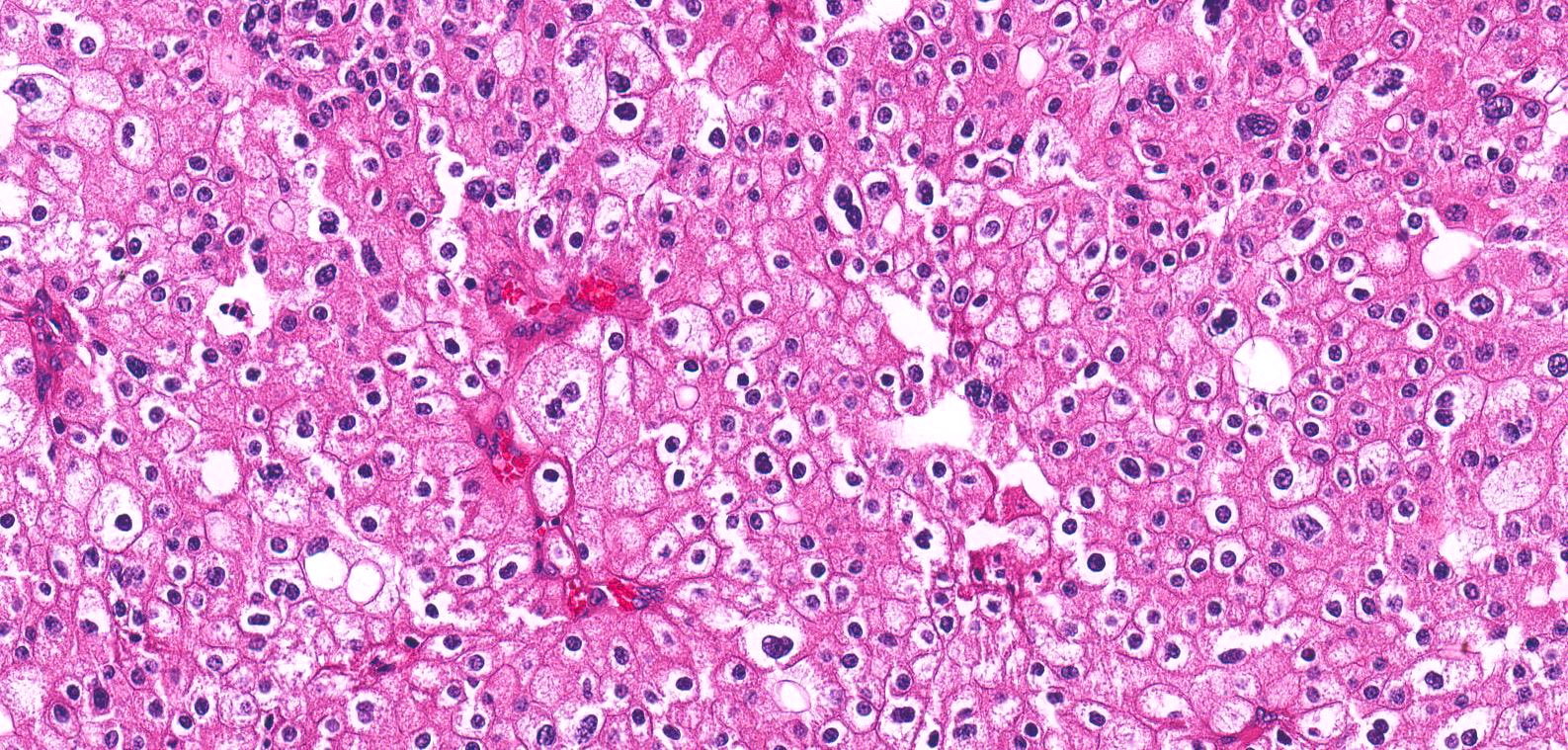
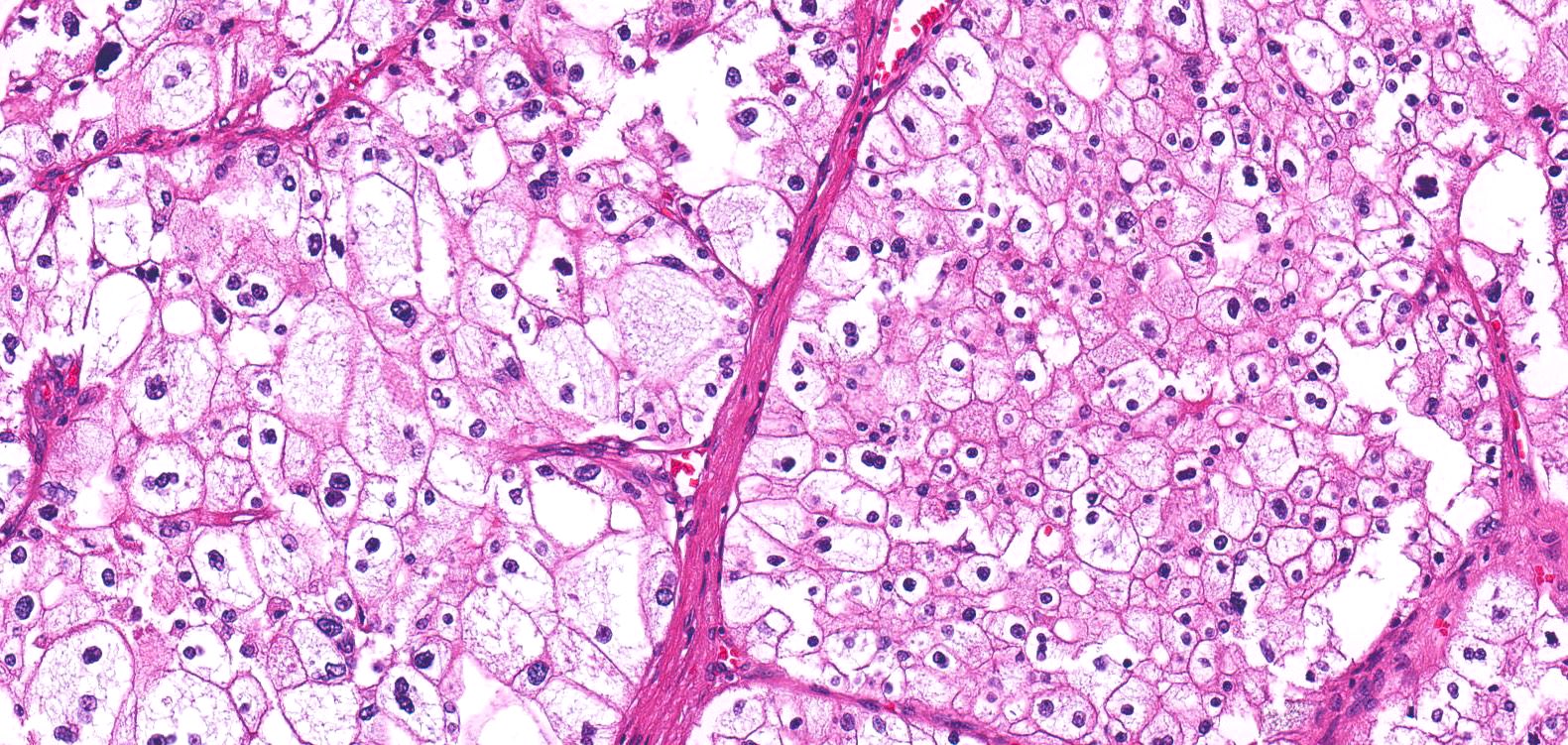
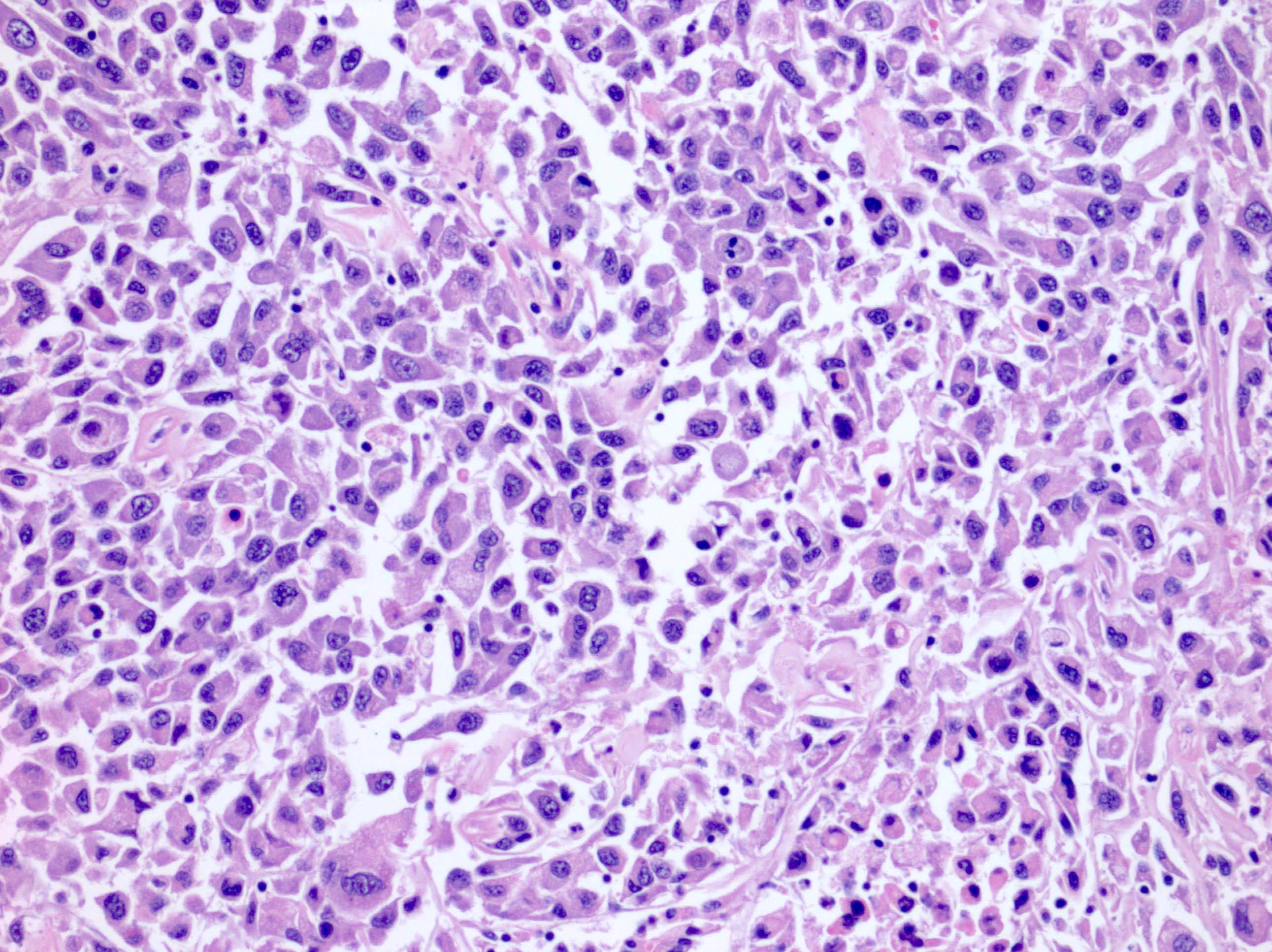
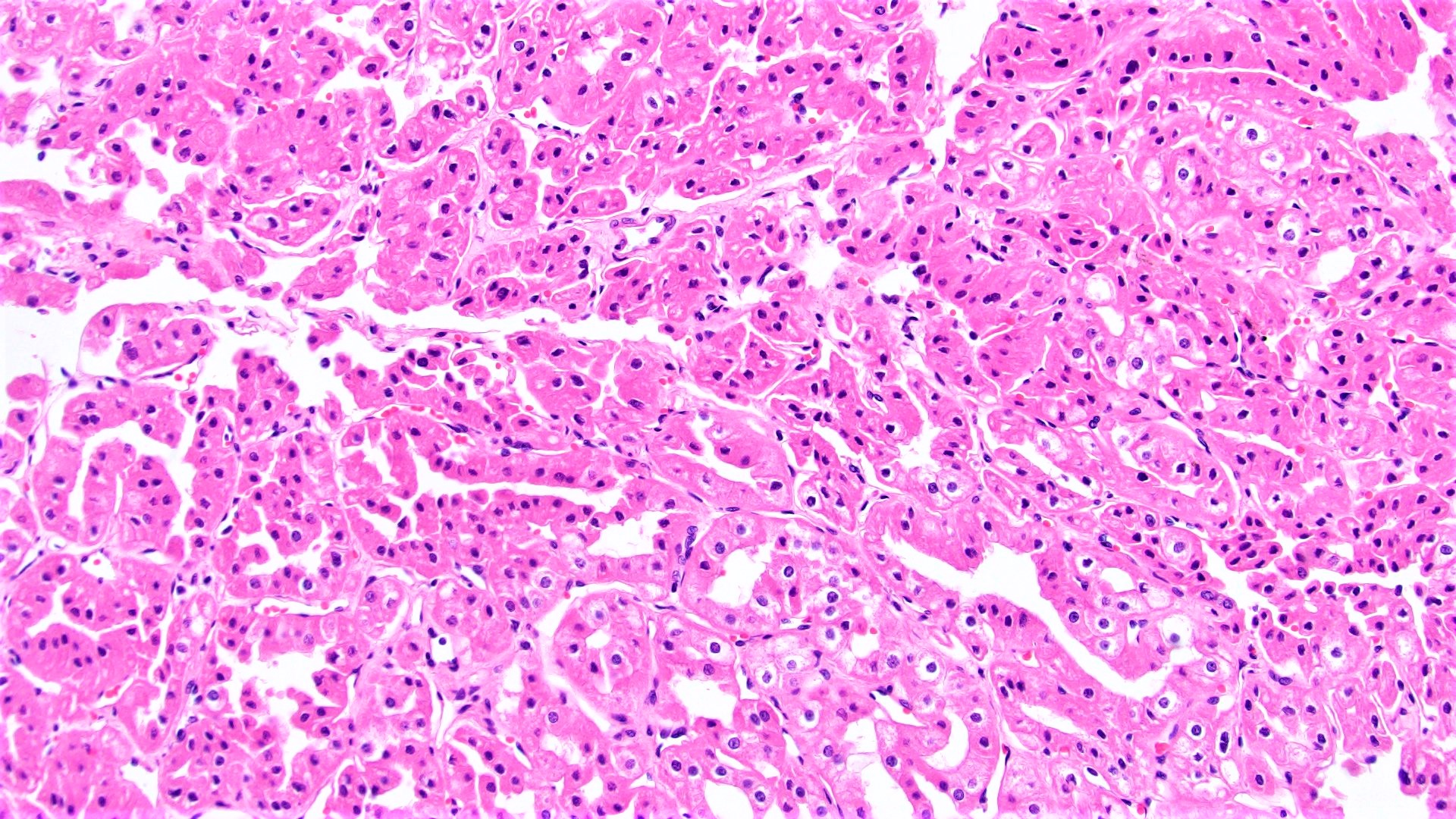
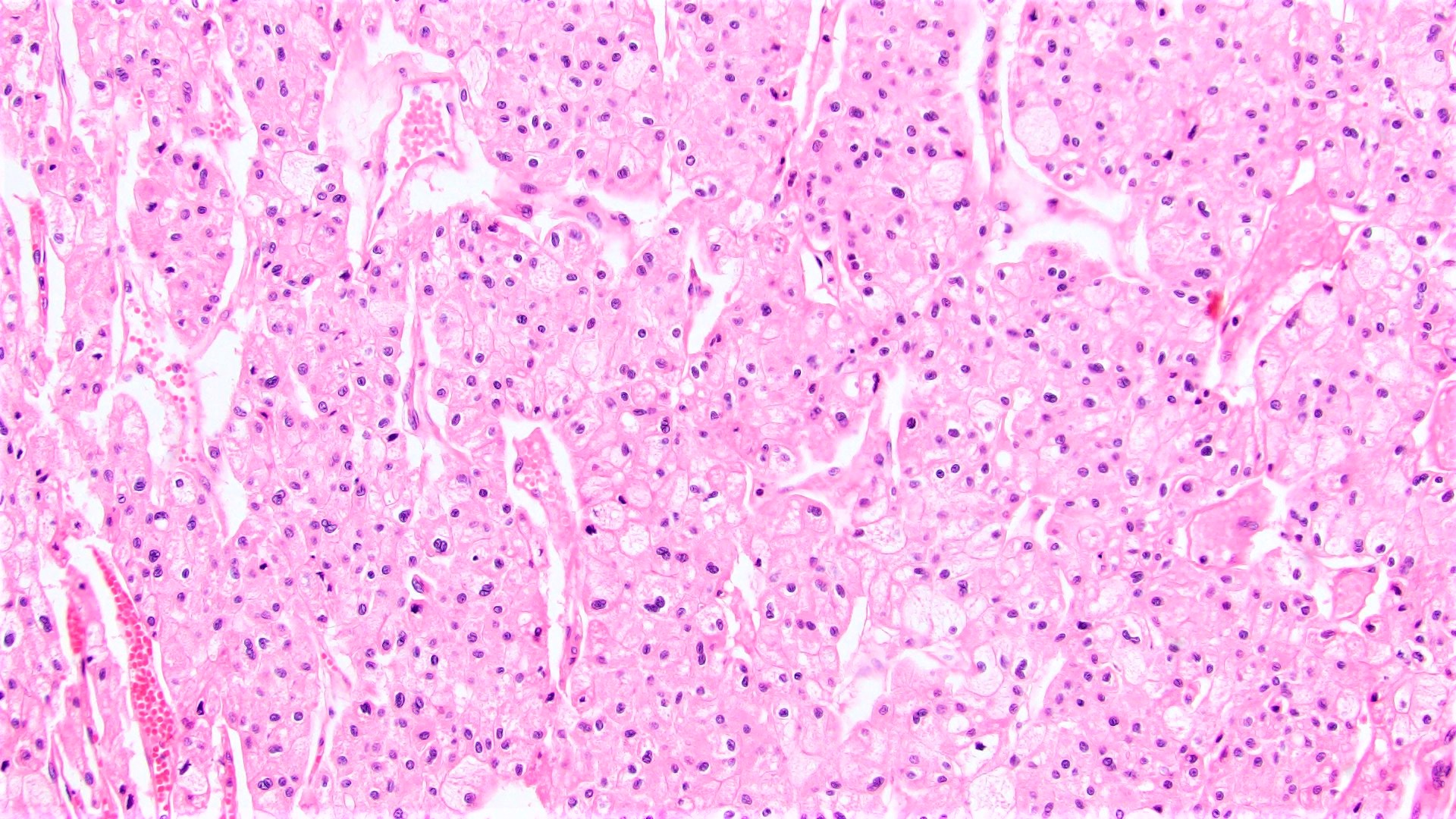
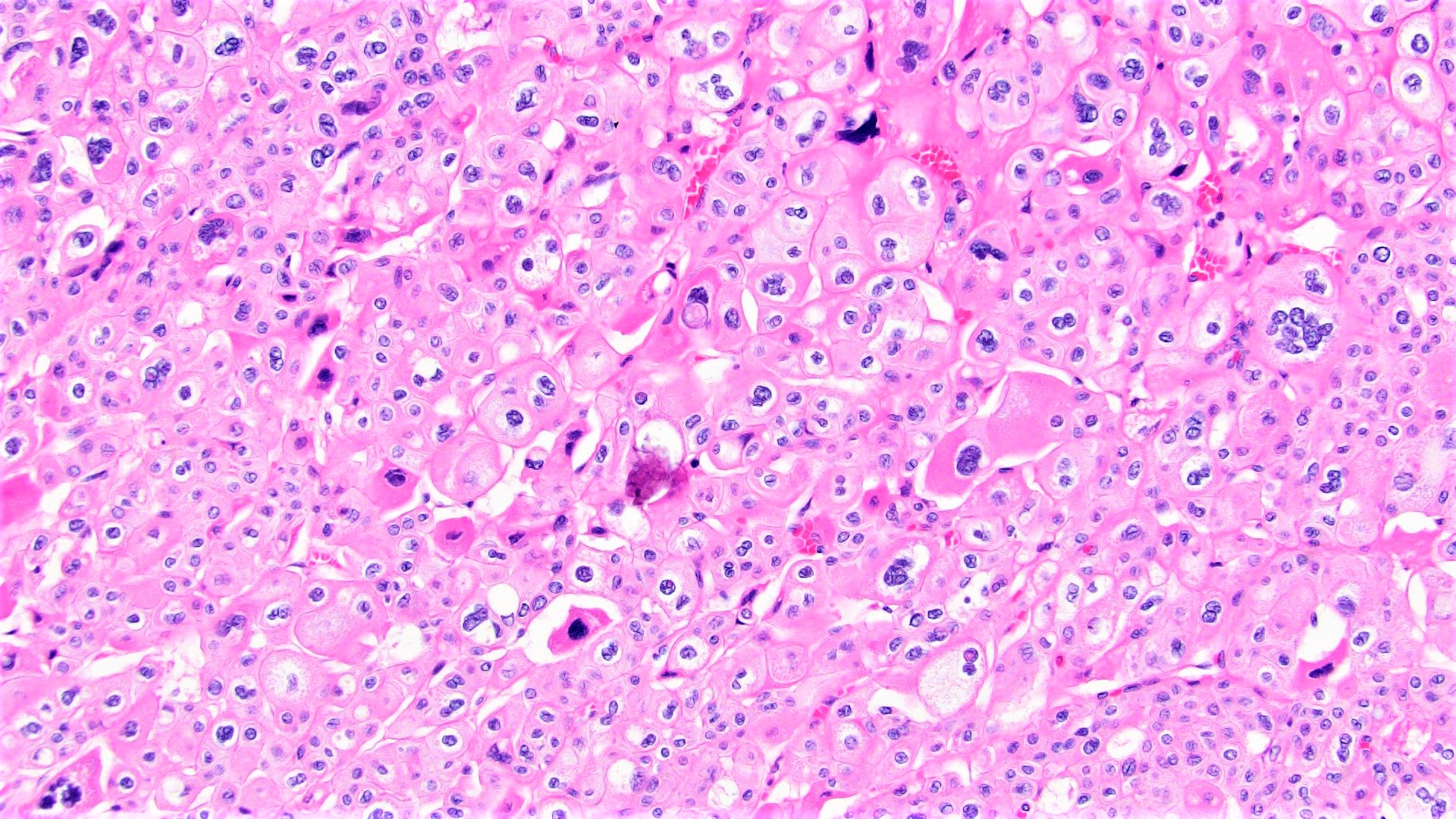
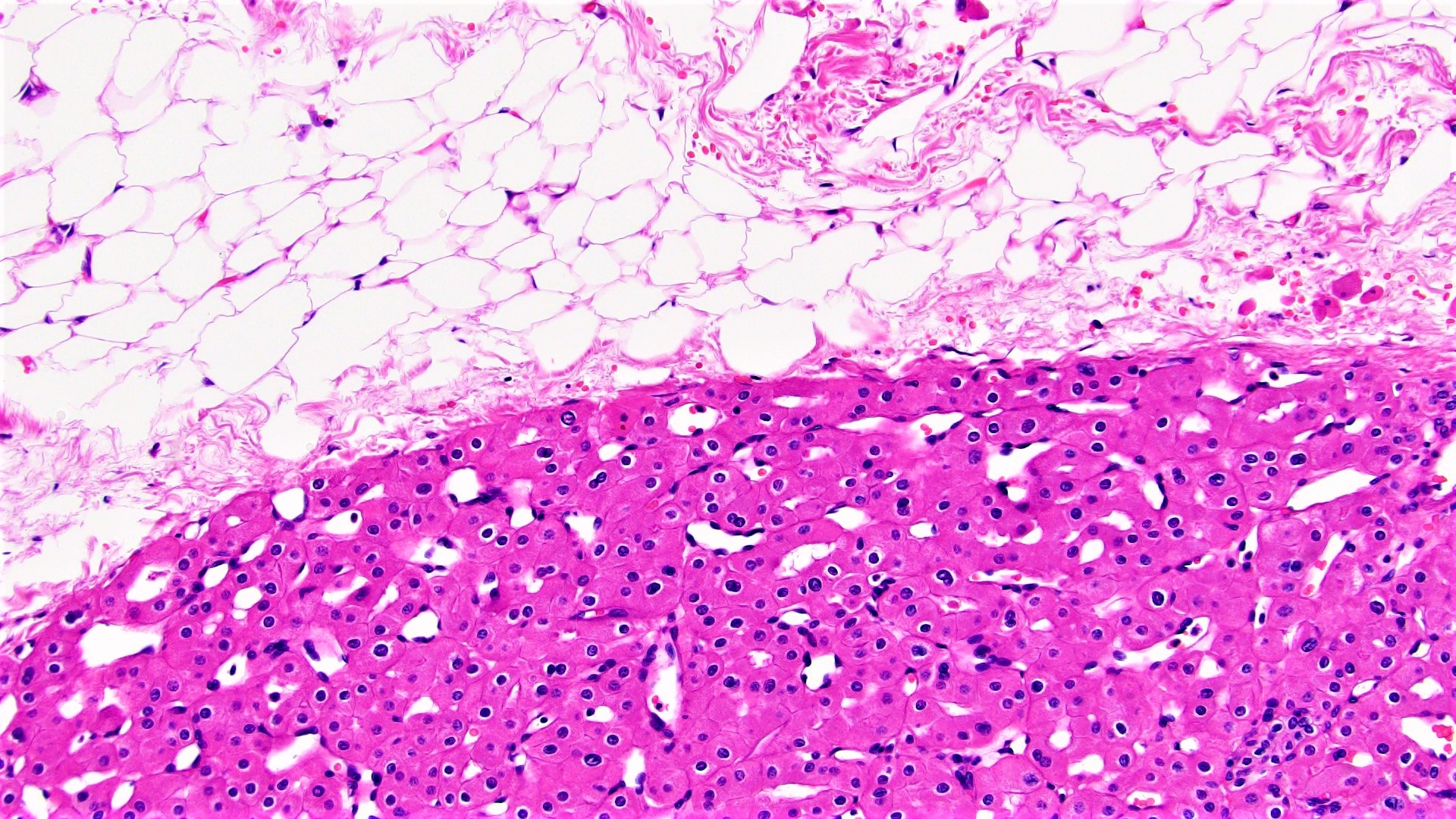
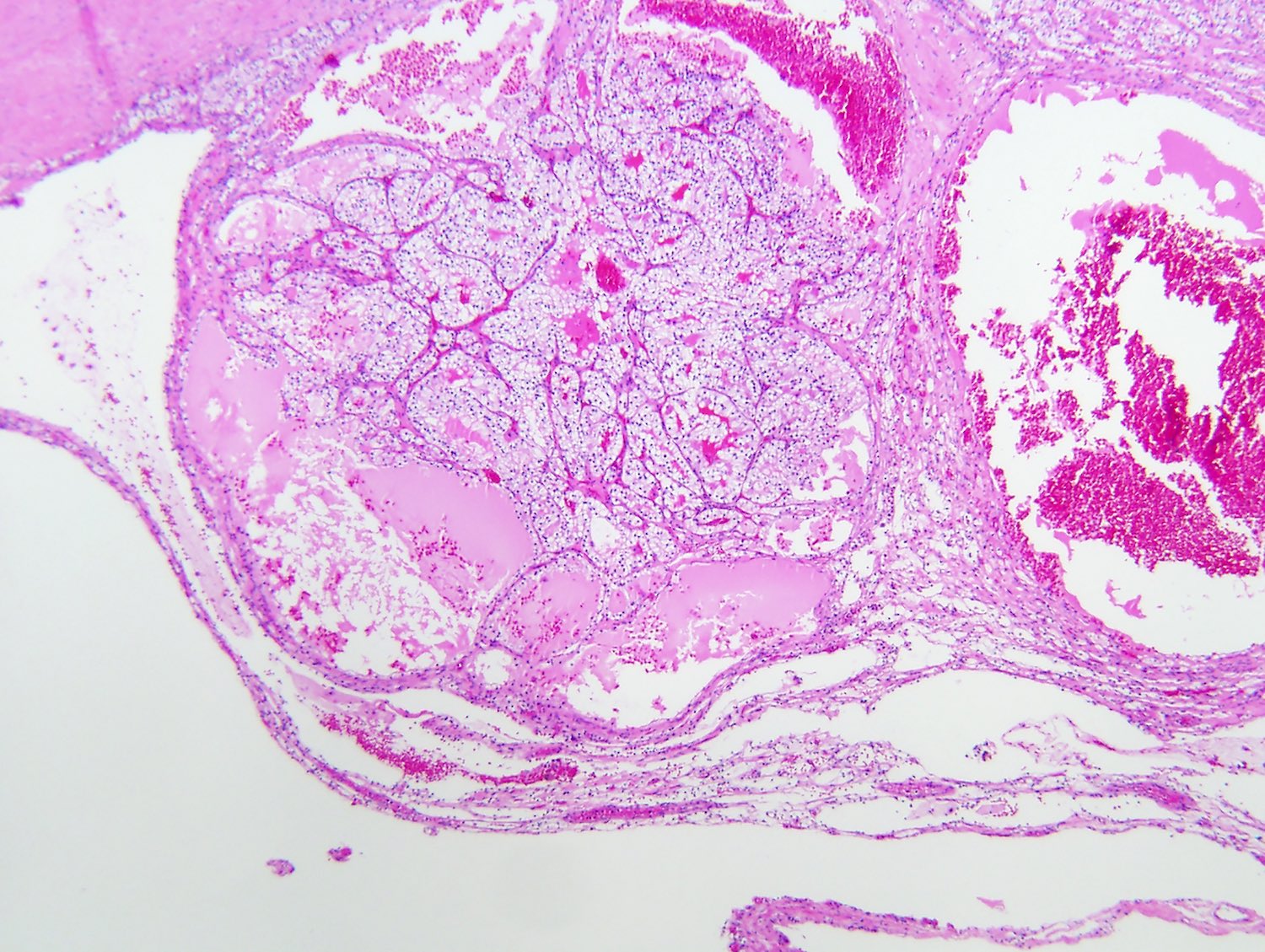
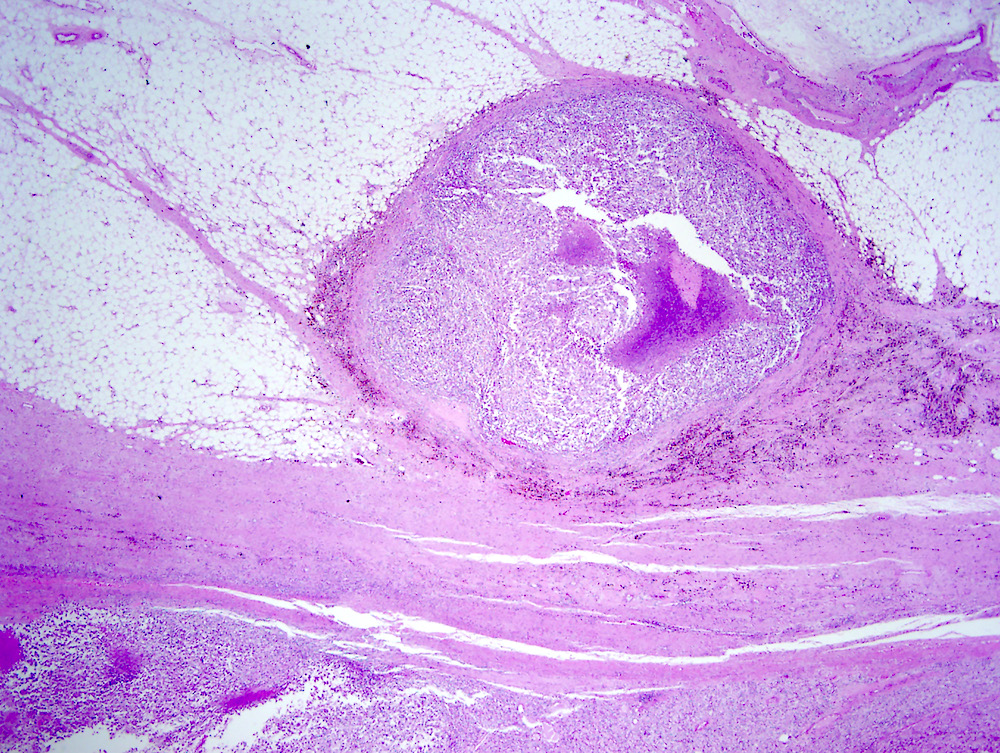
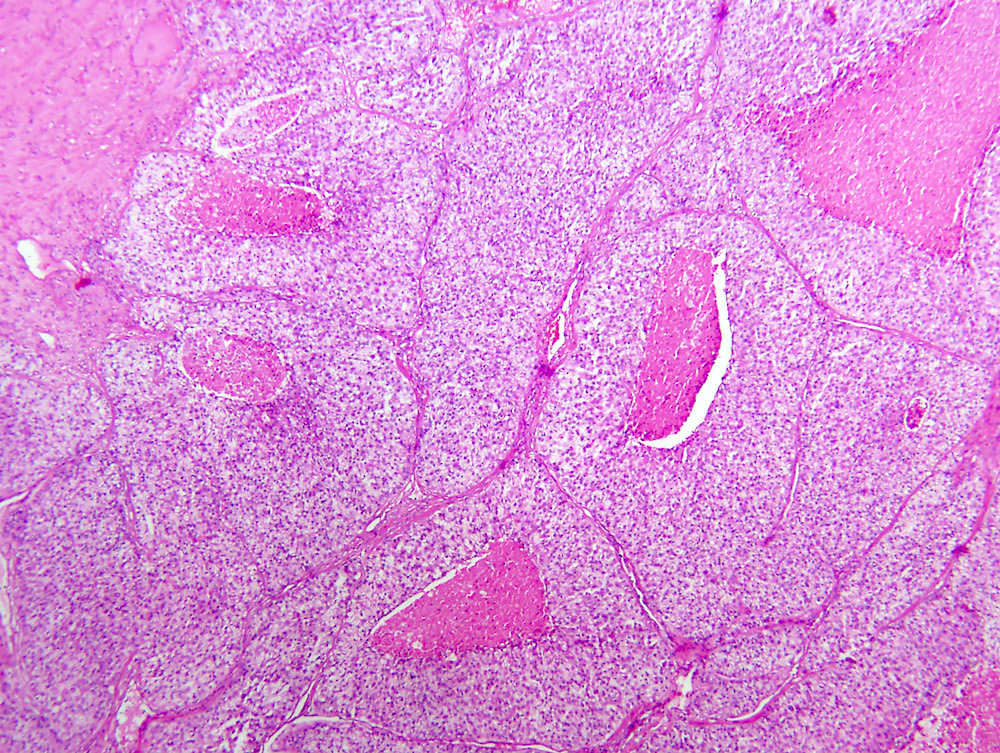
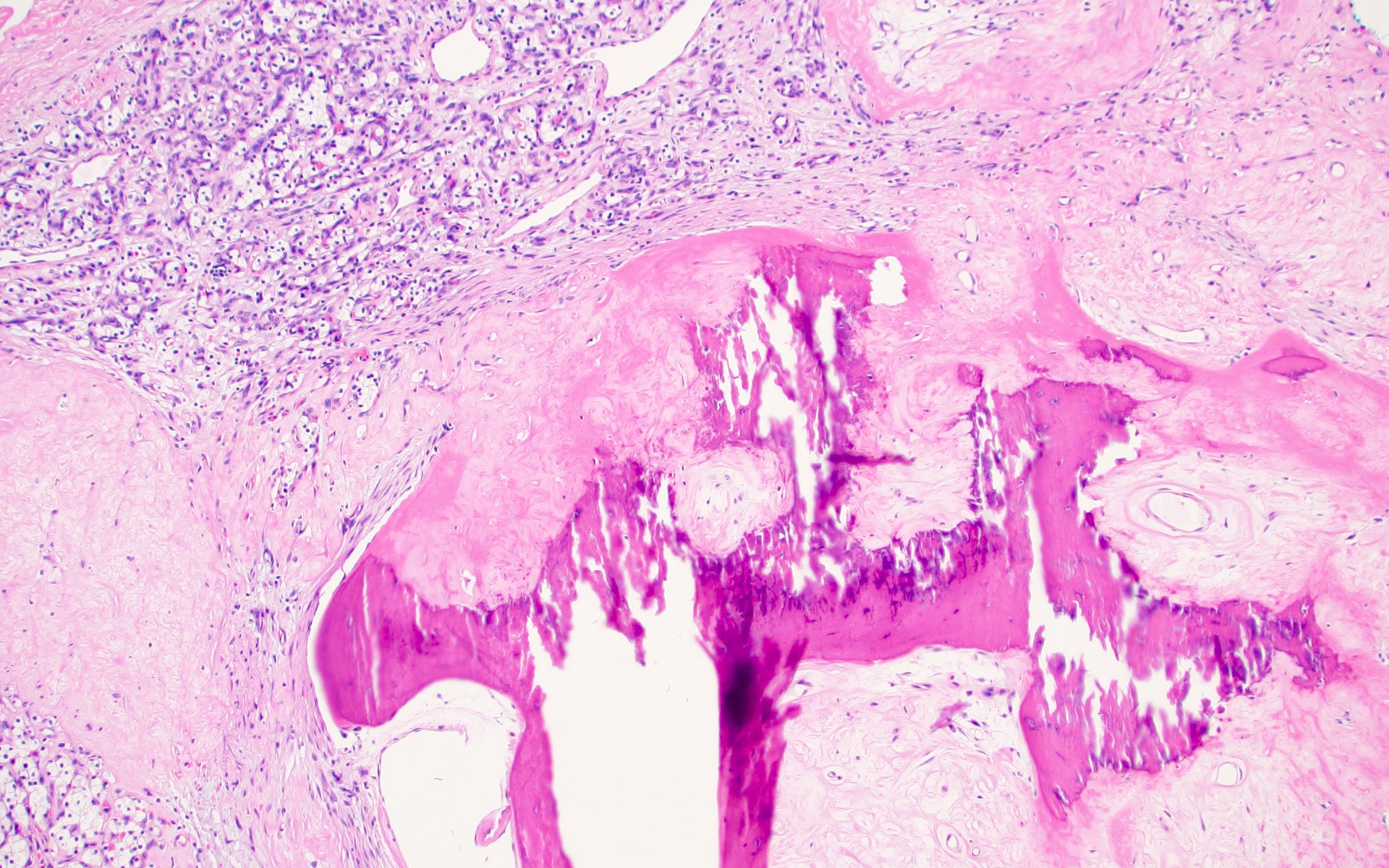
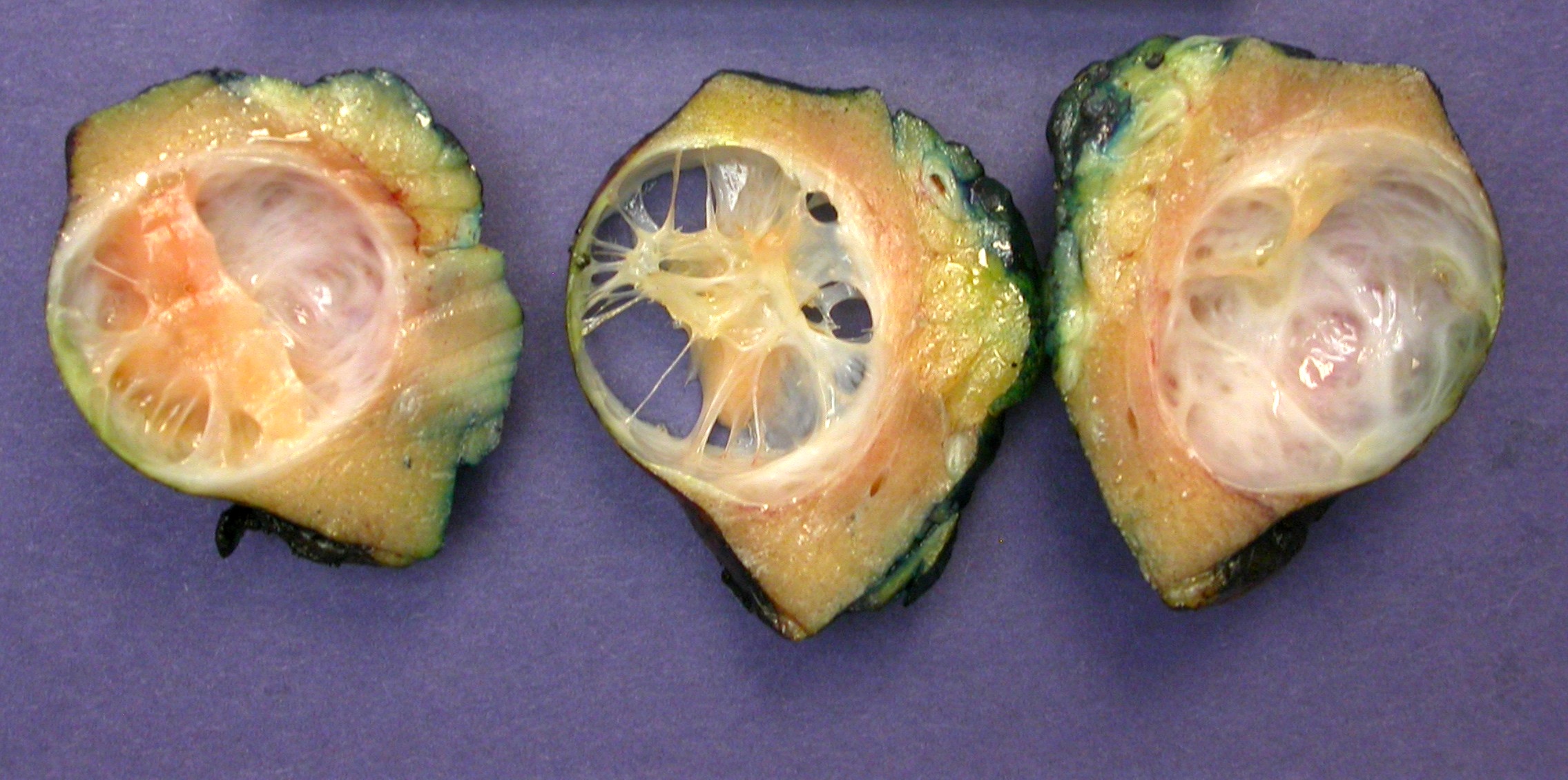
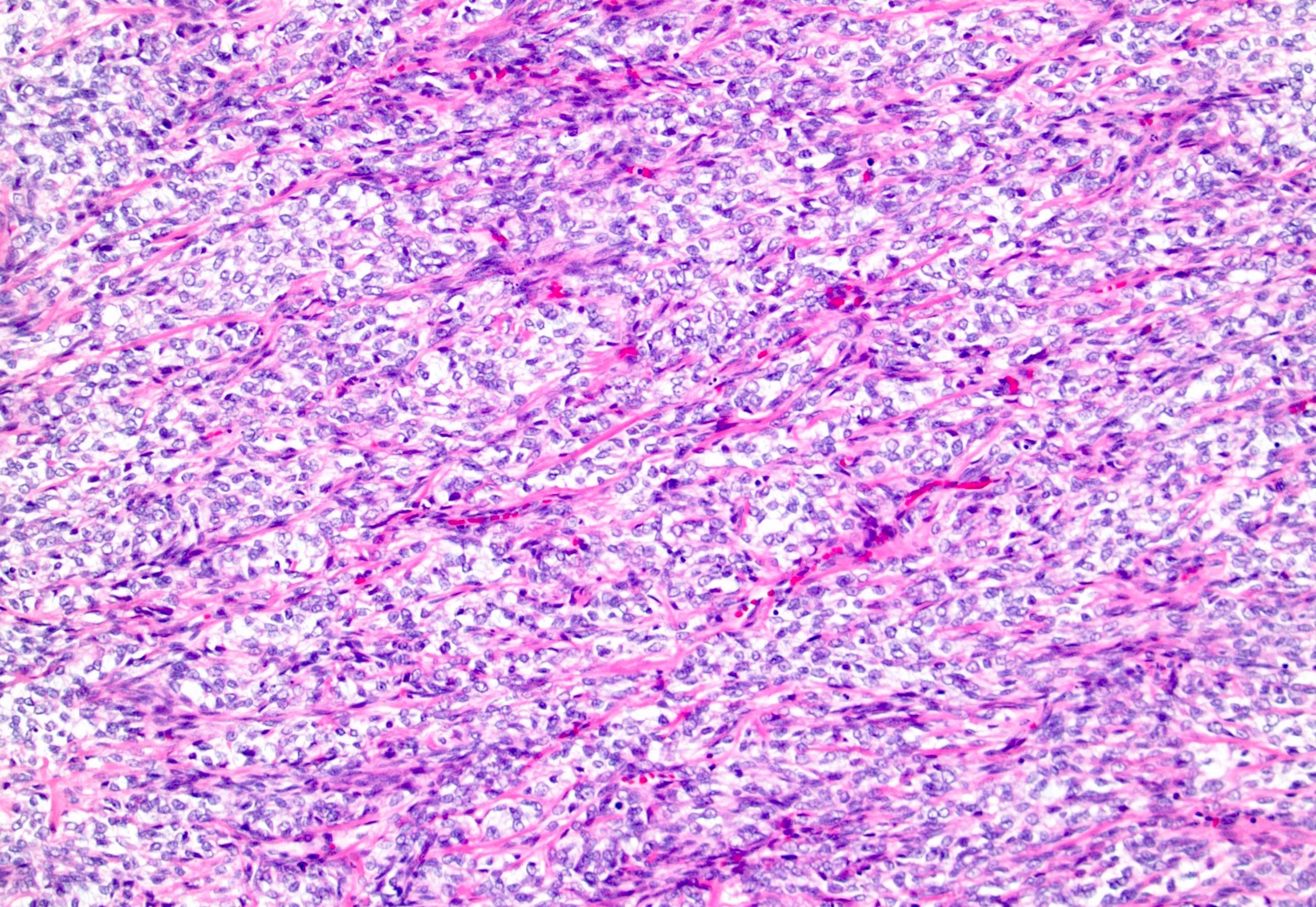
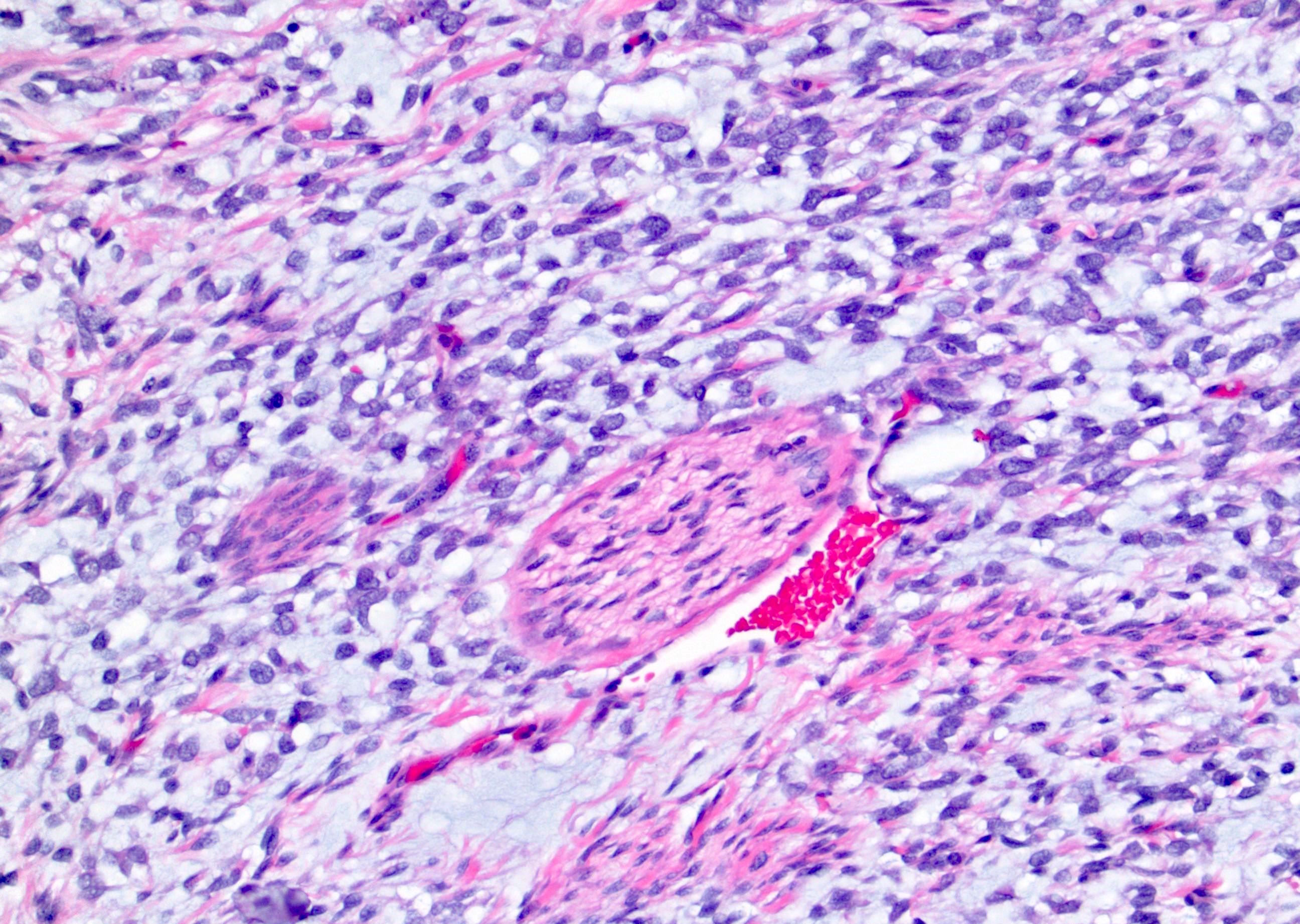
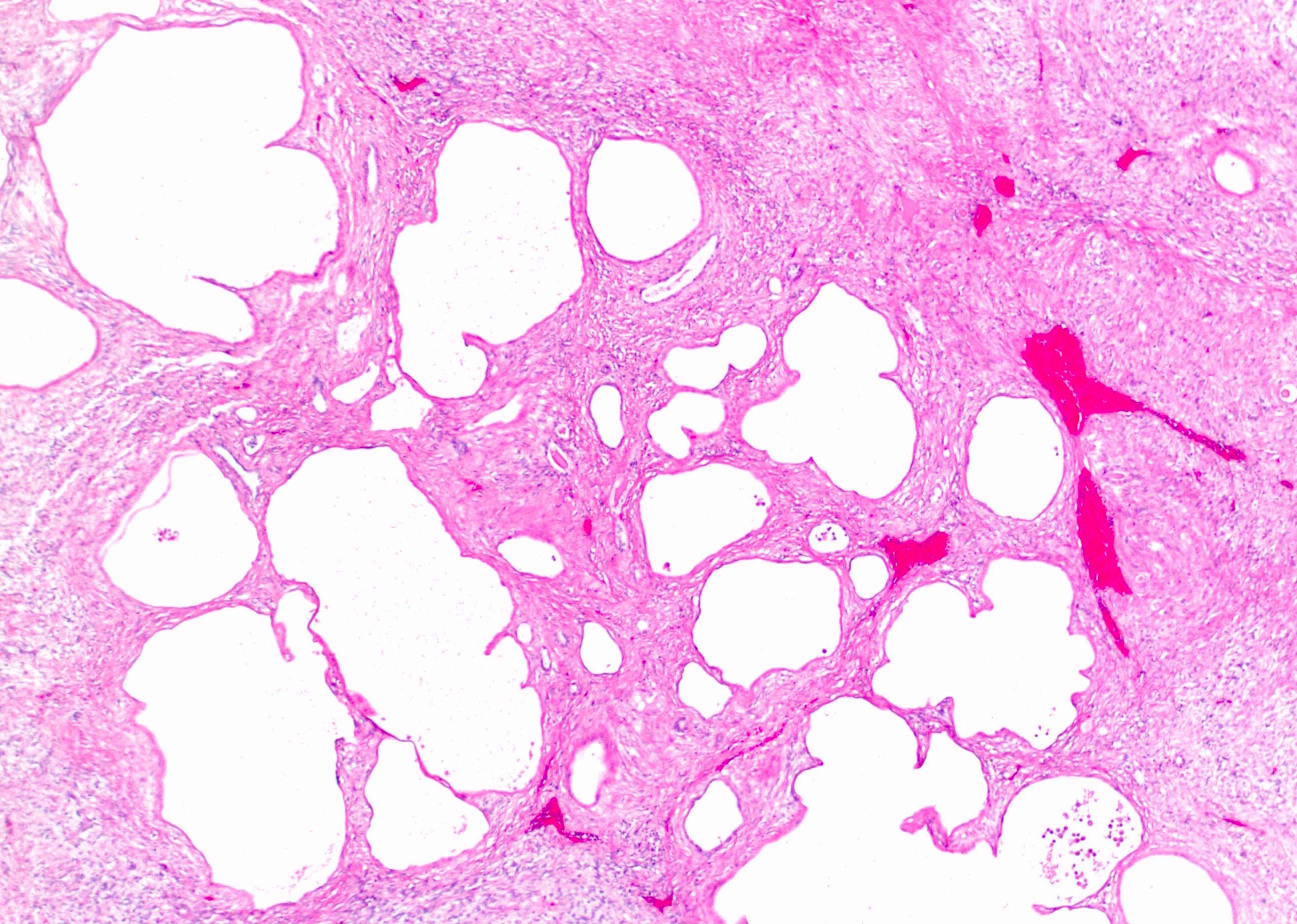
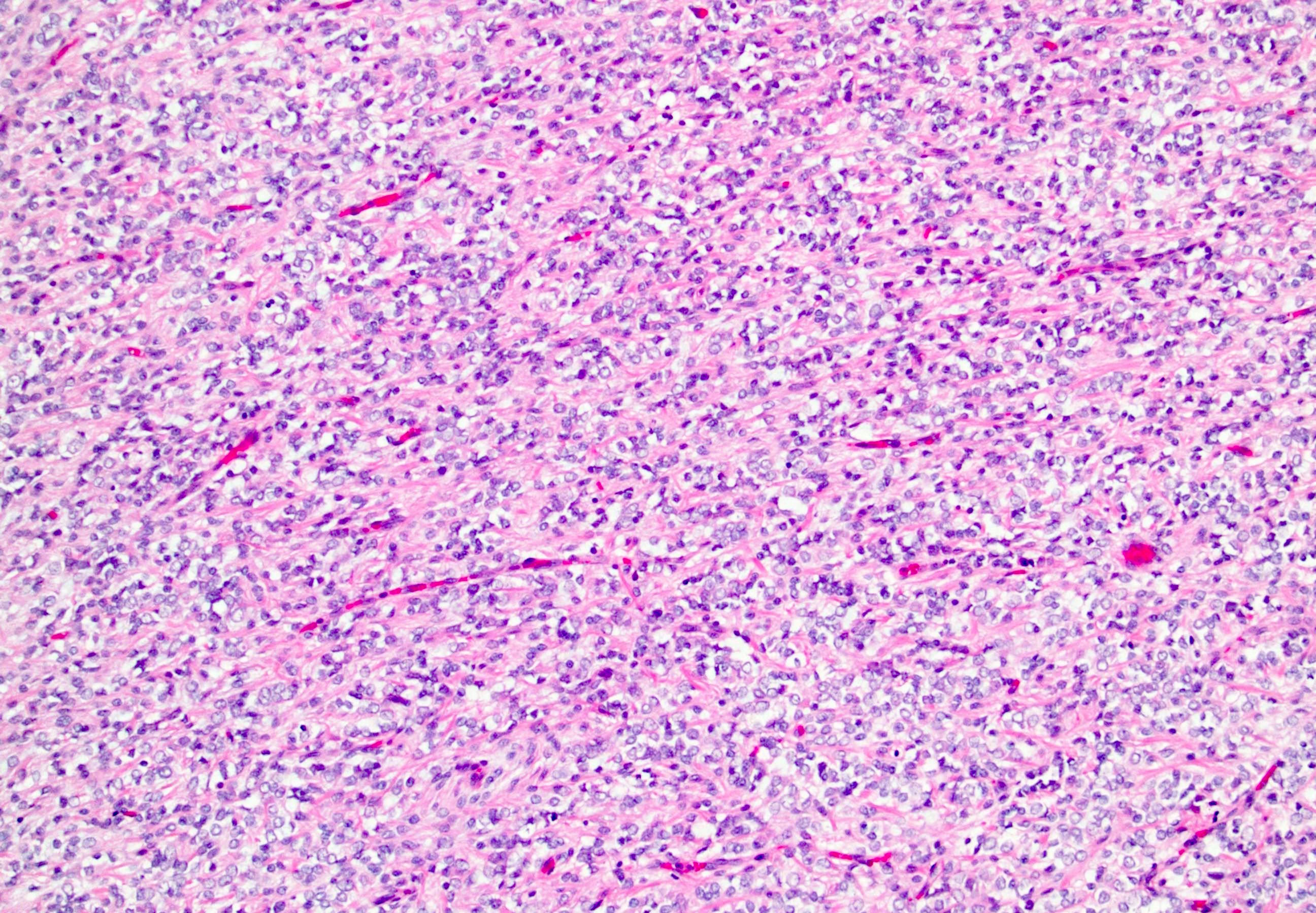
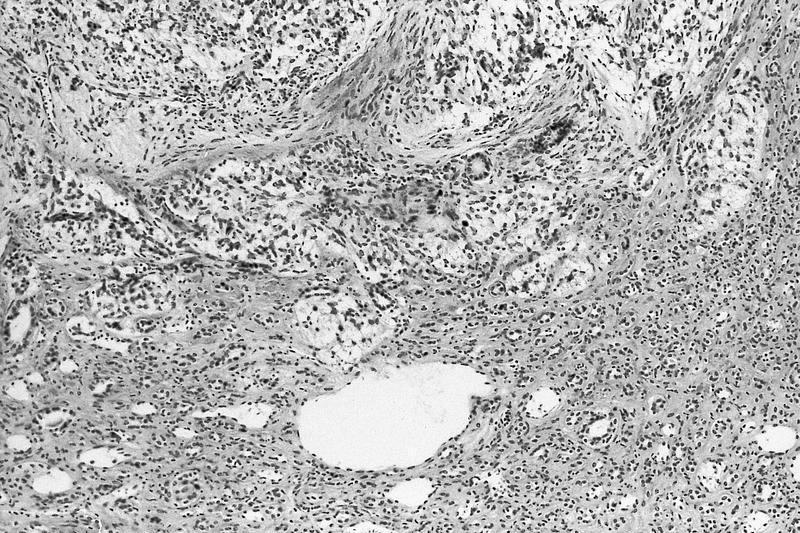
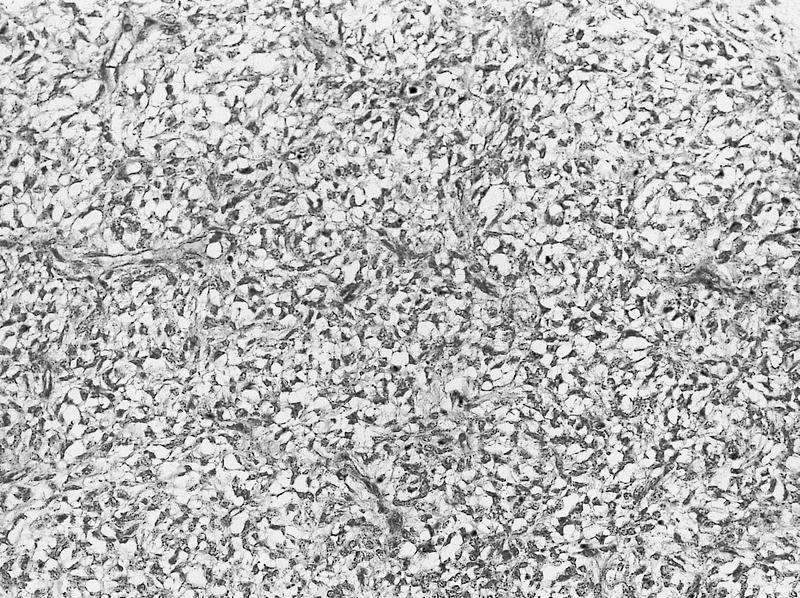
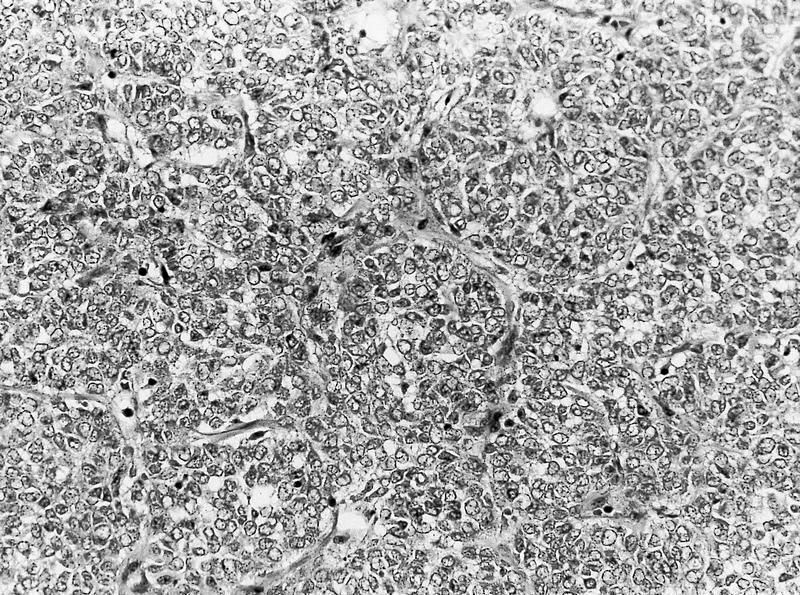
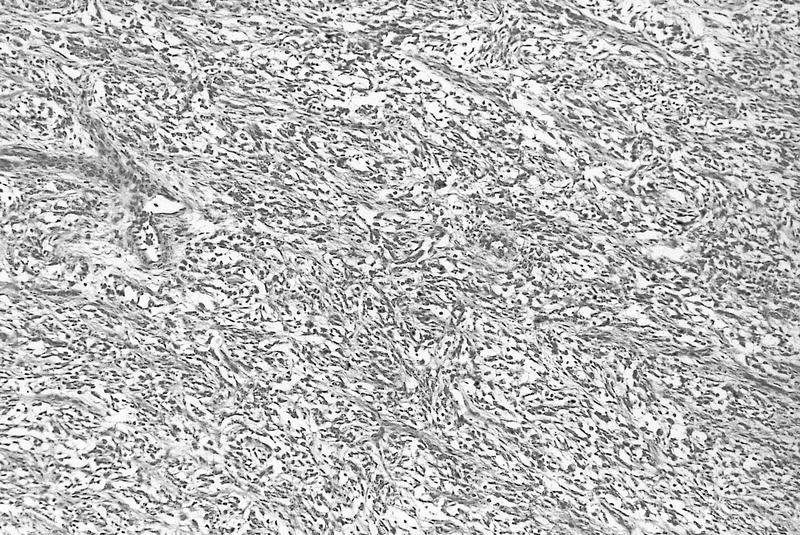
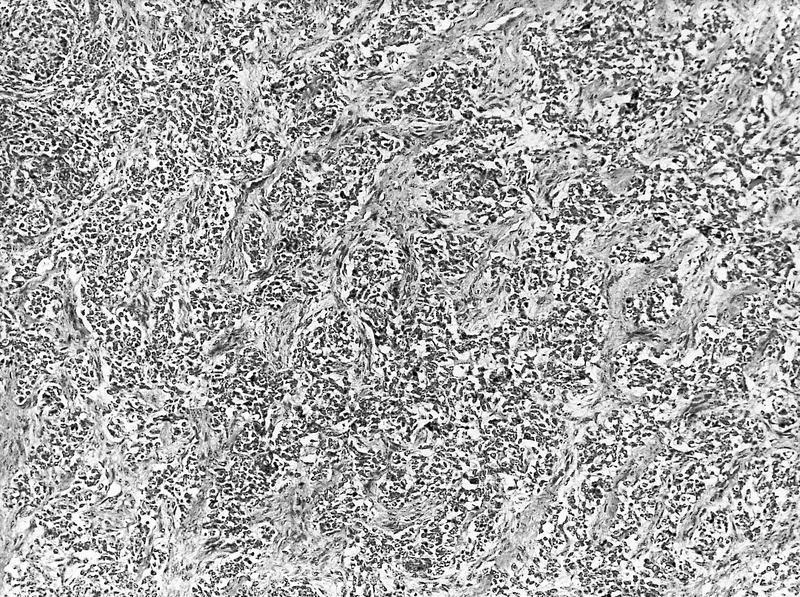
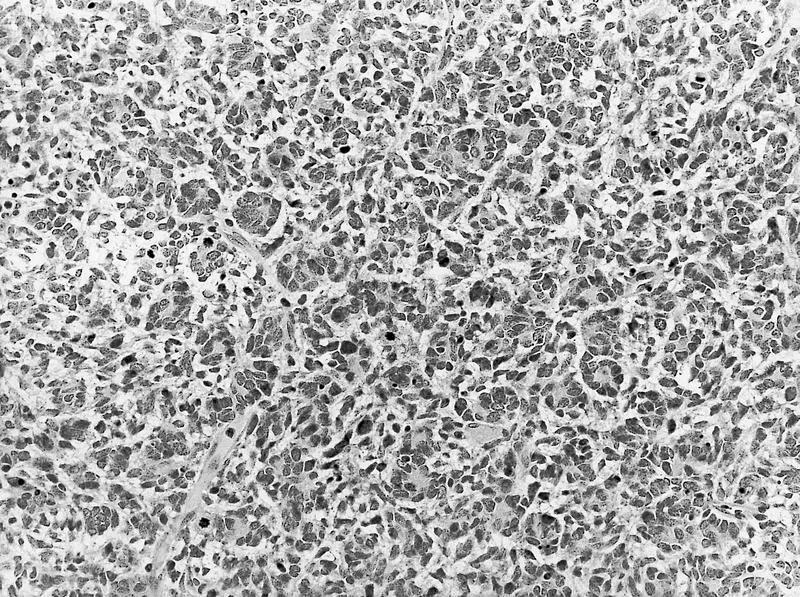
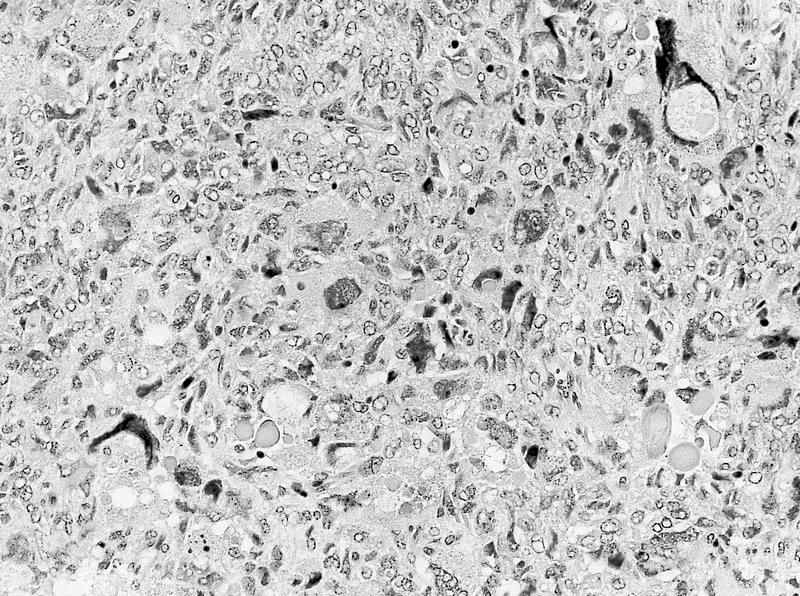
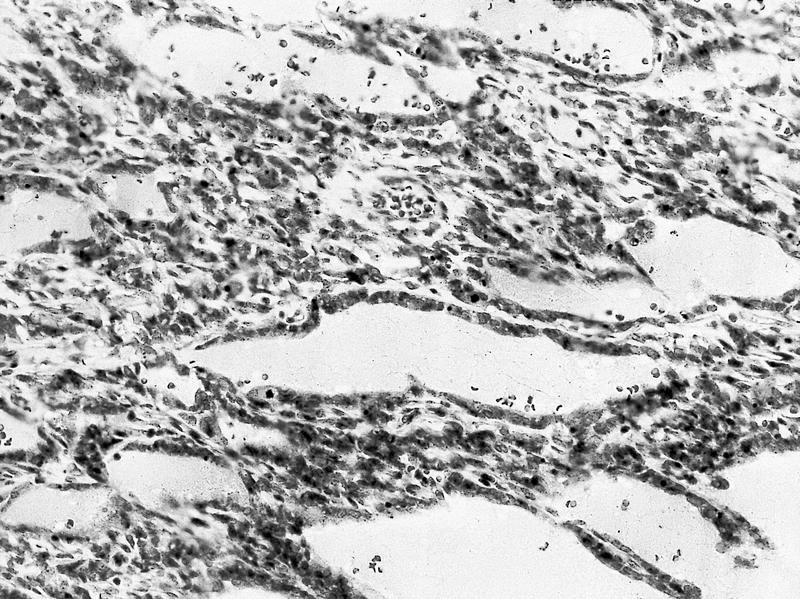
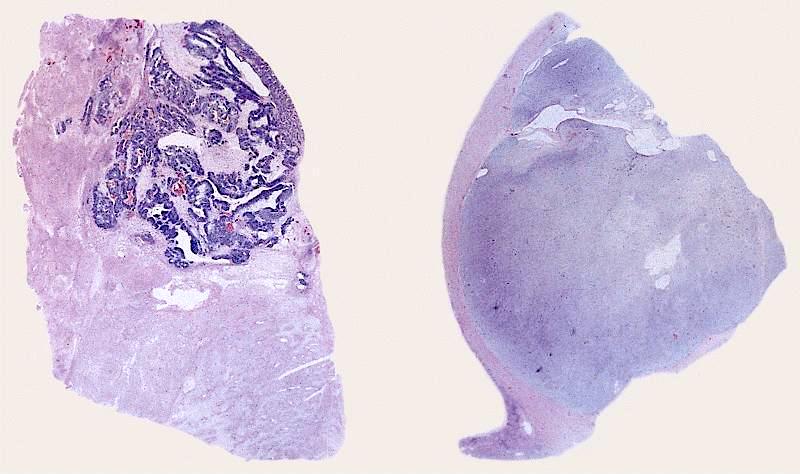
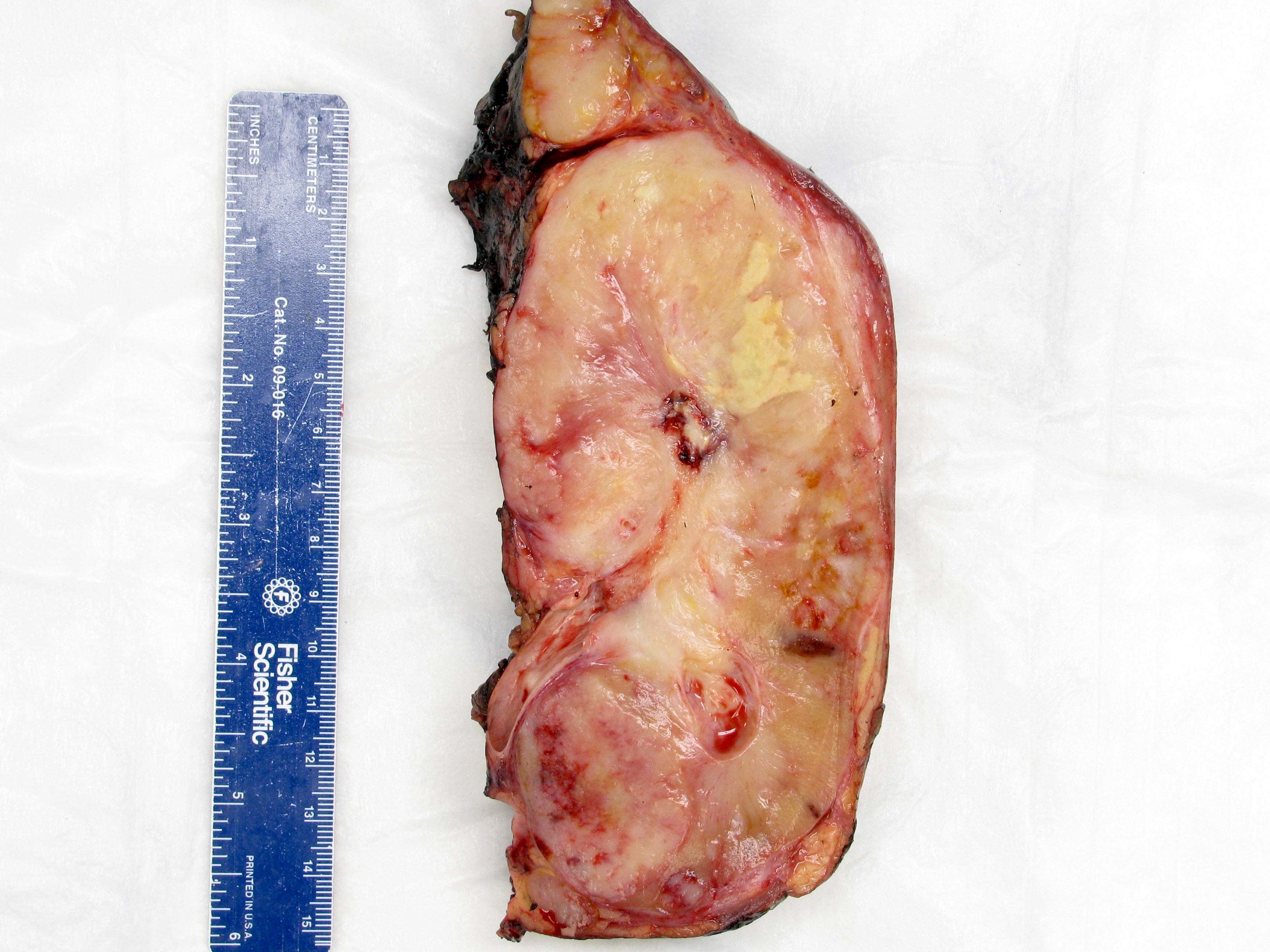
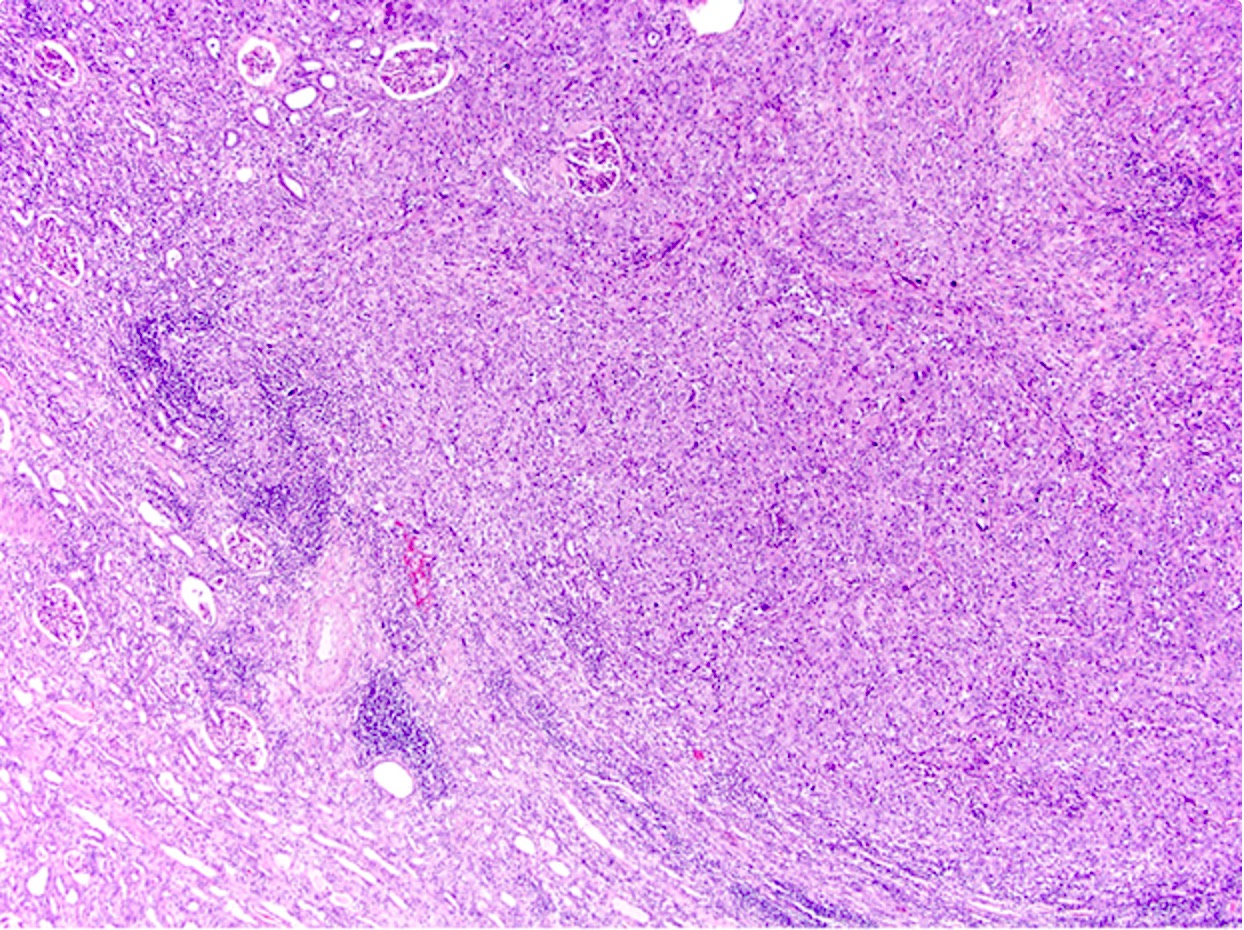
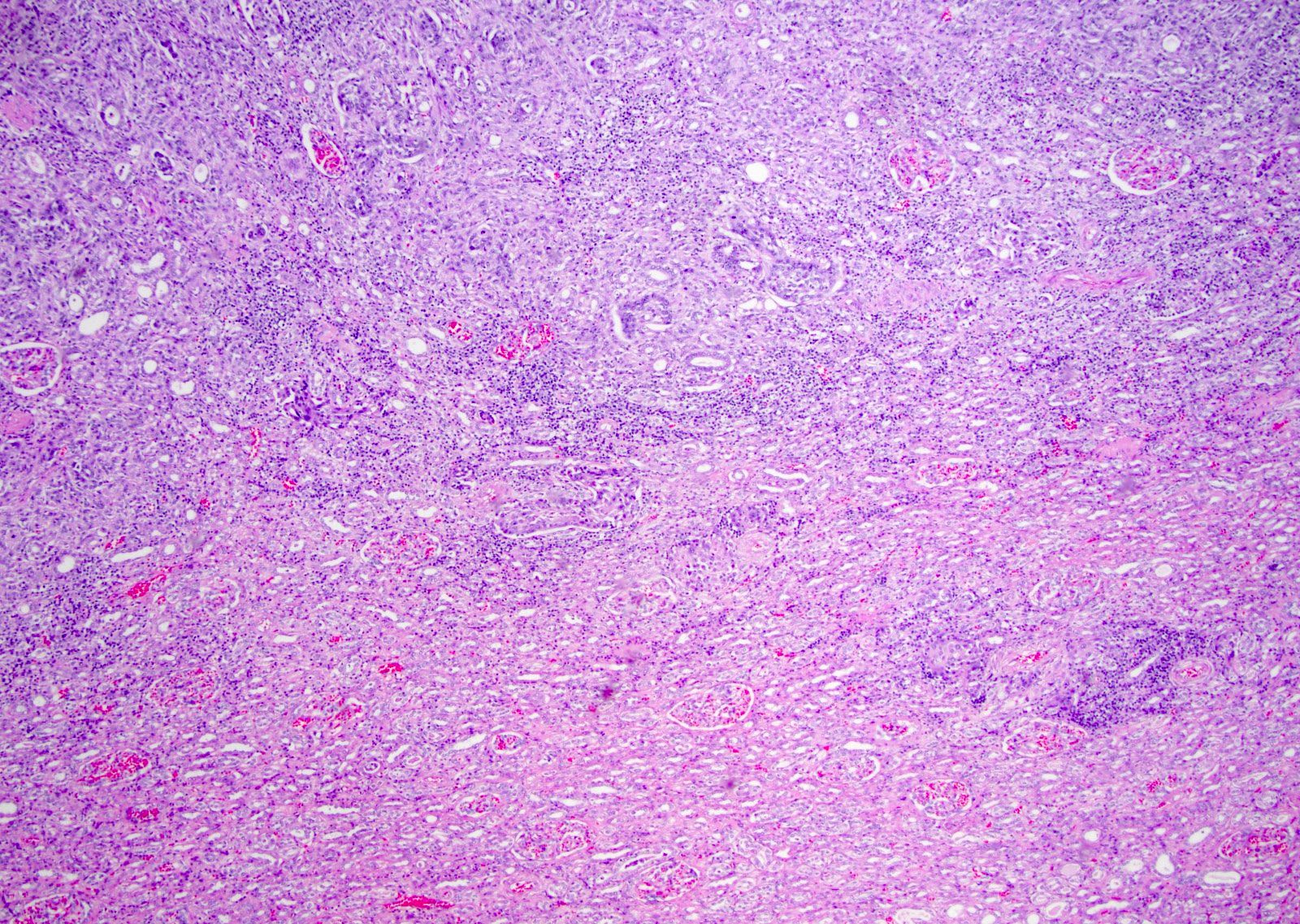
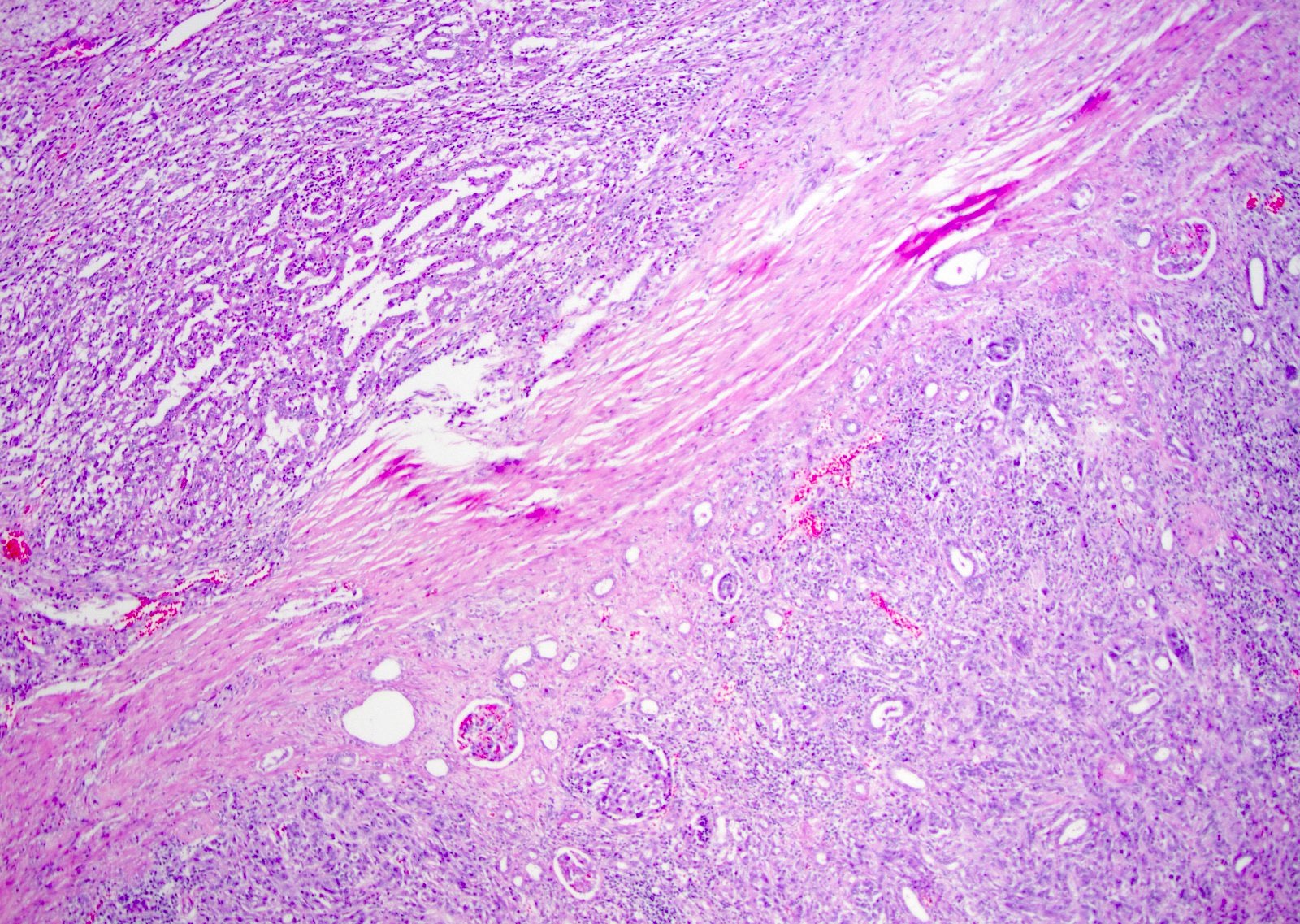
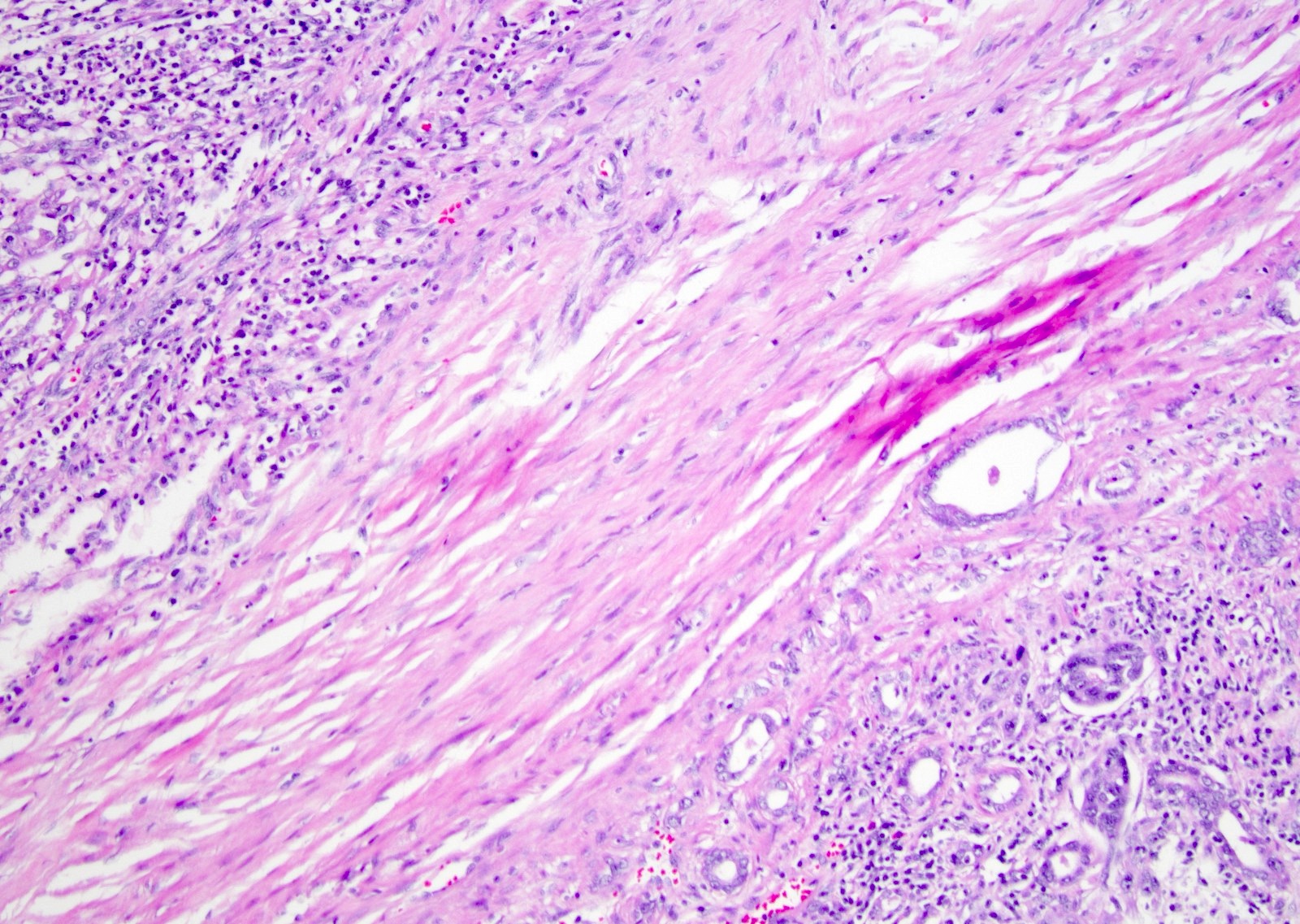
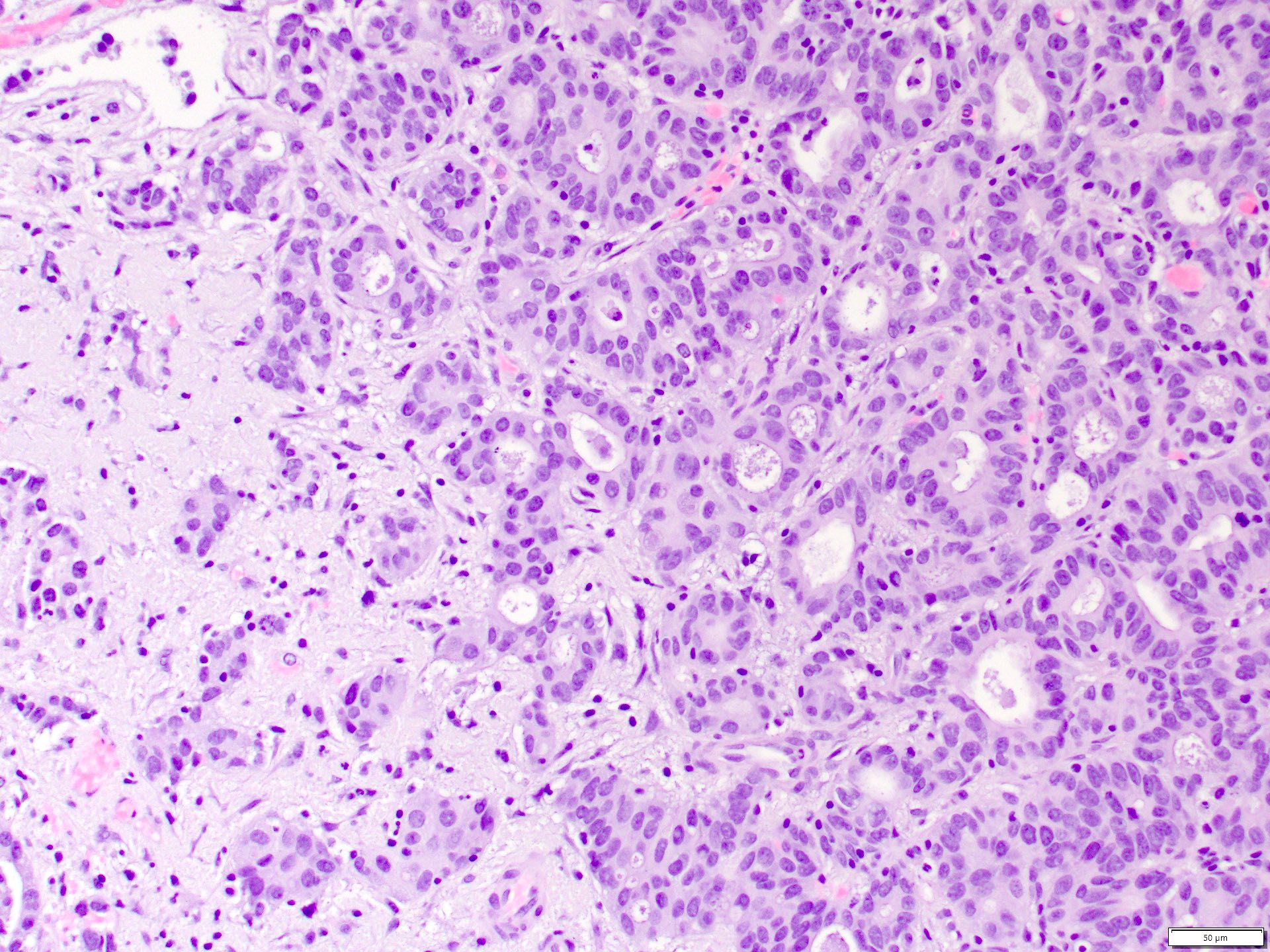
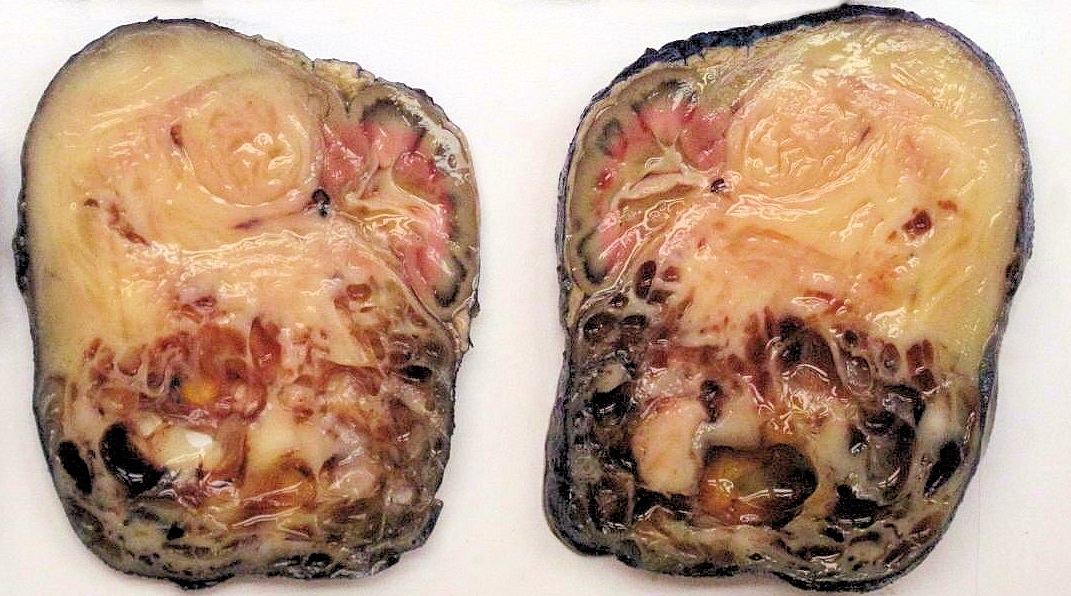
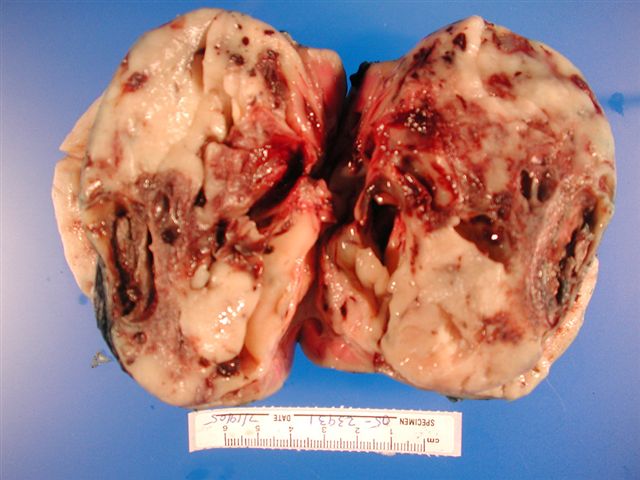
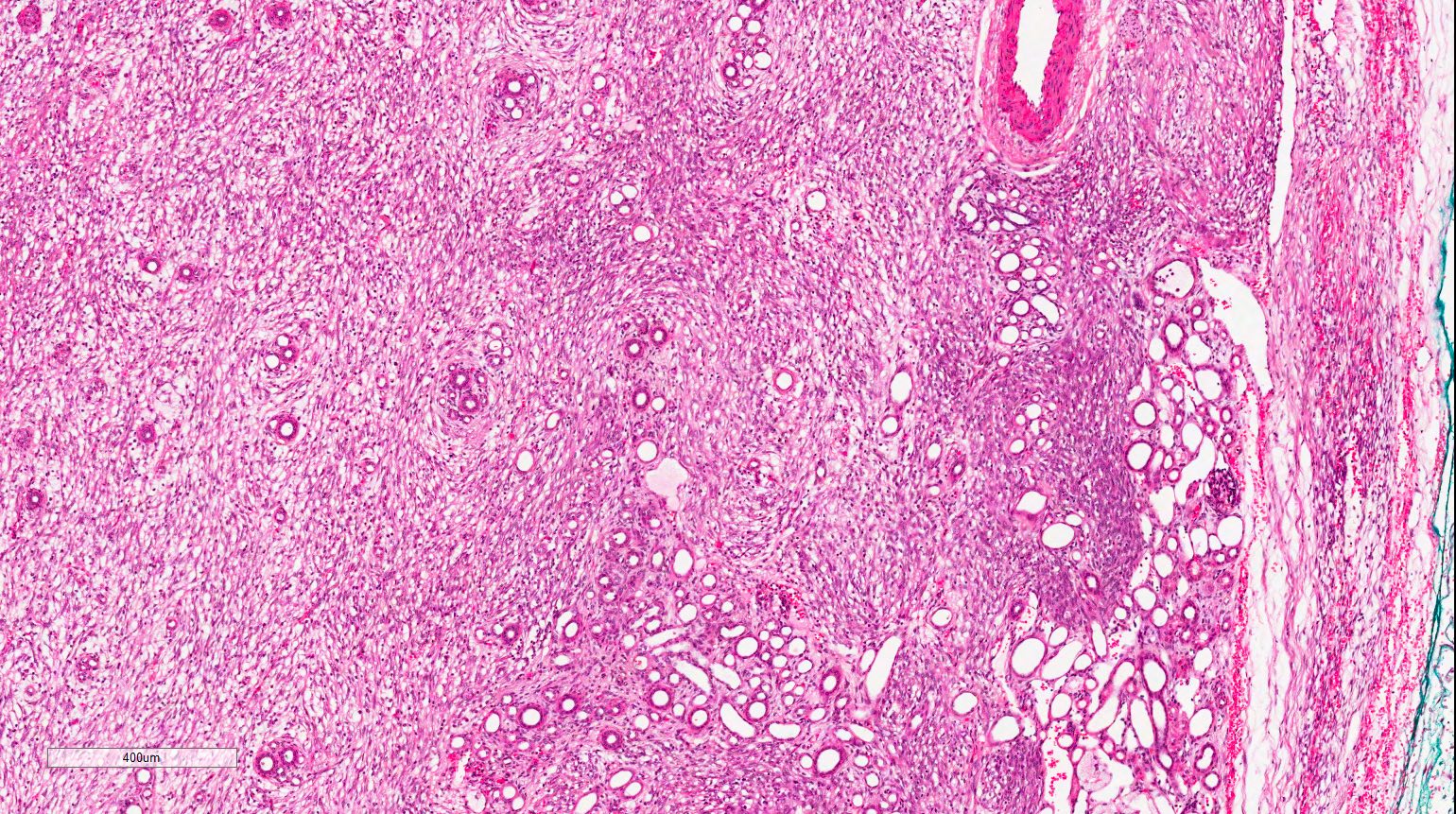
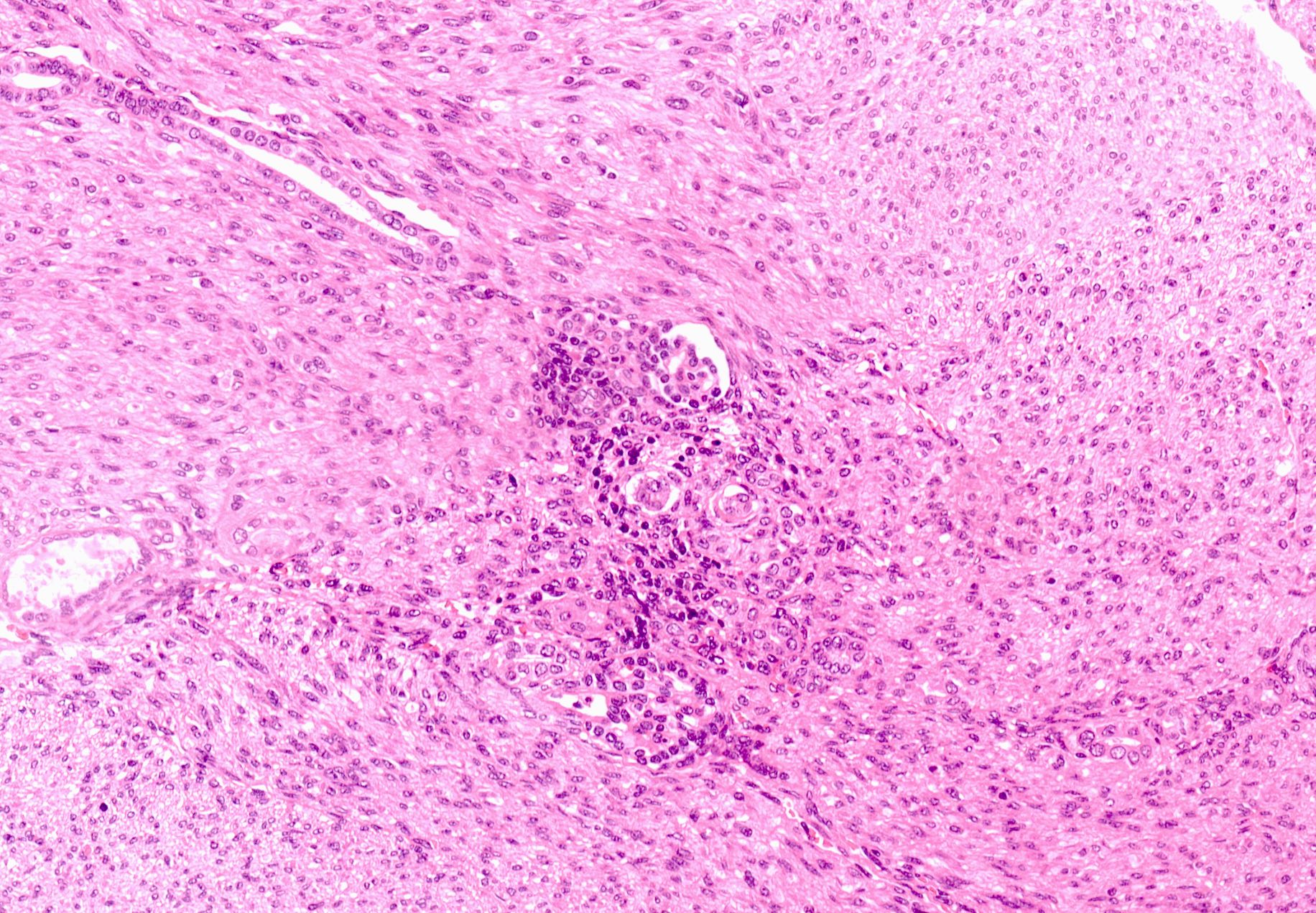
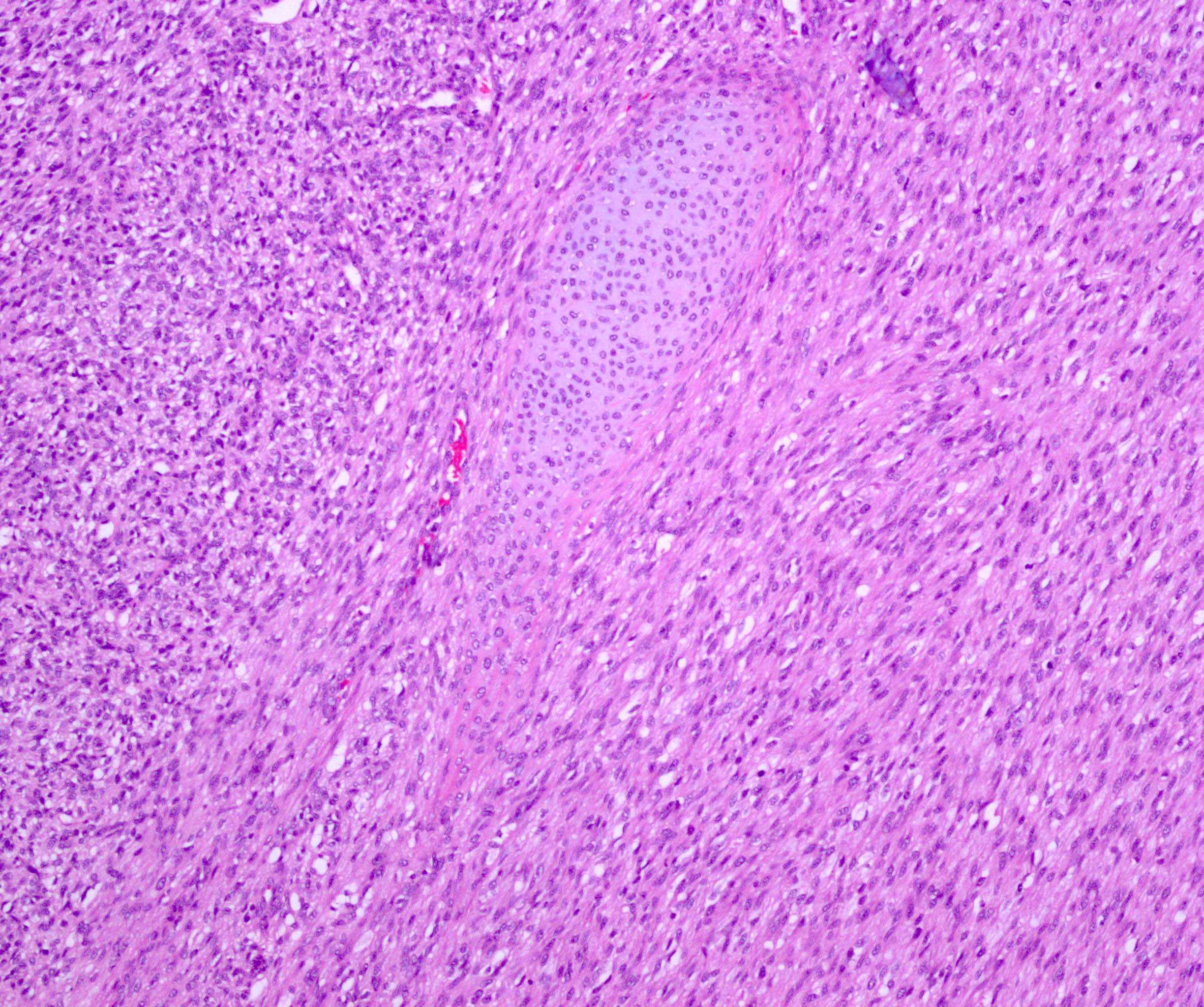
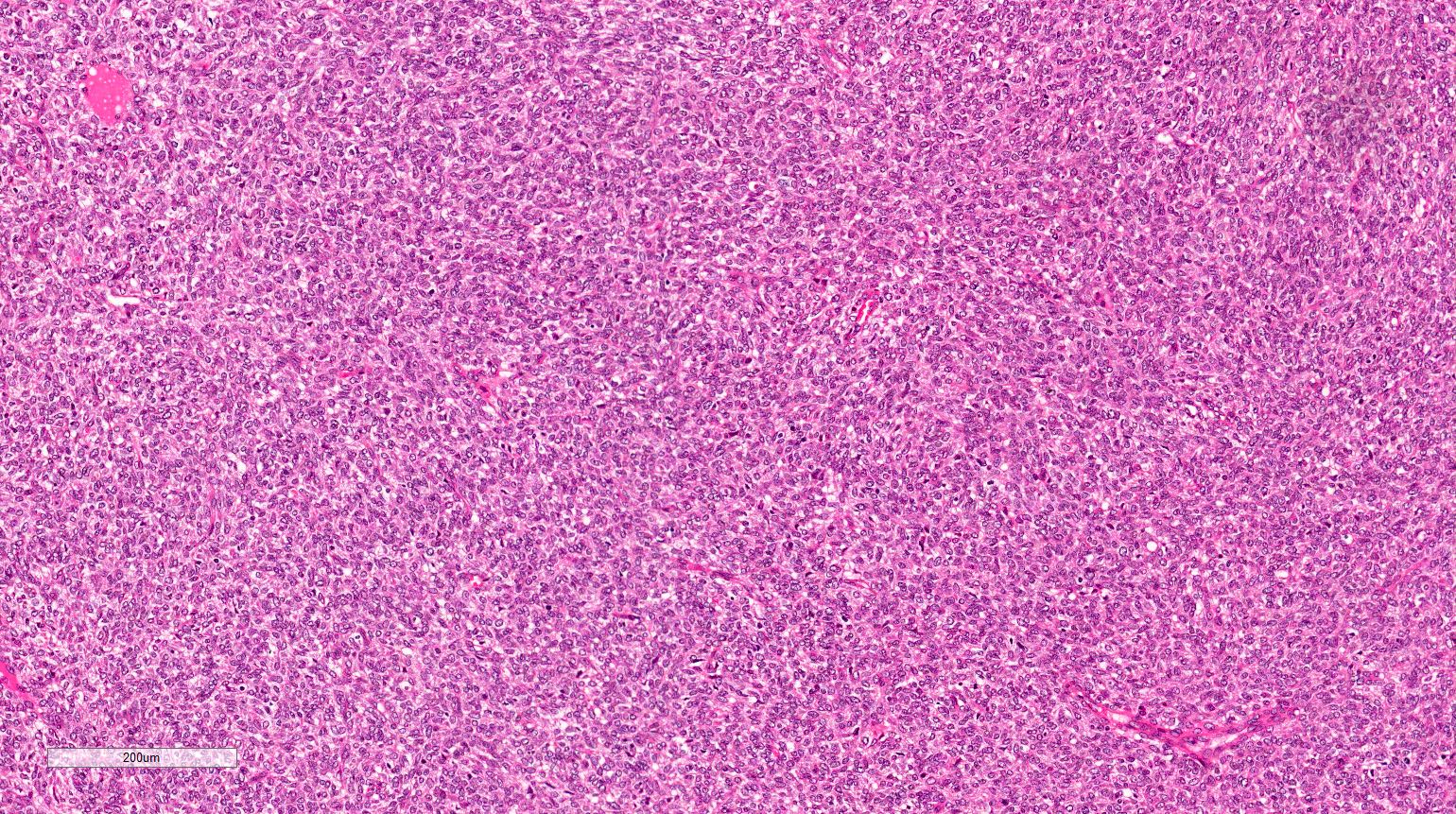
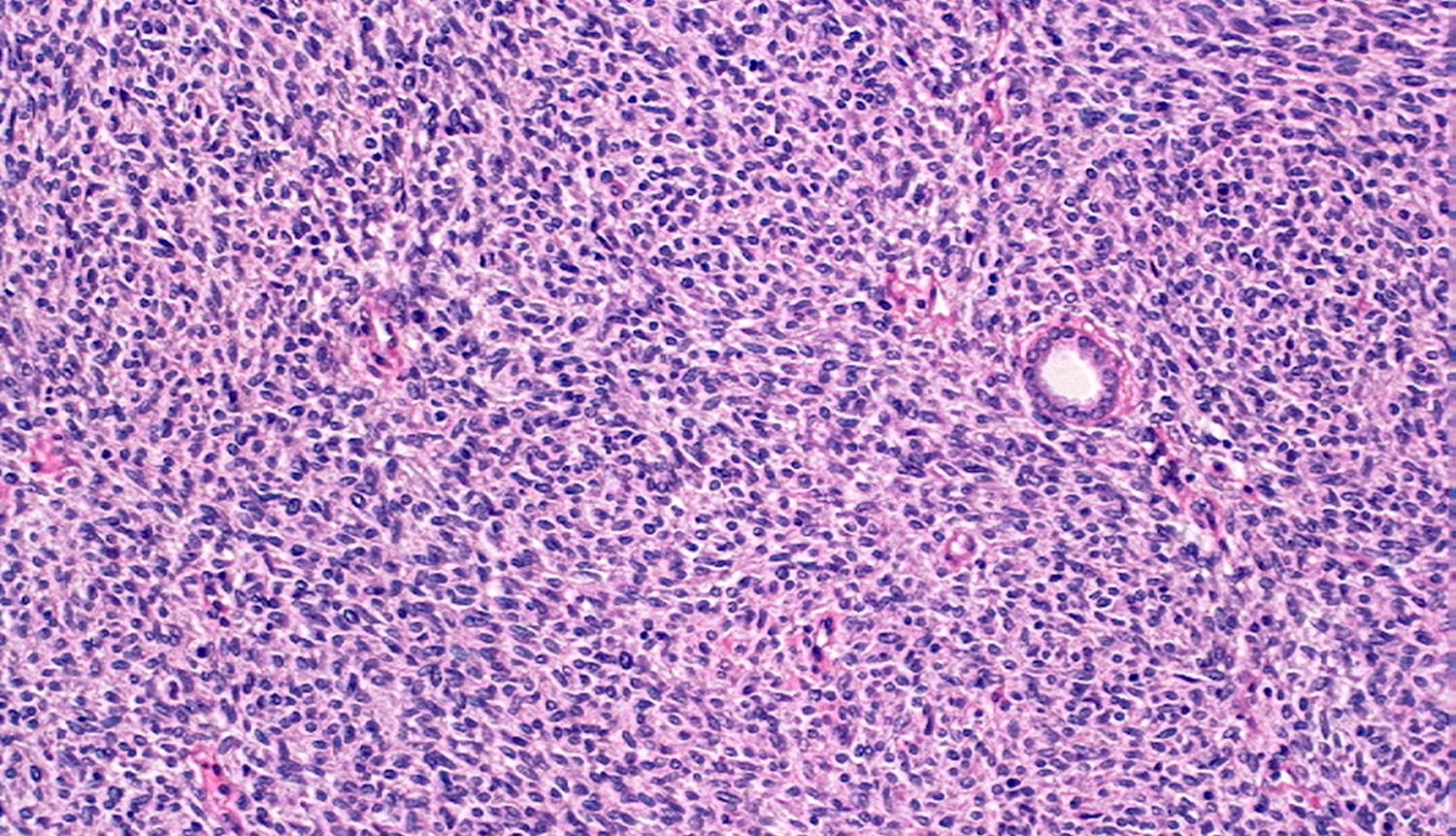
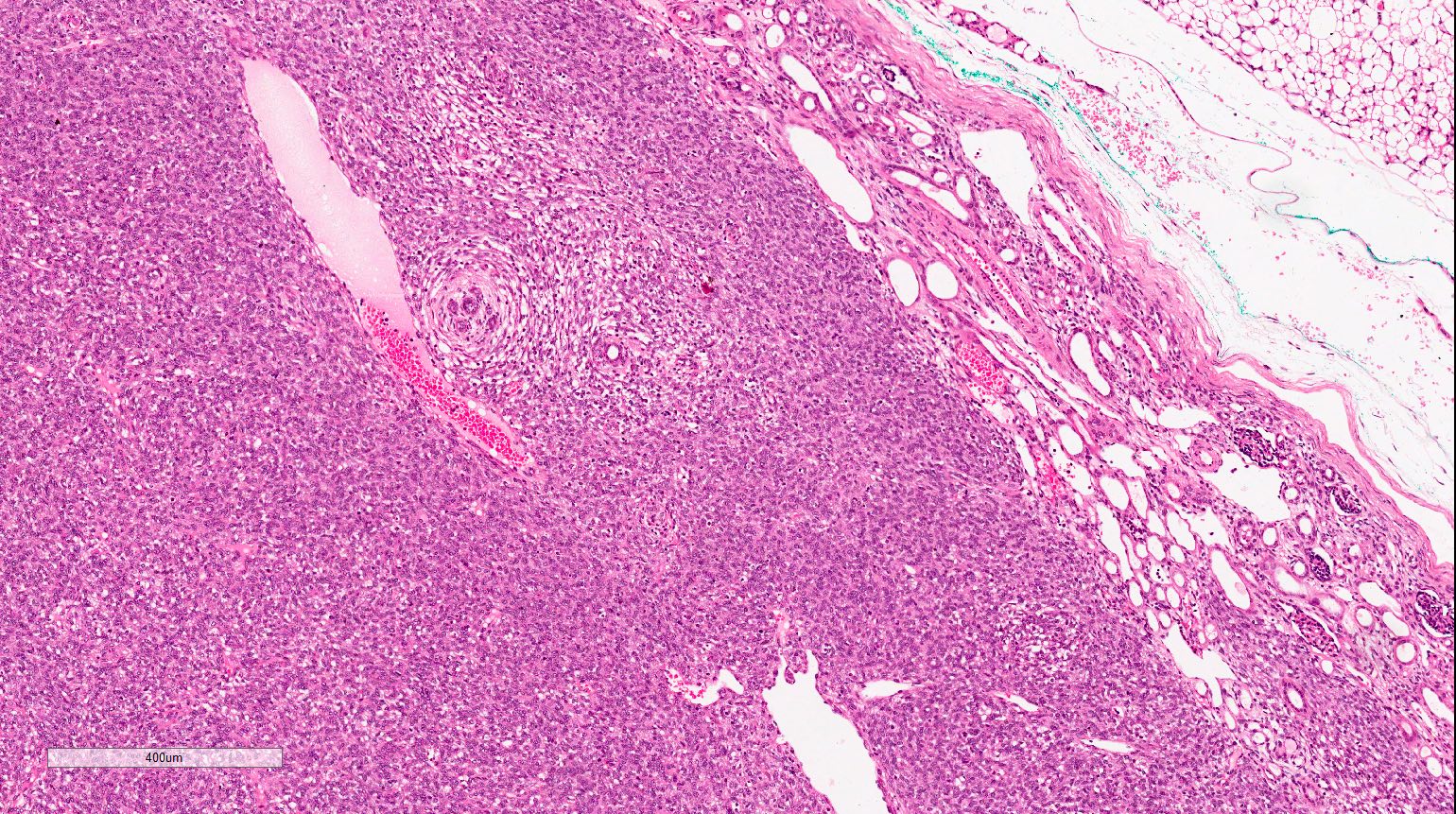
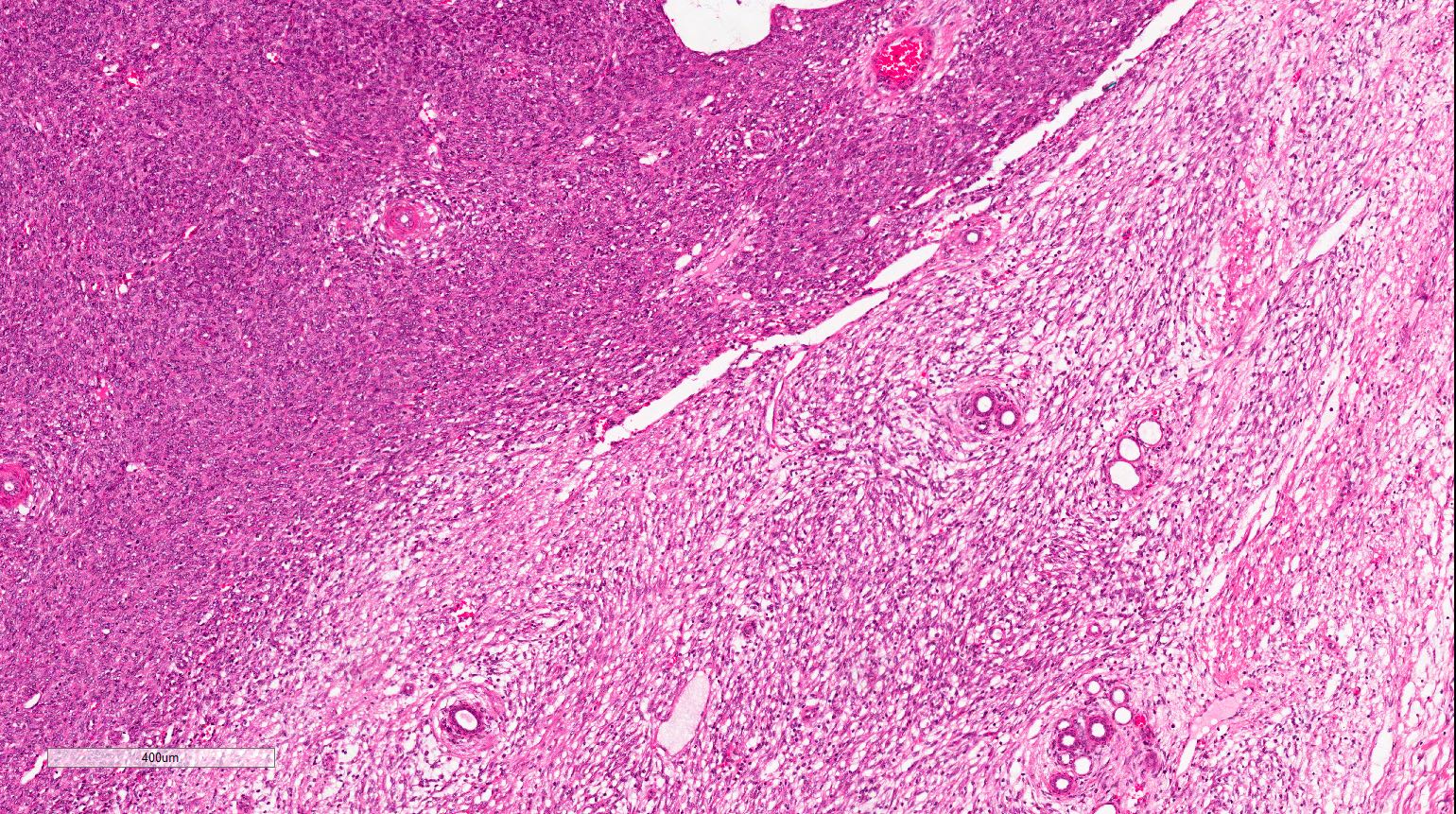
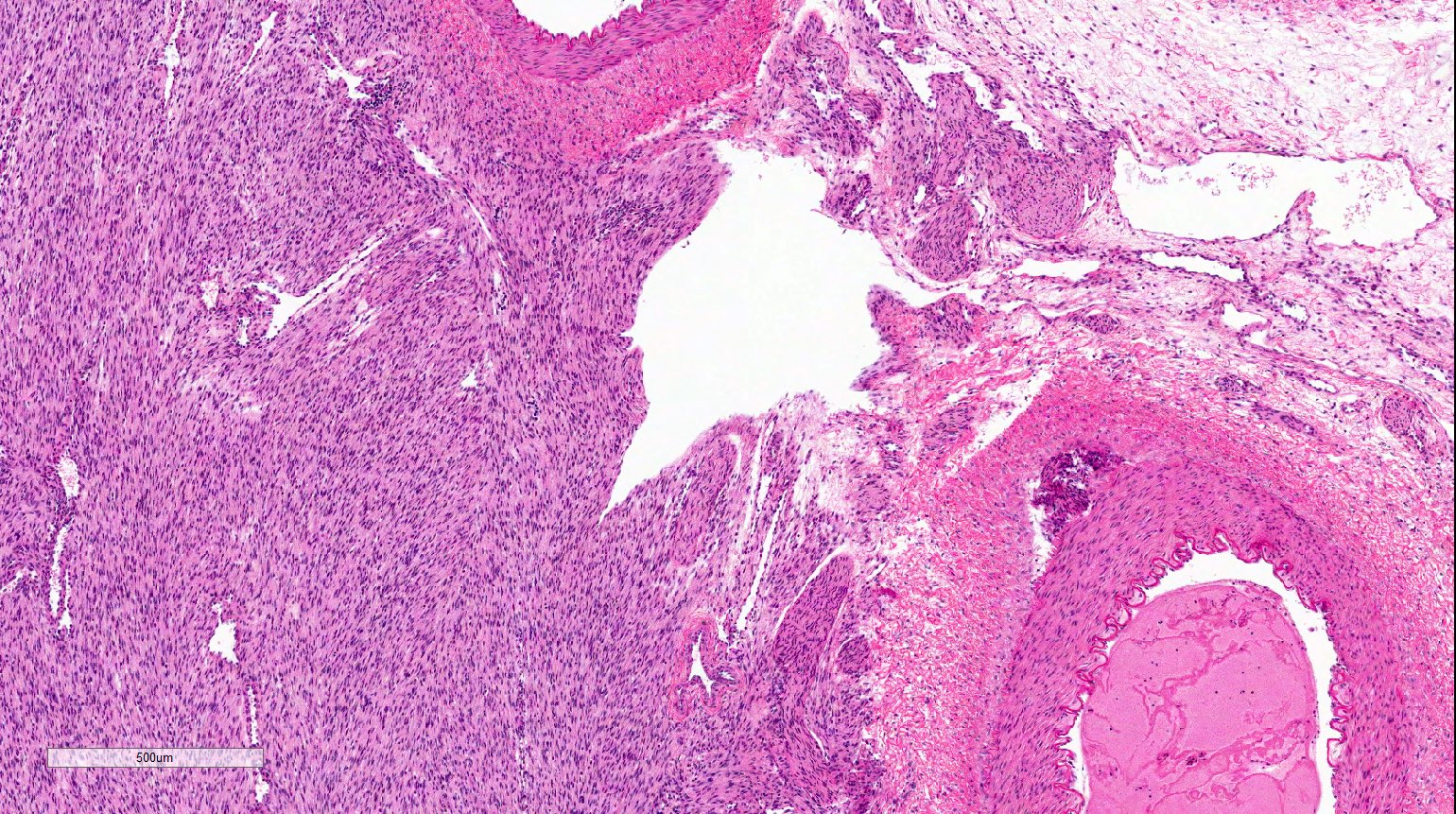
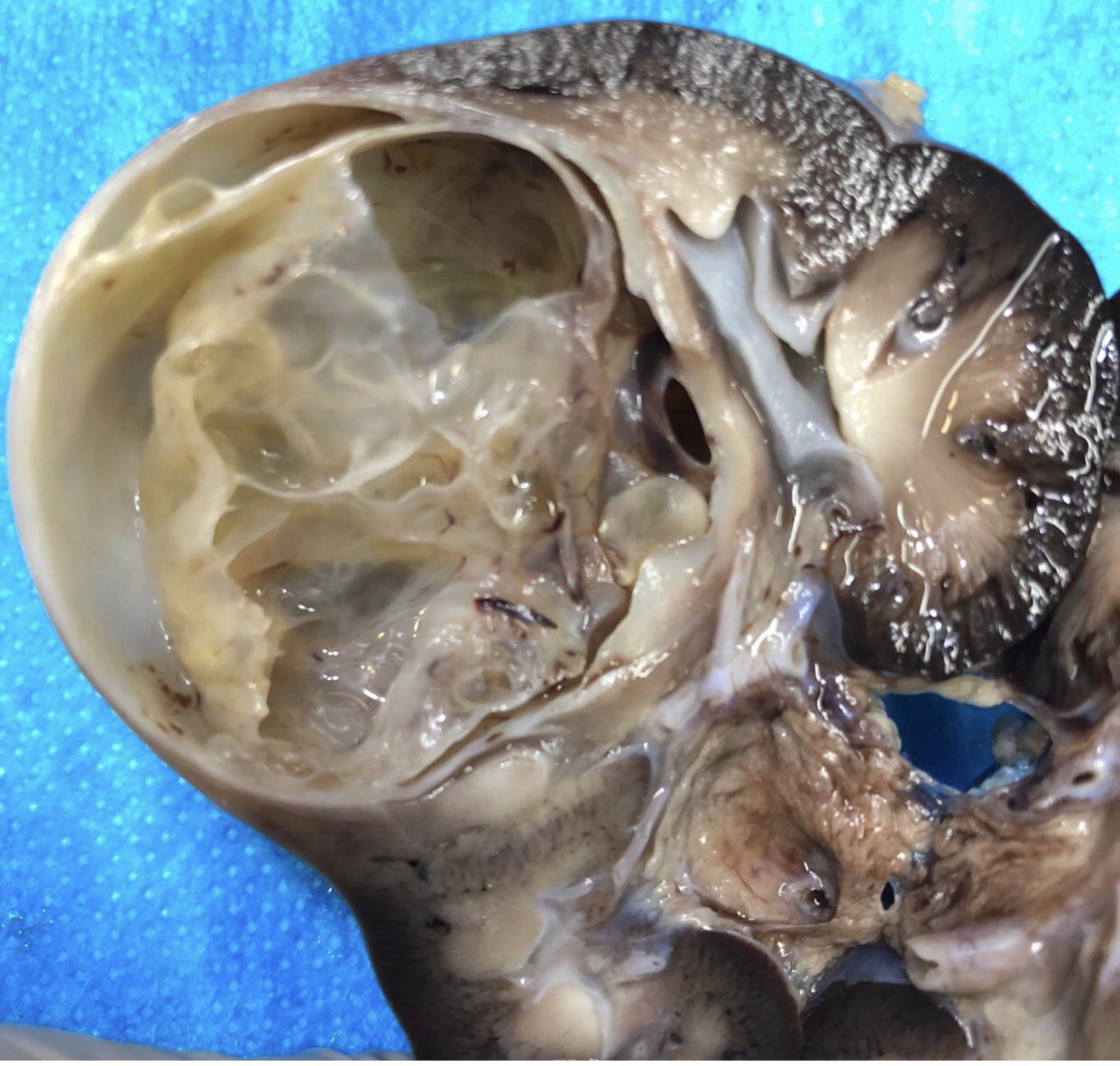
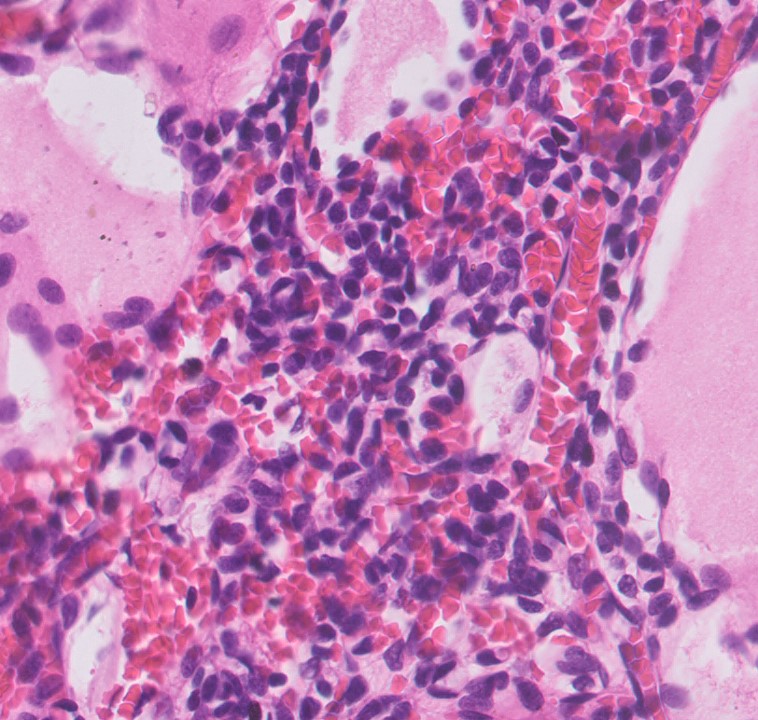
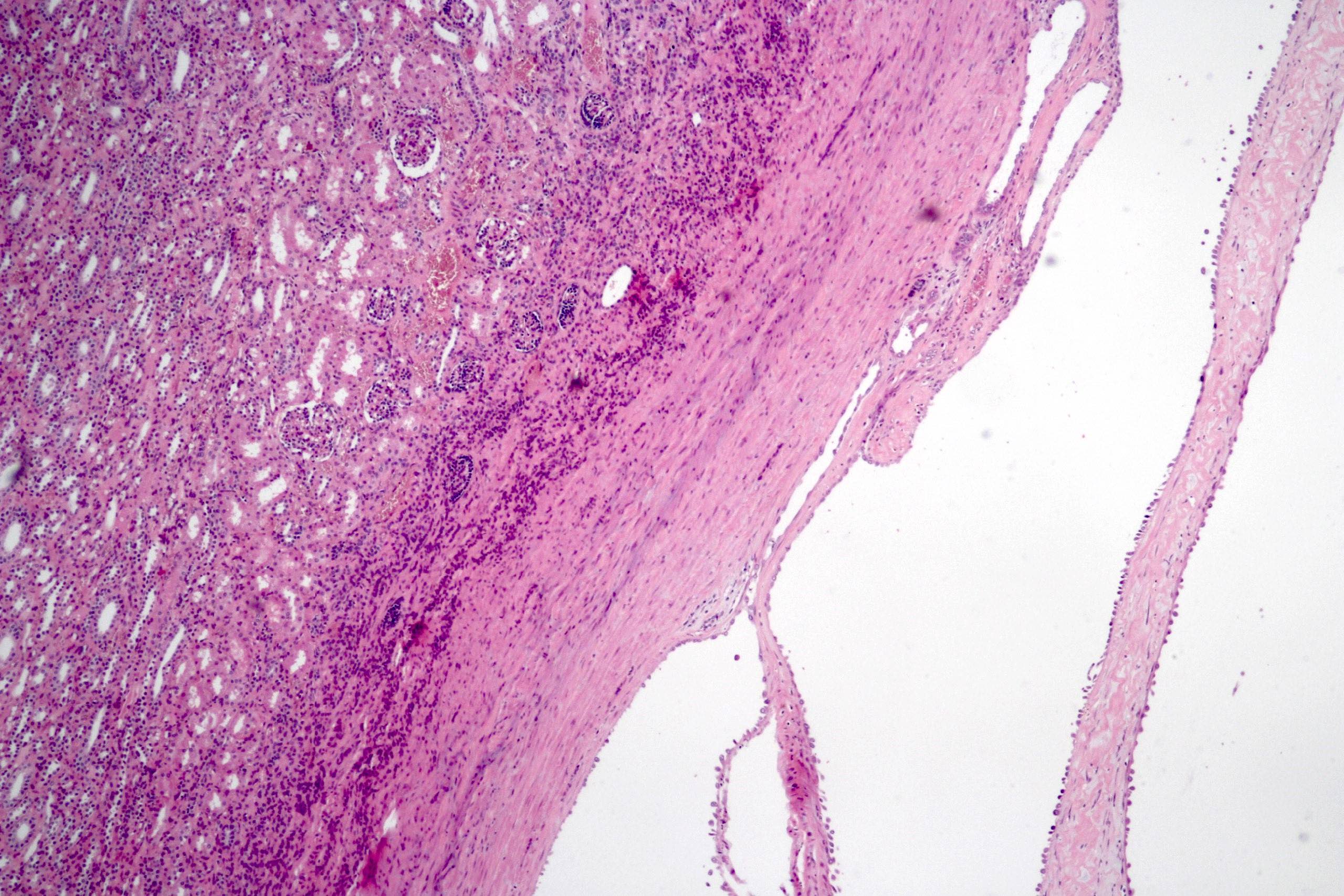
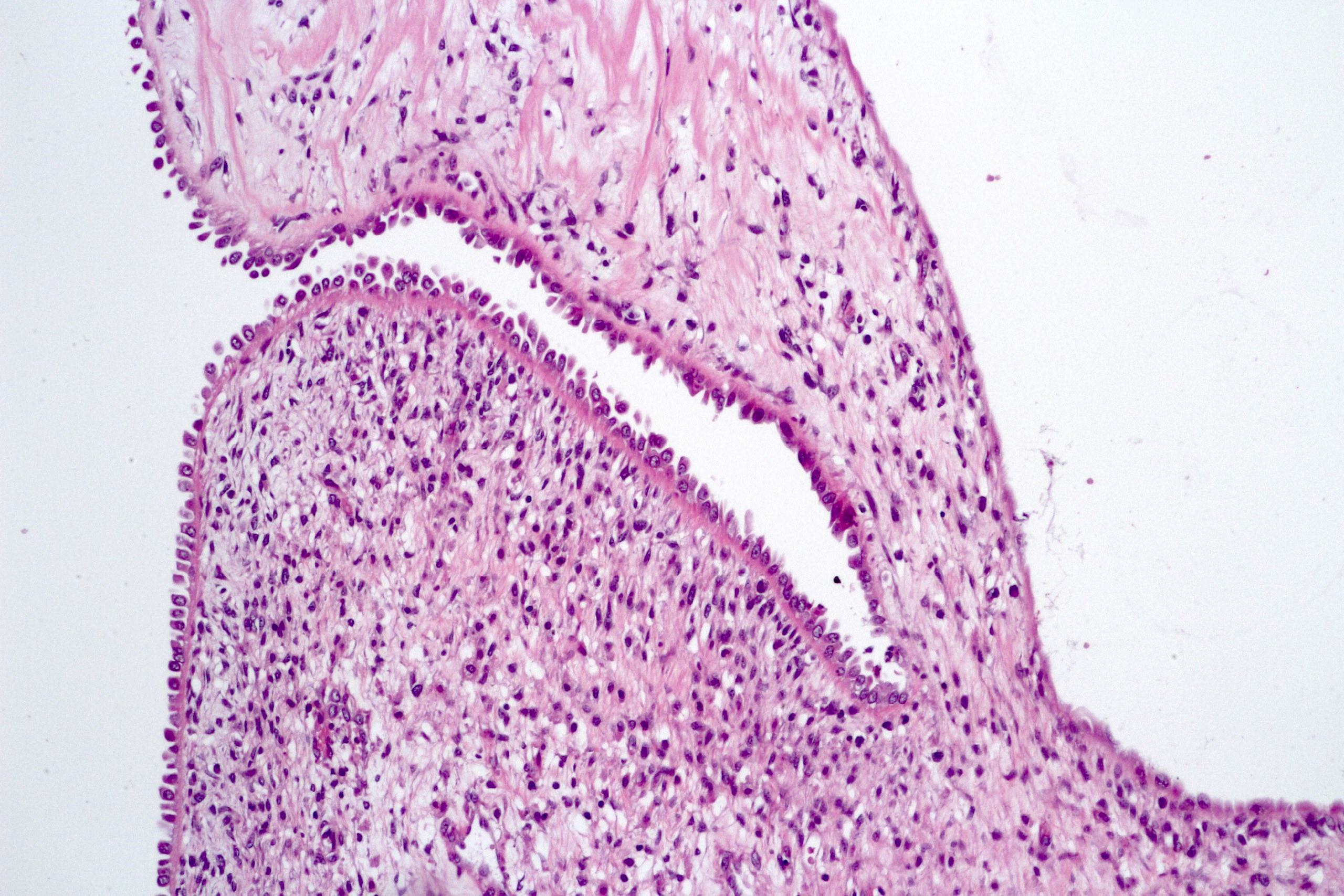
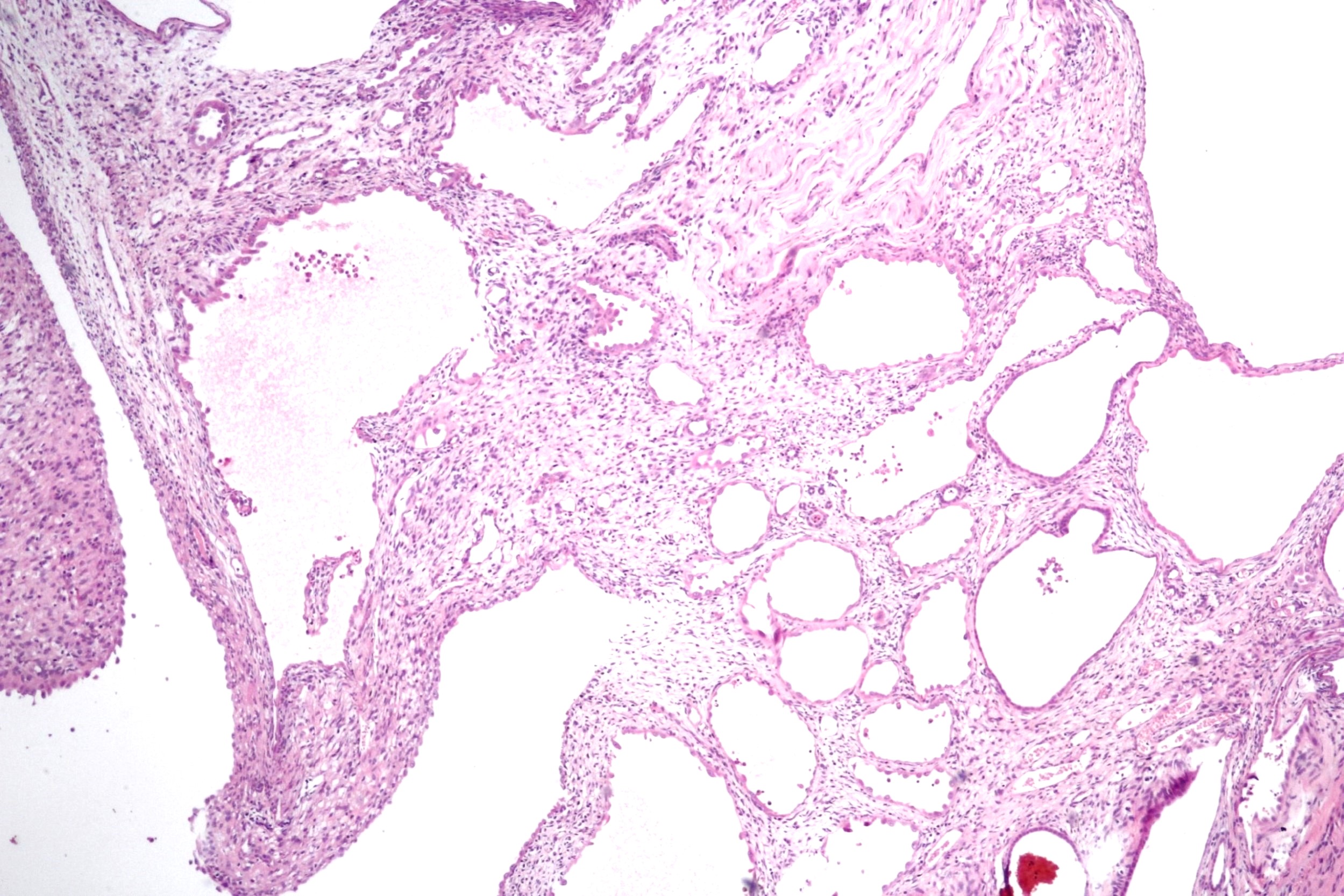
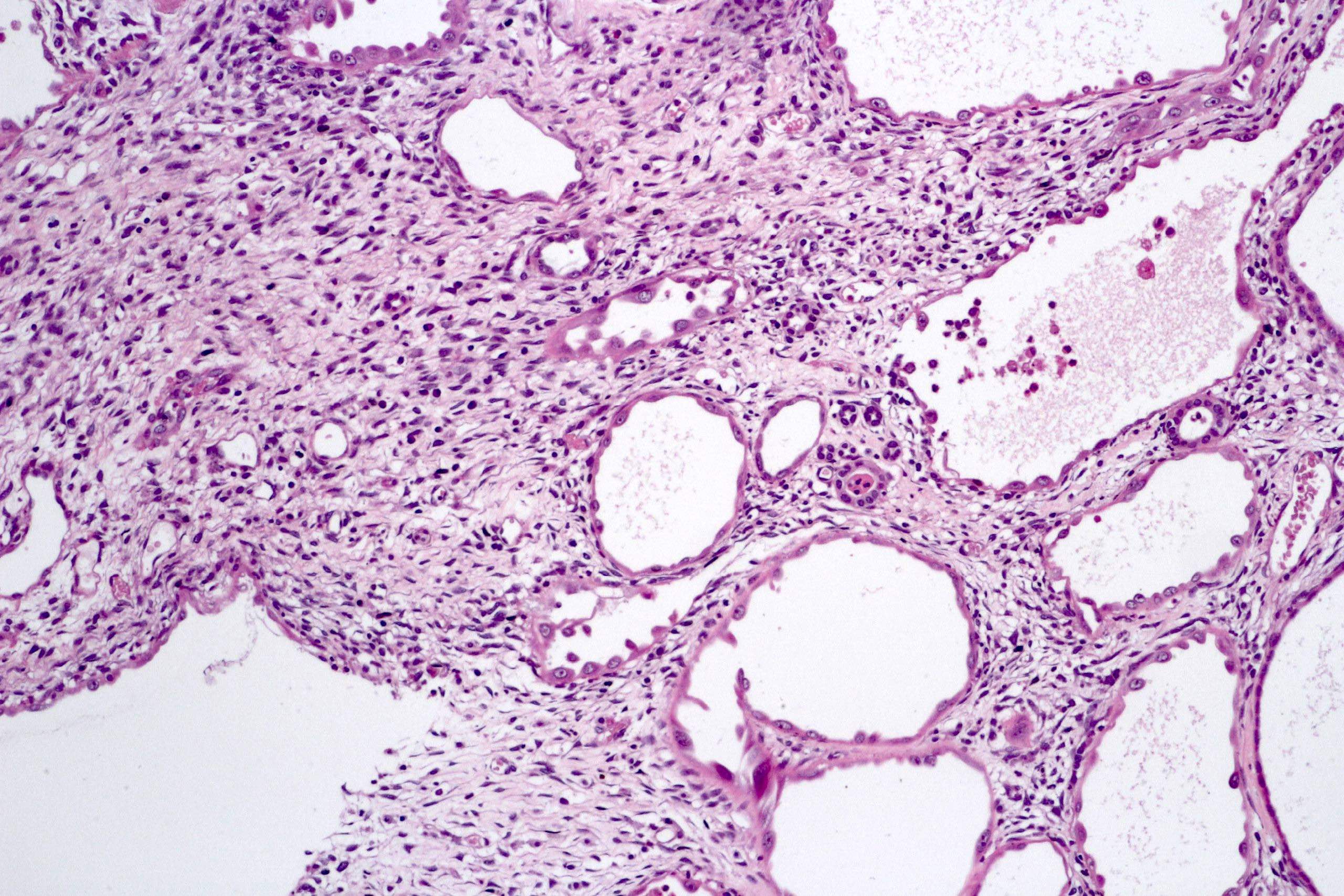
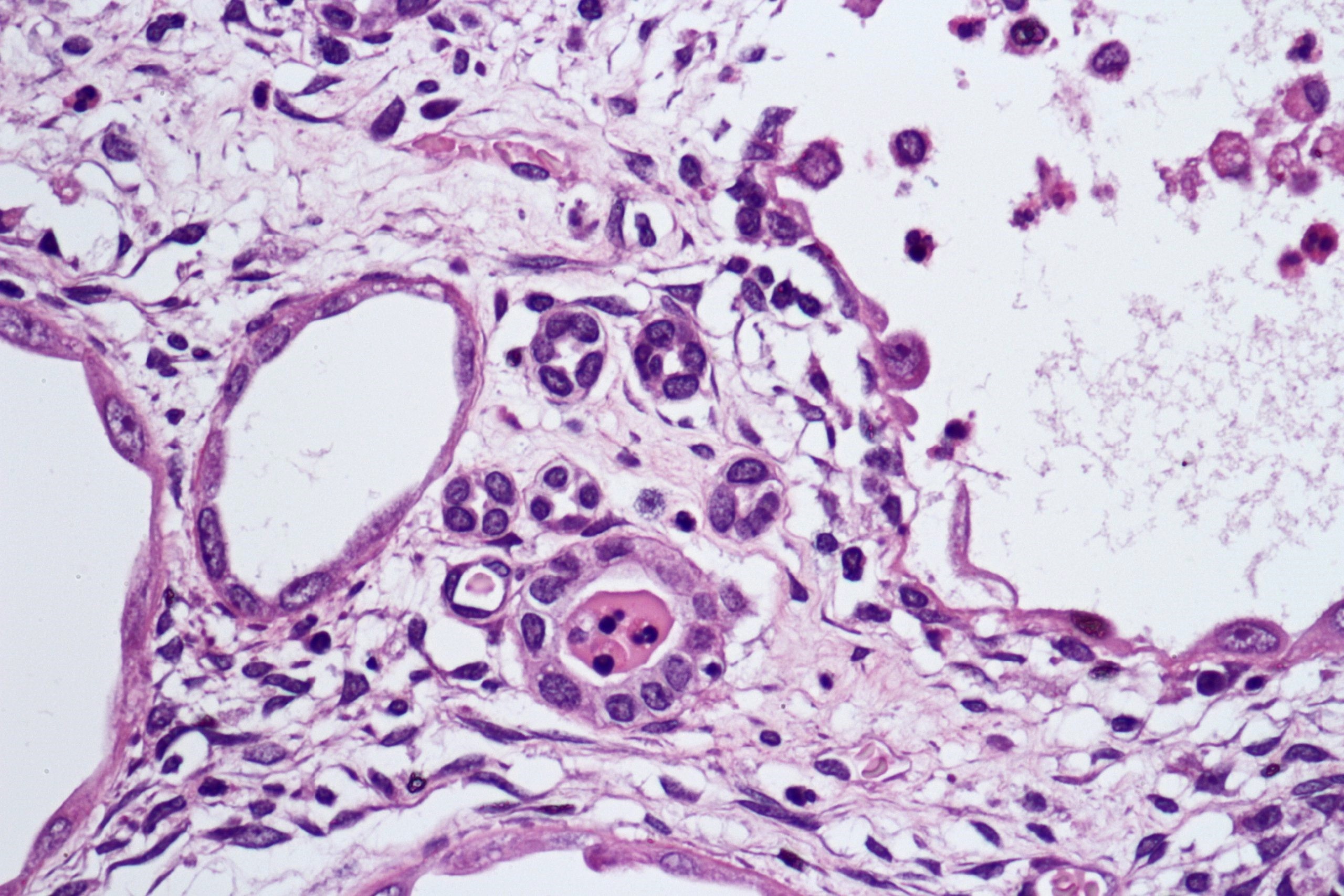
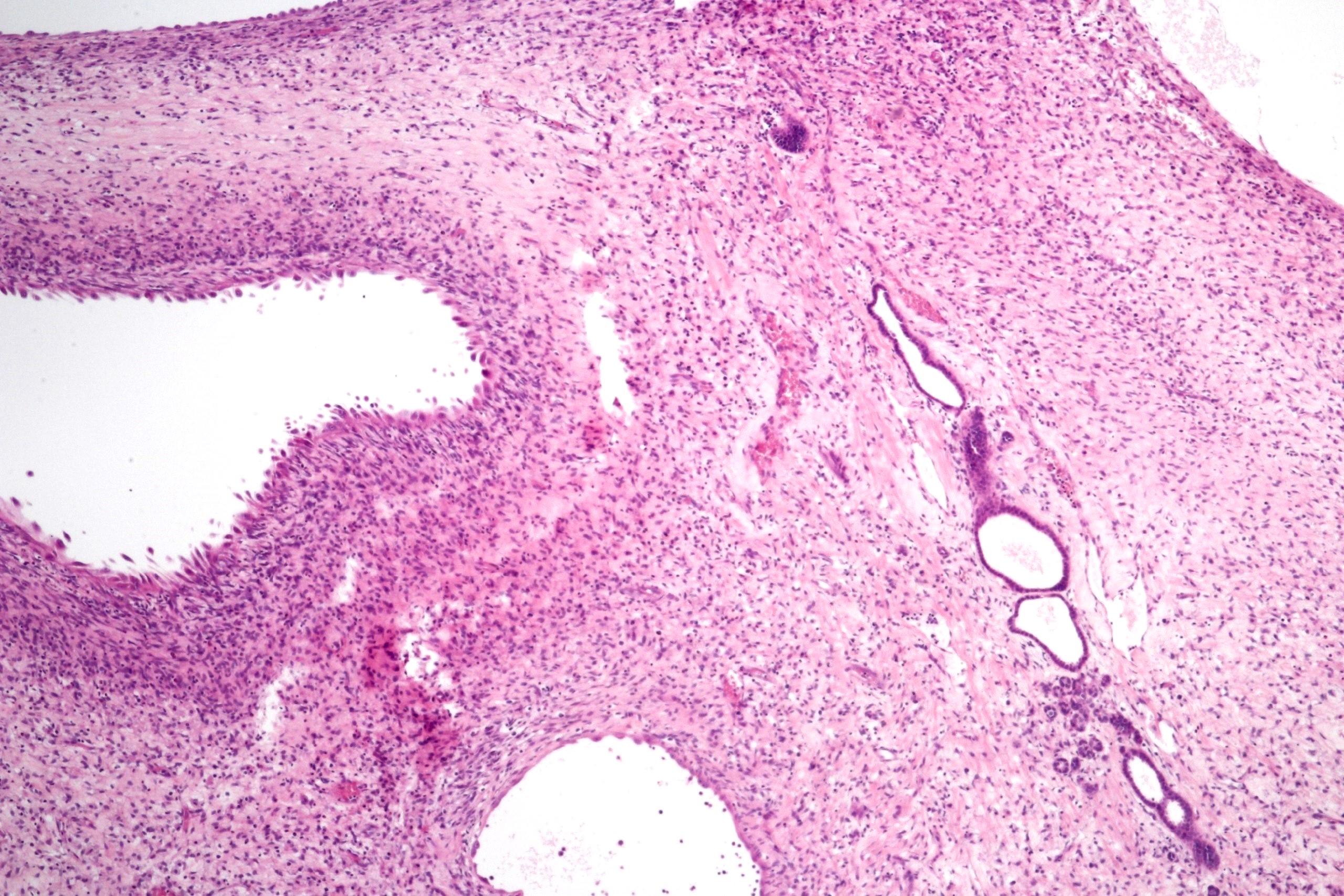
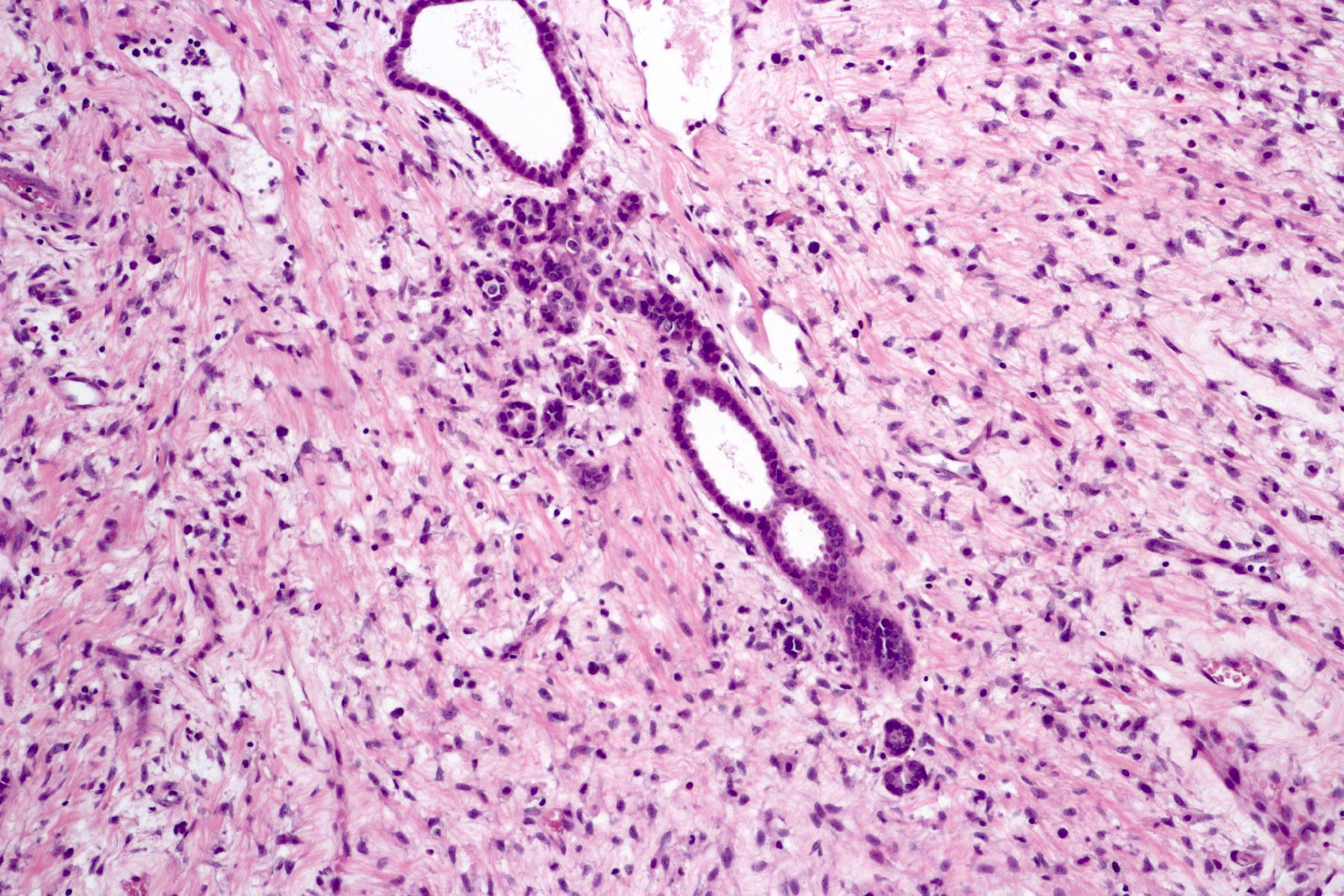
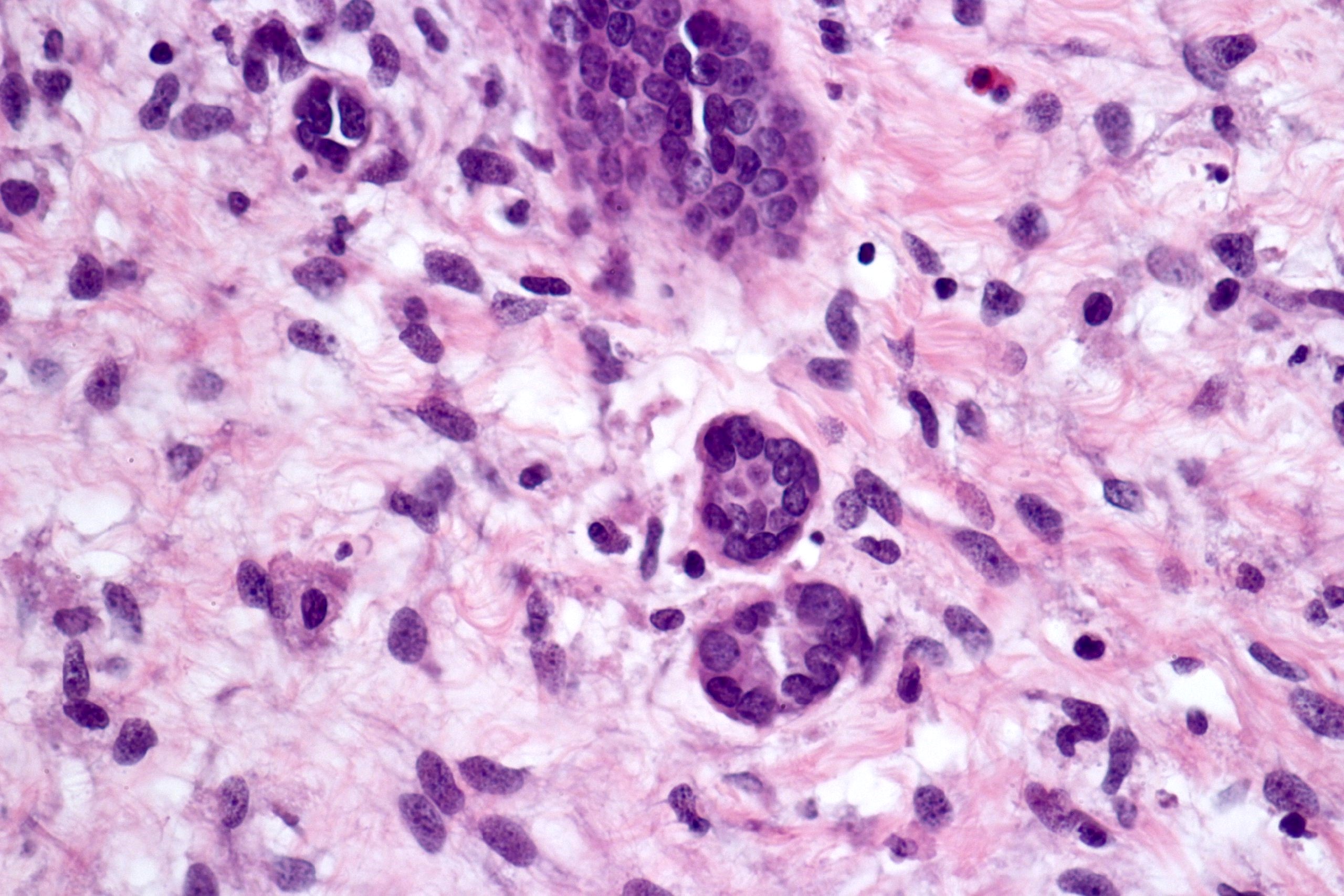
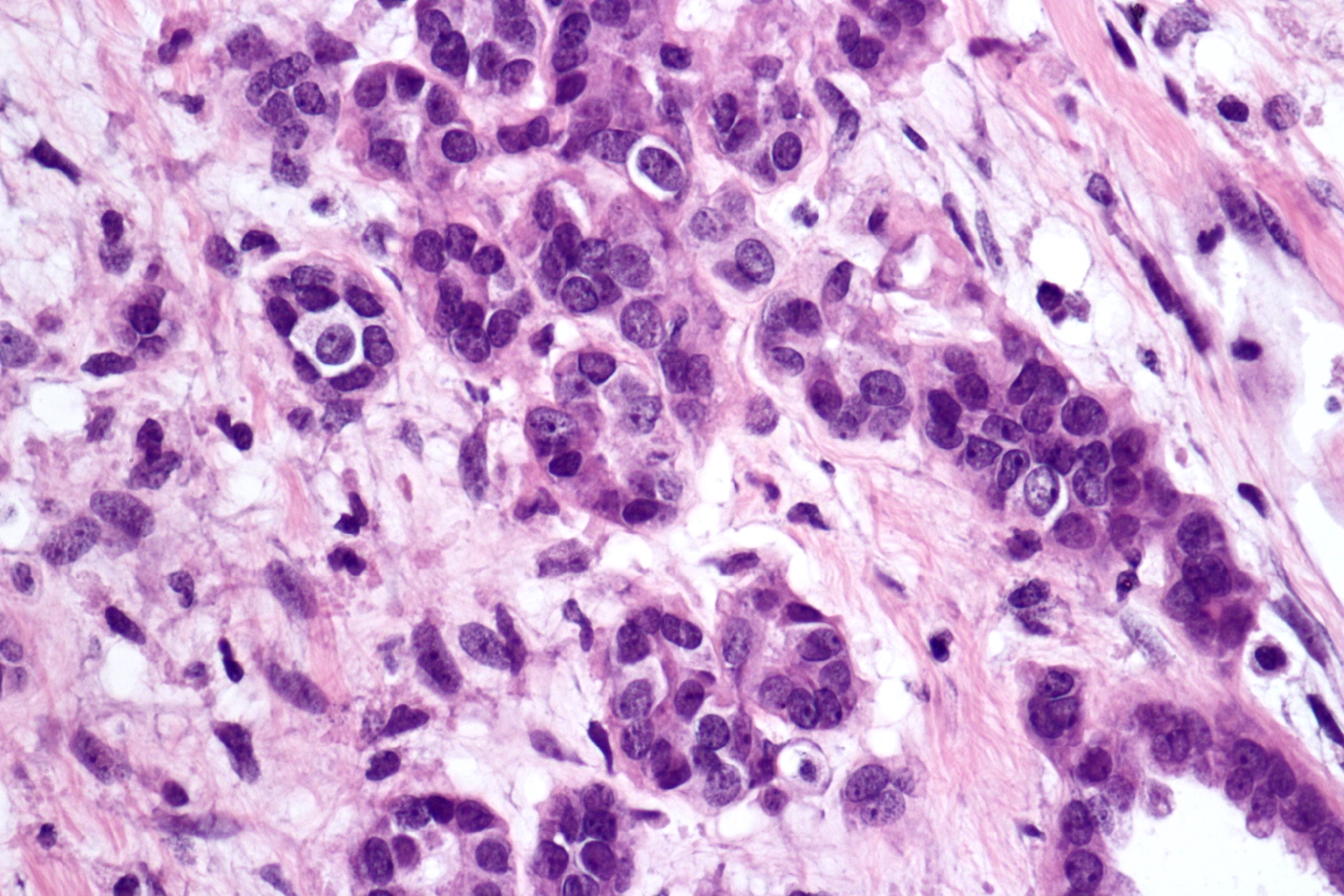
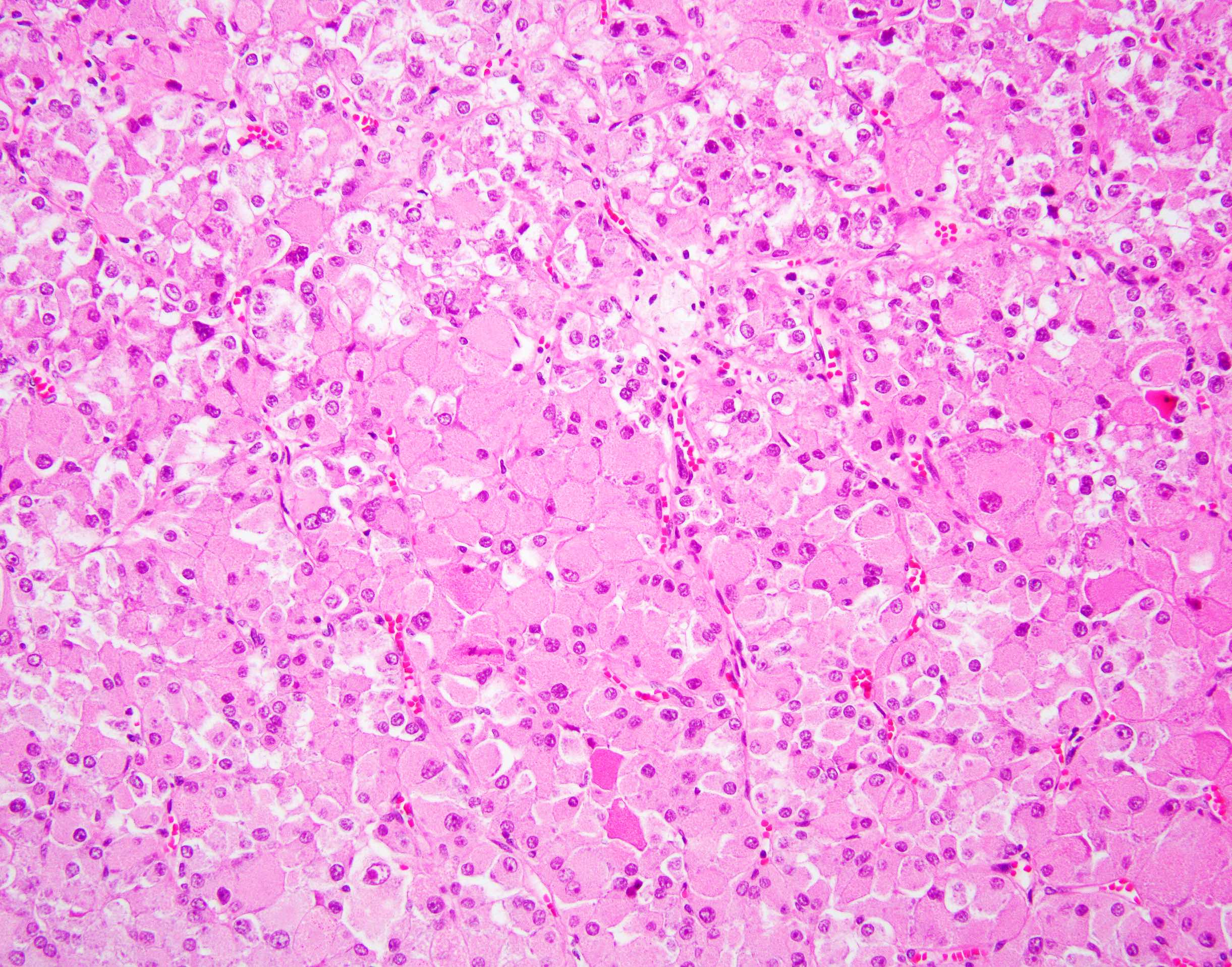
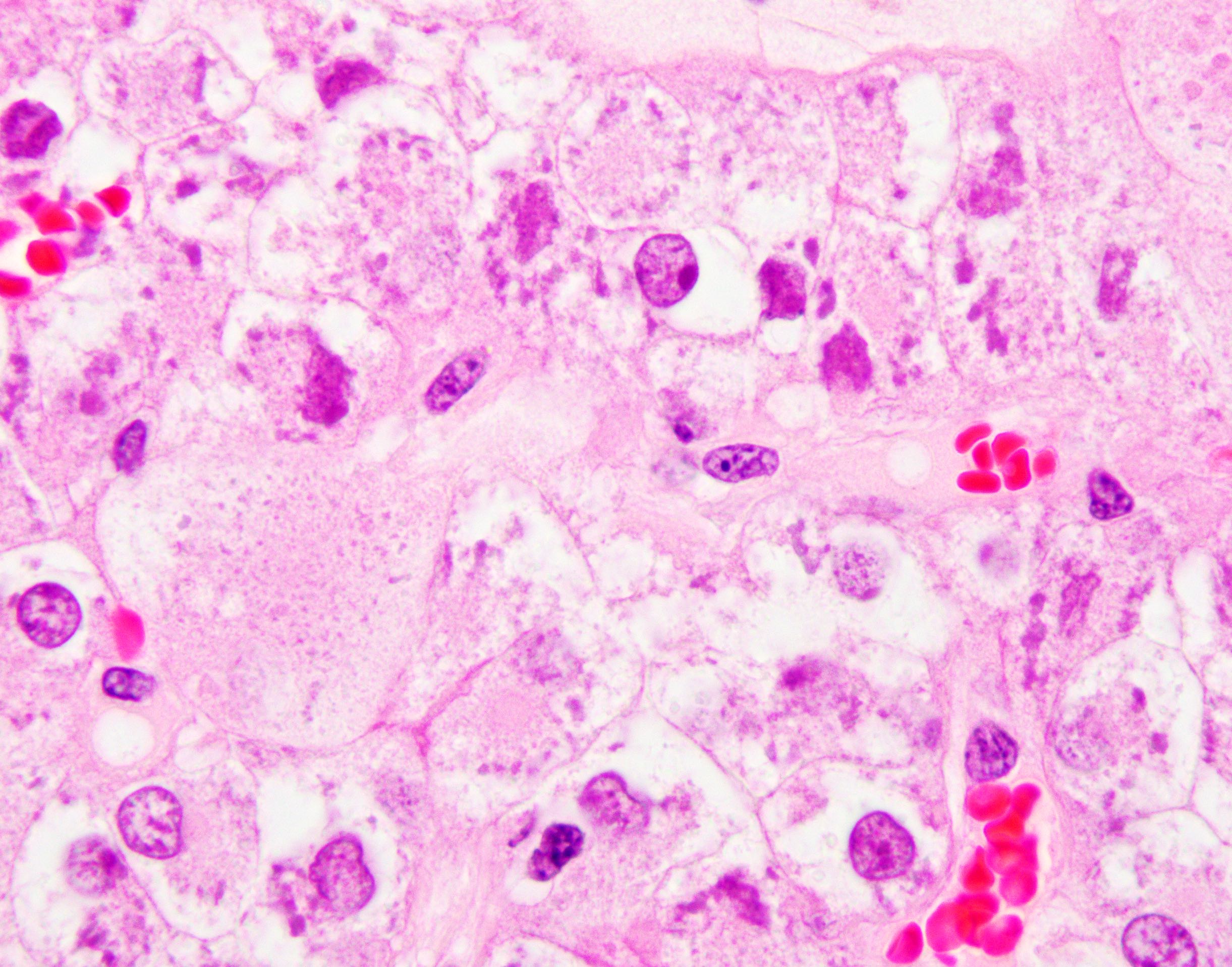
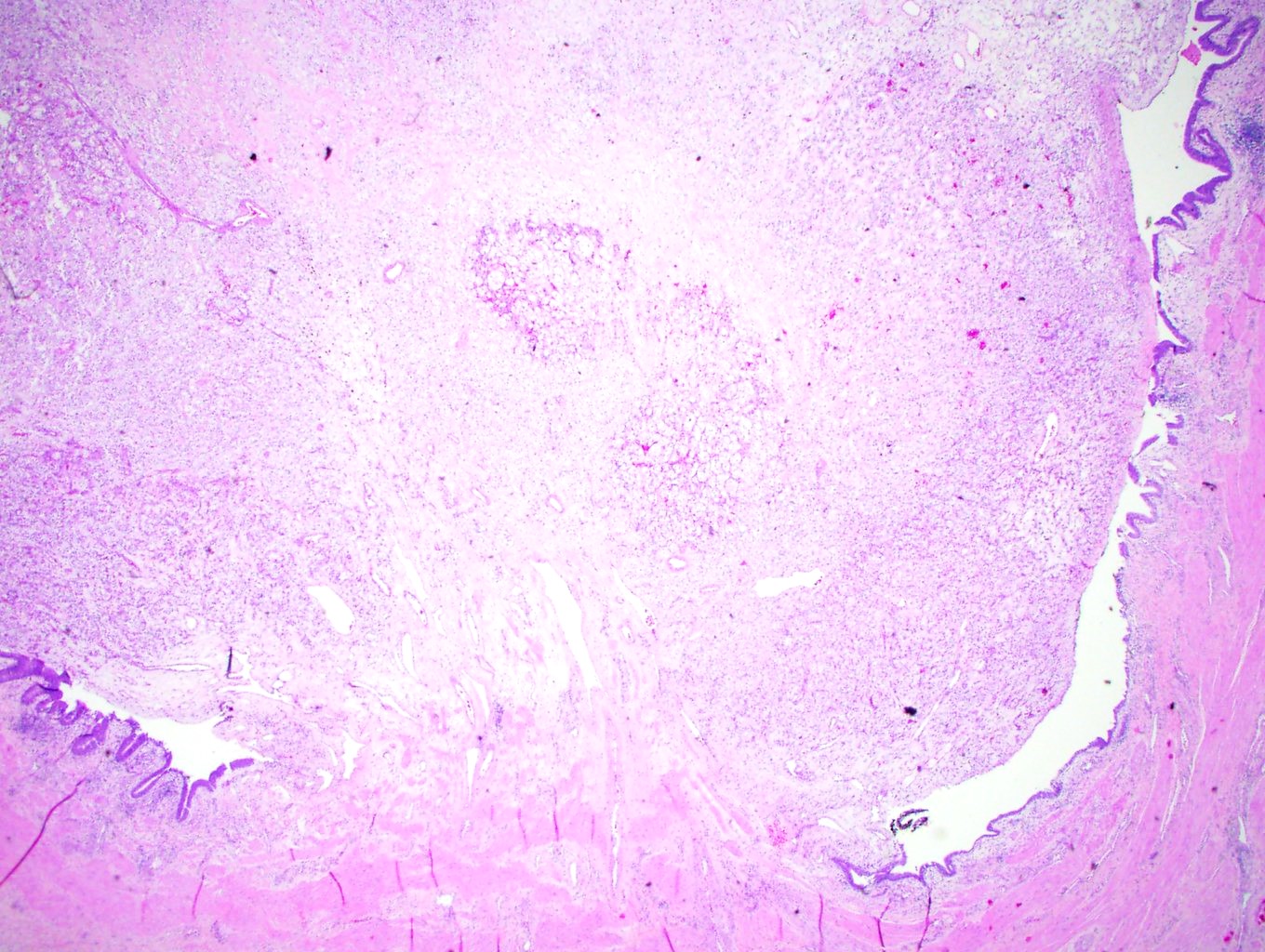
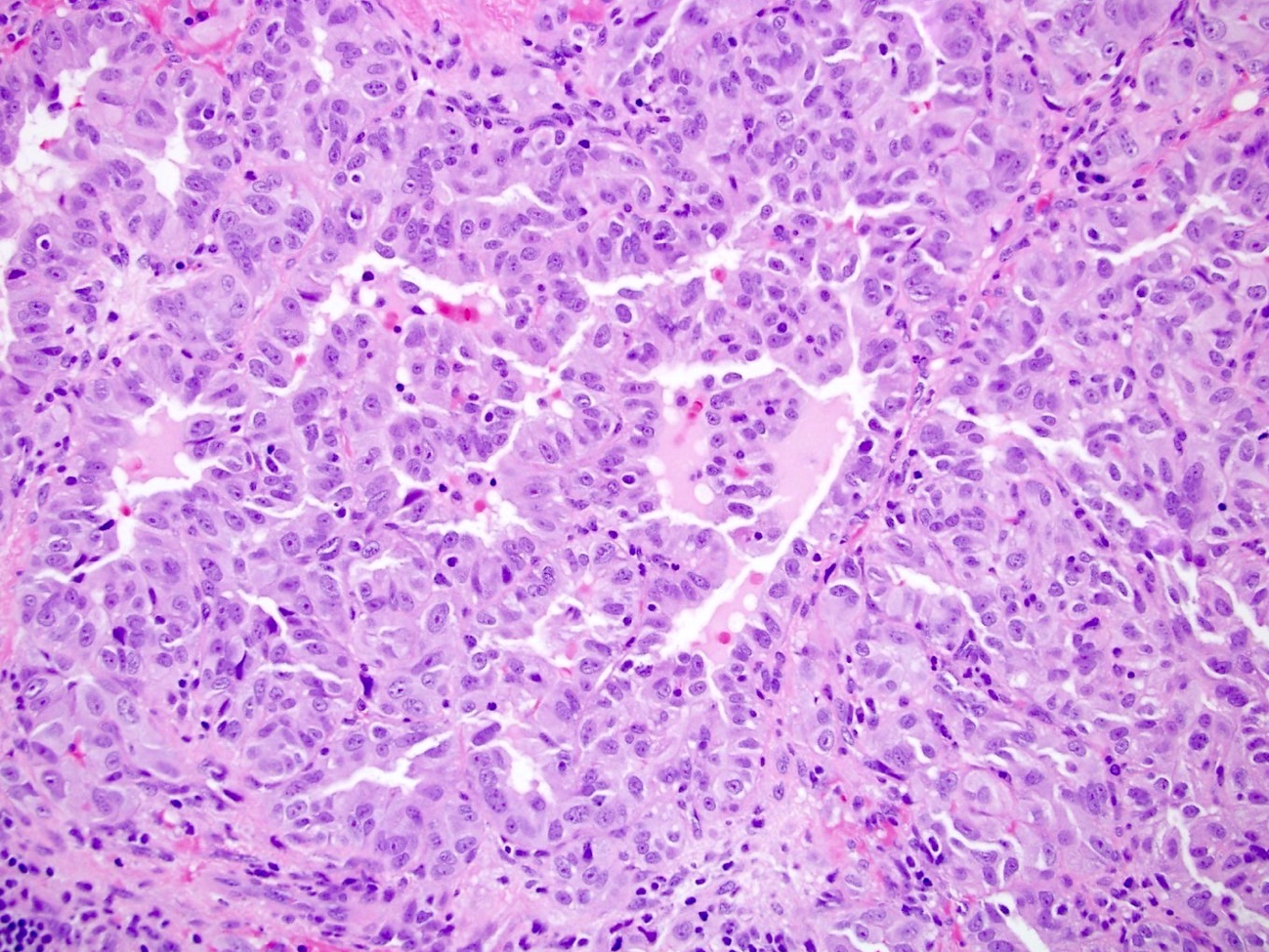
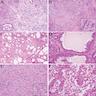
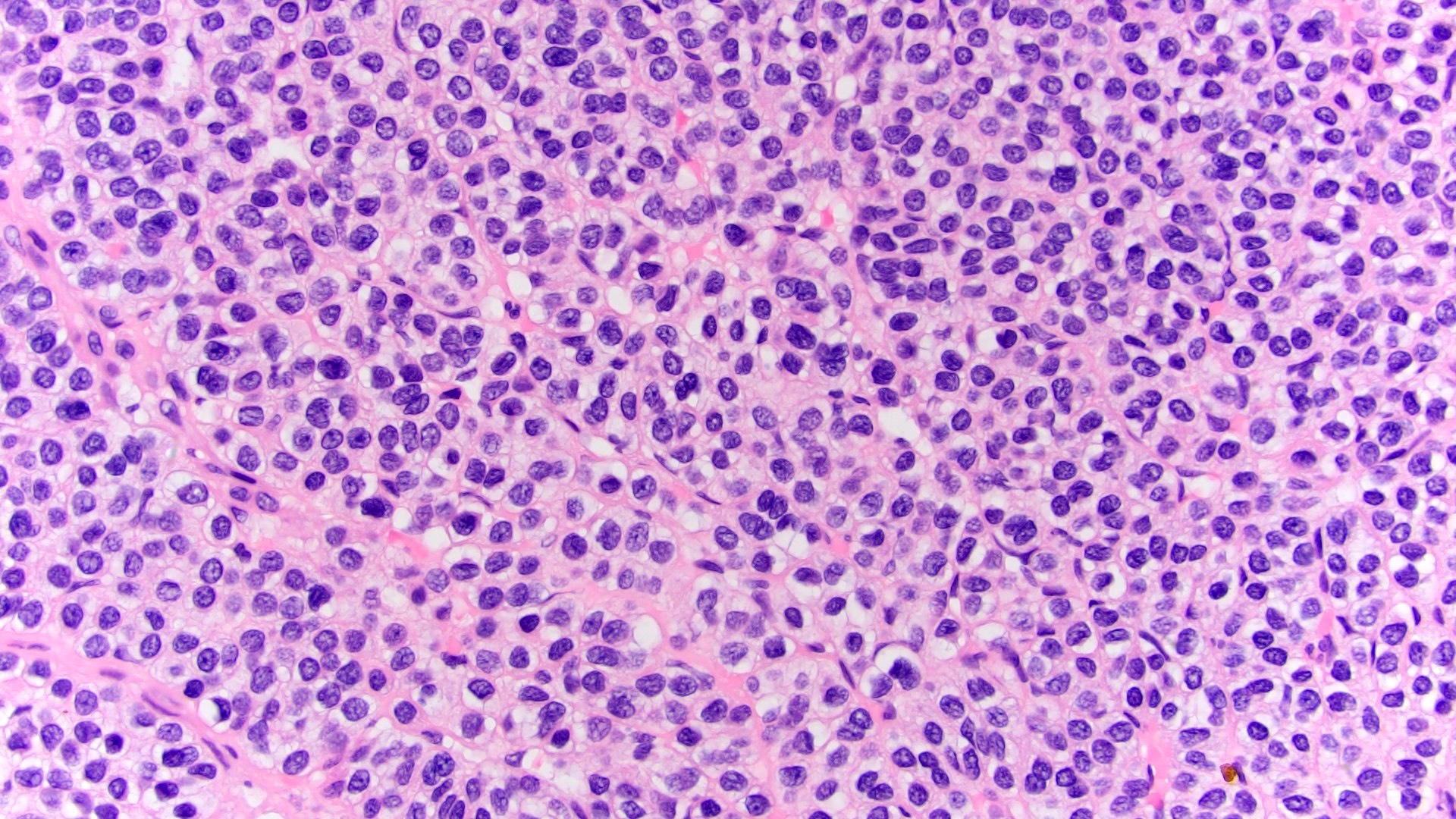
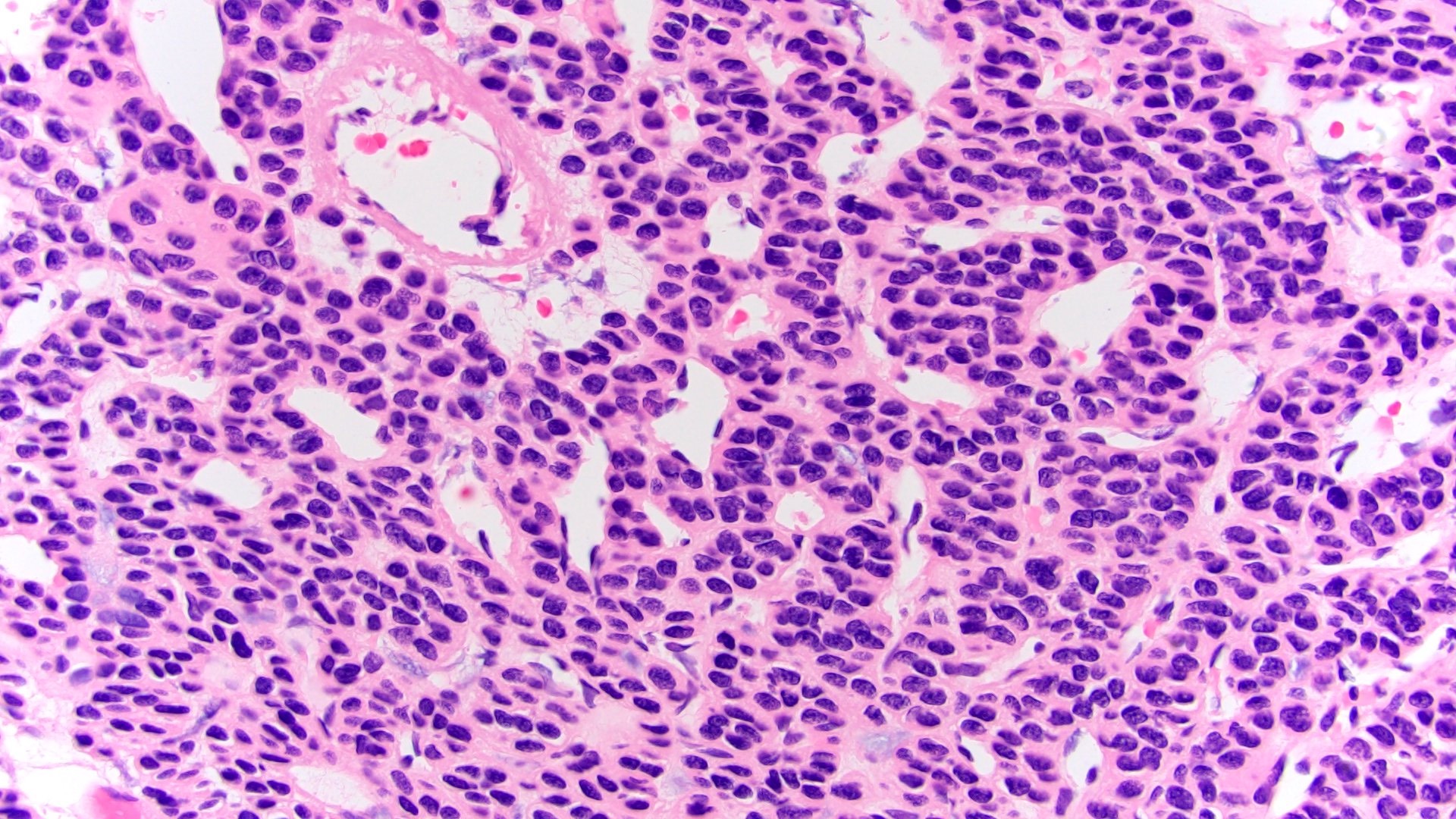
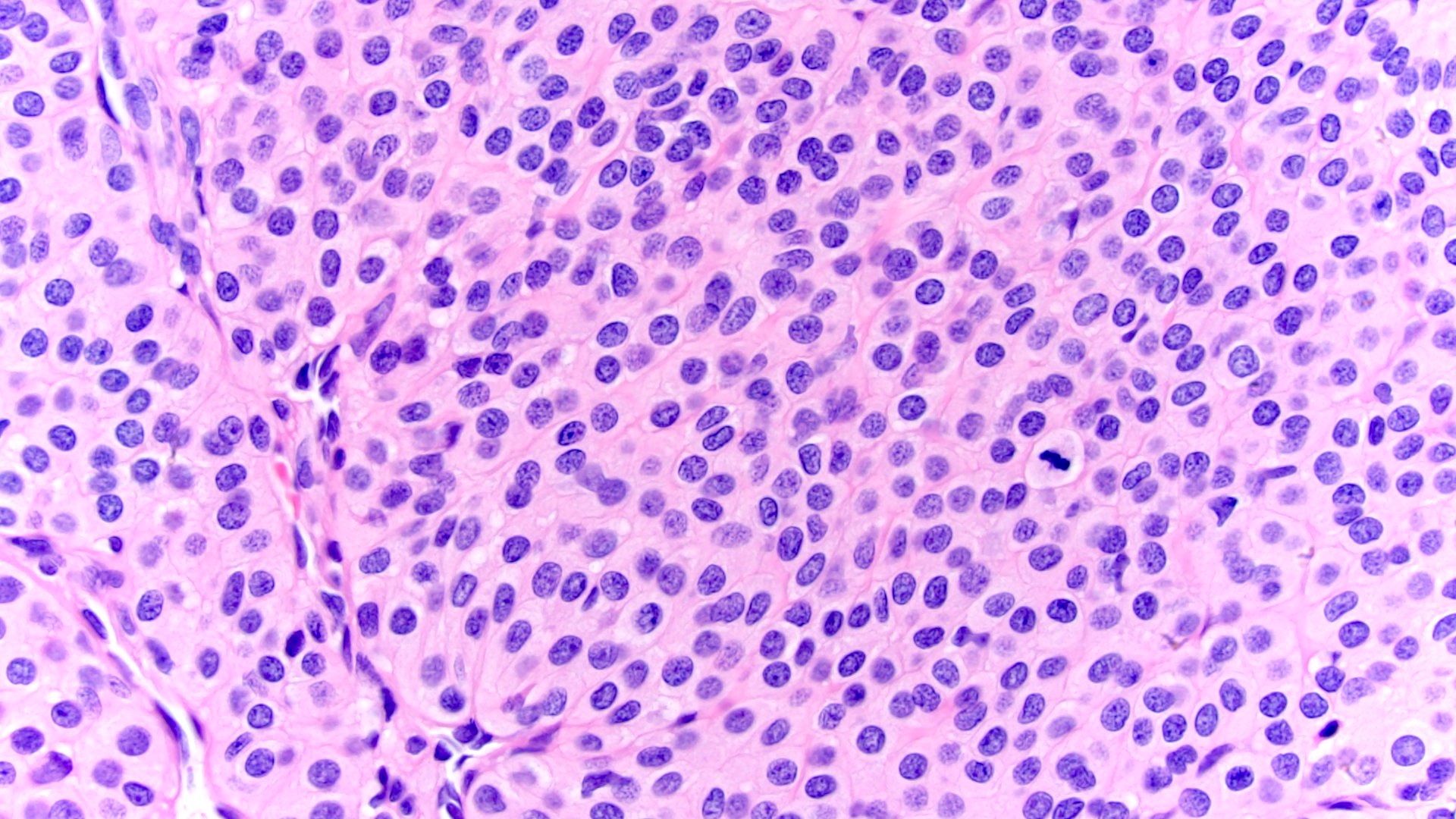
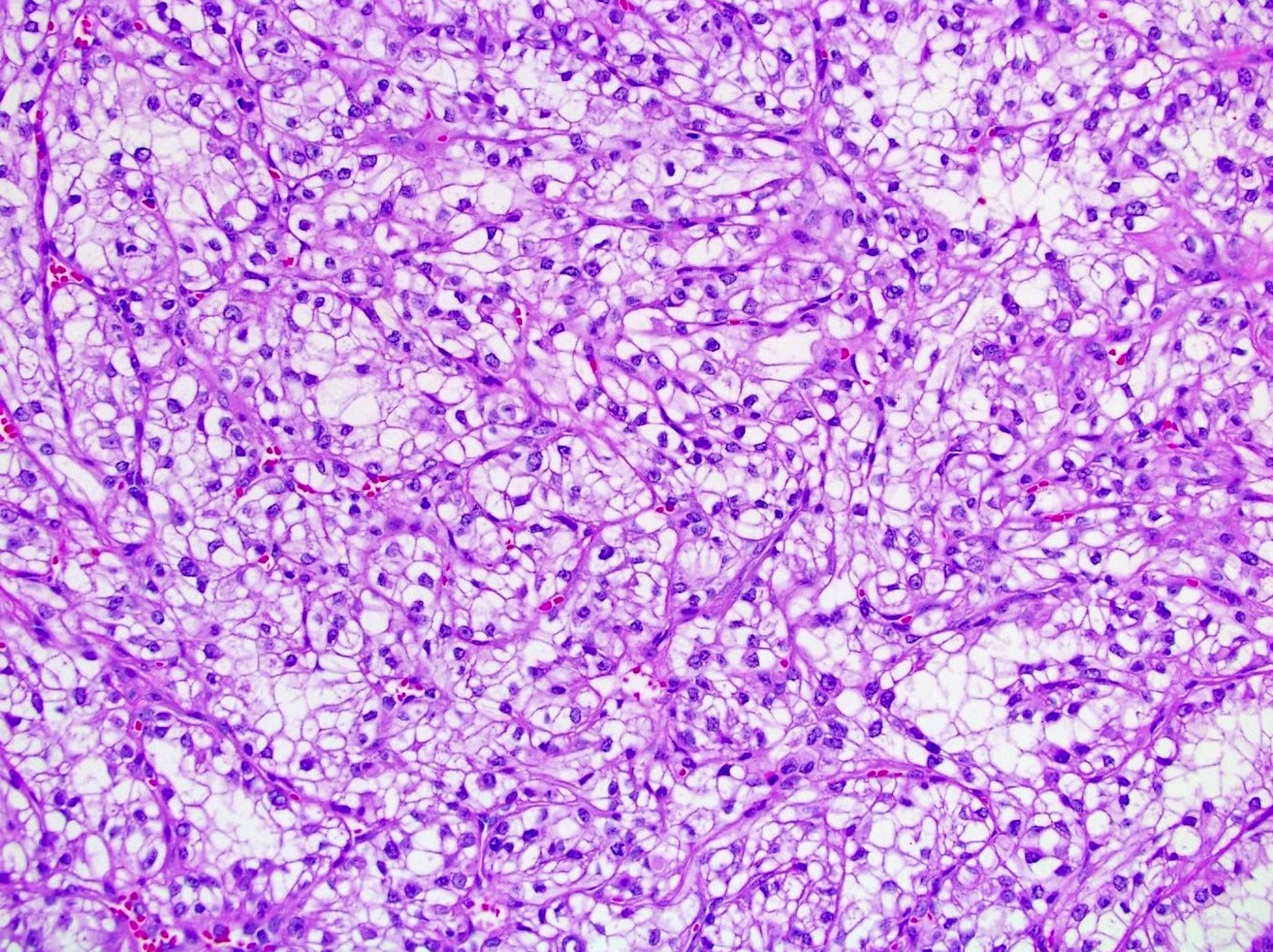
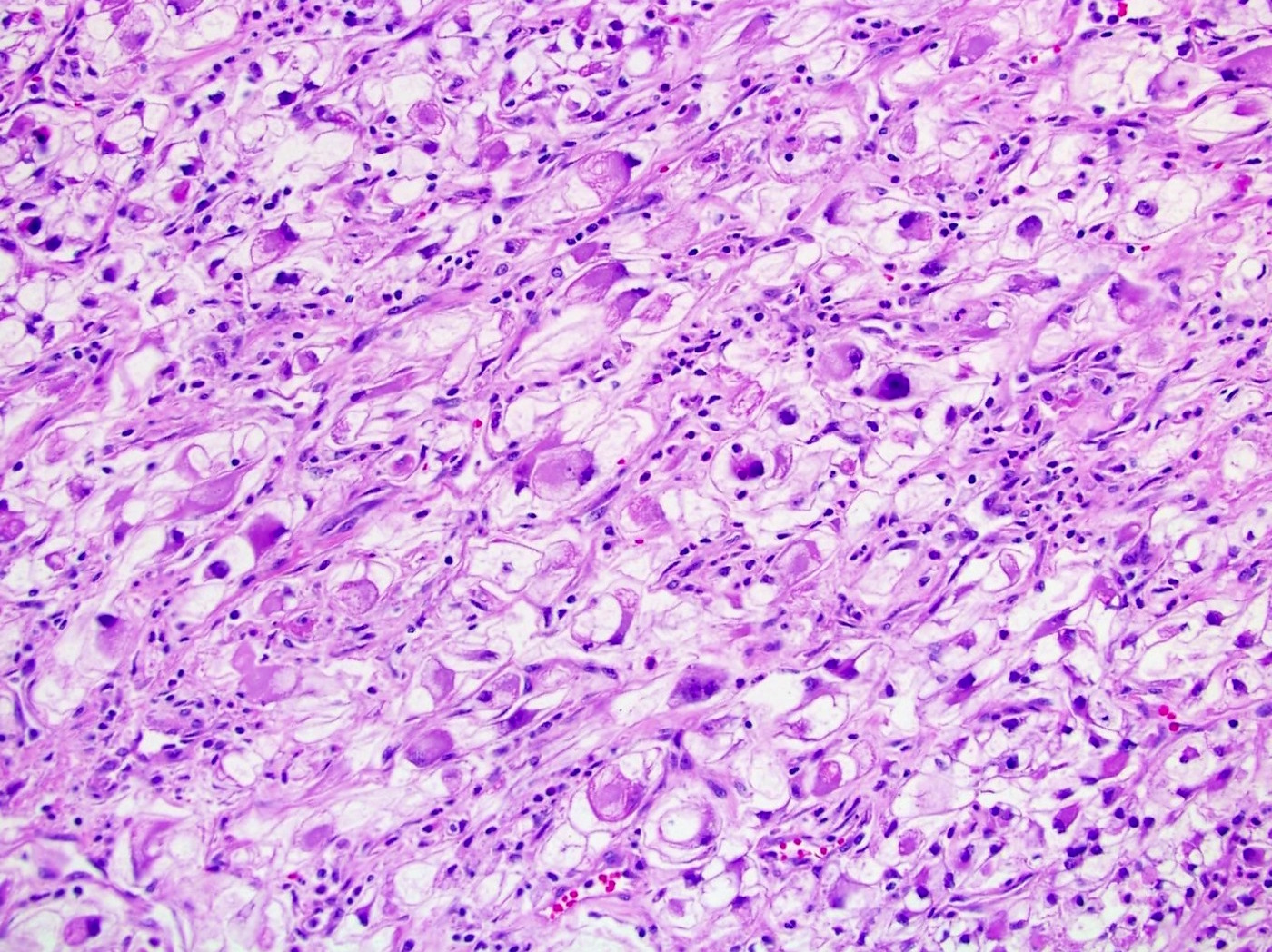
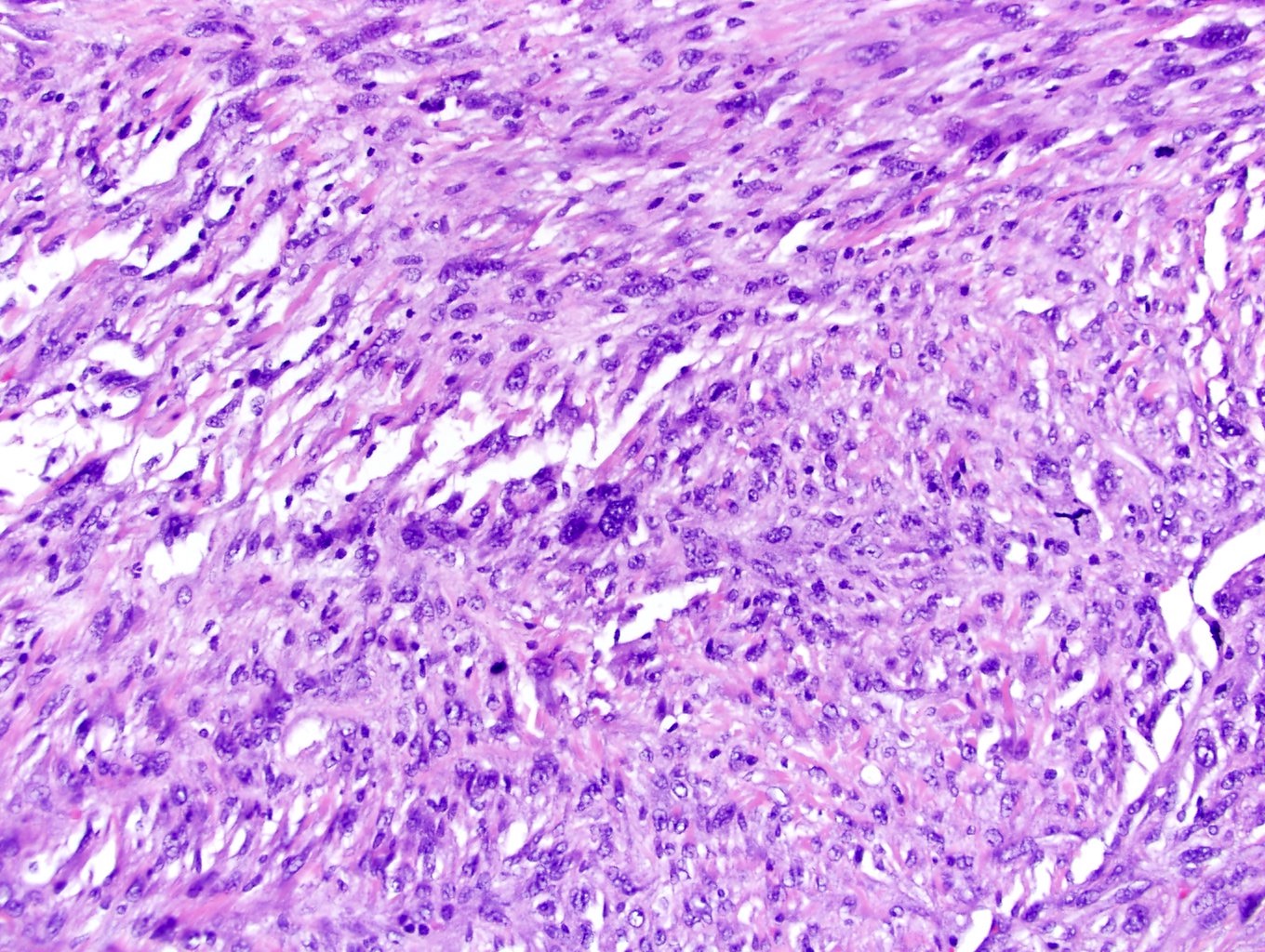
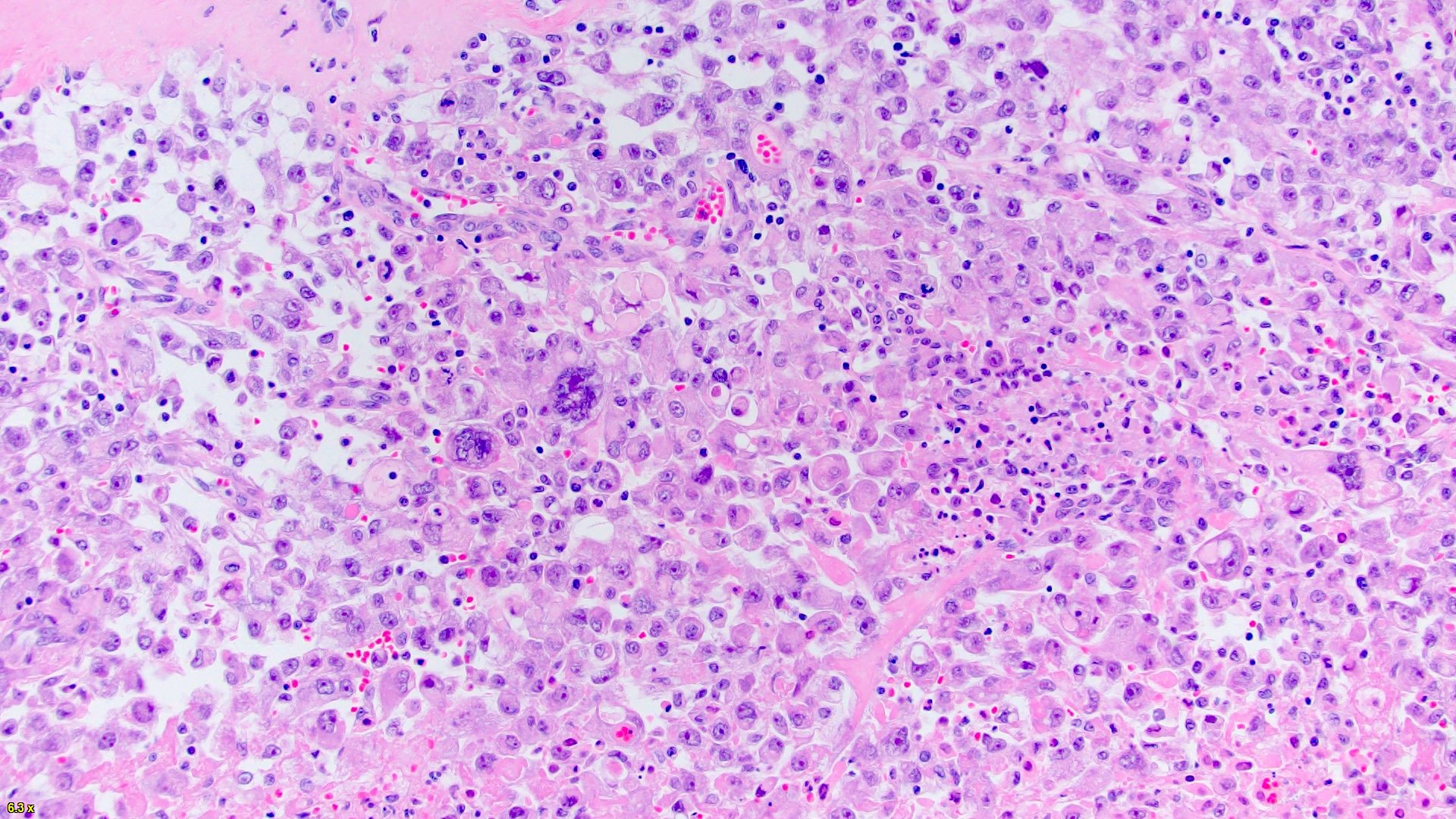
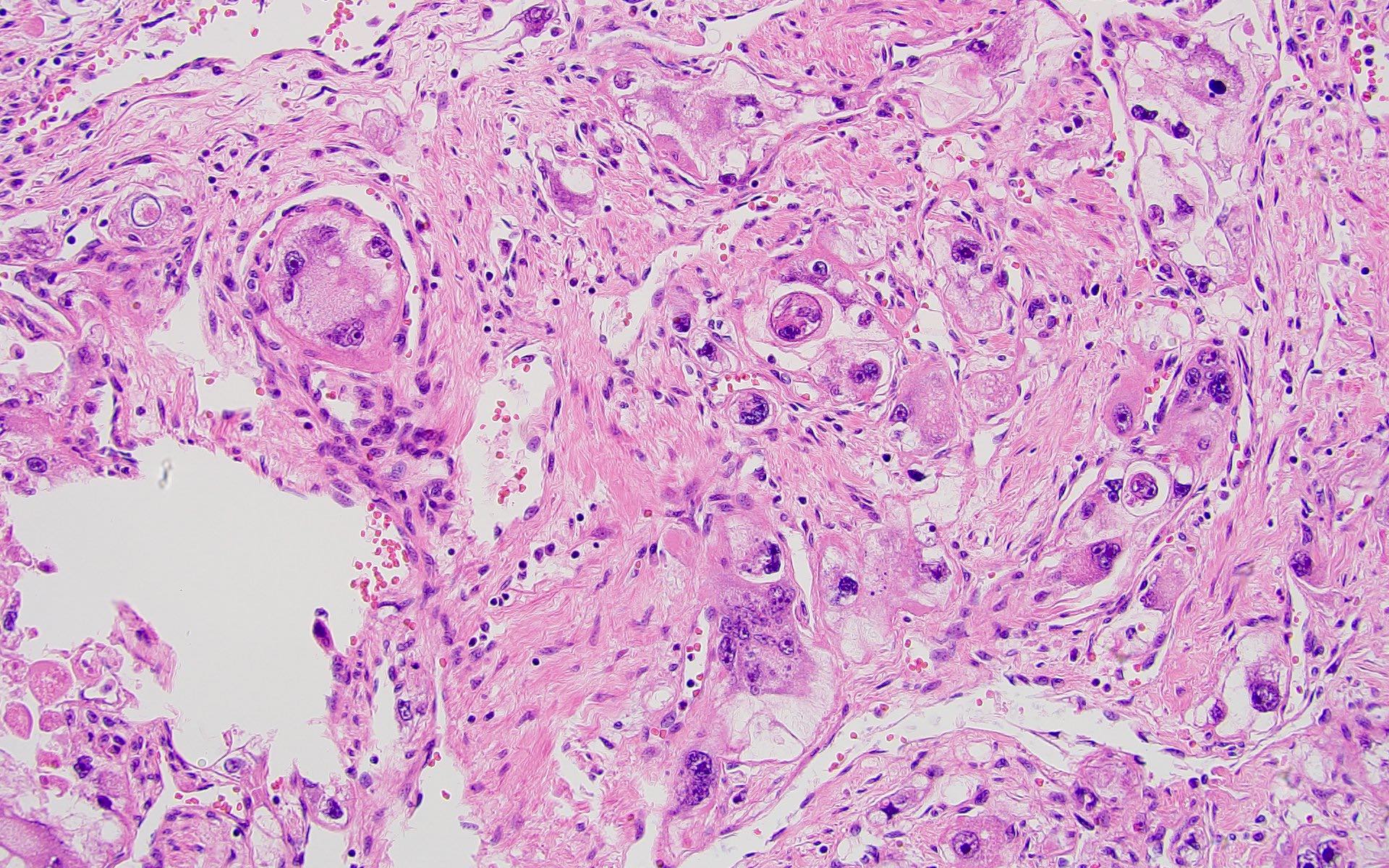
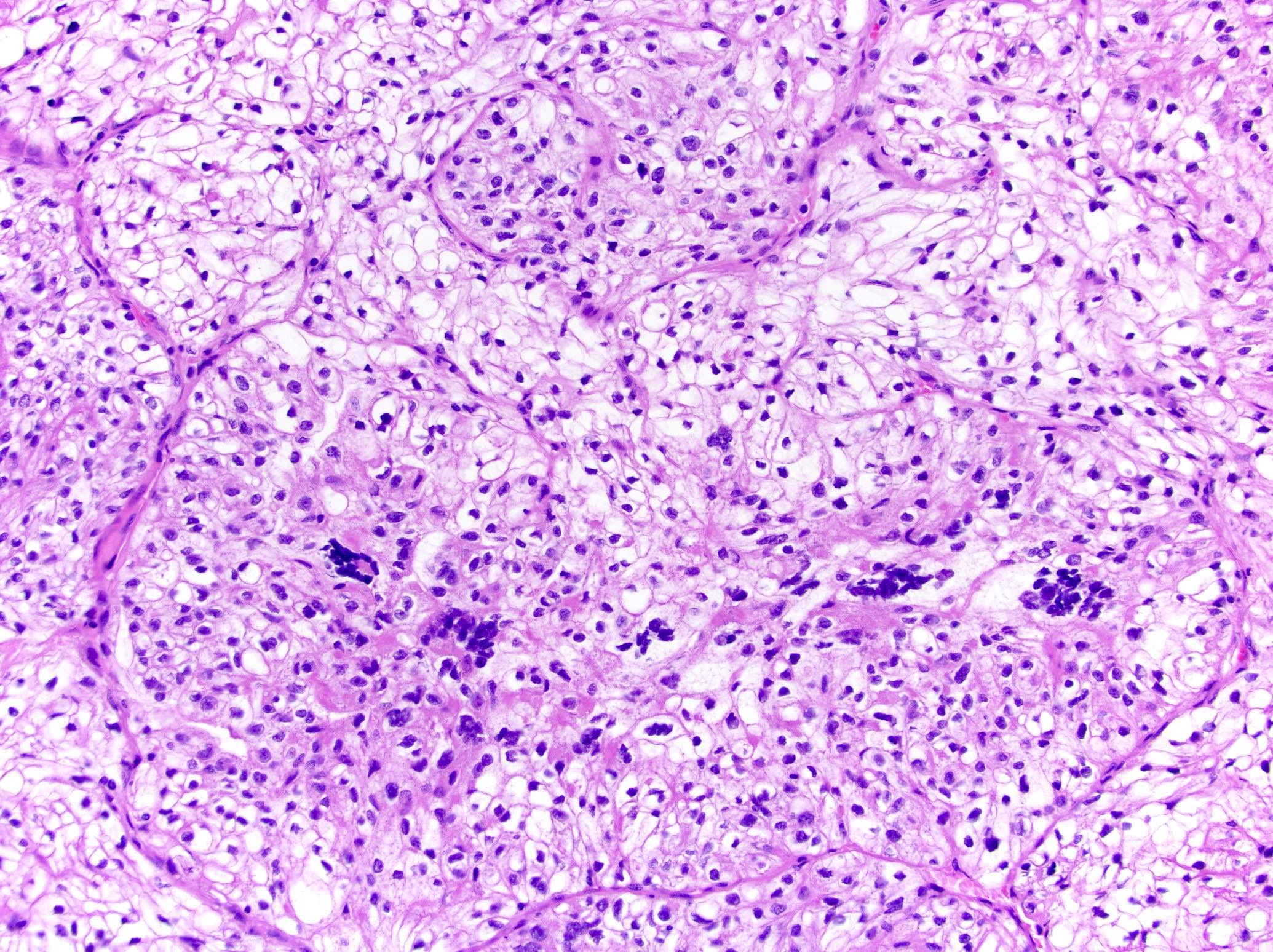
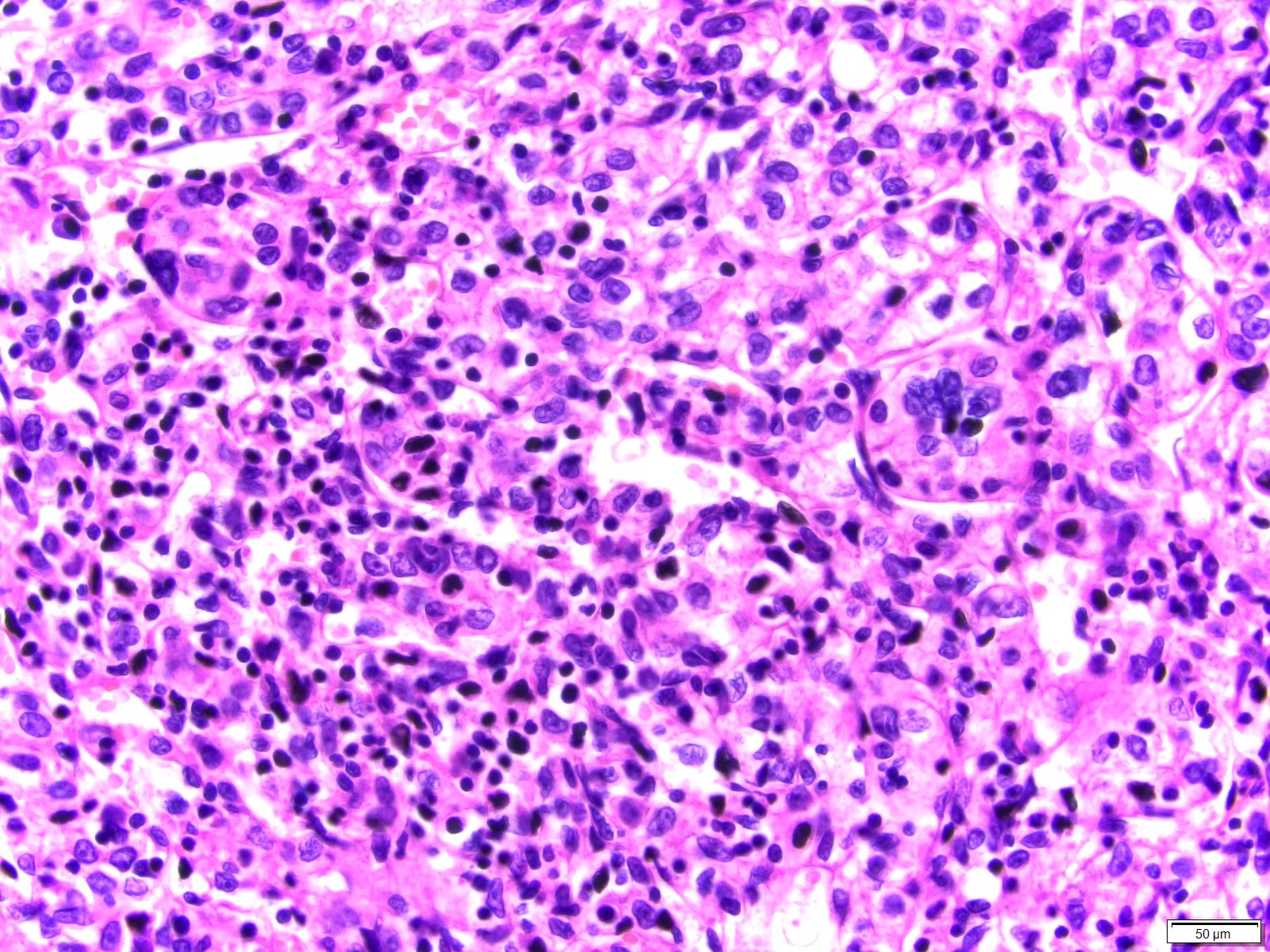
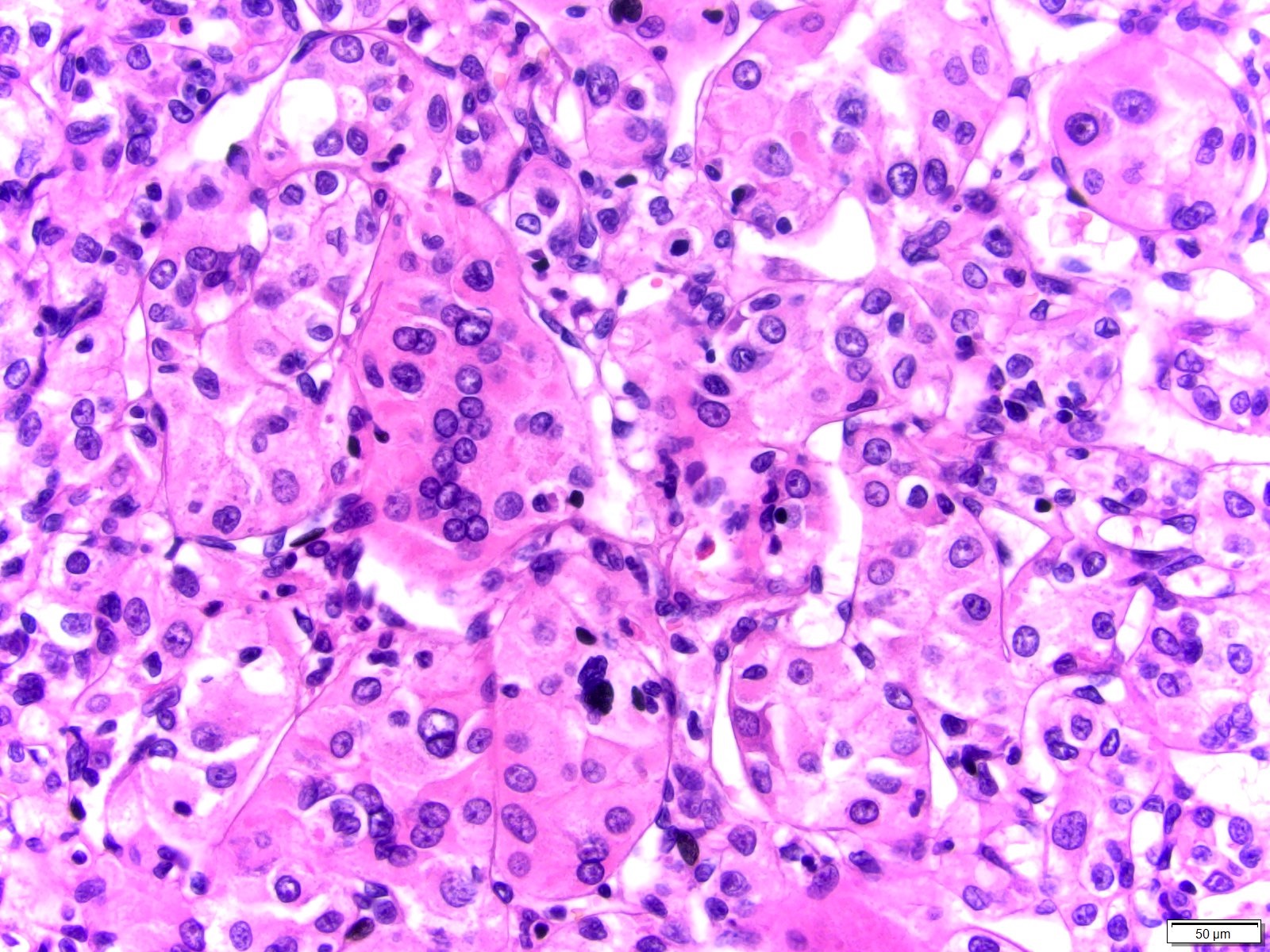
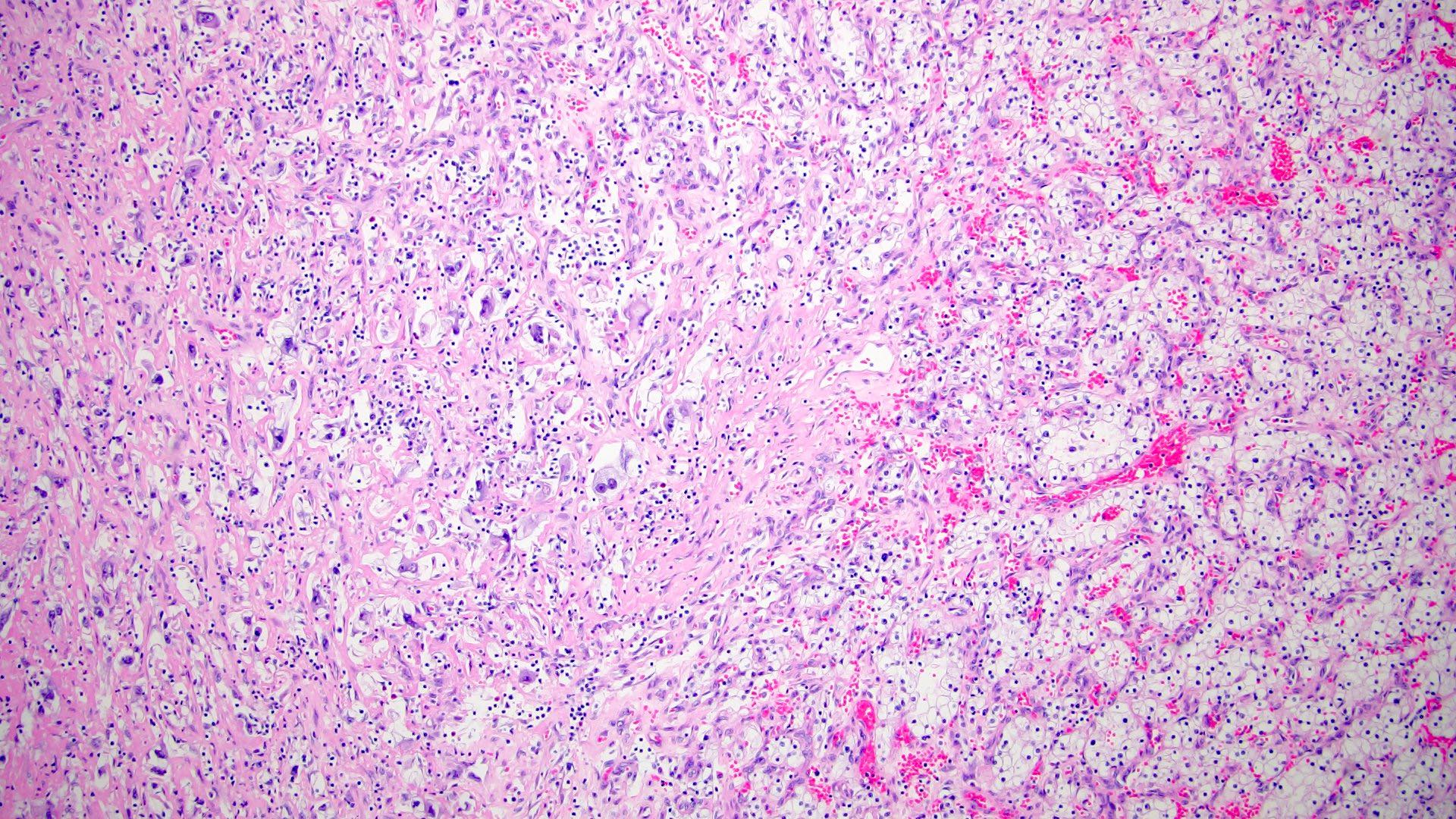
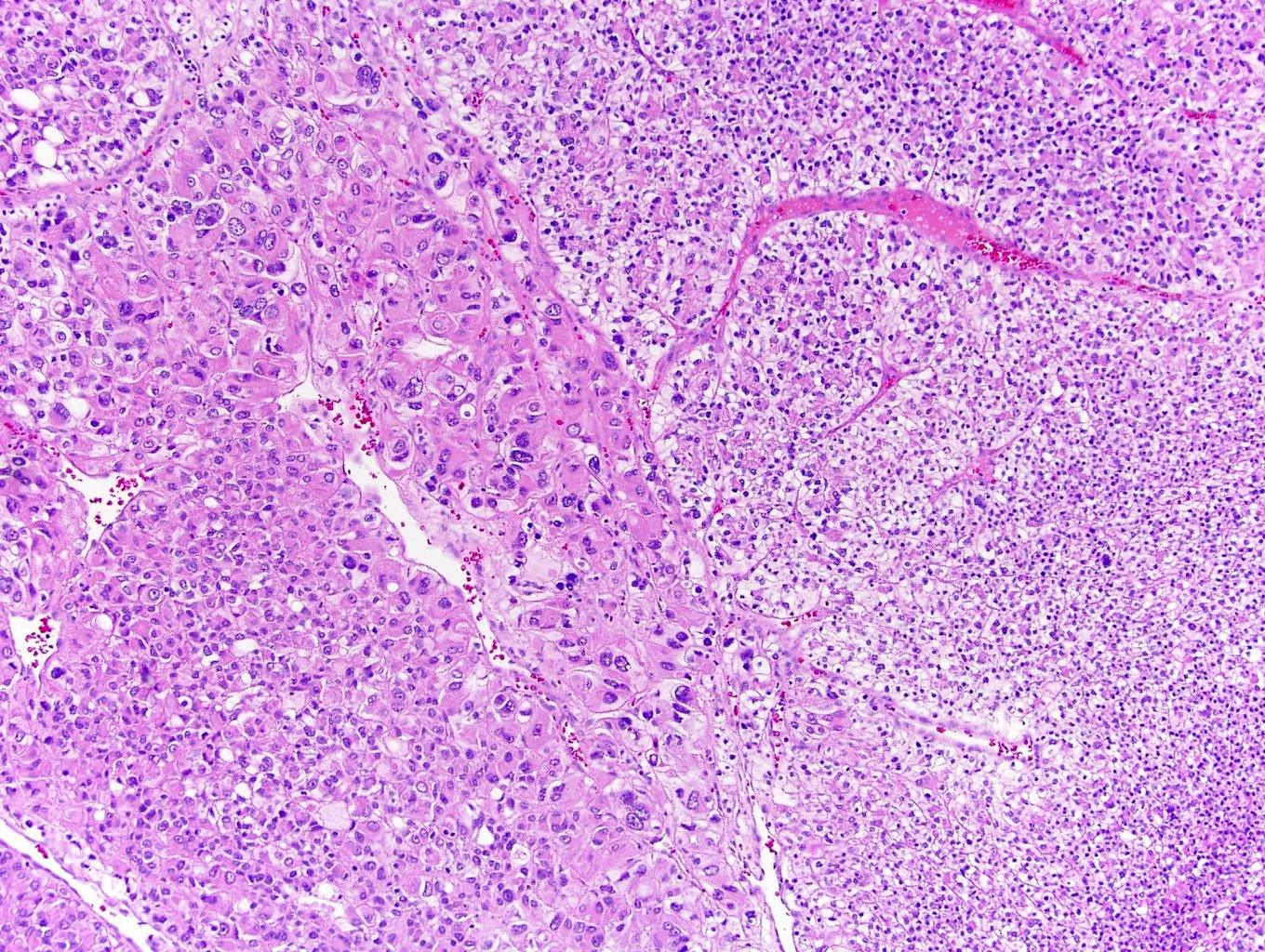
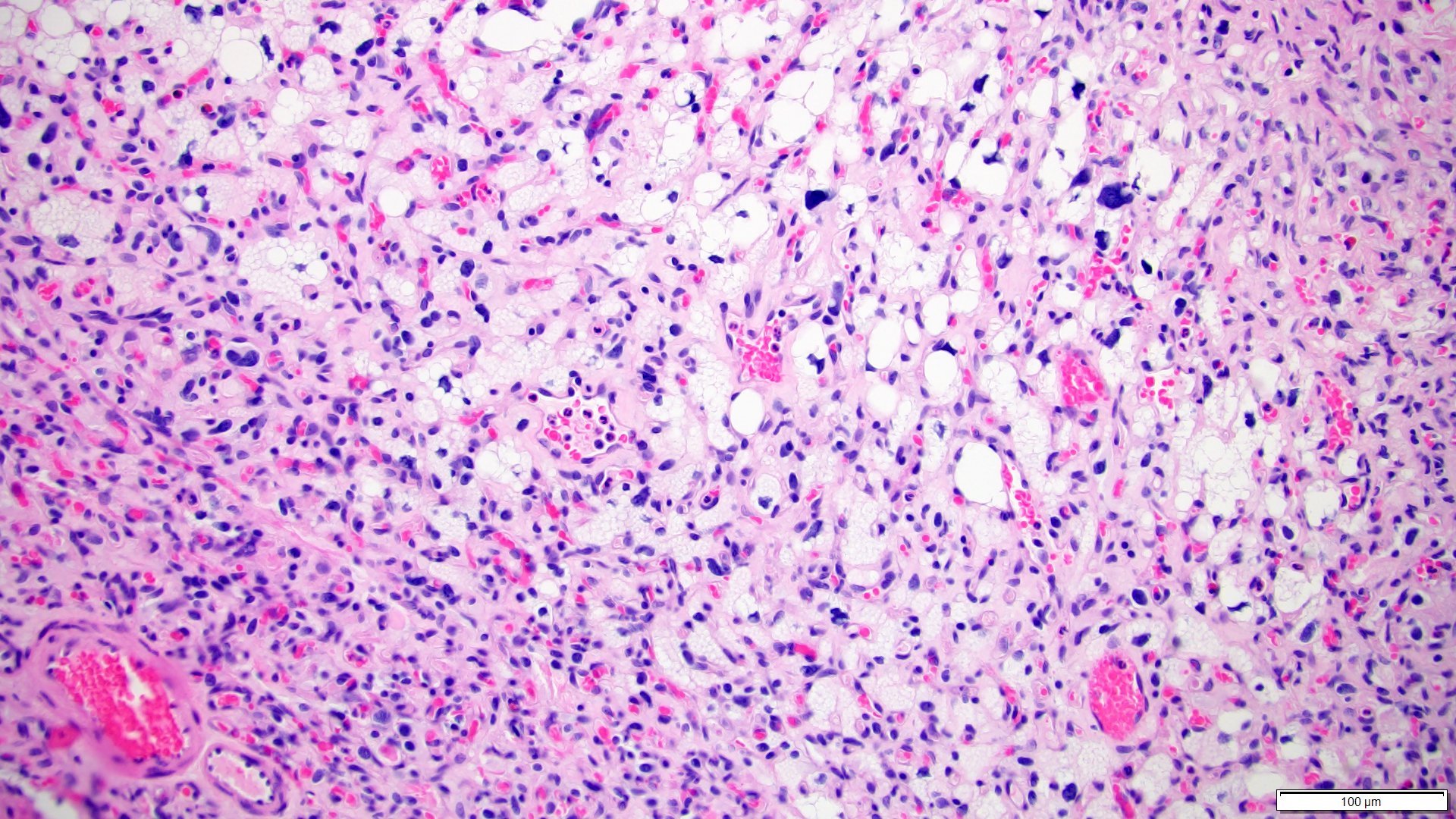
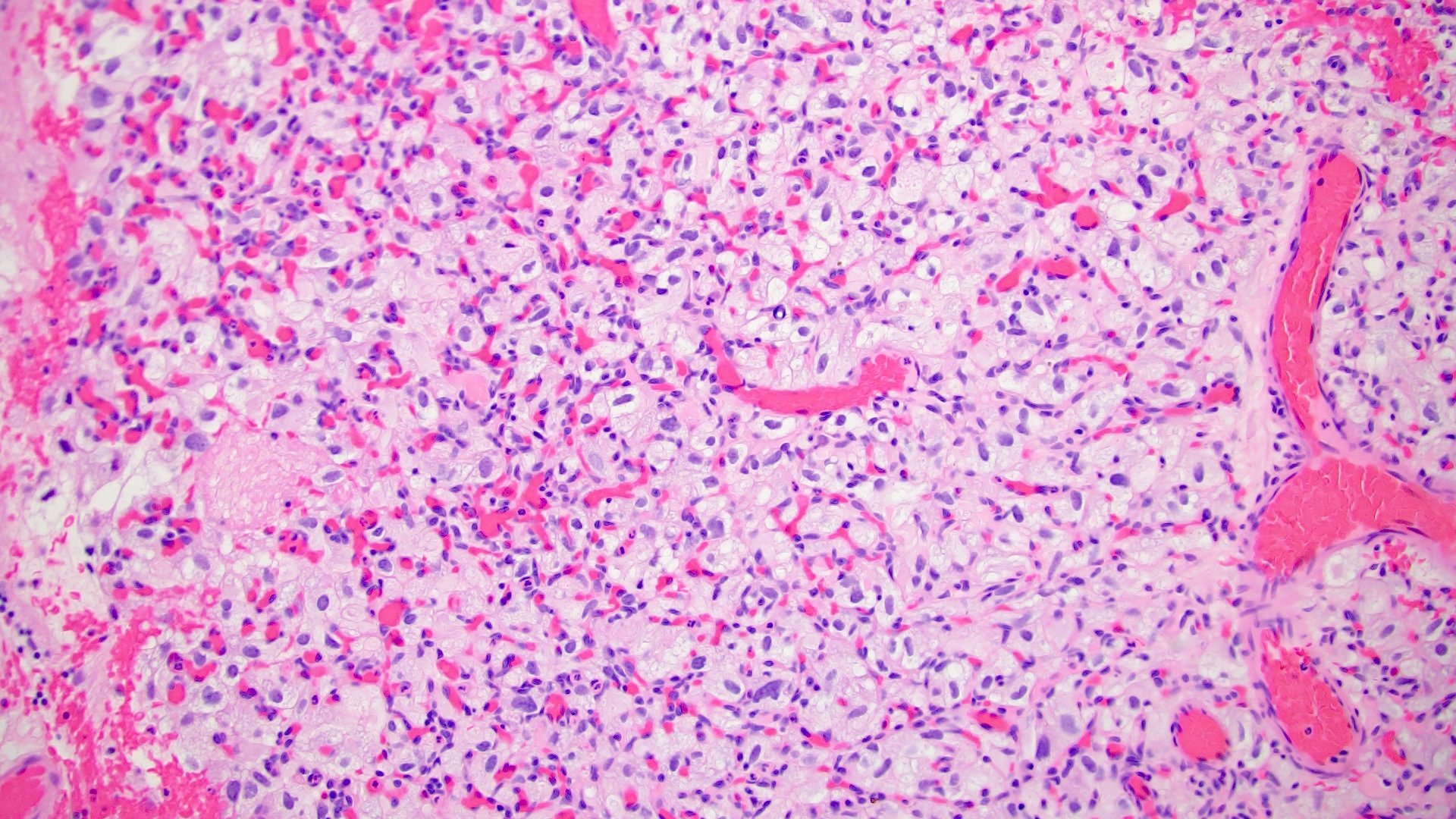
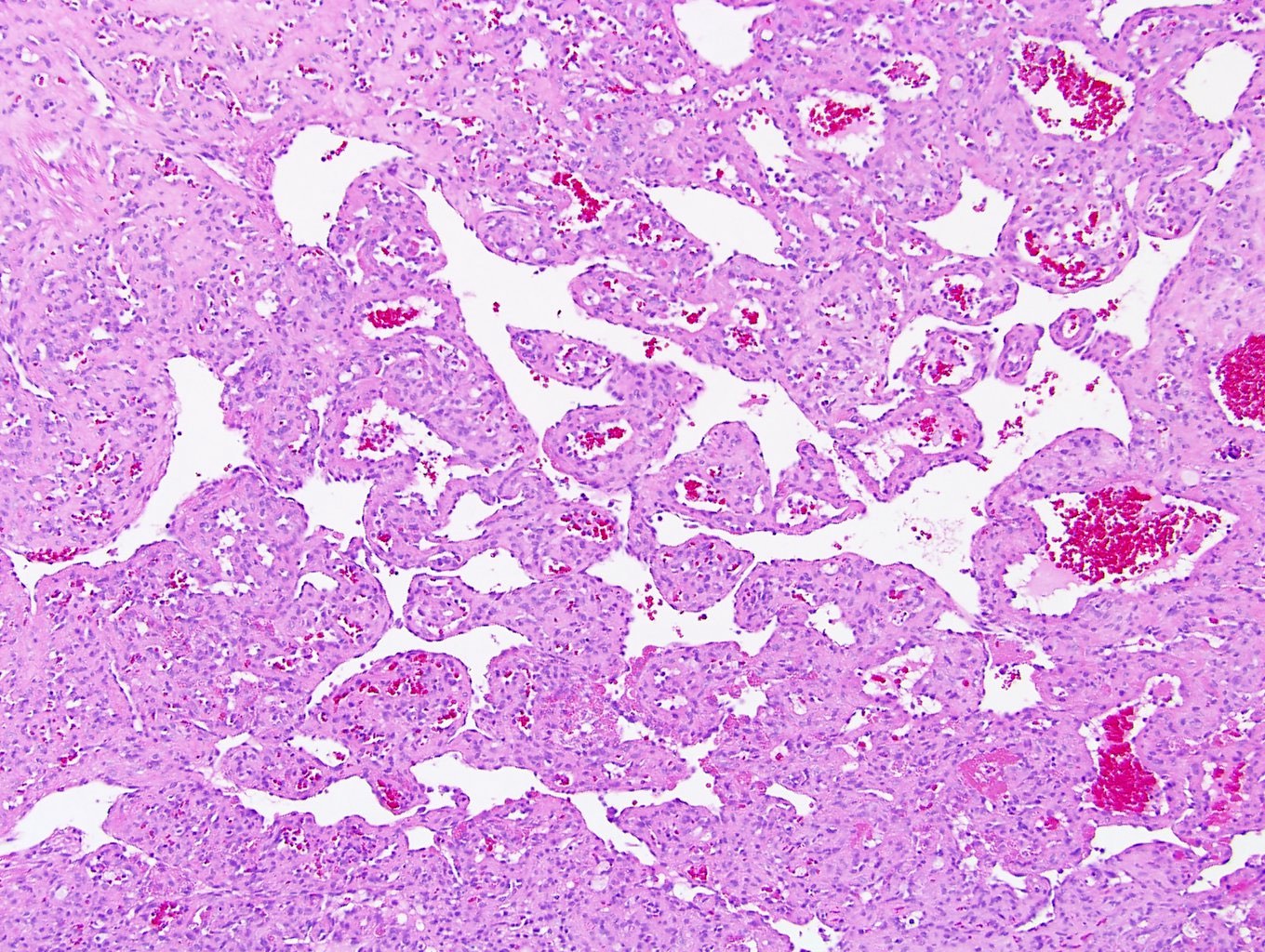
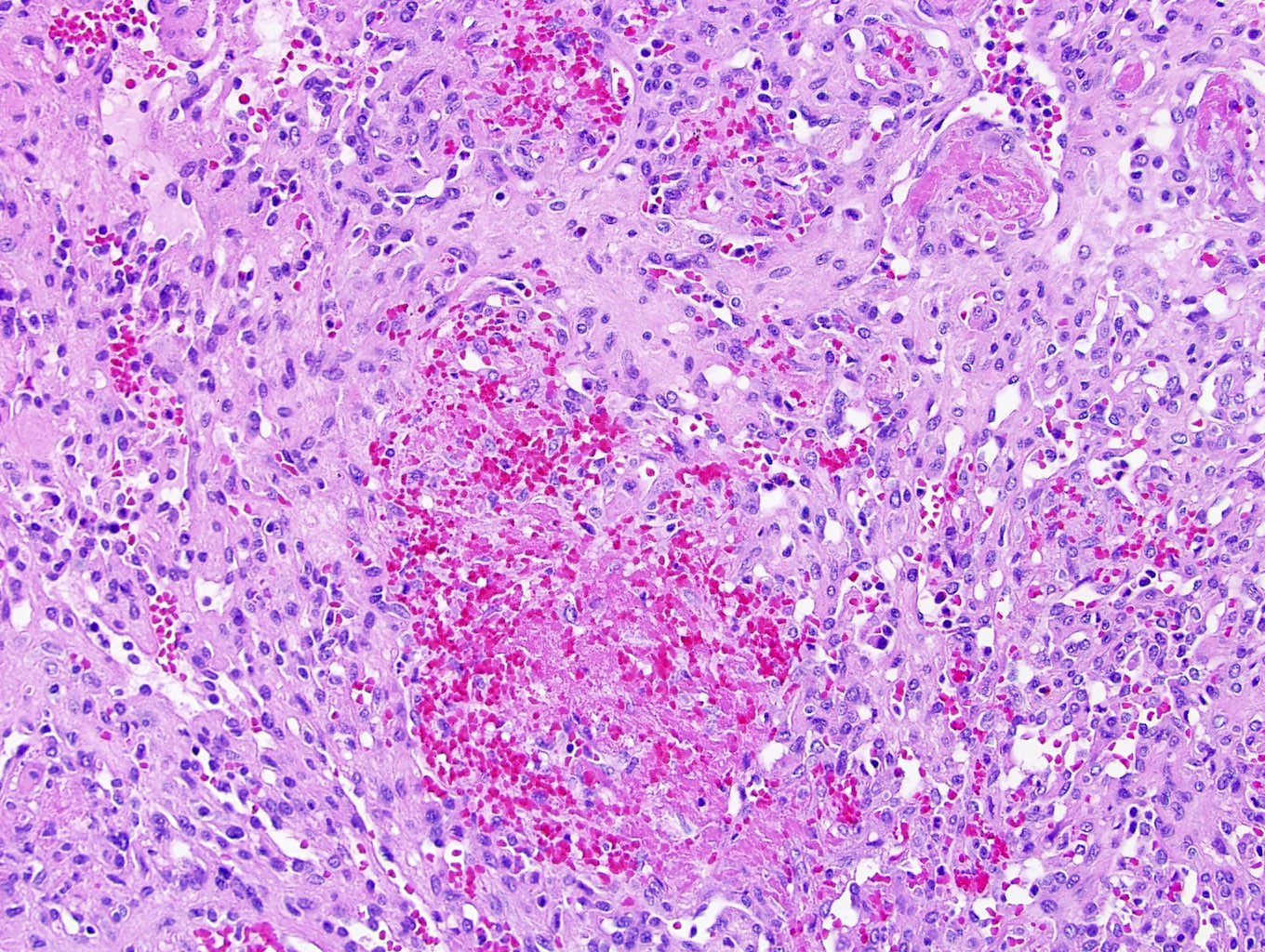
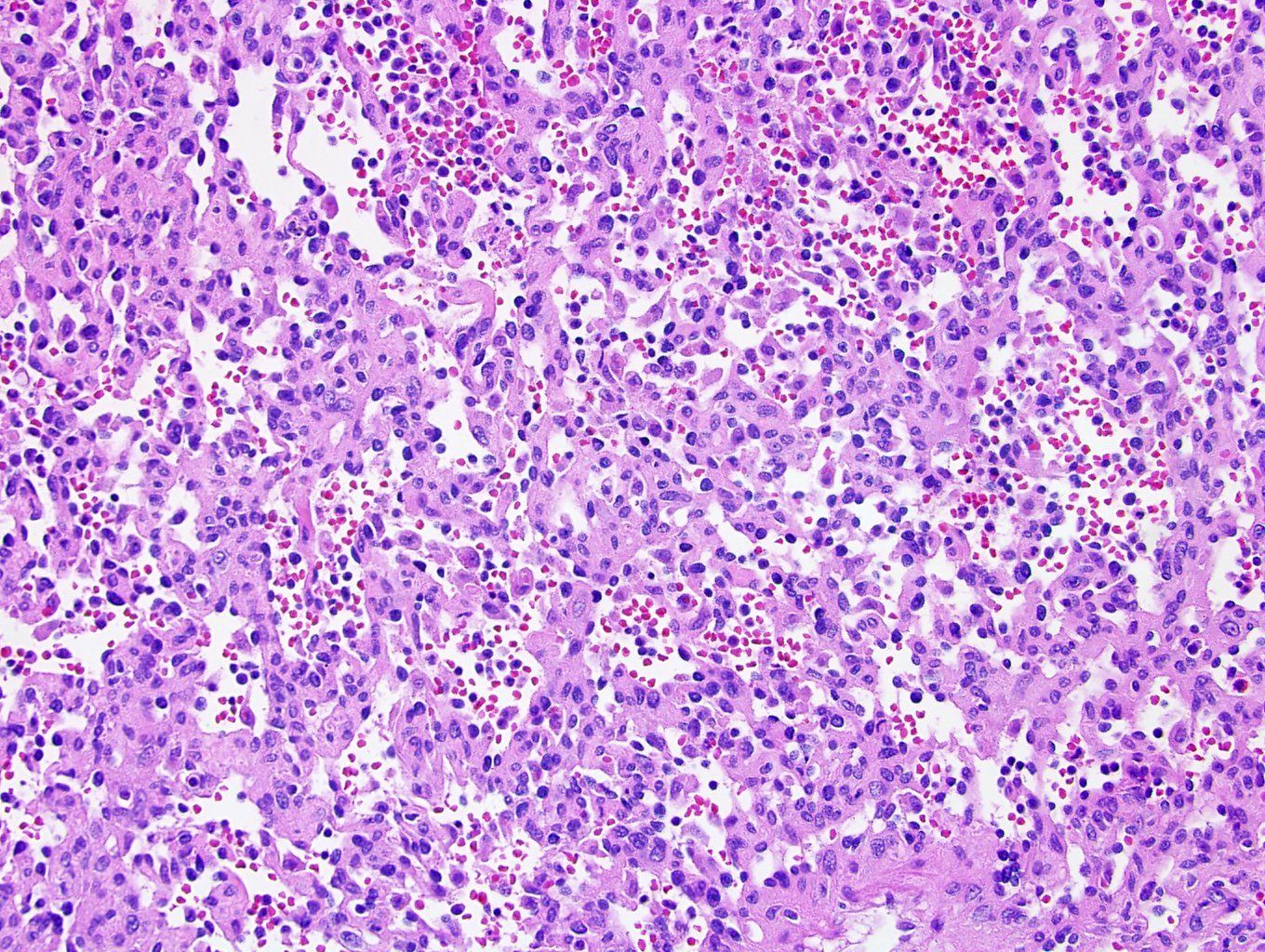
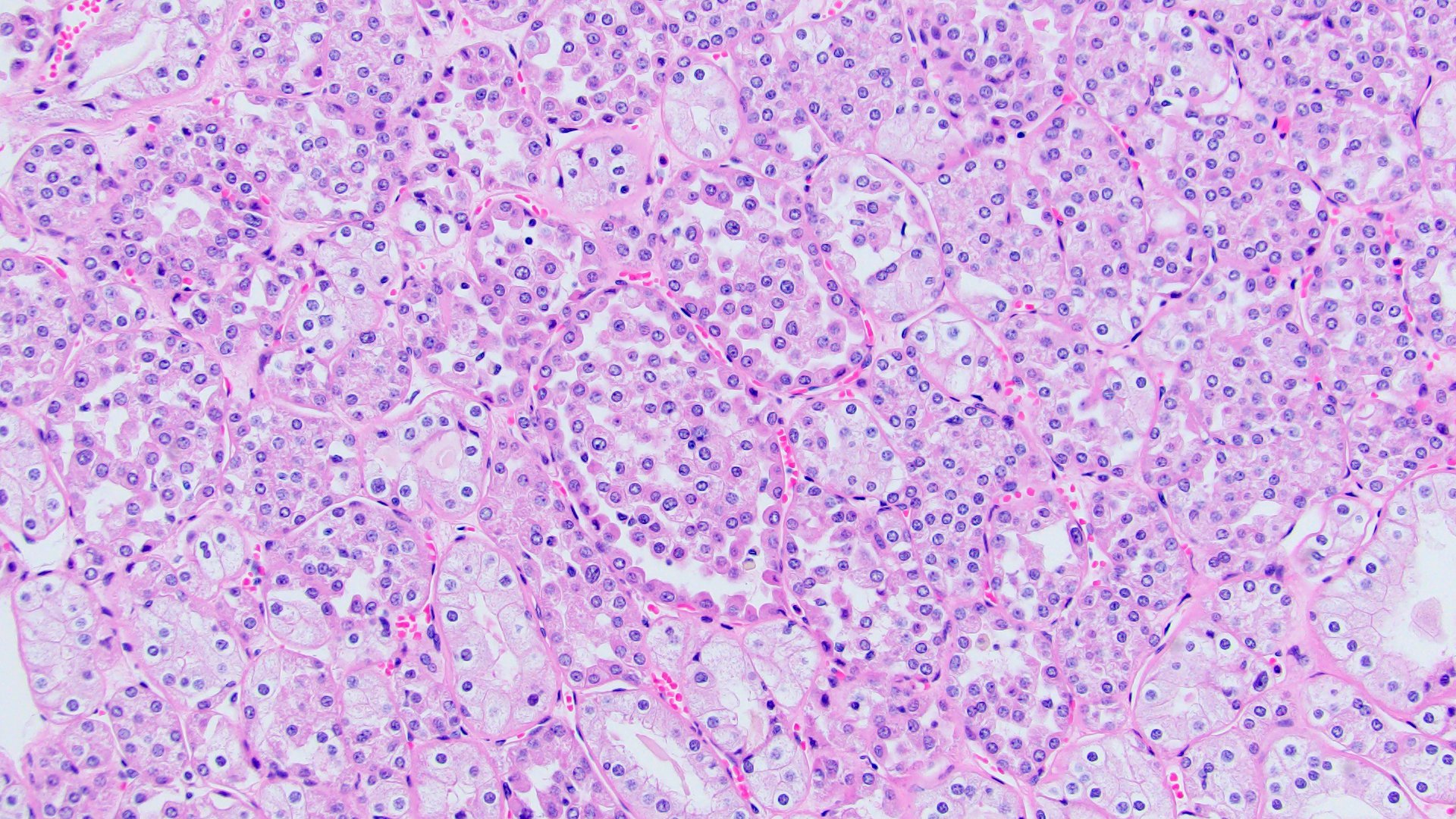
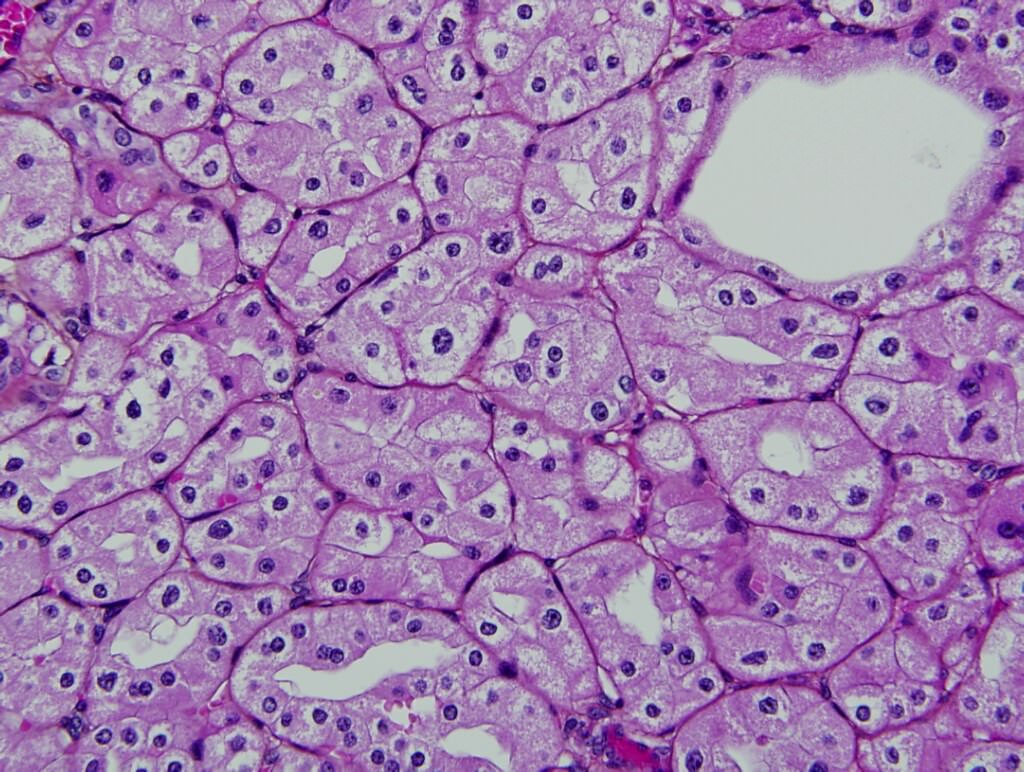
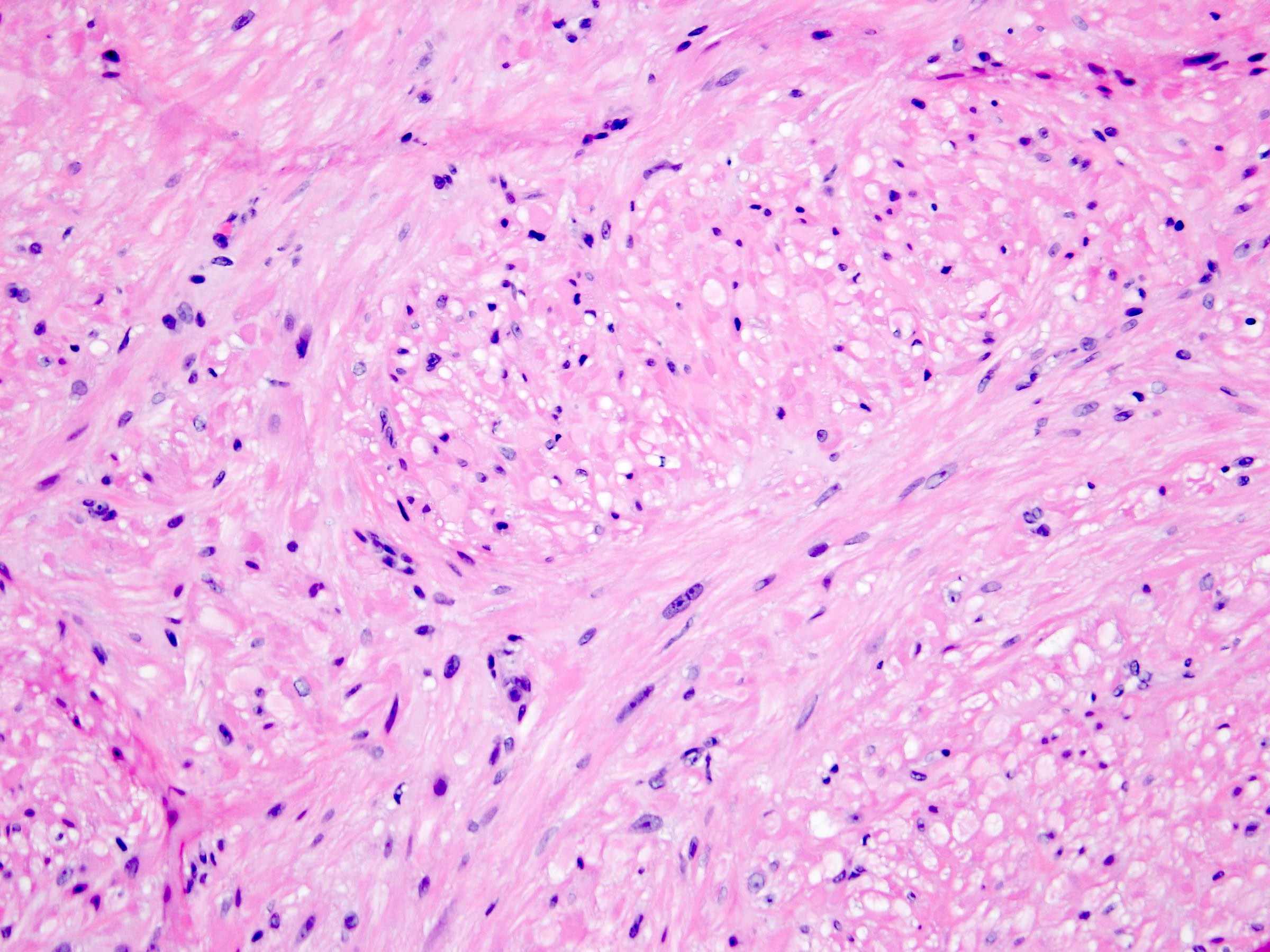
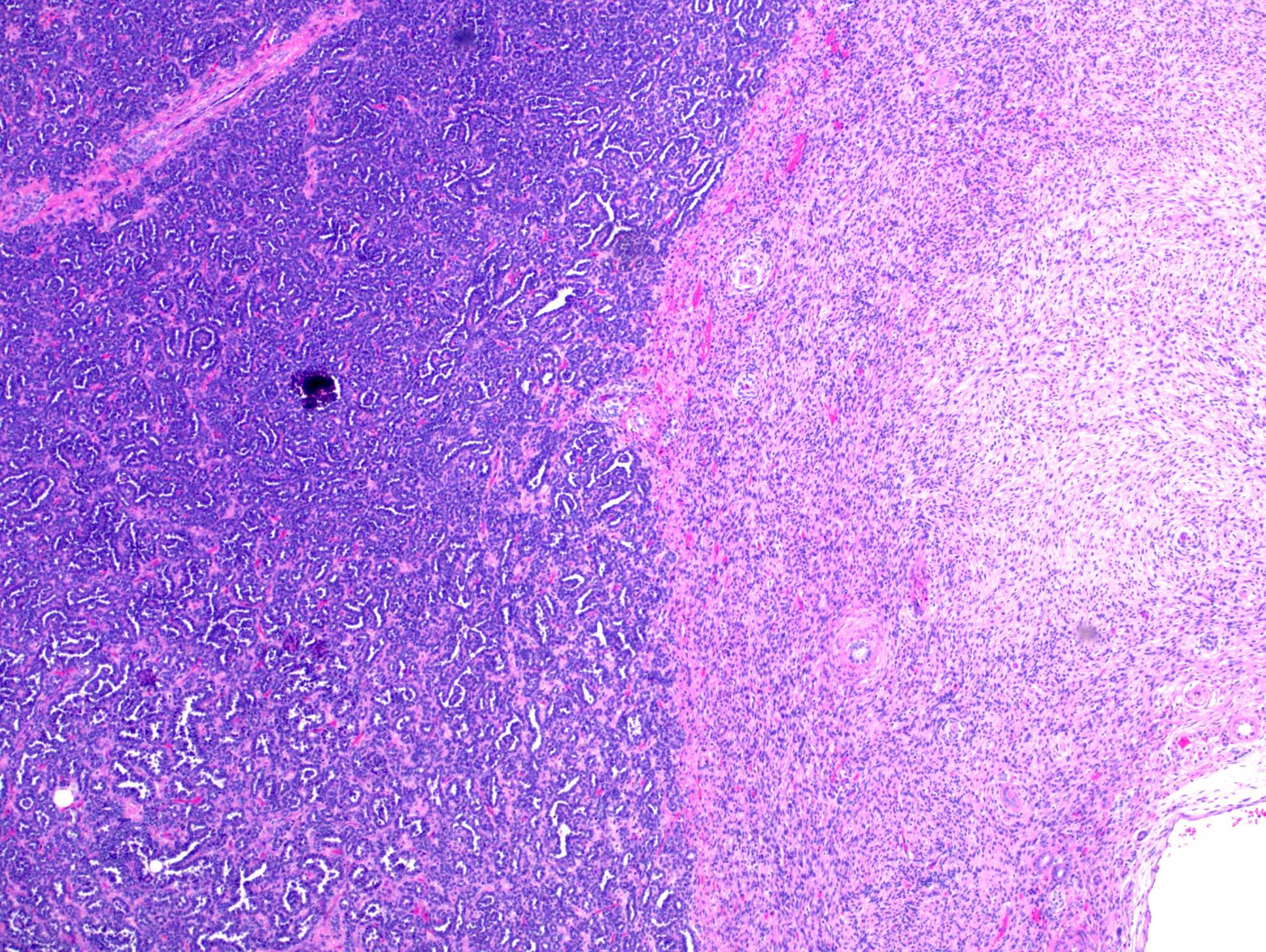
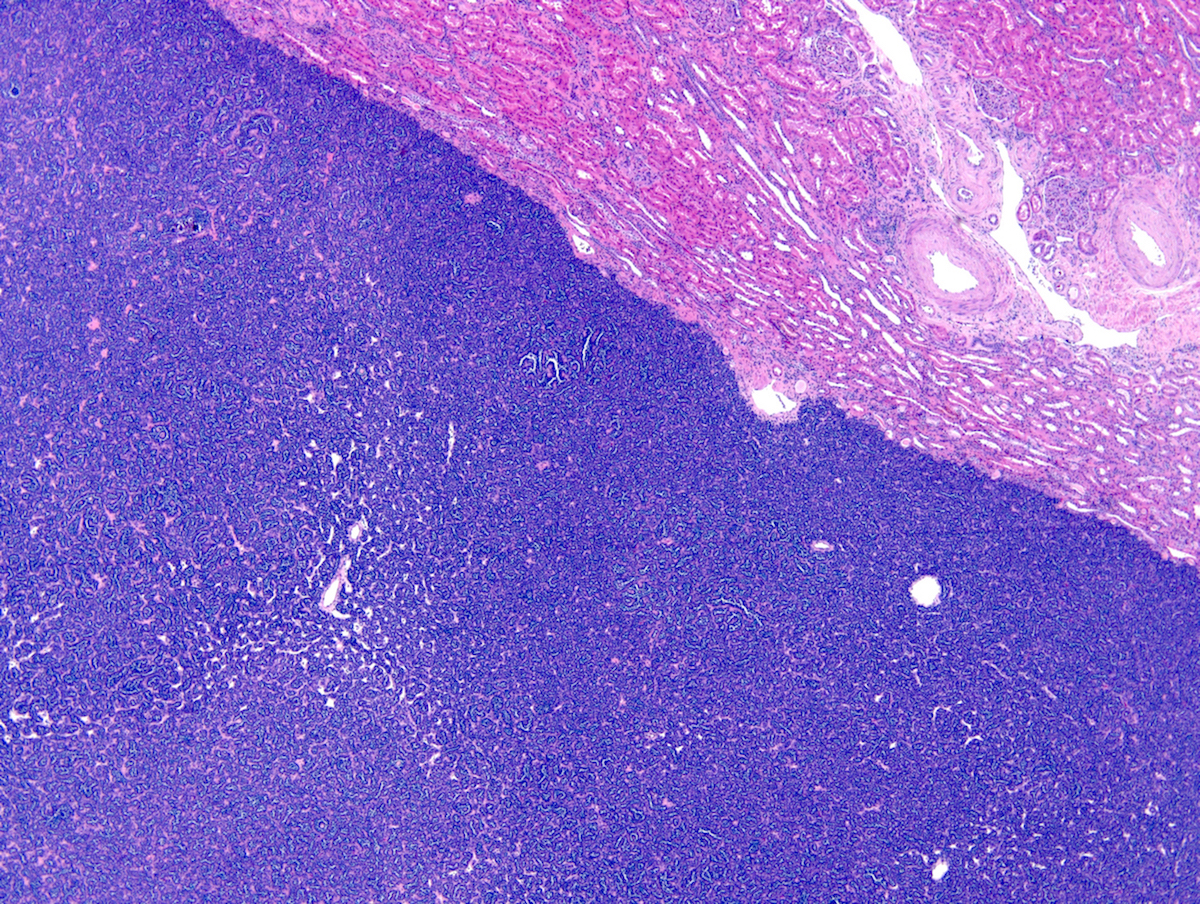
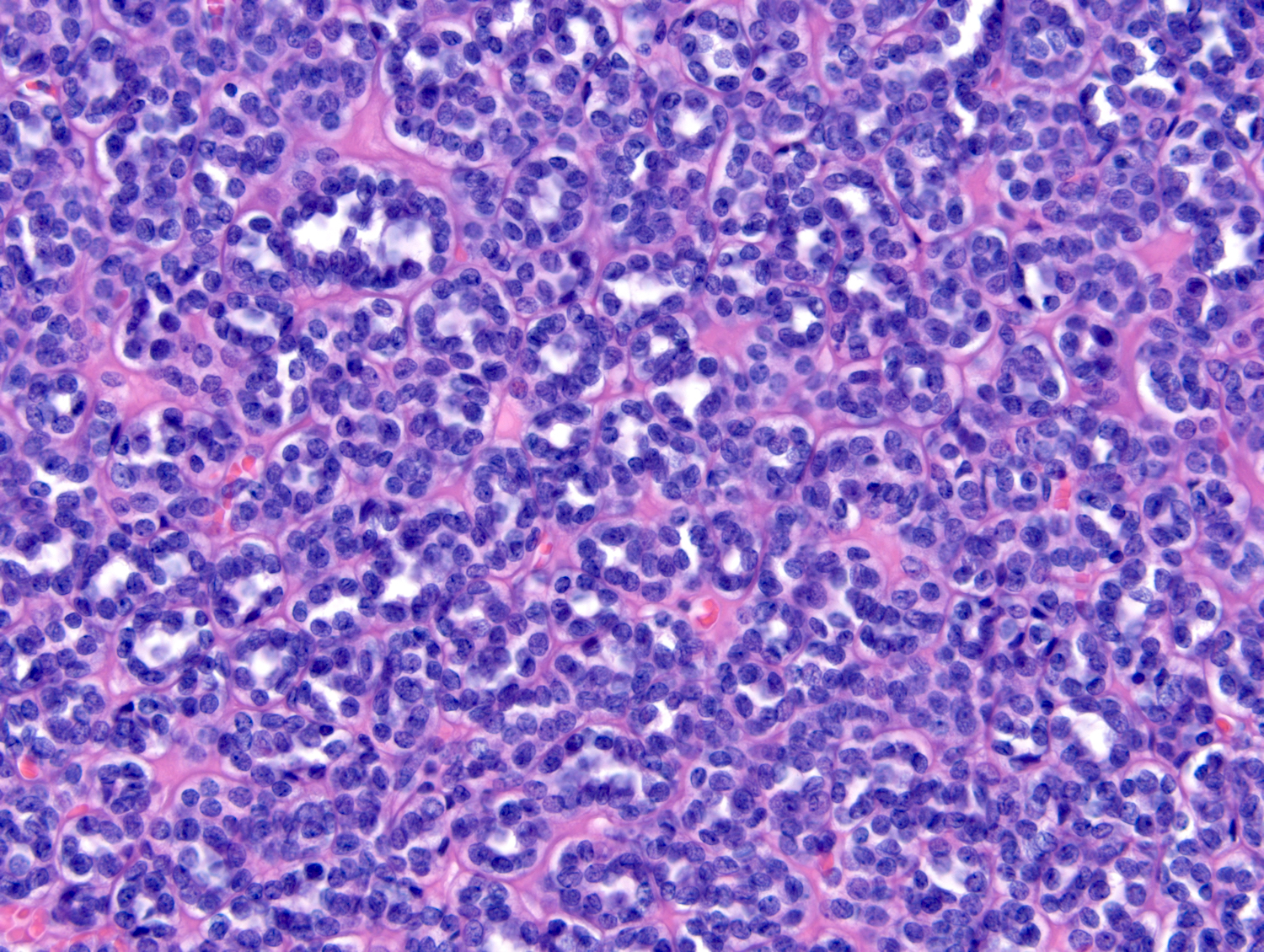
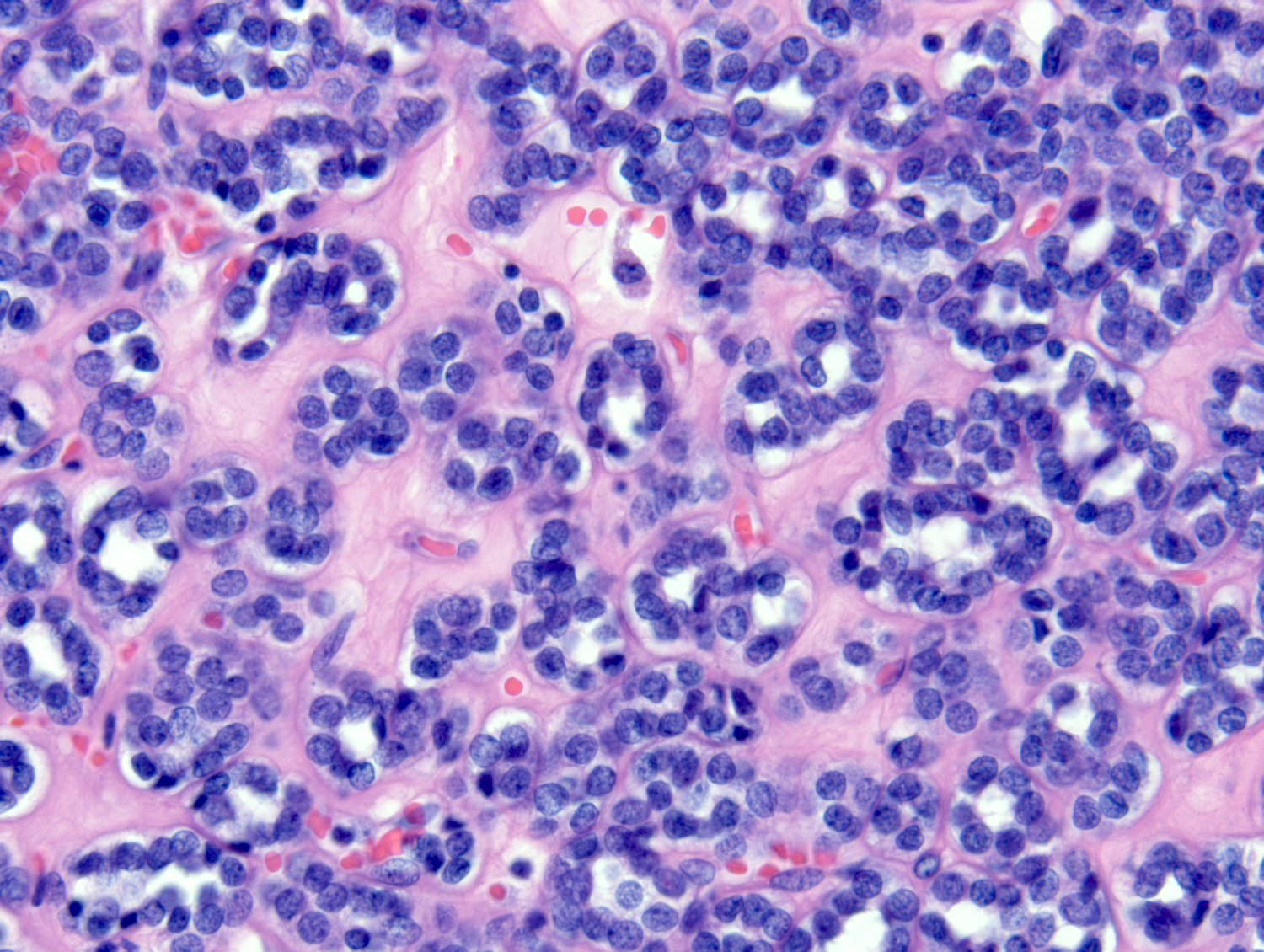
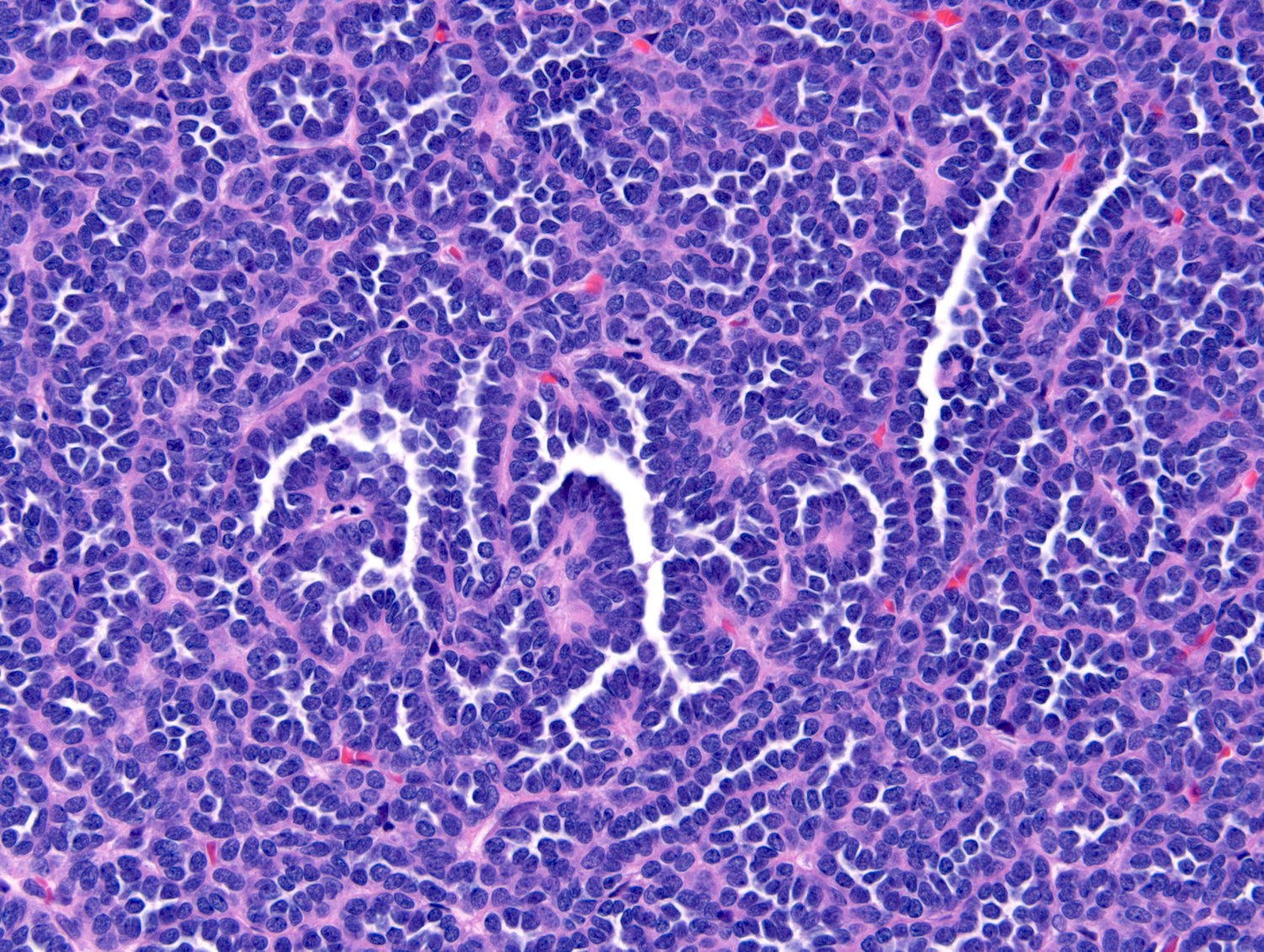
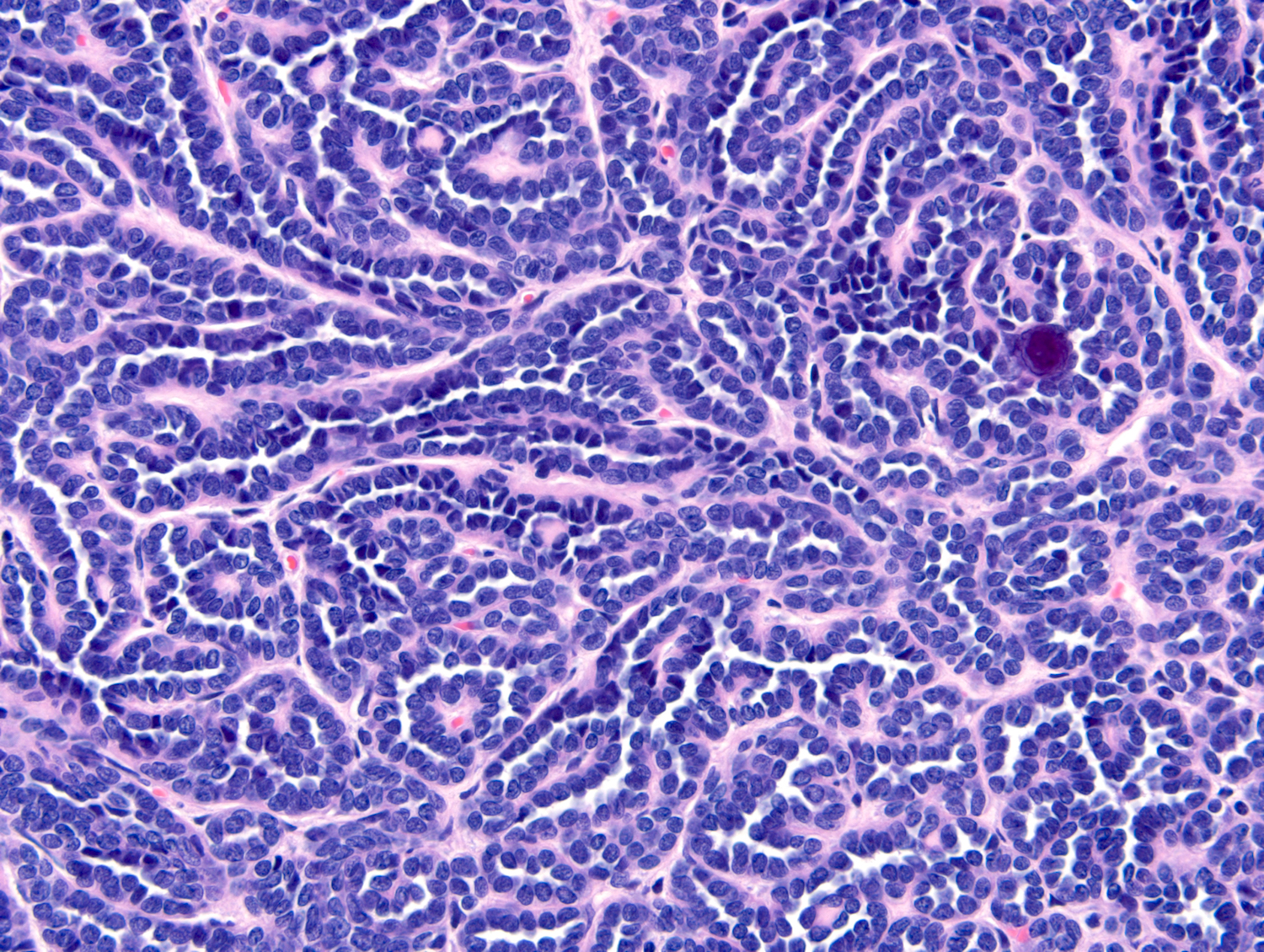
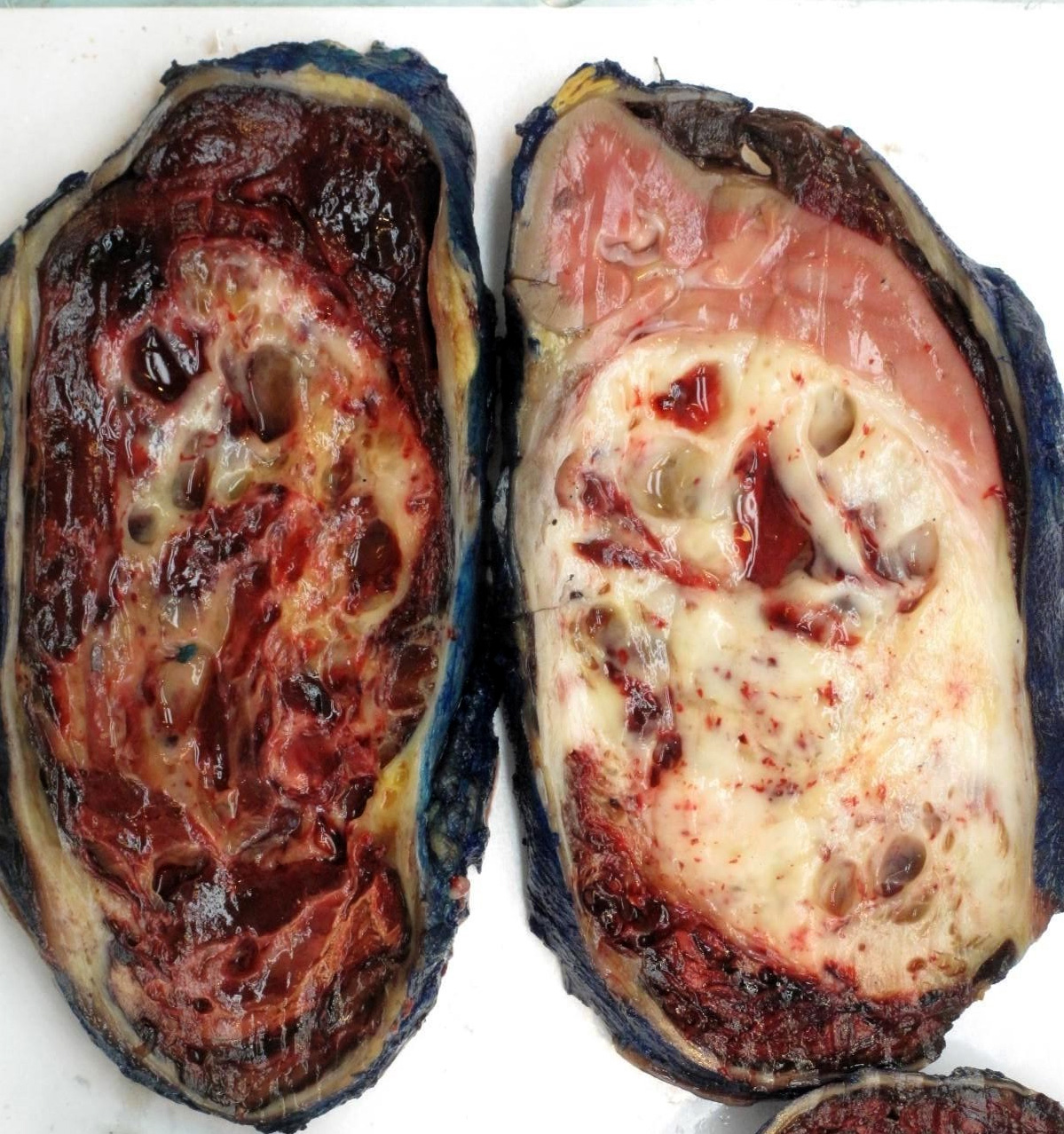
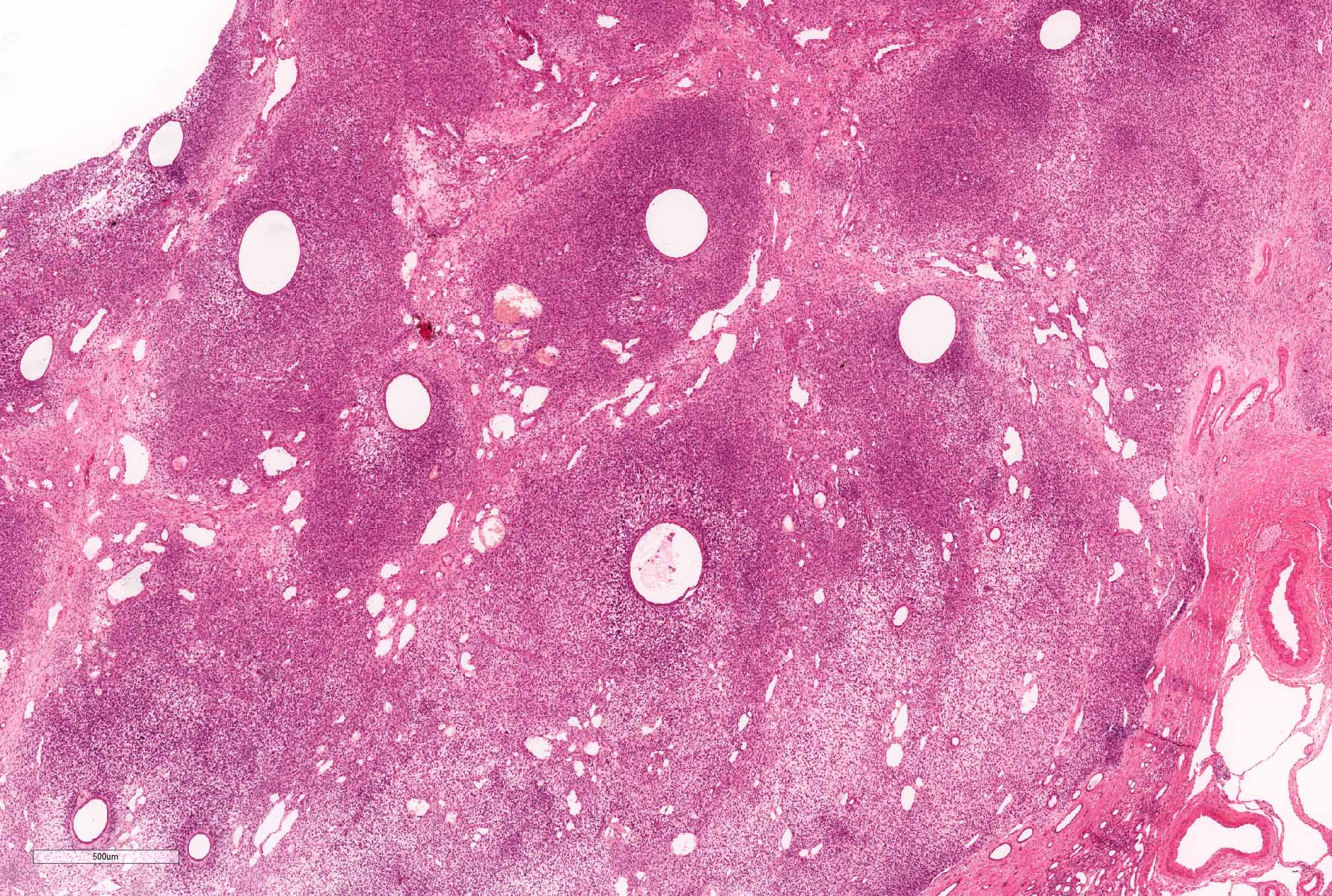
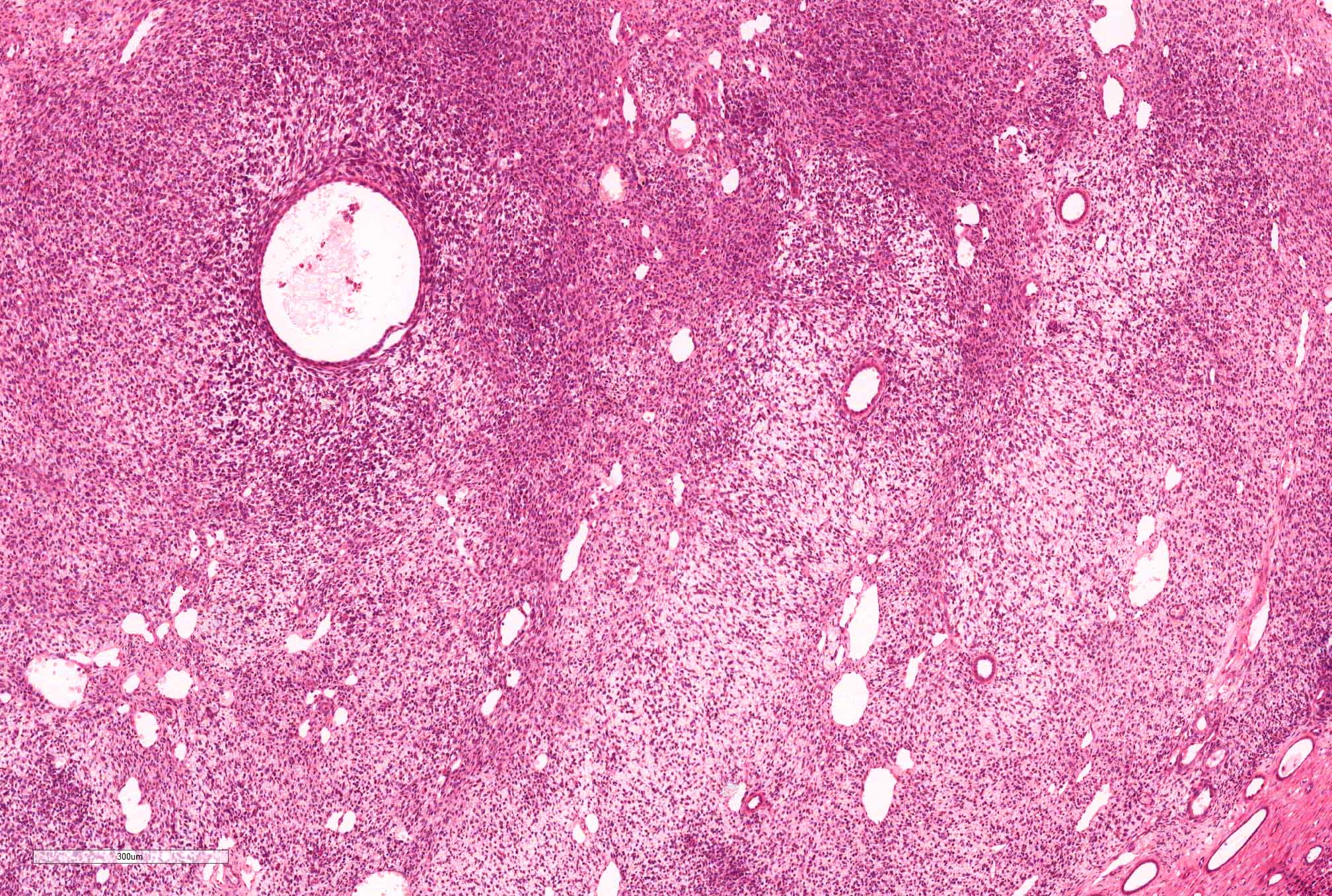
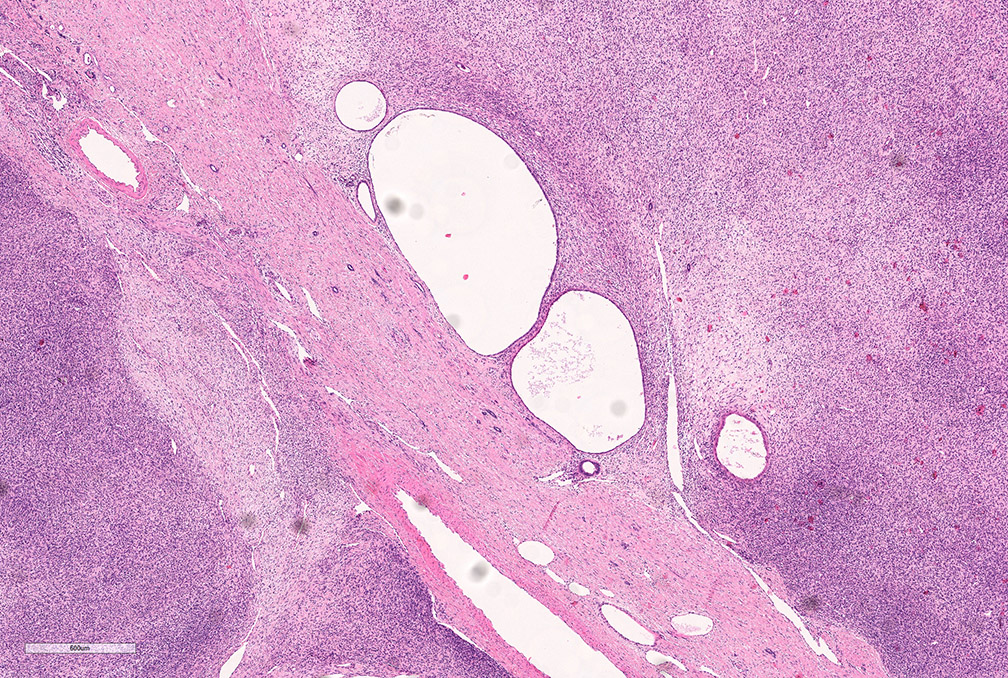
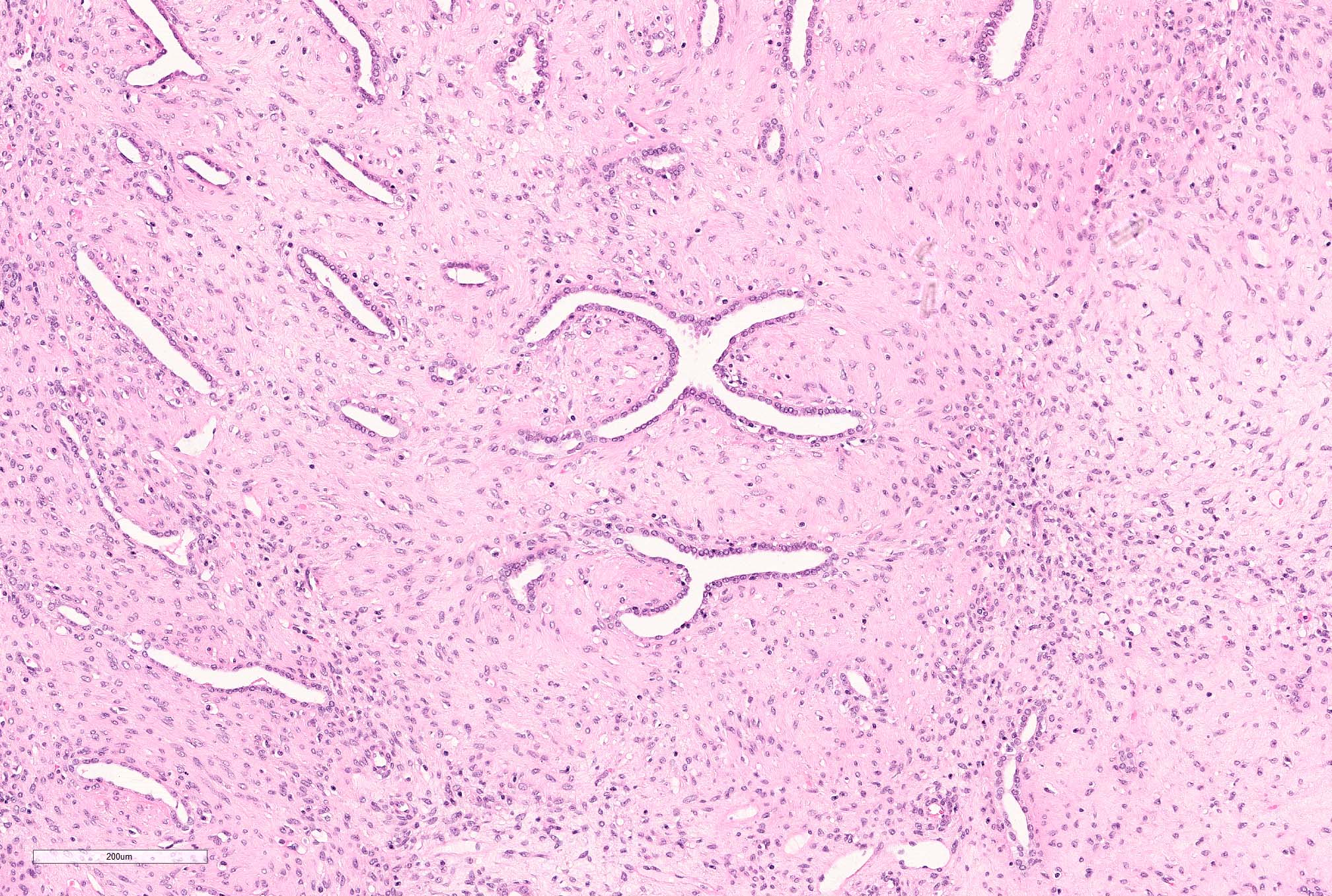
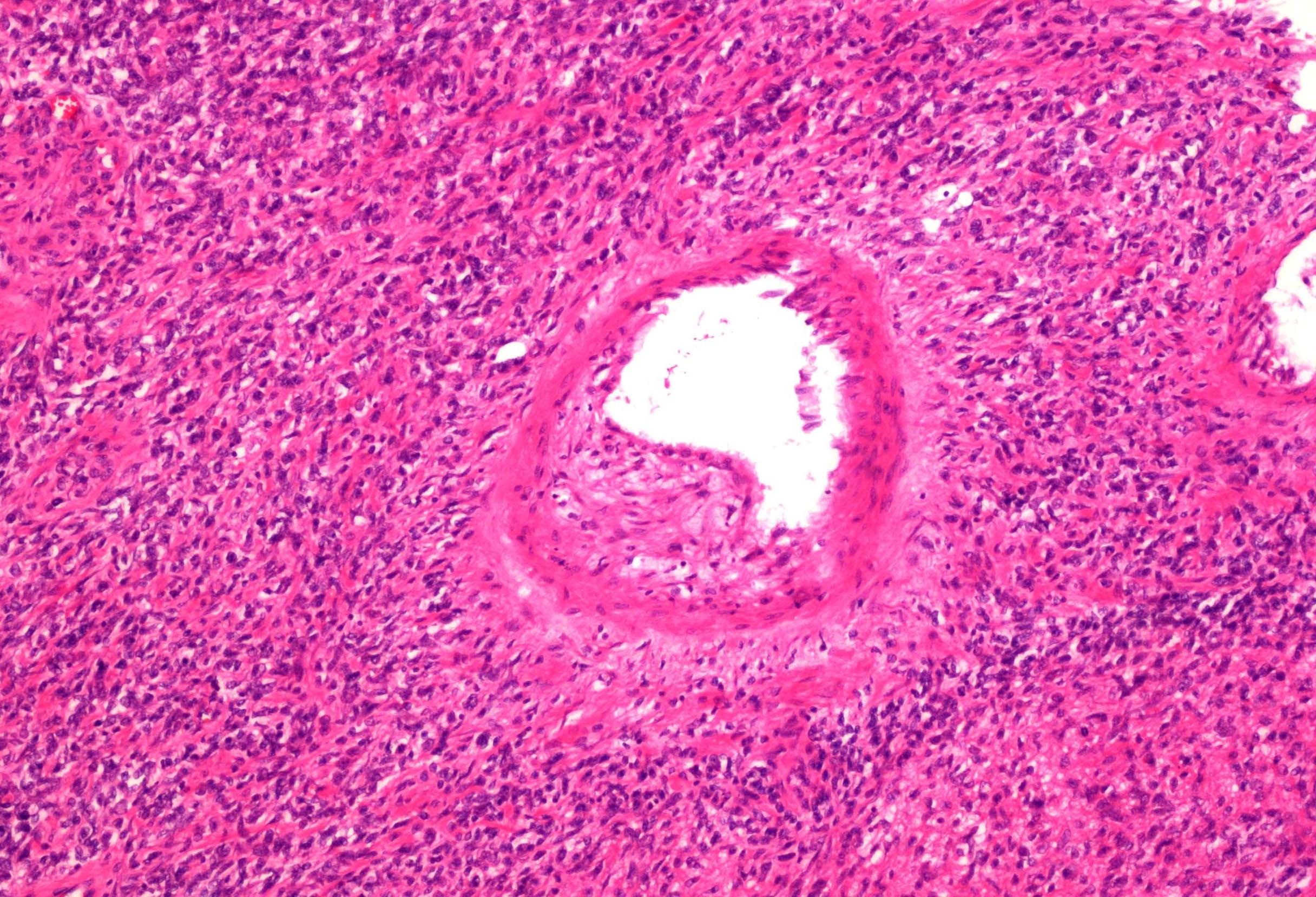
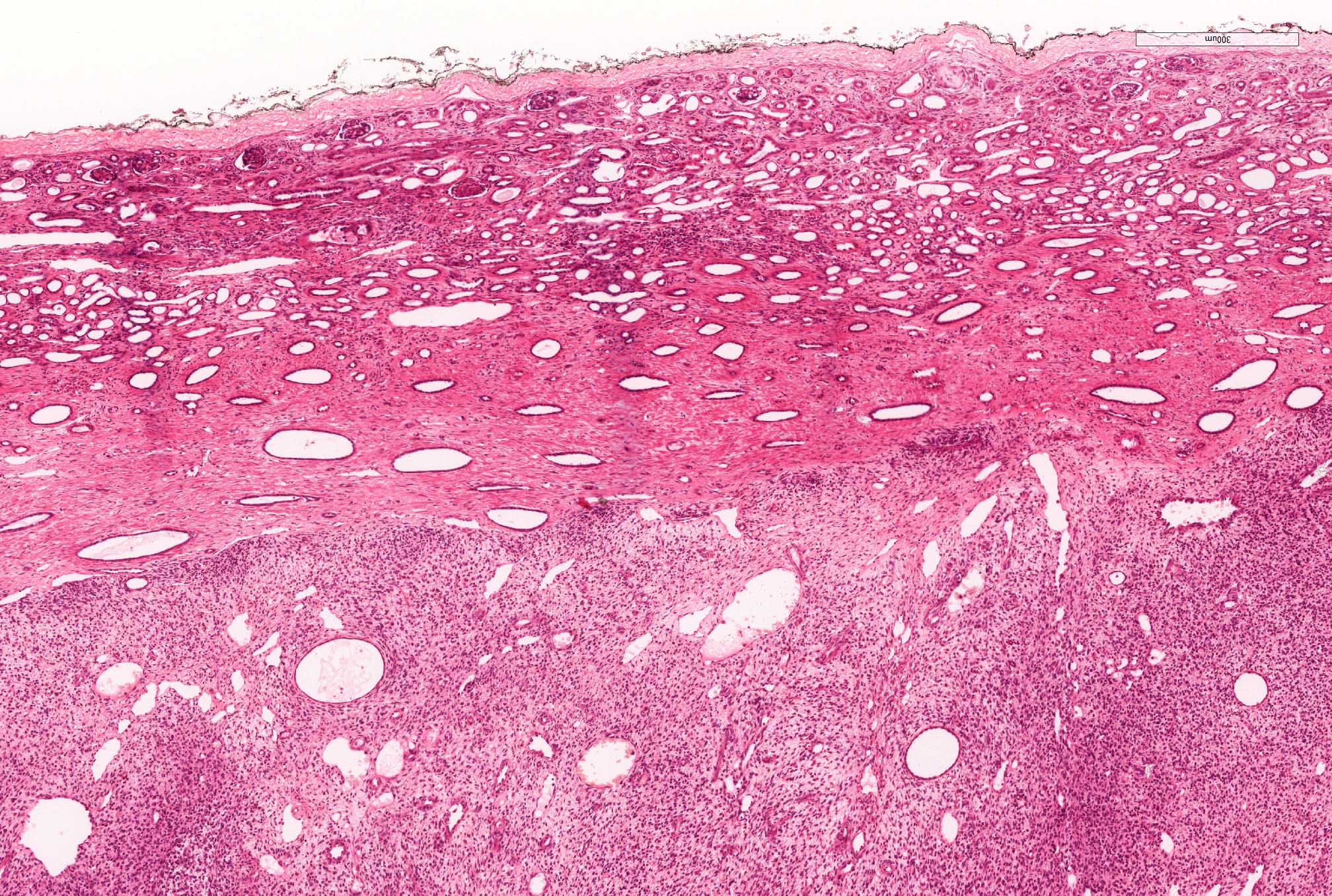
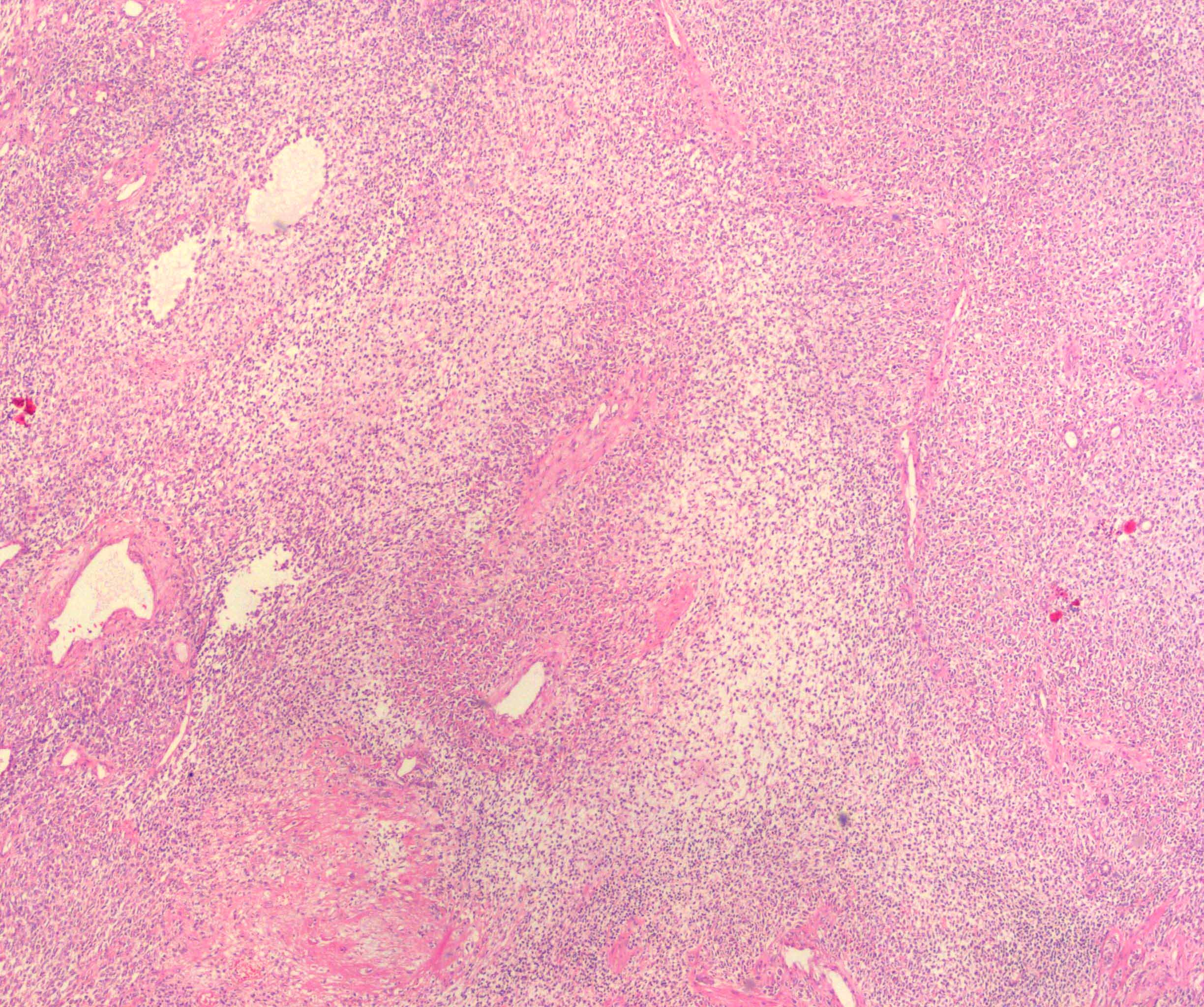
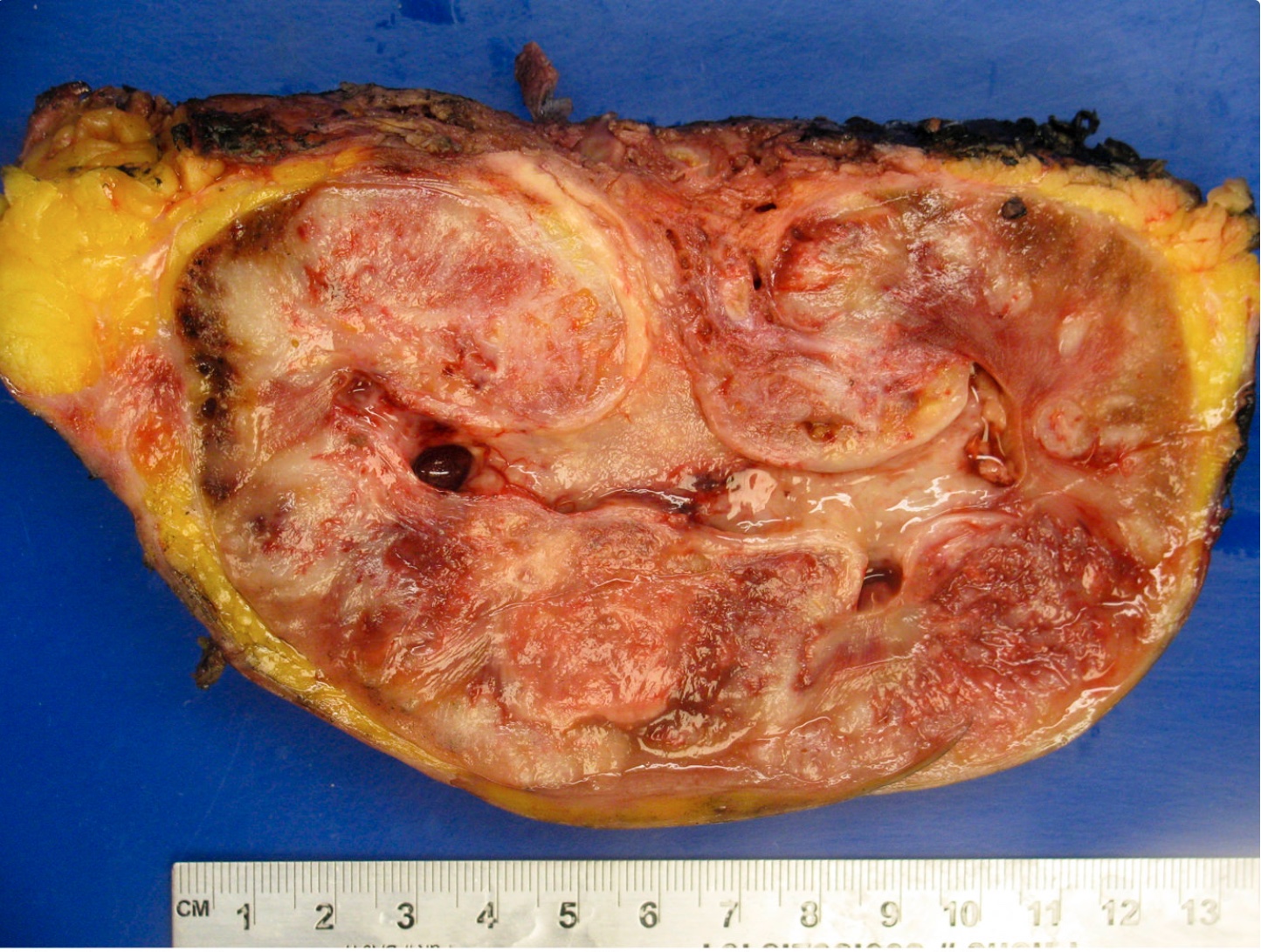
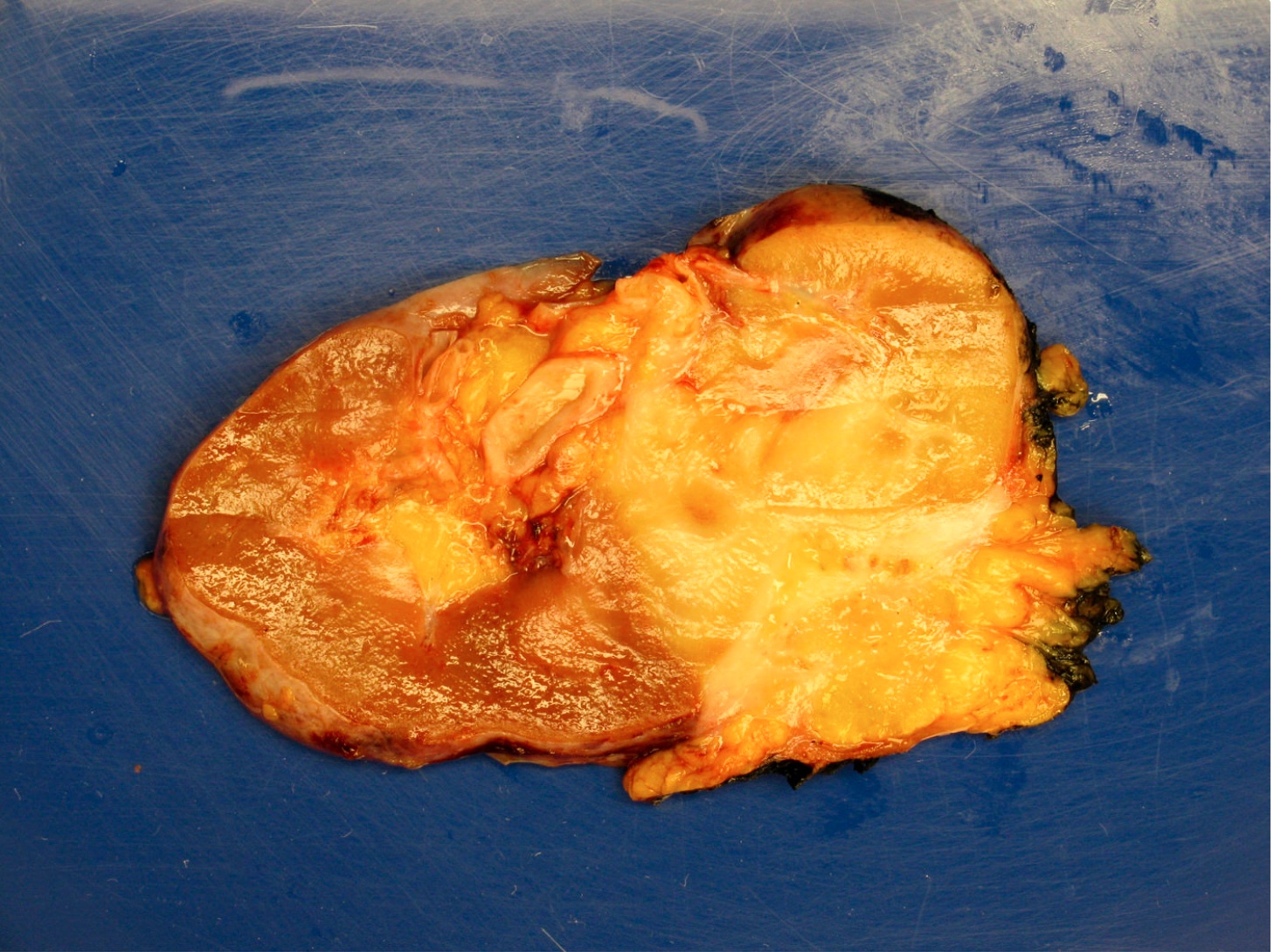
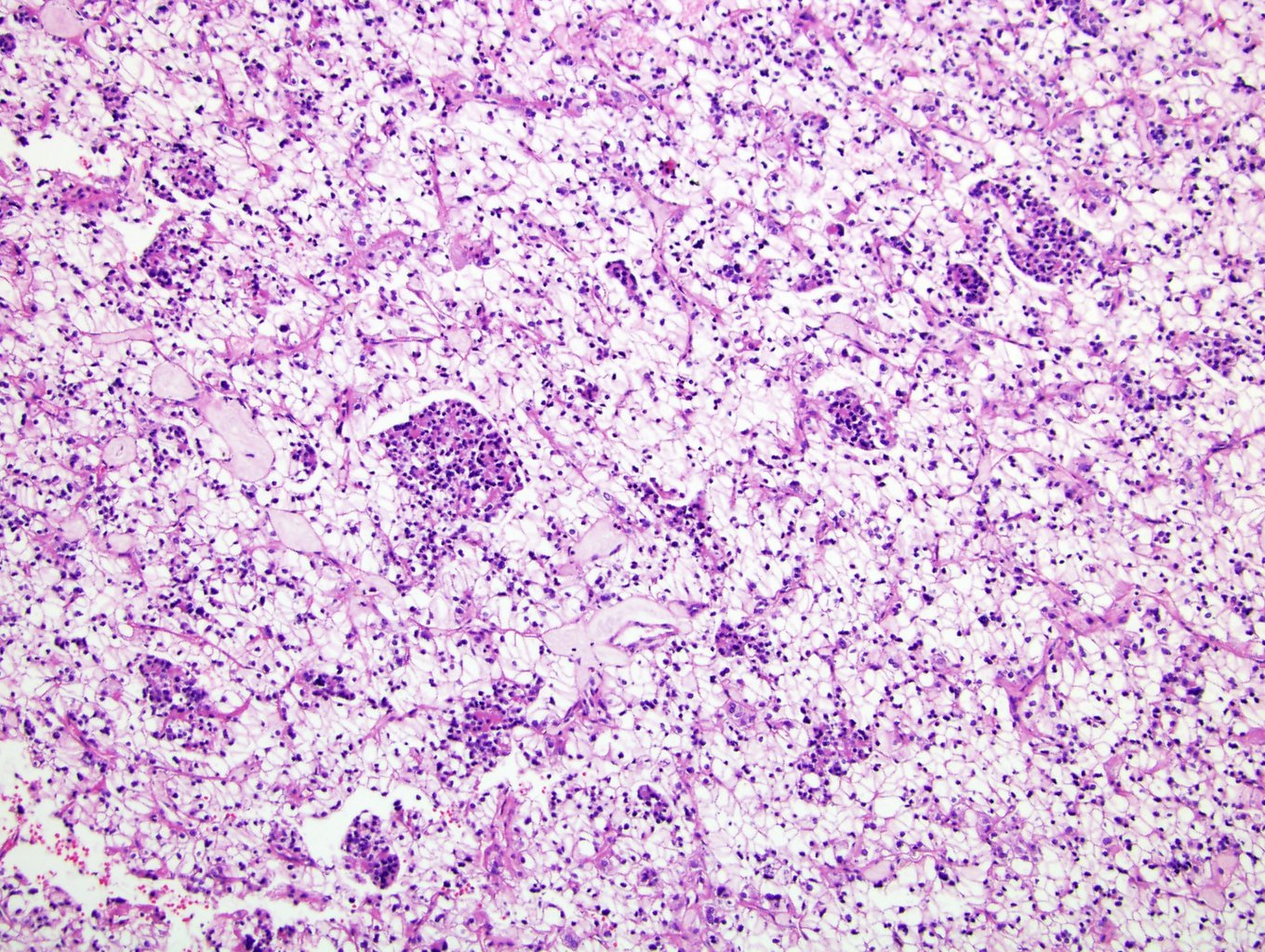
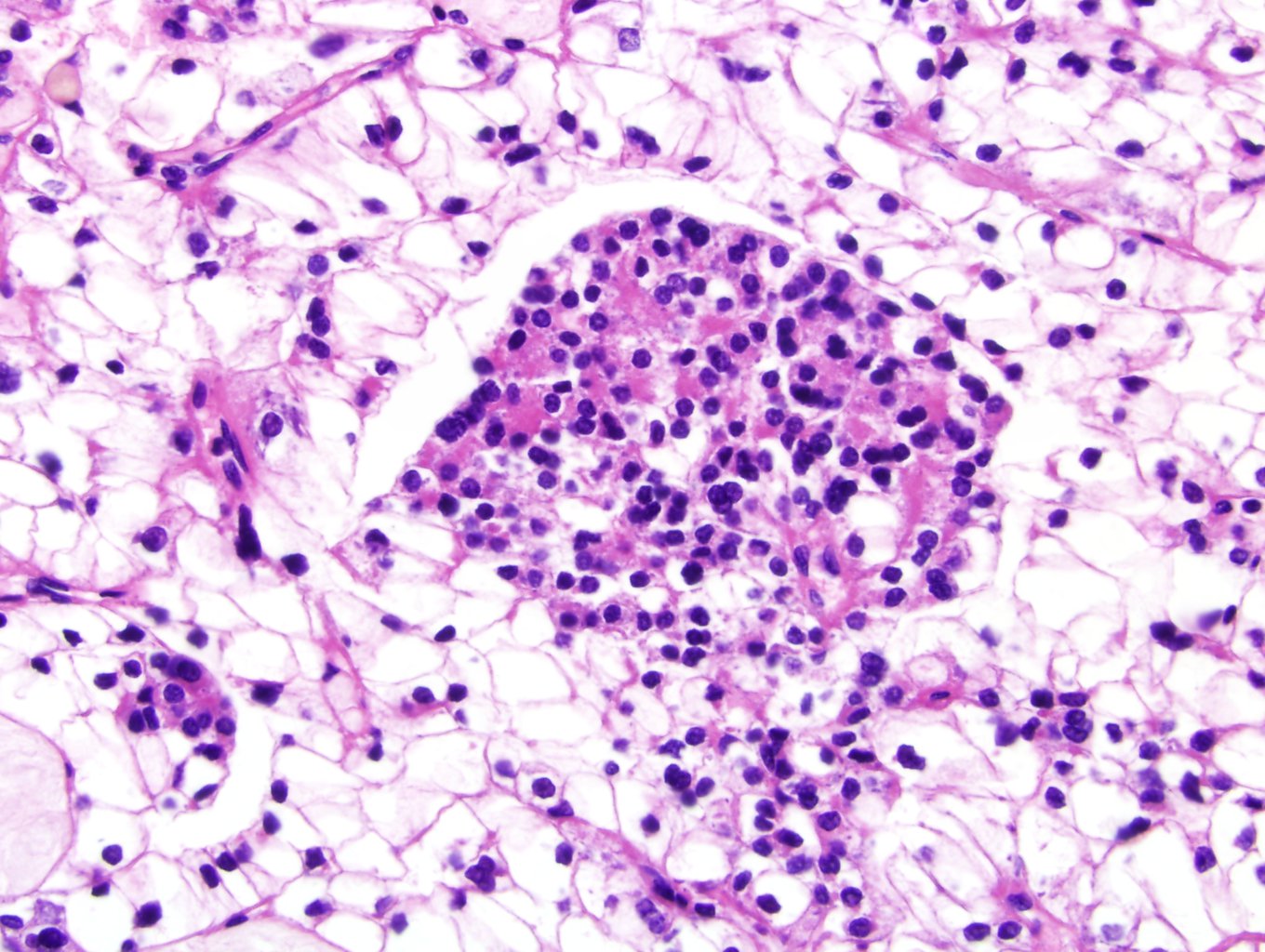
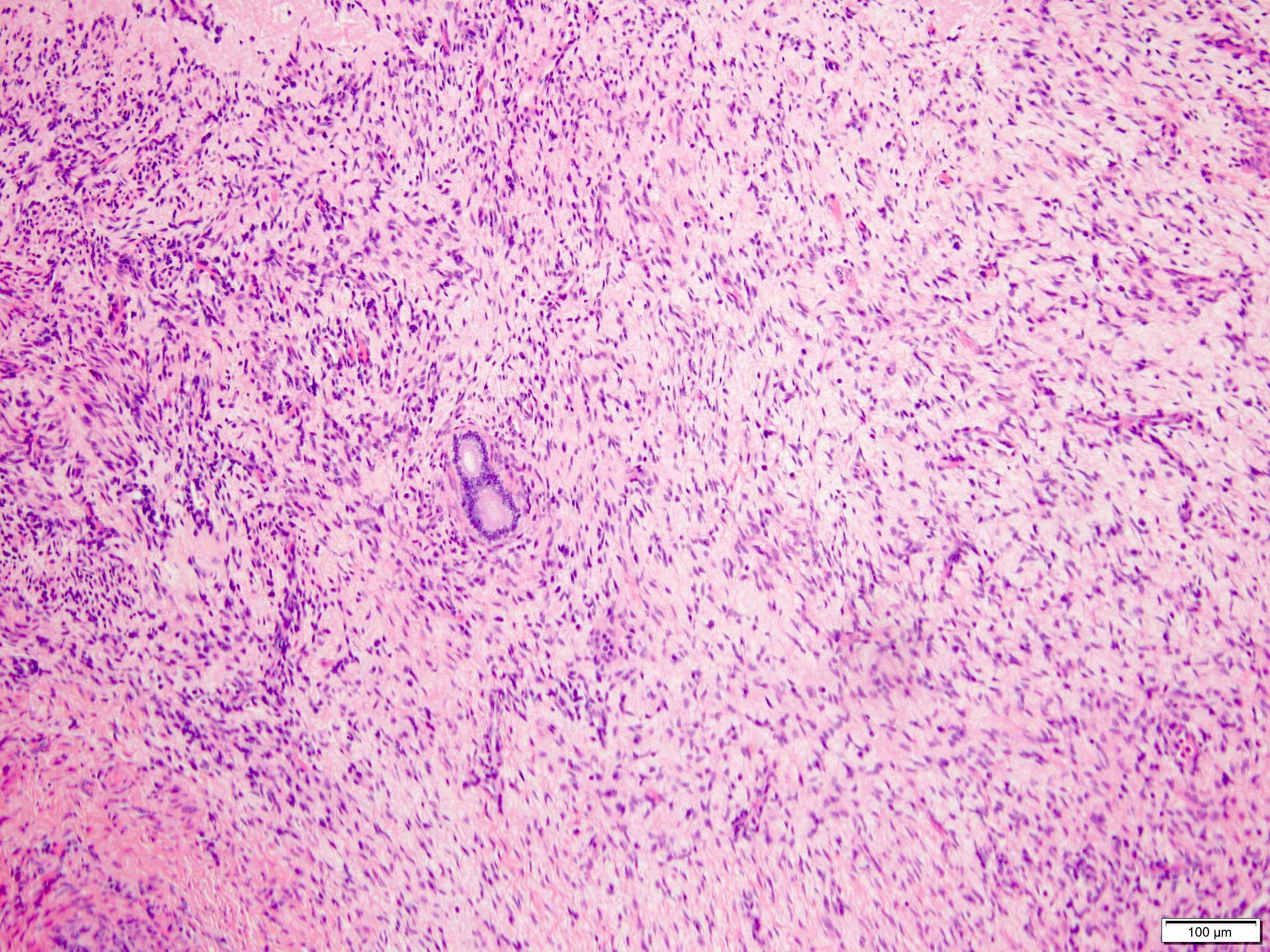
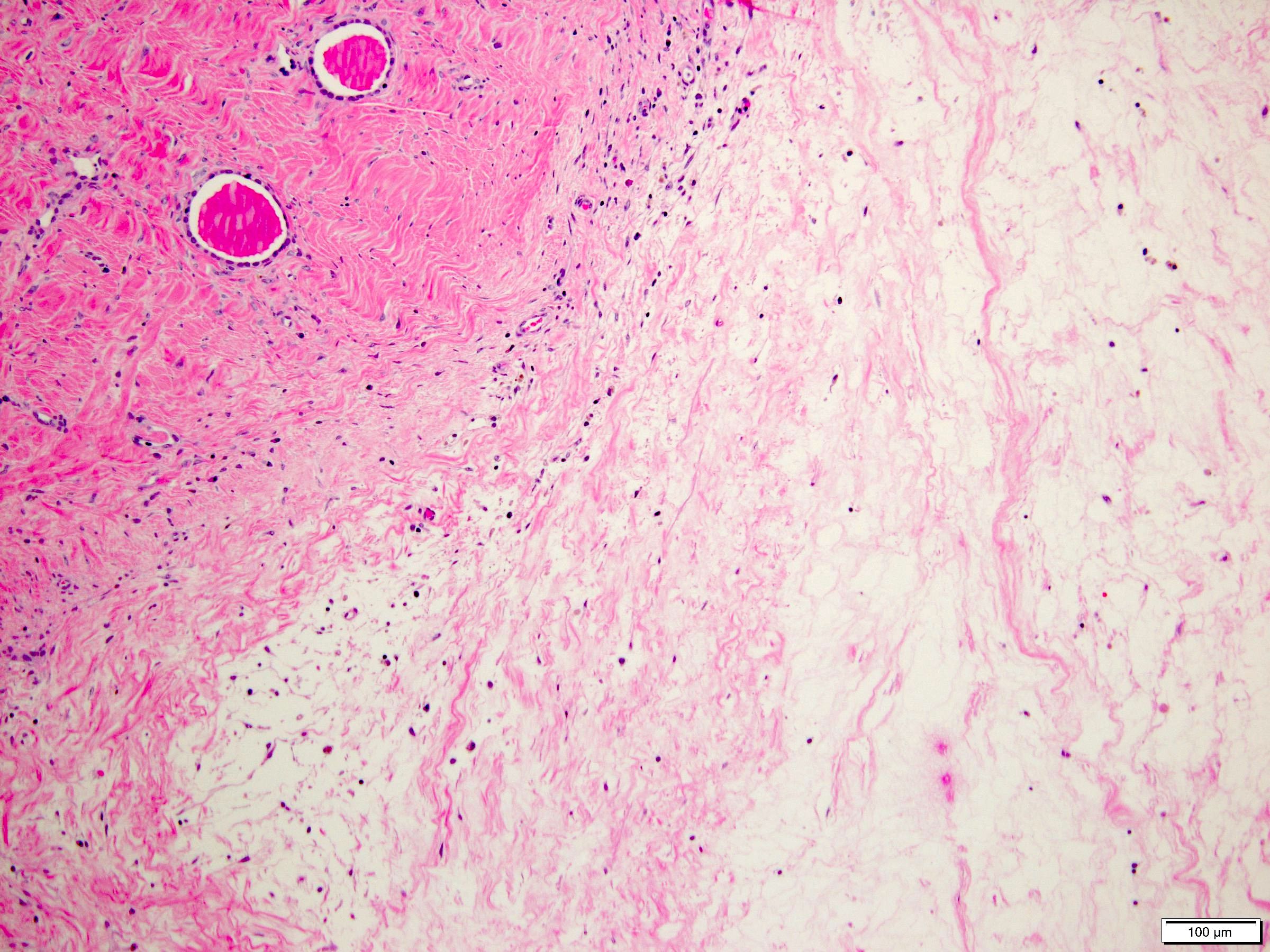
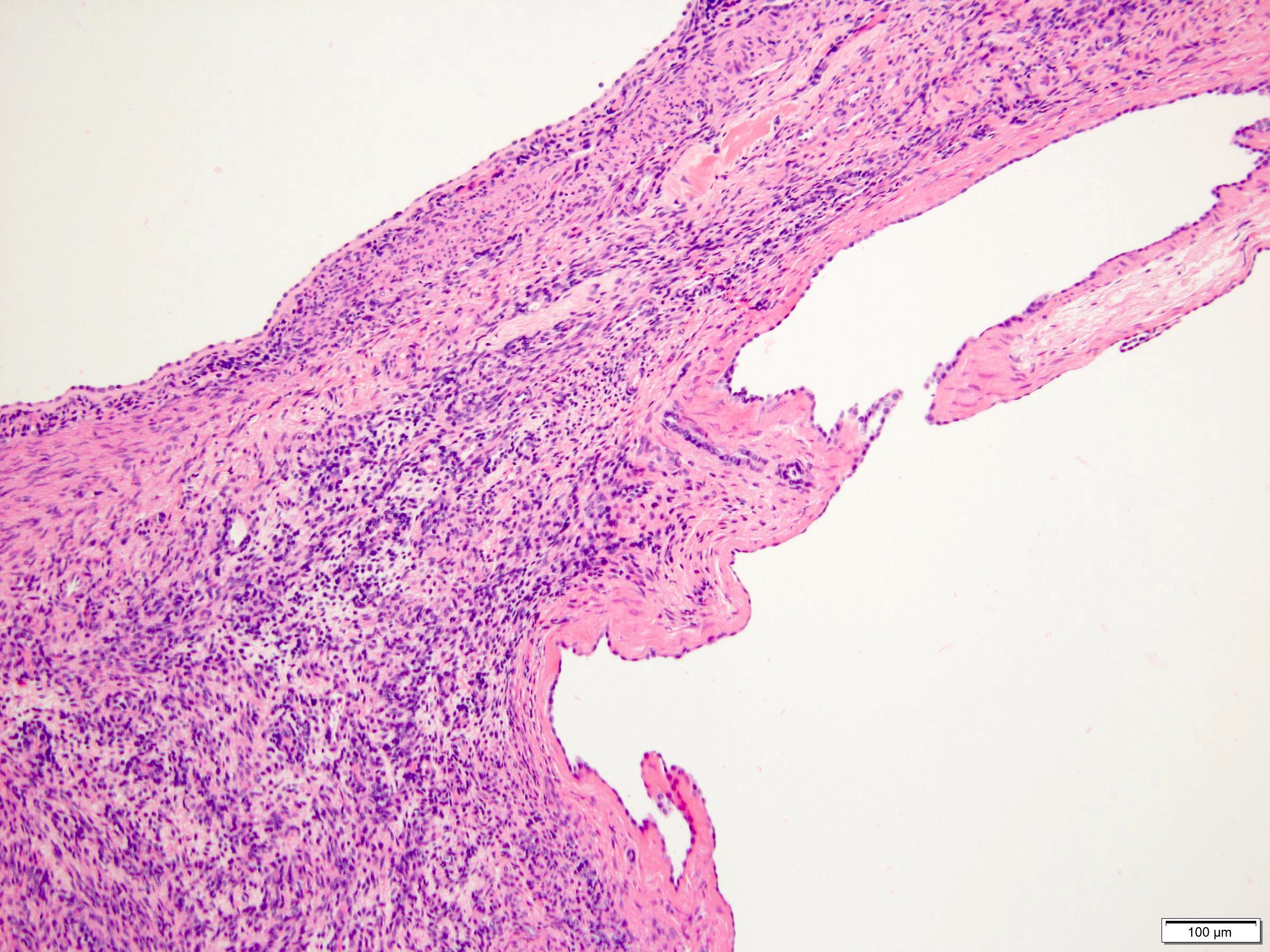
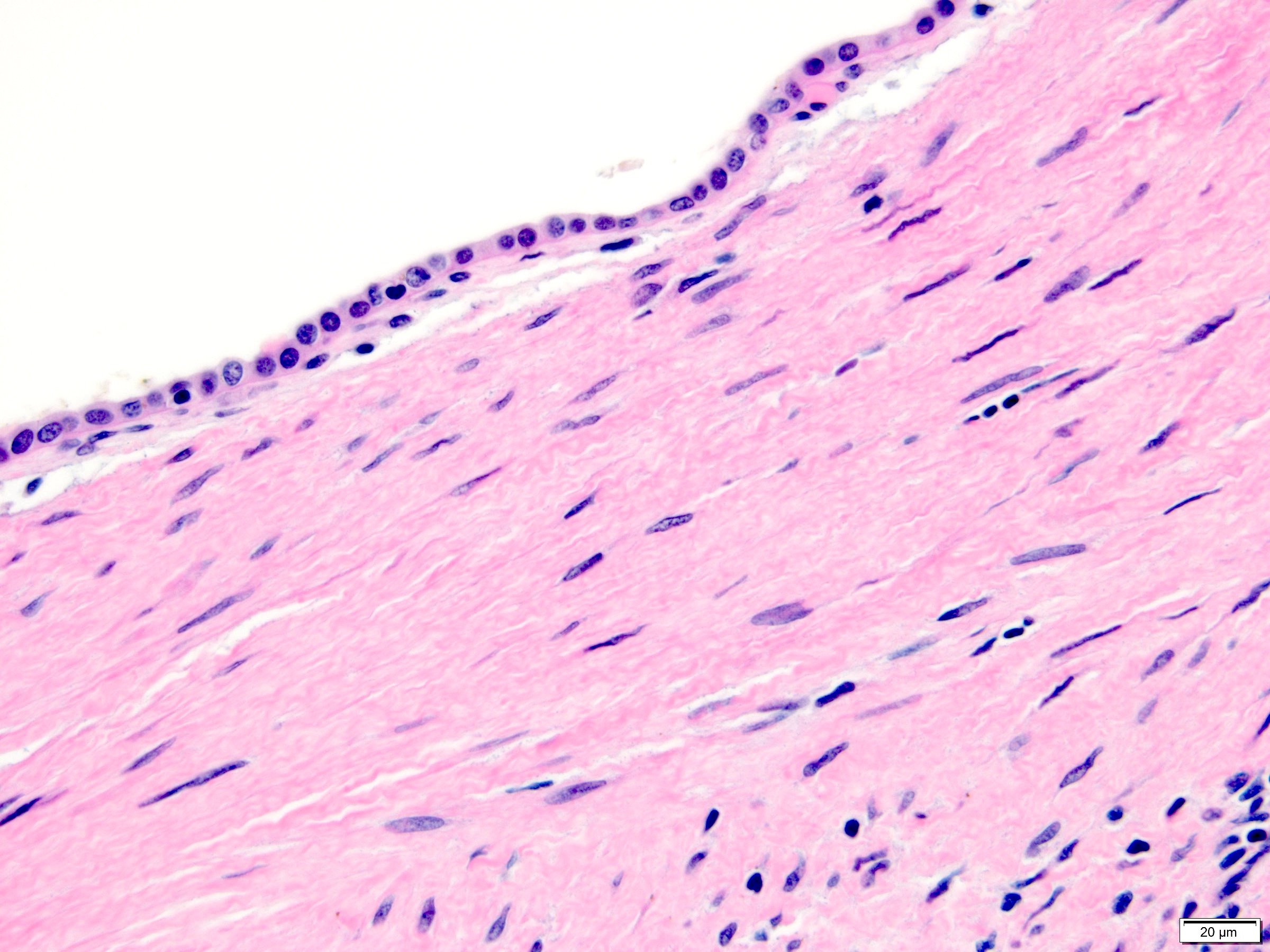
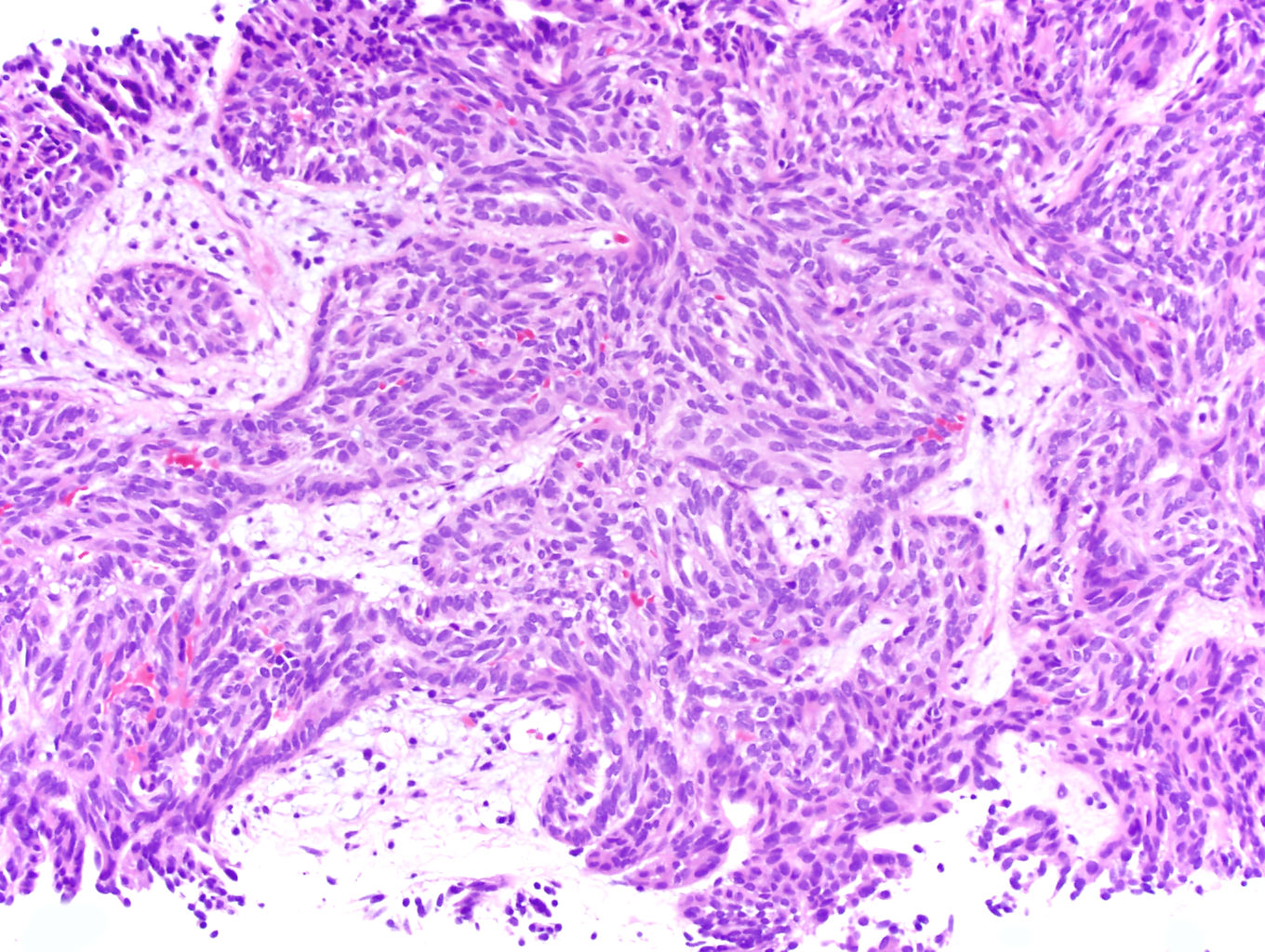
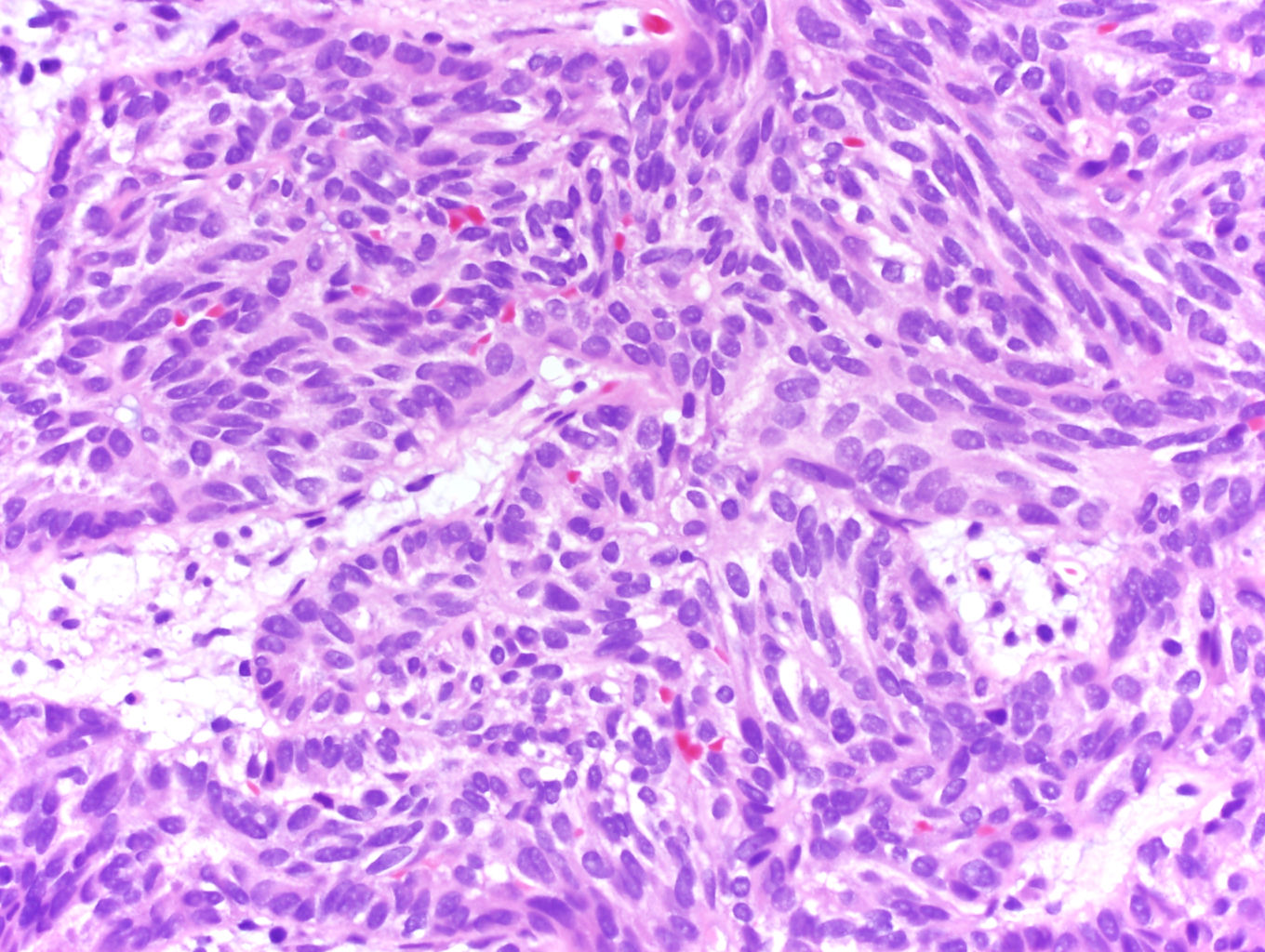
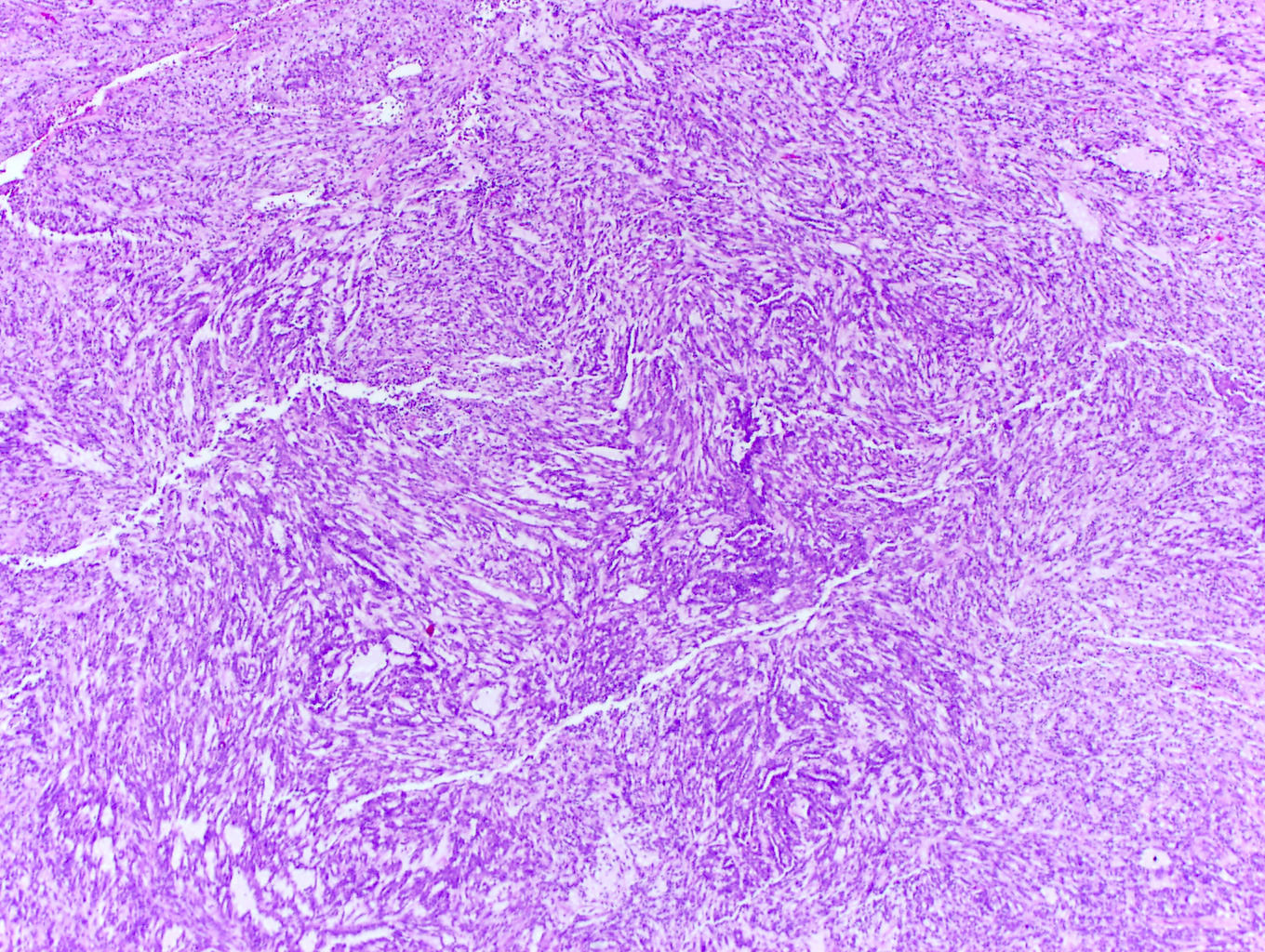
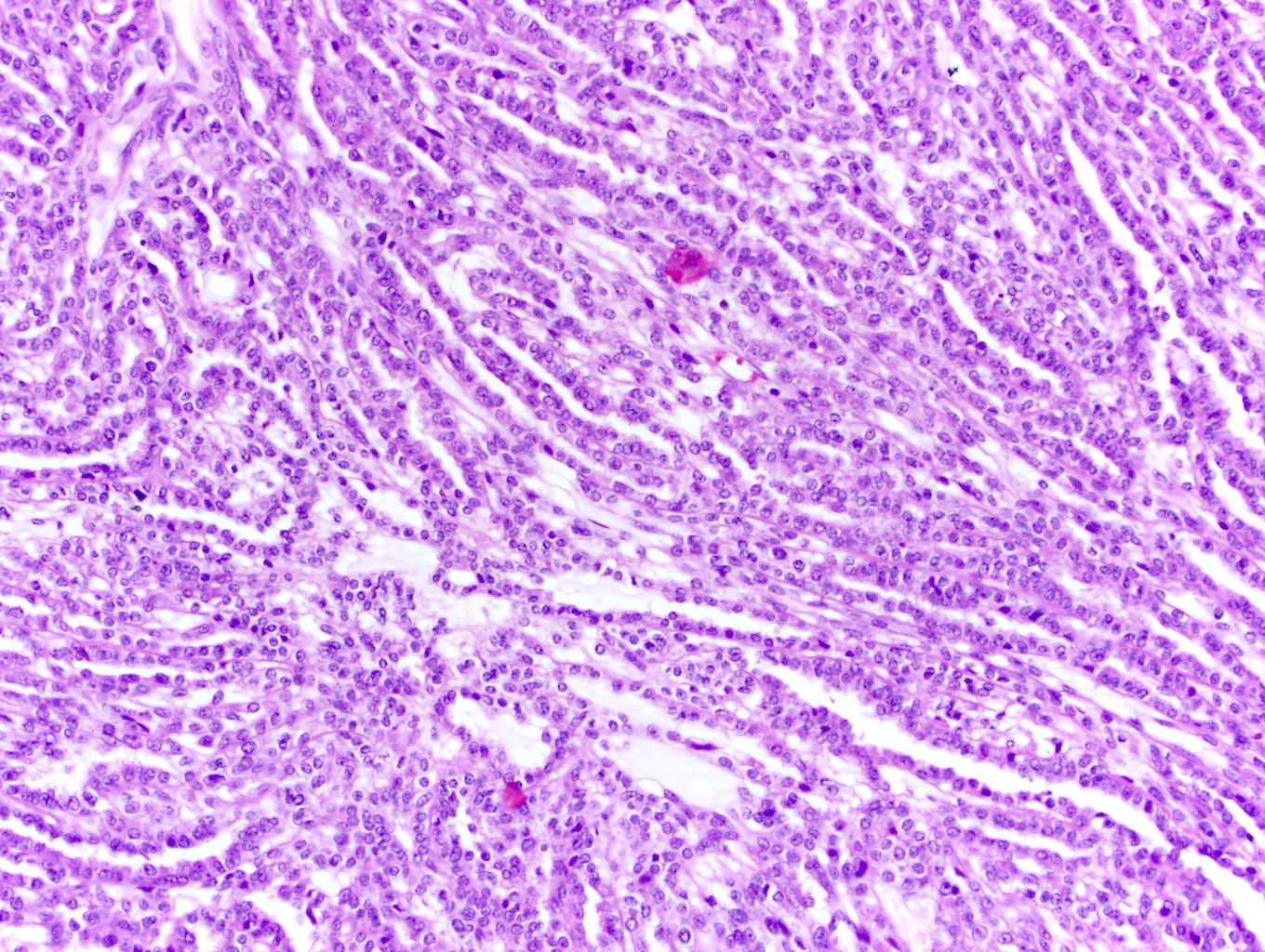
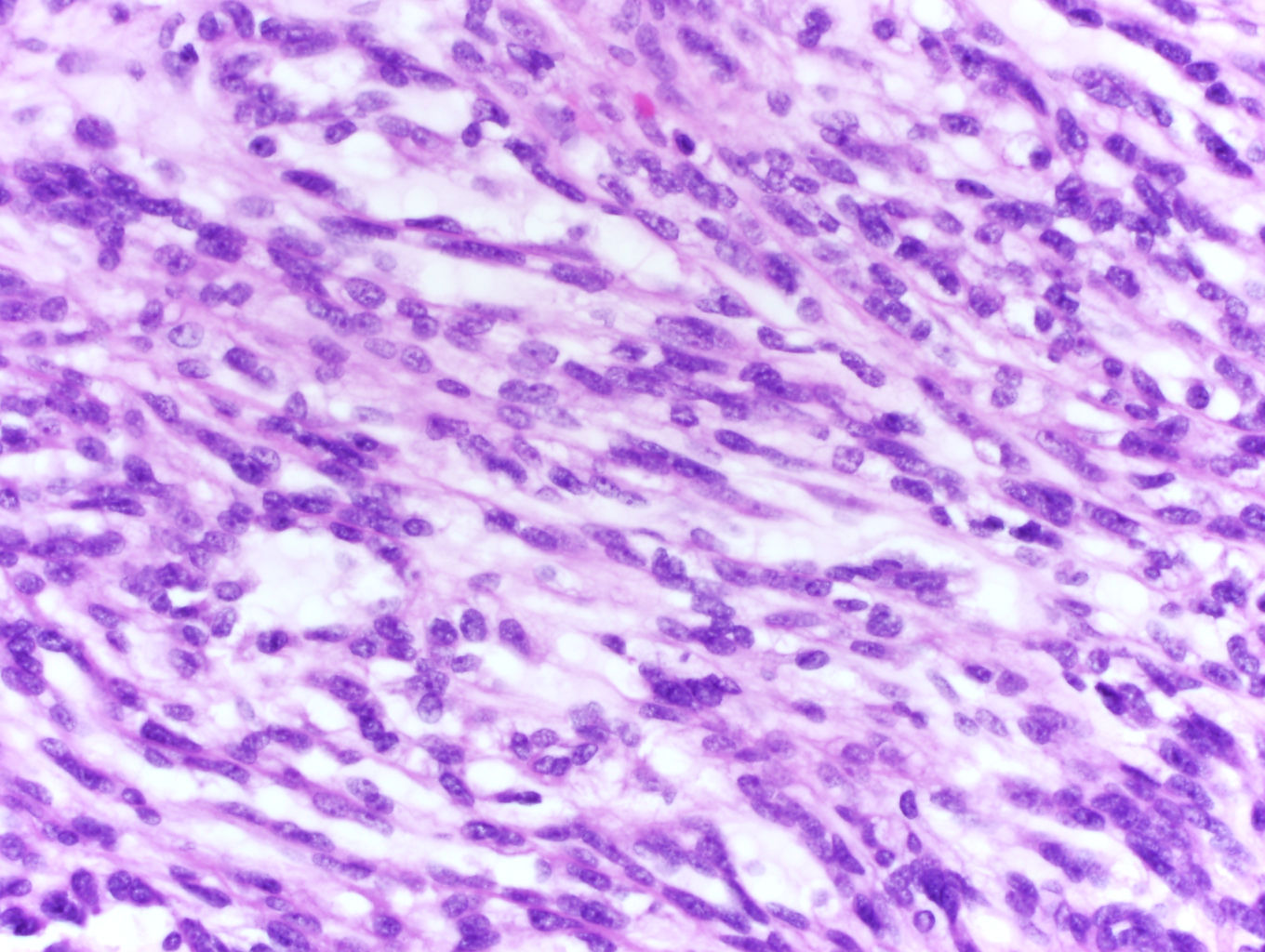
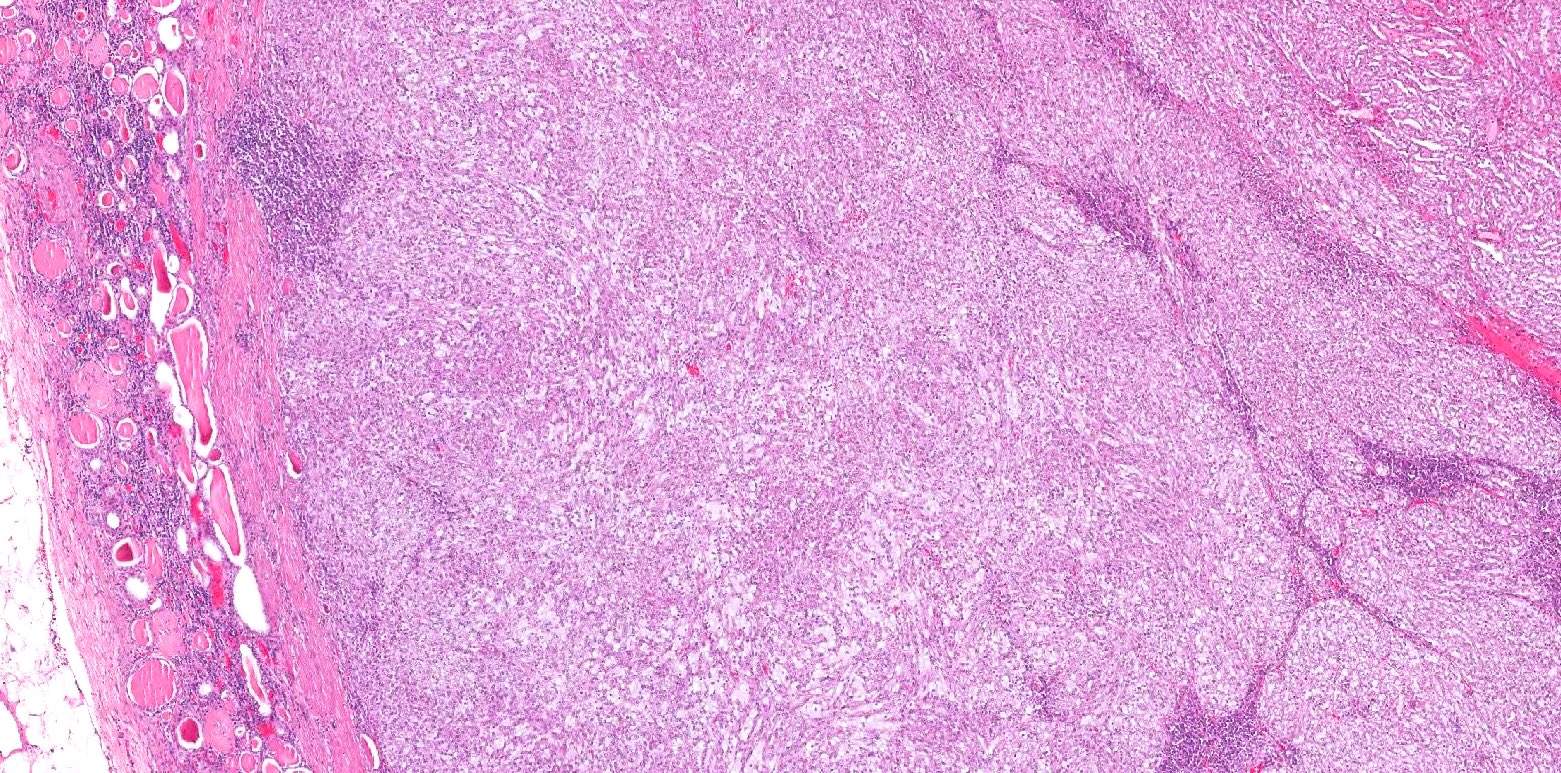
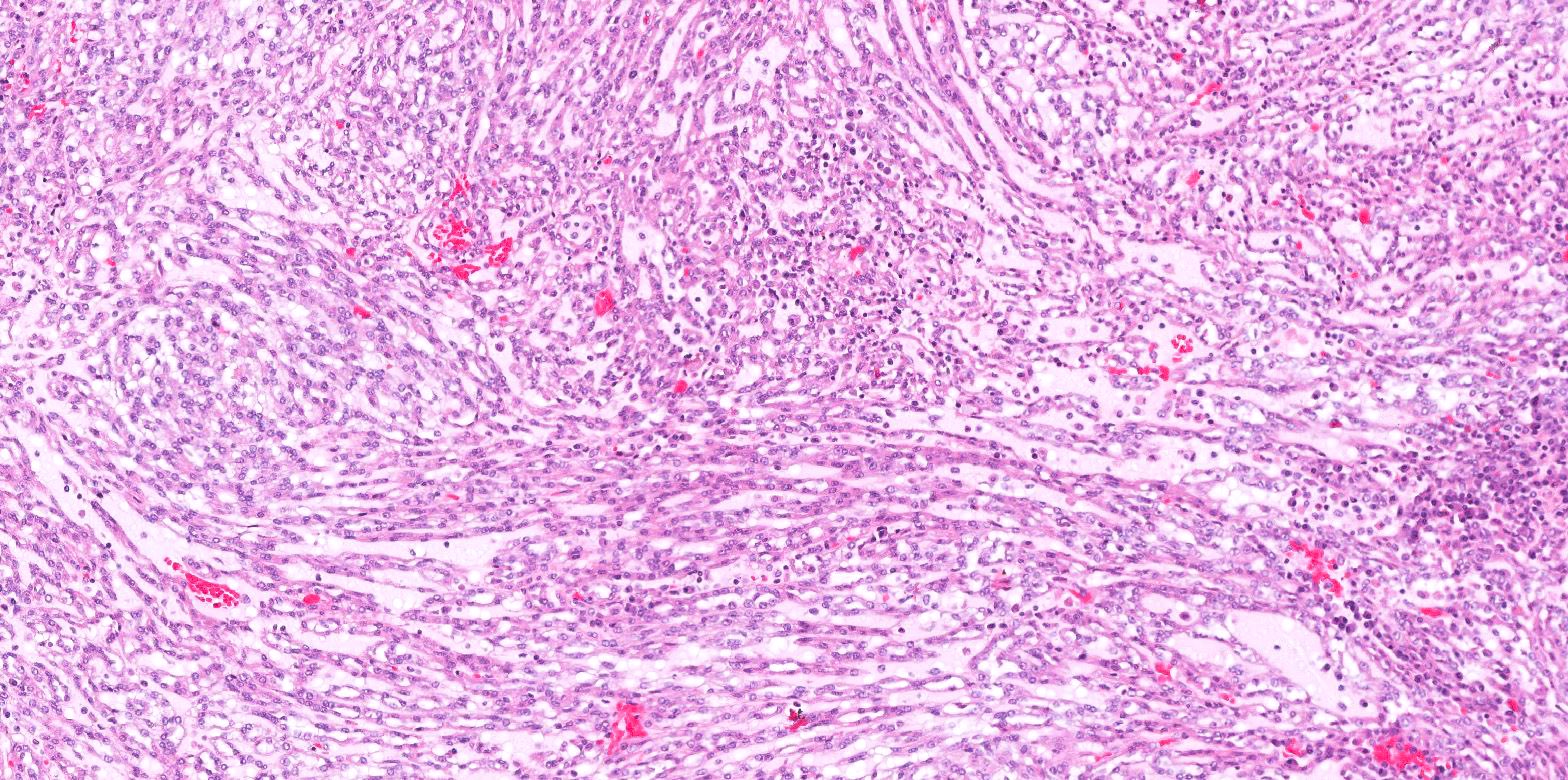
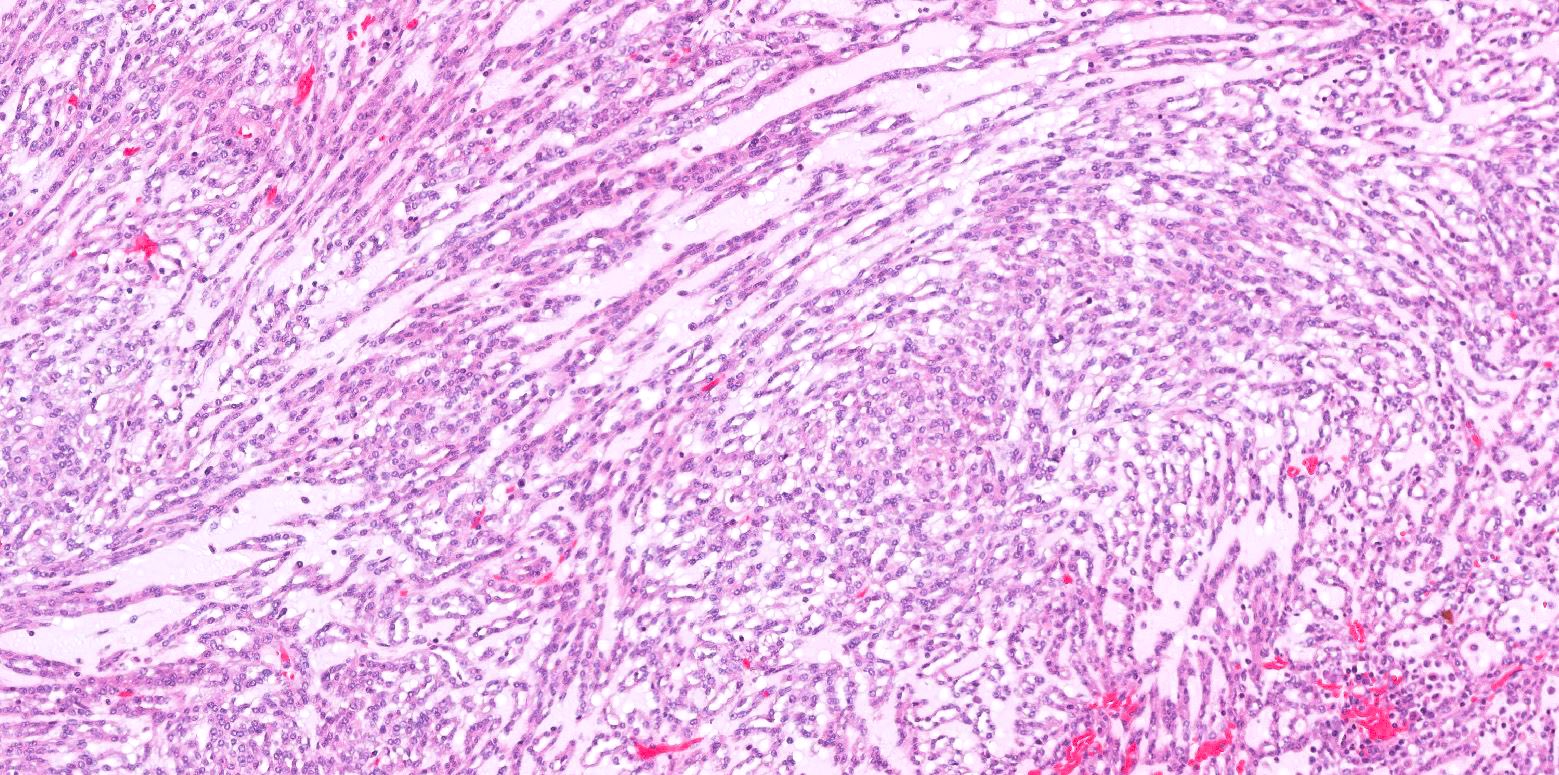
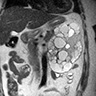
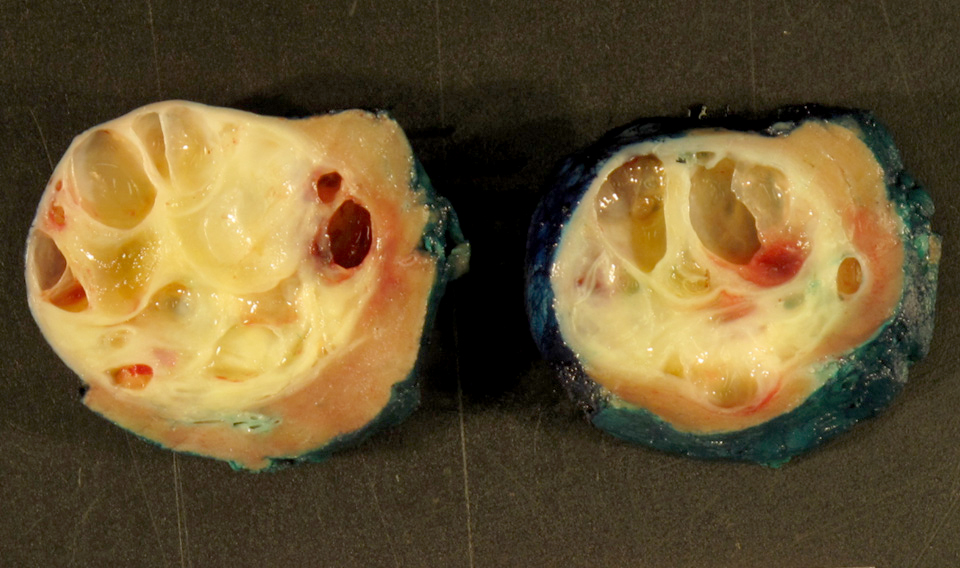
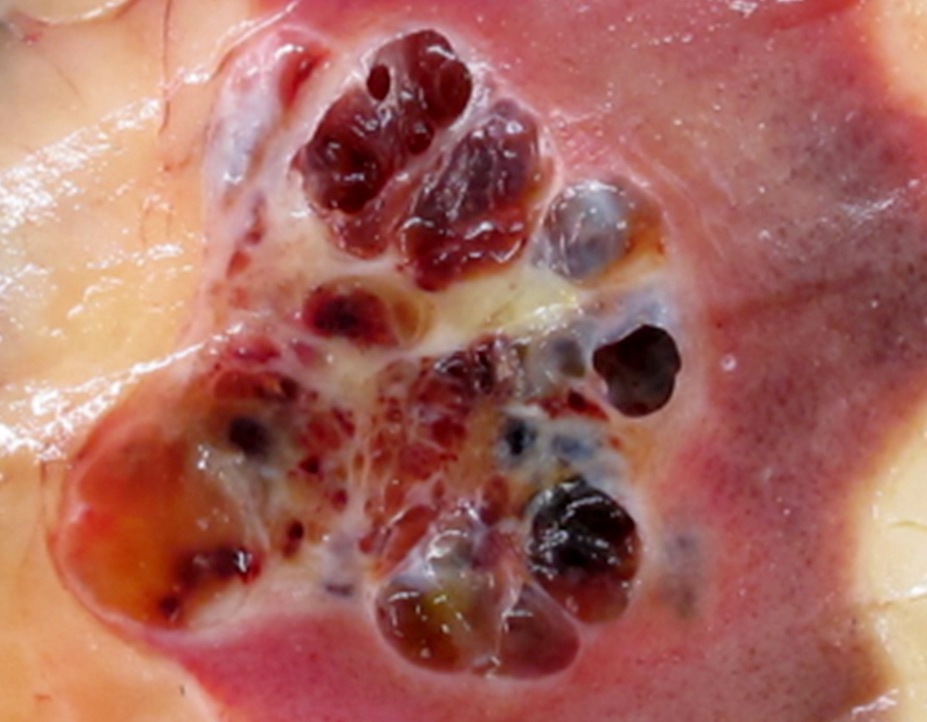
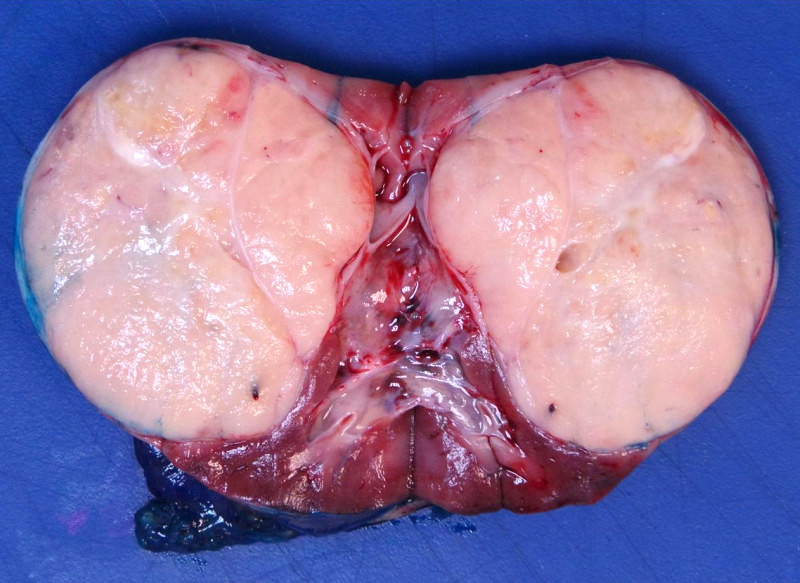
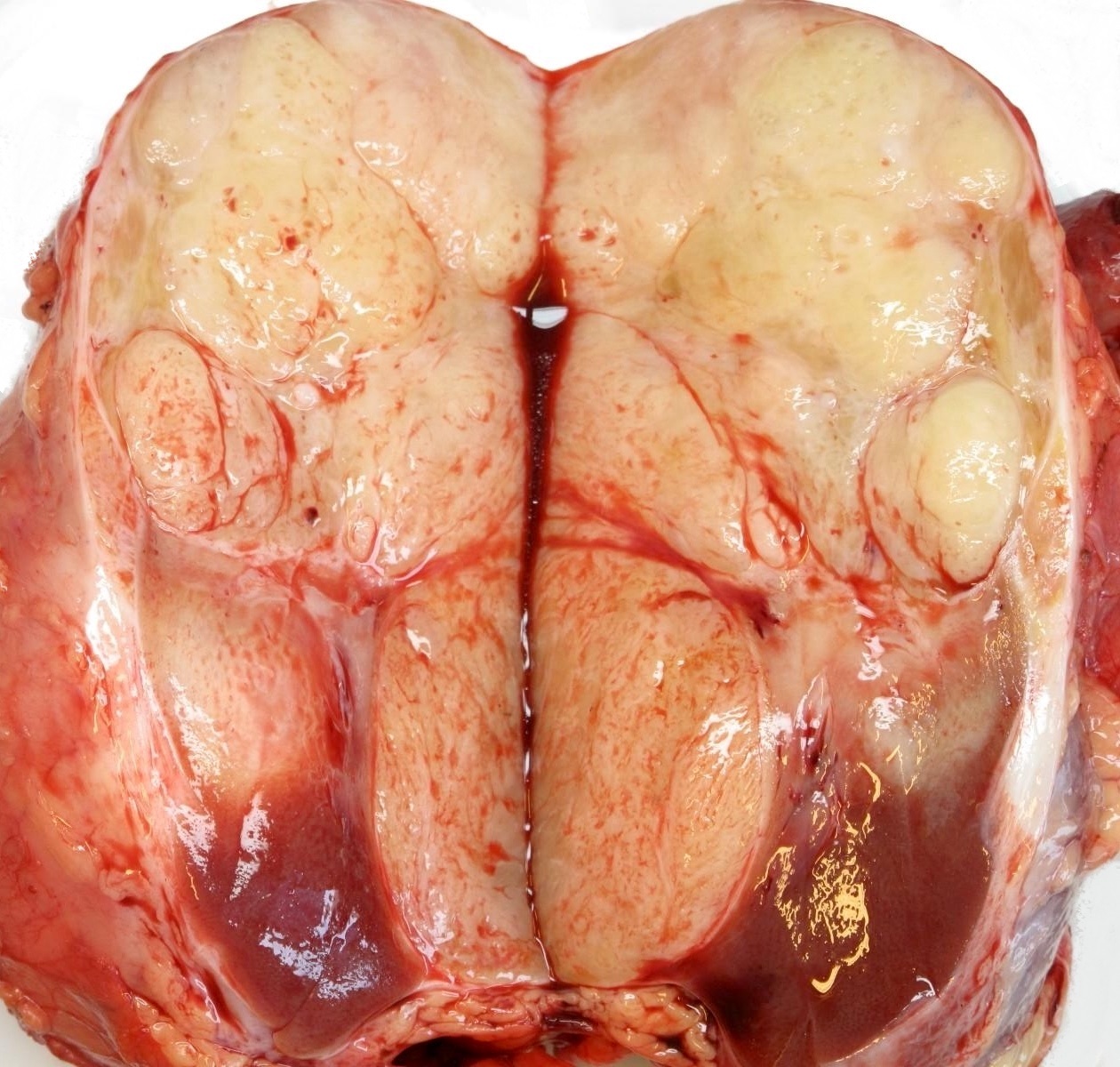
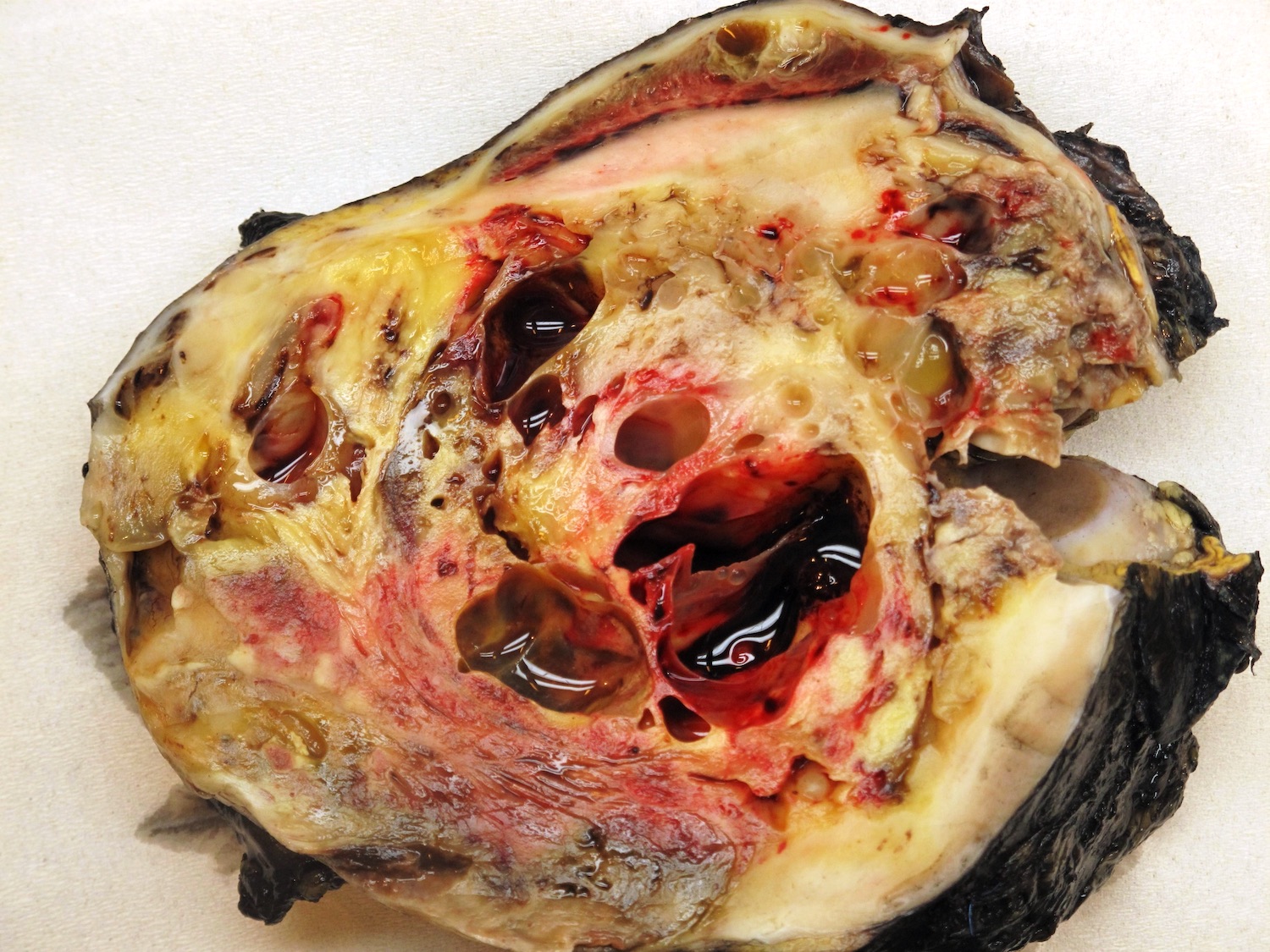
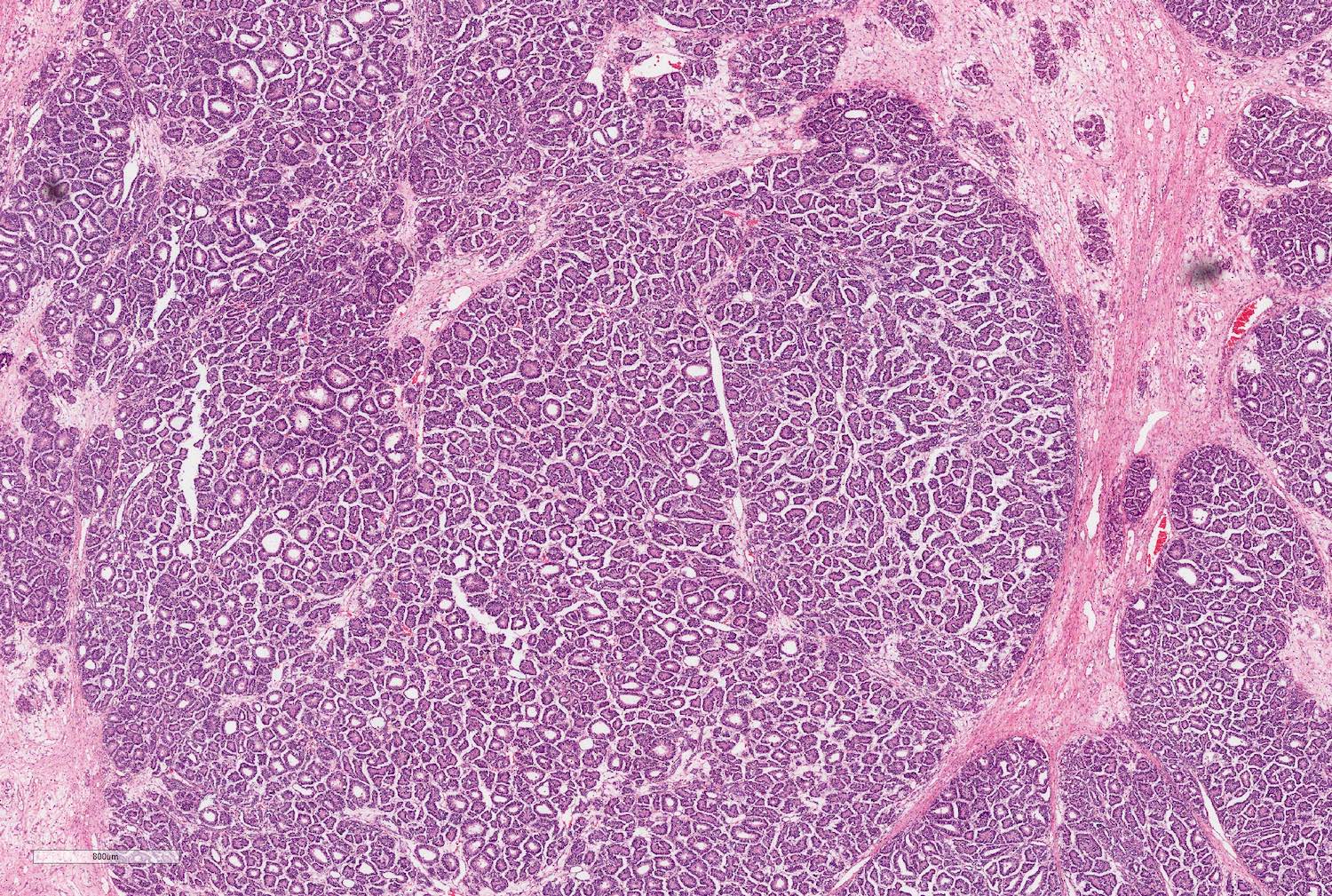
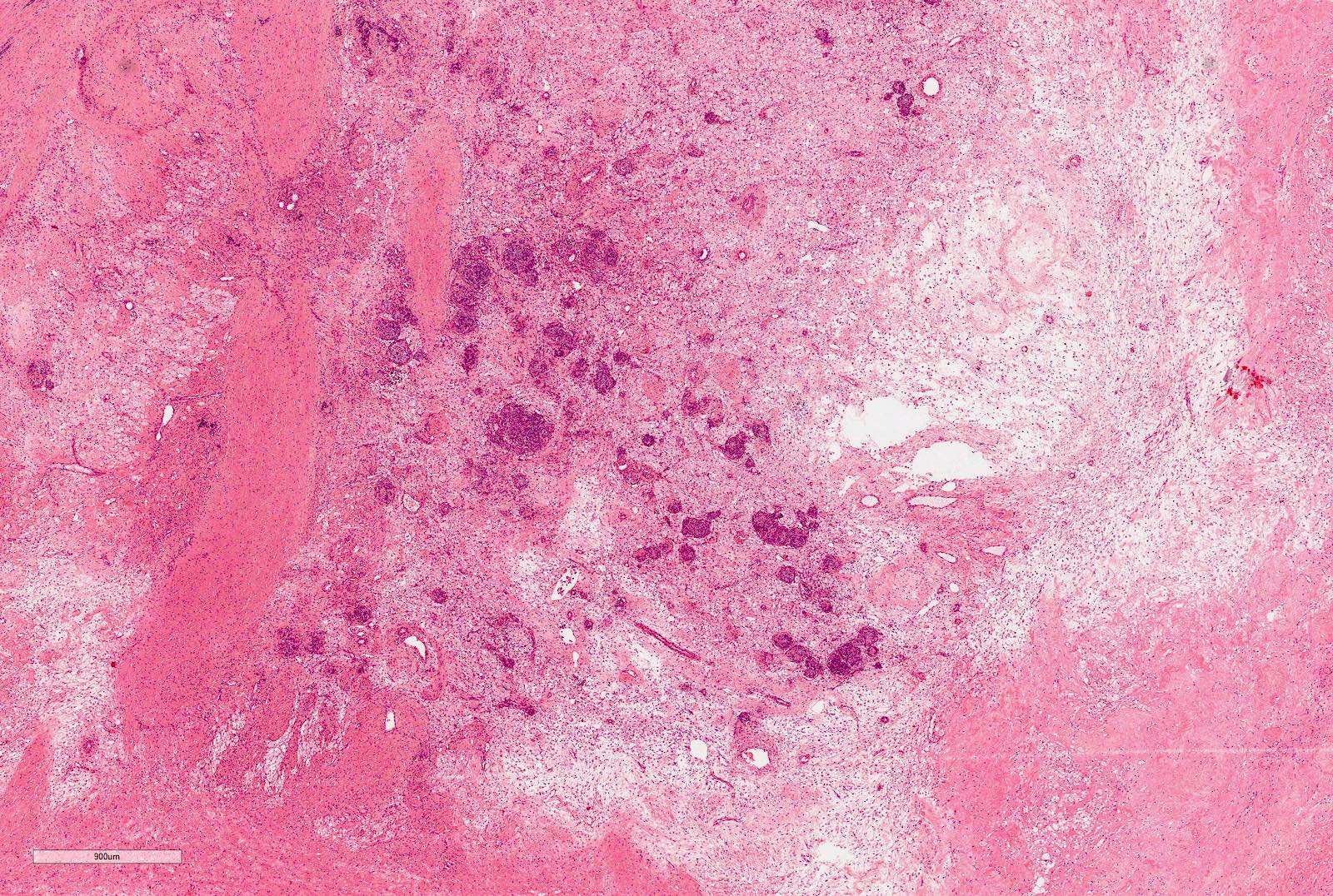
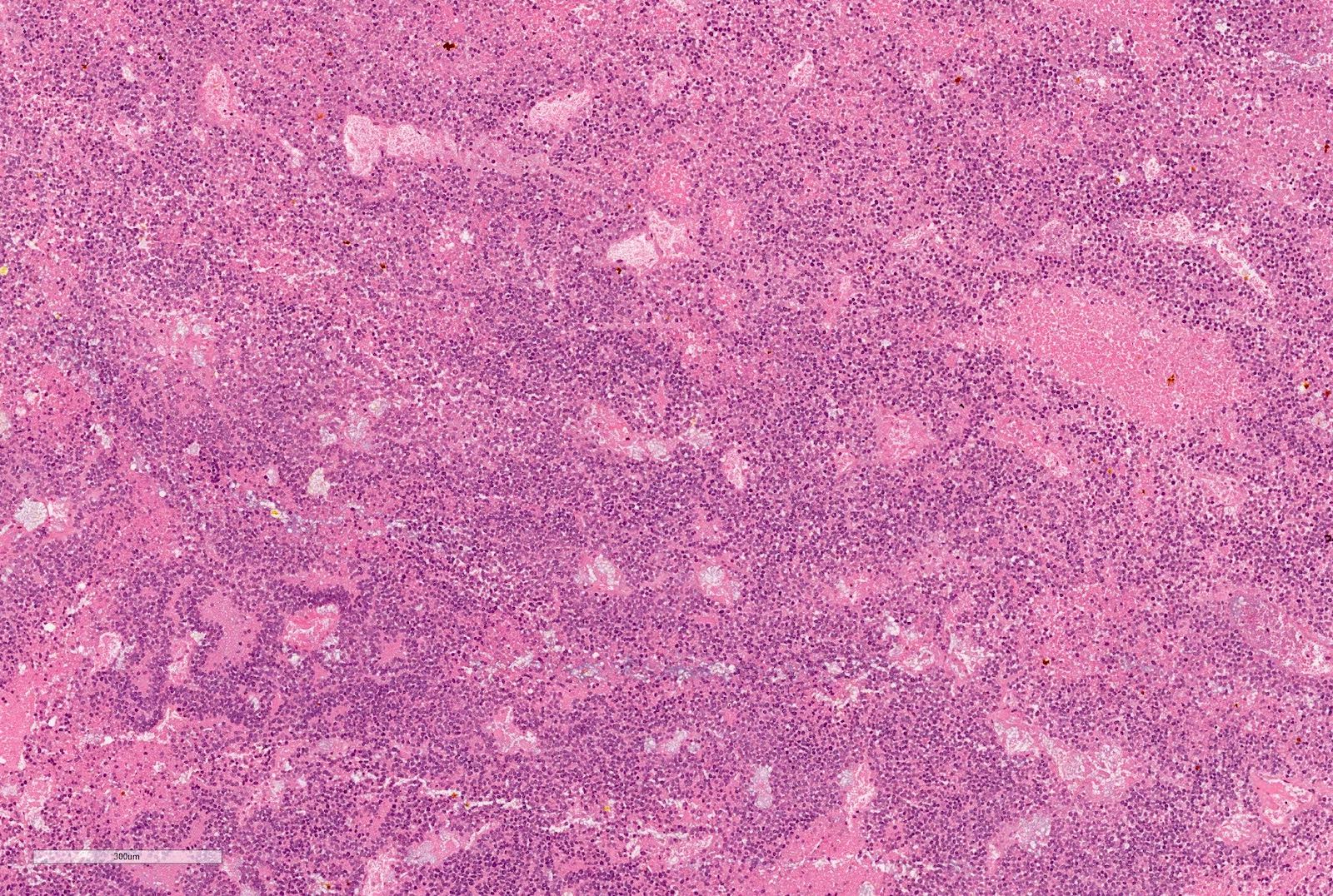
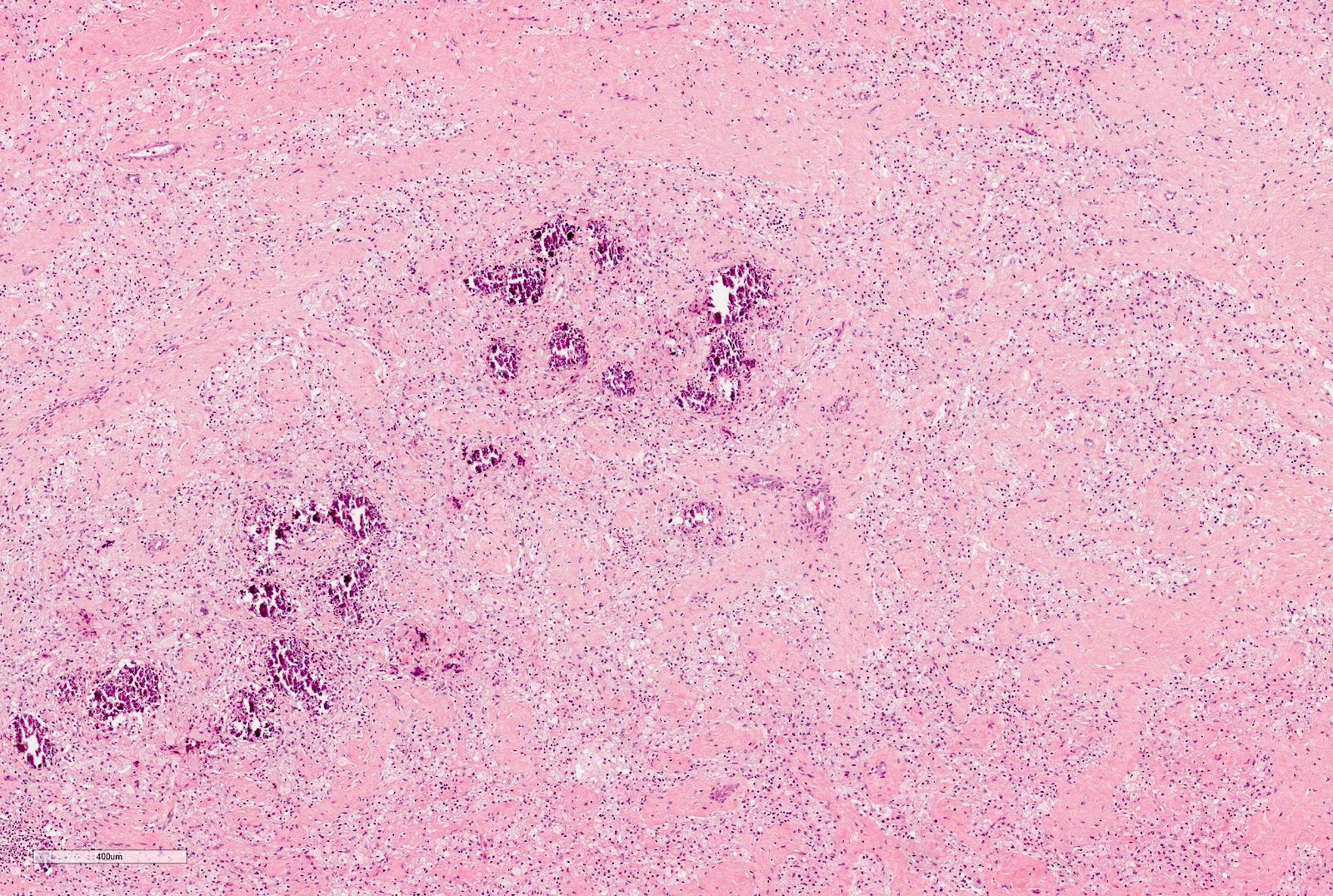
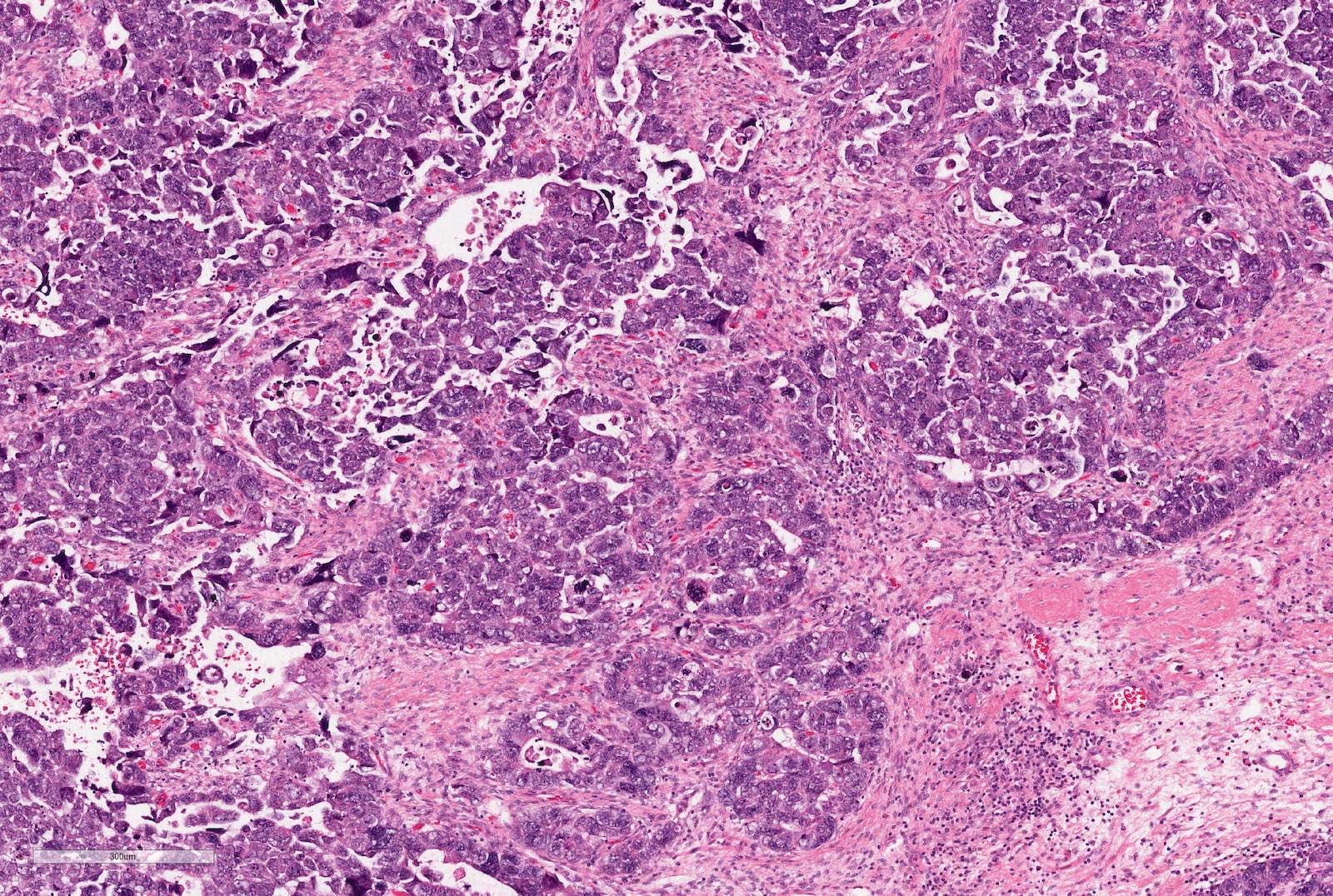
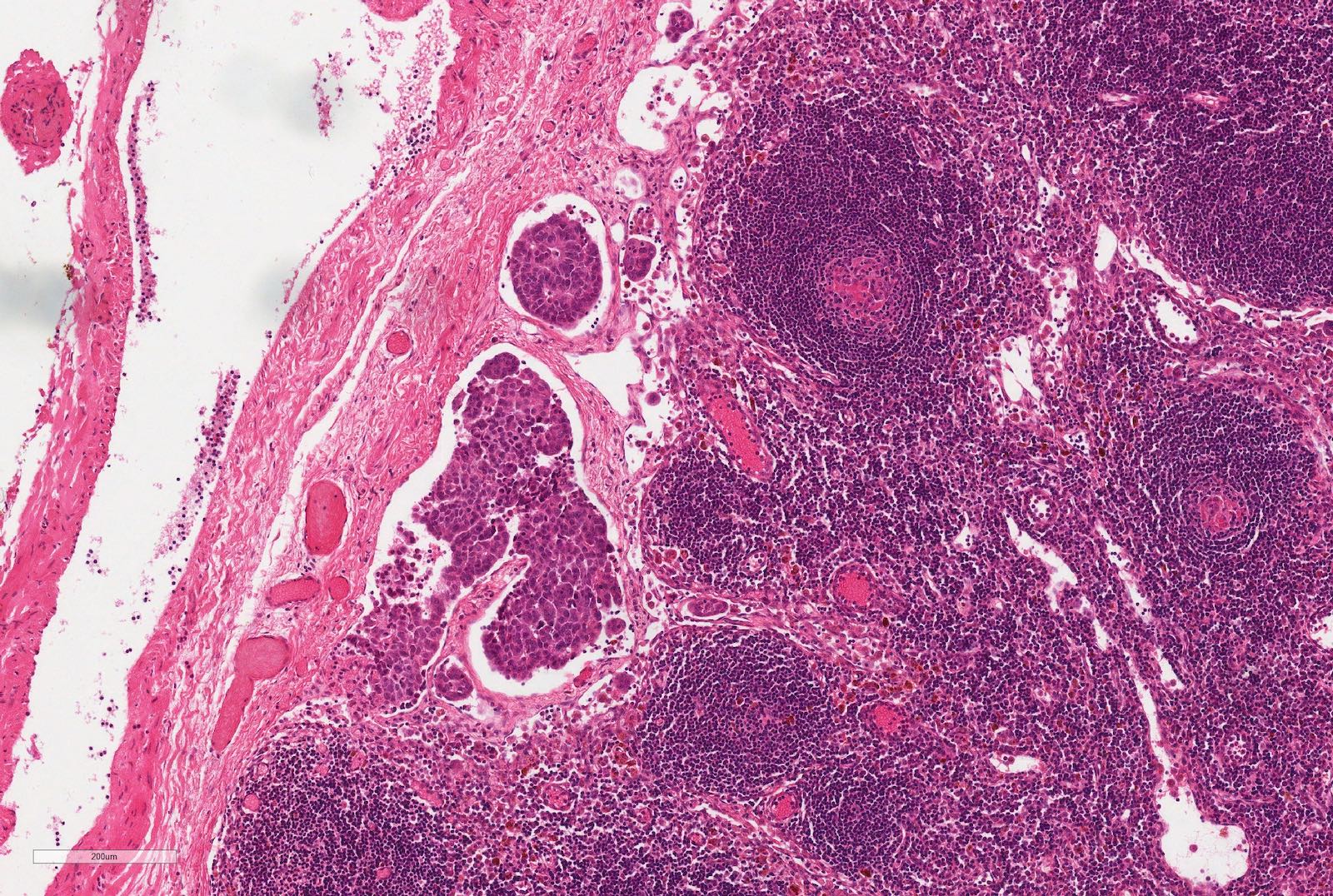
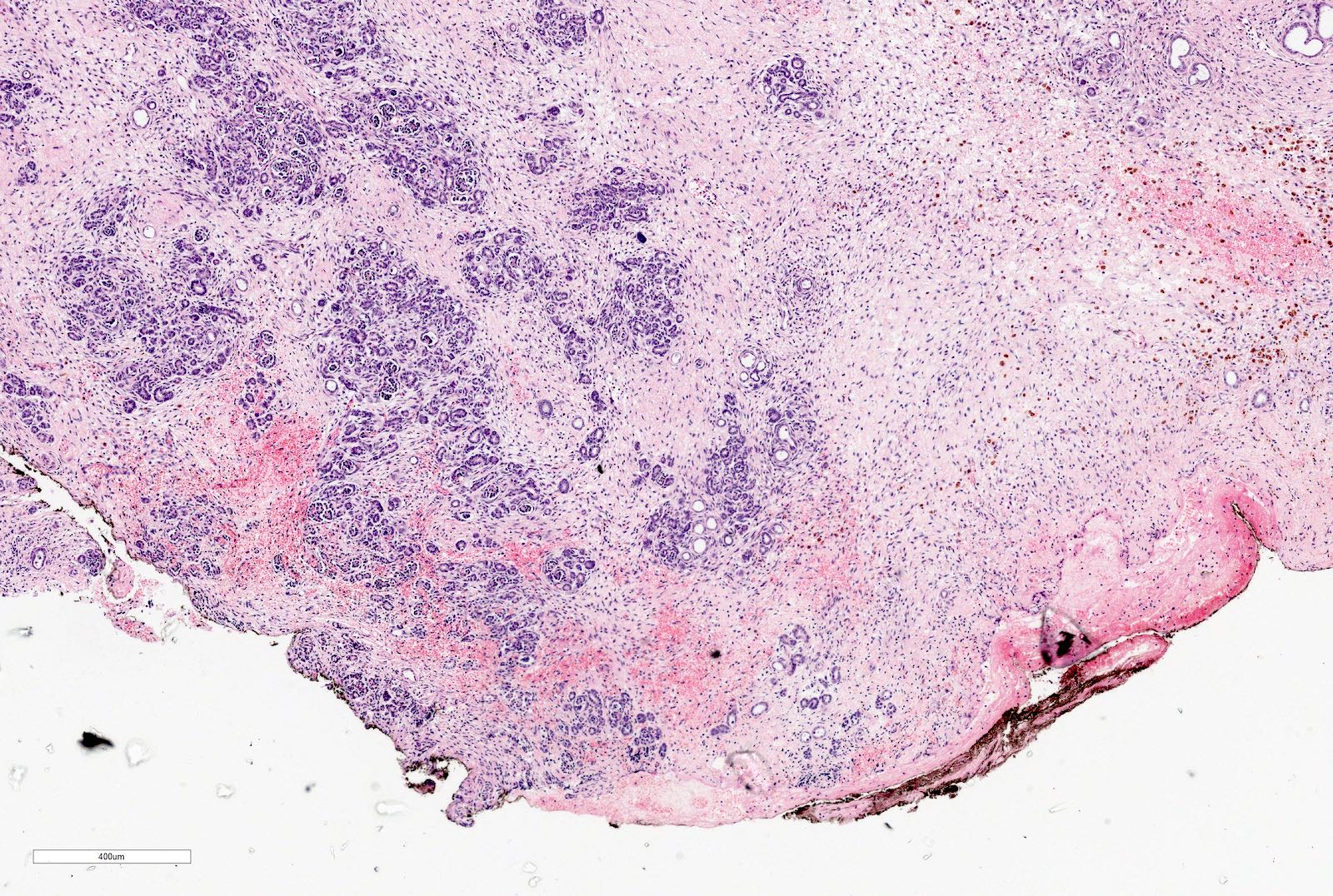
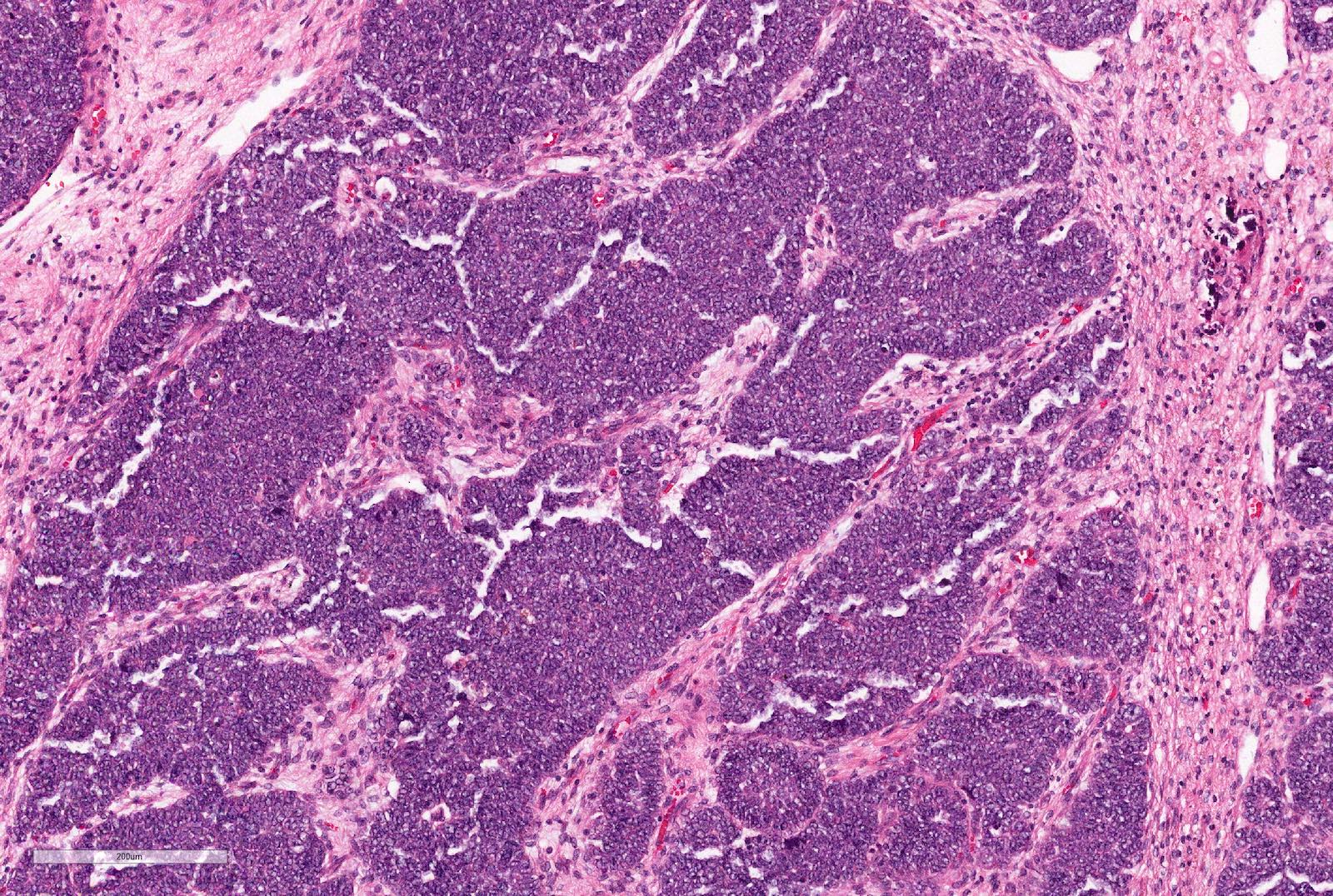
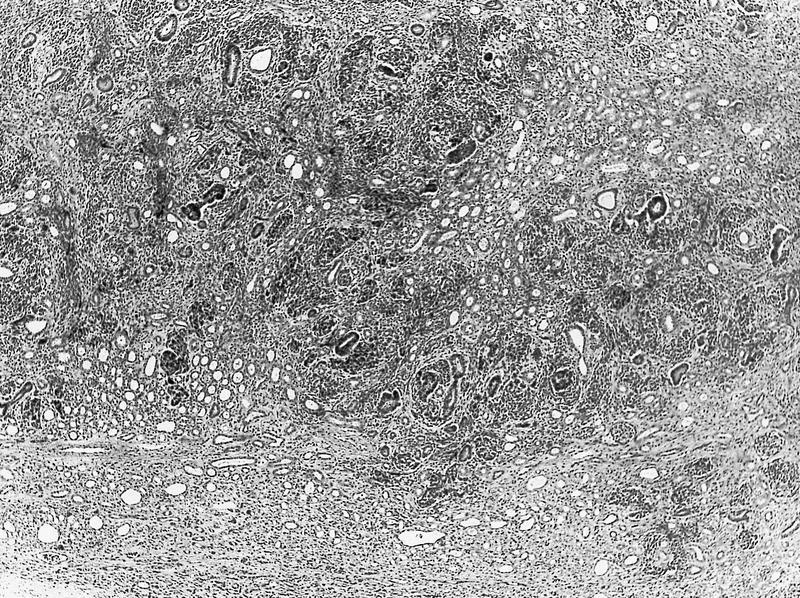
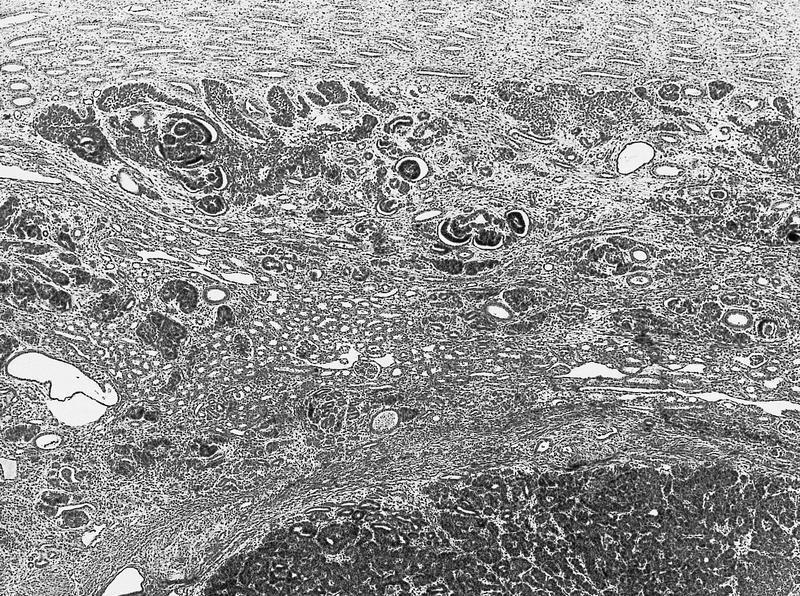
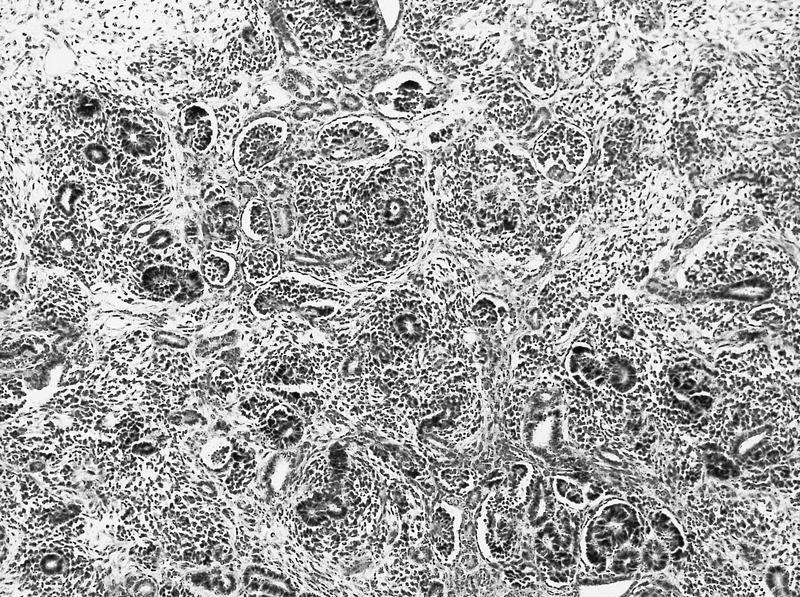
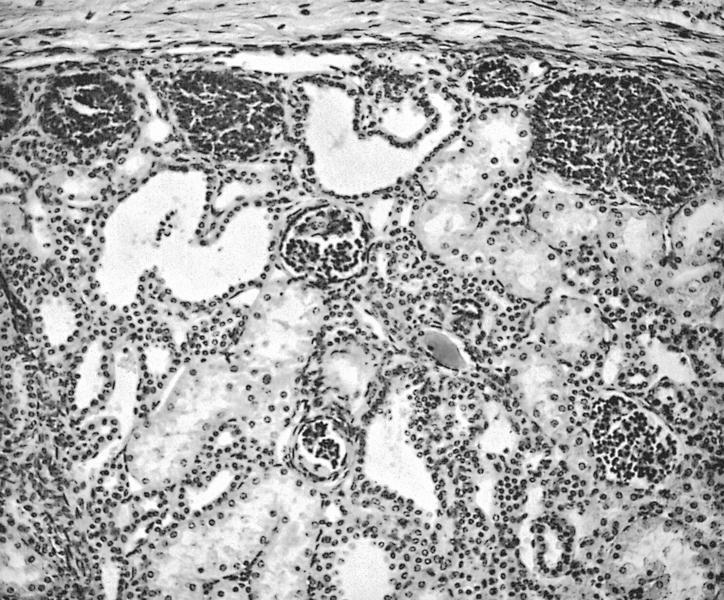
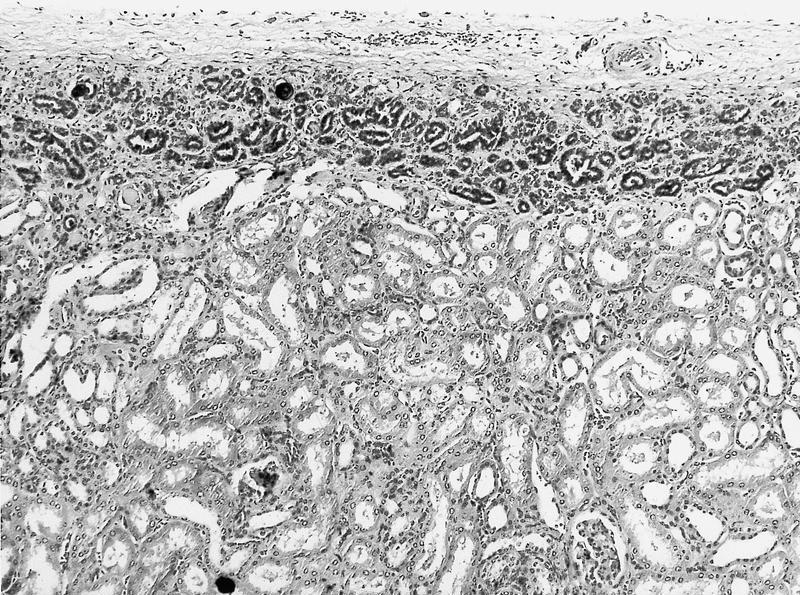
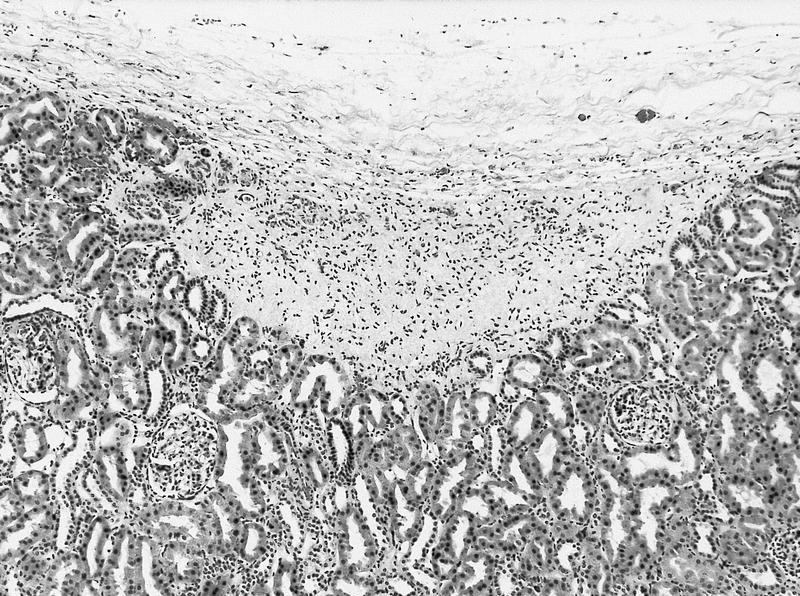
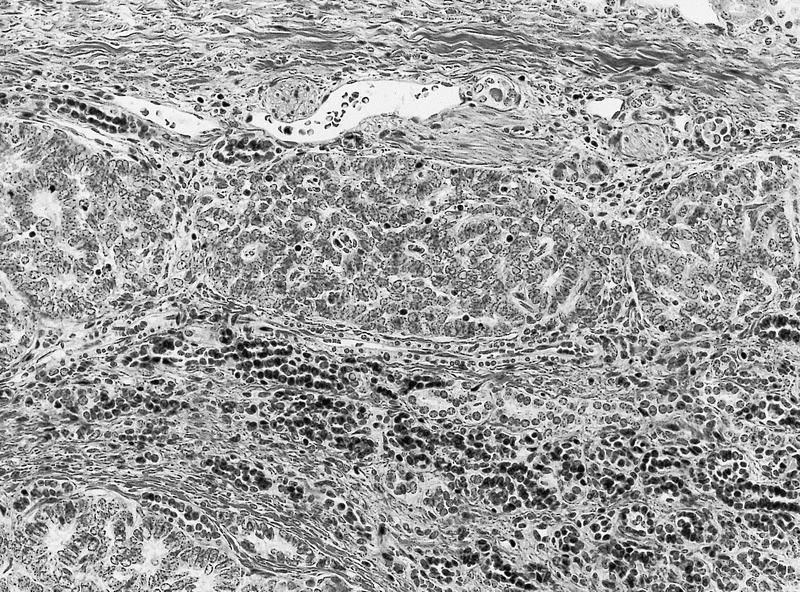
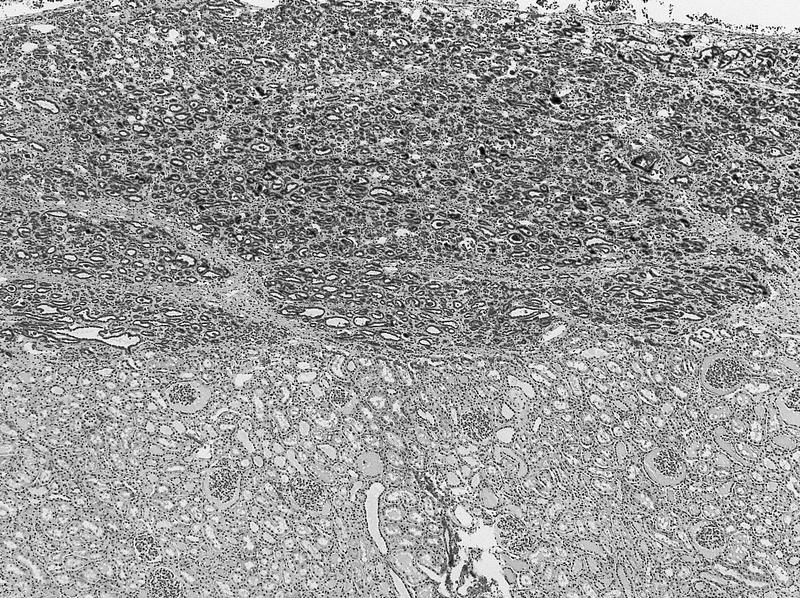
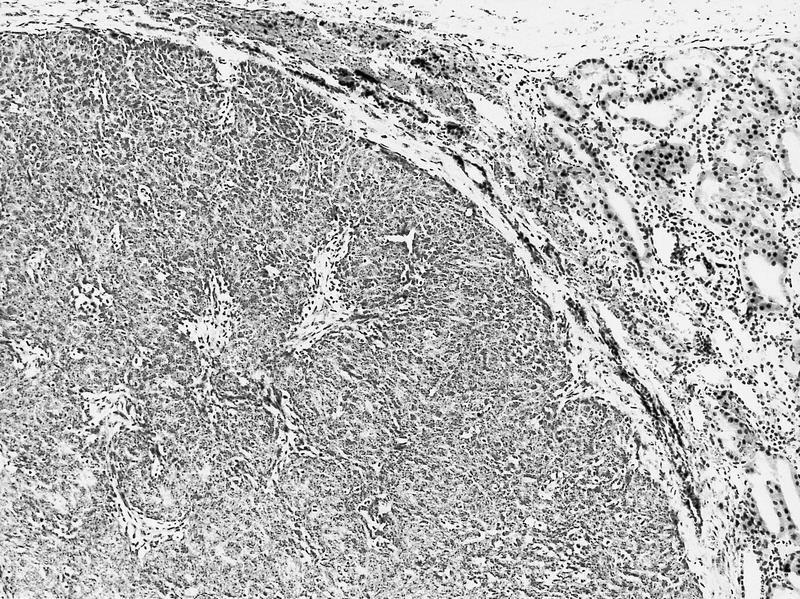
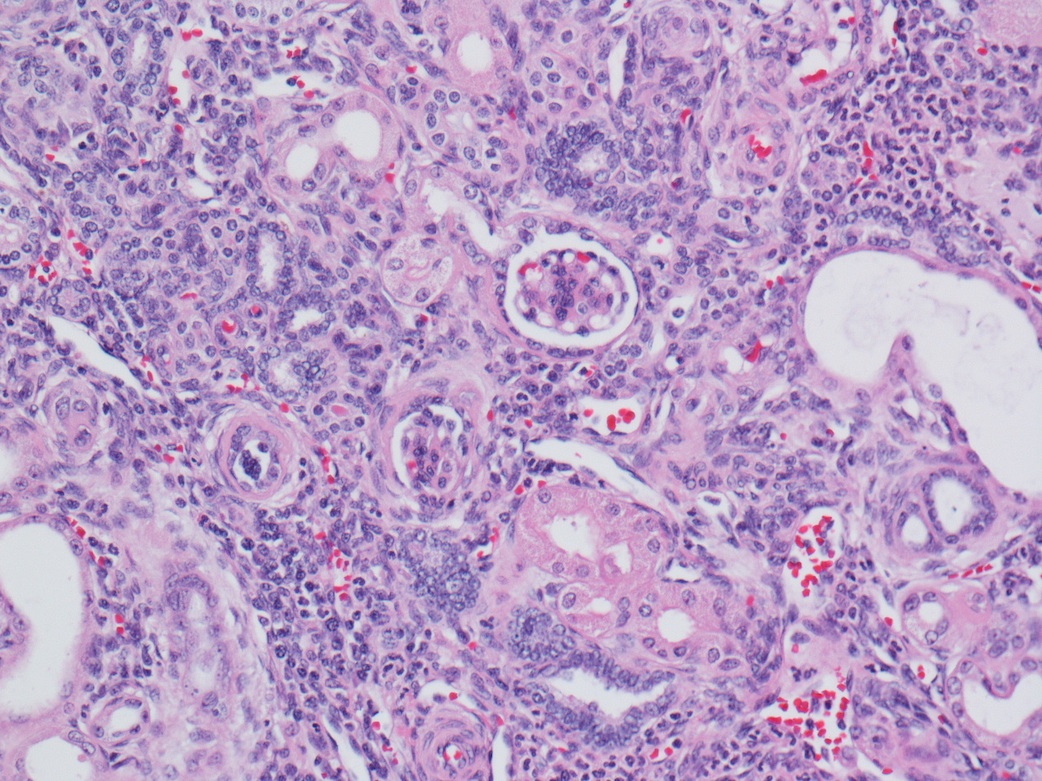
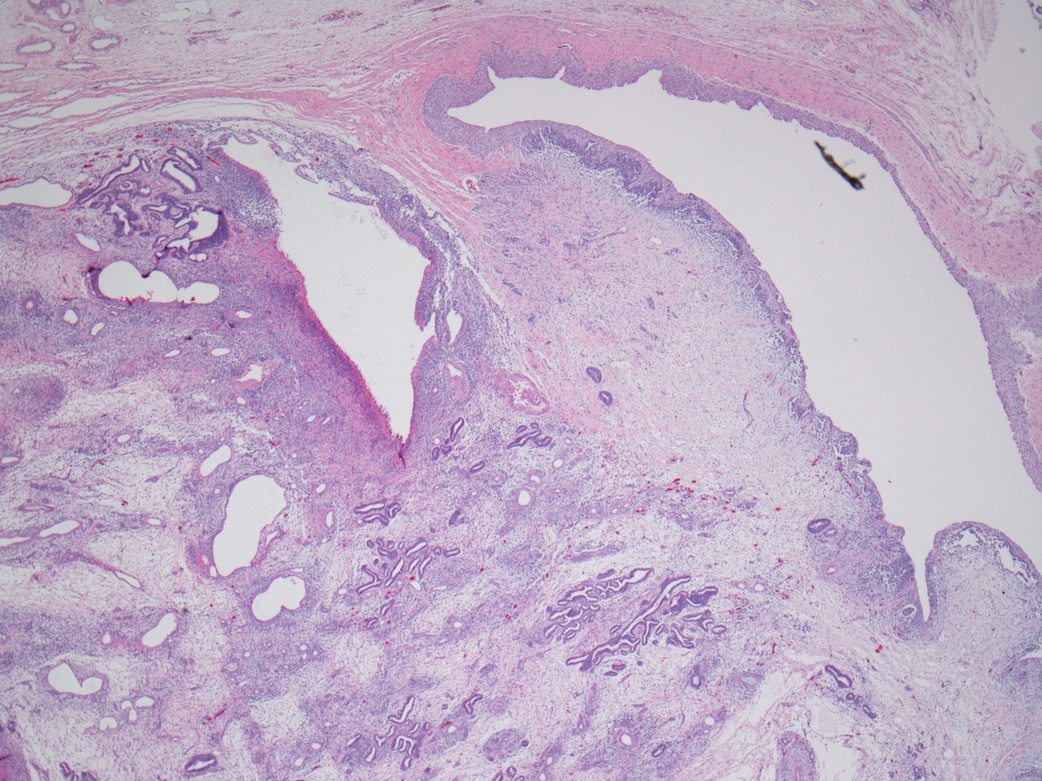
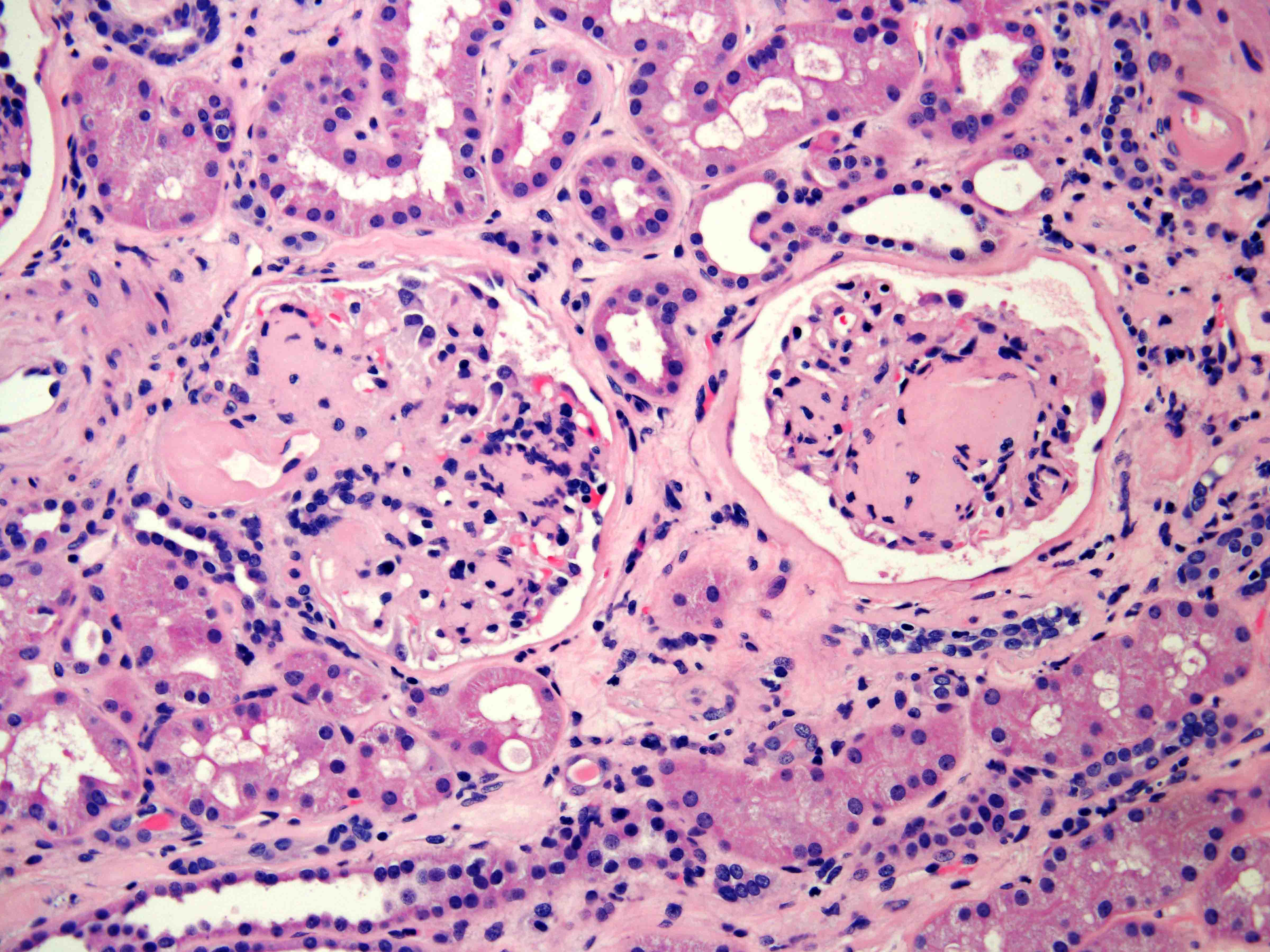
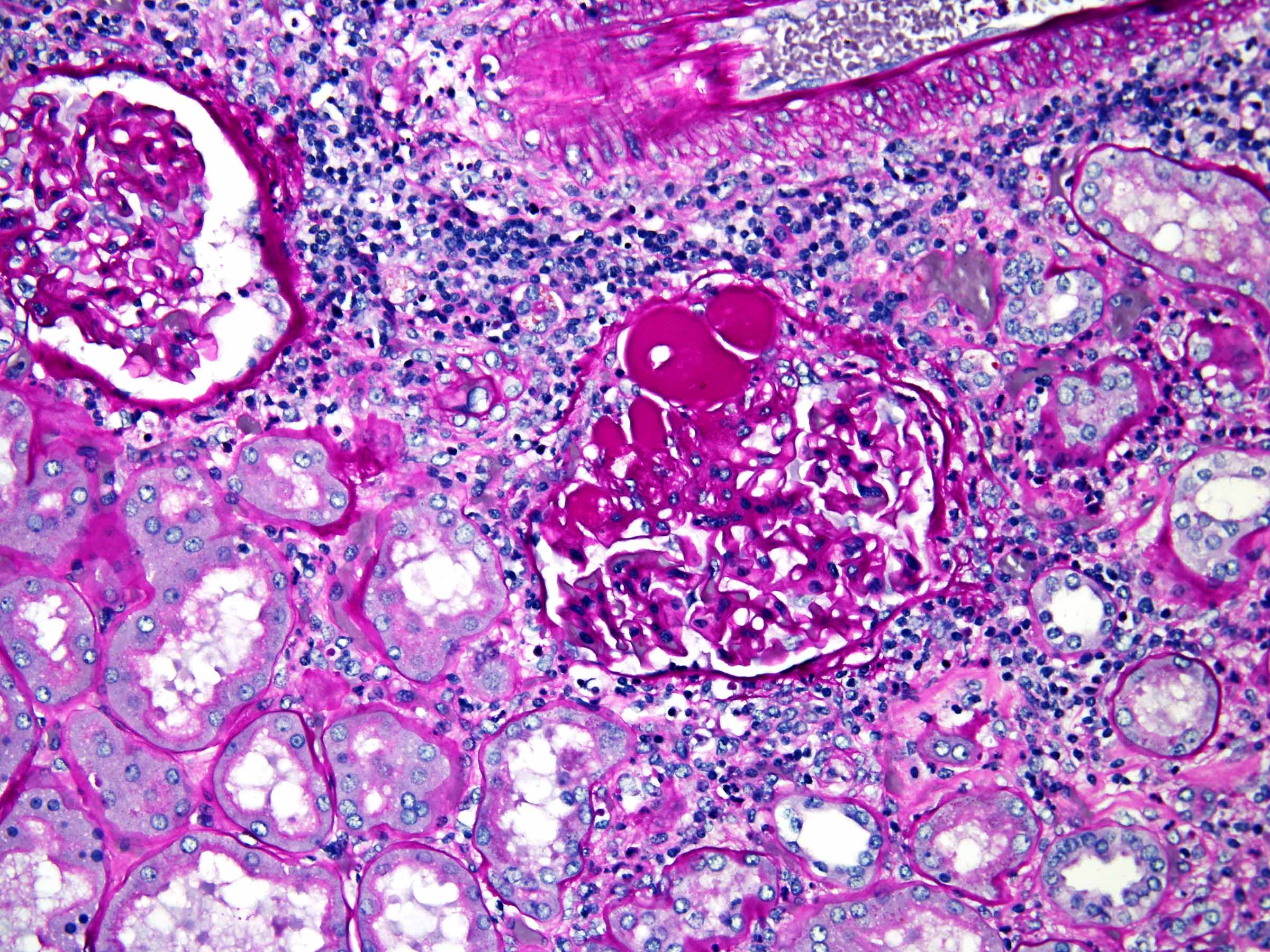
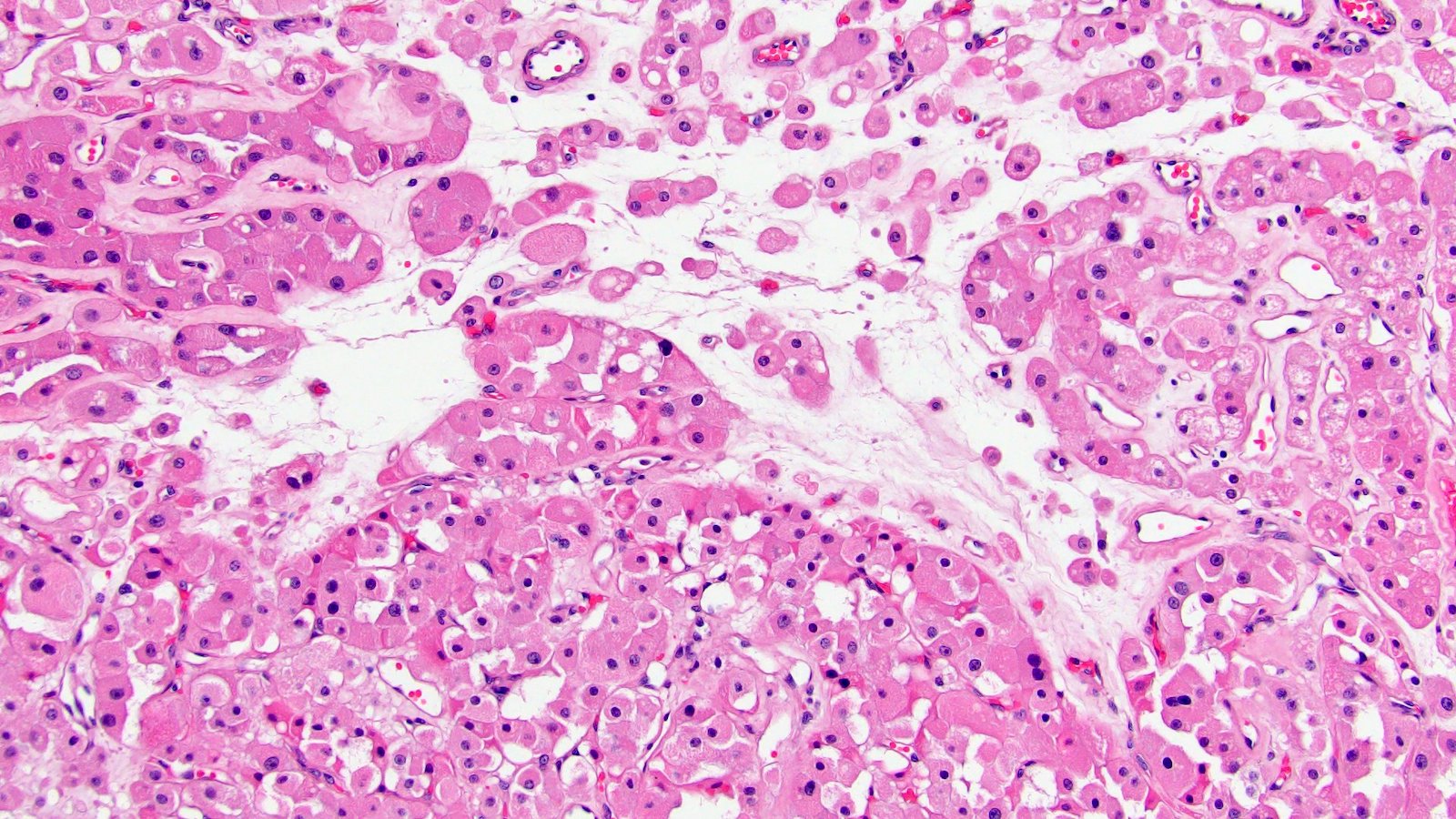
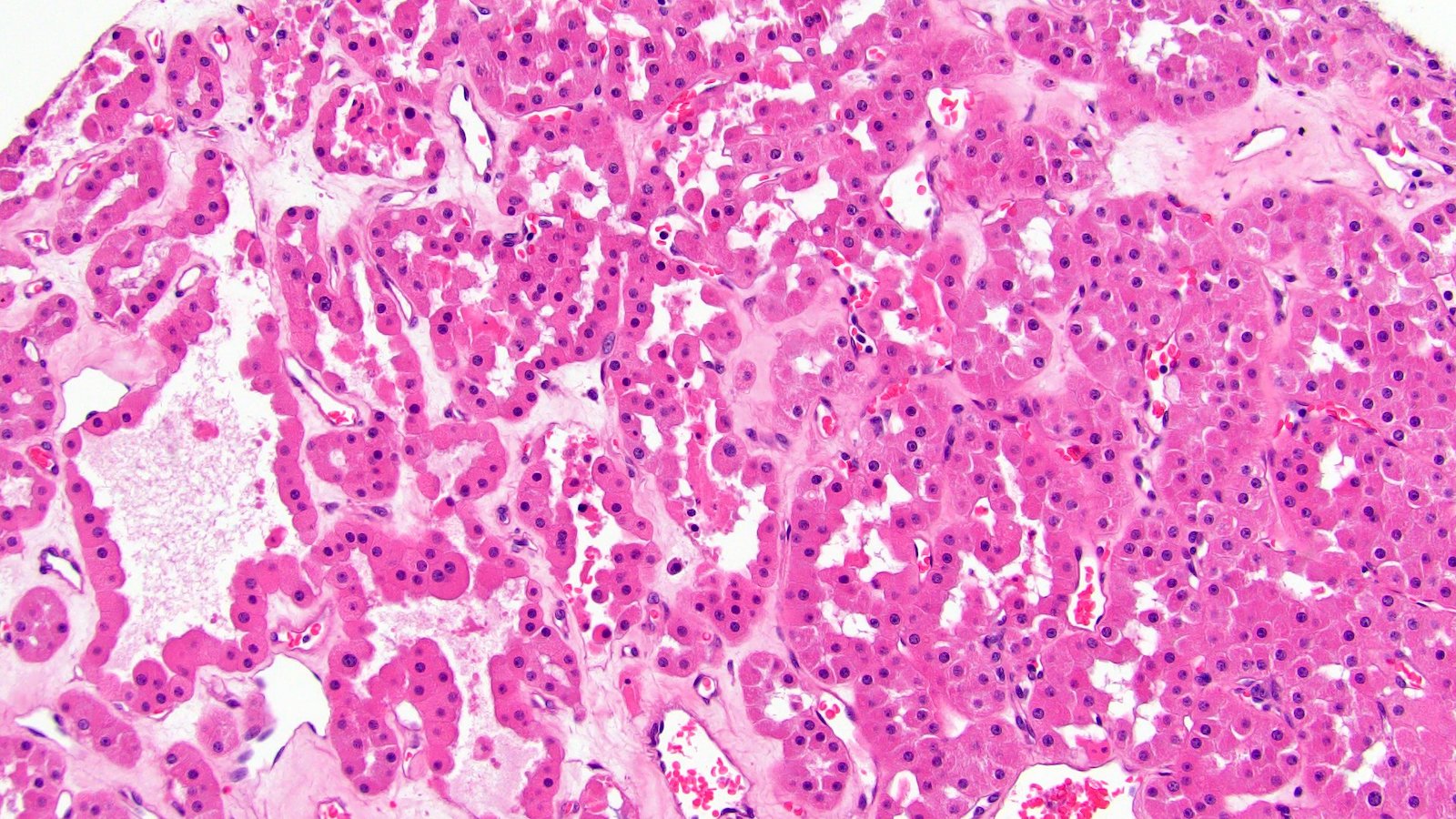
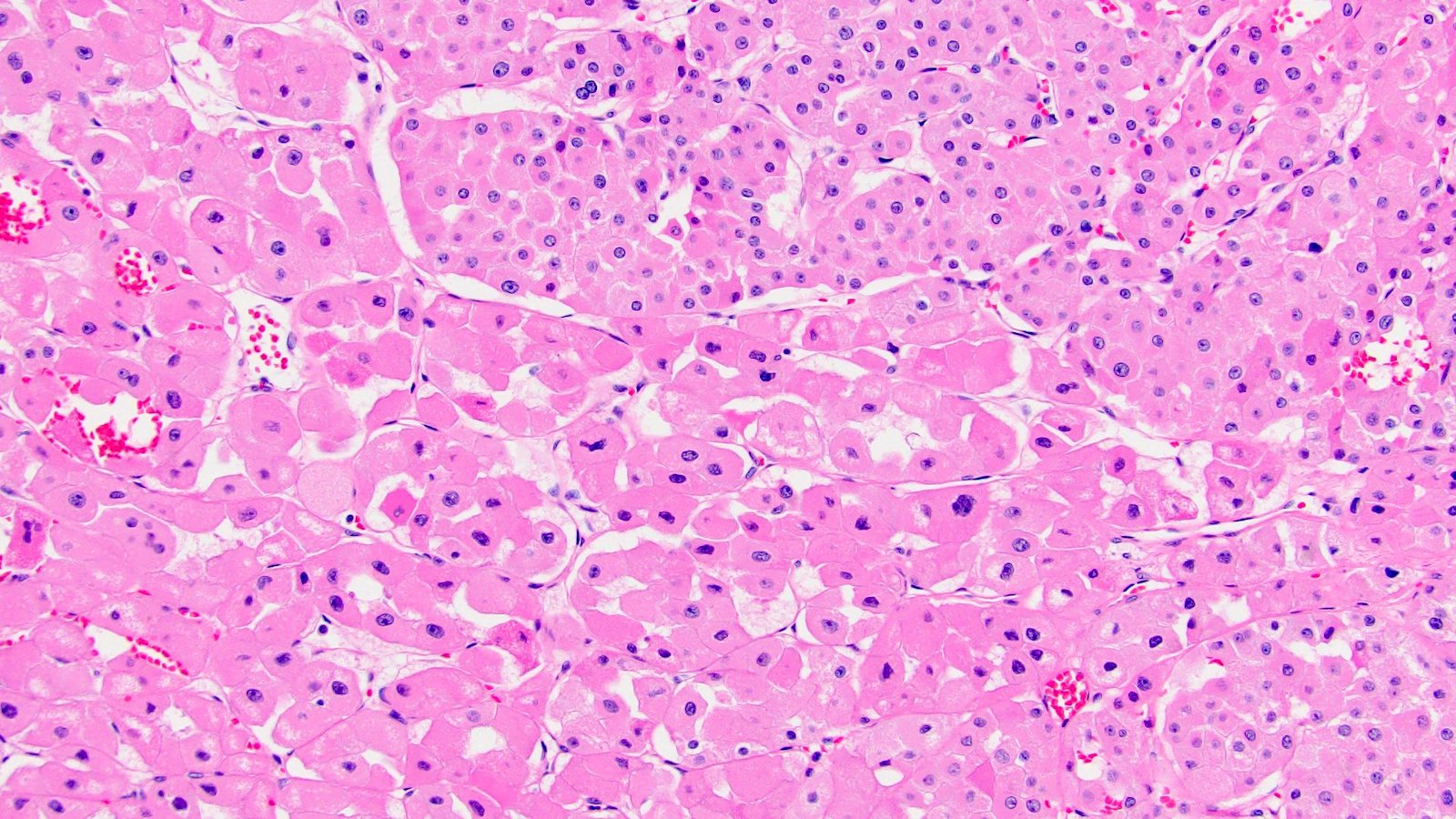
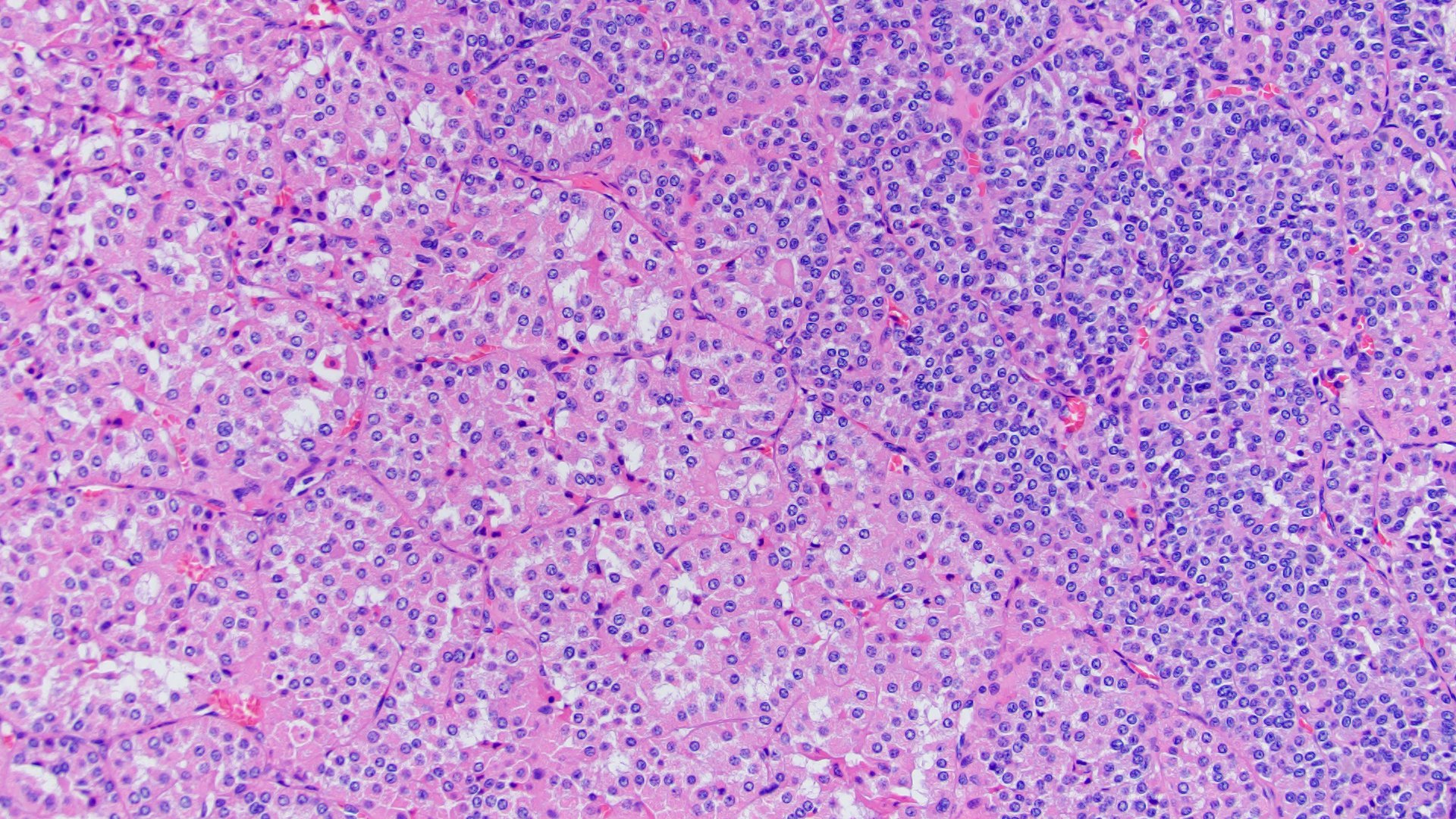
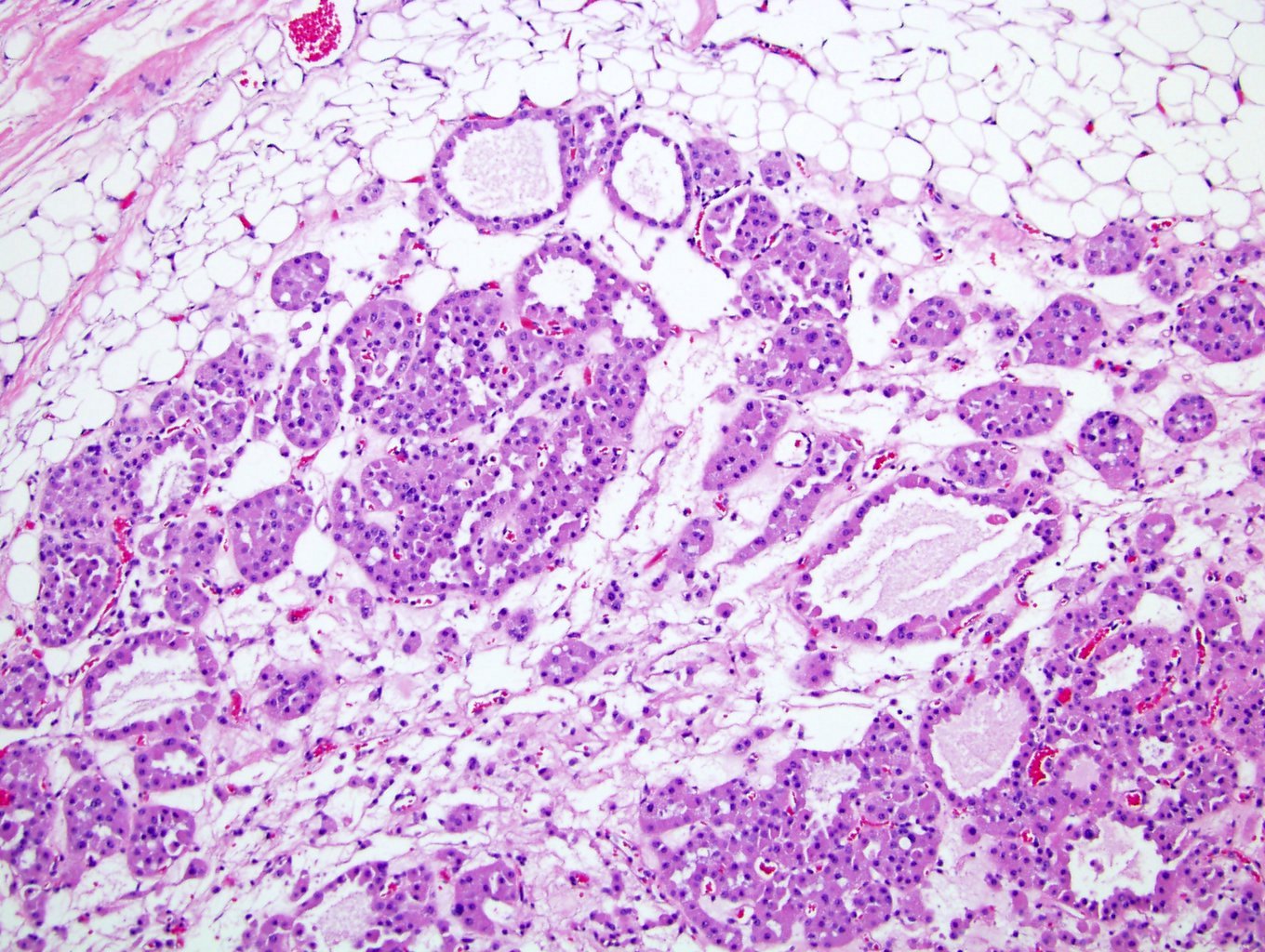
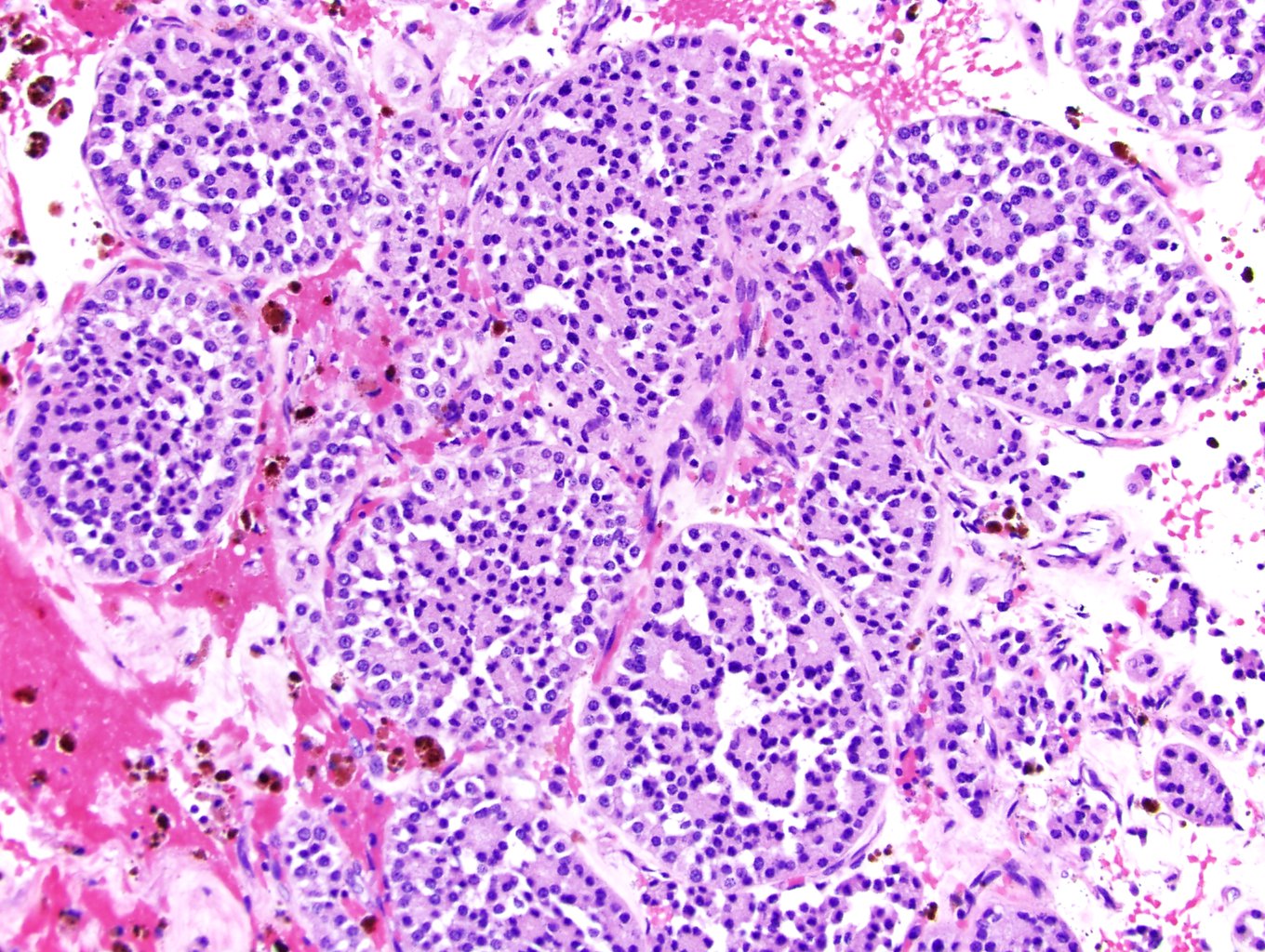
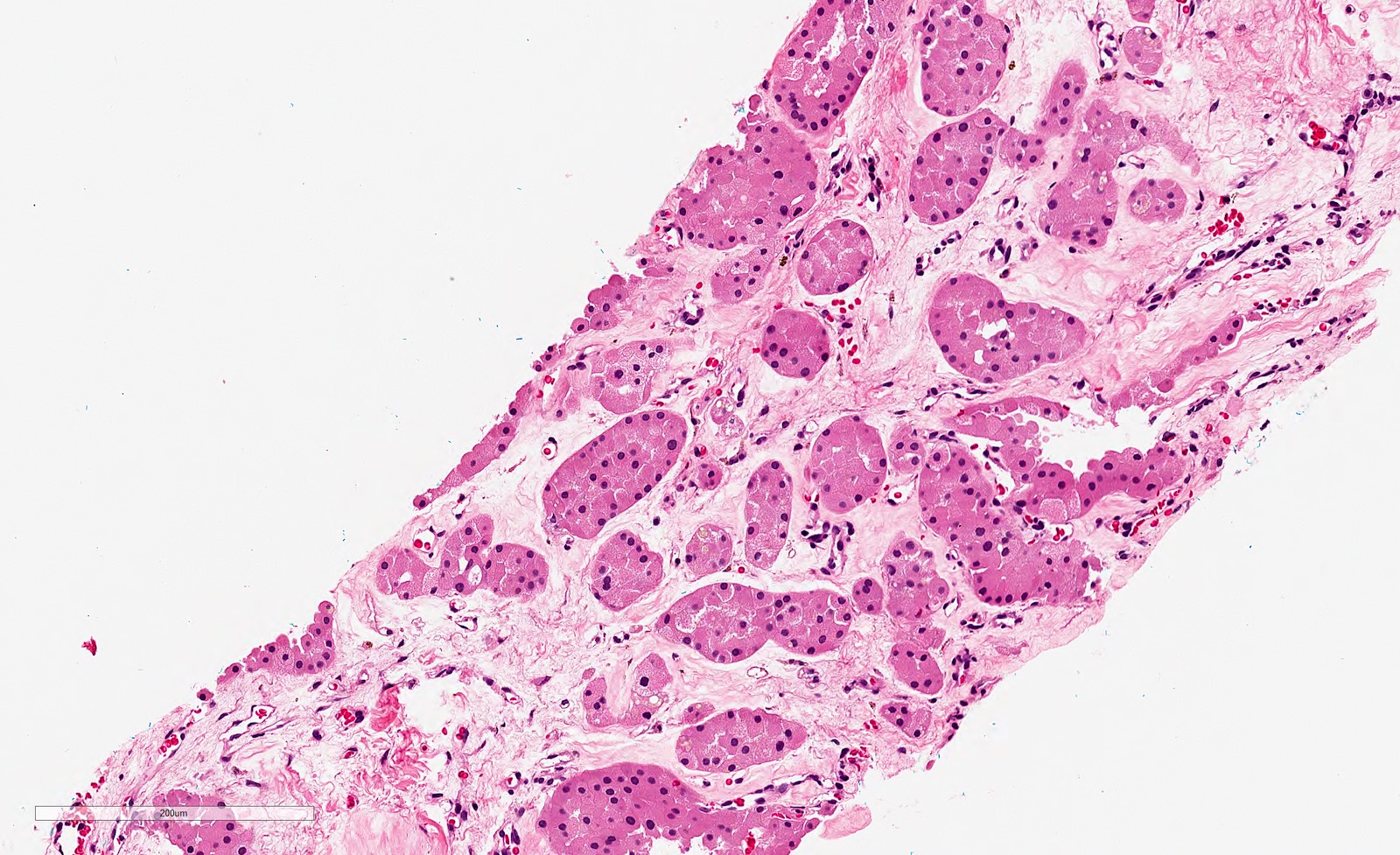
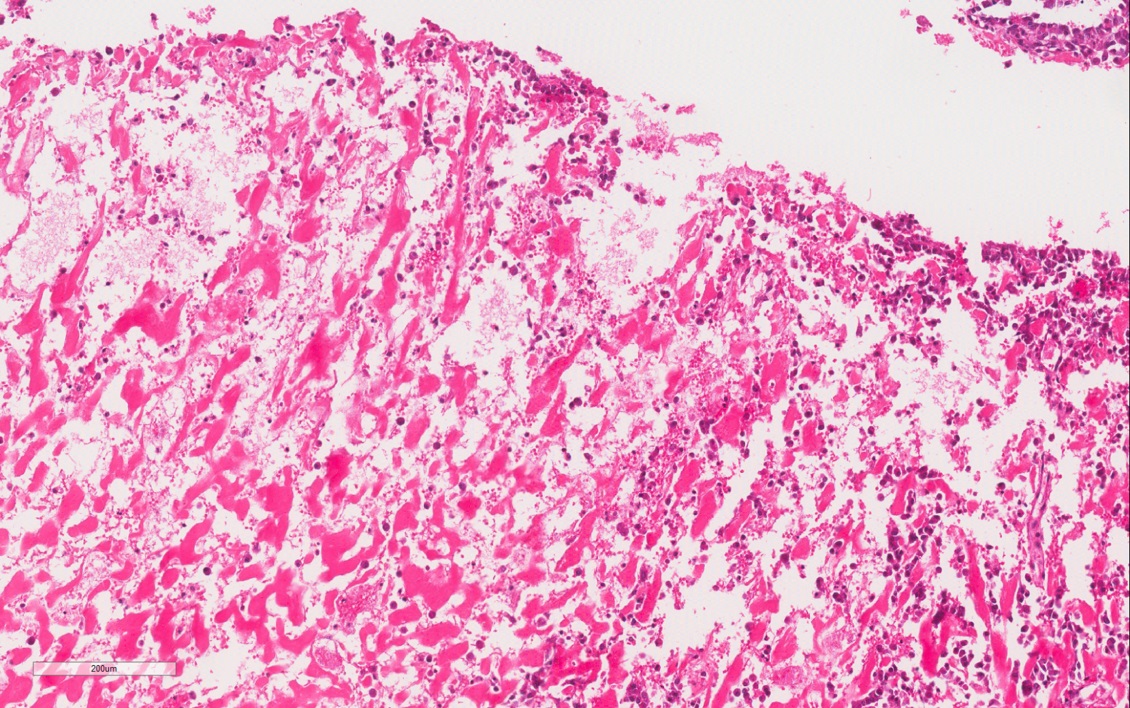
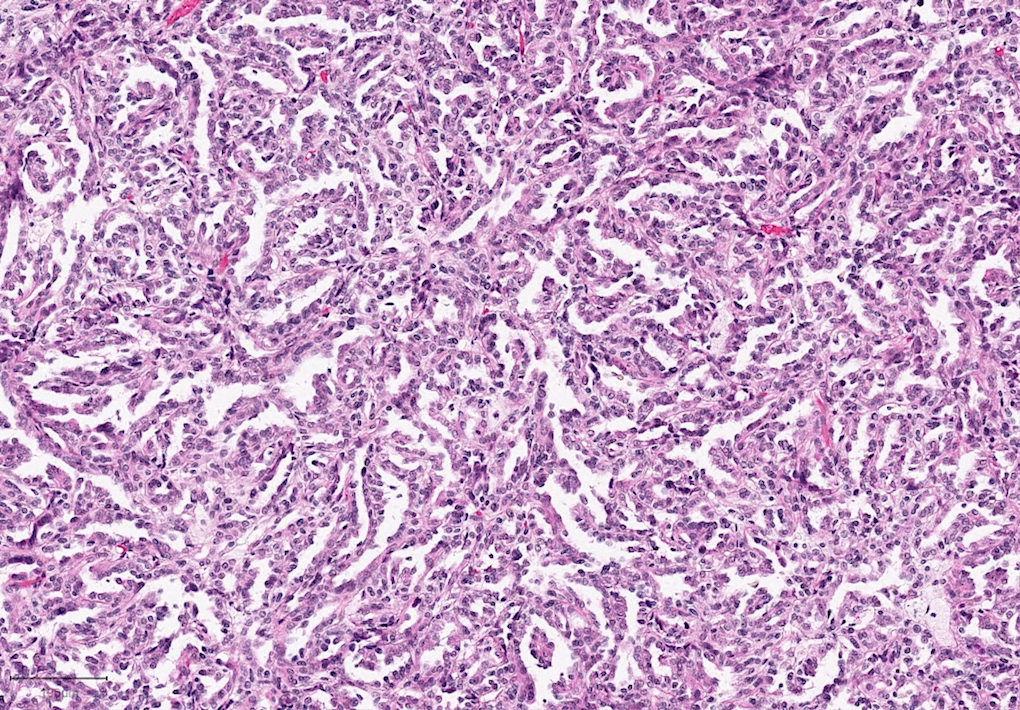
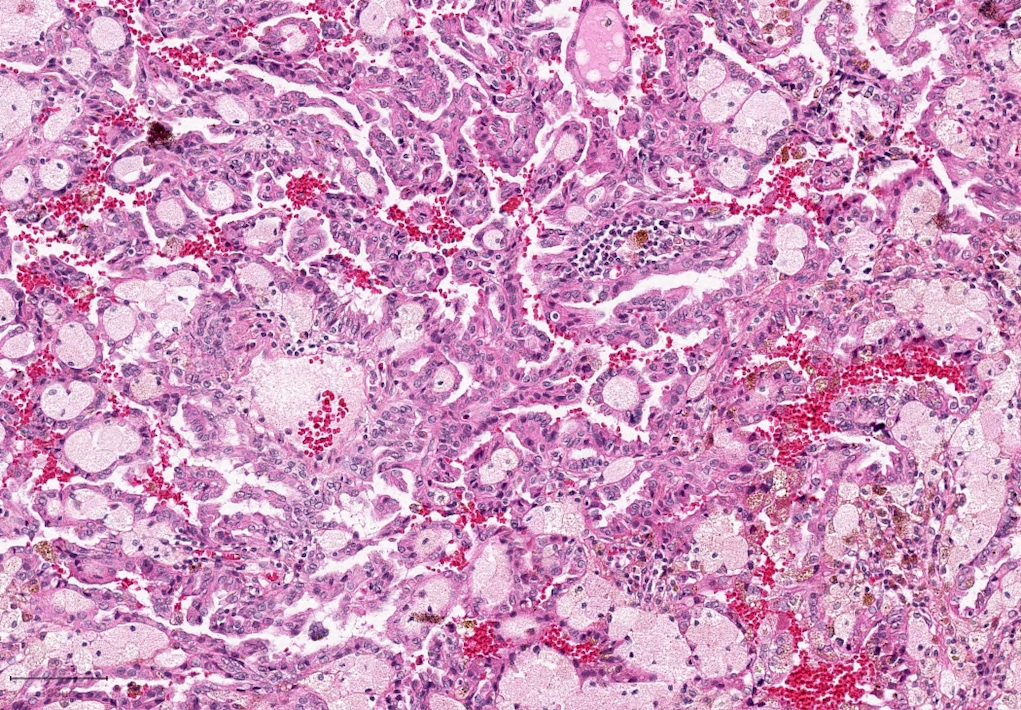
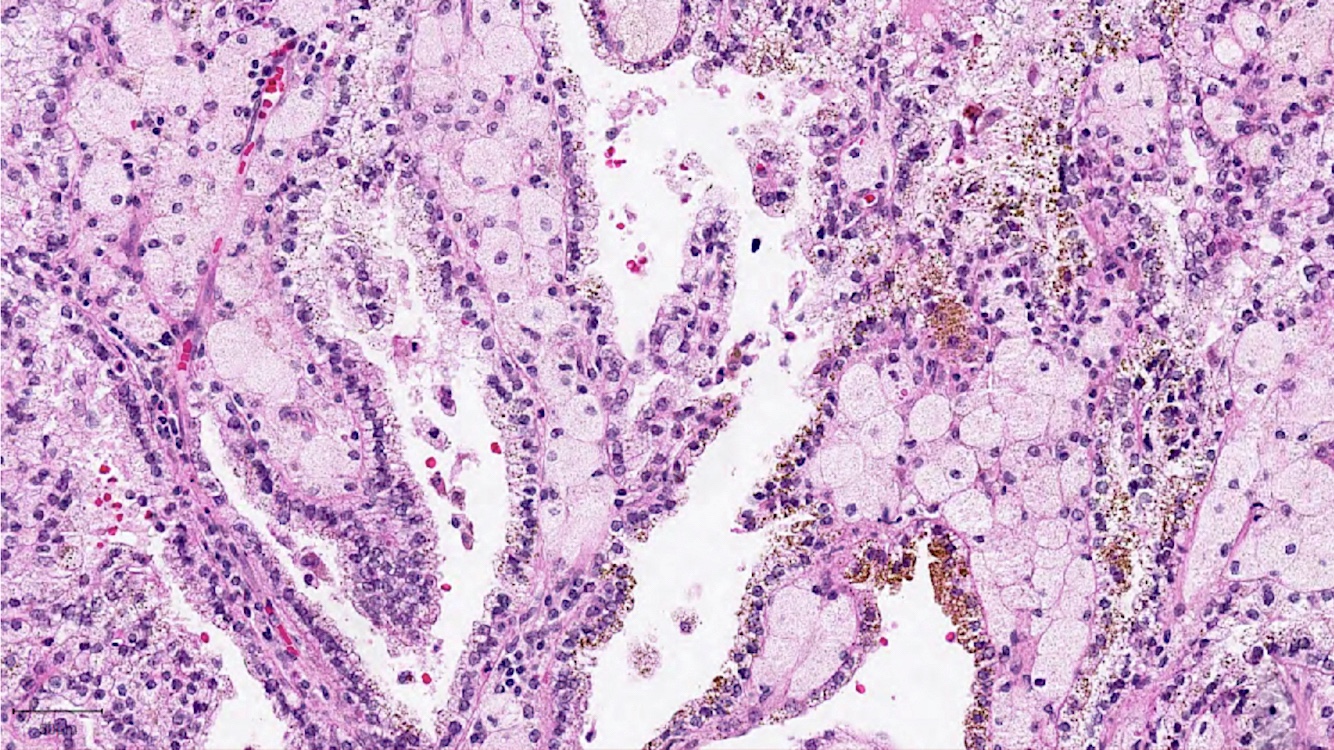
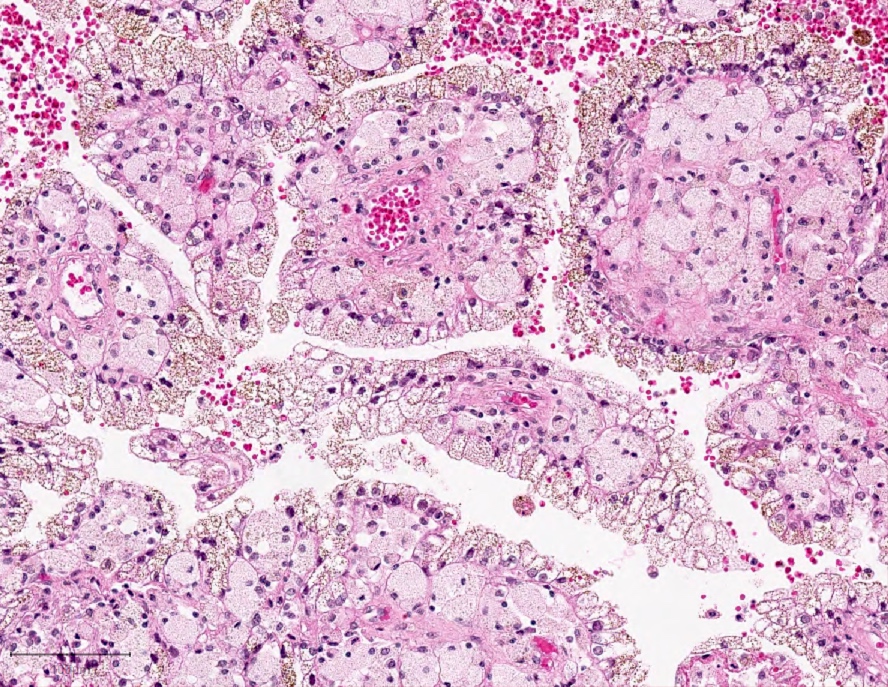
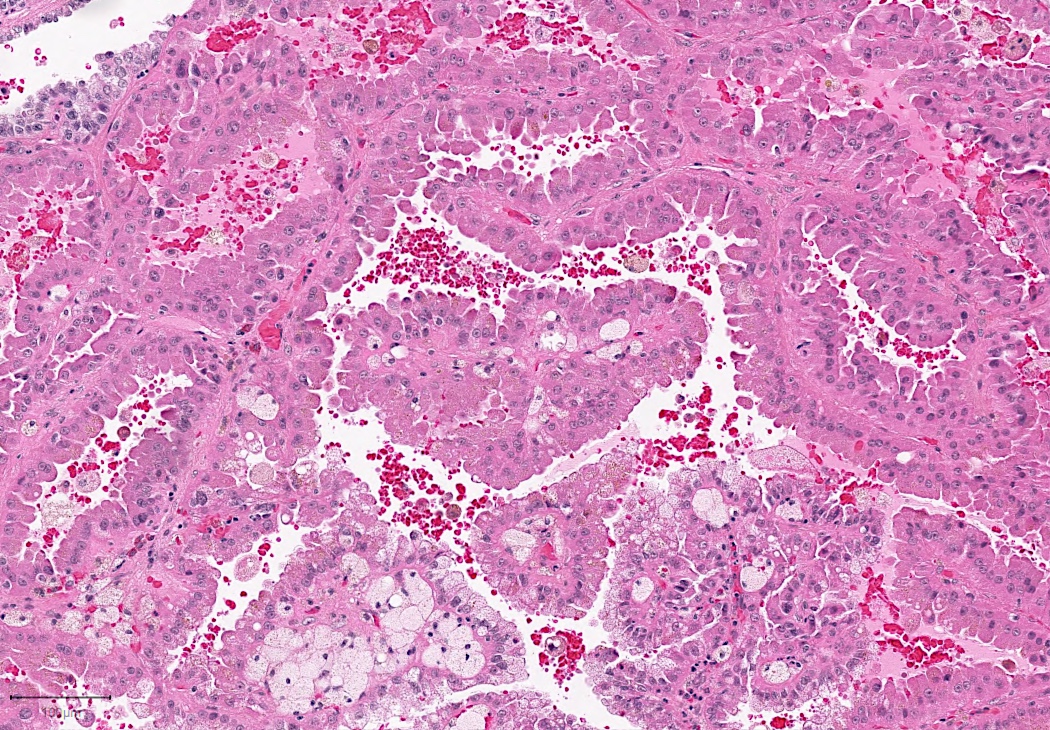
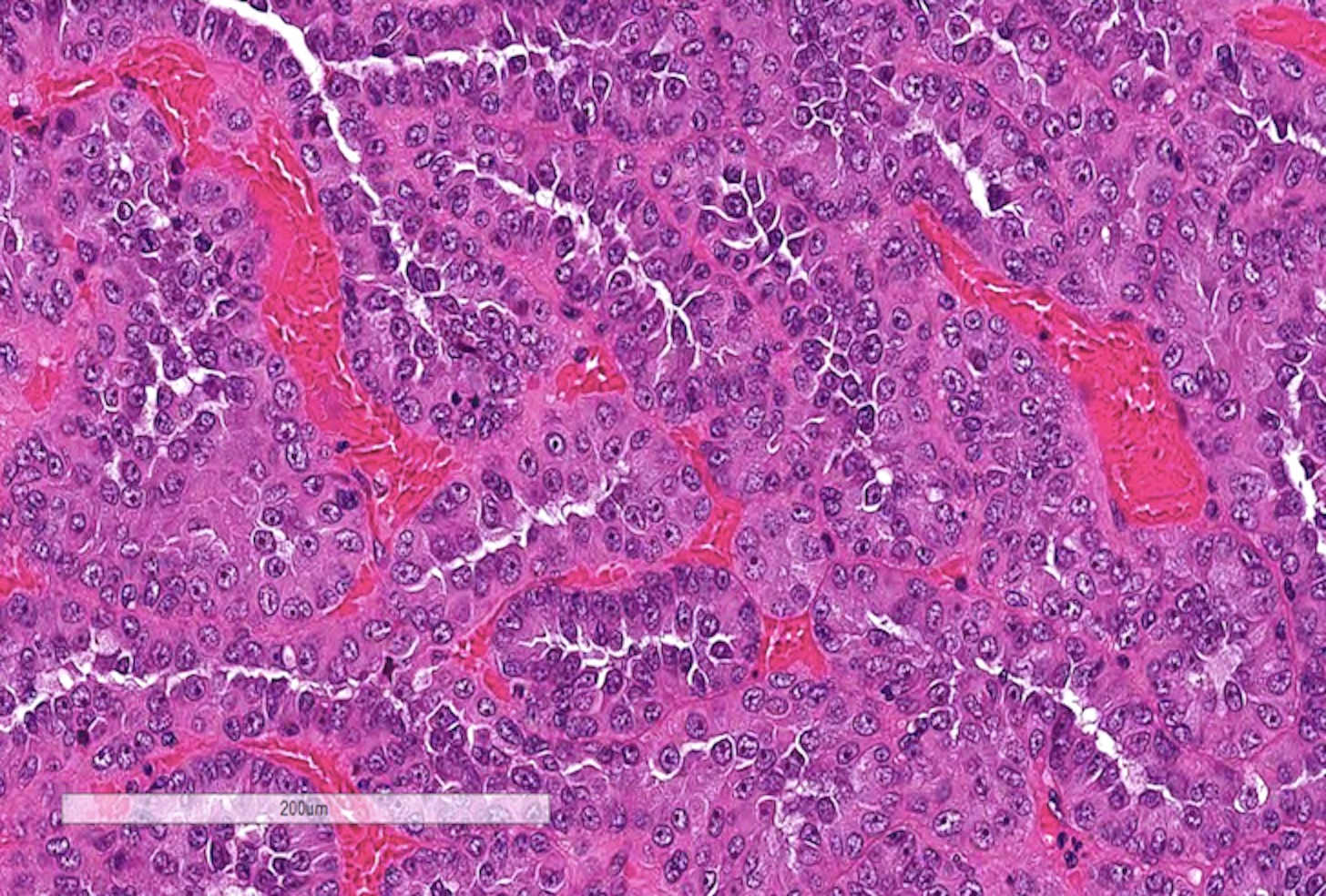
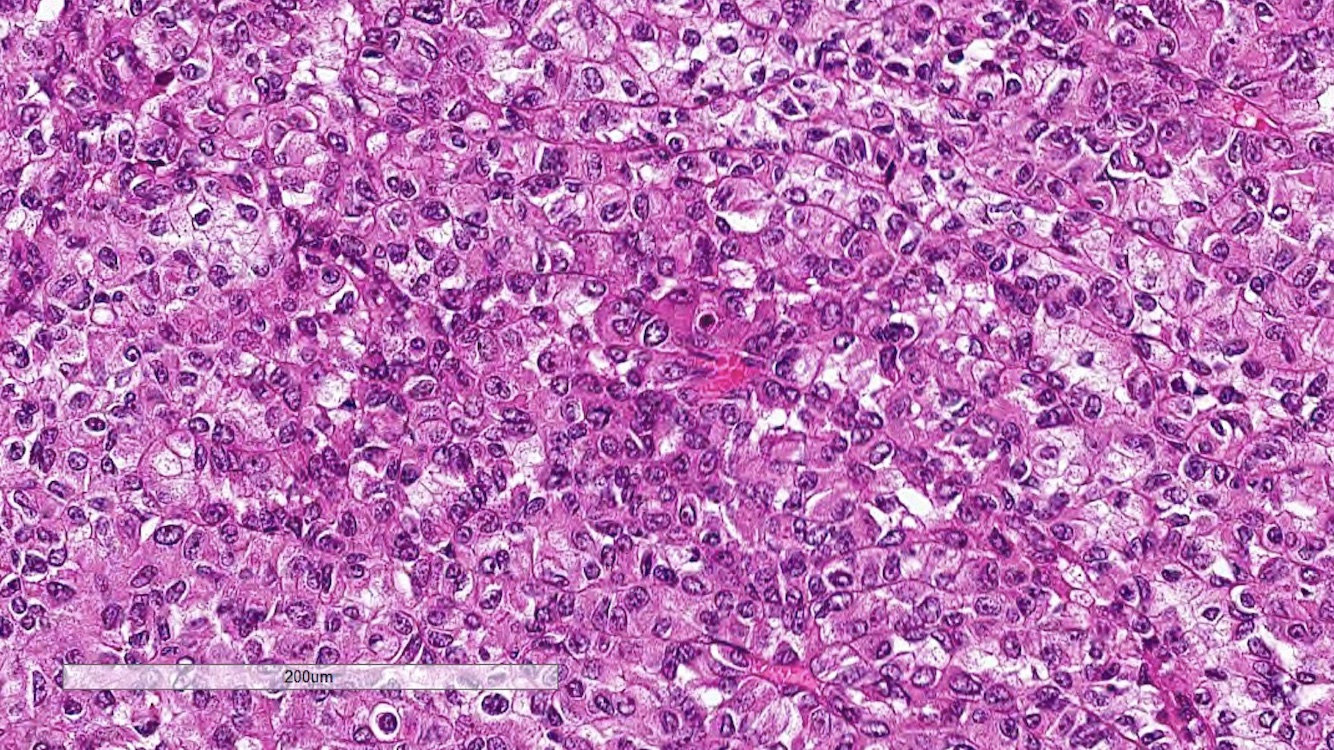
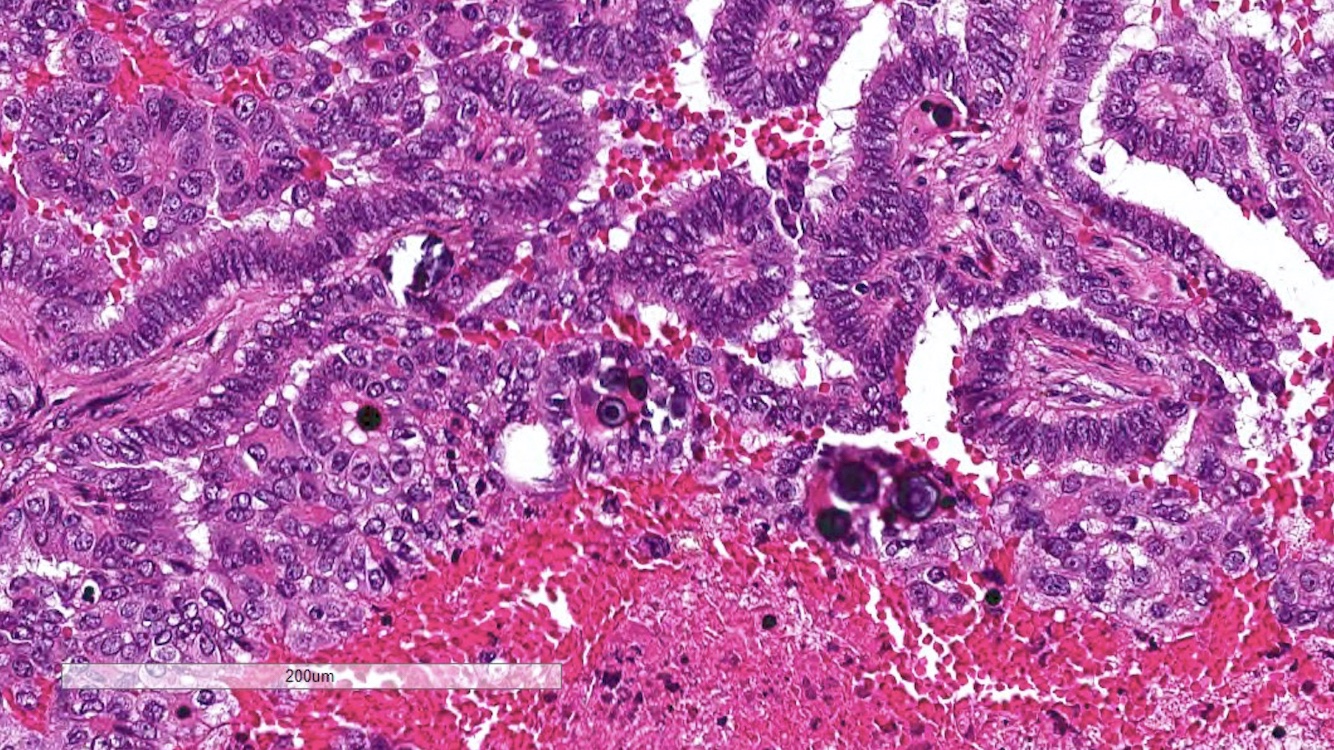
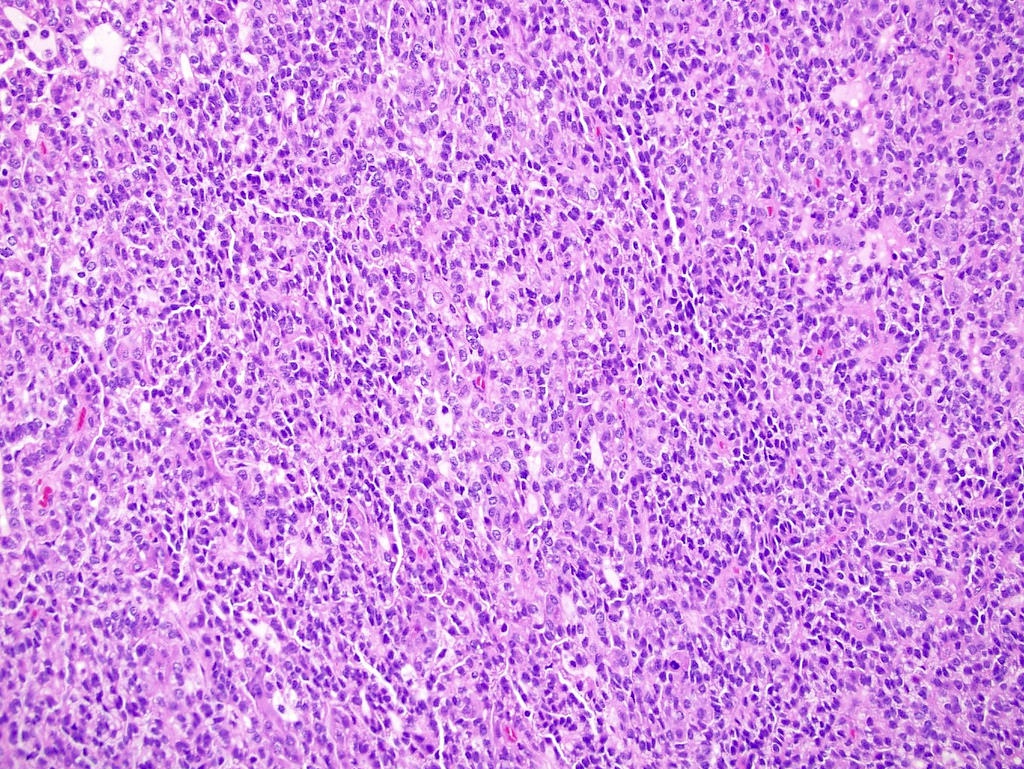
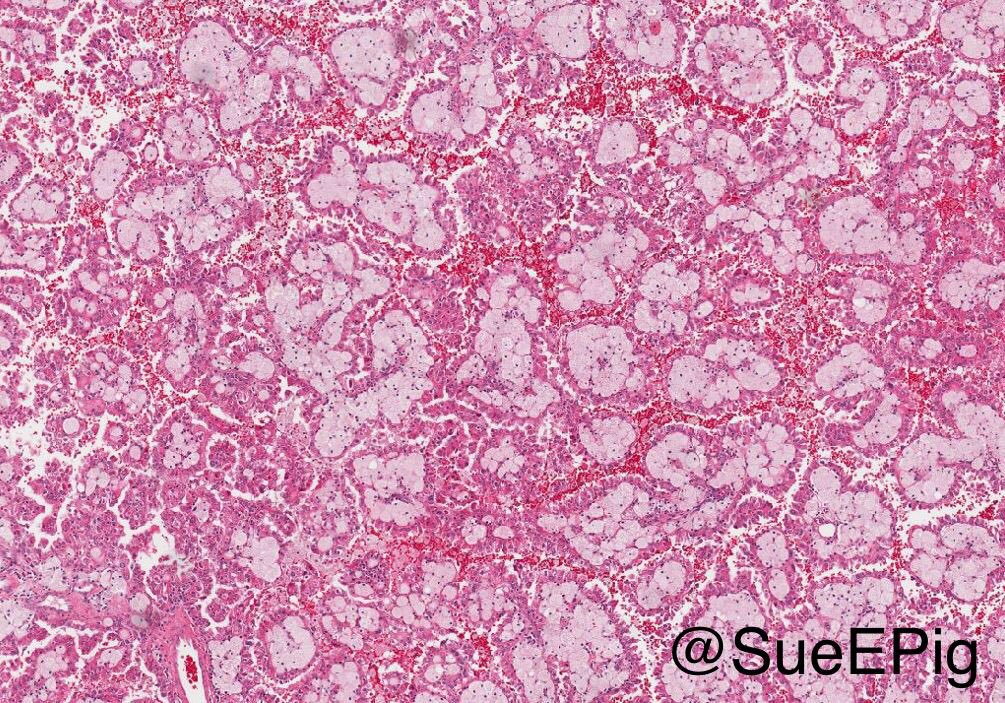
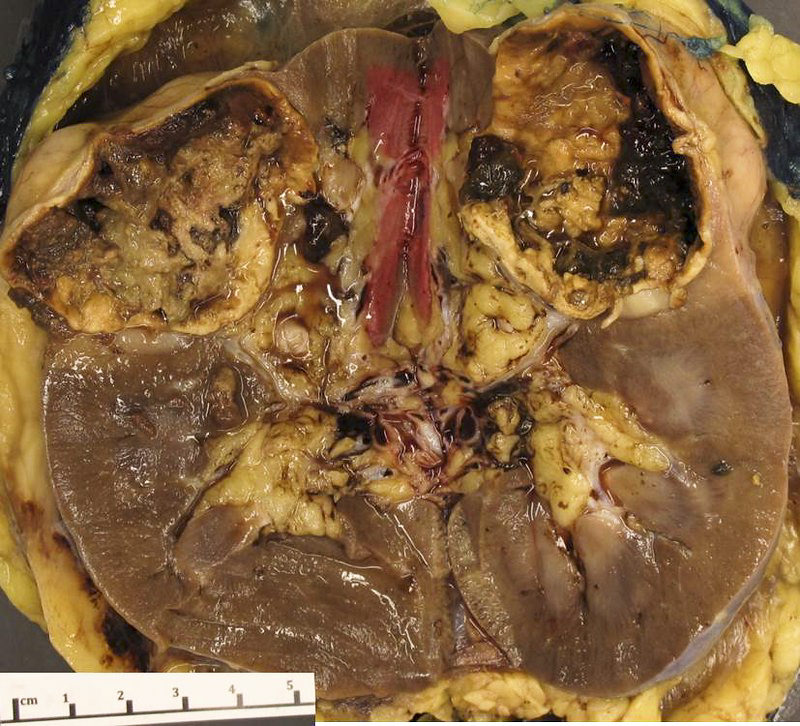
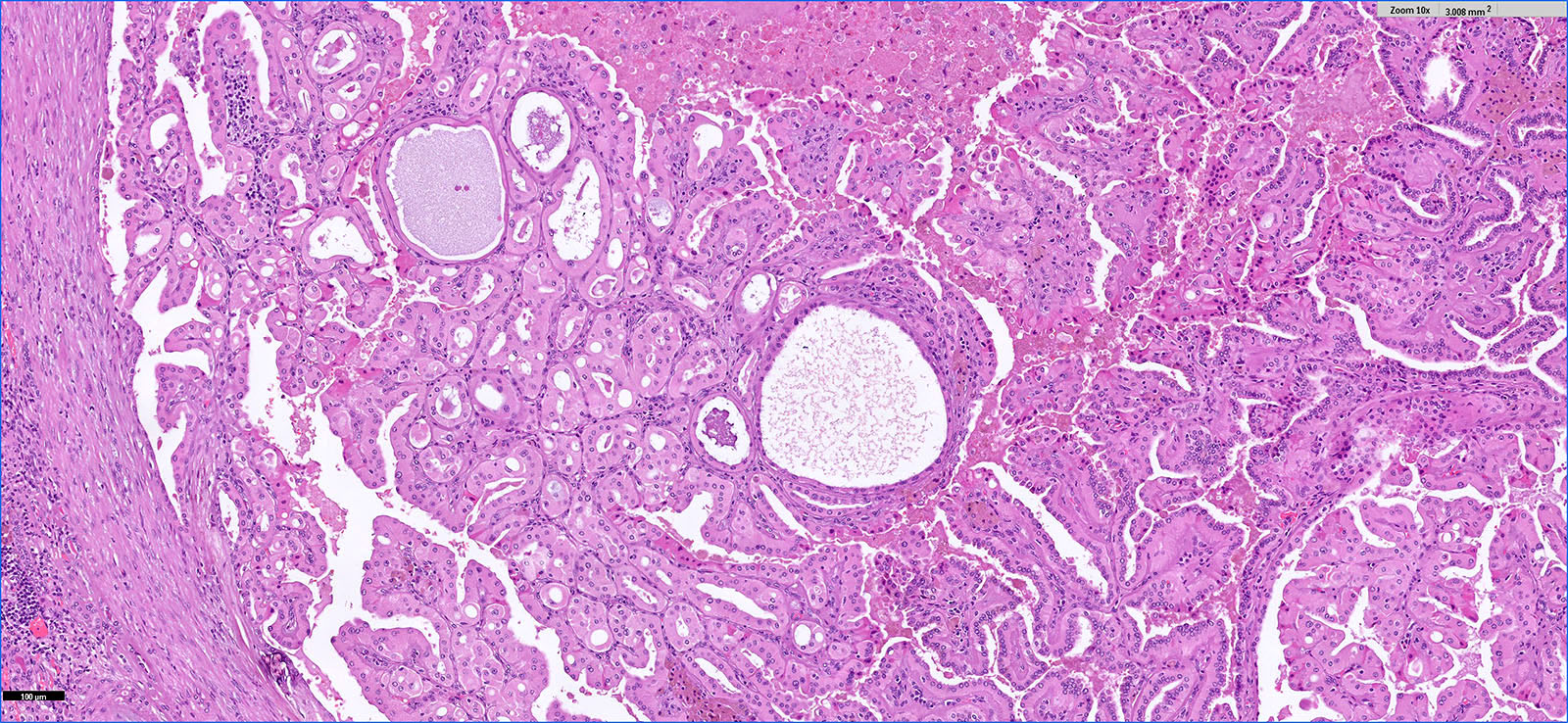
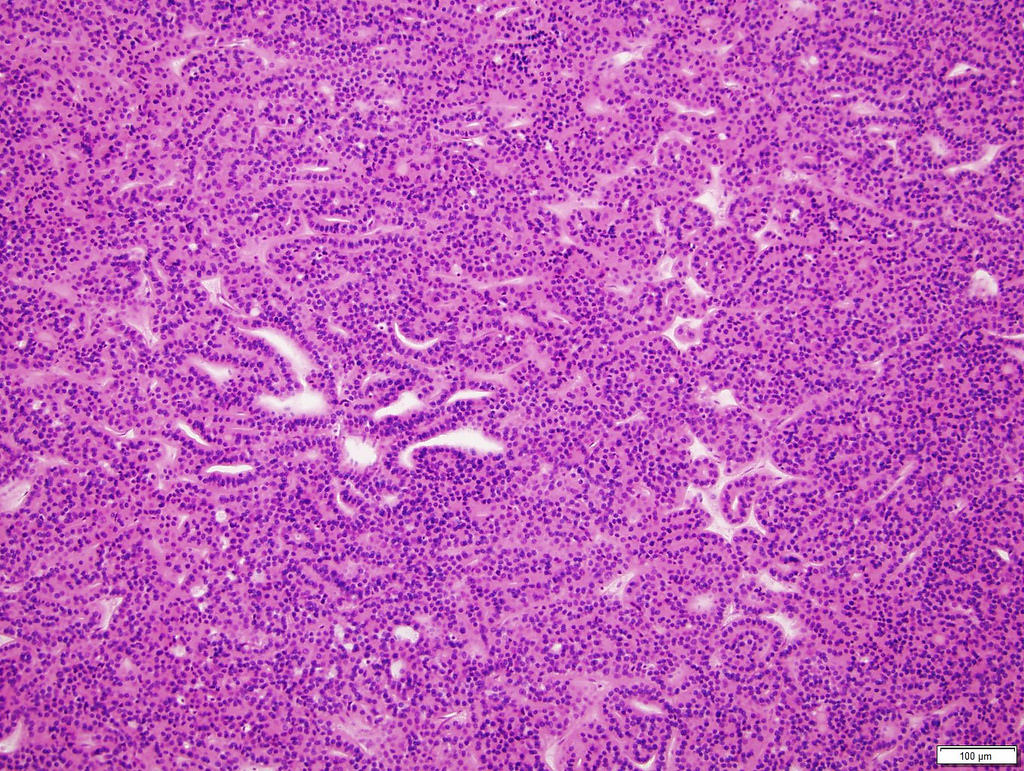
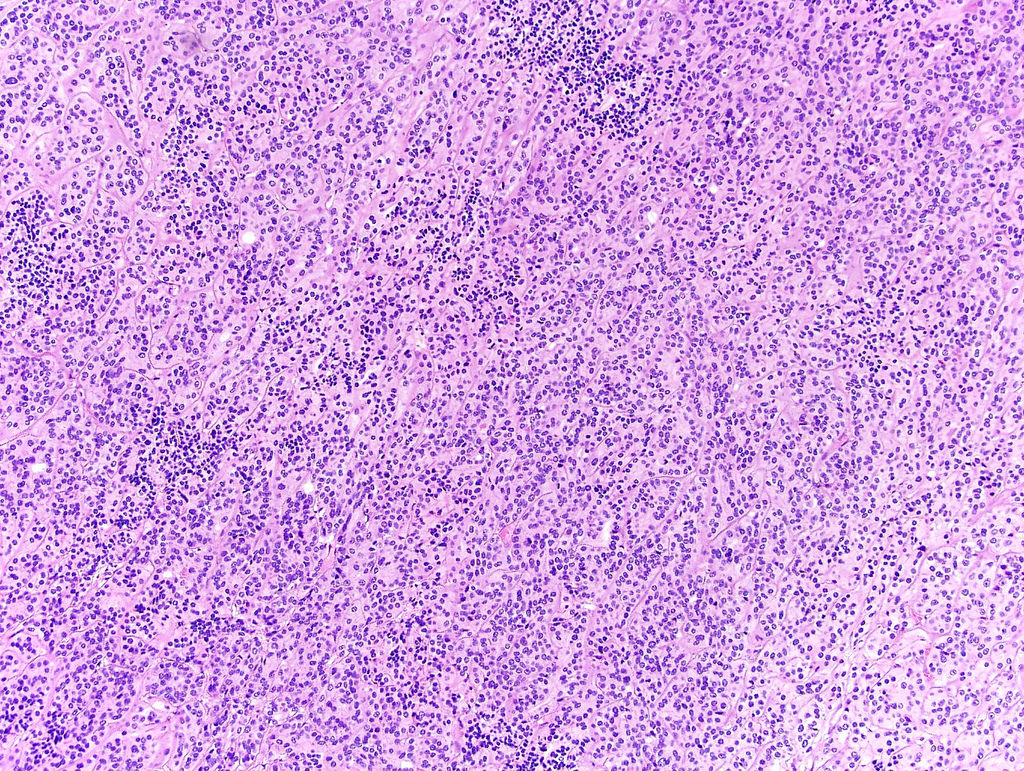
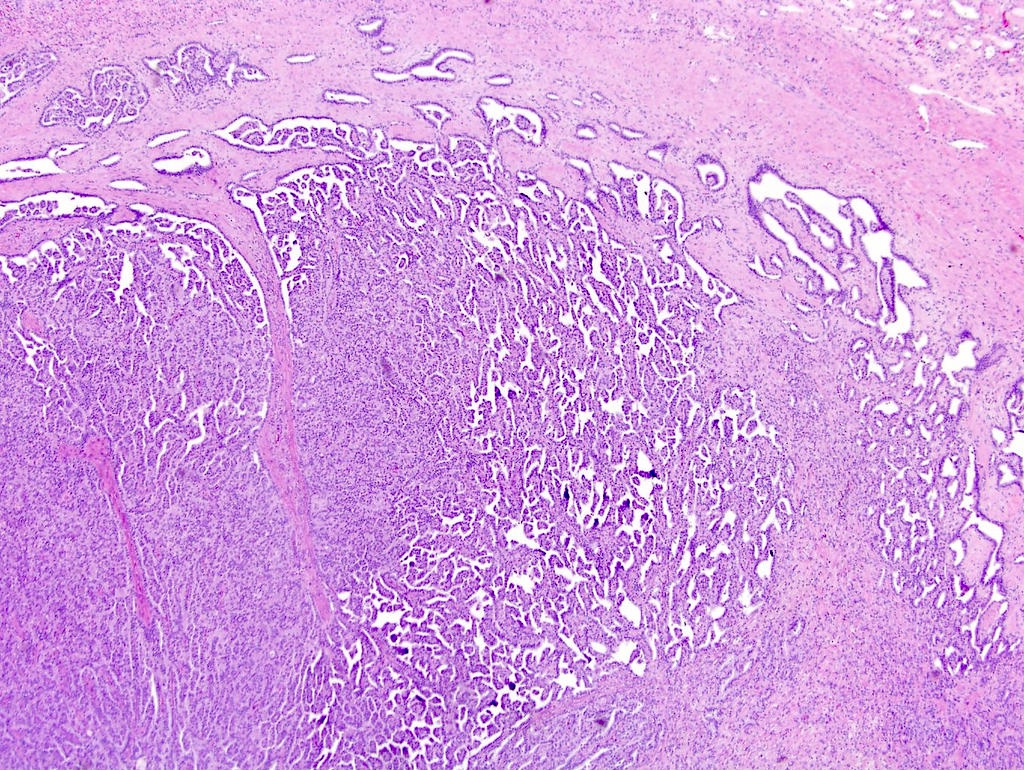
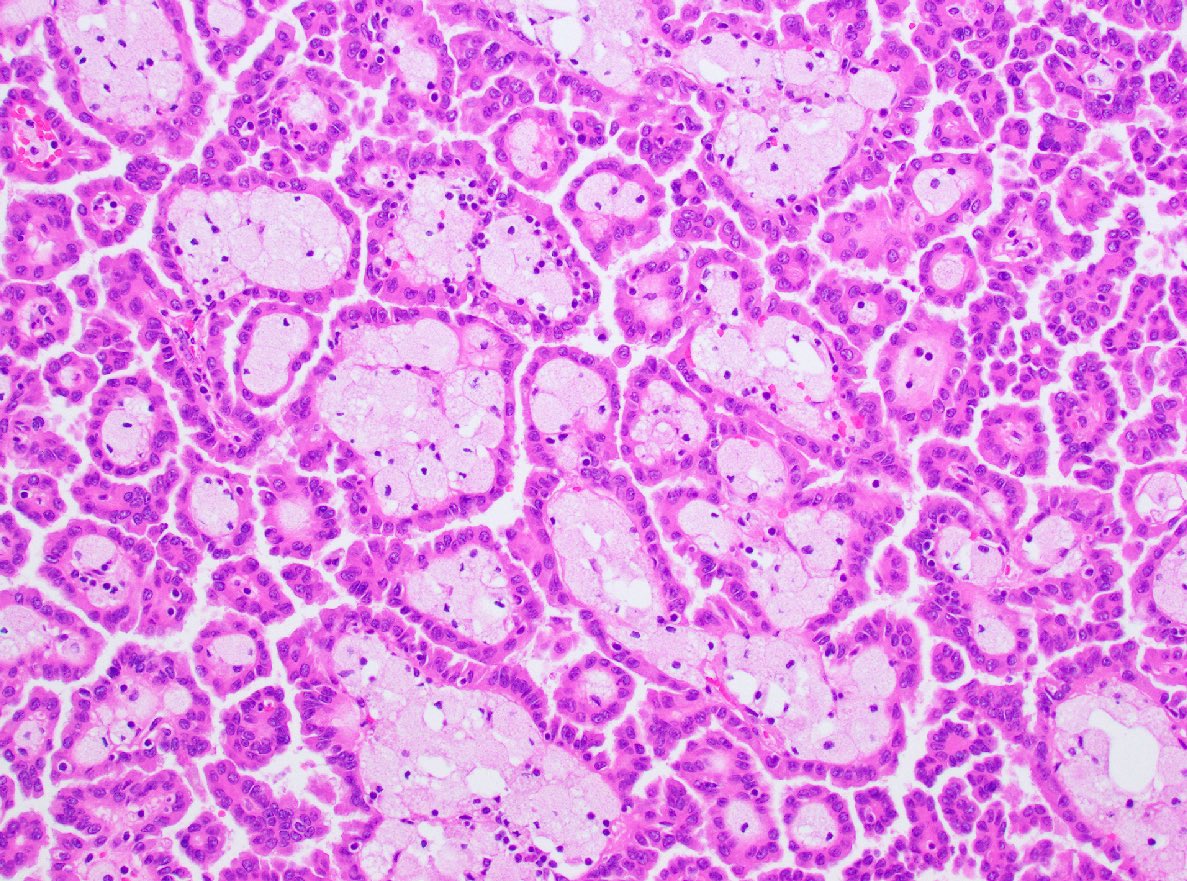
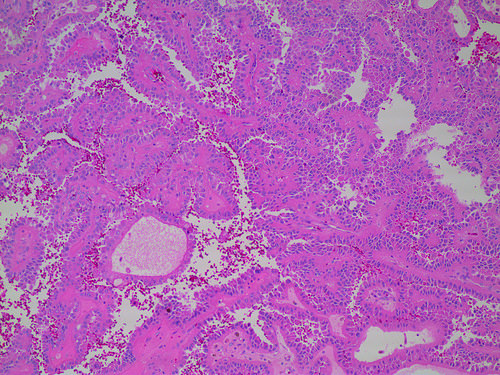
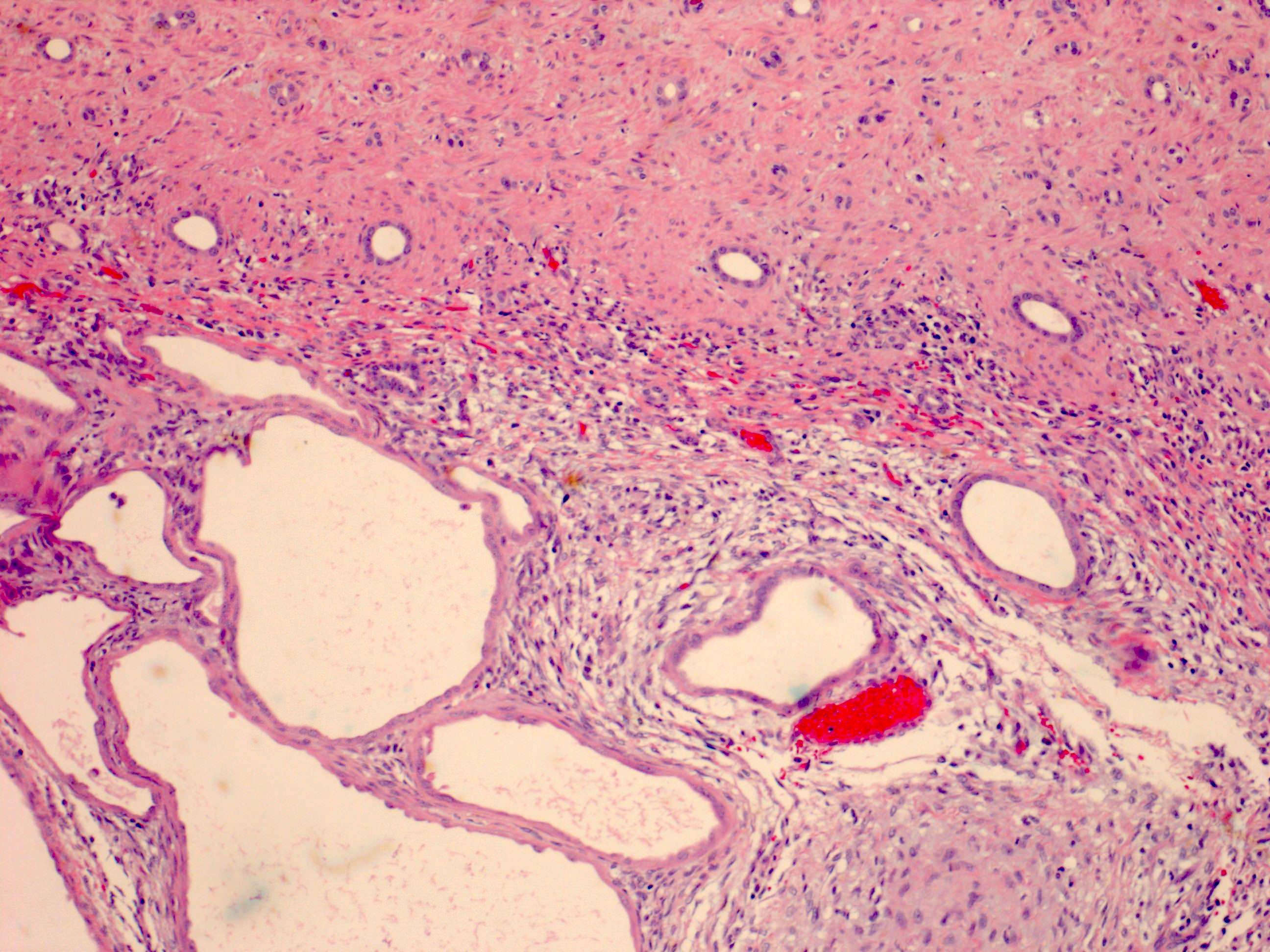
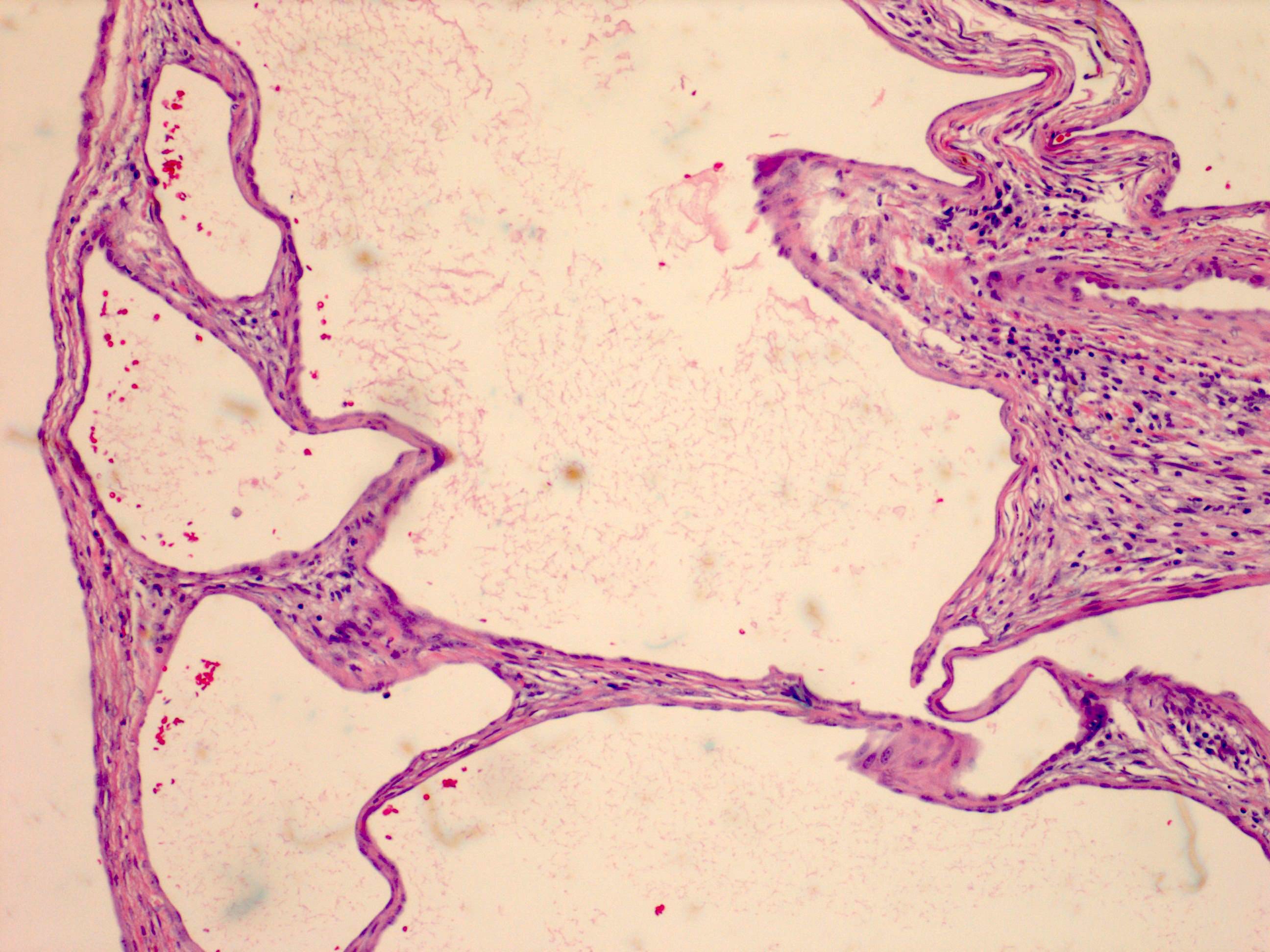
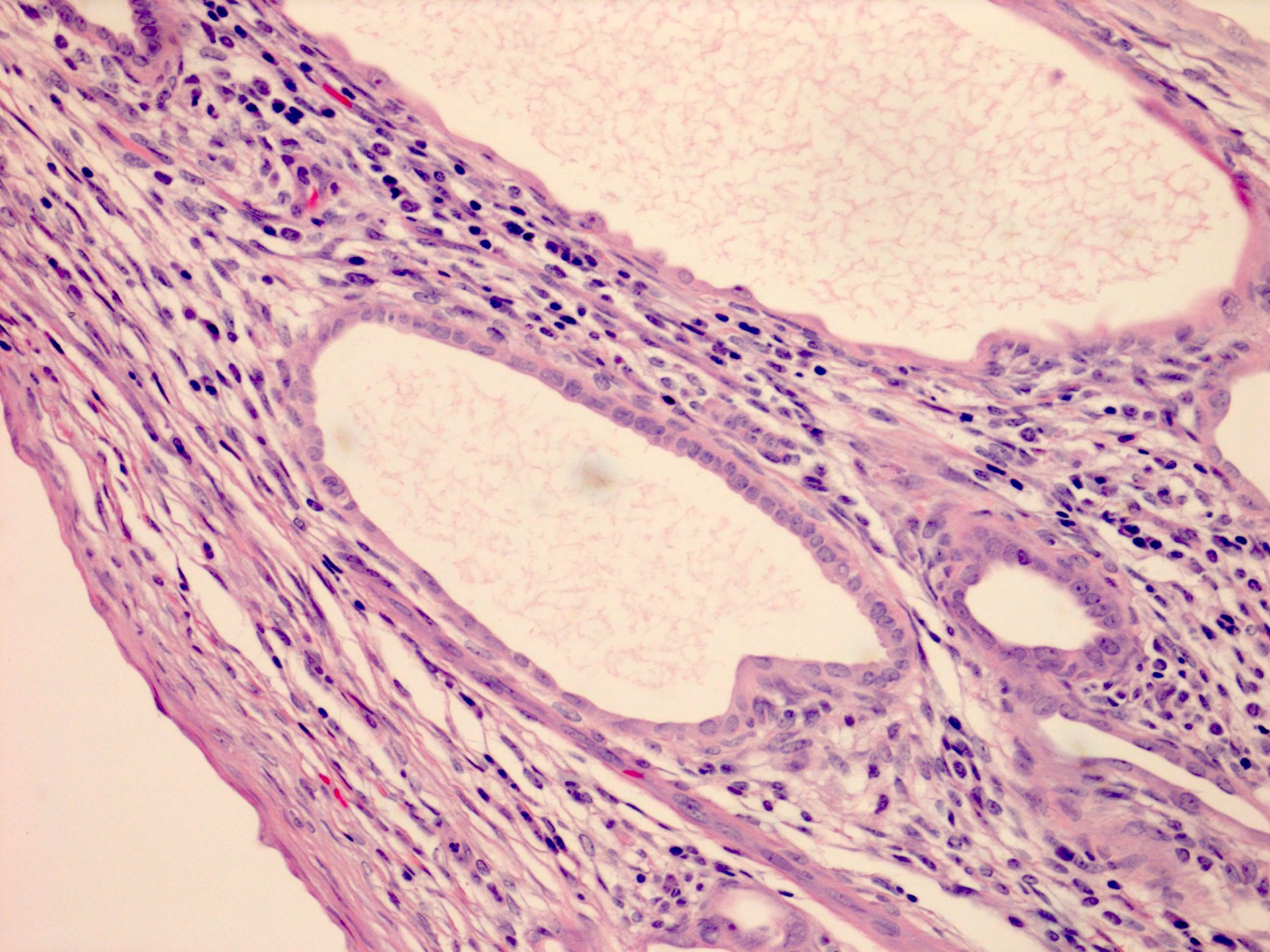
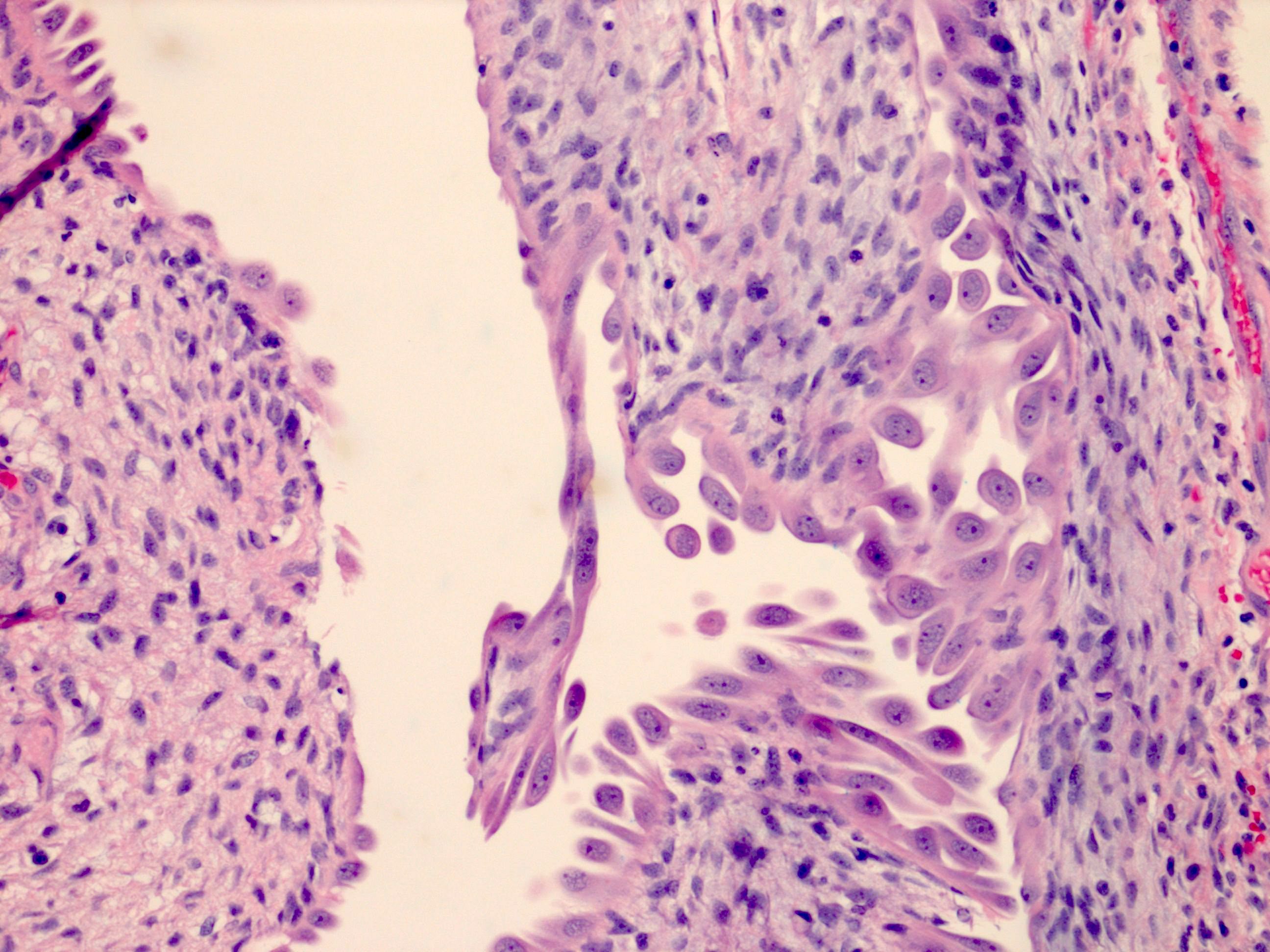
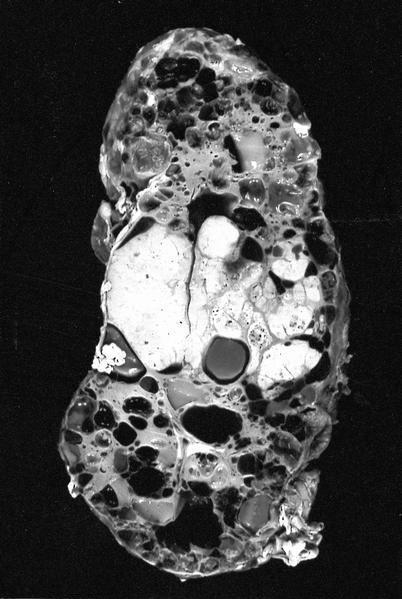
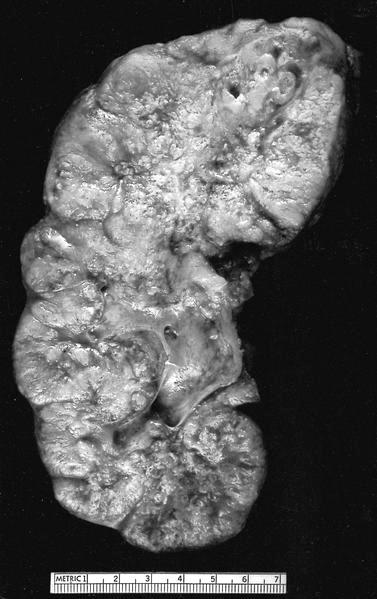
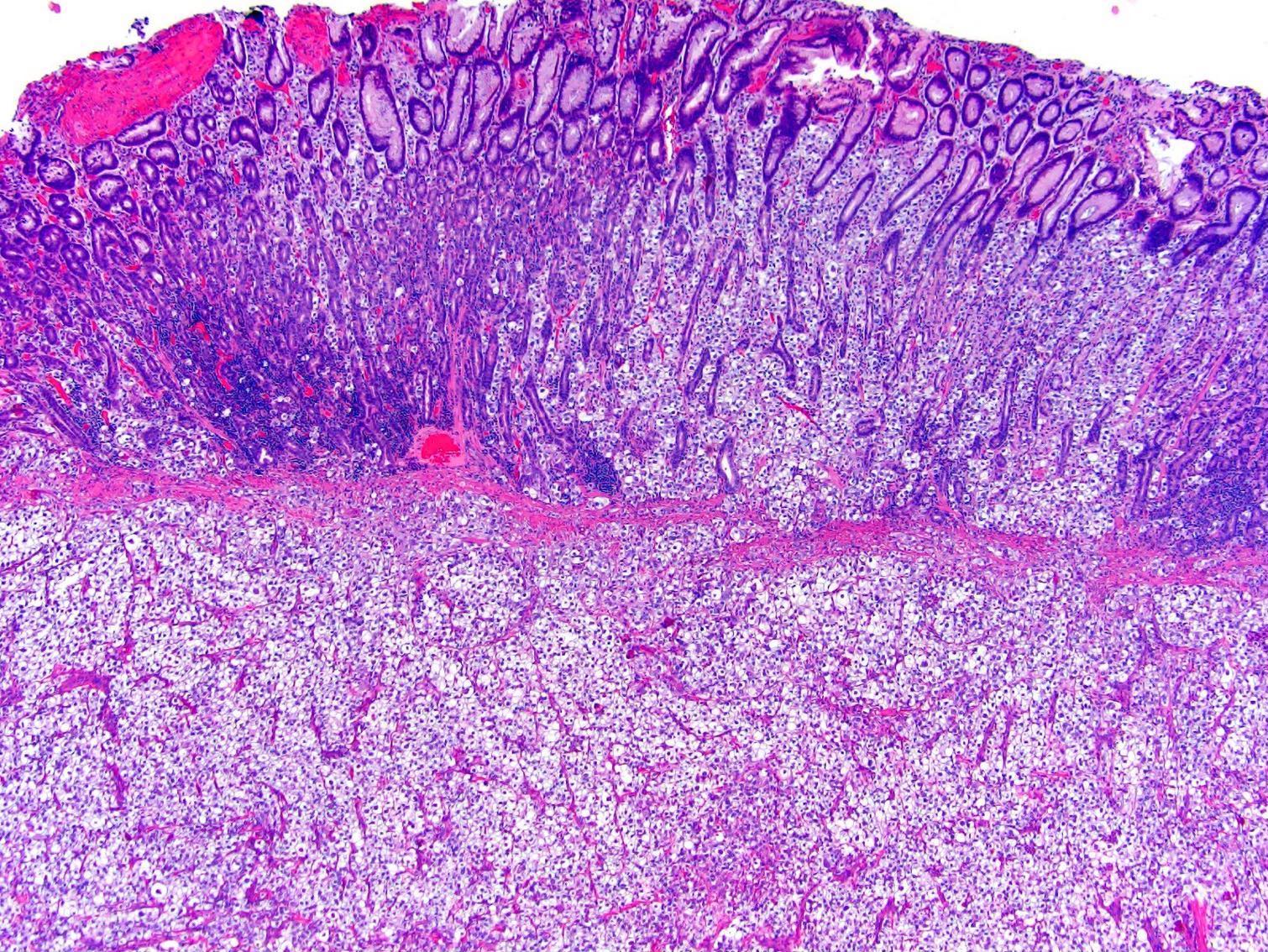
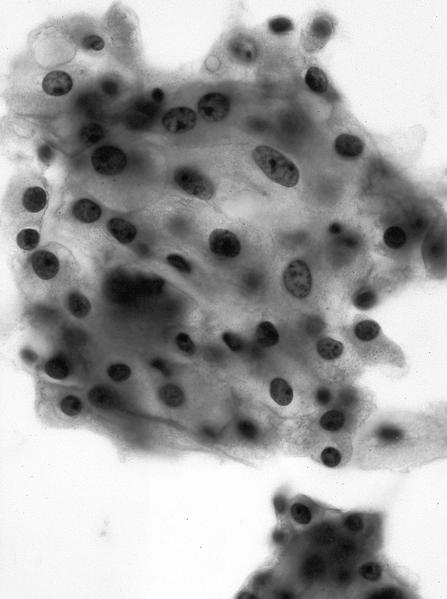
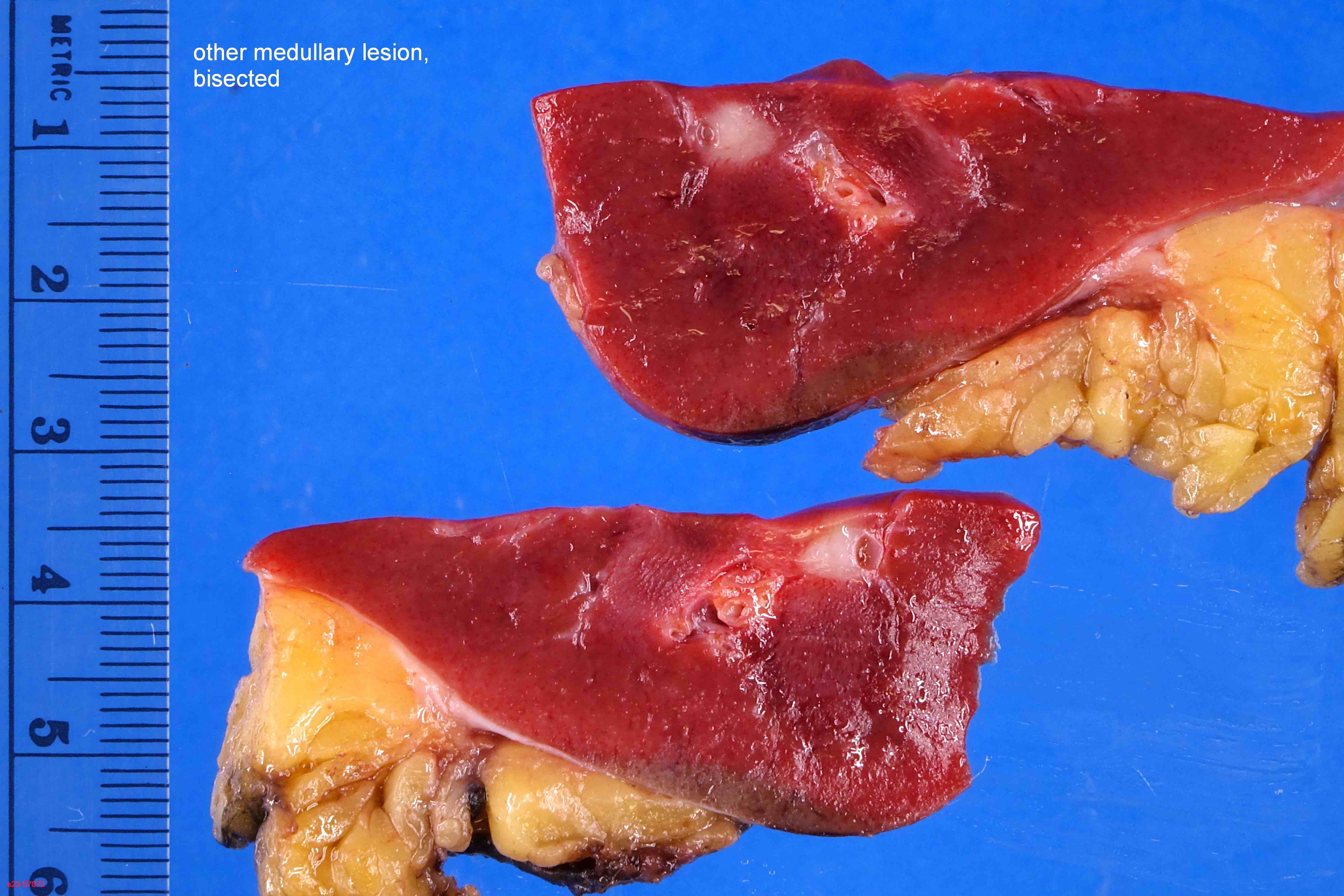
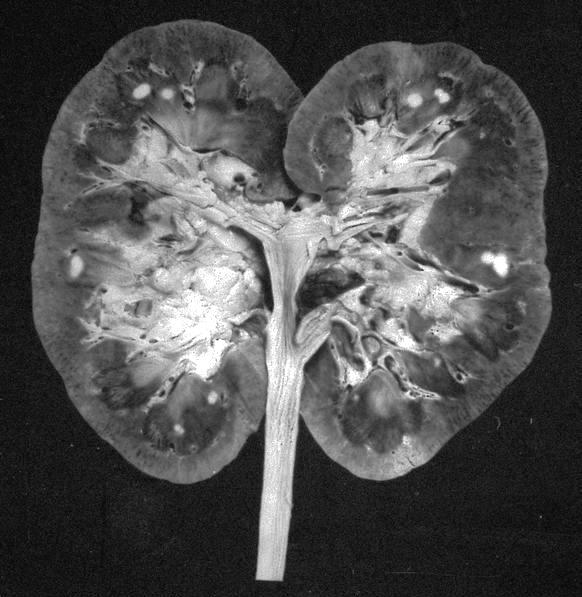
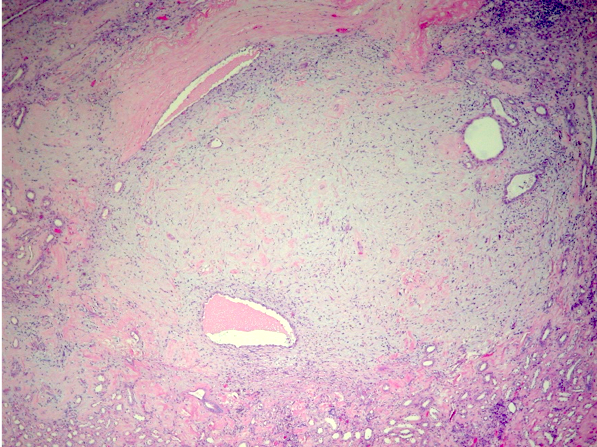
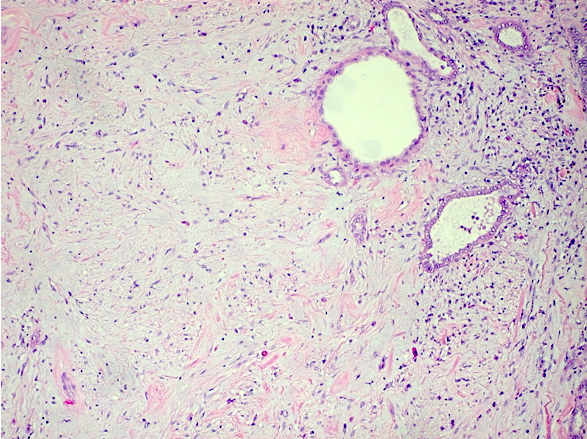
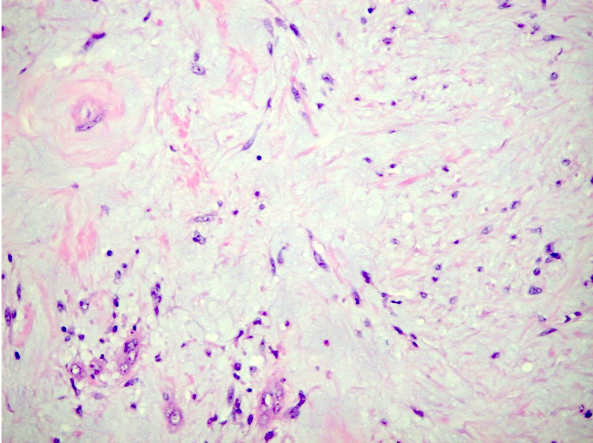
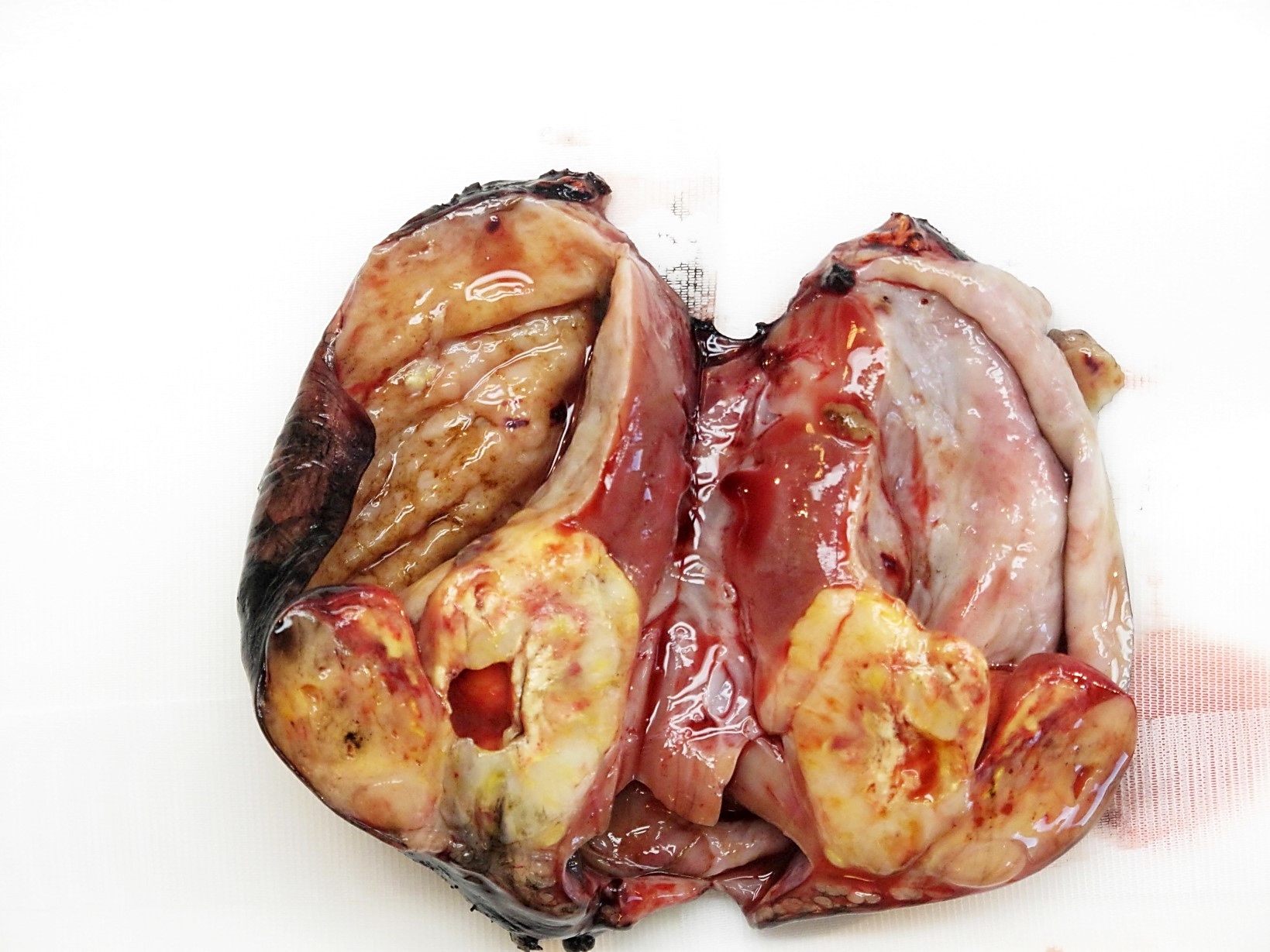
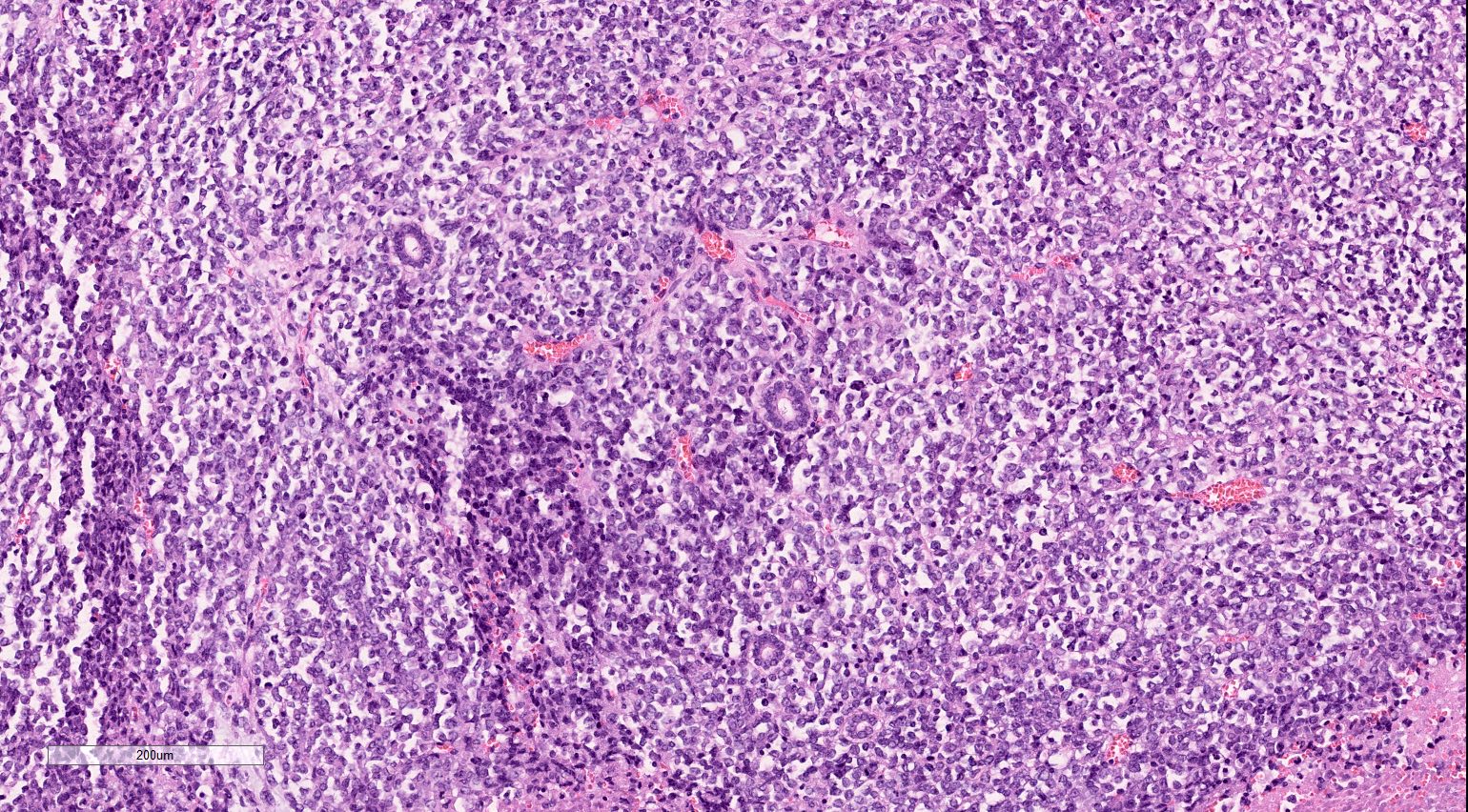
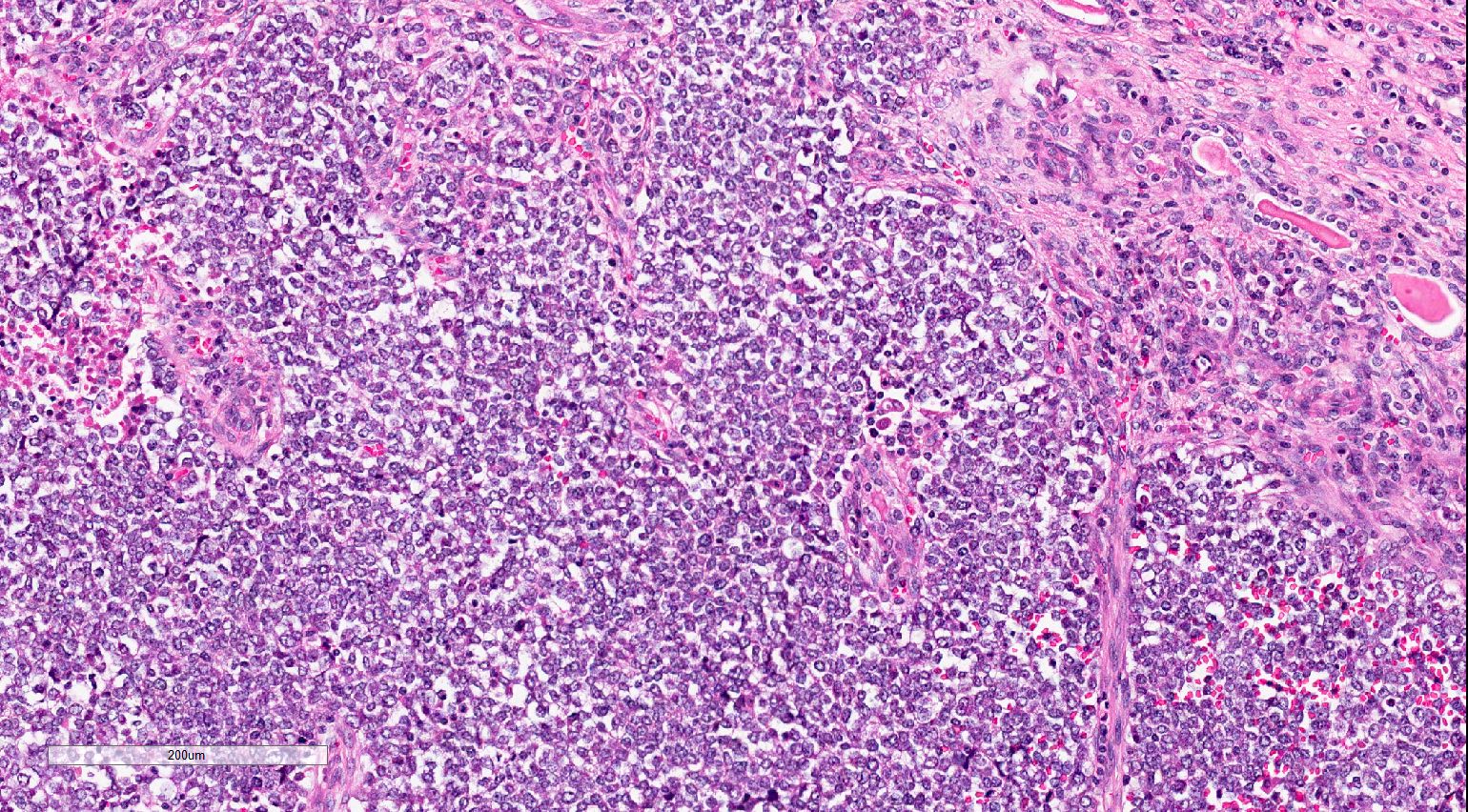
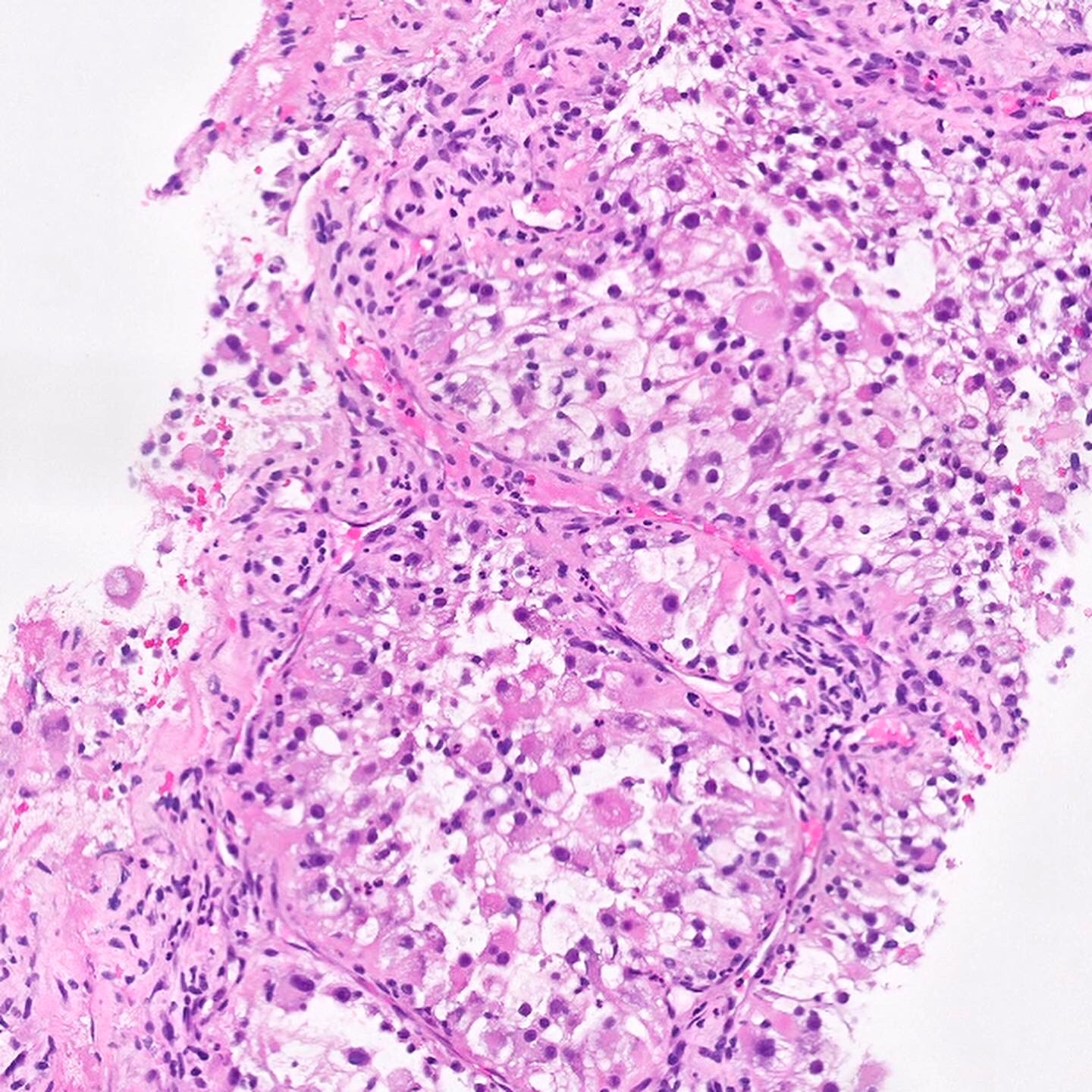
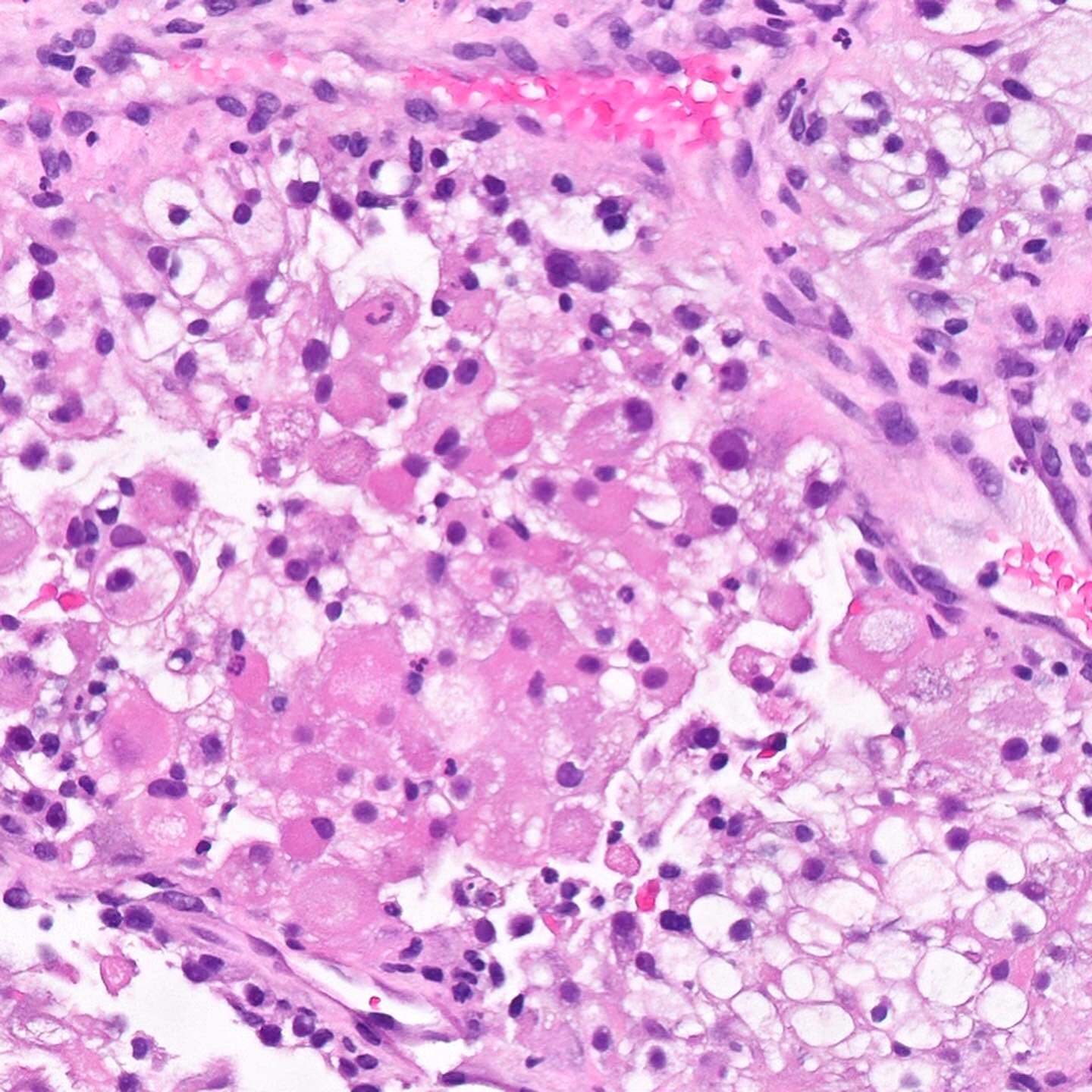
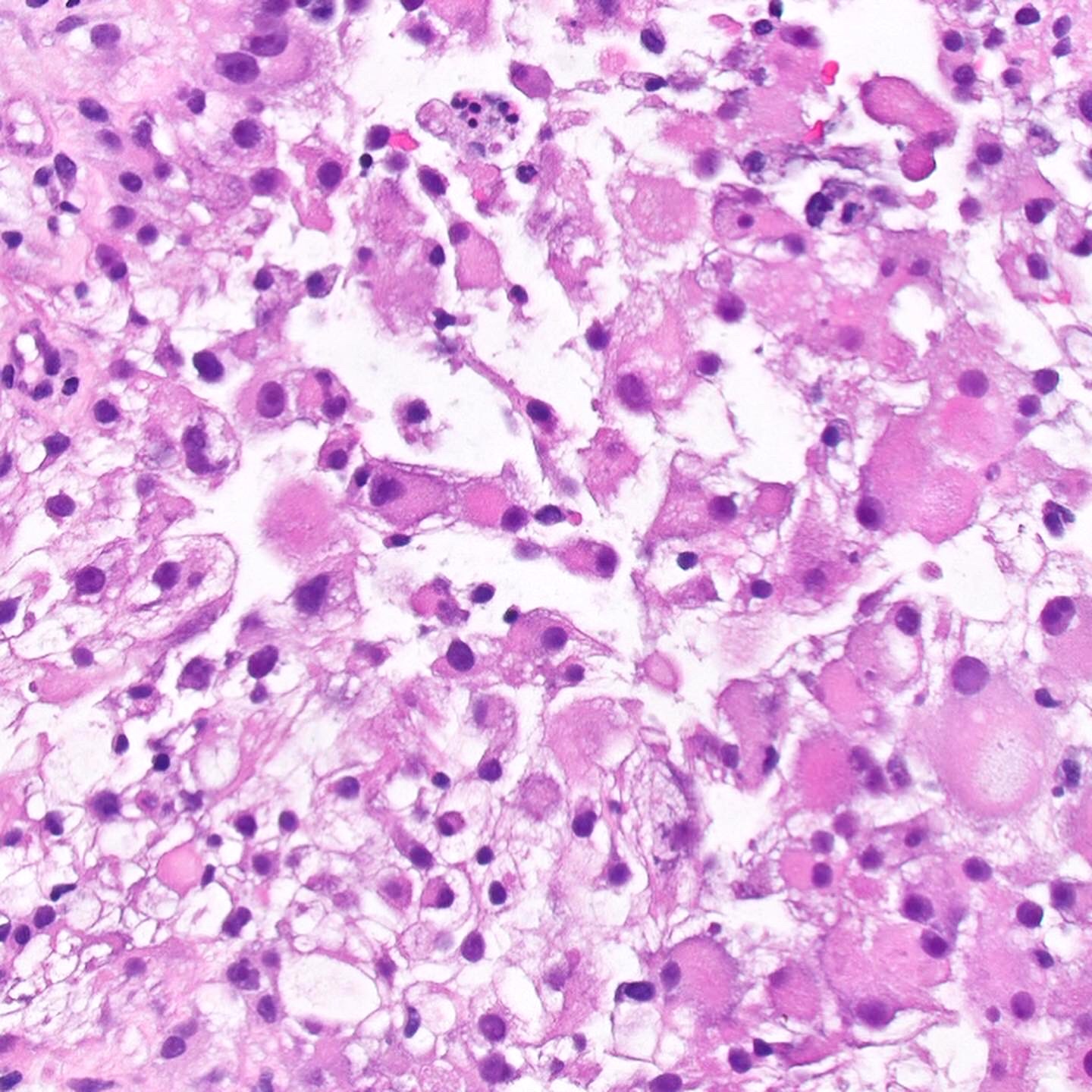
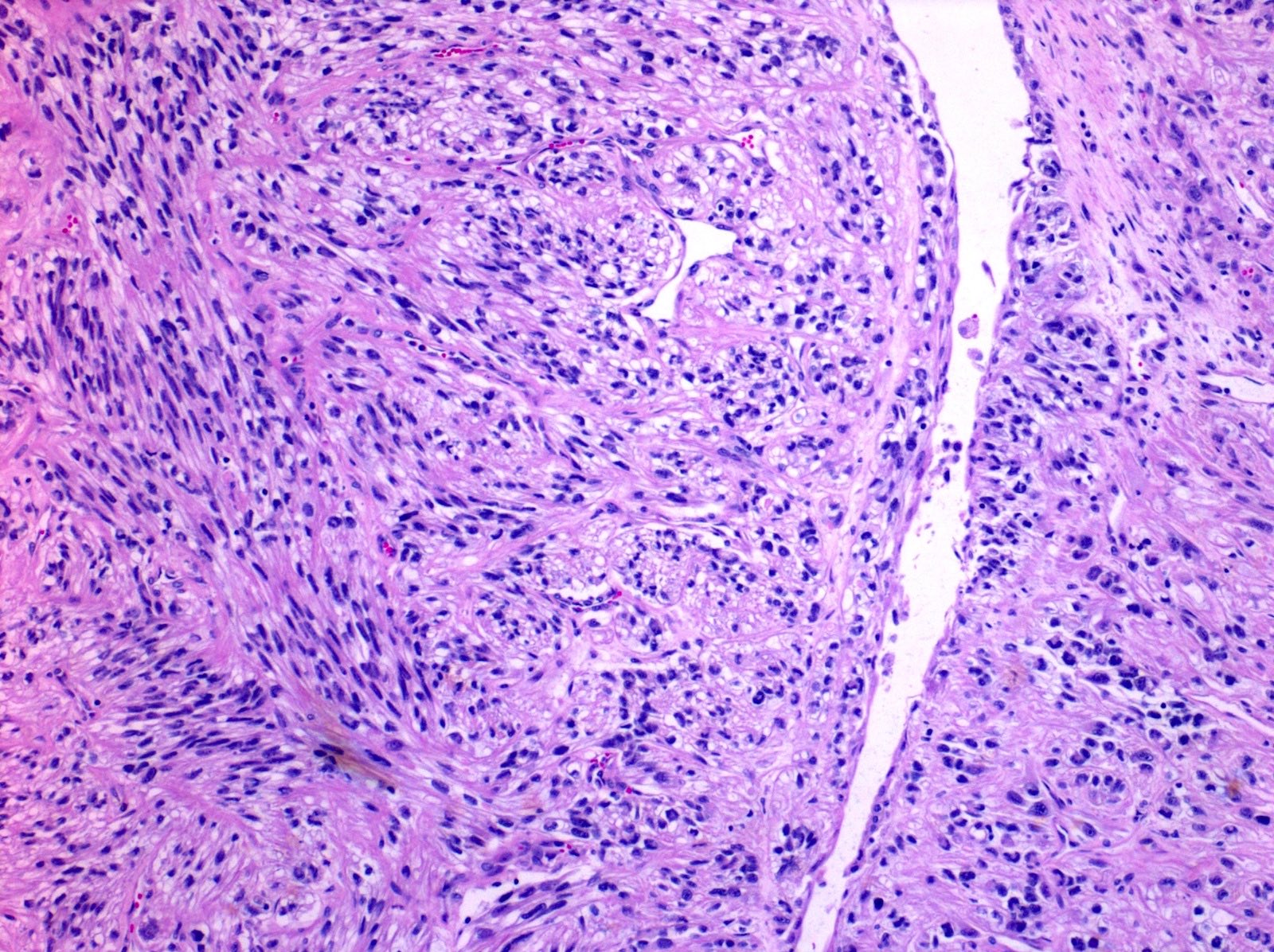
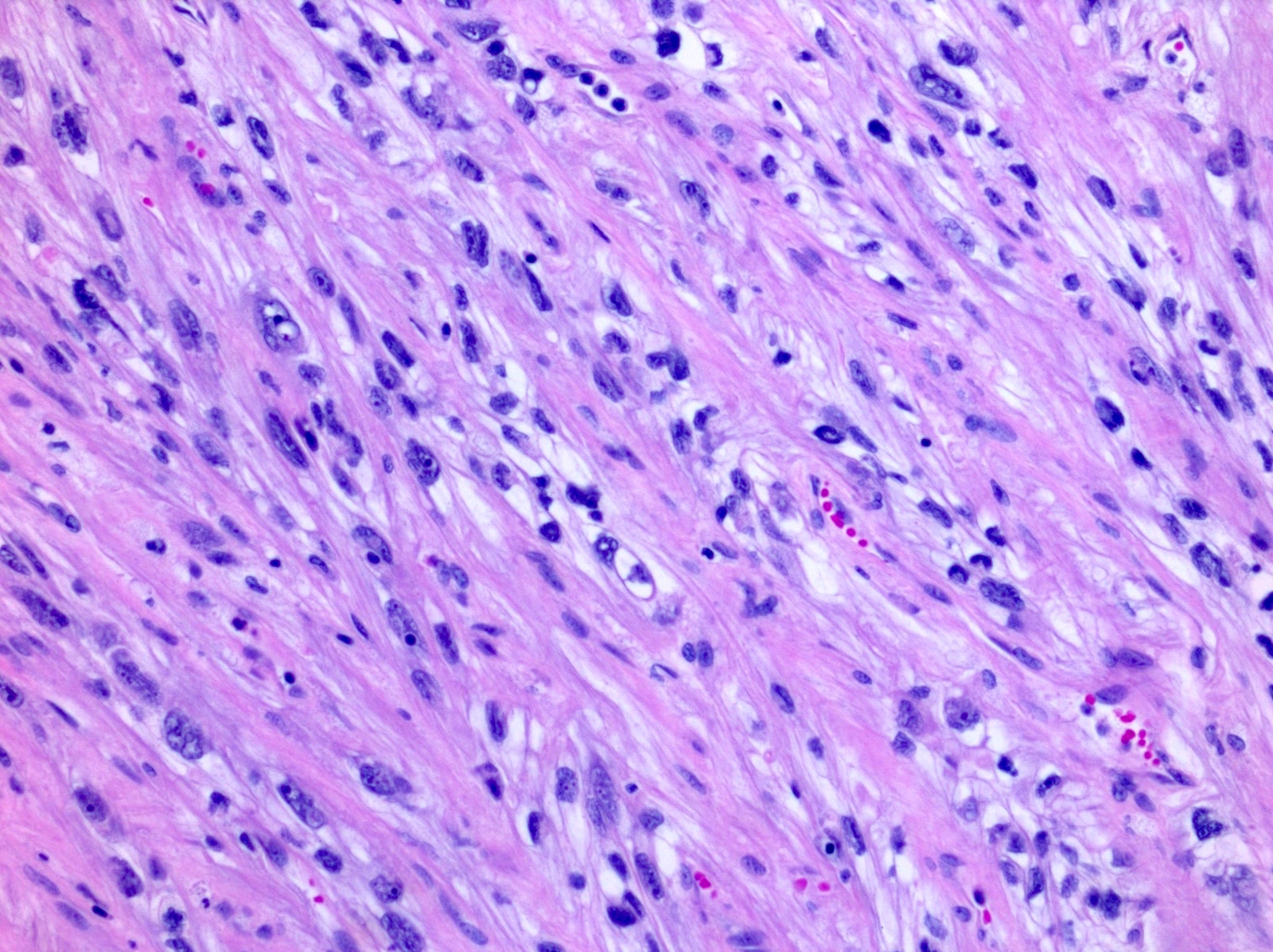
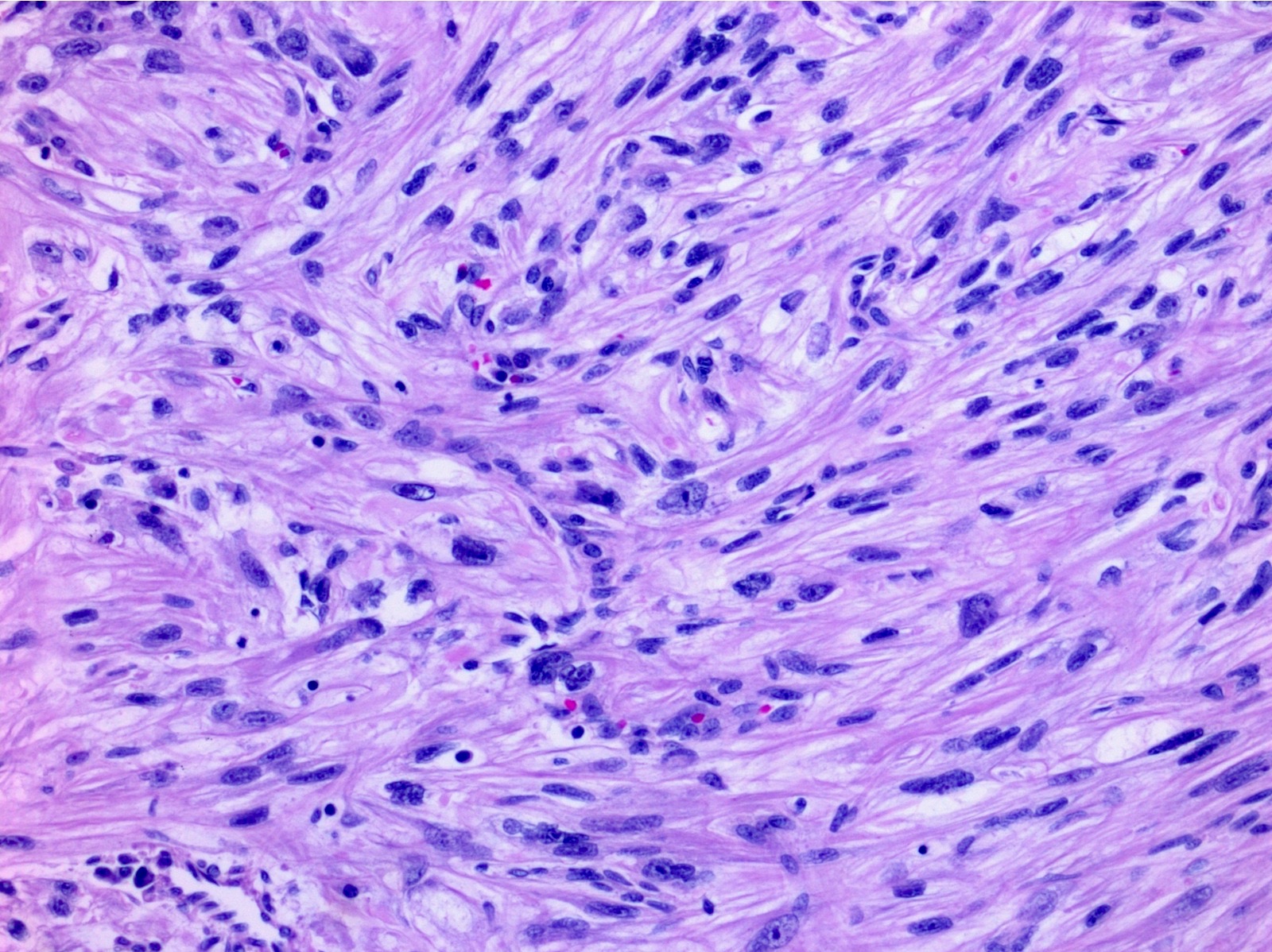
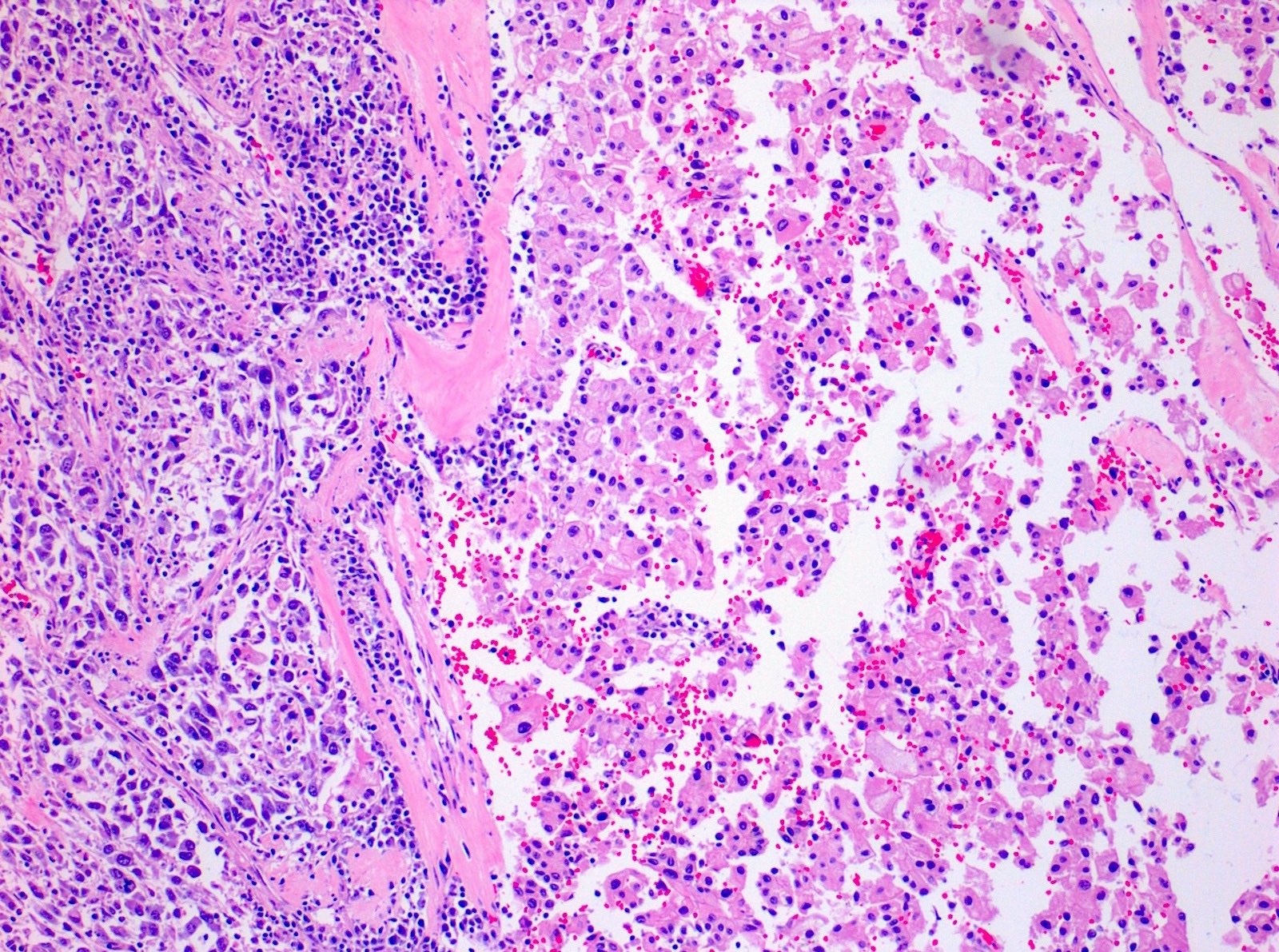
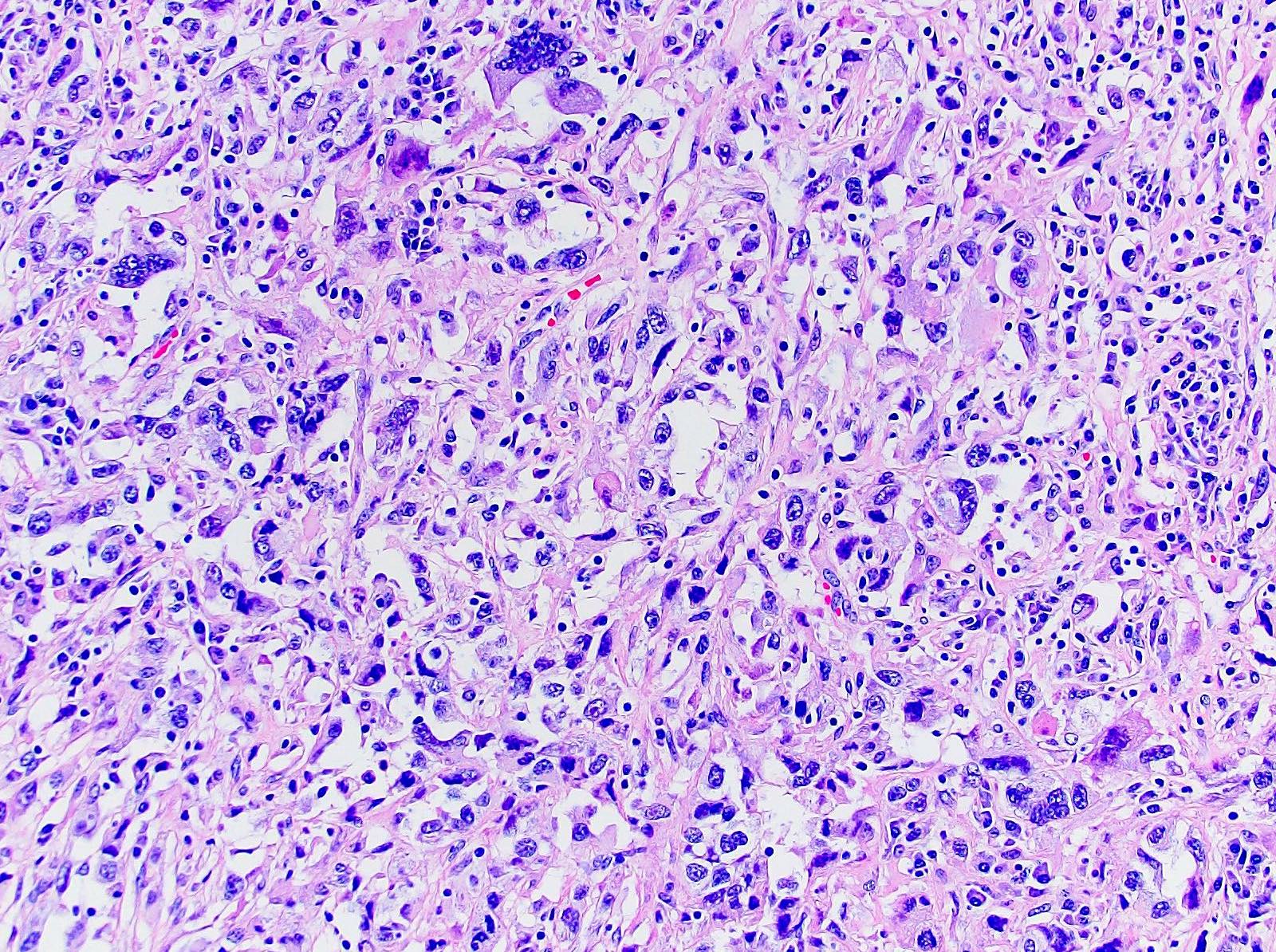
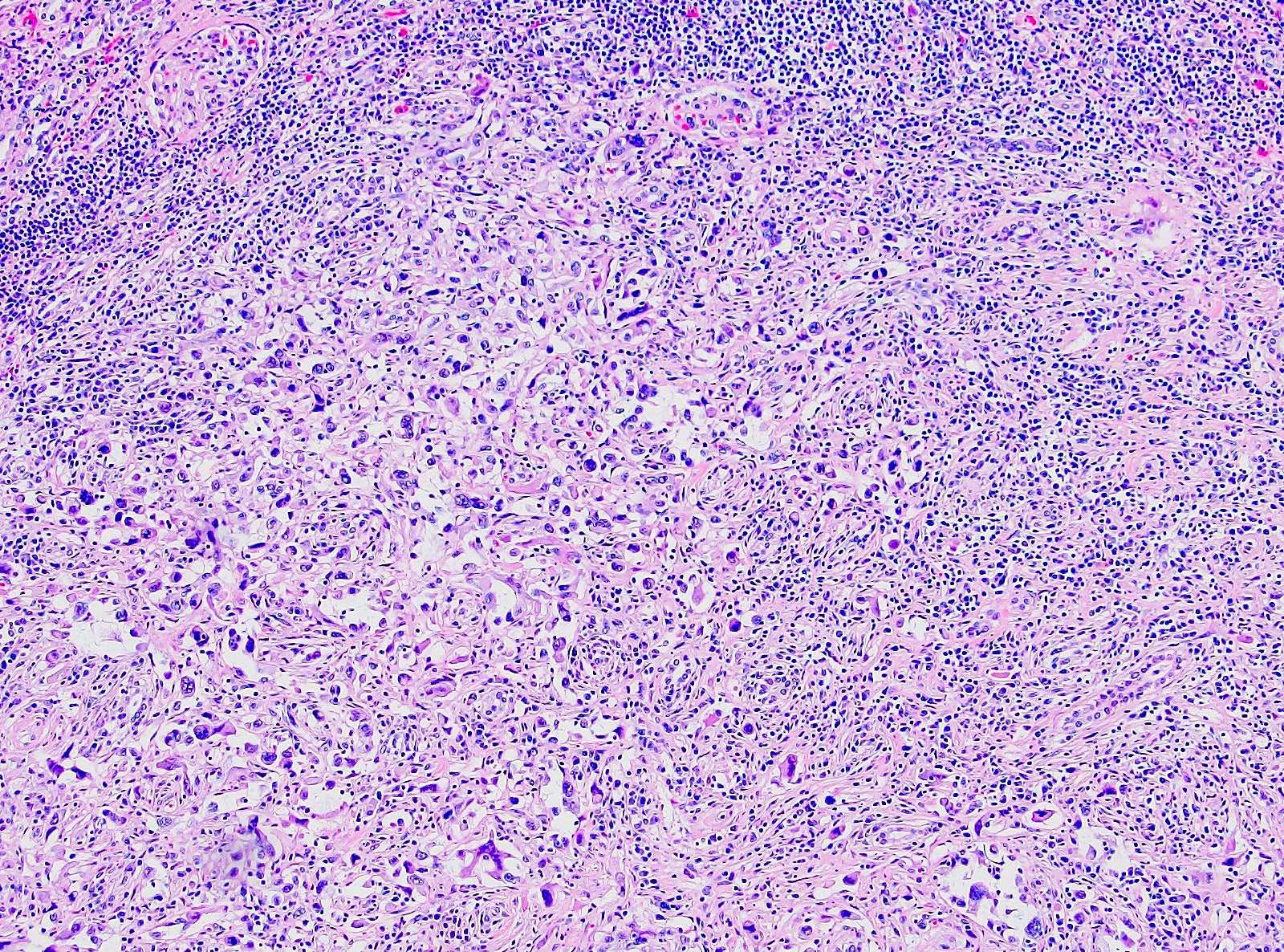
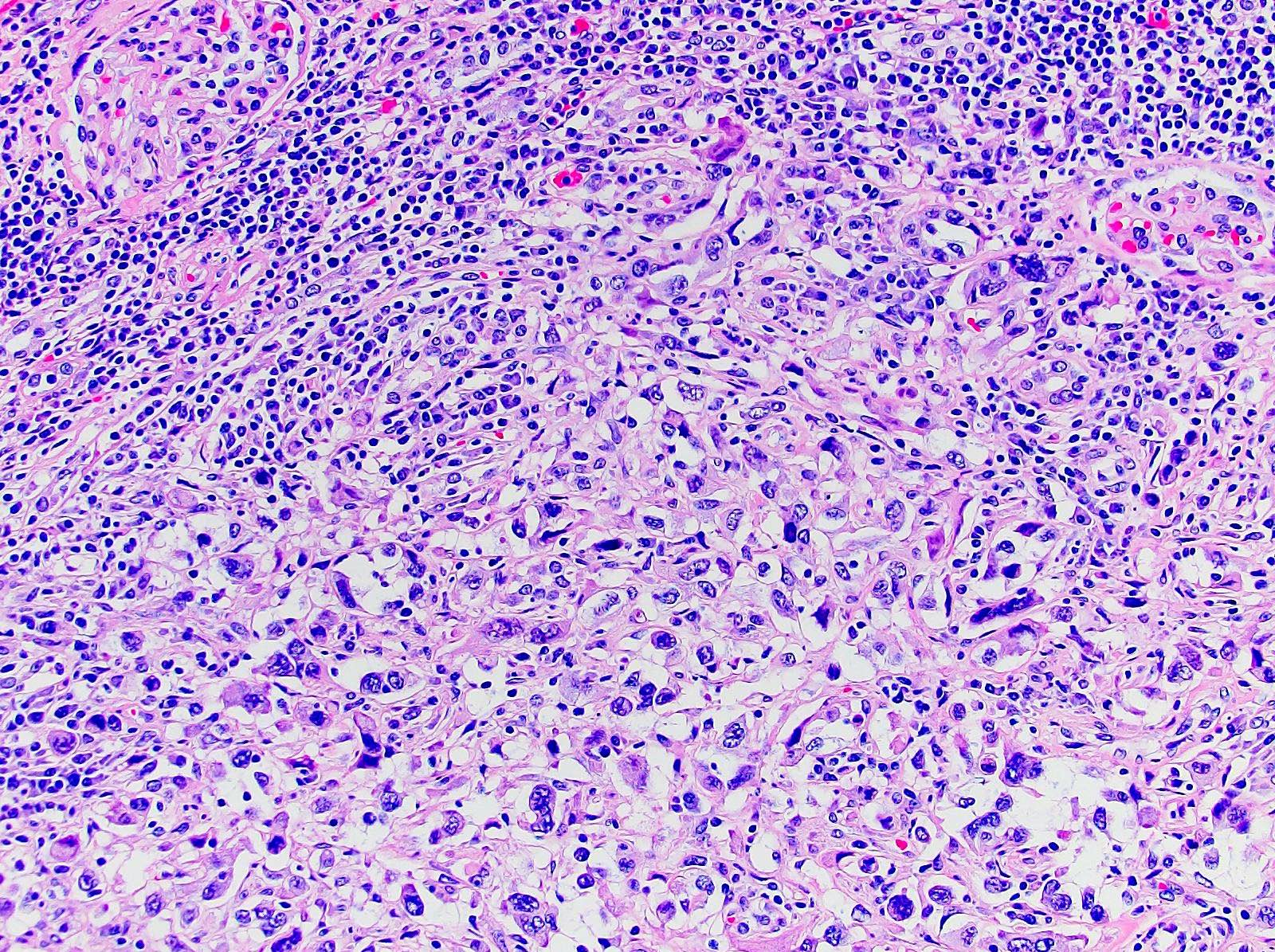
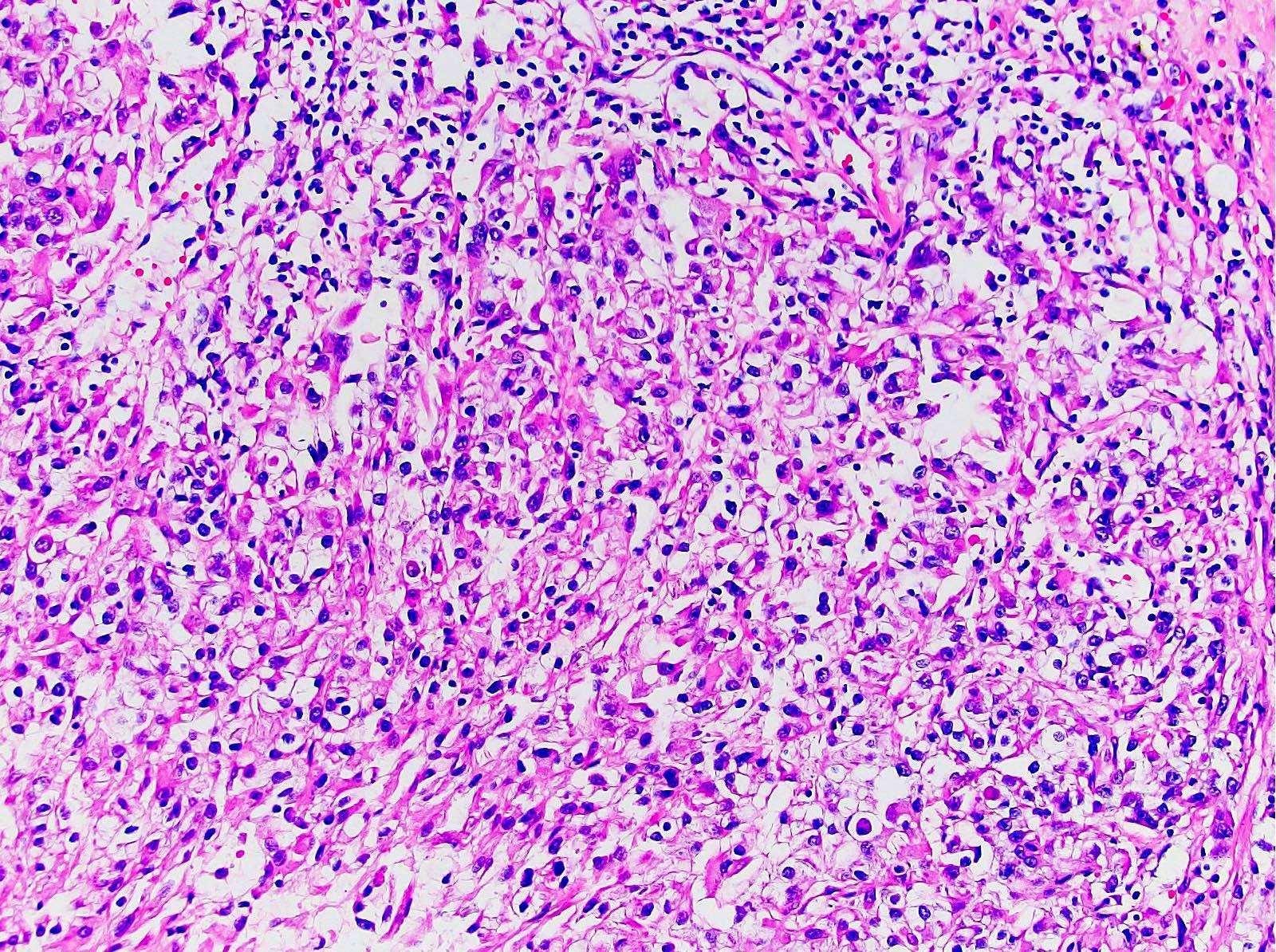
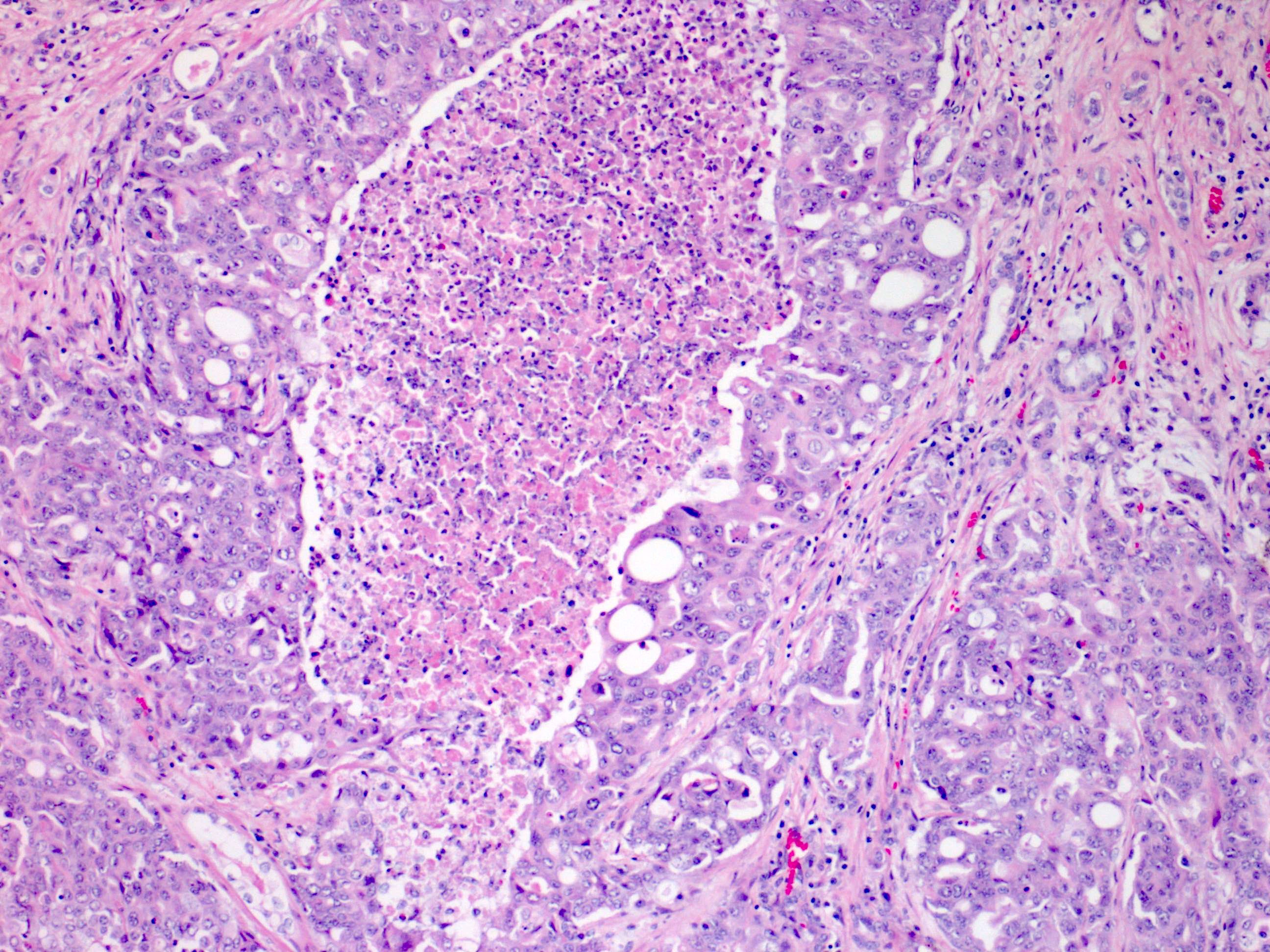
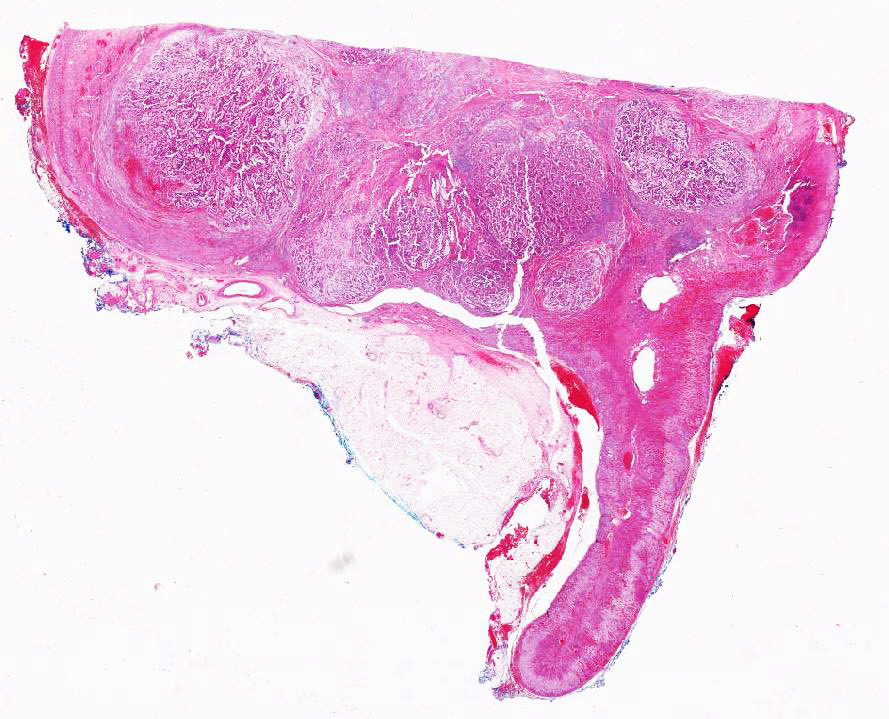
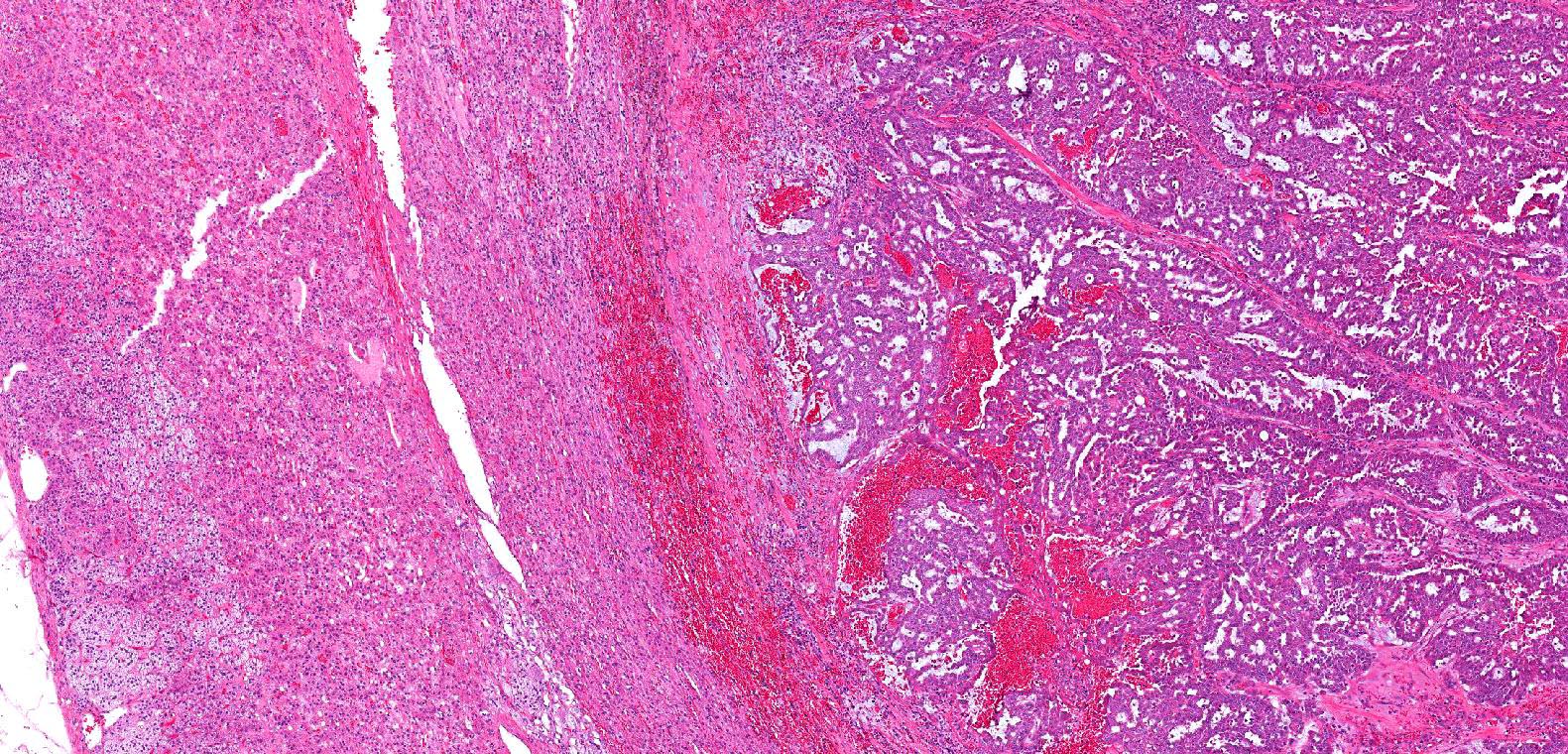
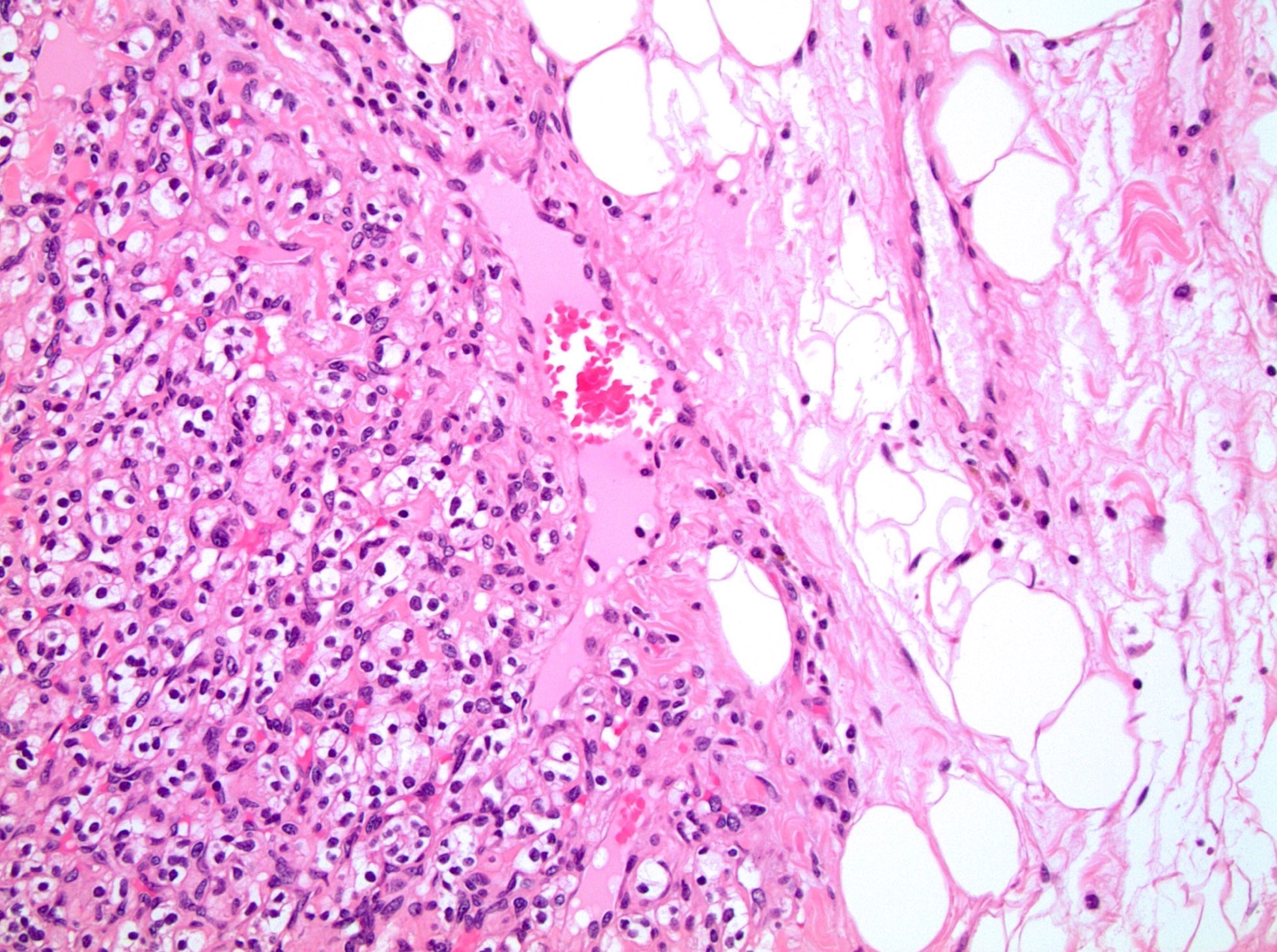
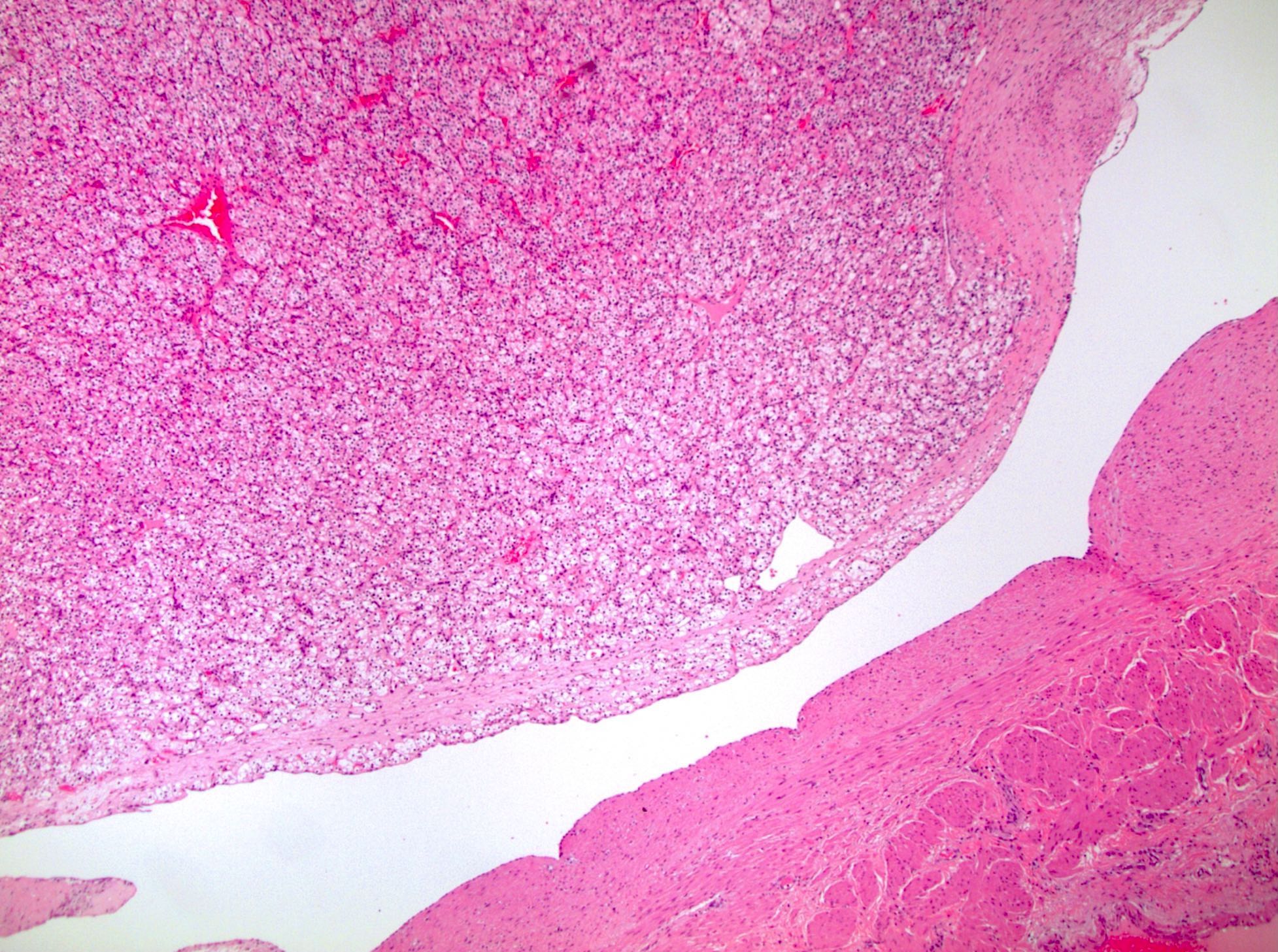
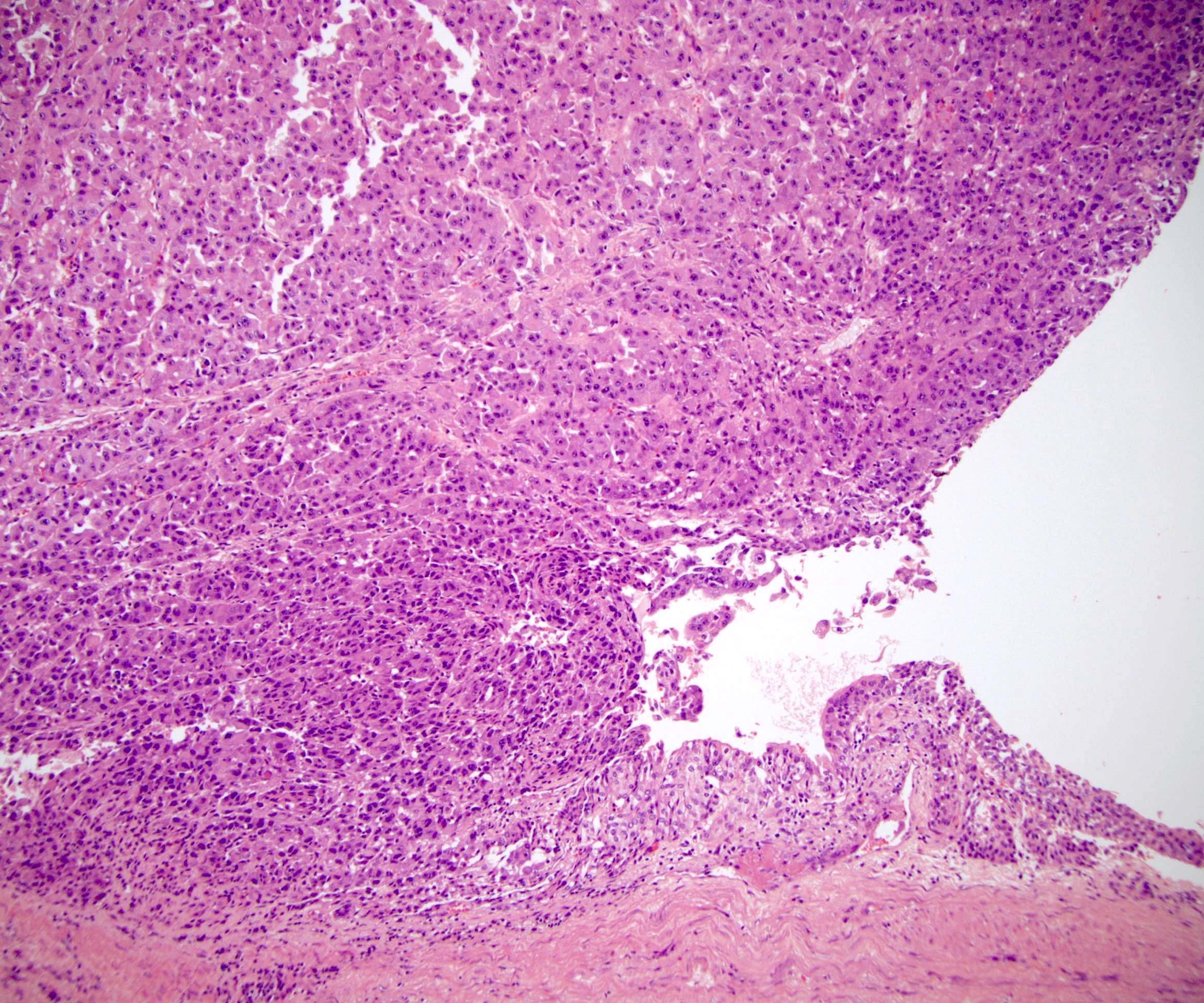
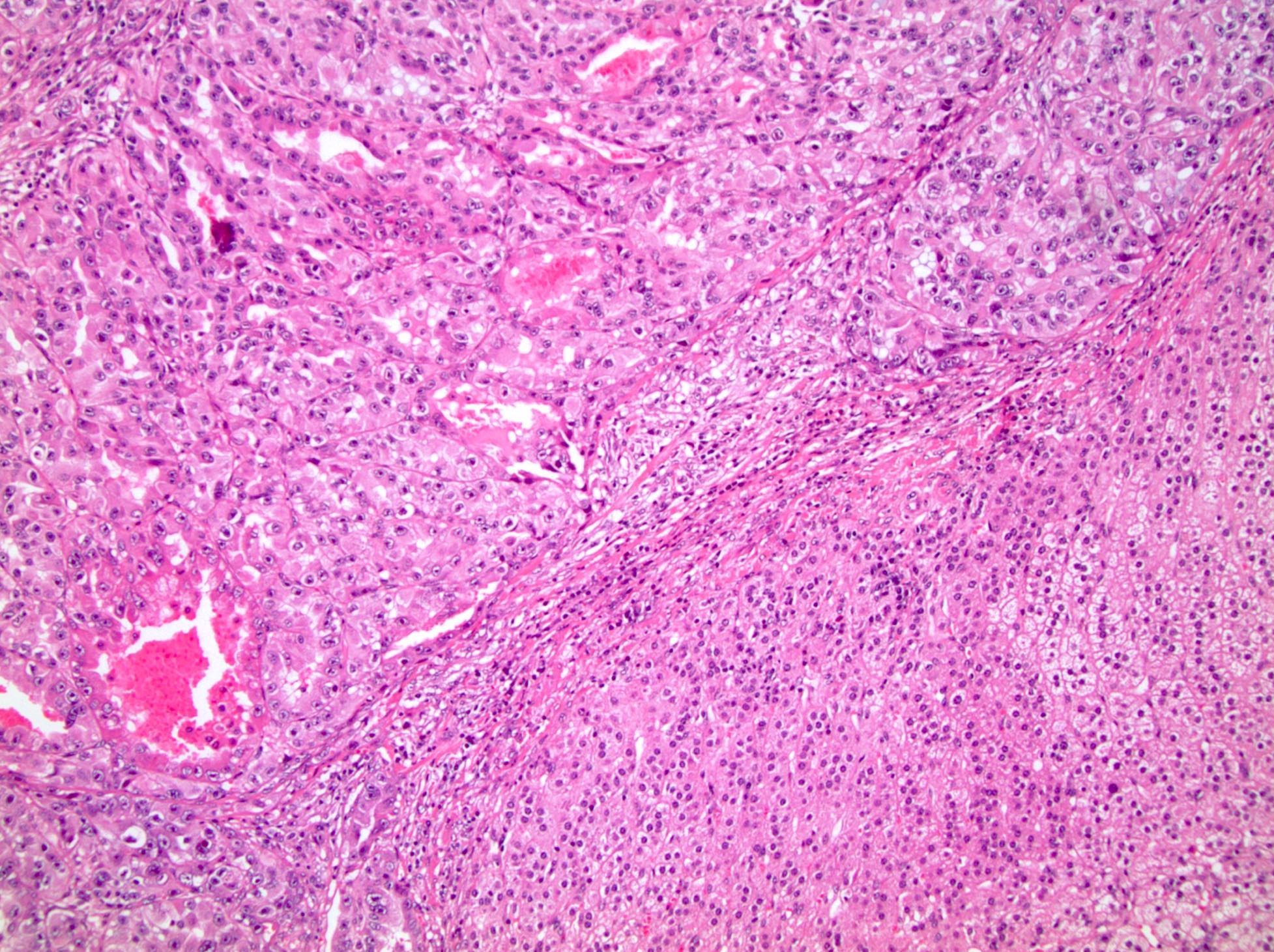
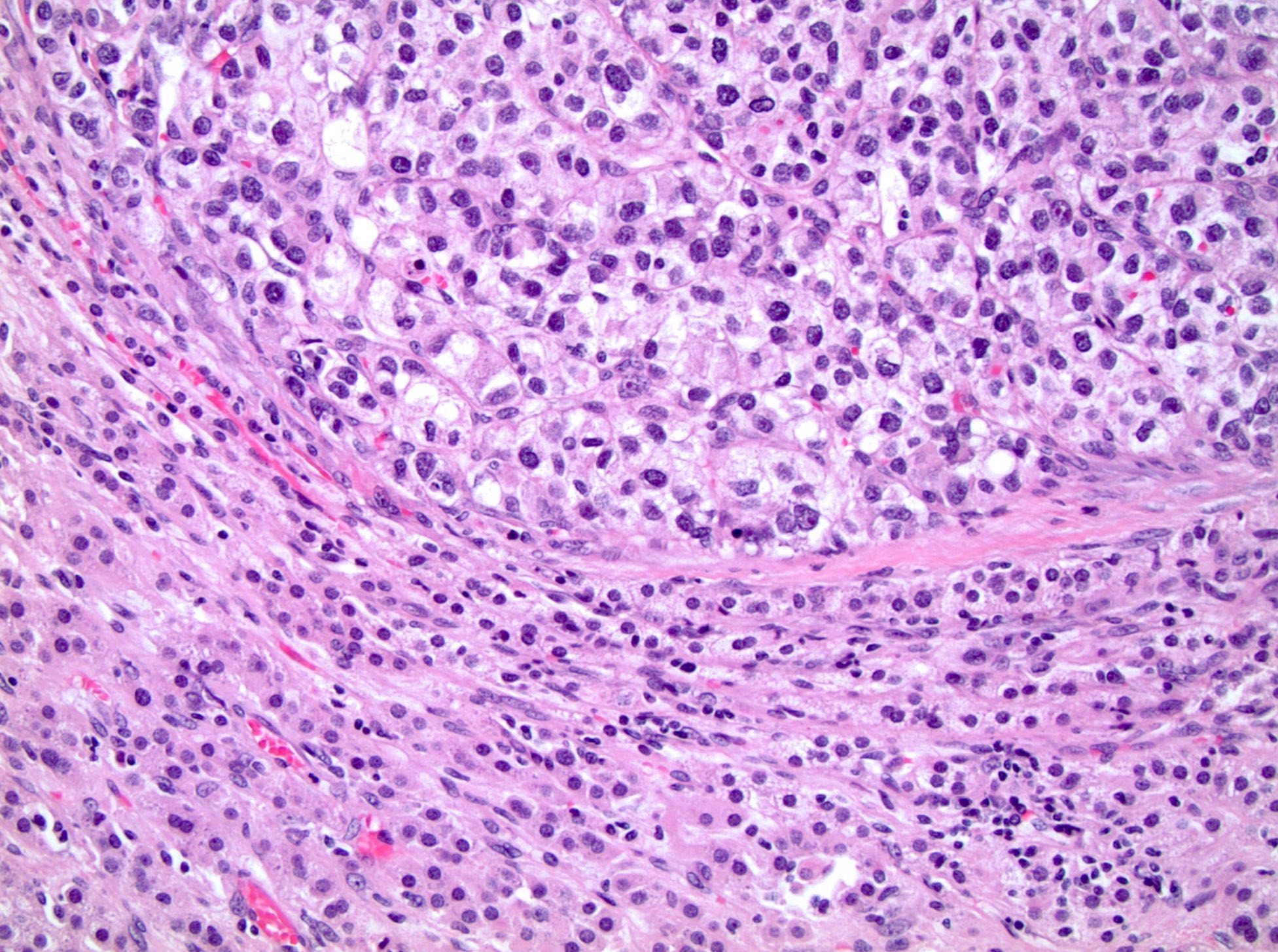
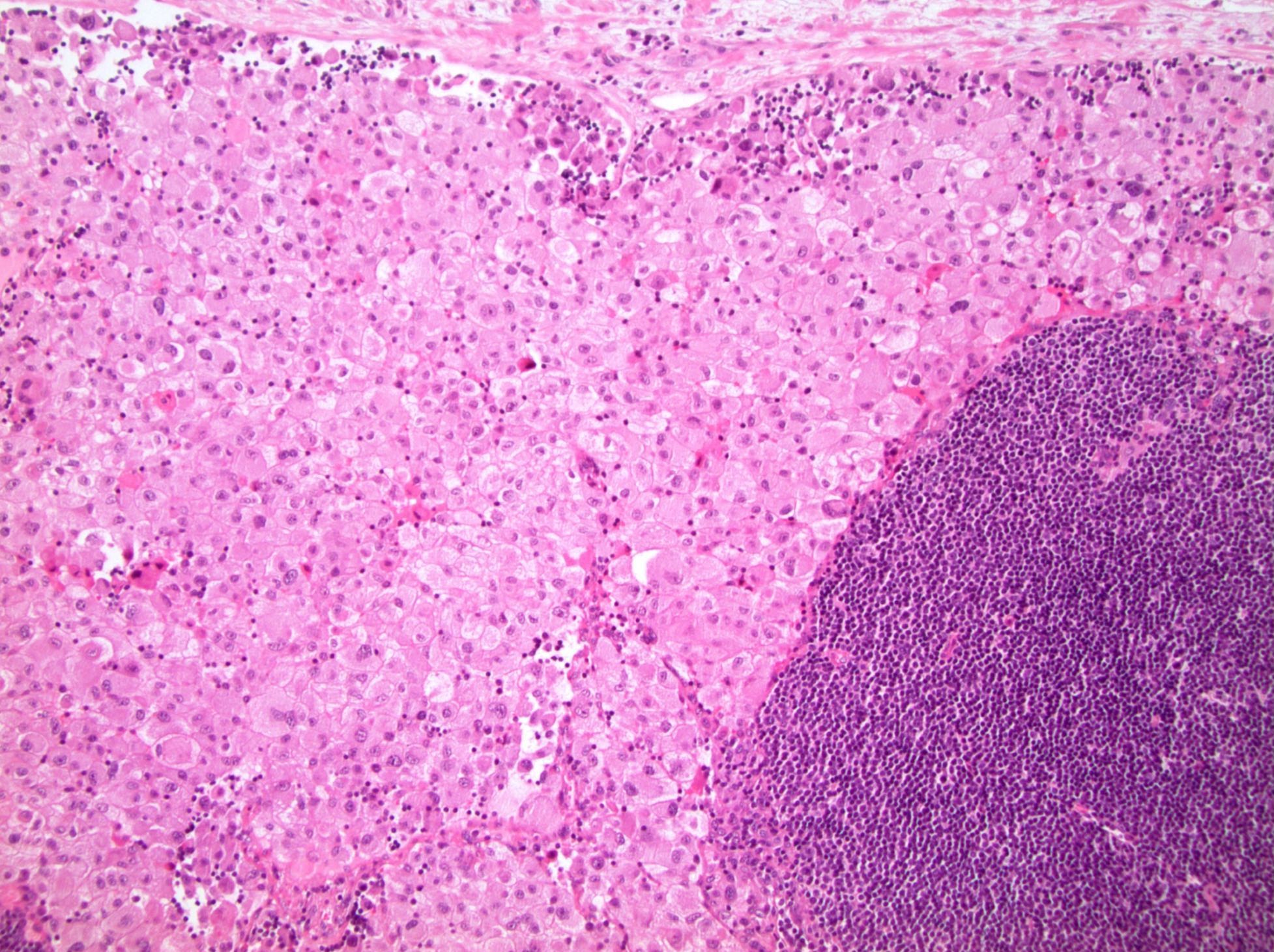
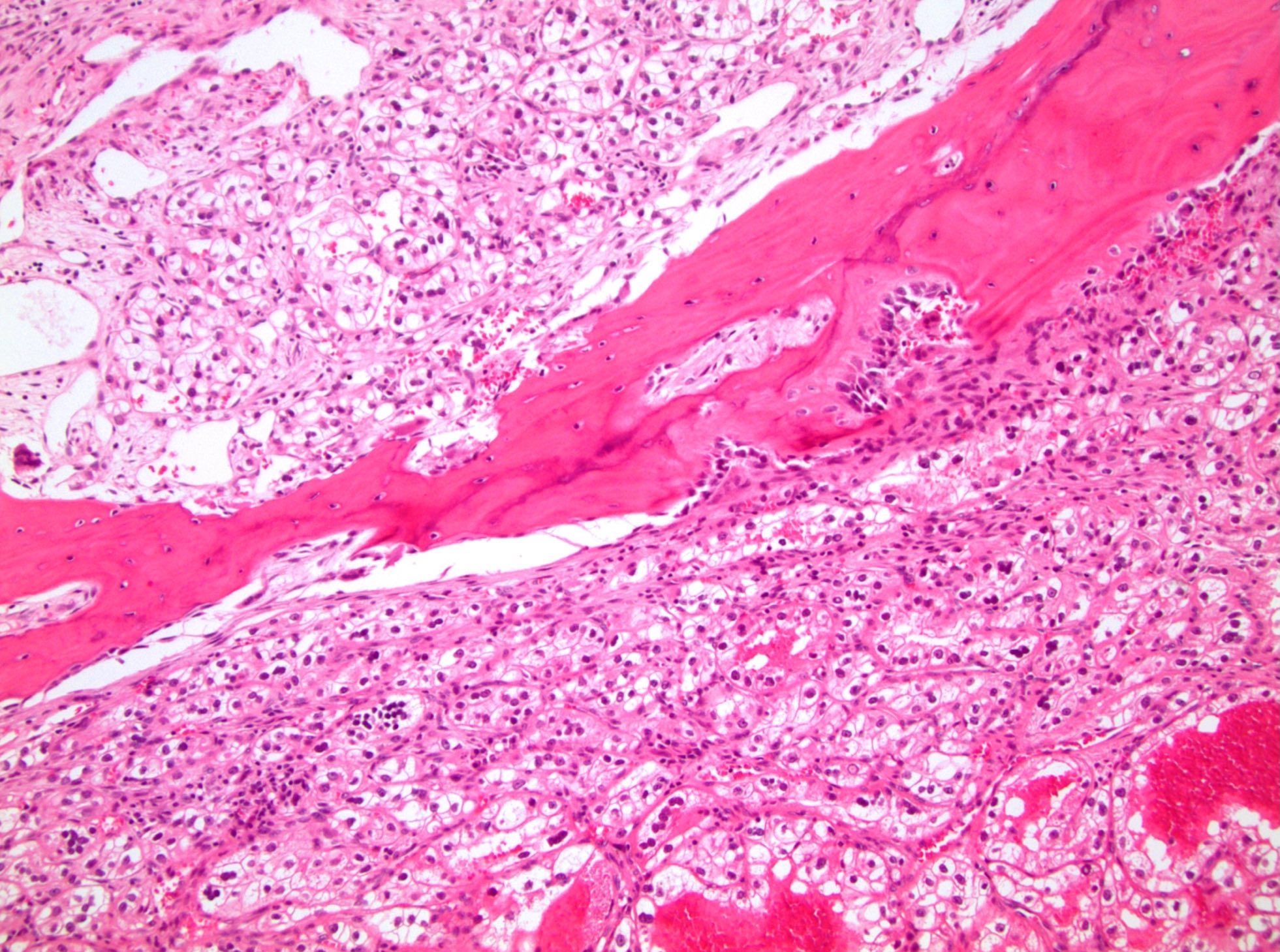
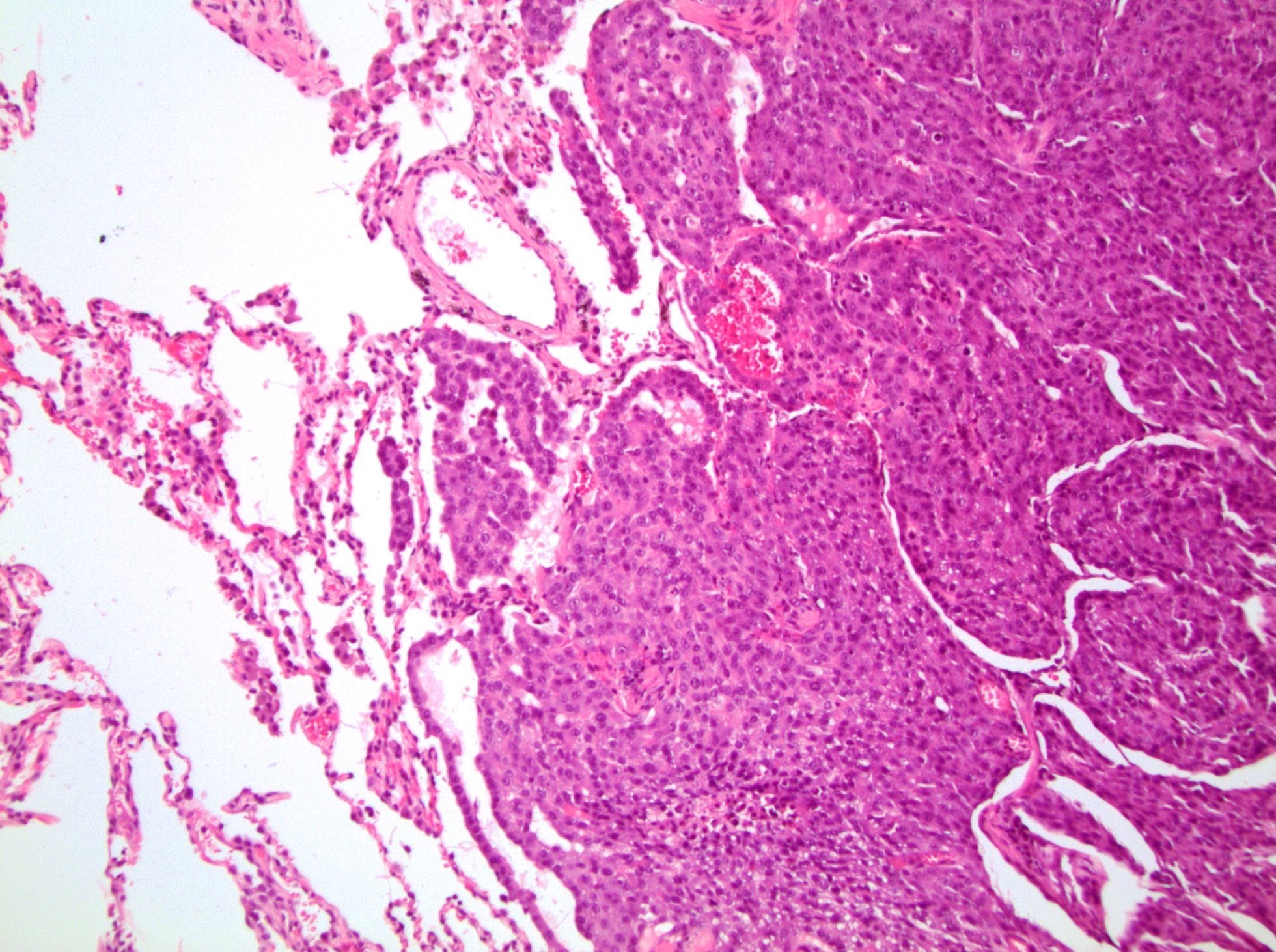
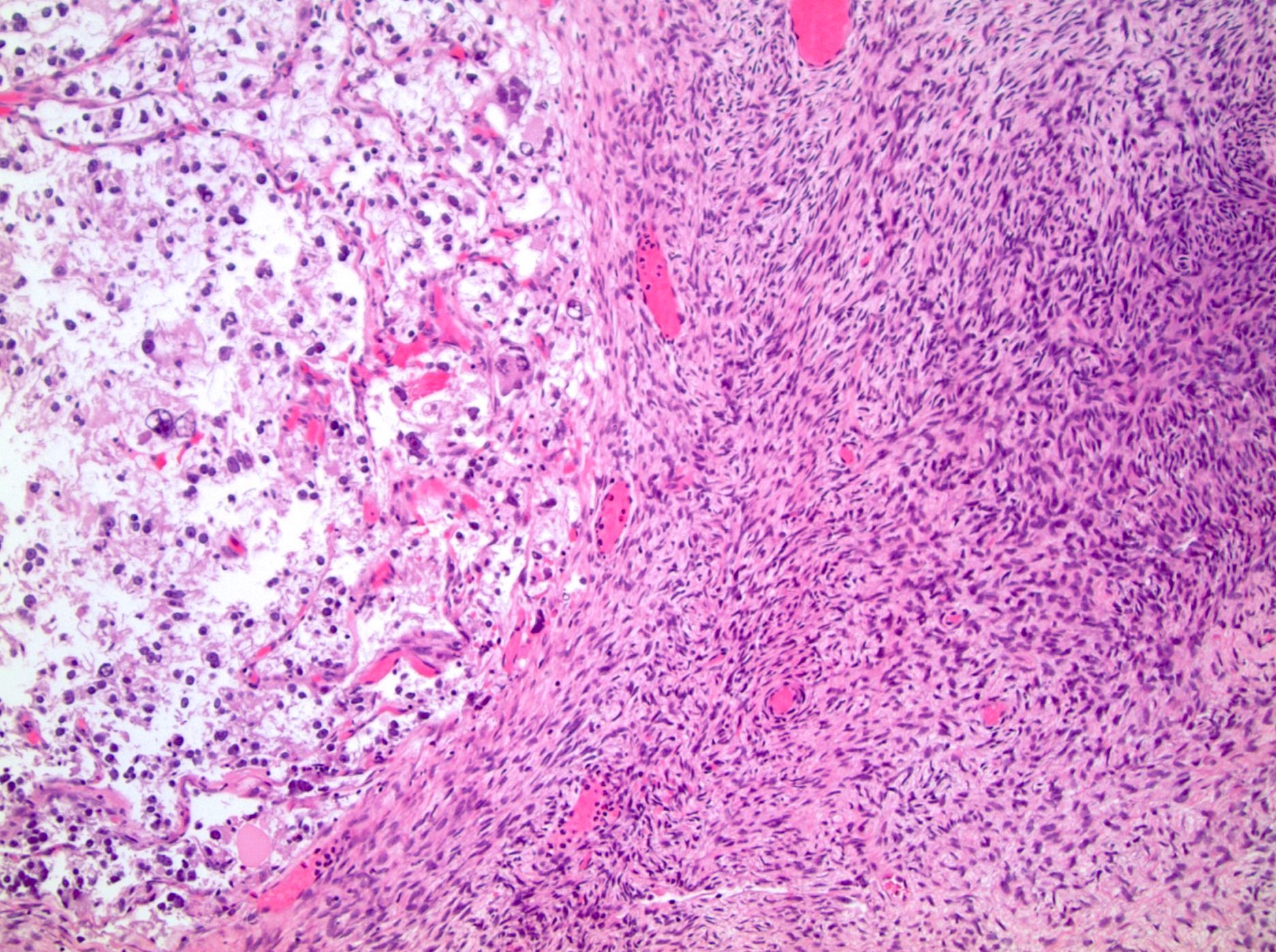
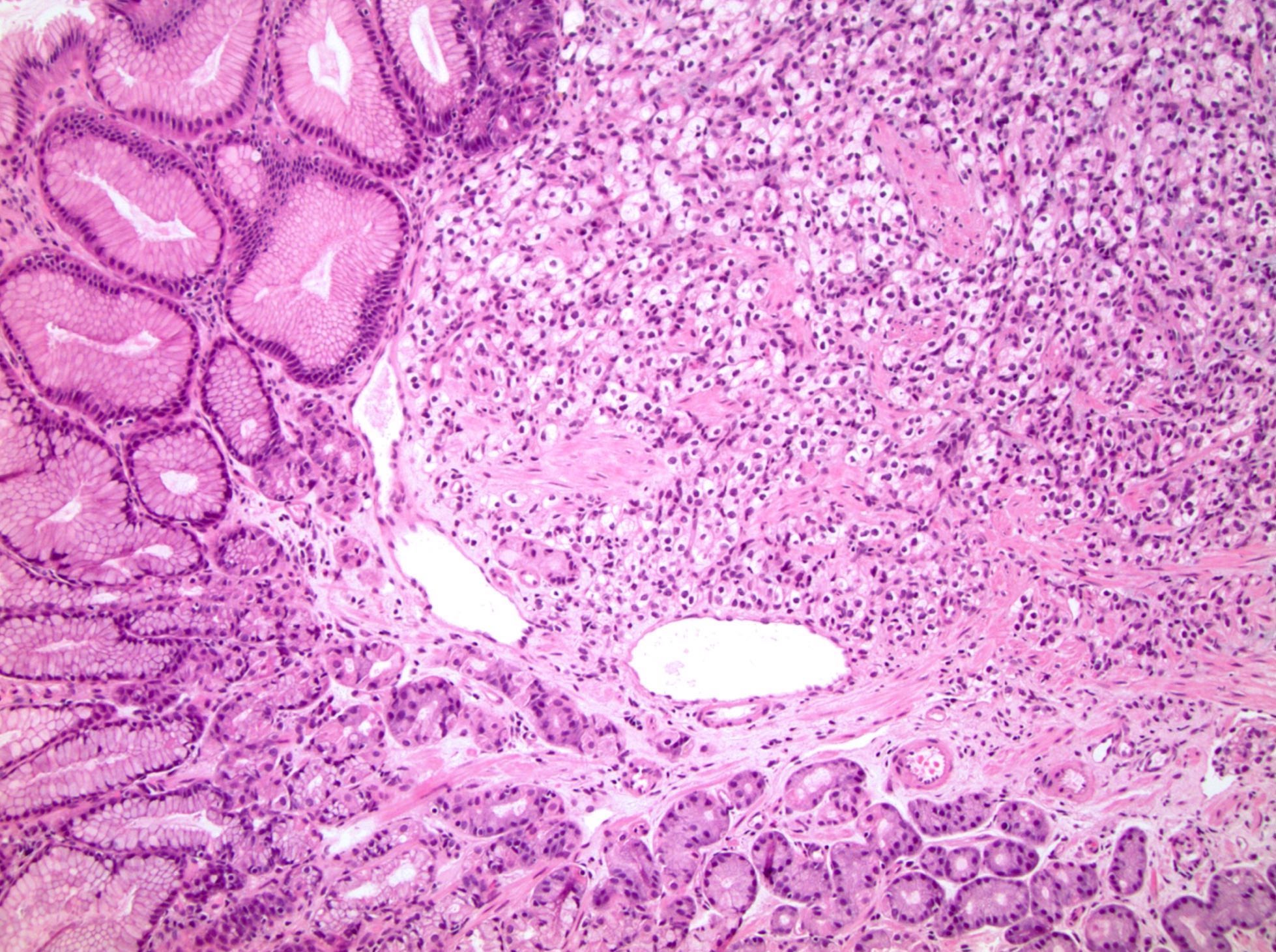
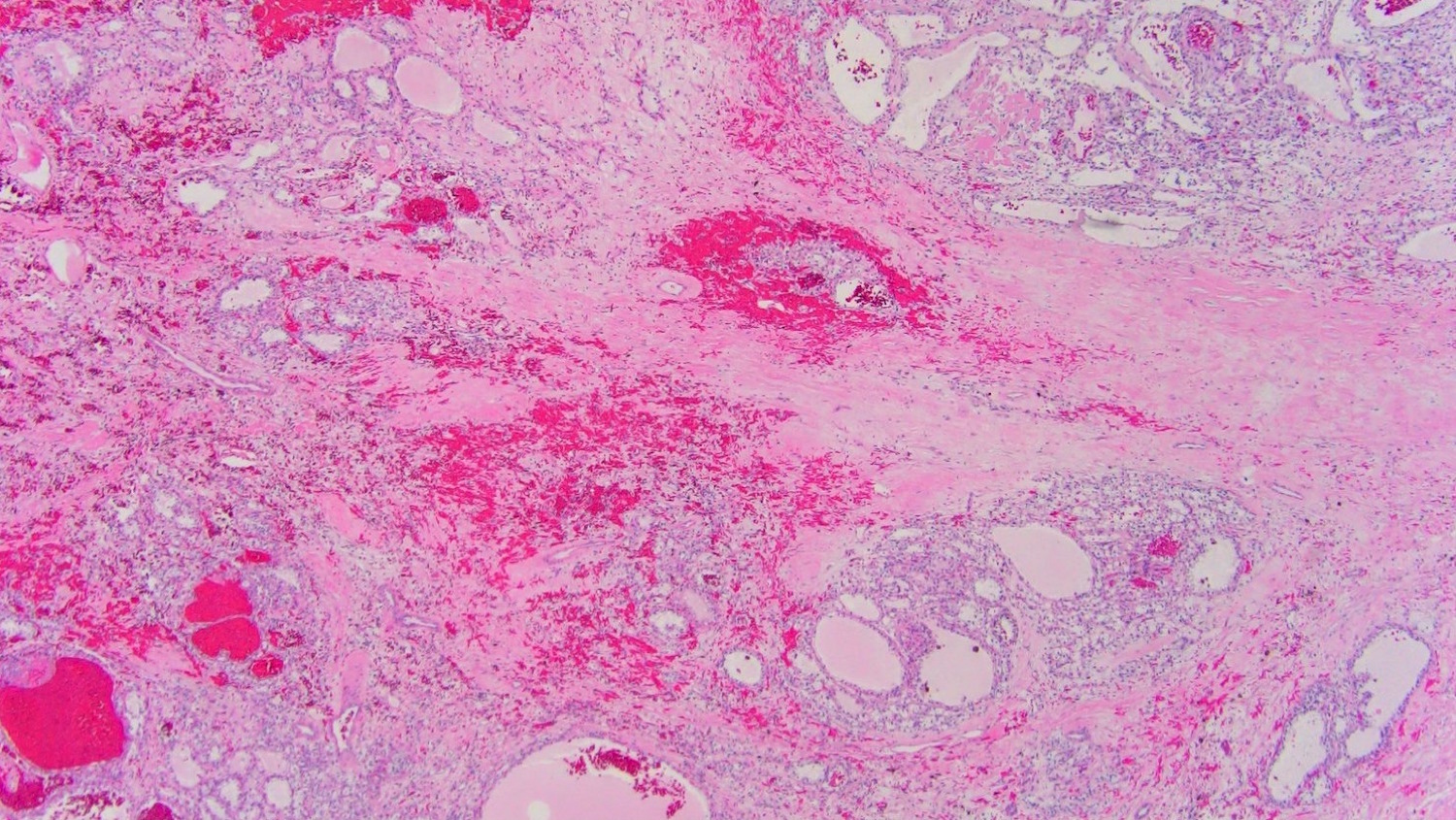
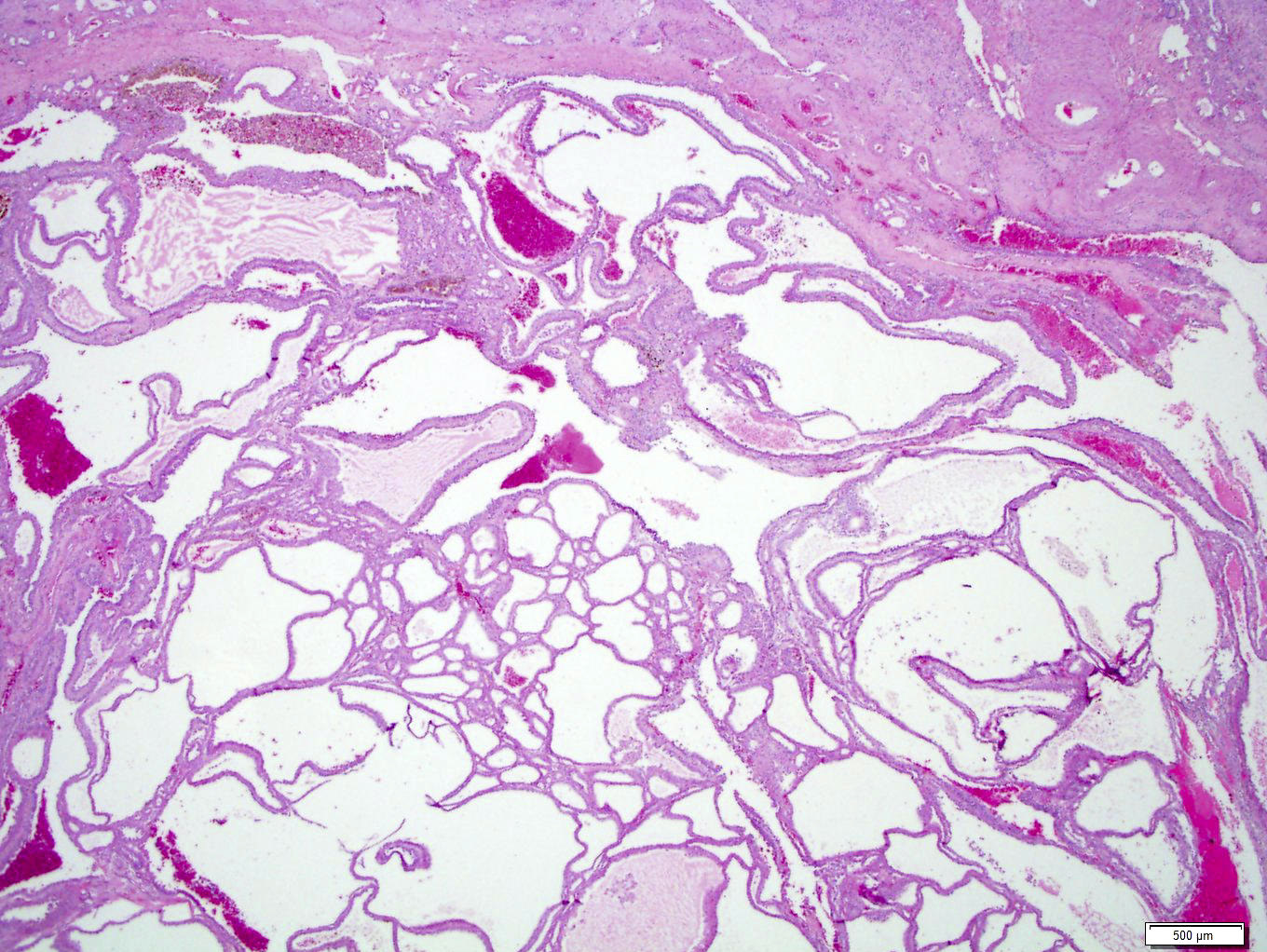
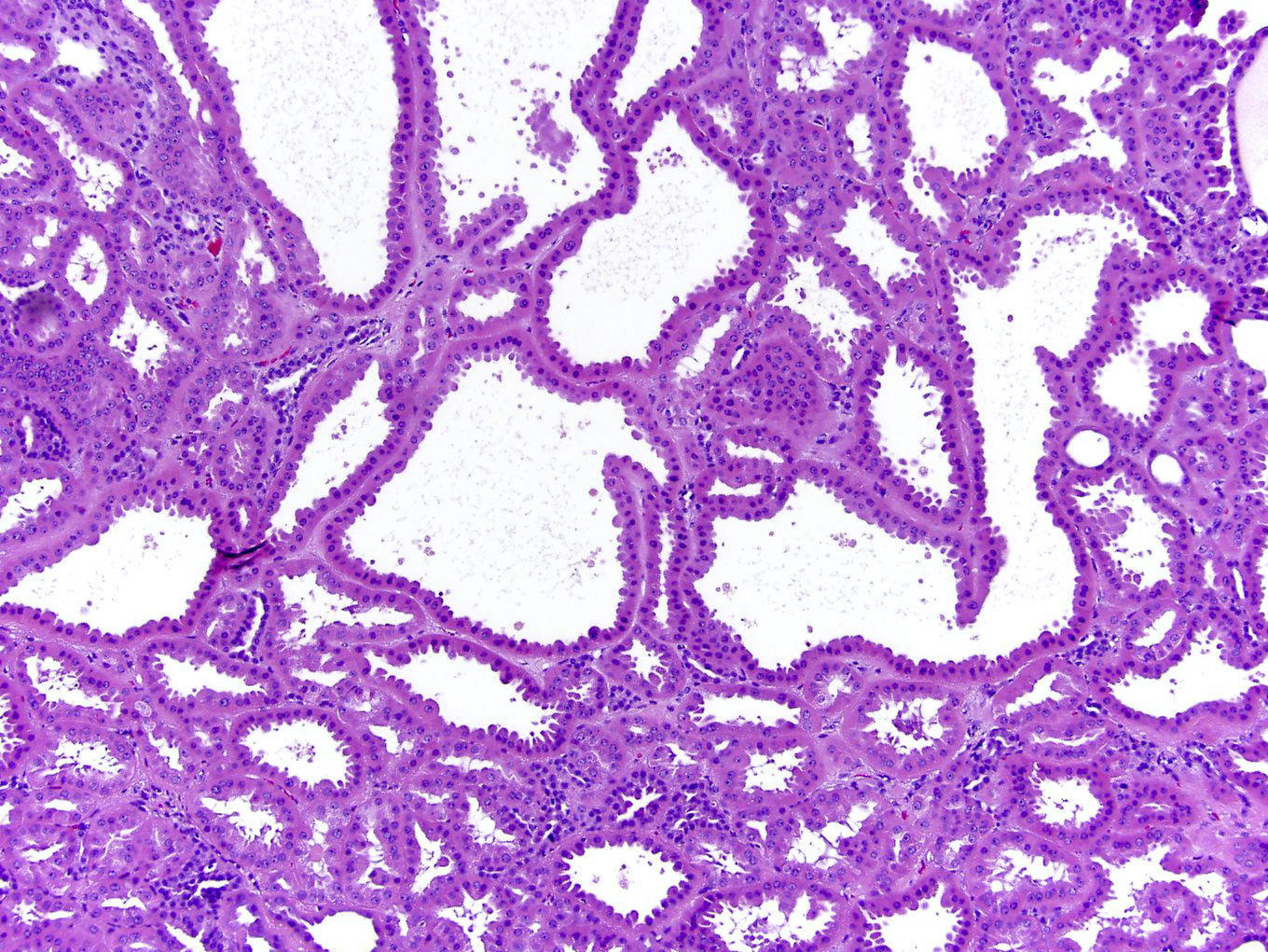
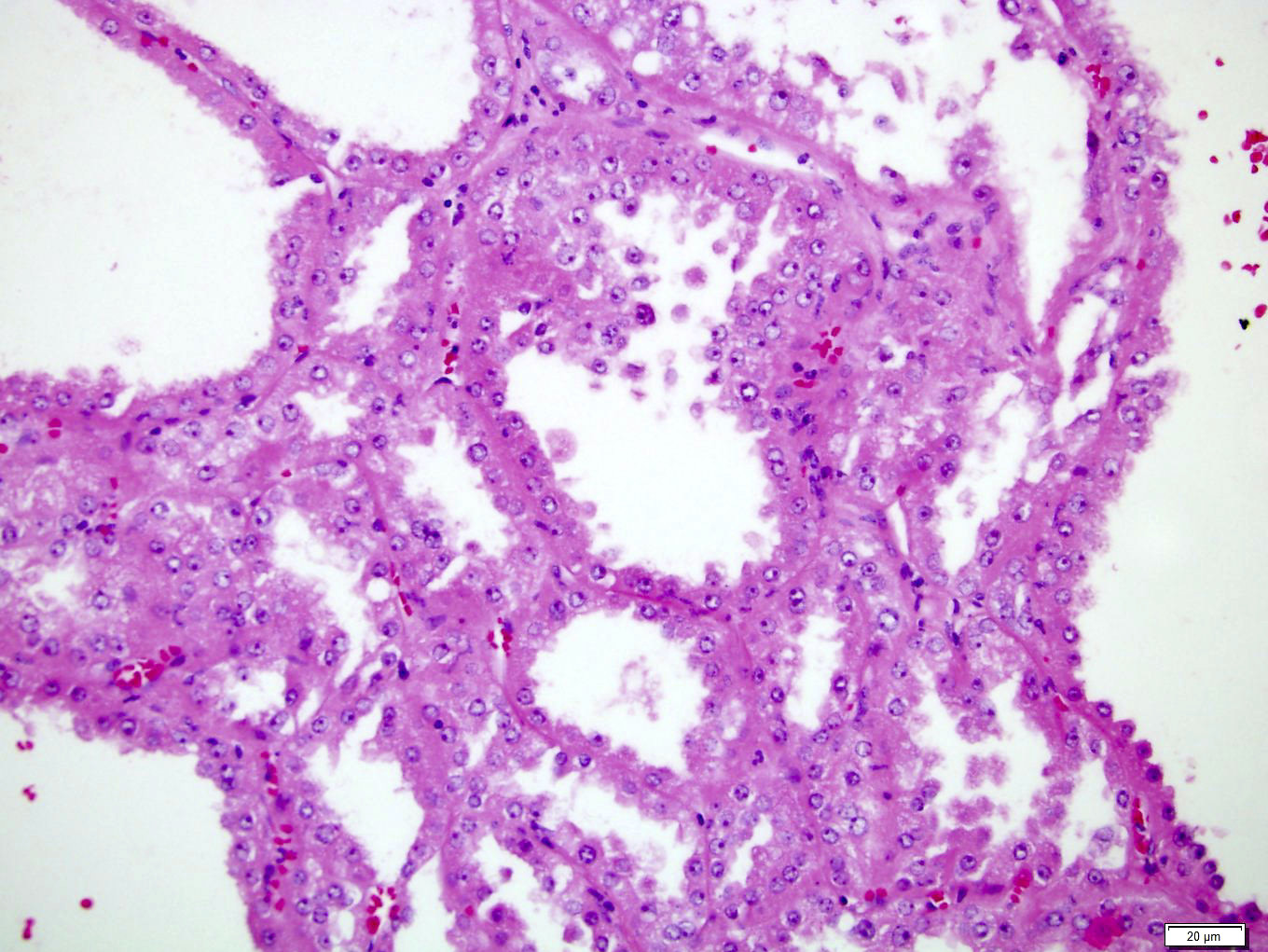
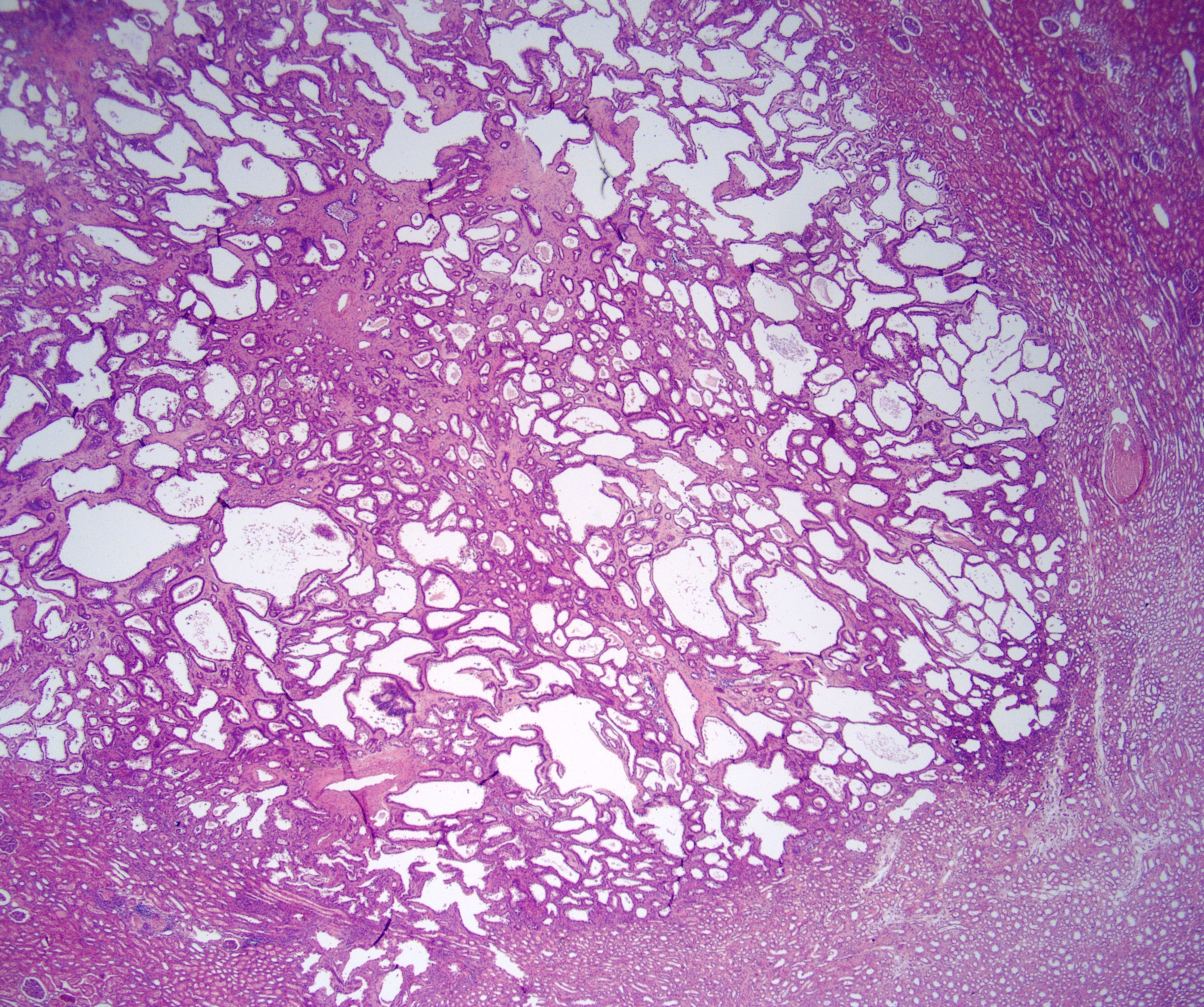
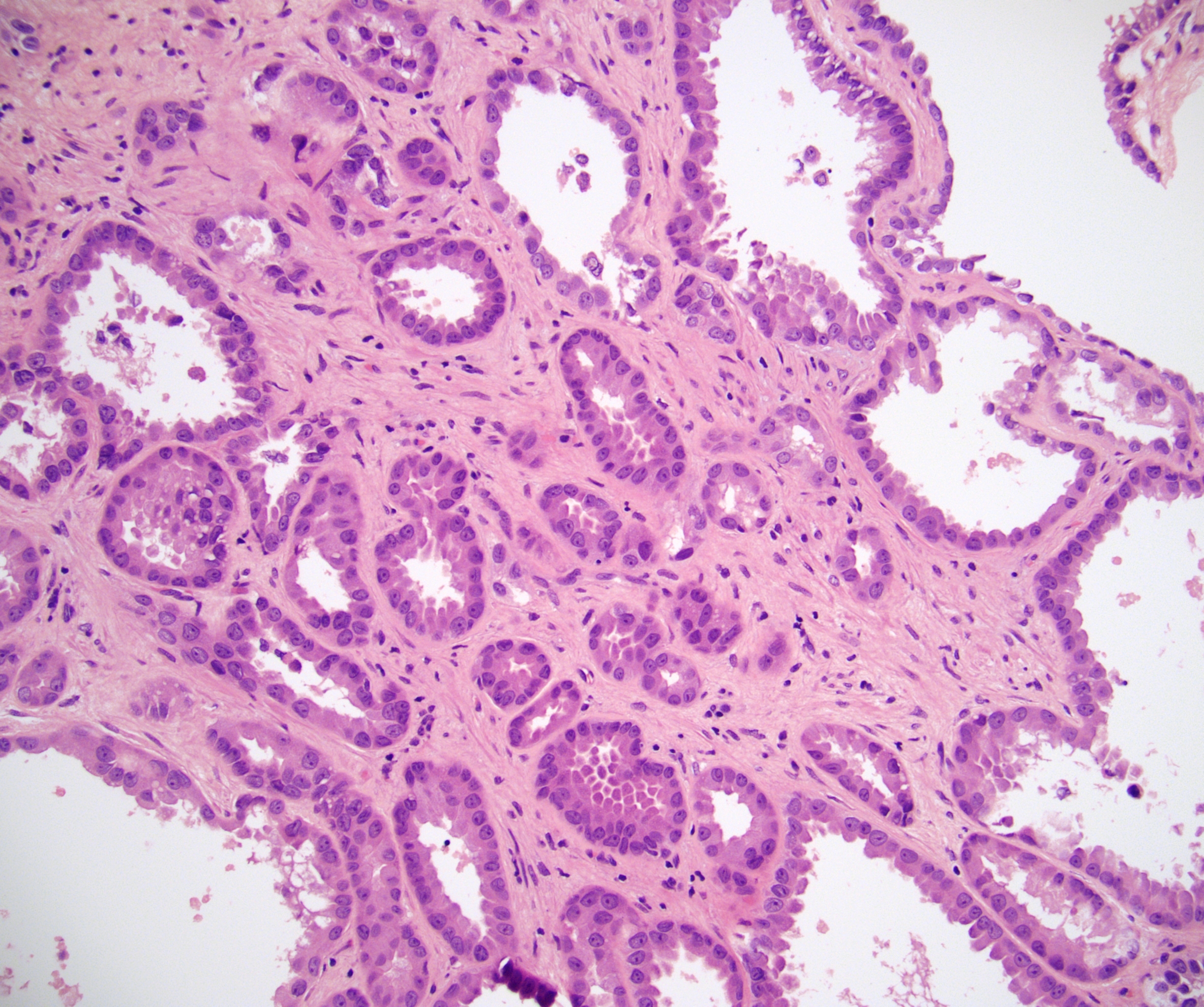
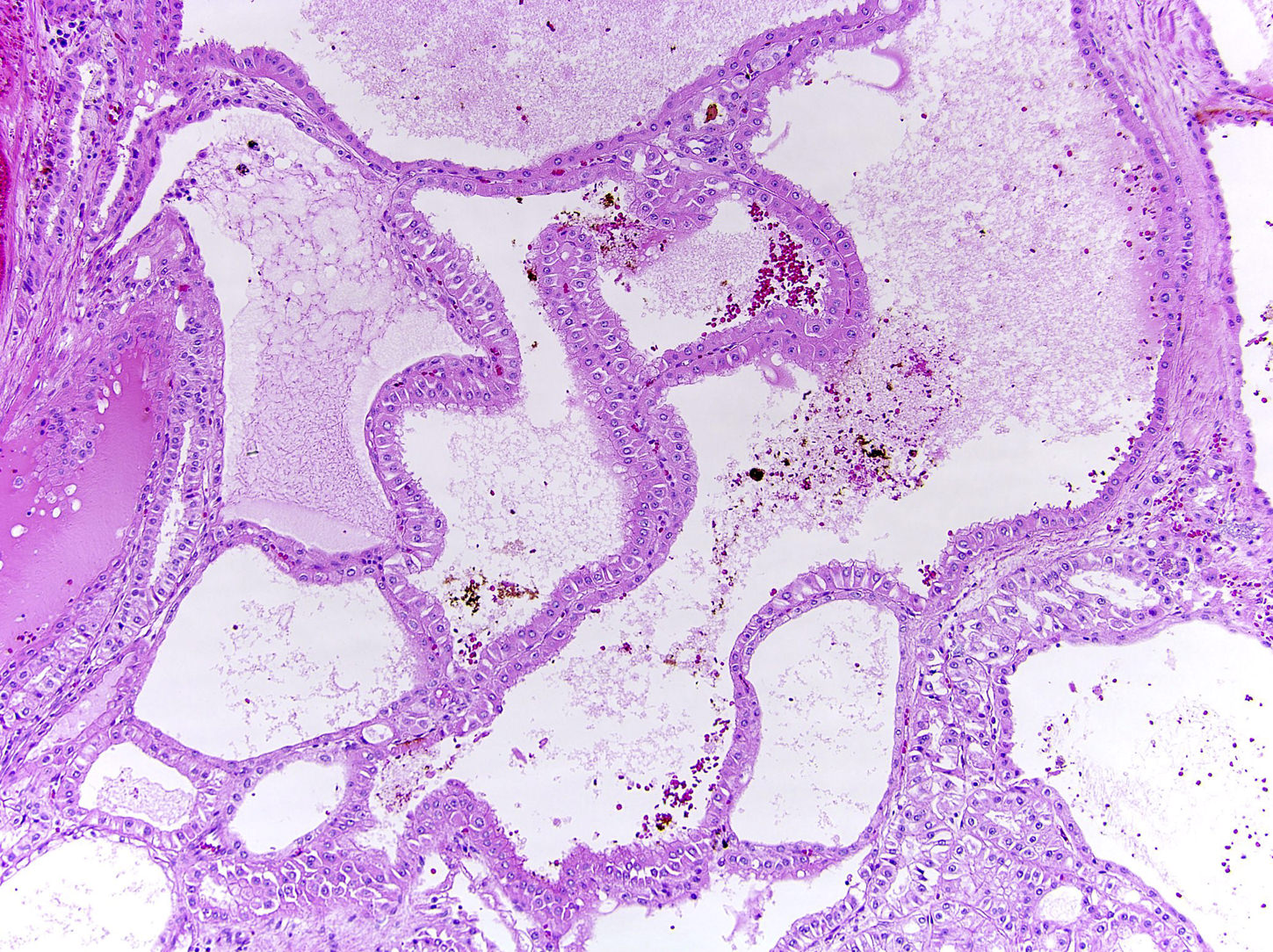
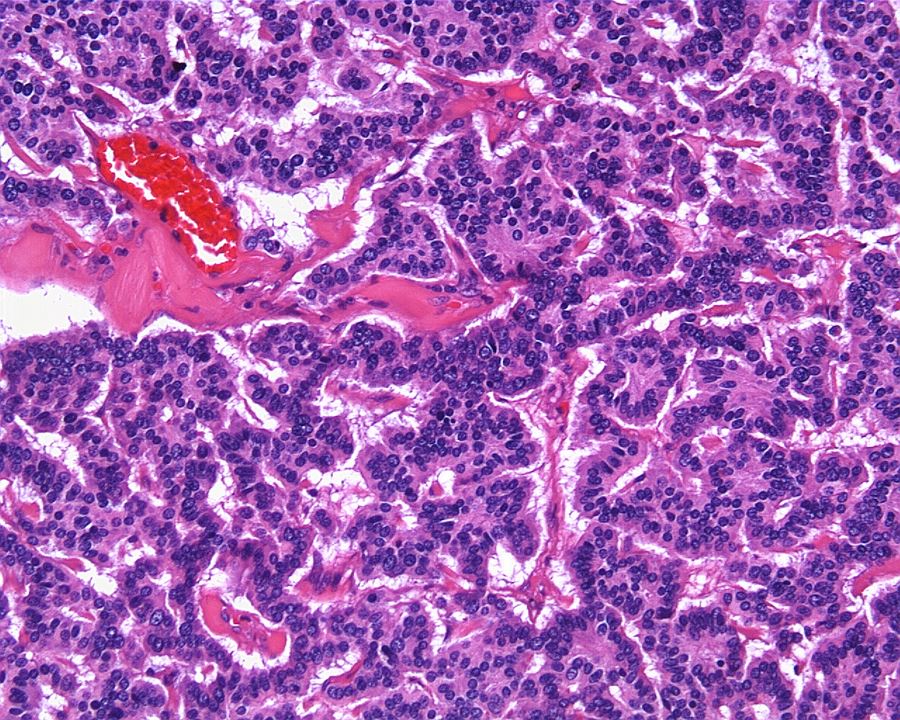
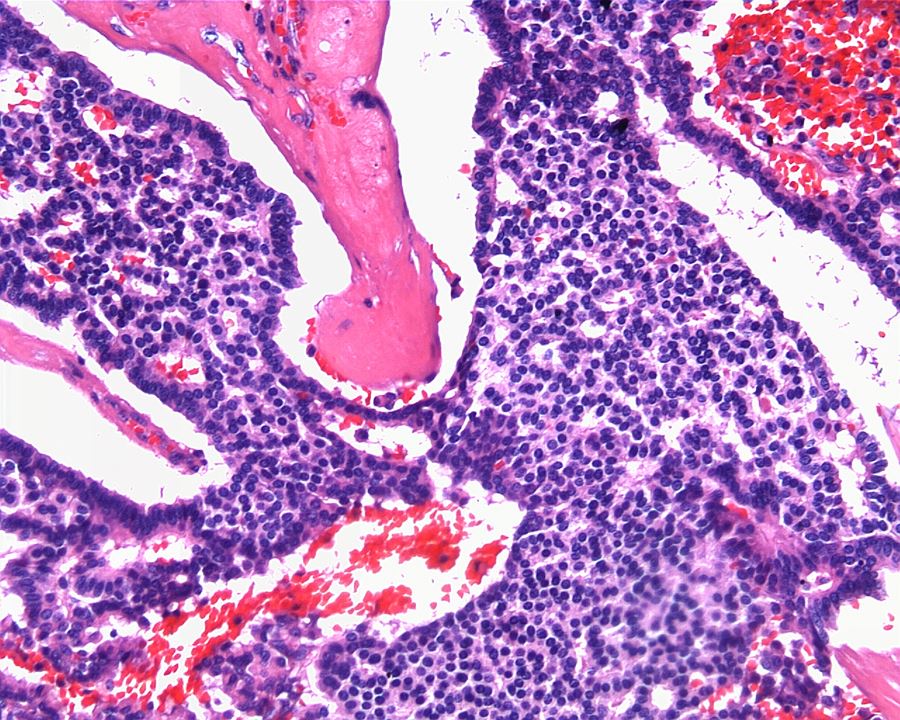
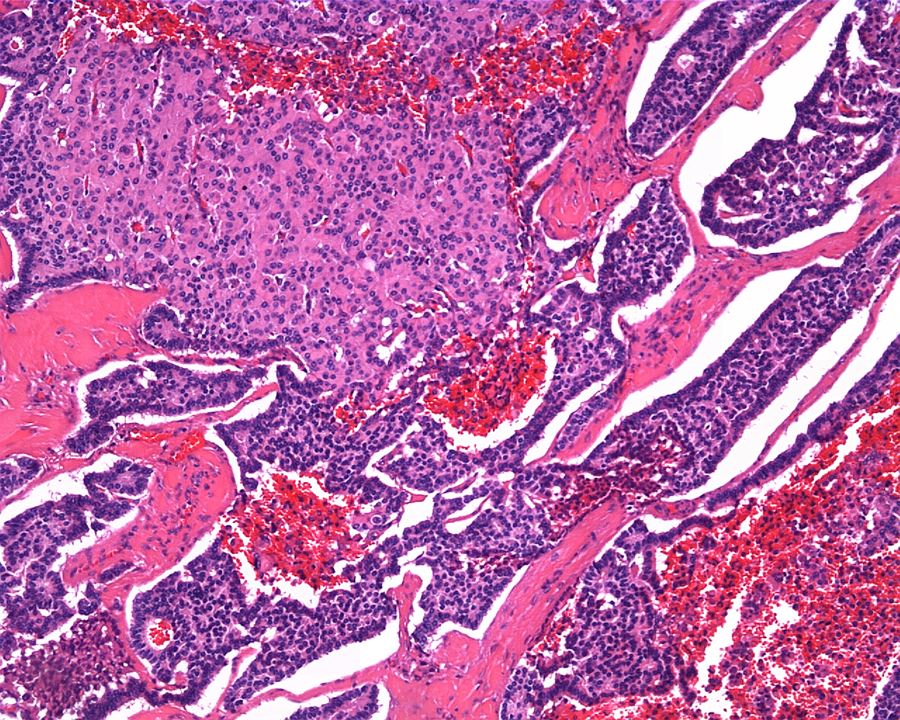
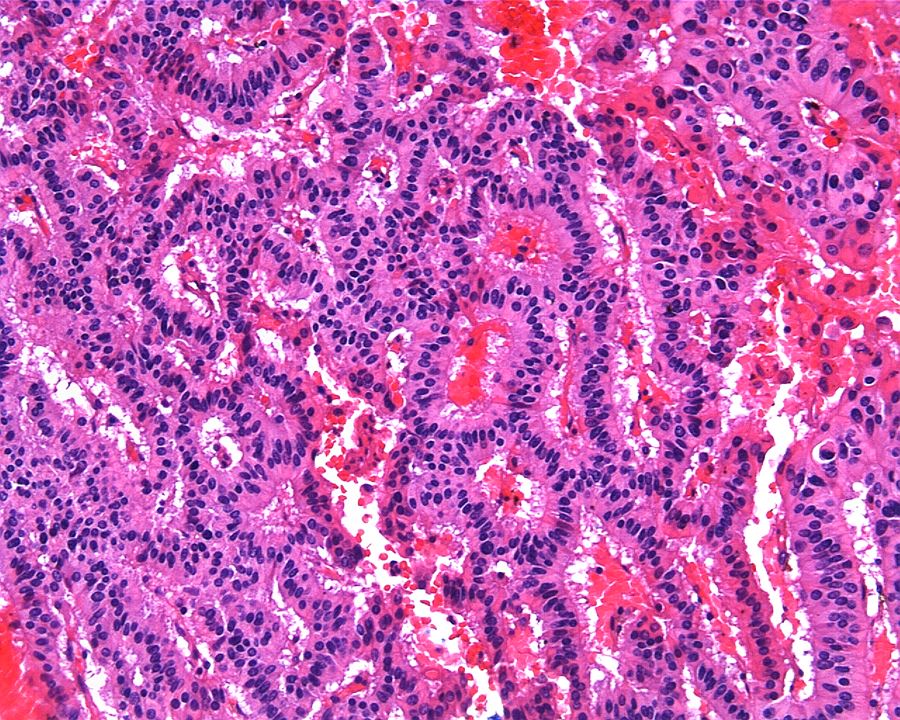
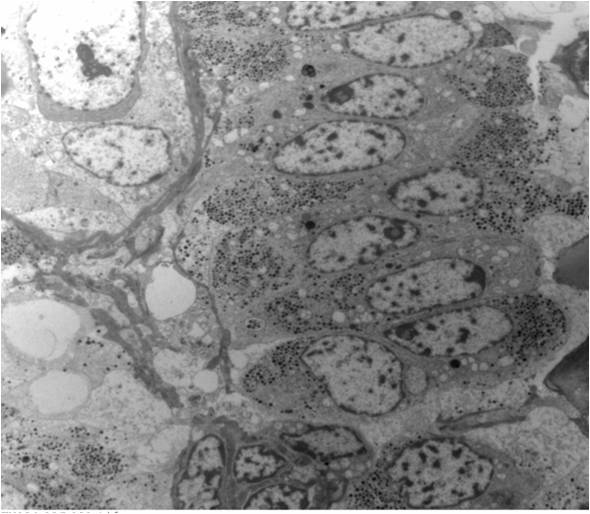
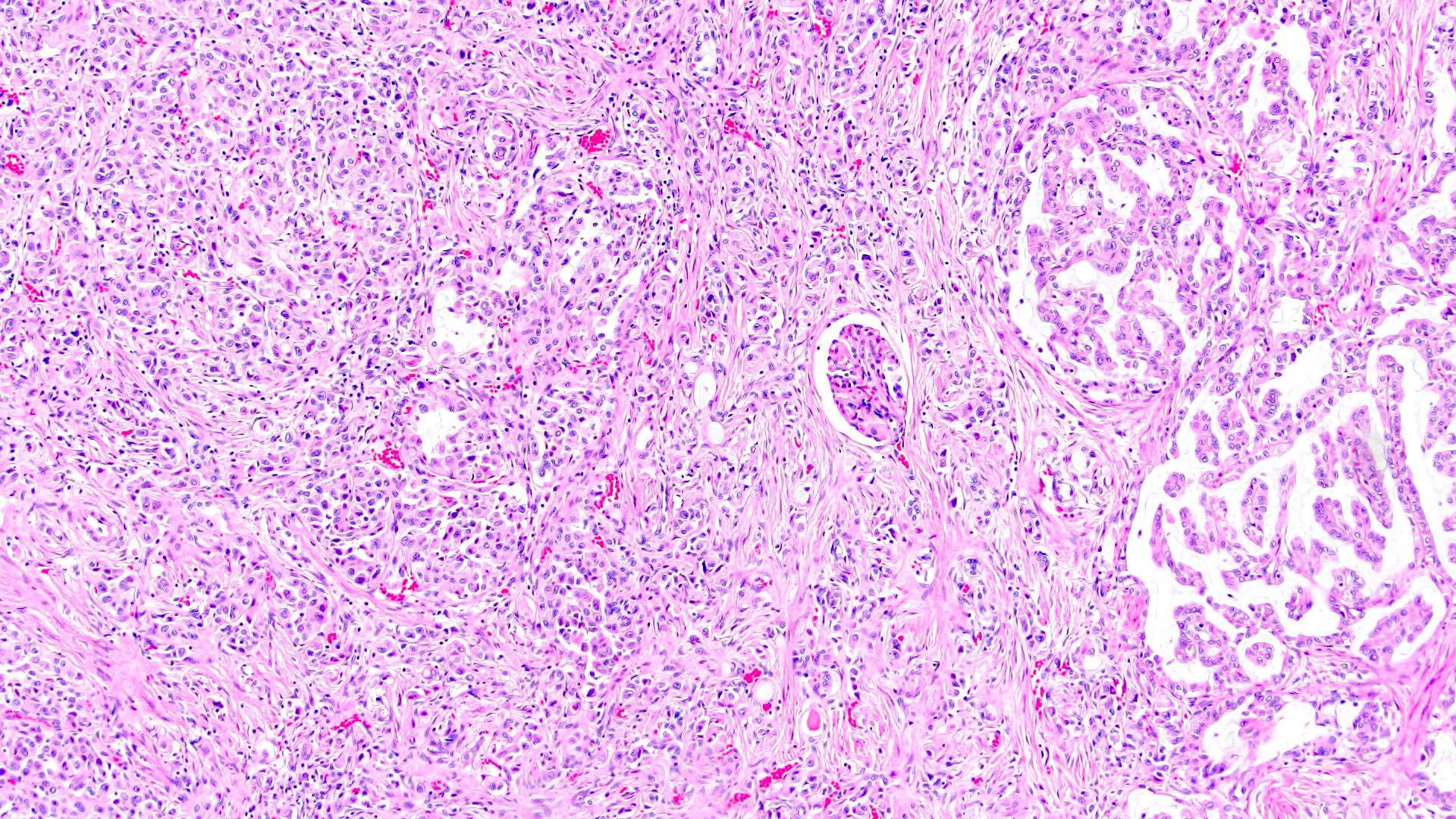
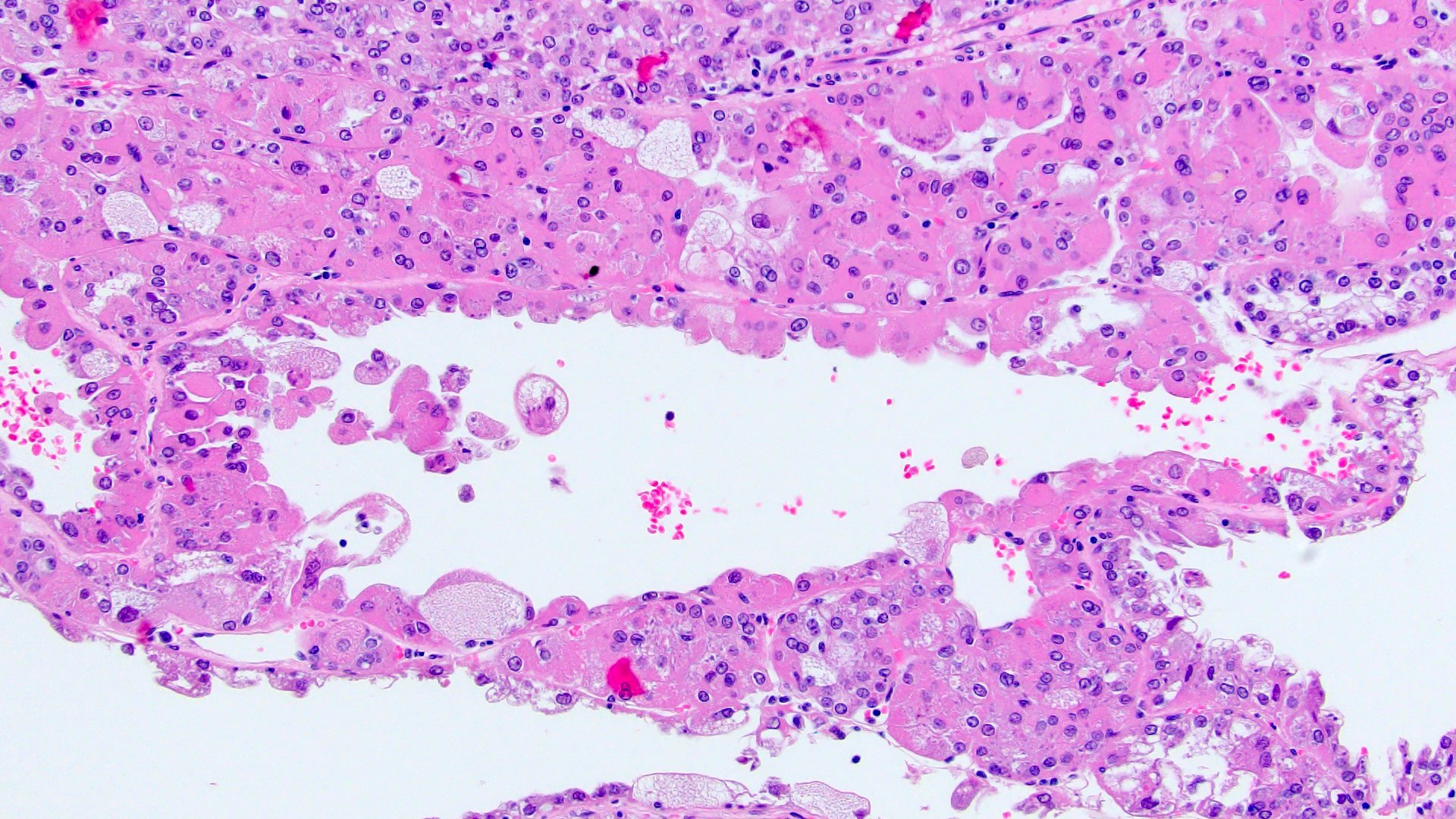
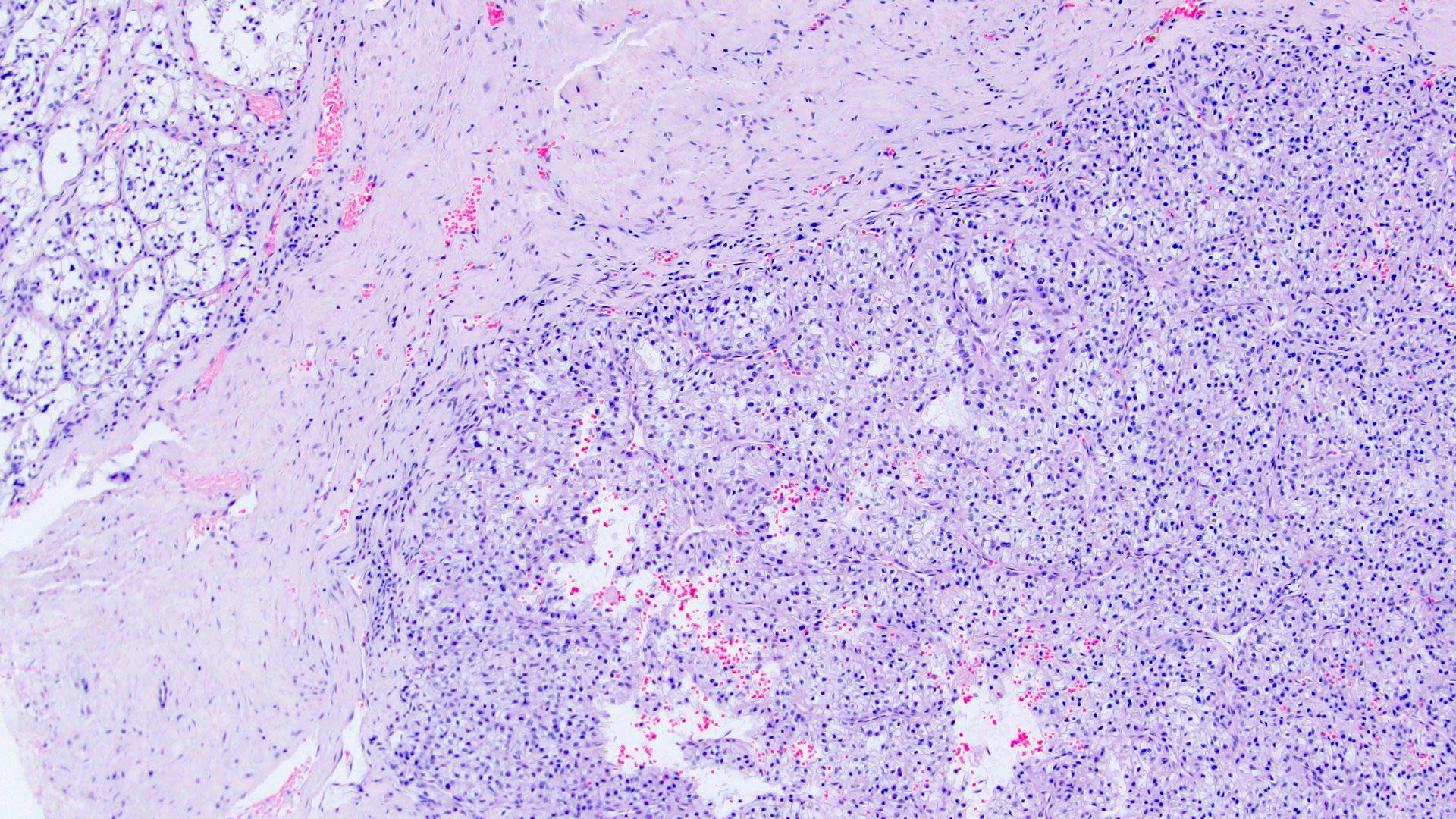
ALK-RCC are medulla based and morphologically resemble medullary renal carcinomas. In adults,
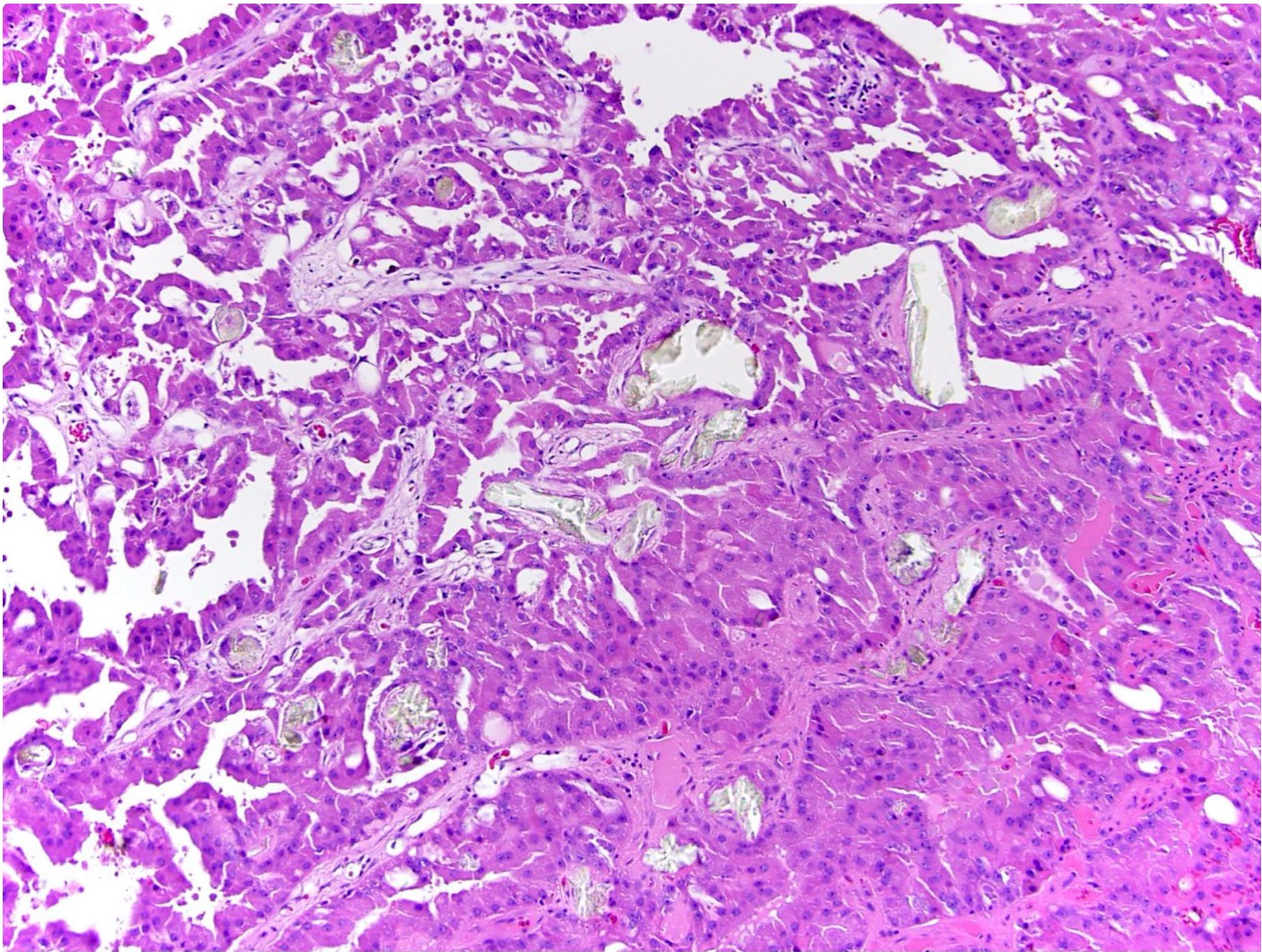
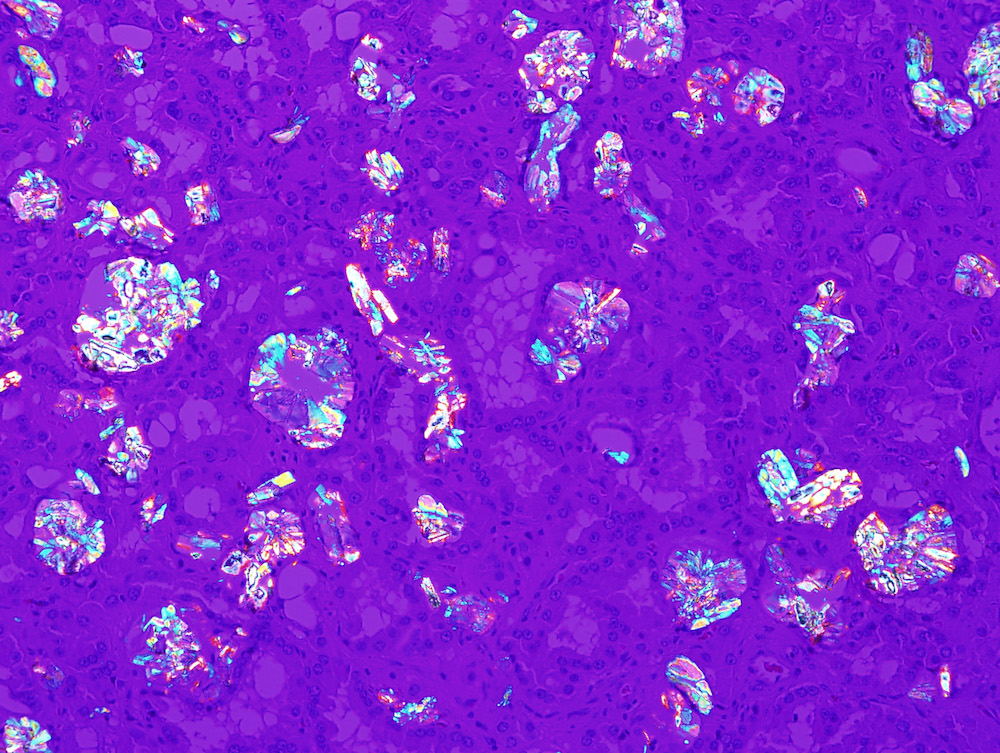
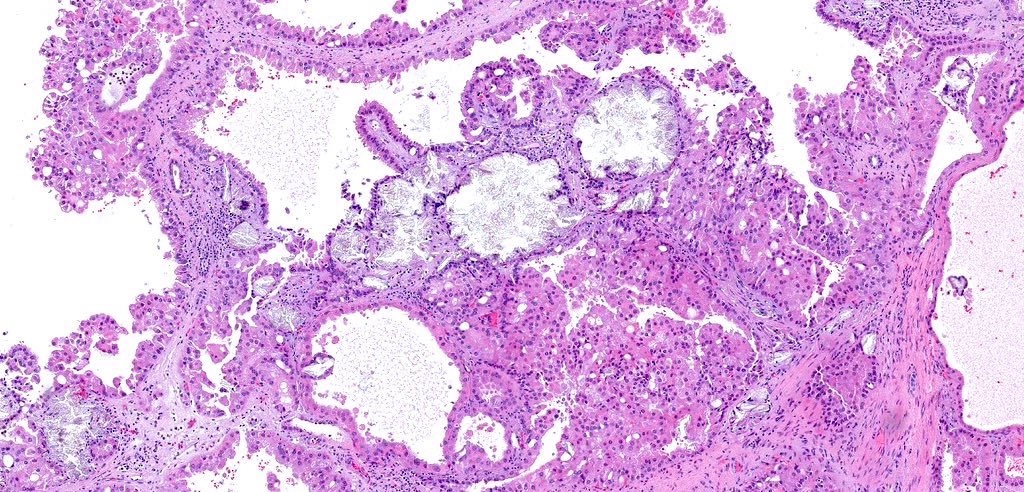
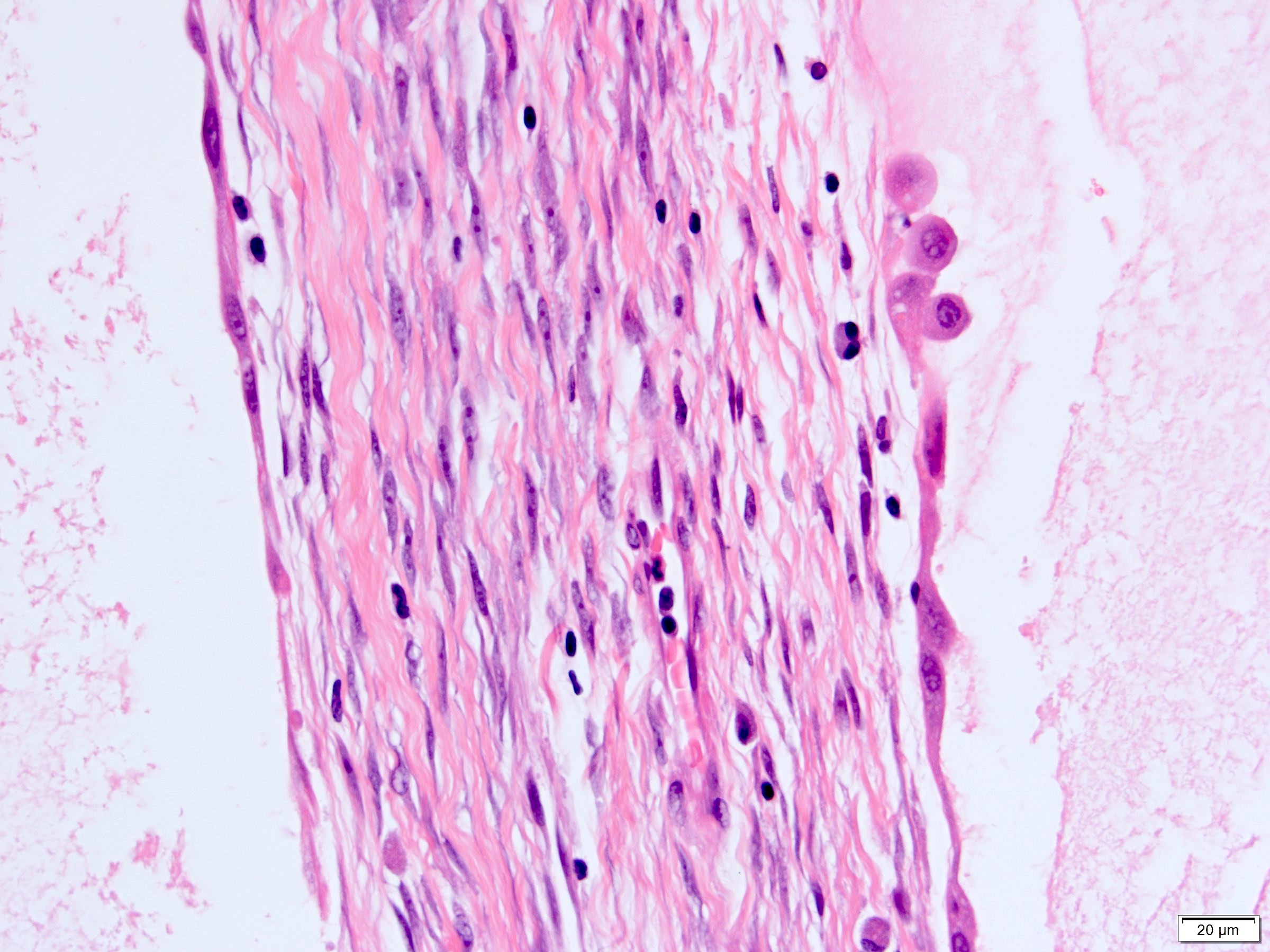
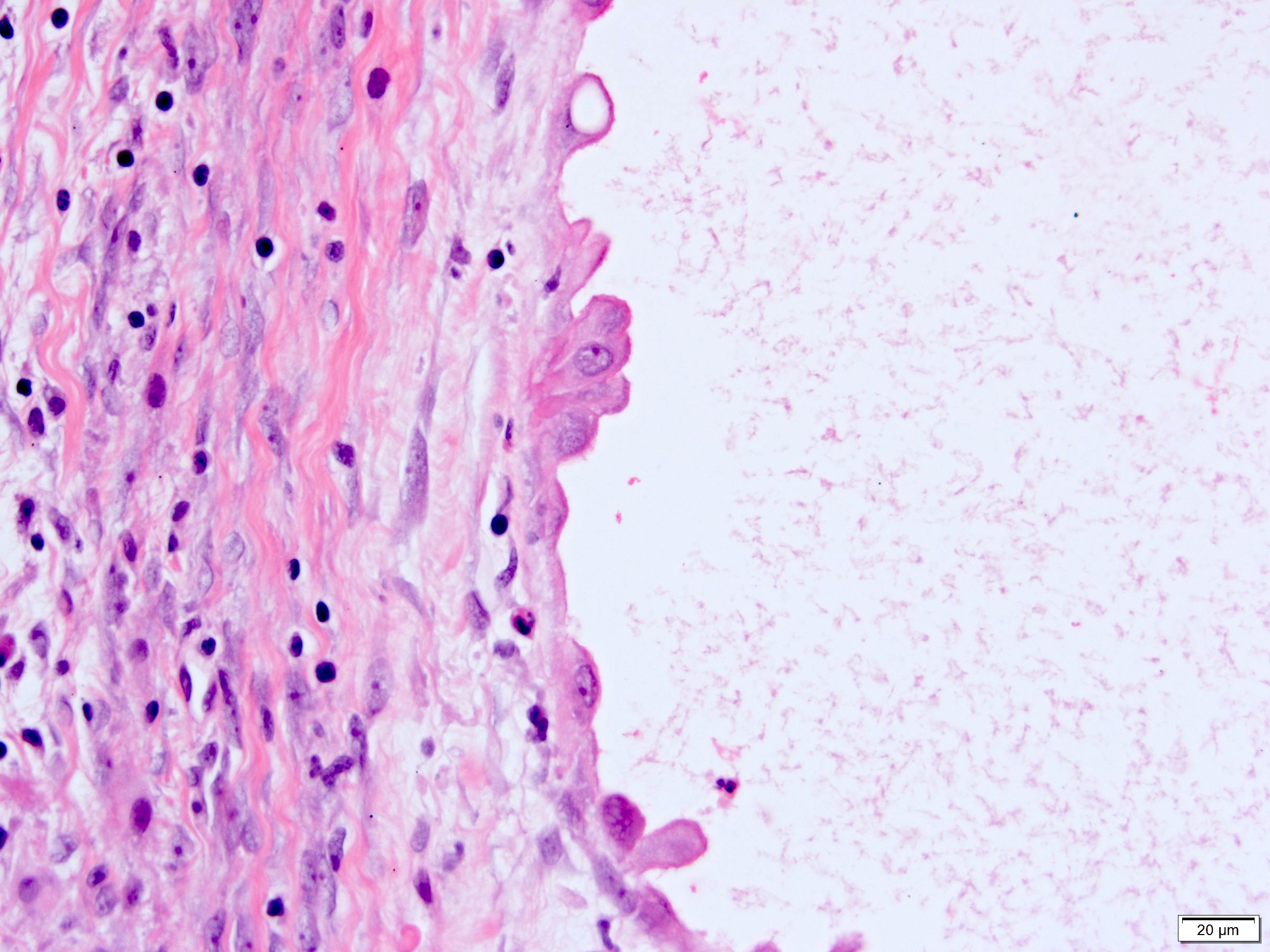
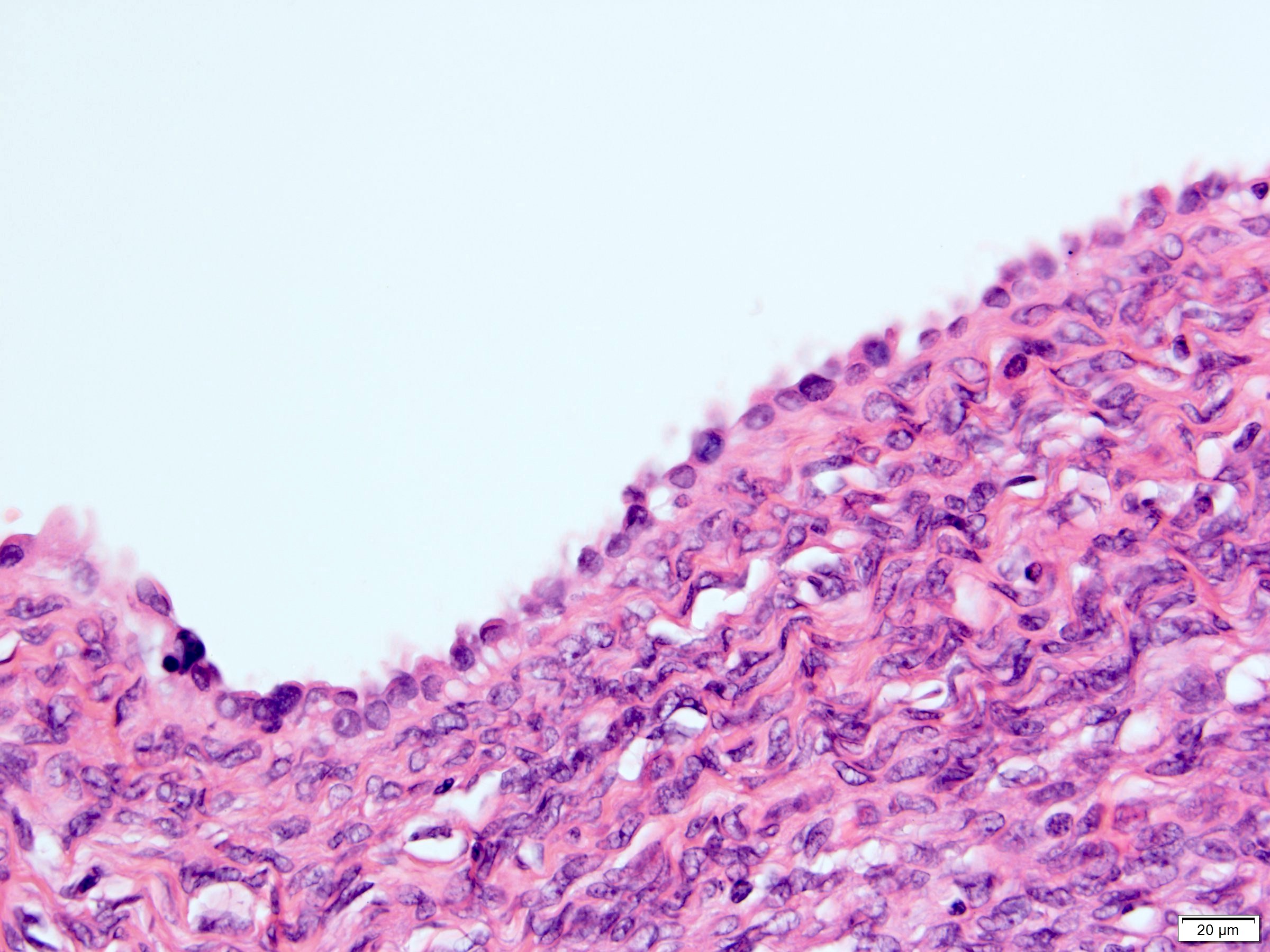
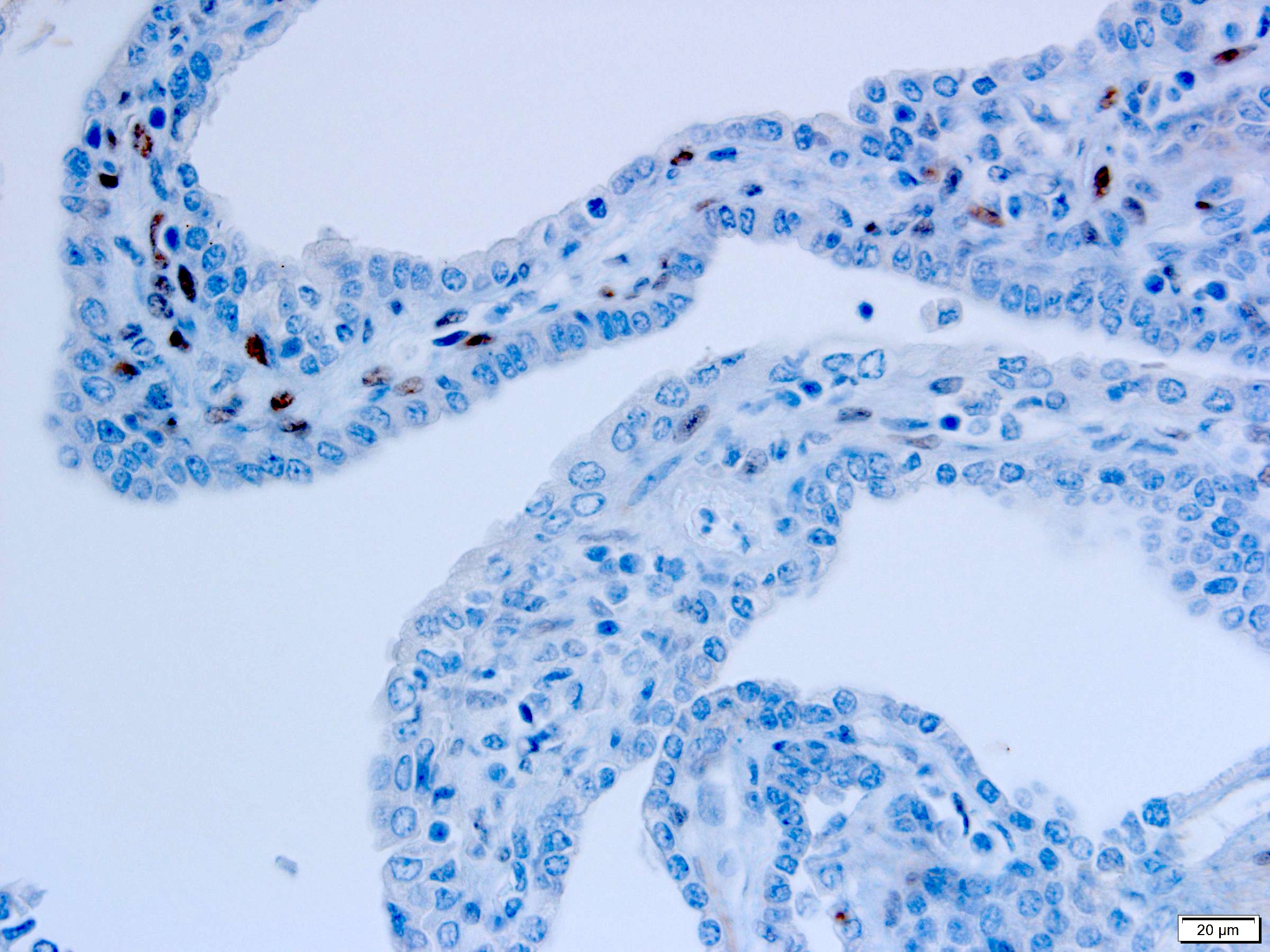
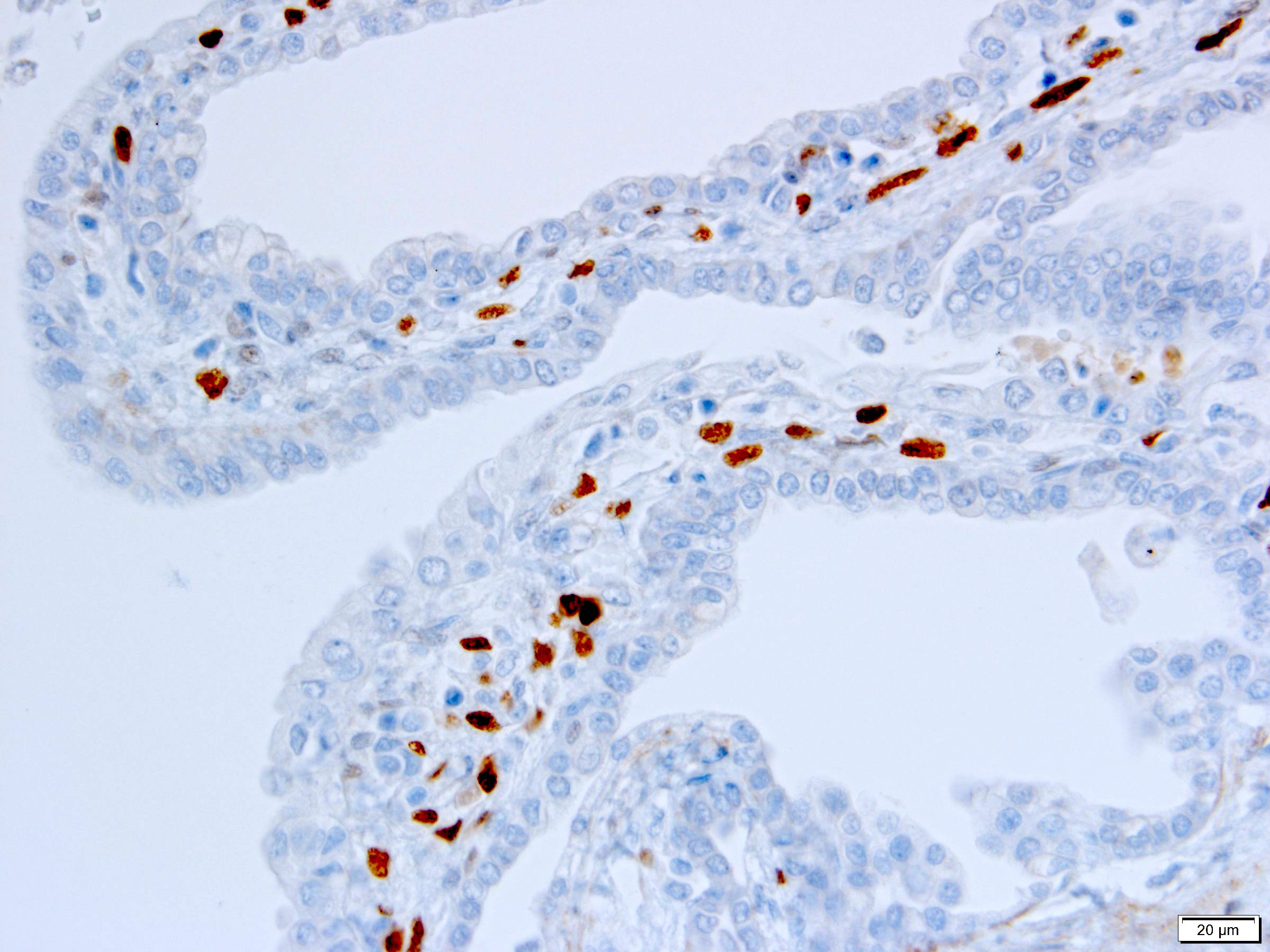
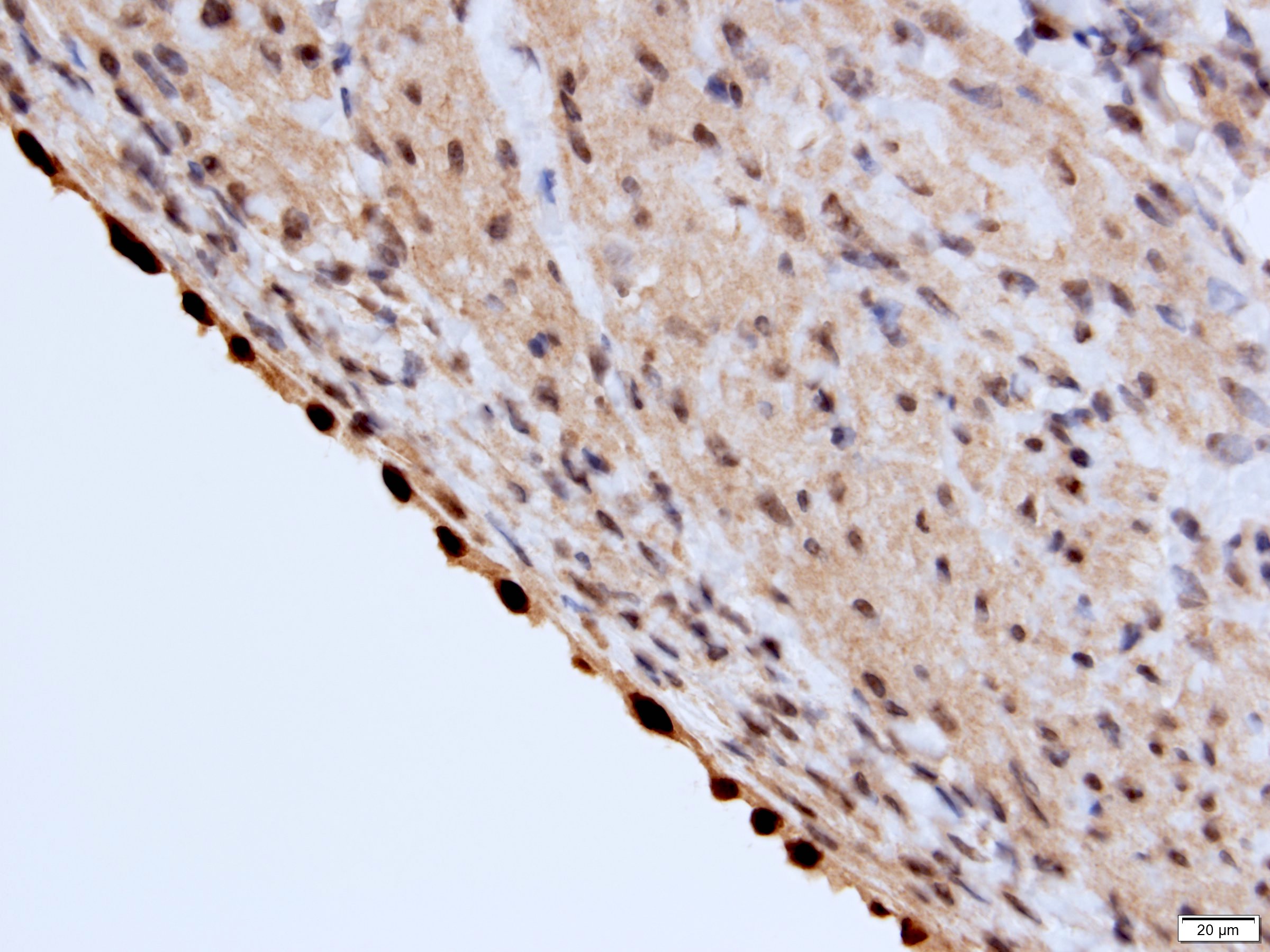
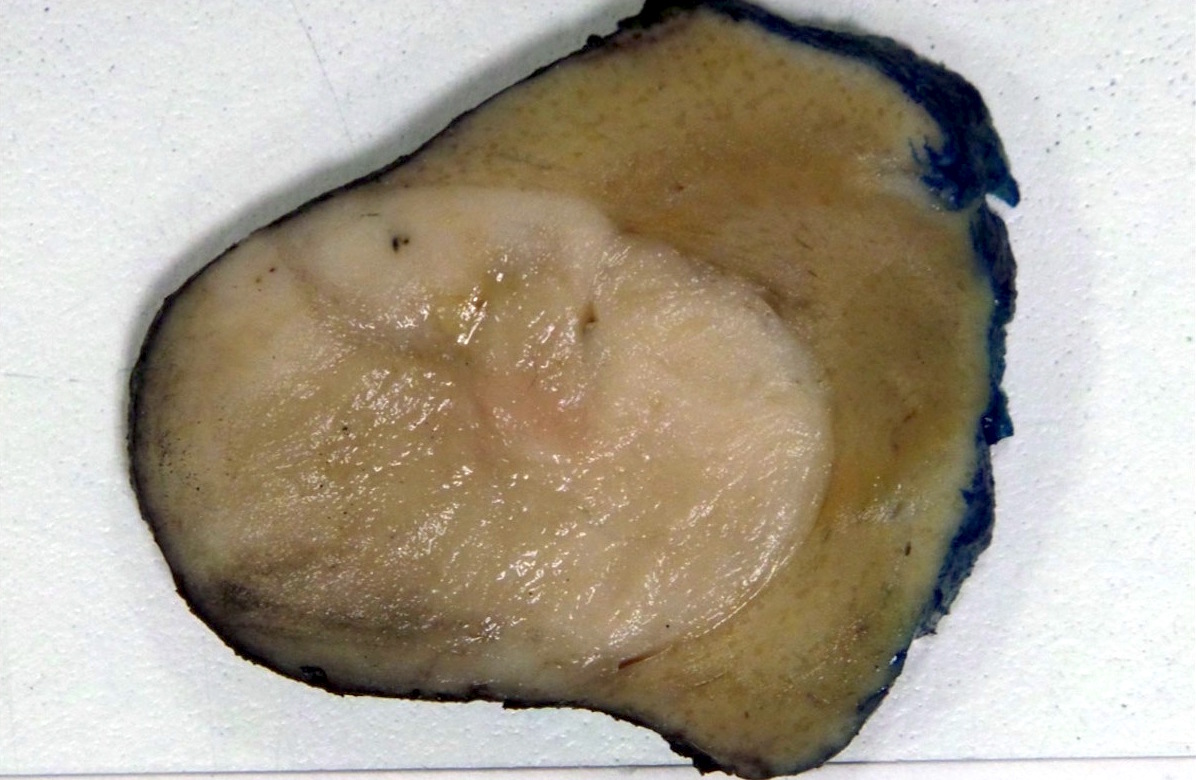
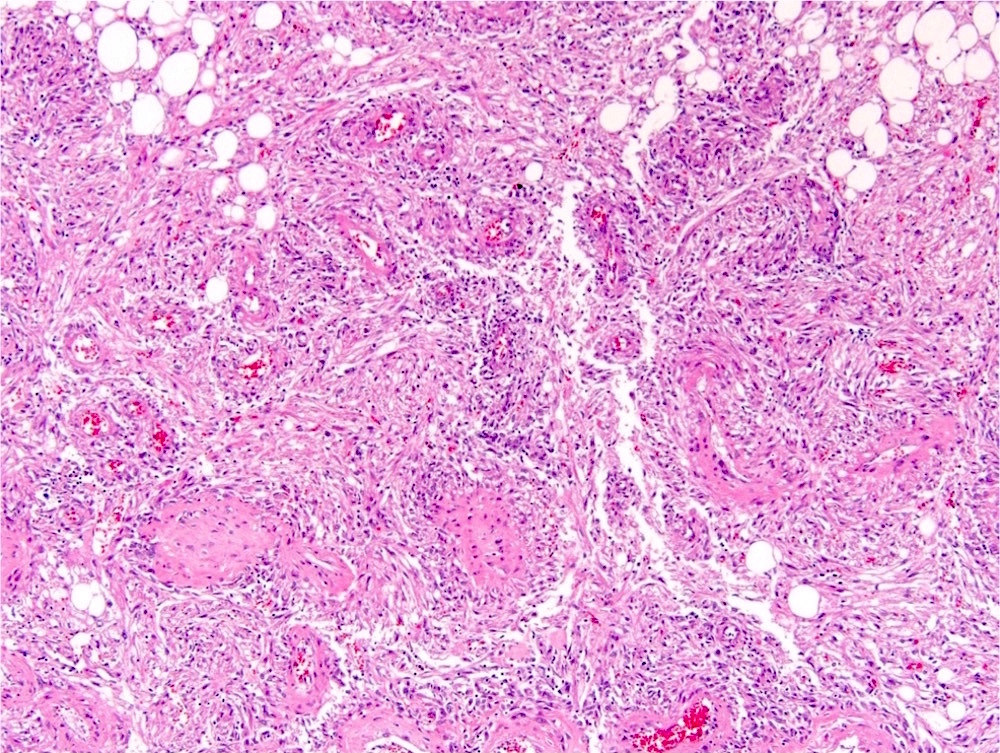
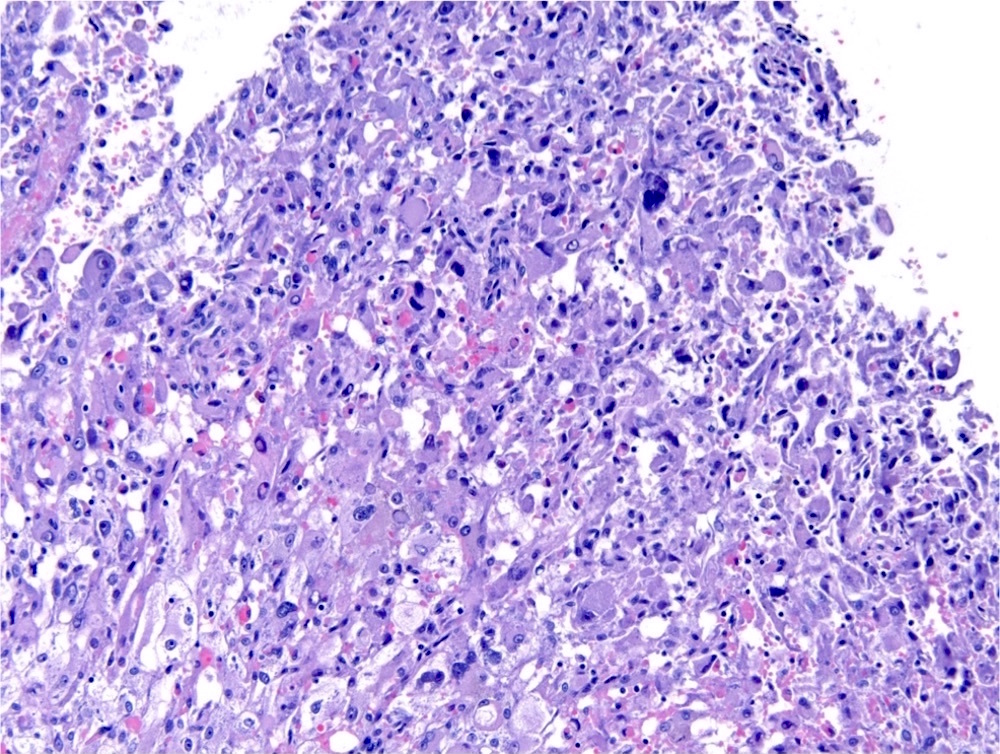
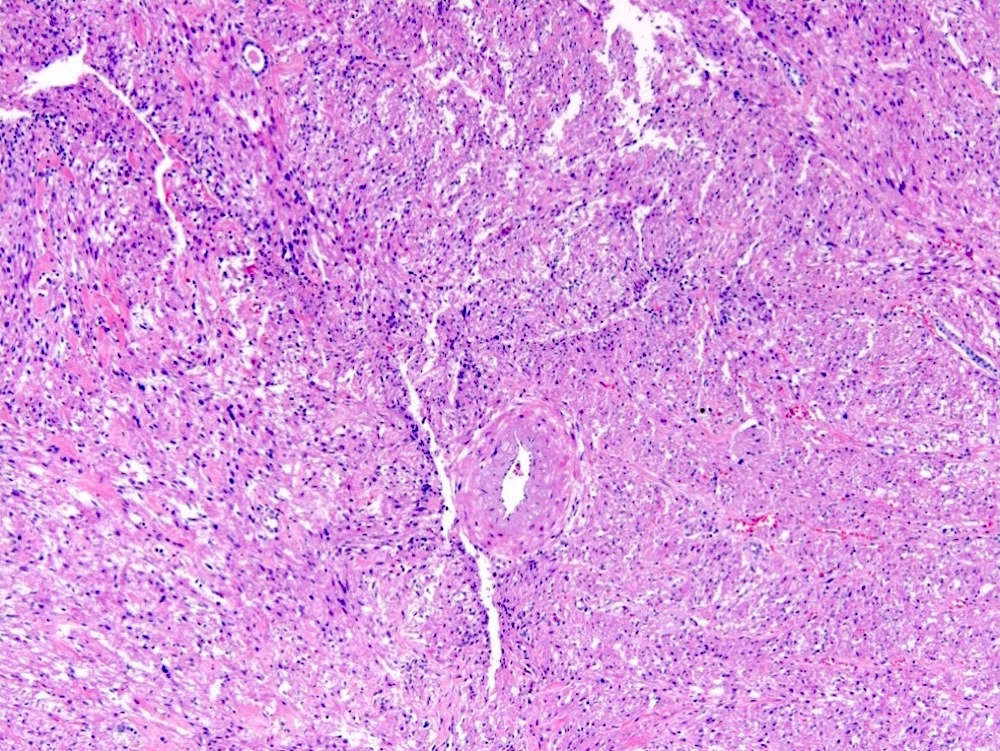
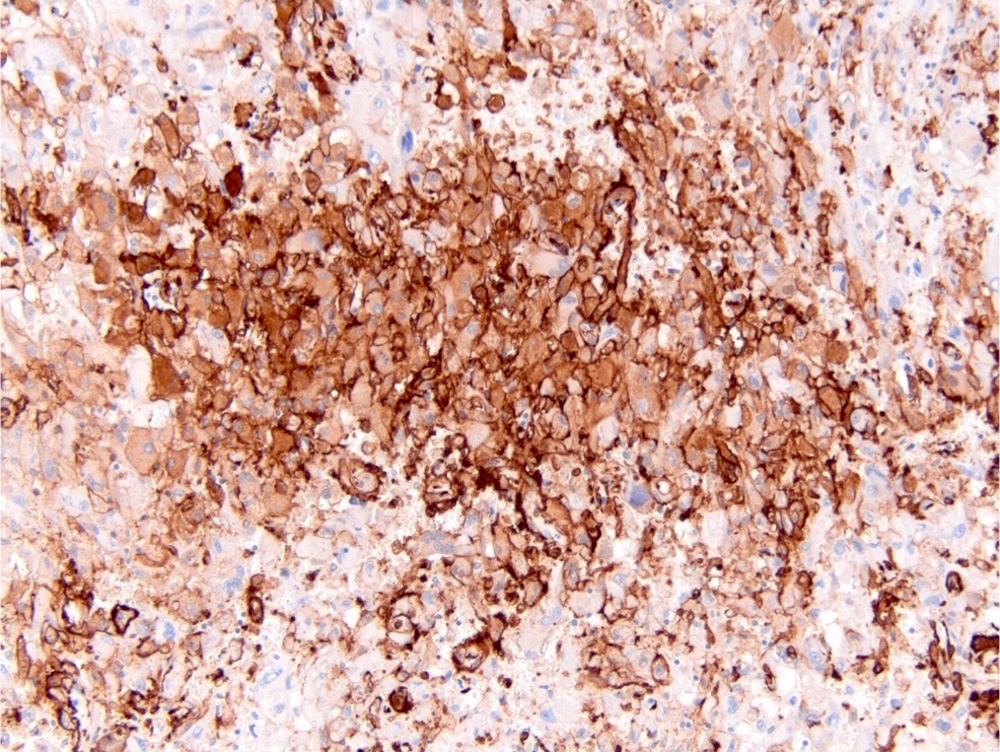
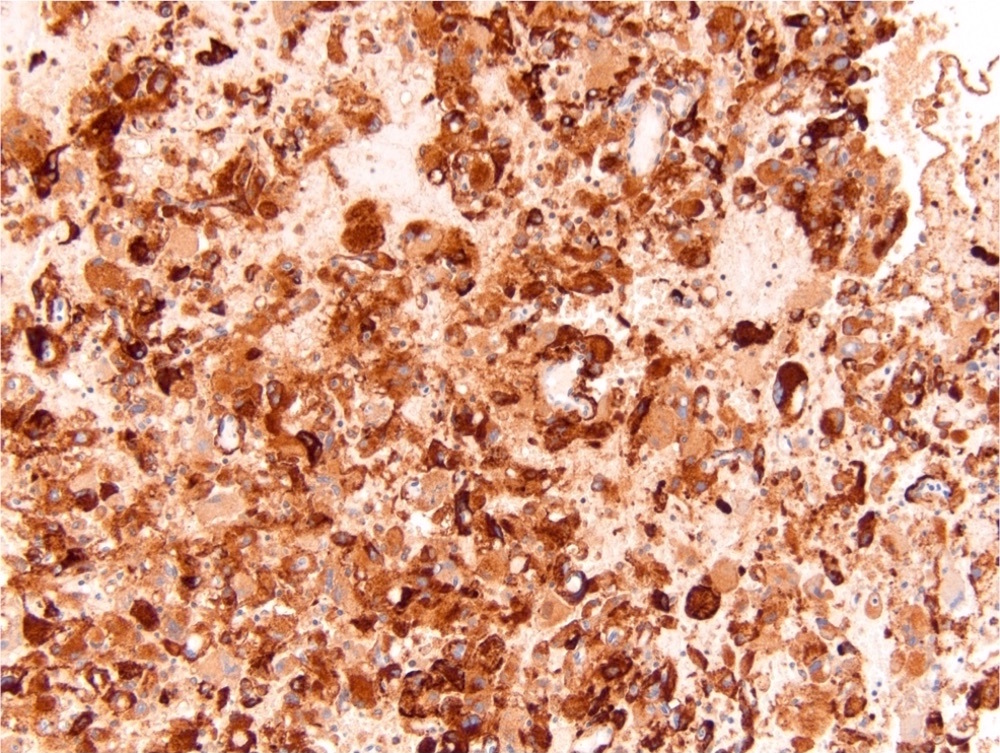
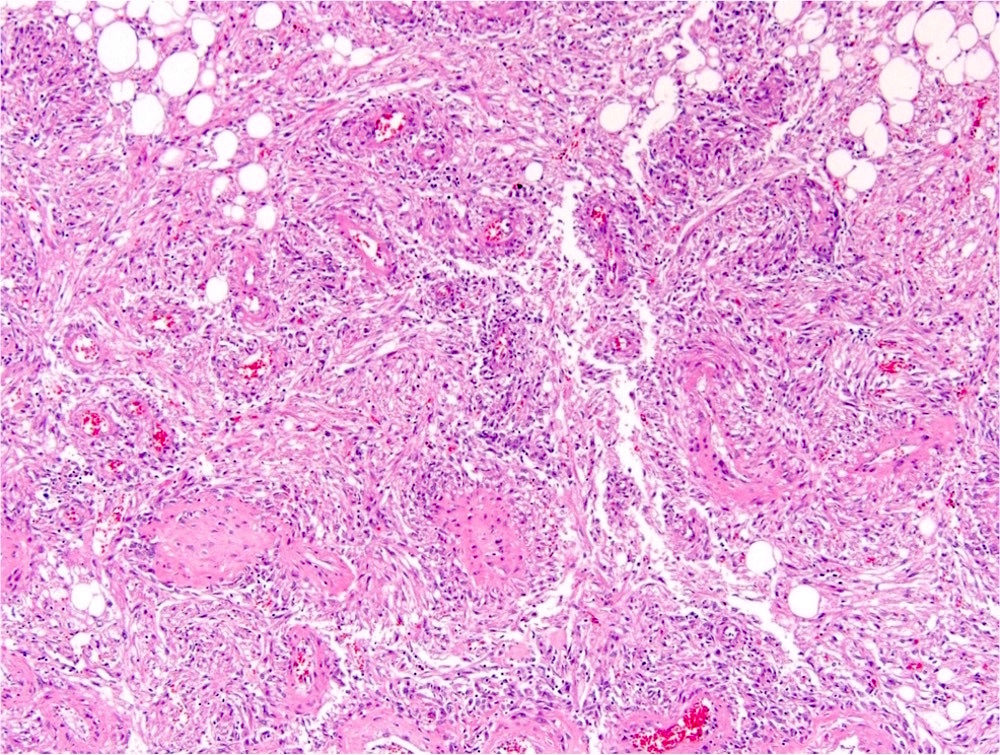
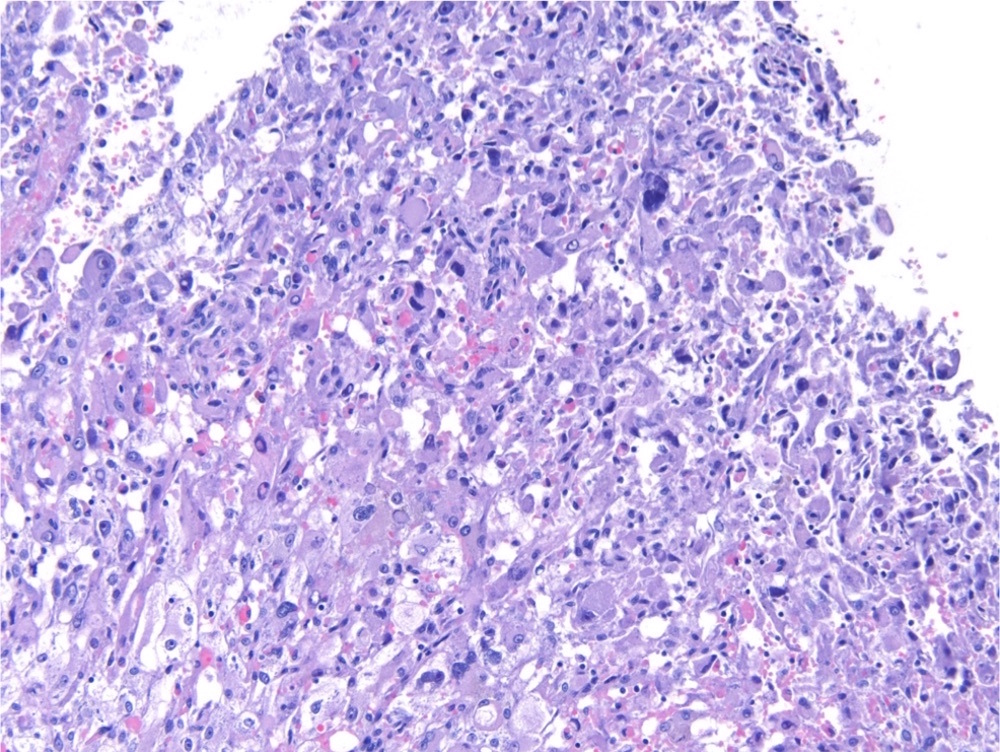
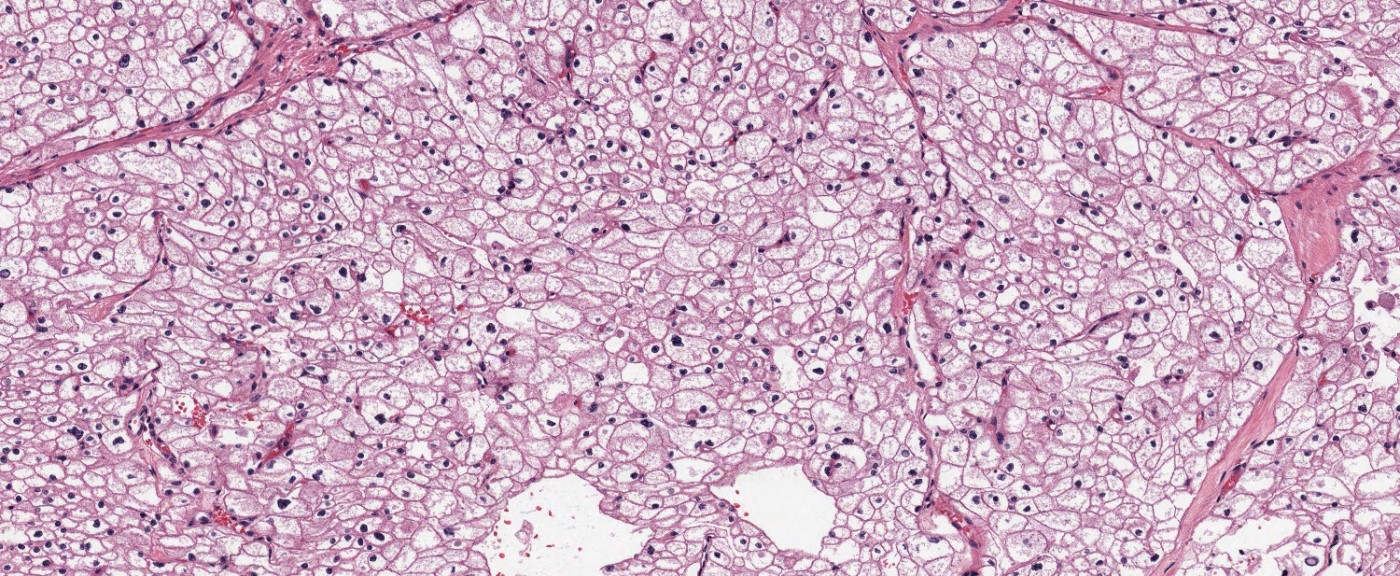
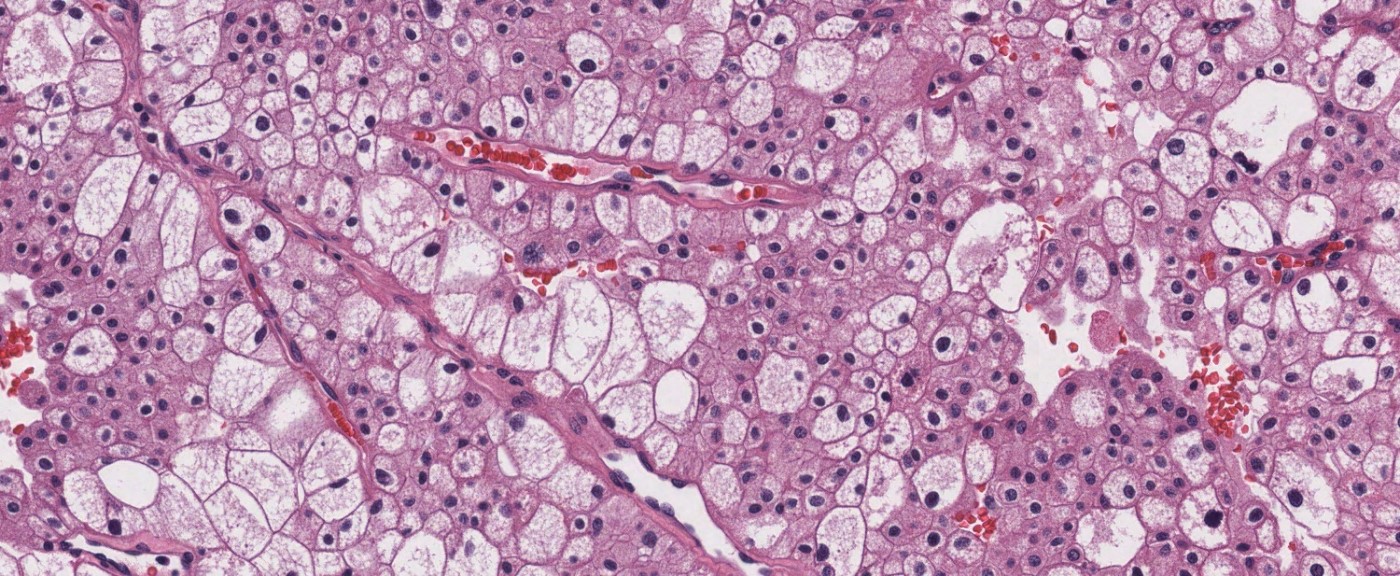
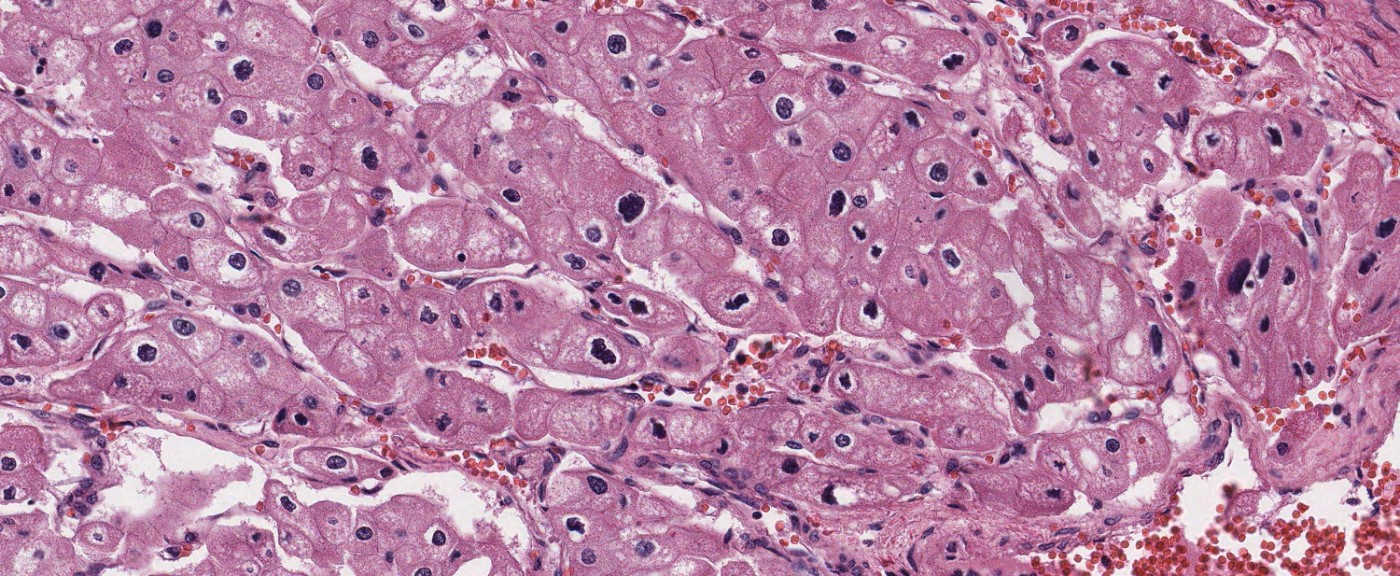
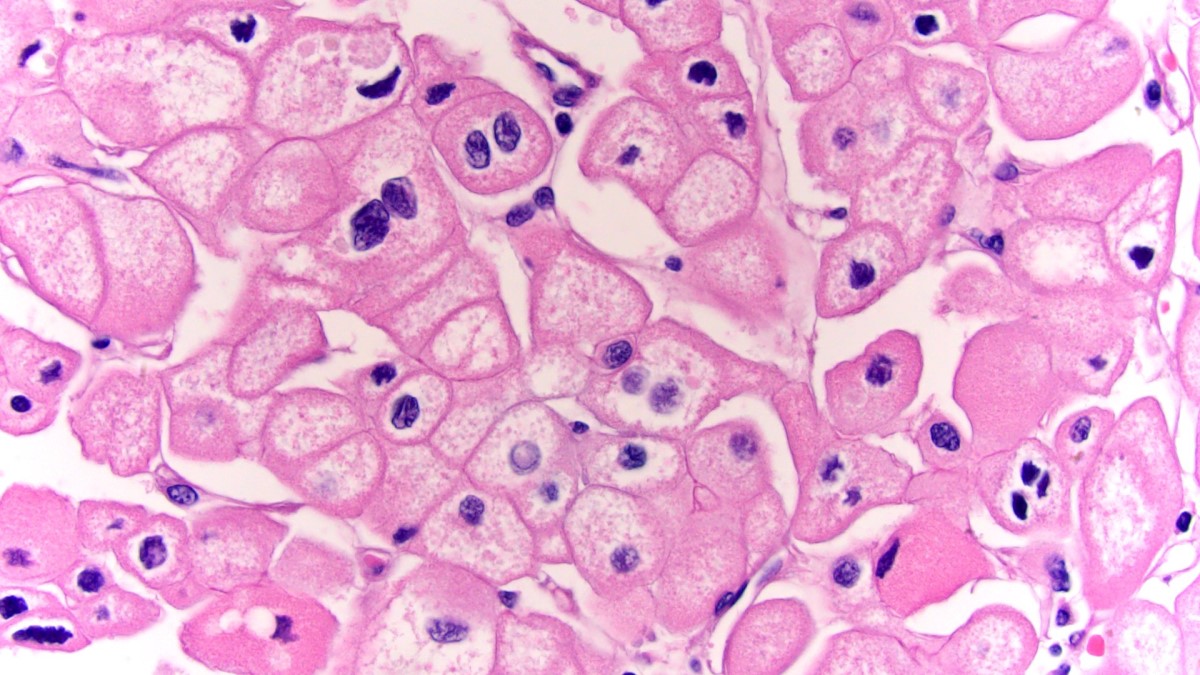
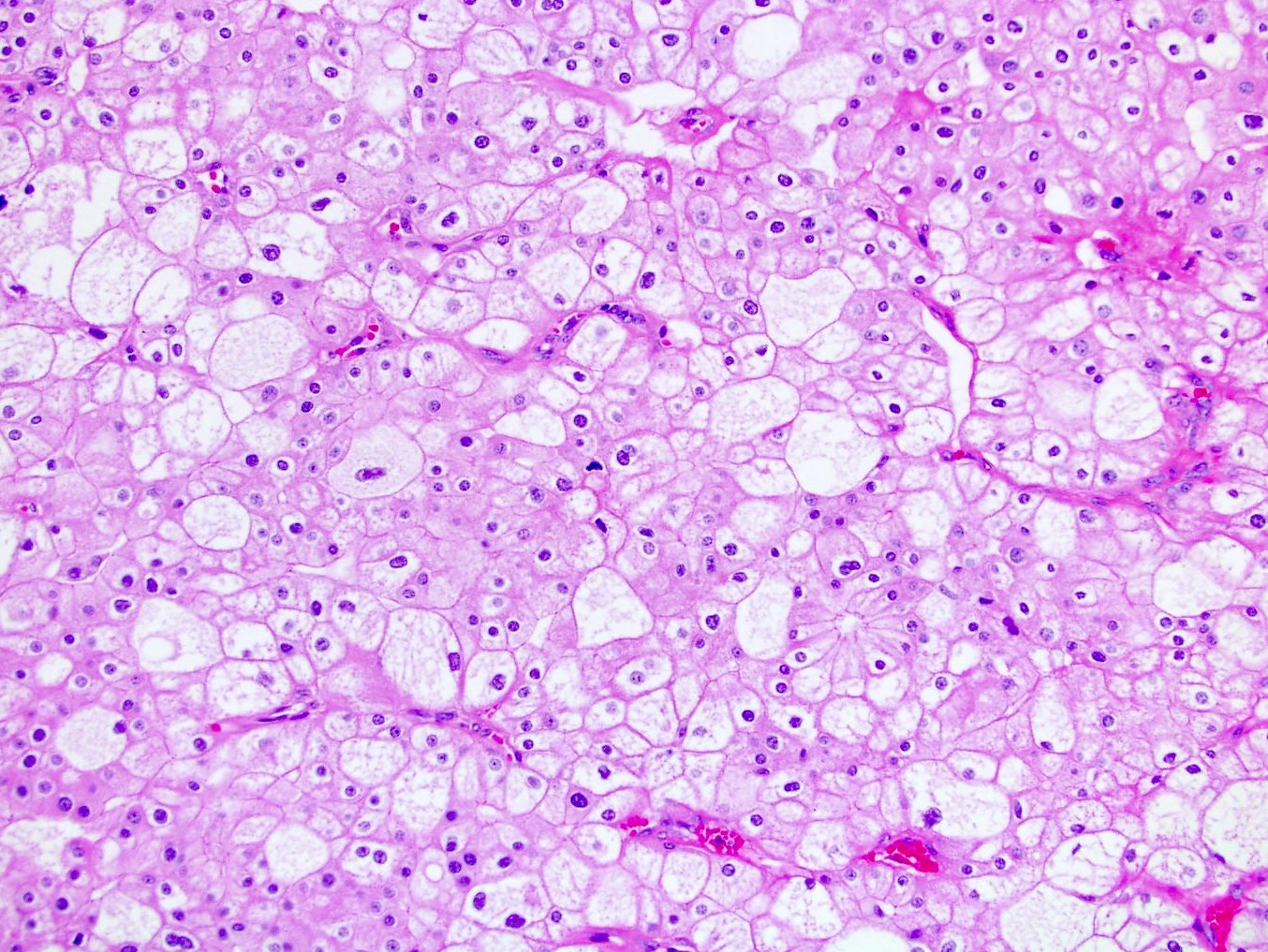
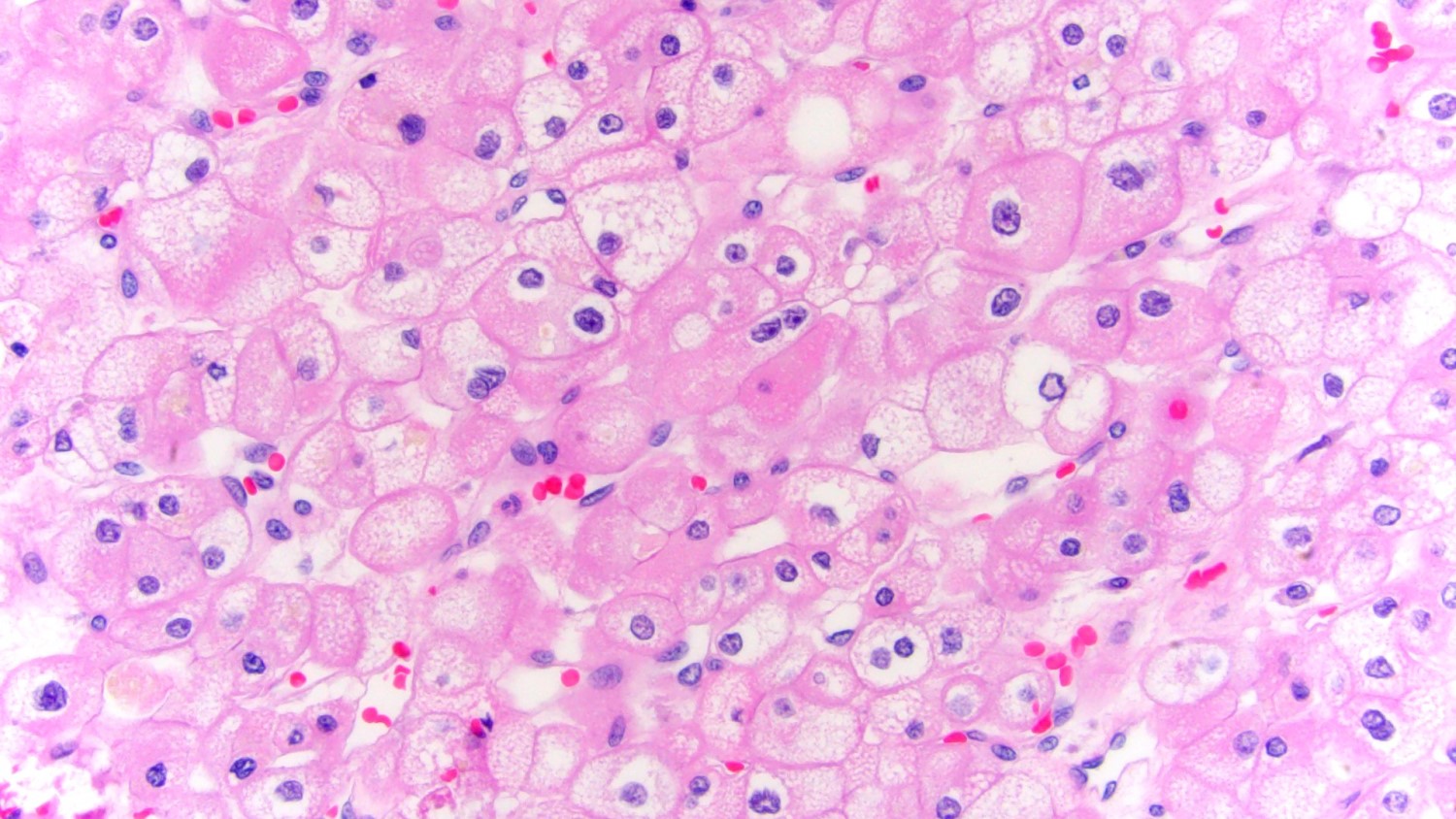
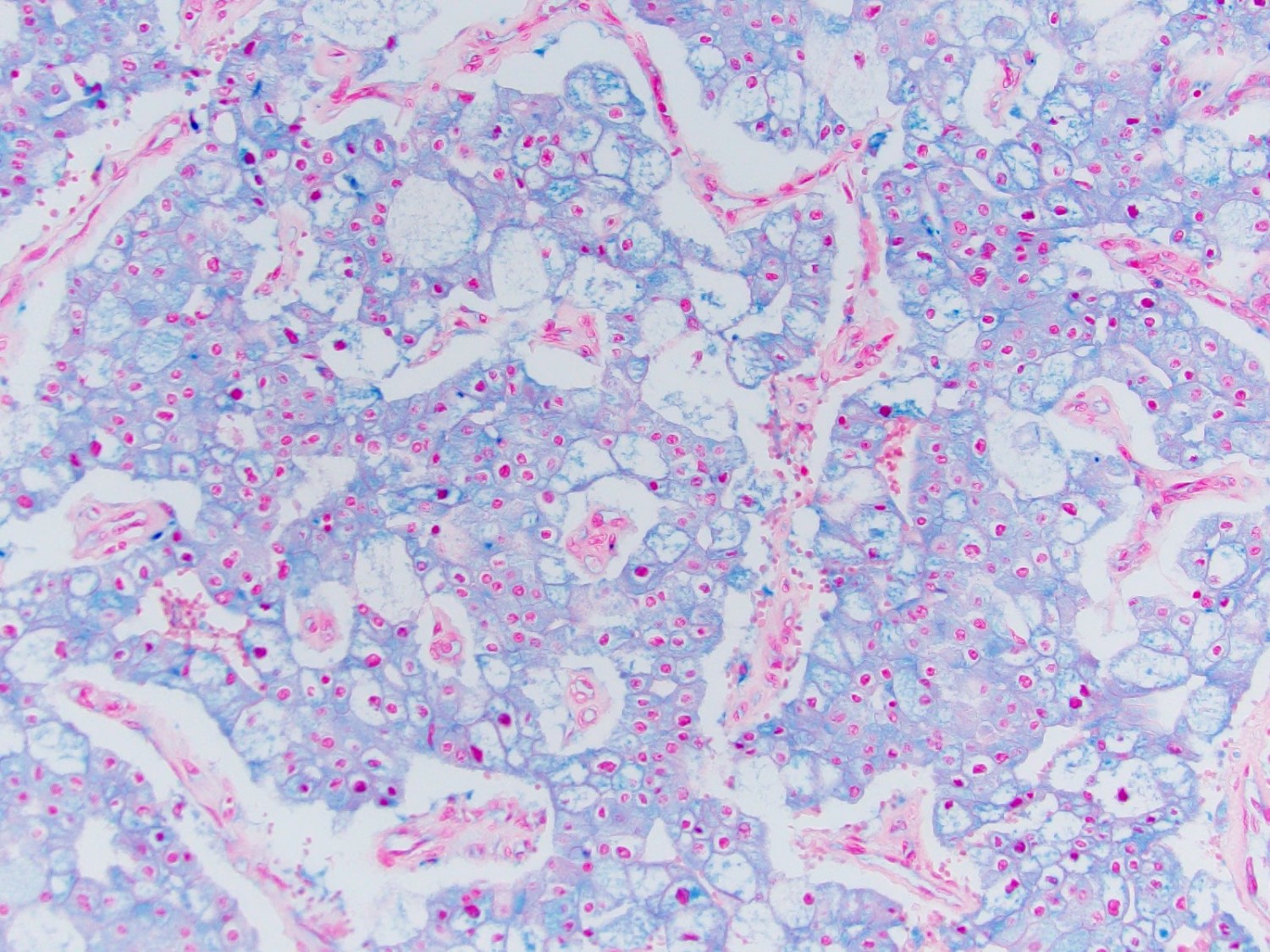
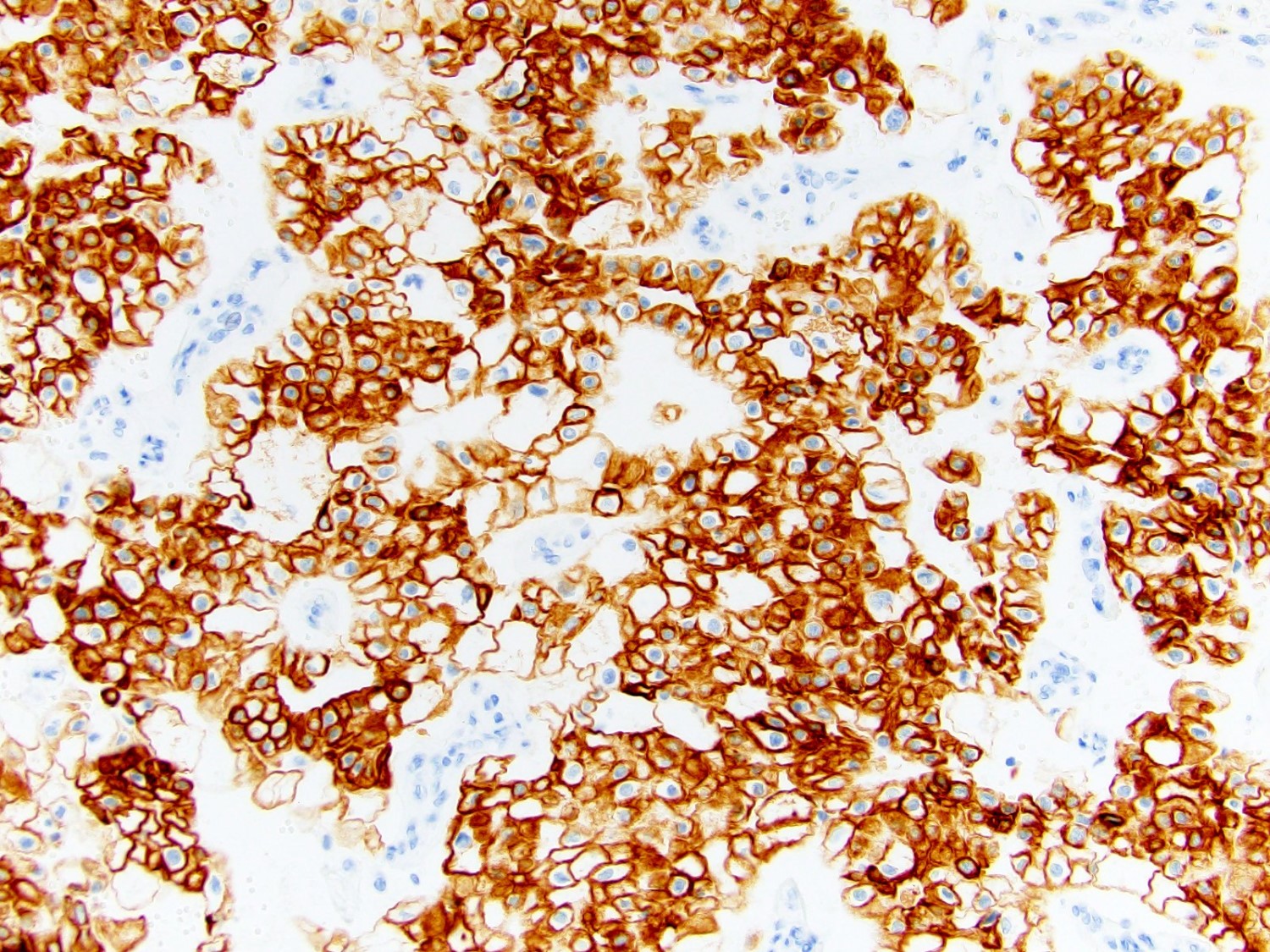
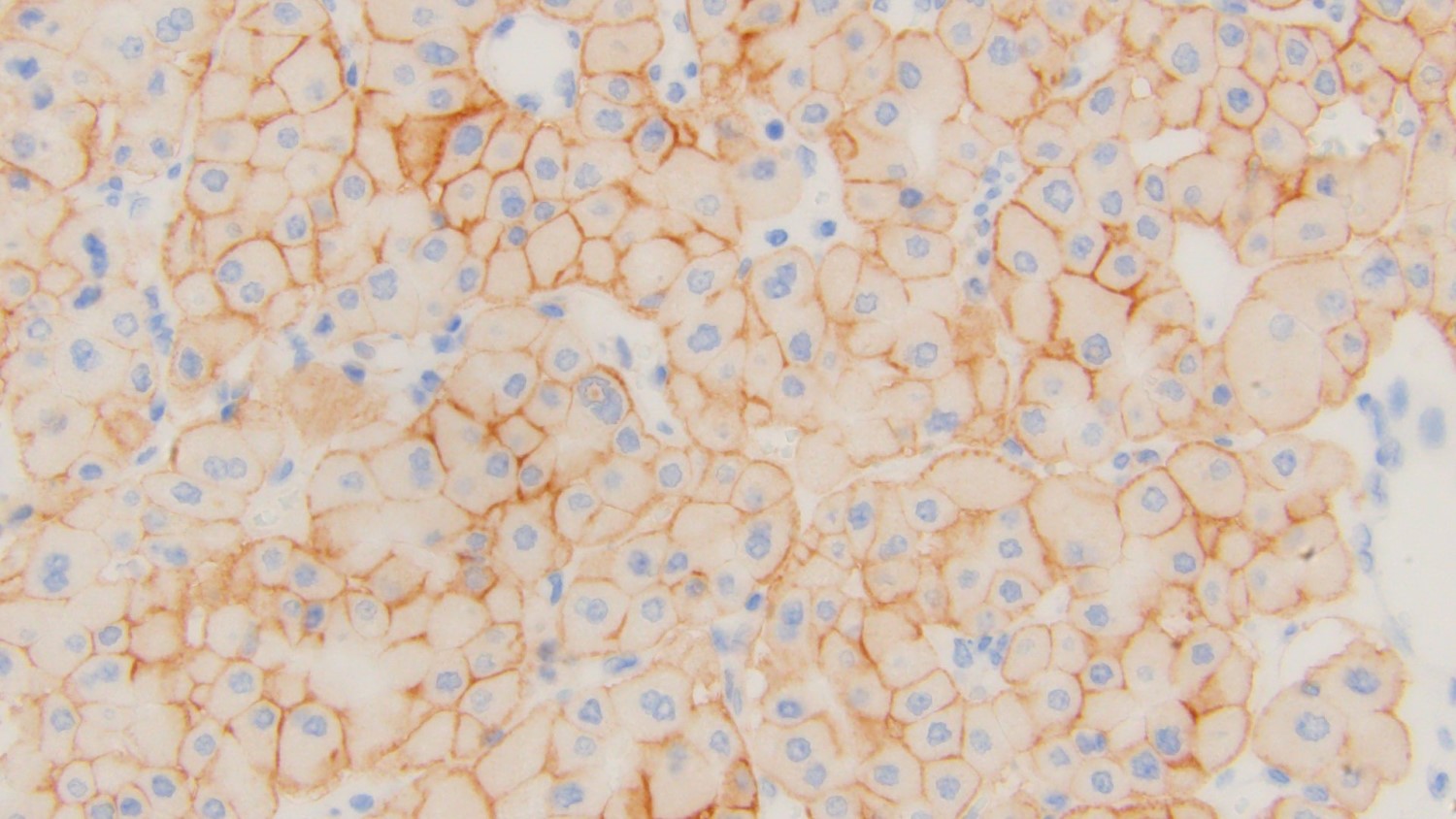
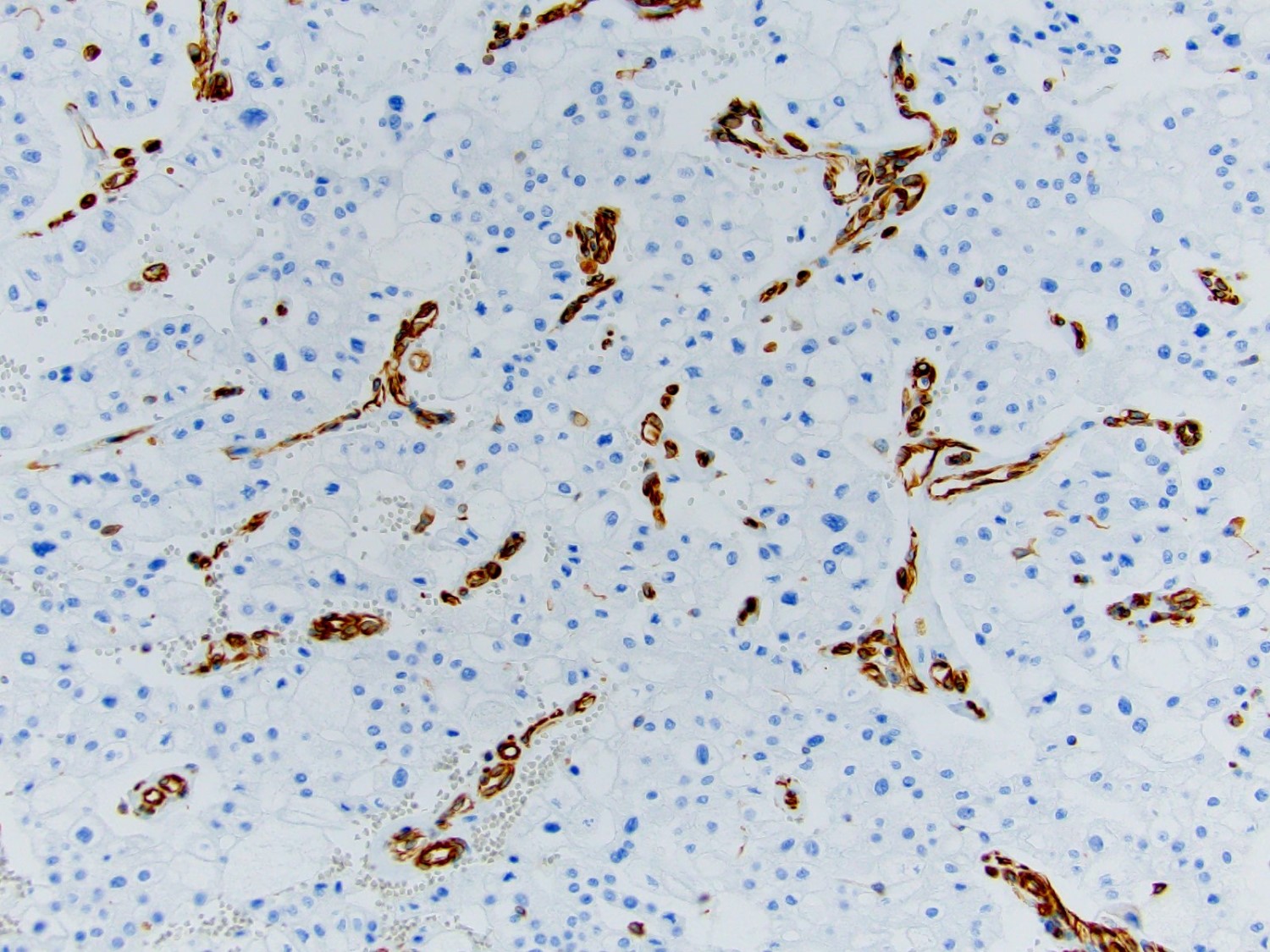
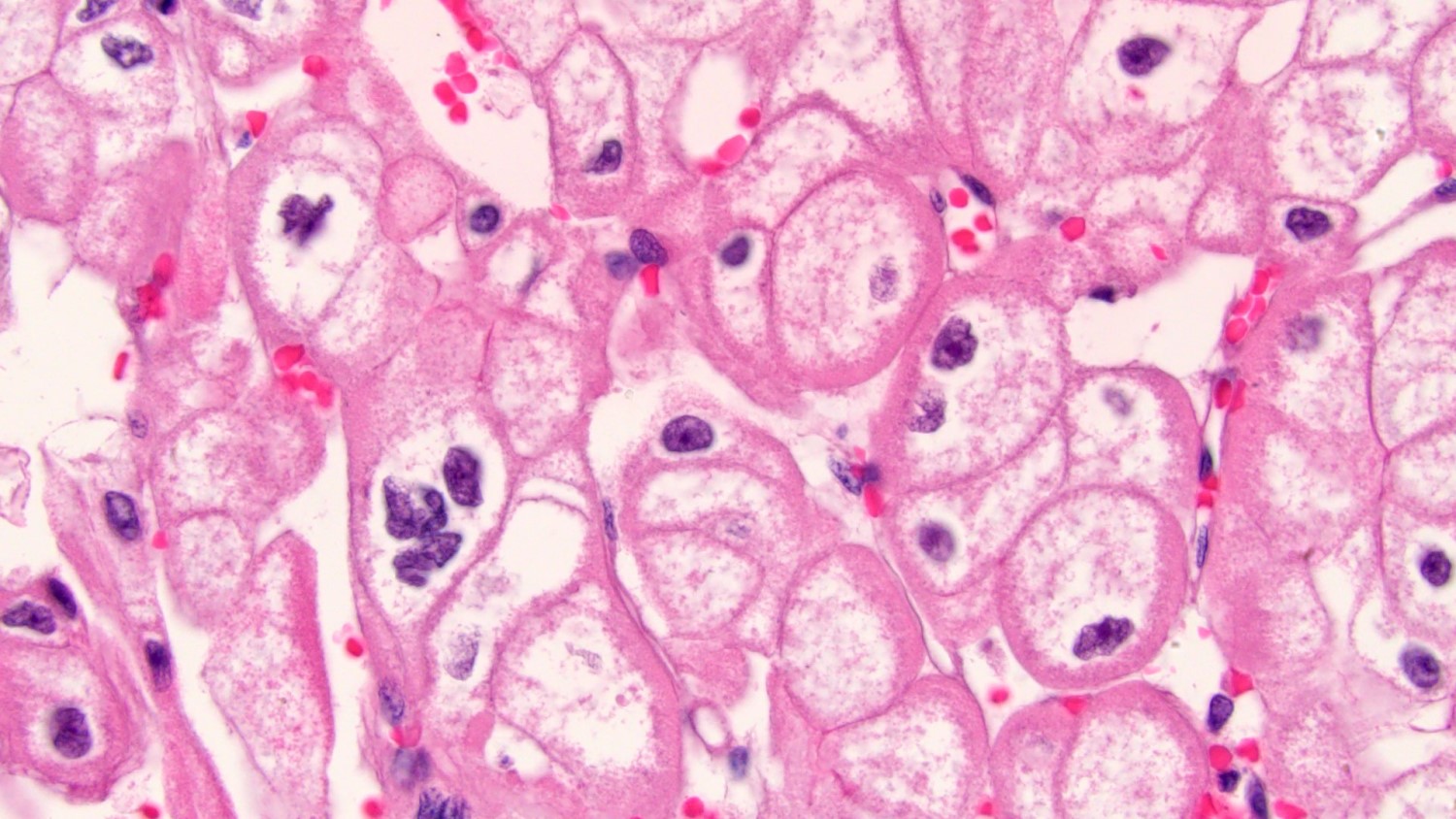
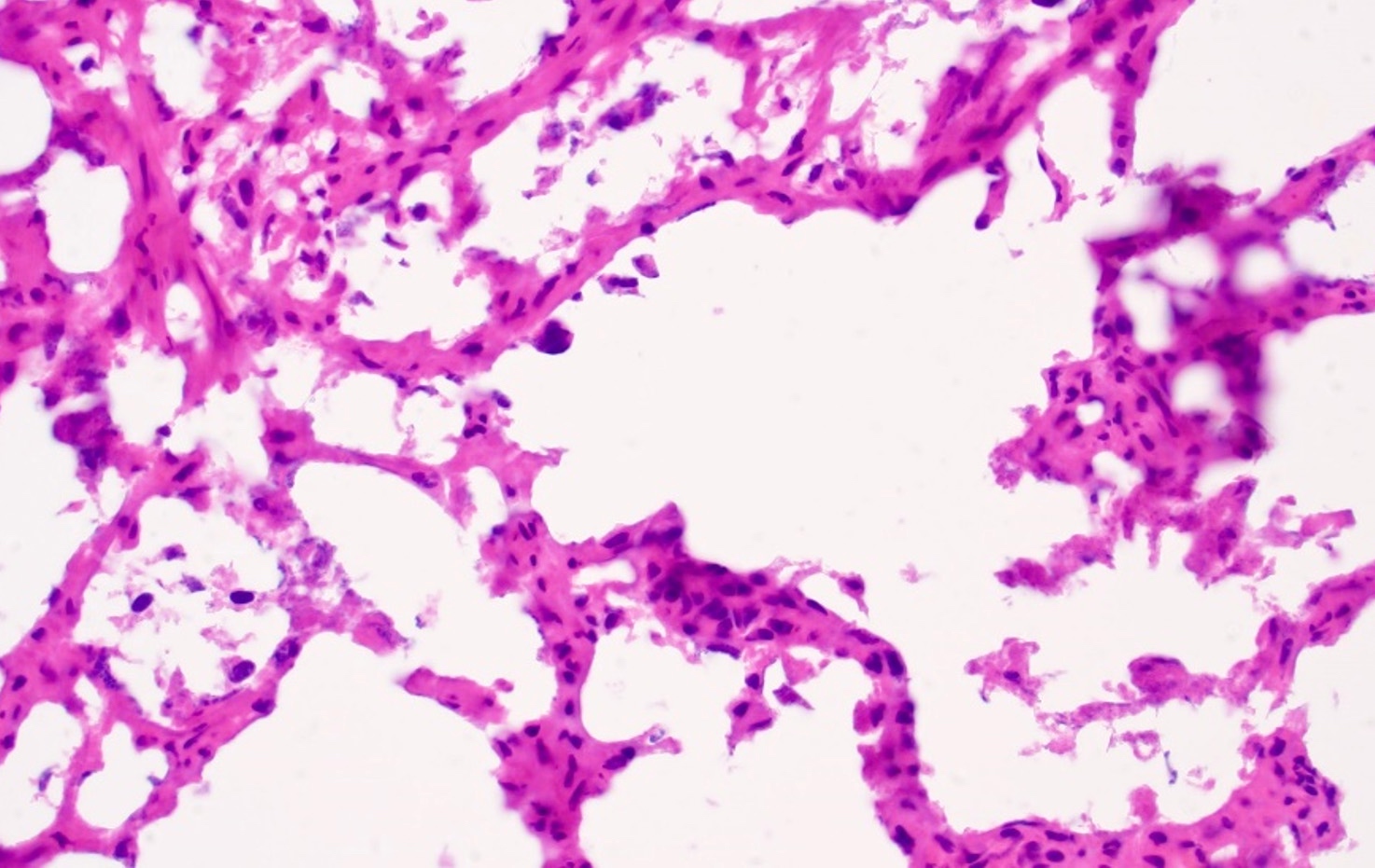
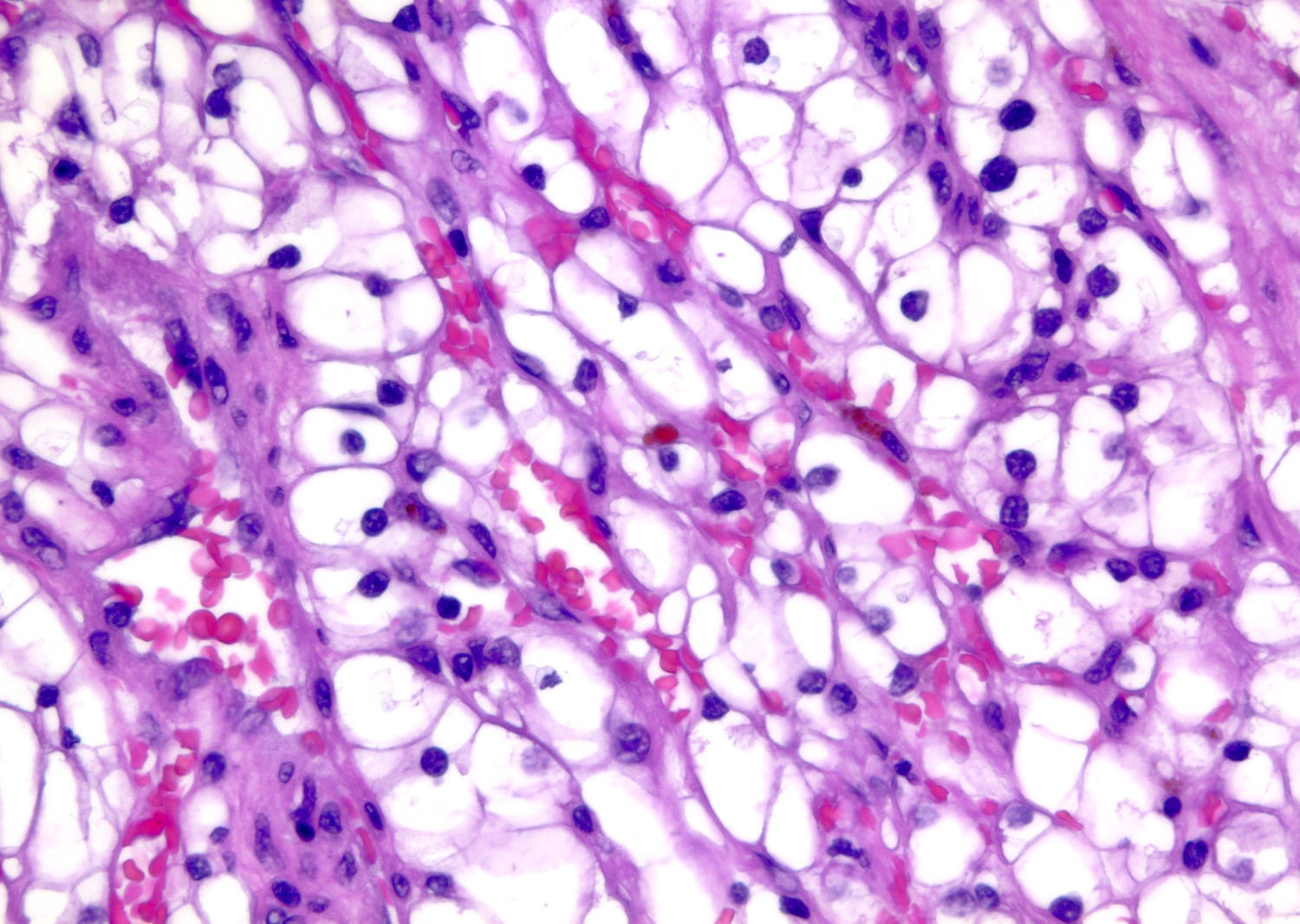
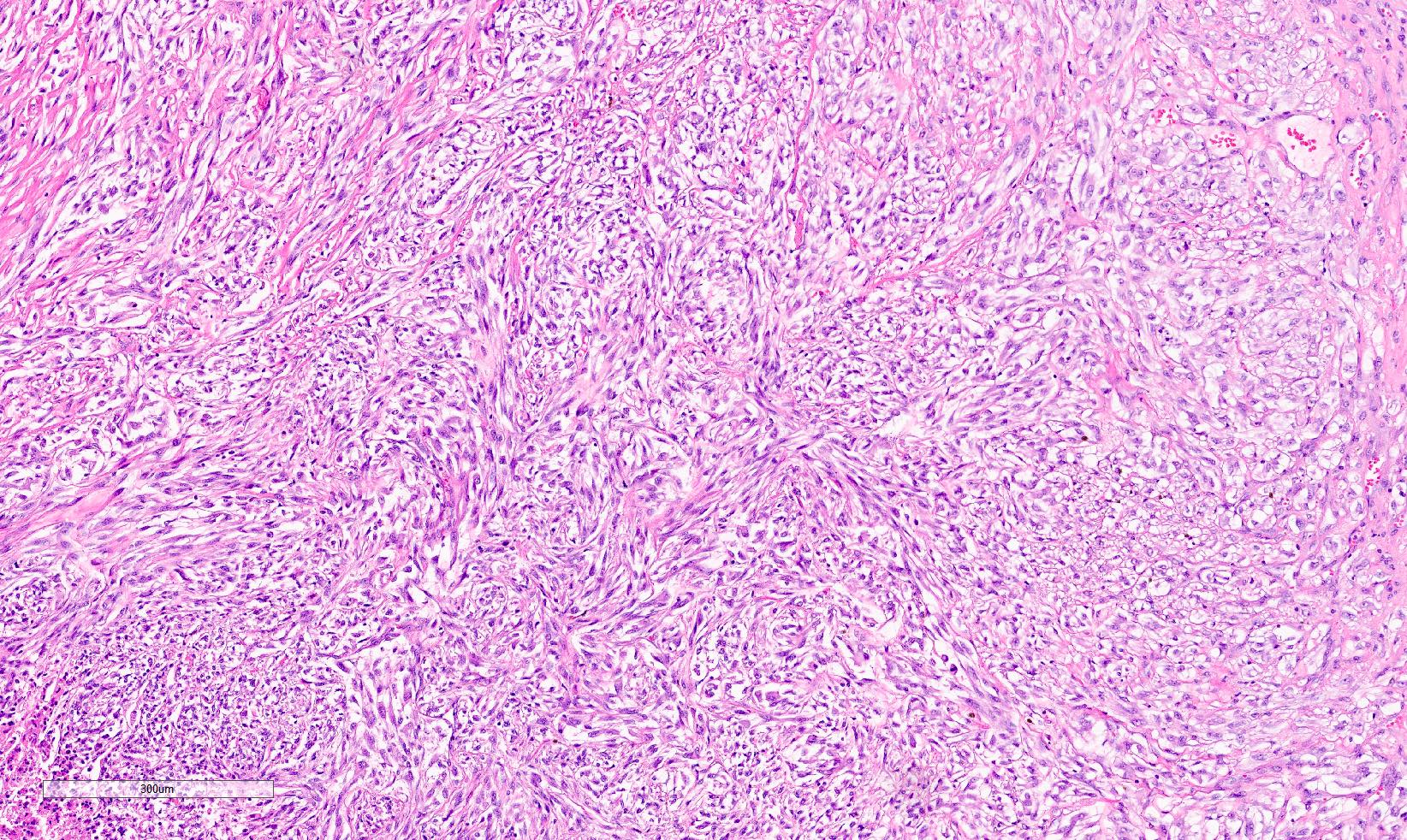
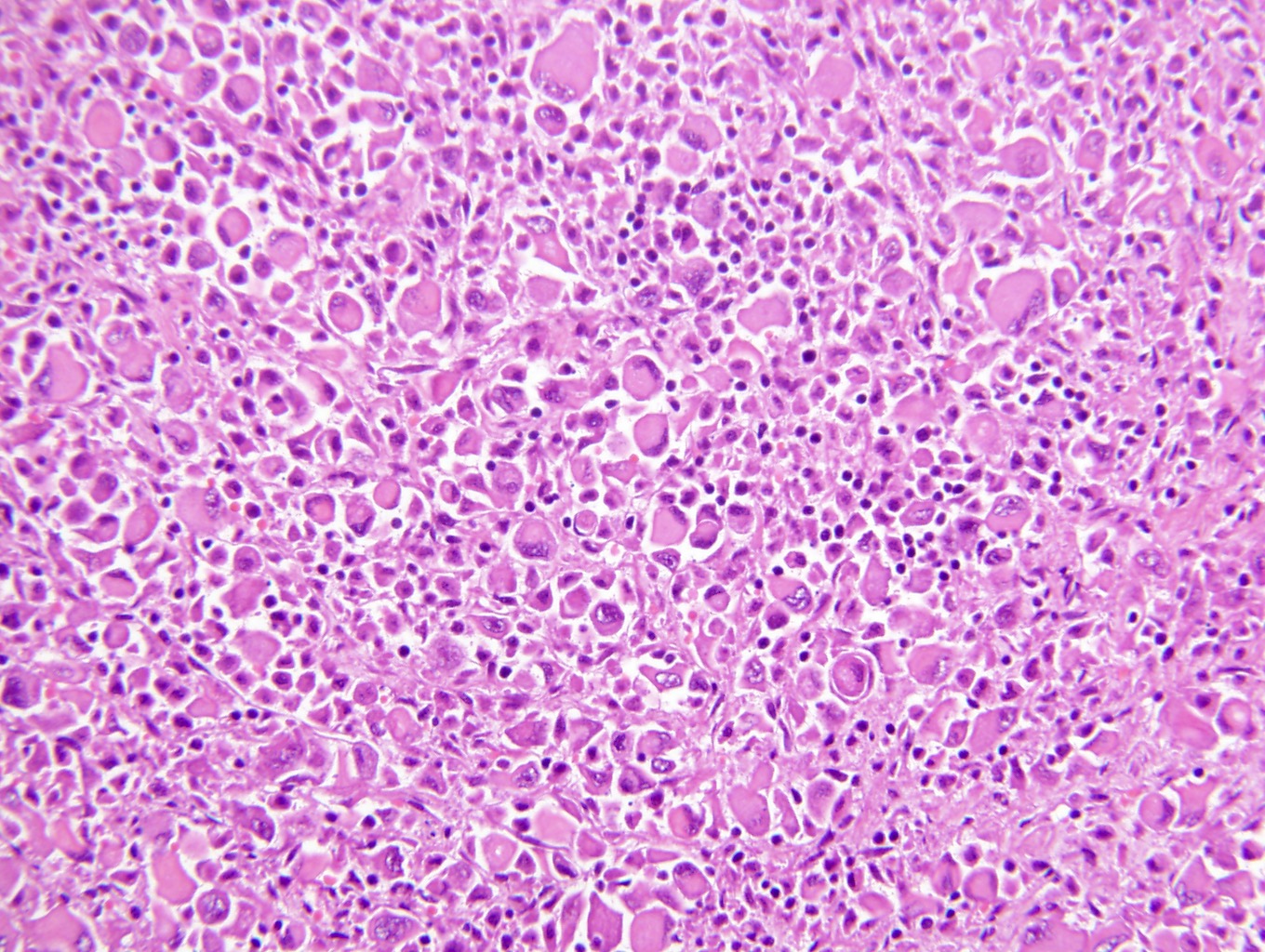
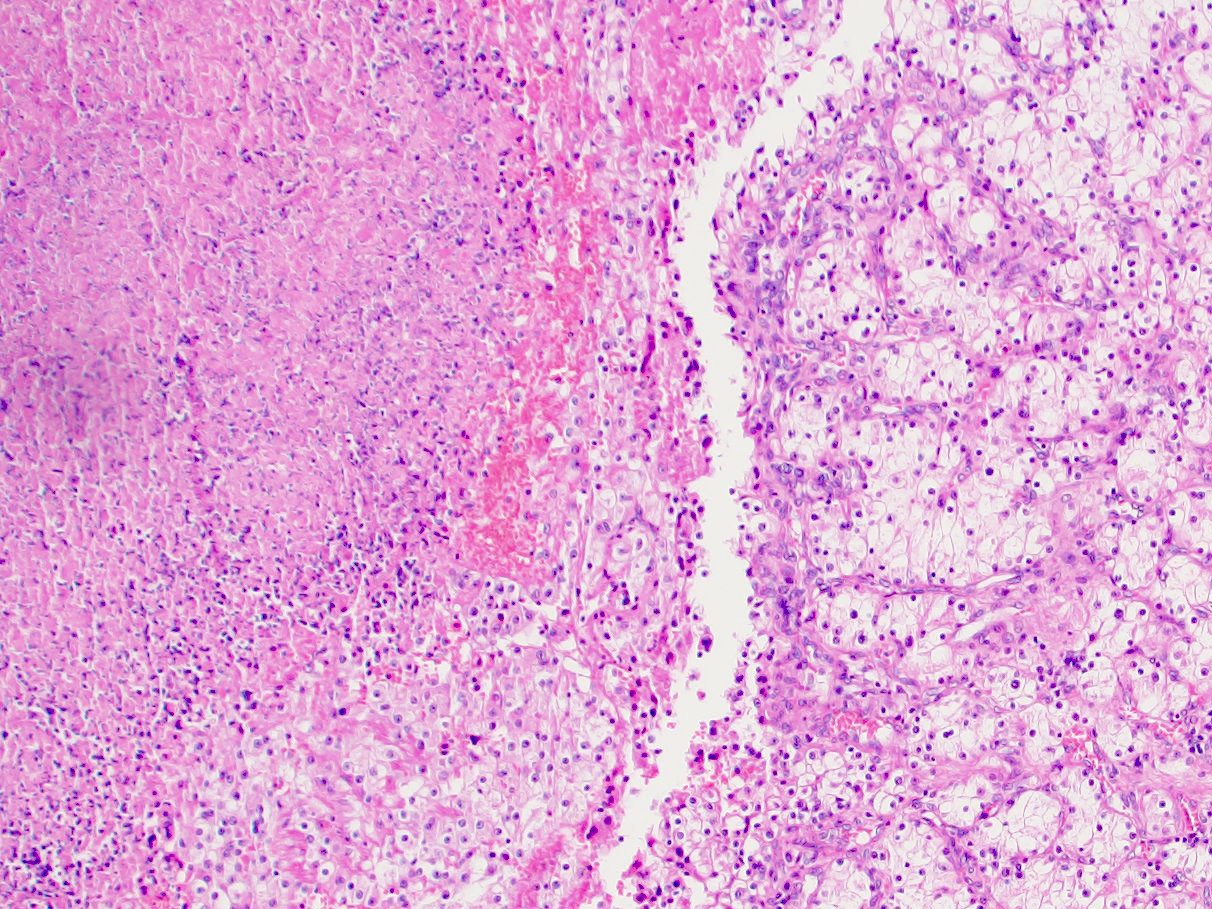
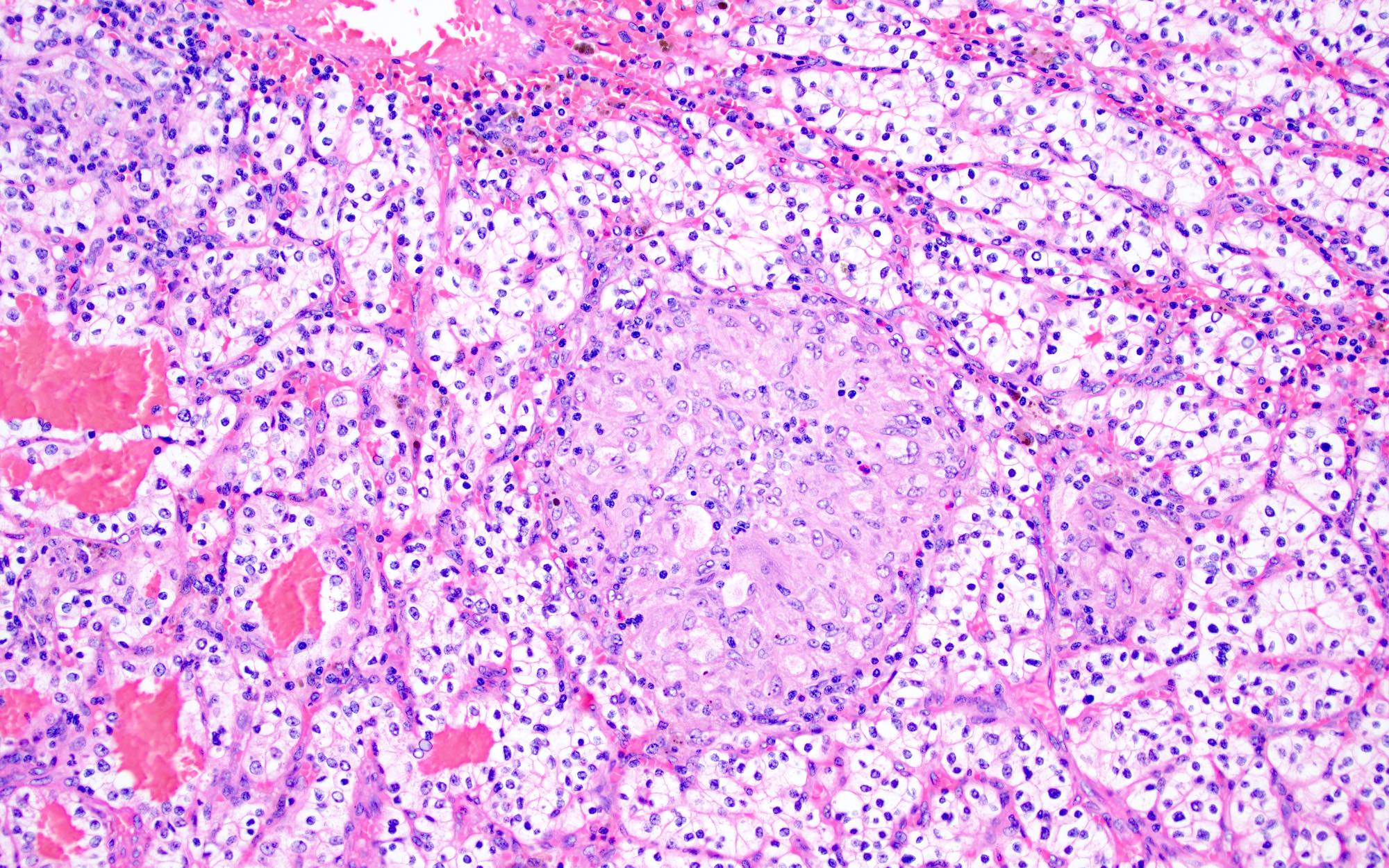
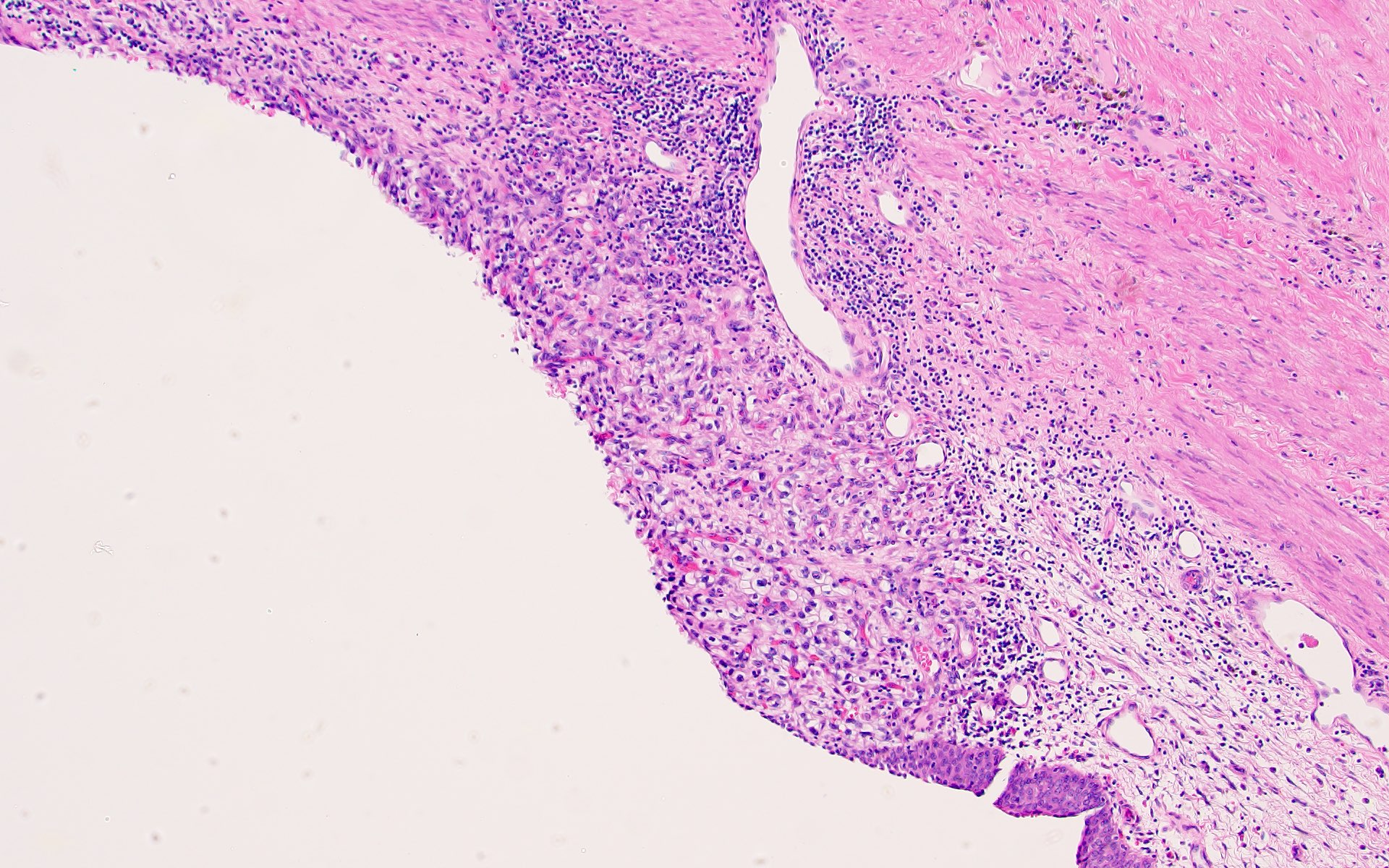
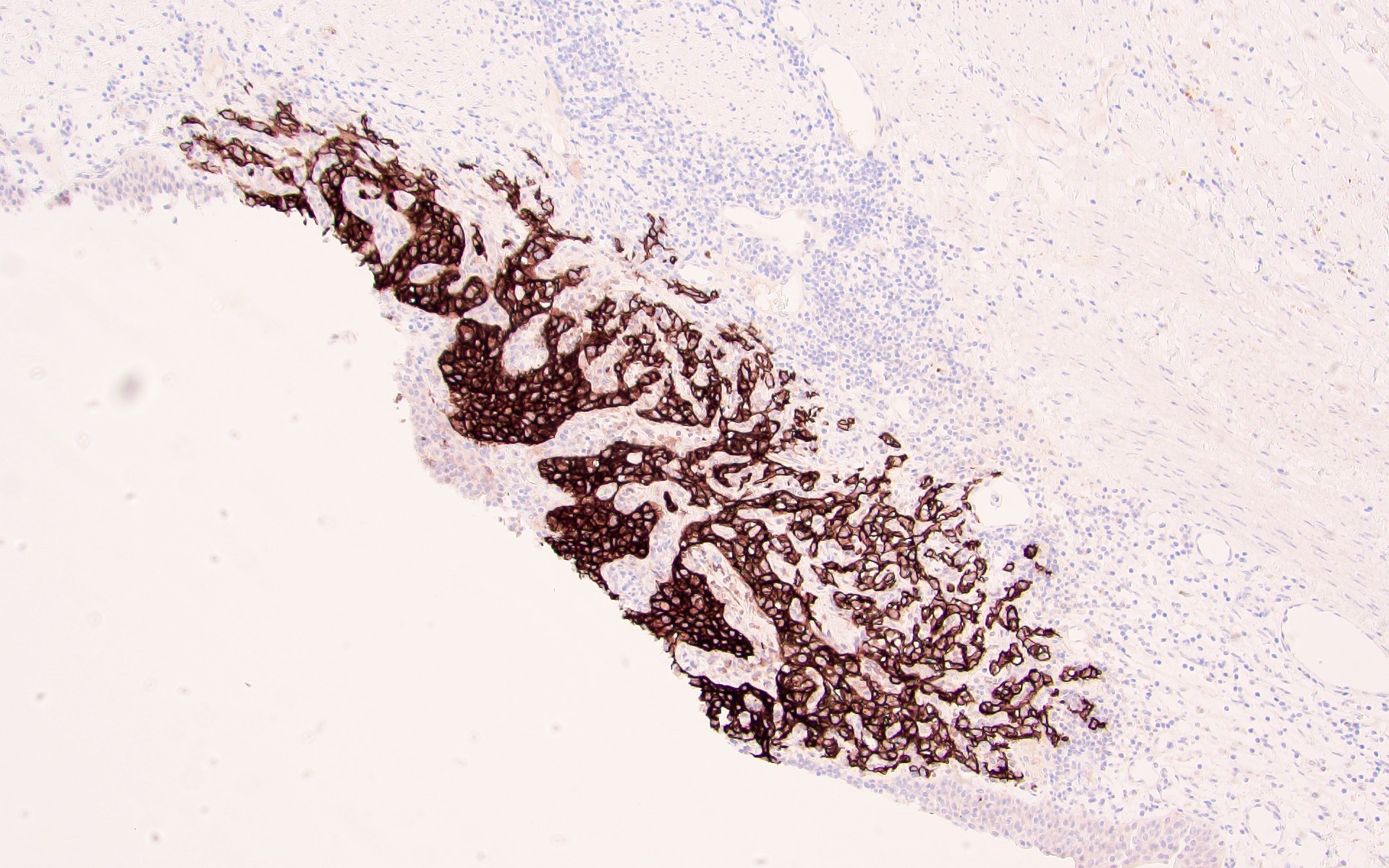
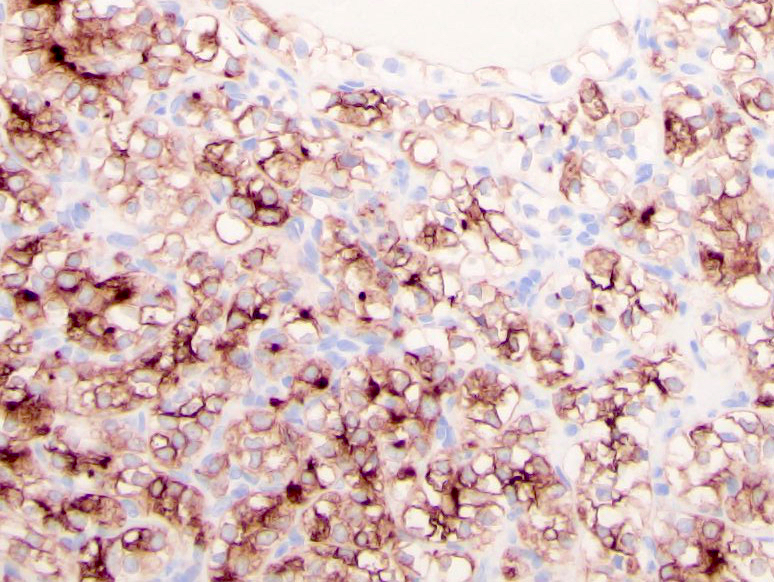
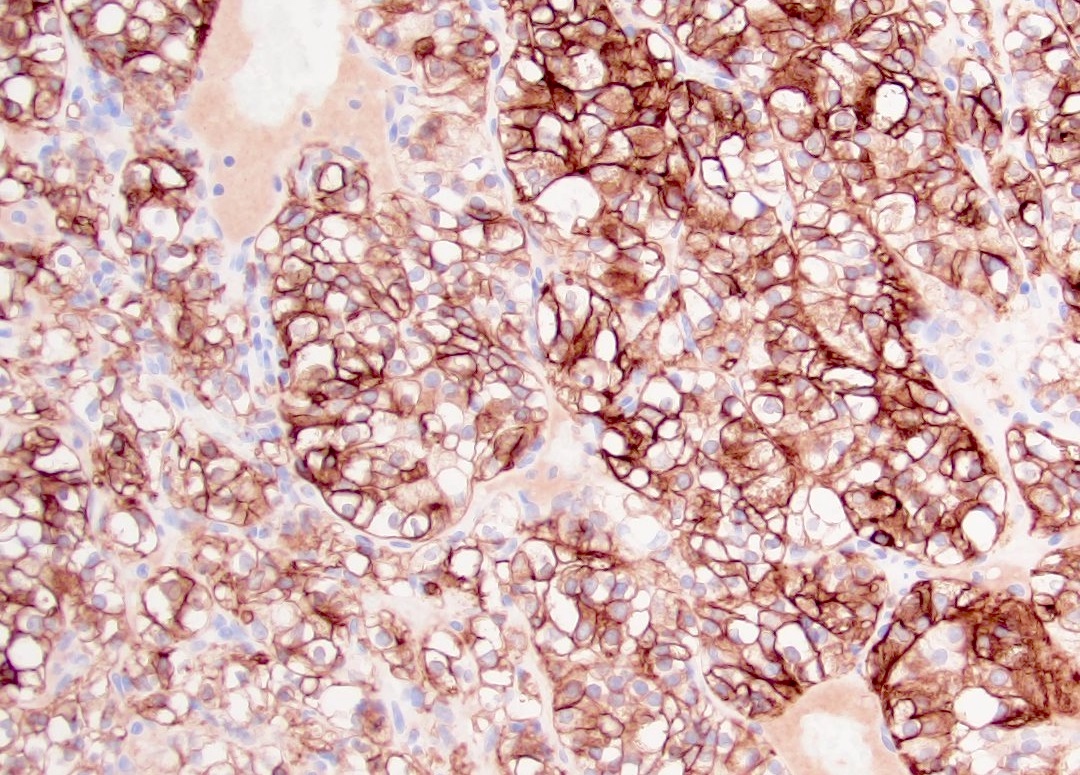
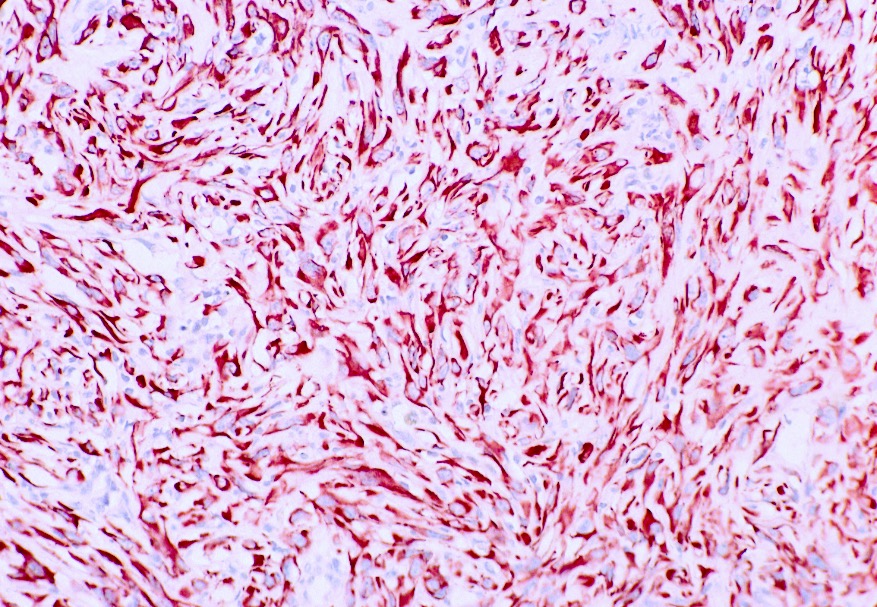
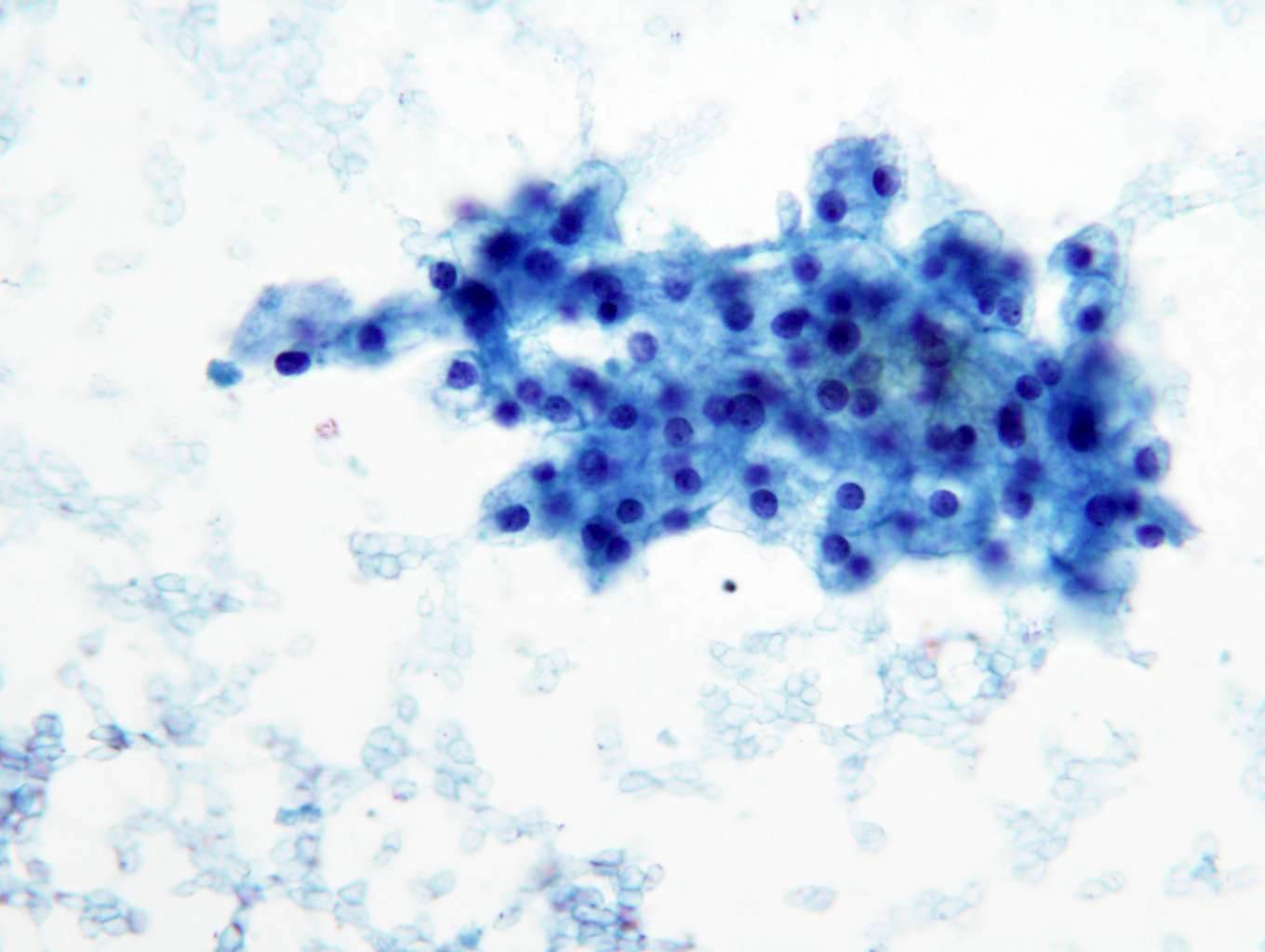
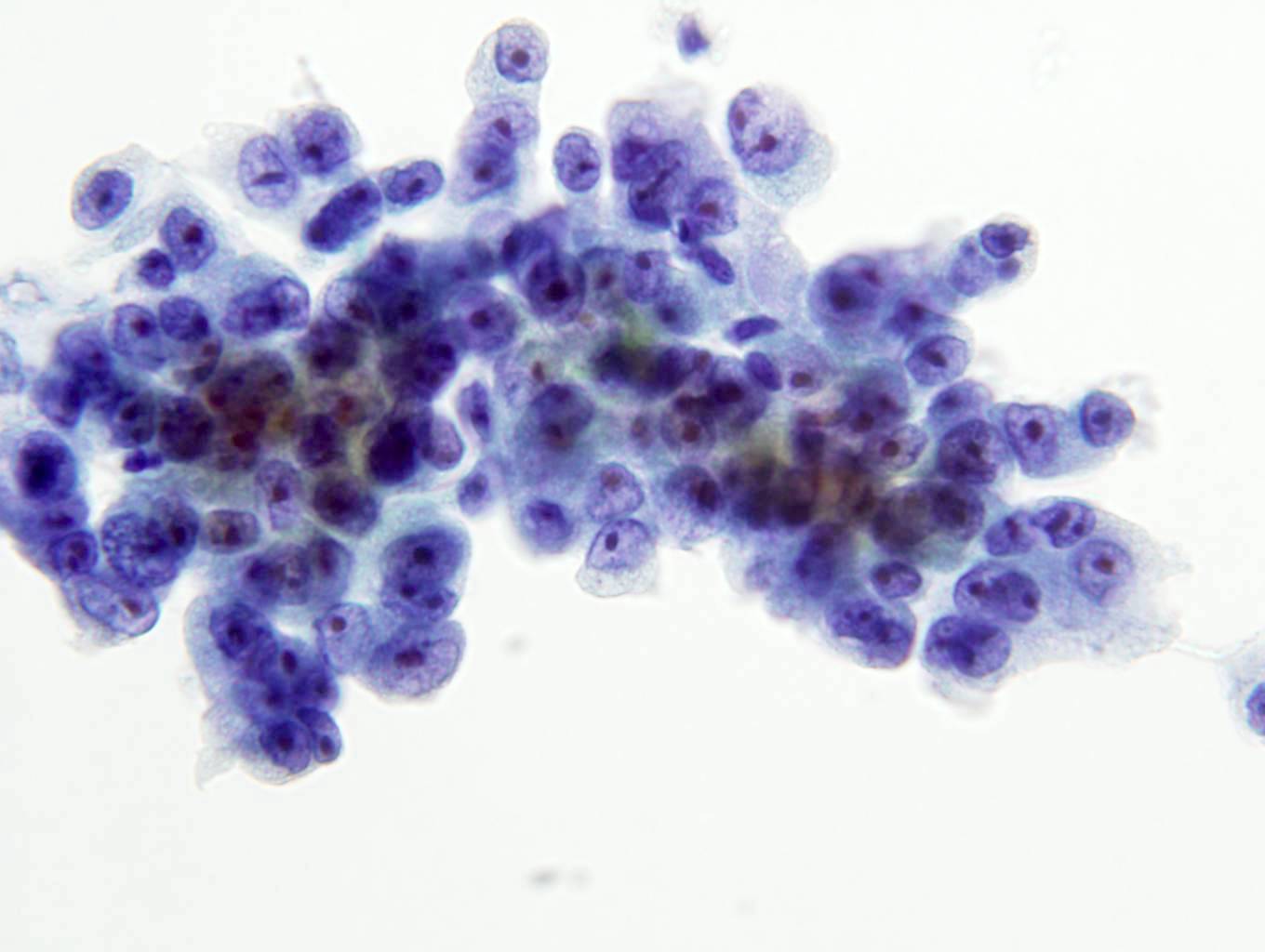
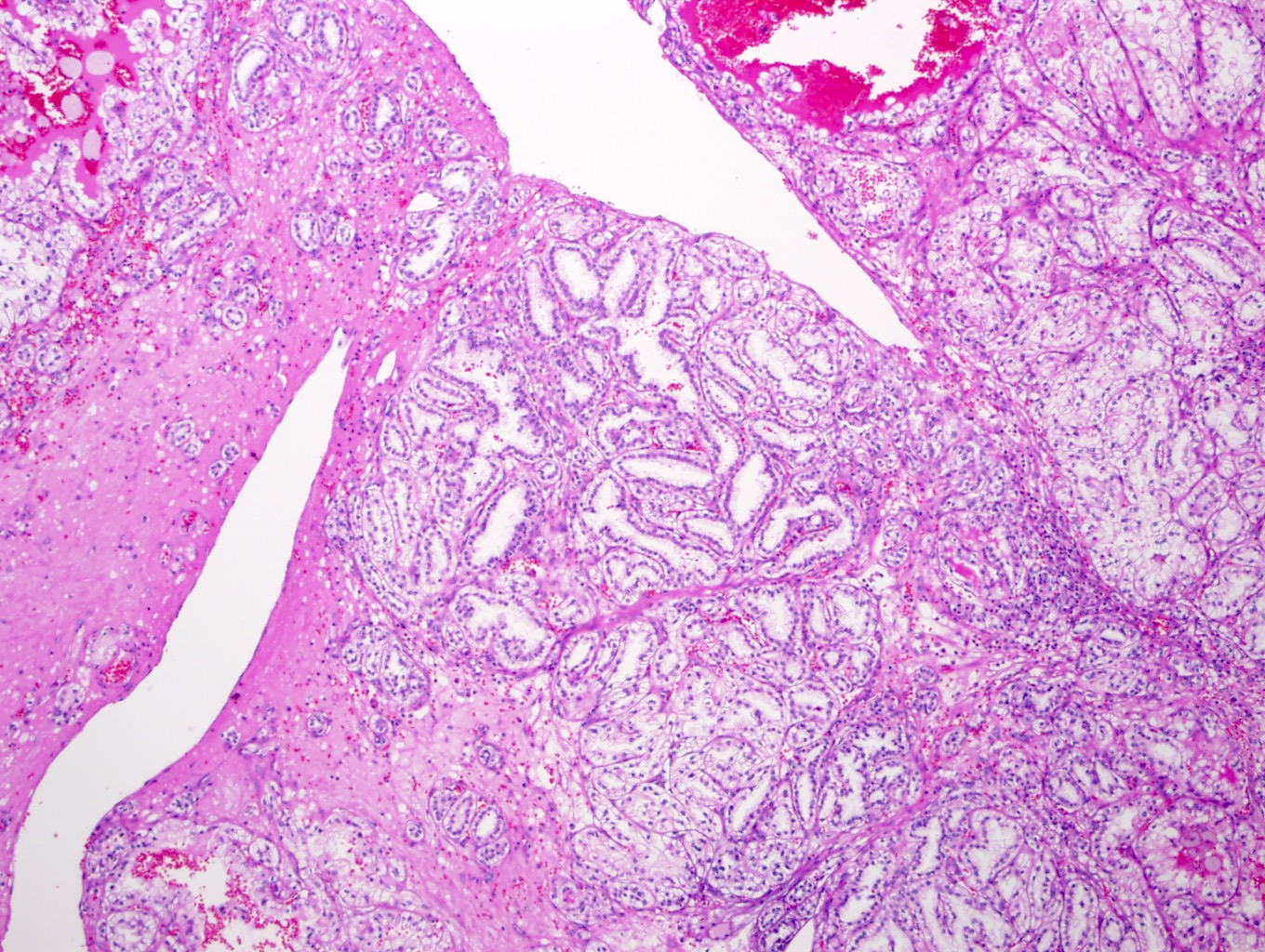
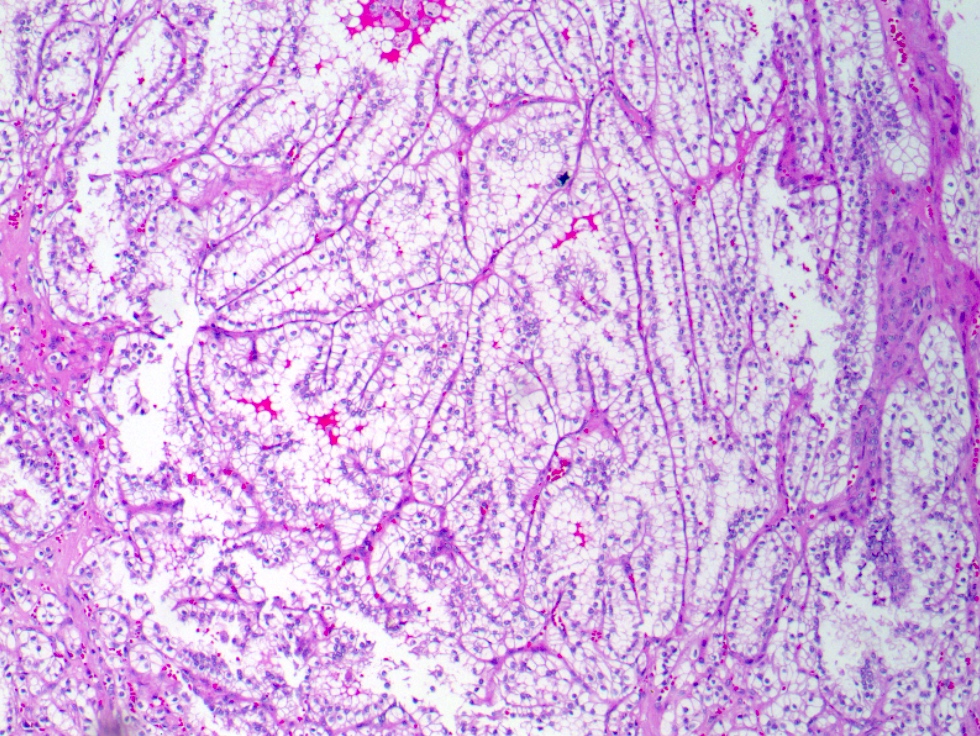
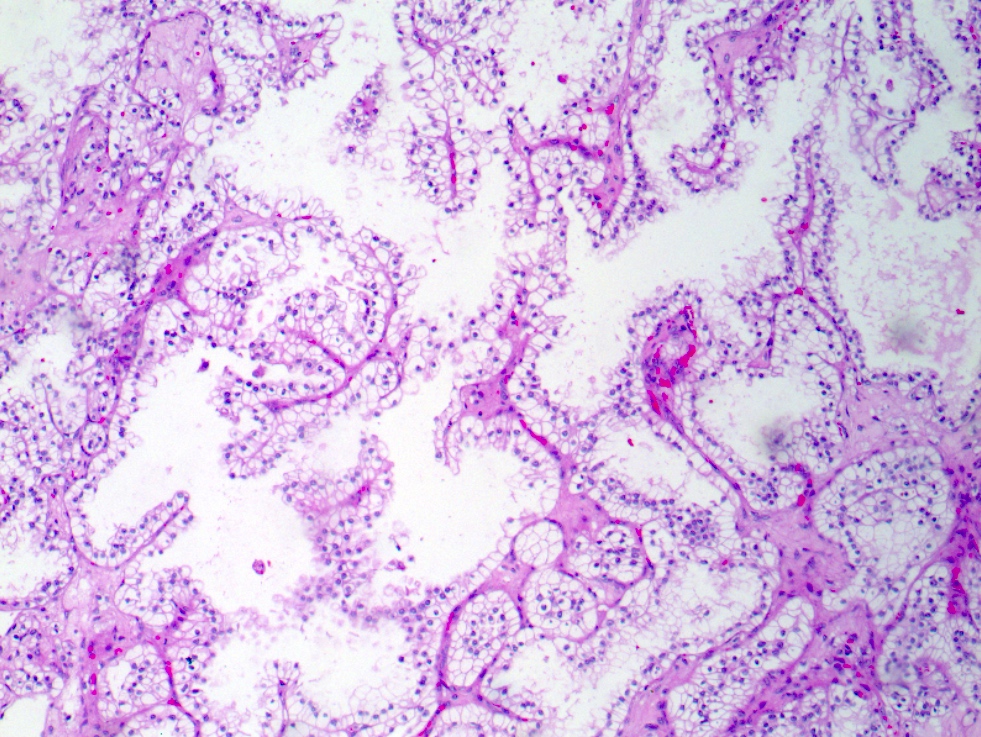
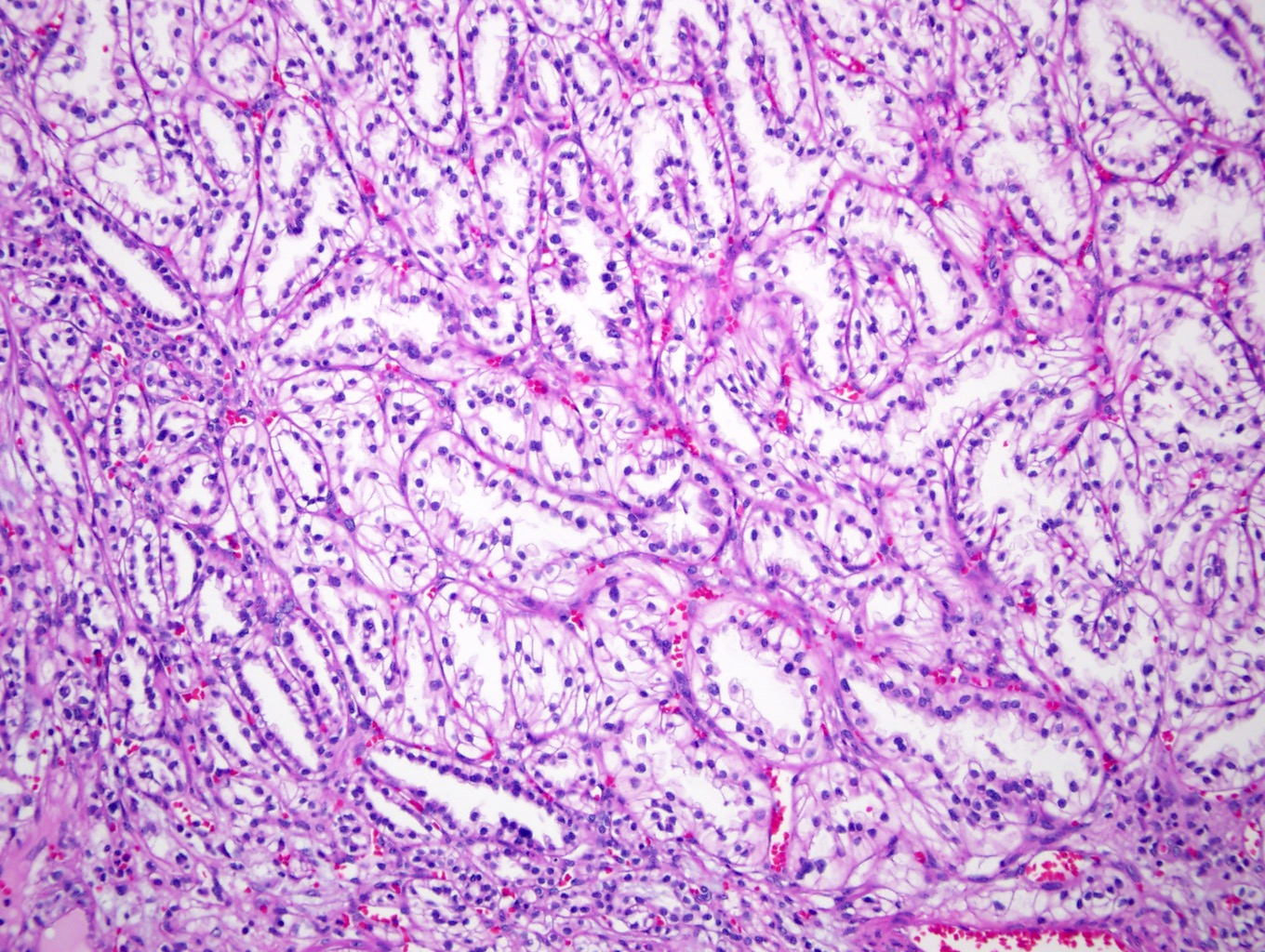
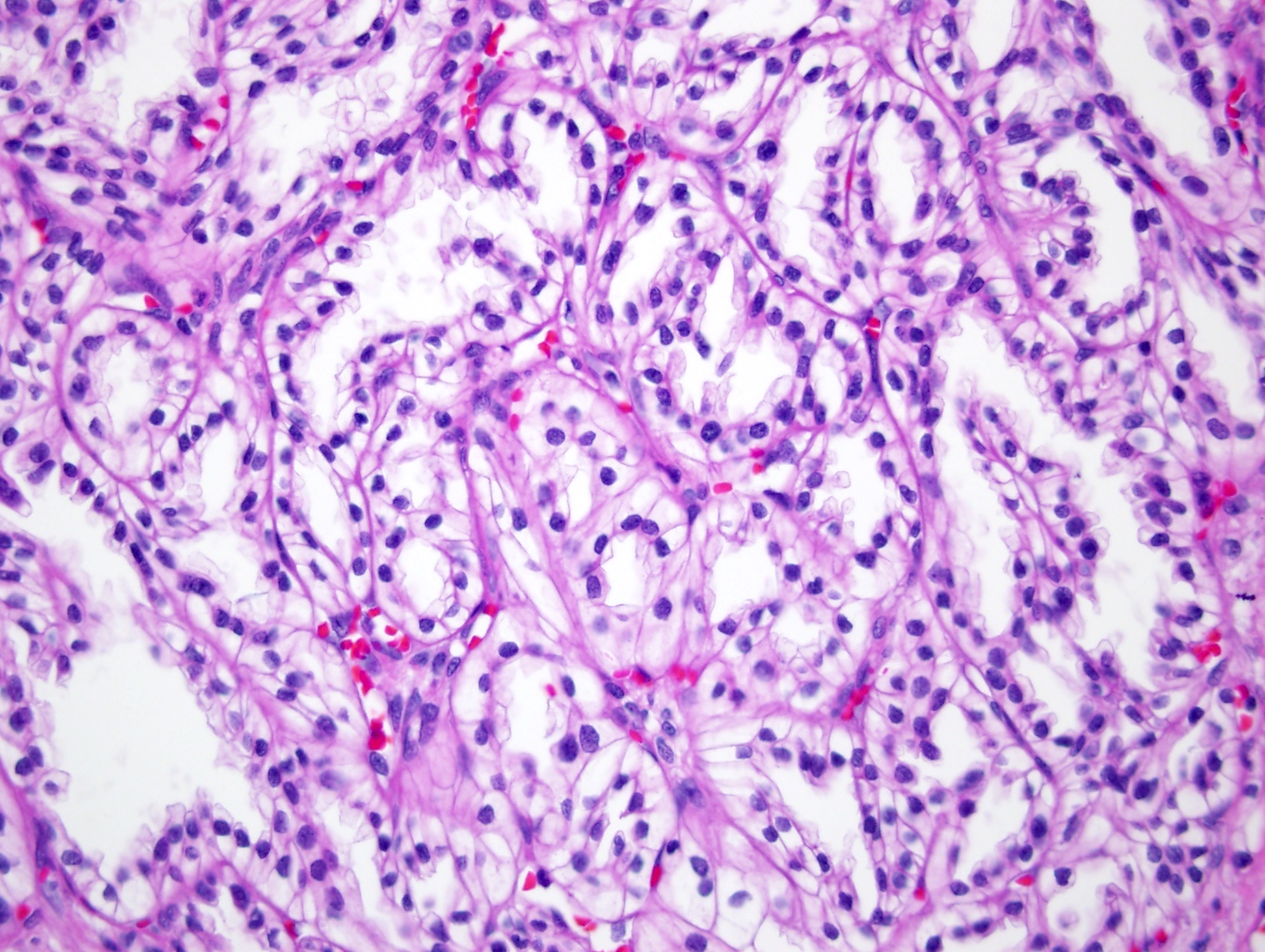
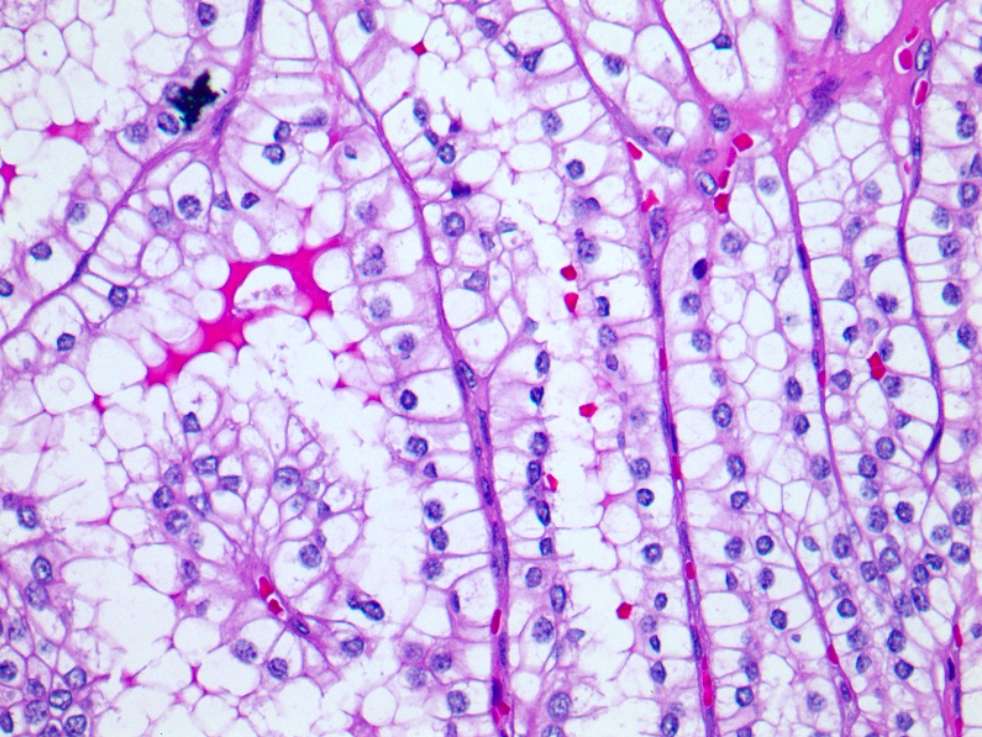
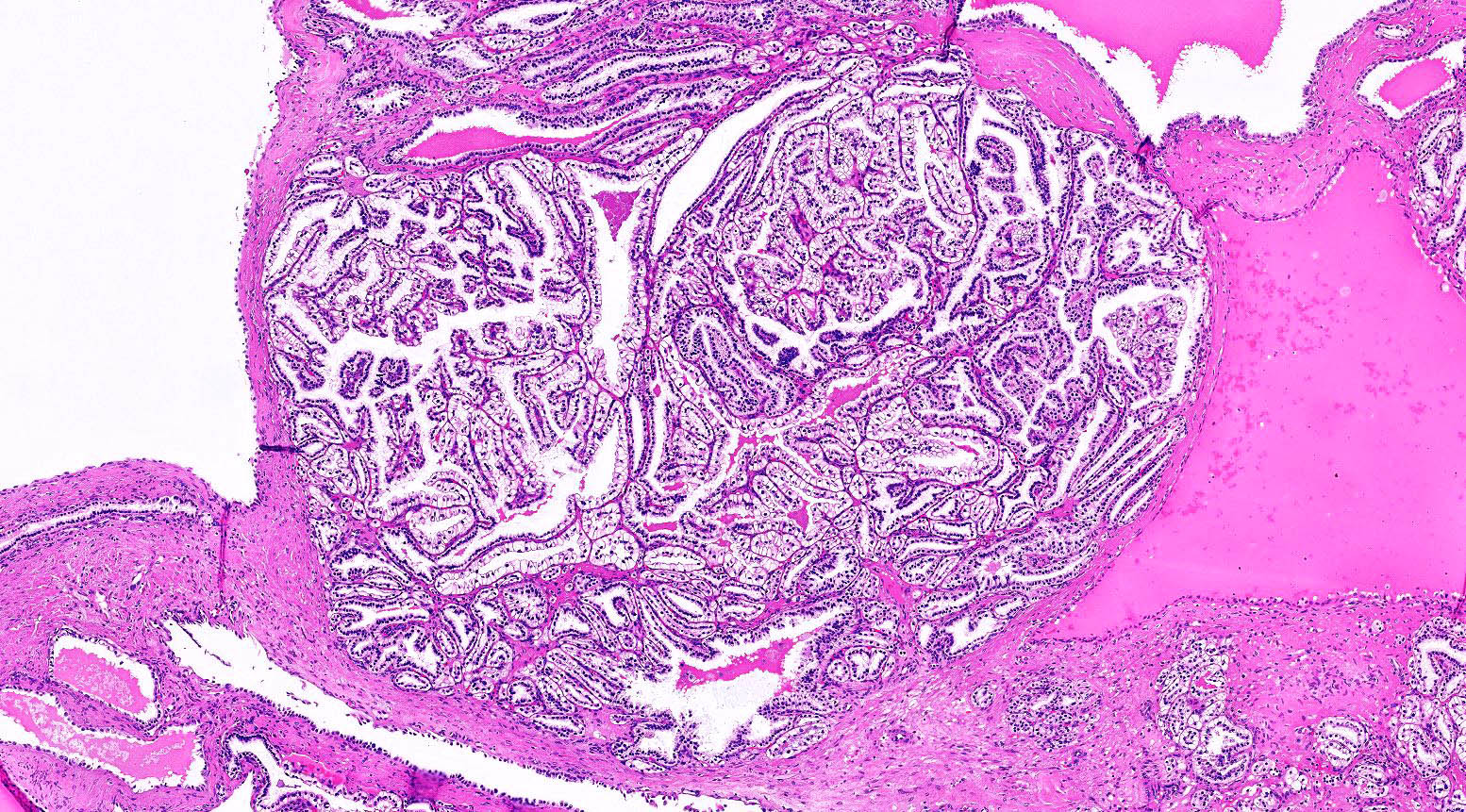
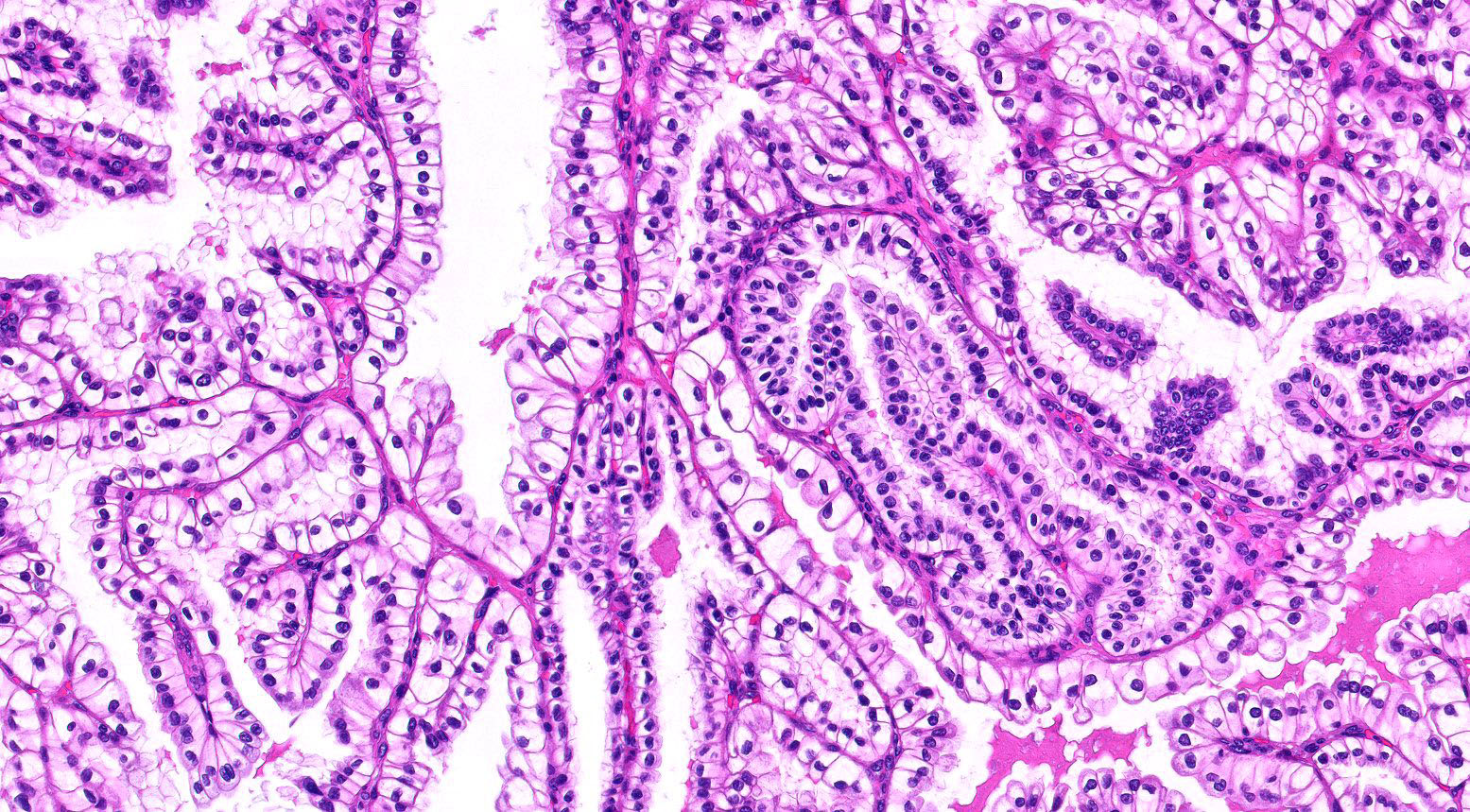
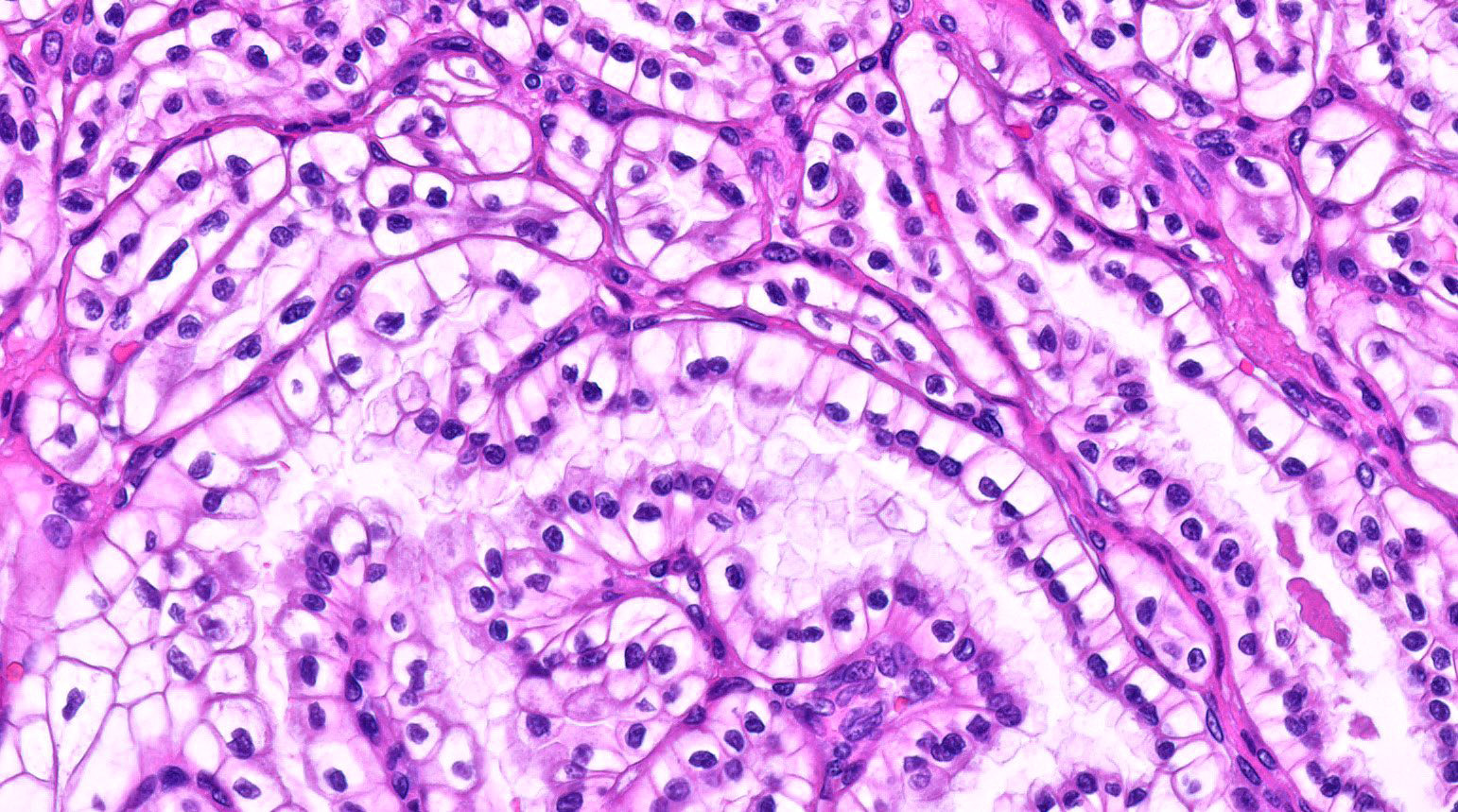
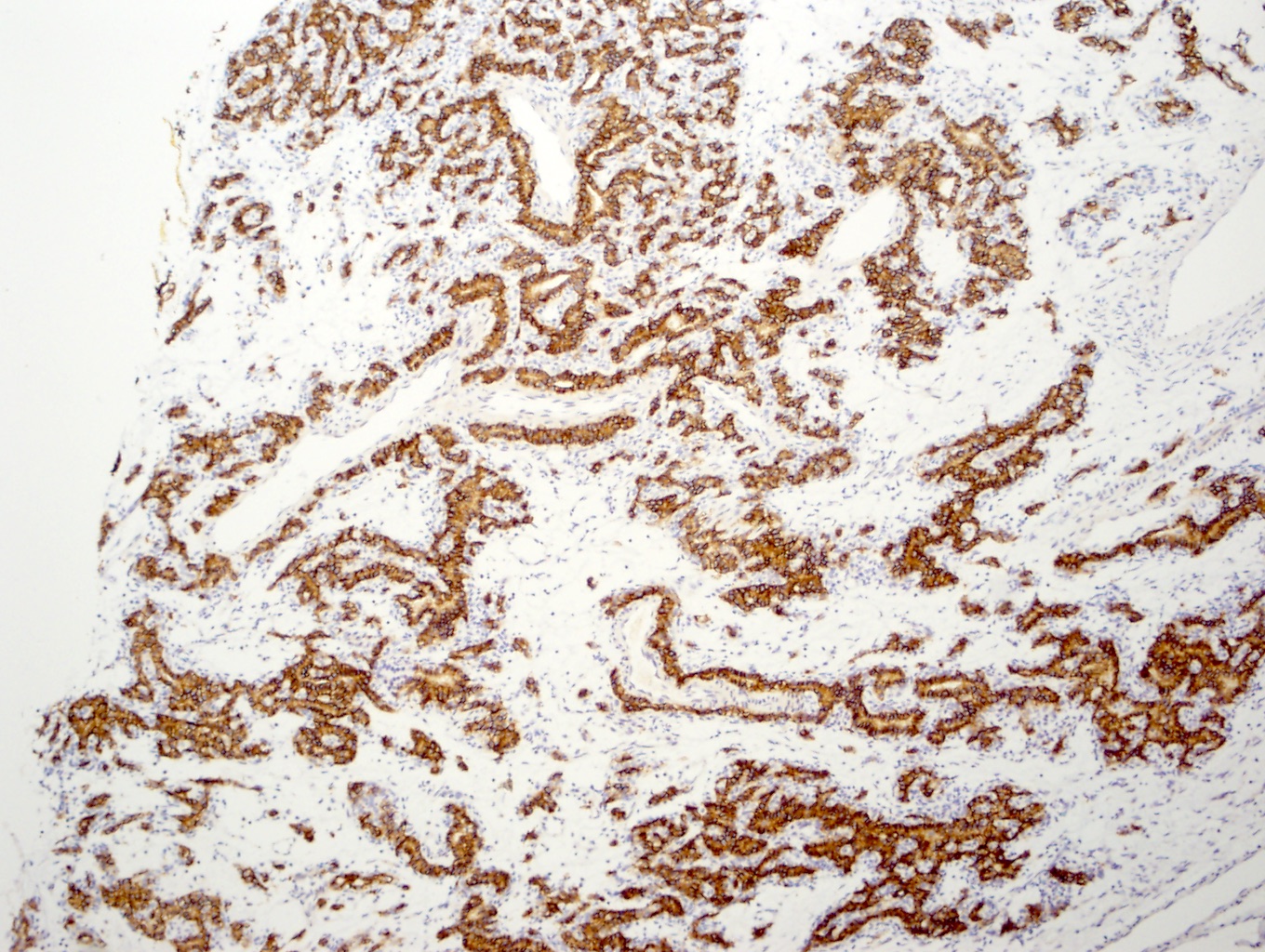
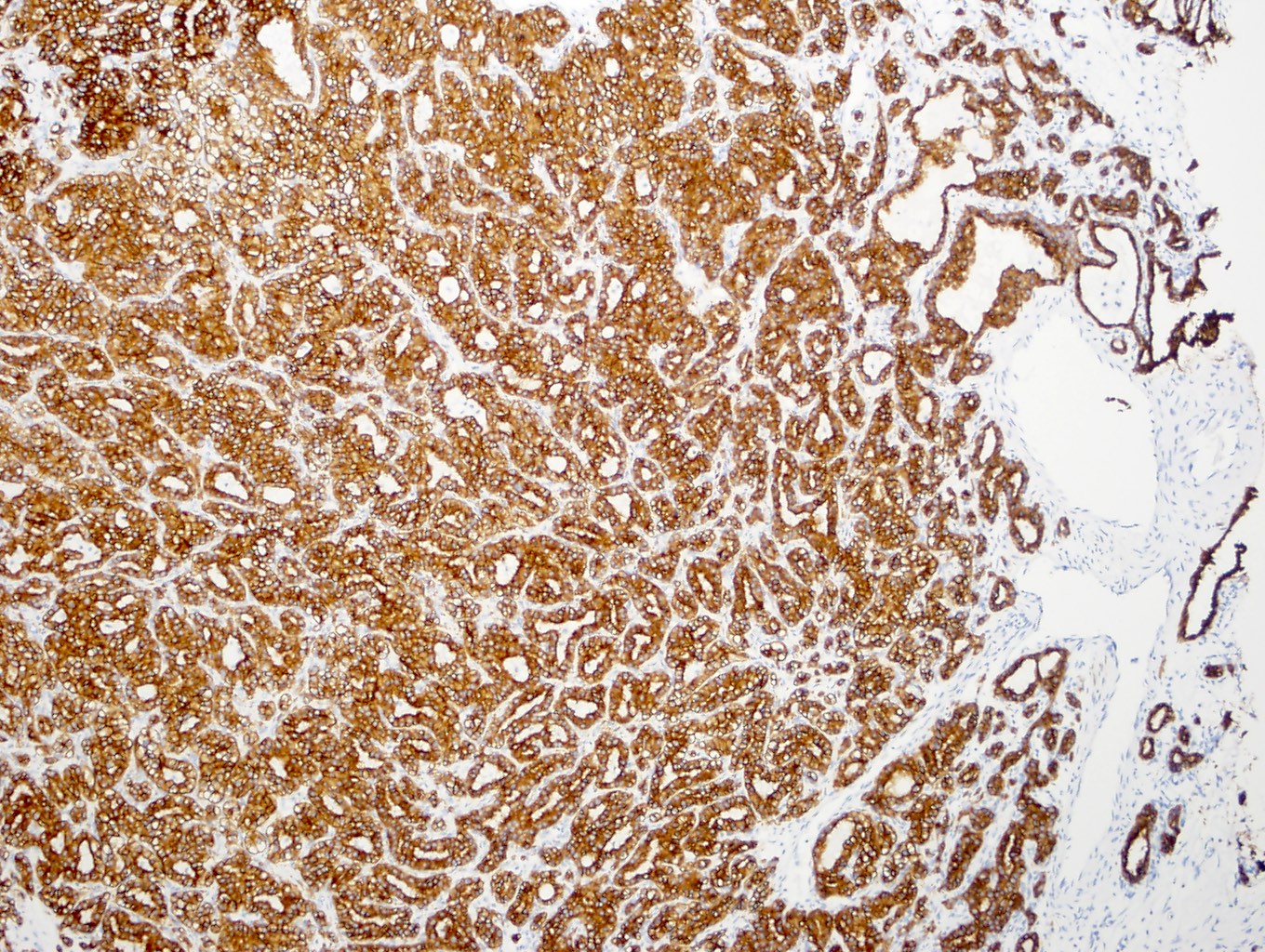
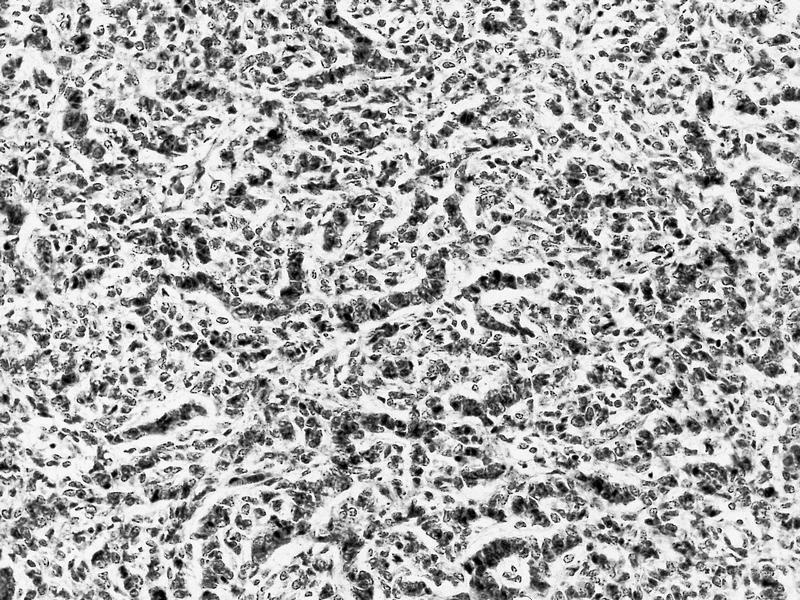
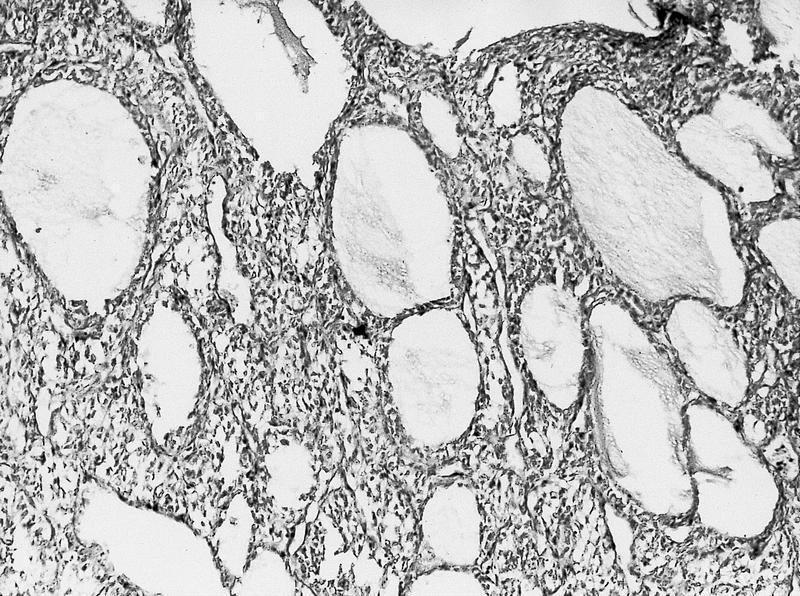
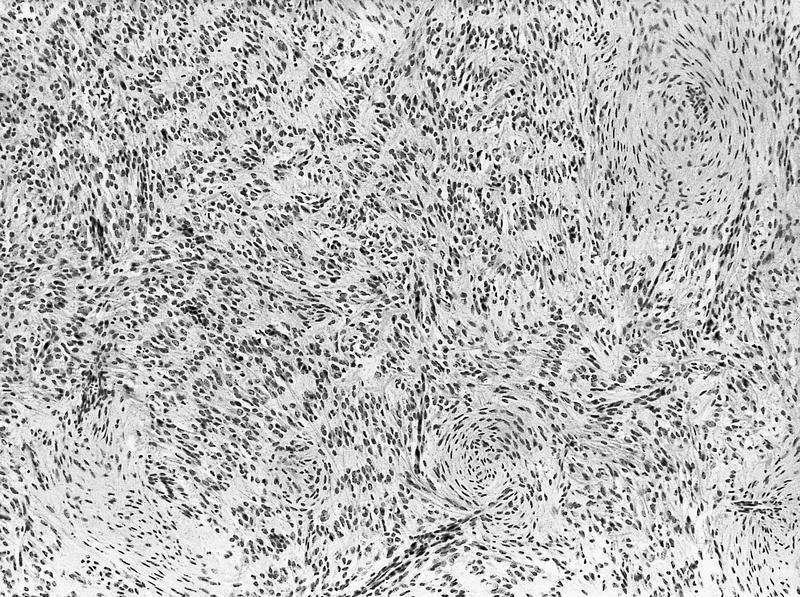
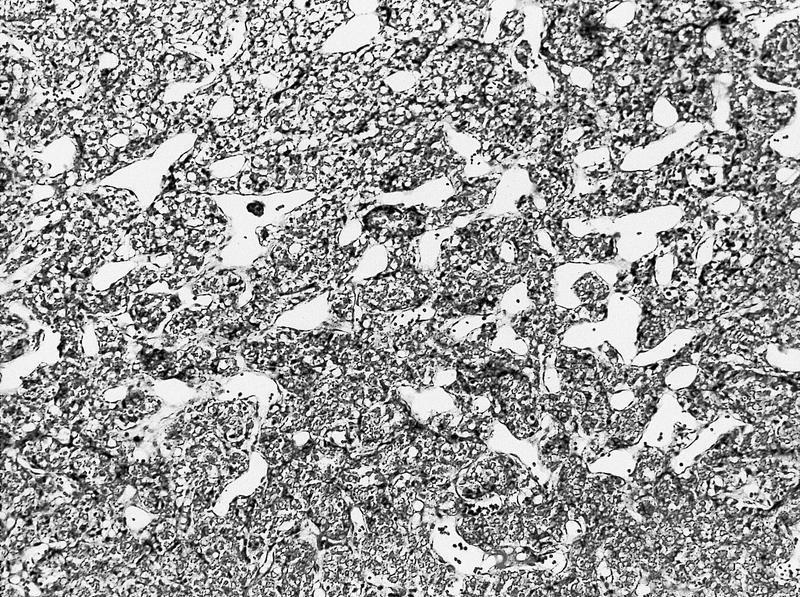
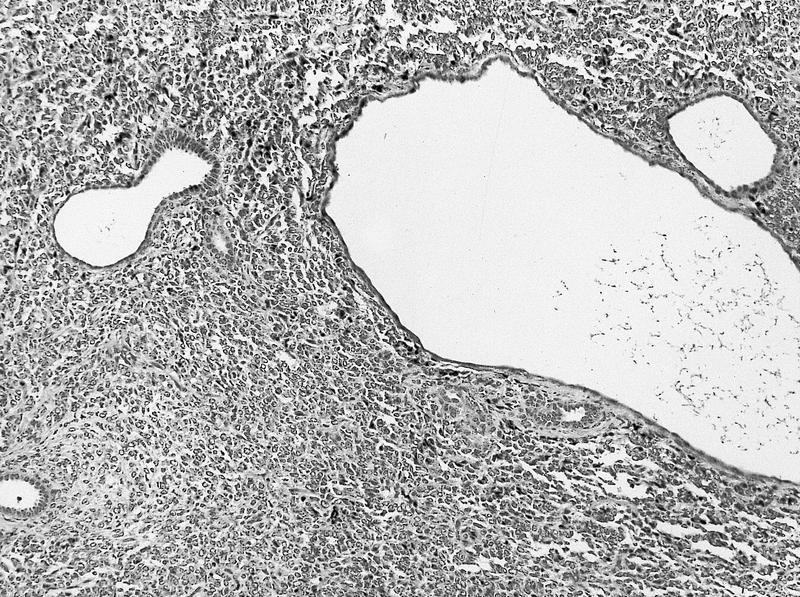
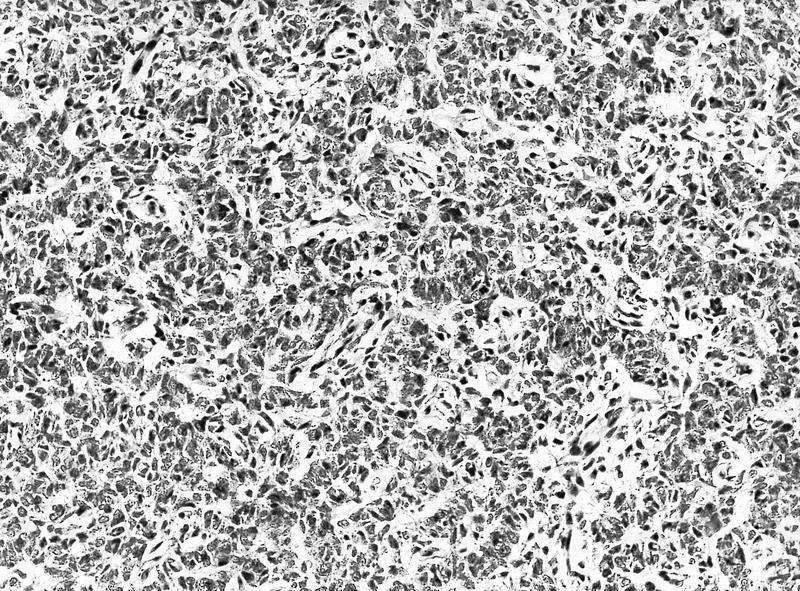
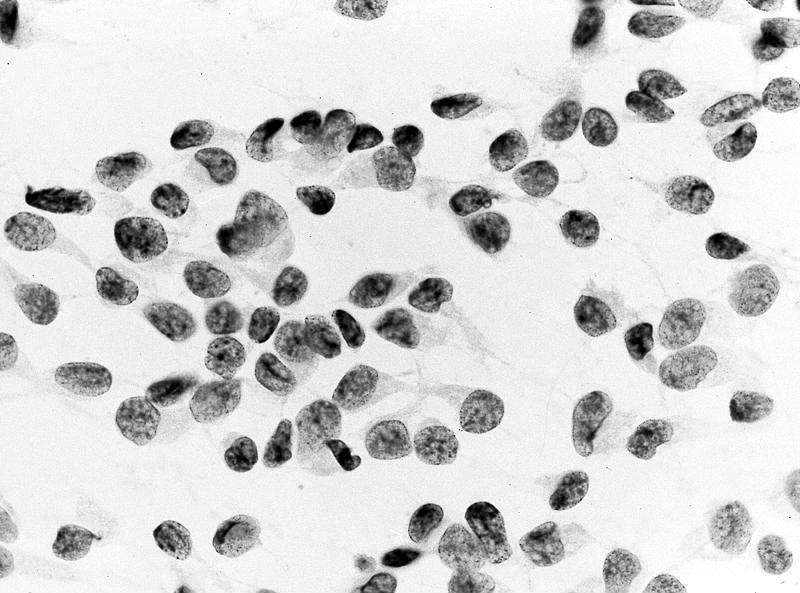
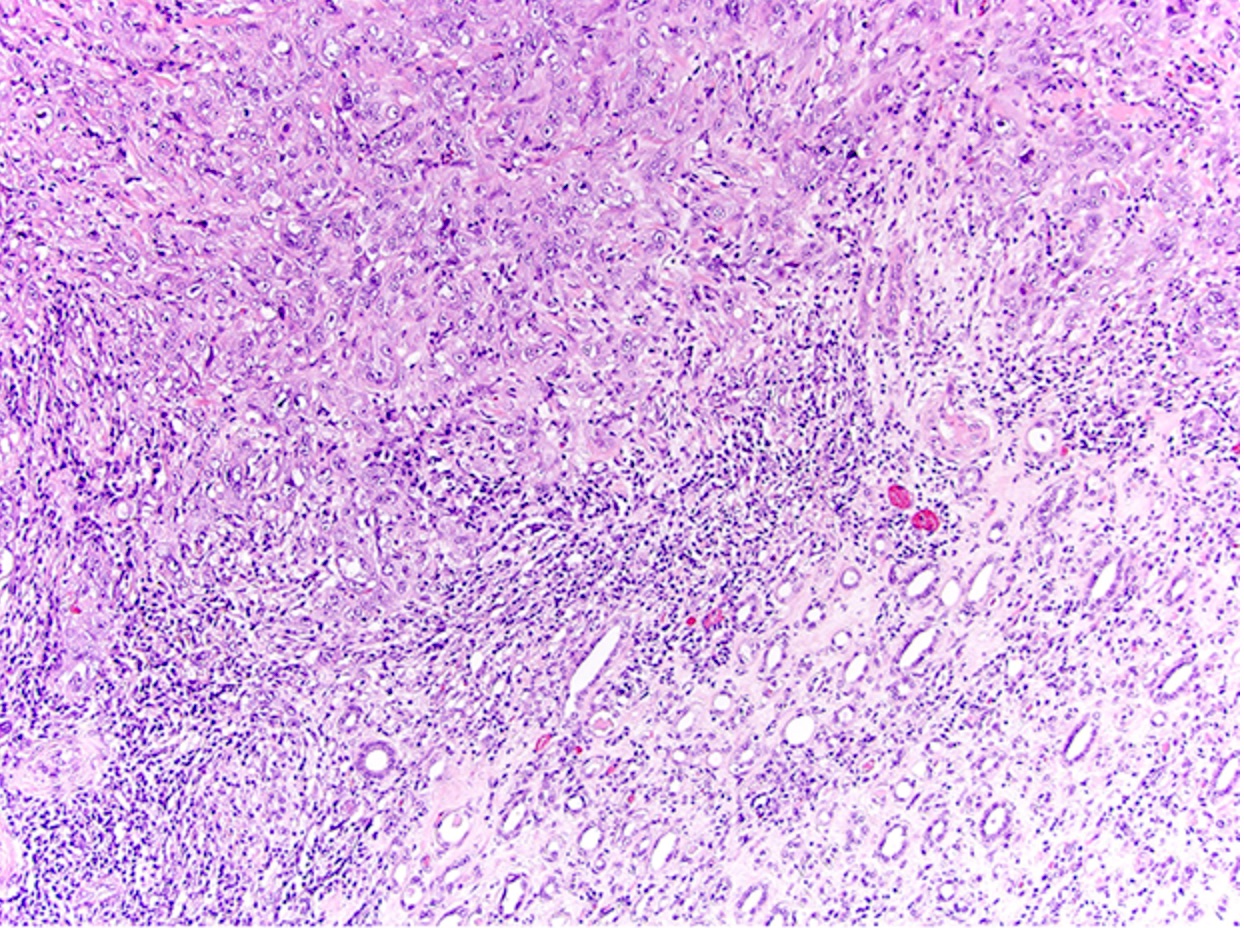
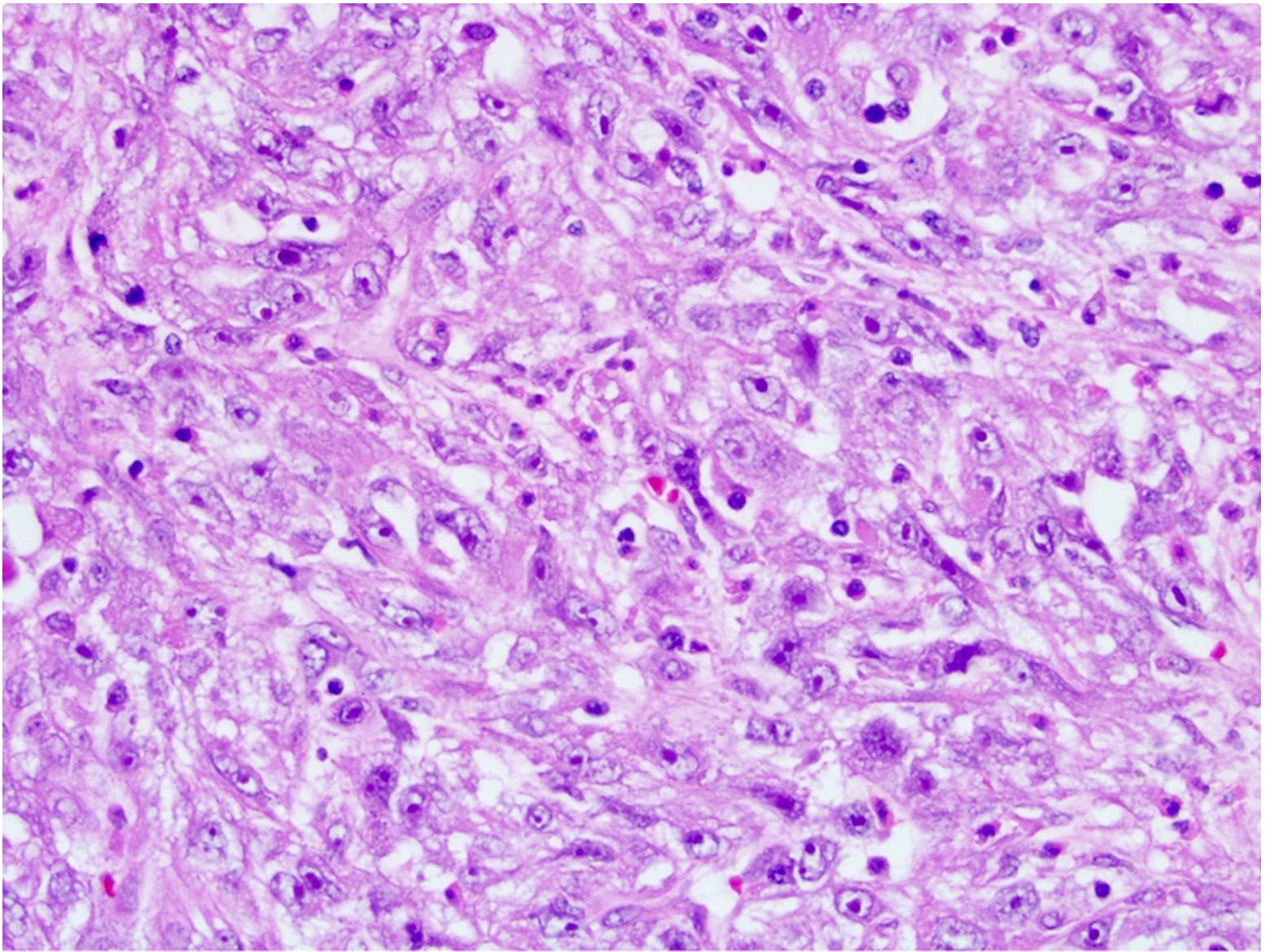
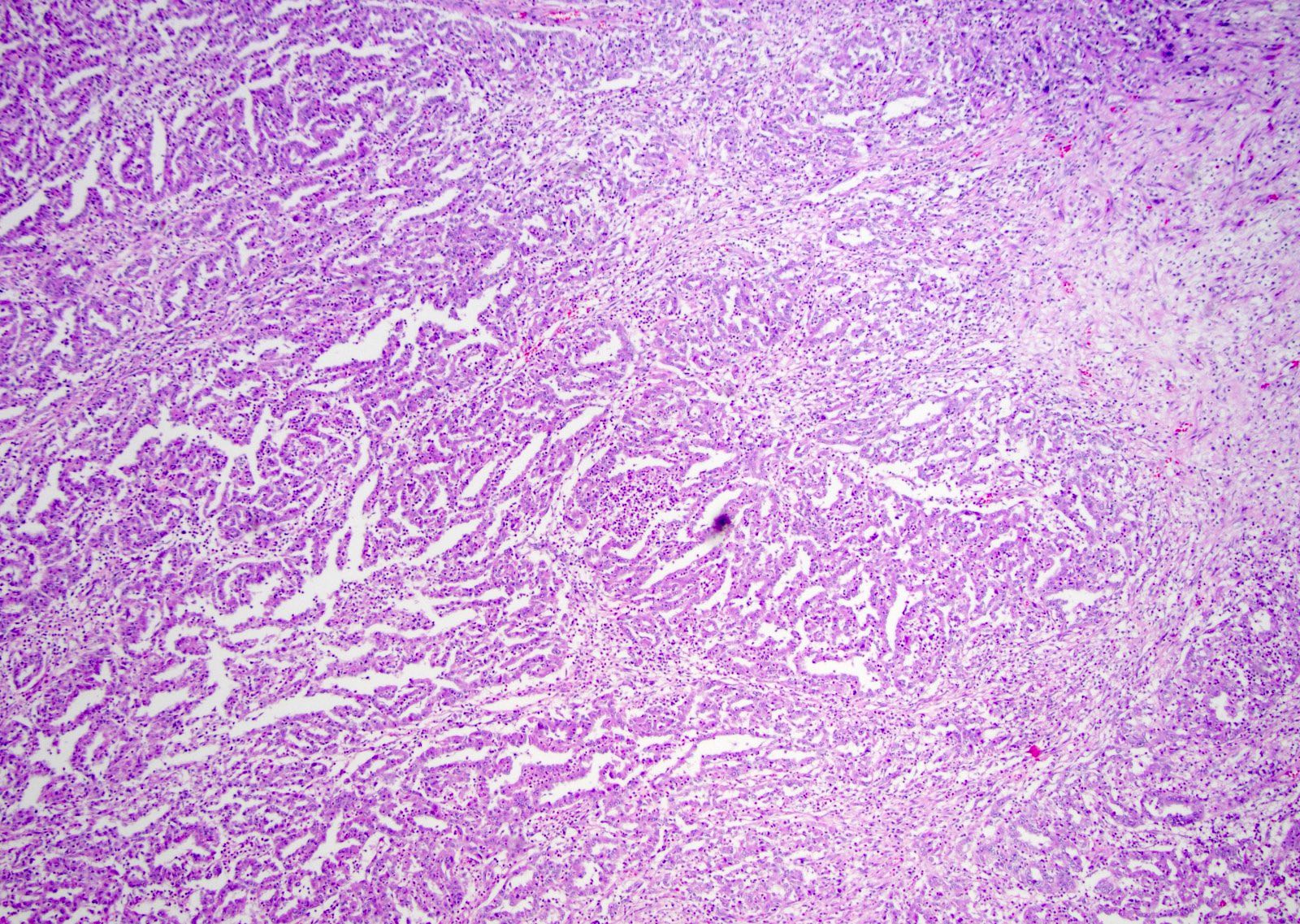
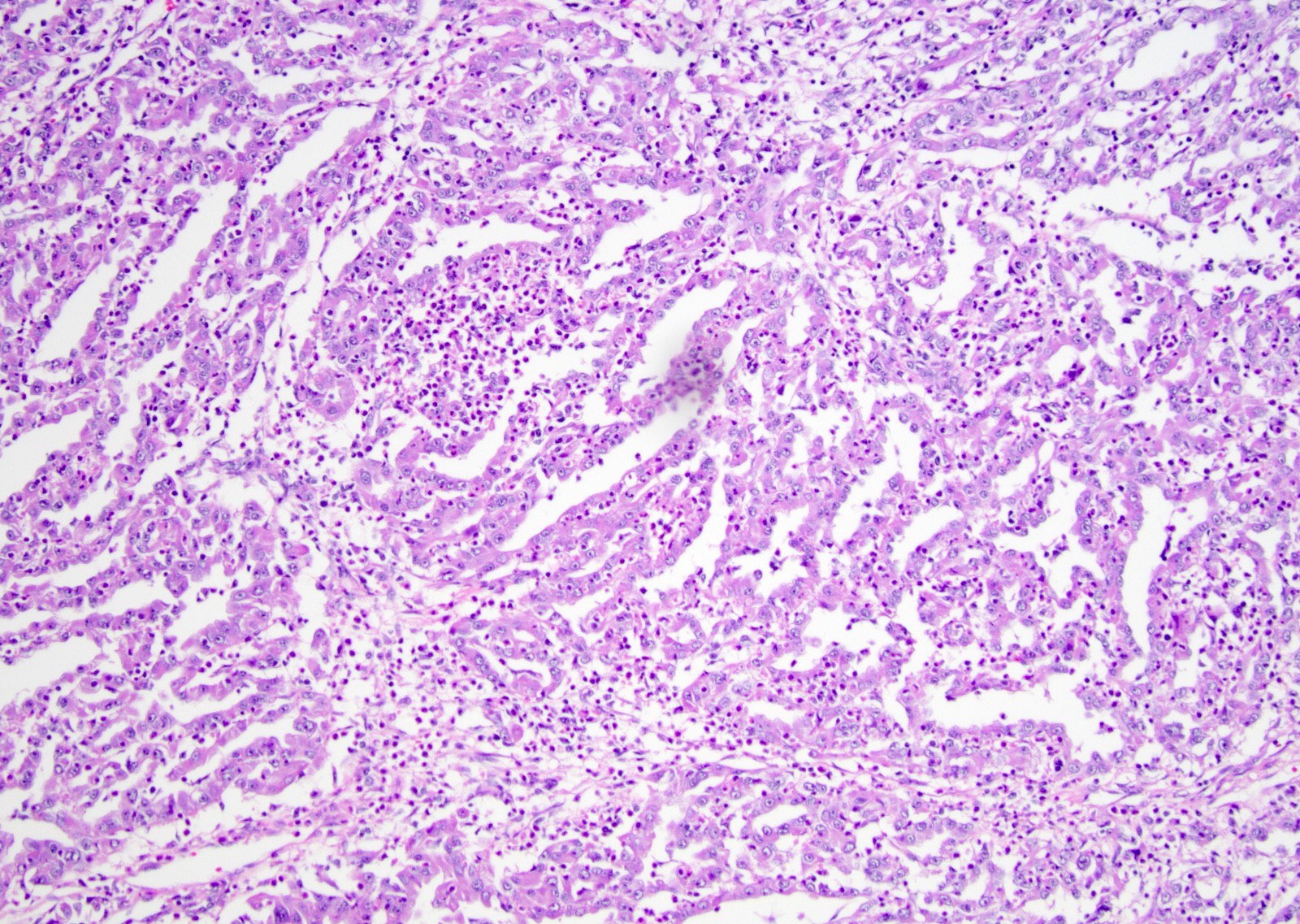
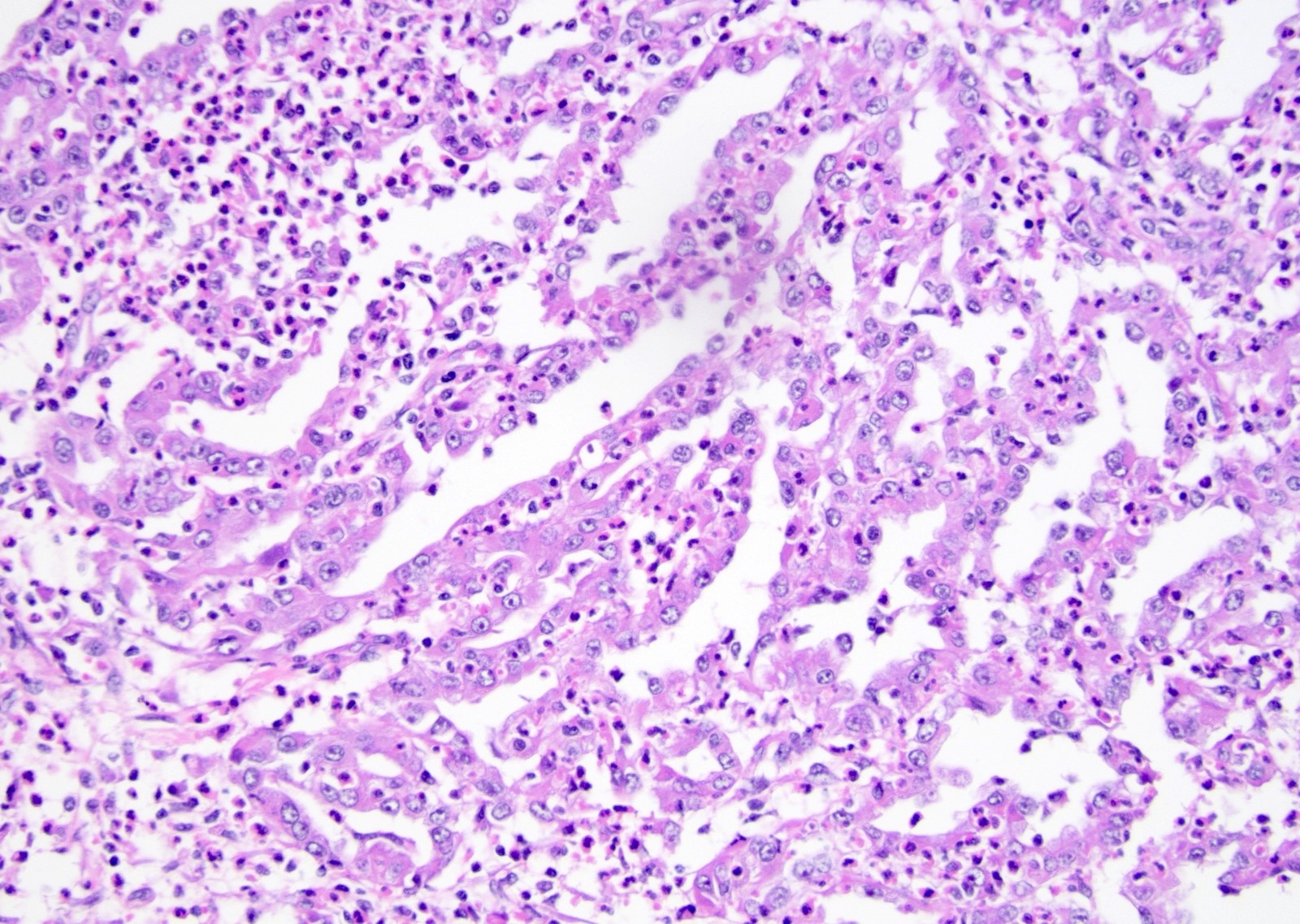
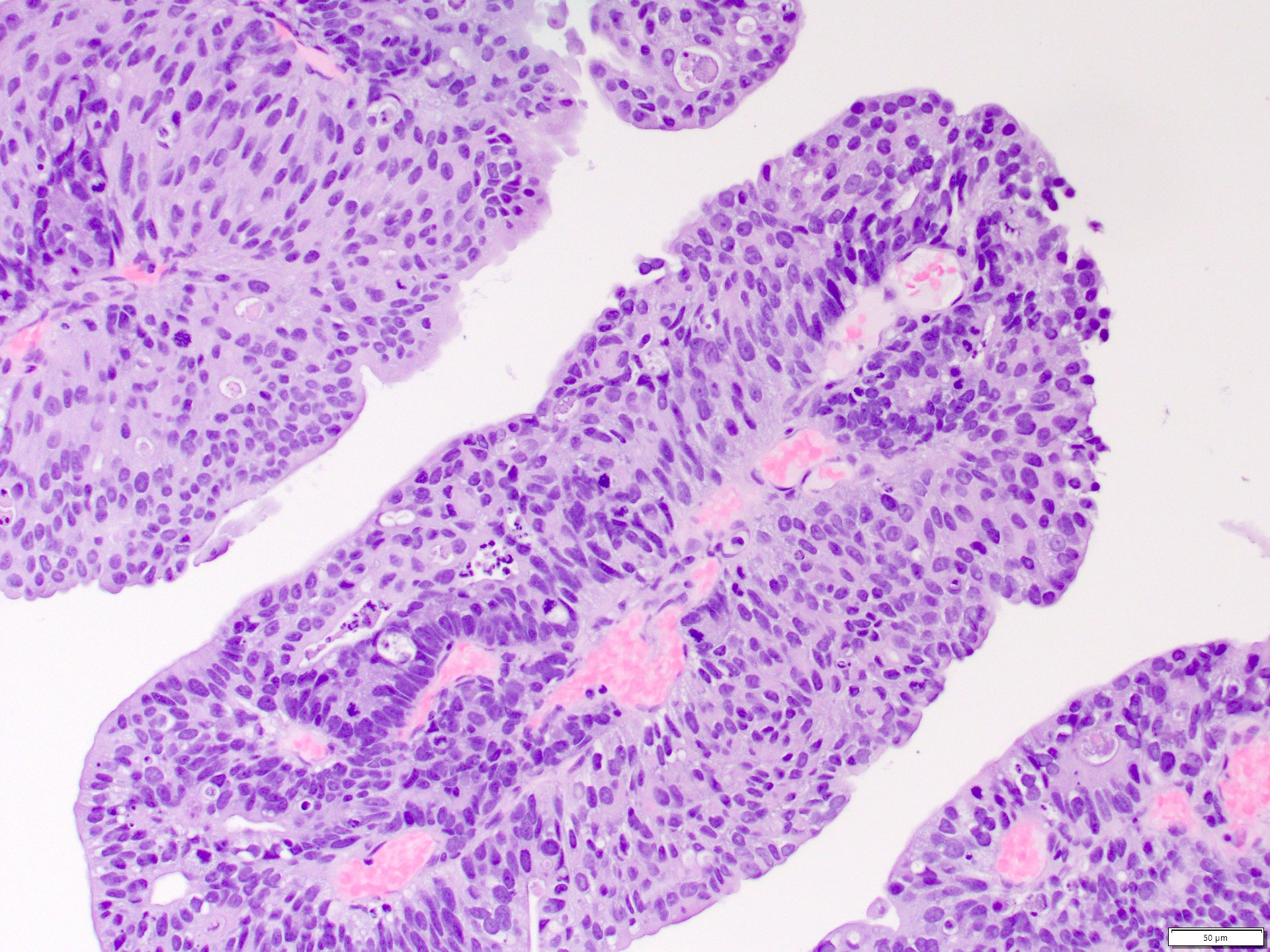
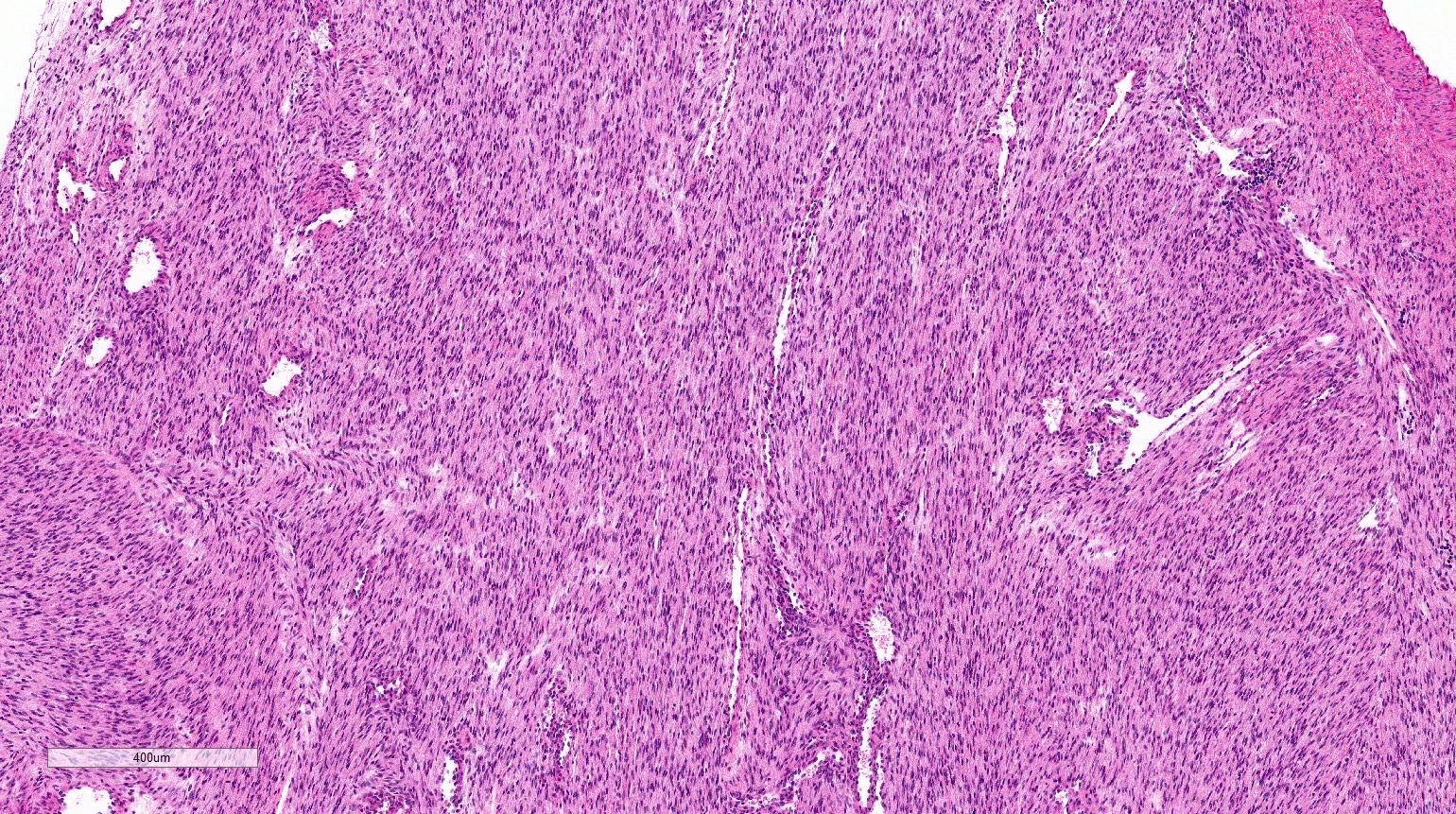
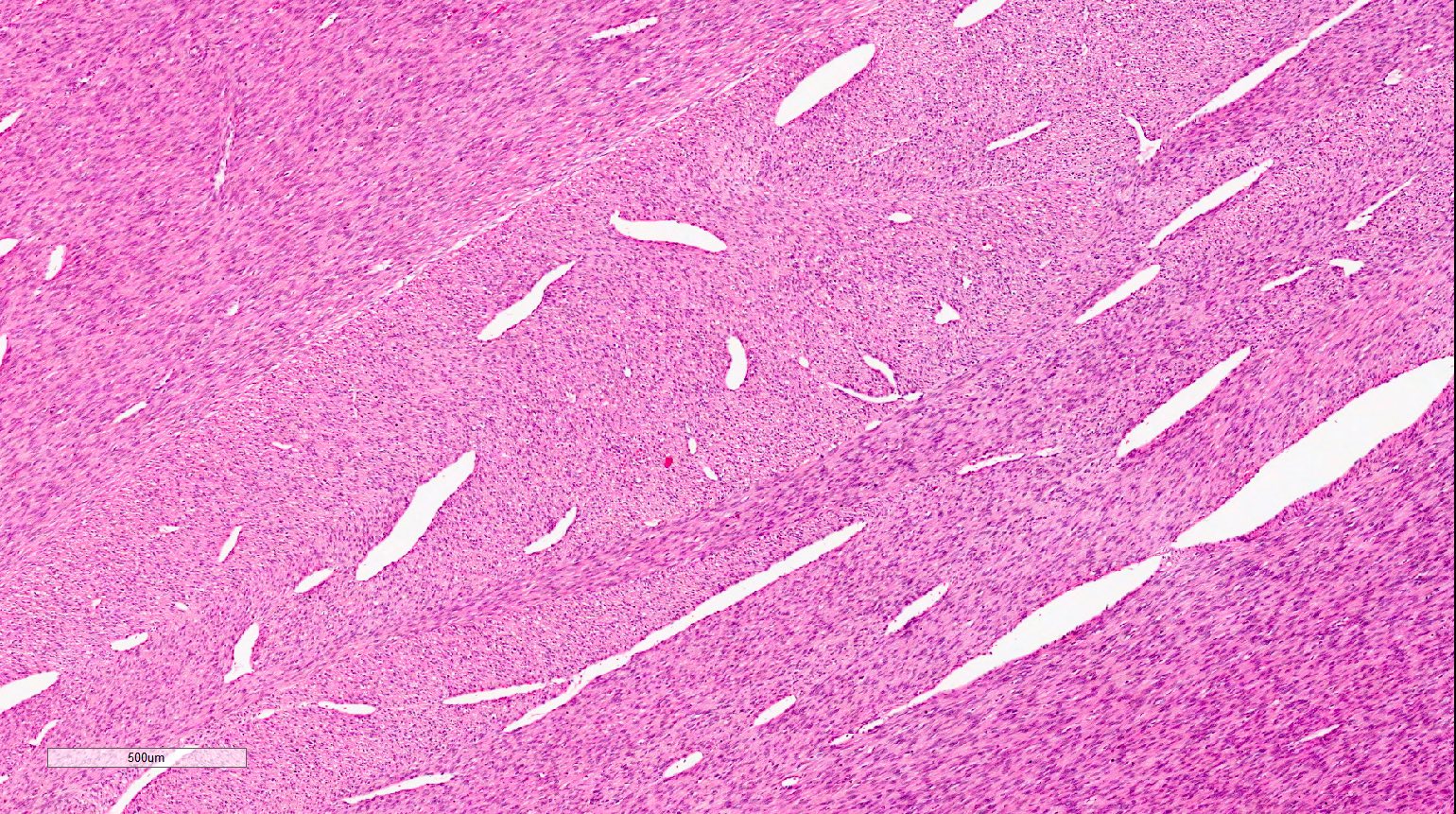
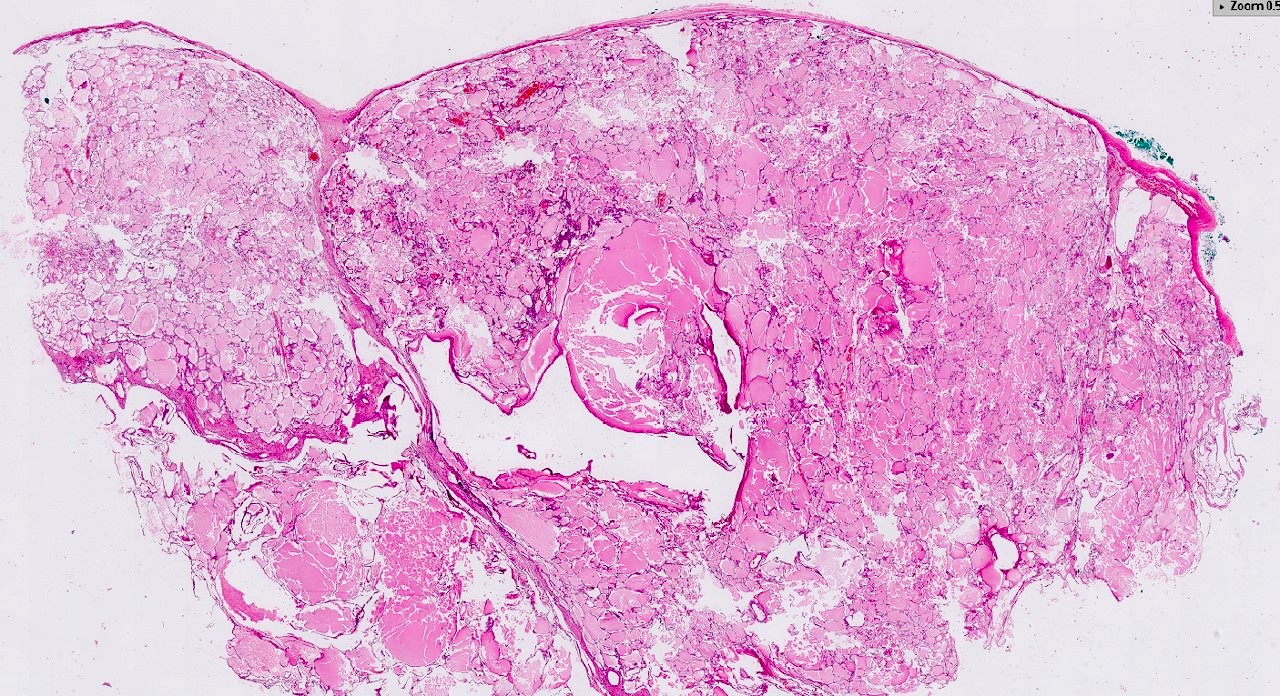
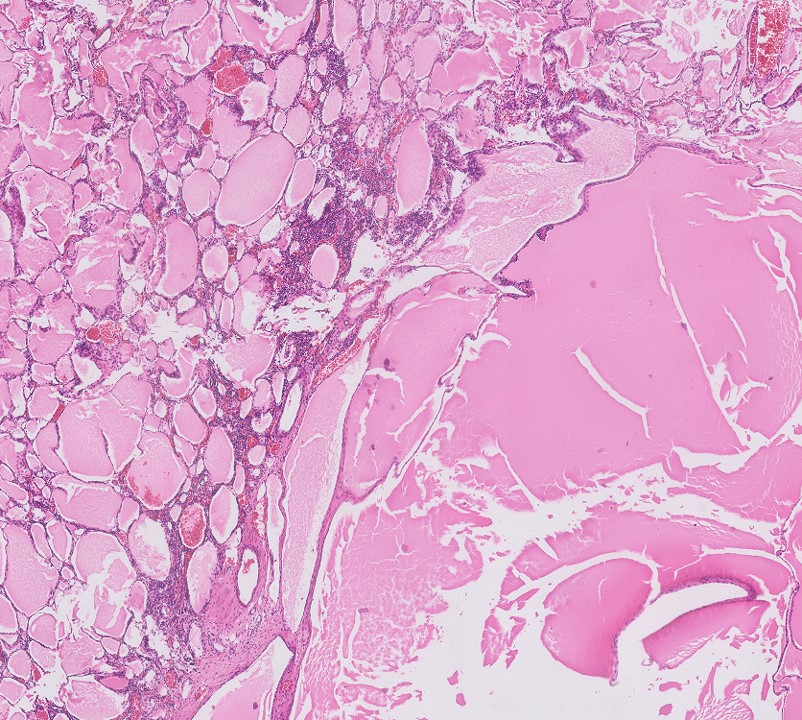
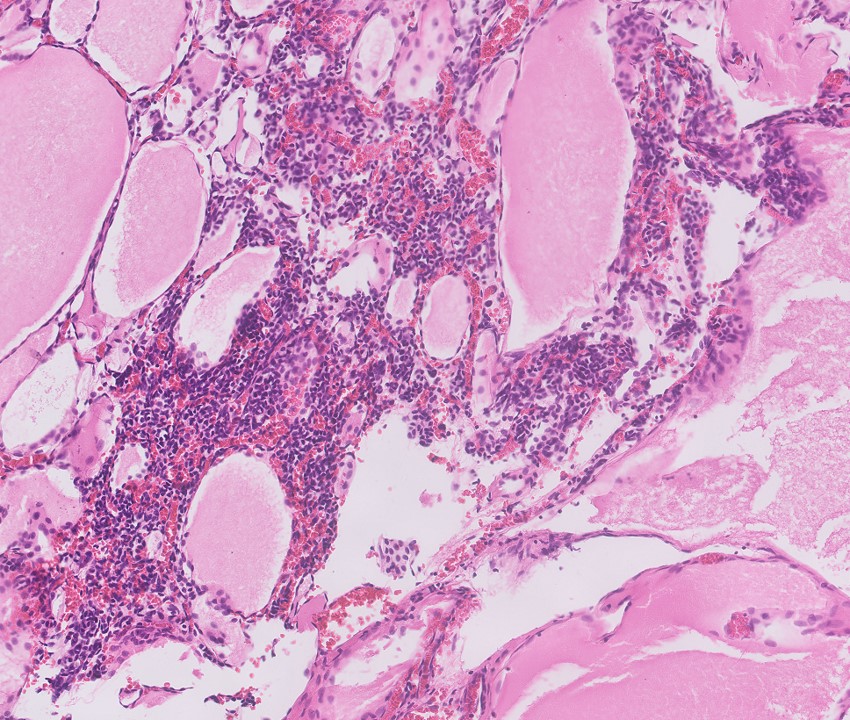
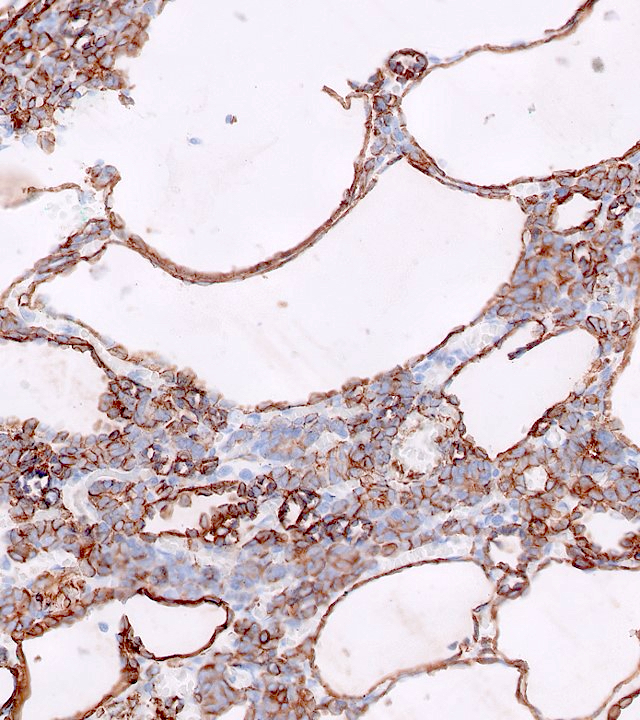
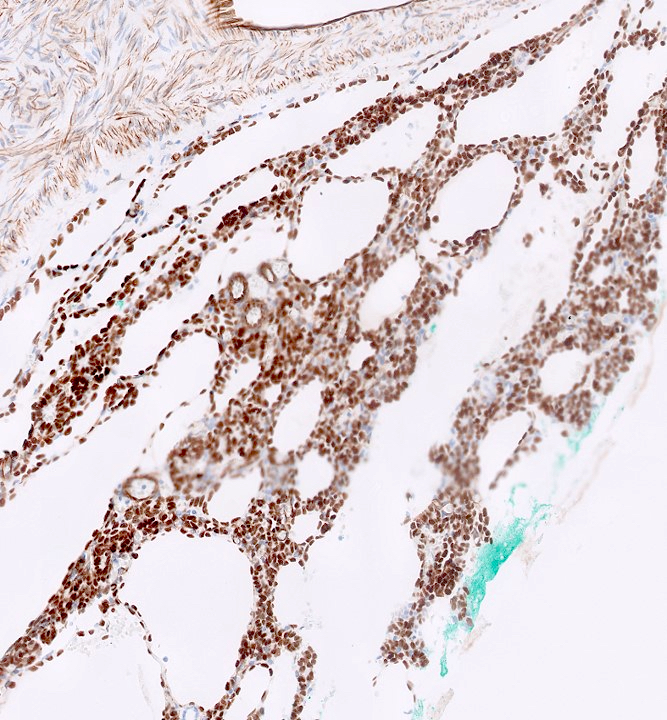
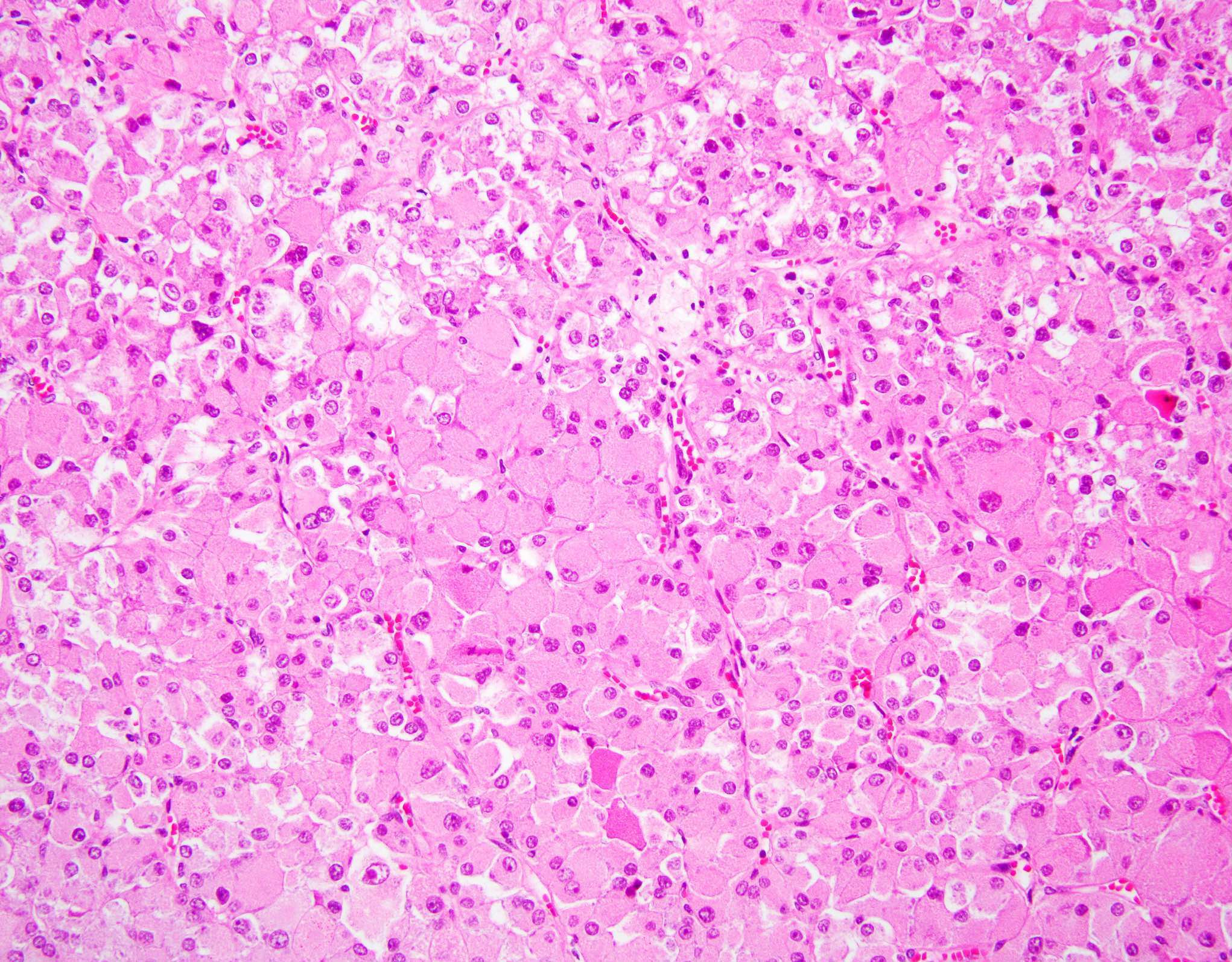
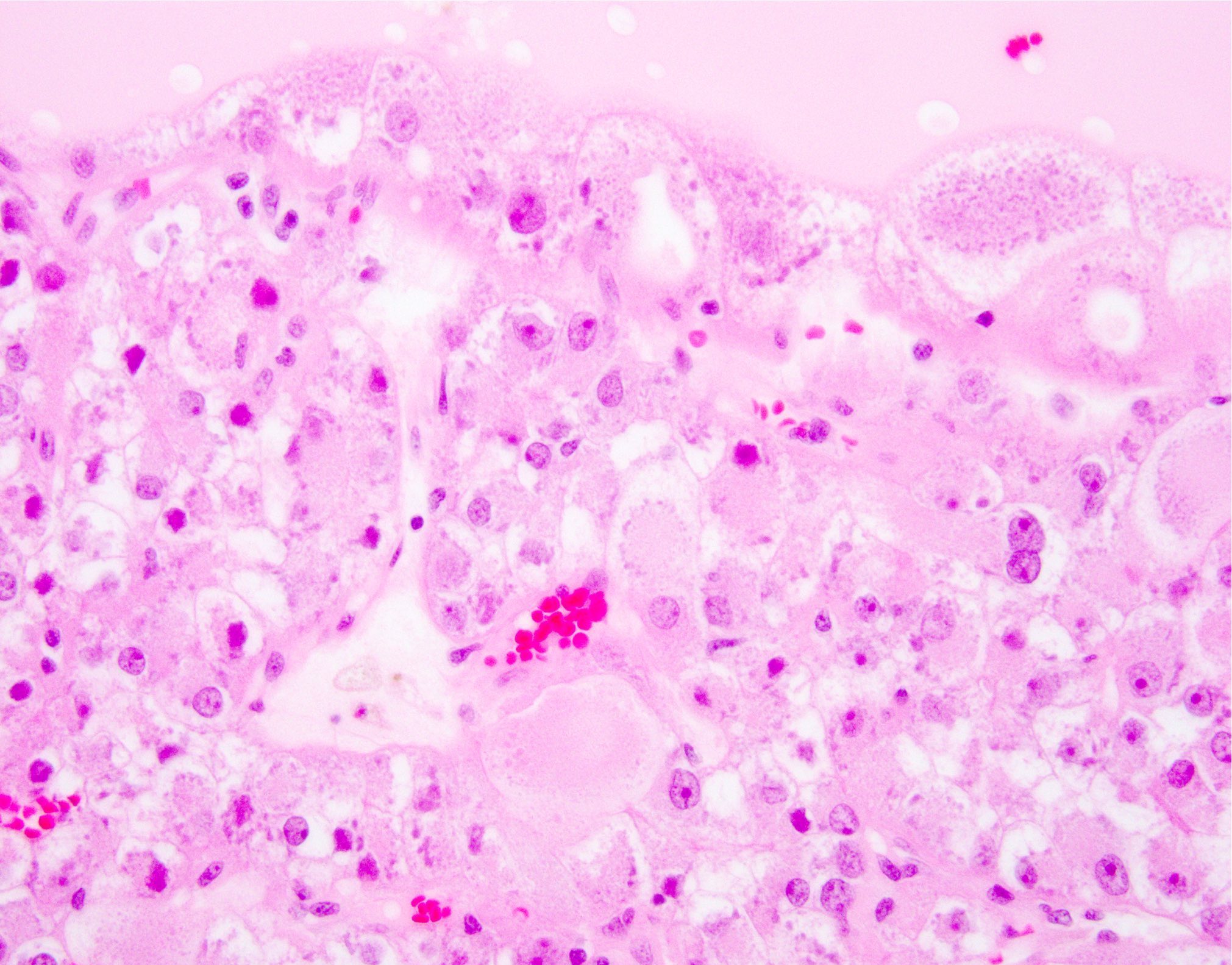
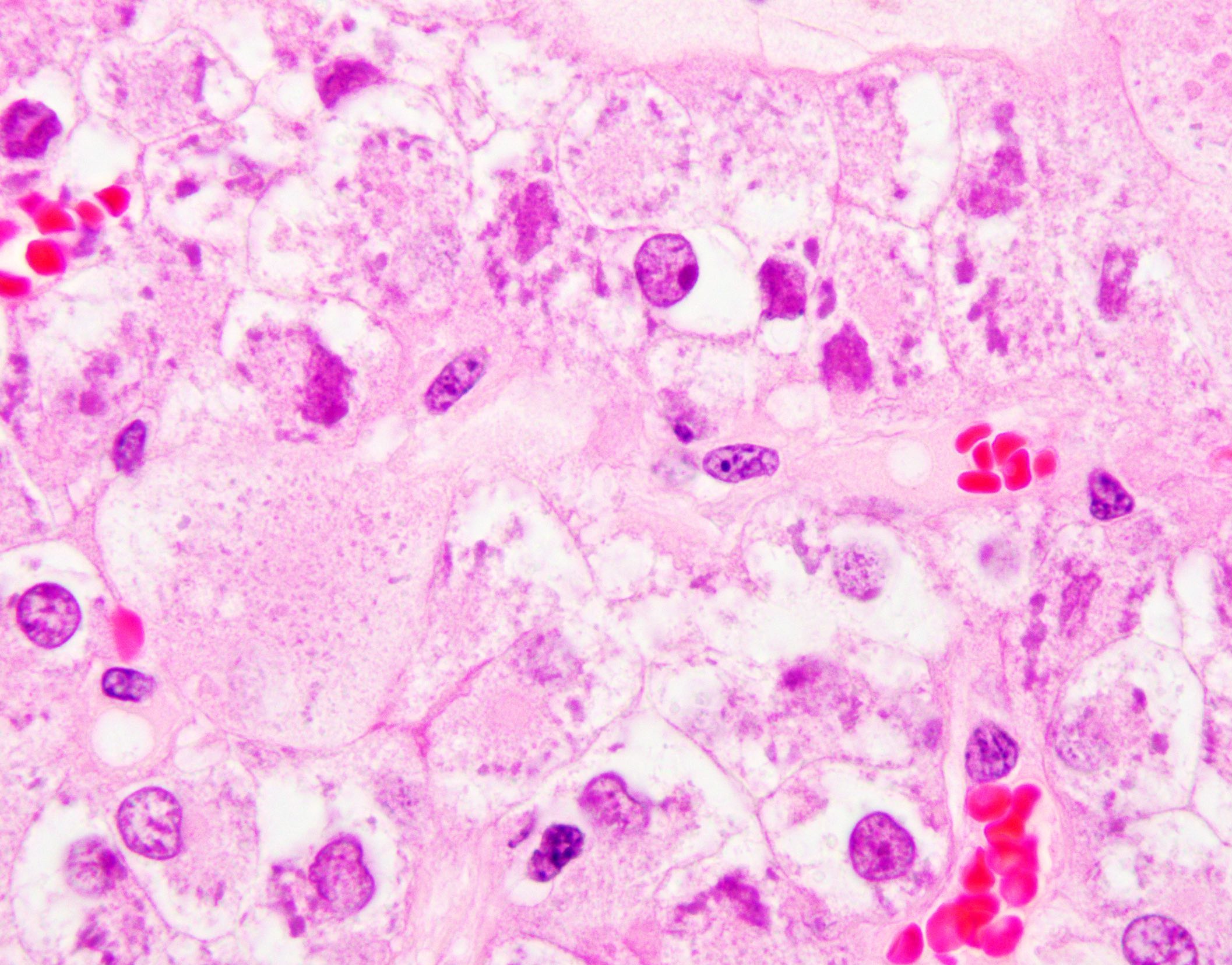
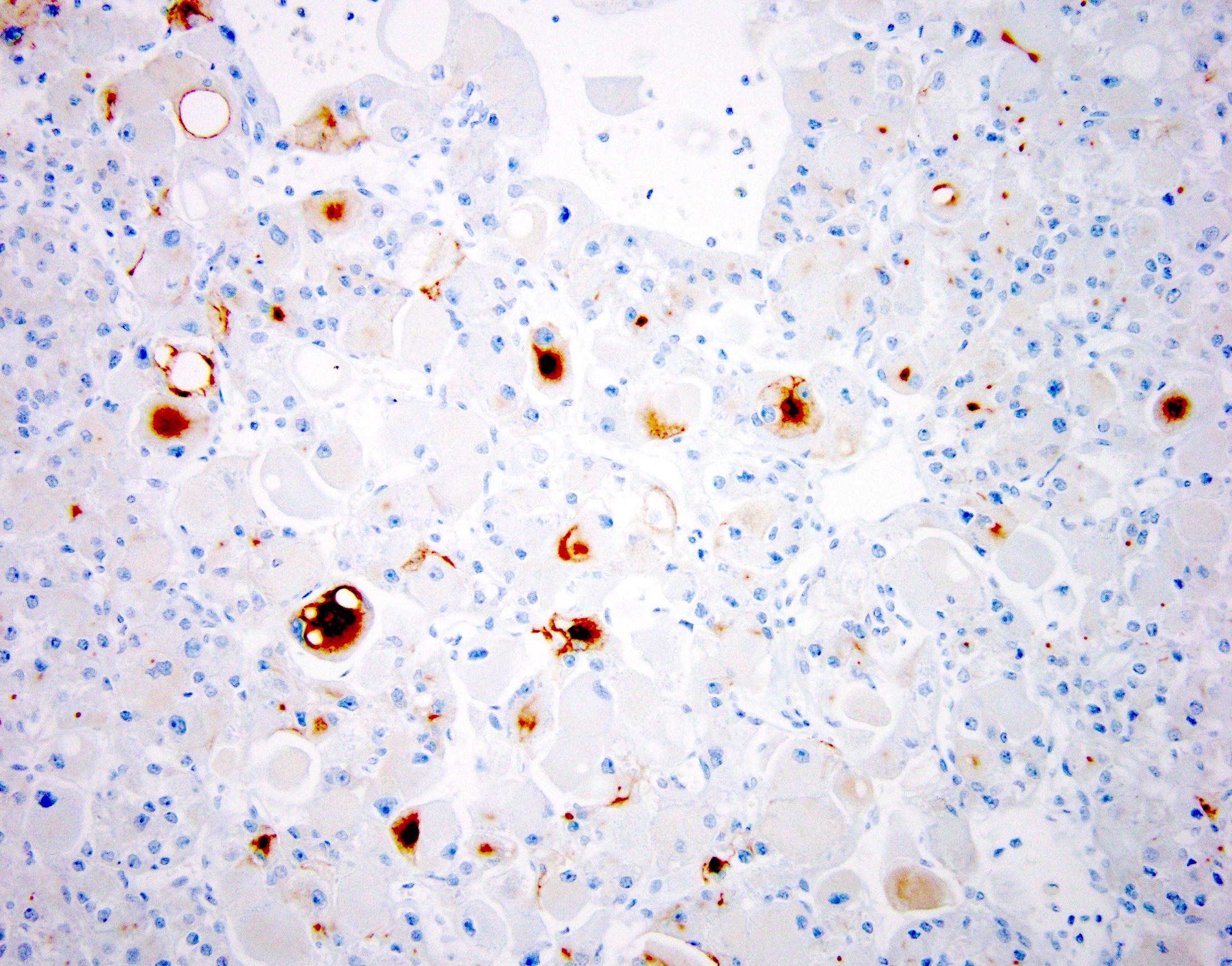
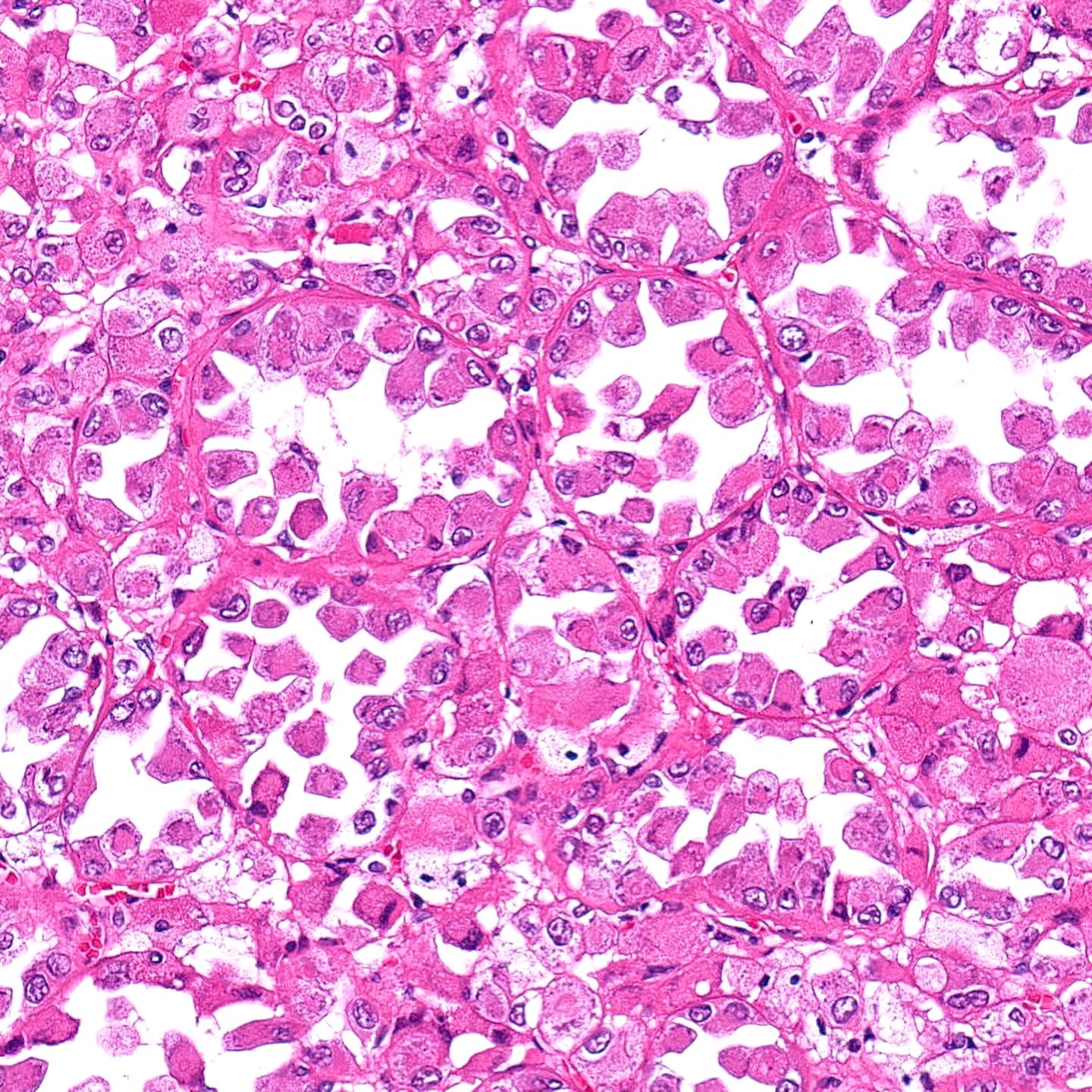
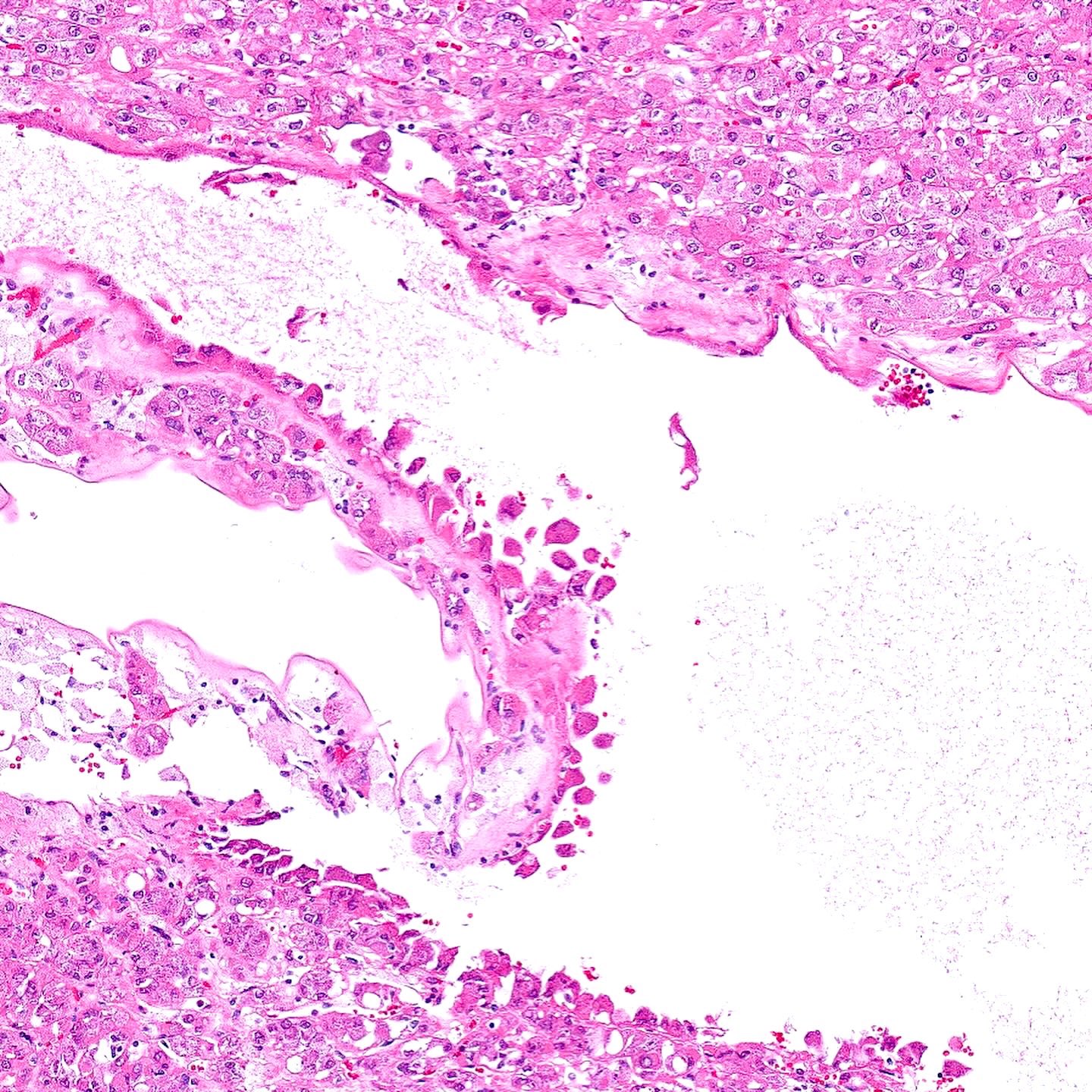
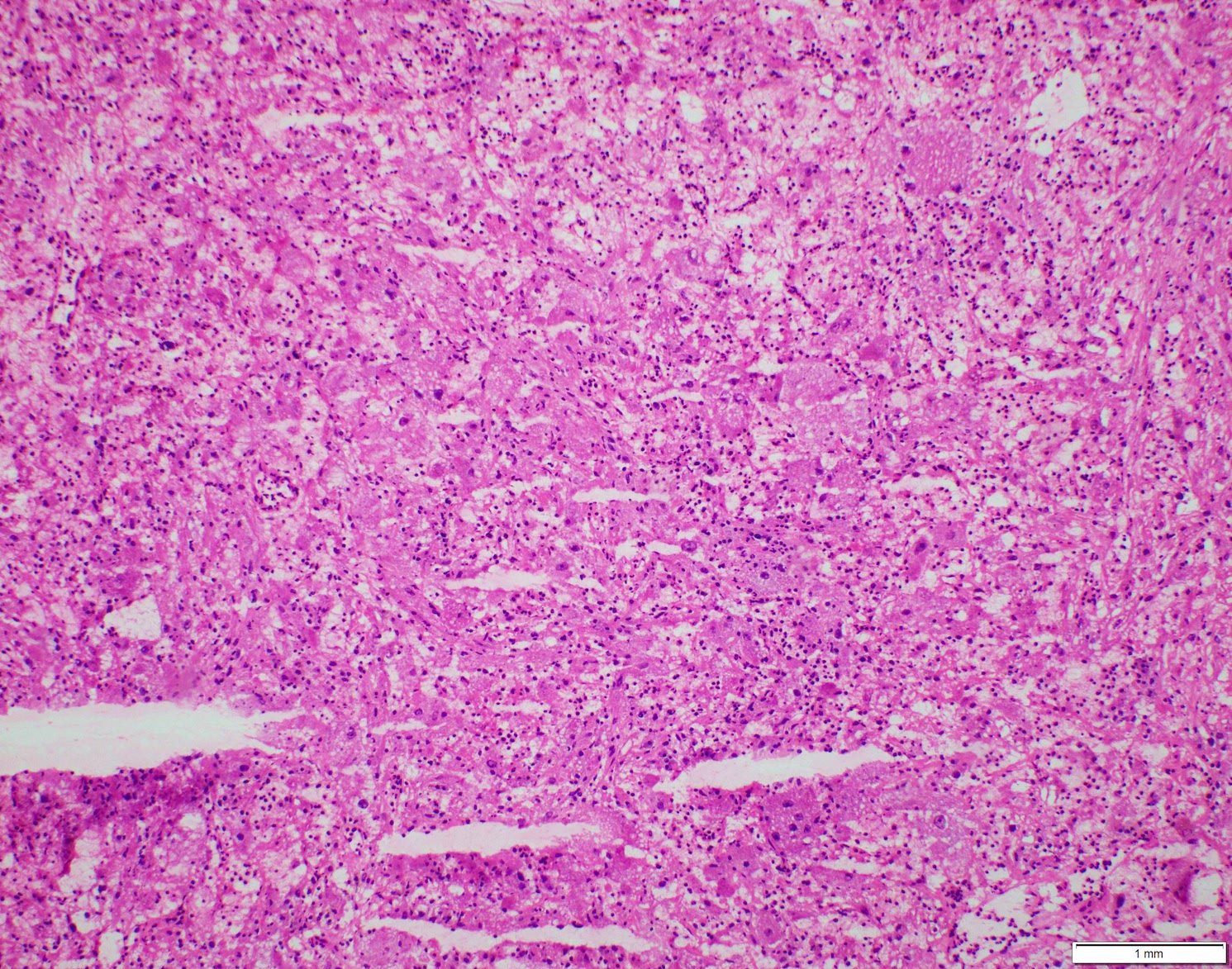
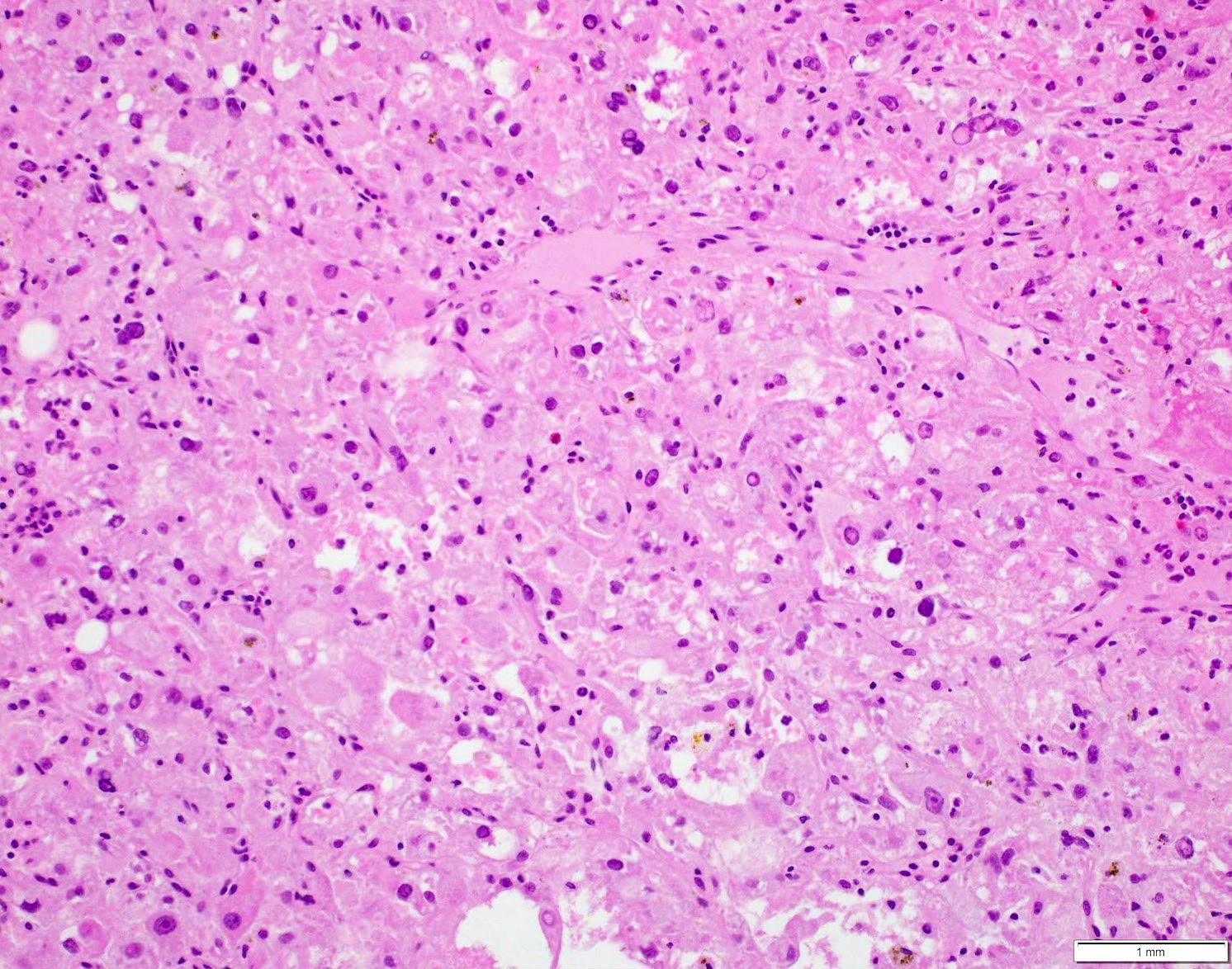
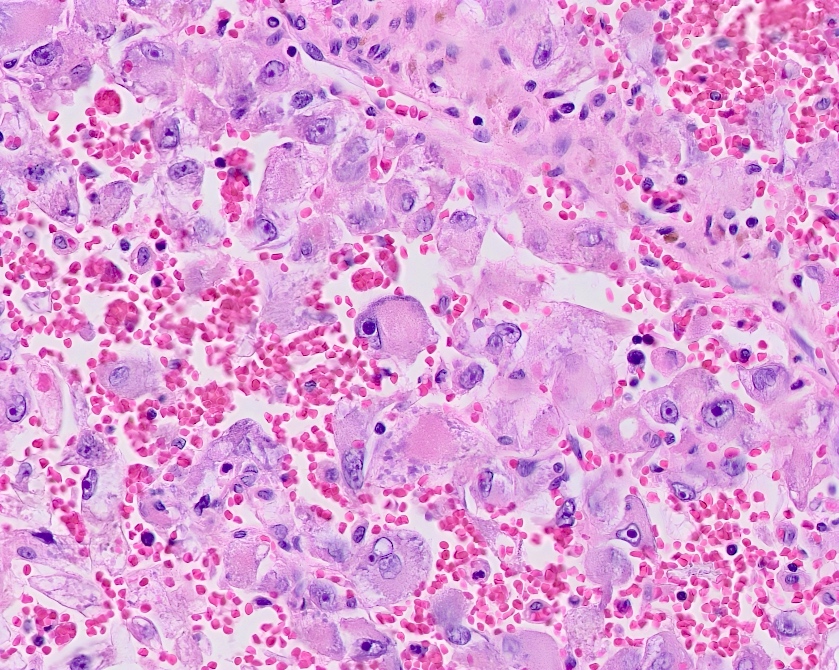
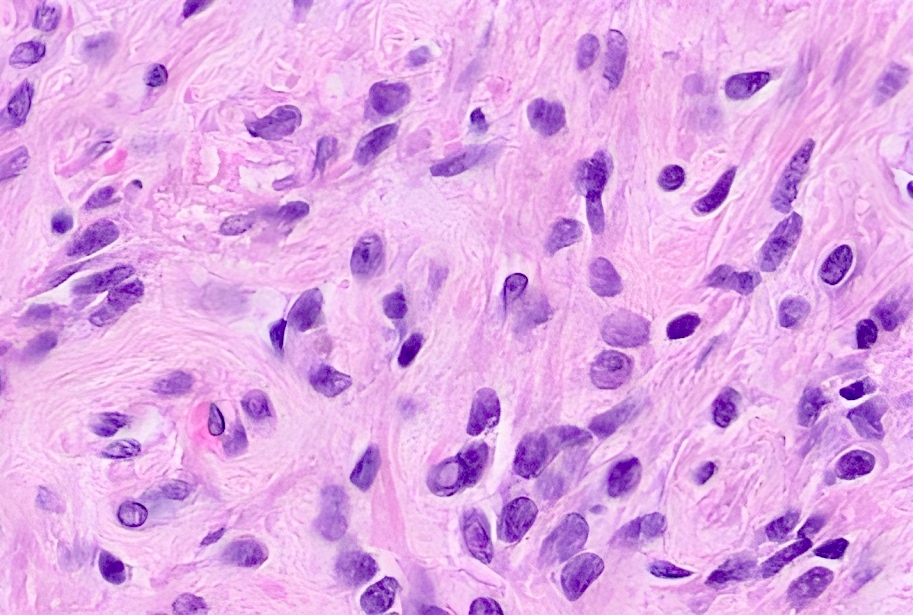
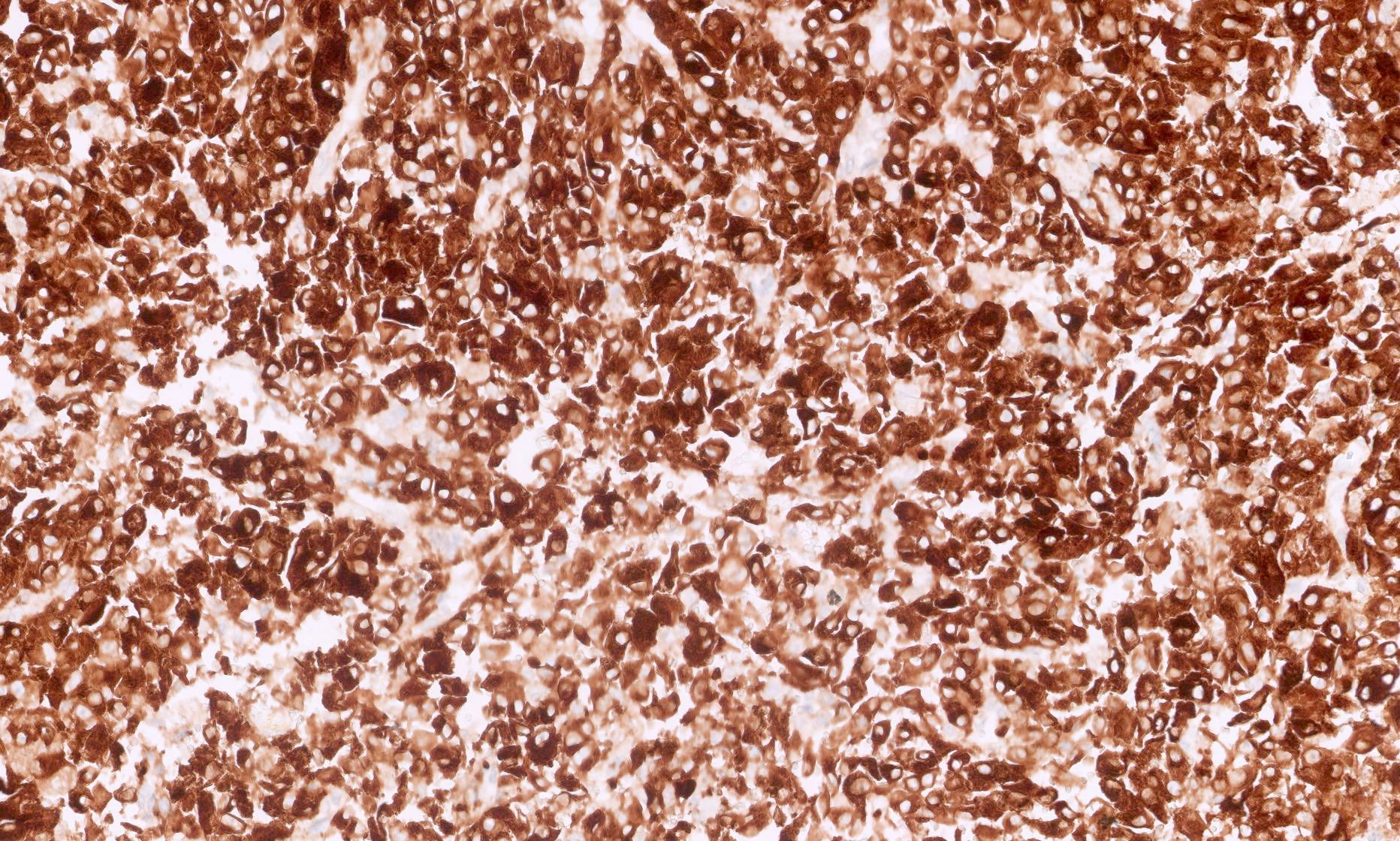
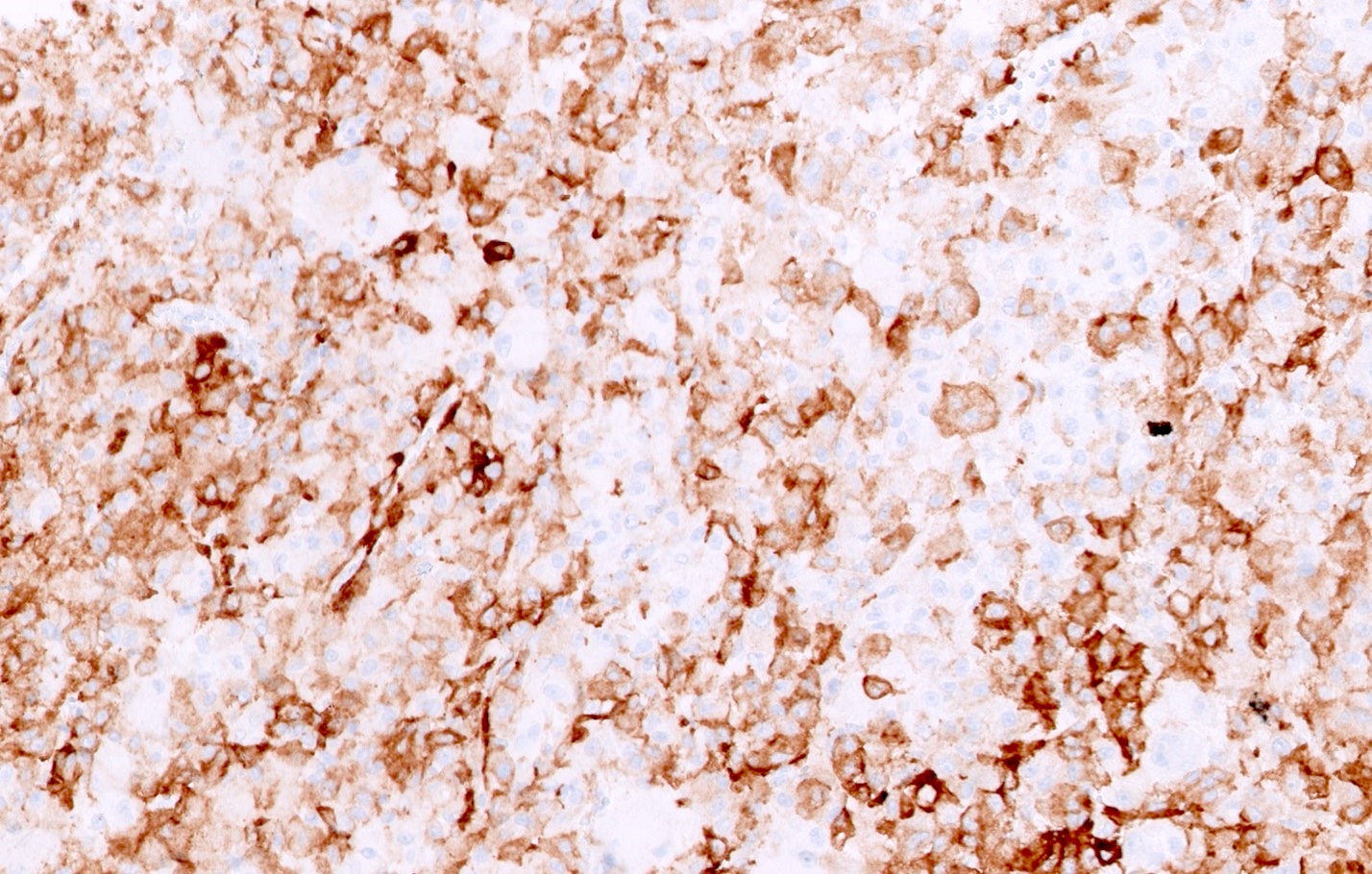
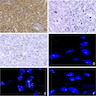
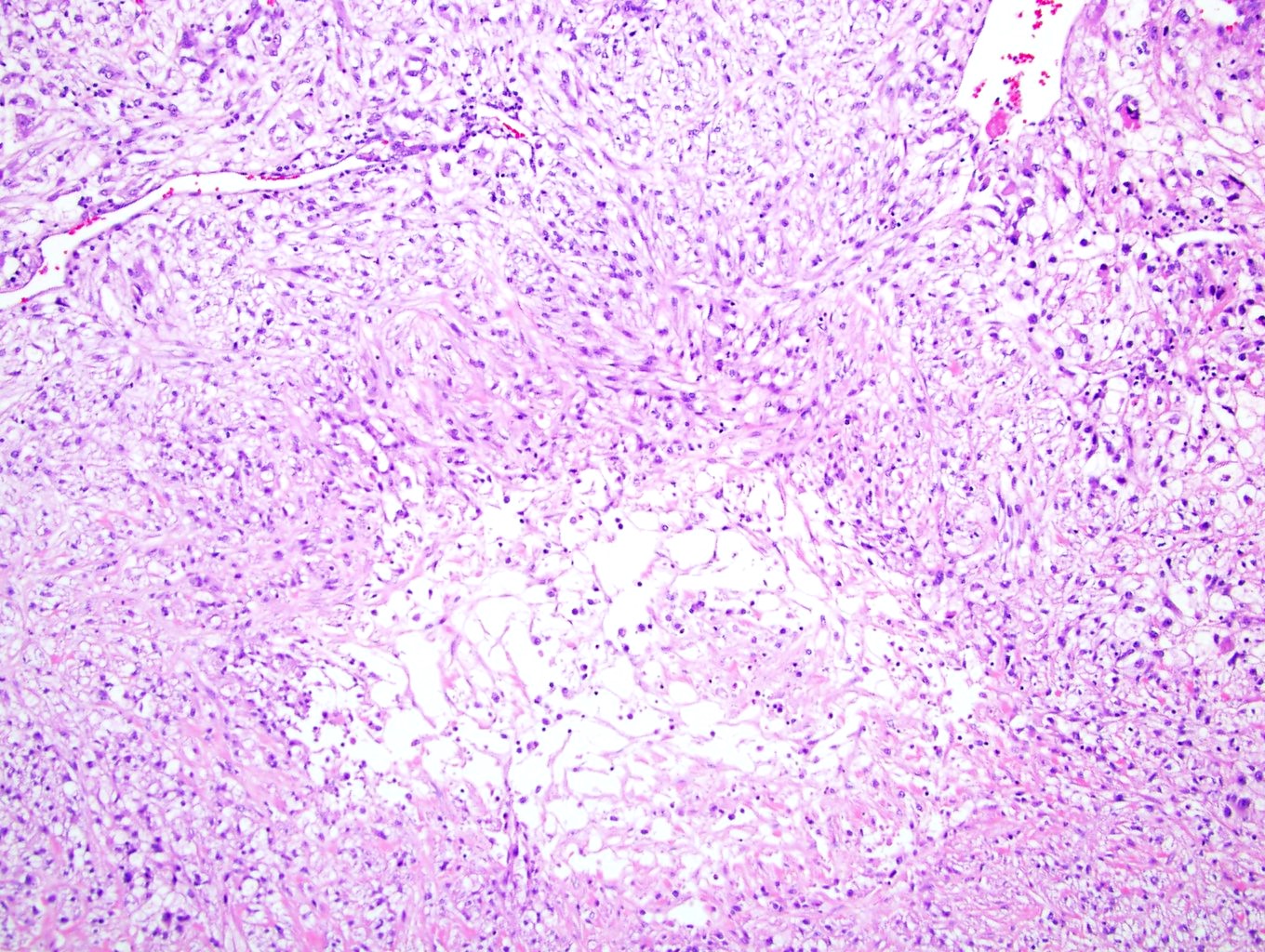
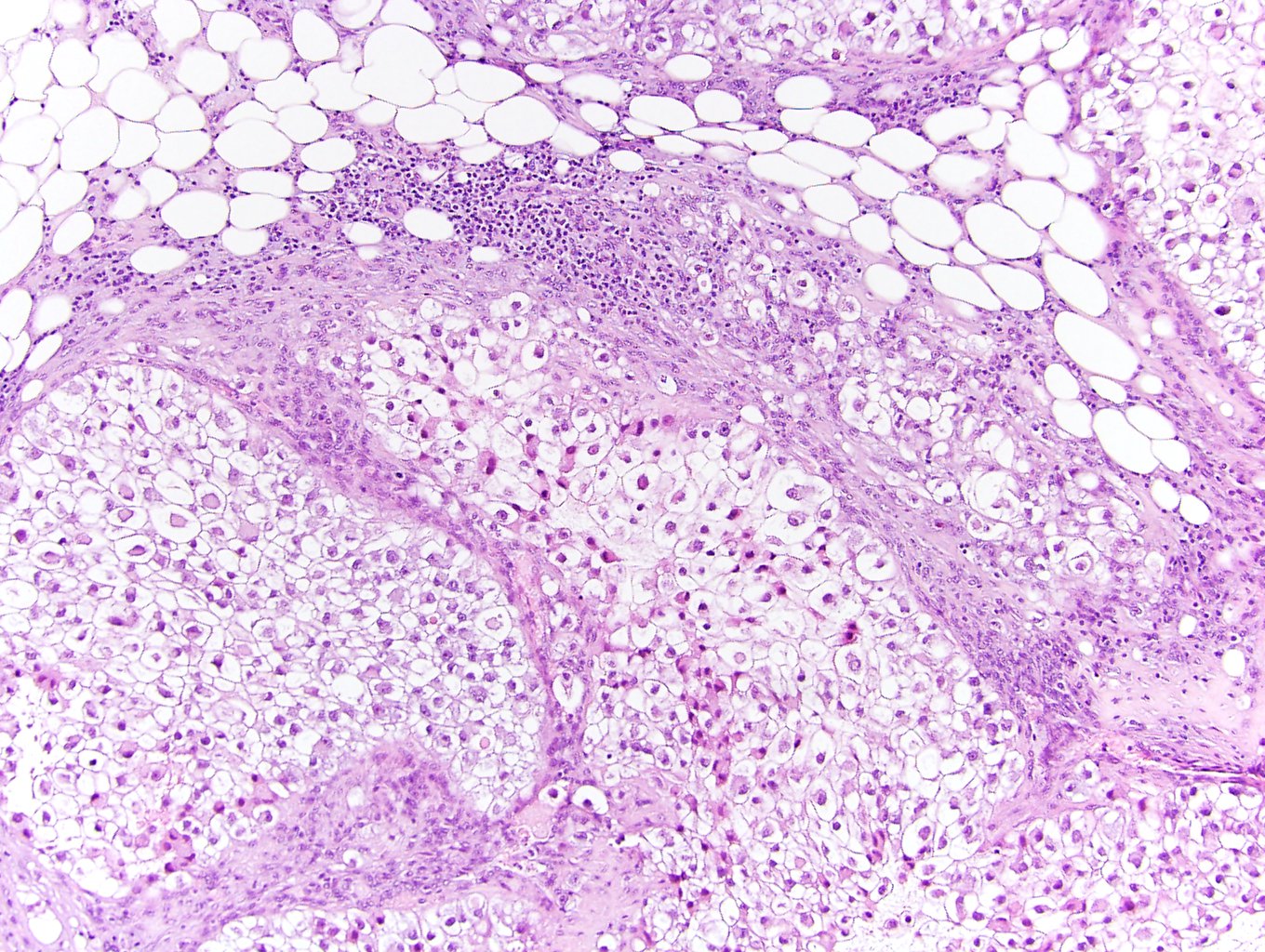
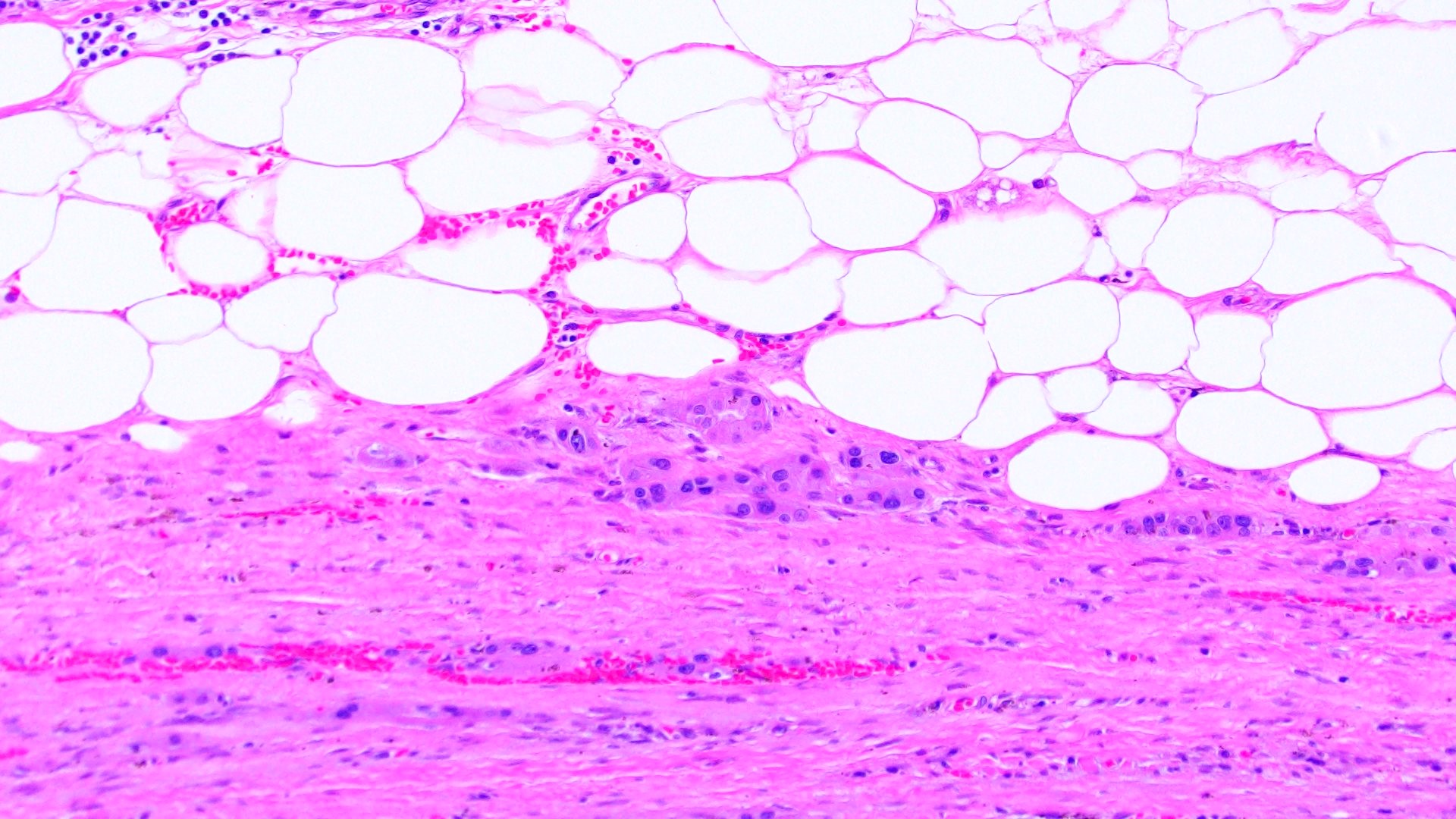
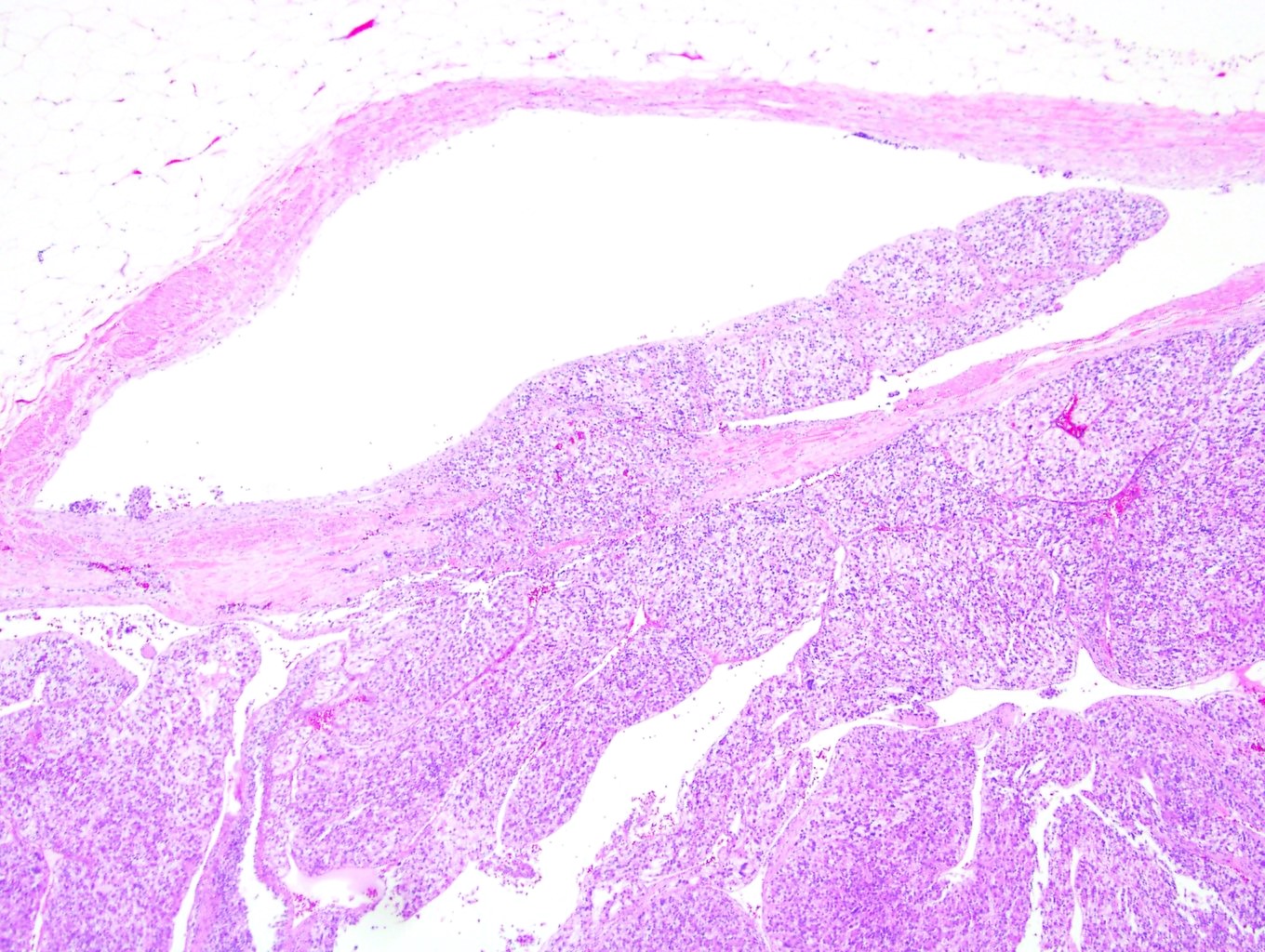
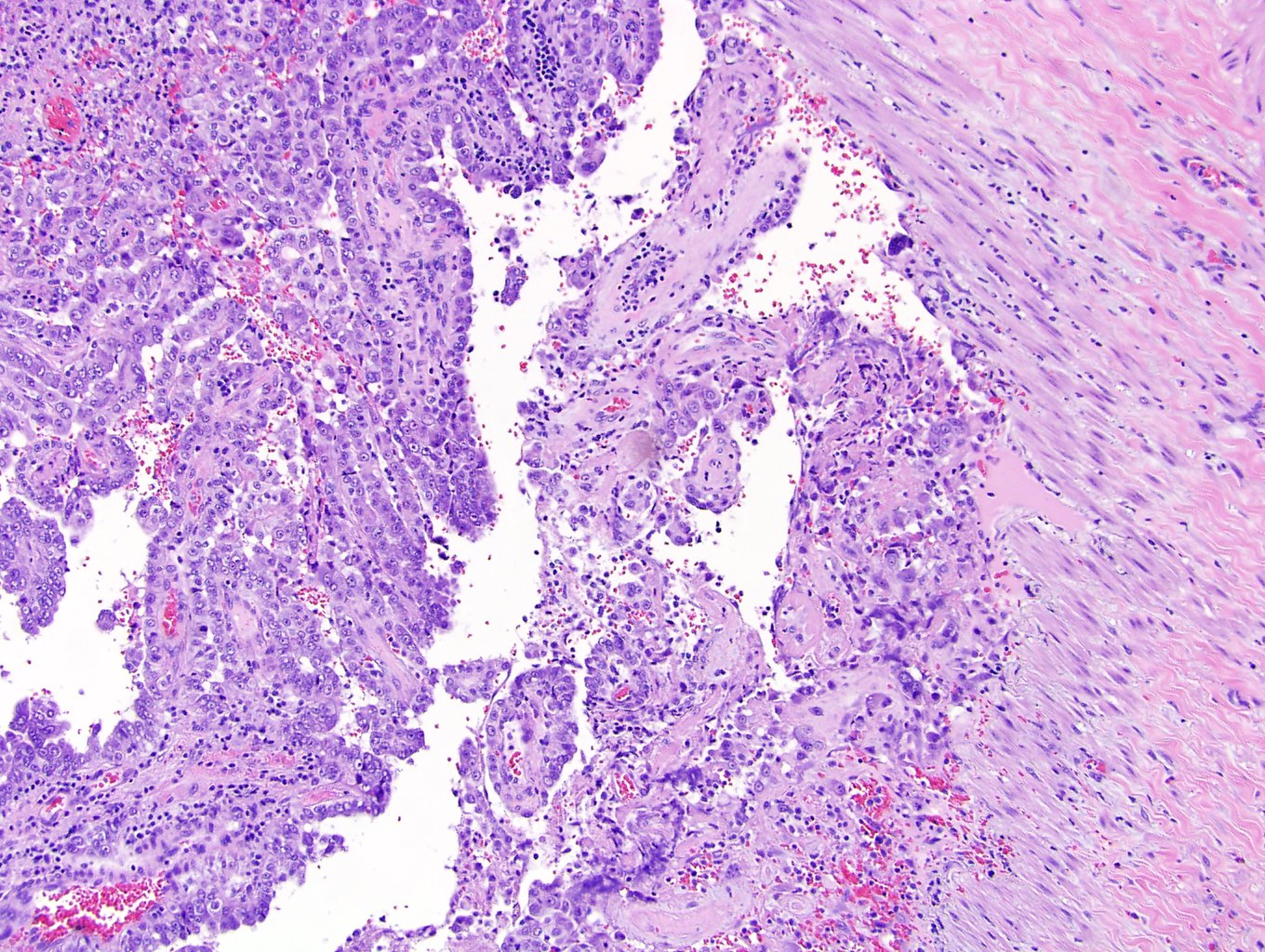
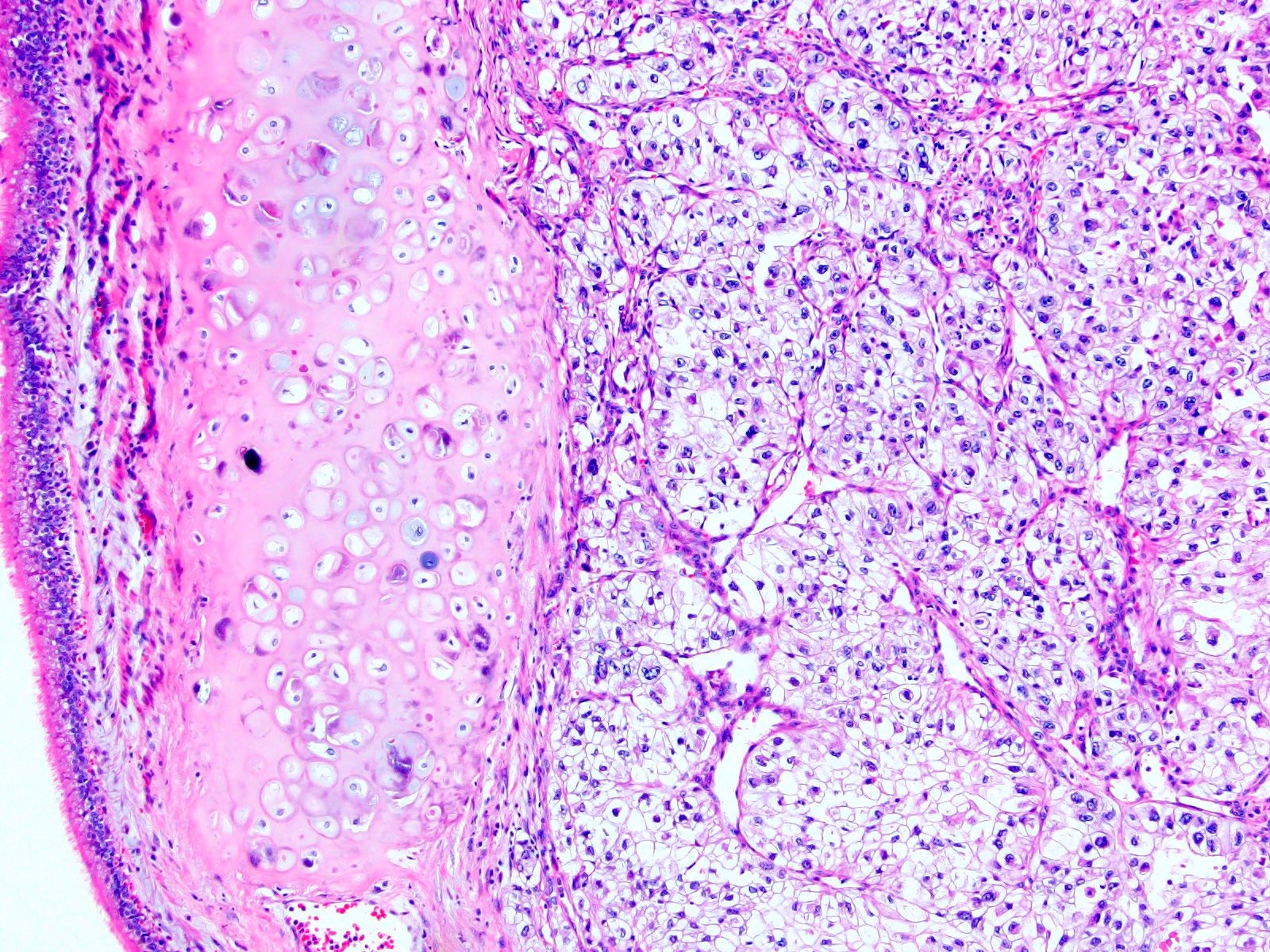
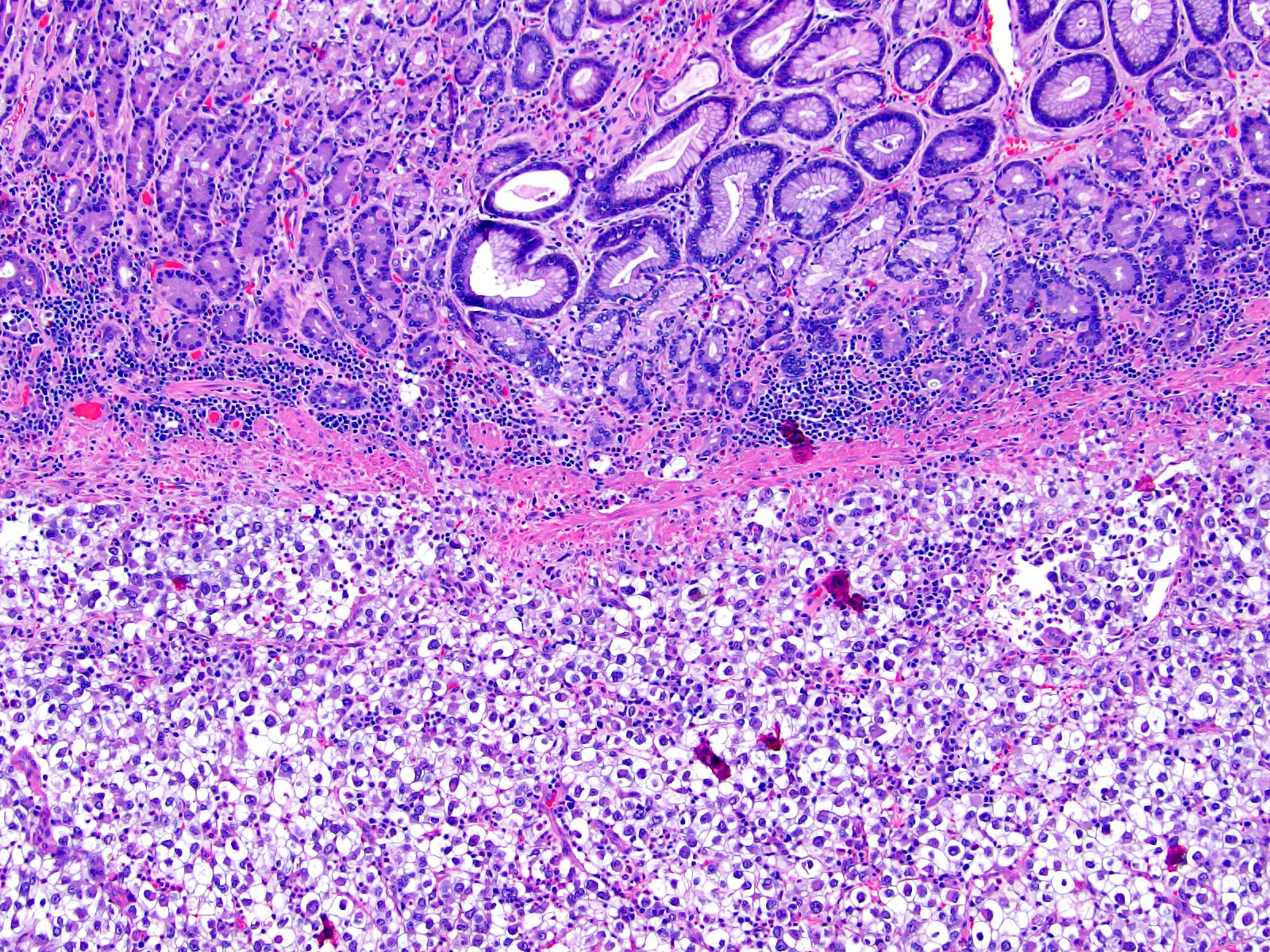
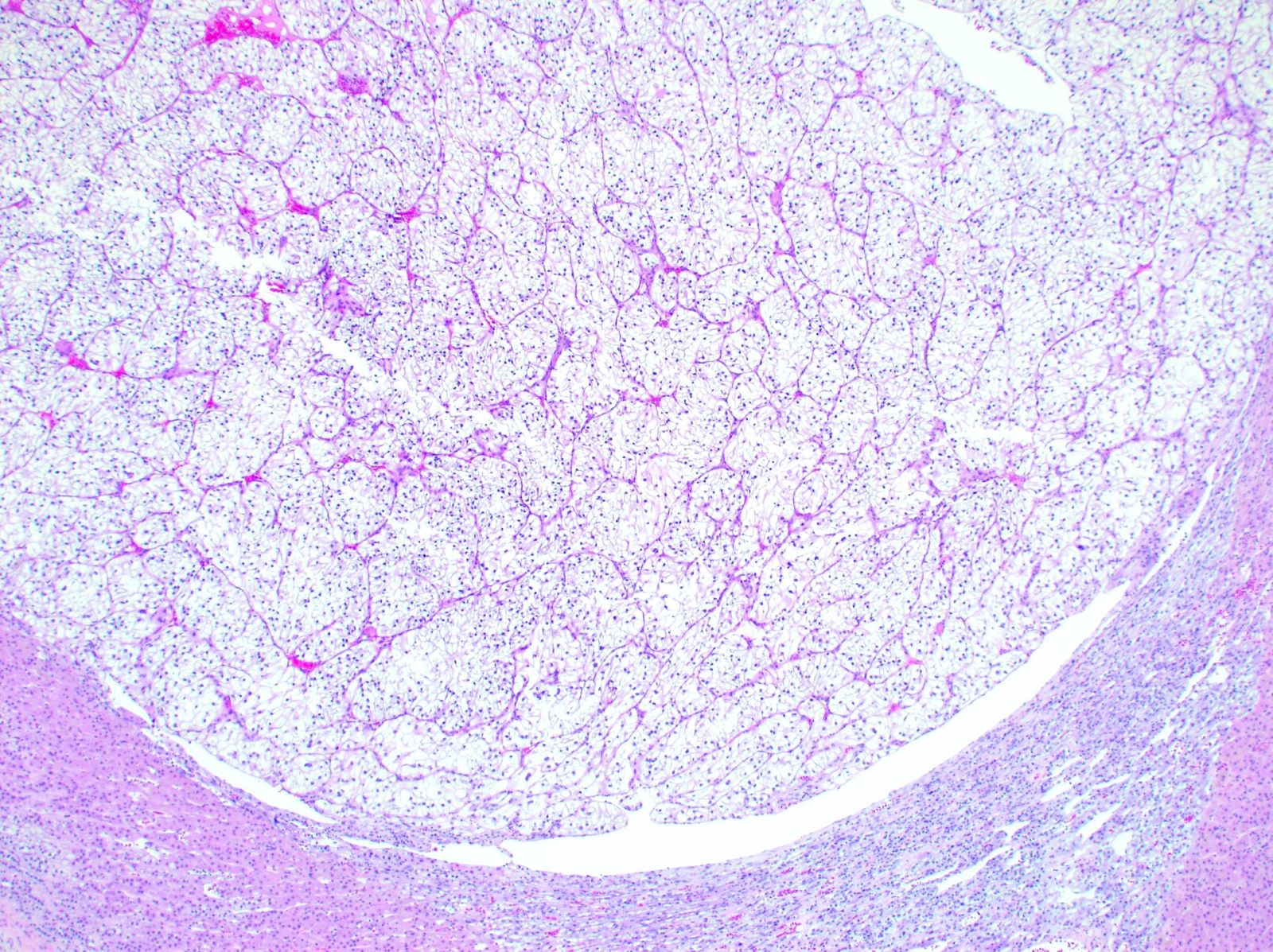
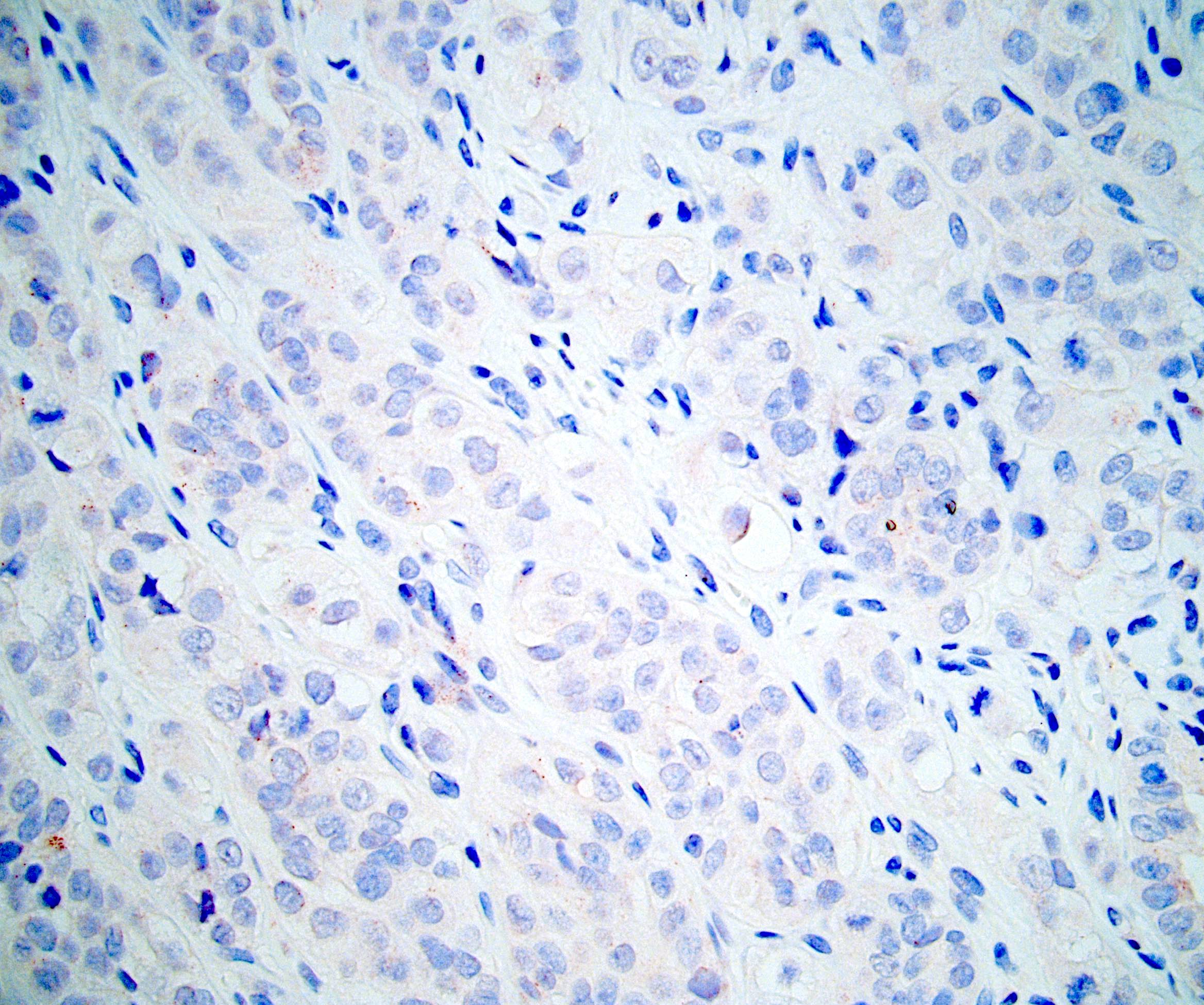
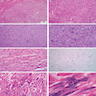
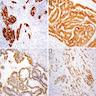
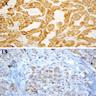
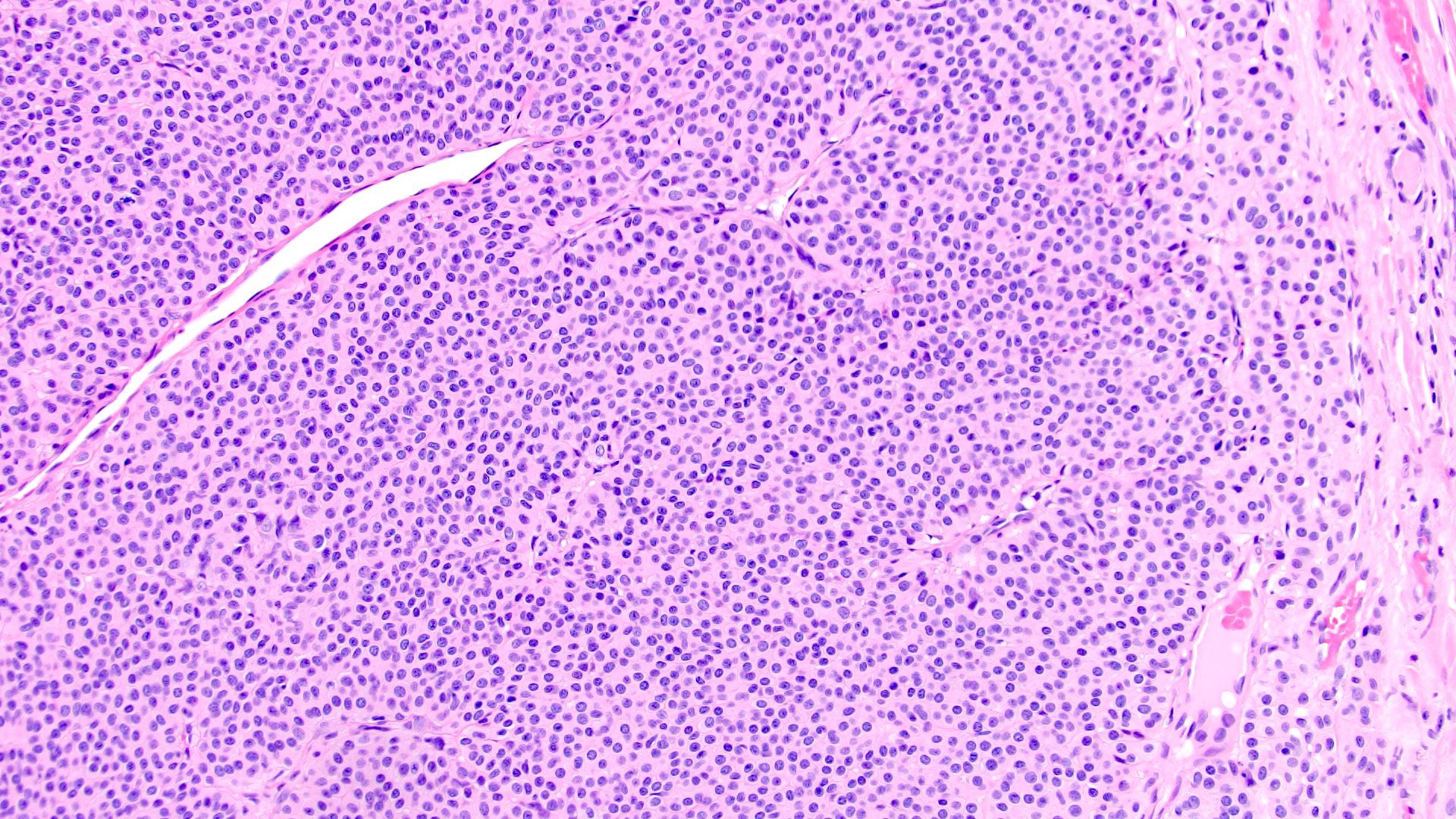
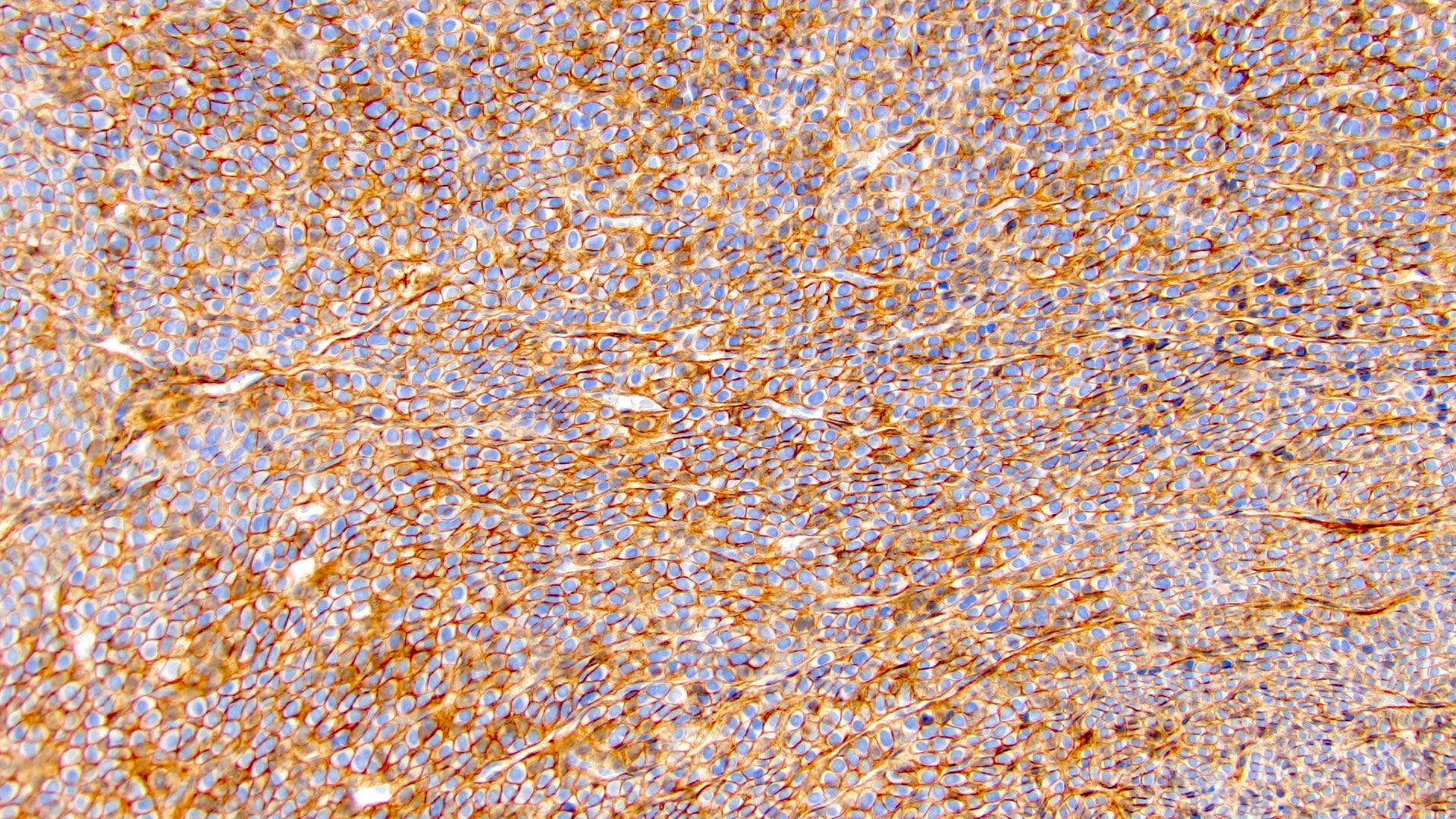
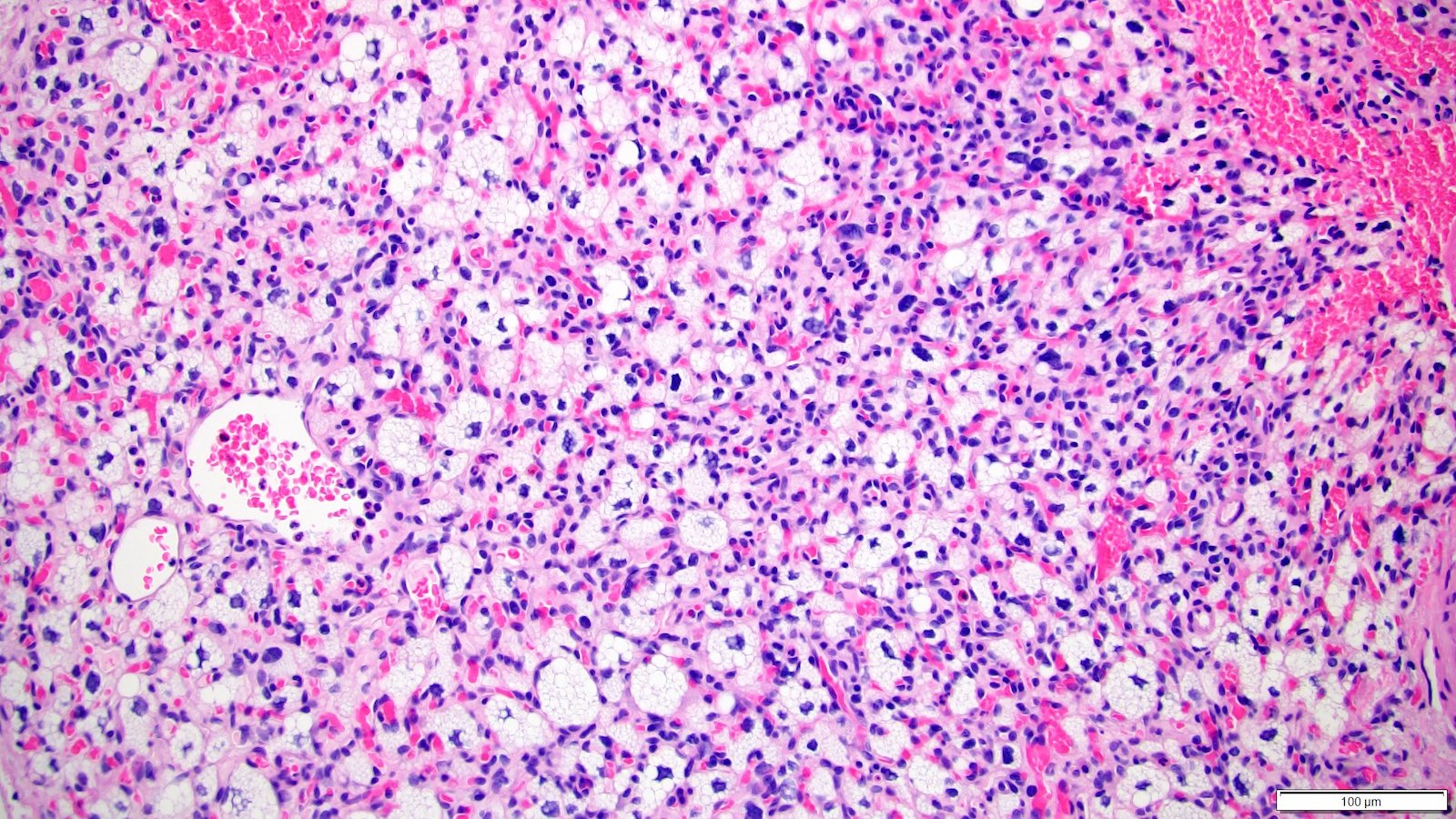
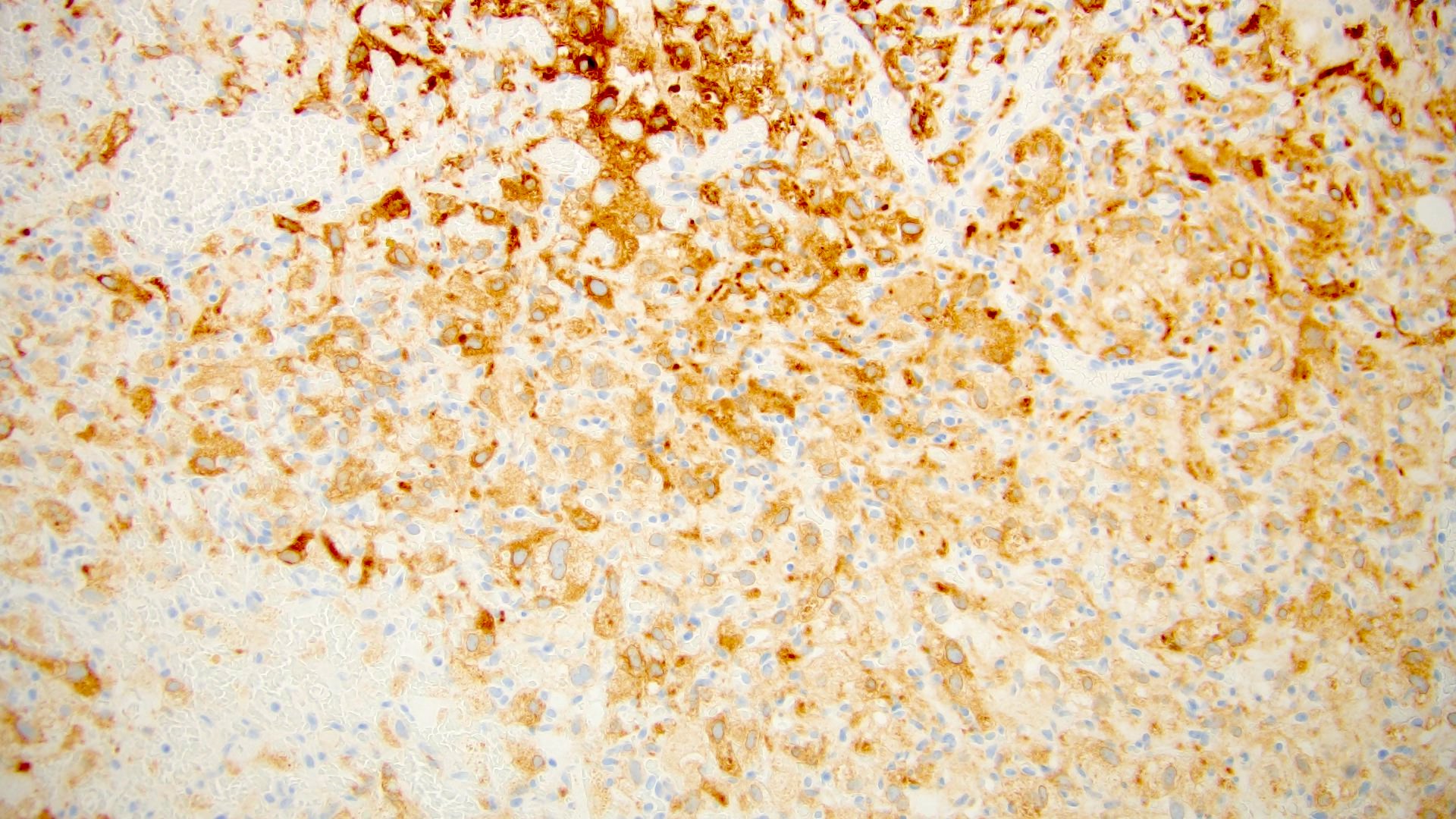
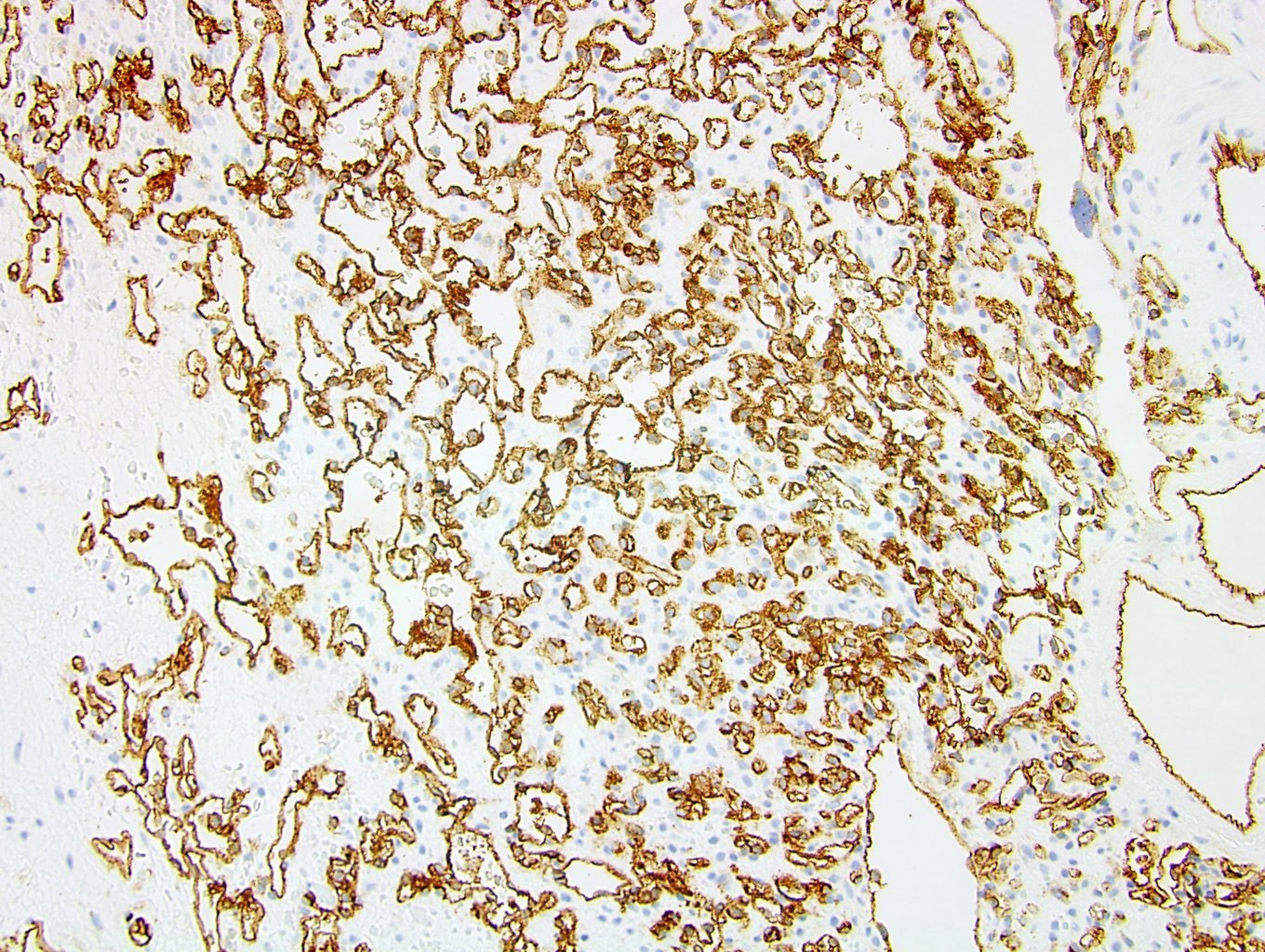
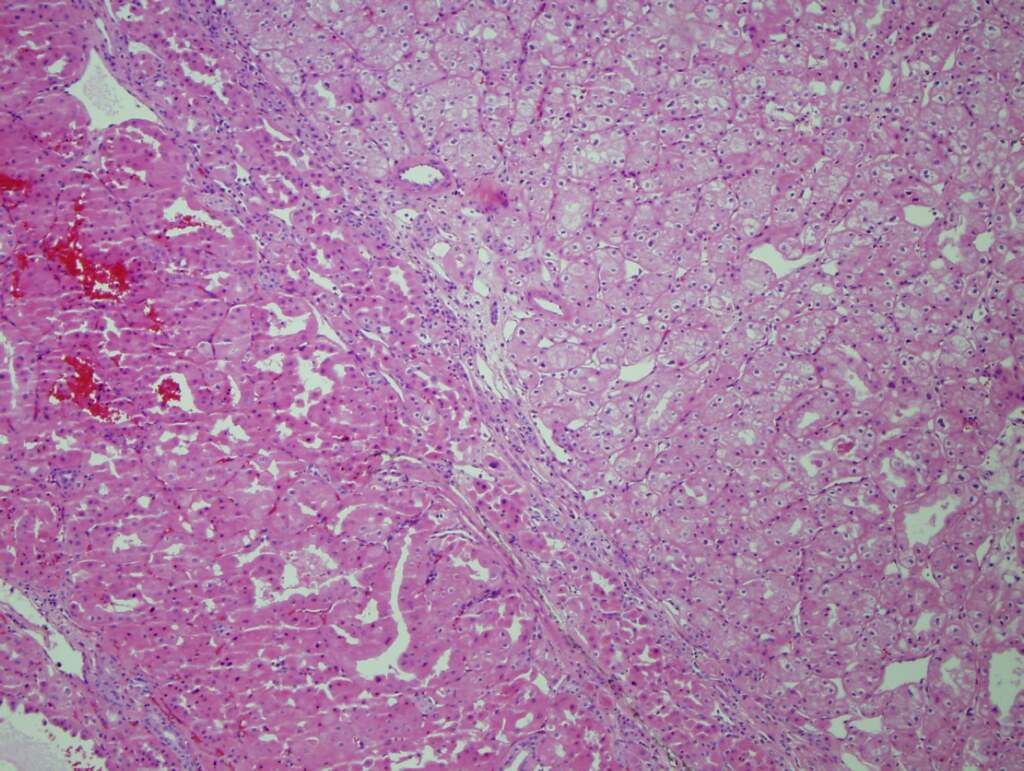
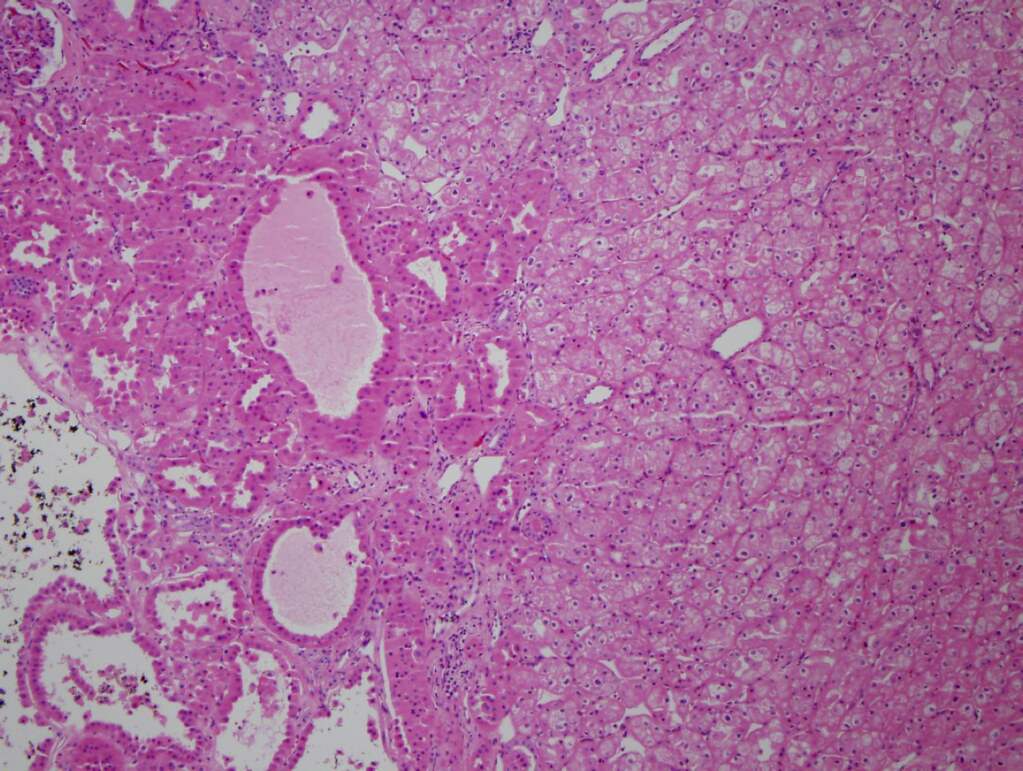
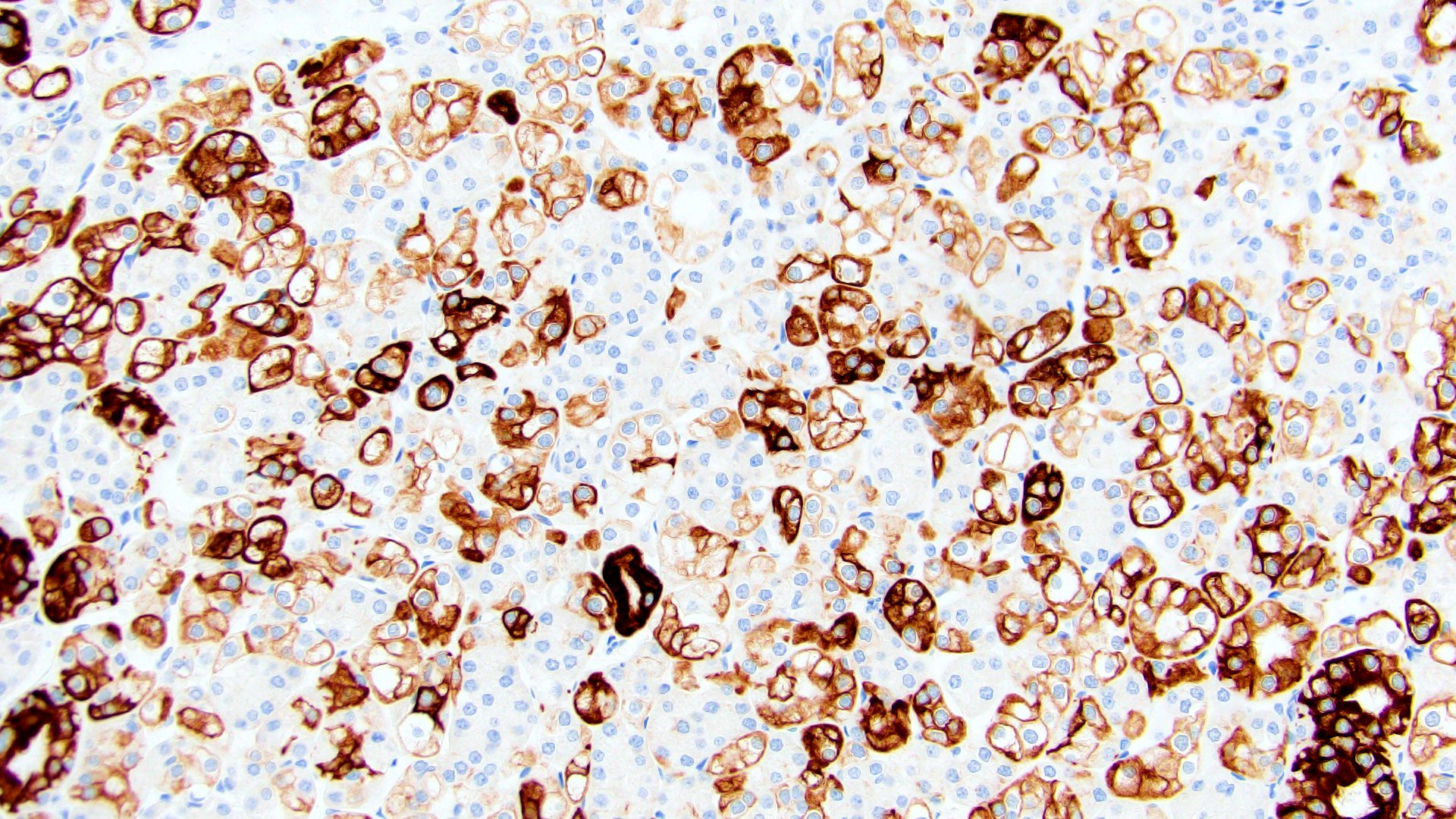
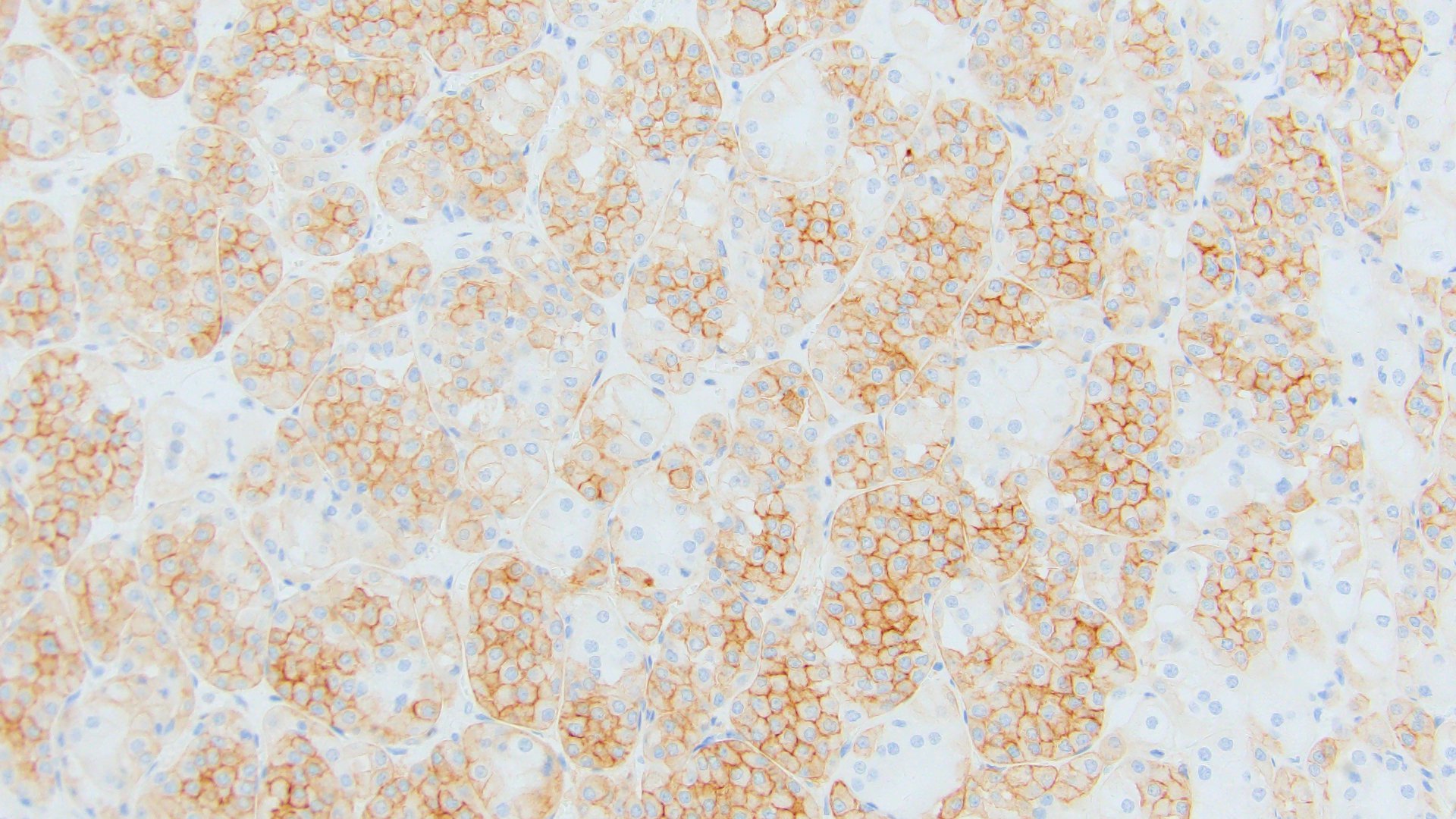
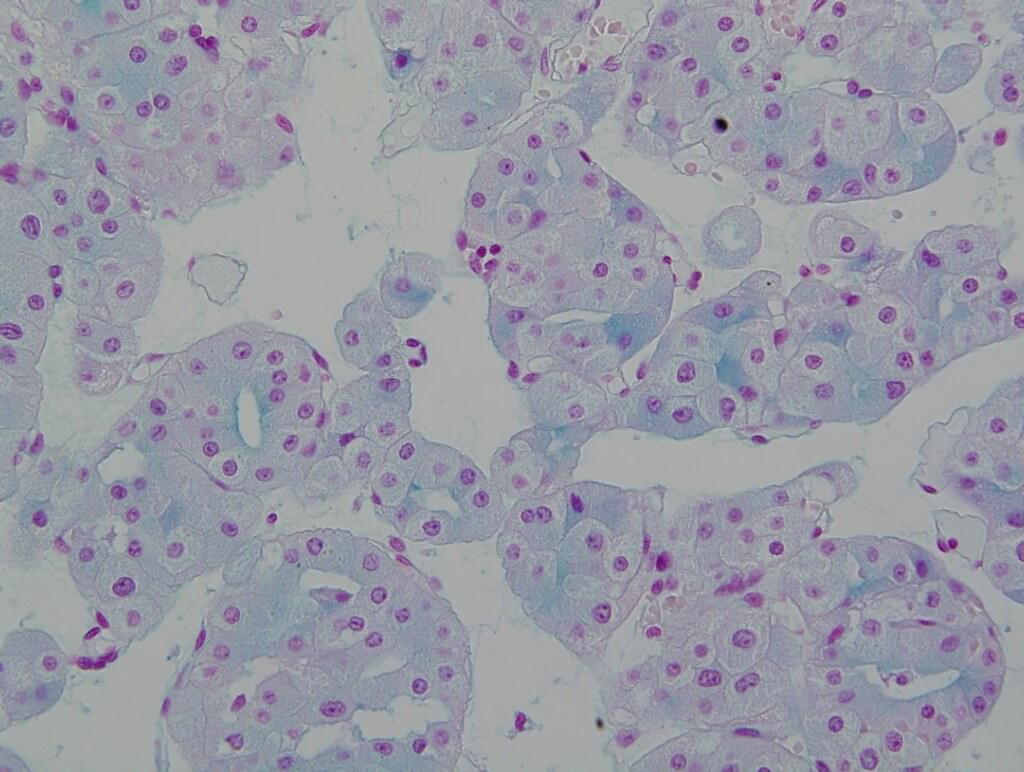
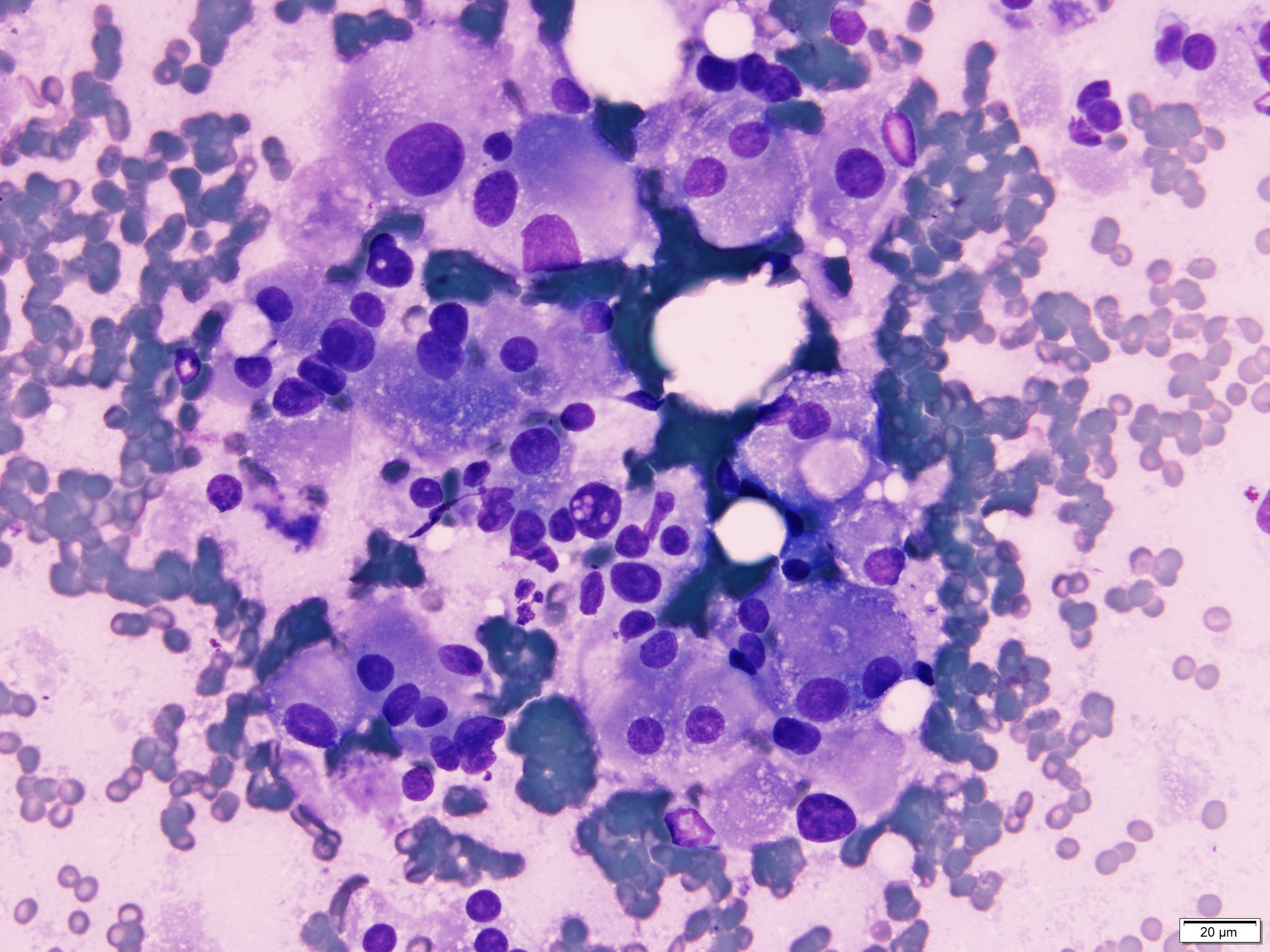
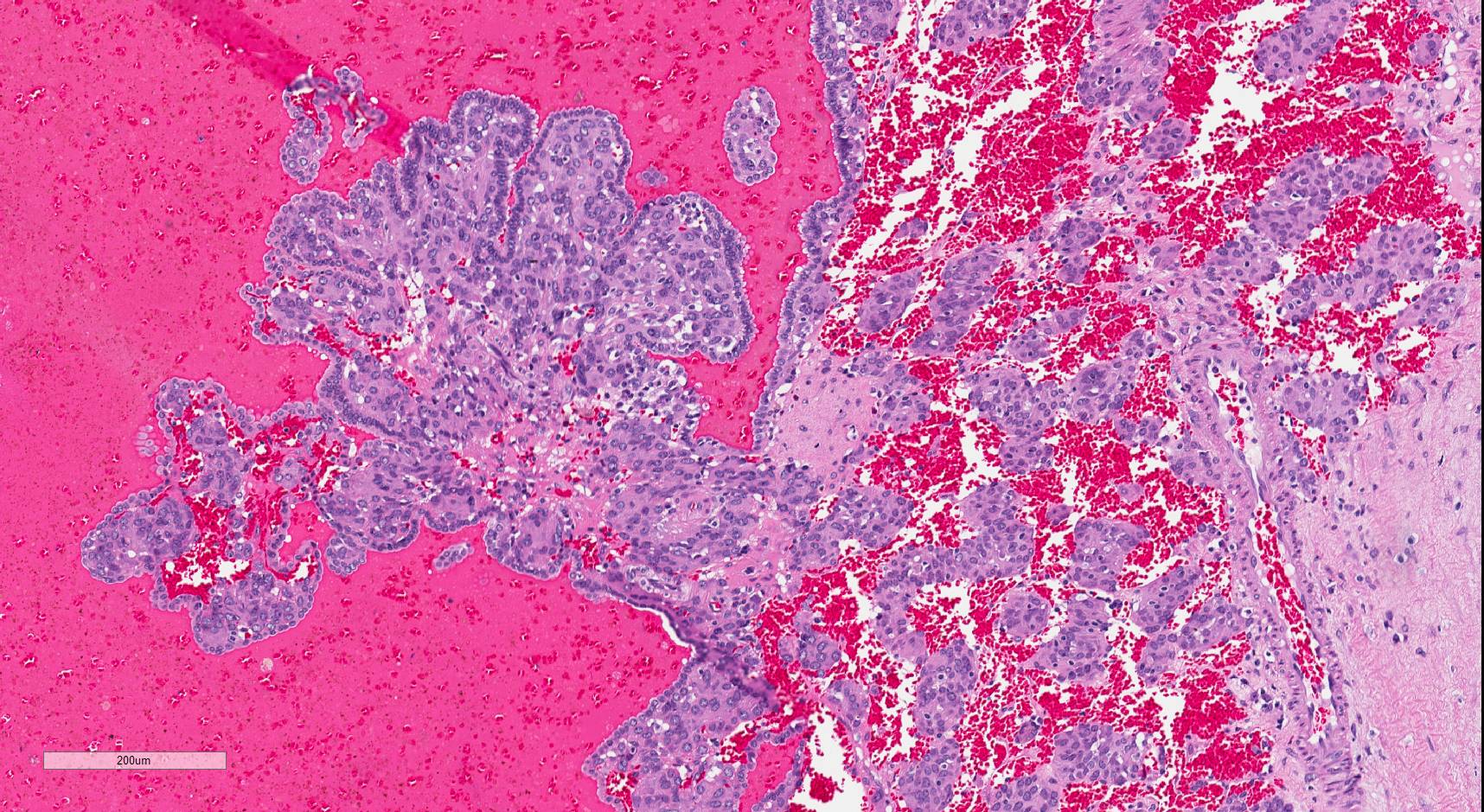
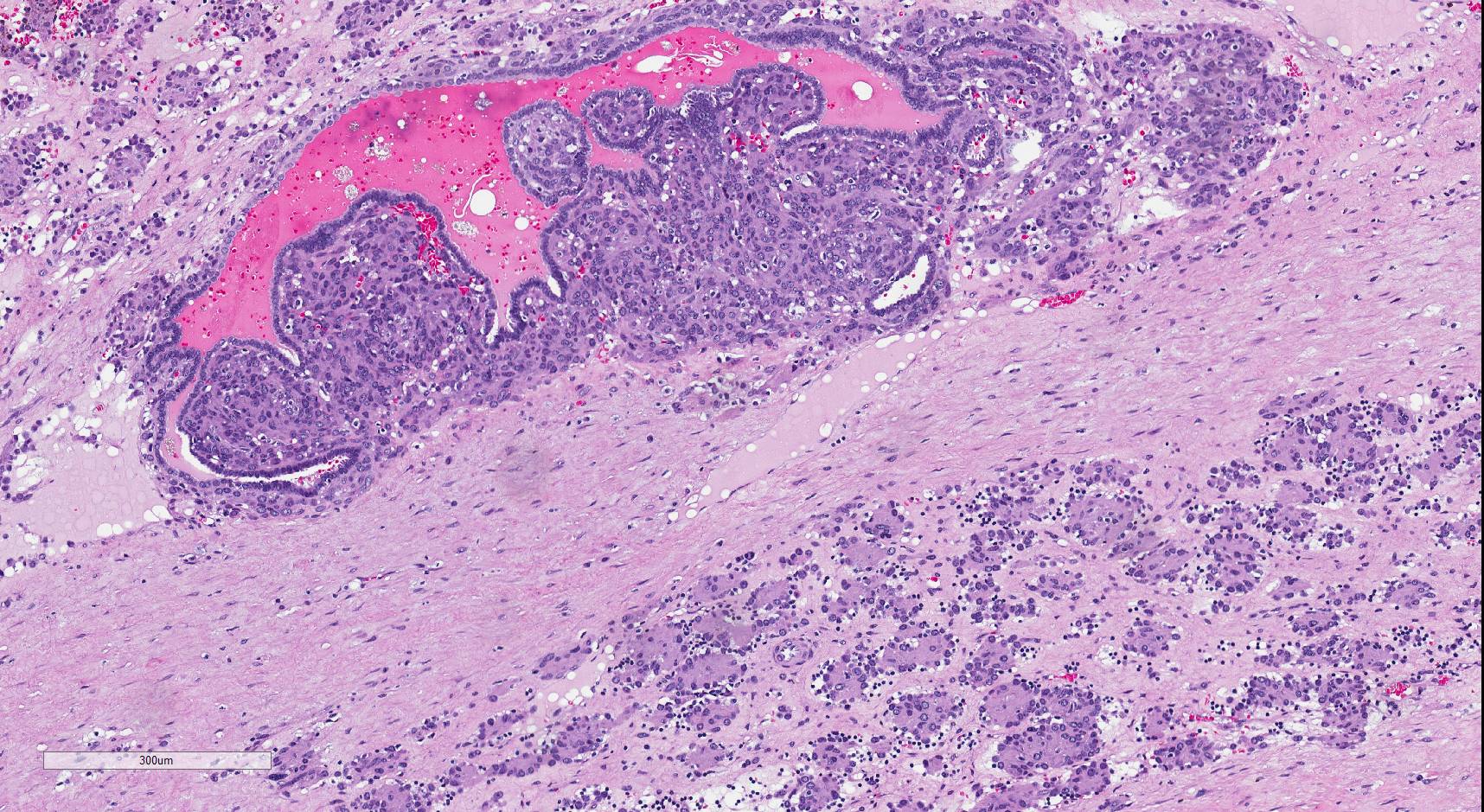
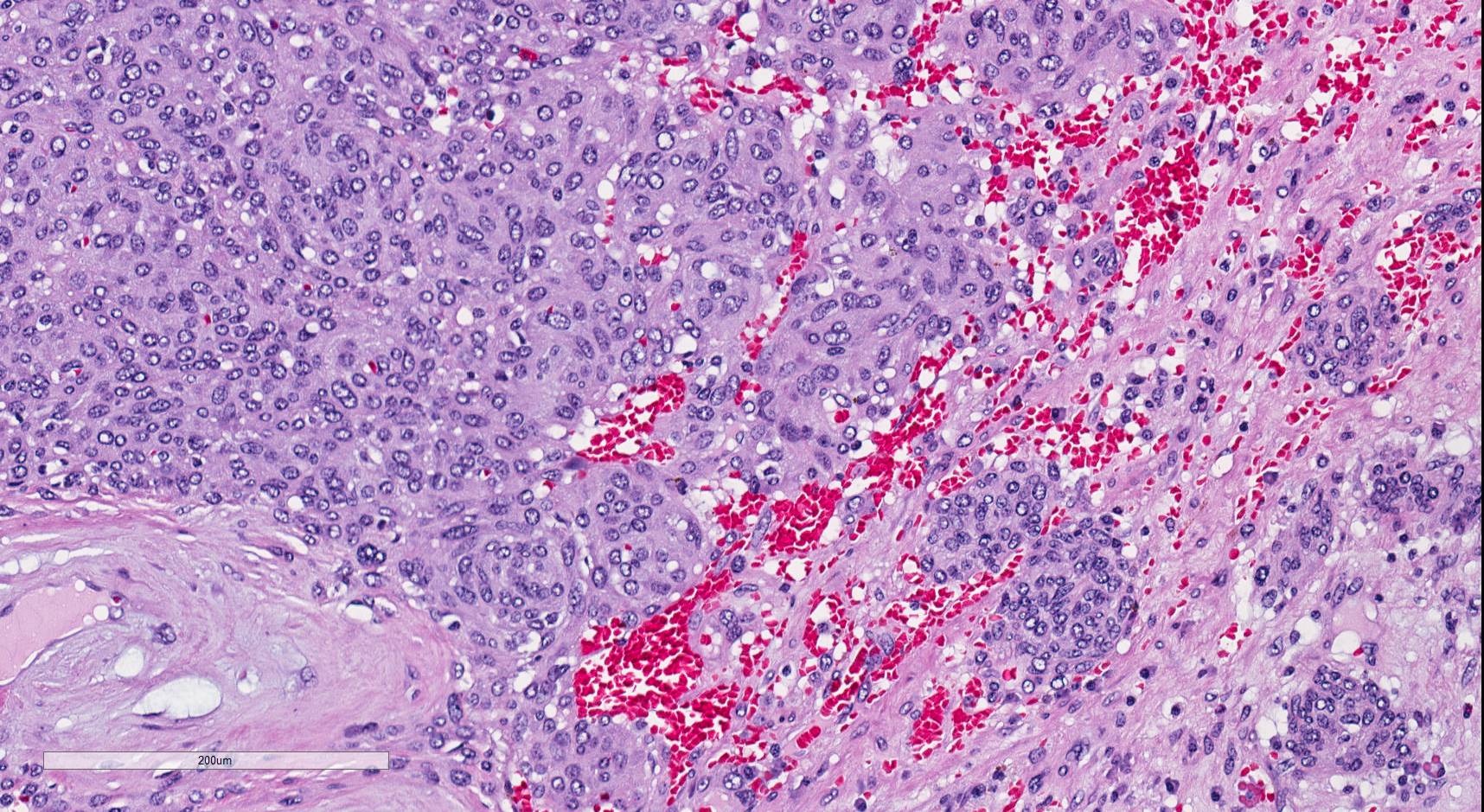
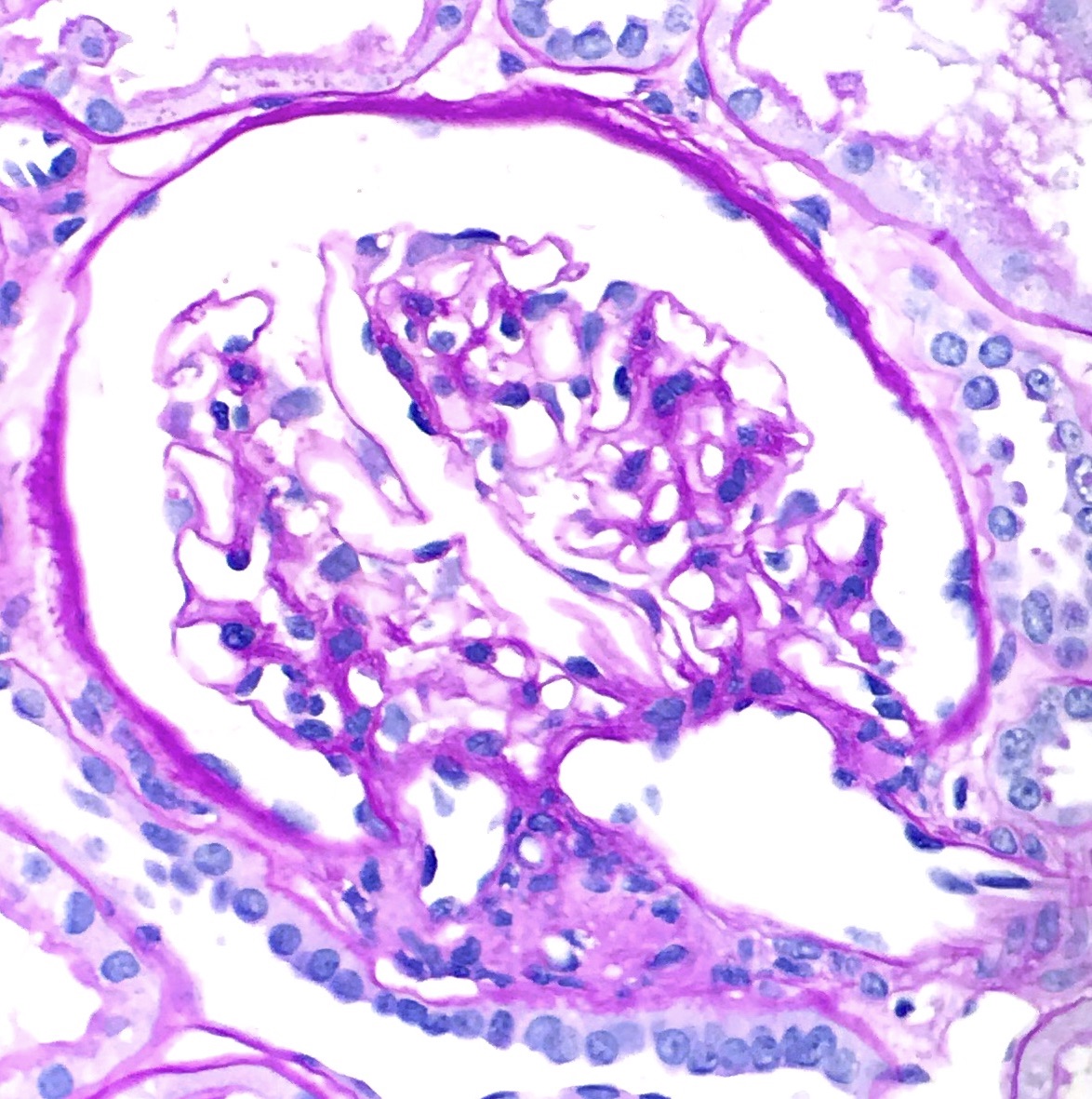
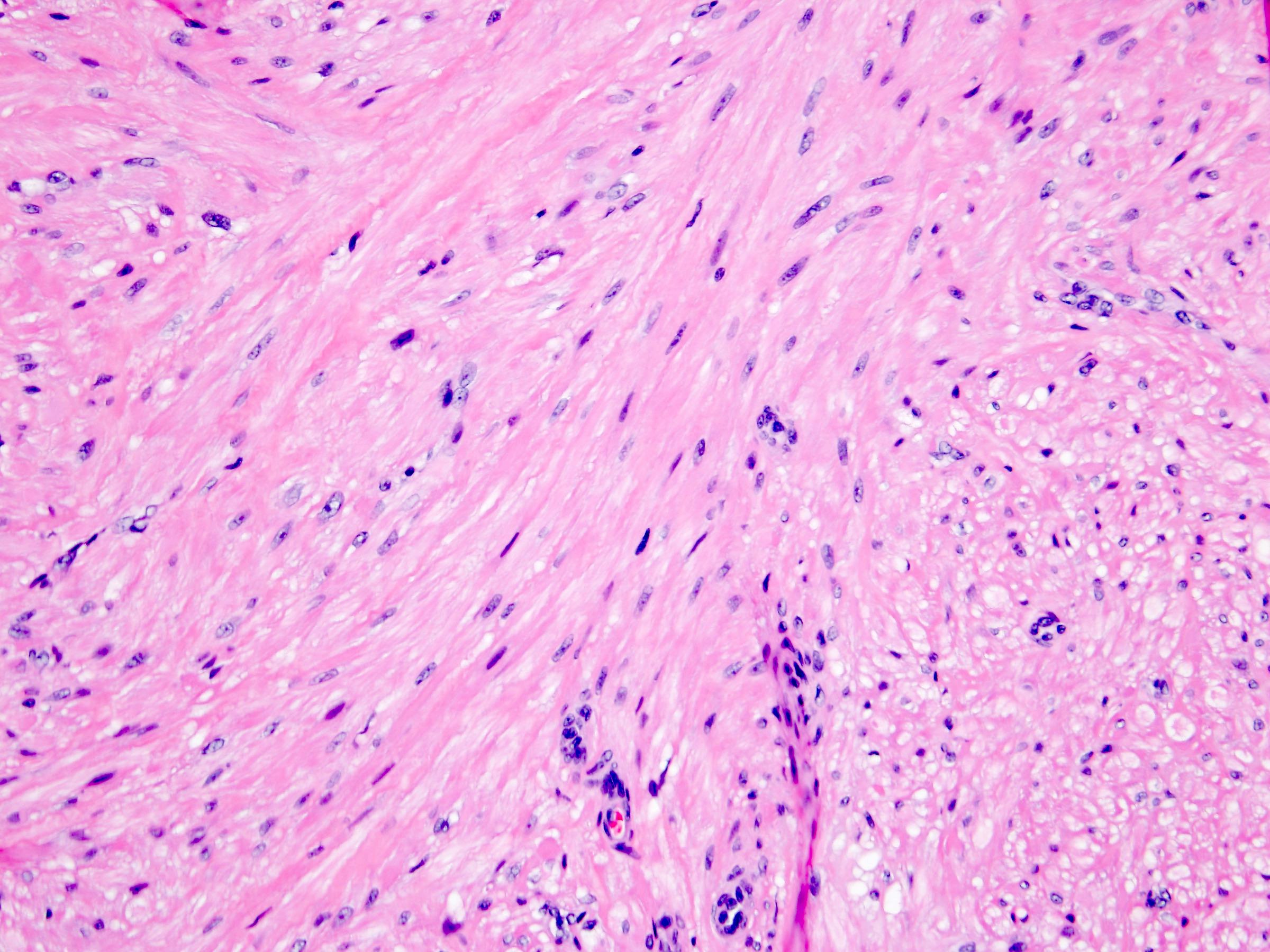
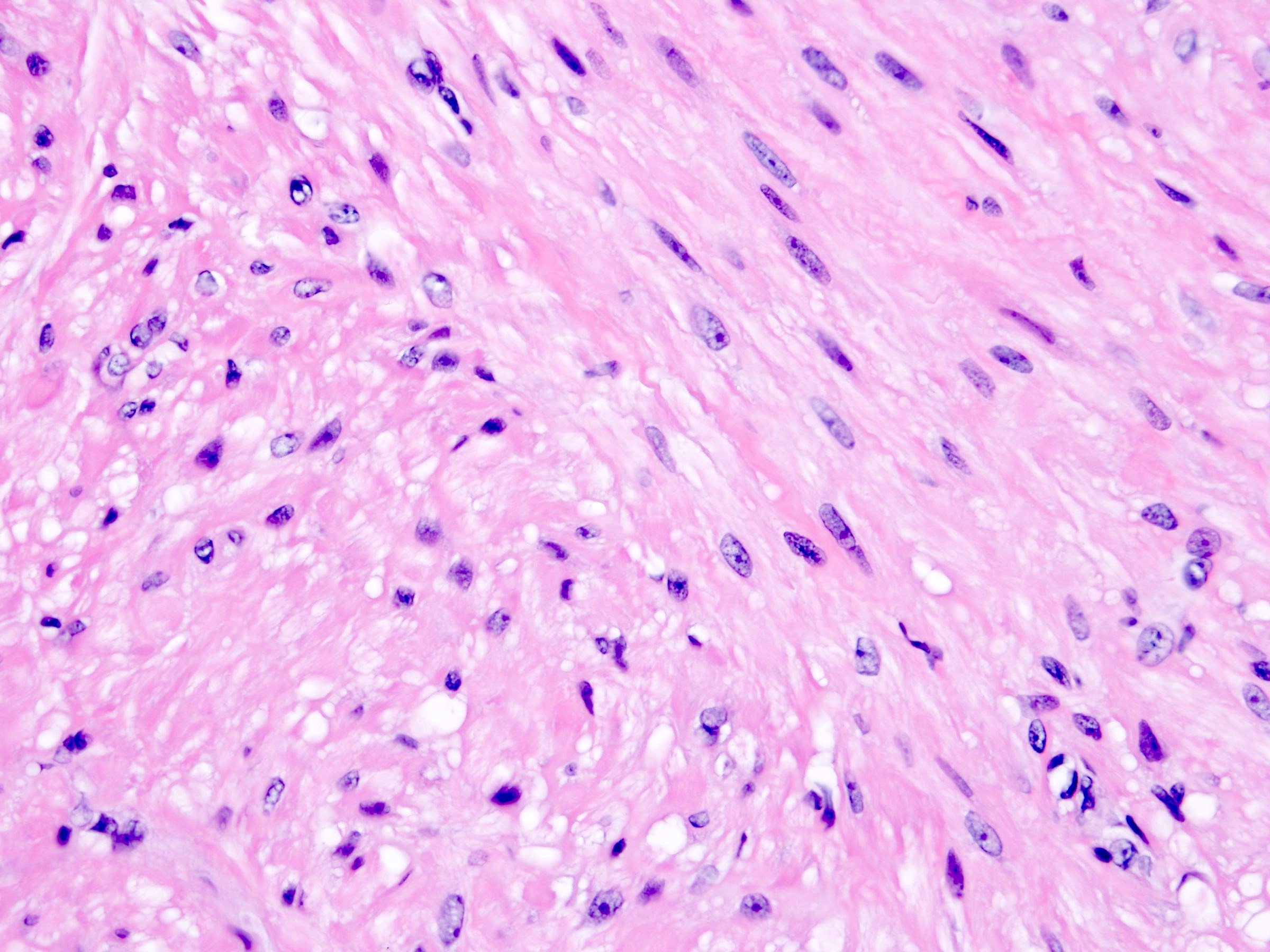
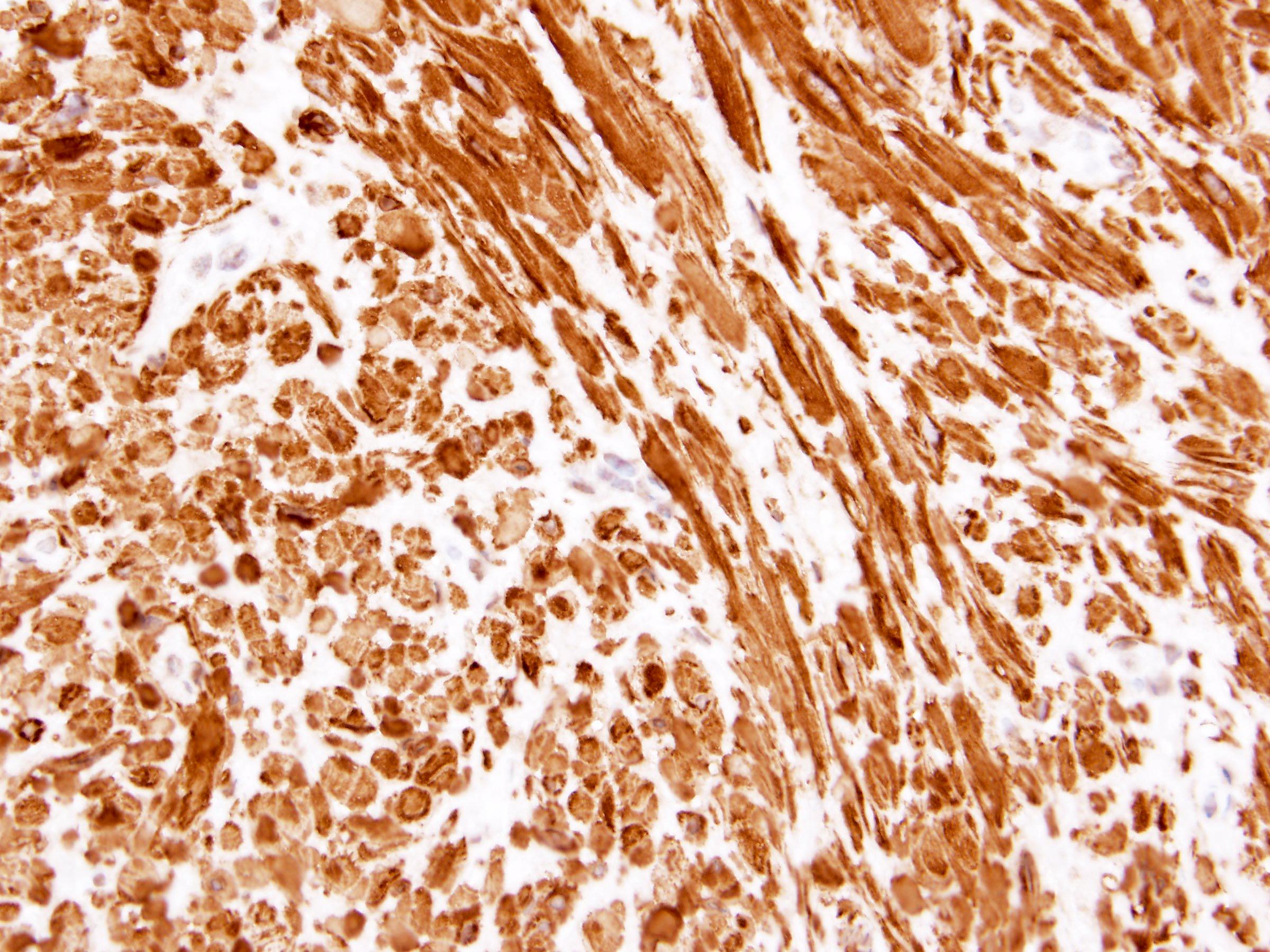
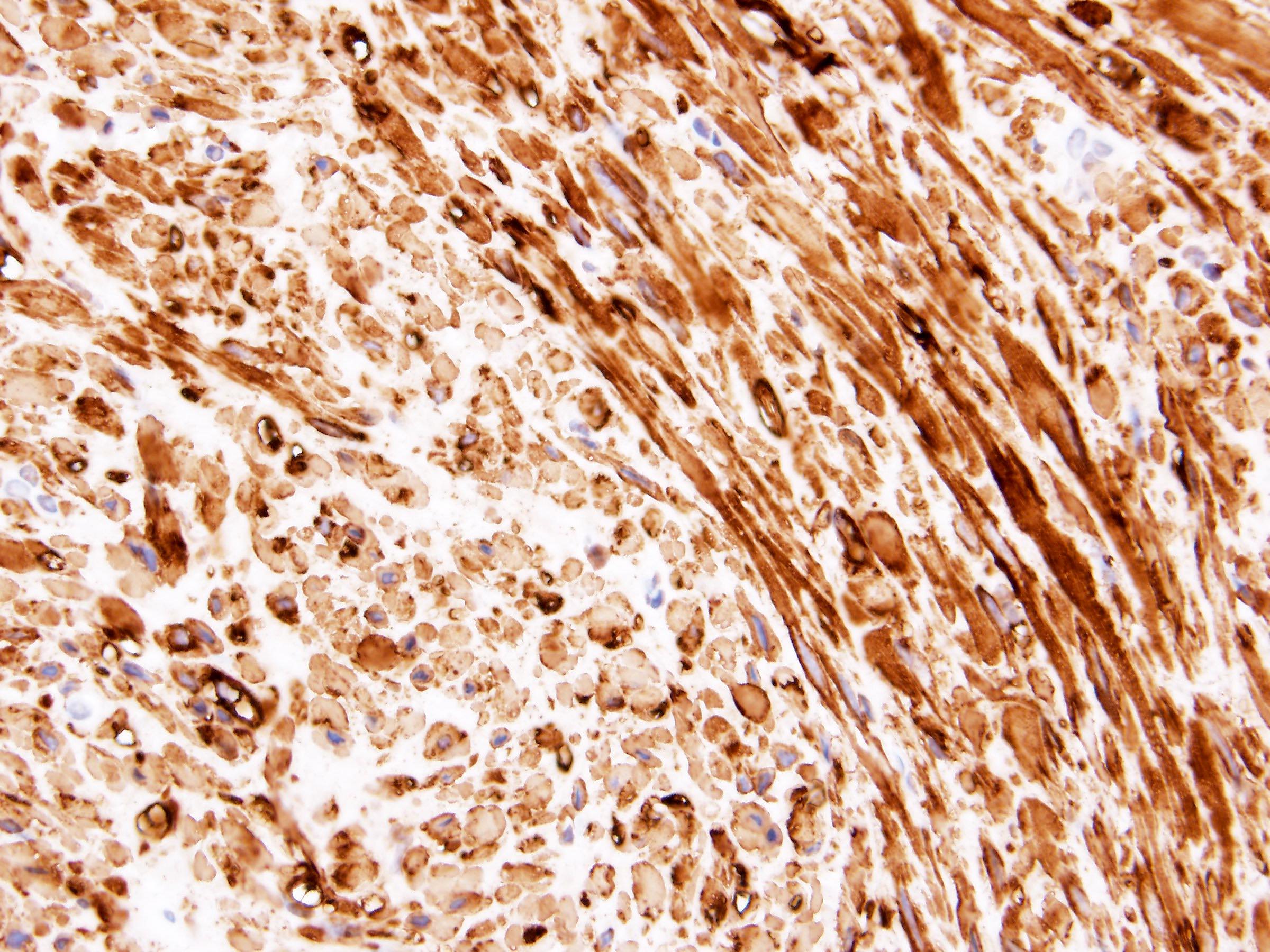
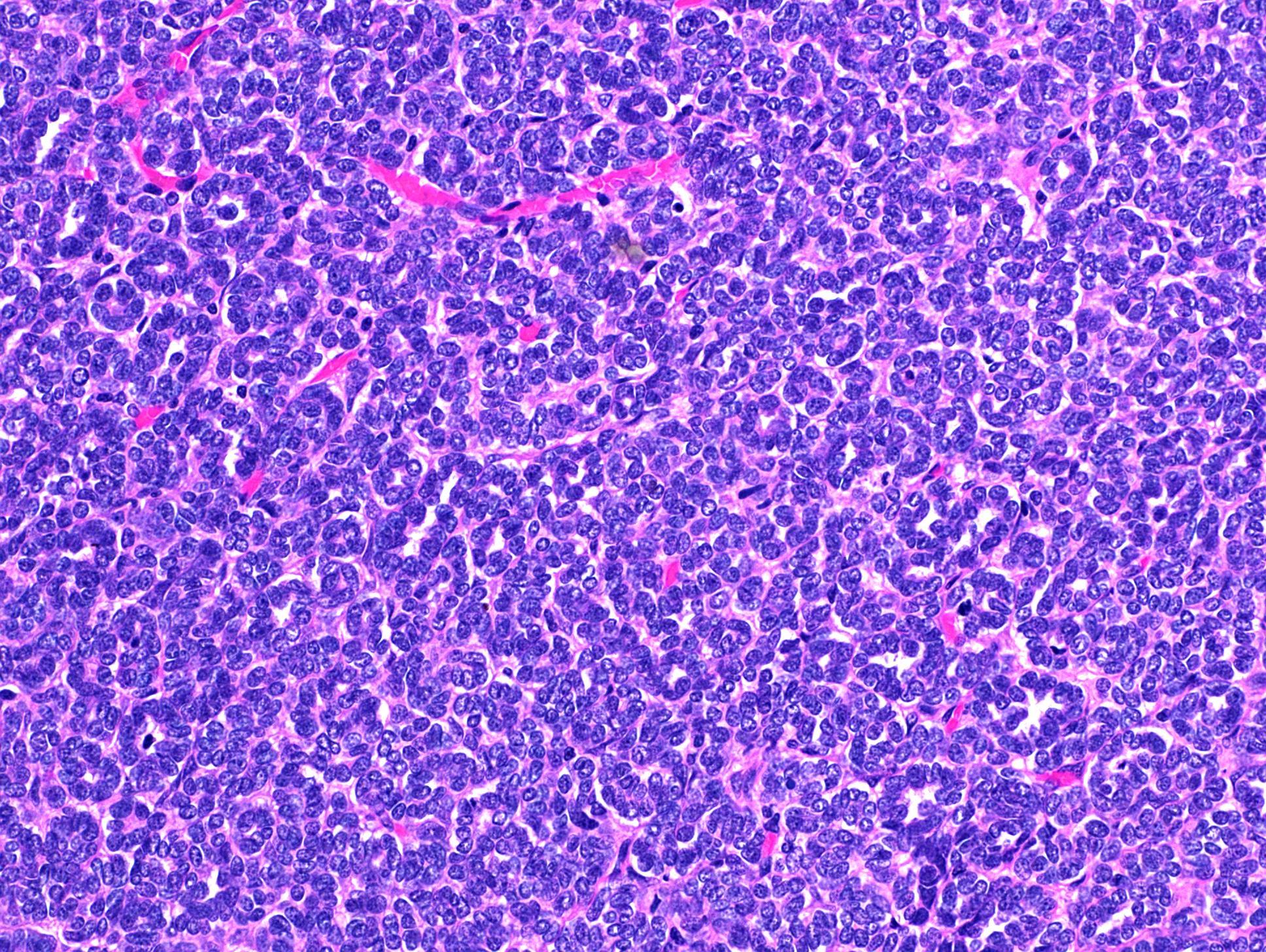
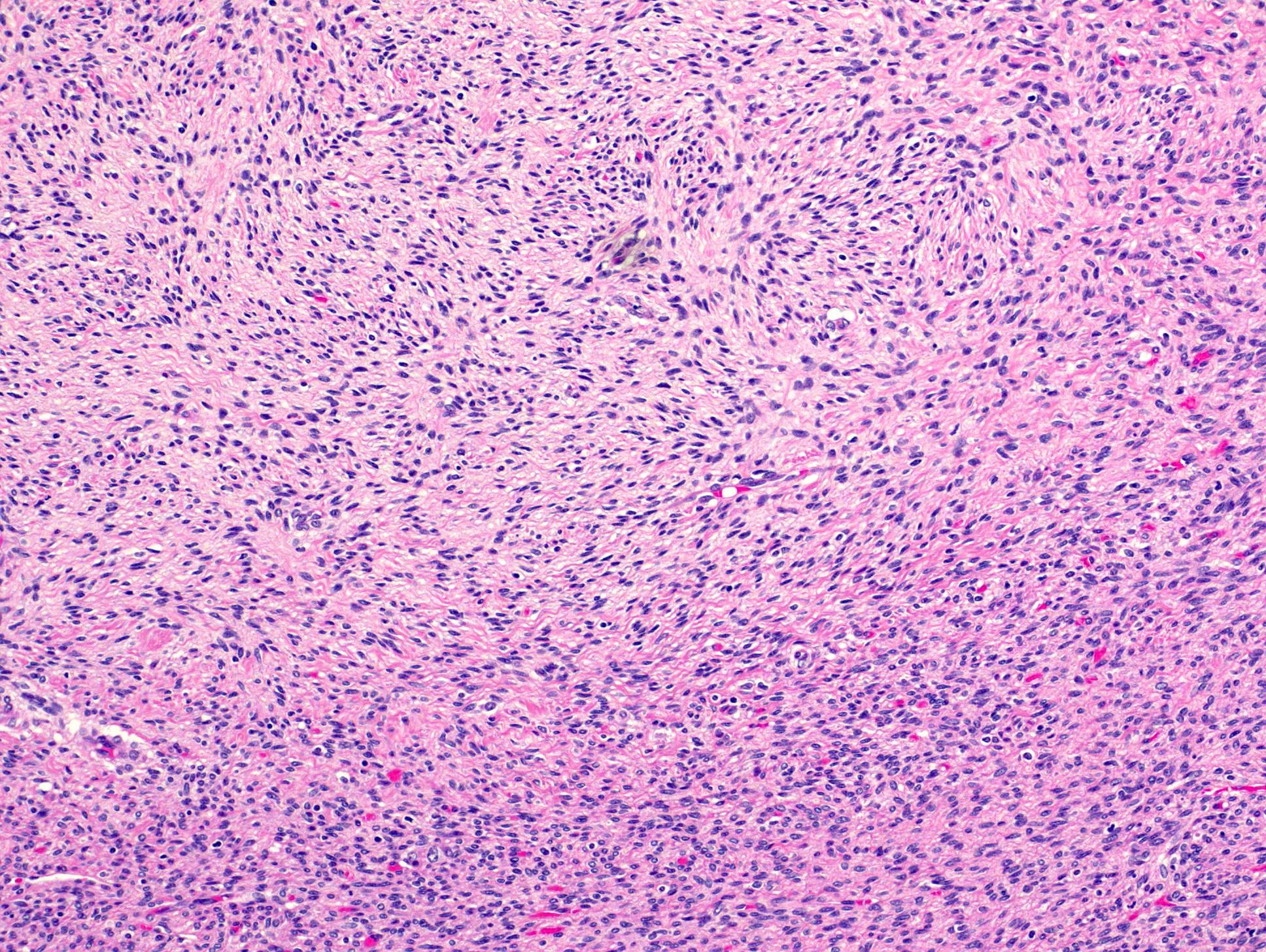
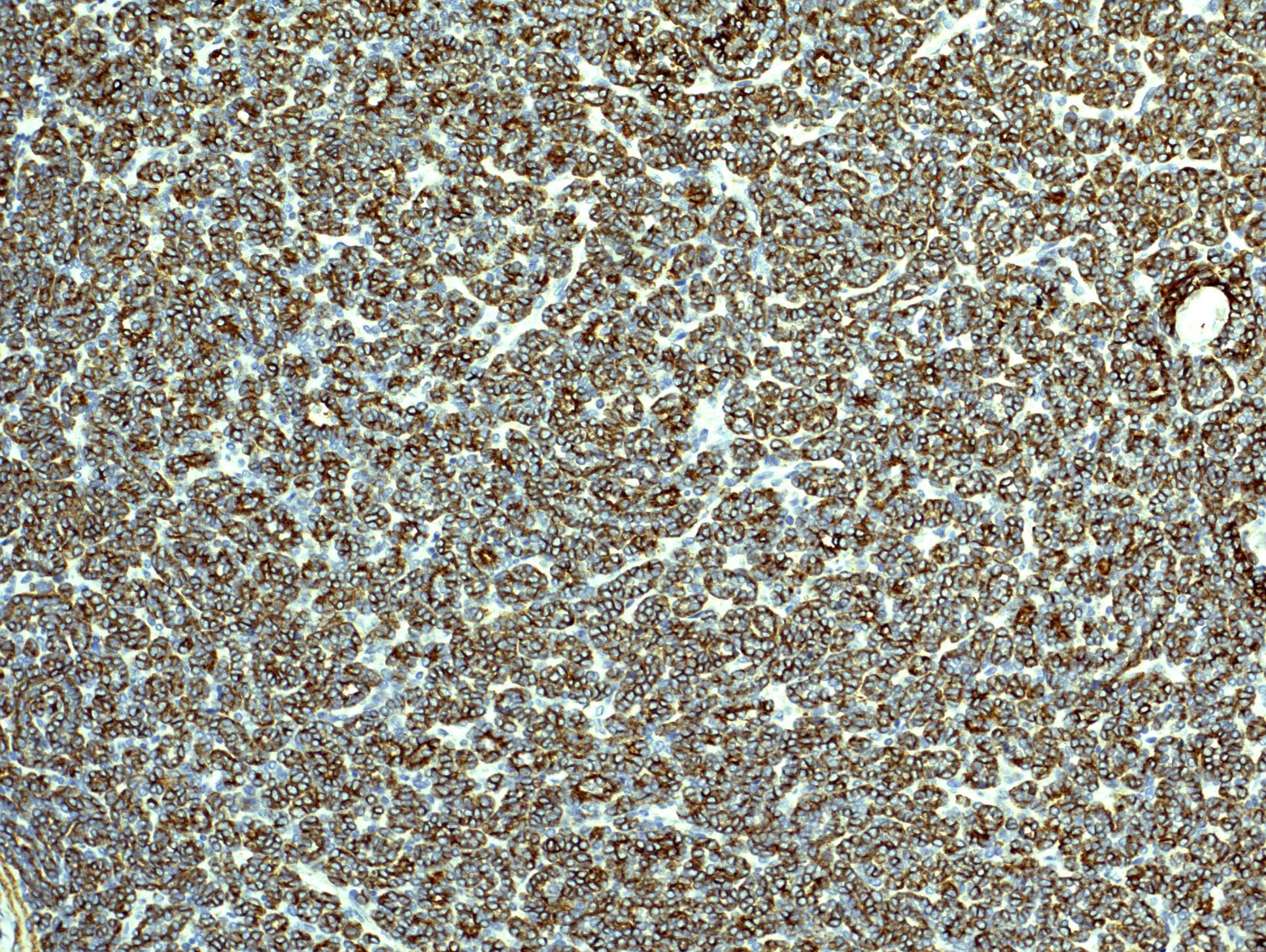
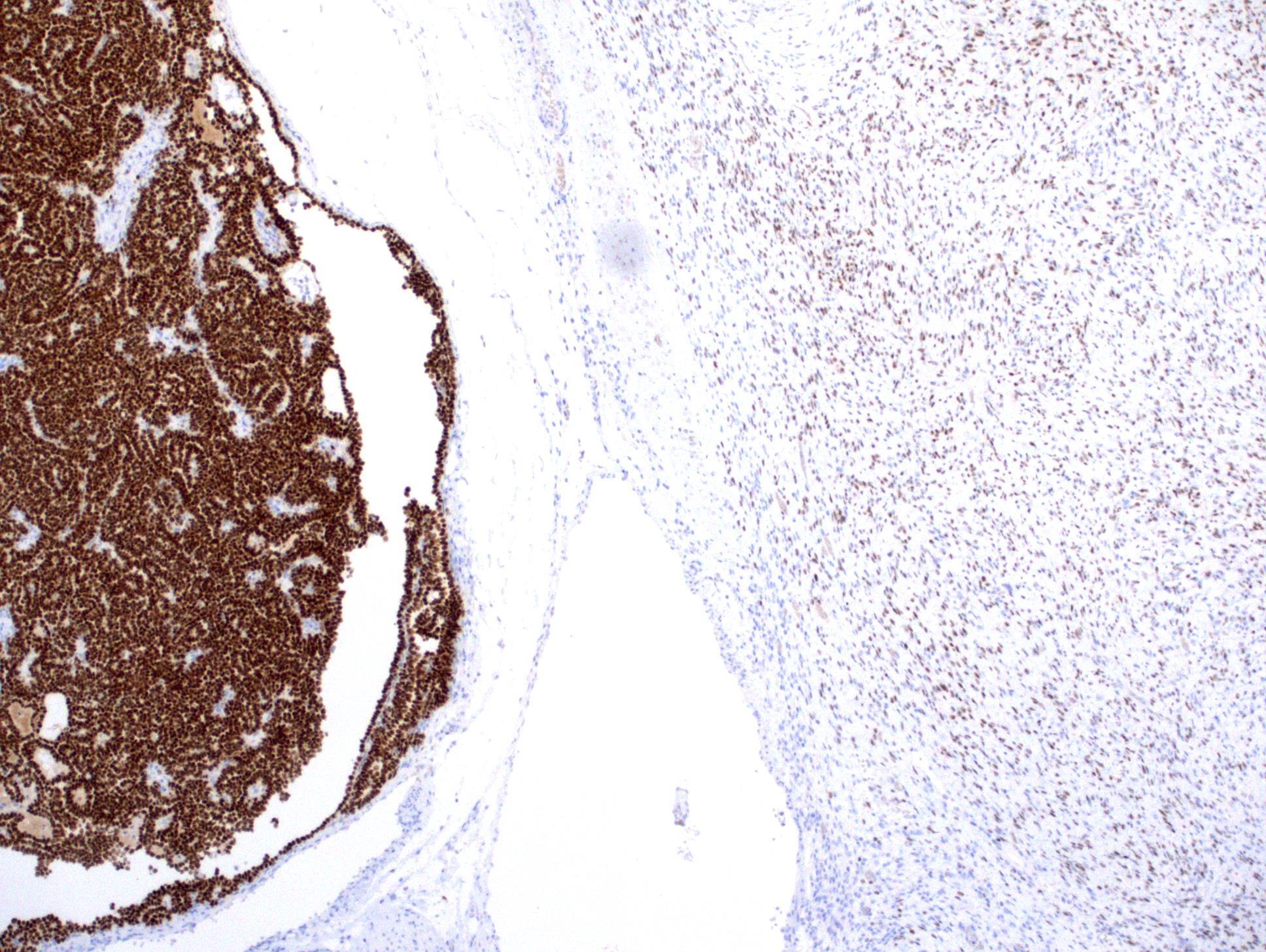
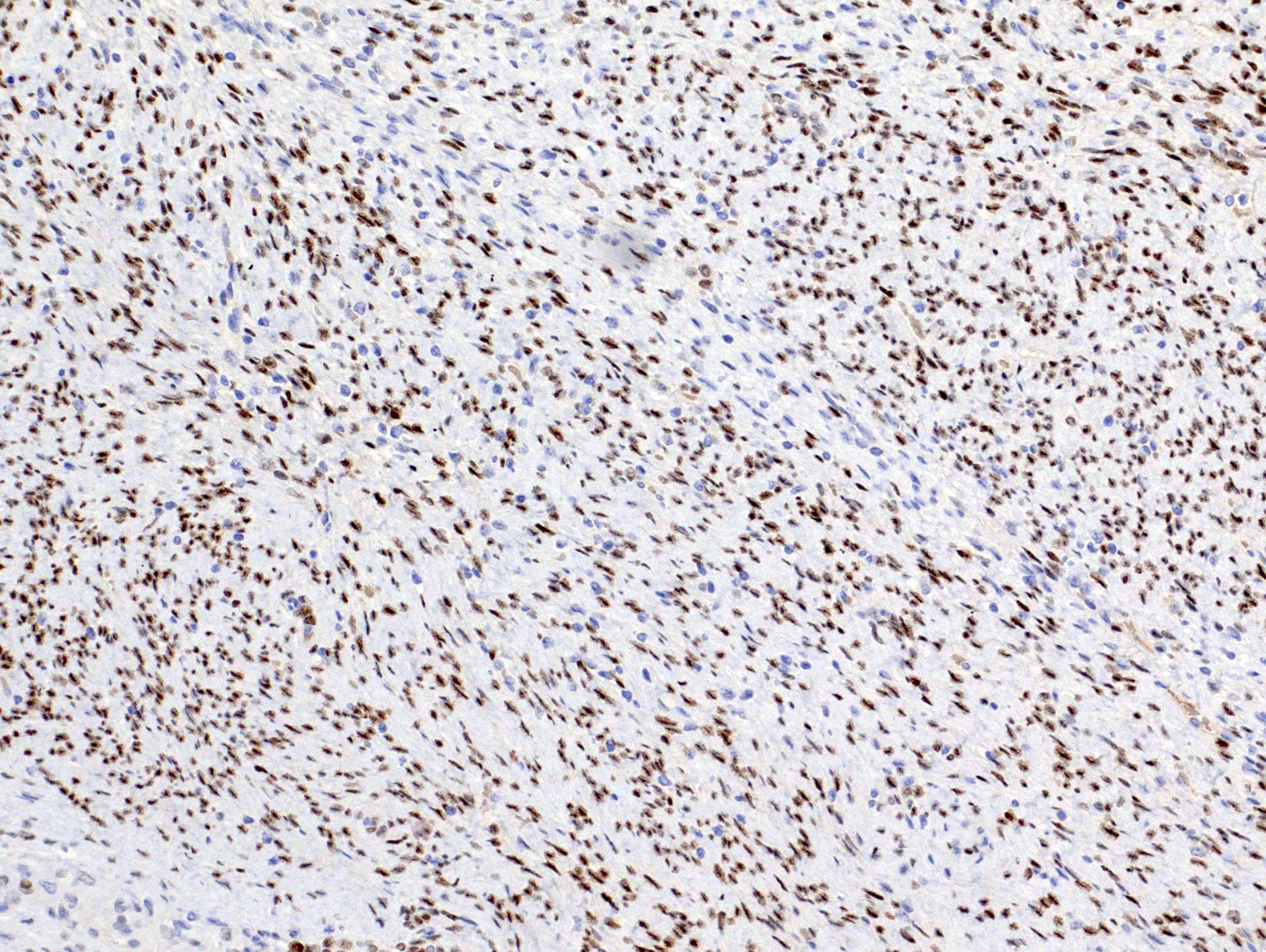
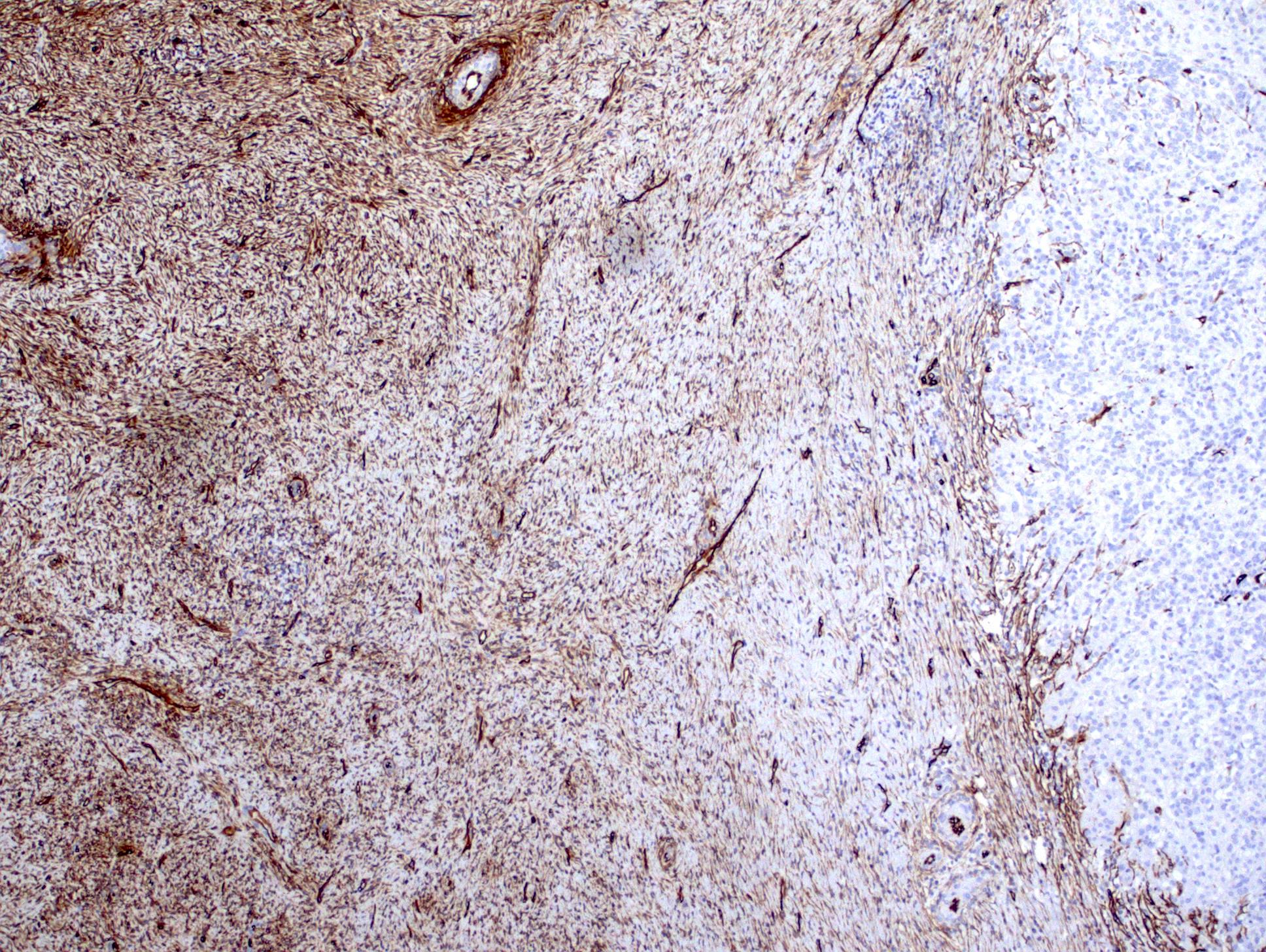
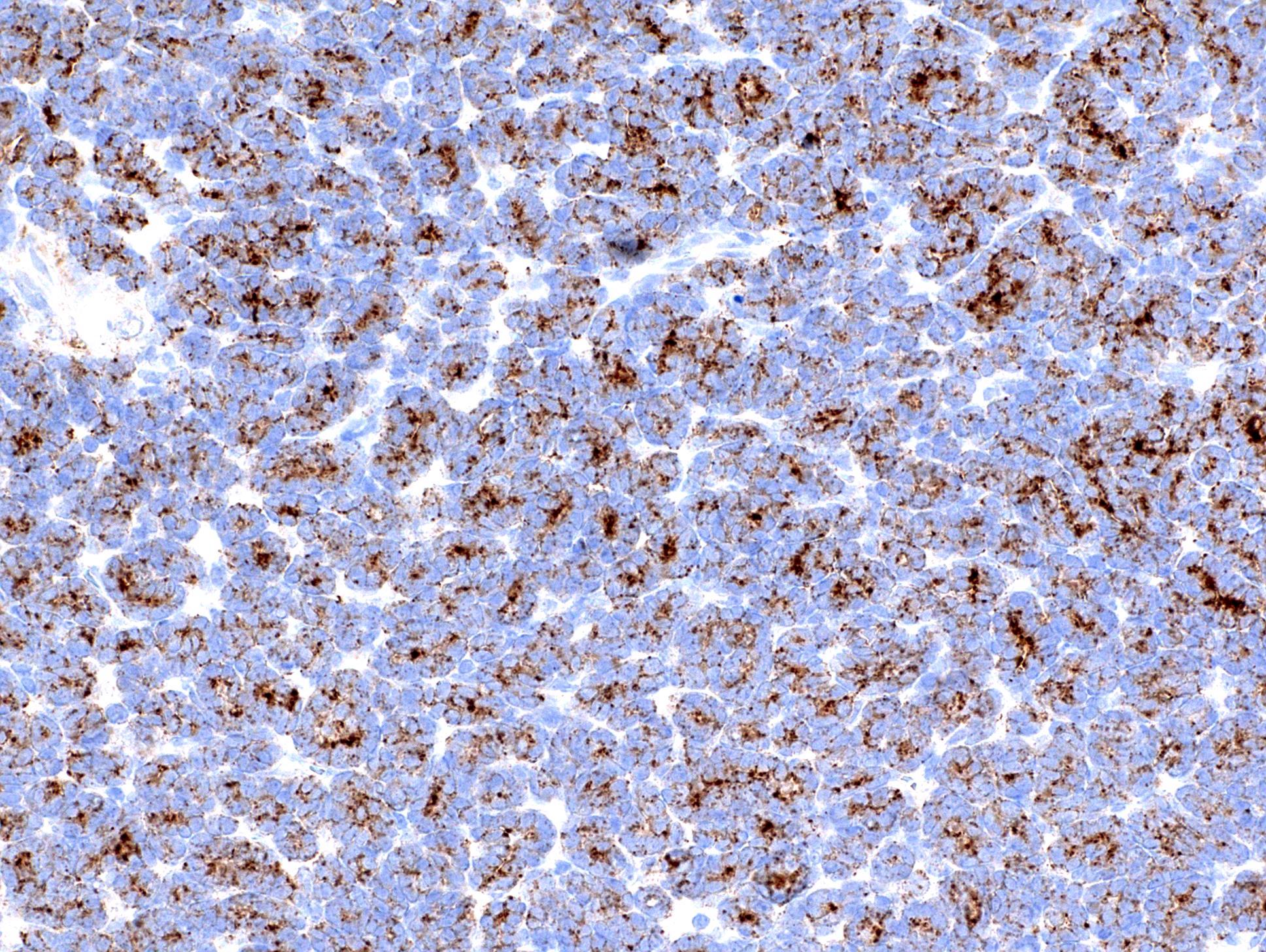
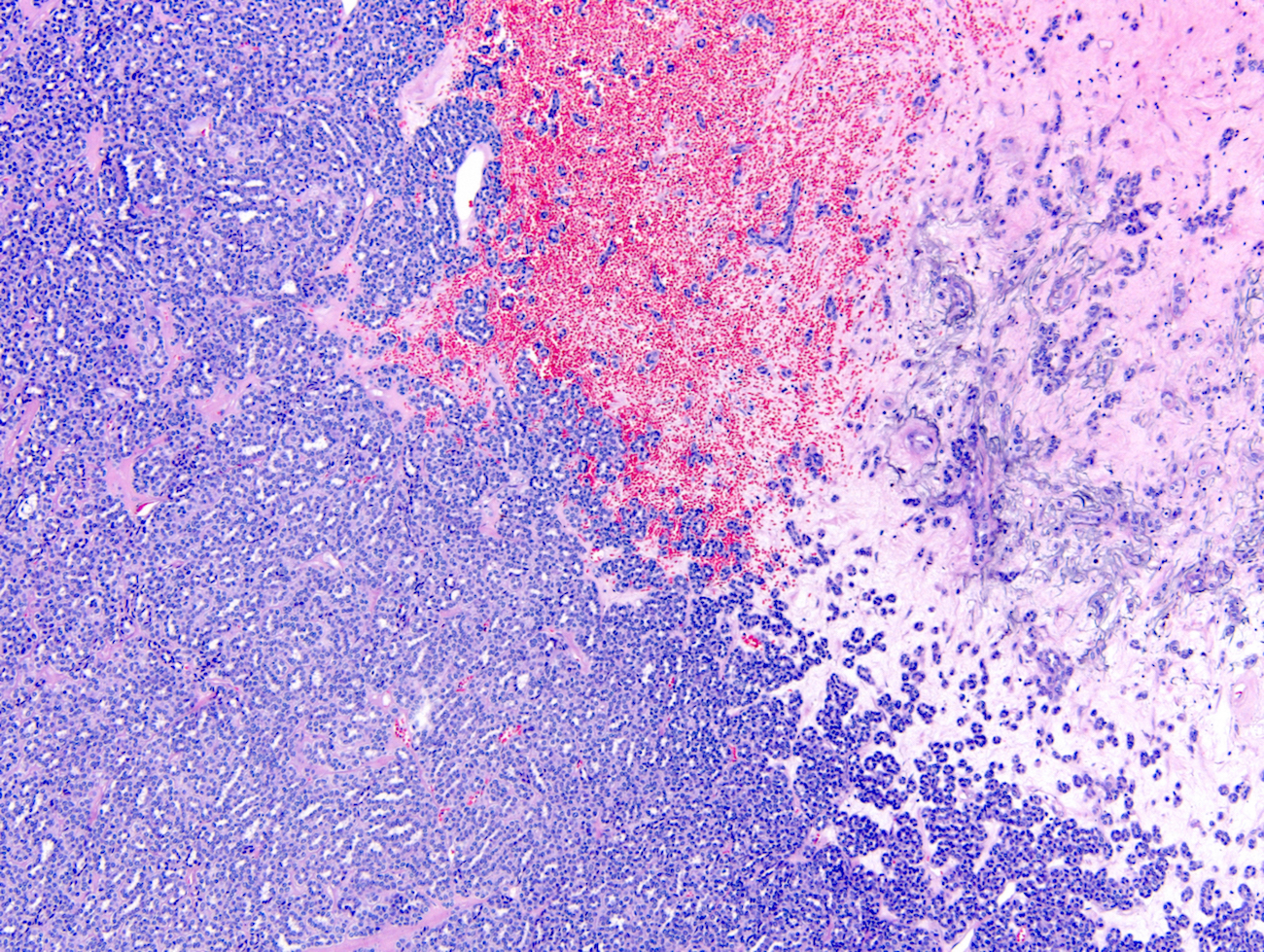
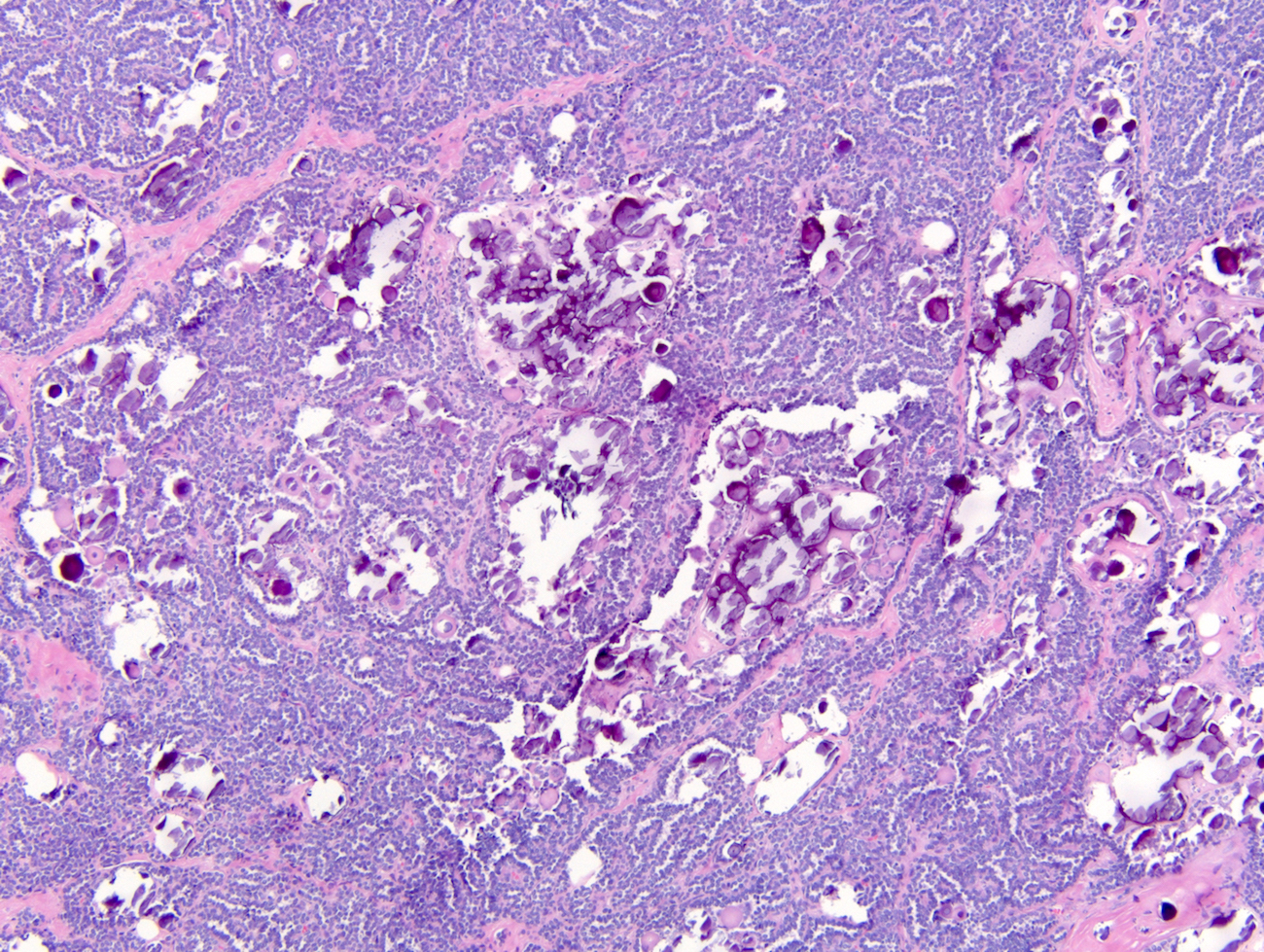
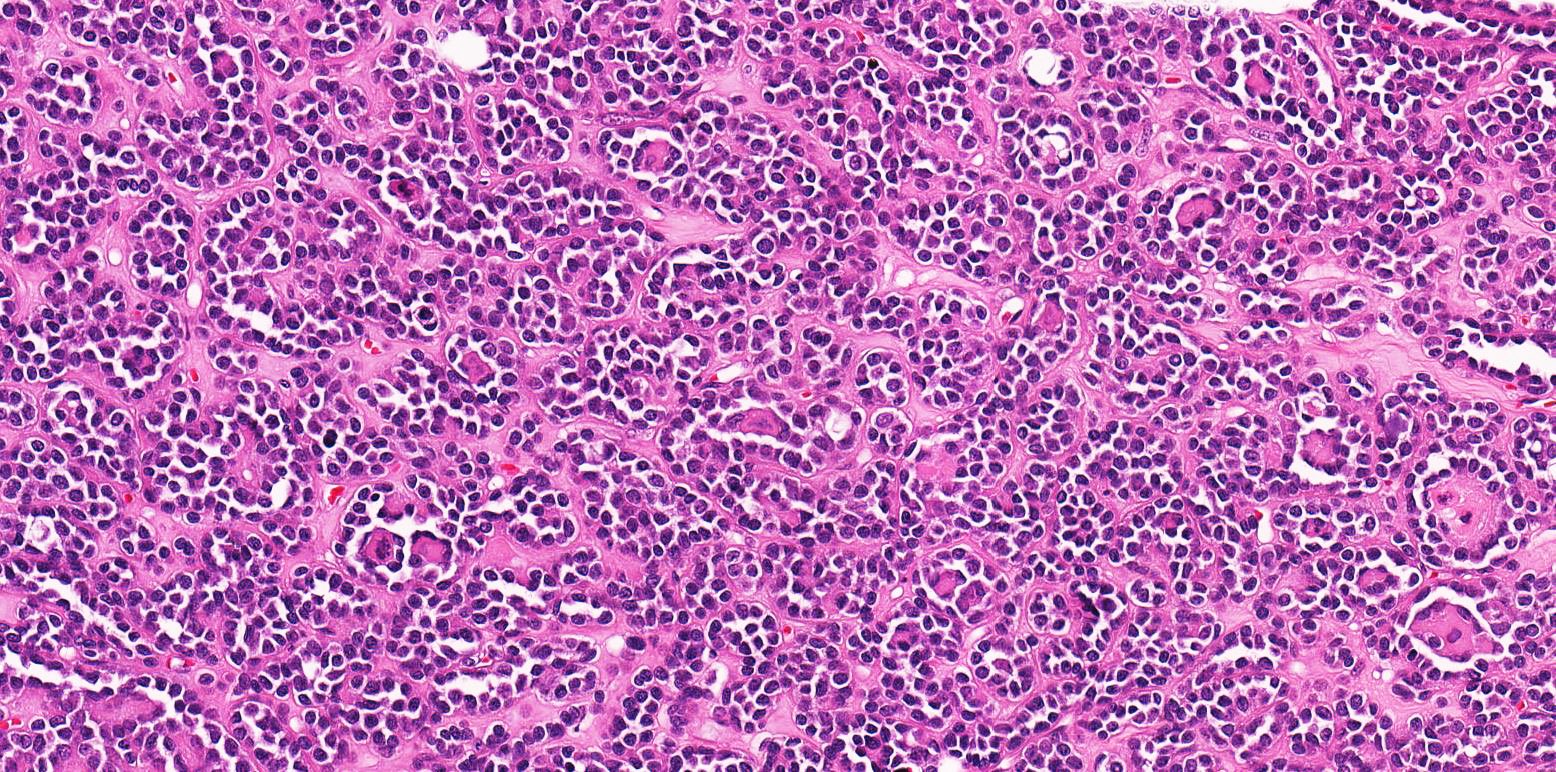
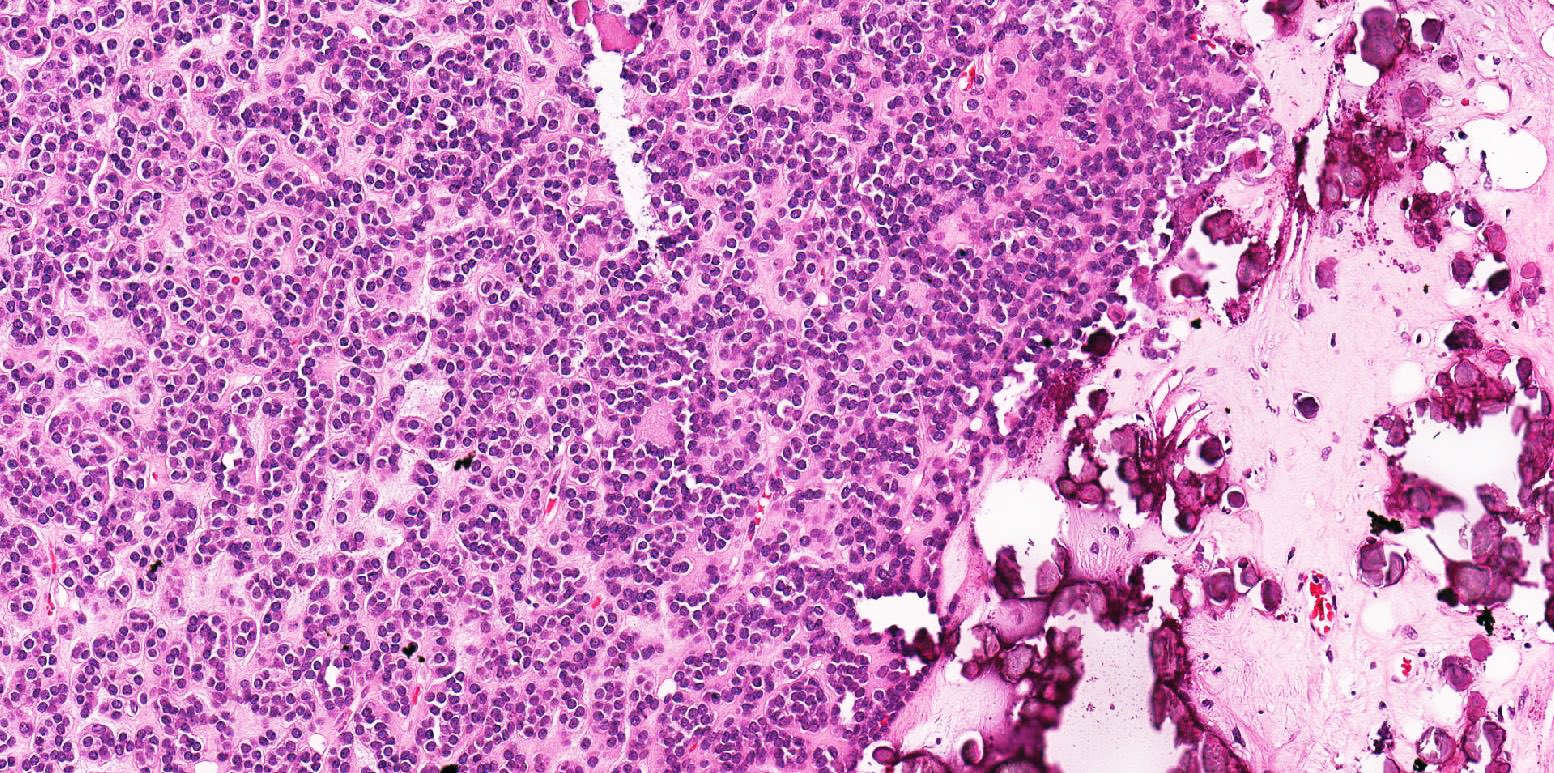
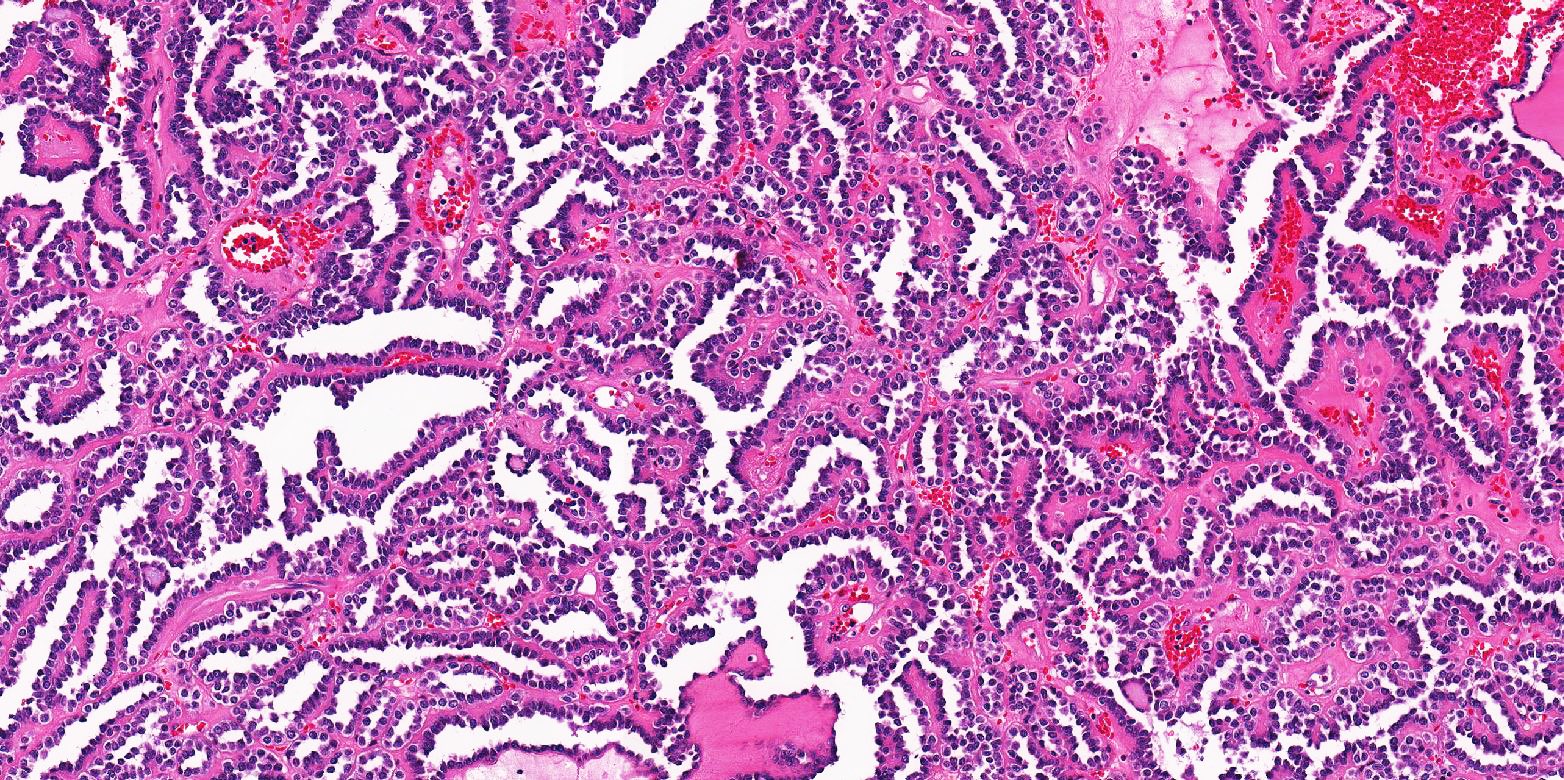
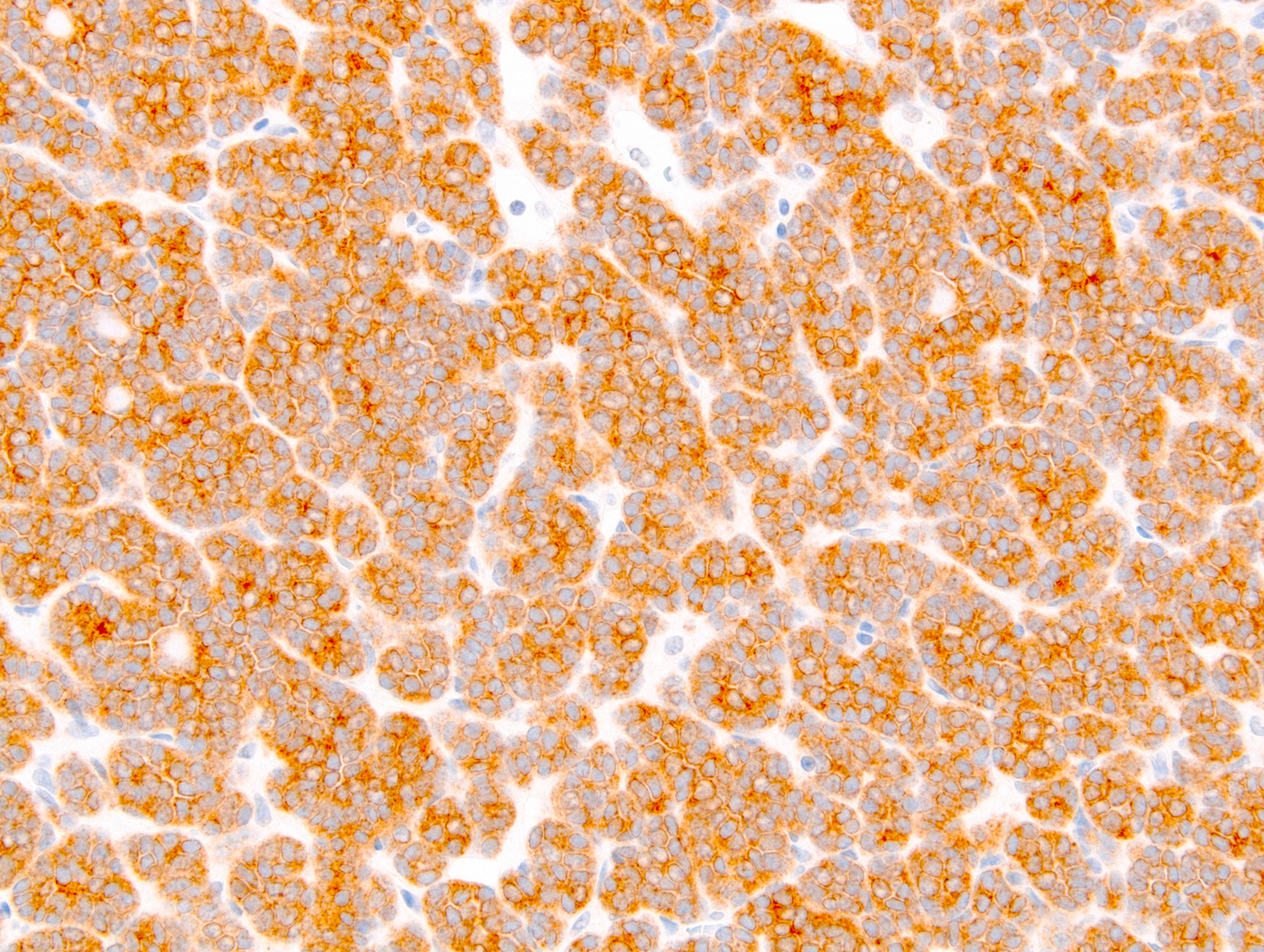
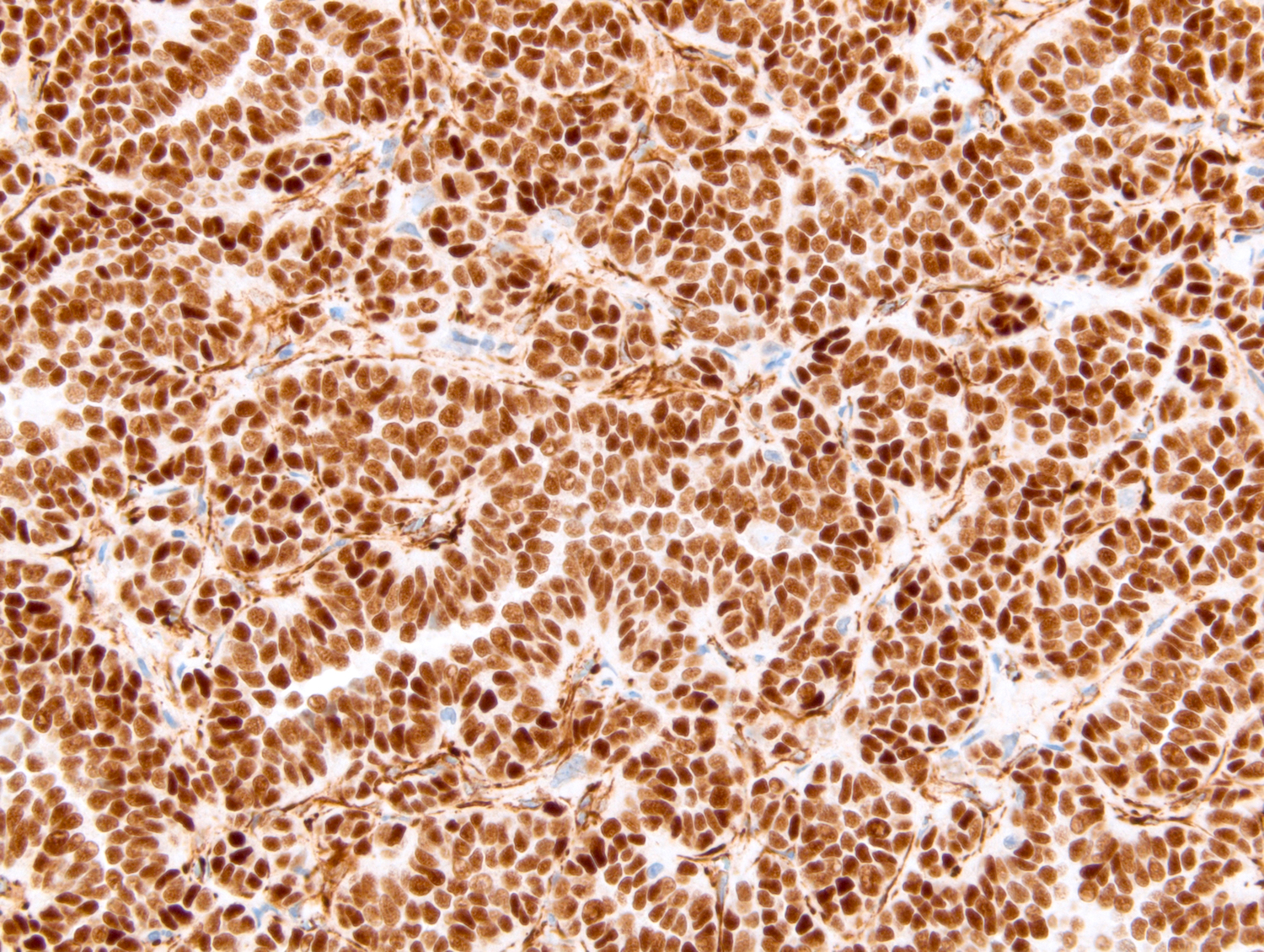
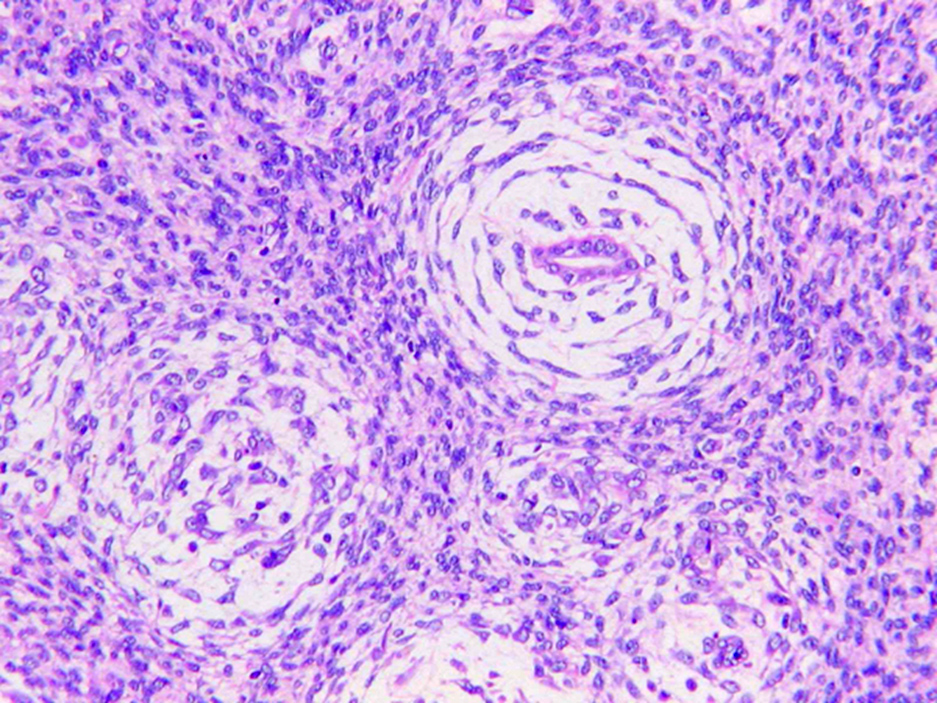
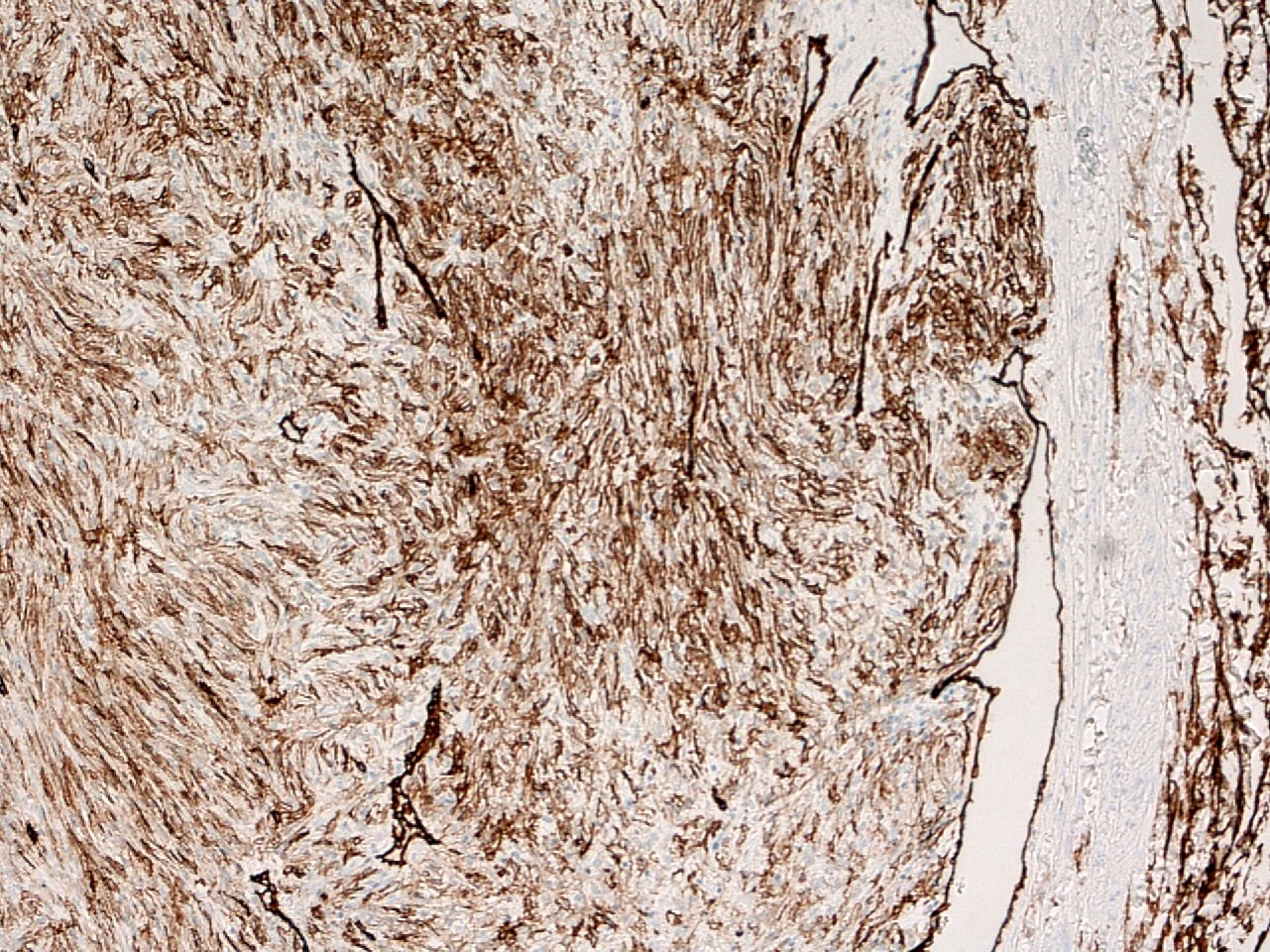
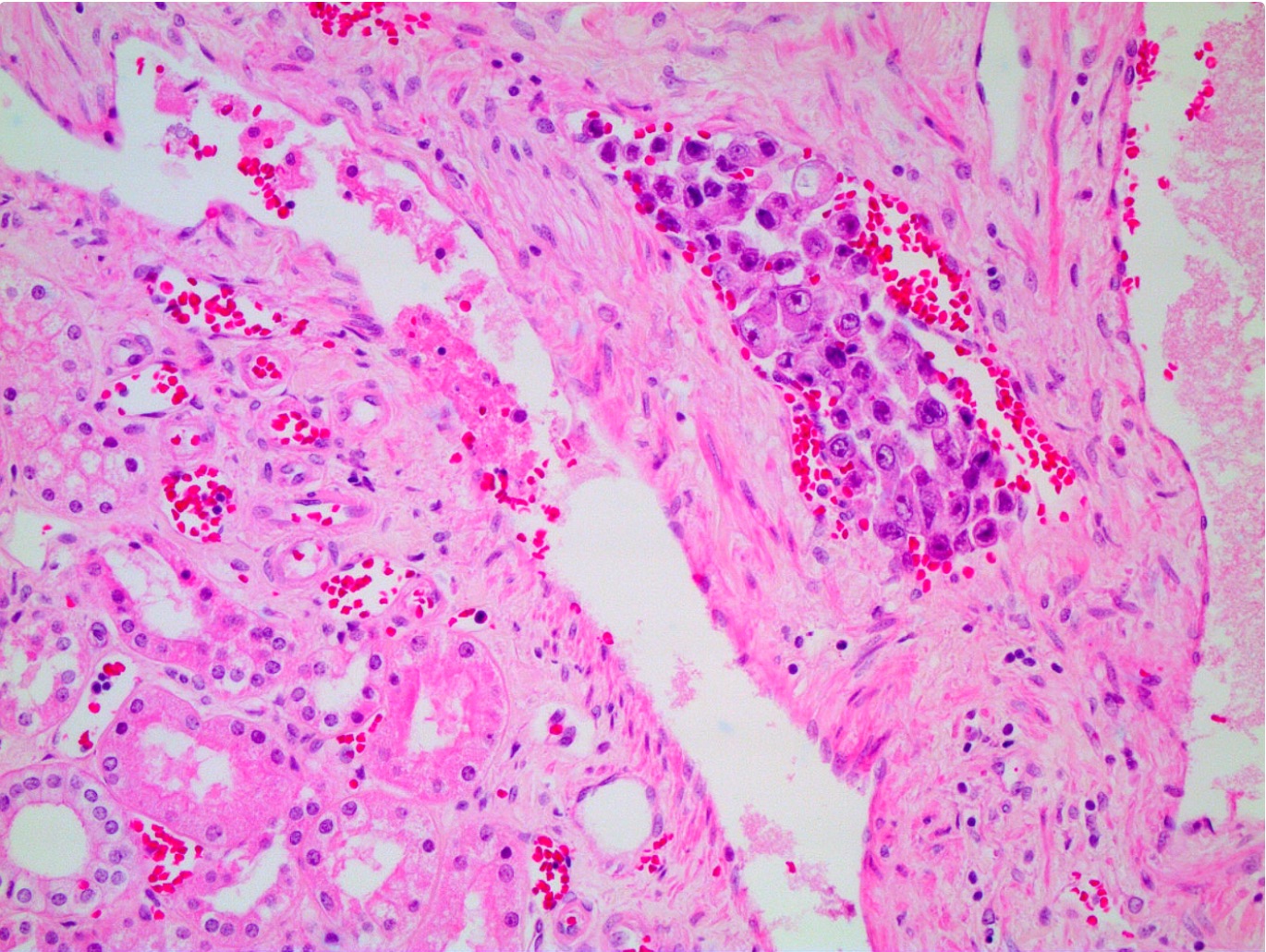
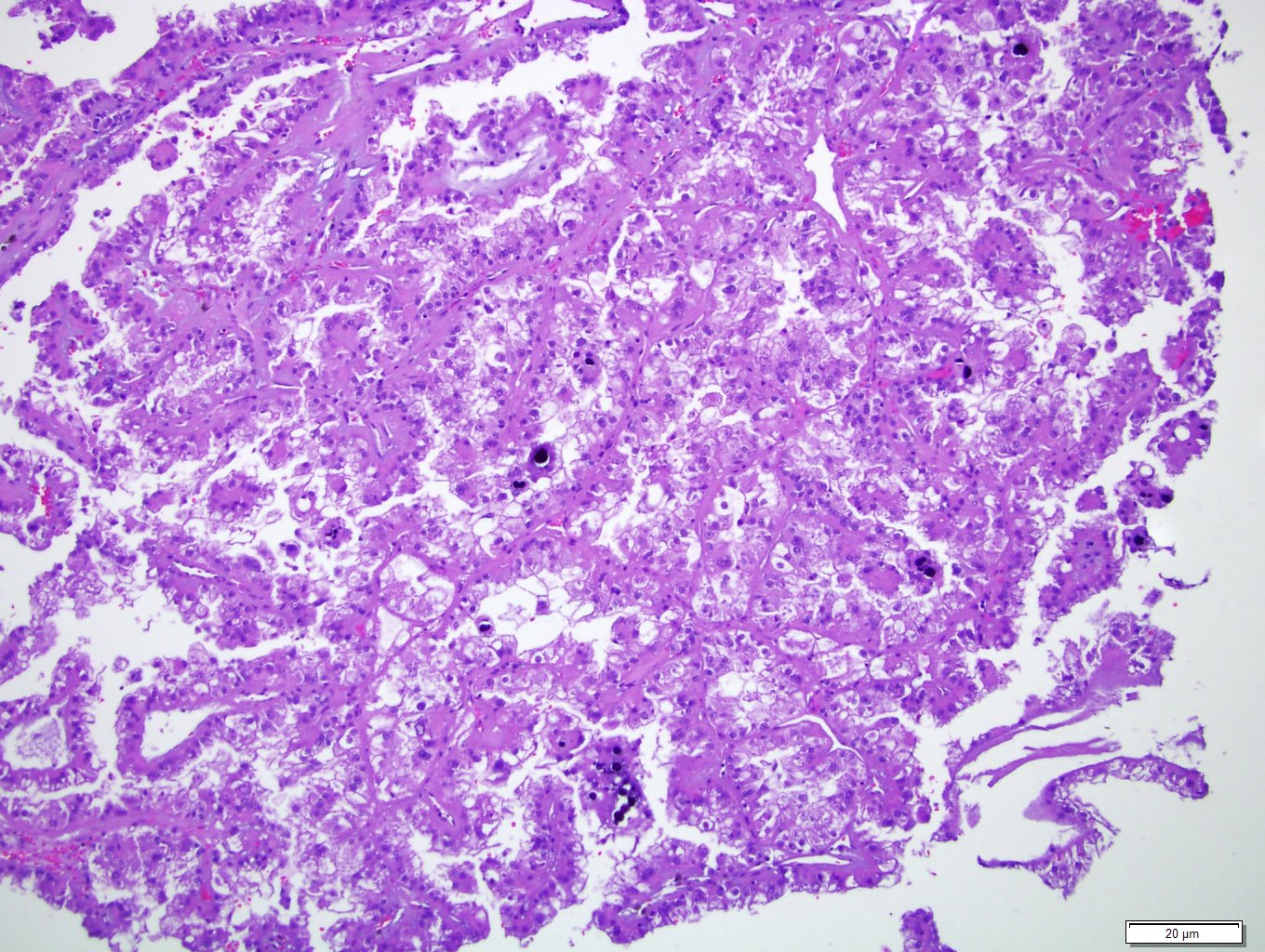
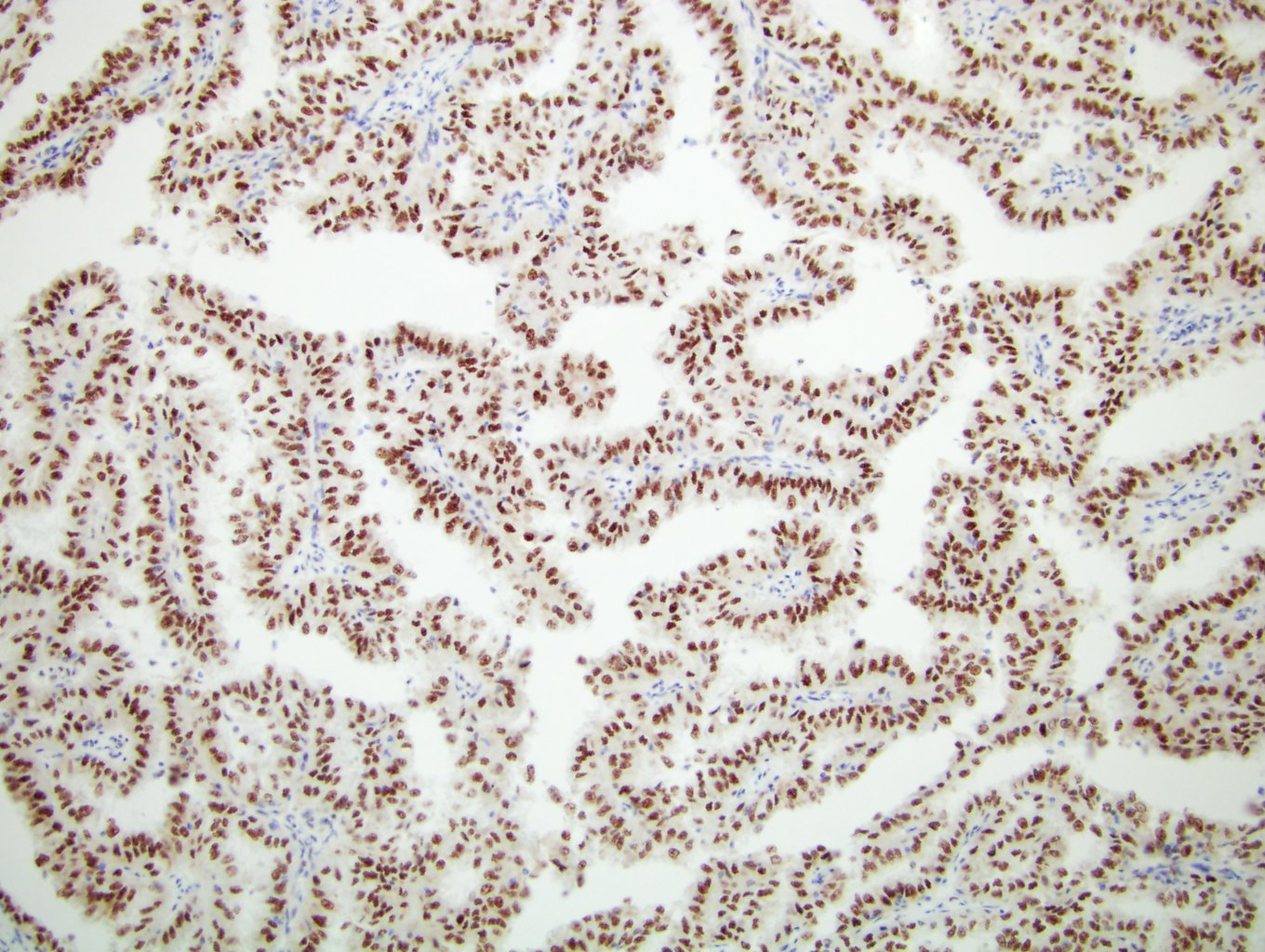
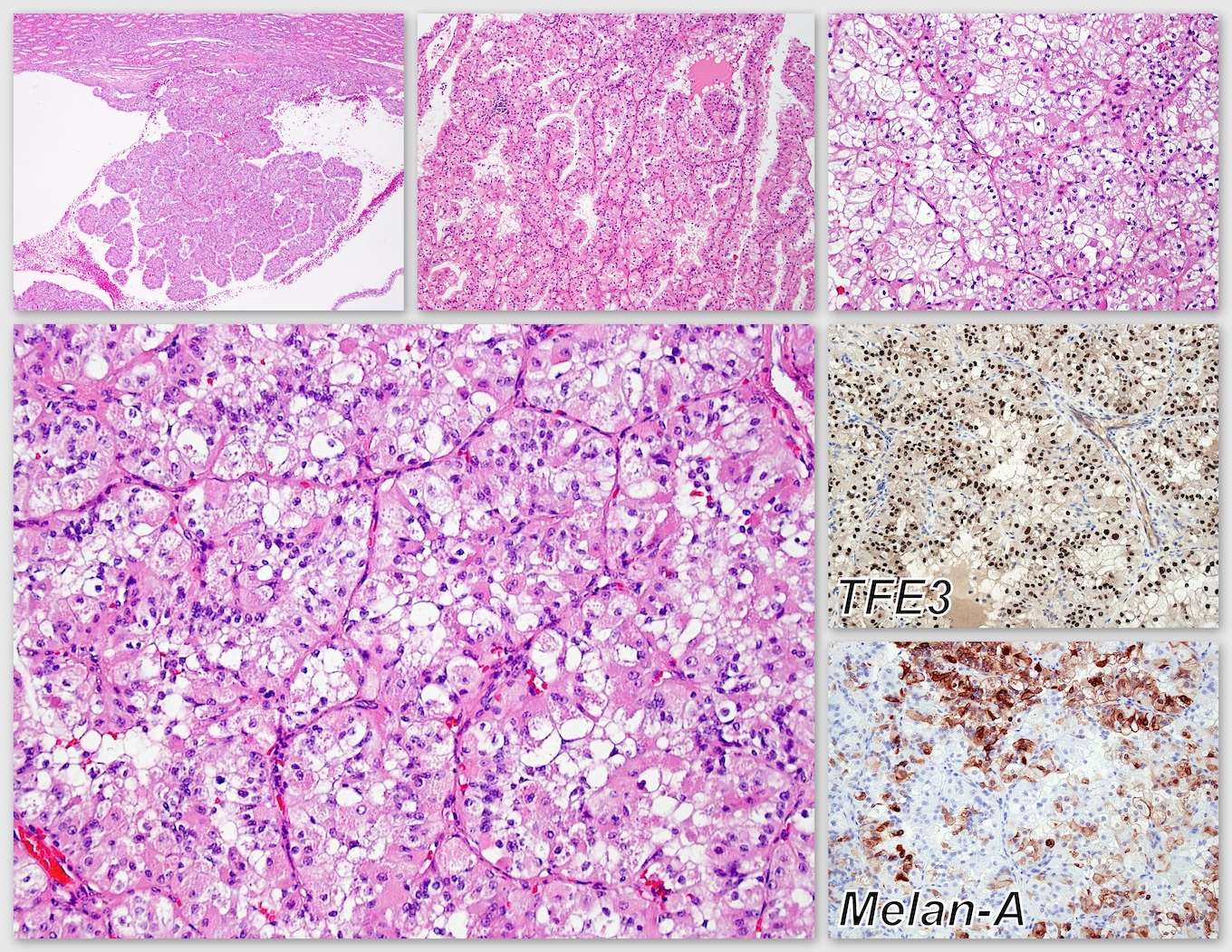
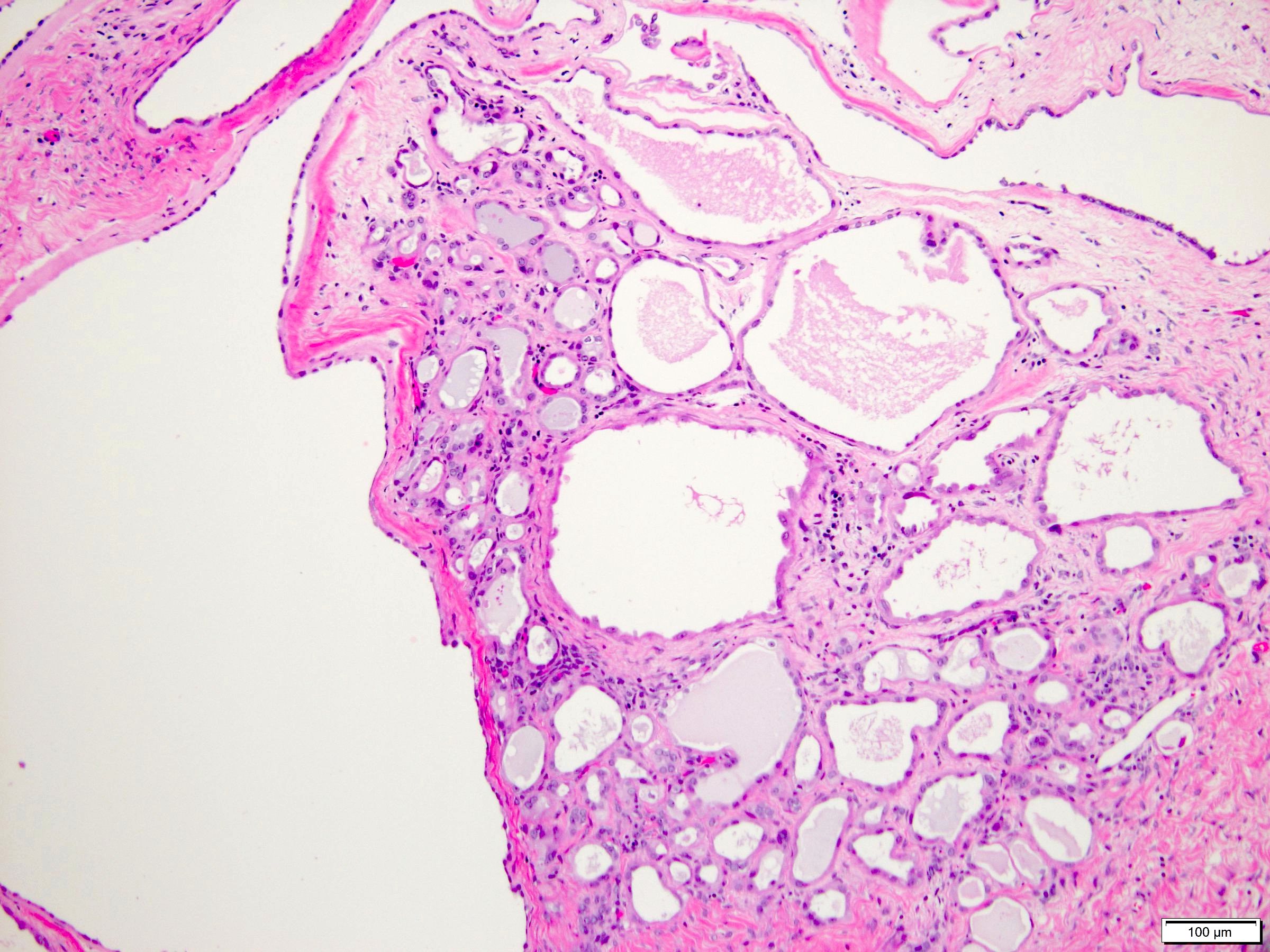
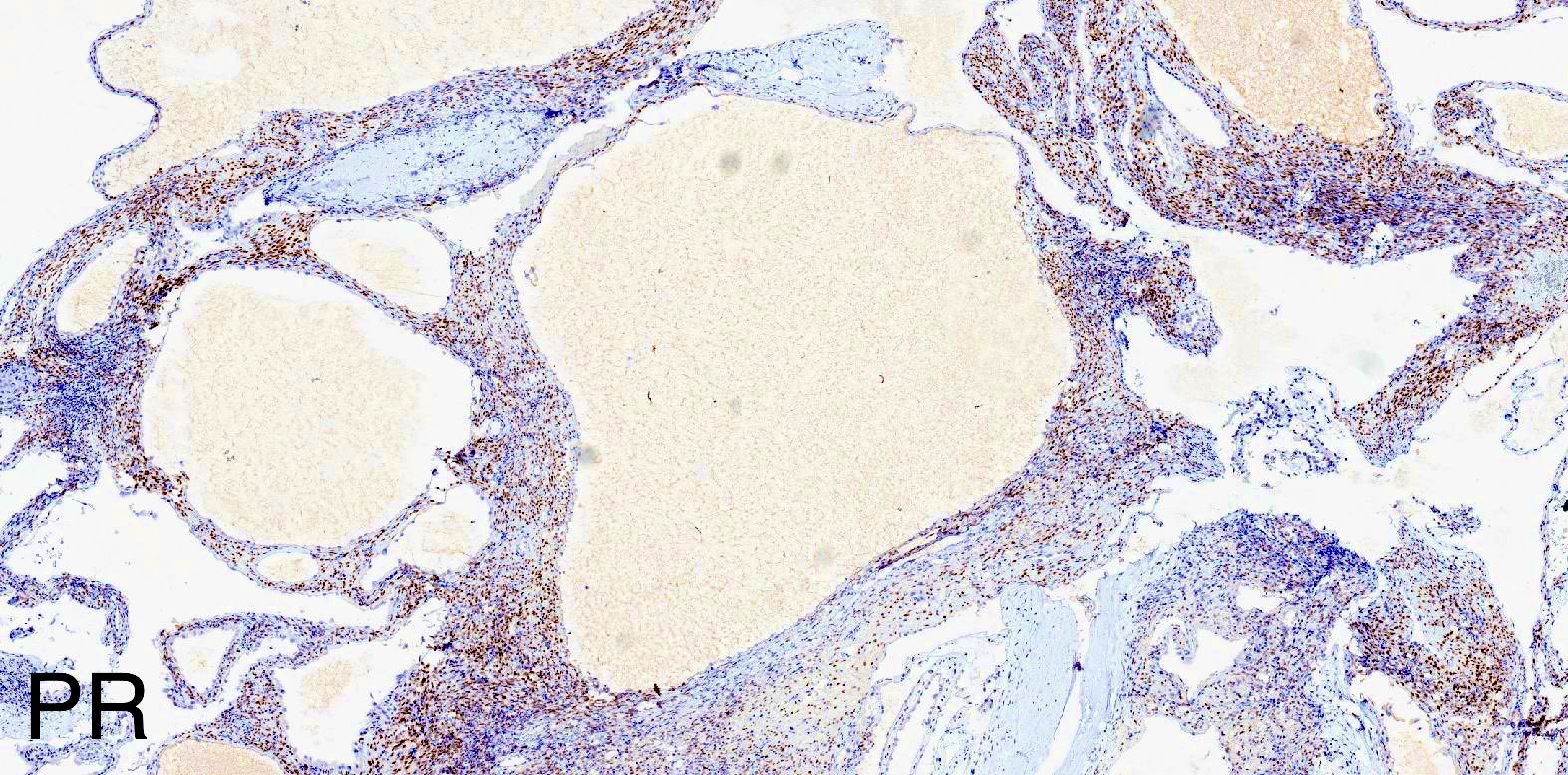
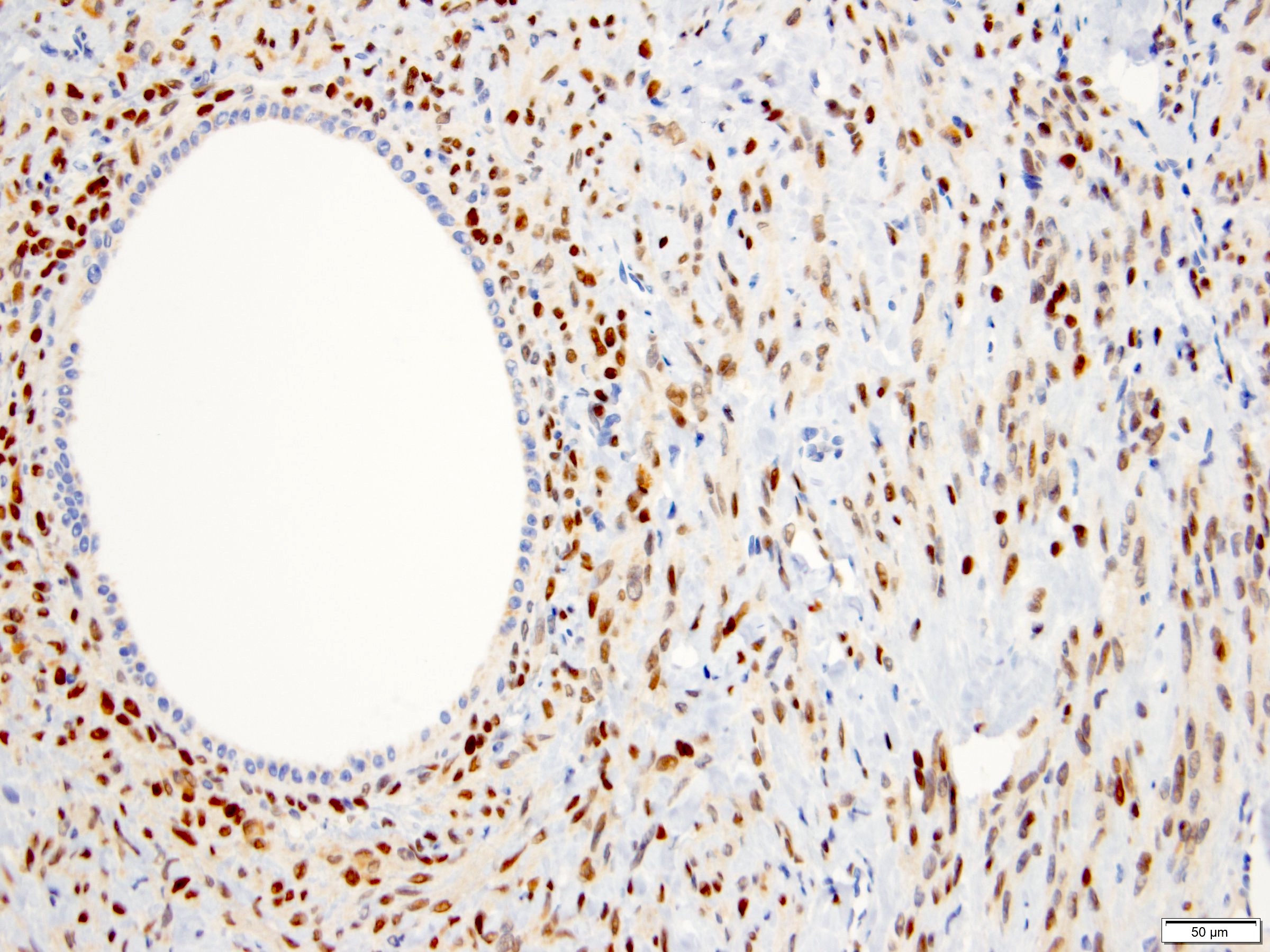
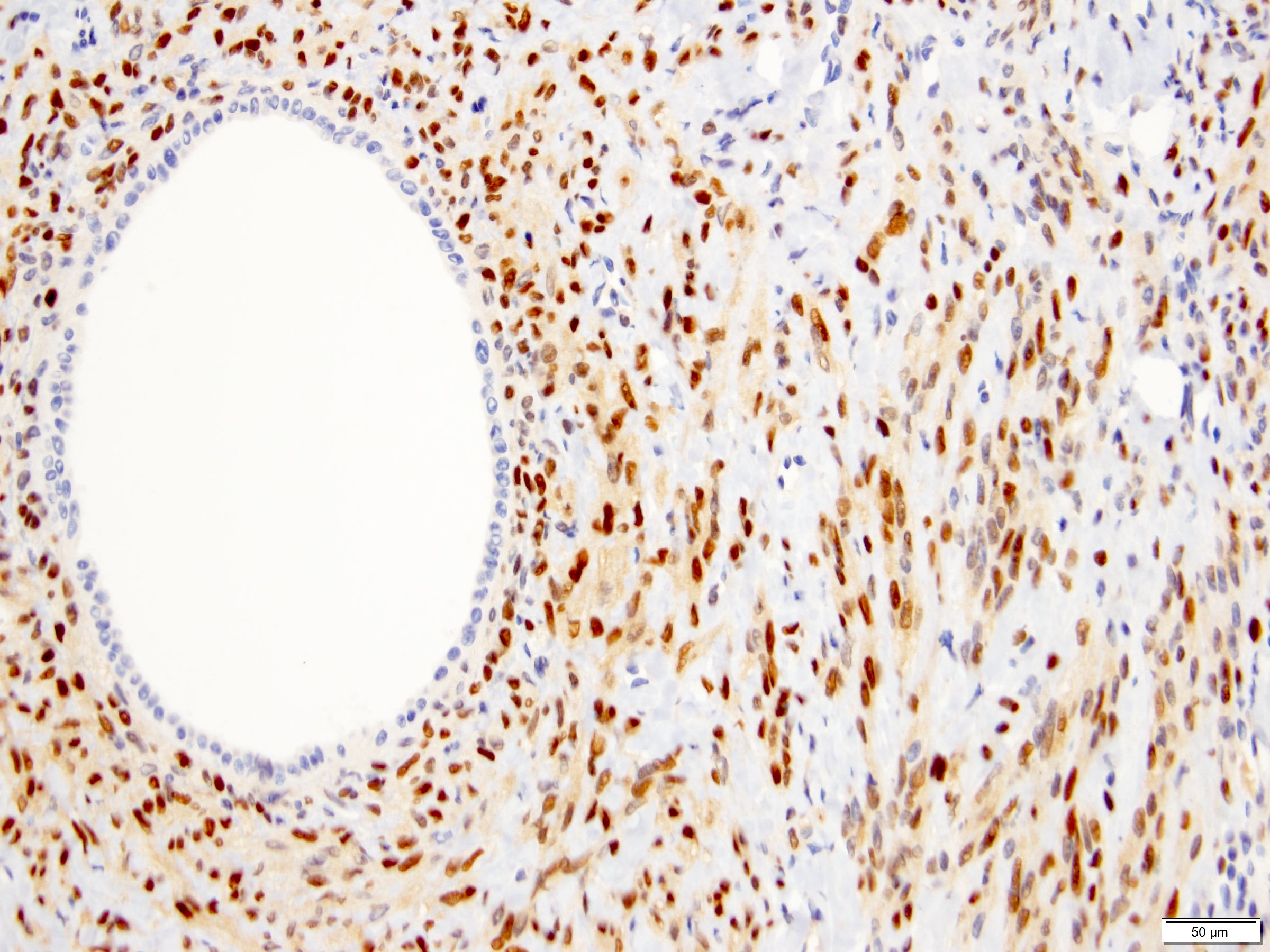
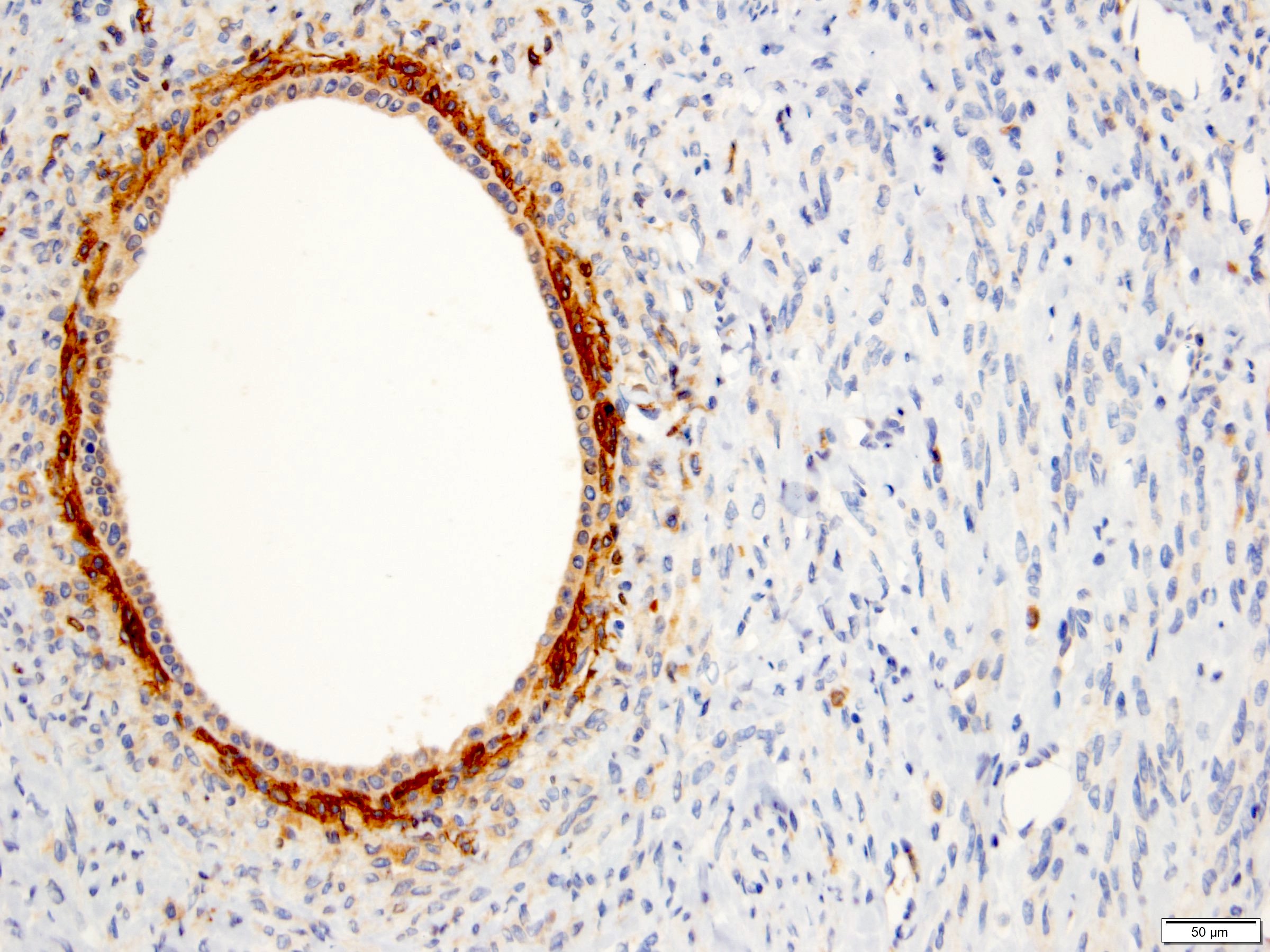
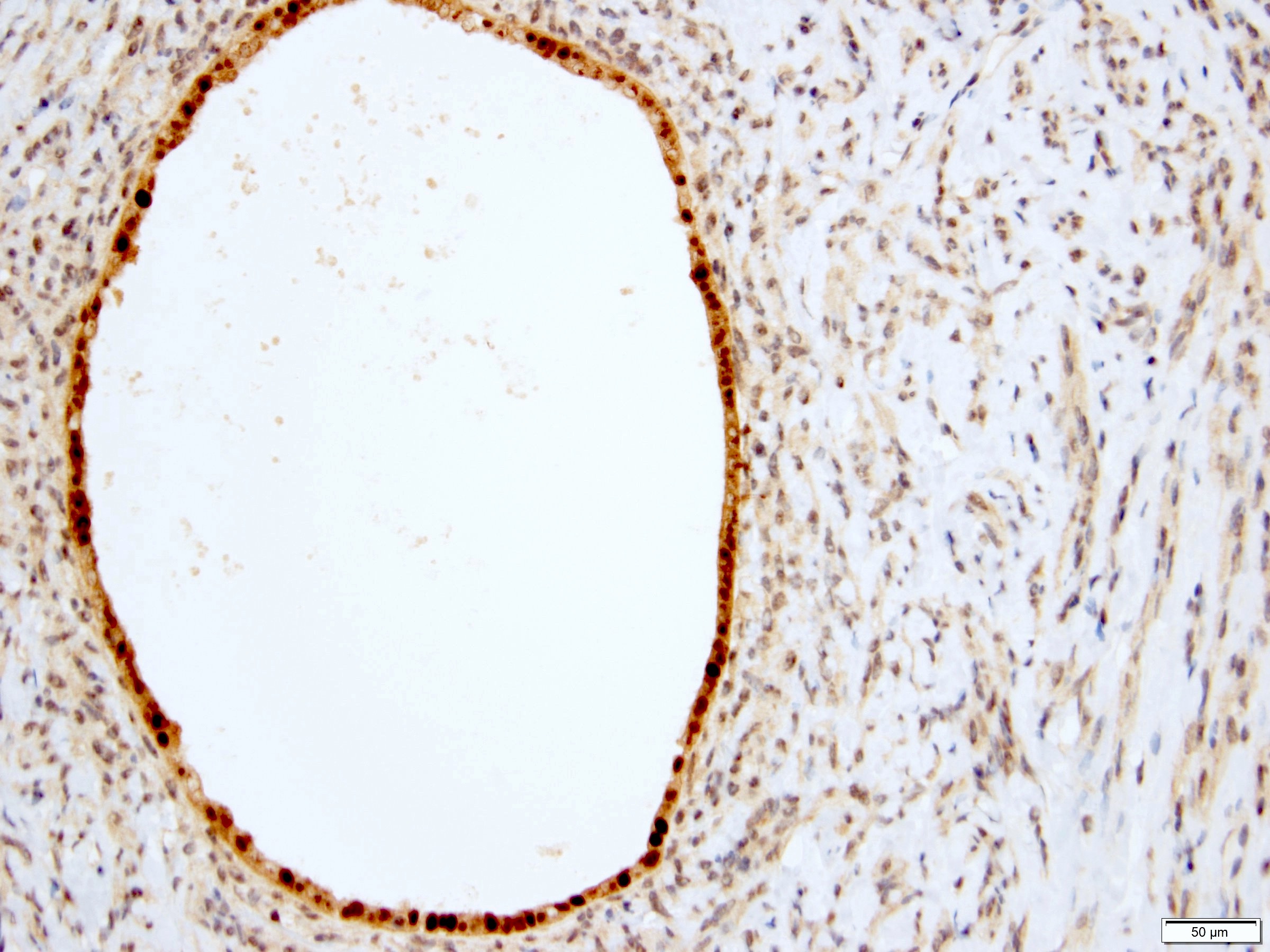
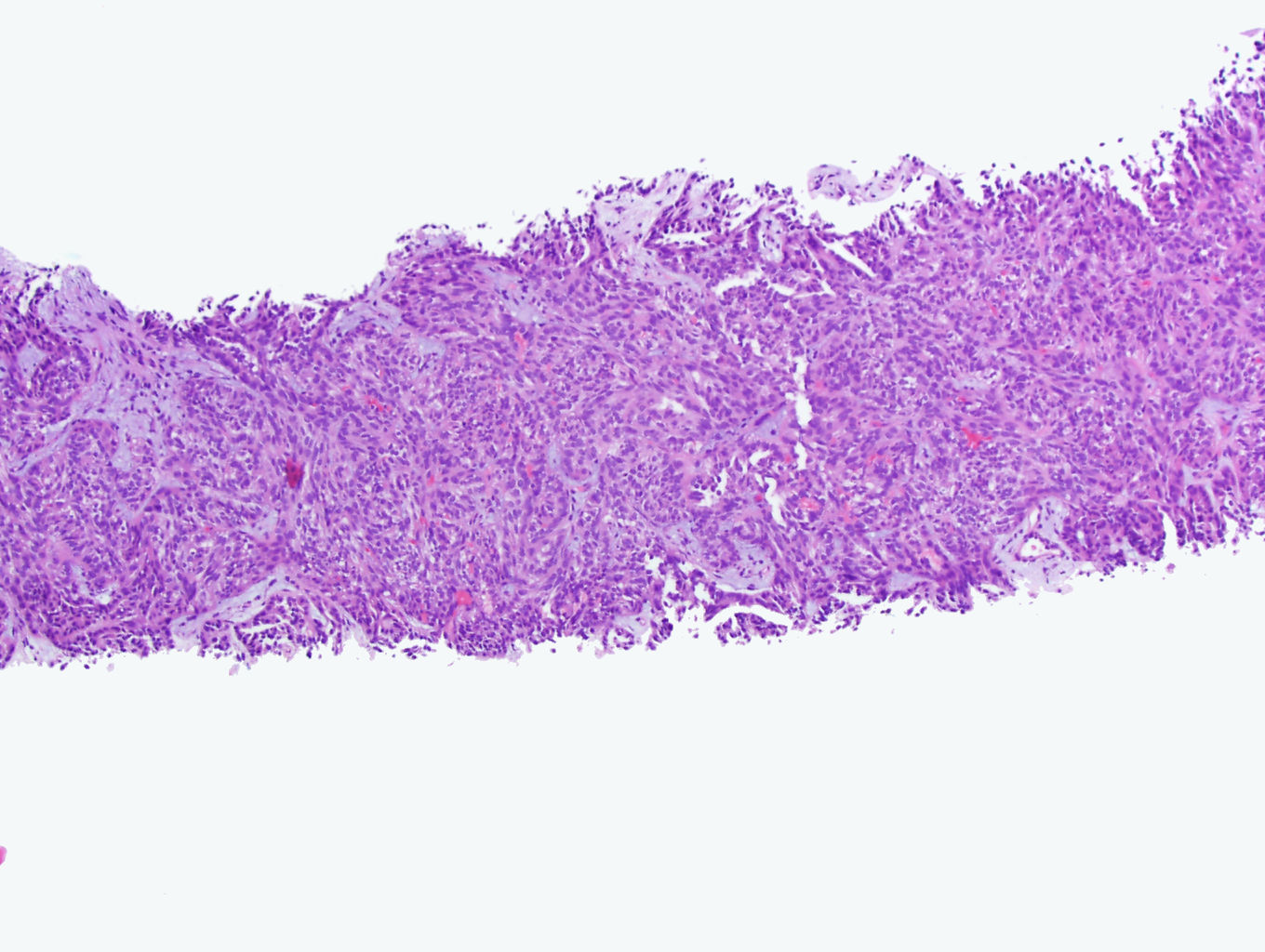
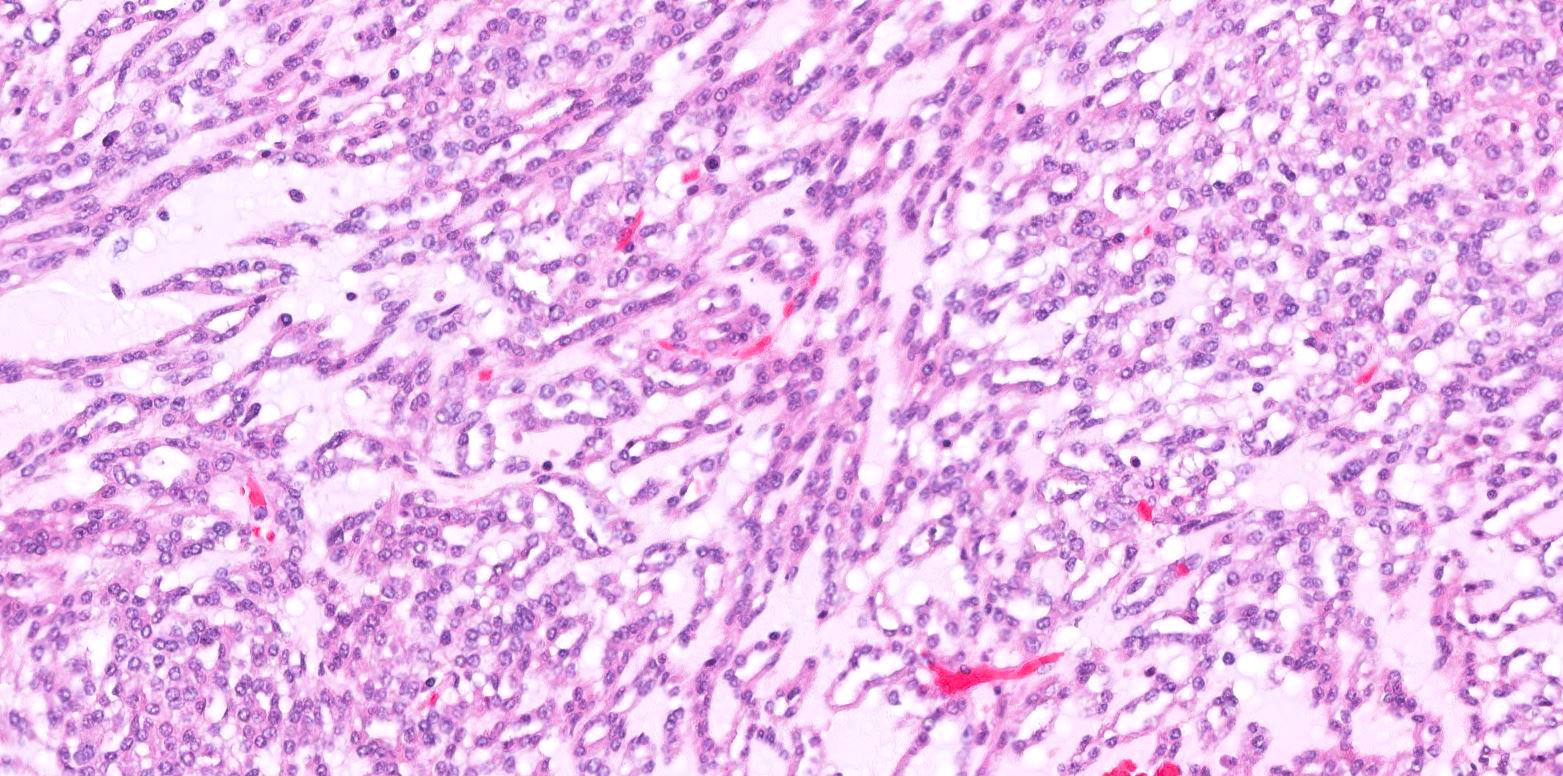
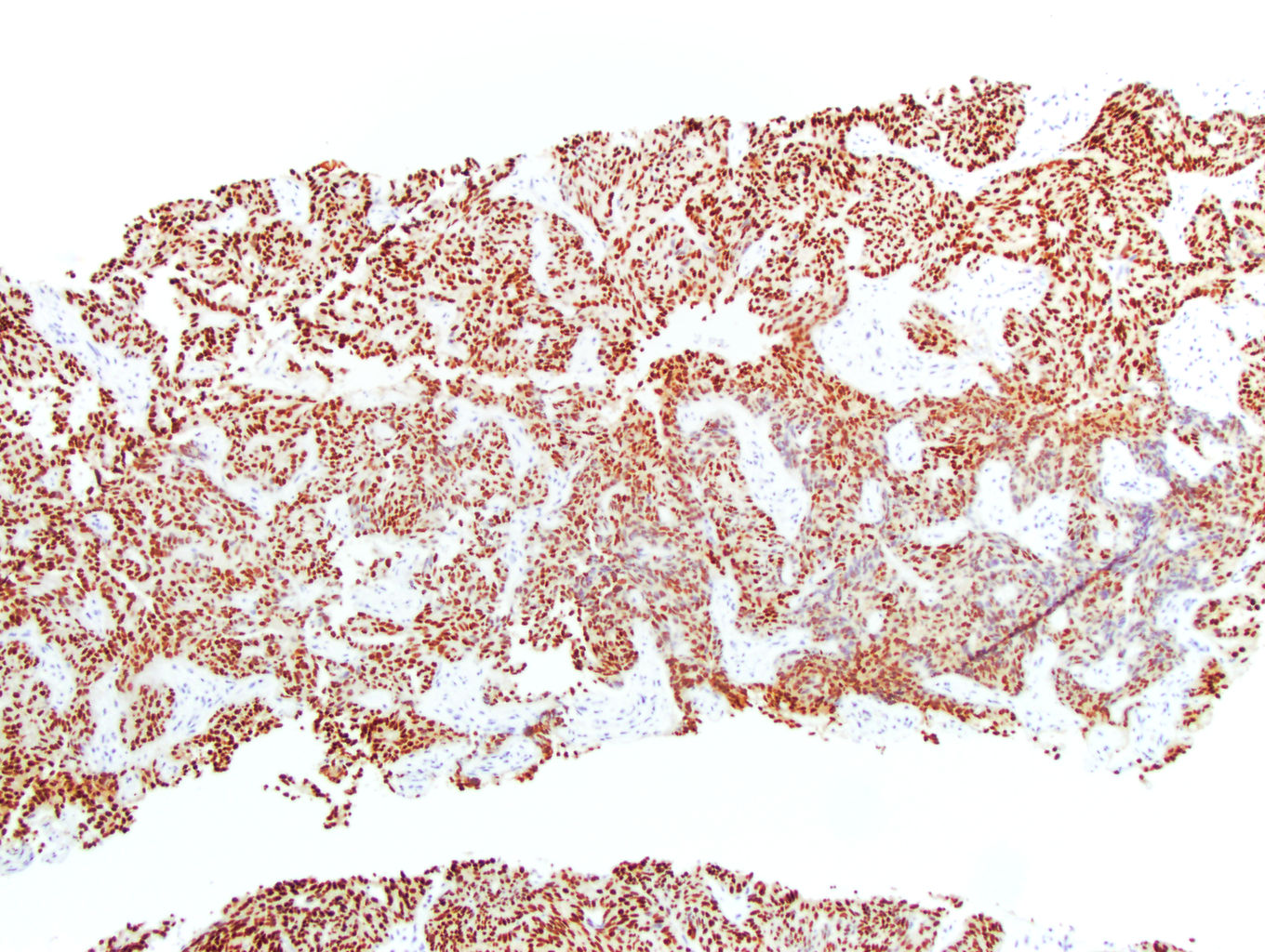
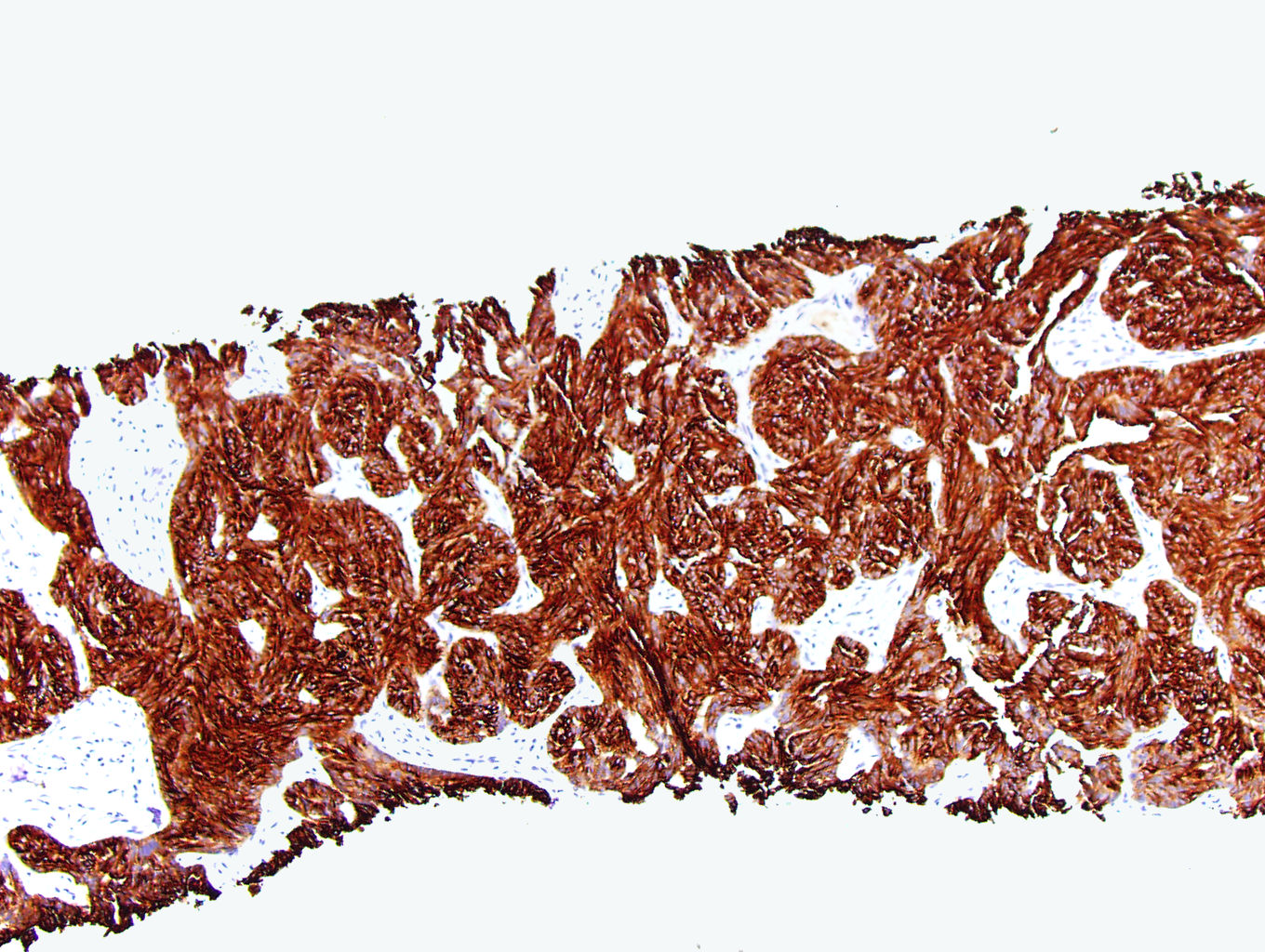
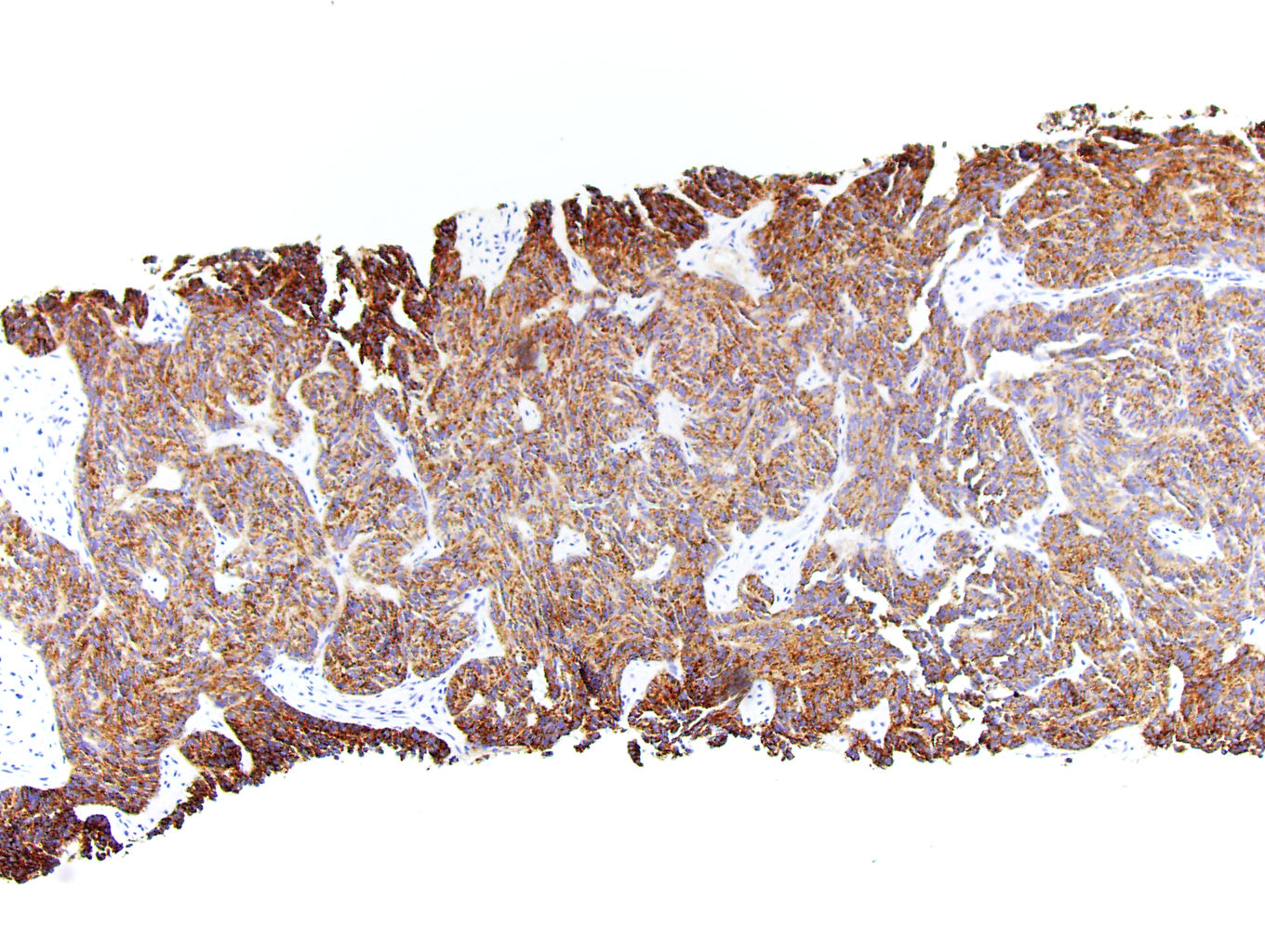
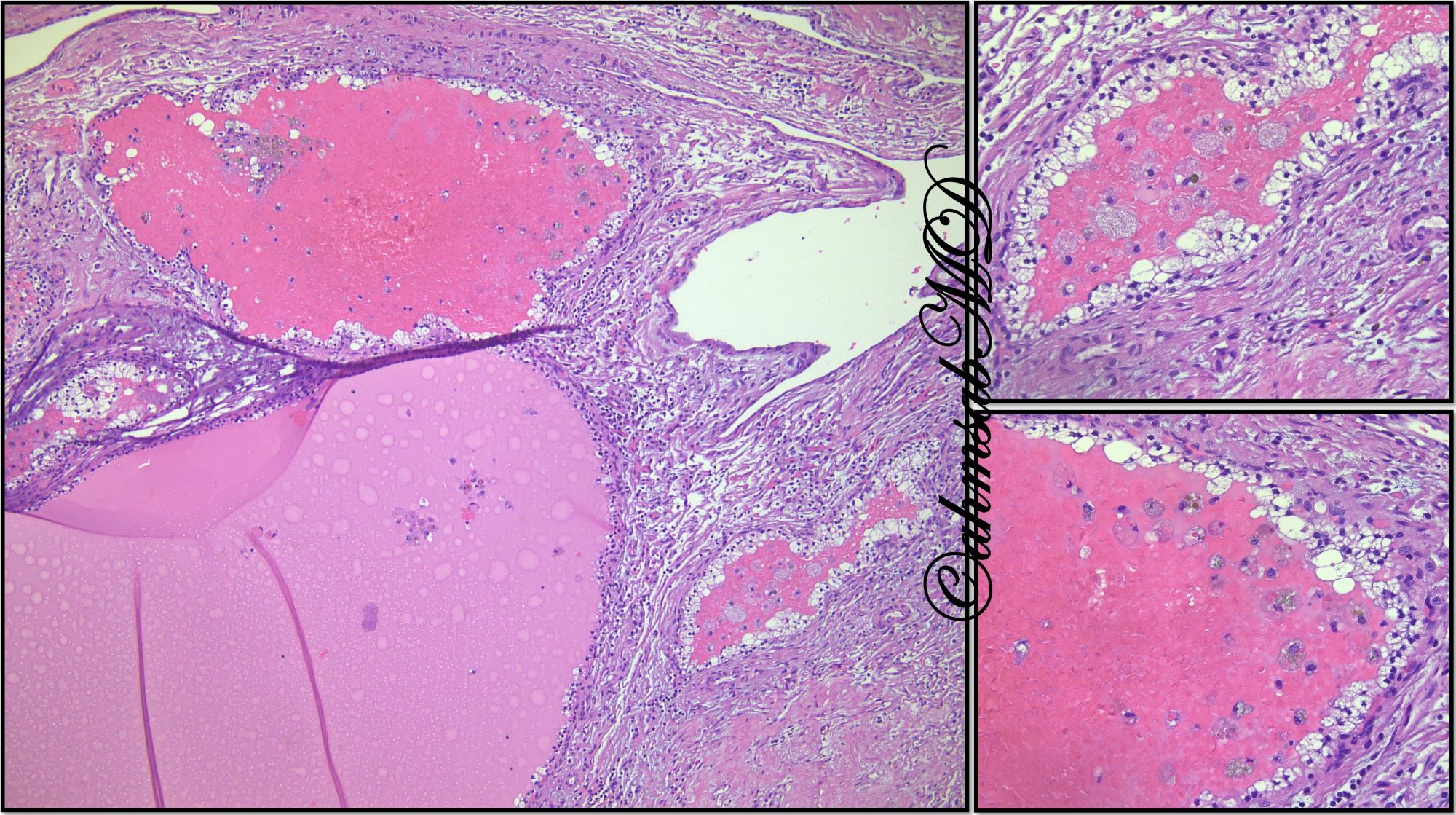
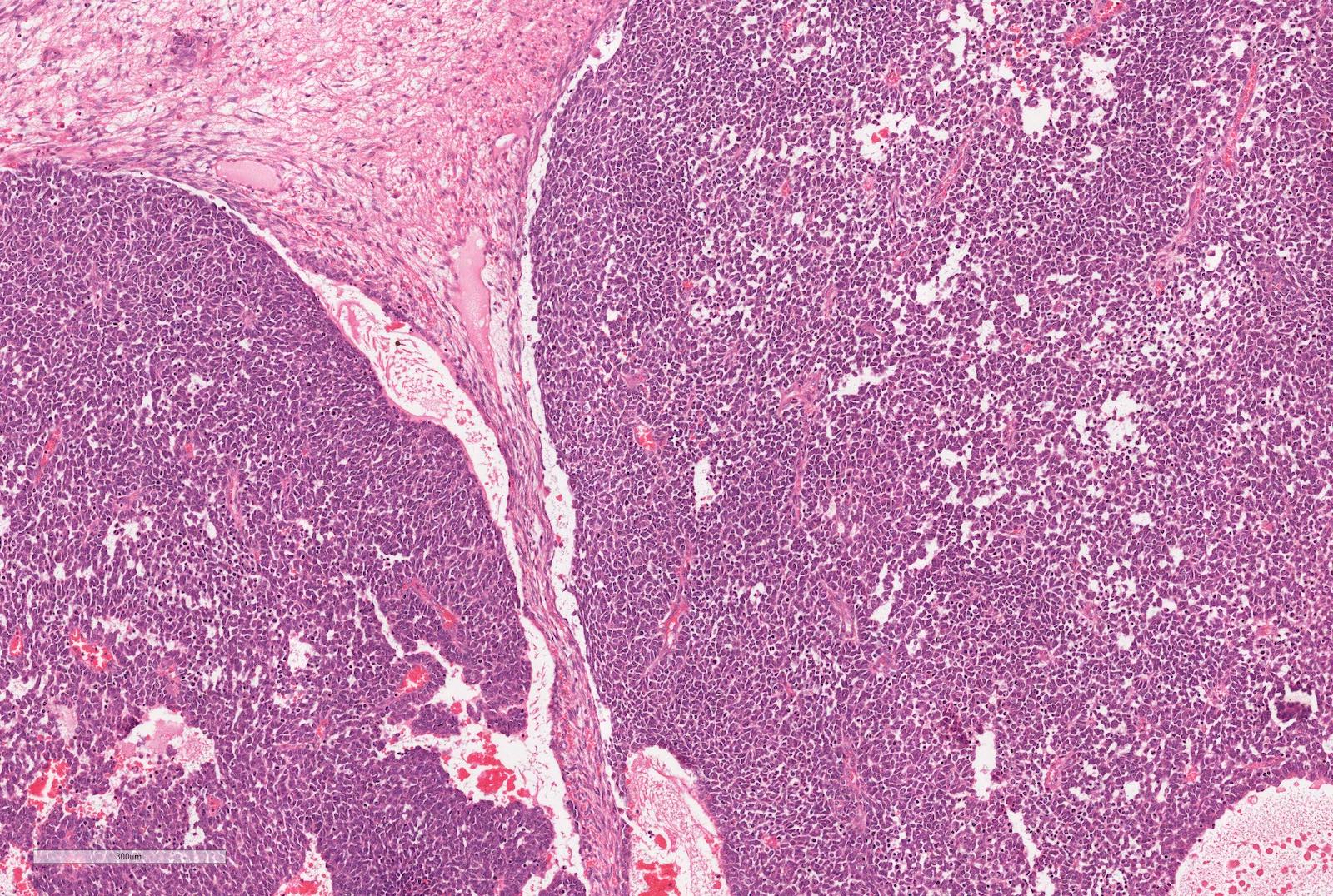
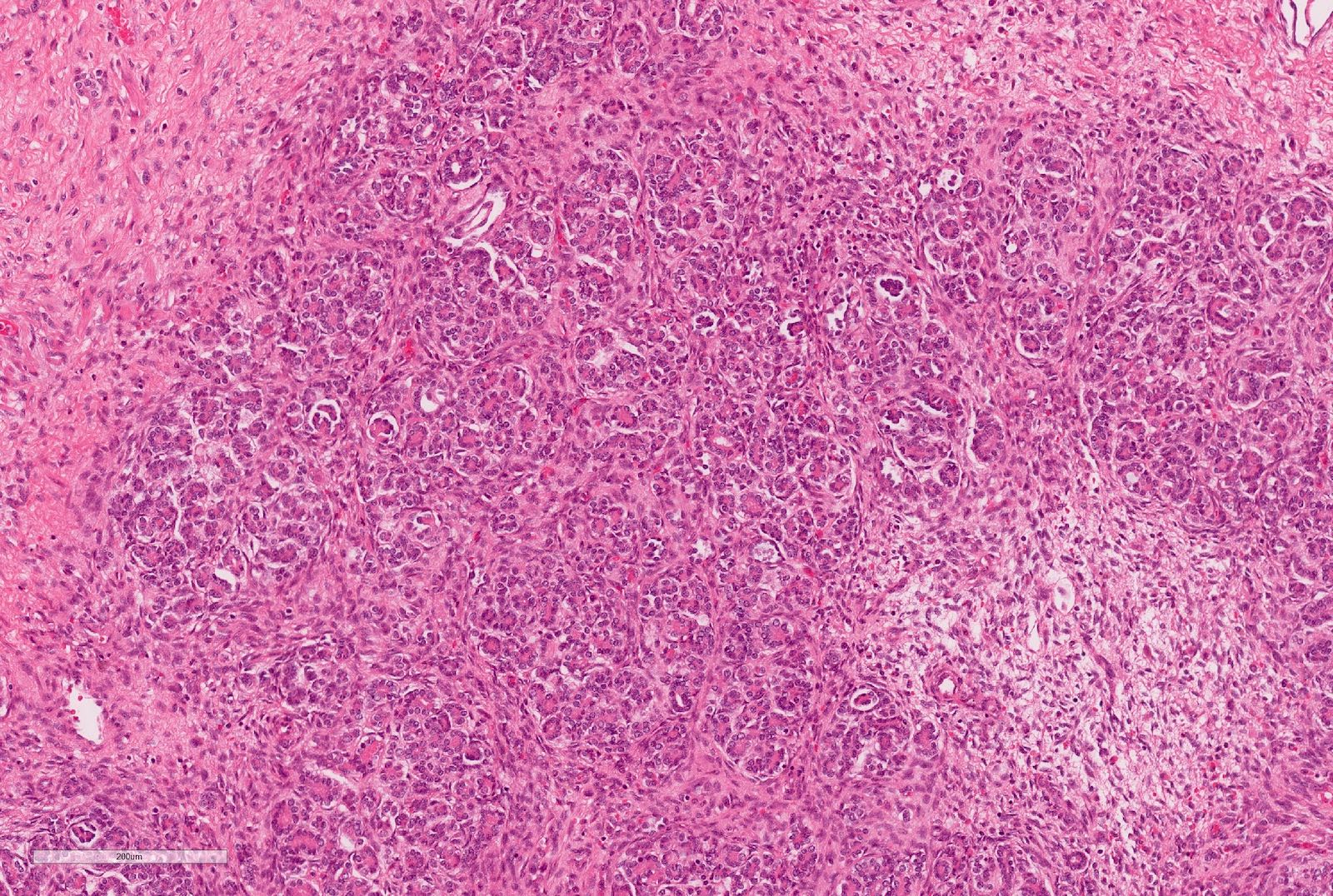
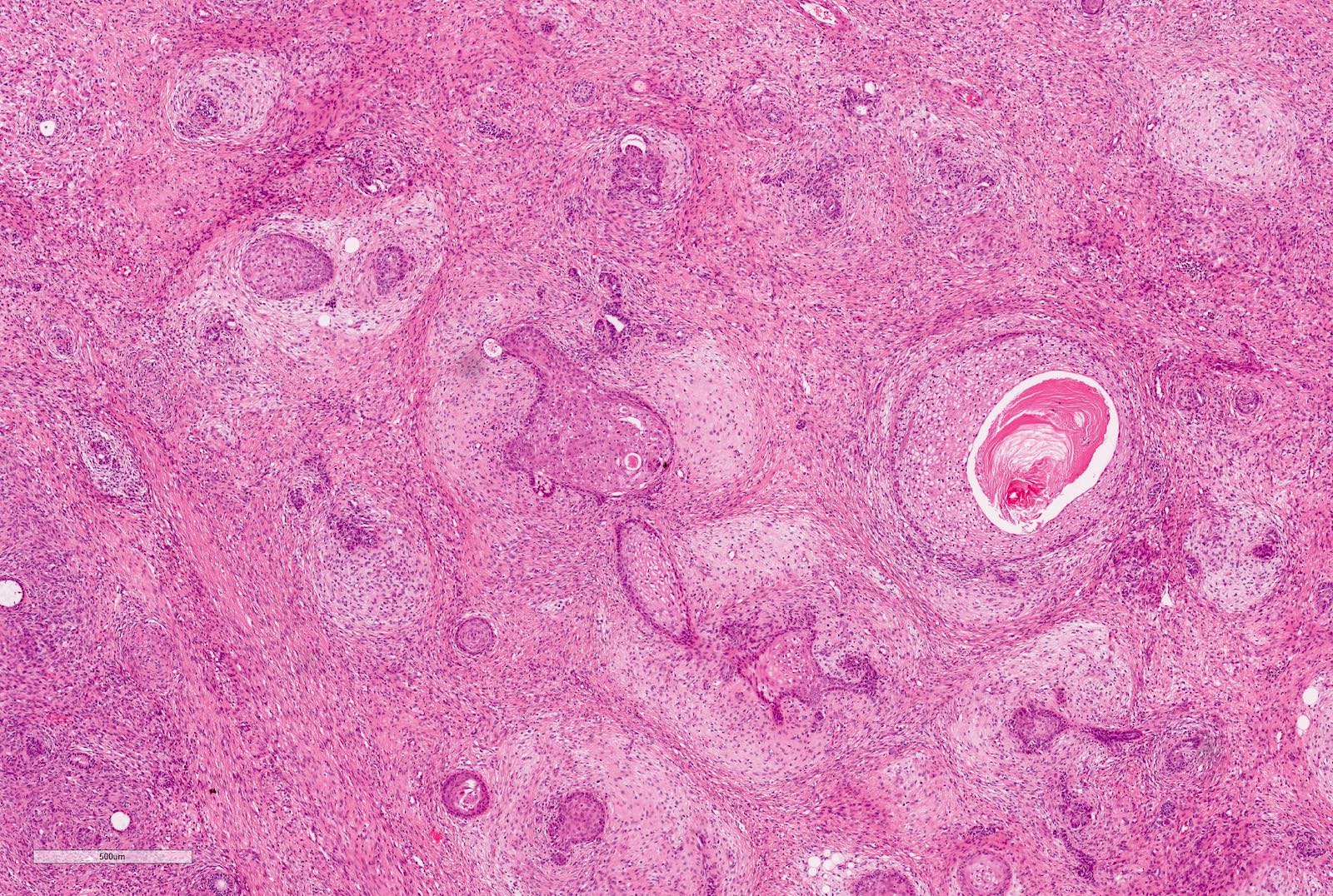
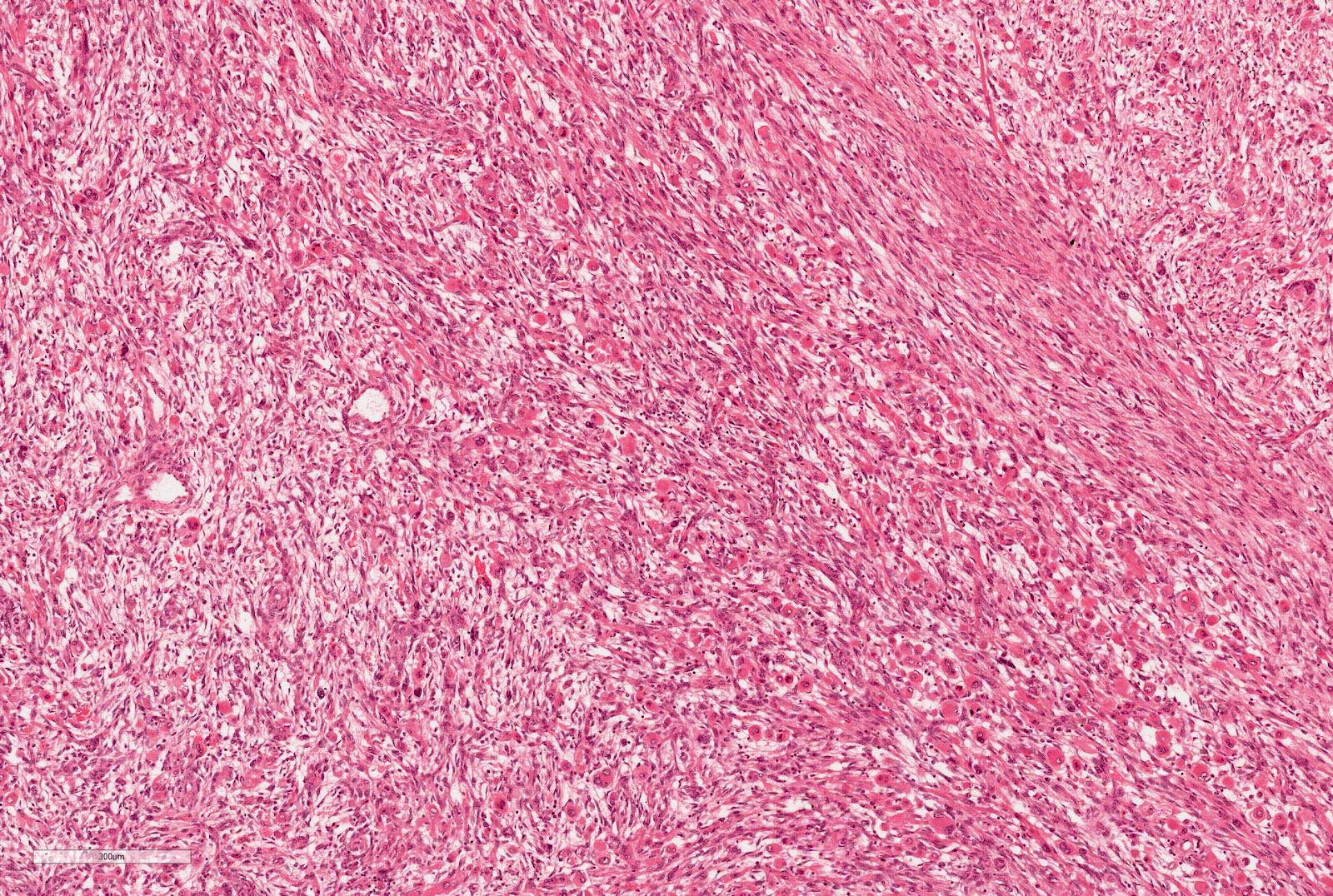
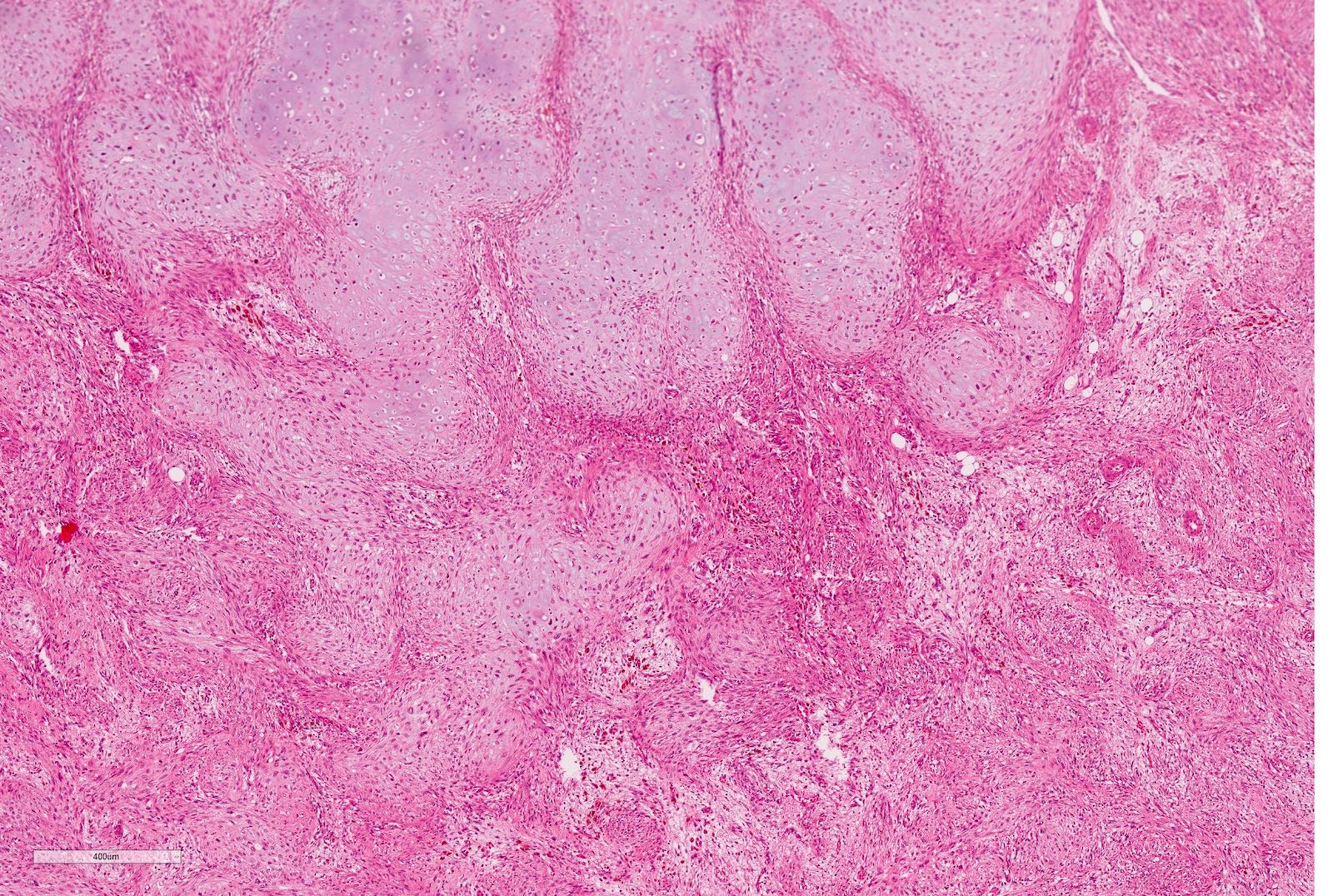
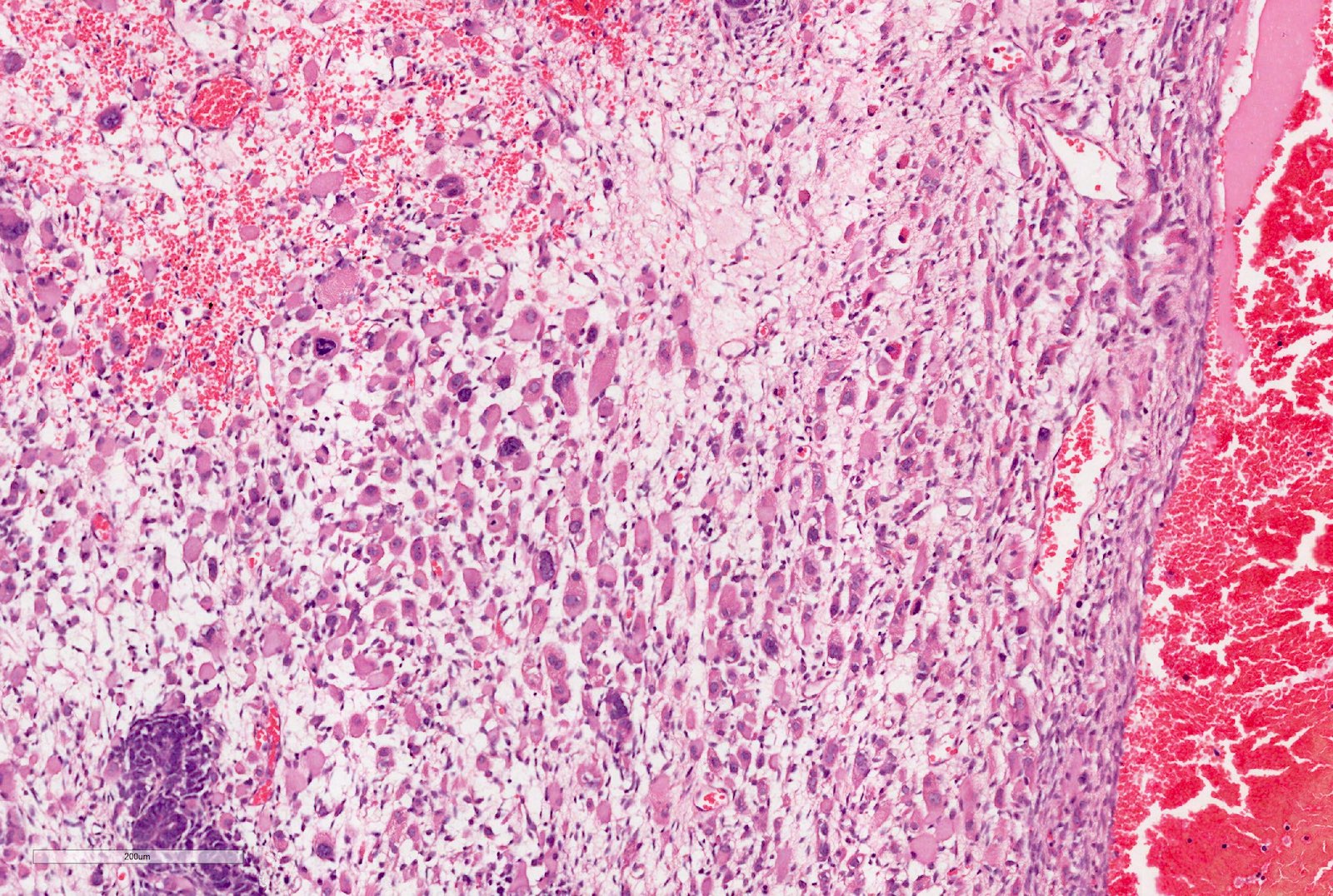
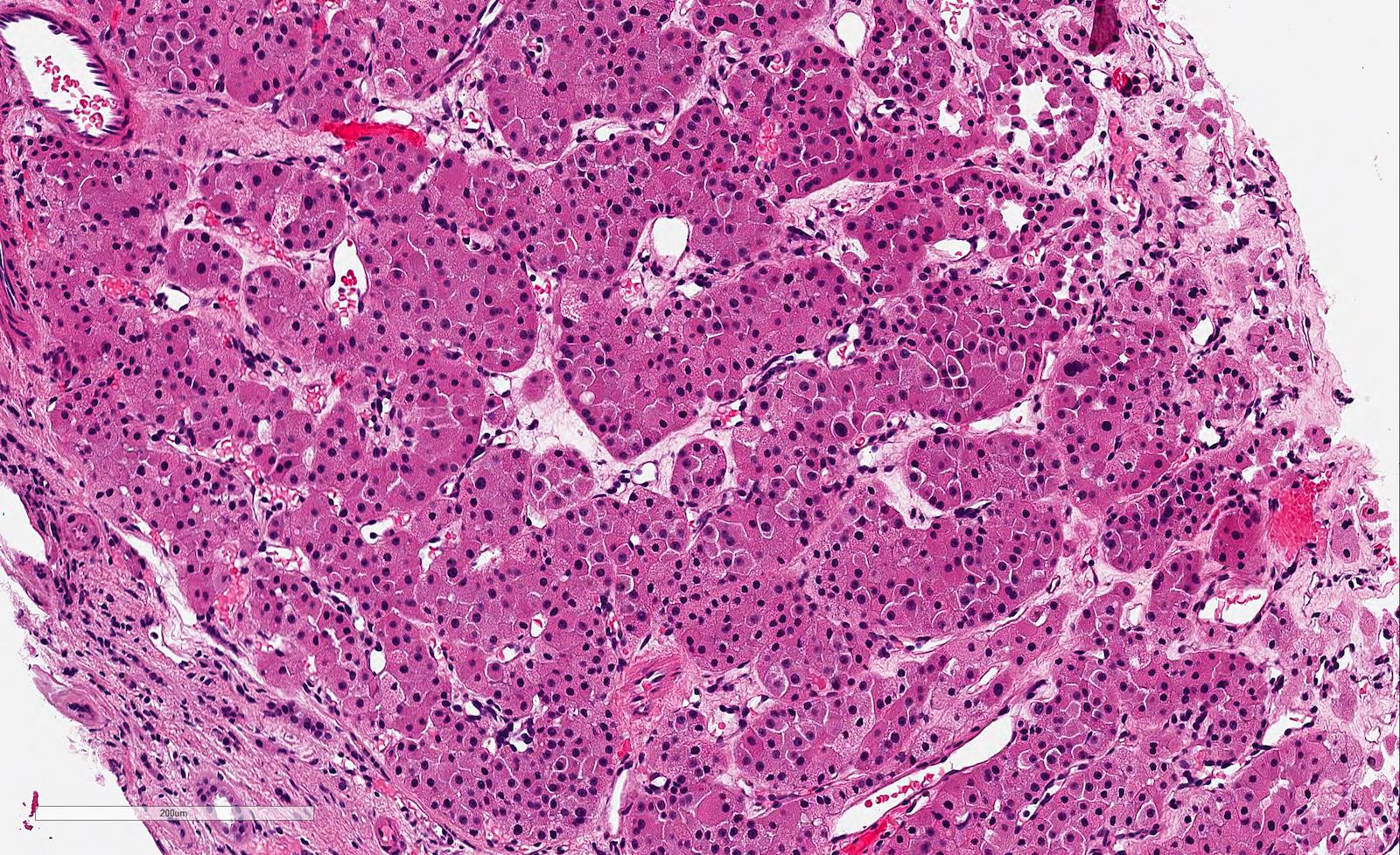
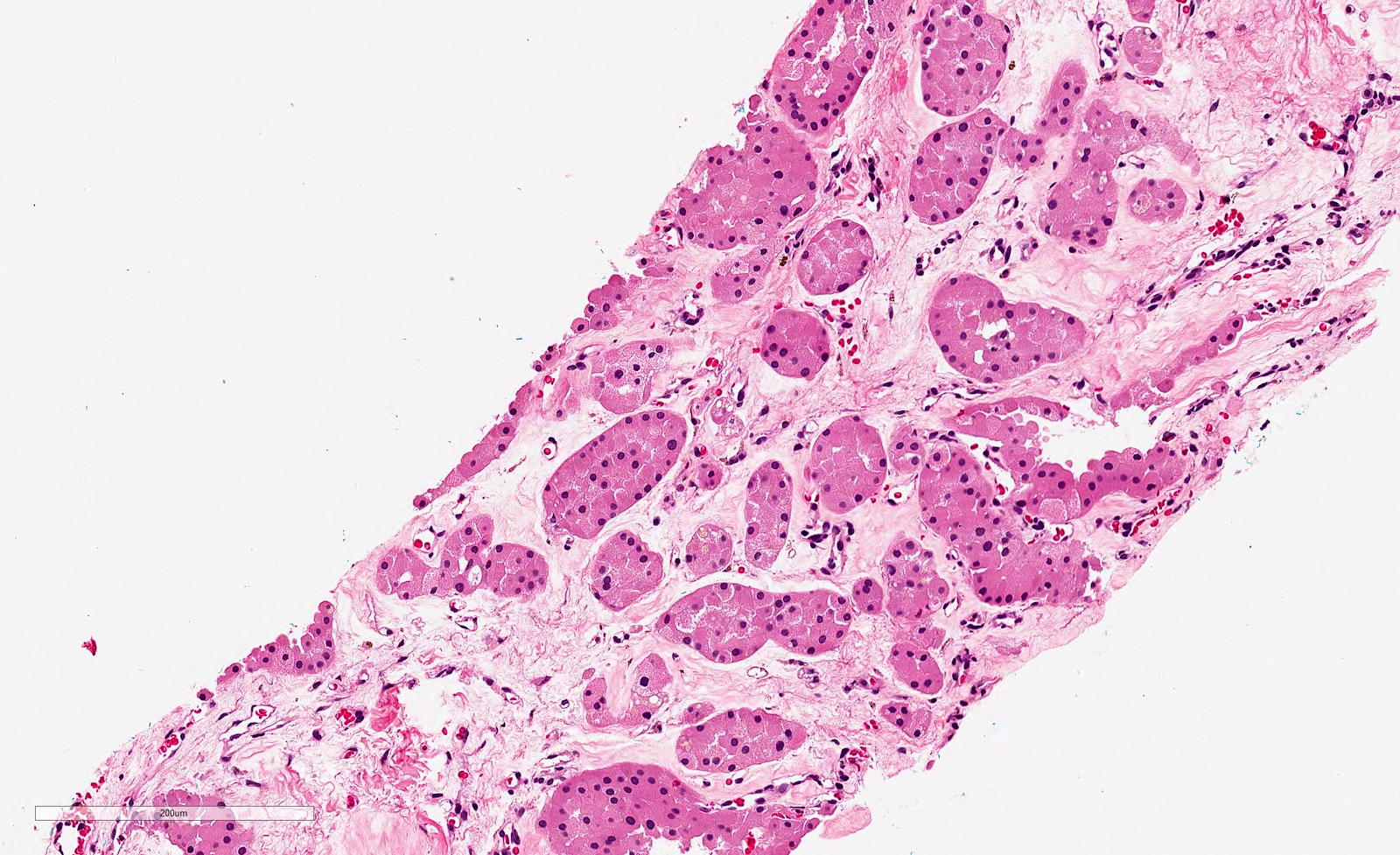
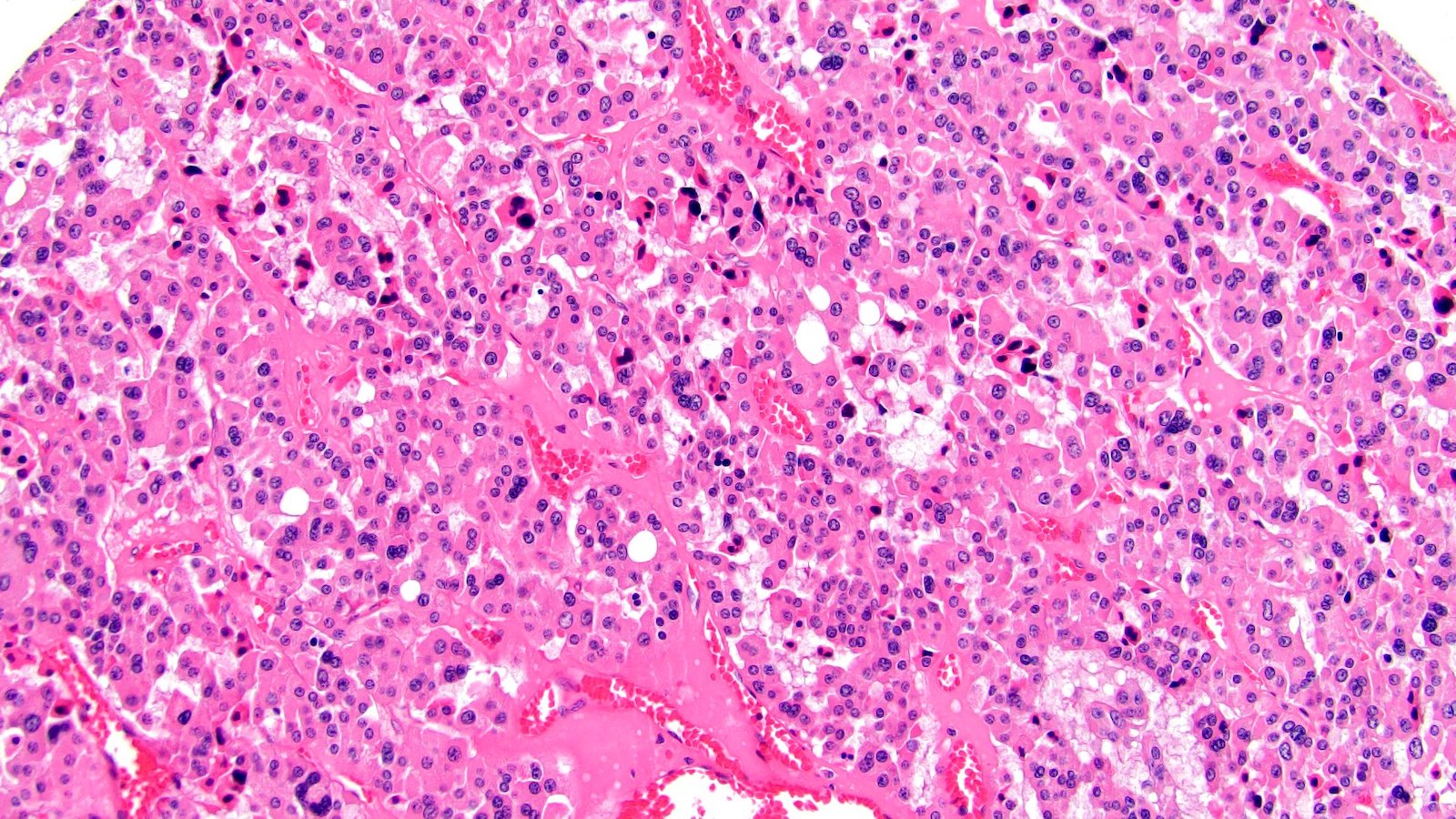
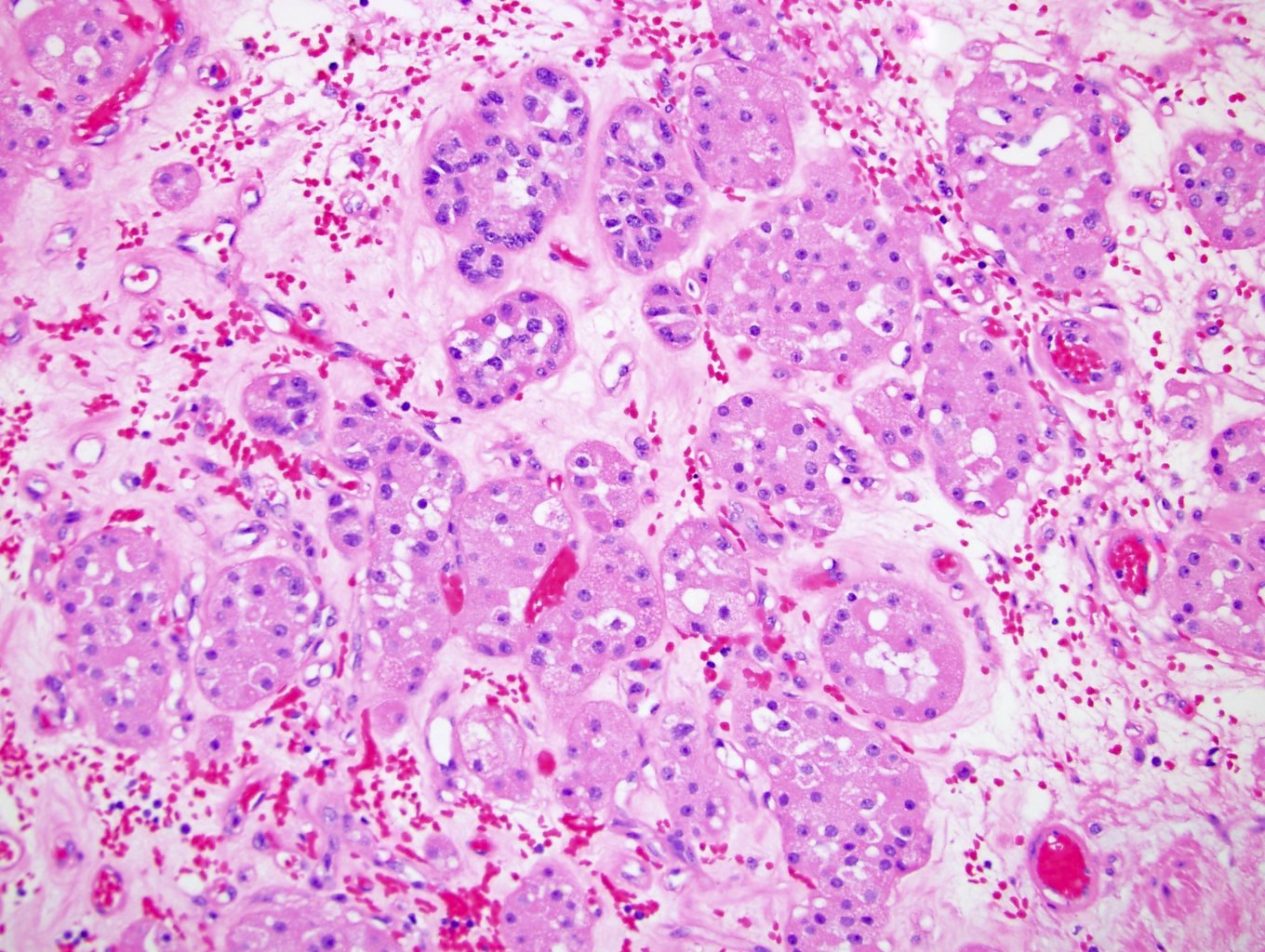
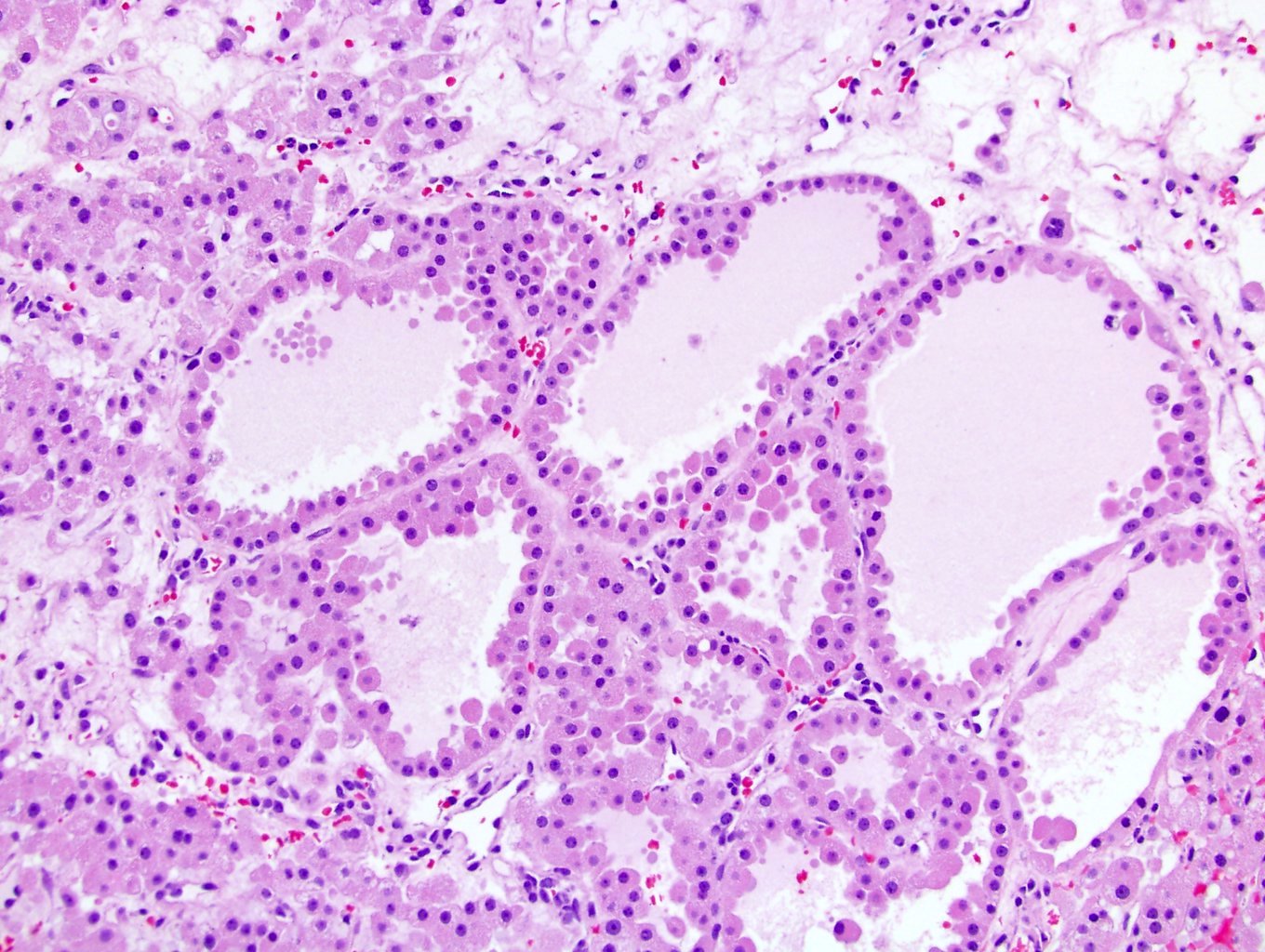
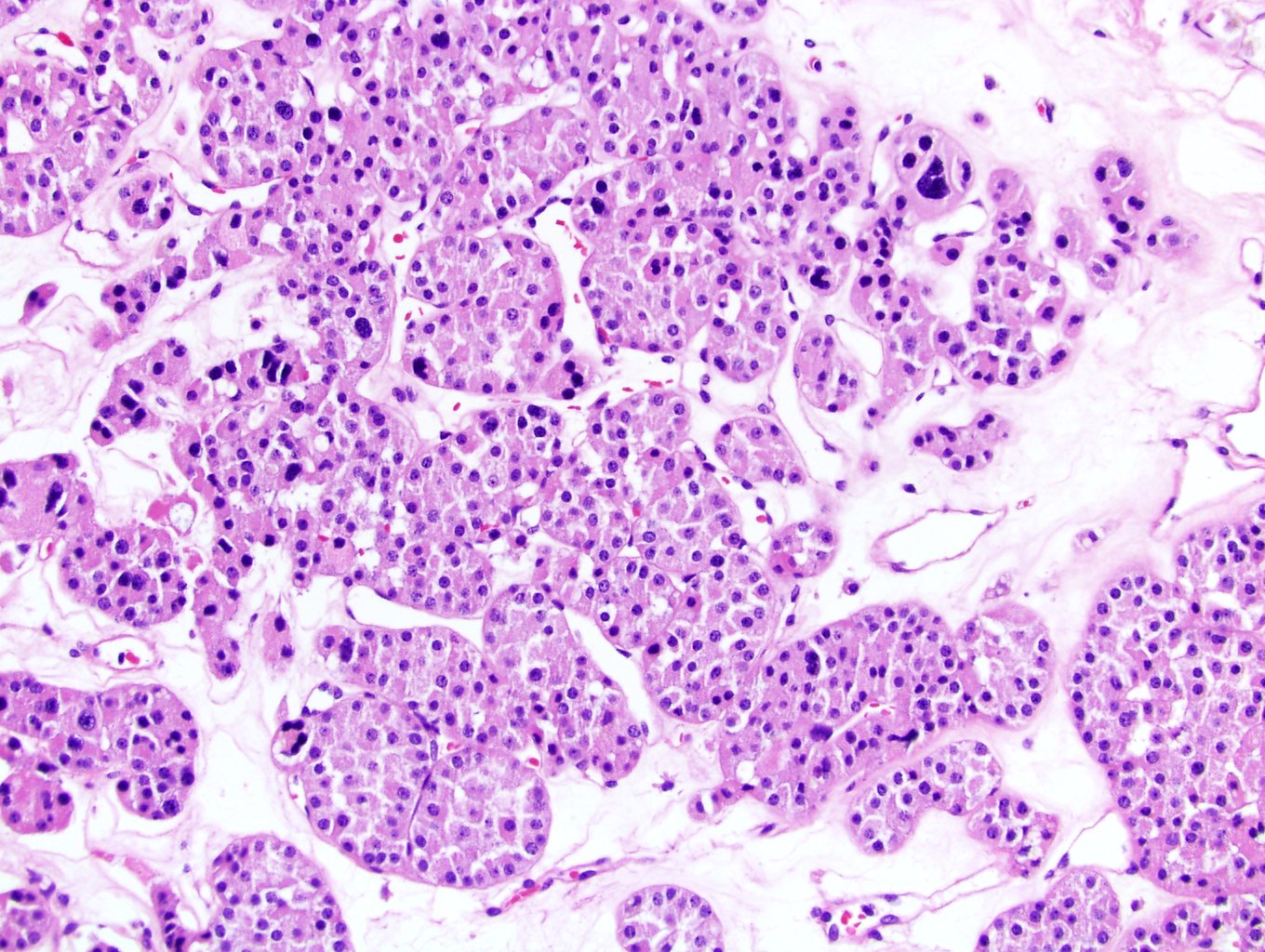
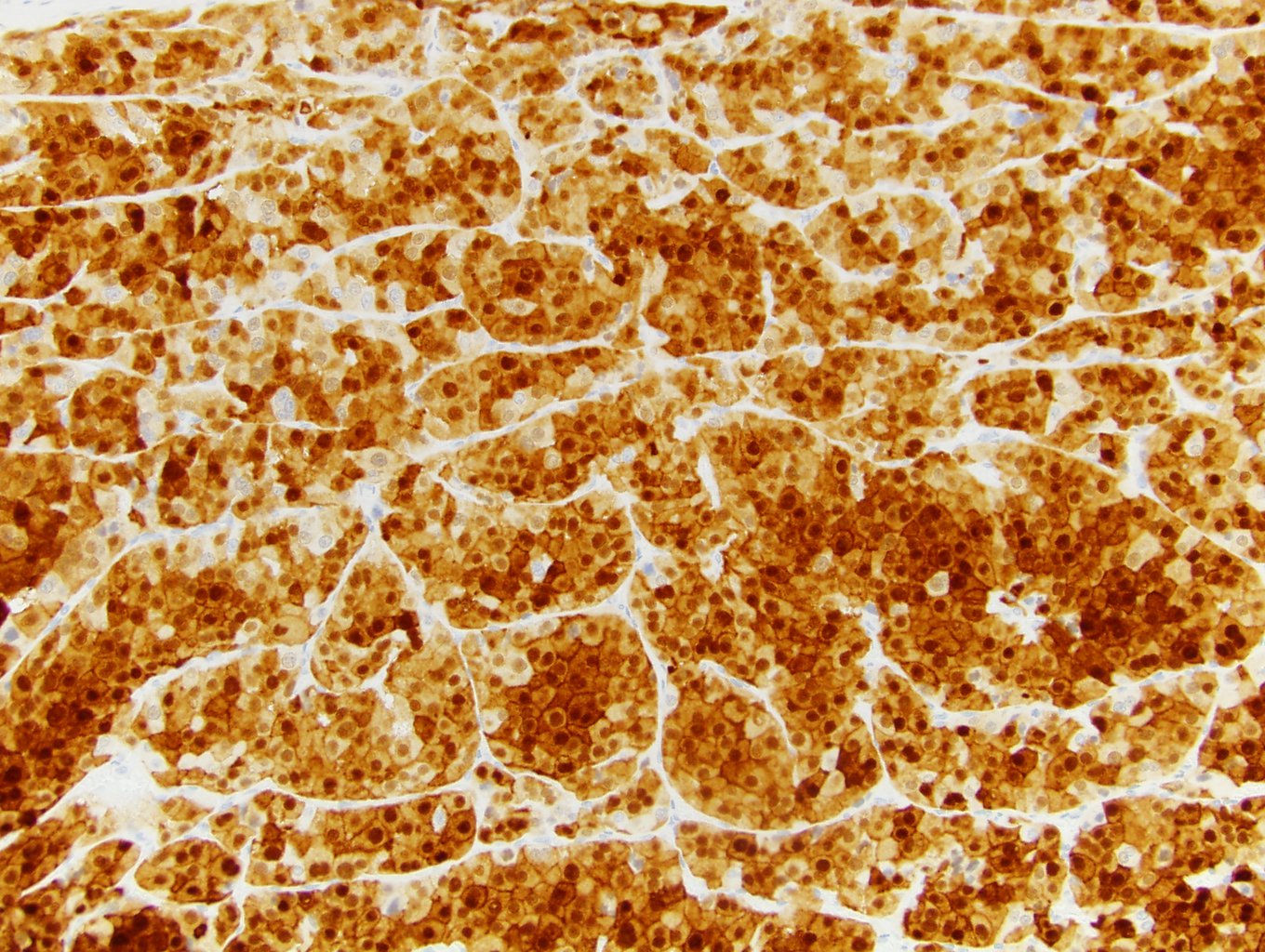
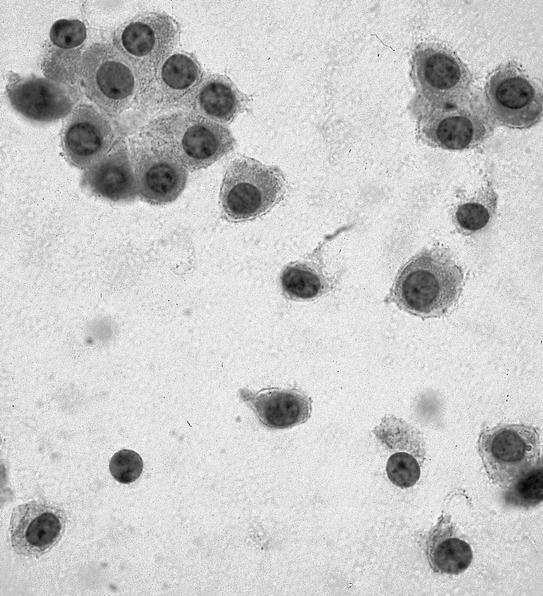
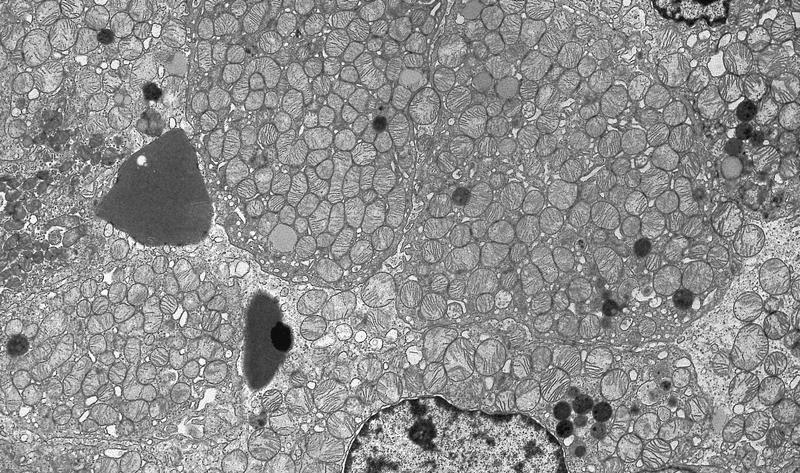
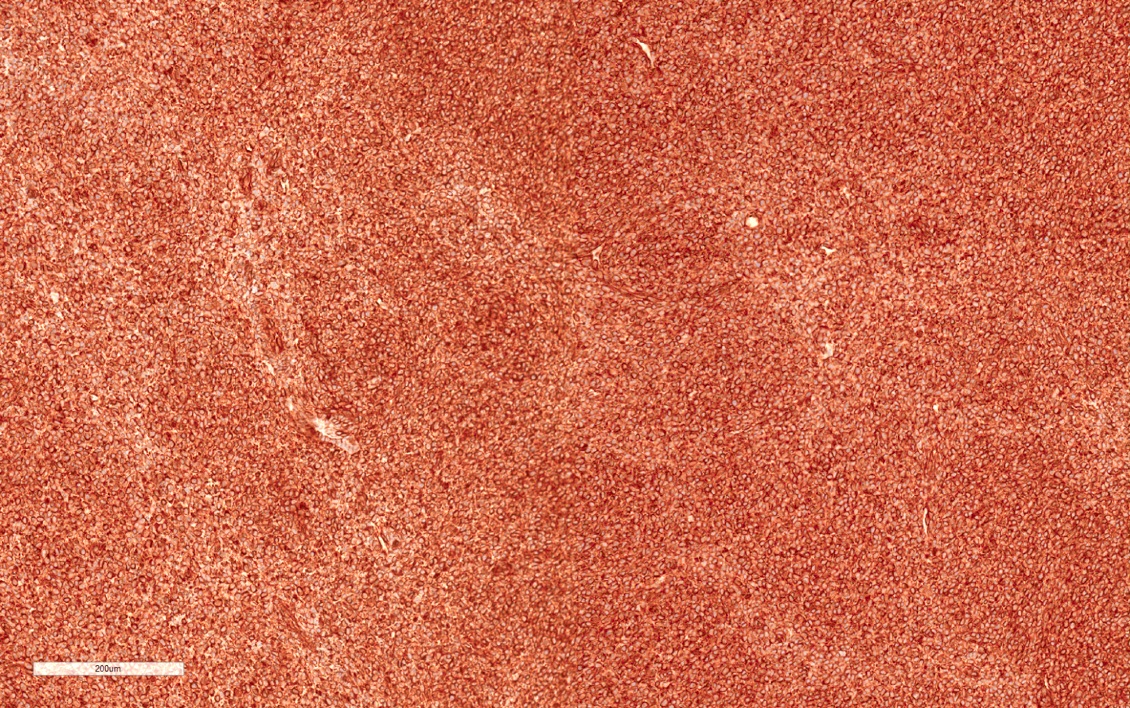
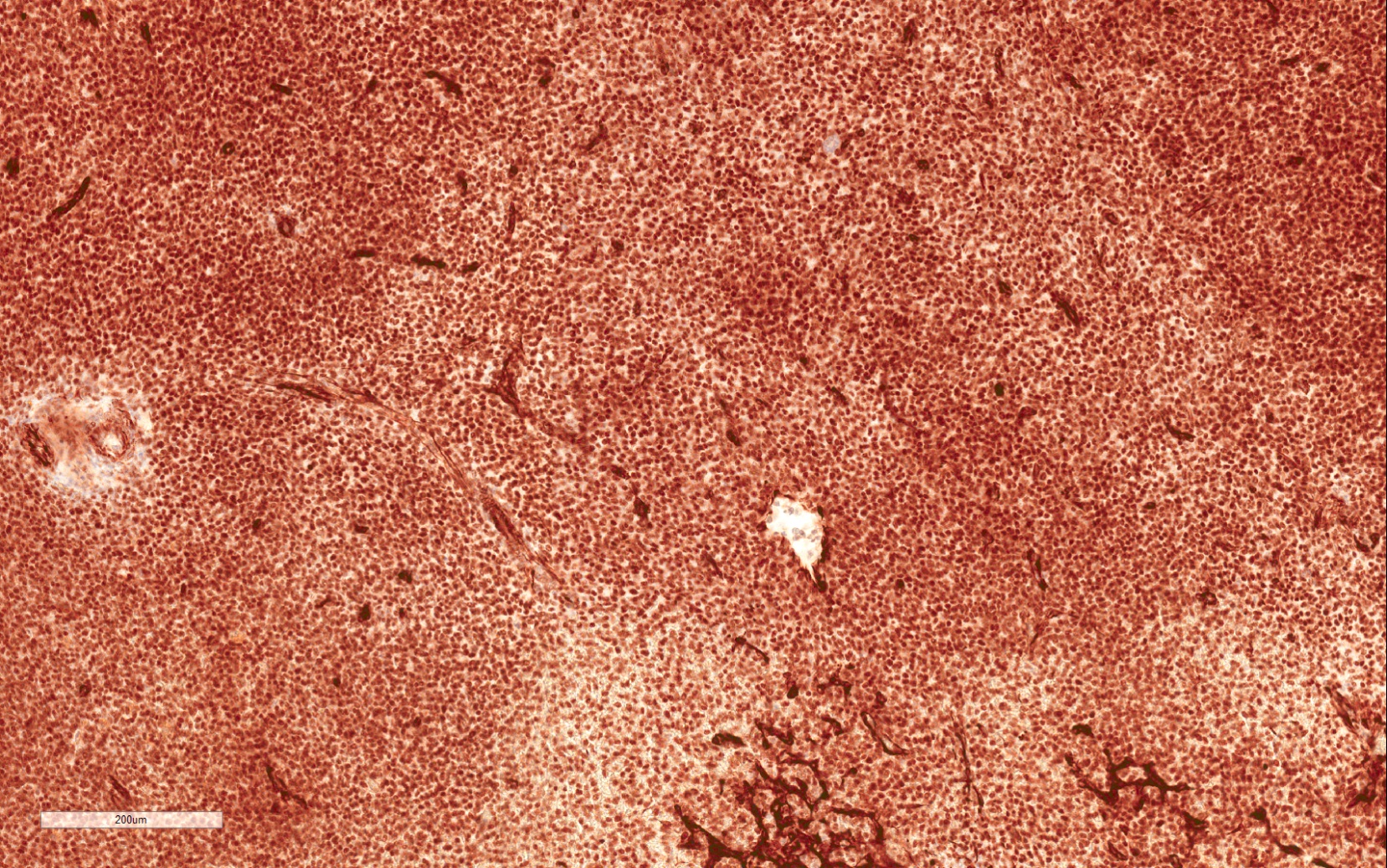
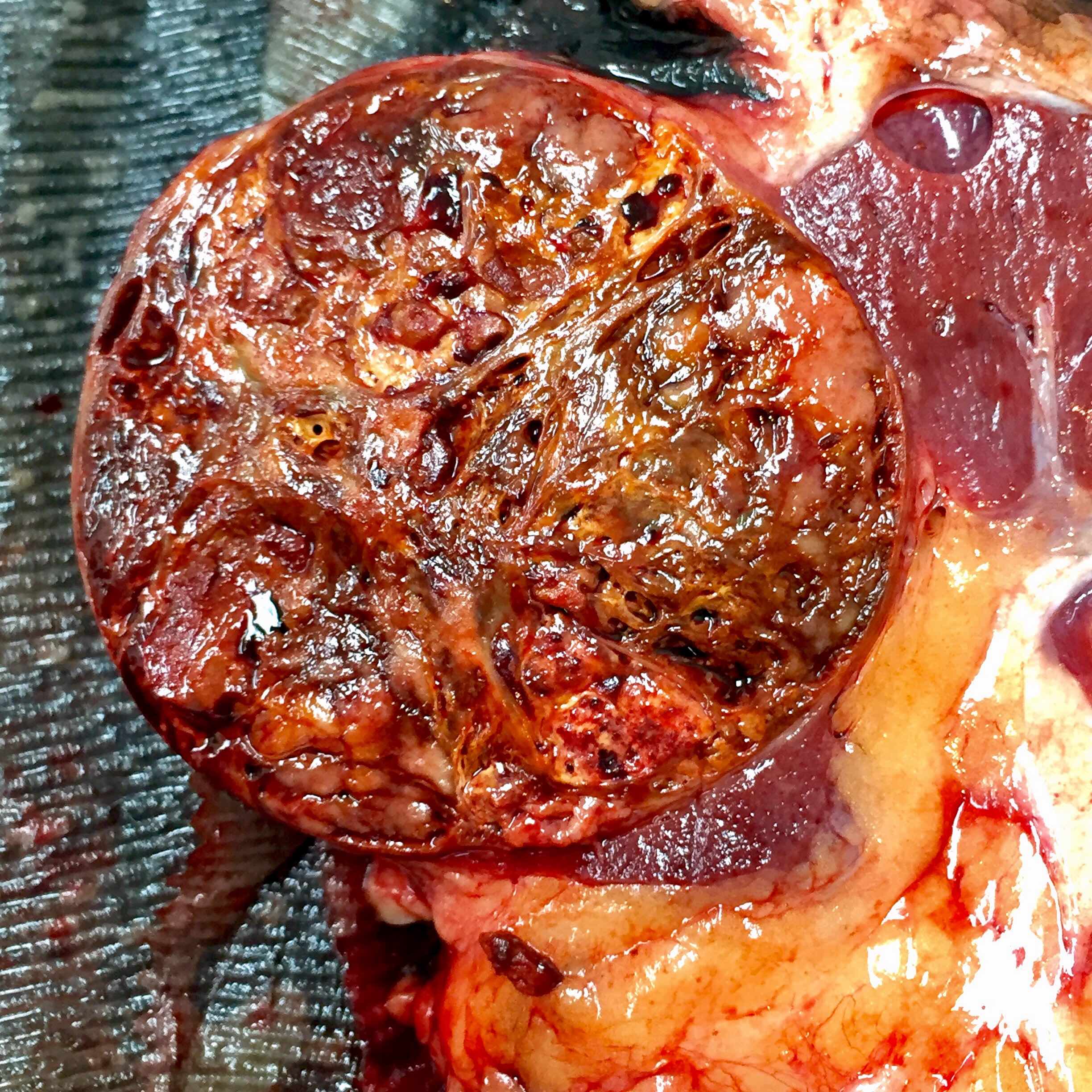
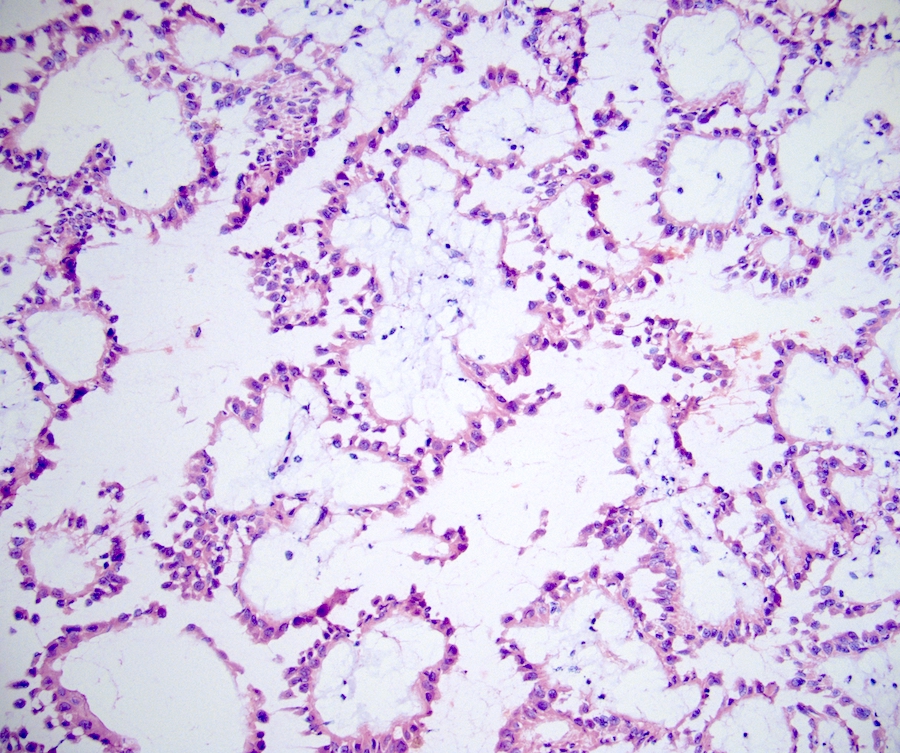
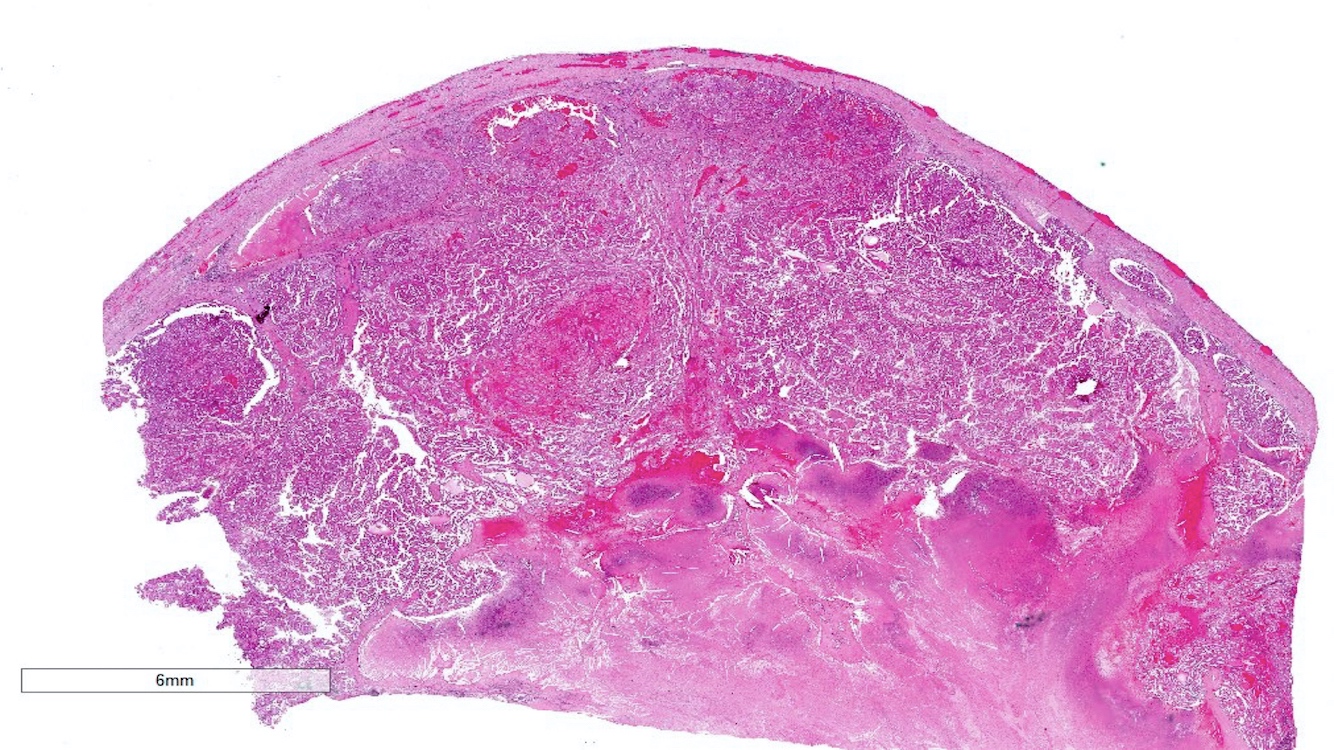
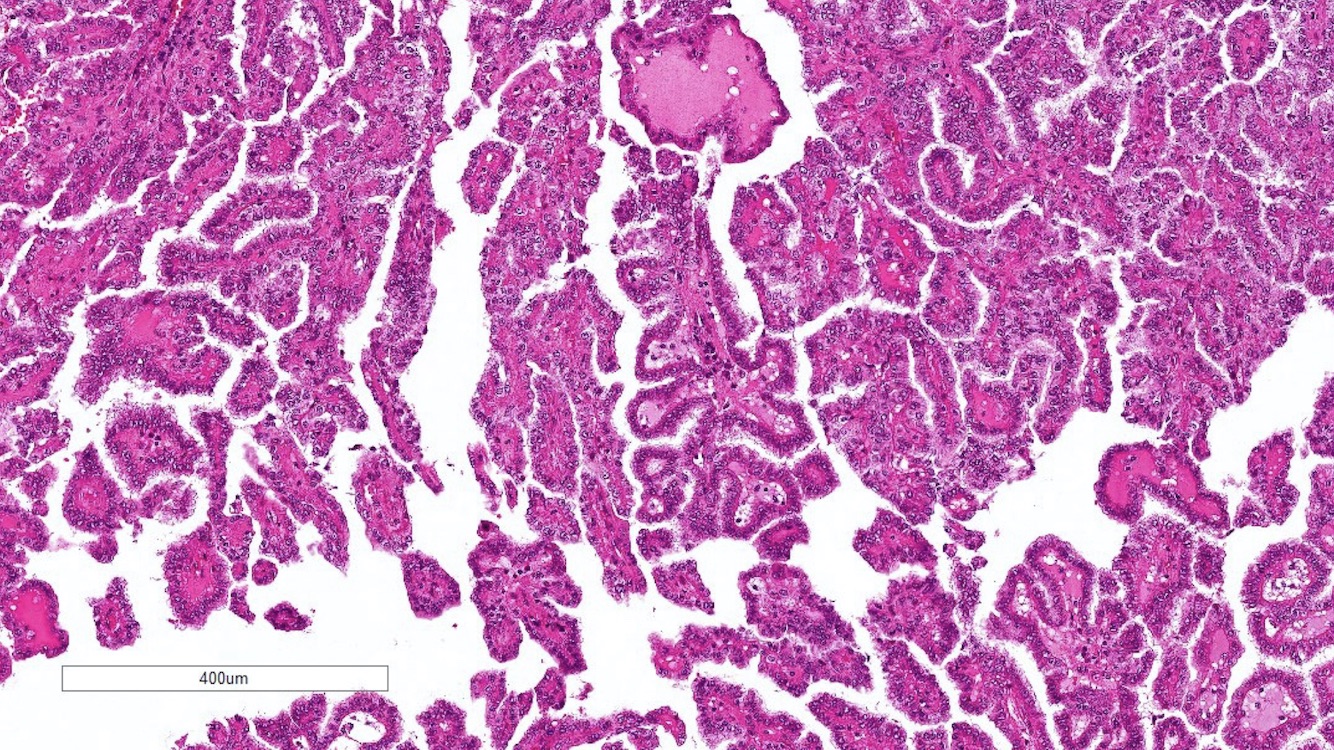
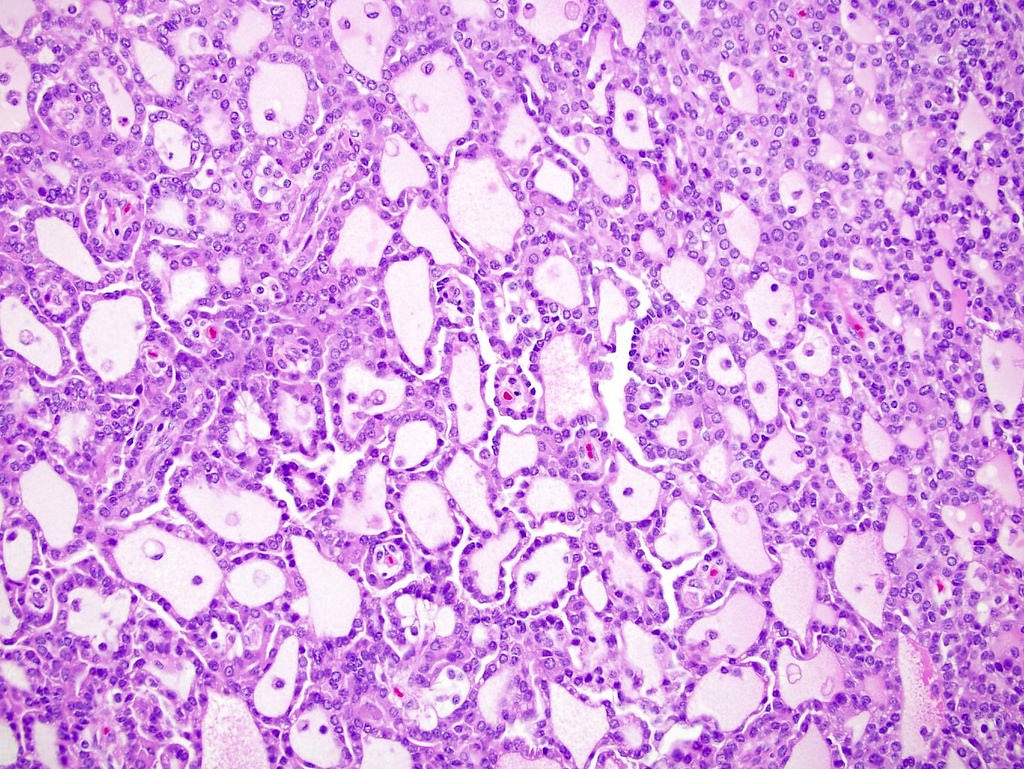
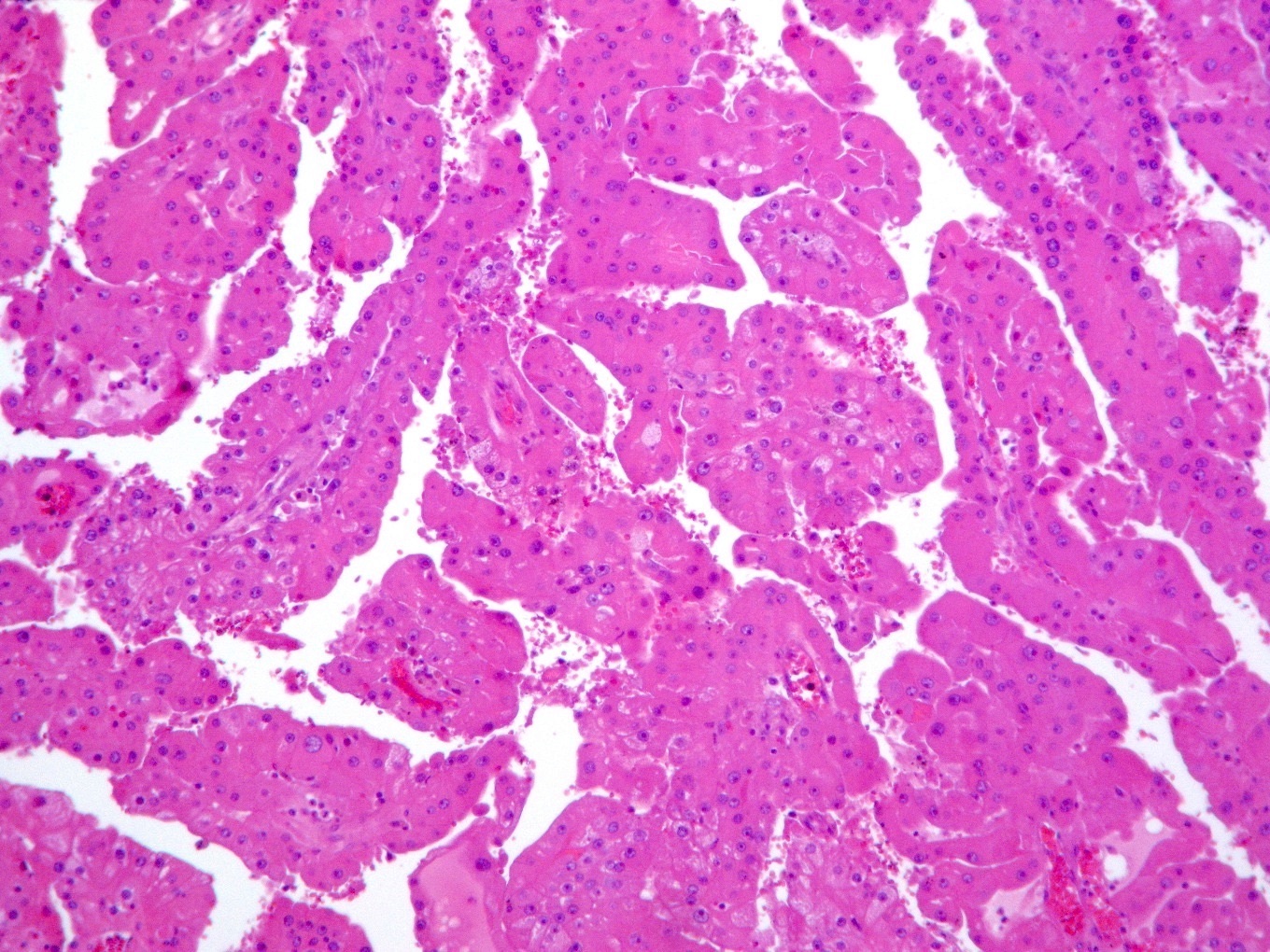
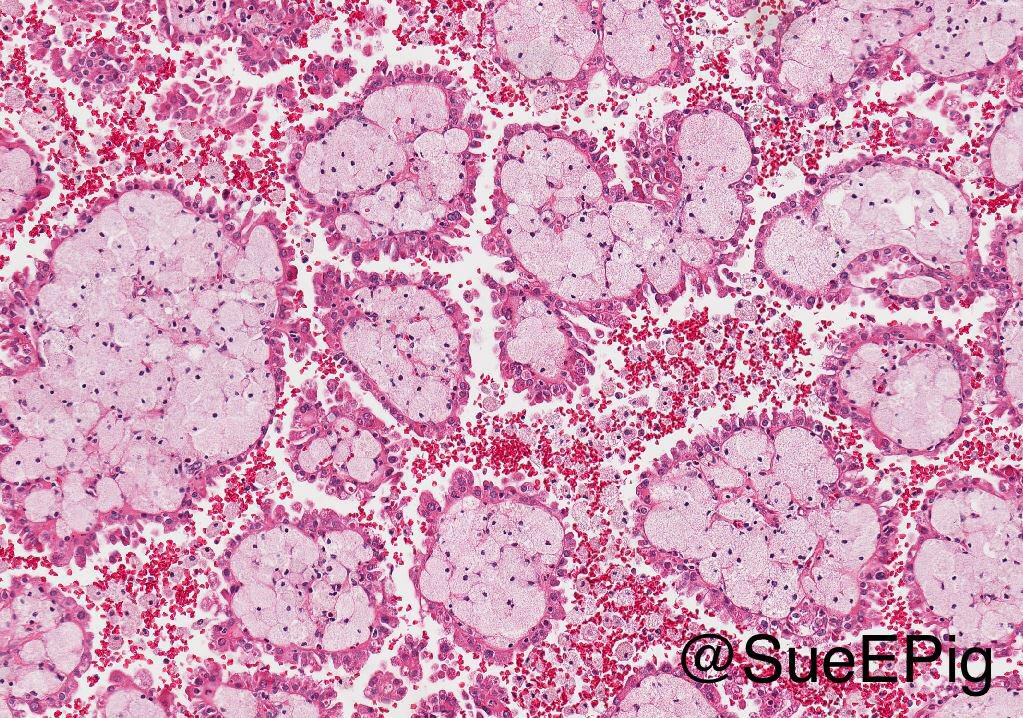
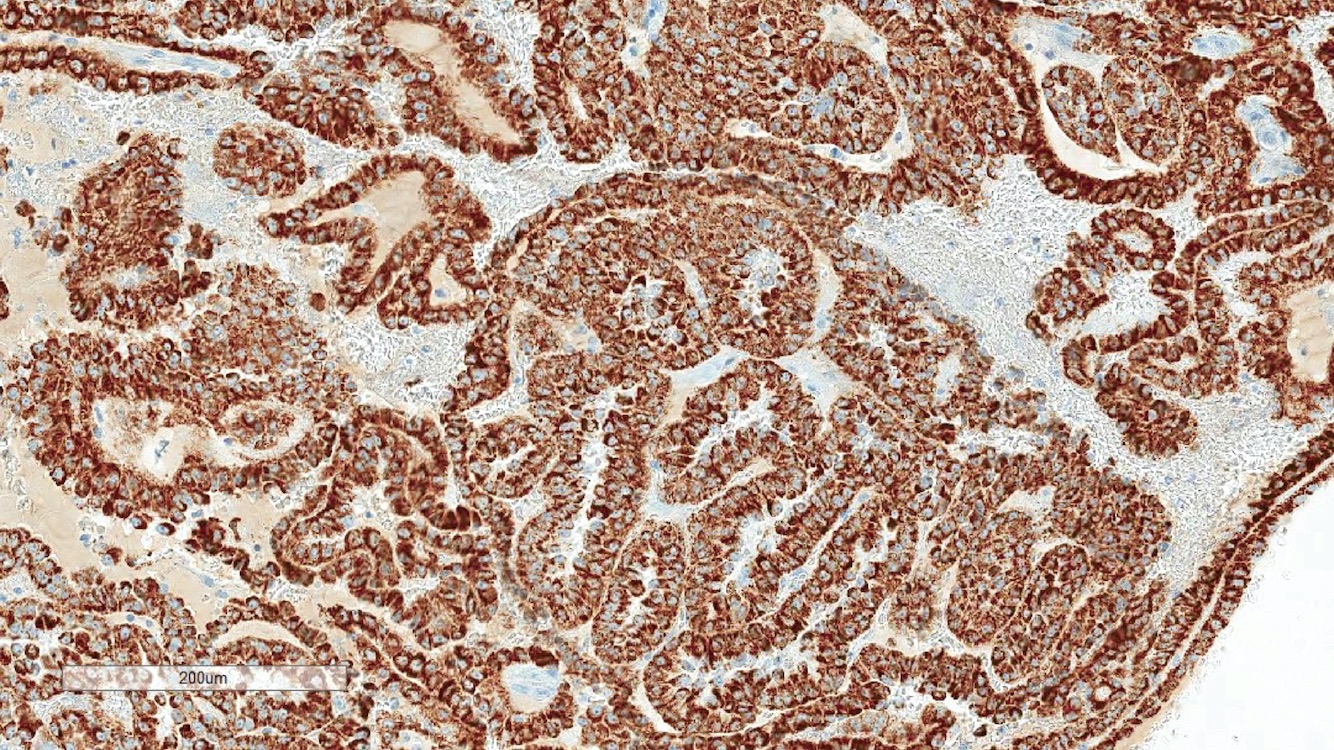
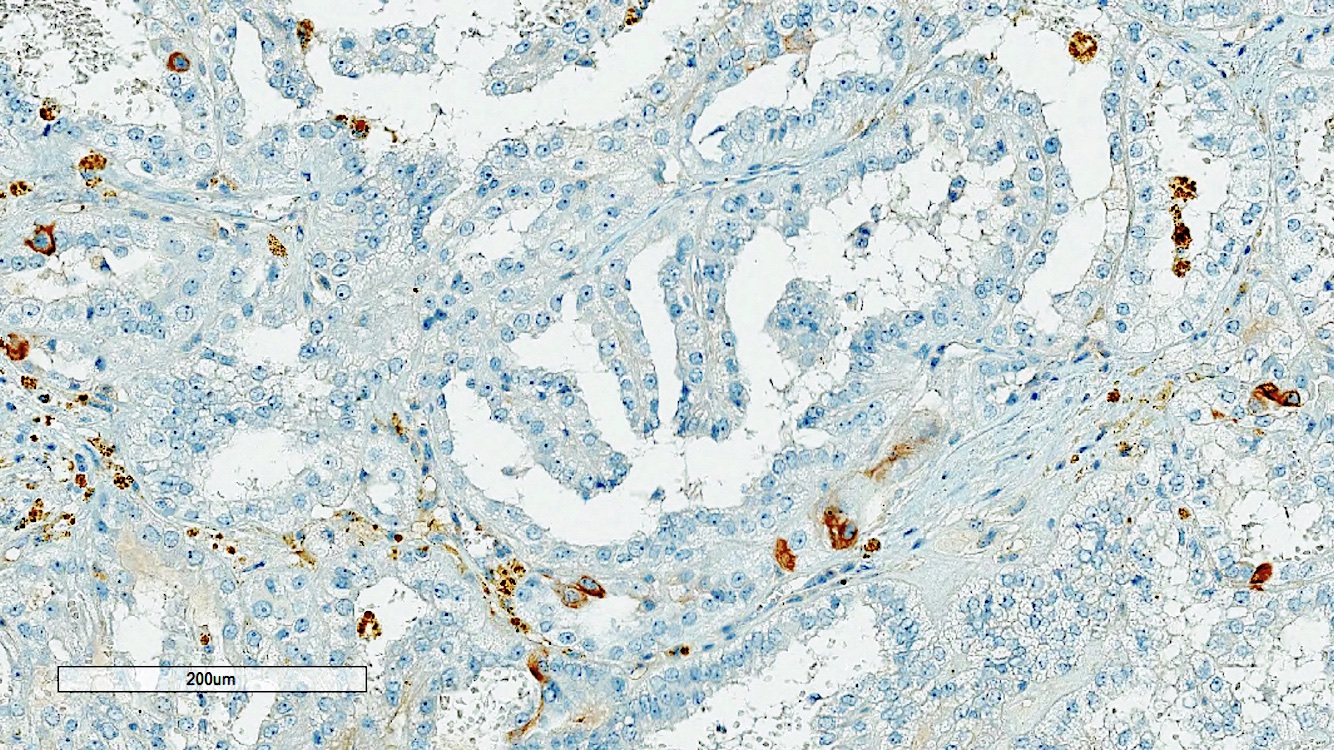
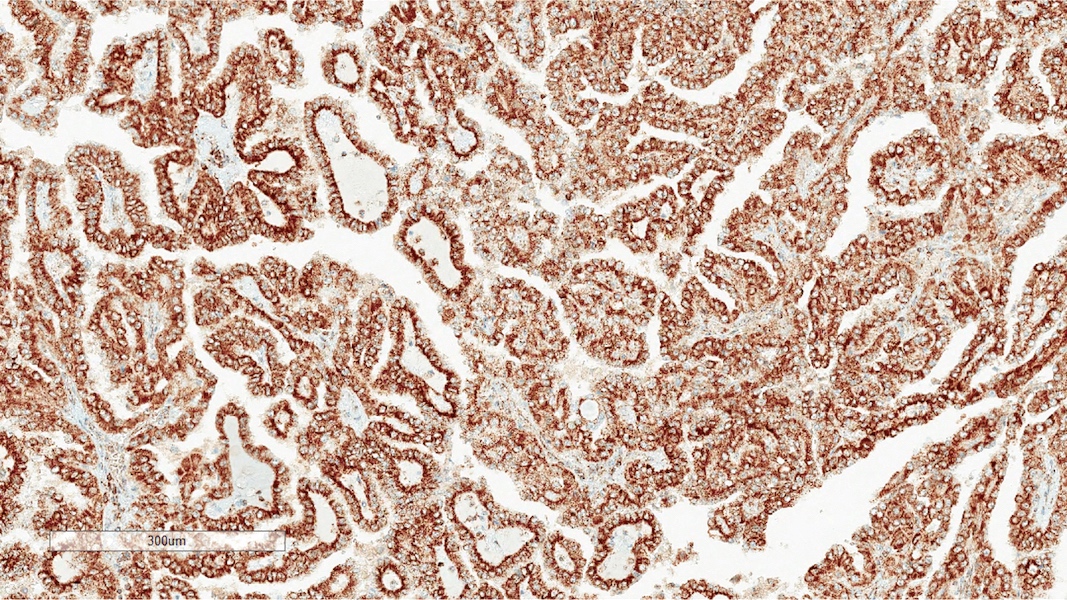
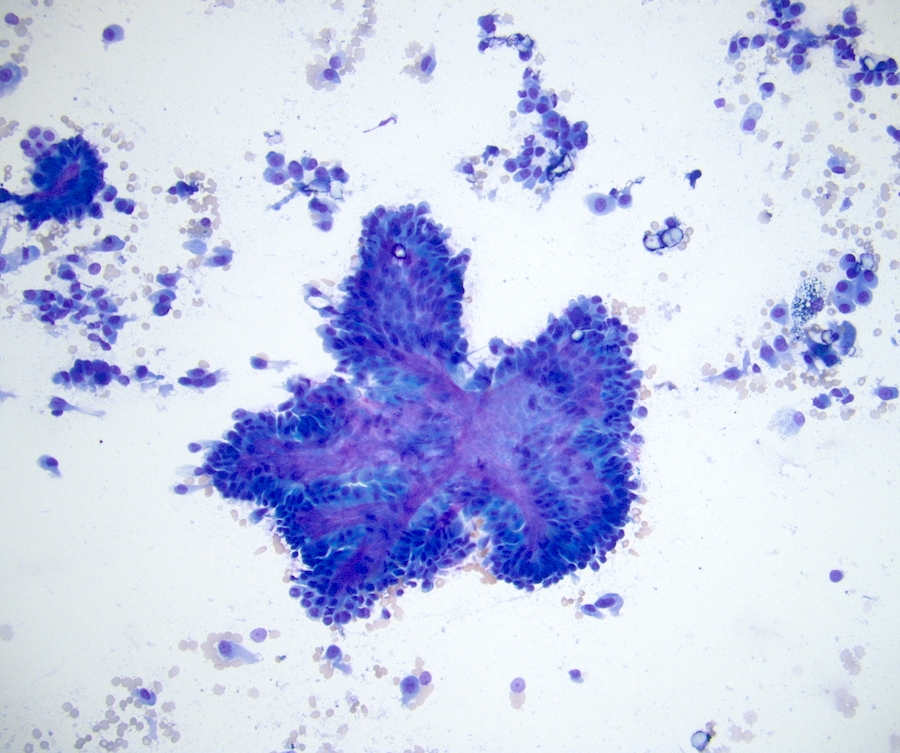
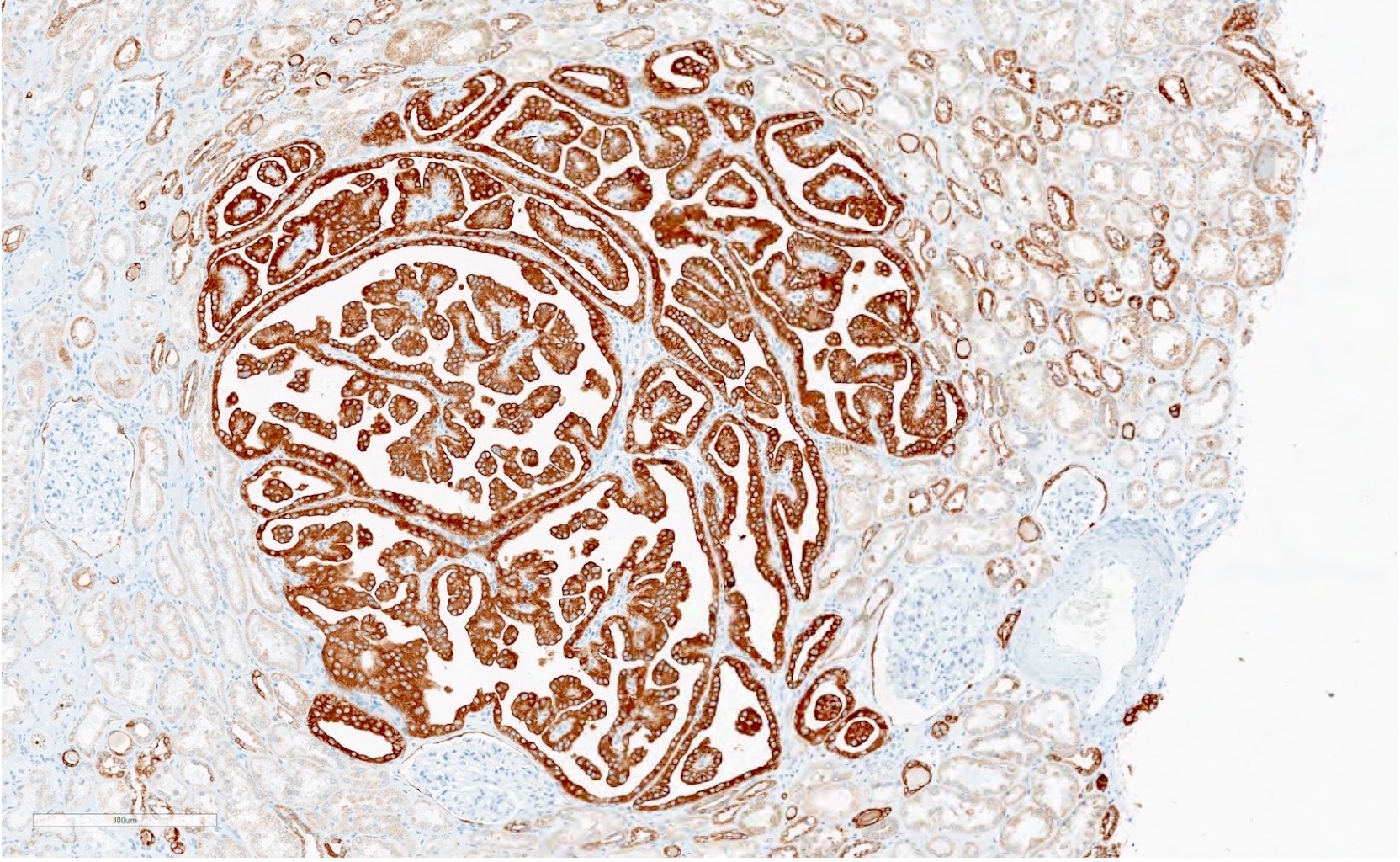
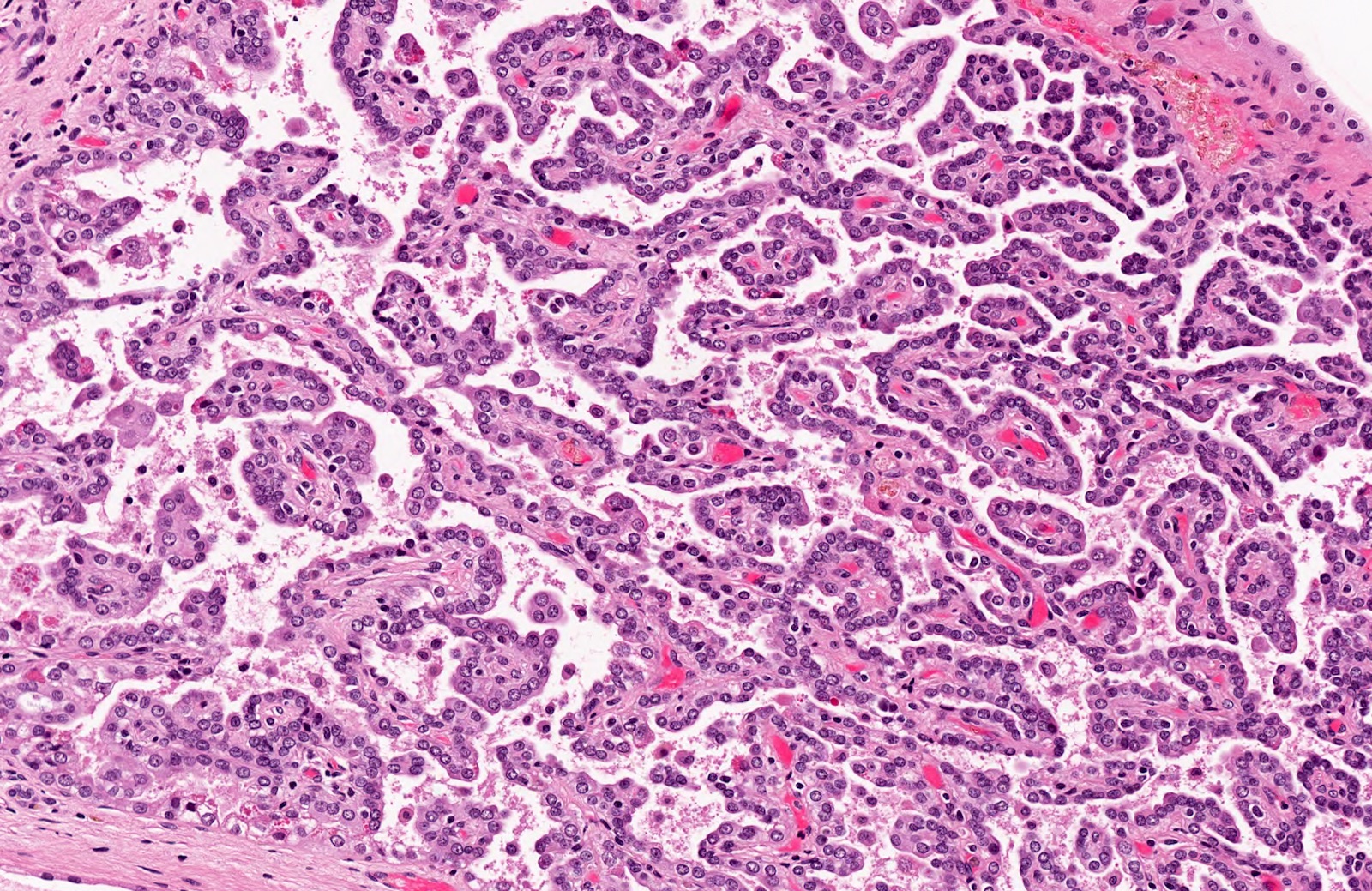
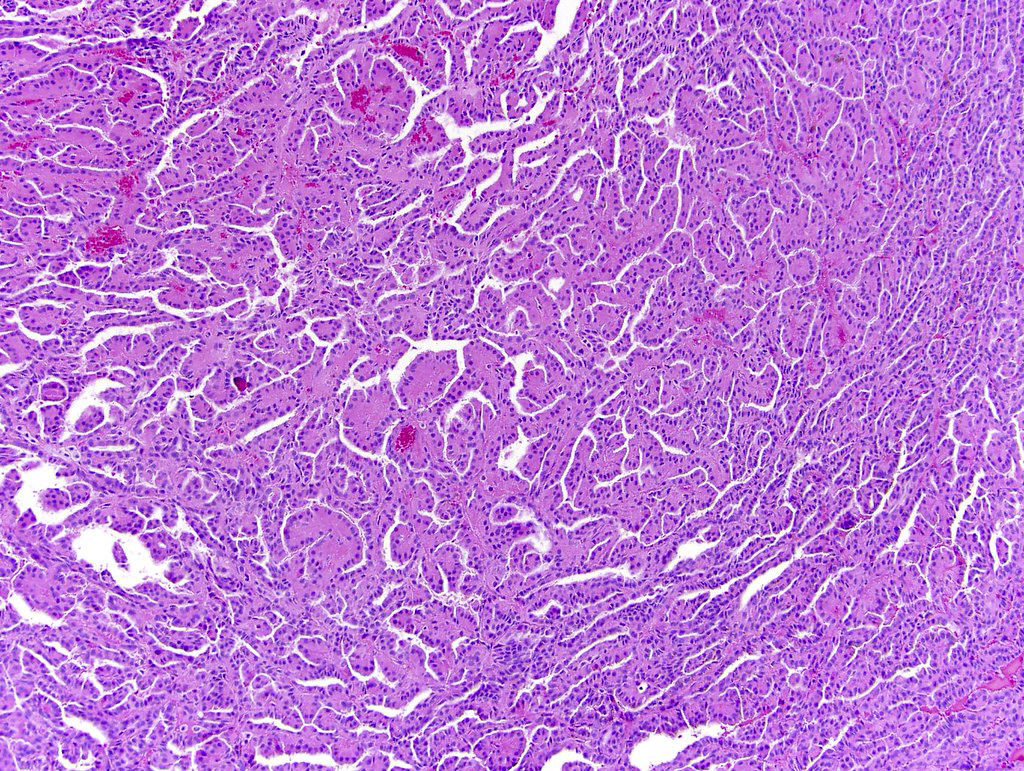
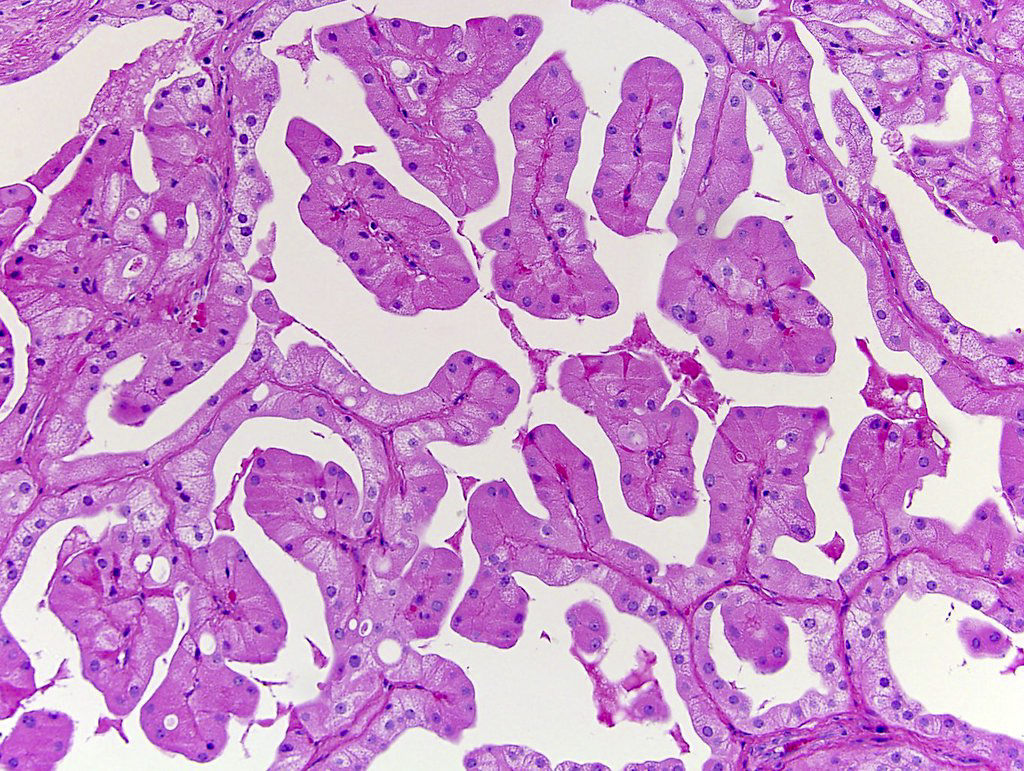
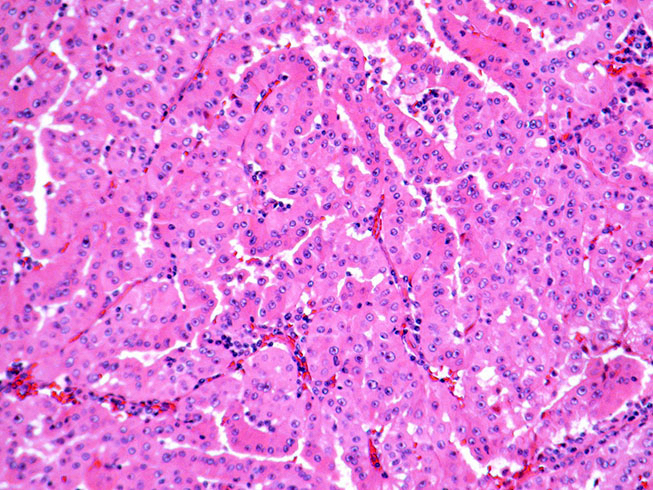
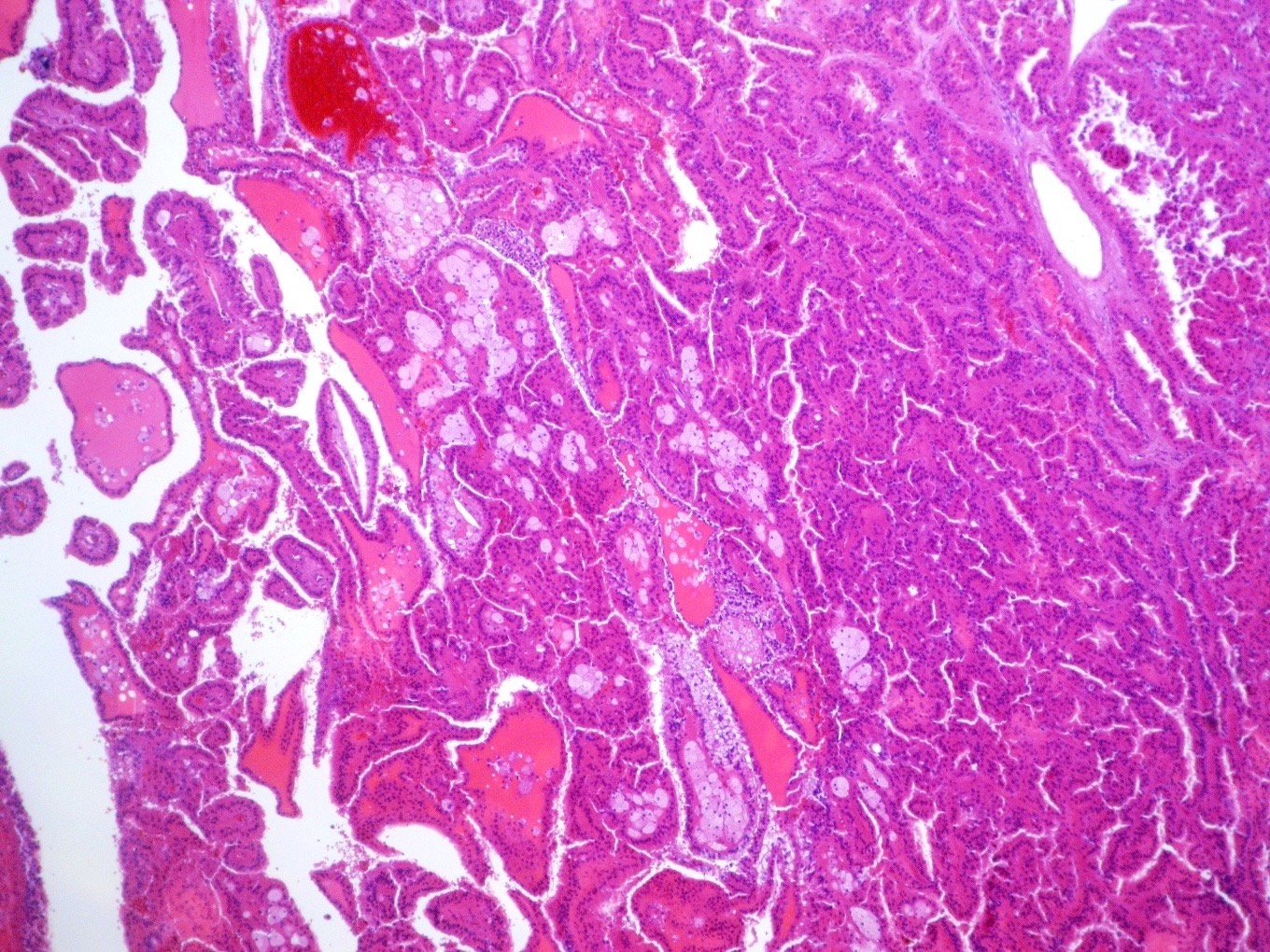
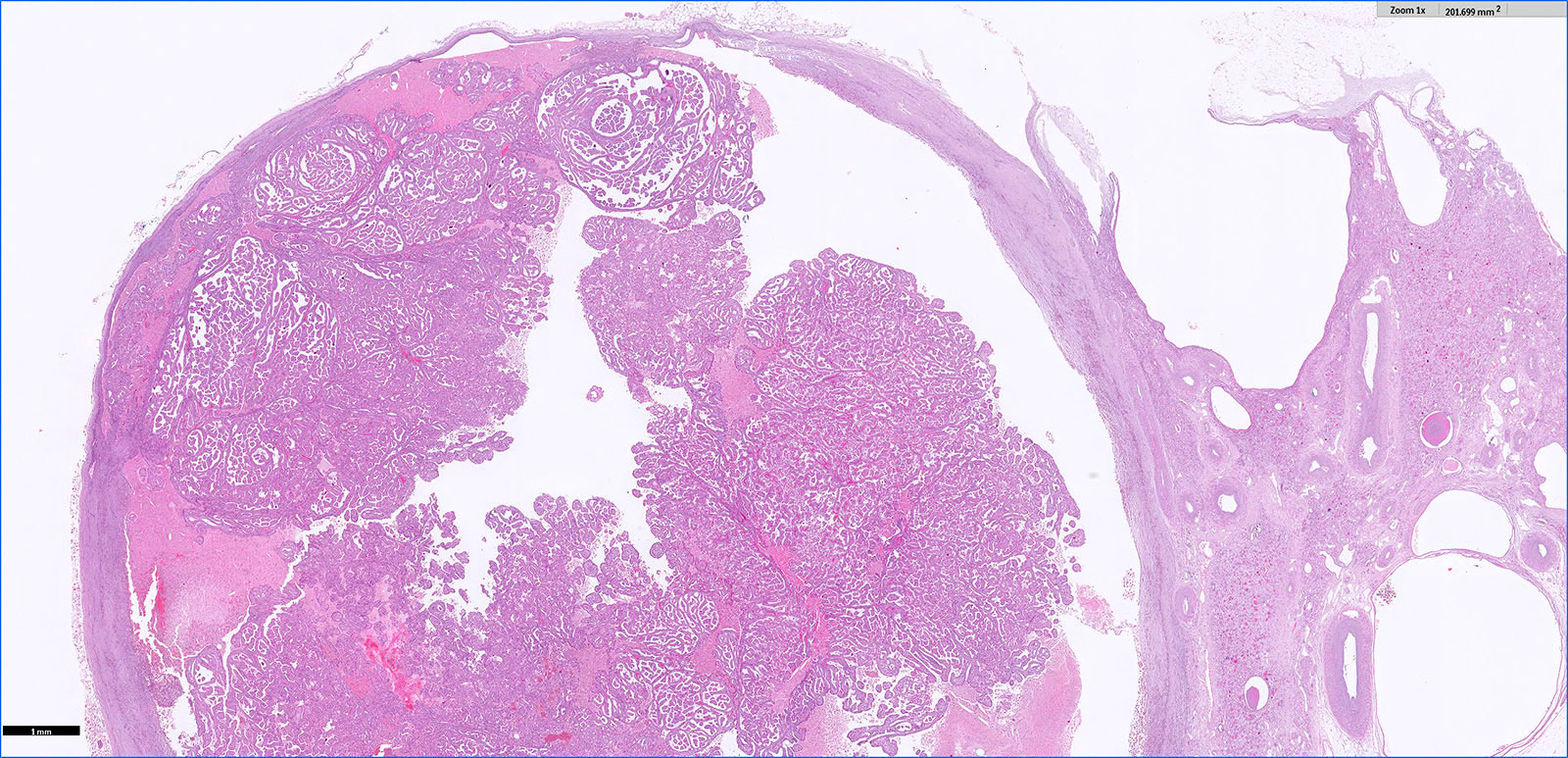
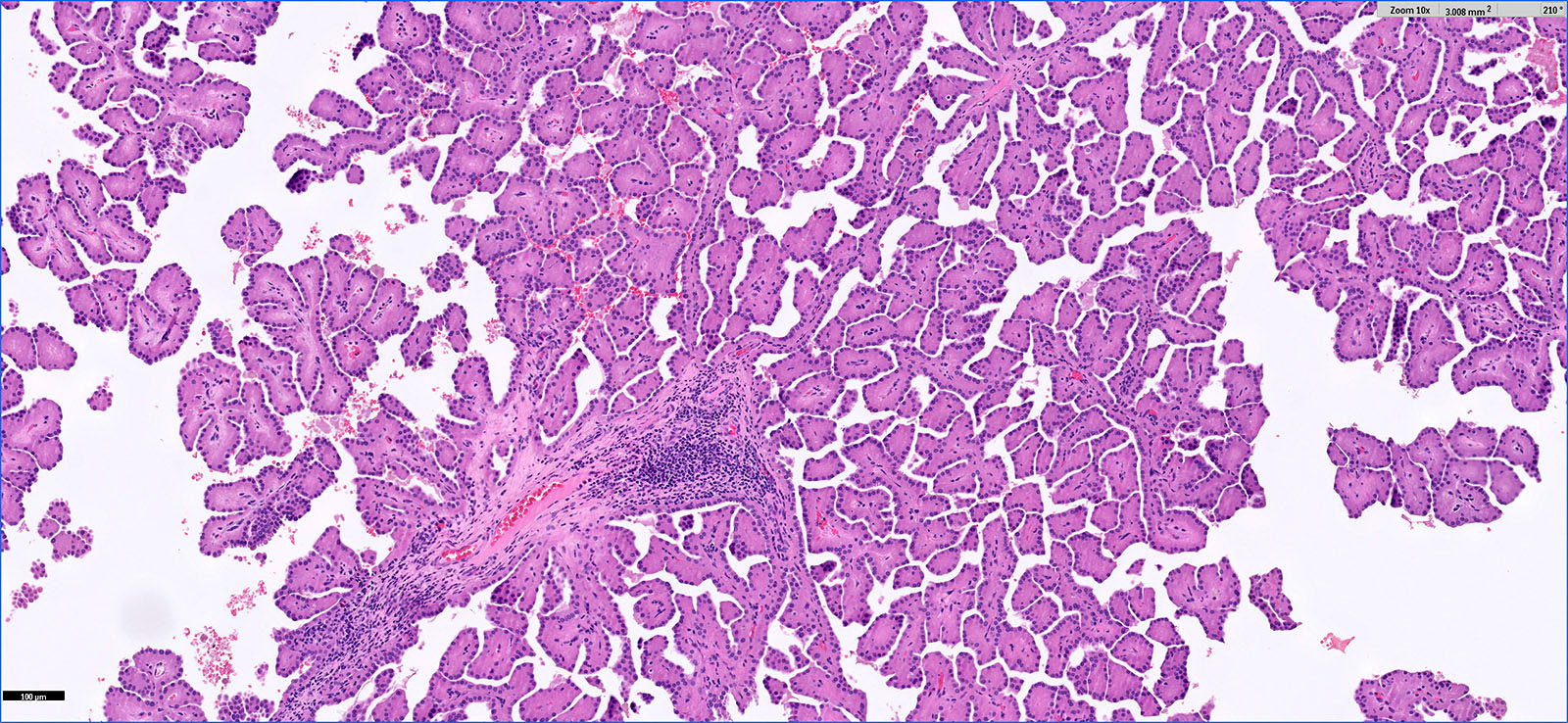
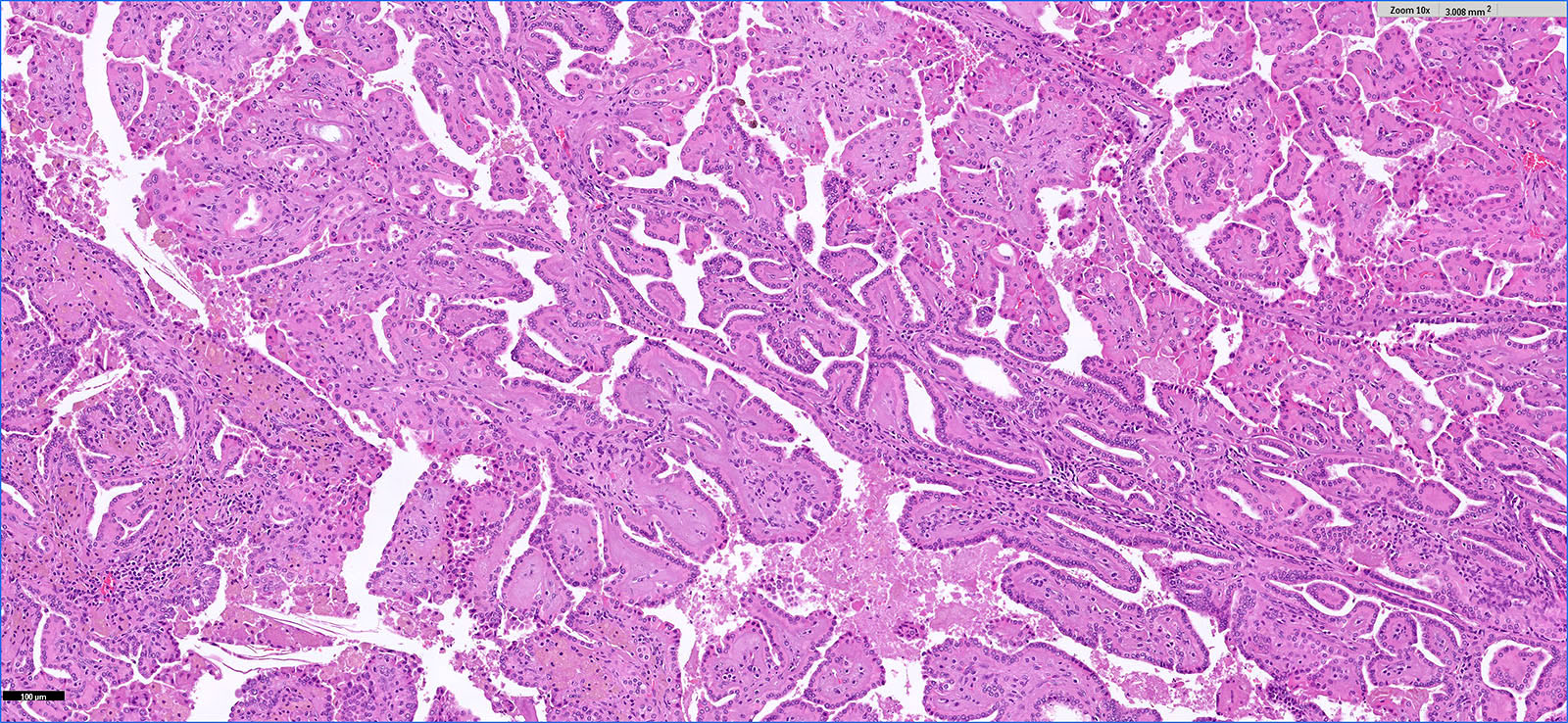
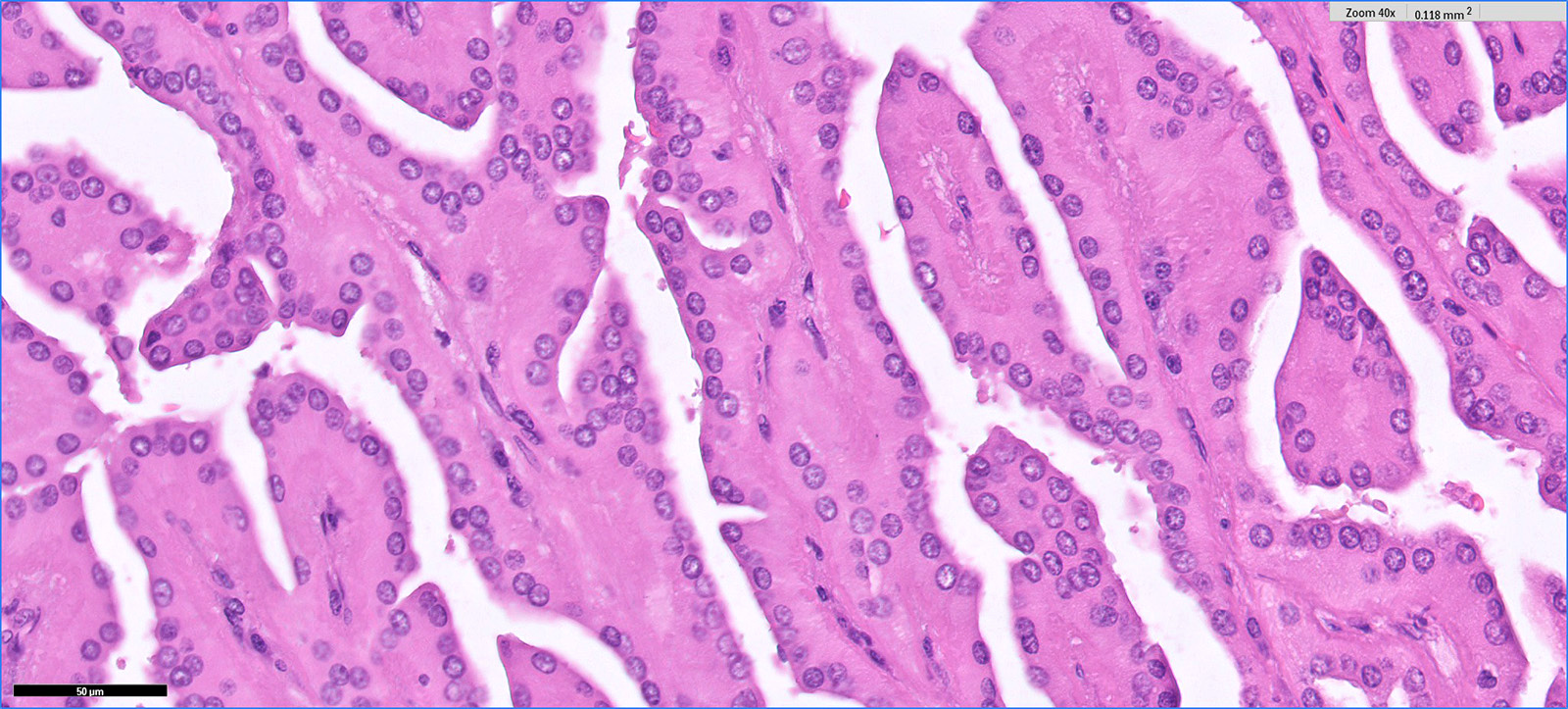
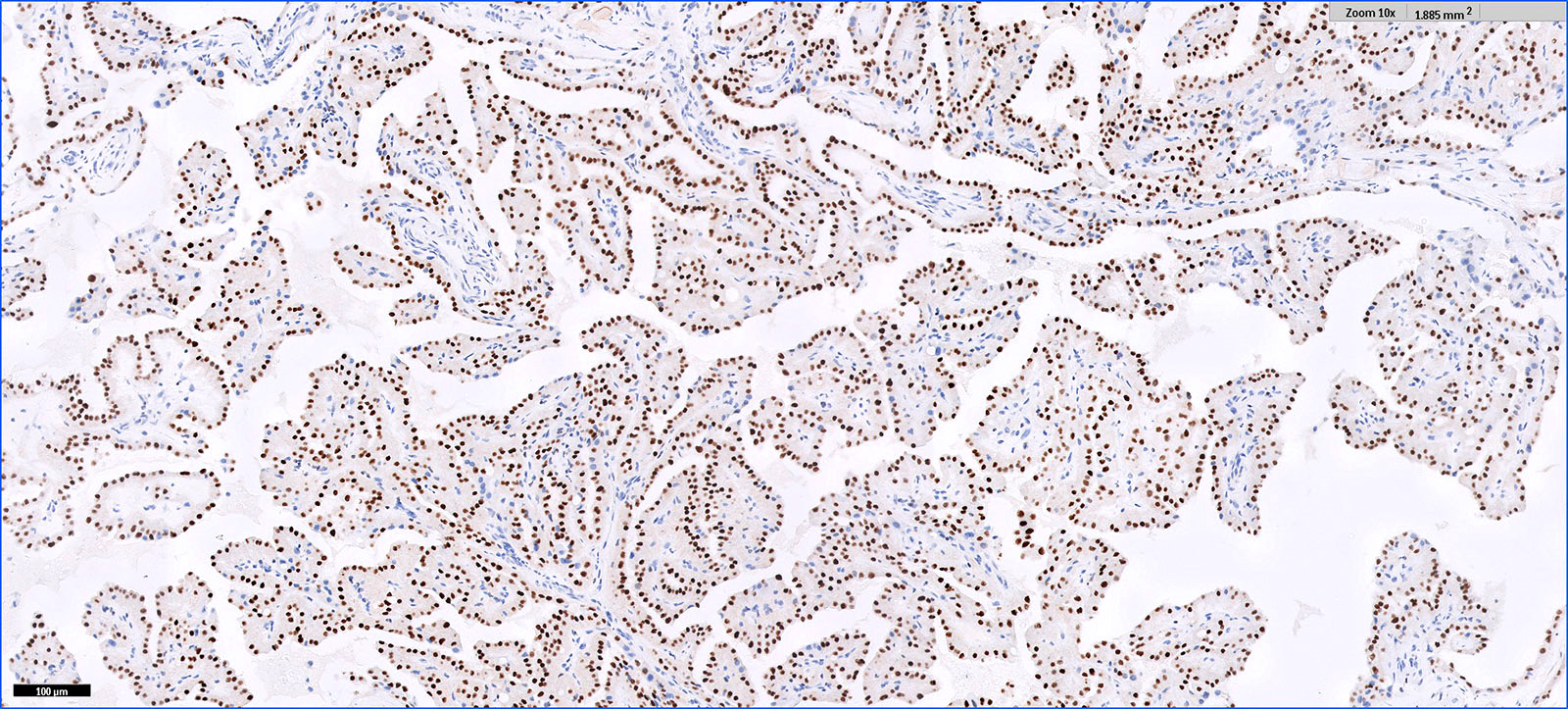
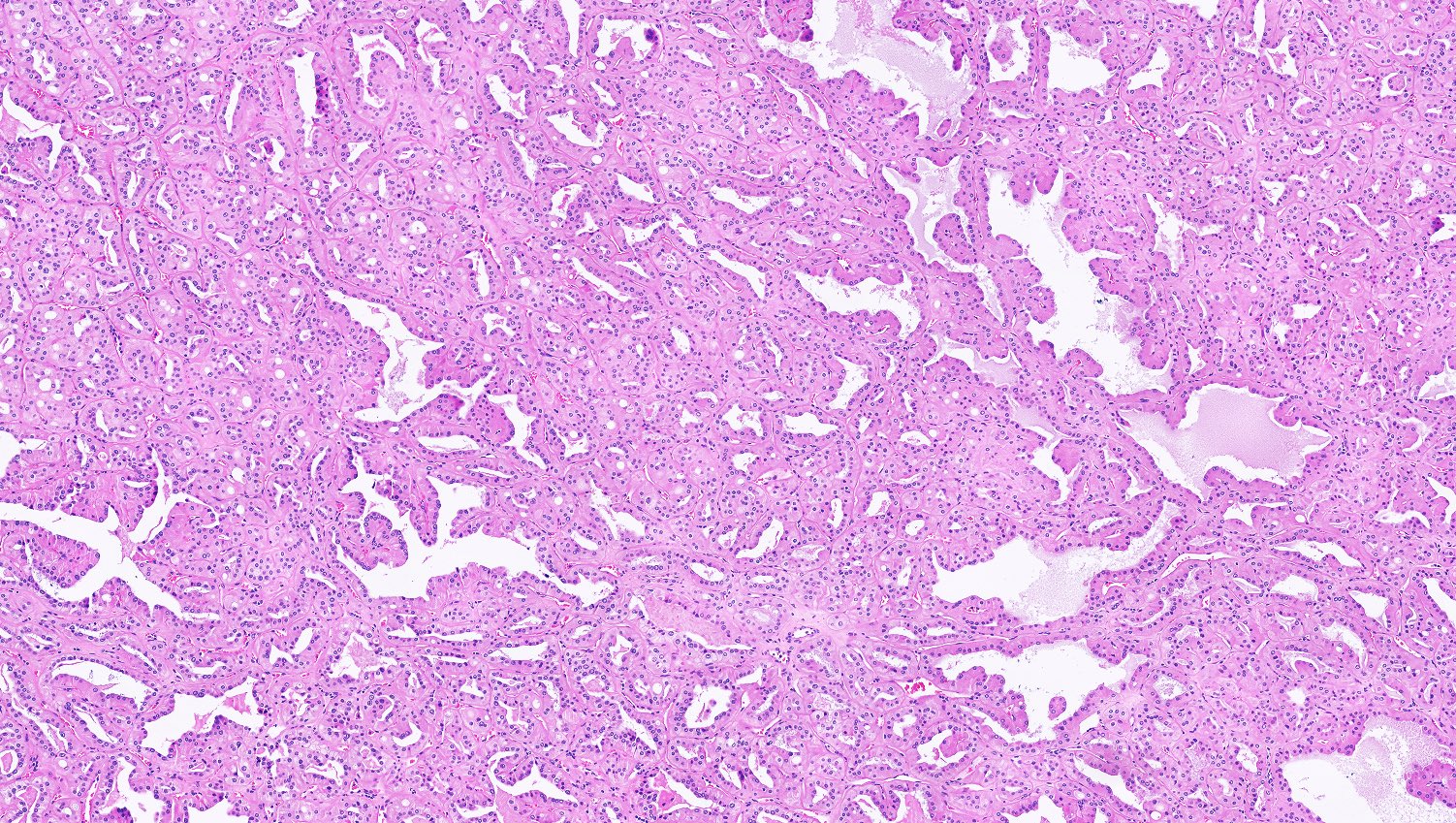
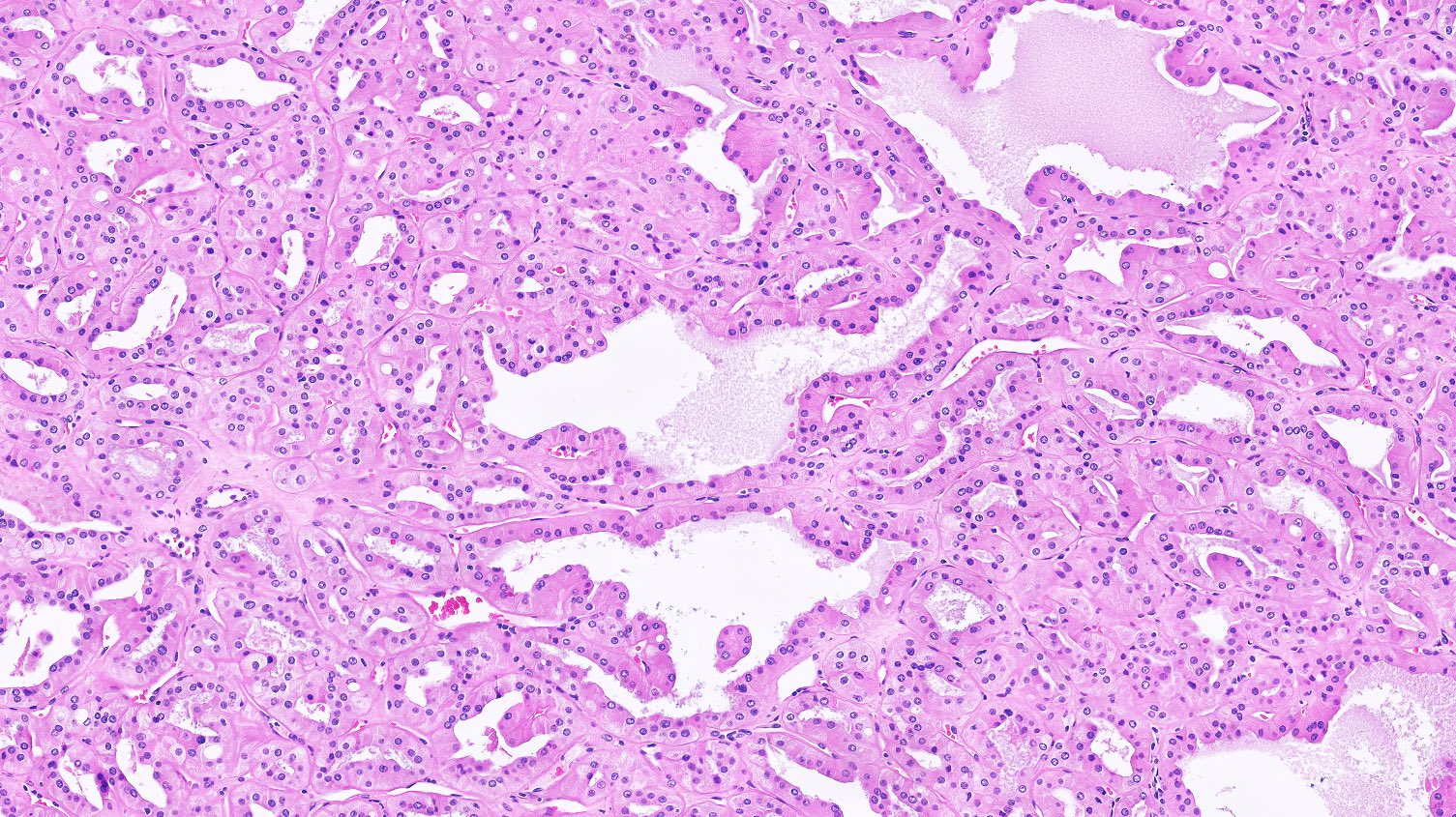
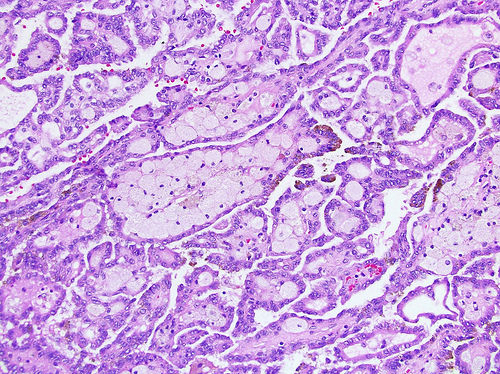
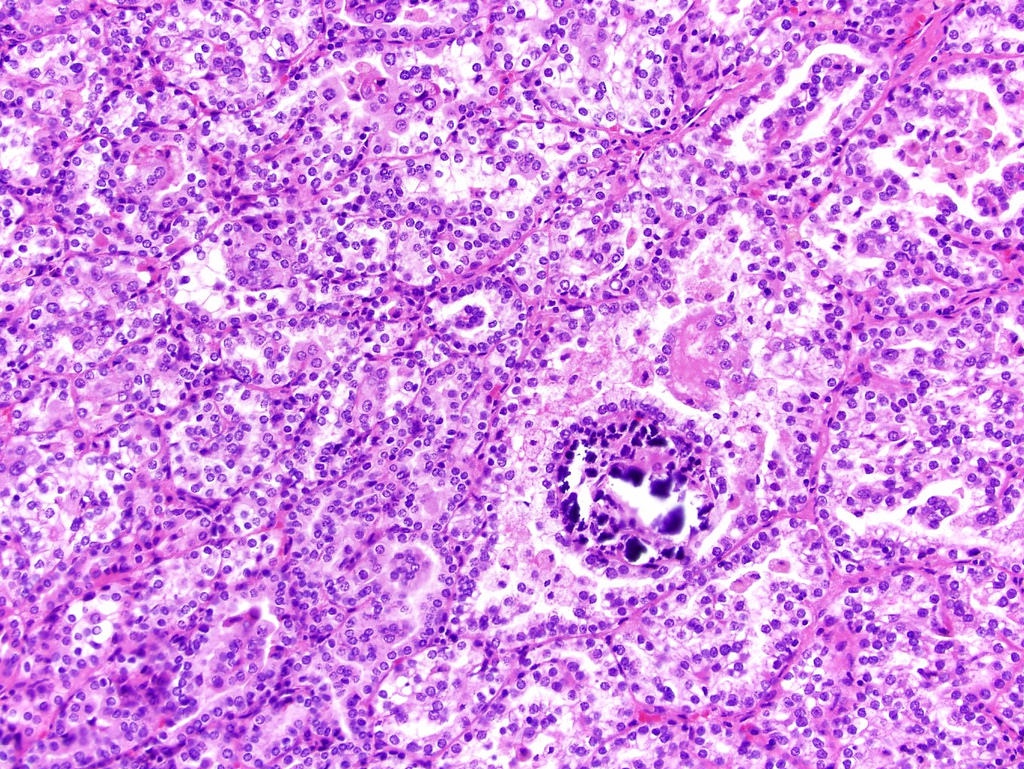
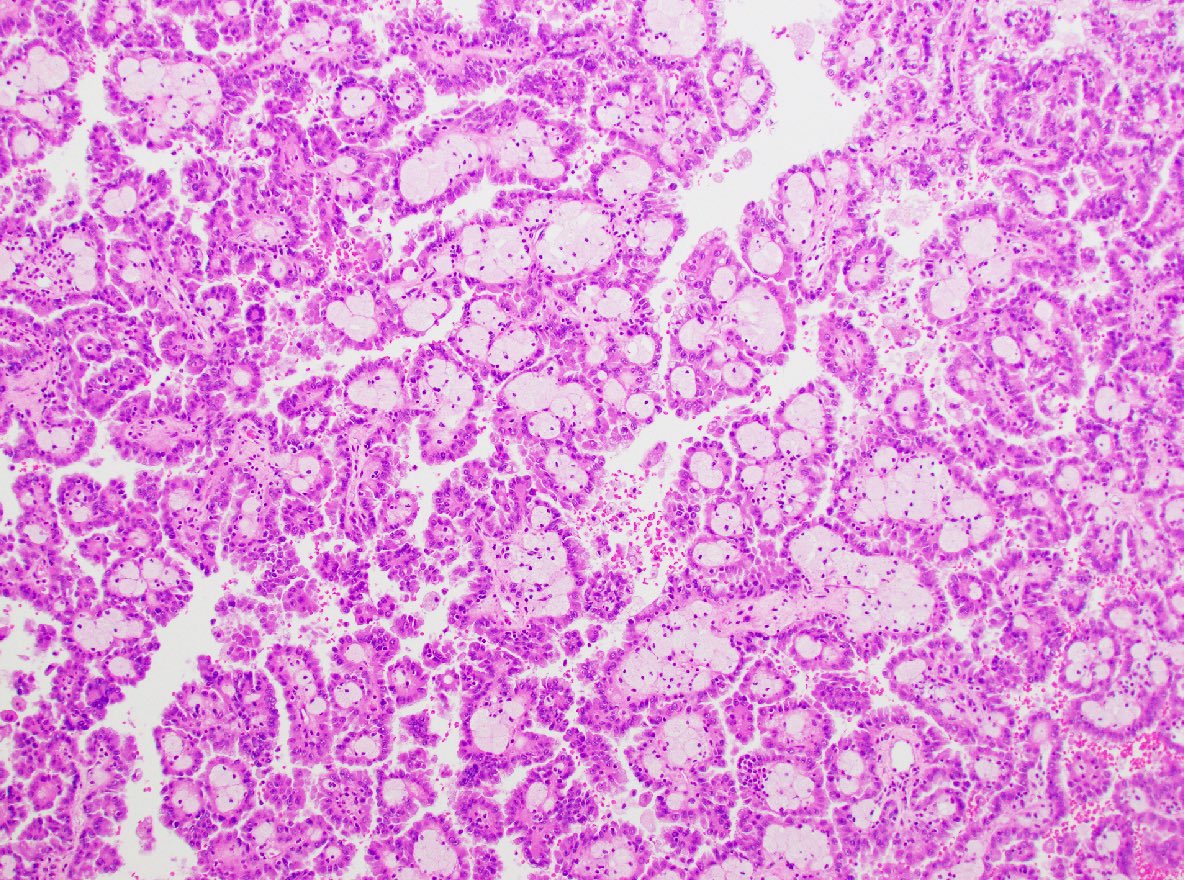
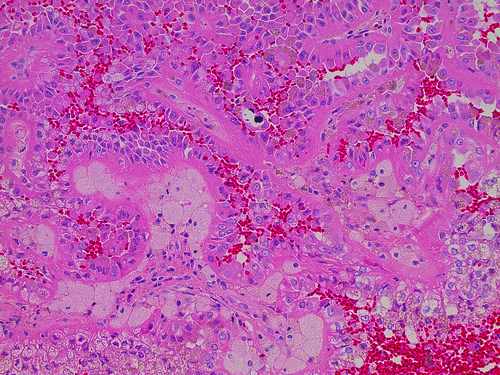
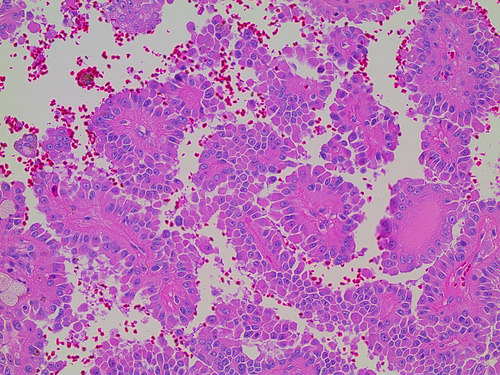
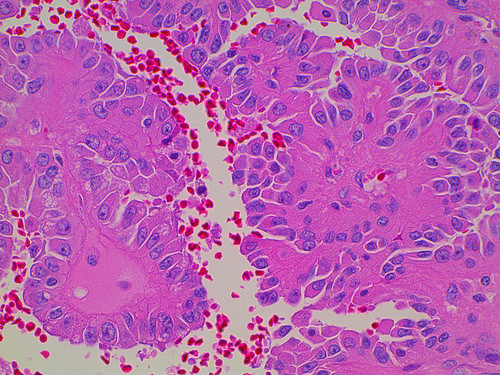
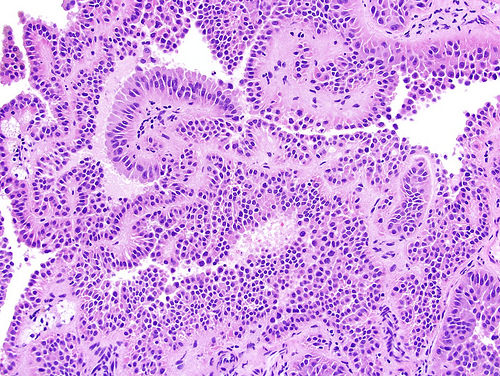
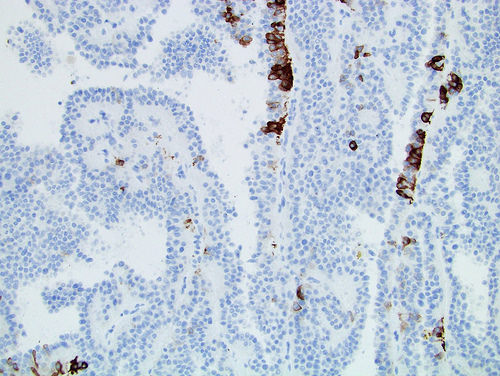
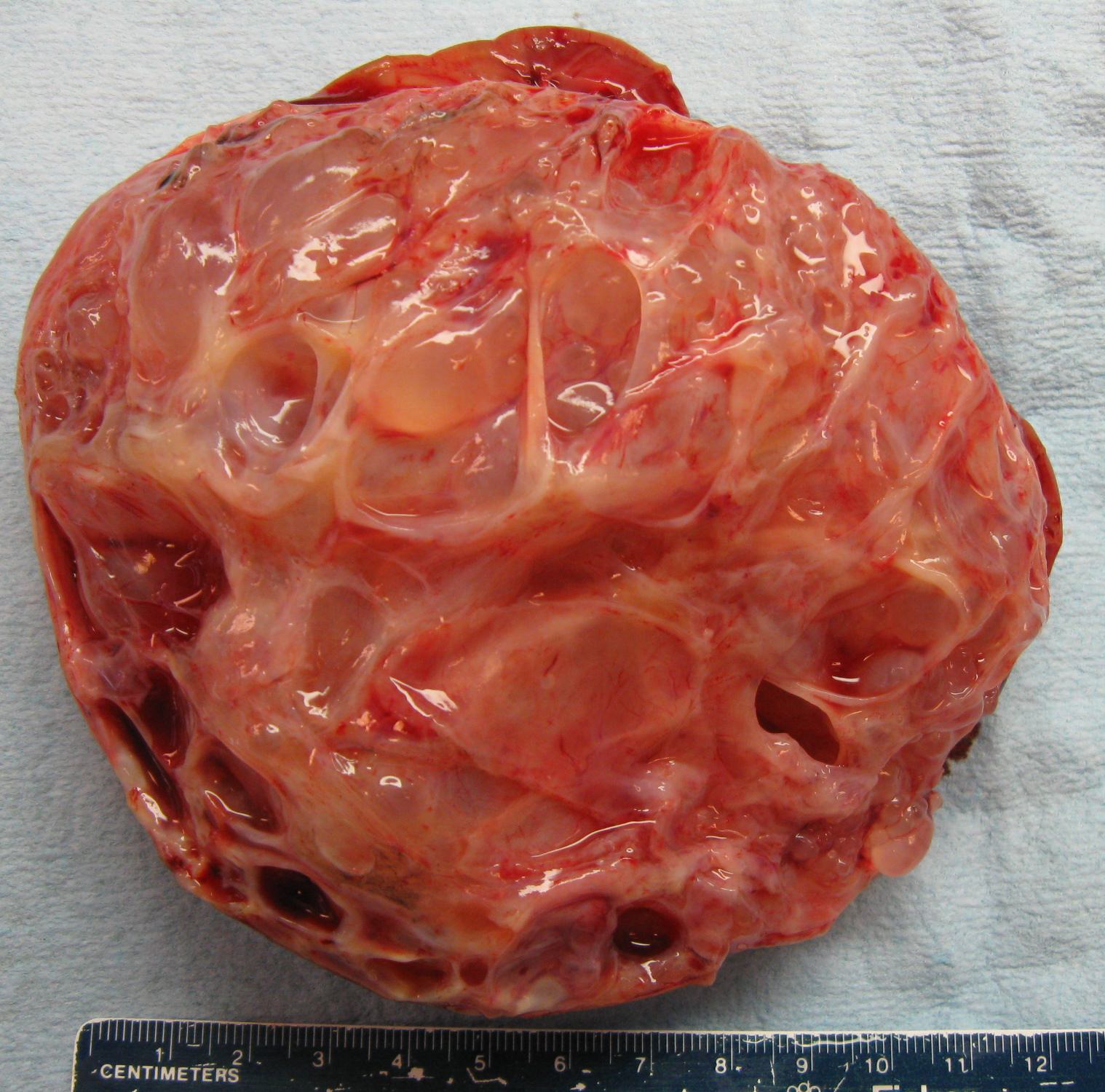
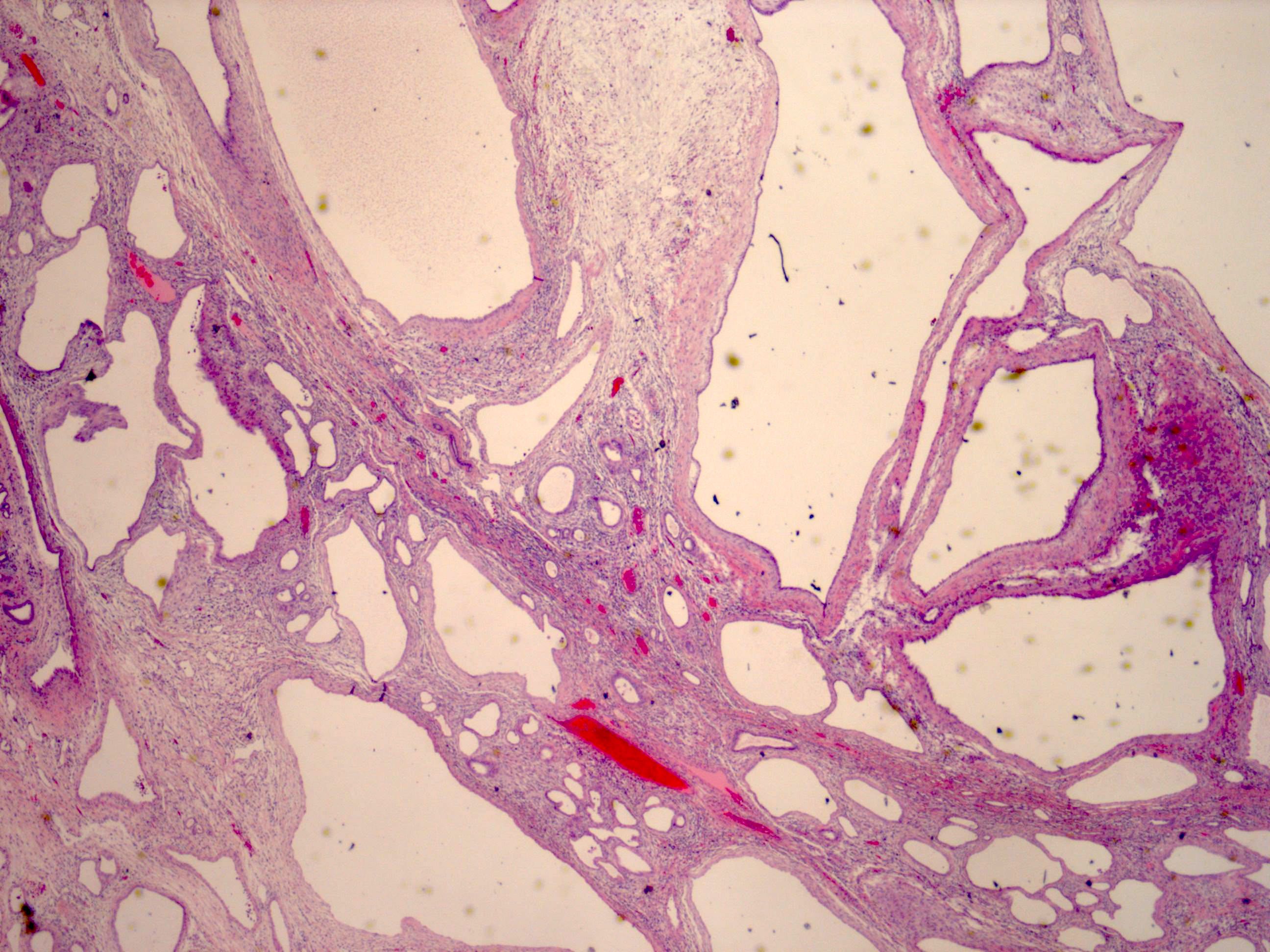
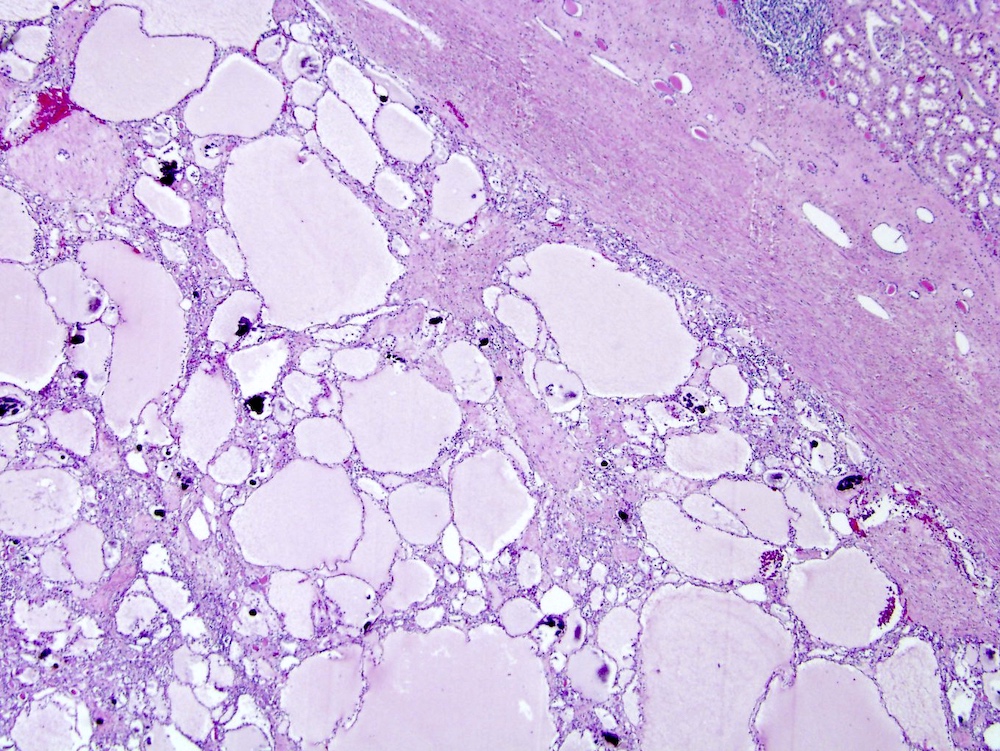
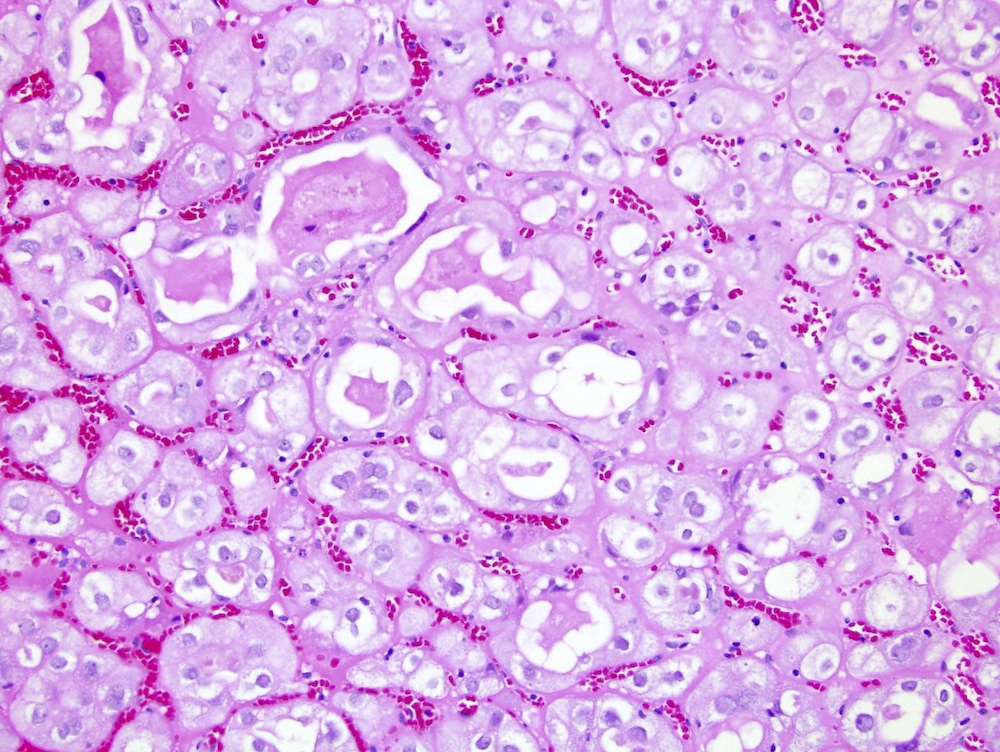
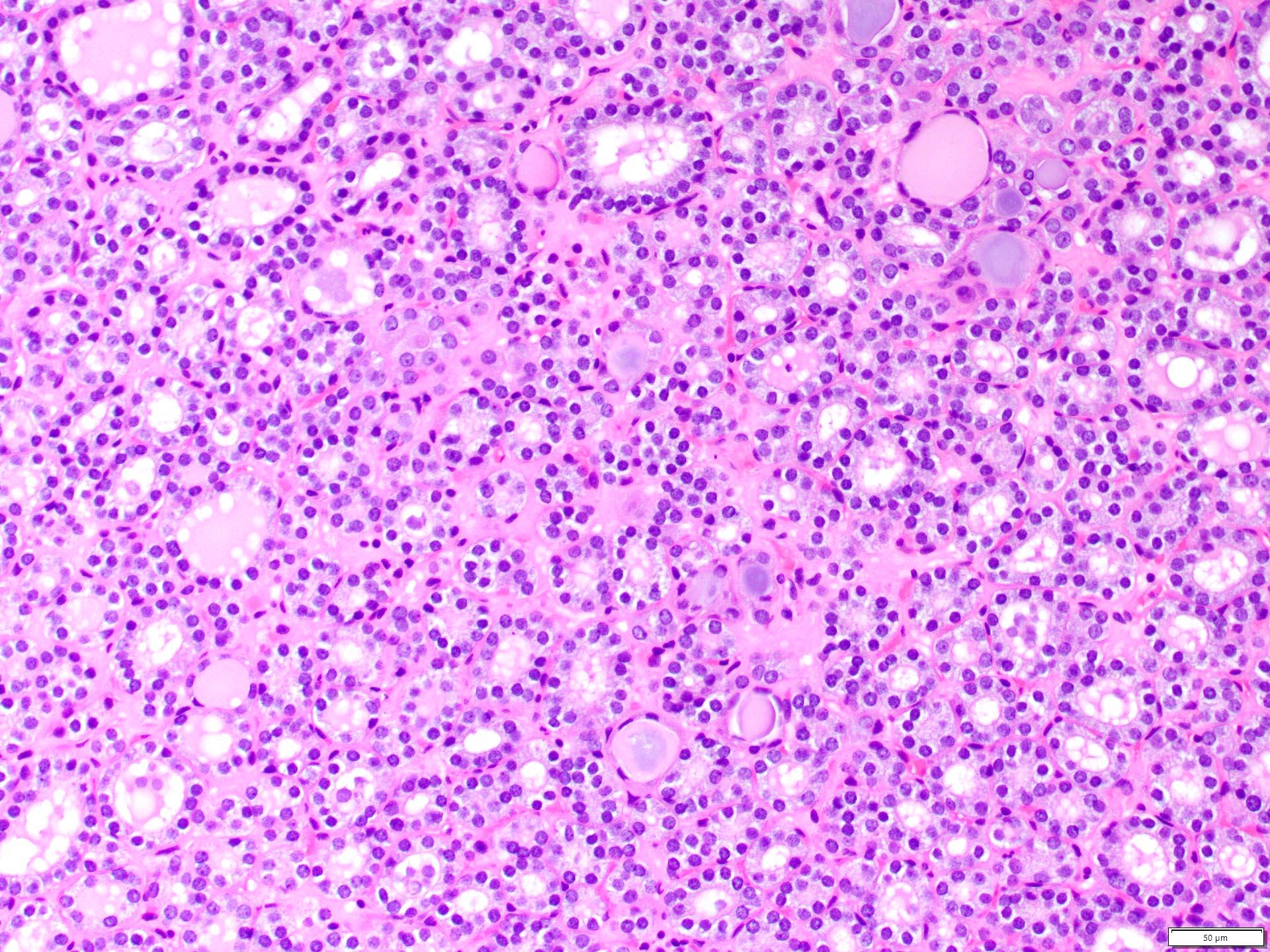
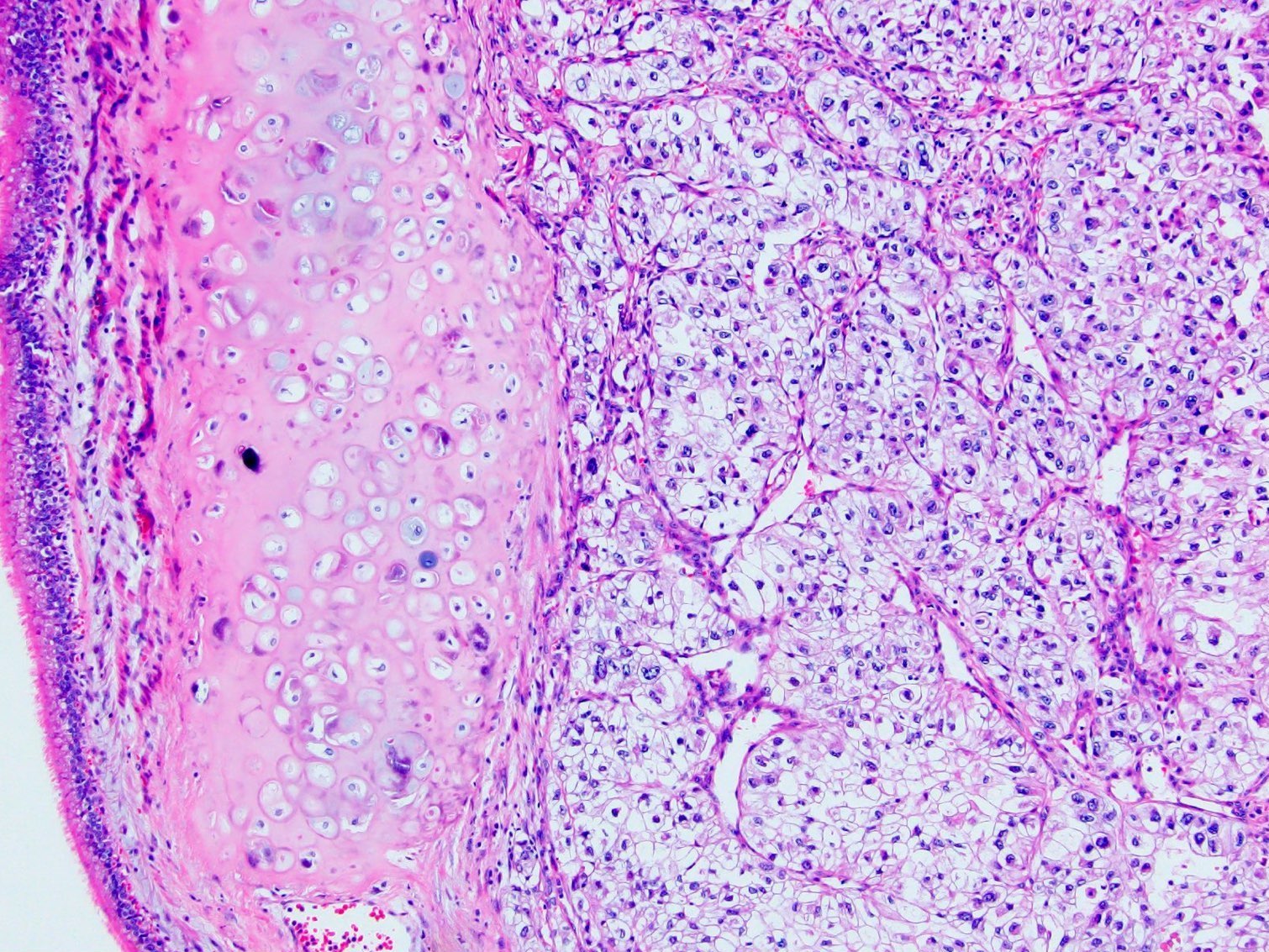
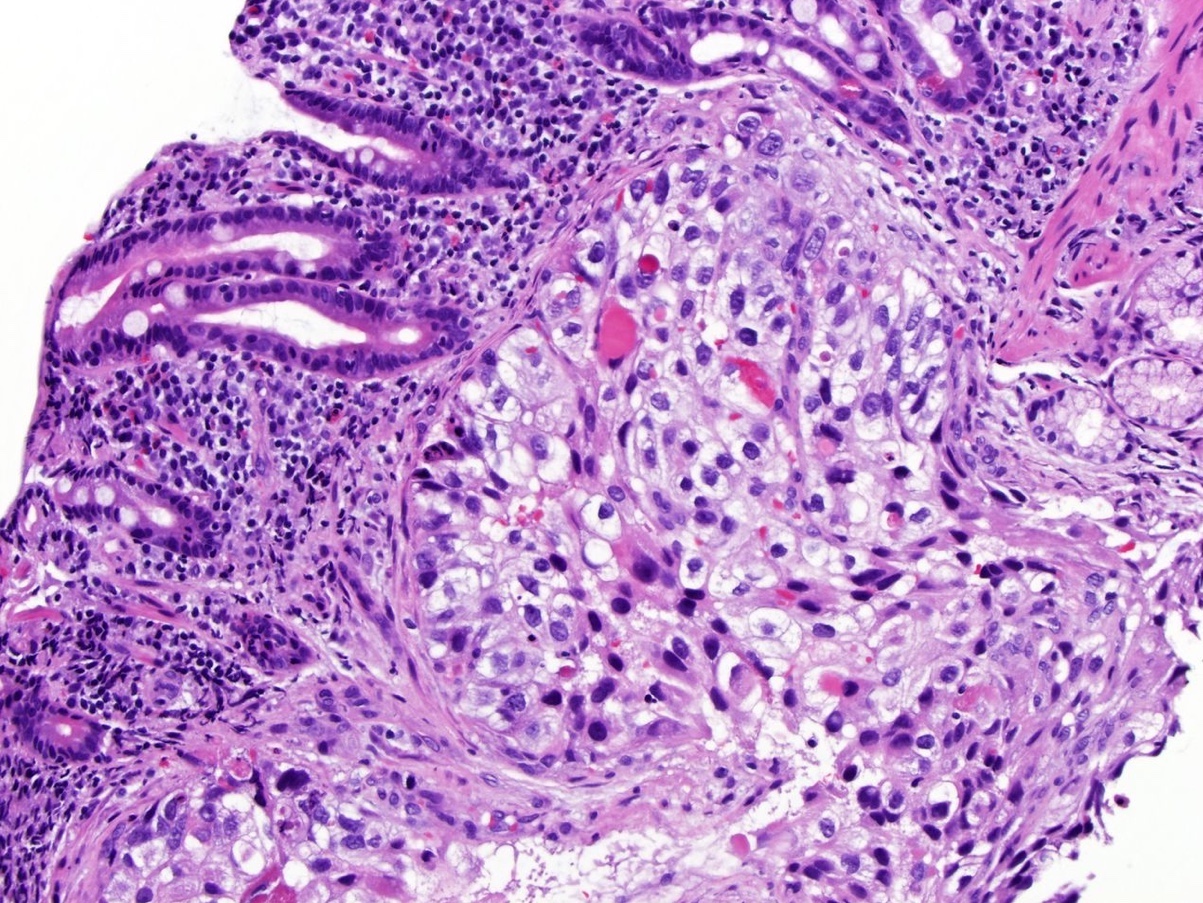
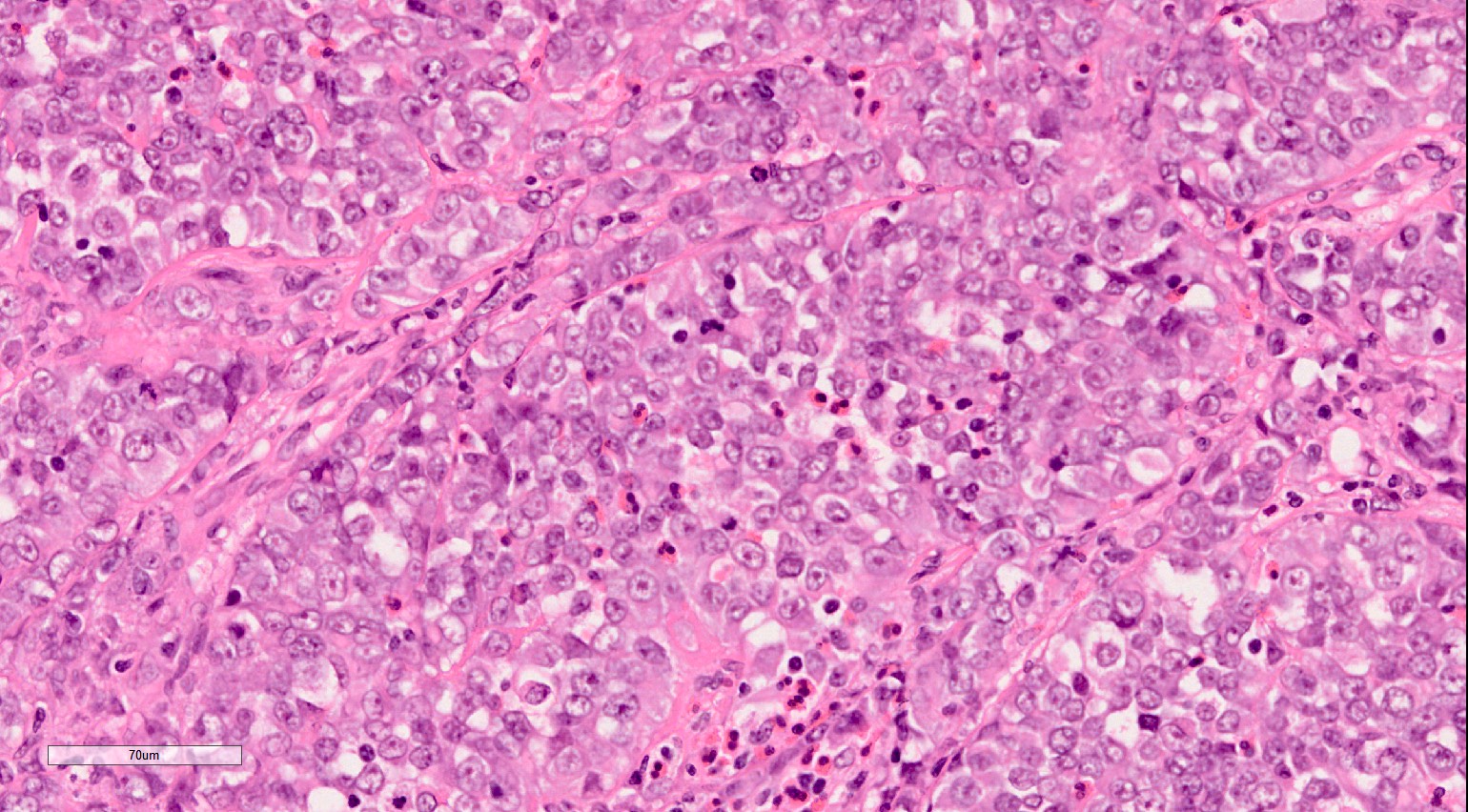
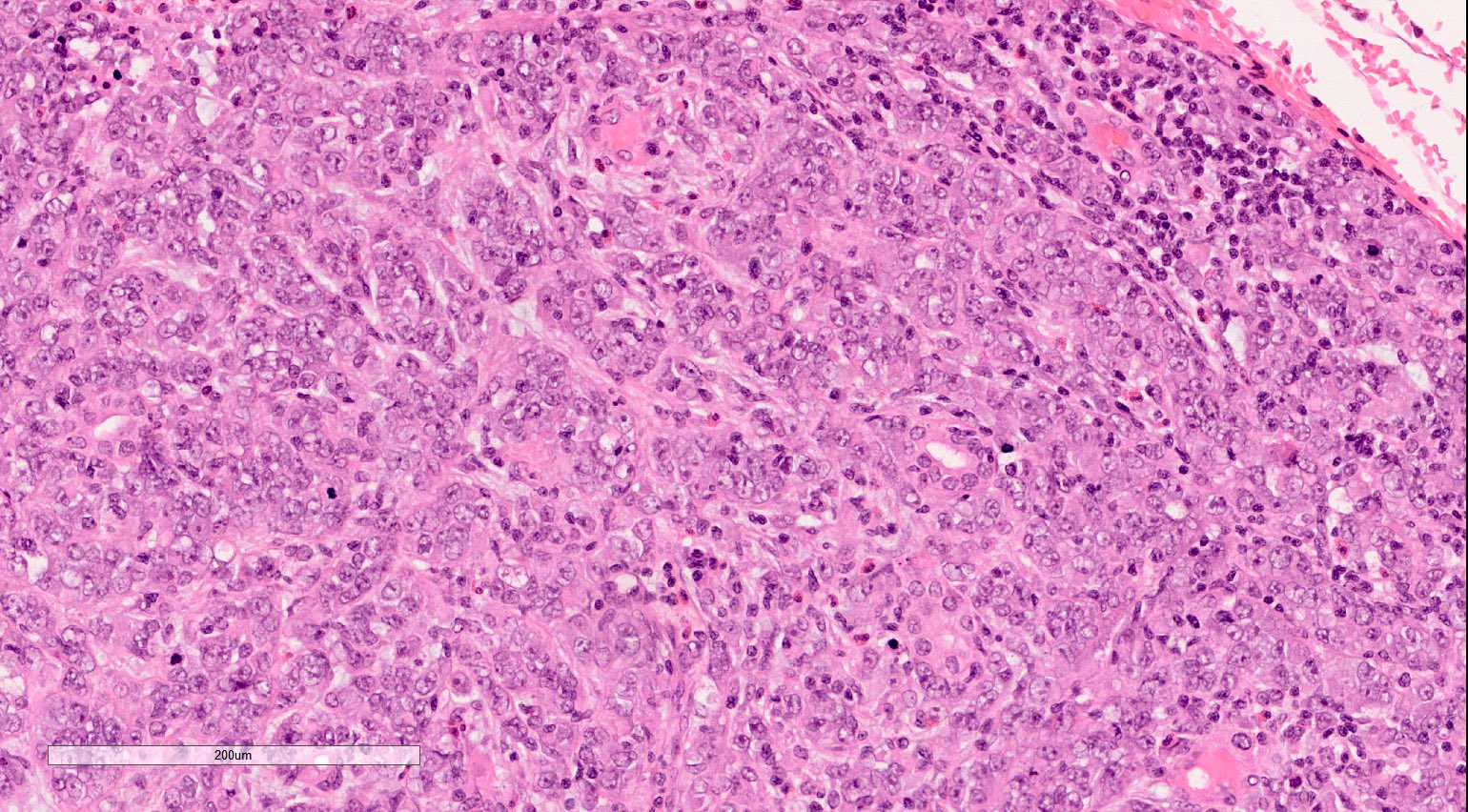
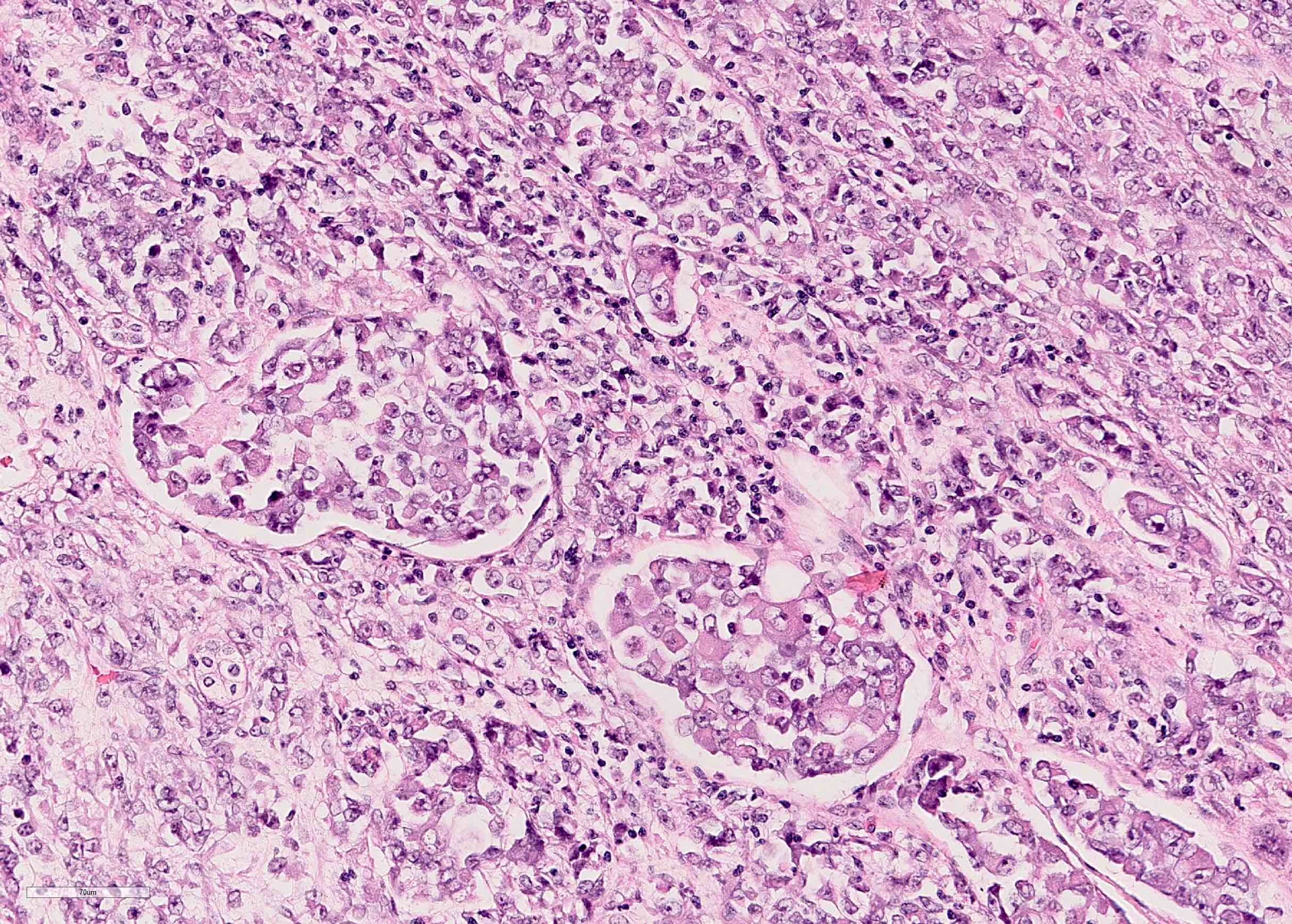
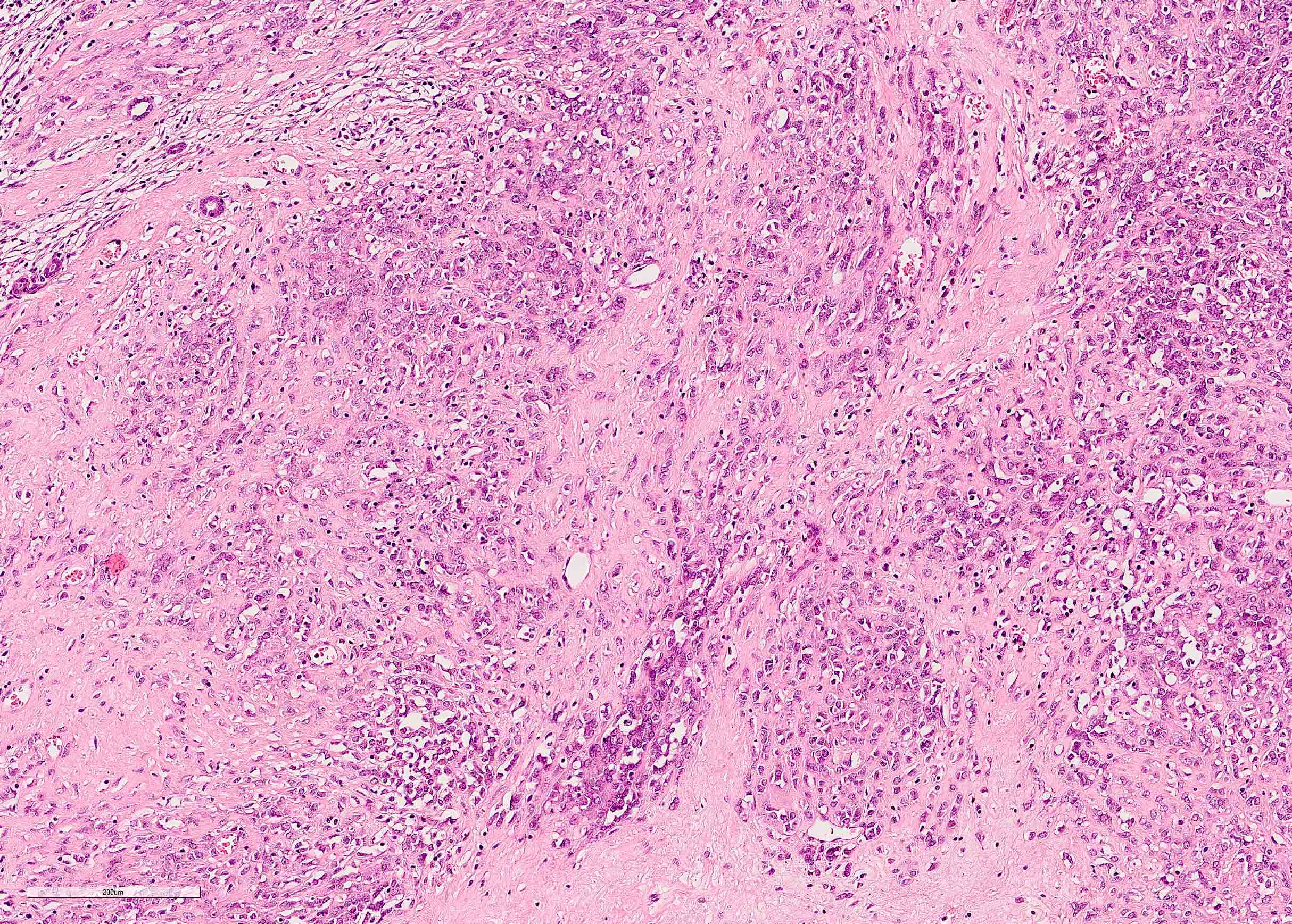
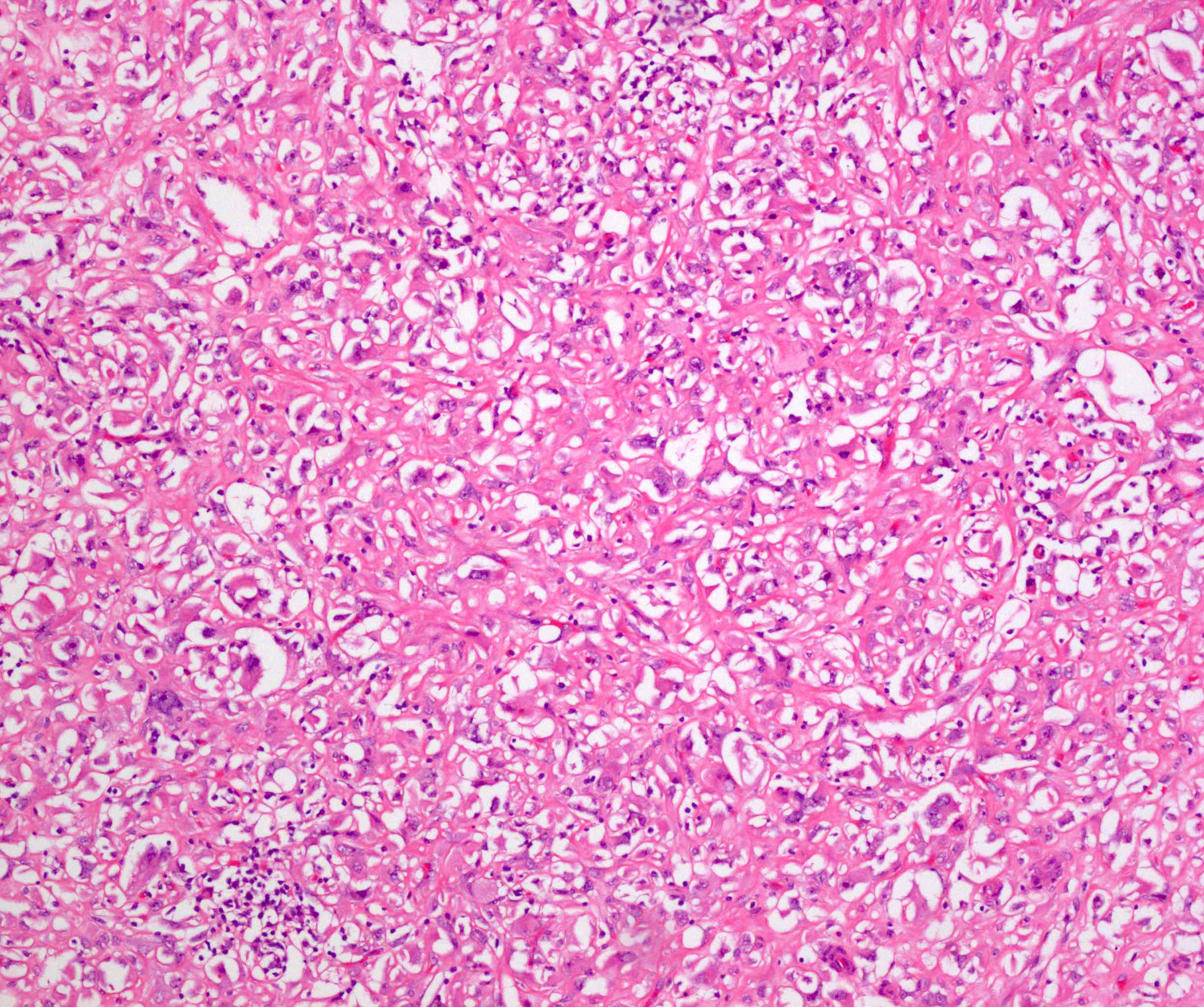
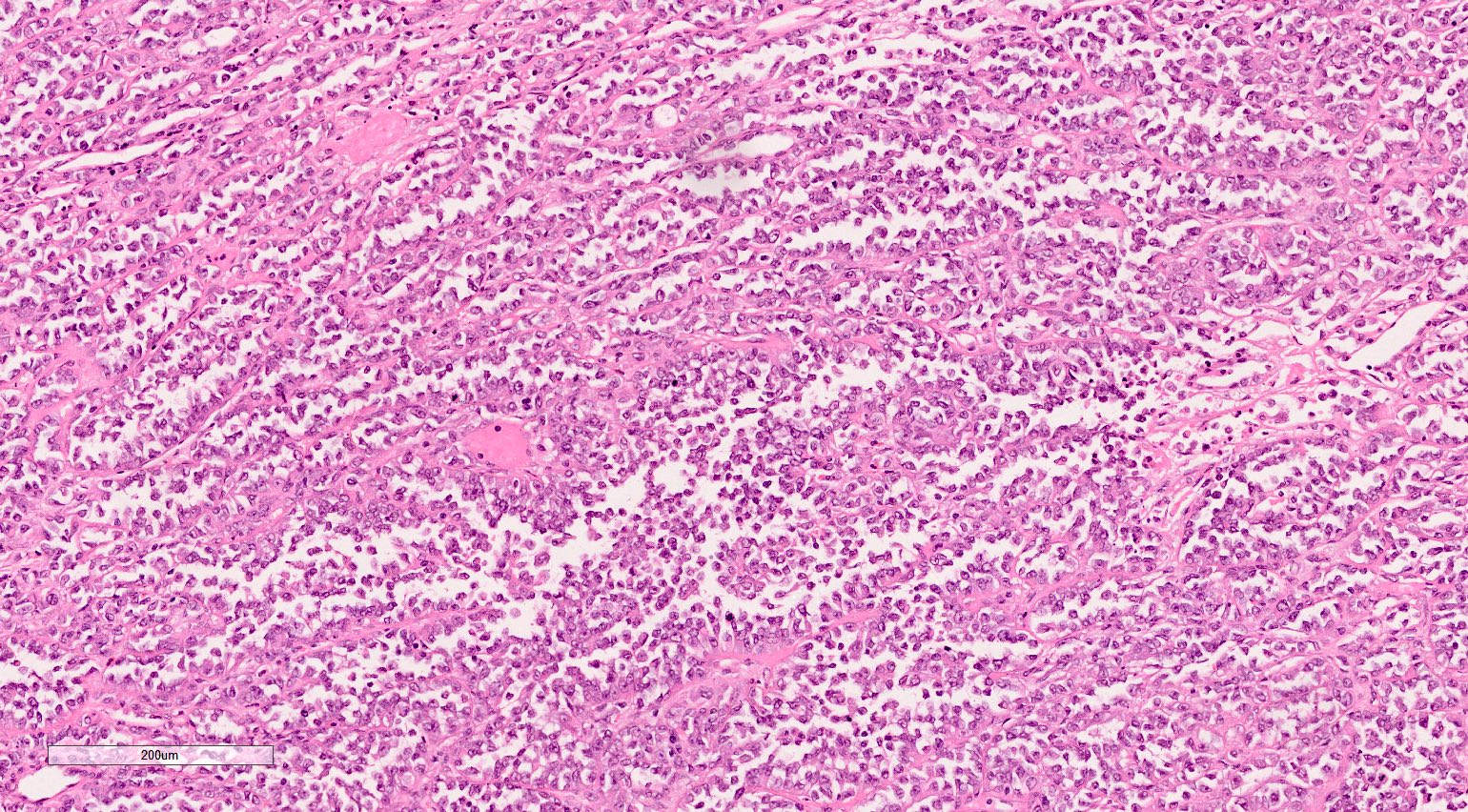
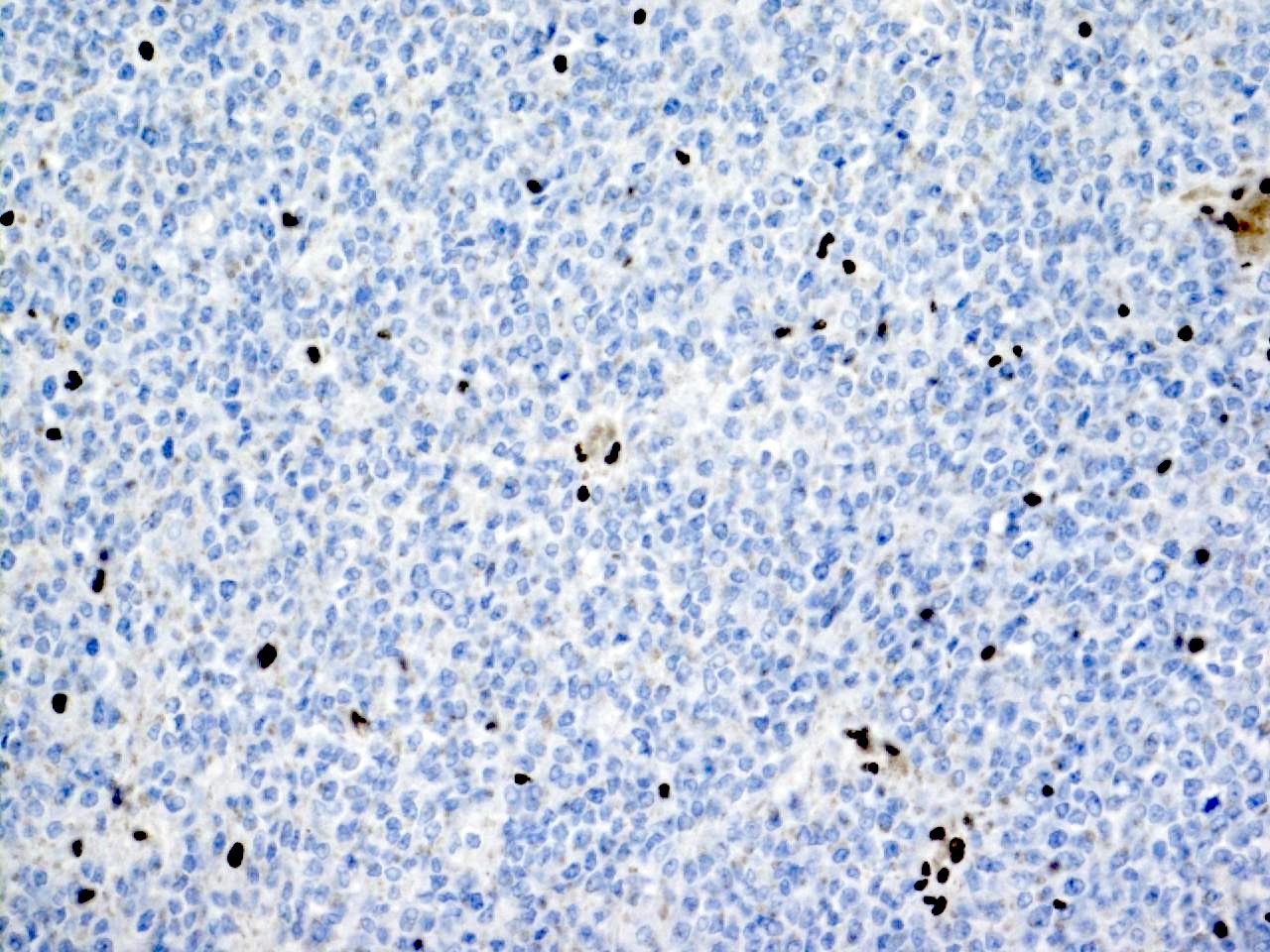
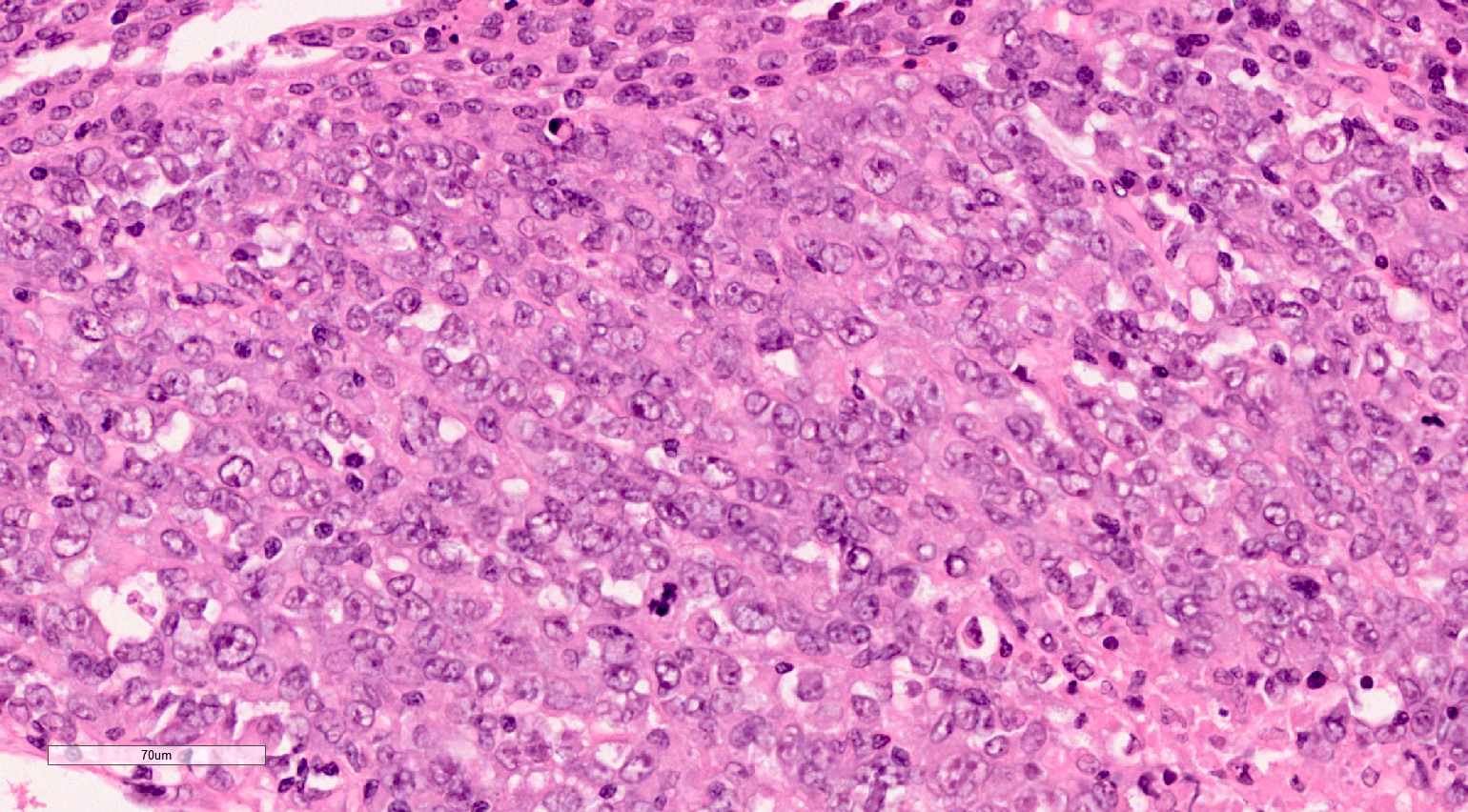
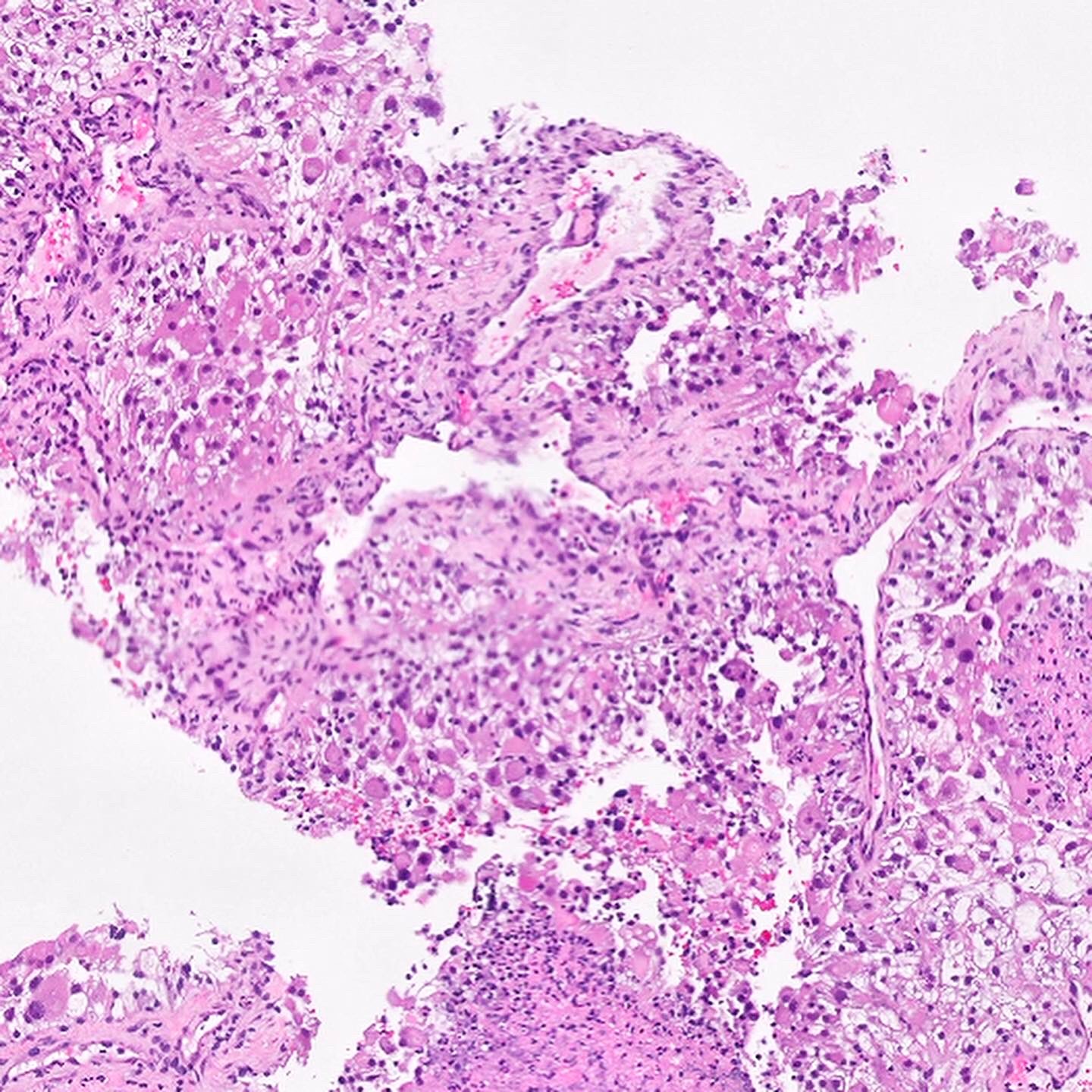
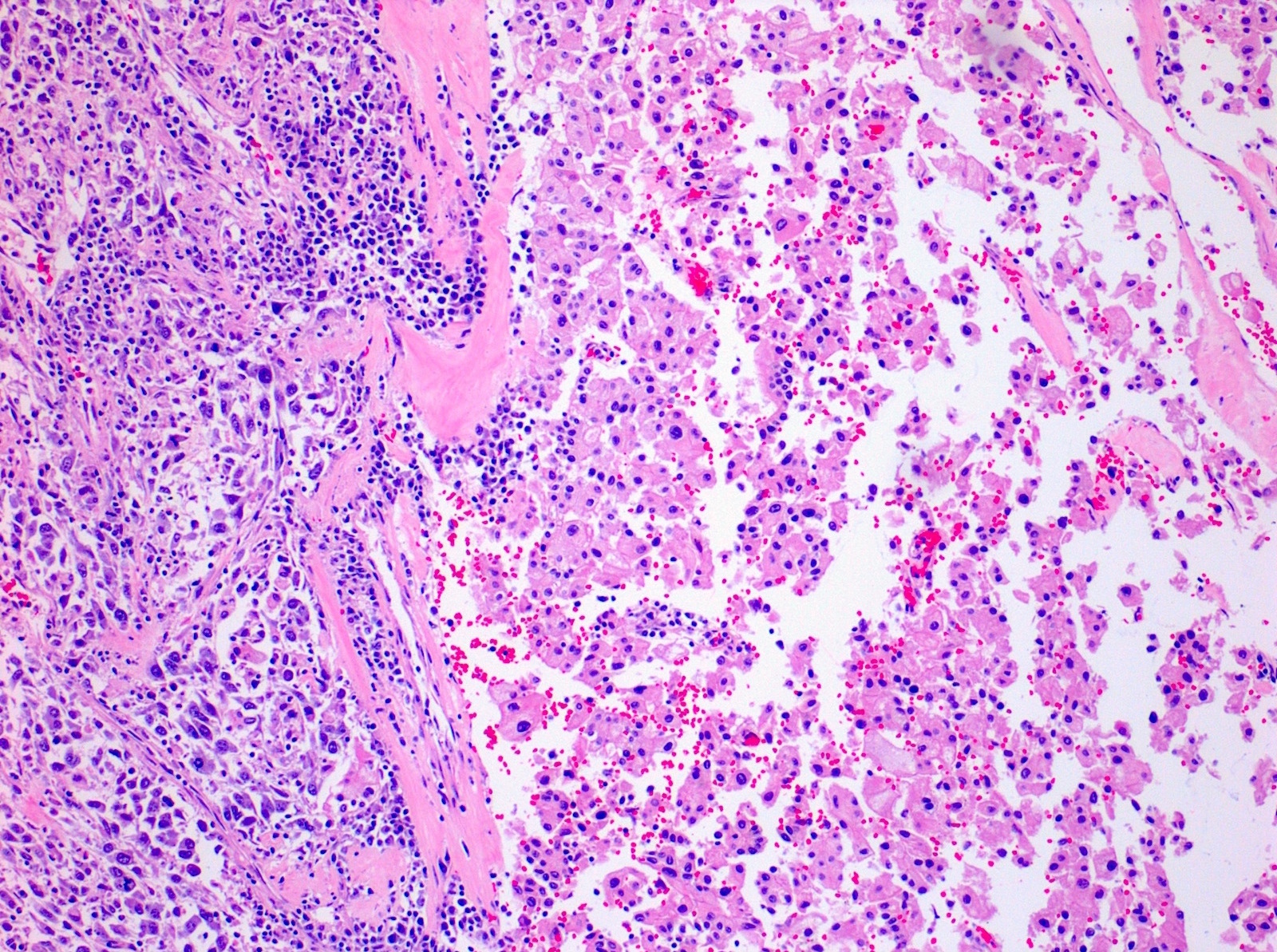
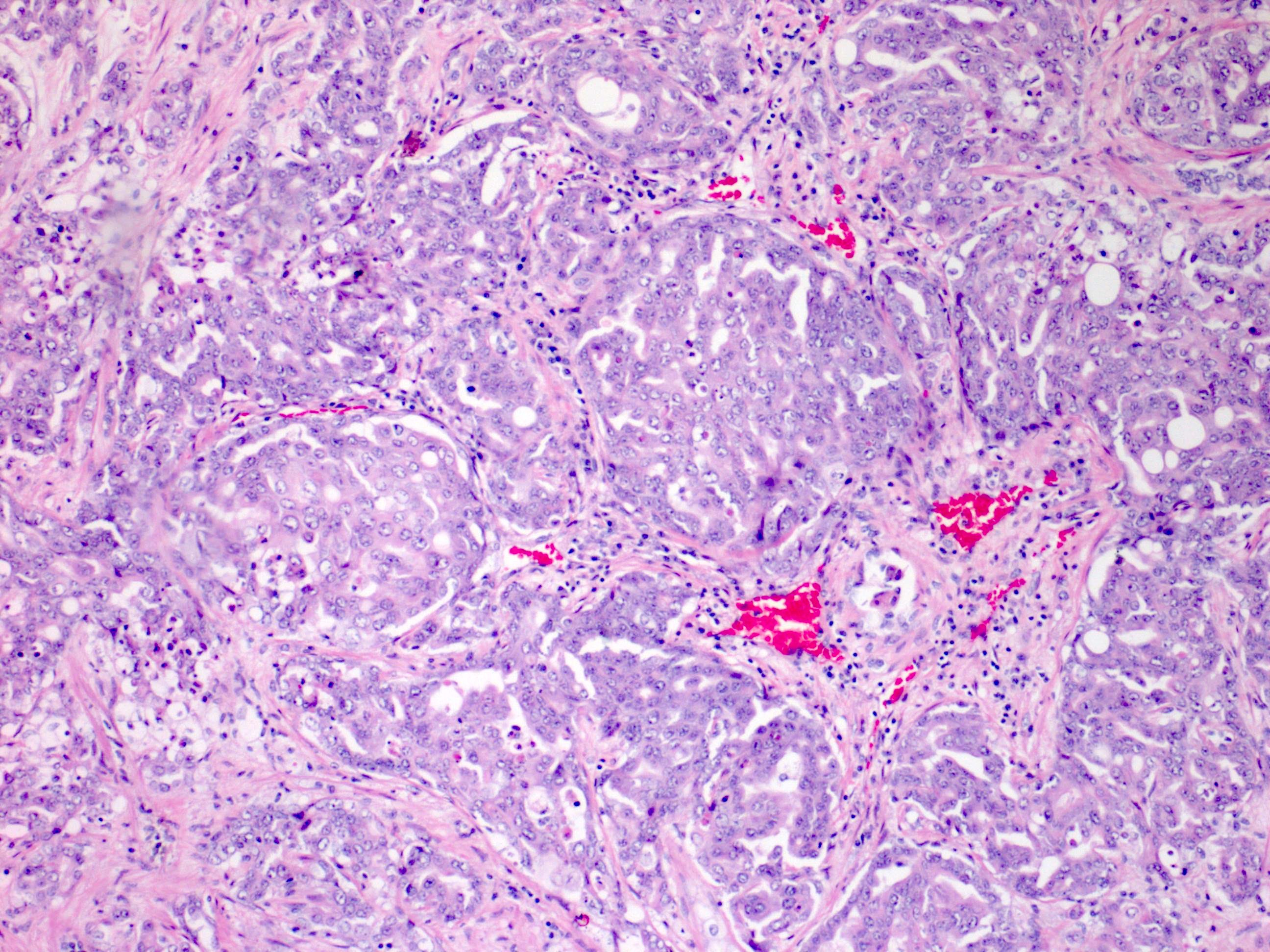
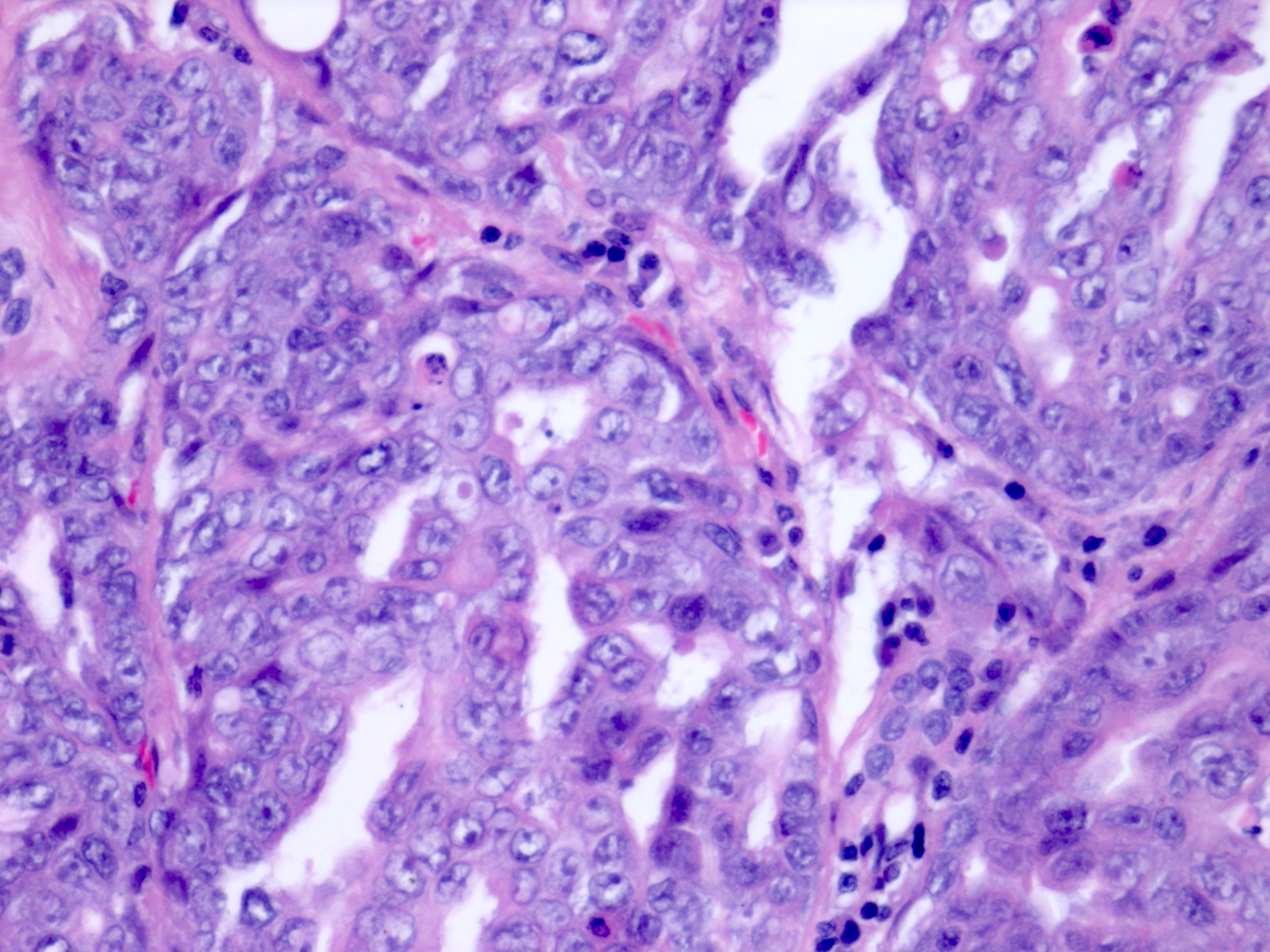
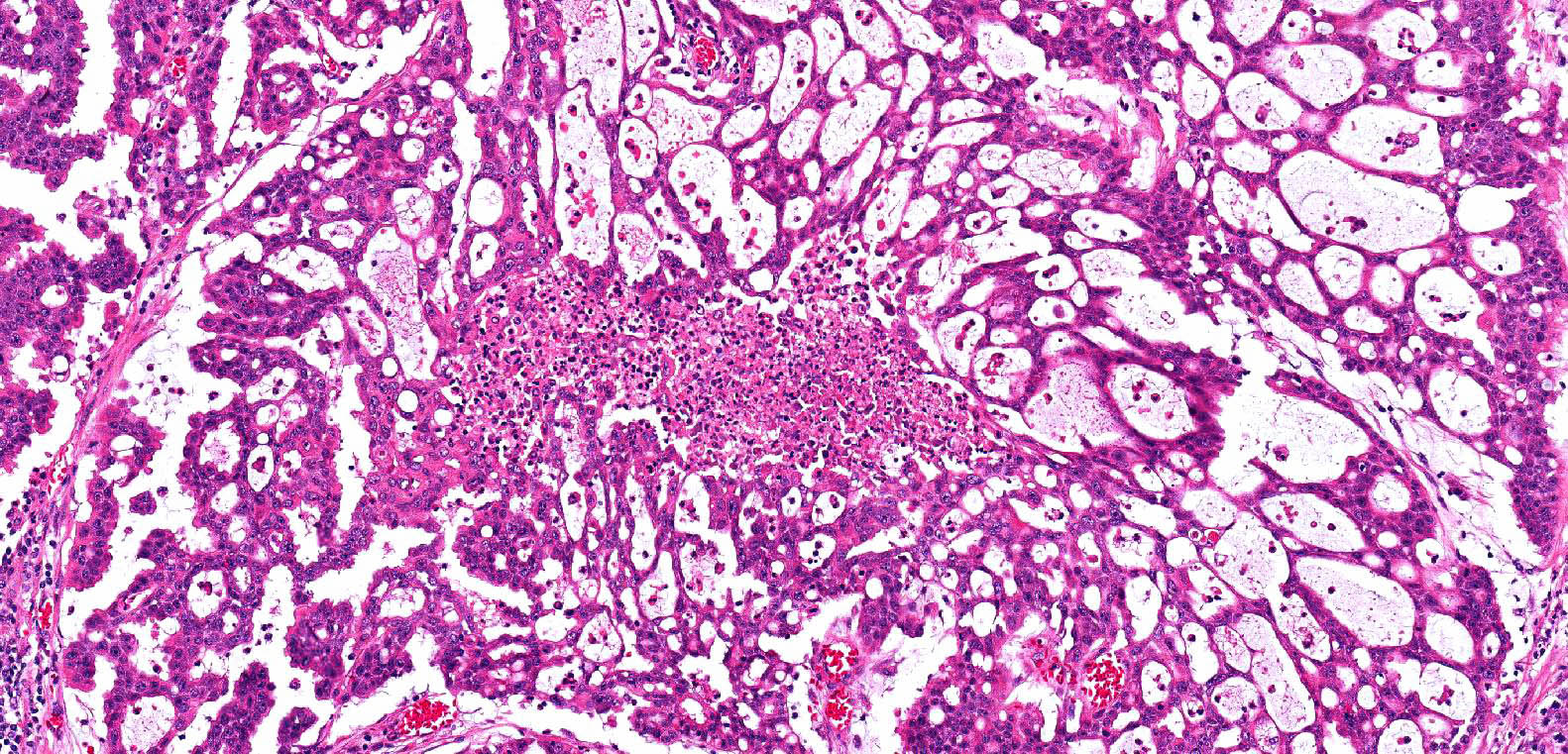
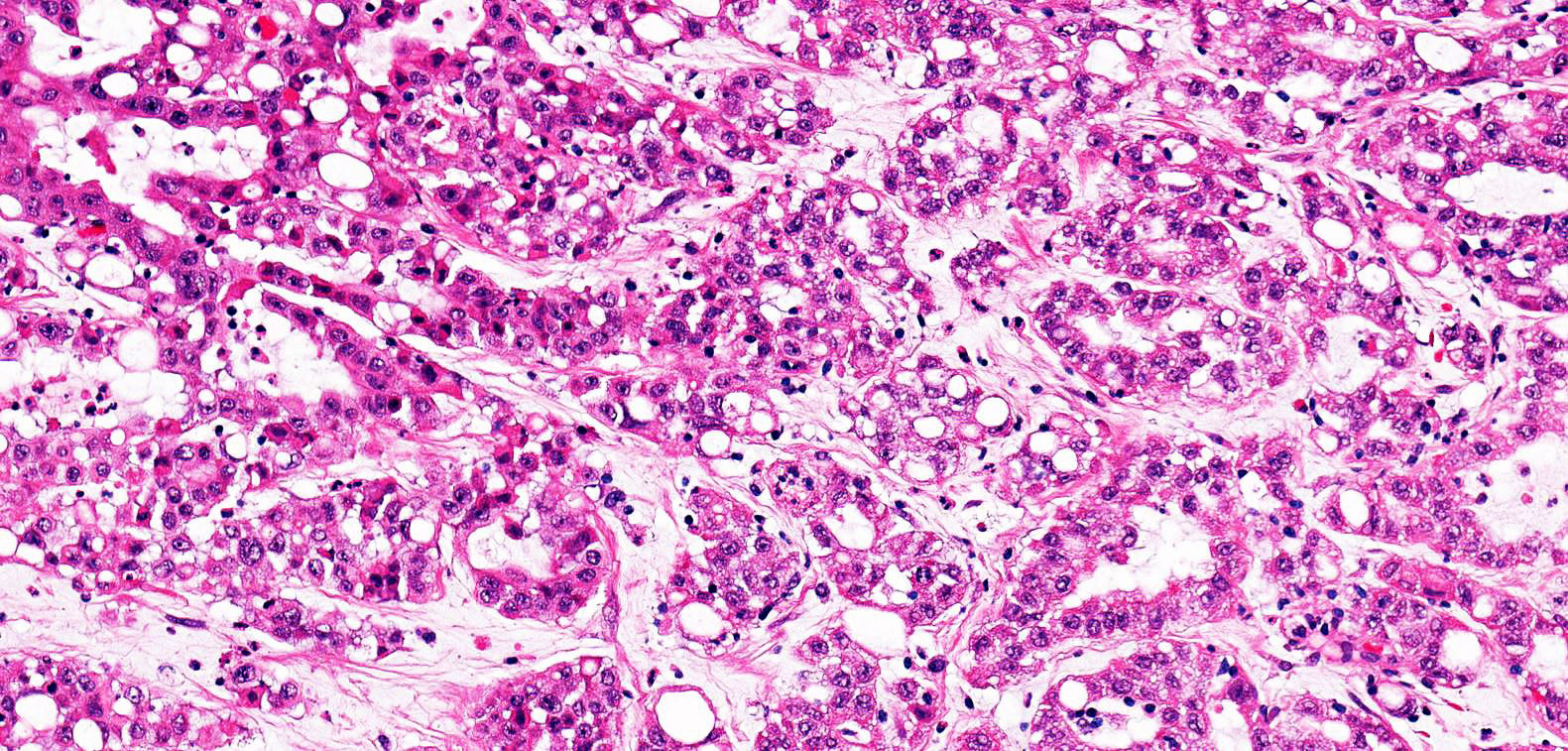
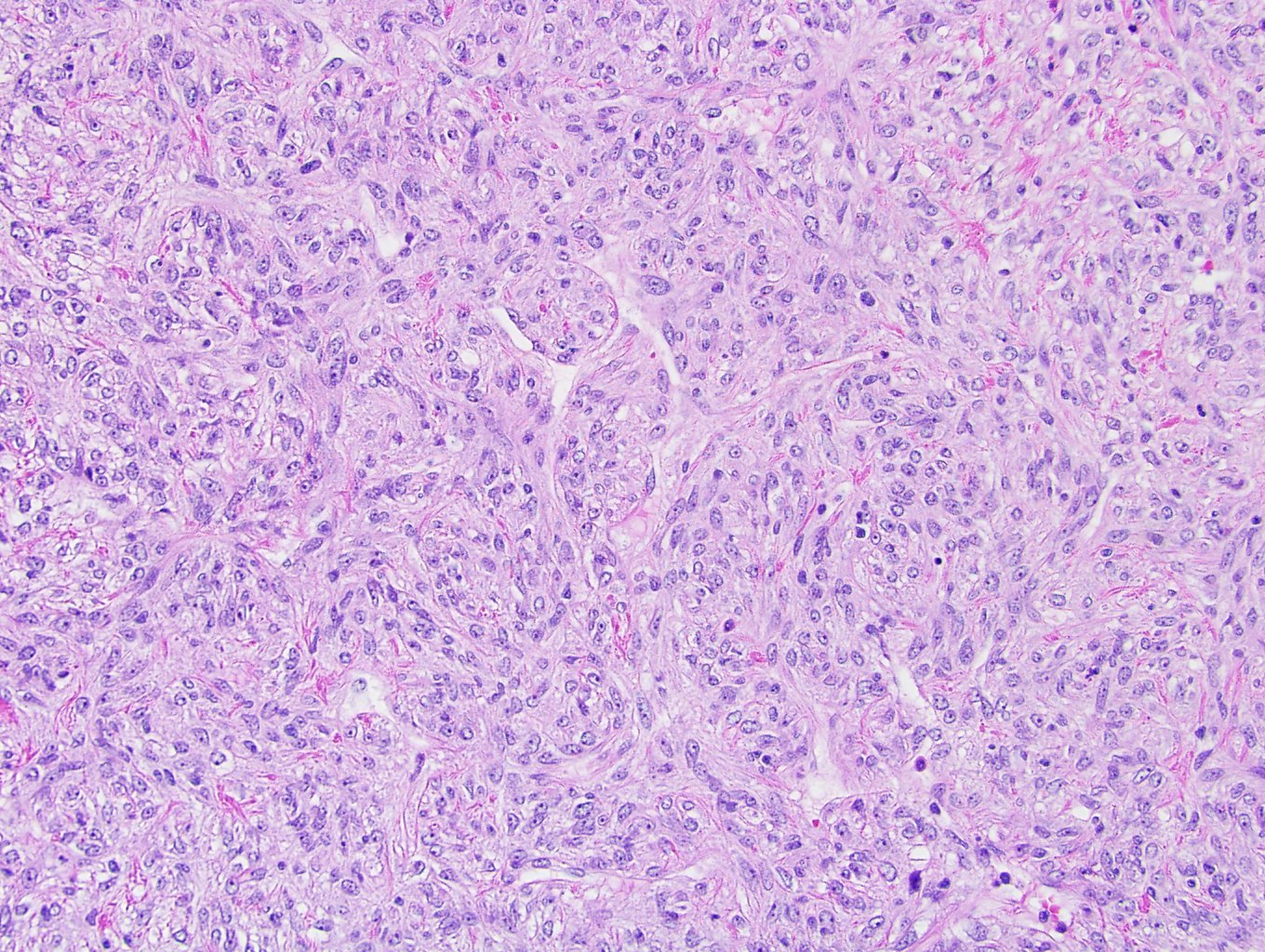
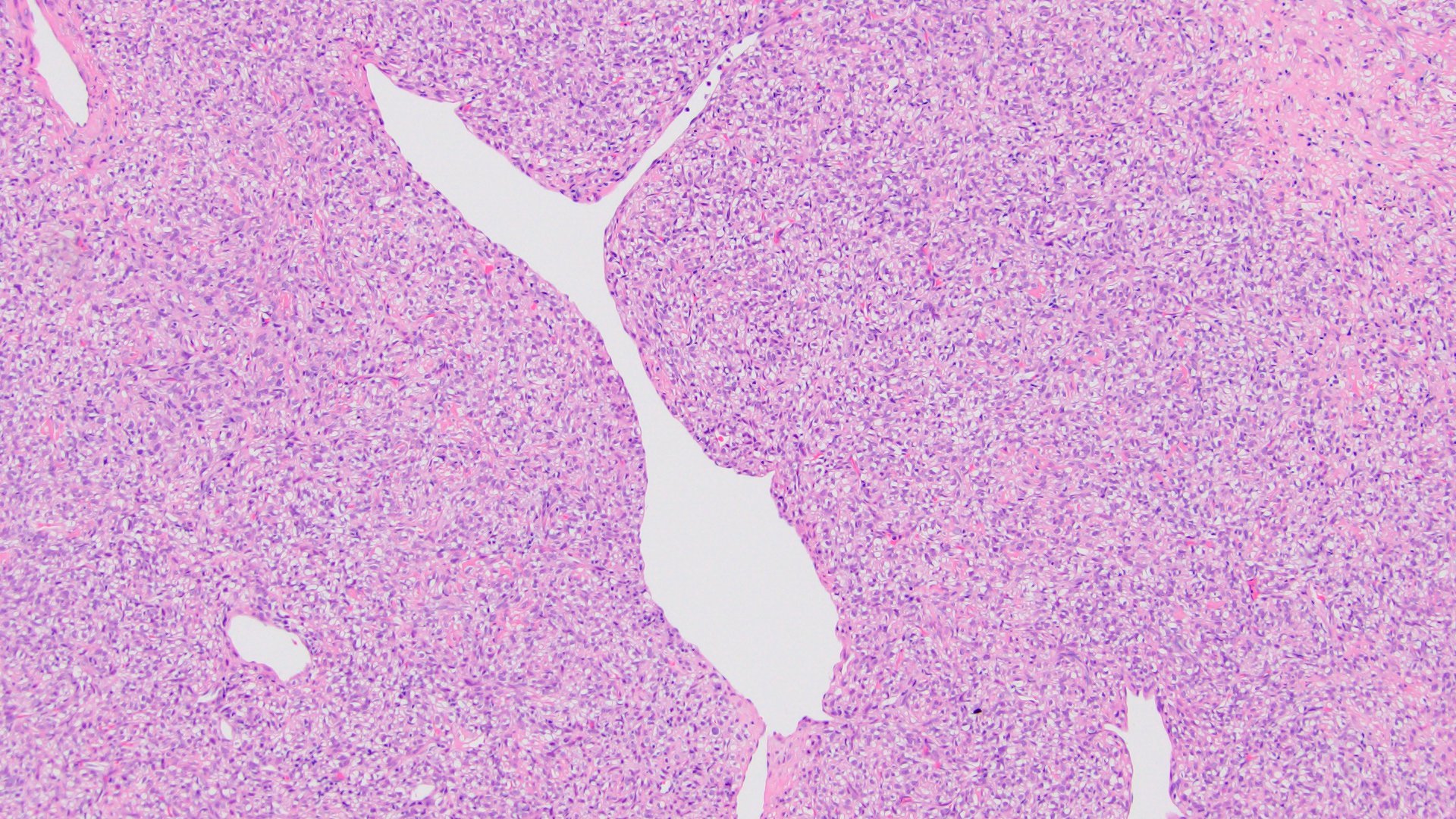
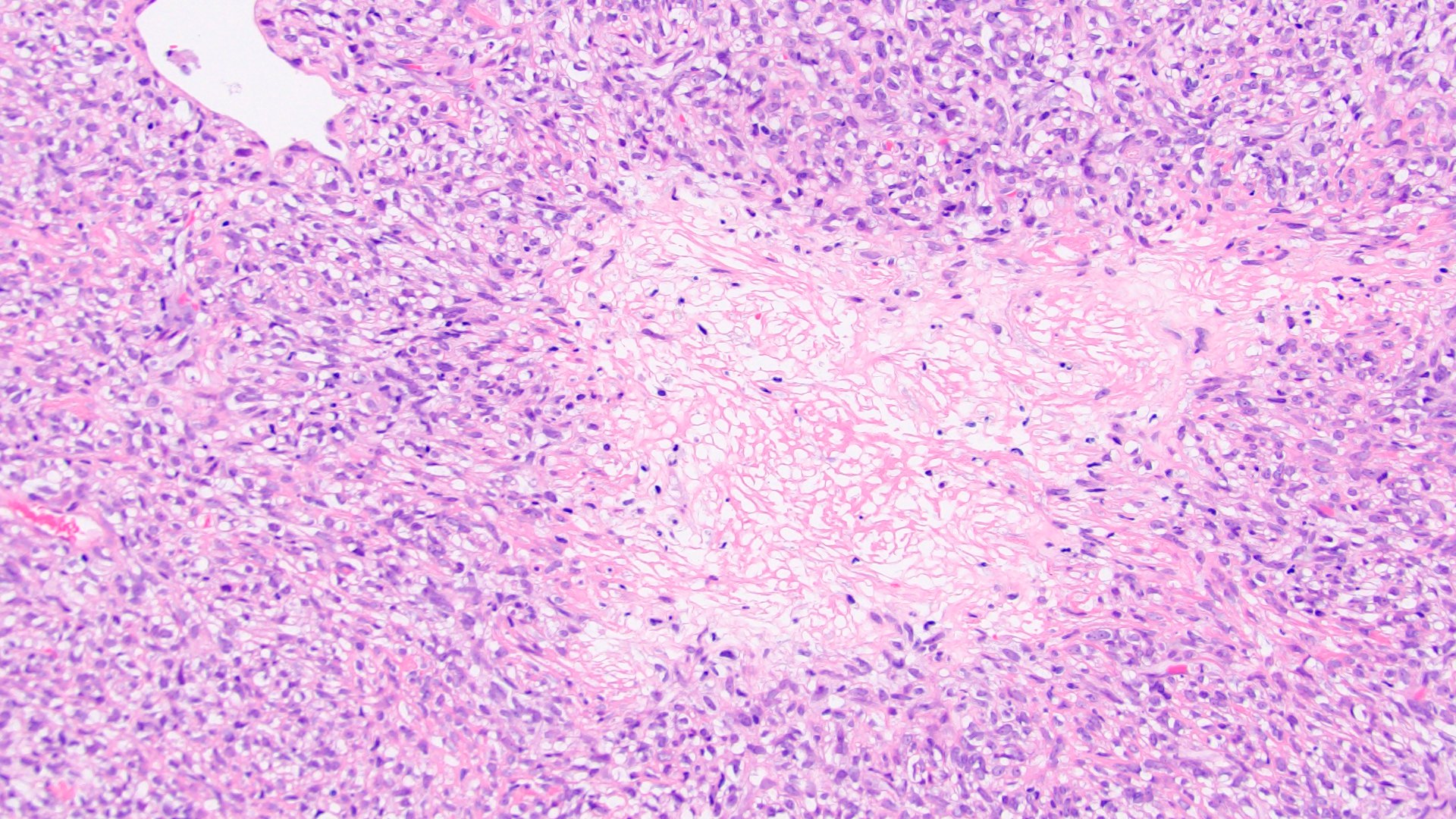
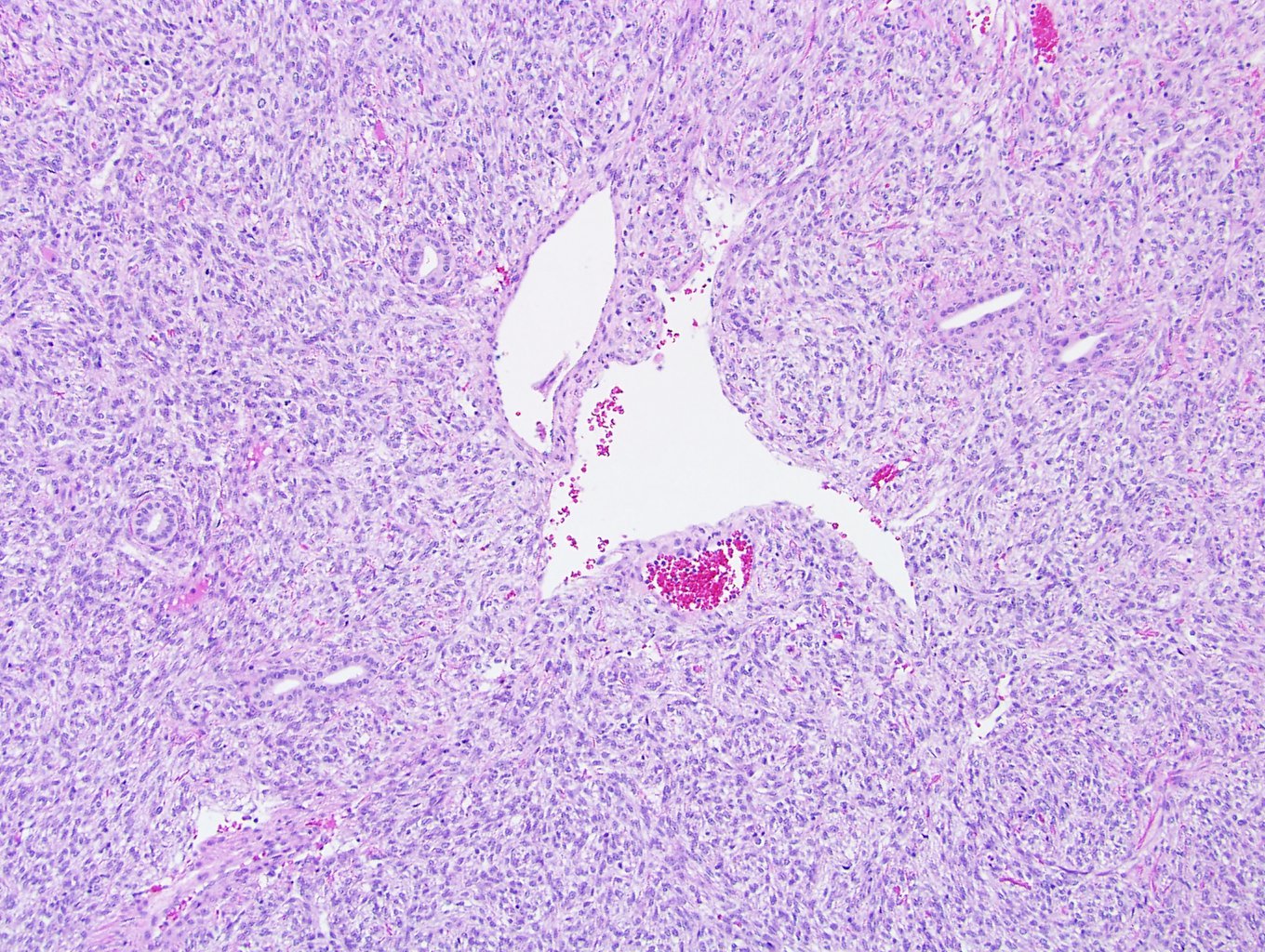
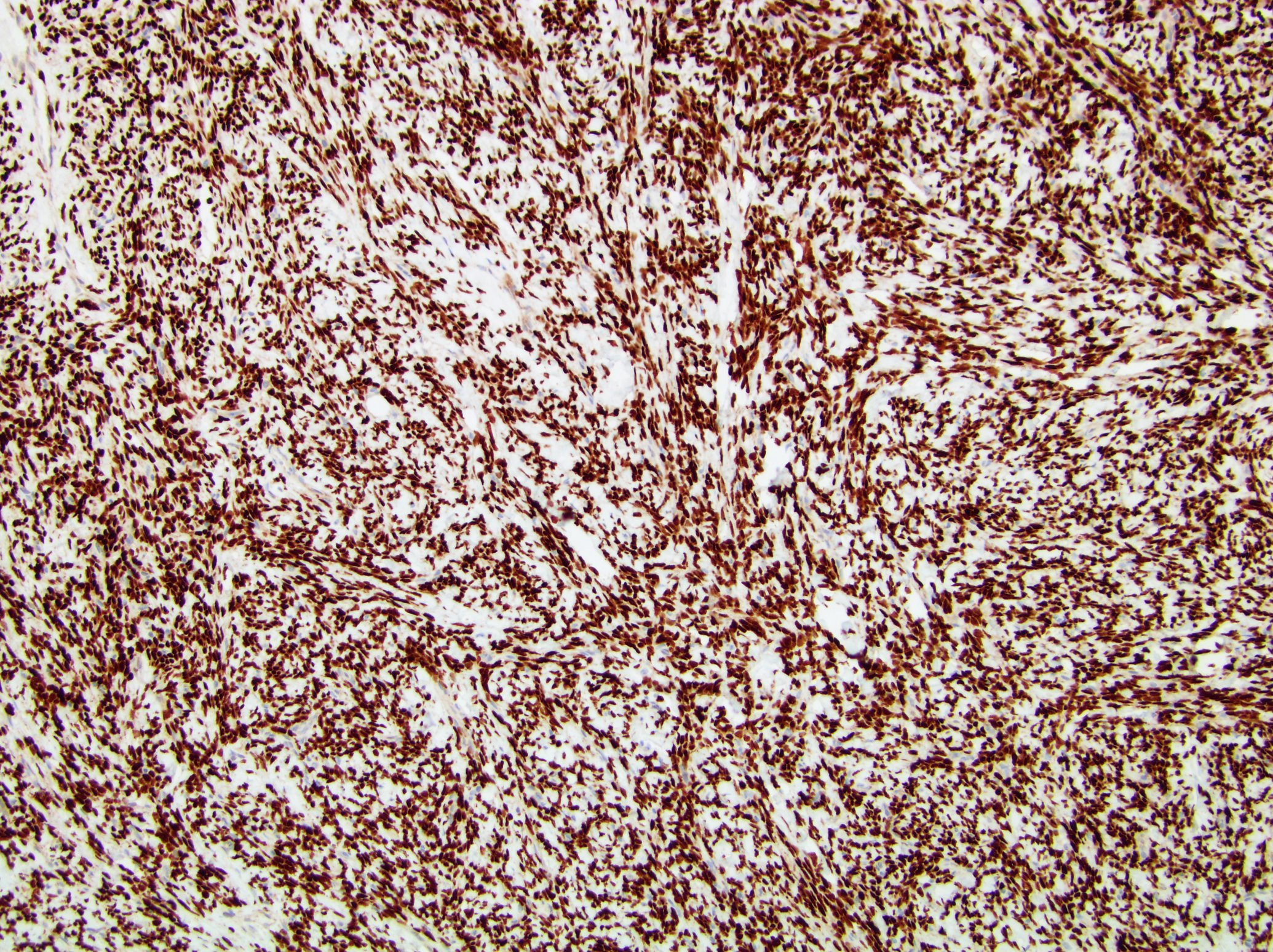
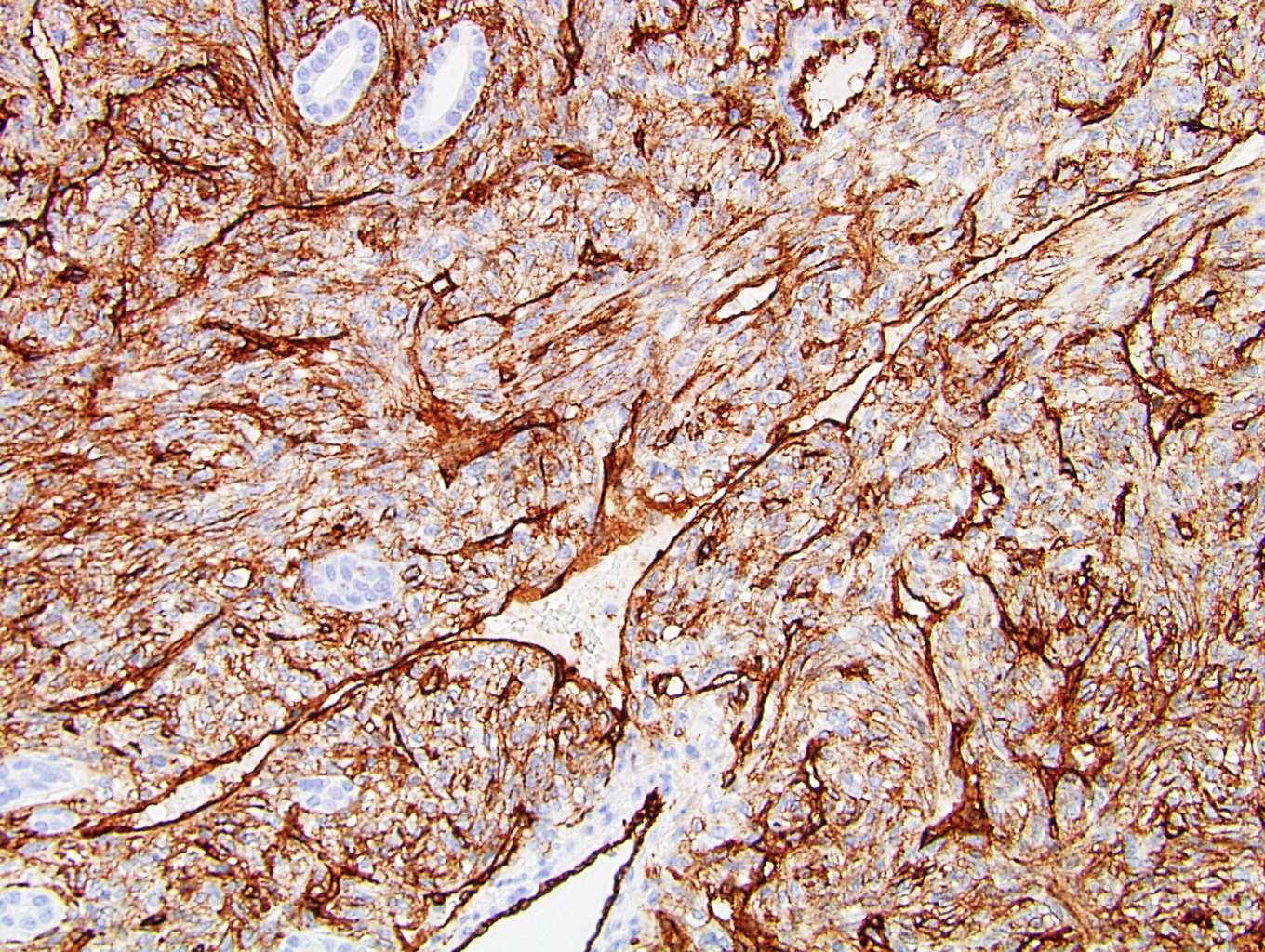
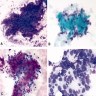
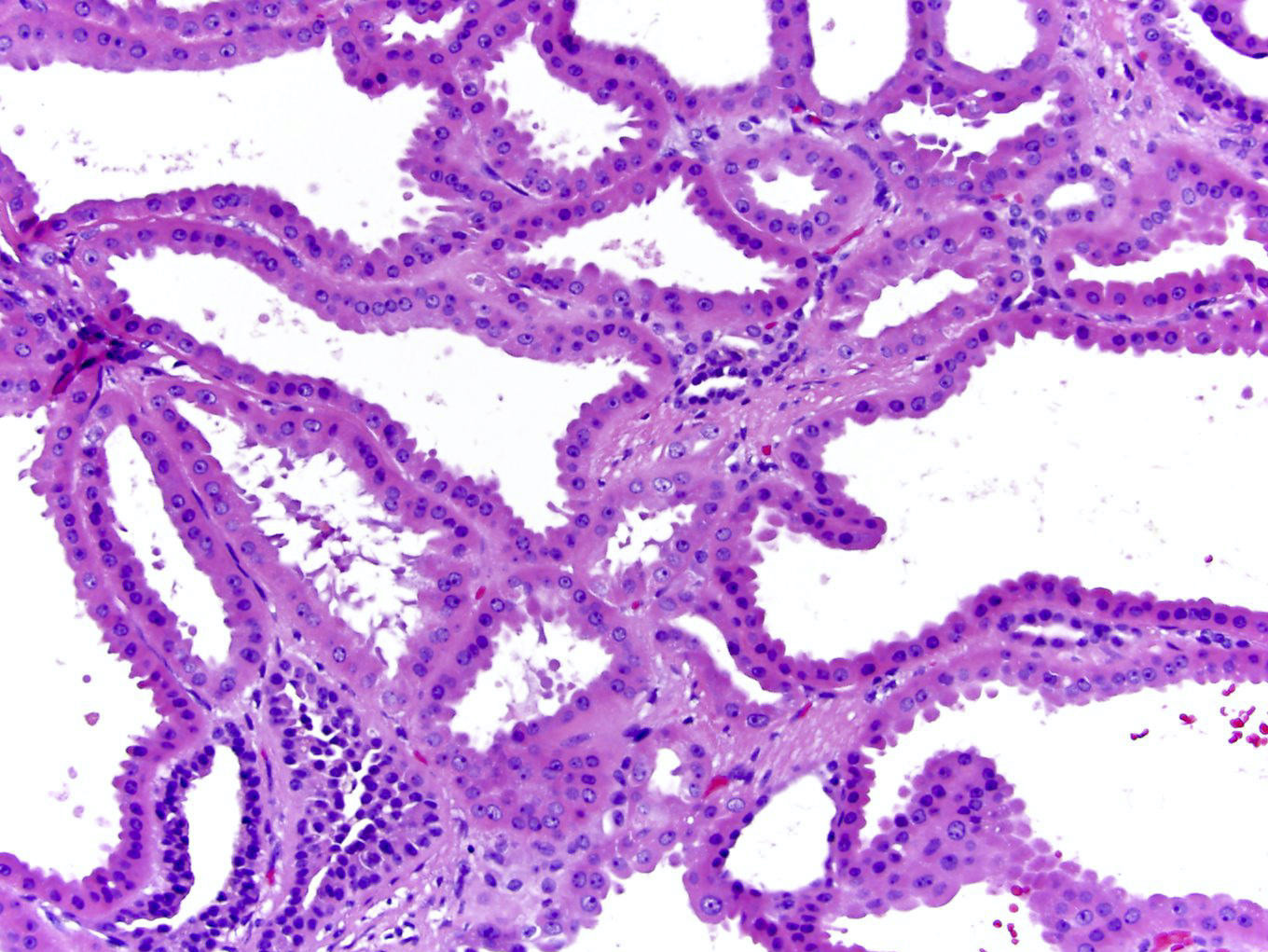
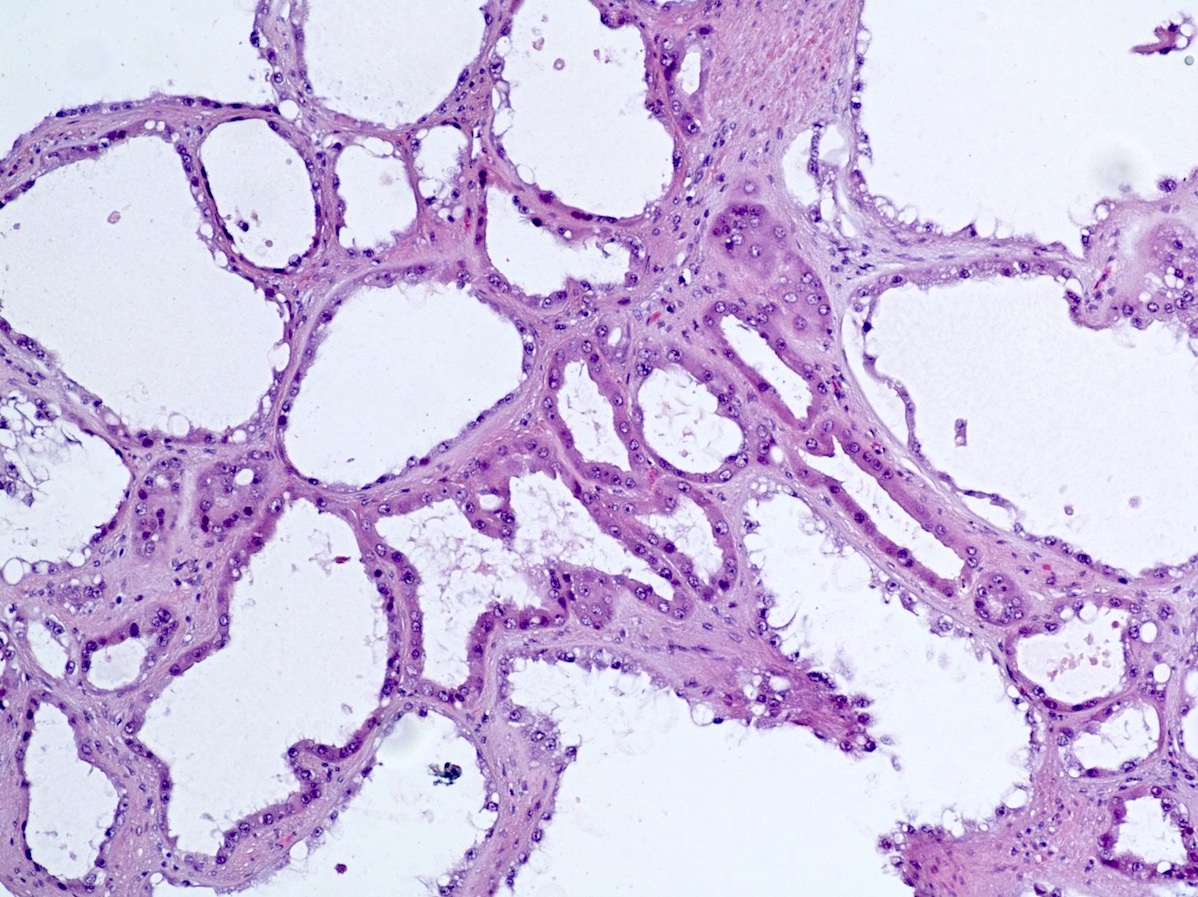
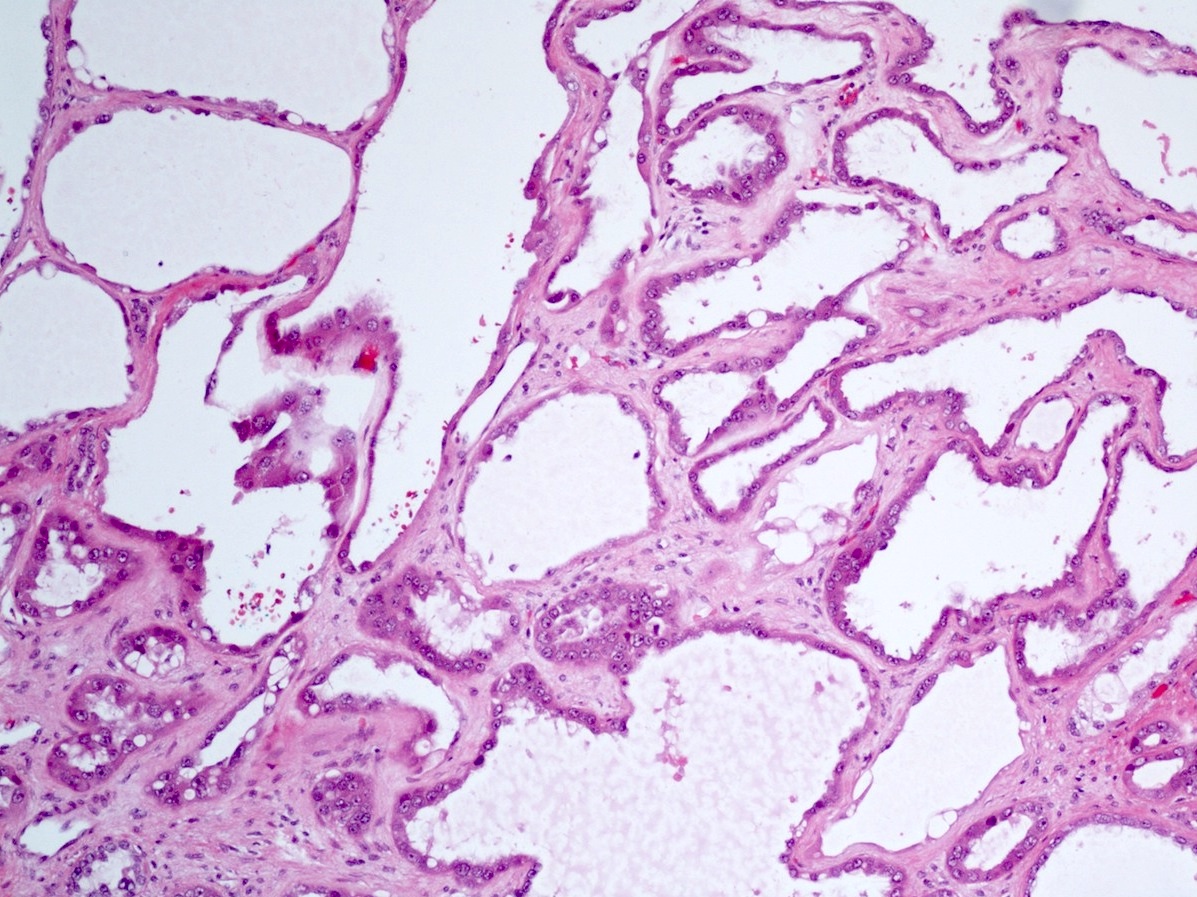
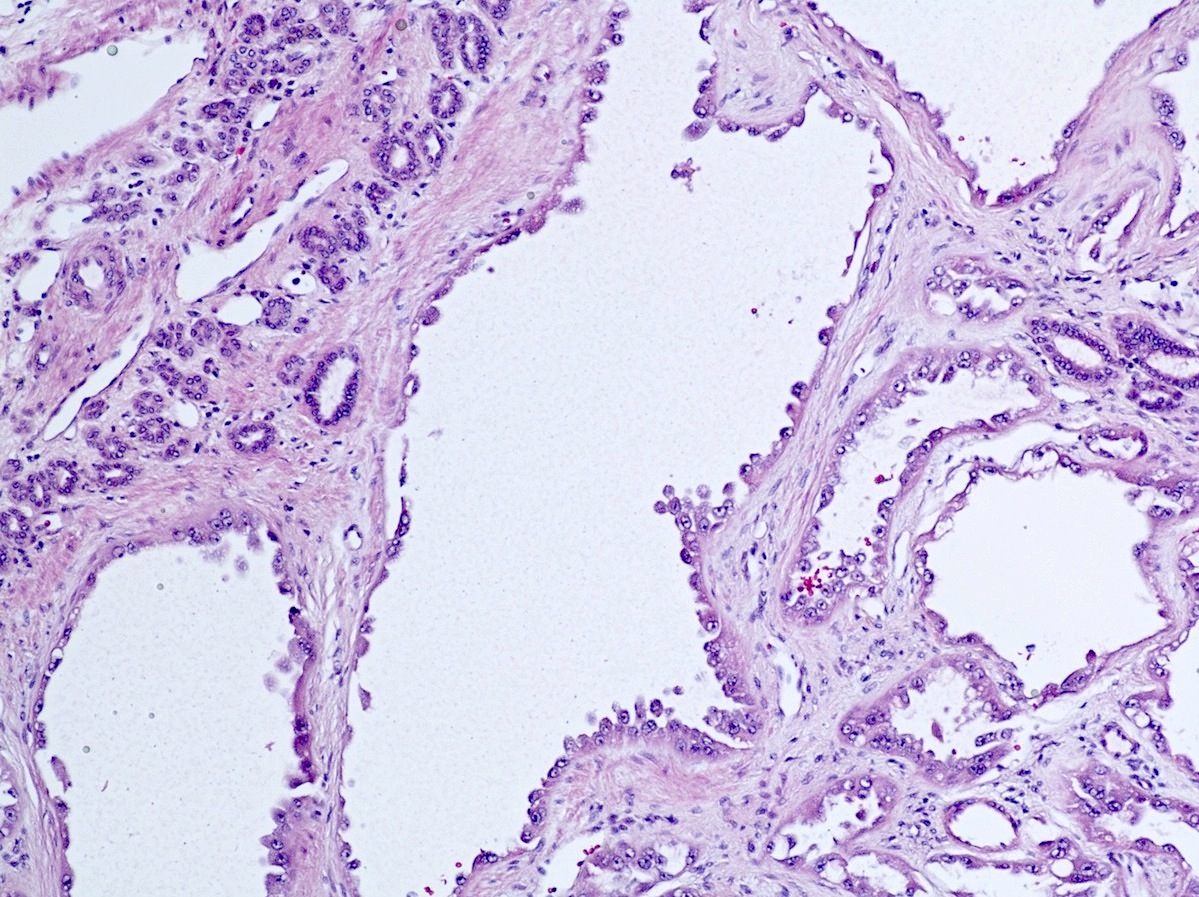
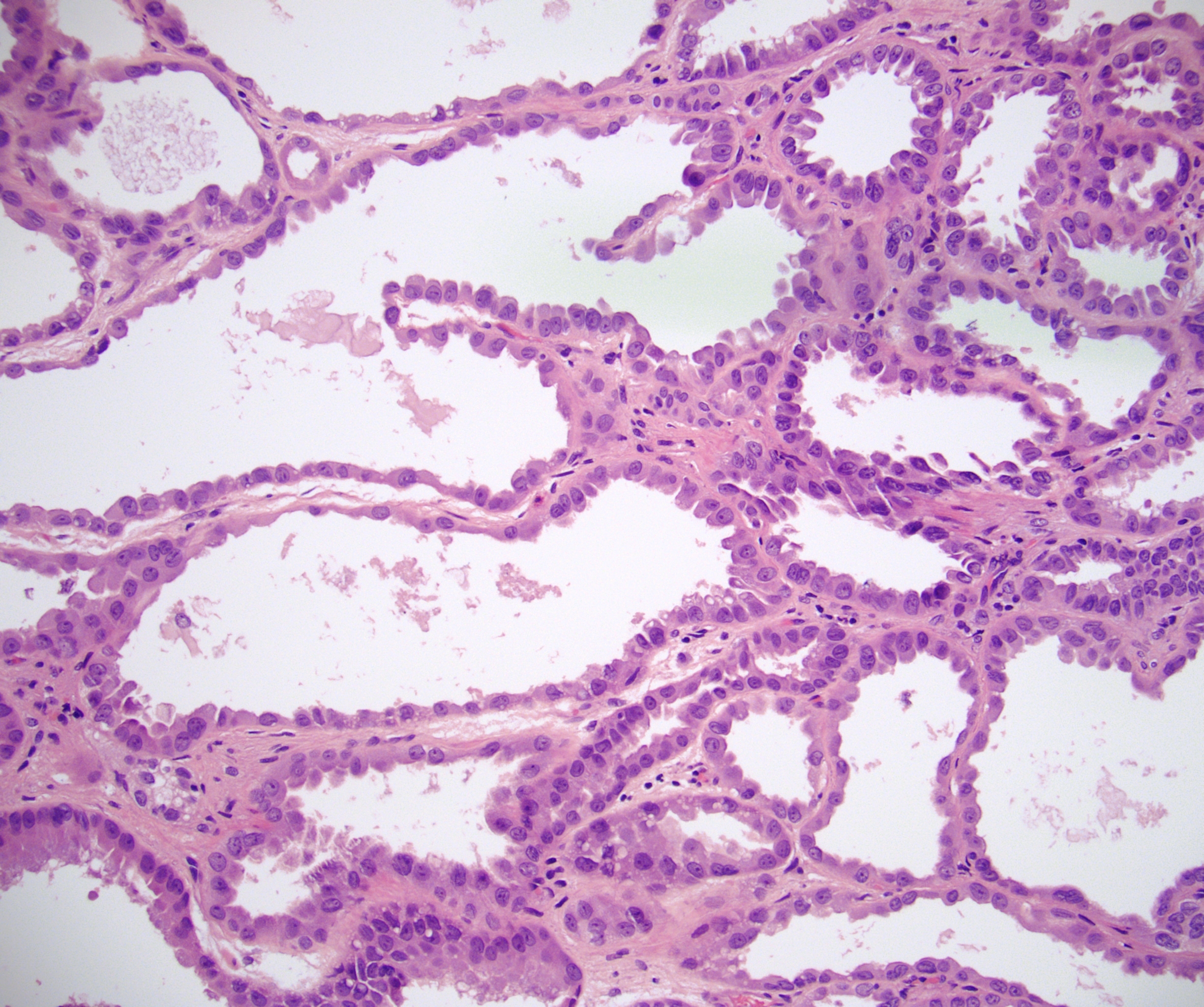
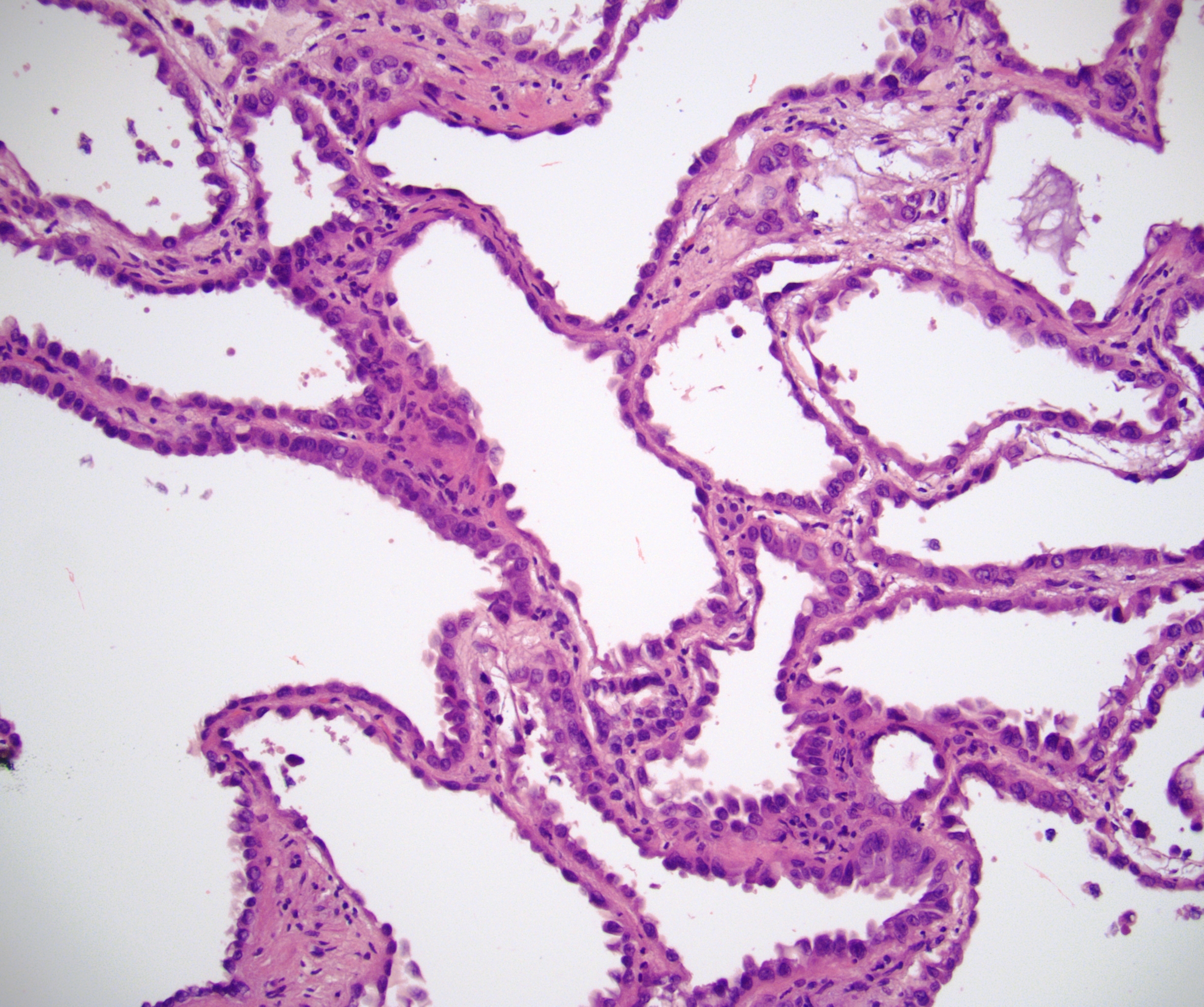
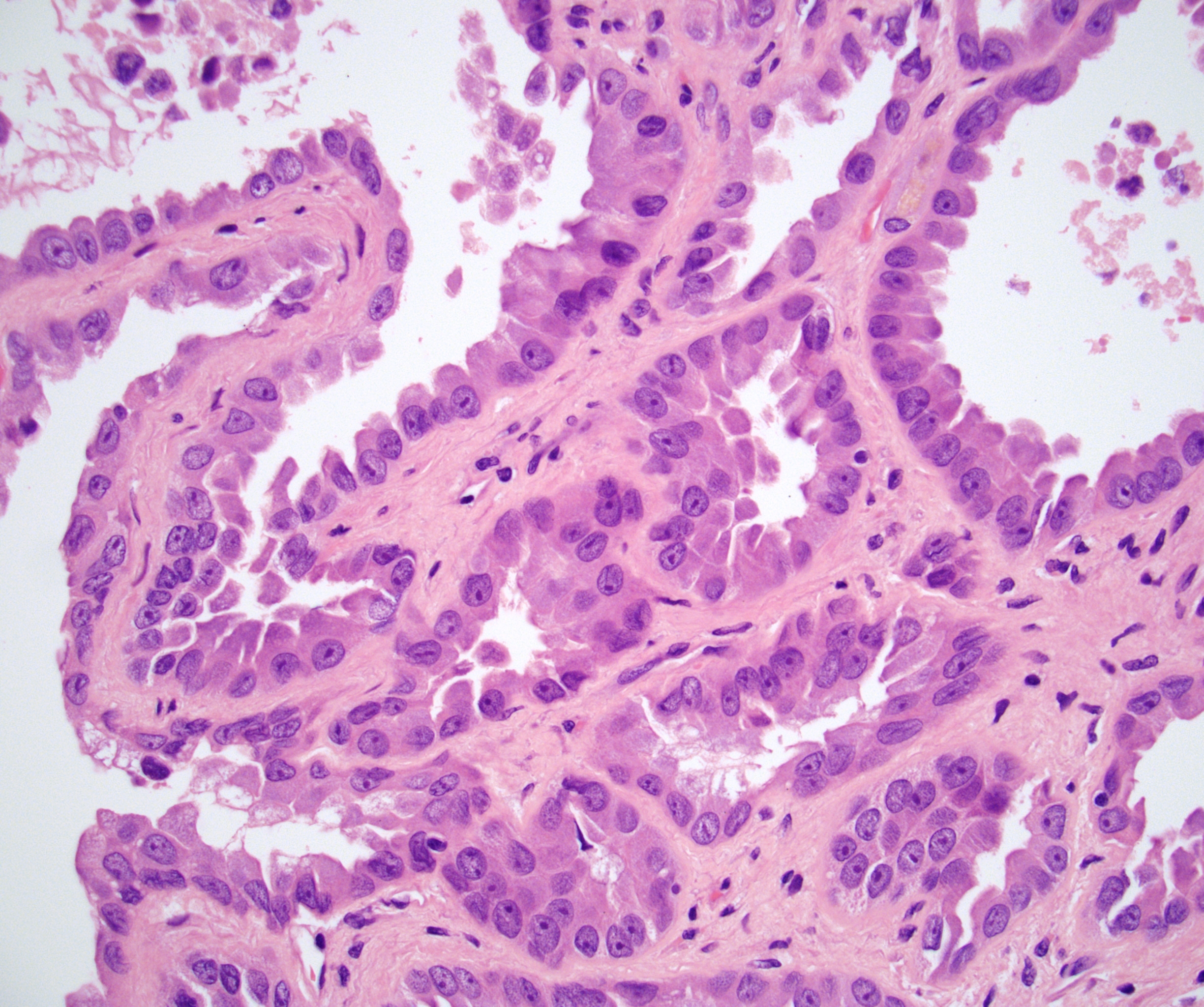
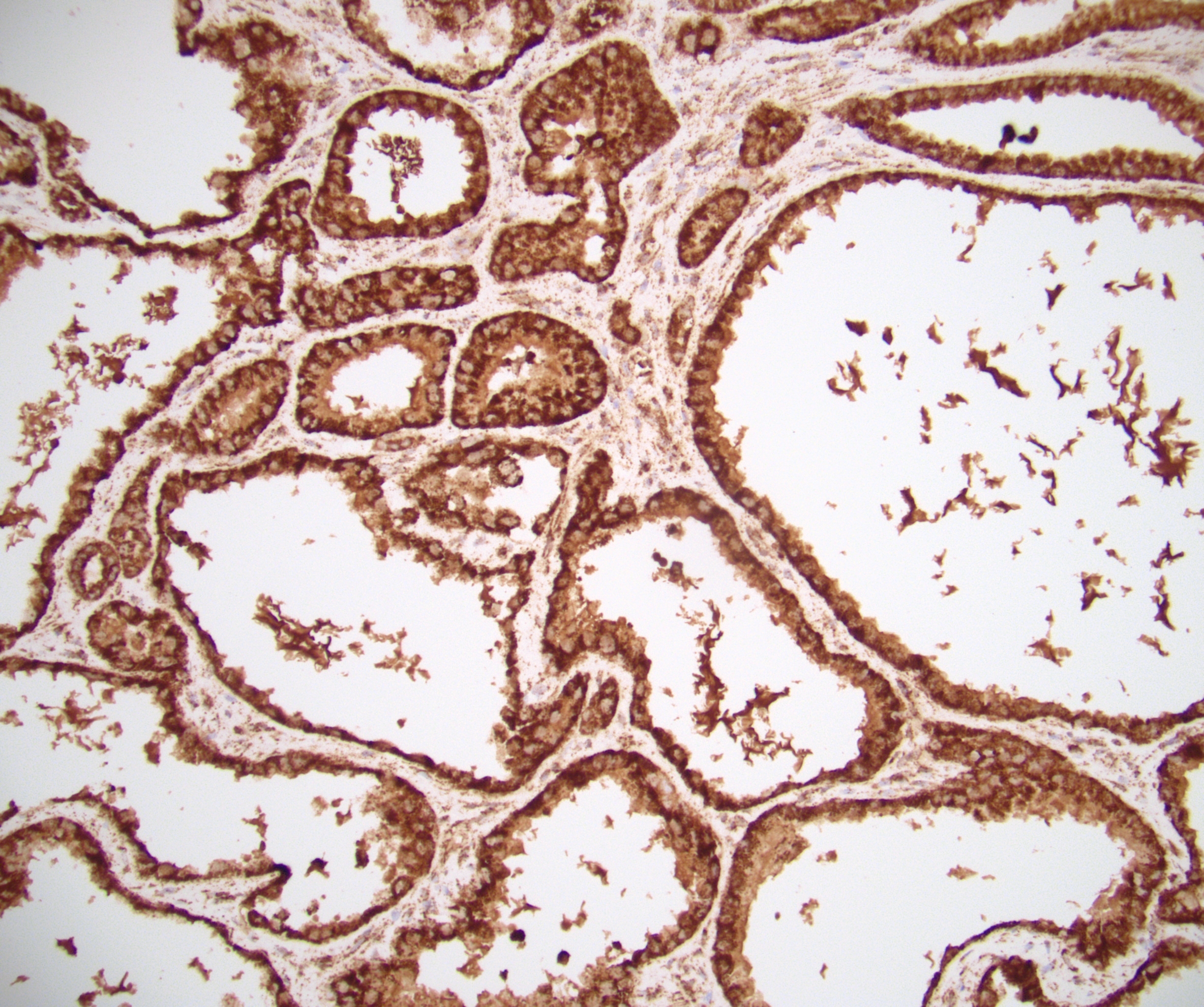
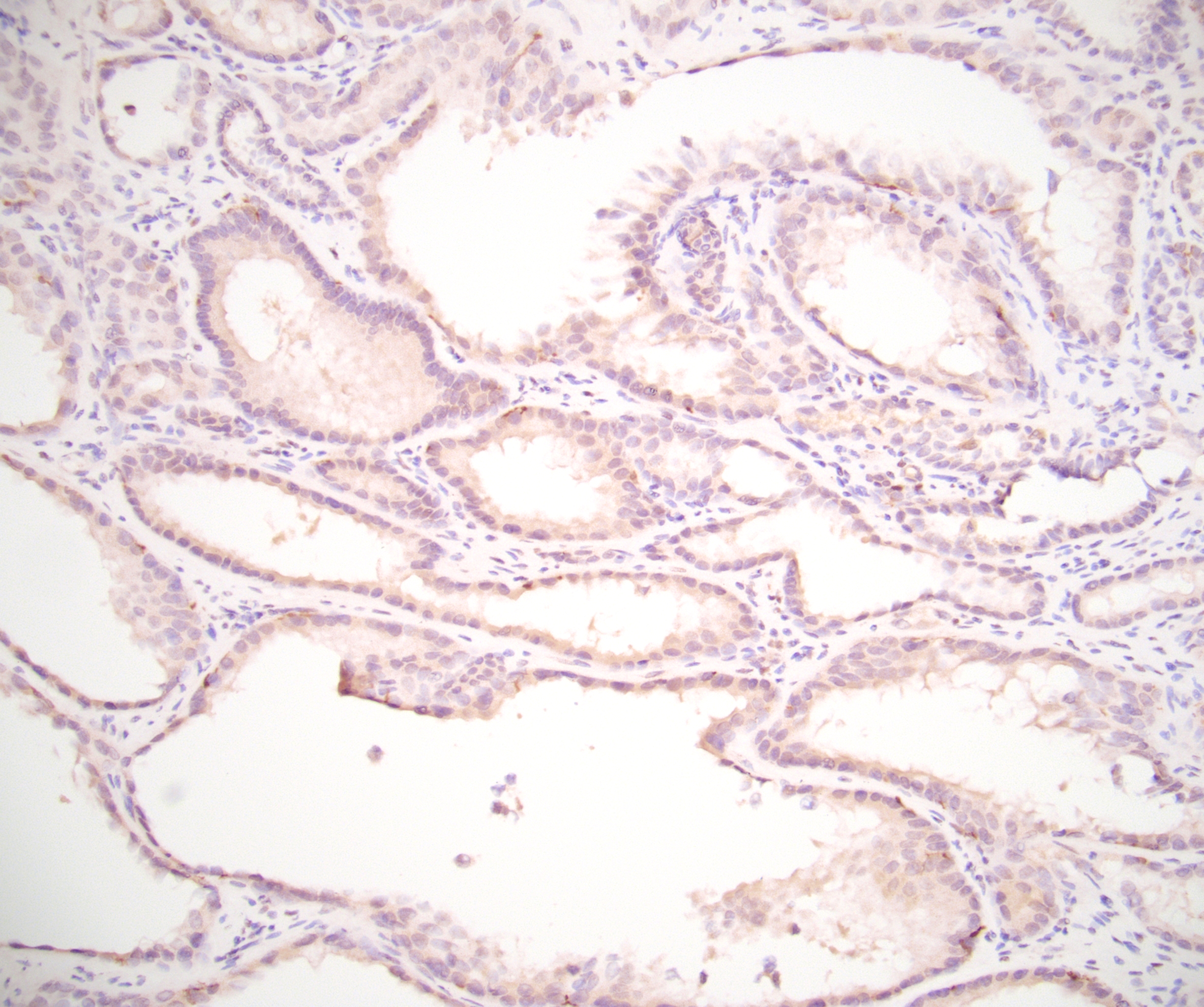
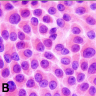
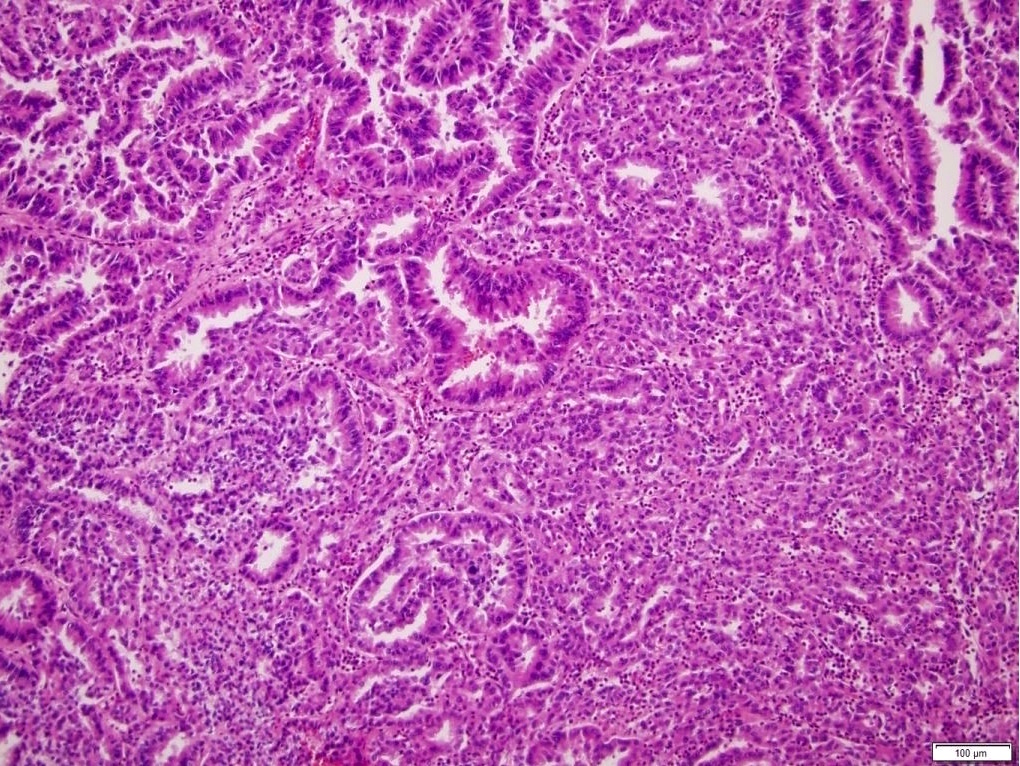
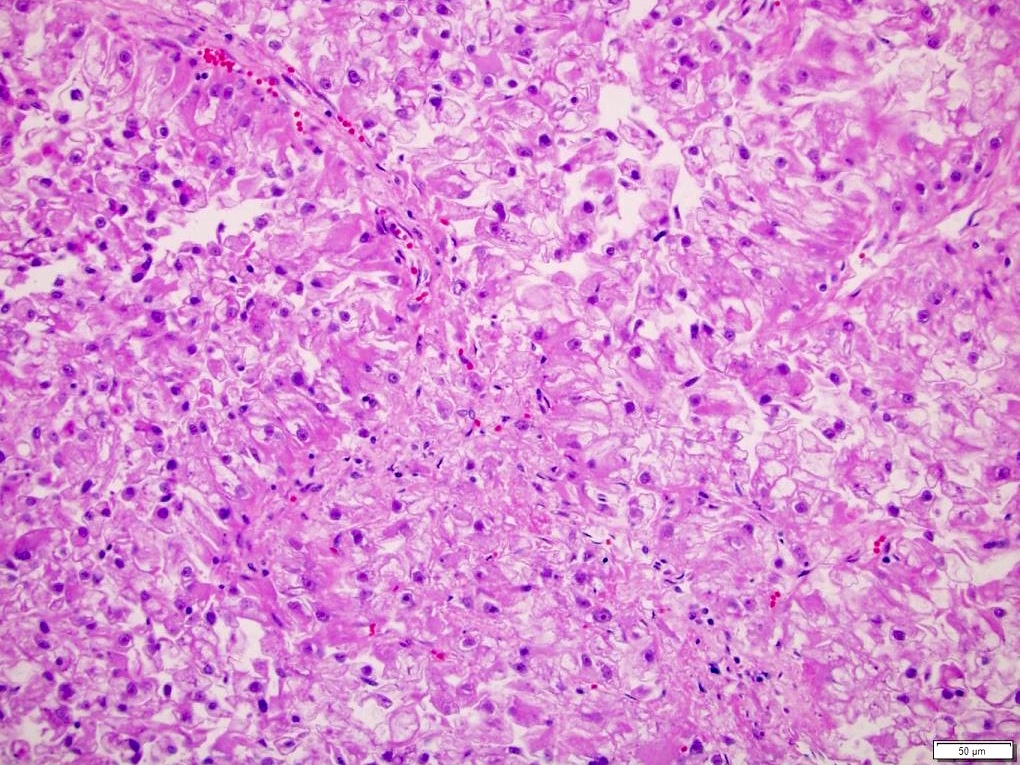
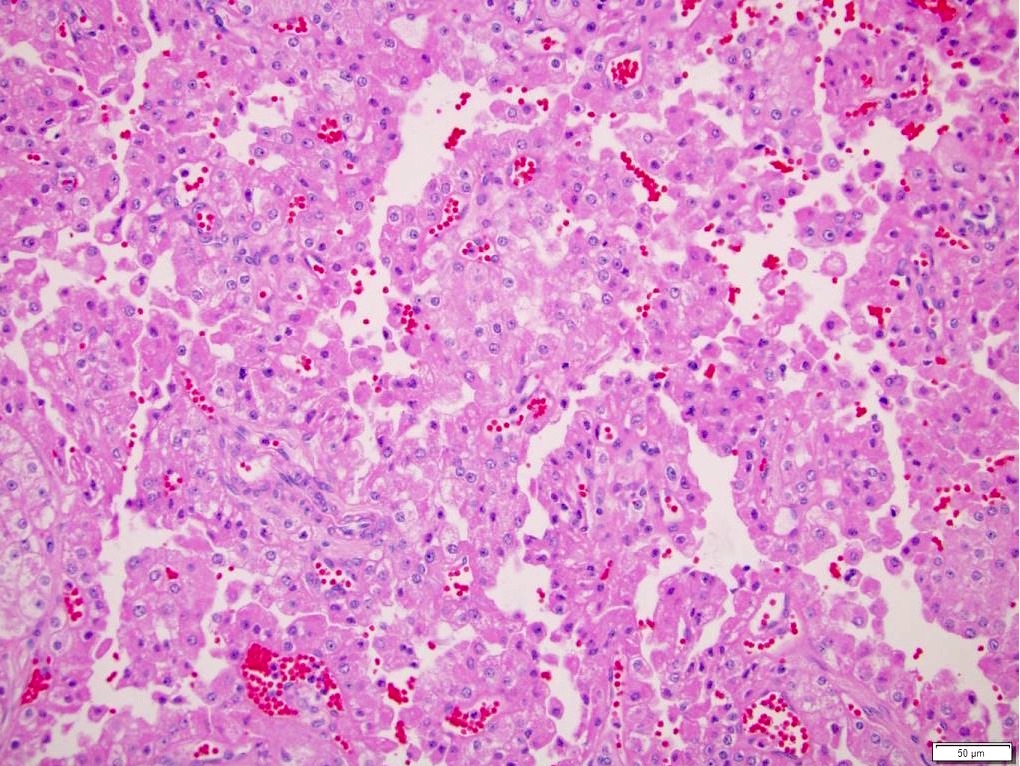
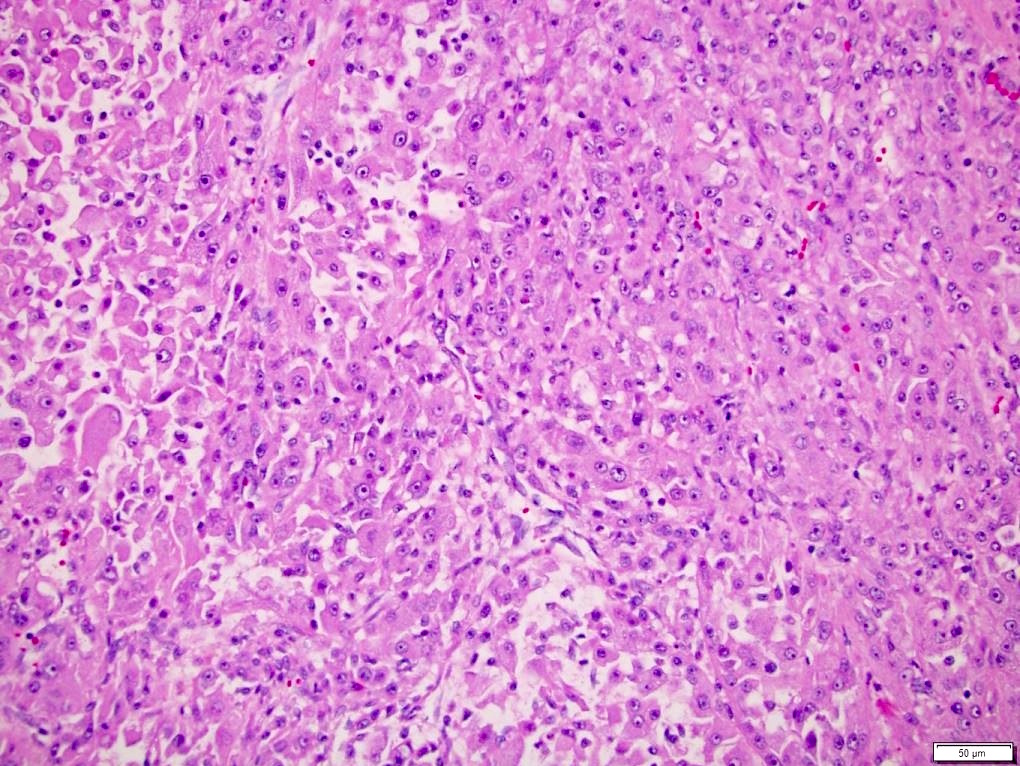
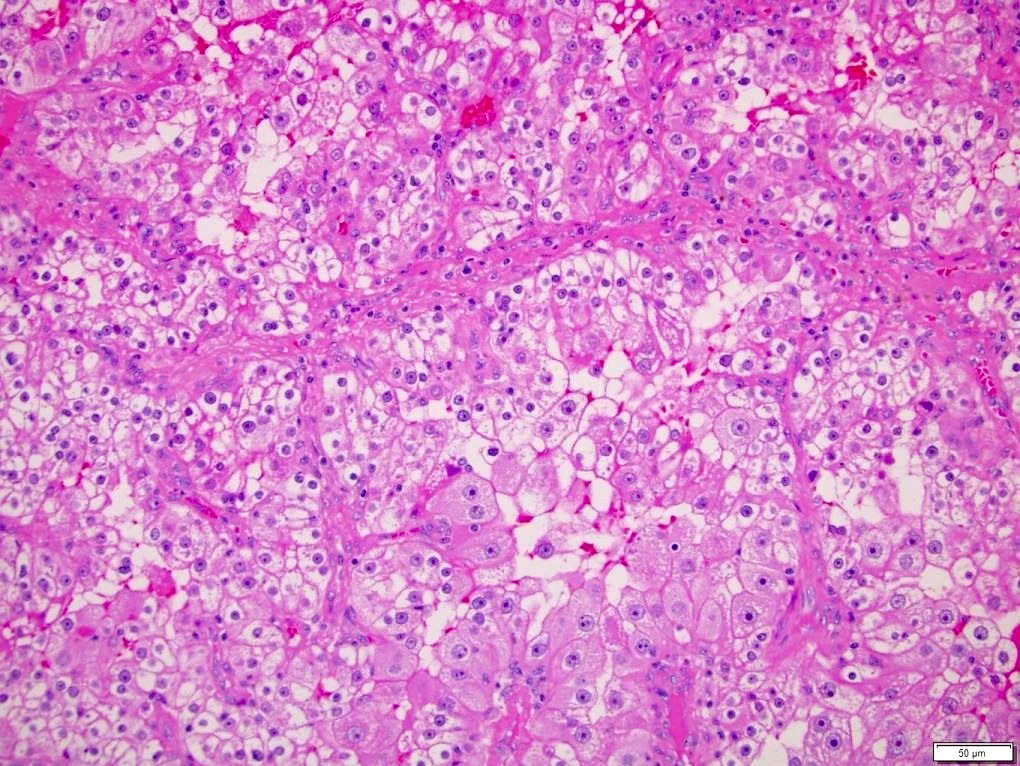
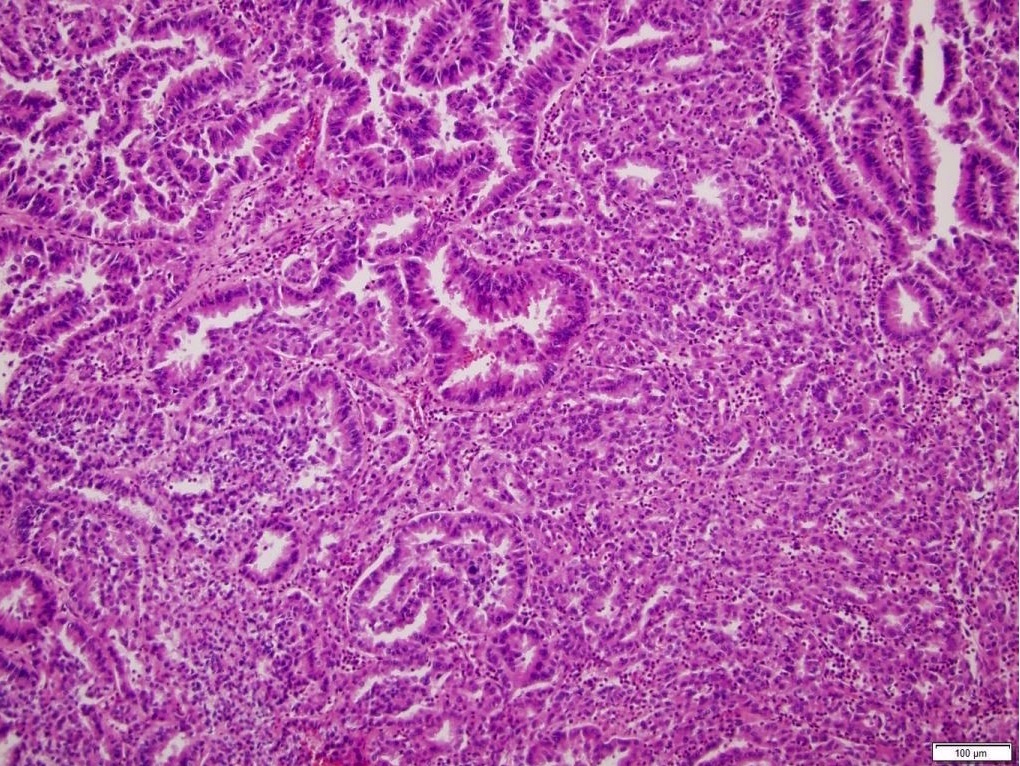
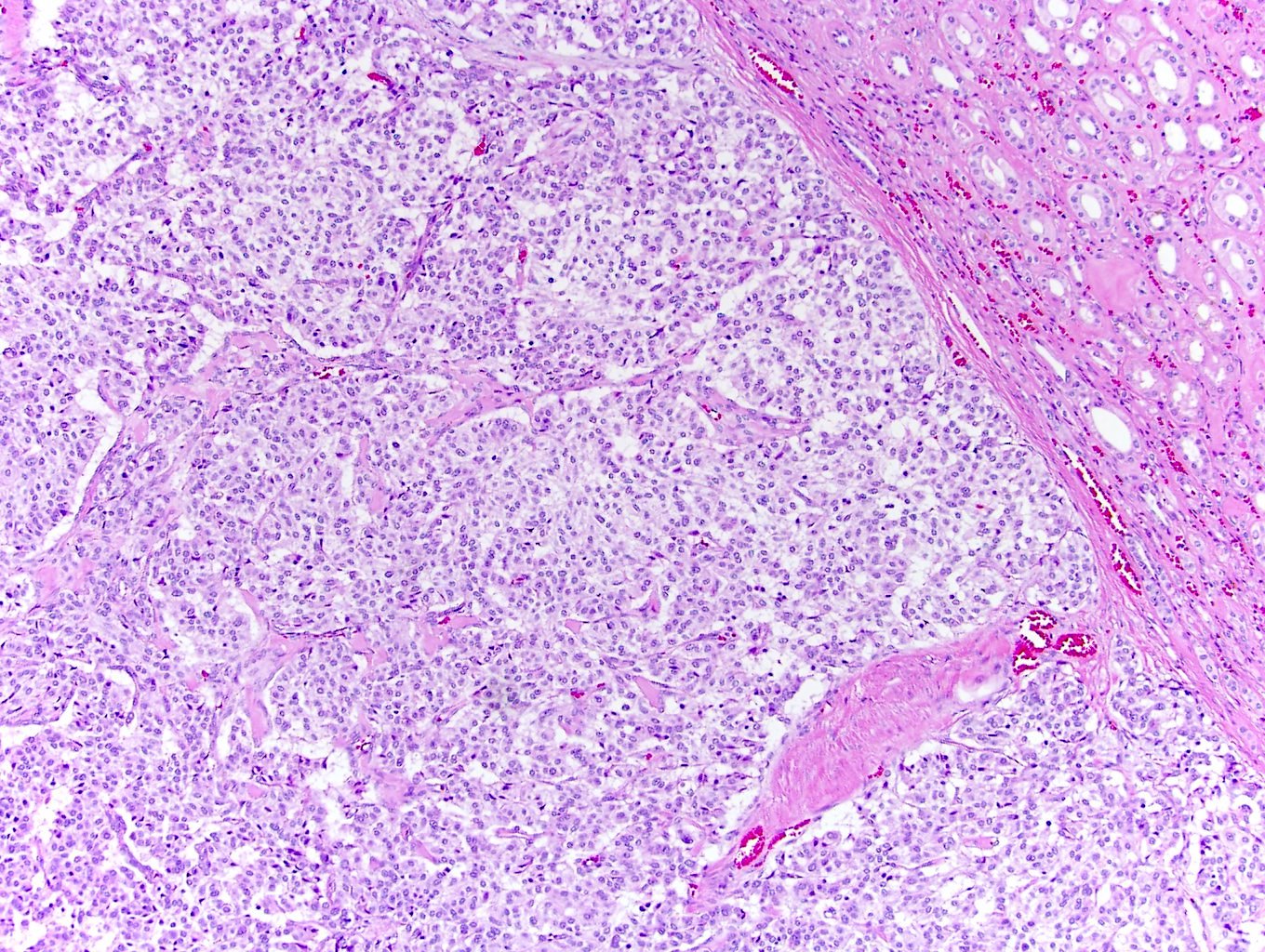
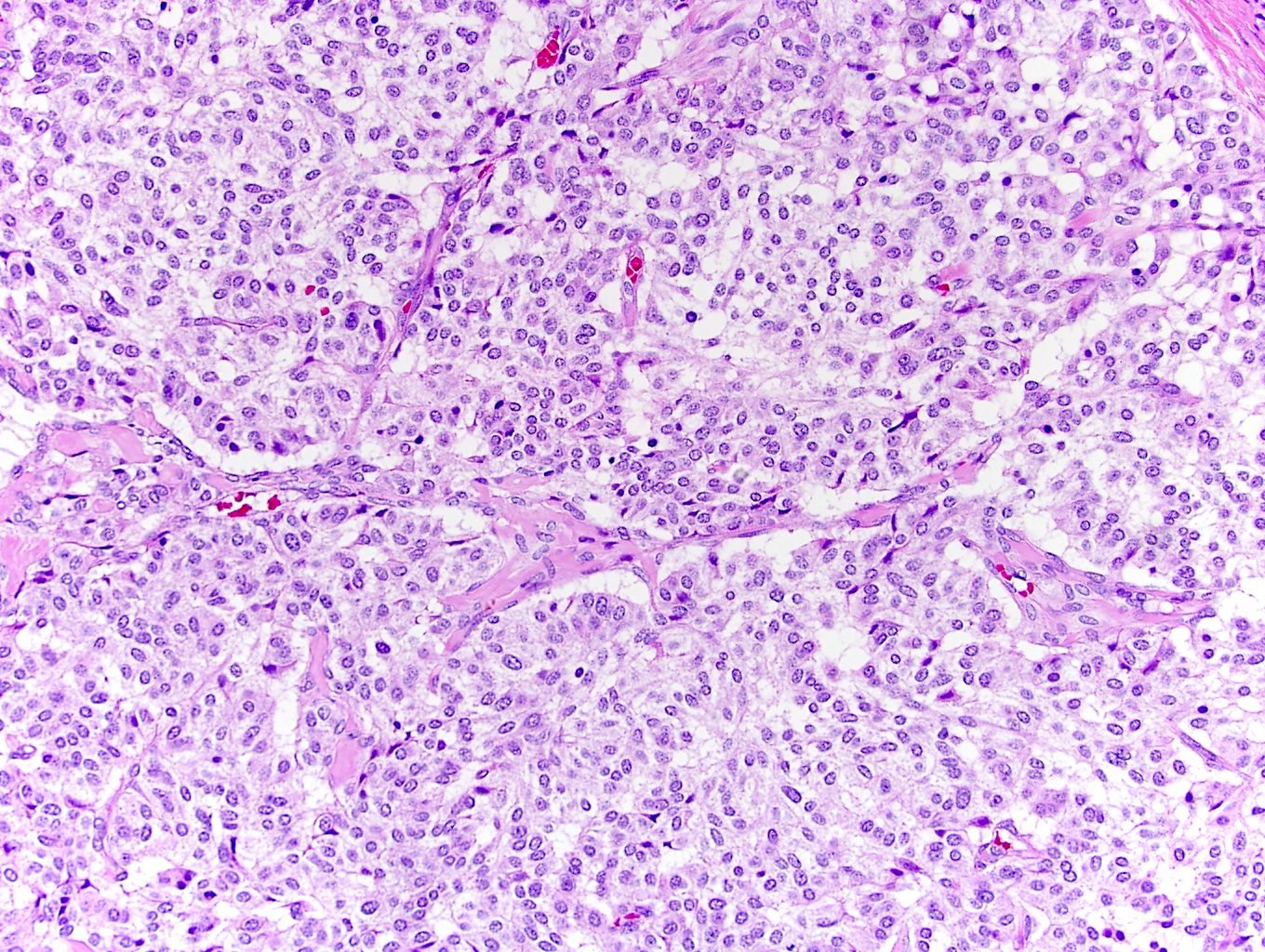
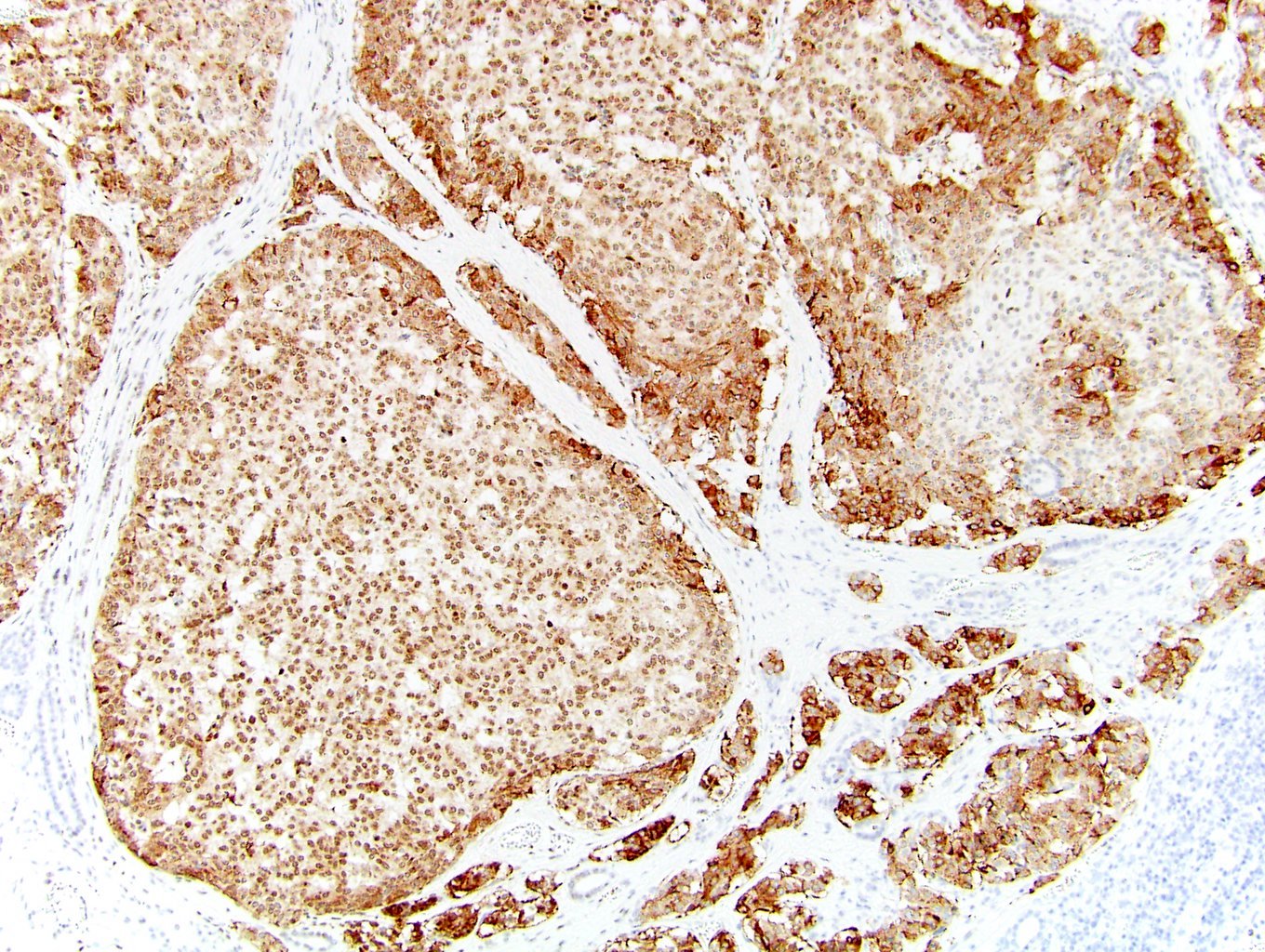
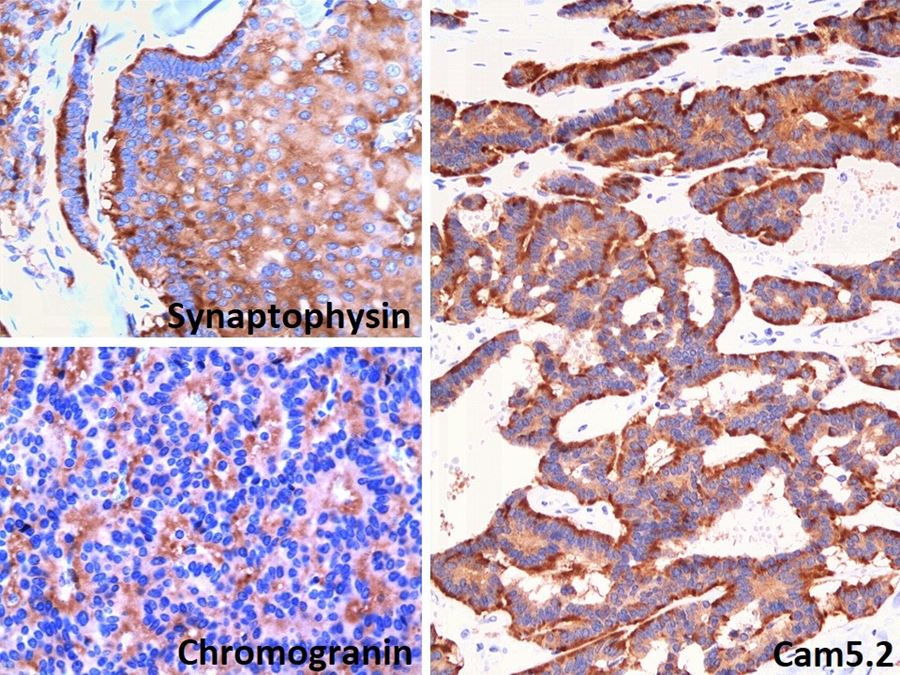
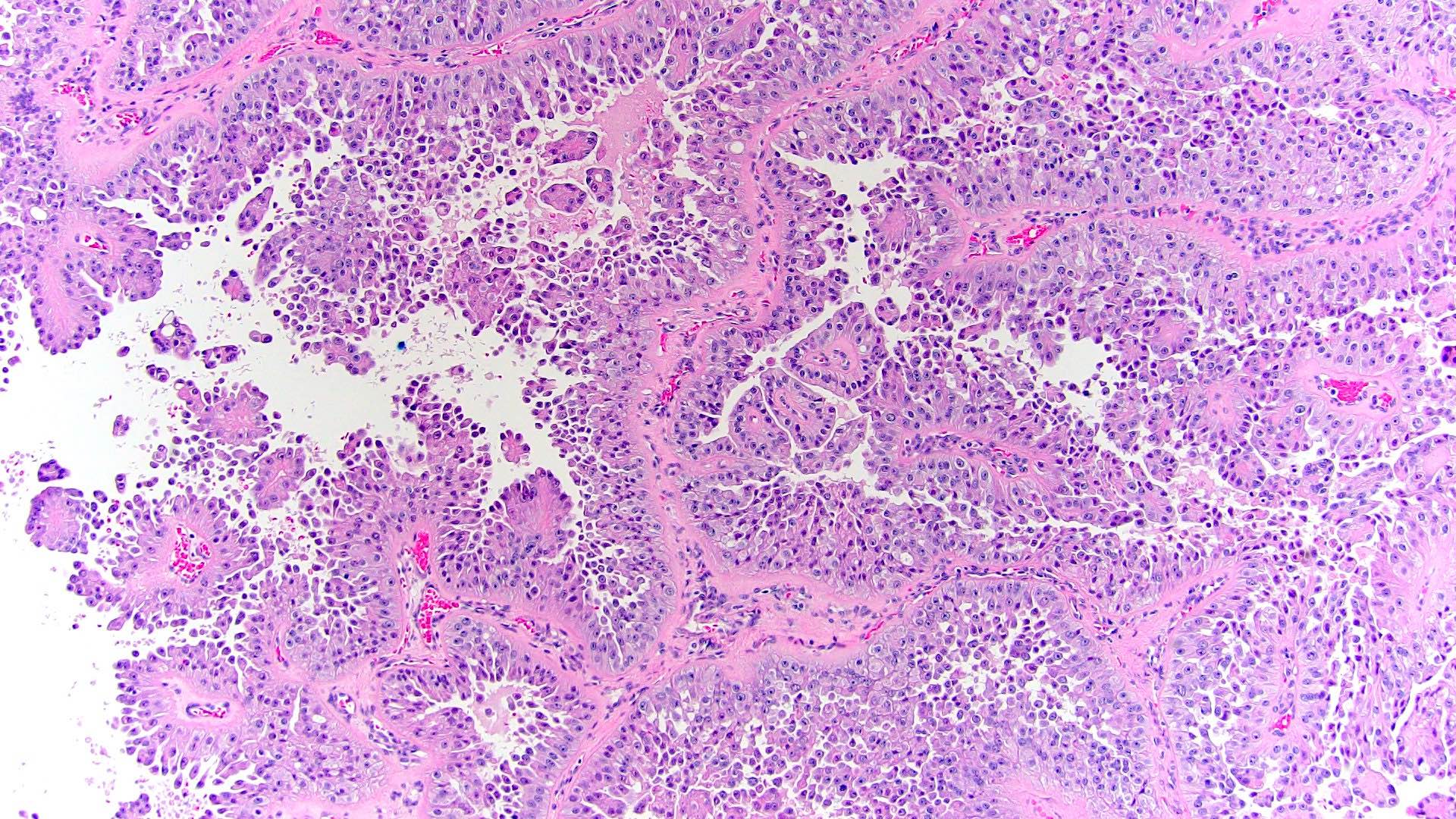
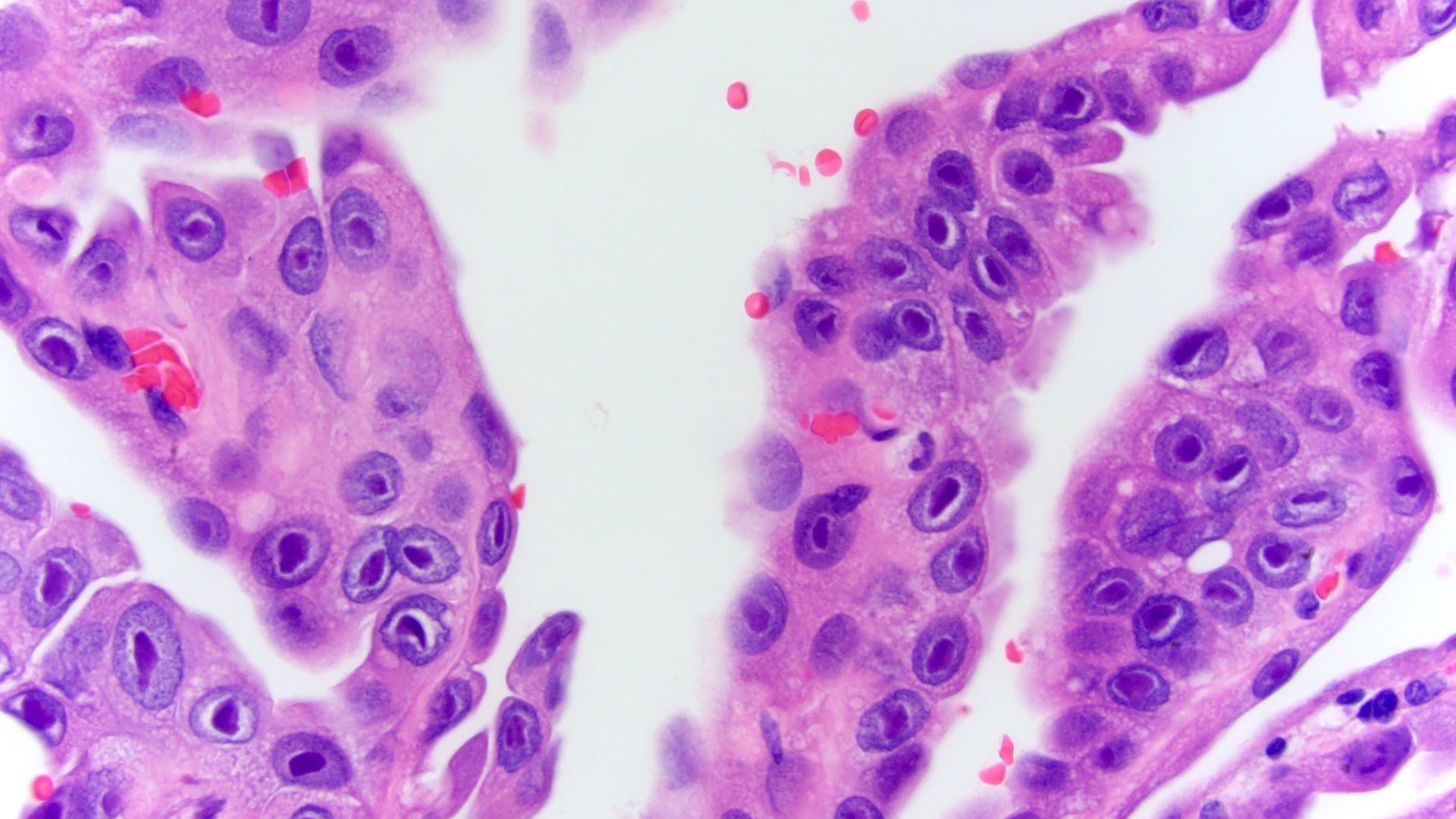
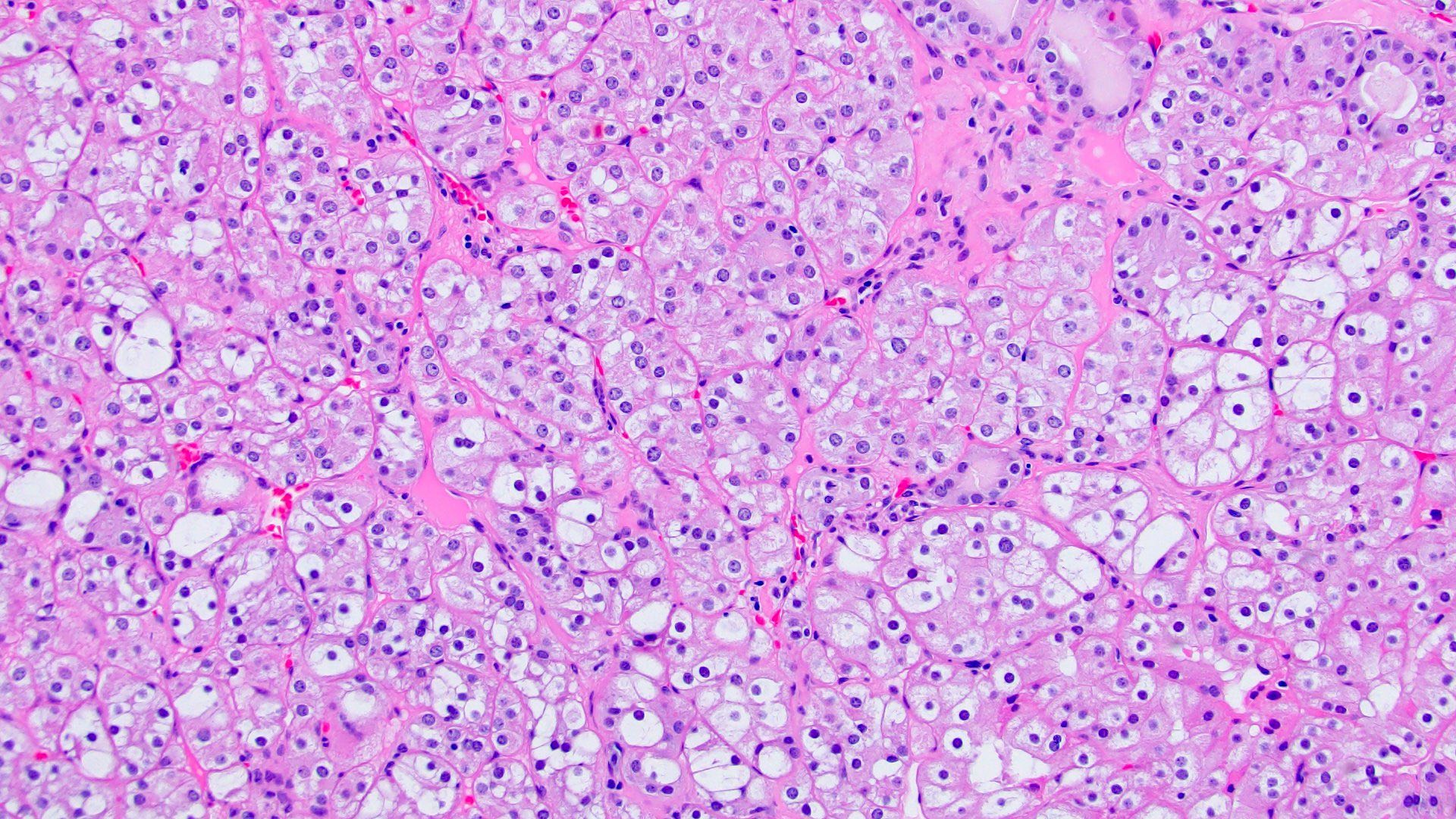
ALK-RCC is very heterogeneous with solid, tubular mucinous, rhabdoid, cribriform and signet ring histology. Tumor cells could be amphophilic, eosinophilic, polygonal rhabdoid and often contain cytoplasmic vacuoles. Immunostaining with ALK is positive (focally or diffusely), whereas INI1 protein is intact.
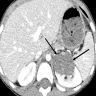
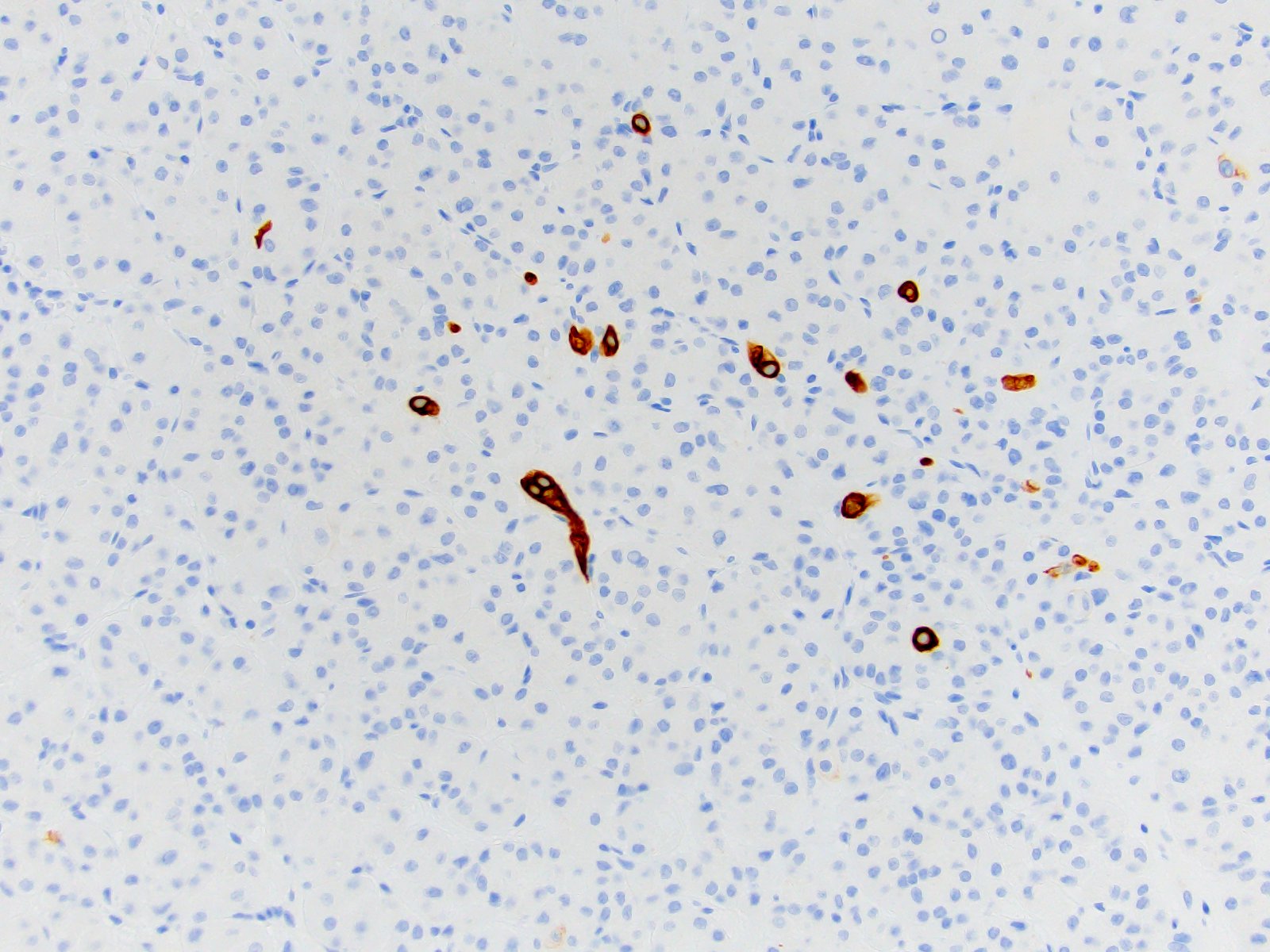
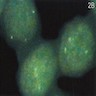
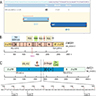
ALK gene rearrangement should be confirmed by FISH or molecular studies. The majority of reported tumors are indolent; however, some cases may pursue an aggressive clinical course.
Comment Here Reference:
ALK translocation