


- Adamantinomatous craniopharyngioma is a histologically benign, partially cystic epithelial neoplasm of the suprasellar or sellar region, resembling ameloblastoma or keratinizing and calcifying odontogenic cyst
- Always WHO grade 1
- Tumor with palisading epithelium, wet keratin and stellate reticulum associated with surrounding gliosis and Rosenthal fibers
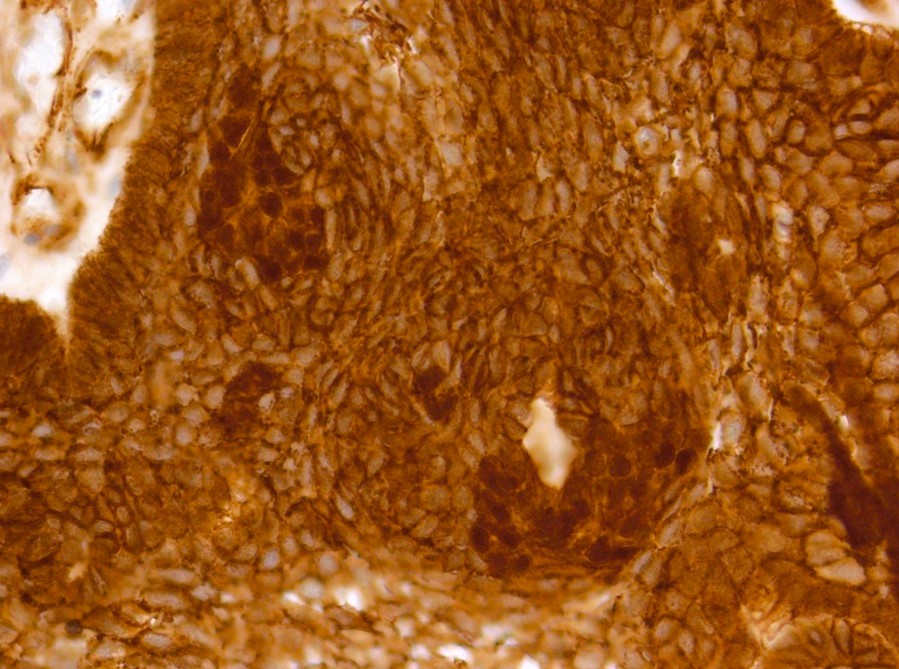
- Shows CTNNB1 mutations and aberrant nuclear expression of beta catenin in up to 95% of cases (Acta Neuropathol Commun 2016;4:20)
- Incidence: low
- Age: bimodal, with peaks at 5 - 15 years and 45 - 60 years; rare neonatal and fetal cases have been reported (Nat Rev Dis Primers 2019;5:75)
- More common than papillary craniopharyngioma (even in adults)
- Incidence rates are similar in males and females and between Caucasians and African Americans (Neurosurg Focus 1997;3:e1)
- Suprasellar: frequently extends into neighboring structures
- Rarely intranasal, sphenoid sinus, cerebellopontine angle and pineal region
- Reference: J Neurol Surg Rep 2016;77:e121
- Possibly arises from neoplastic transformation of ectodermal derived epithelial cell remnants of Rathke pouch and the craniopharyngeal duct
- Misplaced odontogenic rests along pituitary stalk
- Reference: Childs Nerv Syst 2005;21:622
- Insidious, with a delay of approximately 1 - 2 years between initial symptoms and diagnosis (Orphanet J Rare Dis 2007;2:18)
- Visual disturbances are more frequently observed in children
- Endocrine deficiencies:
- Growth hormone (GH) > luteinizing hormone (LH) / follicle stimulating hormone (FSH) > adrenocorticotrophic hormone (ACTH) > thyroid stimulating hormone (TSH)
- Growth failure and delayed puberty in children
- Diabetes insipidus (> adults)
- Headache (most common, due to mass effect or hydrocephalus)
- Cognitive impairment and personality changes (about 50% of patients)
- Rare chemical meningitis with cyst rupture and spillage
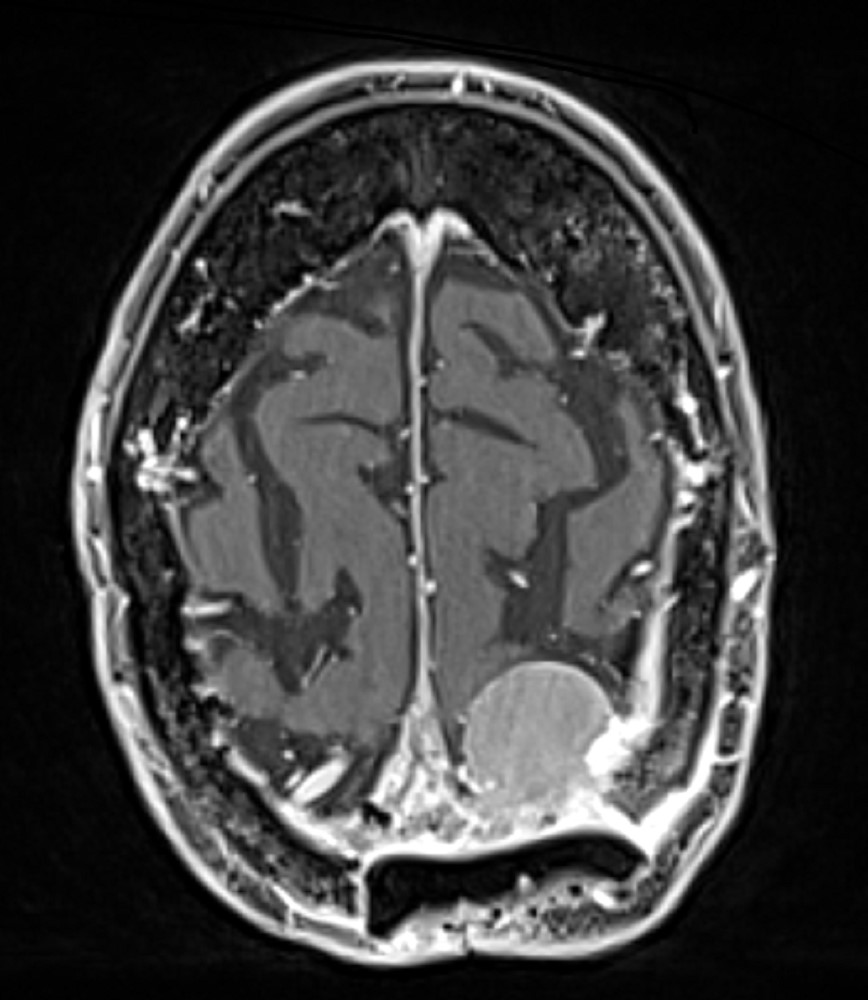
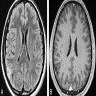
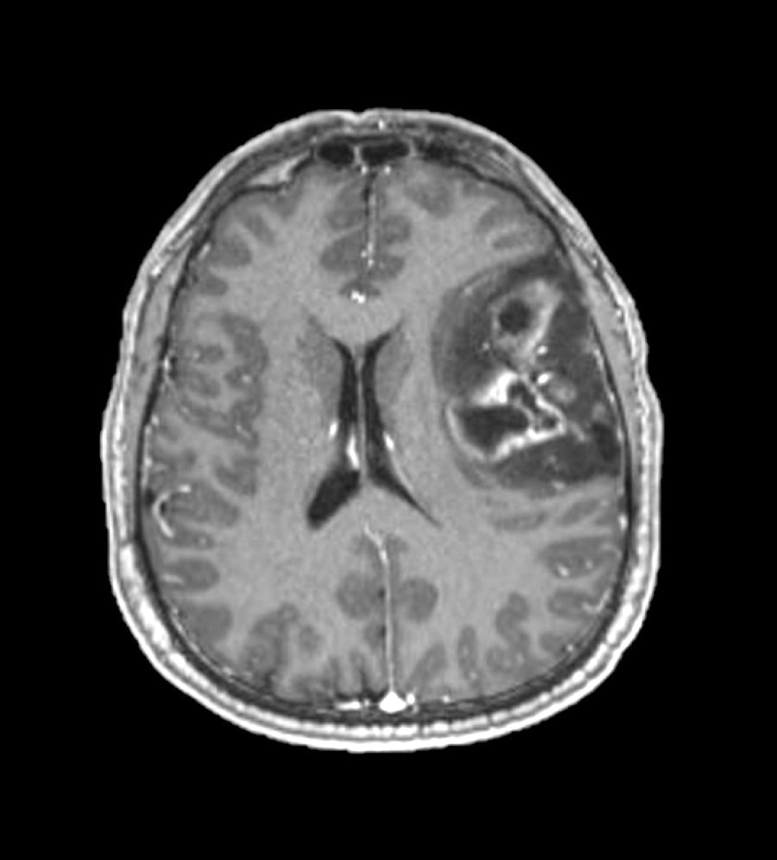
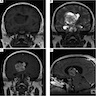
- Best diagnostic clue is preoperative imaging (Front Endocrinol (Lausanne) 2011;2:70)
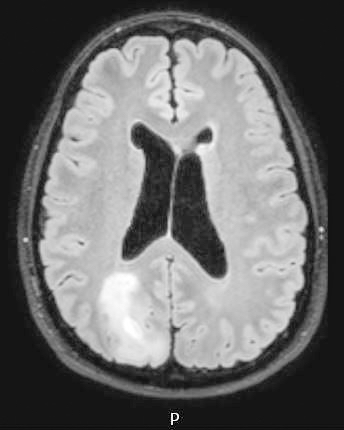
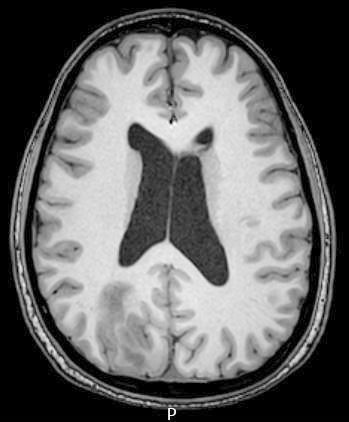
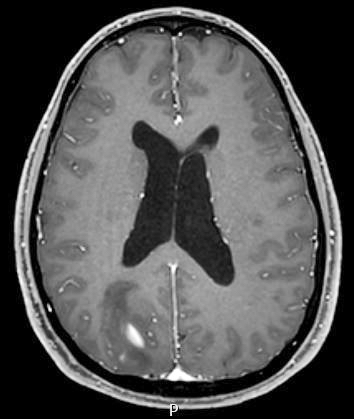
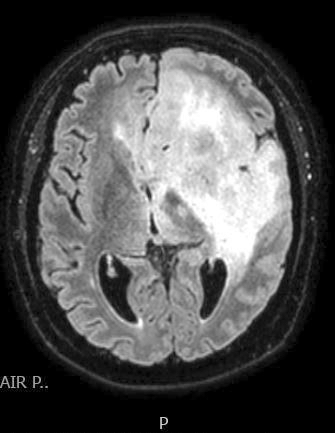
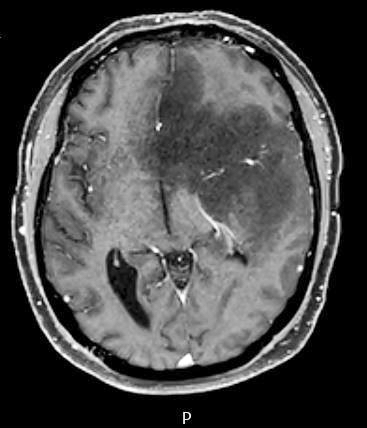
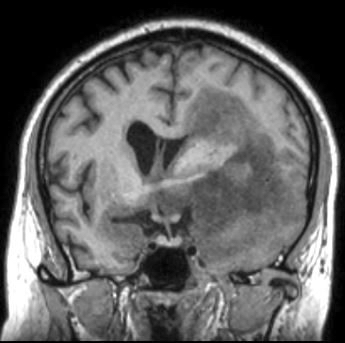
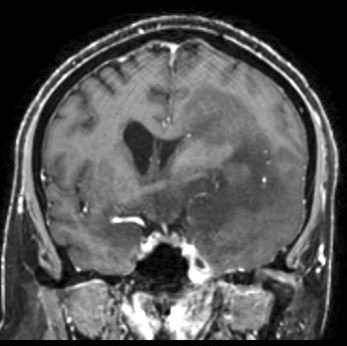
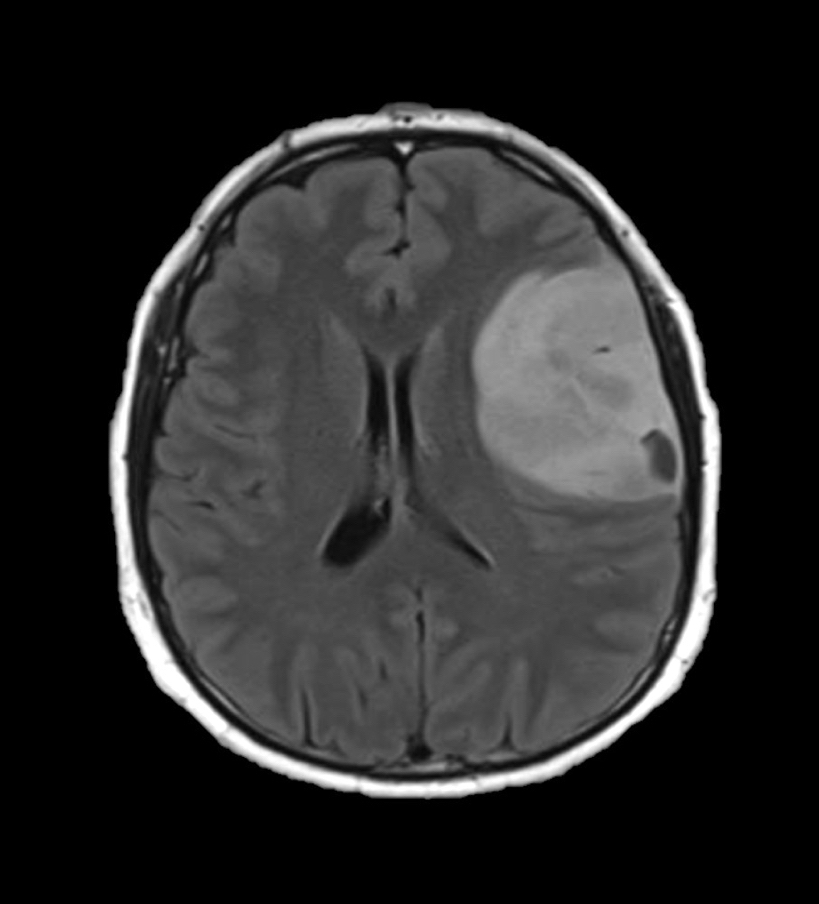
- Usually extra-axial and suprasellar
- Multilobulated and multicystic lesions
- Variable in size, often > 5 cm
- Recurrences may be massive
- MRI:
- High signal intensity on T1 weighted images
- Heterogeneous enhancement
- Fluid levels consistent with cystic components
- CT:
- Better than MRI at showing calcifications
- Same for both types of craniopharyngiomas
- Full pituitary endocrine workup is usually mandatory (Front Endocrinol (Lausanne) 2011;2:70)
- Visual acuity and visual field assessment is also performed to show any deficits and rule out papilledema
- MRI:
- T1: solid regions are hypo- or isointense, cystic regions are hyperintense
- Strong heterogeneous enhancement
- Hyperintense on T2
- CT:
- 9Solid regions and cyst wall enhancement
- Calcifications visible (J Neurol Surg Rep 2016;77:e121)
Images hosted on other servers:
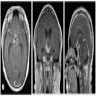
MRI suprasellar mass
- 5 year survival excellent (Front Endocrinol (Lausanne) 2011;2:70)
- Patients may be left with variable endocrinologic deficiencies
- Cystic recurrence common after incomplete excision
- Very rare malignant transformation
- 26 year old man with malignant craniopharyngioma without radiation therapy (J Korean Neurosurg Soc 2017;60:108)
- 35 year old man with adamantinomatous craniopharyngioma (Int Med Case Rep J 2020;13:123)
- 52 year old man with craniopharyngioma presenting with severe hyponatremia, hyponatremia induced myopathy and panhypopituitarism (J Med Case Rep 2017;11:31)
- Gross total excision or subtotal resection followed by radiation therapy (Front Endocrinol (Lausanne) 2011;2:70)
- Anatomic location, size, invasion of the nearby structures and the nature of the tumor determine surgical approach
- Most common indication for surgery is neurologic compromise from tumor mass effect
- In children, hypothalamic and endocrine dysfunction may develop before visual defects are noticed
- Radiotherapy is indicated for treatment of residual tumor or recurrence
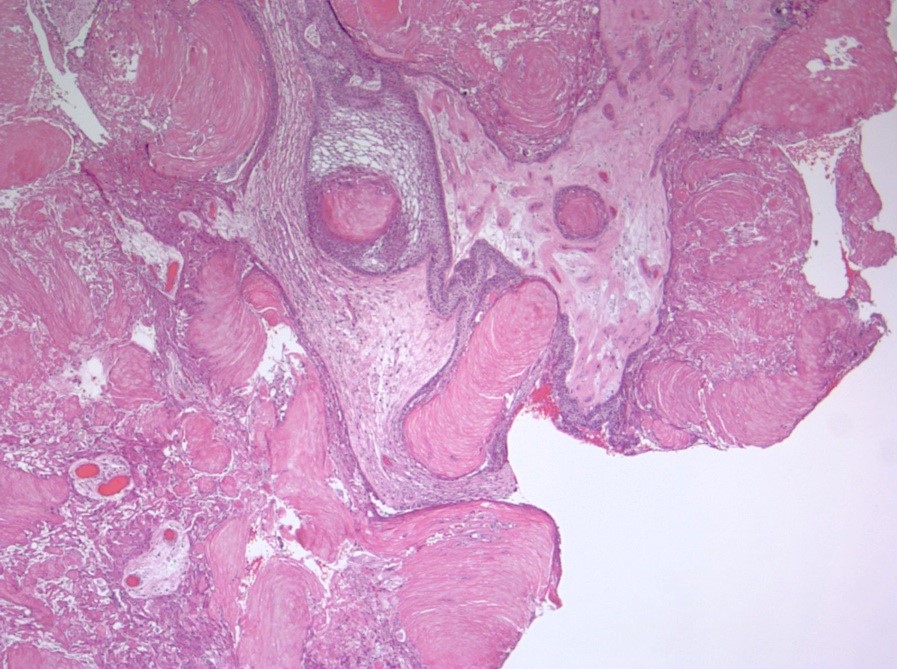
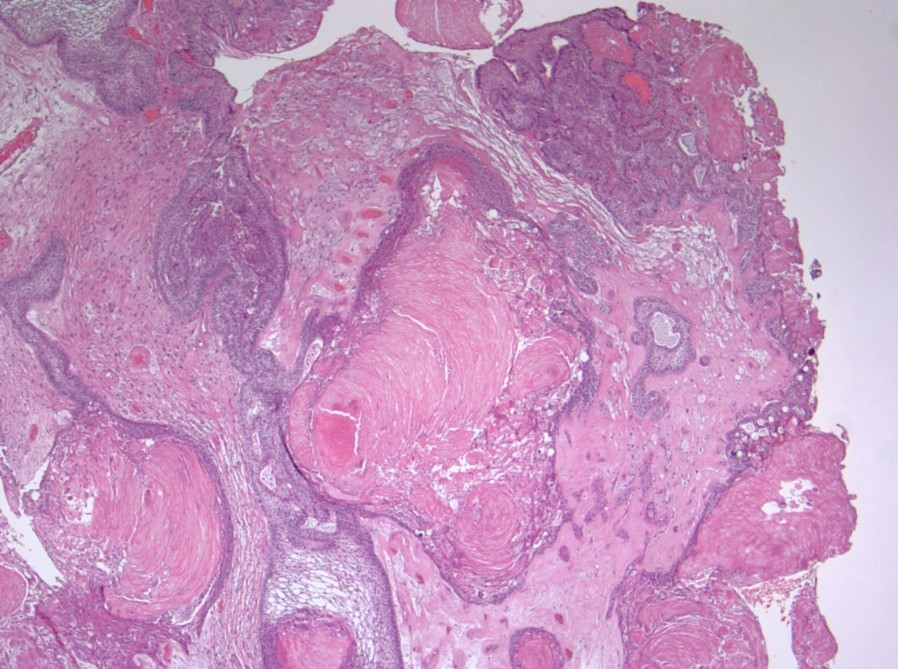
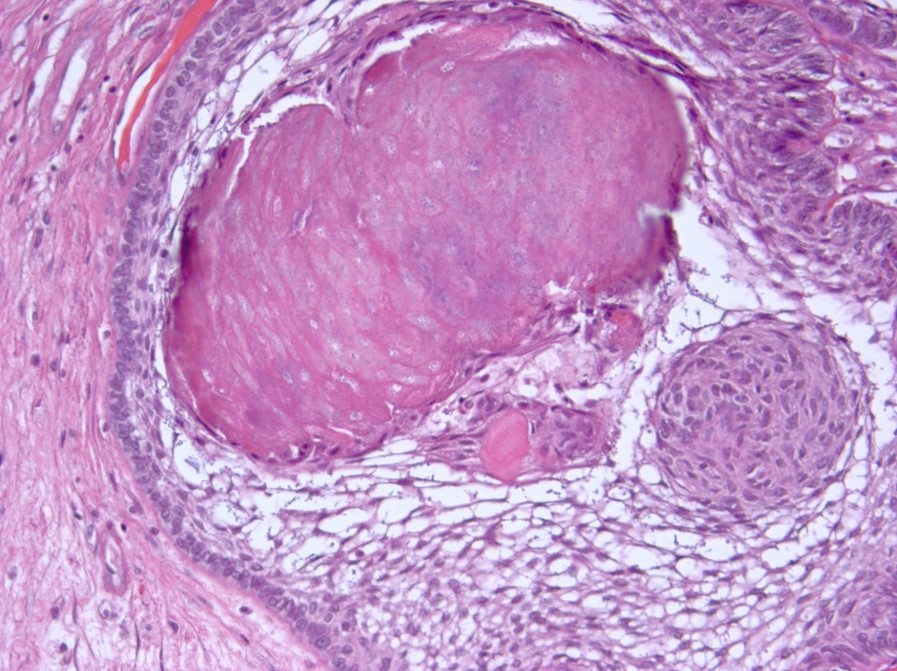
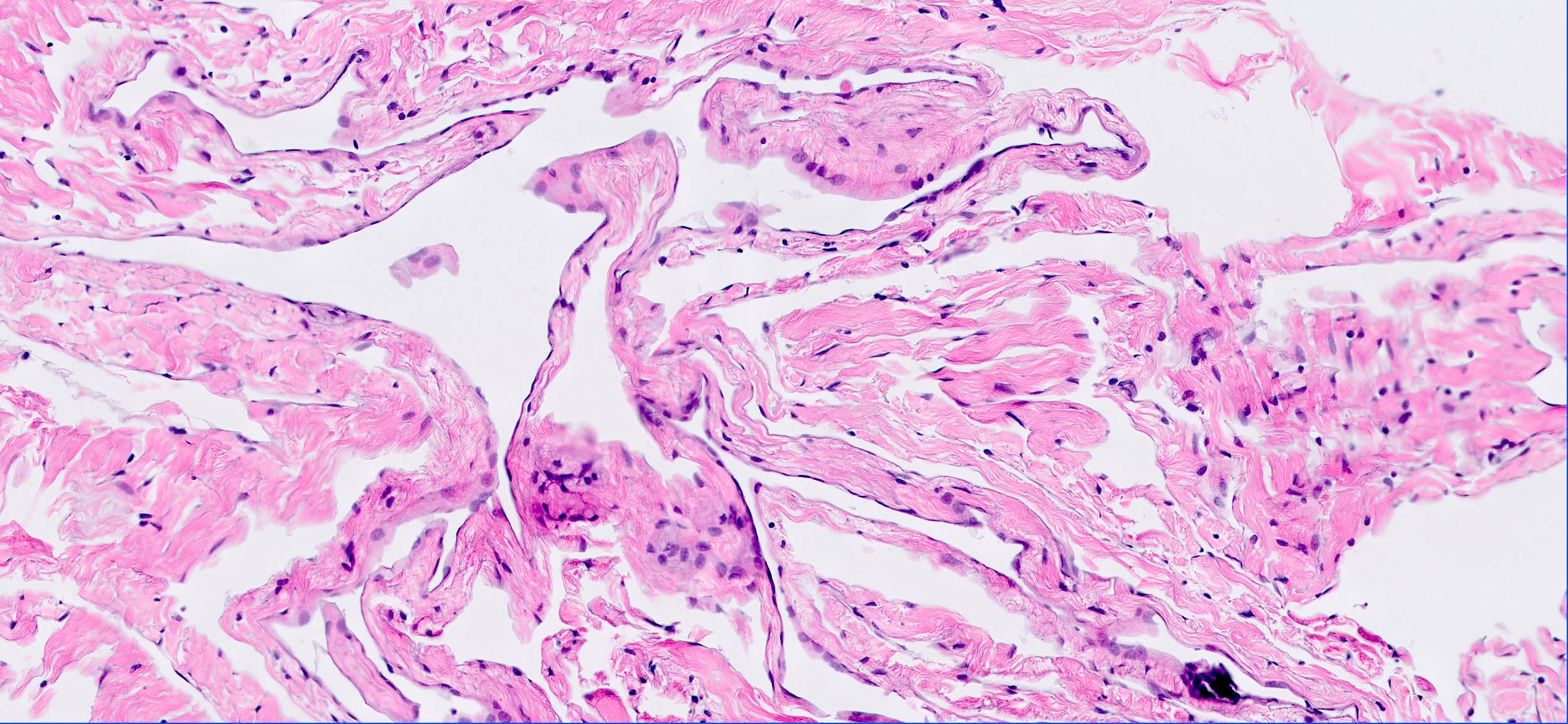
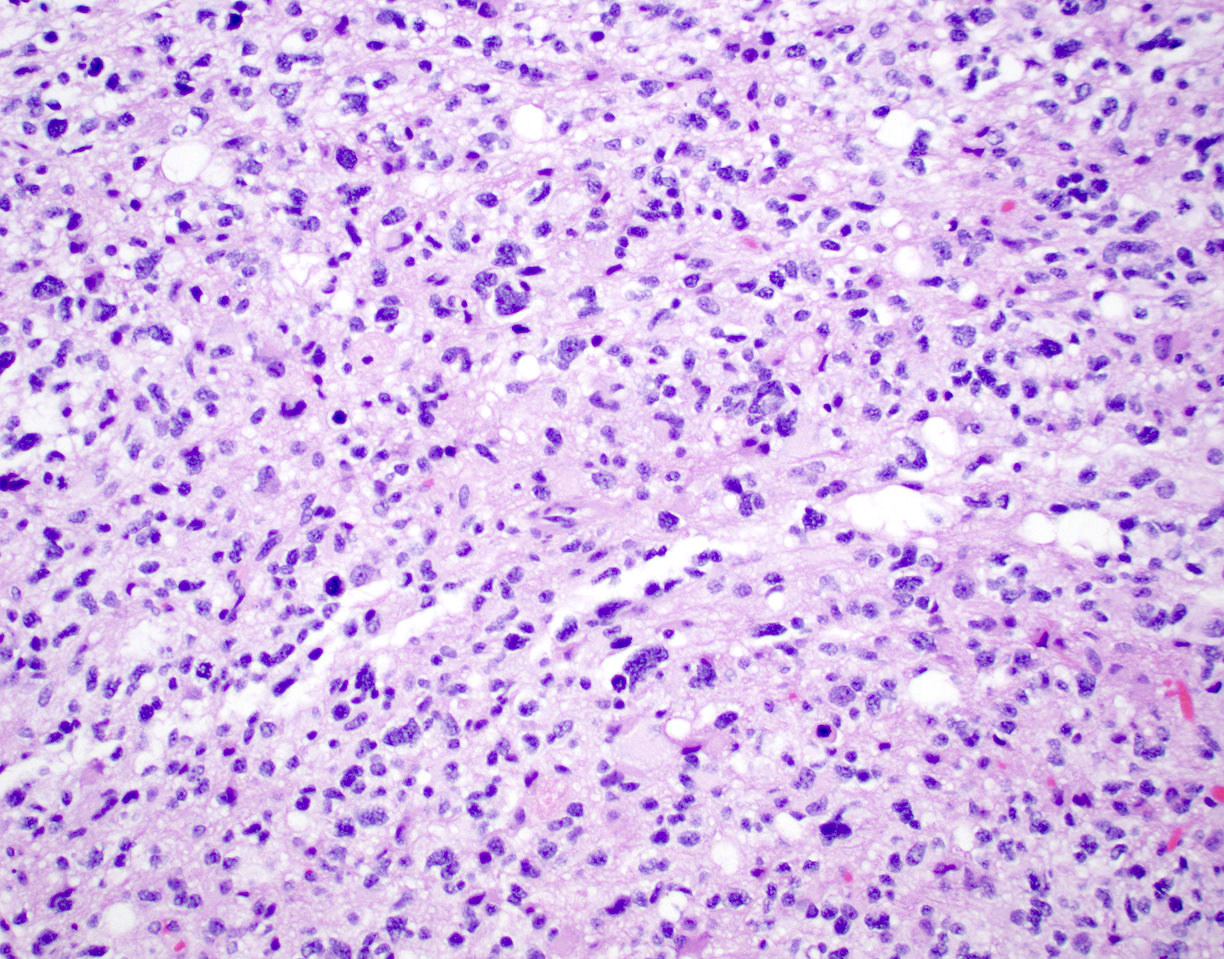
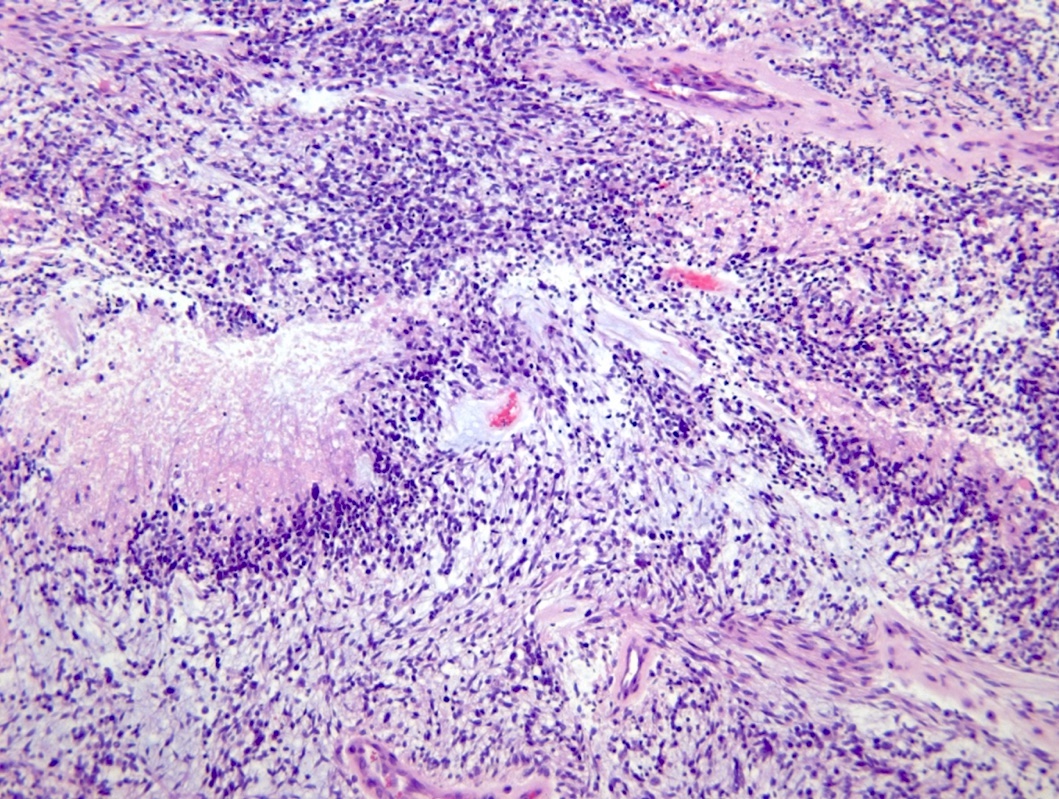
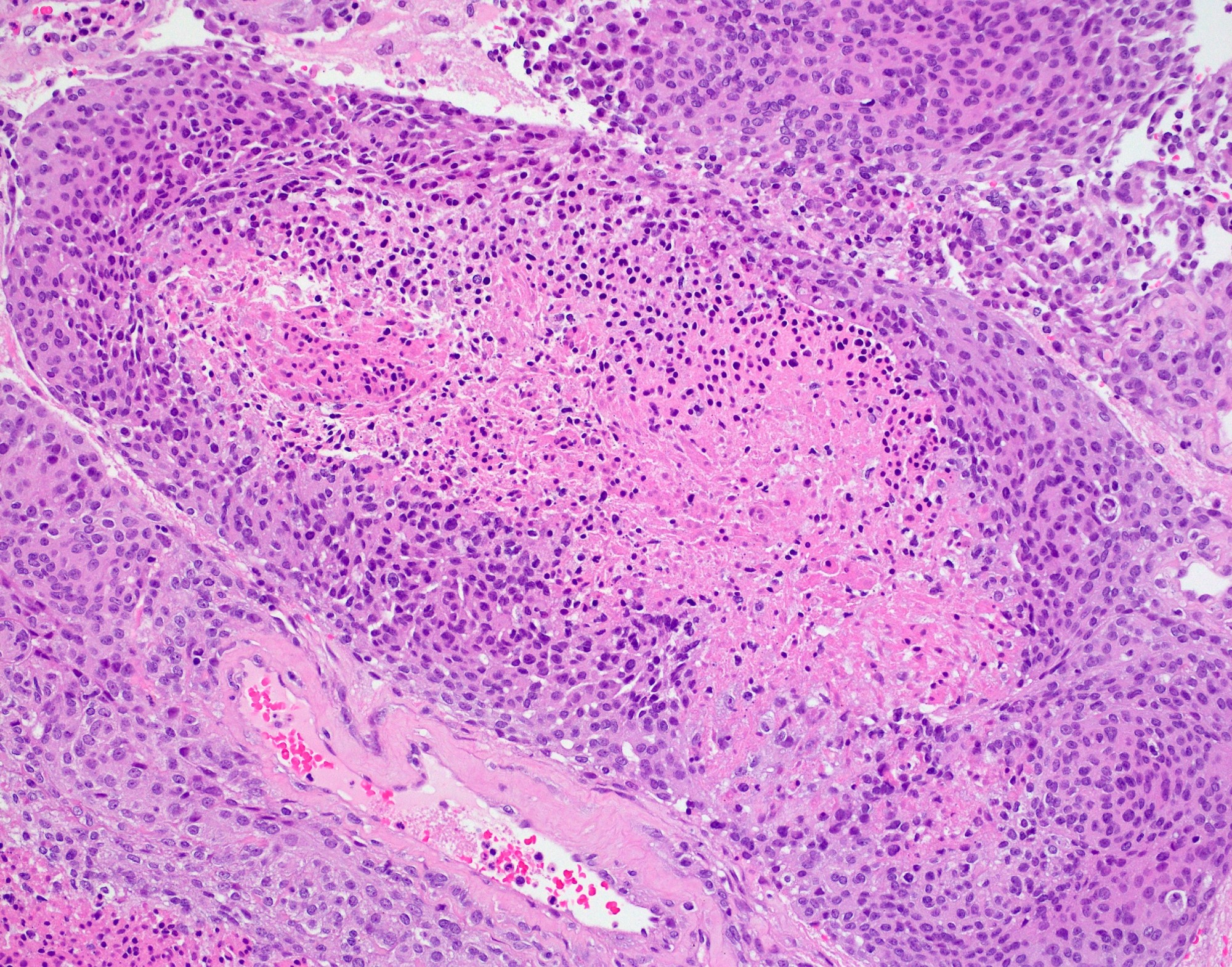
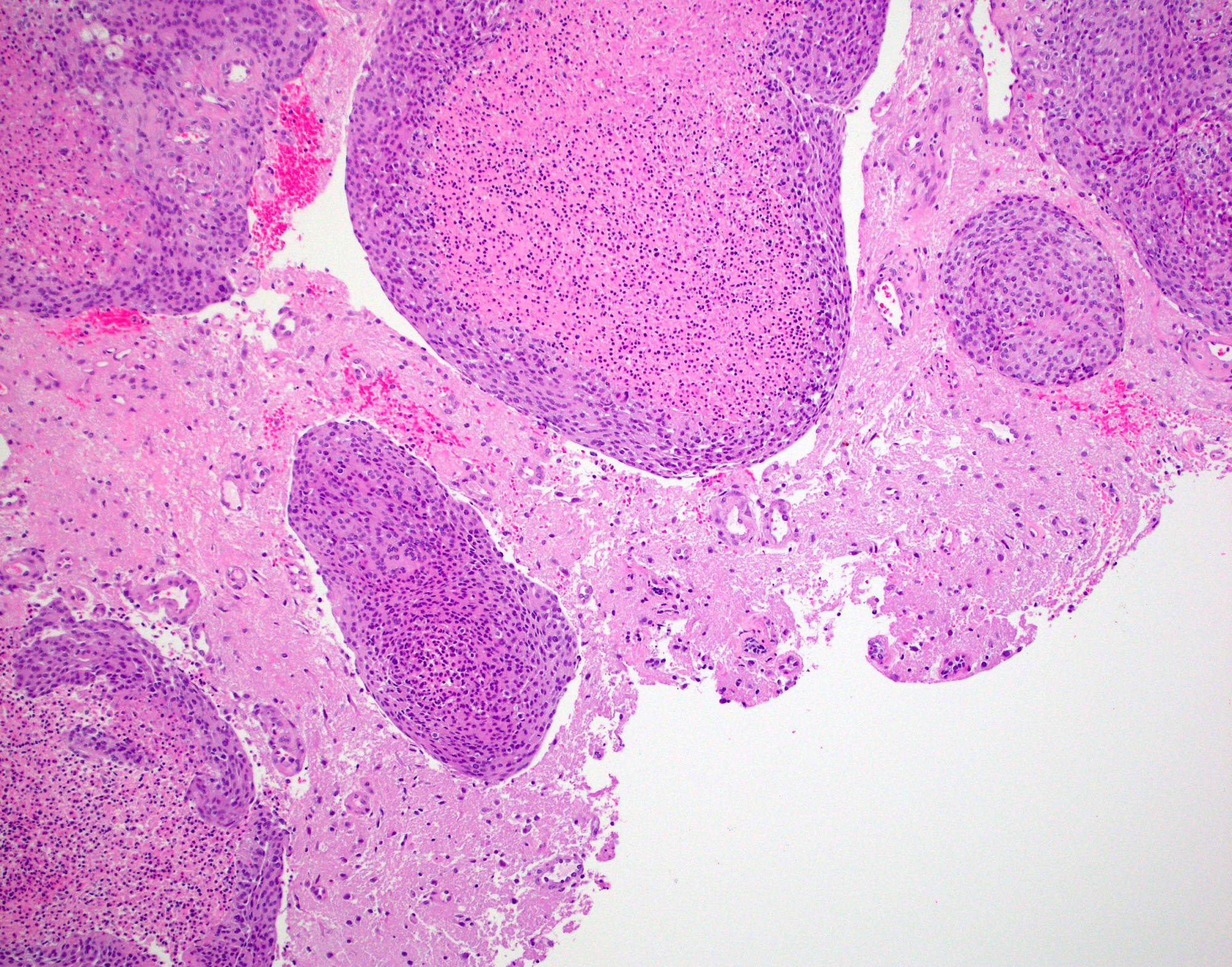
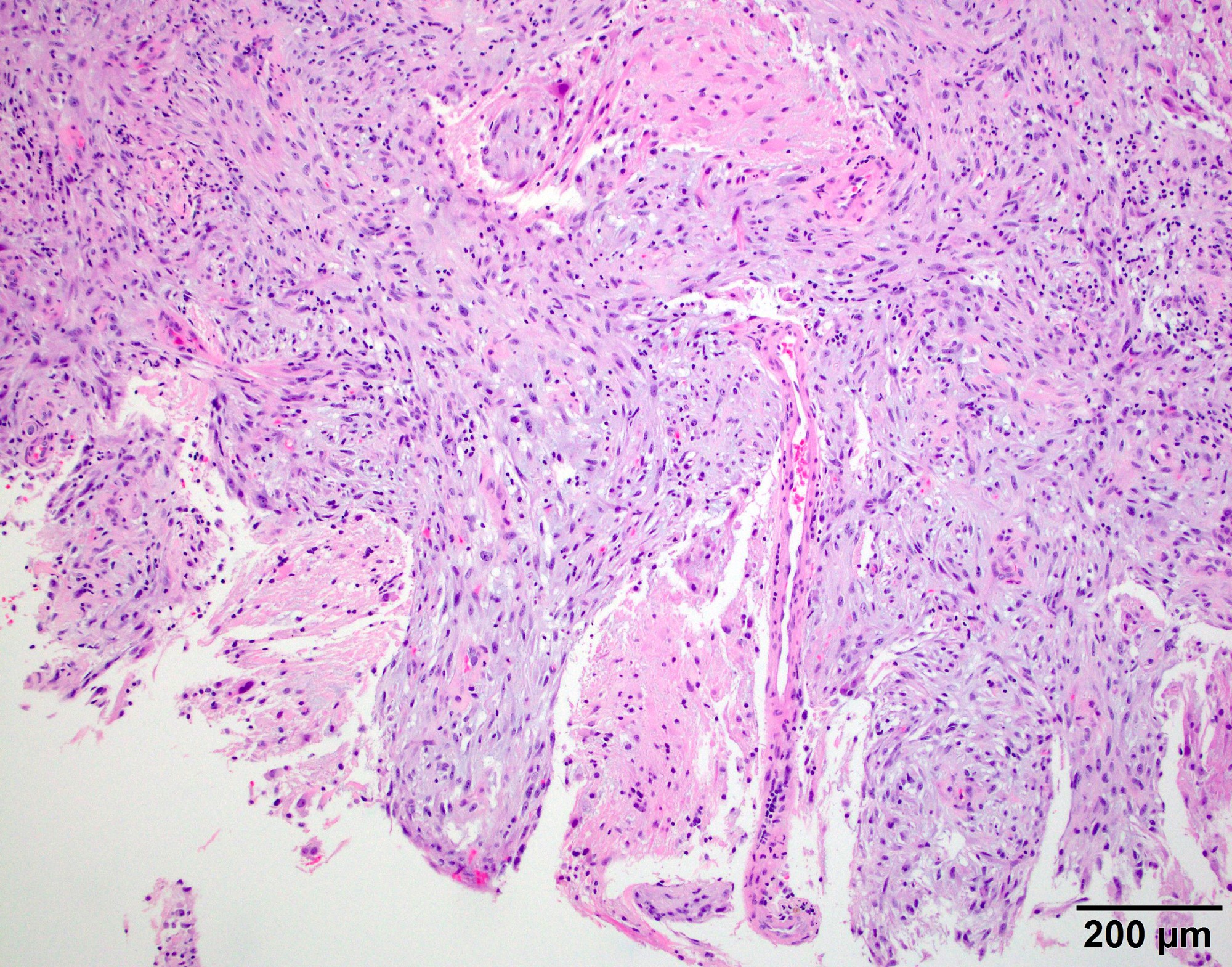
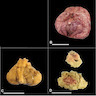
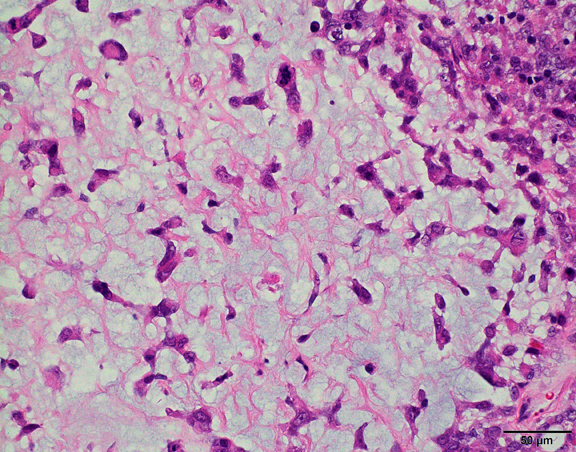
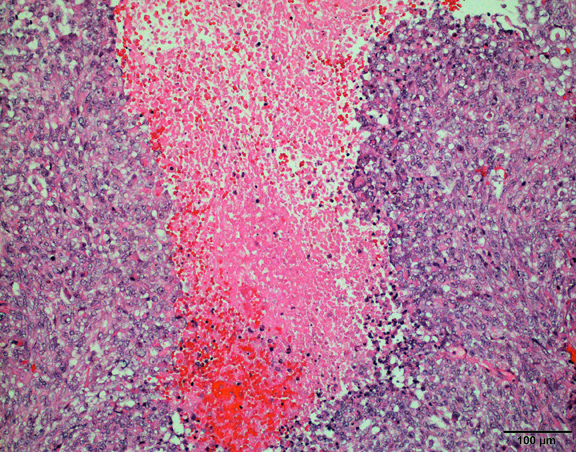
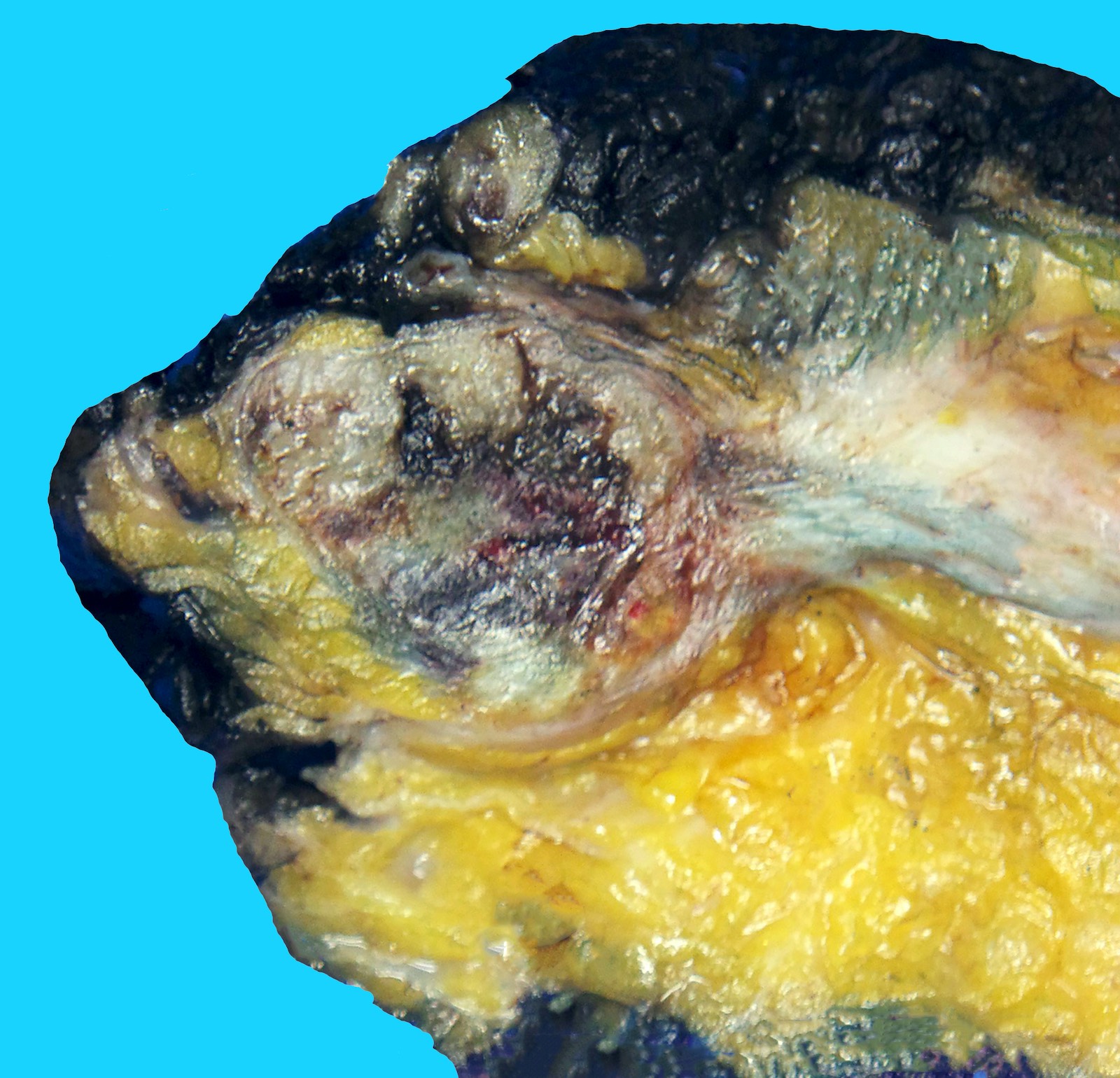
- Lobular and cystic tumor with calcifications
- Cysts with dark "motor oil" fluid composed of cholesterol and hemorrhage
- Irregular tumor interface with adjacent brain
- Can be densely adherent to brain
Images hosted on other servers:
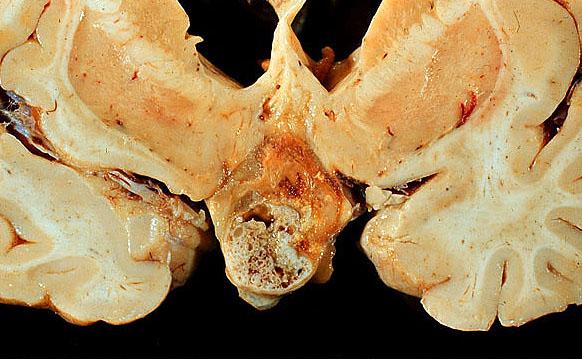
Autopsy image
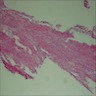
- May appear well circumscribed
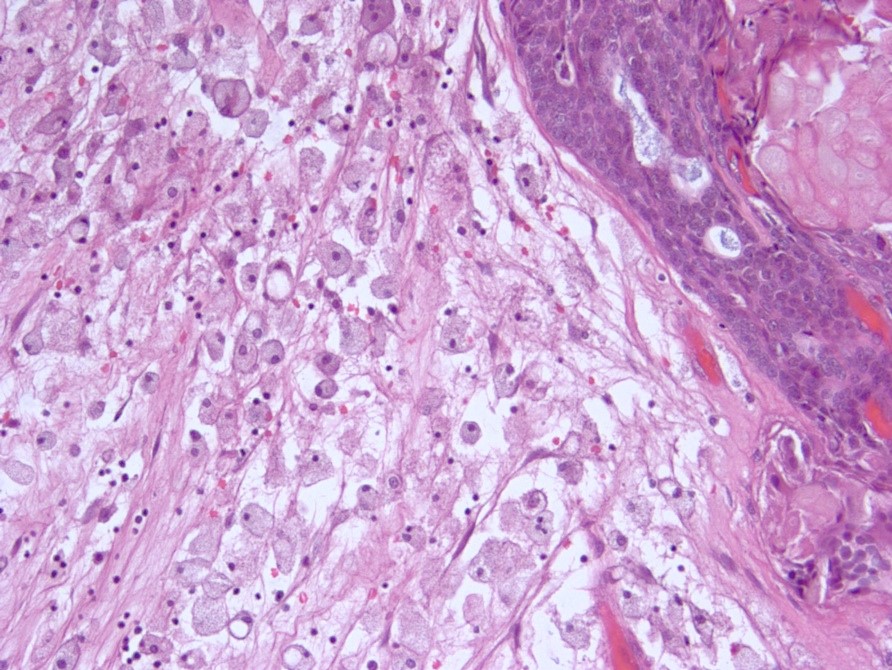
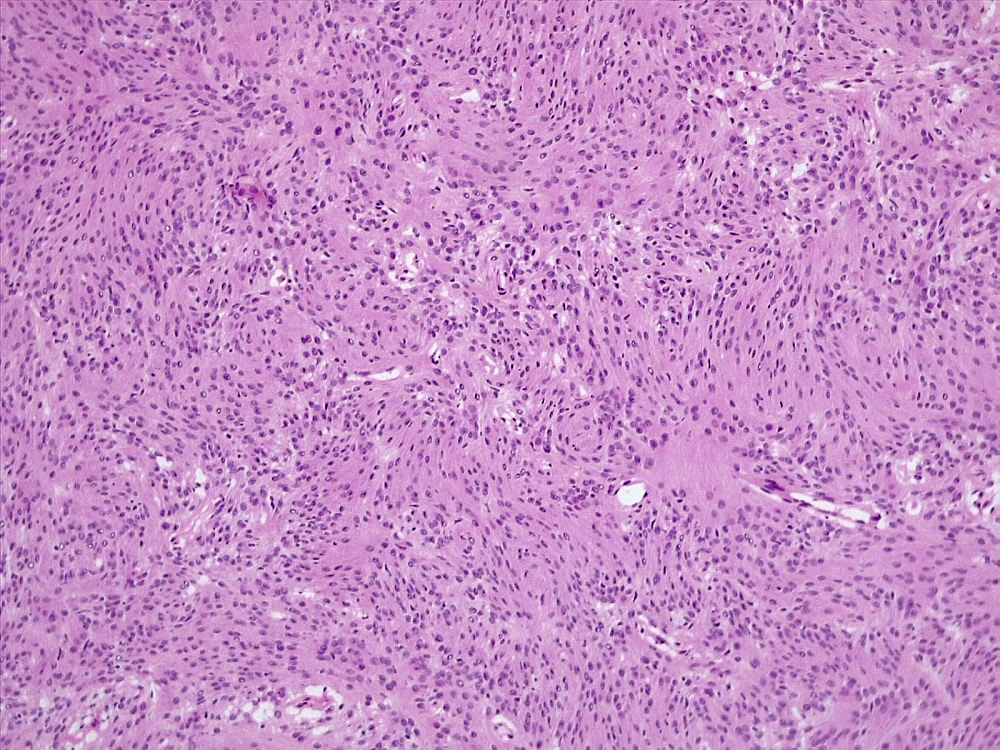
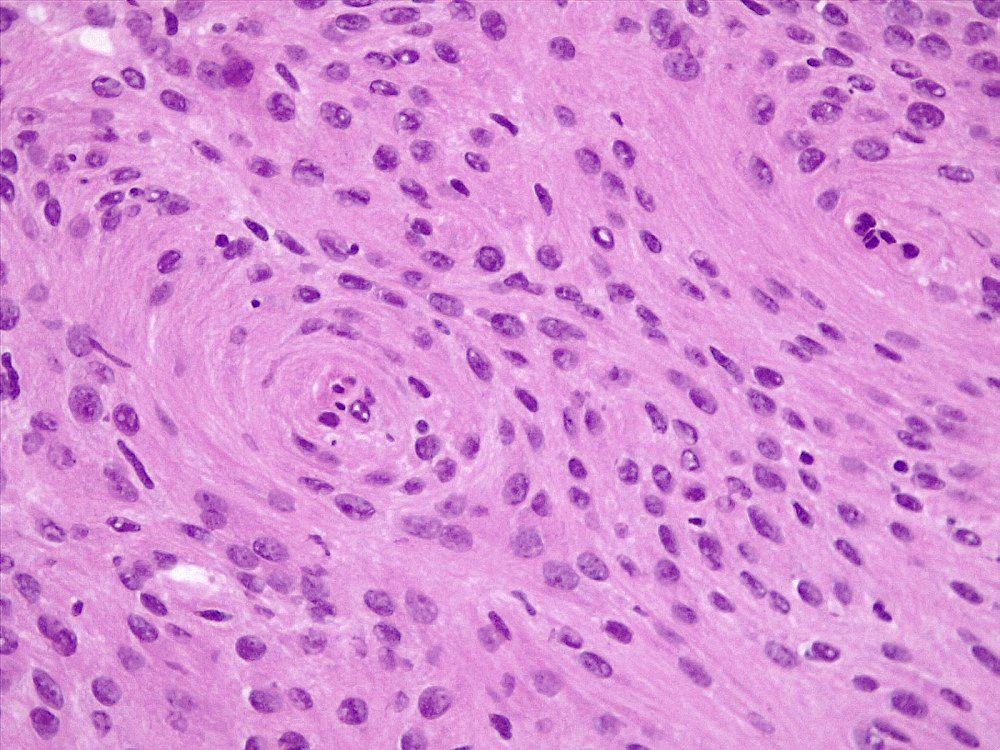
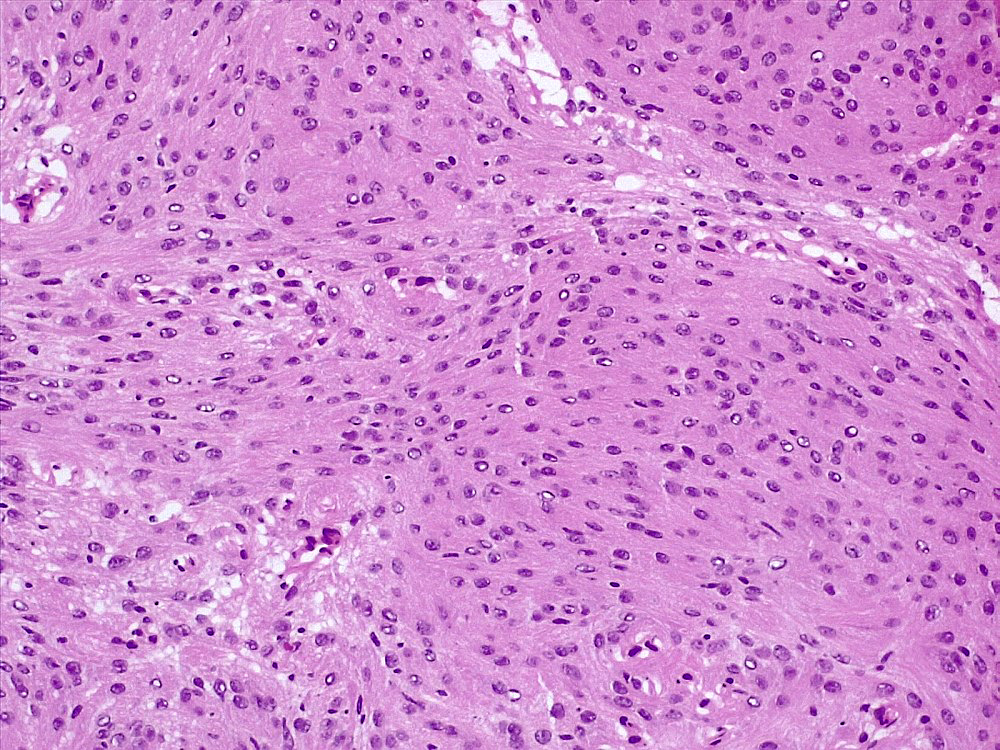
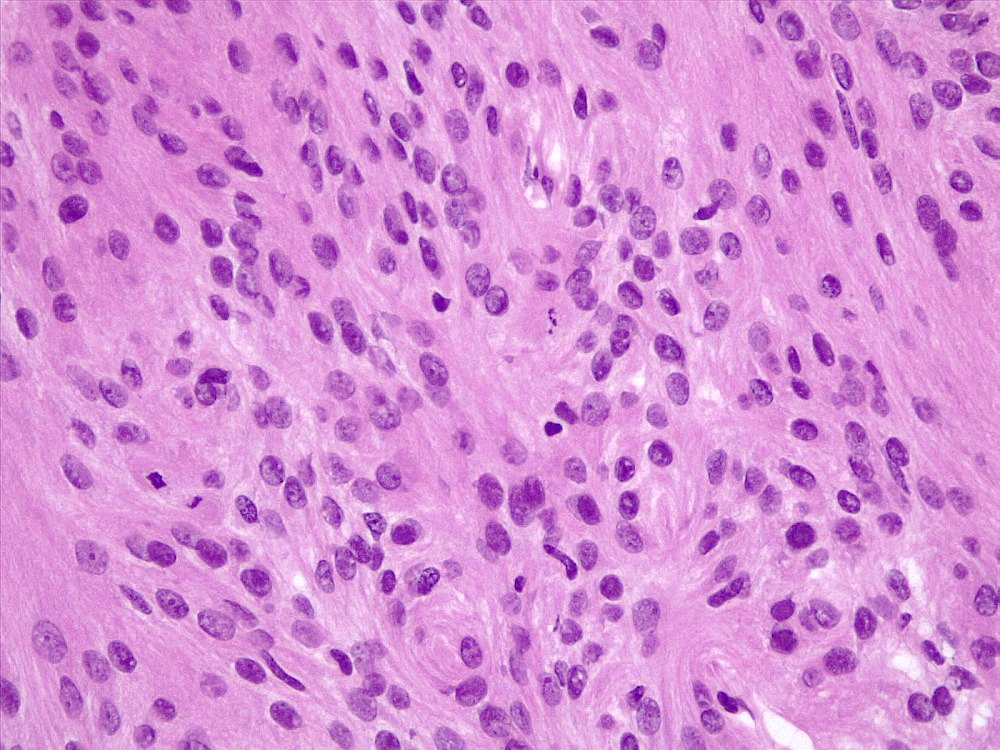
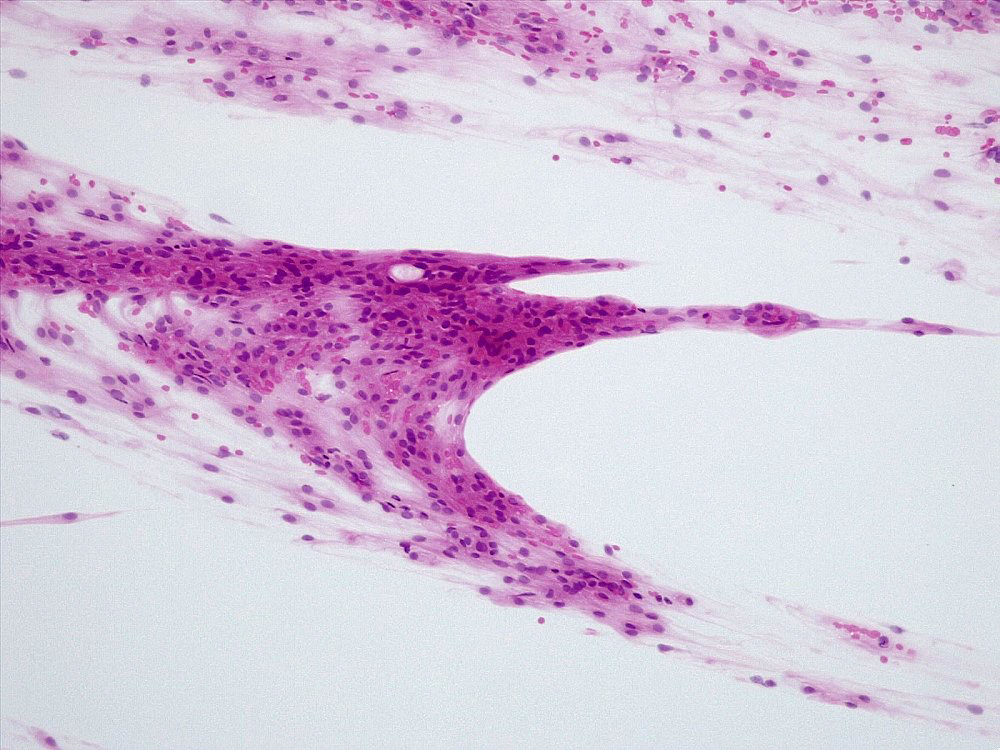
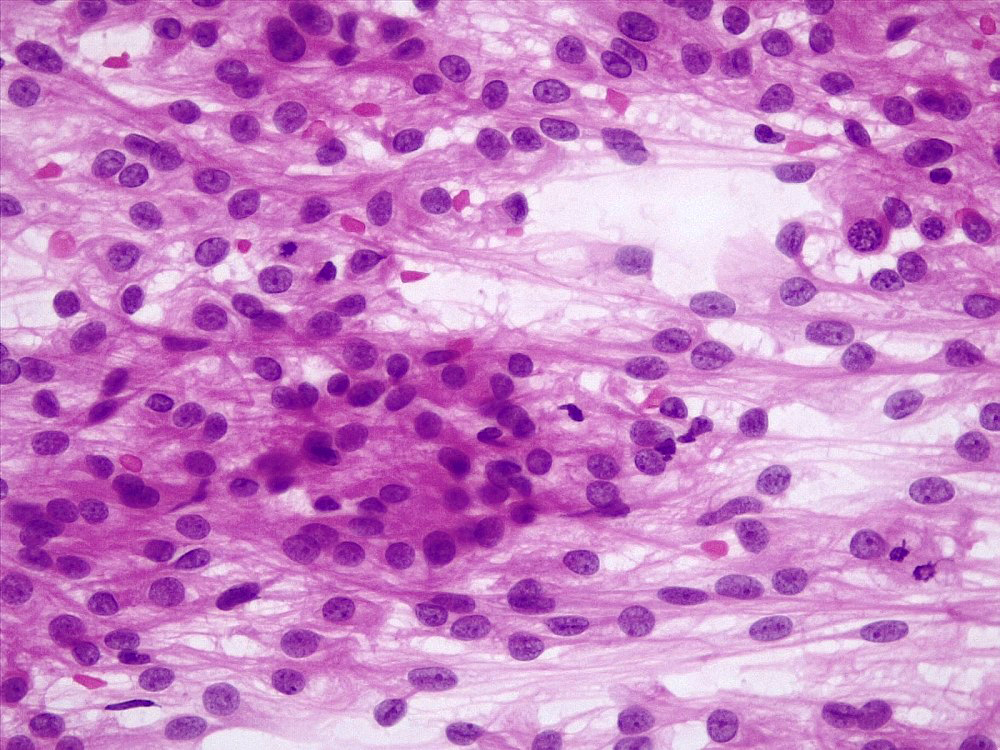
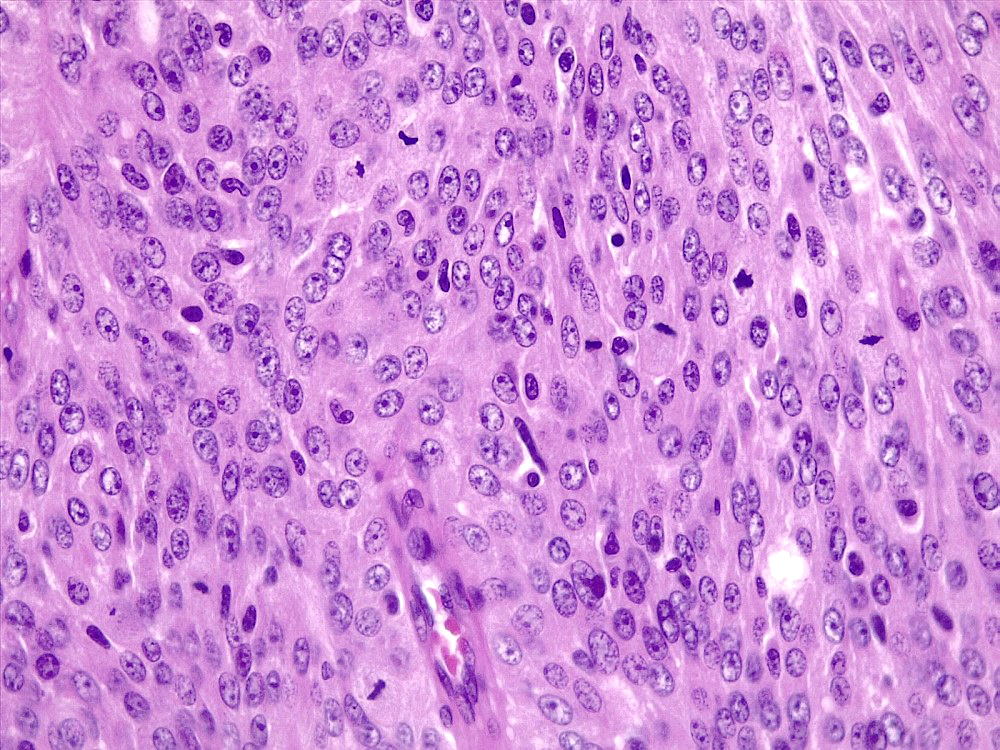
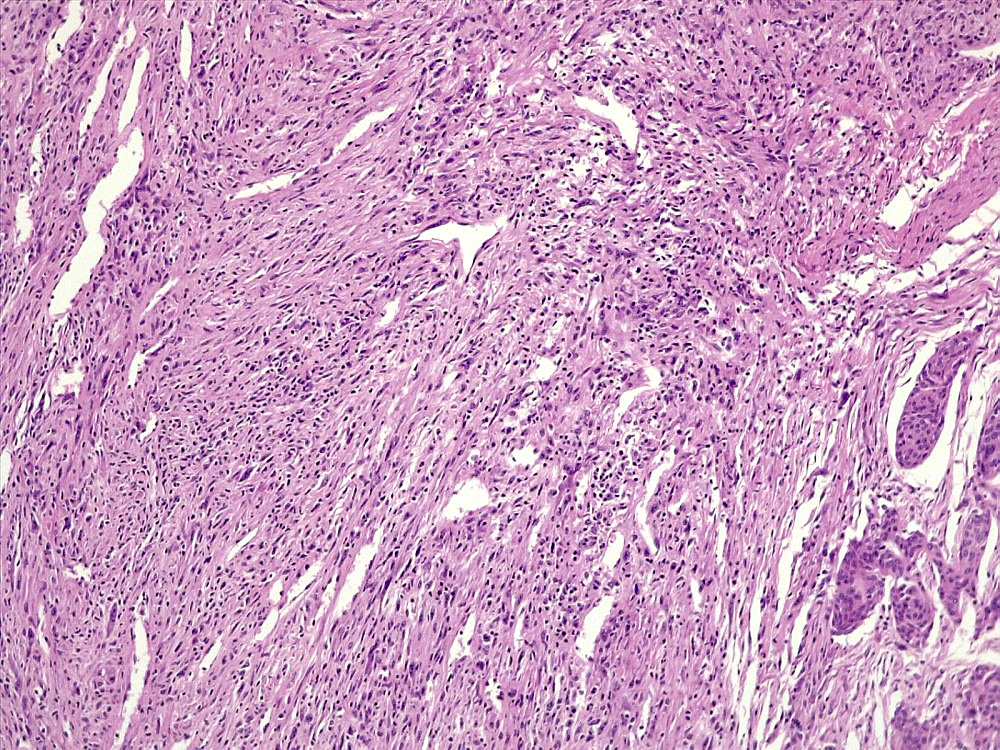
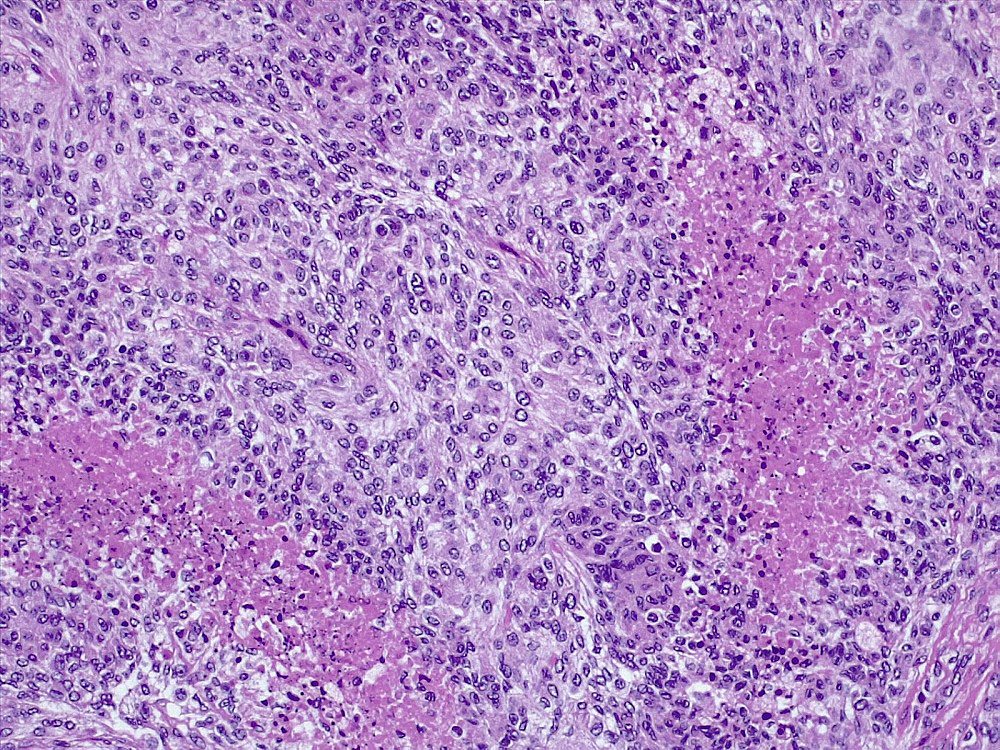
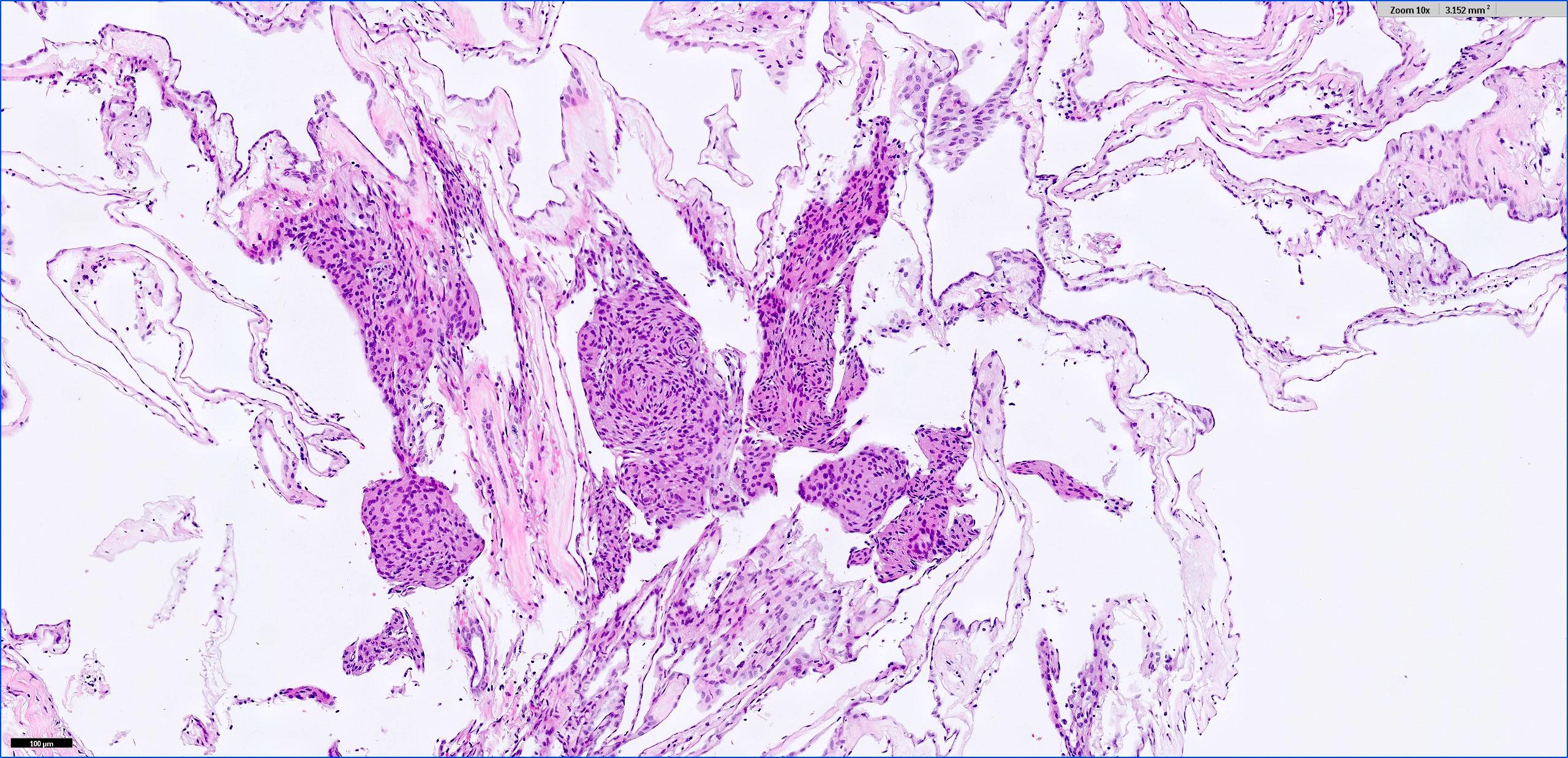
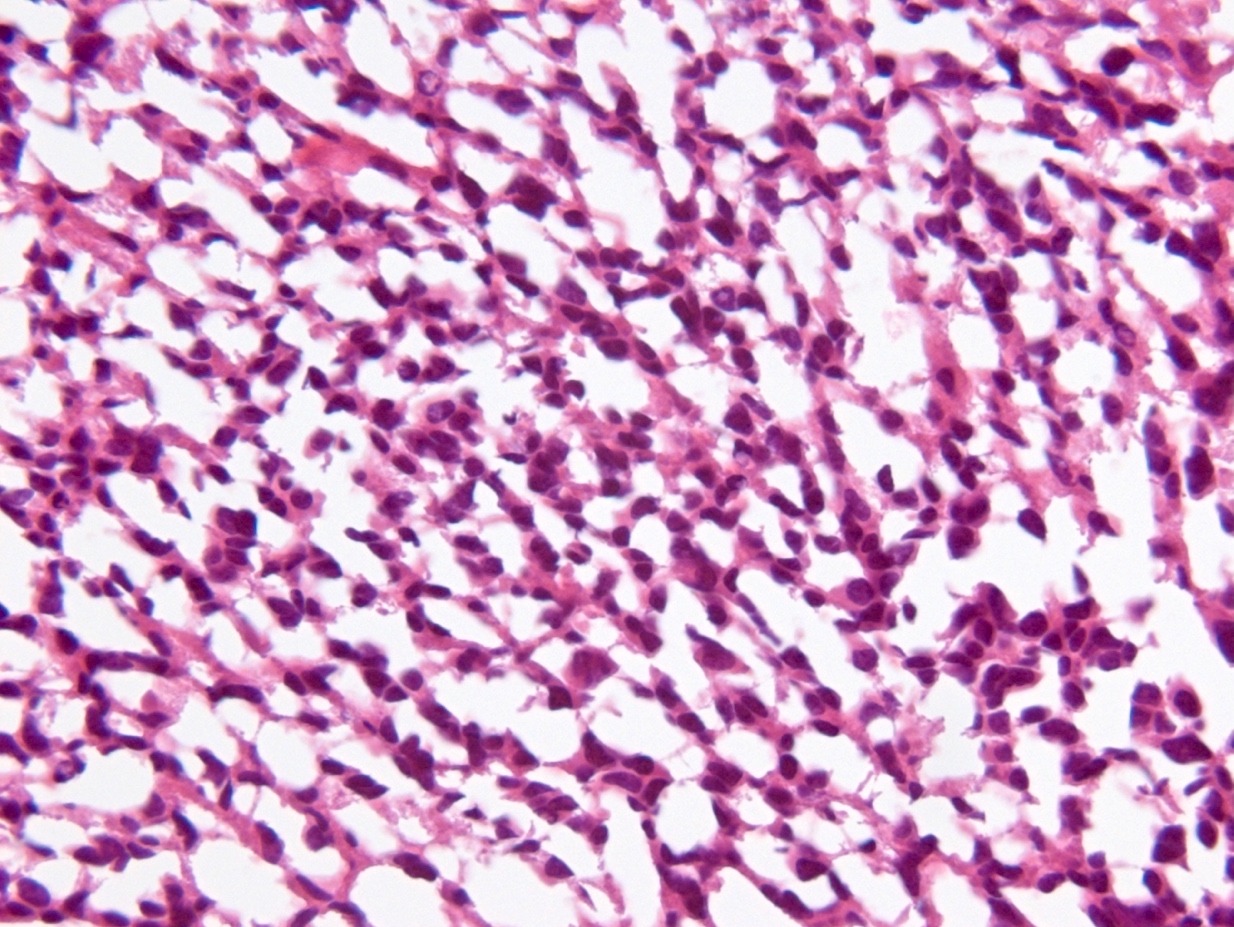
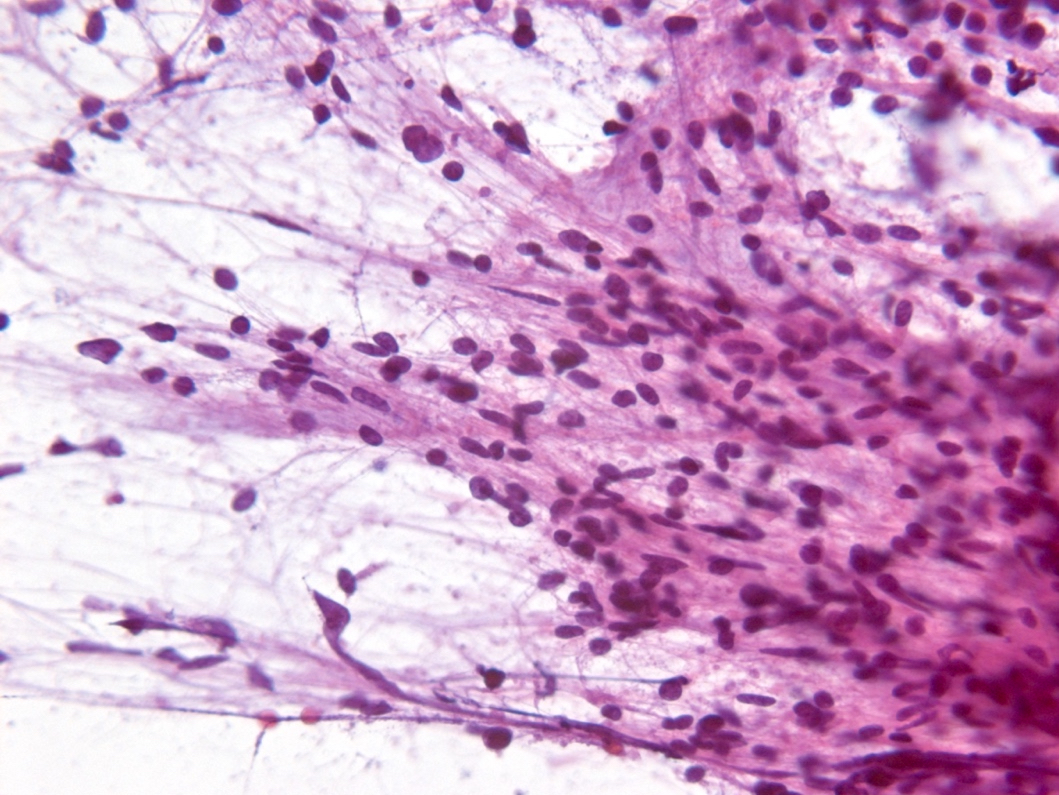
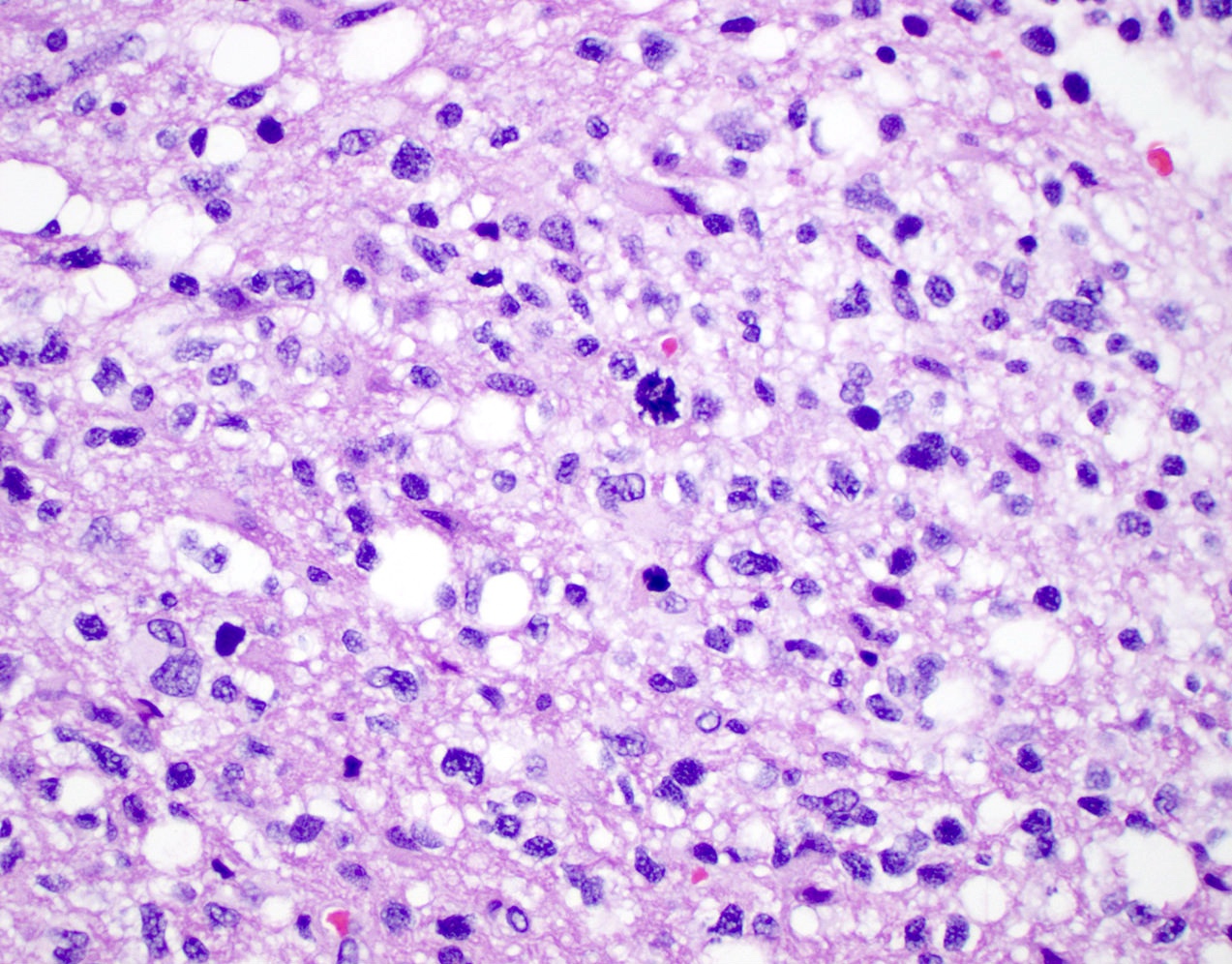
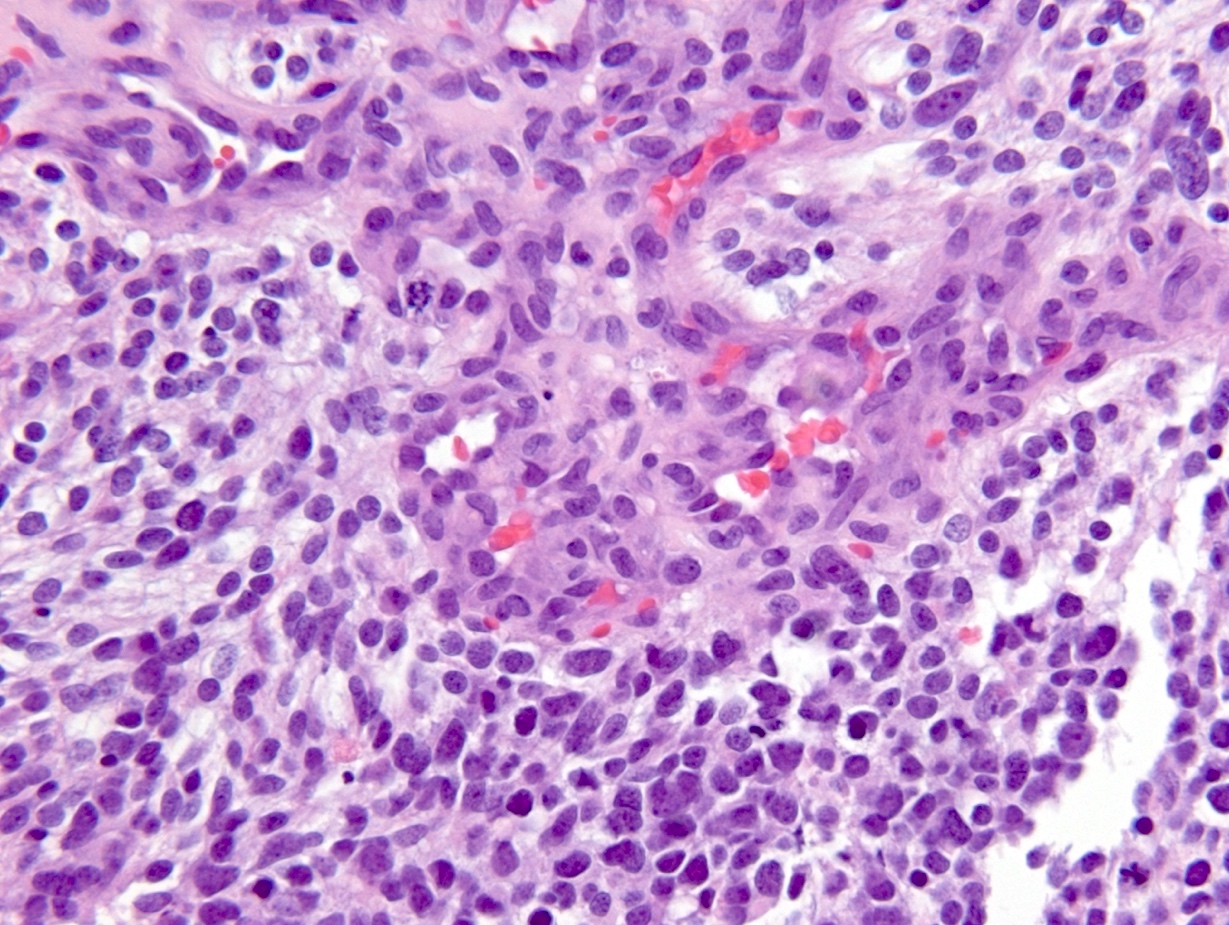
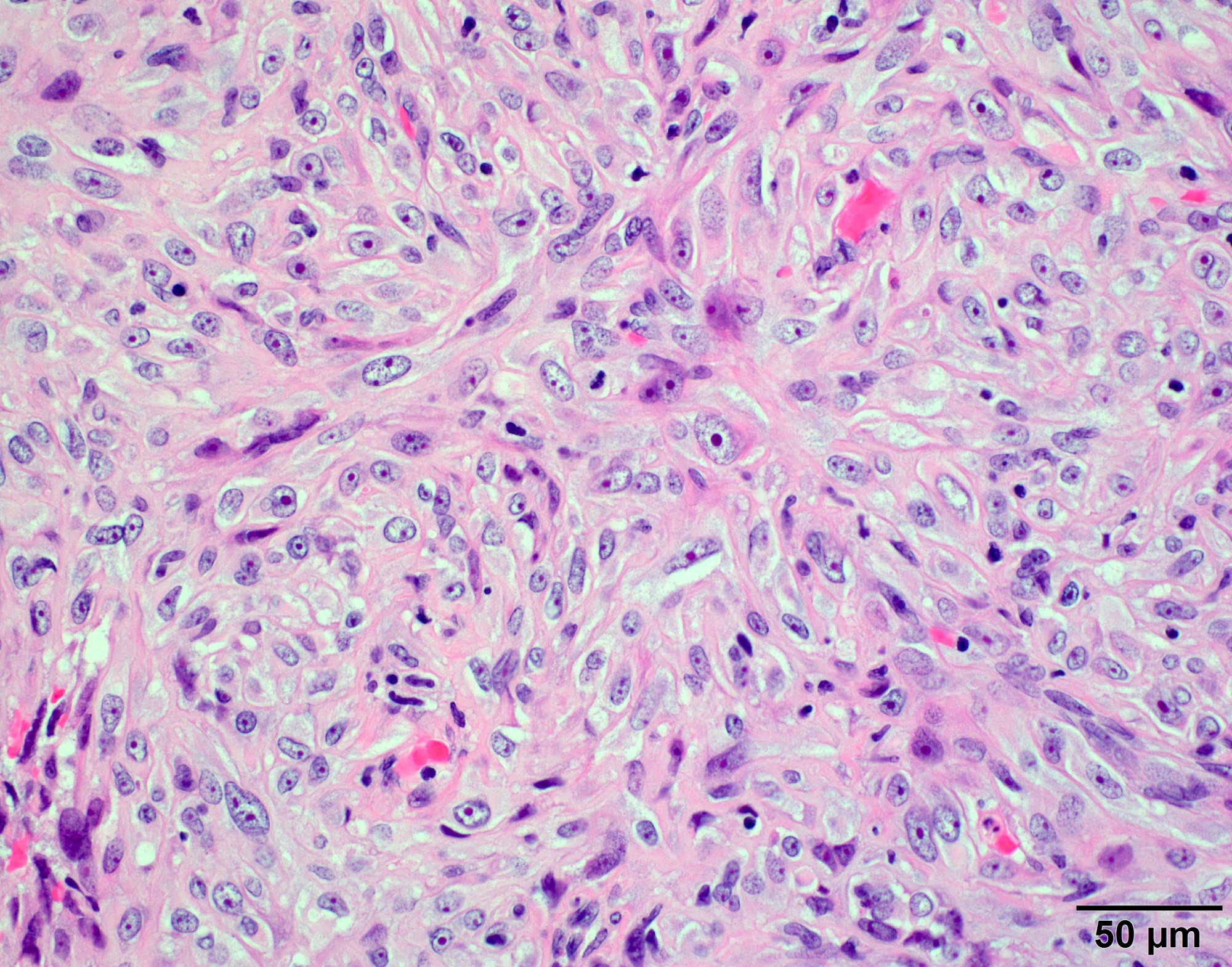
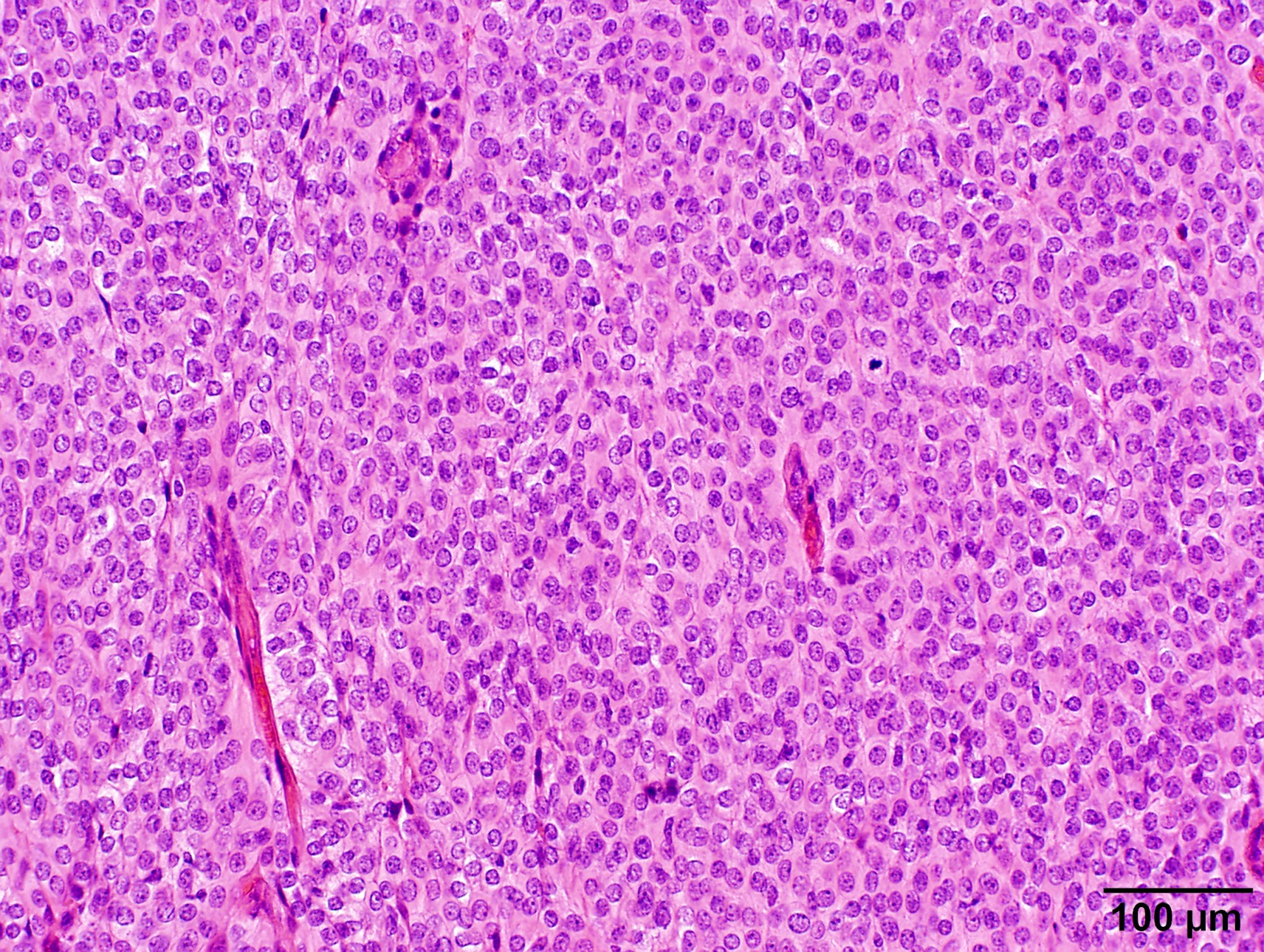
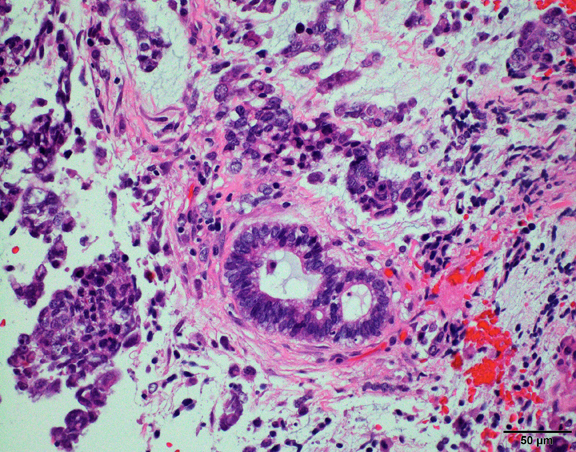
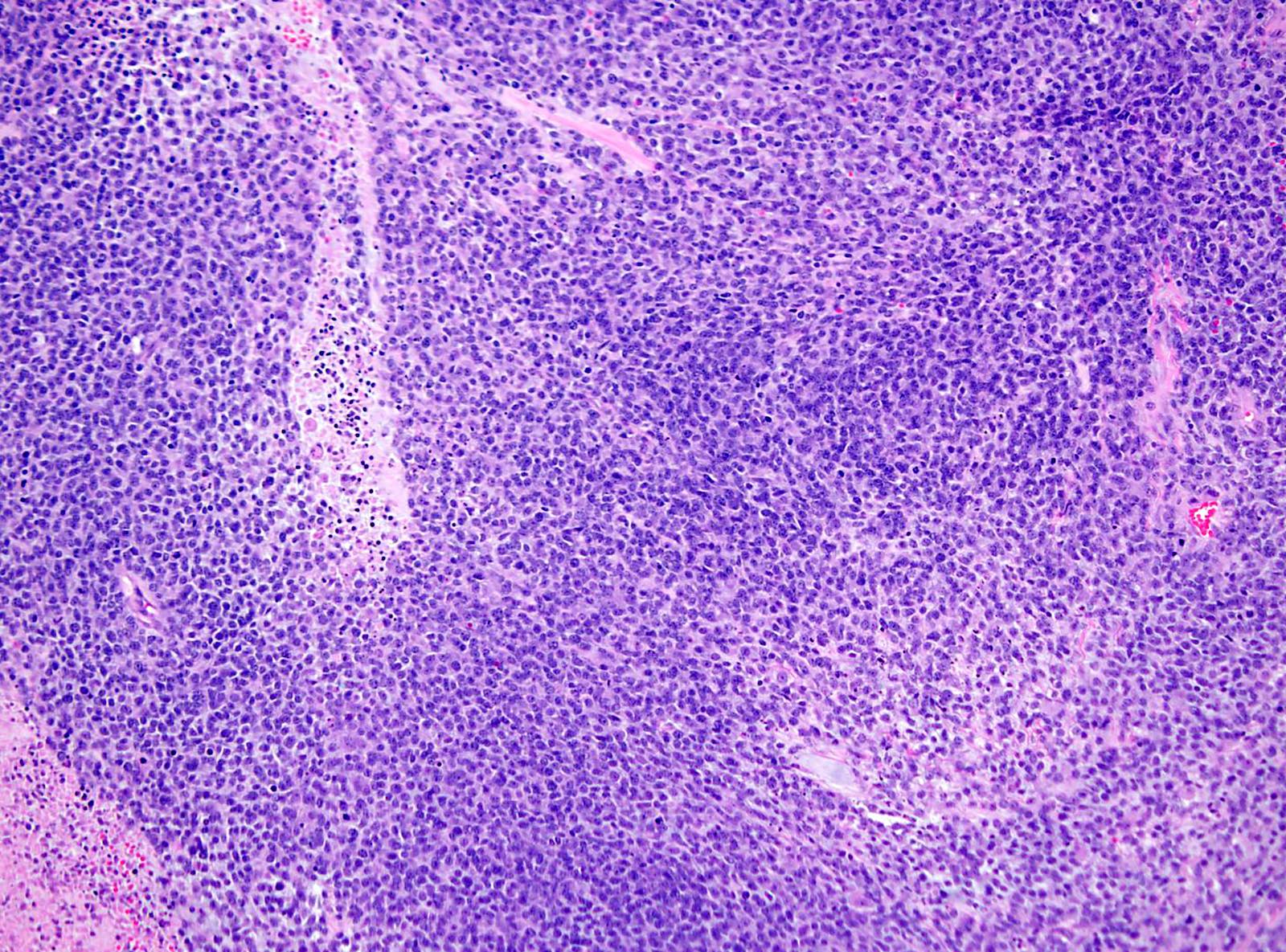
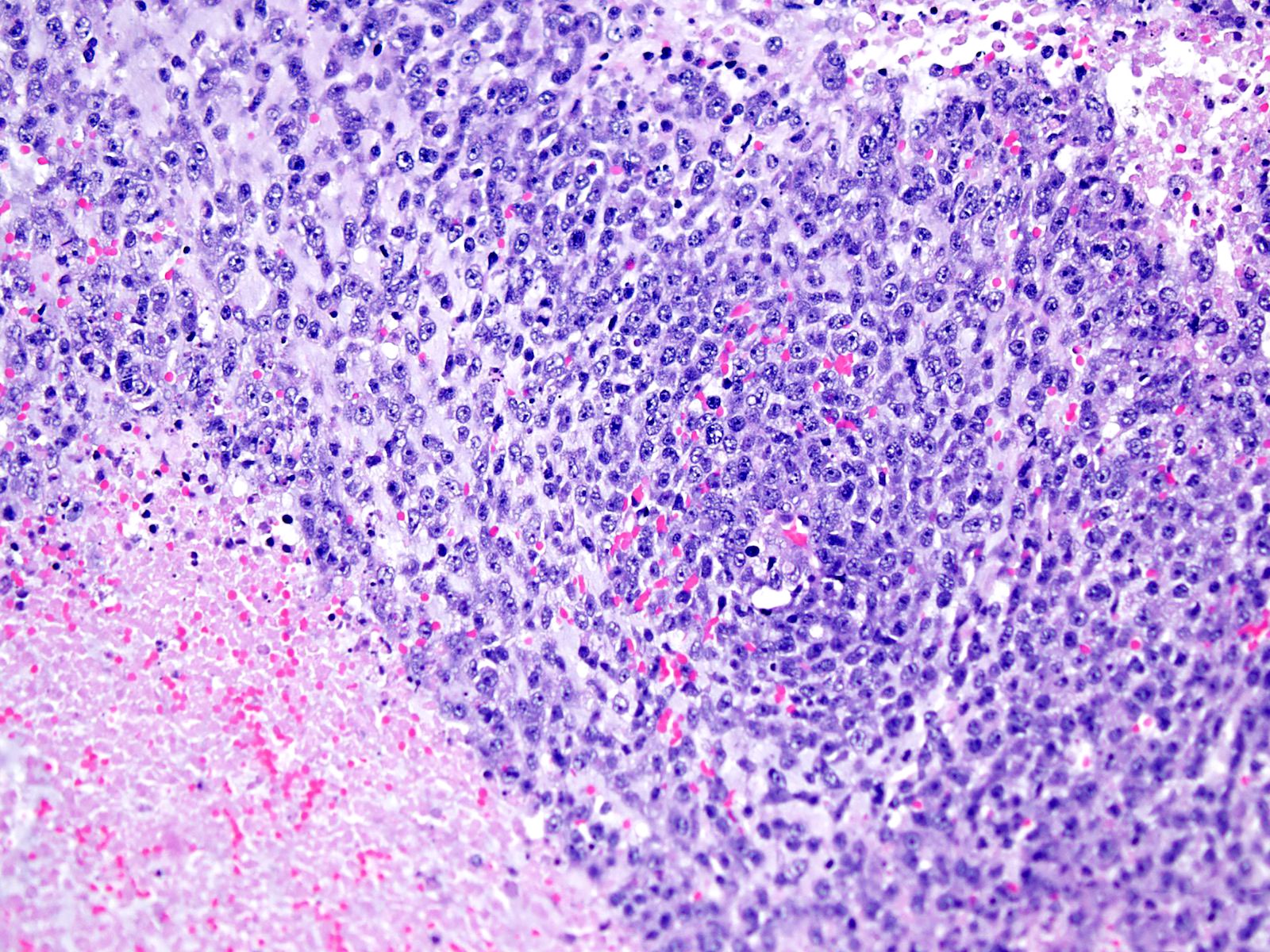
- Cords, lobules, nodular whorls and trabeculae of well differentiated squamous epithelium bordered by palisading columnar epithelium
- Peripheral cells surround looser plumper cells called stellate reticulum
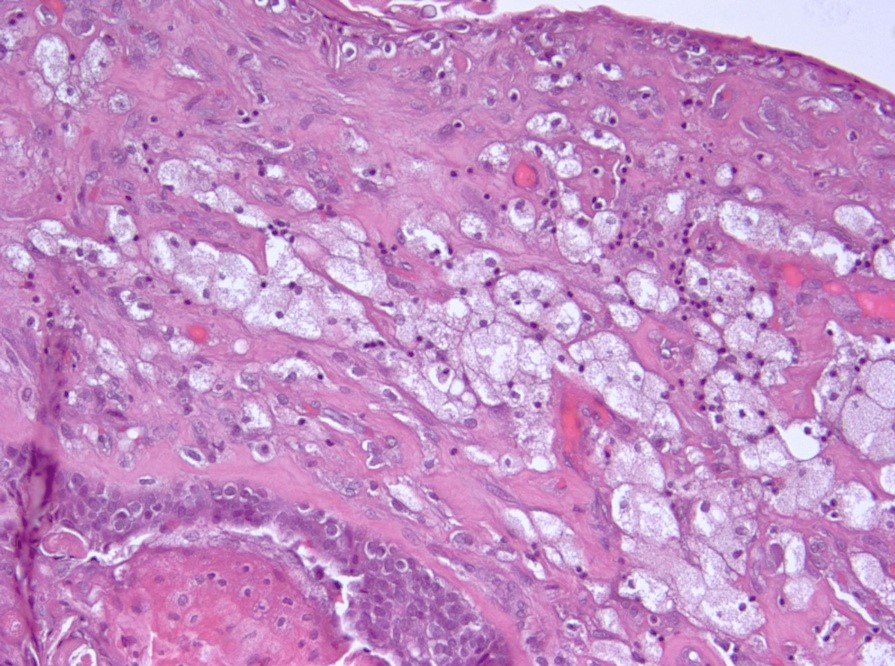
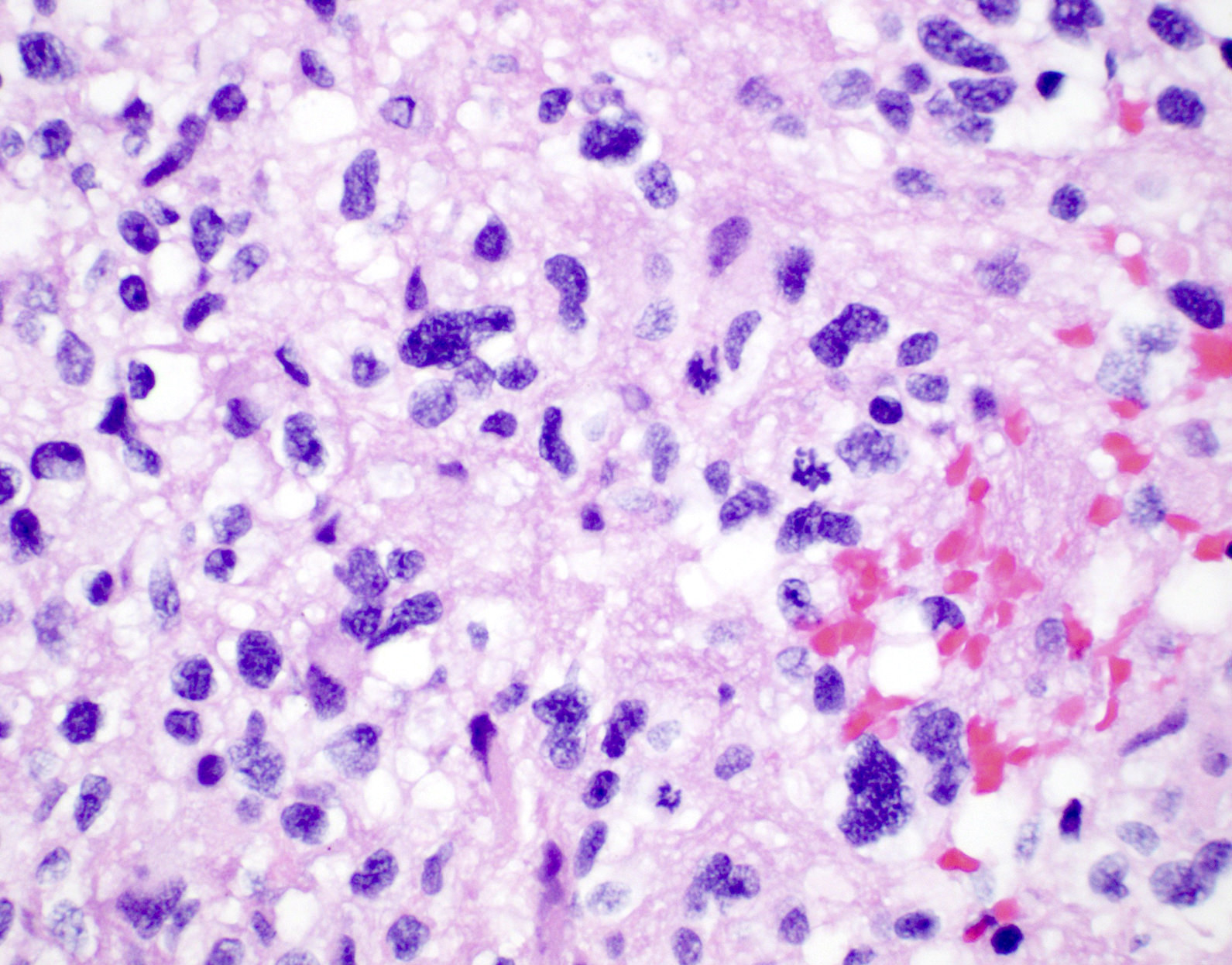
- Nodules of plump, anucleate squamous cells (ghost cells) and wet keratin
- Intralobular whorl-like formations (Int Med Case Rep J 2020;13:123)
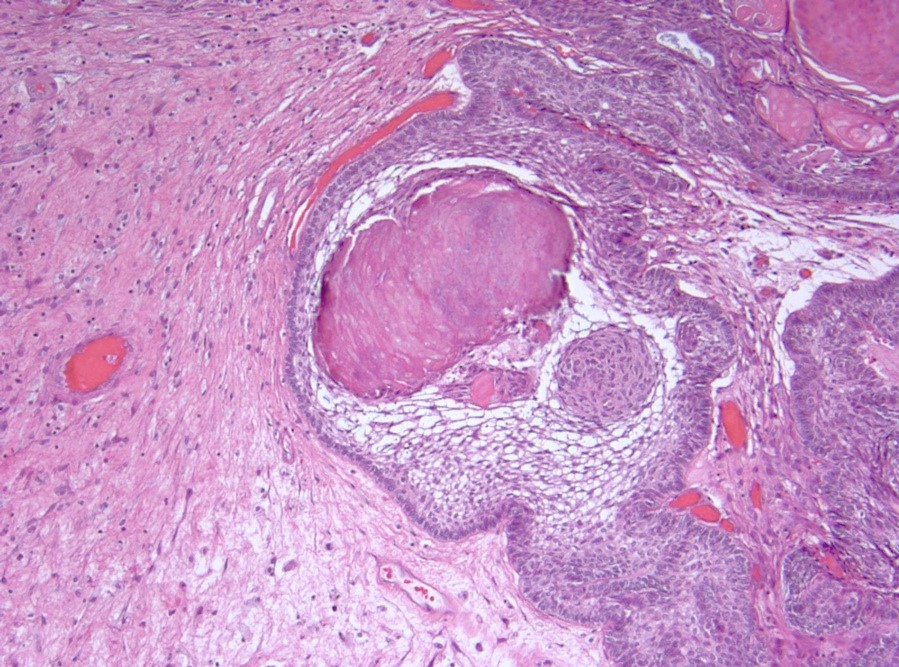
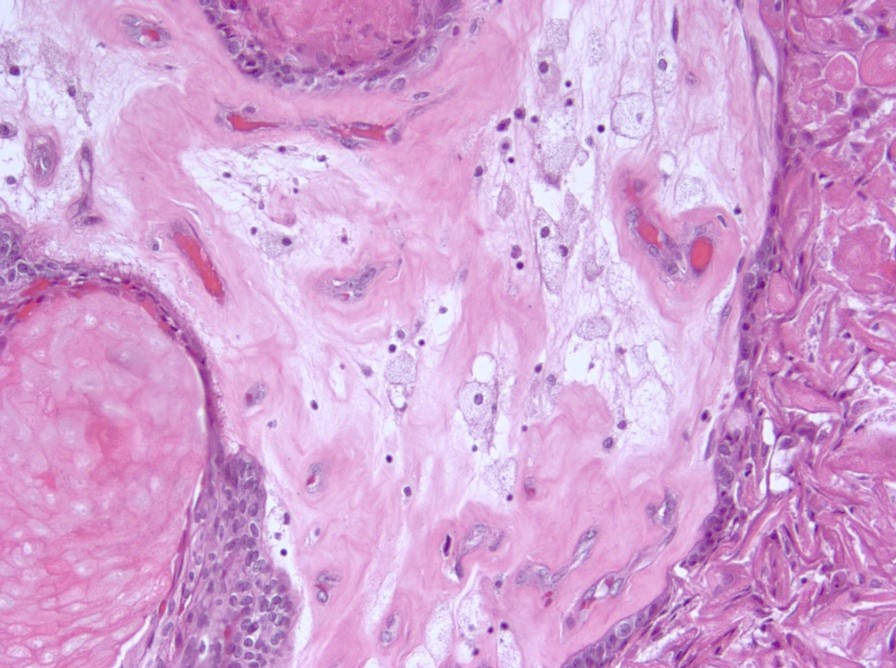
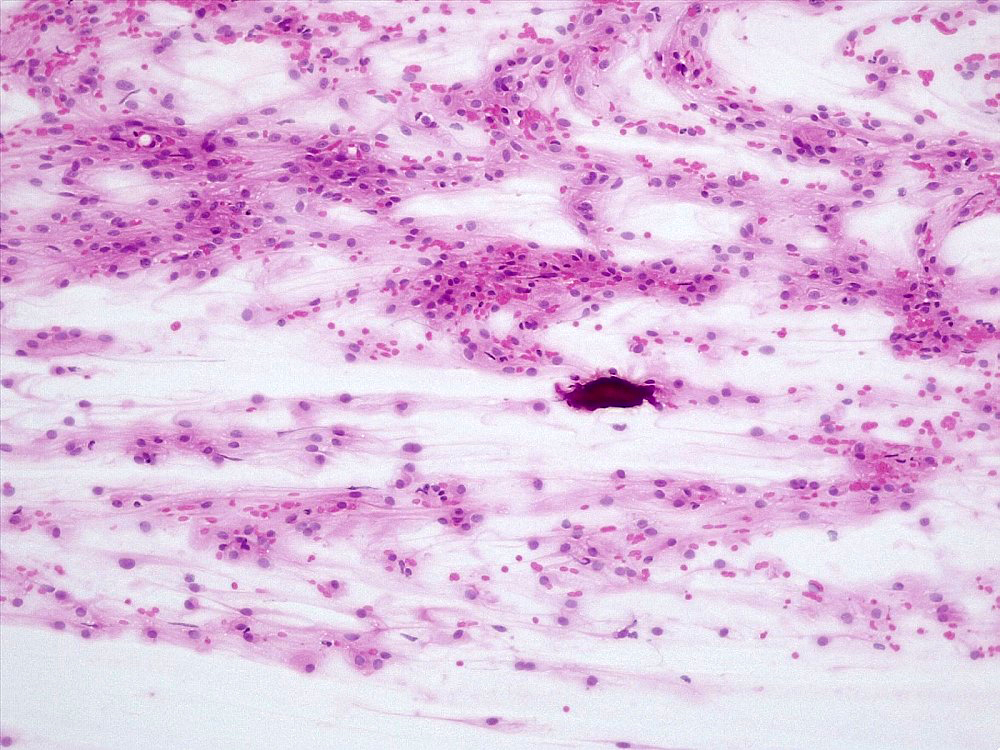
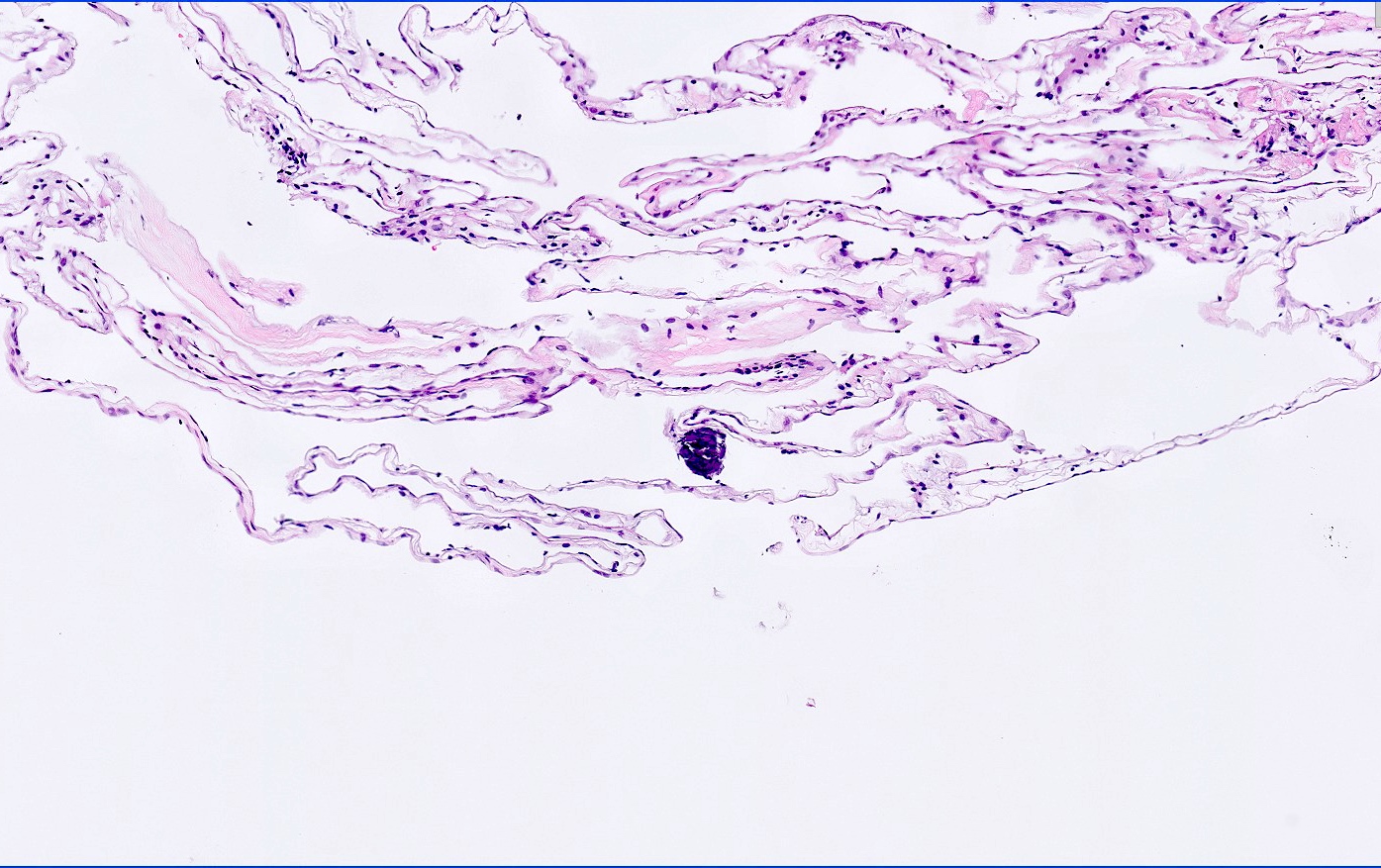
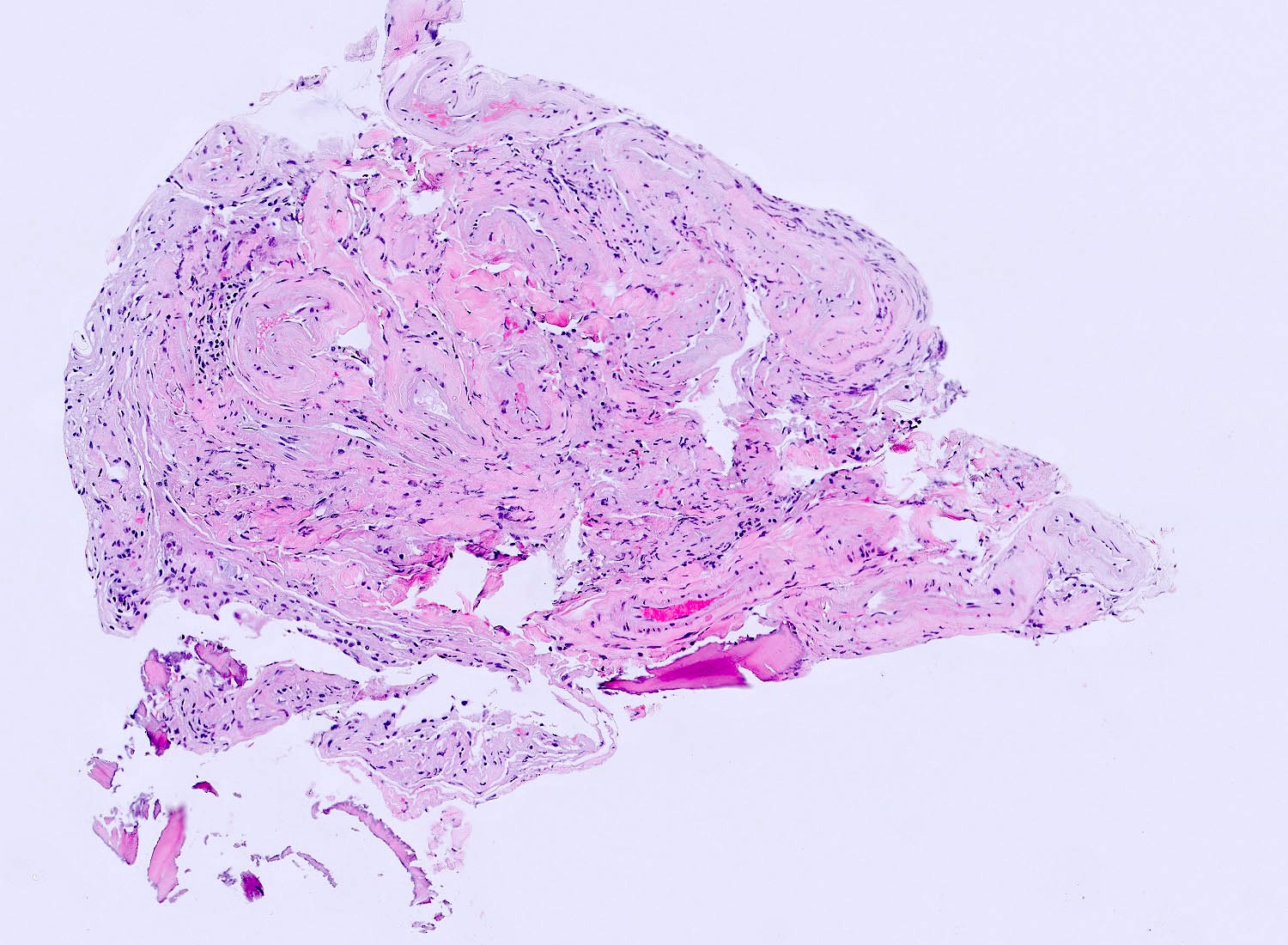
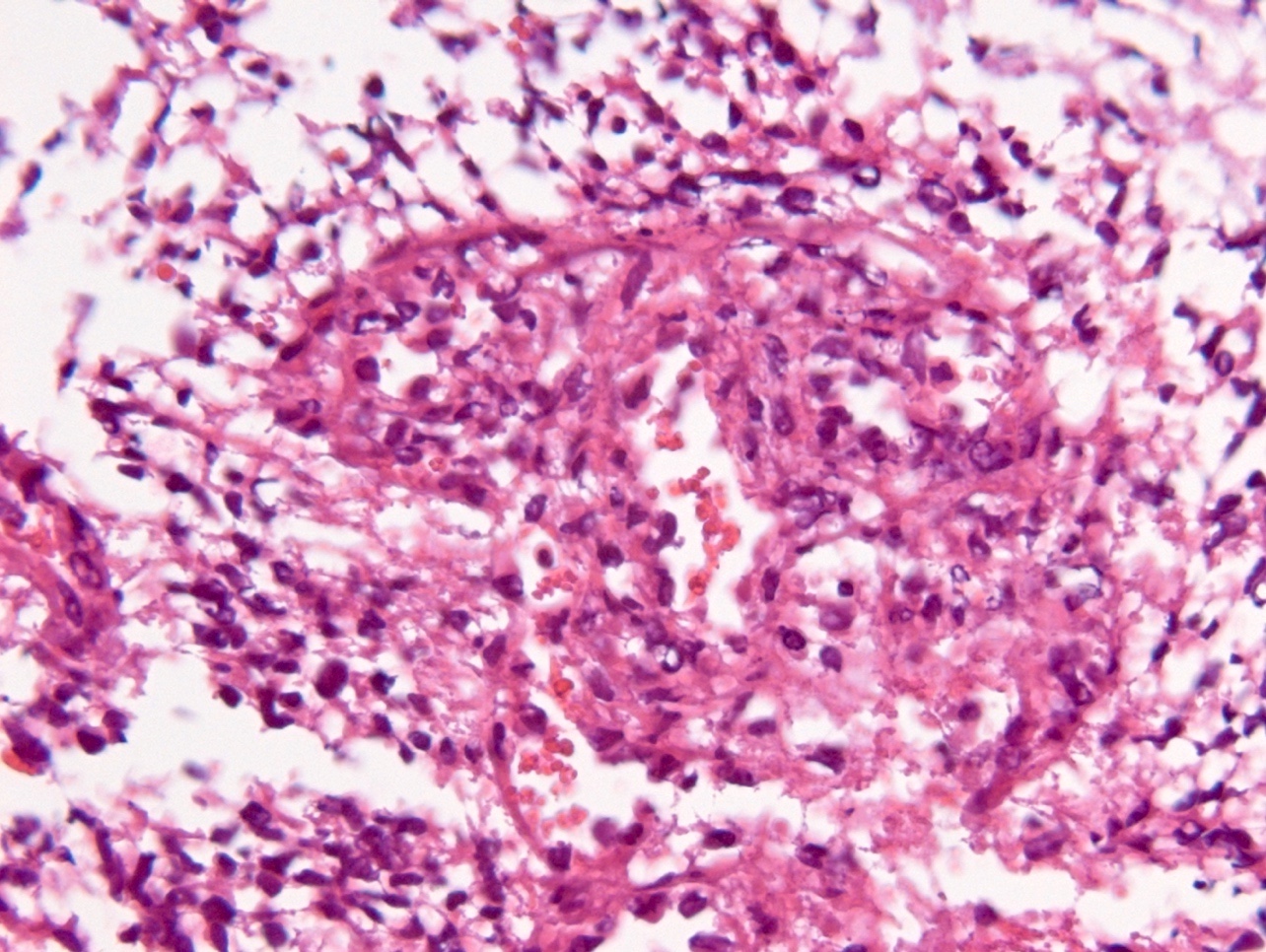
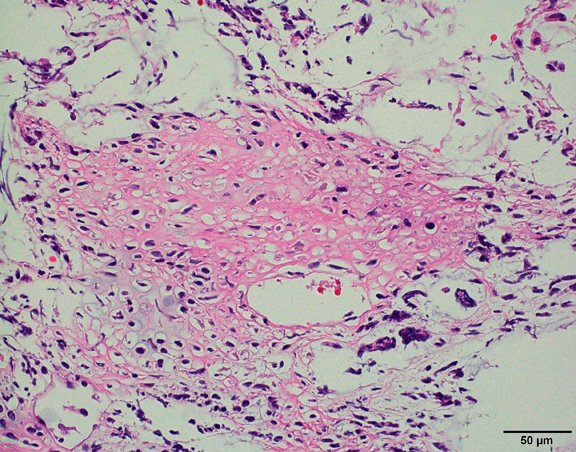
- May have degenerative changes with cystic degeneration, calcifications and xanthogranulomatous reactions with giant cells
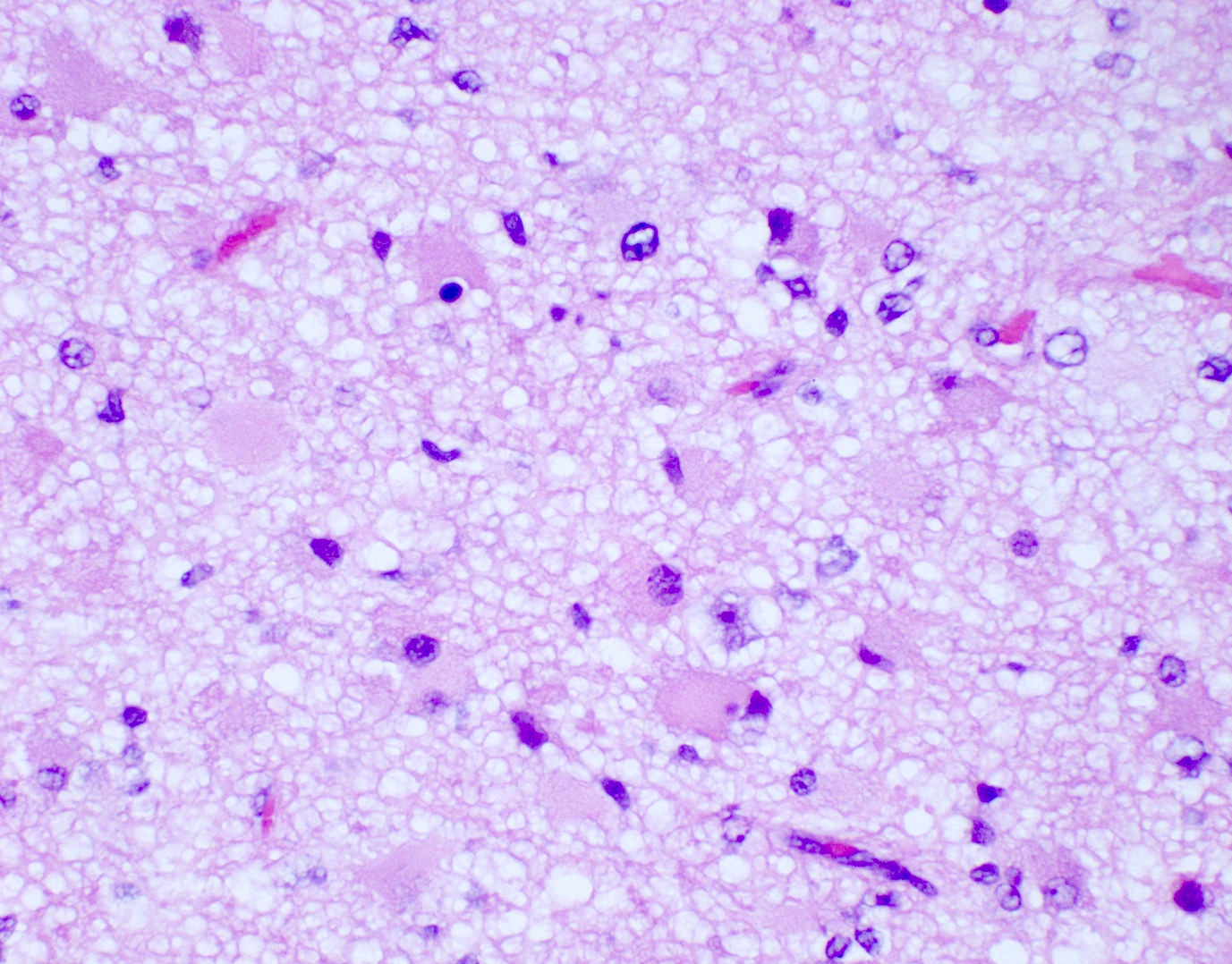
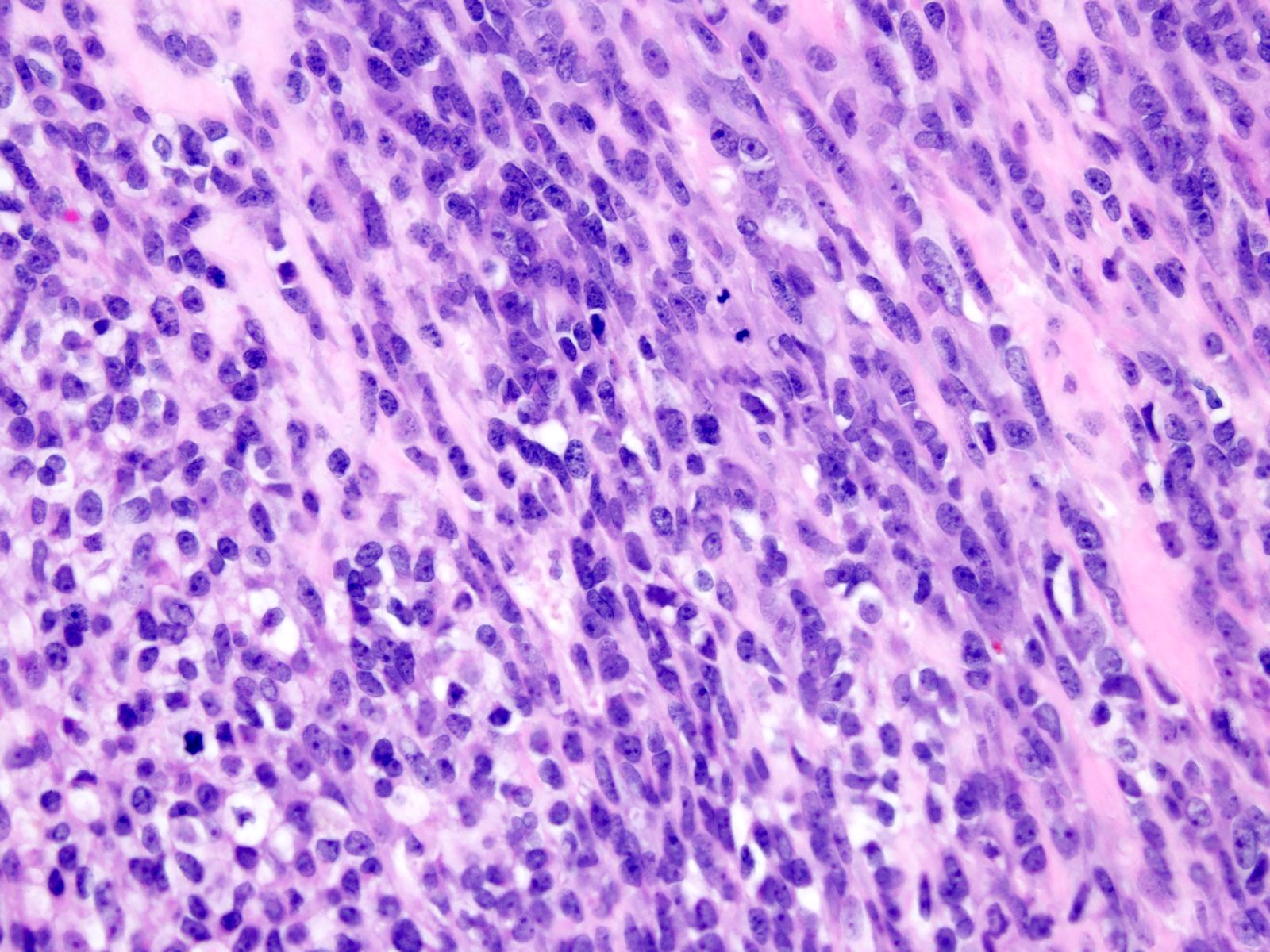
- Piloid gliosis and Rosenthal fibers in adjacent brain
- Rarely, melanin pigment
- Microscopic brain invasion common with tongues of tumor extending into hypothalamic parenchyma
Contributed by Nelli S. Lakis M.D., M.Sc.
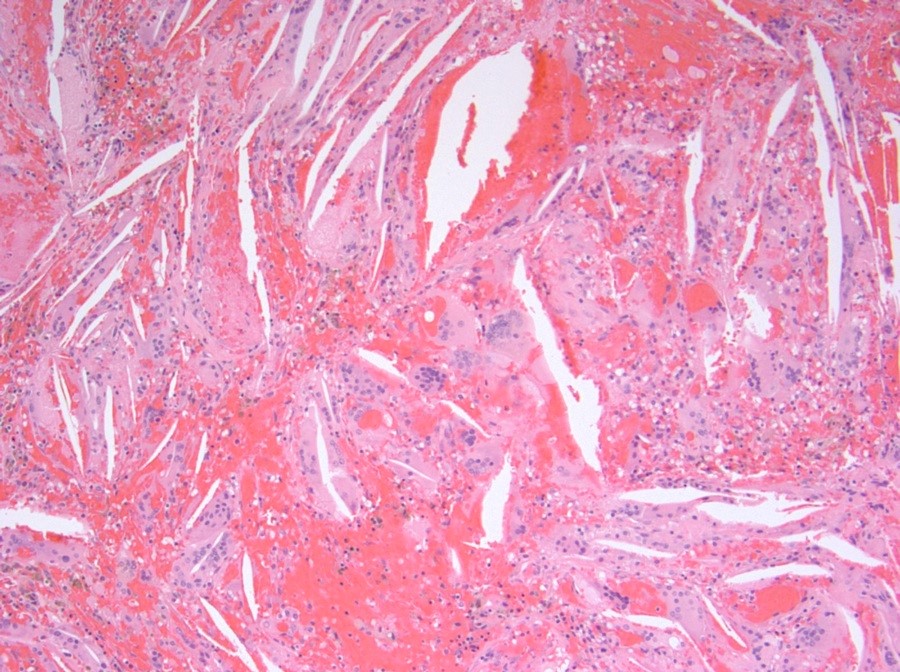
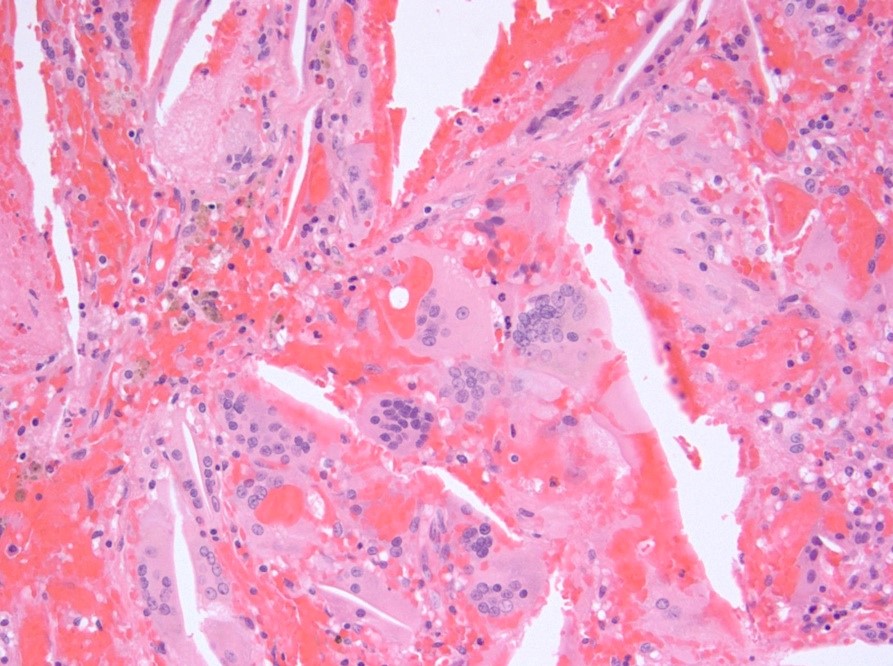
Xanthogranulomatous reaction
3 components
Tumor tongues
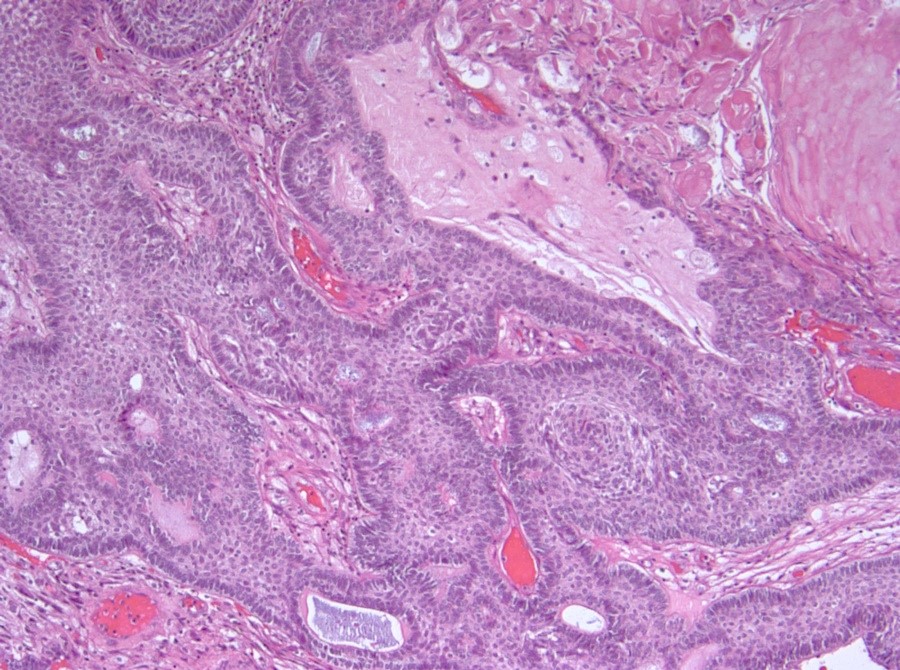
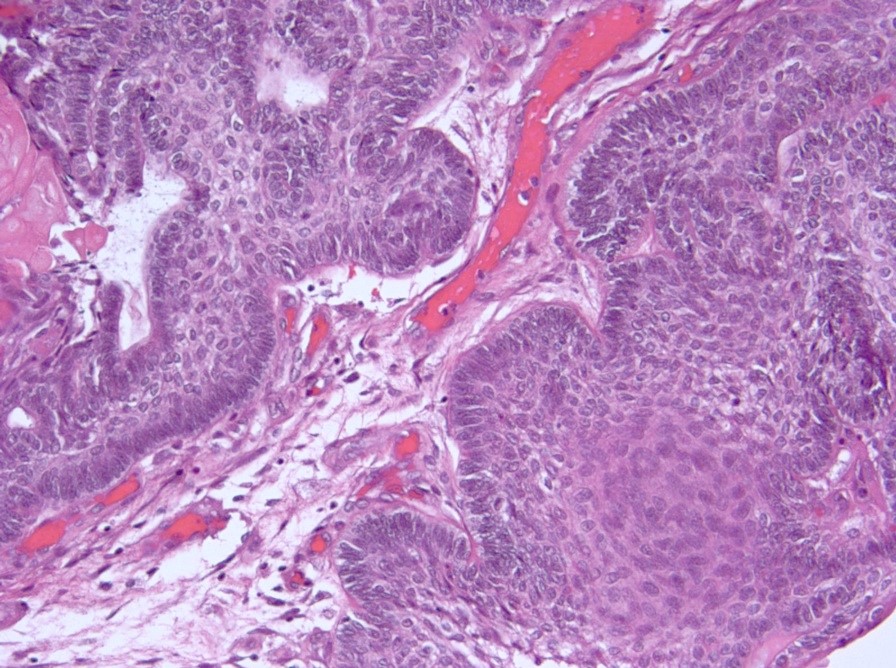
Nuclear palisading
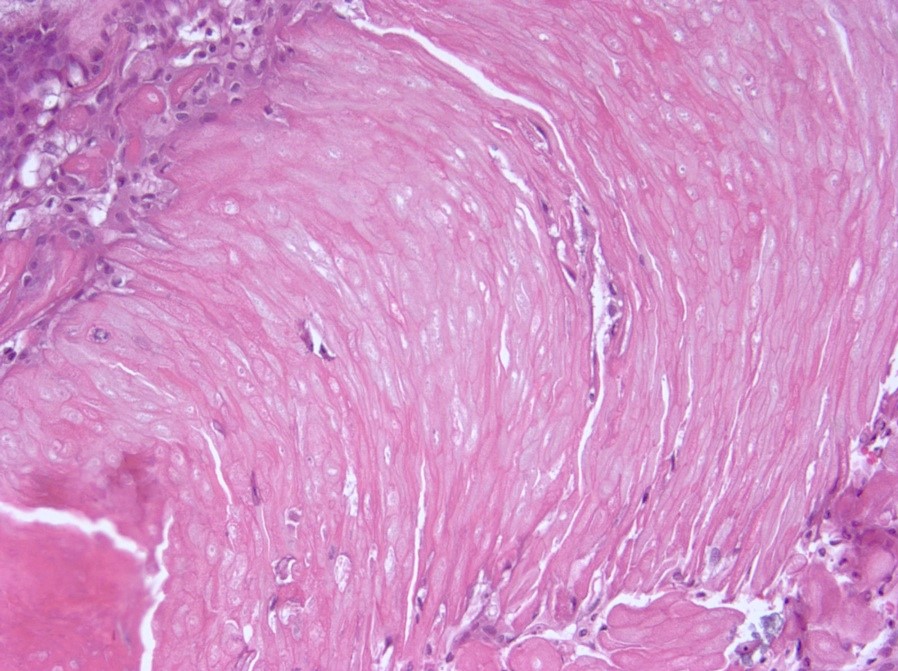
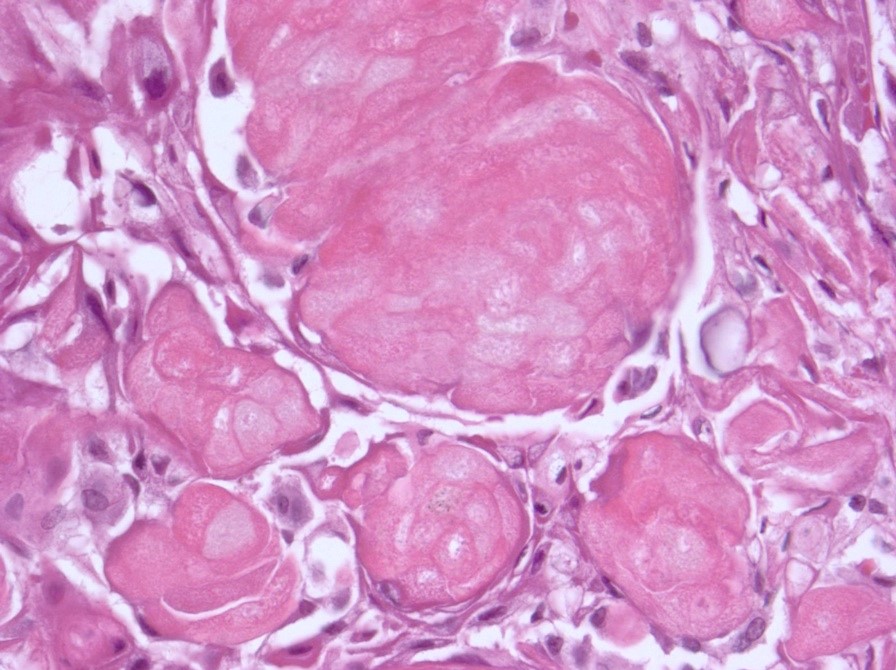
Wet keratin
Cystic degeneration
Fibrosis
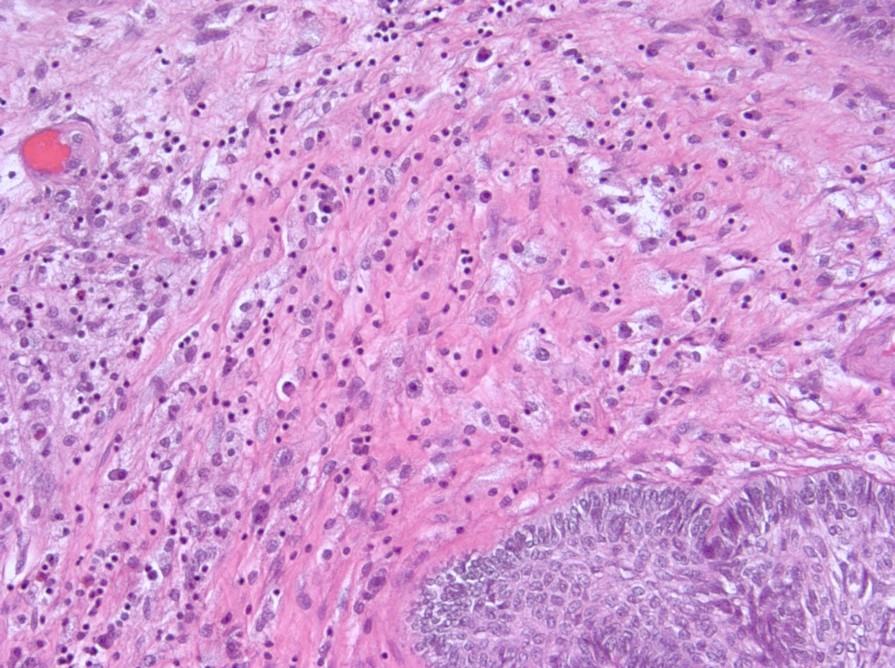
Inflammatory infiltrate
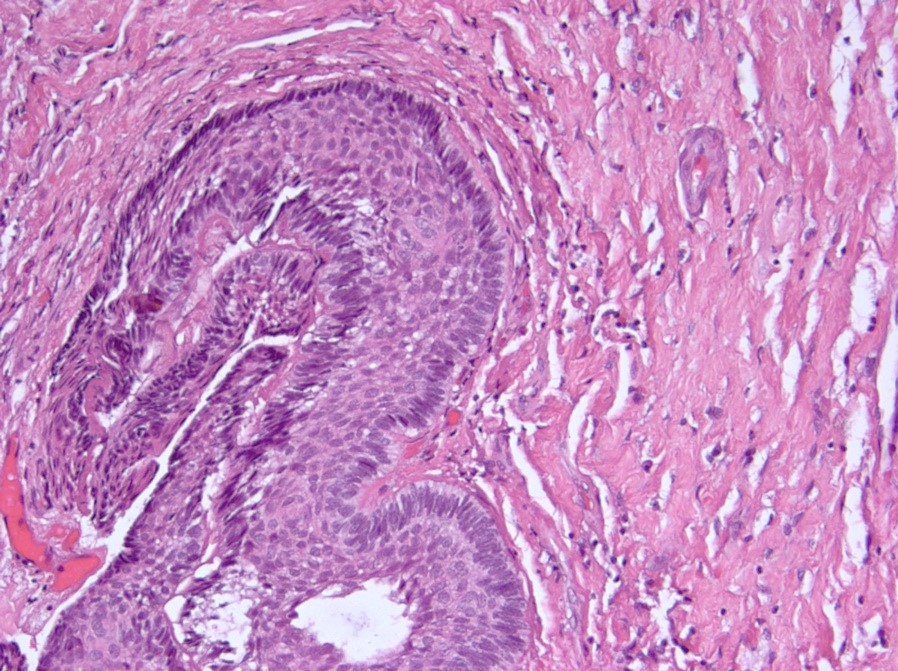
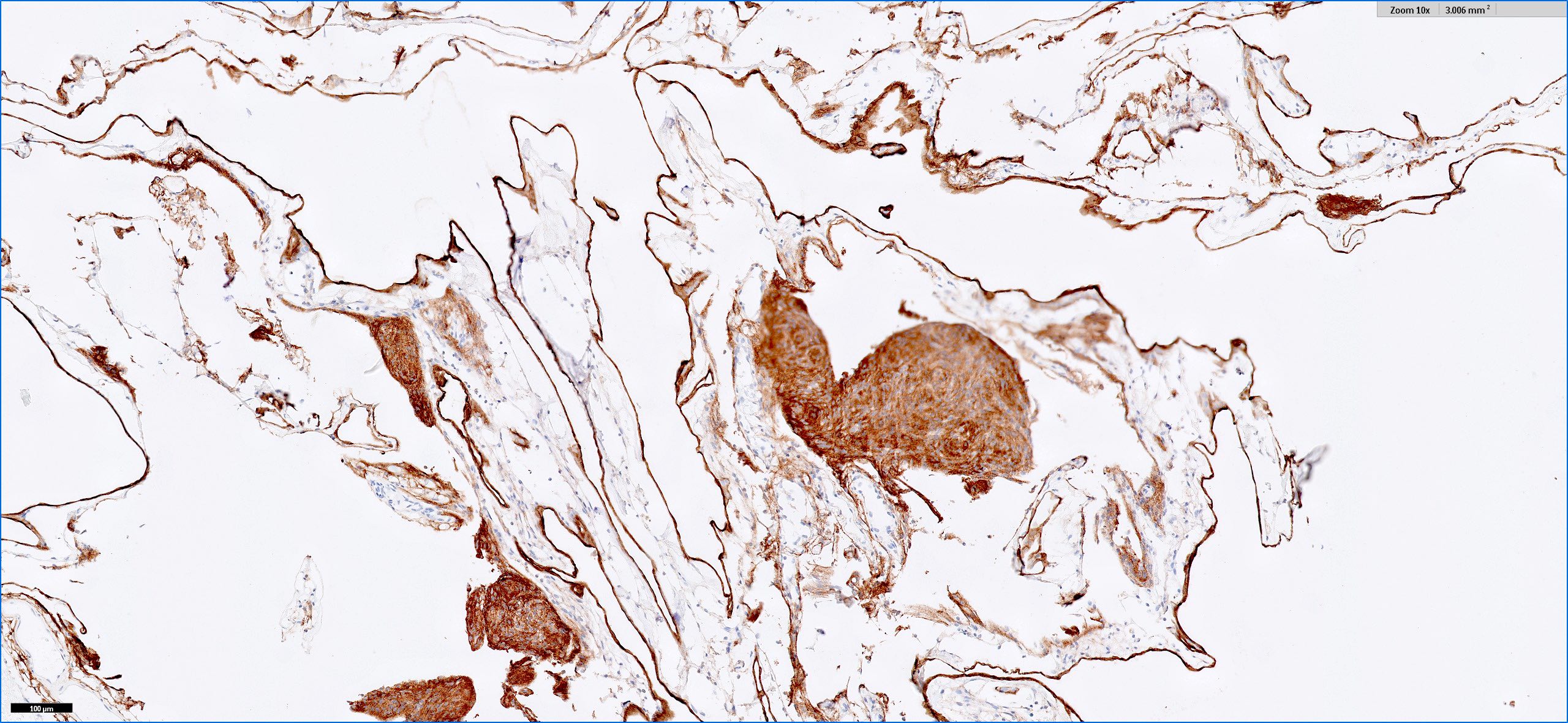
Adamantinomatous
epithelium
Stellate reticulum
Beta catenin
Images hosted on other servers:
23 year old female
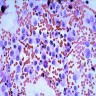
- Cohesive clumps of well differentiated squamous epithelium (Int Med Case Rep J 2020;13:123)
- Nodules of anucleate squamous cells
- Macrophages, amorphous debris and calcifications
- CK7, CK8, CK19, pancytokeratin, CK5/6, EMA, CK14 (Int Med Case Rep J 2020;13:123)
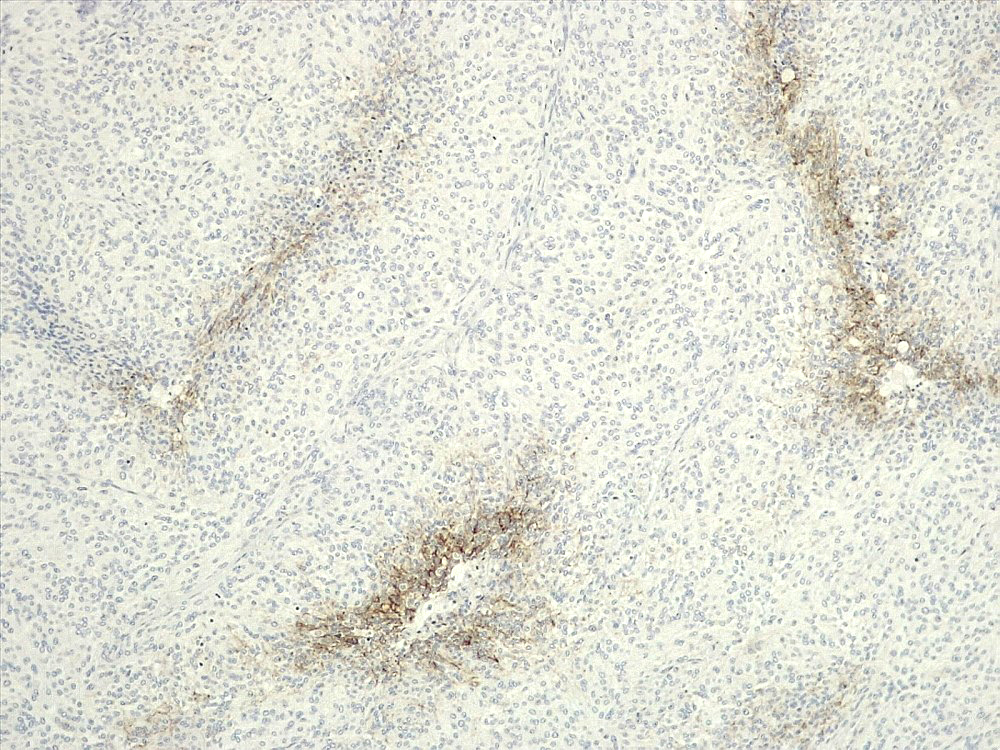
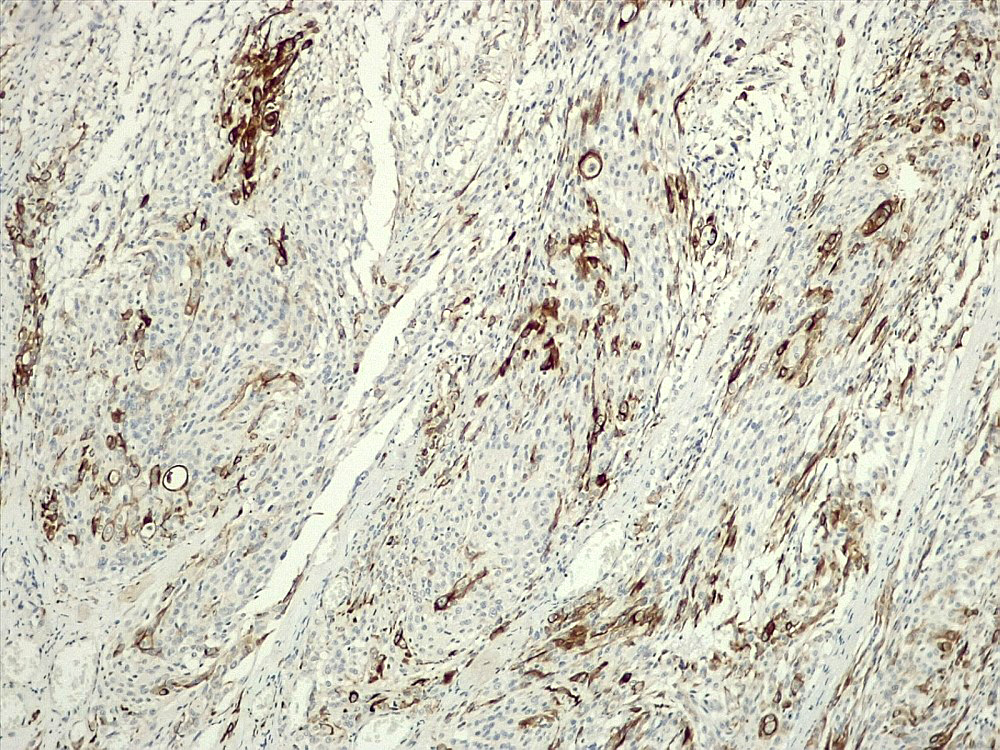
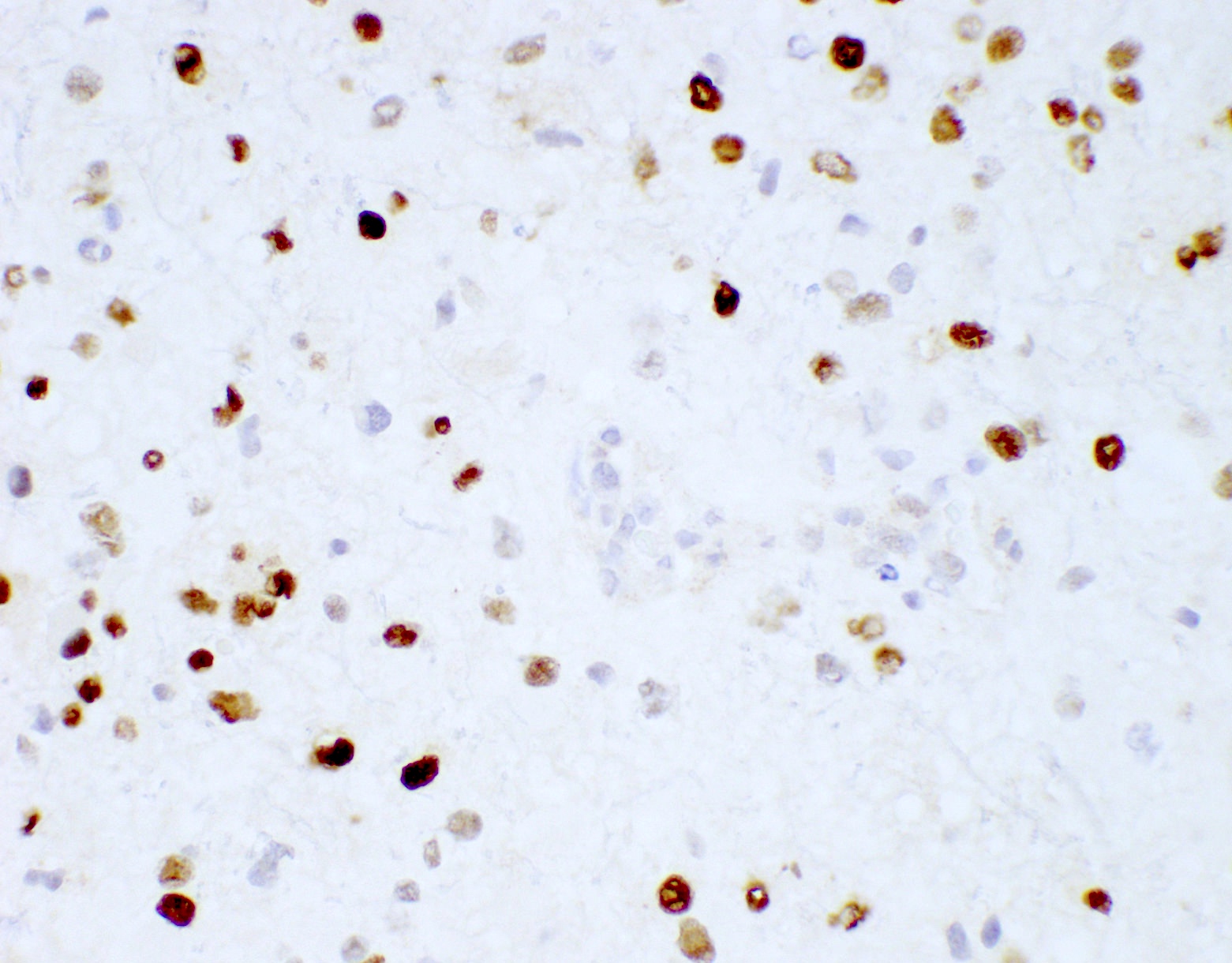
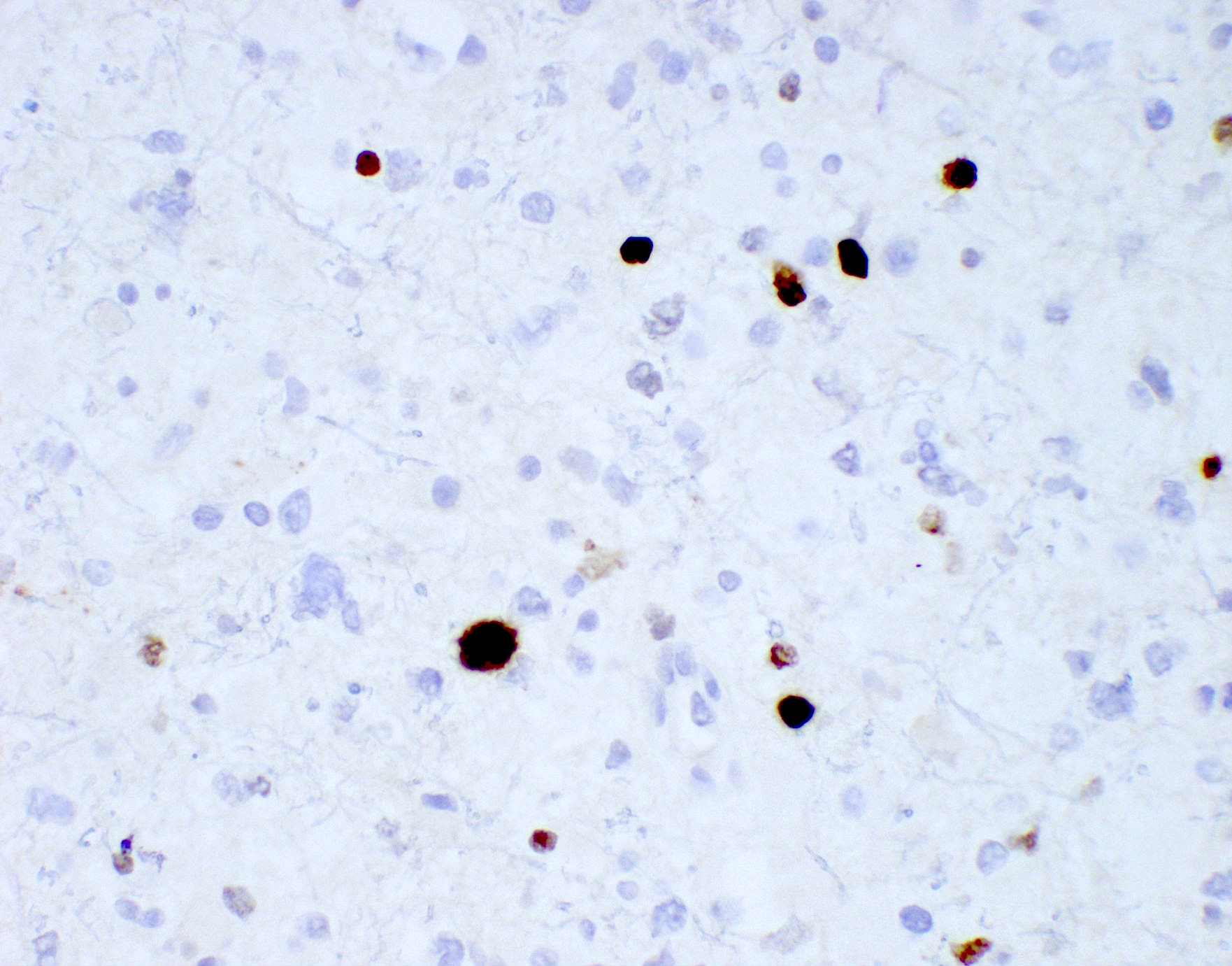
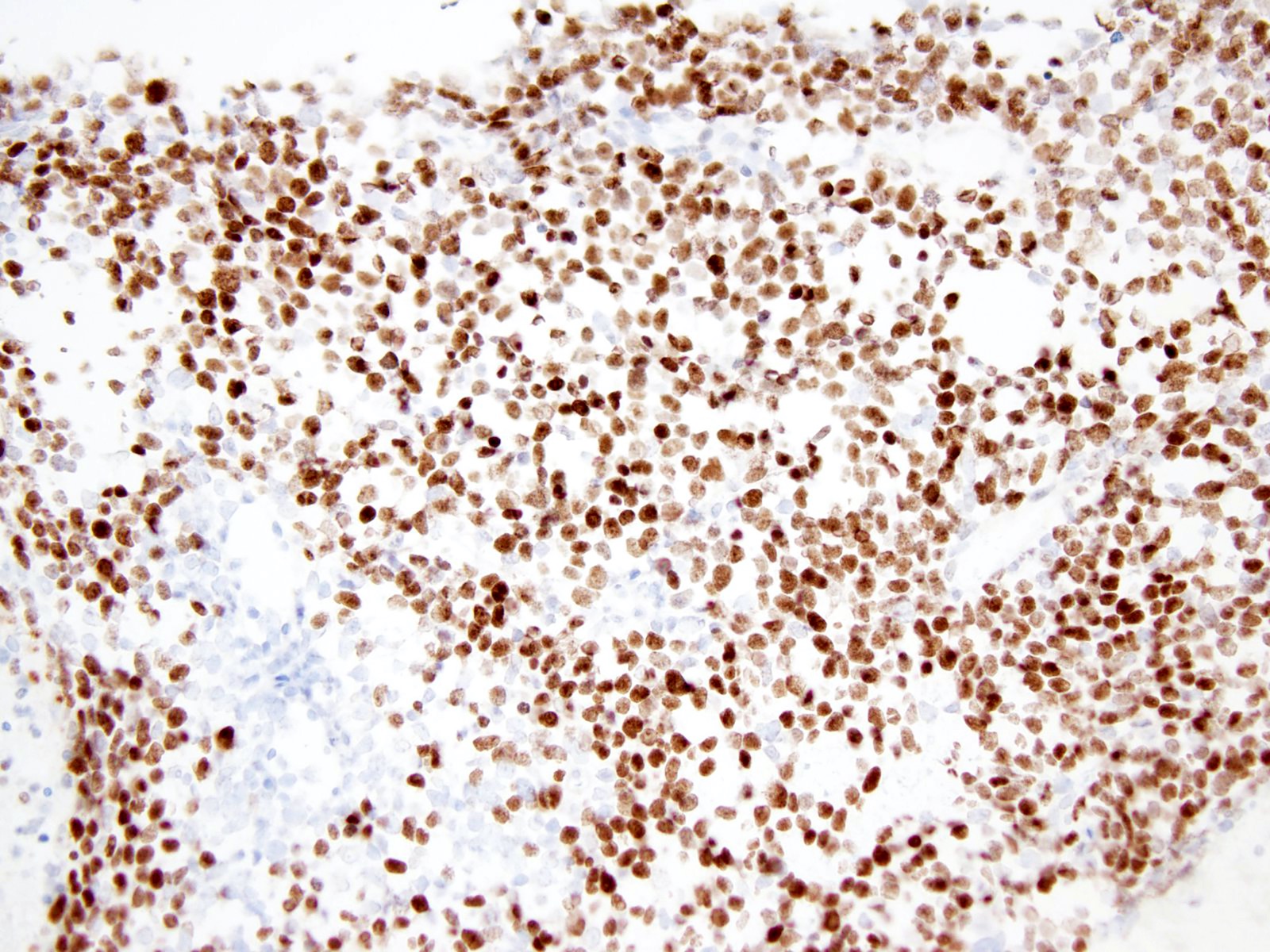
- Beta catenin: in whorls, both intranuclear (translocated) and cytoplasmic
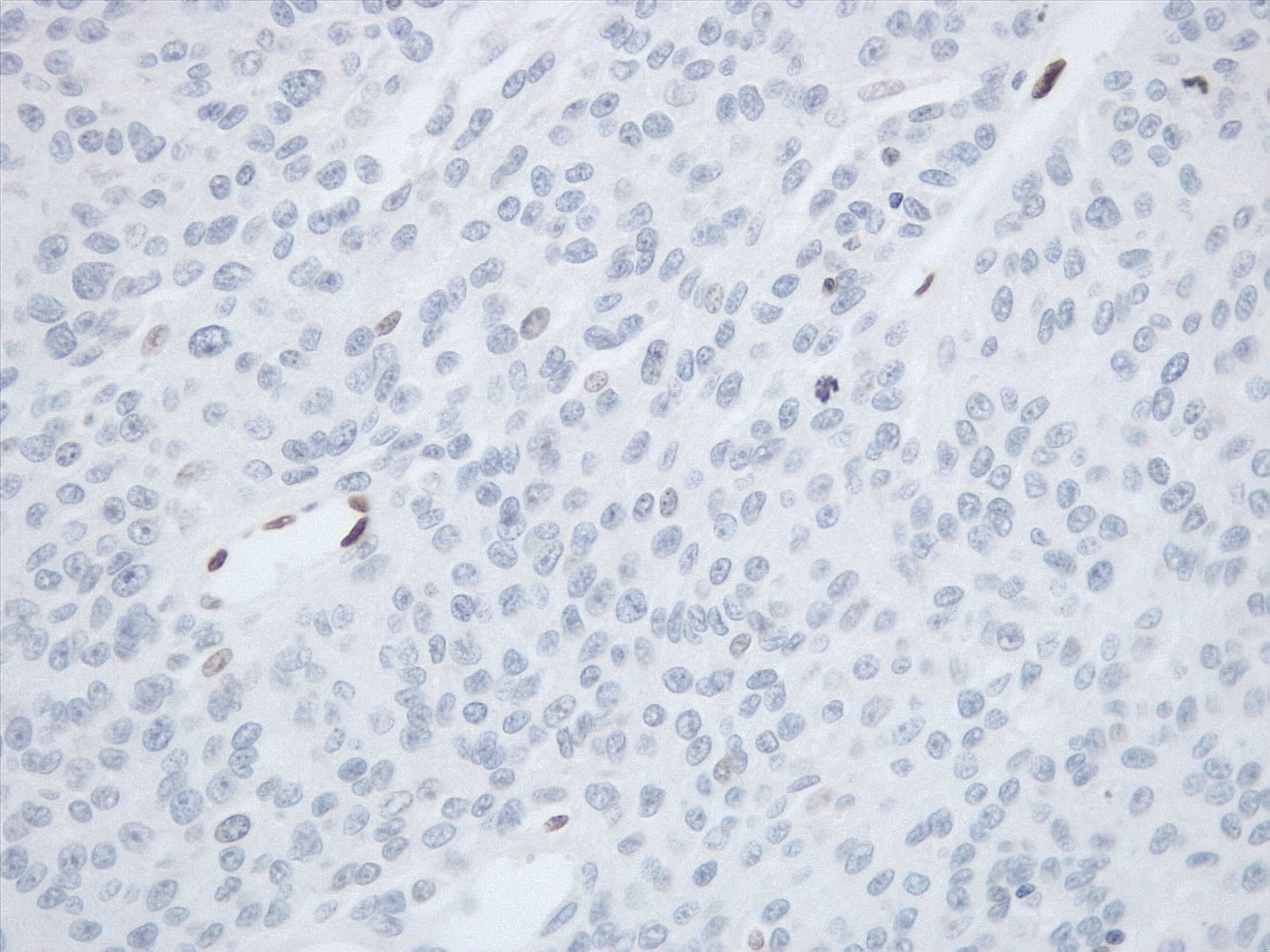
- Ki67 (low, usually concentrated along the peripheral palisading cells)
- Most cases are negative for mutant BRAF (V600E)
- Activating mutations of the WNT pathway gene CTNNB1 encoding beta catenin in almost all cases
- Sellar / suprasellar region, suprasellar mass, endoscopic resection:
- Adamantinomatous craniopharyngioma (see synoptic report)
- Epidermoid cyst:
- Uniloculate with thin layer of keratinizing squamous epithelium and keratohyalin granules
- Papillary craniopharyngioma:
- No palisading, no wet keratin, no calcifications, no "motor oil" cystic fluid, no xanthogranulomatous reaction
- Harbors a BRAF V600E mutation and is negative for beta catenin
- Pilocytic astrocytoma:
- Much greater cellularity than piloid gliosis, biphasic and may have eosinophilic granular bodies
- Rathke cleft cyst with squamous metaplasia:
- Intrasellar, squamous epithelium, as well as ciliated or mucus containing cells, no wet keratin, no calcifications
- Beta catenin negative nuclei
- Xanthogranuloma of sellar region:
- Sellar region cholesterol clefts, lymphoplasmacytic infiltrates, marked hemosiderin deposits, fibrosis, multinucleated giant cells around cholesterol clefts, eosinophilic granular necrotic debris and accumulation of macrophages, no true epithelium and may be associated with Rathke cleft cyst leakage / rupture / hemorrhage (Brain Pathol 2017;27:377)
- Eur J Endocrinol 2016;174:R139, Clin Neuropathol 2003;22:229, J Egypt Natl Canc Inst 2015;27:139, J Neuropathol Exp Neurol 2017;76:126, AJNR Am J Neuroradiol 2015;36:E55, Neuropathol Appl Neurobiol 2015;41:733, eMedicine: Surgery for Craniopharyngiomas Treatment & Management [Accessed 26 April 2021], Radiopaedia: Craniopharyngioma [Accessed 26 April 2021], Radiopaedia: Craniopharyngioma - Papillary [Accessed 26 April 2021], Kleinschmidt-DeMasters: Diagnostic Pathology - Neuropathology, 2nd Edition, 2016
A 16 year old boy comes to the clinic because he has been experiencing headaches and vision disturbances. The headaches have worsened over this past year and also are associated with nausea. His head is imaged and a suprasellar mass with calcifications is identified. The mass is removed surgically and gross histological examination reveals the presence of cystic spaces that are filled with thick dark brown fluid. Which of the following is the most likely diagnosis?
- Adamantinomatous craniopharyngioma
- Epidermoid cyst
- Papillary craniopharyngioma
- Rathke cleft cyst
- BRAF V600E mutation
- Lack of nuclear palisading
- Nodules of anucleate squamous cells
- WHO grade 2
- WHO 2021 definition: a meningioma with overtly malignant cytomorphology that can
- Resemble a carcinoma, melanoma or high grade sarcoma
- Display markedly elevated mitotic activity (≥ 12.5 mitoses/mm2, ≥ 20 mitoses/10 high power fields [HPF] of each 0.16 mm2)
- Harbor a TERT promoter mutation or
- Have homozygous CDKN2A / CDKN2B deletion
- CNS WHO grade 3
- 1 - 3% of meningiomas
- De novo (primary) or progression from a lower grade (1 or 2) meningioma (secondary) (Neuro Oncol 2018;20:1113)
- Meningioma with overtly malignant cytomorphology that can
- Resemble a carcinoma, melanoma or high grade sarcoma
- Display markedly elevated mitotic activity (≥ 12.5 mitoses/mm2, ≥ 20 mitoses/10 HPF of each 0.16 mm2)
- Harbor a TERT promoter mutation or
- Have homozygous CDKN2A / CDKN2B deletion
- CNS WHO grade 3
- Shows either histologic or immunohistochemical evidence of meningothelial differentiation
- At least shows focal meningothelial whorls, psammoma bodies or nuclear pseudoinclusions
- Immunohistochemistry: epithelial membrane antigen (EMA)+ (even focal), SSTR2A+, possible focal CK AE1 / AE3+, STAT6-
- Recurrence in 50 - 94%
- Also called:
- Malignant meningioma
- Meningothelial sarcoma (not preferred)
- ICD-O: 9530/3 - meningioma, malignant
- Any age, mainly adults 45 - 85 years (Neuro Oncol 2015;17:1166)
- Higher incidence in females but equal incidence in the 2 sexes at 75 - 85 years (Neuro Oncol 2015;17:1166)
- Cerebral or spinal meninges; cerebral ventricles (Neurosurg Rev 2020;43:513)
- Originates from arachnoid border cells or dural based cells (Acta Neurochir (Wien) 2021;163:57)
- The driver genetic event in most cases is NF2 inactivation by mutation or chromosome 22q loss
- Progression from lower grade meningioma follows pTERT mutation, CDKN2A / CDKN2B homozygous deletion, loss of chromosomes 9p, 1p, 6q, 10, 14q, 18q, gains of chromosomes 1q, 9q, 12q, 15q, 17q, 20q (Clin Cancer Res 2010;16:4155, Brain Pathol 2002;12:183, Brain Pathol 2014;24:184)
- Risk factors may be similar to risk factors for meningioma: ionizing radiations; neurofibromatosis type 2 (Eur J Epidemiol 2020;35:591)
- Neurological deficits depending on tumor location
- Common headaches, weakness and seizures (World Neurosurg 2021;149:e877)
- Based on imaging (CT; MRI) / biopsy / resection specimen
- MRI: contrast enhancing mass with possible necrotic areas (Neurochirurgie 2021;67:193)
- MRI: contrast enhancing dural tail sign at the perimeter
Contributed by Valeria Barresi, M.D, Ph.D.
Extra-axial, dural mass
Images hosted on other servers:

MRI
- Recurrence rate of 50 - 94%
- 10 year relative survival of 59.6% (Neuro Oncol 2020;22:iv1)
- Favorable prognostic factors: age 20 - 44 years and spinal location (Neuro Oncol 2021;23:iii1)
- Unfavorable prognostic factors: TERT promoter mutation; CDKN2A / CDKN2B homozygous deletion; H3K27me3 immunohistochemical loss (J Neuropathol Exp Neurol 2020;79:754)
- 2 year old boy with anaplastic meningioma (CNS Oncol 2016;5:131)
- 51 year old man with anaplastic intraventricular meningioma (World J Surg Oncol 2014;12:238)
- 55 year old man with secondary anaplastic meningioma (Neurochirurgie 2021;67:193)
- Surgery followed by fractioned radiotherapy, experimental chemotherapy or peptide receptor radionuclide therapy (Lancet Oncol 2016;17:e383)
- Dural based and widely variable in size
- May be well circumscribed or readily adherent to brain parenchyma
- Gross necrosis can be present
- Differential diagnosis versus other tumor types: at least focal presence of psammoma bodies, meningothelial whorls or nuclear pseudoinclusions
- Differential diagnosis versus CNS WHO grade 1 meningiomas: presence of mitoses
Contributed by Valeria Barresi, M.D, Ph.D.
Lobular architecture
Whorls
Nuclear pseudoinclusions
Mitoses
Whorls
Psmmoma body
Mitoses
- May have frank malignant cytology resembling a carcinoma, melanoma or high grade sarcoma
- Mitotic index: ≥ 12.5 mitoses/mm2, ≥ 20 mitoses/10 HPF of each 0.16 mm2
- At least focal meningothelial whorls and nuclear pseudoinclusions are useful to establish meningothelial origin
- Psammoma bodies may be present (World Neurosurg 2021;149:e877)
- Necrosis and brain invasion may be present
Contributed by Valeria Barresi, M.D, Ph.D.
Mitoses
Malignant morphology
Whorls
Necrosis
EMA
CK AE1 / AE3
H3K27me3
- Smear with tumor cells in large clumps and wide bridge of tissue in between
- Meningothelial lobules, balls or whorls (Diagn Cytopathol 2012;40:104, Acta Cytol 1998;42:1104, J Pathol Transl Med 2019;53:104)
- Thick cytoplasmic bridges among cells groups
- Psammoma bodies (Diagn Cytopathol 2012;40:104)
- Mitoses
- Reference: Adv Anat Pathol 2007;14:303
- EMA (may be focal) (89%)
- CK AE1 / AE3 (focal) (75%) (Hum Pathol 2004;35:1413)
- Vimentin (100%)
- SSTR2A (100%) (J Neuropathol Exp Neurol 2017;76:289)
- GFAP
- SOX10 (J Neuropathol Exp Neurol 2017;76:289)
- S100 (25% positive)
- HMB45
- CD99 (15% positive)
- BCL2 (weak or focal) (31% positive) (Hum Pathol 2004;35:1413)
- CD34
- Inhibin
- Progesterone receptor (20% positive)
- STAT6
- pTERT mutation (Neuro Oncol 2018;20:1009)
- CDKN2A / CDKN2B homozygous deletion (Brain Pathol 2002;12:183)
- NF2 mutation / deletion
- Loss of chromosomes 9p,1p, 6q, 10, 14q, 18q (Brain Pathol 2010;20:751, Acta Neuropathol Commun 2020;8:171)
- Gains of chromosomes 1q, 9q, 12q, 15q, 17q, 20q (Brain Pathol 2002;12:145, Proc Natl Acad Sci U S A 1997;94:14719)
- Brain, parasagittal mass:
- Diagnosis meningioma, subtype anaplastic, CNS WHO grade 3 (see comment)
- Comment: Meningothelial neoplasia showing focal meningothelial whorls, patternless architecture, brain invasion and spontaneous necrosis. Mitotic index: 15 mitoses/mm2.
- Meningioma, subtype atypical:
- Has mitotic index < 12.5 mitoses/mm2
- Metastasis of carcinoma:
- Shows widespread and not focal cytokeratin immunostaining
- Solitary fibrous tumor (SFT):
- Meningeal melanoma:
- Sarcoma:
- Absence of at least focal meningothelial whorls / nuclear pseudoinclusions / psammoma bodies
- Typically SSTR2A-
A dural based mass is found at the brain convexity. Histological examination shows a tumor with malignant morphology, mitotic index of 15 mitoses/mm2, EMA+, focal CK AE1 / AE3+, SSTR2A+, STAT6-. Which is the most likely diagnosis?
- Anaplastic meningioma
- Melanoma
- Metastasis of carcinoma
- Solitary fibrous tumor
SSTR2A is the most sensitive and specific marker for meningioma, while STAT6 is the most sensitive and specific marker for solitary fibrous tumor. Focal CK AE1 / AE3 immunostaining can be found in anaplastic meningioma, whereas metastatic carcinoma features widespread CK AE1 / AE3 immunostaining.
Comment Here
Reference: Anaplastic meningioma
- Anaplastic meningioma
- Choroid plexus carcinoma
- Ependymoma
- Glioblastoma
Anaplastic meningiomas can also be found in the cerebral ventricles. The presence of meningothelial whorls nuclear inclusions and EMA staining indicate meningothelial derivation. Other entities should be considered in the differential diagnosis, including glioblastoma (which is GFAP+, Olig2+), ependymoma (which is GFAP+, Olig2- and exhibits EMA dot-like staining) and choroid plexus carcinoma (which is more common in children and features widespread, strong CK AE1 / AE3 immunostaining).
Comment Here
Reference: Anaplastic meningioma
- Also called epiphysis, pineal body
- Between superior colliculi at base of brain; 100 - 180 mg
- Develops at month 2 of gestation as diverticulum in diencephalic roof of third ventricle
- Replaced by connective tissue after puberty
- Produces melatonin, which helps regulate circadian rhythms
Images hosted on other servers:

Location in brain
- Shaped like a pine cone, midline, attached to posterior end of roof of third ventricle in front of cerebellum, 1 cm long, red gray
Images hosted on other servers:

Local anatomy (horse)
- Loose neuroglial stroma with nests of pineocytes containing well defined neurosecretory (melatonin) granules
- Also astrocytes
- Has features of photoreceptors and concretions ("brain sand")
- Synaptophysin
- Retinal S antigen
- Superficial cerebrocortical tumour with features of infiltrating astrocytoma and ependymoma
- Relationship to ependymoma is unclear
- Primarily children and young adults
- Wide age range (2 to 70 years, mean 17 years)
- Affect both sexes equally
- Seizures are characteristic
- WHO grade 1
- Criteria for higher grade lesions are unclear
- Well delineated, solid, hyperintense, nonenhancing cortical lesions
- Stalk-like extension to adjacent ventricle is diagnostic
Images hosted on other servers:
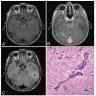
Axial MR images
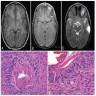
Axial T1 and T2 weighted MR images
Hyperintense cortically based mass
- Generally favorable with rare recurrence
- 3 cases: children 10, 10 and 13 years old (J Neurosurg Pediatr 2009;3:197)
- Surgical excision
- Role of chemo or radiotherapy is unclear
- Ill defined, firm
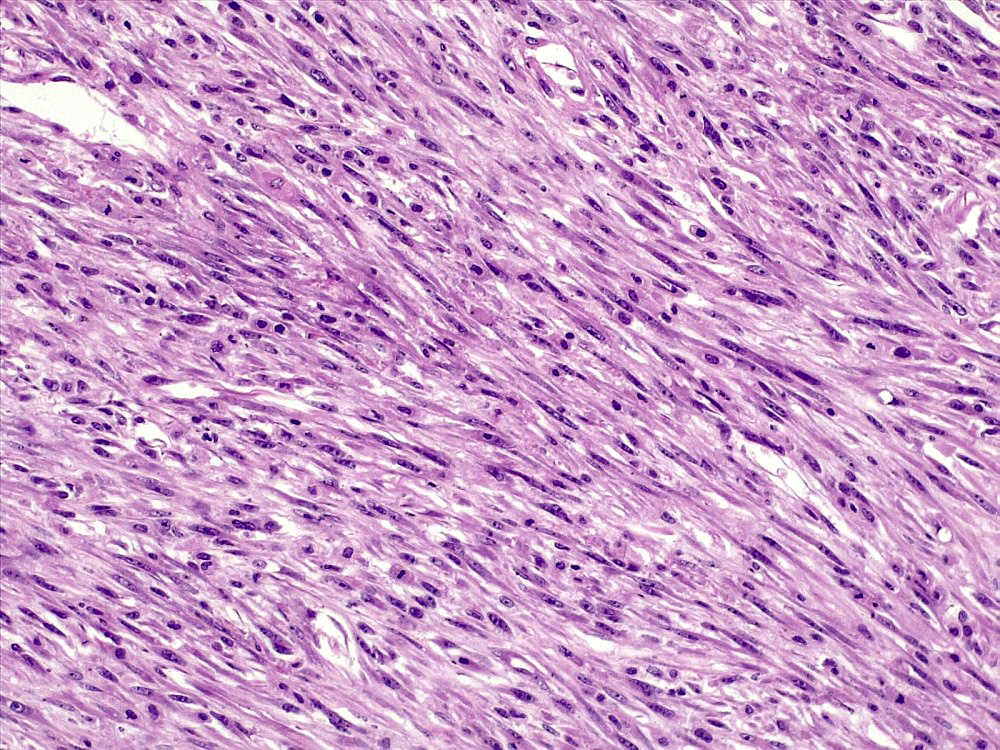
- Infiltrative, monomorphous, bipolar spindled cells arranged in angiocentric pattern about cortical blood vessels
- Also ependymoma-like pseudorosettes, subpial palisading, accumulation of tumor cells, miniature schwannoma-like nodules
- Usually no mitoses, no vascular proliferation, no necrosis
- Mitotically active lesions are associated with increased risk of recurrence
- Astroblastoma:
- Discrete borders, epithelioid cells, vascular sclerosis
- Astrocytoma, infiltrating:
- No pseudorosettes or subpial palisading
- Ependymoma:
- Enhancing, discrete borders
- Arachnoid cysts are nonneoplastic, intracranial cerebrospinal fluid (CSF) filled spaces lined with arachnoid membranes (Cureus 2018;10:e2458)
- 1% of intracranial masses (Neurosurgery 1997;41:951)
- Nonneoplastic, intracranial CSF filled spaces lined by meningothelial cells and an outer collagenous membrane
- Most primary developmental arachnoid cysts occur in the middle frontal fossa due to the splitting of arachnoid membranes (J Neuropathol Exp Neurol 1981;40:61)
- Meningothelial cells are positive for epithelial membrane antigen (EMA)
- Meningeal cyst
- Arachnoid cysts are classified as primary developmental cysts or secondary cysts (Cureus 2018;10:e2458)
- 50 - 65% of primary developmental cysts present in the middle cranial fossa / Sylvian fissure (Pediatr Neurosurg 1996;25:165)
- Arachnoid cysts in the middle cranial fossa are found more frequently in men than in women (Pediatr Neurosurg 1996;25:165)
- Arise within both cranial and spinal meninges
- Most are supratentorial and found in the middle fossa (J Neurosurg 2013;118:222)
- Other sites include retrocerebellar, convexity, cerebellopontine angle and spinal cord (J Neurosurg 2013;118:222)
- Primary developmental cysts occur due to the splitting of arachnoid membranes in utero, resulting in abnormal collections of cerebrospinal fluid (CSF) (J Neuropathol Exp Neurol 1981;40:61)
- Secondary cysts are less common and often occur after trauma, infection or surgery (Case Rep Orthop 2015;2015:250710)
- Mutation of the FOXC2 gene has been reported in familial forms
- Depends on size and location
- Headaches are the most common symptom (J Neurol Neurosurg Psychiatry 2007;78:1129)
- Other symptoms include hydrocephalus, intracranial hypertension, dizziness, nausea, vomiting, mental status changes, ataxia, seizures and hearing loss (Childs Nerv Syst 2015;31:77)
- Arachnoid cysts can be asymptomatic (Childs Nerv Syst 2015;31:77)
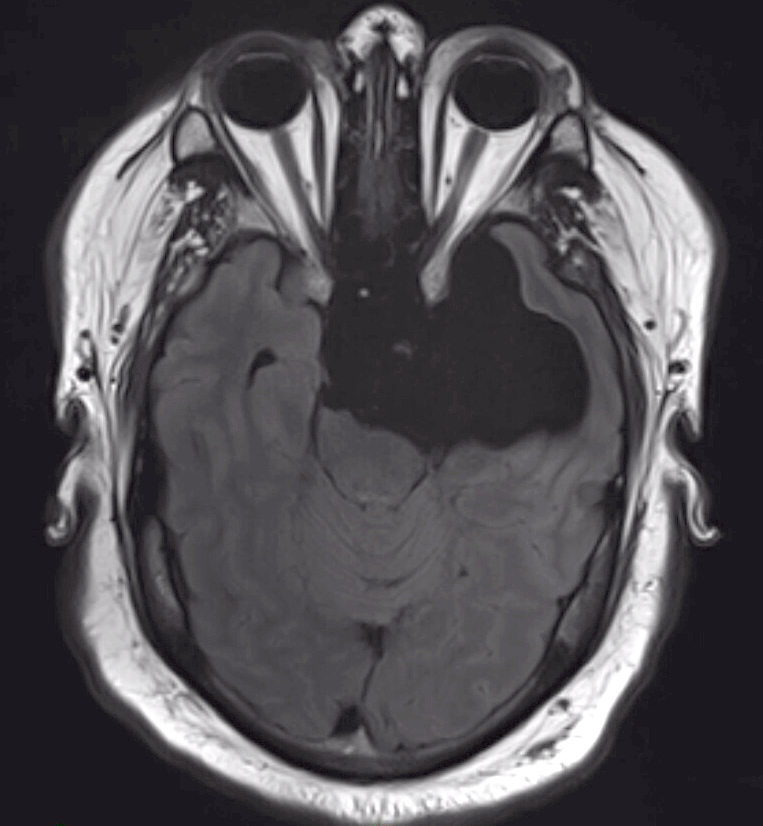
- Computed tomography (CT) and magnetic resonance imaging (MRI) for radiologic assessment
- Surgical resection is required for a definitive diagnosis
- MRI is the diagnostic procedure of choice
- Arachnoid cysts show low signal intensity on diffusion weighted imaging (DWI) and fluid attenuated inversion recovery (FLAIR) (Tani Girisim Radyol 2003;9:418)
- No enhancement
Contributed by Saman Seyed Ahmadian, M.D.
T2 FLAIR MRI

T2 MRI
Images hosted on other servers:
T2 MRI
- 22 year old man with a spinal epidural arachnoid cyst (Case Rep Orthop 2015;2015:250710)
- 36 year old woman with a well delineated cystic sellar lesion with suprasellar extension (Einstein (Sao Paulo) 2019;17:eAI4269)
- 45 year old woman with a pontomedullary junction arachnoid cyst (Asian J Neurosurg 2022;17:389)
- Surgery if symptomatic
- Variable in size; can be very large with mass effects
- Thin, transparent wall with a clear, colorless fluid
- Cyst is distinct from leptomeninges and dura
- Reference: Love: Greenfield's Neuropathology, 9th Edition, 2015
- Cyst wall is composed of a single layer of meningothelial cells and an outer collagenous membrane
- Meningothelial cells often partially denuded and may not always be recognizable
- Rare foci of meningothelial hyperplasia with or without psammoma bodies
- Focal inflammation (rare)
- Reference: Love: Greenfield's Neuropathology, 9th Edition, 2015
Contributed by Saman Seyed Ahmadian, M.D.
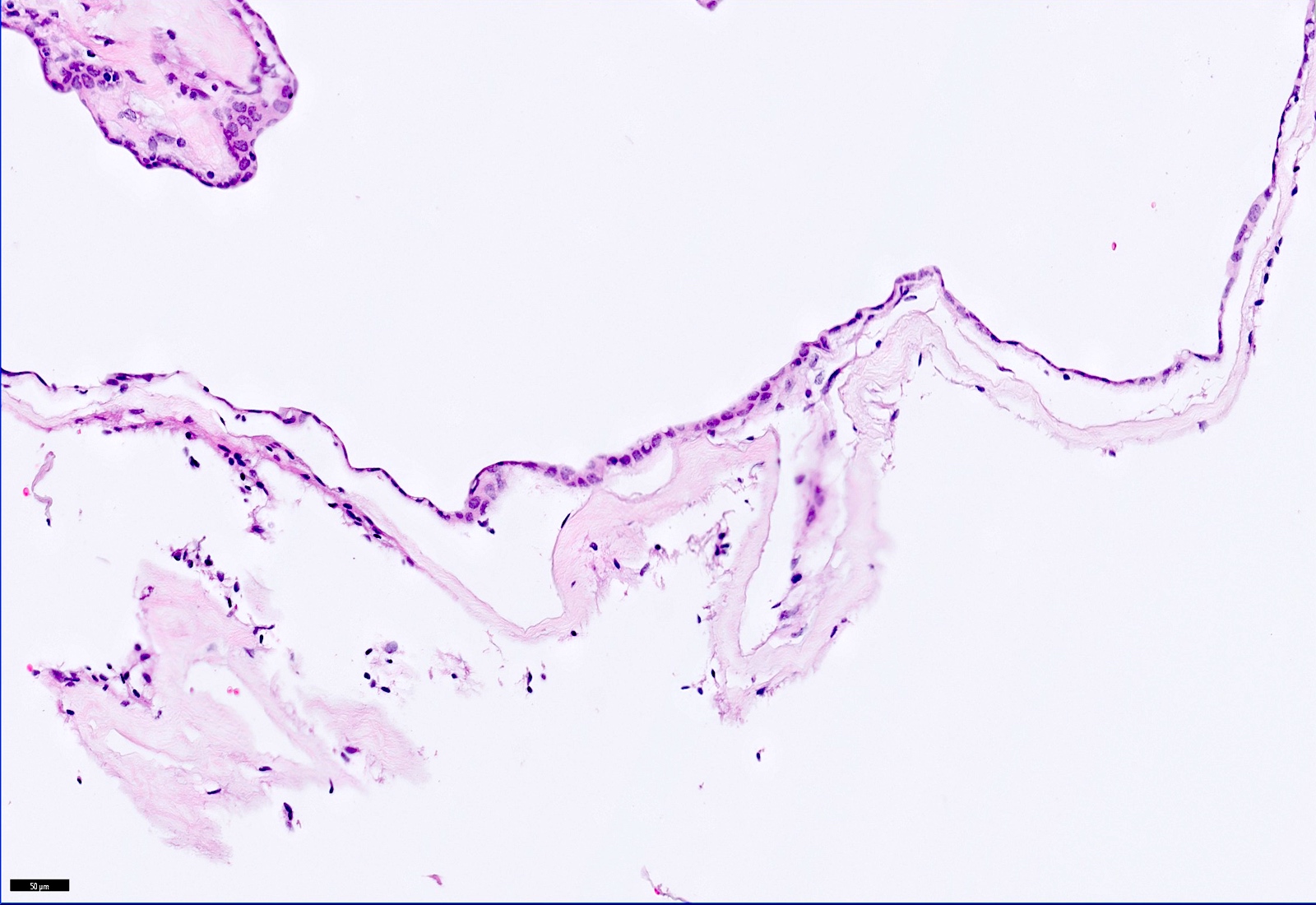
Cyst lining
Meningothelial hyperplasia
Calcification
No apparent meningothelial cells
EMA
Images hosted on other servers:

Arachnoid cyst, resection
- Meningothelial cells are positive for EMA
- Cytokeratin, GFAP, transthyretin and synaptophysin
- Tendency of meningothelial tissues to cleave along the dura - arachnoid interface layer (J Neuropathol Exp Neurol 1979;38:434)
- Mutation of the FOXC2 gene has been reported in familial forms (PLoS One 2013;8:e80548)
- Cyst wall, excision:
- Arachnoid cyst (see comment)
- Comment: The histologic section shows a cystic lesion composed of a single layer of meningothelial cells with an outer layer of delicate fibrous tissue. The meningothelial cells are positive for EMA by immunohistochemistry, which confirms the diagnosis.
- Epidermoid cyst:
- Squamous cyst lining
- Lamellar keratin debris
- Positive for keratin
- Endodermal cyst:
- Pseudostratified epithelium cyst lining with bronchogenic (ciliated) or gastrointestinal (glands) differentiation
- Positive for keratin
- Ependymal cyst:
A 71 year old patient with altered mental status had a 7.6 cm cystic lesion in the left frontoparietal convexity. The cystic lesion was excised. What immunohistochemistry confirms the diagnosis?
- Cytokeratin
- EMA
- GFAP
- Synaptophysin
Comment Here
Reference: Arachnoid cyst
- Rare ( < 3% of primary brain gliomas), compact glial neoplasm with perivascular pseudorosettes formed of GFAP+ cells arranged around central, often sclerotic, blood vessels
- Controversial entity - relationship with ependymoma is not clear
- Usually children and young adults, median age 11 years, range 1 - 58 years
- Features of astrocytoma and ependymoma; expresses nonfibrillar form of GFAP (so PTAH-)
- No WHO grade assigned for astroblastoma or anaplastic astroblastoma
- Discrete supratentorial cerebral, often superficial, contrast enhancing mass
- Cystic change is common
Images hosted on other servers:
Huge well demarcated mass in frontal lobe
- Anaplastic histology predicts poor prognosis (J Neurooncol 1998;40:59)
- Newborn boy with large cerebral mass (Case #312)
- 6 year old girl with intraventricular astroblastoma (J Neurosurg Pediatr 2008;1:152)
- 8 year old girl with tumor in frontoparietal lobe (Neuropathology 2006;26:72)
- 10 year old girl with recurrent low grade astroblastoma with signet ring-like cells and high proliferative index (Fetal Pediatr Pathol 2013;32:284)
- 15 year old girl with headache and diplopia (J Korean Med Sci 2004;19:772)
- Resection (adequate for well differentiated tumors), more aggressive treatment needed for malignant tumors
- Well circumscribed, peripheral, cerebral hemispheric masses
- Firm, often cystic
- Well circumscribed with discrete pushing borders, occasionally infiltrative in high grade lesions
- Perivascular pseudorosettes resembling ependymoma but with thick processes from cell body to adventitia of vessel
- Also vascular hyalinization, little fibrillar background
- Limit diagnosis to tumors in which these features predominate (other tumors have these features focally)
- High grade astroblastomas have hypercellular and mitotically active regions, often with vascular proliferation or necrosis with pseudopalisading; rare features are signet ring cells (Neuropathology 2002;22:200)
Case #312

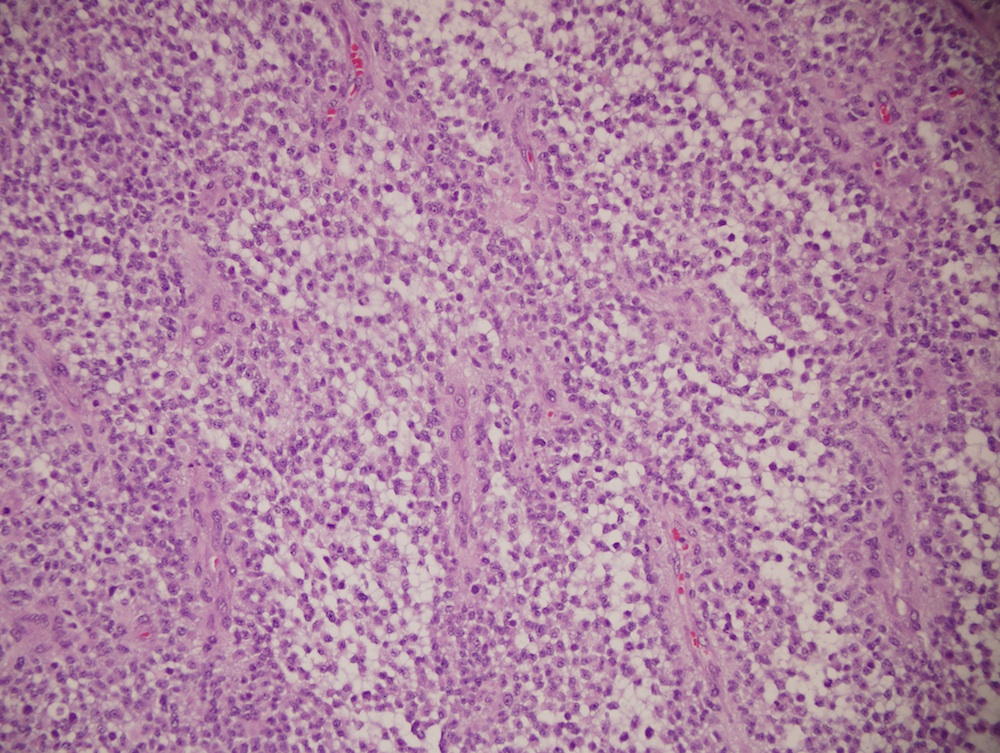
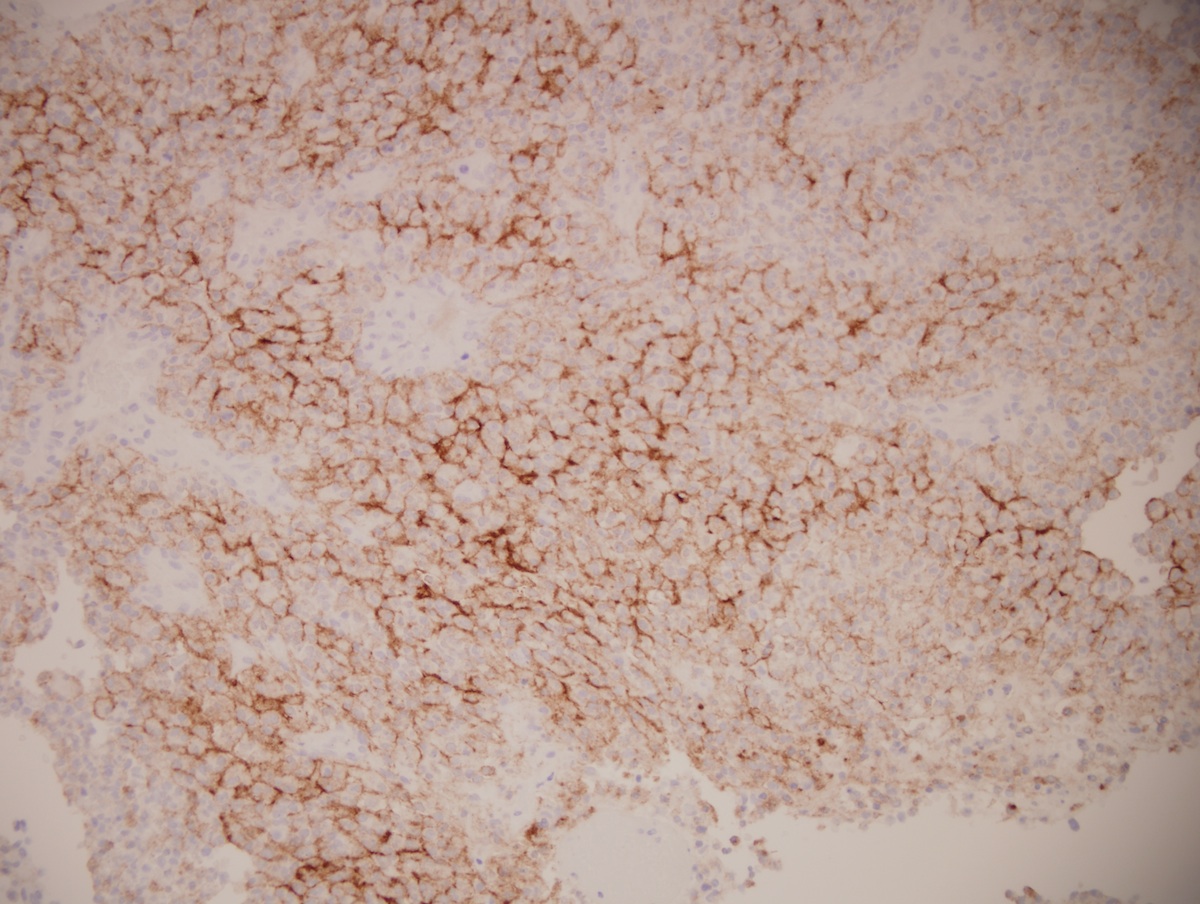
EMA
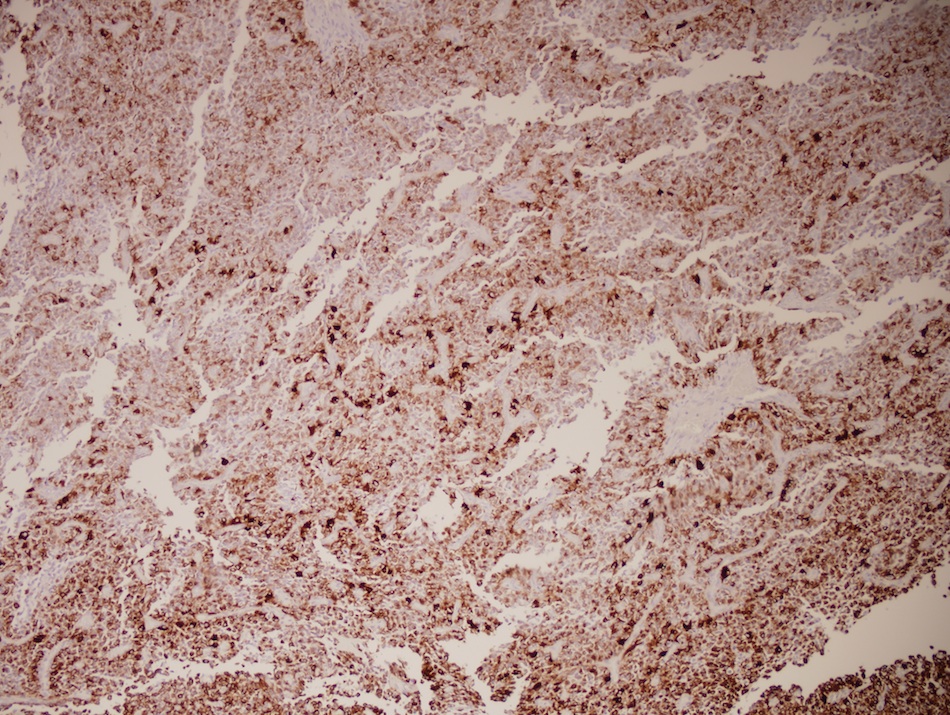
GFAP
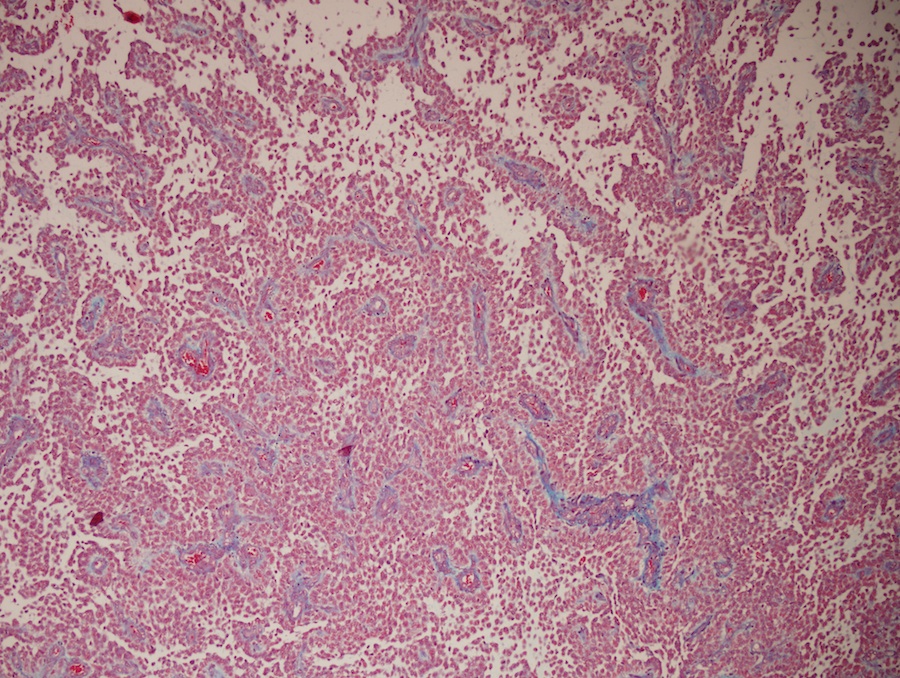
Trichrome
- PTAH, synaptophysin, cytokeratin
- Abundant intermediate filaments forming bundles in tumor cytoplasm, membrane junctions and external lamina when cells are in contact with collagen fibers (Surg Neurol 1991;35:116)
- +20q, +19 (Brain Pathol 2000;10:342)
- Ependymoma: more fibrillar, nuclei smaller and less pleomorphic, true rosettes, less sclerosis (AJNR Am J Neuroradiol 2002;23:243, Childs Nerv Syst 2005;21:211)
- Pilocytic astrocytoma: biphasic piloid areas with Rosenthal fibers alternating with spongy microcystic areas with eosinophilic granular bodies
- Pleomorphic xanthoastrocytoma: fascicular pattern, pleomorphic cells, lipidized cells, eosinophilic granular bodies, perivascular lymphocytes
- IDH1 / IDH2 mutated, diffusely infiltrating glioma, most often with concurrent TP53 or ATRX mutations and without 1p / 19q codeletion
- Can be graded CNS WHO grade 2, 3 or 4
- IDH1 codon 132 or IDH2 codon 172 mutated, diffusely infiltrating glioma without 1p / 19q codeletion and usually with TP53 or ATRX mutations
- In the absence of 1p / 19q codeletion, a component that morphologically resembles oligodendroglioma is compatible with this diagnosis
- Can be designated CNS WHO grade 2, 3 or 4 depending on presence of mitotic activity, nuclear atypia, pleomorphism, necrosis, microvascular proliferation or CDKN2A / CDKN2B homozygous deletion
- Significant proliferative activity is consistent with a CNS WHO grade 3 diagnosis
- Presence of either necrosis, microvascular proliferation or CDKN2A / CDKN2B homozygous deletion is consistent with a CNS WHO grade 4 diagnosis
- Astrocytoma, IDH mutant, CNS WHO grade 2; previously designated diffuse astrocytoma, IDH mutant
- Astrocytoma, IDH mutant, CNS WHO grade 3; previously designated anaplastic astrocytoma, IDH mutant
- Astrocytoma, IDH mutant, CNS WHO grade 4; previously designated glioblastoma, IDH mutant
- Astrocytoma, IDH mutant, CNS WHO grade 4; historically referred to as secondary glioblastoma
- ICD-11:
- 2A00.0Y & XH6PH6 - other specified gliomas of brain & astrocytoma, NOS
- 2A00.0Y & XH2HK4 - other specified gliomas of brain & diffuse astrocytoma, IDH mutant
- Age of diagnosis is typically younger than glioblastoma, IDH wild type, with higher grade tumors occurring more often in older patients (third or fourth decade for grade 2 or 3, versus fifth decade for grade 4) (Acta Neuropathol 2015;129:867)
- M > F, within grade 2 and grade 3 tumors (N Engl J Med 2015;372:2481)
- Majority of tumors are sporadic
- A small percentage are associated with familial cancer syndromes, such as neurofibromatosis, tuberous sclerosis and Li-Fraumeni syndrome (Neuro Oncol 2014;16:896)
- Occurs throughout the CNS but is preferentially located in the cerebral hemispheres, especially the frontal lobes (AJNR Am J Neuroradiol 2012;33:1349)
- Cell of origin is unknown, although the commonality of IDH mutation across IDH mutant astrocytoma and oligodendroglioma suggests a common histogenesis in these tumors; similarly, single cell sequencing of IDH mutant gliomas suggests this as well (Acta Neuropathol 2009;118:469, Science 2017;355:eaai8478)
- Majority of tumors are sporadic
- A small percentage are associated with familial cancer syndromes, such as Li-Fraumeni syndrome and Ollier disease (Cancer Res 2003;63:6643, Brain Tumor Pathol 2018;35:202)
- Commonly, gradual onset of symptoms
- May present incidentally in work up for headache or following trauma (Lancet Oncol 2017;18:e315)
- Seizures are a common symptom in cerebral hemispheric lesions
- Changes in behavior or personality, especially in frontal lobe tumors
- Uncommonly, site dependent neurological deficits
- Manifestations of increased intracranial pressure
- Often present after a clinical history of a few months of neurologic symptoms
- MRI with contrast is the preferred imaging modality
- Diagnosis is by biopsy or surgical resection
- CT:
- Expanding, intra-axial, poorly defined mass of low density
- Variable calcification may be seen
- Contrast enhancement and central hypodensity due to necrosis, occur with higher grades
- MRI:
- T1 hypodensity and T2 hyperintensity
- T2 hyperintensity with relative FLAIR sequence hypointensity (T2 FLAIR mismatch) is a relatively suggestive indication of IDH mutant astrocytoma (Clin Cancer Res 2017;23:6078)
- Distortion and enlargement of involved areas, including associated cortical ribbon
- Contrast enhancement is typically present in higher grade tumors (J Neurooncol 2019;141:327)
- Ring-like enhancement around central necrosis typical of grade 4
Contributed by John DeWitt, M.D., Ph.D. and Meaghan Morris, M.D., Ph.D.
MRI axial T1 FLAIR
MRI axial T1 precontrast
MRI axial T1 postcontrast
MRI axial T1 FLAIR
MRI axial T1 postcontrast
MRI coronal T1 precontrast
MRI coronal T1 postcontrast
Bright on FLAIR
Contrast enhancing
- Within IDH mutant astrocytoma, younger age is correlated with improved prognosis and survival (Acta Neuropathol 2018;136:153)
- Extent of resection and the presence or absence of residual tumor post surgery also correlates with survival (Neuro Oncol 2014;16:81)
- Proliferative activity, including mitotic count and Ki67 index, are not clearly correlated with differences in prognosis within grade 2 and grade 3 astrocytoma, IDH mutant (J Neuropathol Exp Neurol 2019;78:1002, Brain Pathol 2020;30:541, Oncotarget 2016;7:21190)
- Astrocytoma, IDH mutant tumors containing any CNS WHO grade 4 features (necrosis, microvascular proliferation or CDKN2A / CDKN2B homozygous deletion) have a significantly worse prognosis than CNS grade 2 or 3 tumors (Acta Neuropathol 2018;136:153)
- Even within histologic CNS WHO grade 4 tumors, the presence of CDKN2A / CDKN2B homozygous deletion may portend a worse prognosis (Neuropathol Appl Neurobiol 2019;45:108)
- 12 year old girl with constitutional mismatch repair deficiency syndrome and concomitant IDH wild type glioblastoma and IDH1 mutant anaplastic astrocytoma (Neuropathol Appl Neurobiol 2018;44:233)
- 28 year old man with widely metastatic IDH1 and TP53 mutant glioblastoma with atypical molecular features (Diagn Pathol 2019;14:16)
- 28 year old woman with symptoms of postpartum depression (J Med Case Rep 2018;12:374)
- 40 year old man with long term daily temozolomide with dose dependent efficacy in MGMT promotor methylation negative, recurrent, high grade astrocytoma (Cancer Chemother Pharmacol 2017;80:1043)
- Complete resection as extensively as is safely possible (Neuro Oncol 2015;17:868)
- Chemotherapy (temozolomide) or radiation therapy
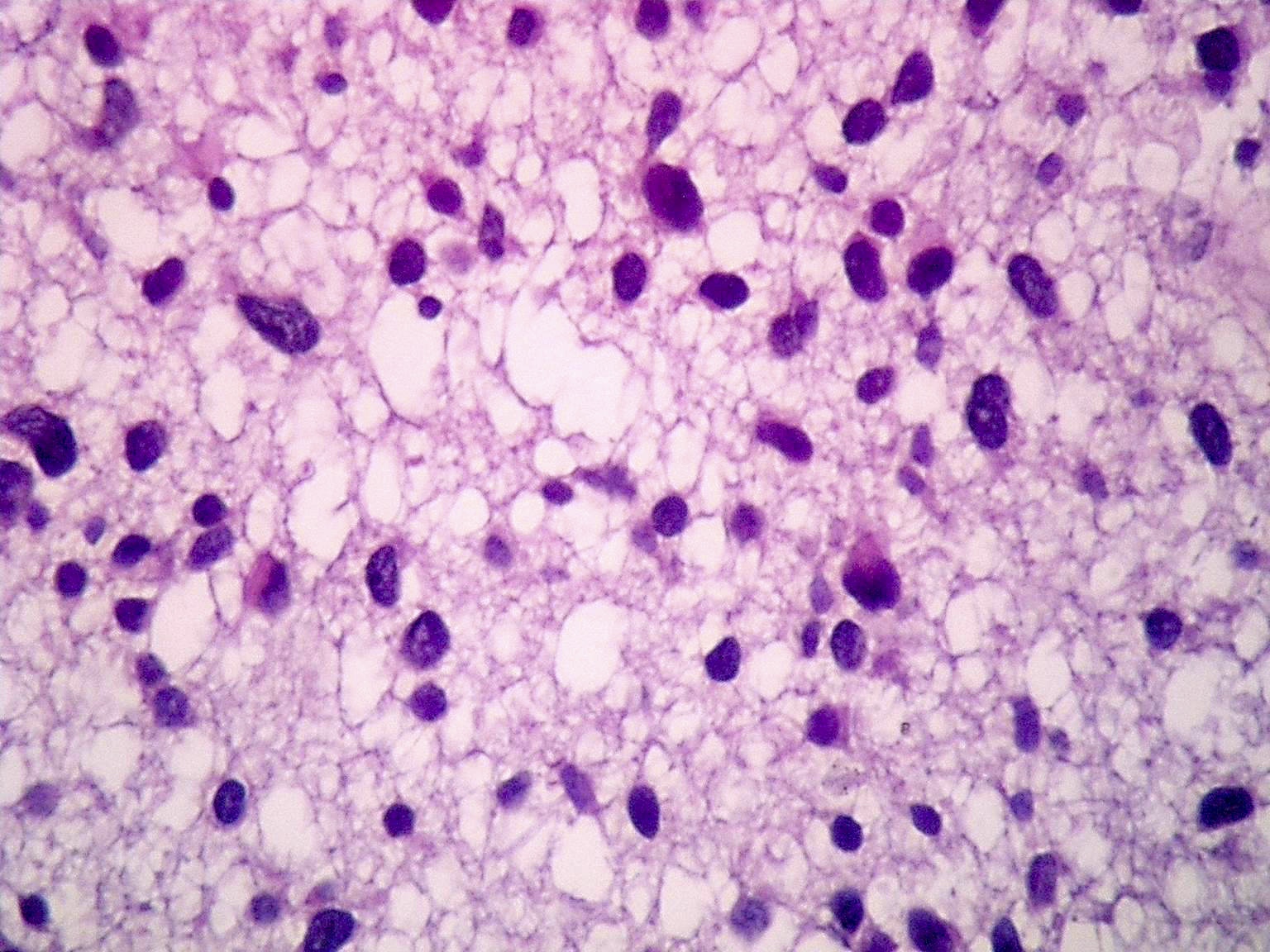
- Ill defined neoplasm with blurring of gray-white junction and expansion of the infiltrated brain areas
- Variable textures: firm, soft, gelatinous, granular, depending on CNS WHO grade (cellularity, necrosis)
- Variable microcystic change imparting a spongy appearance
- Variable calcification with a gritty sensation
- Typically invades surrounding brain without overt tissue destruction
- Expands native structures and can give a mass-like appearance on cut section
- Areas of granularity or softening may be present
- Soft gray-tan tissue with variable yellow-tan necrotic material
- Often fragmented
- Interface of tumor with brain parenchyma is indistinct
Images hosted on other servers:
Glioma with cystic spaces in the cerebral hemisphere
- Smear done at the time of frozen section will show astrocytic appearing tumor cells with oblong irregular nuclei with varying degrees of atypia and glial processes
- High grade nuclear features and mitotic activity may be observed on frozen section but necrosis or microvascular proliferation (features of glioblastoma) should not be present
- Cellular morphology can be highly variable
- Often predominantly tumor cells with oval hyperchromatic nuclei in a fibrillary background
- Variably present, larger cells and pleomorphism
- Variable quantity of cells with eccentric nuclei and glassy eosinophilic cytoplasm (gemistocytes)
- Some show predominantly small cells with little pleomorphism and scant cytoplasm
- Sections are hypercellular showing infiltrating neoplastic cells with edema
- Variably present mitotic figures, necrosis and microvascular proliferation
- Vascular thromboses and myxoid background may be present
- Smear most commonly shows predominantly smaller cells with fine fibrillar processes, elongated nuclei, nuclear atypia and may show mitotic figures

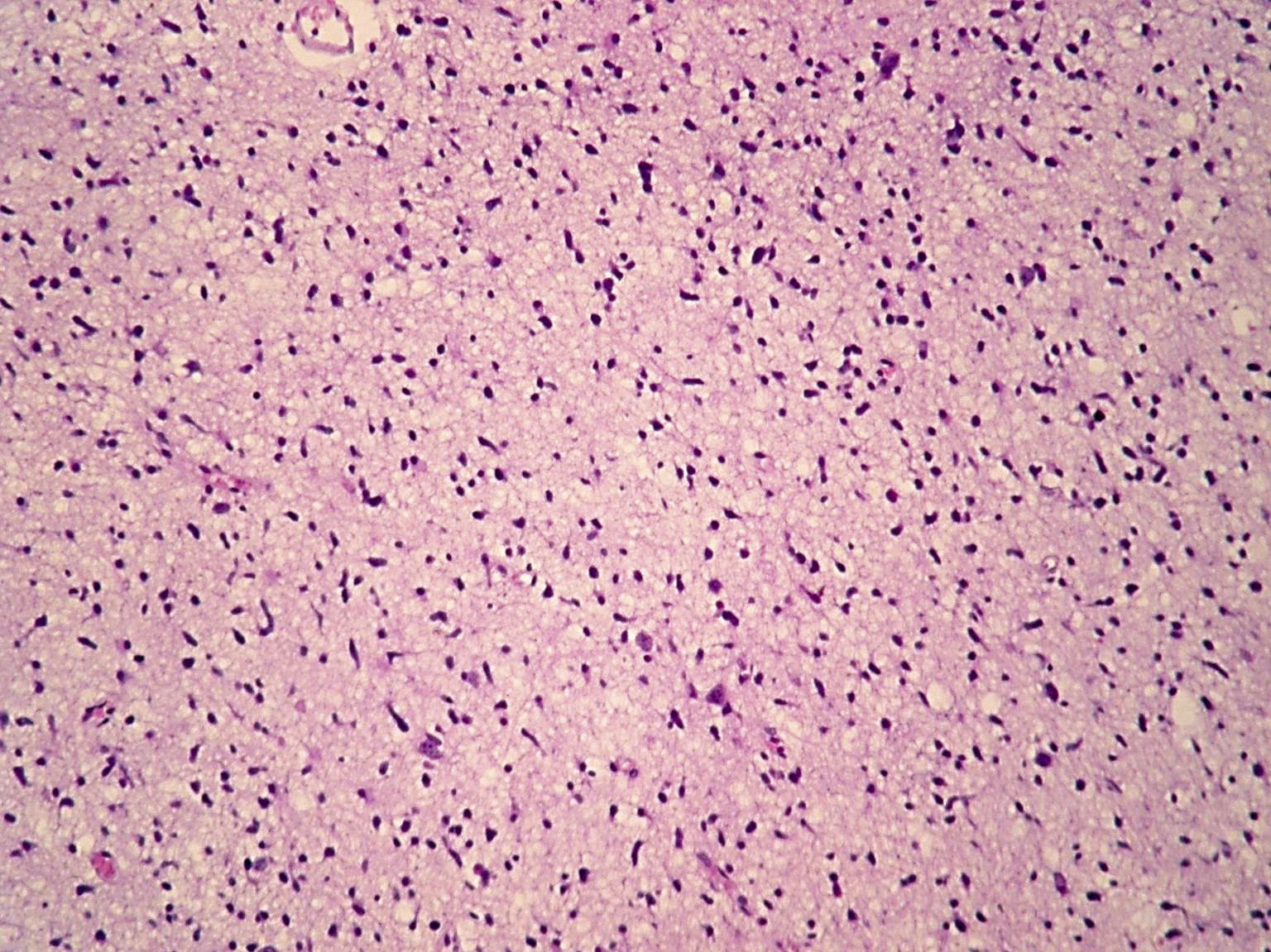
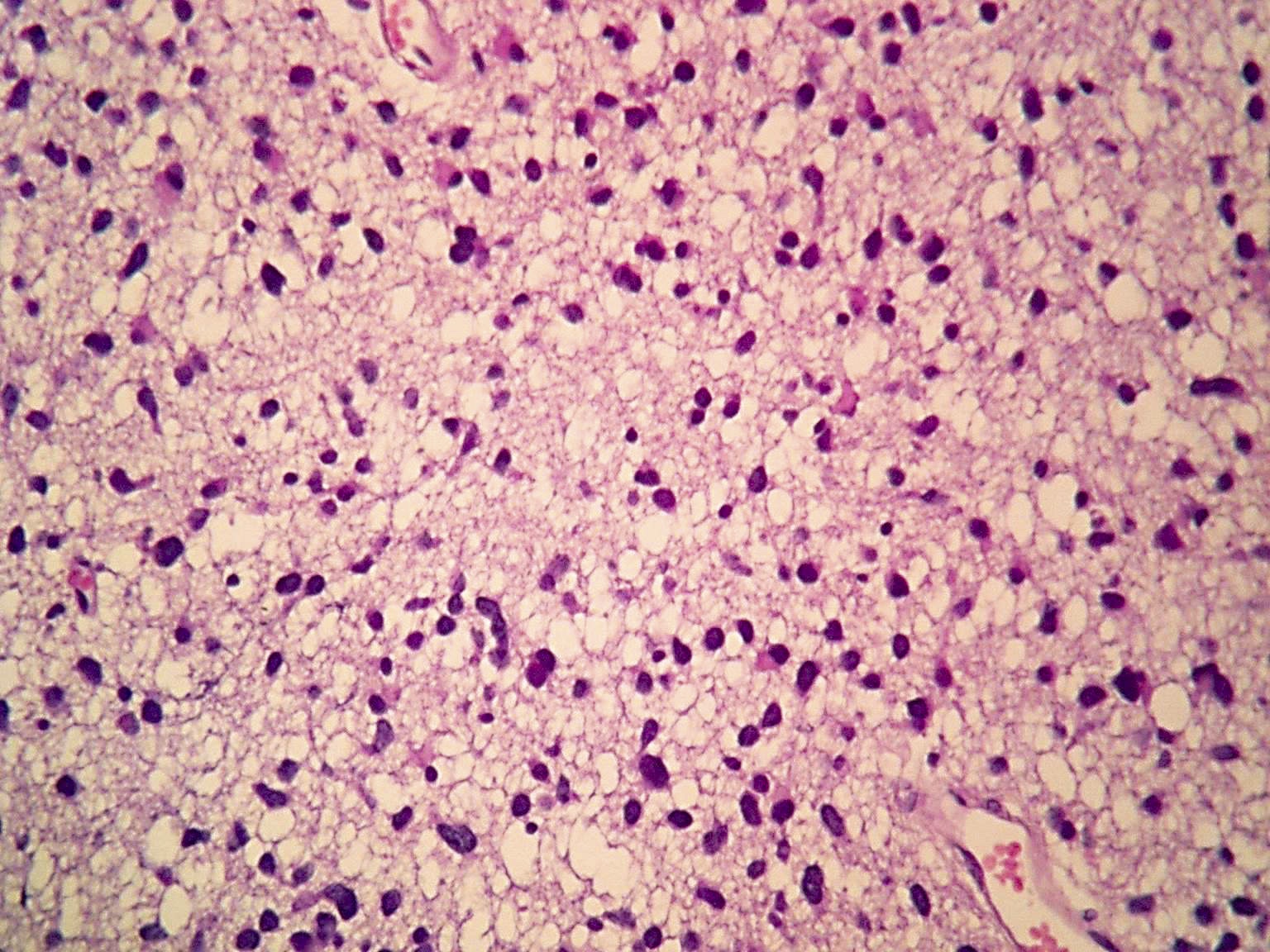
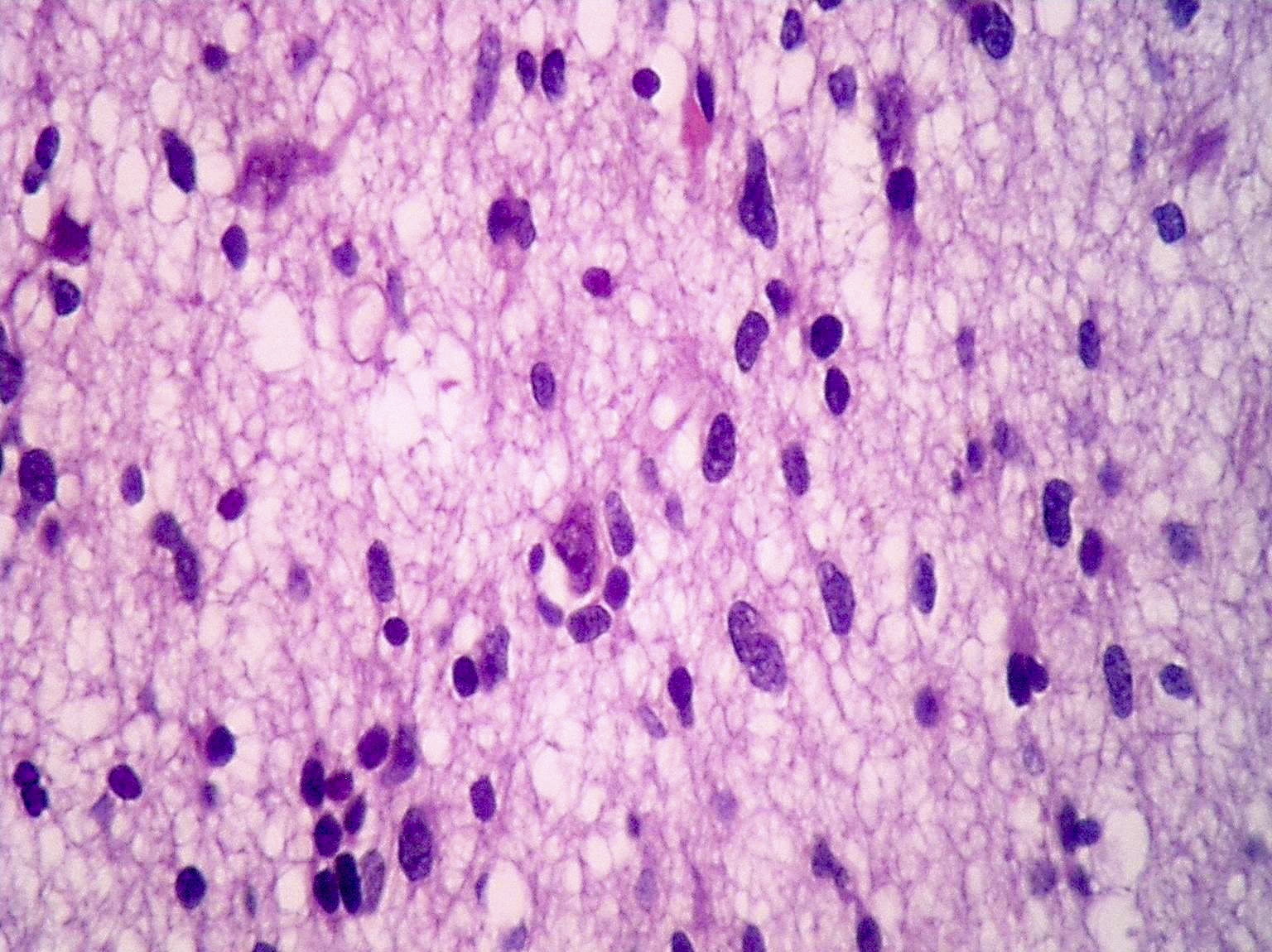
- Diffusely infiltrating tumor cells with oval to elongated astrocytic nuclei and varying appearance of tumor cytoplasm and fibrillar glial processes (Acta Neuropathol 2015;129:789)
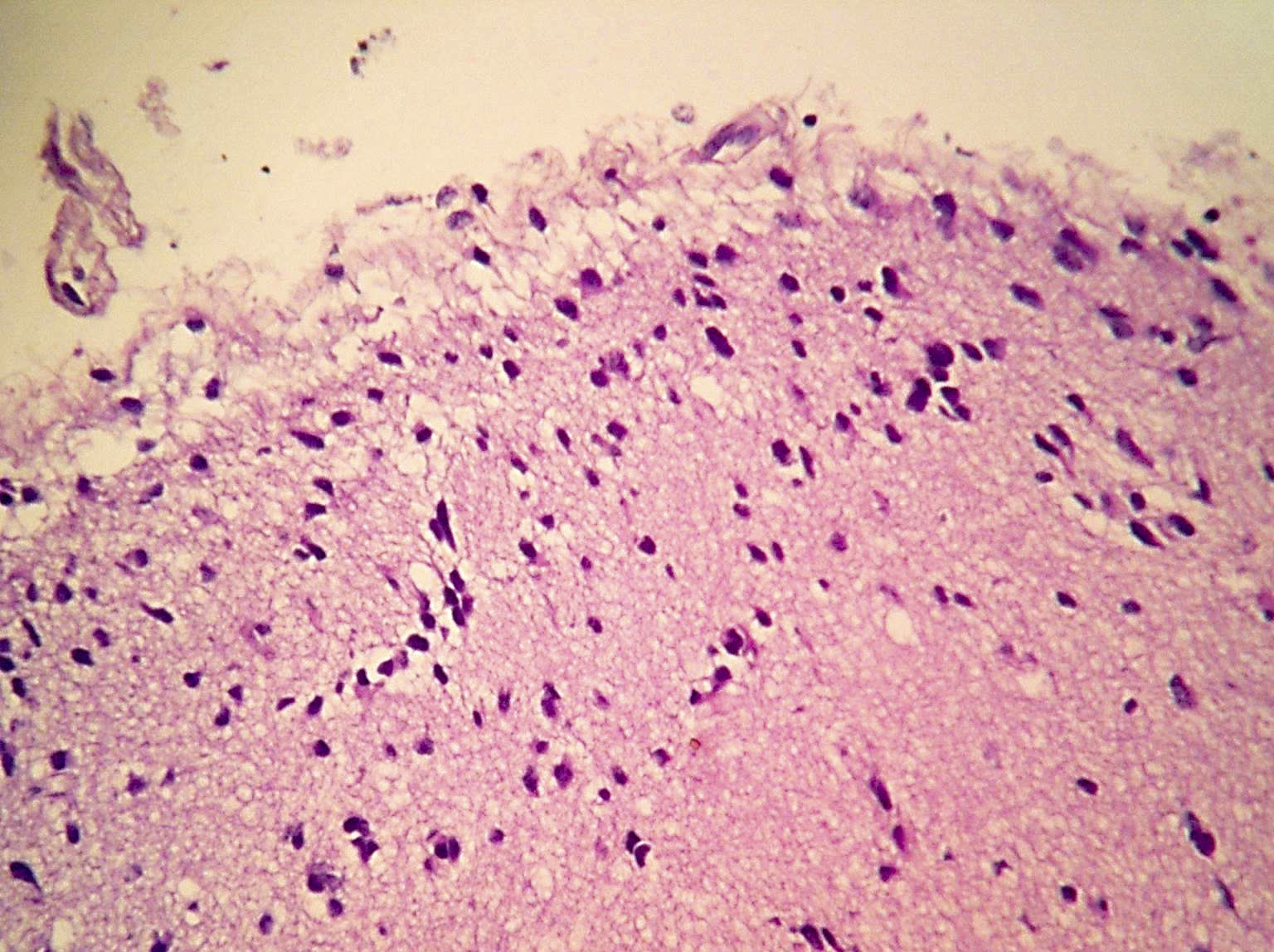
- At the periphery, tumor cells may infiltrate in a diffuse single cell pattern, often with entrapped neurons and axons
- Cellular morphology is variable, even within a single tumor
- Commonly there is a mix of cells with elongated nuclei and fine fibrillar processes, cells with eccentric nuclei and glassy eosinophilic cytoplasm (gemistocytes), larger pleomorphic cells and small cells with scant cytoplasm
- May show oligodendroglioma-like areas
- Myxoid background and microcyst formation may be present
- Variable mitotic activity, cellularity and nuclear atypia depending on CNS WHO grade
- In small biopsy specimens, the presence of 1 mitosis may be sufficient for a CNS WHO grade 3 diagnosis, while the presence of a few mitotic figures in a large resection would not be sufficient for grade 3 designation (Acta Neuropathol 2020;139:603)
- Presence of necrosis or microvascular proliferation would be consistent with a CNS WHO grade 4 designation
Contributed by Eman Abdelzaher, M.D., Ph.D., John DeWitt, M.D., Ph.D. and Meaghan Morris, M.D., Ph.D.
CNS WHO grade 2 astrocytoma, NOS
Subpial accumulation
CNS WHO grade 2 astrocytoma, NOS

Perineuronal satellitosis
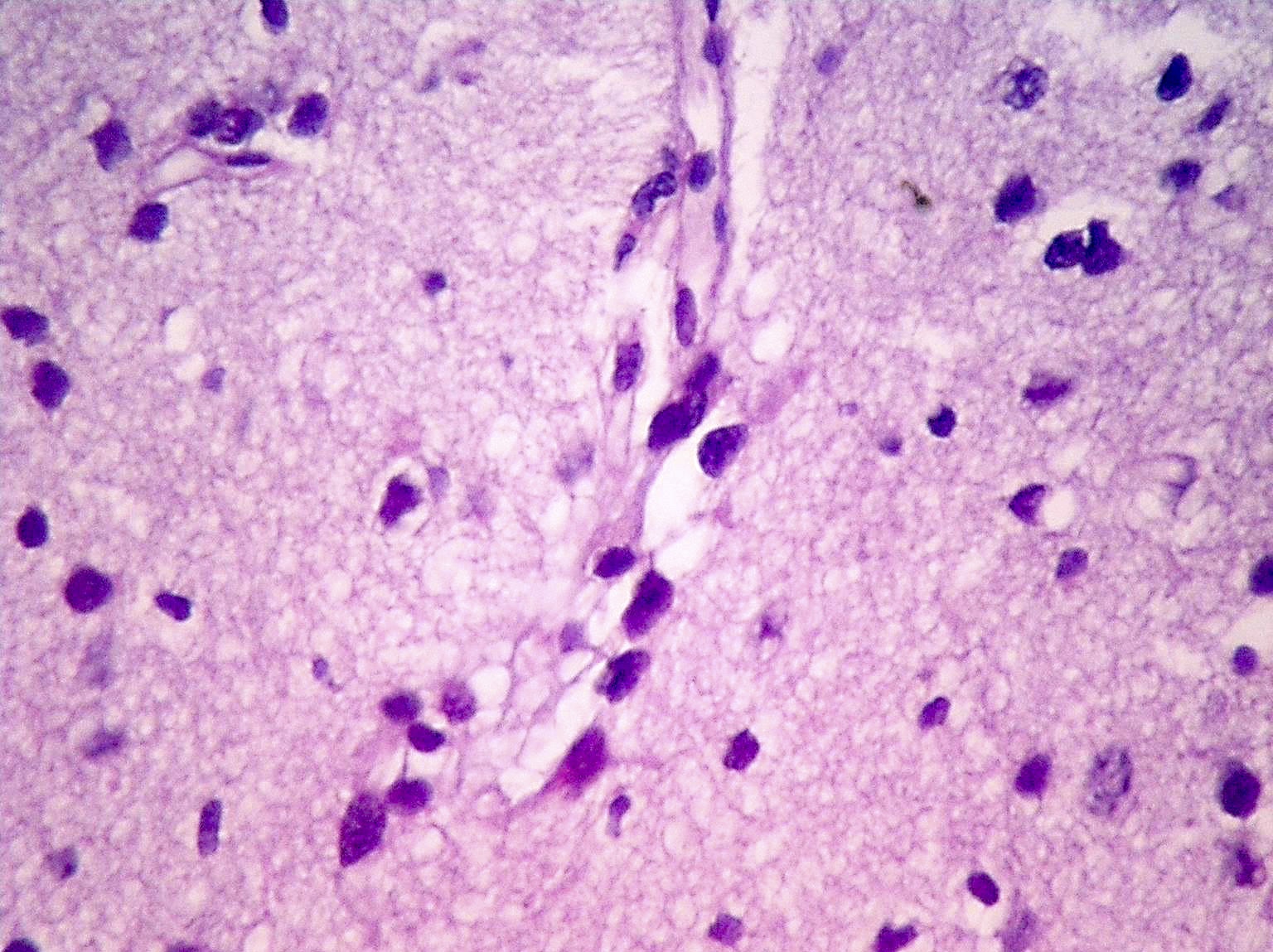
Perivascular accumulation
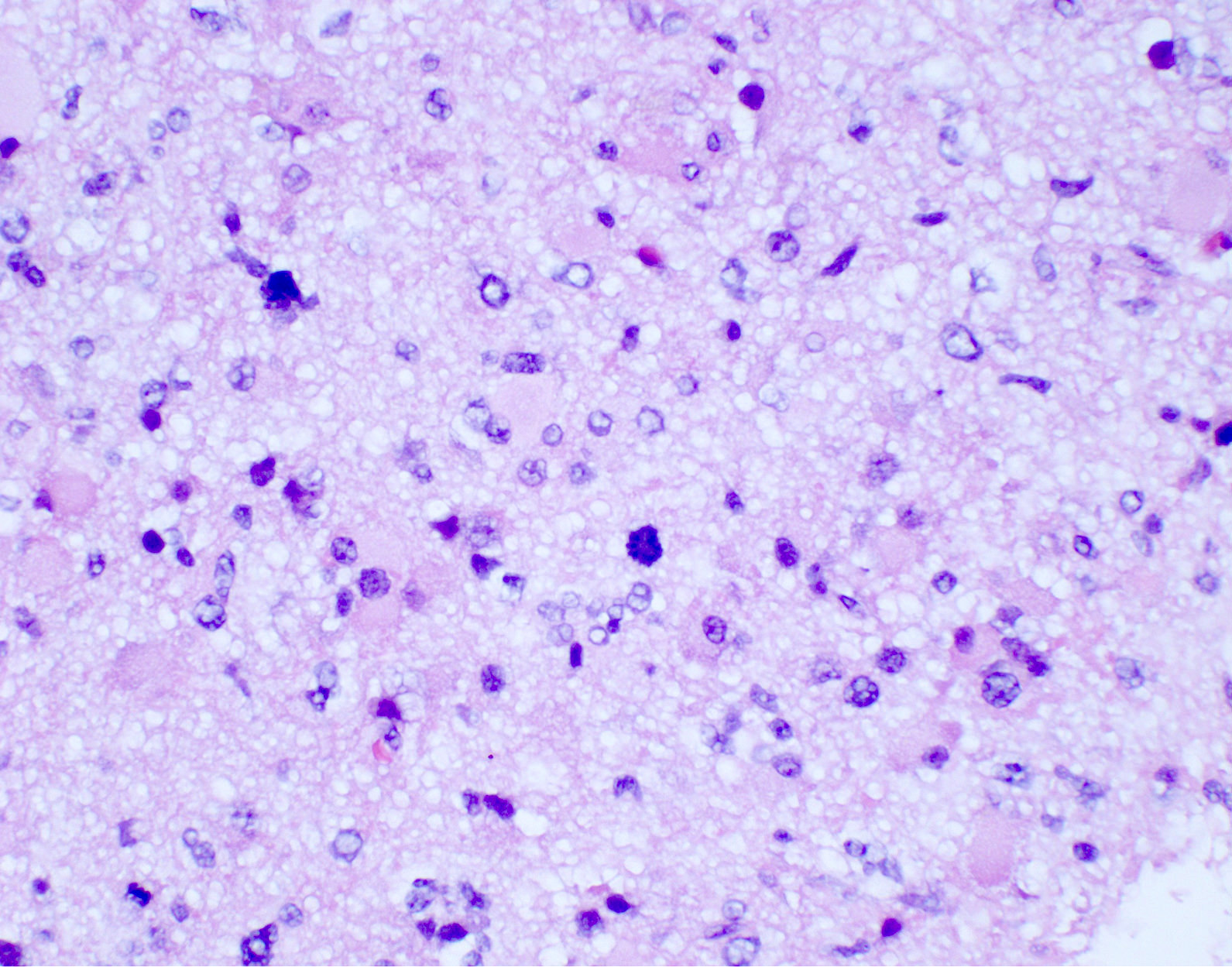
Mitotically active
Pleomorphic hypercellular astrocytic appearance
Gemistocytic histology
Variable morphology and necrosis
Microvascular proliferation

Entrapped neurons

R132H IDH1

ATRX
p53
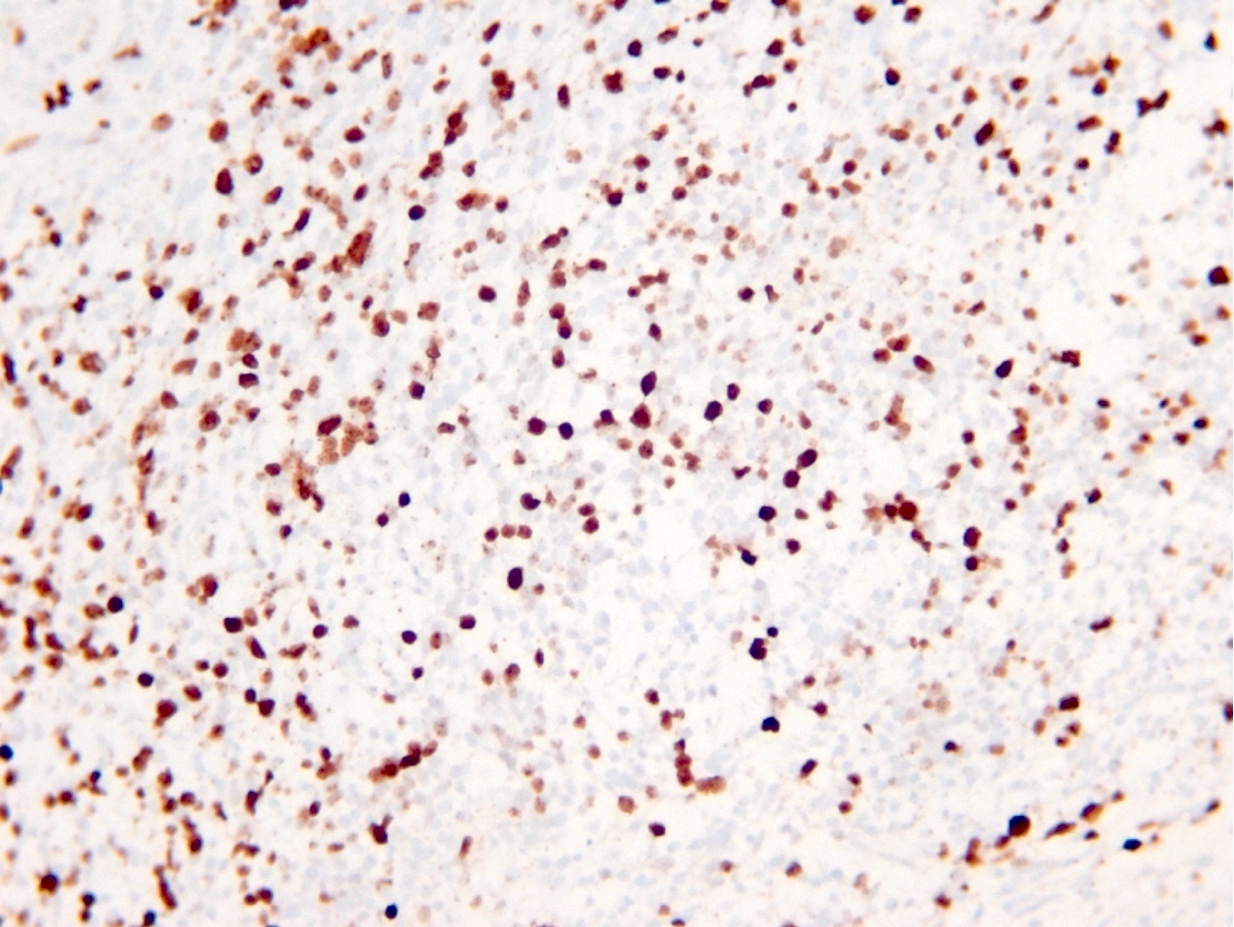
Ki67
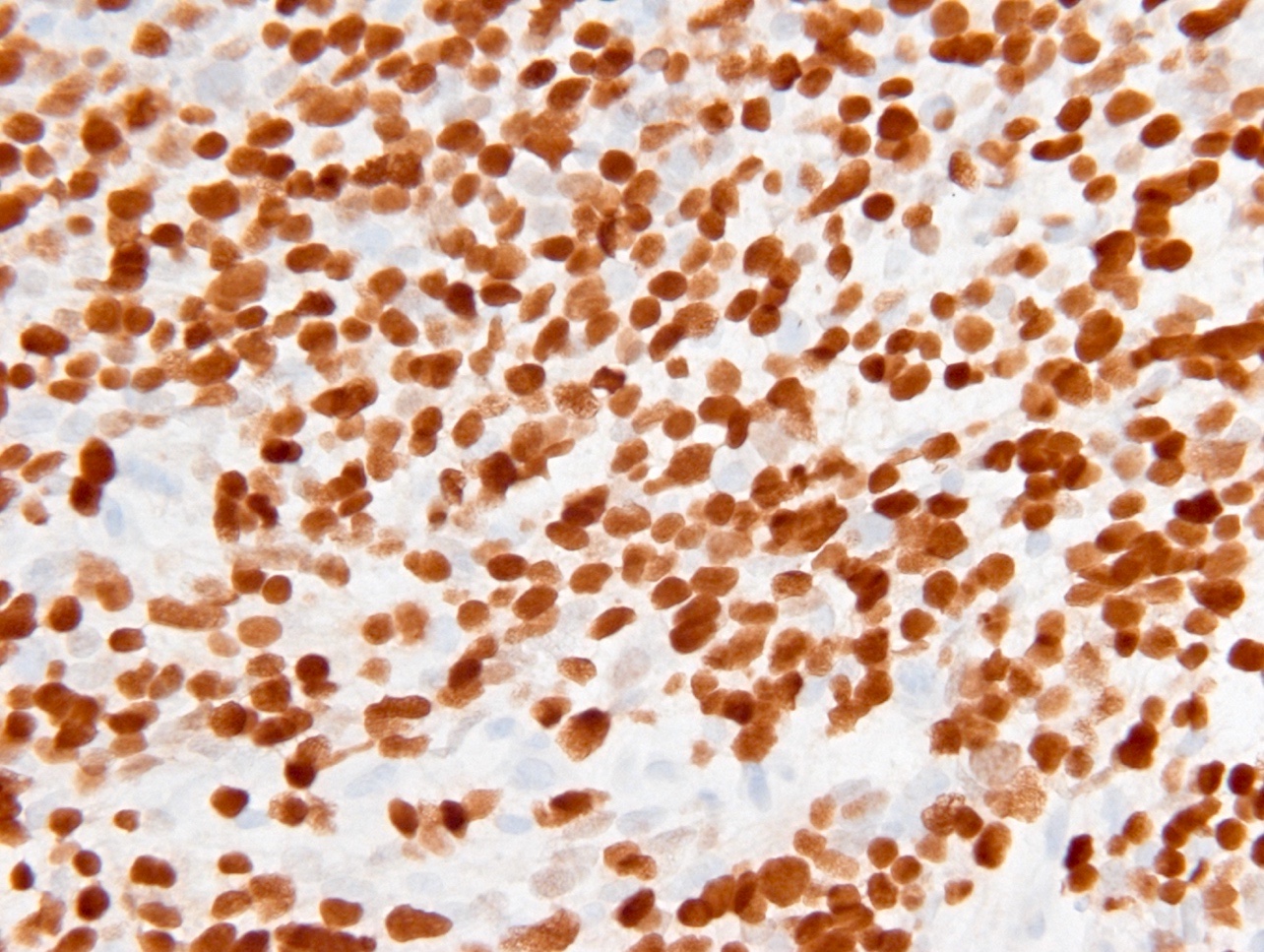
Olig2

IDH

ATRX

p53
Ki67
- GFAP (variable)
- Olig2
- IDH1
- Marker of infiltrating gliomas, both astrocytic or oligodendroglial (Curr Neurol Neurosci Rep 2013;13:345)
- Recognizes only the most common mutation (IDH1 R132H, which accounts for about 90% of all glioma associated IDH mutations) (Neuro Oncol 2014;16:1478)
- In the absence of immunohistochemical evidence for IDH mutation, other IDH1 / IDH2 mutations must be diagnosed by mutational analysis (Neuro Oncol 2014;16:1478)
- Rare in children < 14 years (Childs Nerv Syst 2011;27:87)
- p53
- Ki67: increasing proliferation index with increasing grade
- Neurofilament (highlights entrapped axons)
- ATRX
- Loss of nuclear ATRX is typical of diffuse astrocytomas, not oligodendrogliomas or reactive gliosis (Front Oncol 2017;7:236)
- Strong nuclear expression in nonneoplastic vasculature and cells serves as an internal control
- Keratins (although cocktails may show cross reactivity)
- CAM5.2, CD45, CD20, MelanA
- IDH1 or IDH2 mutation necessary for the diagnosis, with R132H IDH1 variant seen in > 90% of cases (Acta Neuropathol 2009;118:469)
- Tumors negative for R132H IDH1 immunohistochemistry require IDH1 / IDH2 sequencing to determine IDH status (Neuro Oncol 2017;19:1640)
- p53 mutation and ATRX promoter mutation nearly always present (typically absent in oligodendroglioma, IDH mutant and 1p19q codeleted) (N Engl J Med 2015;372:2481)
- Gain of chromosome 7
- Usually lack TERT promoter mutations
- Presence of CDKN2A / CDKN2B homozygous deletion portends a worse prognosis and is sufficient for a CNS WHO grade 4 designation regardless of histologic features present (Acta Neuropathol 2018;136:153)
- Brain, frontal lobe, biopsy:
- Integrated diagnosis: astrocytoma, IDH mutant, CNS WHO grade 4
- Histological diagnosis: astrocytoma with elevated proliferative activity and necrosis
- WHO histological grade: 4
- Molecular information:
- IDH1: mutant (R132H immunohistochemistry)
- ATRX: nuclear expression lost (consistent with mutant)
- p53: many positive cells (immunohistochemistry; consistent with mutant)
- Normal brain:
- Demyelinating disease:
- Pilocytic astrocytoma:
- Circumscribed and contrast enhancing, histologically compact biphasic architecture (alternating piloid and spongy areas)
- IDH1 negative
- Reactive gliosis:
- Evenly distributed hypertrophic astrocytes, IDH1 negative
- Glioblastoma, IDH wild type:
- High grade infiltrative glial neoplasm with astrocytic differentiation, nuclear atypia, pleomorphism, elevated mitotic activity and necrosis or microvascular proliferation
- IDH mutation is not present by immunohistochemistry or sequencing
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted:
- Infiltrating glioma composed of round cells resembling oligodendrocytes, hyperchromatic rounded nuclei, perinuclear halos, fine branching vasculature, scattered calcifications
- Grade 3 tumors may have increased mitotic activity, variable microvascular proliferation and variable necrosis
- IDH1 / IDH2 mutation present by immunohistochemistry or sequencing and whole arm codeletion of chromosomes 1p and 19q must be present by molecular testing
- ATRX alterations and TP53 mutations are typically absent (Acta Neuropathol 2012;124:615, N Engl J Med 2009;360:765)
- Lymphoma:
- Parenchymal lymphomas in the central nervous system are typically diffuse large B cell lymphomas, which lack the fine fibrillar cell processes typical of glial cells
- Diffuse large B cell lymphoma in the central nervous system often shows a perivascular tumor distribution
- Positive for CD45 and CD20 but negative for GFAP and Olig2 by immunohistochemistry
- Metastatic disease:
- Codeletion of chromosomes 1p and 19q is a characteristic molecular alteration
- IDH mutant astrocytomas have a significantly better prognosis than IDH wild type tumors
- IDH protein expression is detected in astrocytic tumors only
- Malignant progression to higher grades does not occur
- Retained nuclear ATRX is typical of IDH mutant astrocytomas
Comment Here
Reference: Astrocytoma, IDH mutant

Which of the following is true about the entity in the figure above presenting in a 50 year old man?
- If R132H IDH1 immunohistochemistry is negative, no further testing is necessary
- If R132H IDH1 immunohistochemistry is positive, ATRX staining of tumor cells is expected to be lost
- If R132H IDH1 immunohistochemistry is positive, ATRX staining of tumor cells is expected to be retained
- If R132H IDH1 immunohistochemistry is positive, patient survival is similar to glioblastoma, IDH wild type
- If R132H IDH1 immunohistochemistry is positive, p53 staining of tumor cells is expected to be weak and scattered
Comment Here
Reference: Astrocytoma, IDH mutant
- CDKN2A / CDKN2B homozygous deletion
- Microvascular proliferation
- Mitoses and pleomorphism in the absence of necrosis, microvascular proliferation or CDKN2A / CDKN2B homozygous deletion
- Necrosis
- Necrosis, microvascular proliferation or CDKN2A / CDKN2B homozygous deletion
Comment Here
Reference: Astrocytoma, IDH mutant
- A meningioma of intermediate aggressiveness between benign and malignant forms, comprising 5 - 15% of meningiomas
- WHO grade 2
- Diagnostic criteria: fulfilling either 1 of 2 major criteria or 3 of 5 minor criteria
- Major criteria:
- 4 - 19 mitotic figures/10 high power fields
- Brain invasion
- Minor criteria:
- Increased cellularity
- Small cells with high N/C ratio
- Large and prominent nucleoli
- Patternless or sheet-like growth (loss of lobular architecture)
- Foci of spontaneous or geographic necrosis
- Major criteria:
- Invasion of dura, bone or soft tissue does not affect grading
- Pleomorphic or atypical nuclei do not affect grade
- Ki67 is not a true diagnostic criteria; however, it is usually greater than 4% and up to 20%
- Atypical meningiomas have an intermediate recurrence rate between benign and malignant meningiomas
- 29 - 52% recur (versus 7 - 25% of classic meningiomas and 50 - 94% of anaplastic meningiomas) (Louis: WHO Classification of Tumours of the Central Nervous System, 4th Edition, 2016)
- Molecular genetic and epigenetic signatures of atypical meningiomas are becoming increasingly important in predicting prognosis and new targeted therapy in progressive tumors
- ICD-10: D32.9 - benign neoplasm of meninges, unspecified
- Similar to meningioma, overall
- Intracranial, intraspinal or intraorbital
- Arising from the meningothelial cells or the arachnoid layer
- Risk factors include male gender and prior surgery (Louis: WHO Classification of Tumours of the Central Nervous System, 4th Edition, 2016)
- Some risk factors may be similar to benign meningioma:
- Ionizing radiation (Acta Neuropathol 2017;134:155)
- Hormone replacement therapy or oral contraceptives (J Clin Oncol 2008;26:279, J Neurosurg 2013;118:649)
- Germline mutations in NF2 or SMARCB1 predispose to multiple meningiomas (Neurogenetics 2012;13:1, J Med Genet 2011;48:93)
- Clinical presentation of atypical and anaplastic meningioma is similar to their benign counterpart
- Common symptoms include headaches, seizures and focal neurological deficit due to tumor compression (Neurosurg Clin N Am 2016;27:239)
- Diagnose by imaging and pathology of biopsy / resection specimen
- Extra-axial mass with dural tail
- Uniformly contrast enhancing
- Extensive peritumoral edema is associated with brain invasion (Neuro Oncol 2020 Aug 13 [Epub ahead of print])
- Several benign meningioma variants, including angiomatous, microcystic, secretory and lymphoplasmacyterich meningiomas may also have prominent peritumoral edema (J Neurooncol 2013;111:49)
- Extent of surgery and WHO grading
- DNA methylation profiling may better predict tumor recurrence and prognosis than histologic classification (Lancet Oncol 2017;18:682)
- 36 and 70 year old women with optic nerve seeding of atypical meningiomas presenting with subacute visual loss (J Neurosurg 2013;119:494)
- 44 year old man with atypical primary meningioma in the nasal septum with malignant transformation and distant metastasis (BMC Cancer 2012;12:275)
- Elderly man with metastatic atypical meningioma (J Clin Neurosci 2000;7:69)
- Gross total resection
- Postsurgical radiation is often offered for atypical meningiomas, especially after a subtotal resection (J Neurooncol 2013;115:241)
- Stereotactic radiosurgery
- Rubbery, well circumscribed mass firmly attached to the inner surface of the dura
- Brain invasive meningiomas readily adherent to adjacent brain tissue
- Reference: Perry: Practical Surgical Neuropathology - A Diagnostic Approach, 1st Edition, 2010
- Similar to meningioma
- May have histology of any grade 1 variant meningioma with increased mitoses (4 - 19/10 high power fields)
- Mitotic rate is defined as the highest count over 10 consecutive high power fields (1 high power field = 0.16 mm²)
- May have increased cellularity or areas of small cell collections
- May have sheet-like growth pattern
- May have areas of spontaneous necrosis
- May have macronucleoli
- Brain invasion is defined as irregular projections of tumor cells into adjacent CNS parenchyma without an intervening layer of leptomeninges at the tumor to brain interphase (Am J Surg Pathol 1997;21:1455)
Contributed by Chunyu "Hunter" Cai, M.D., Ph.D.
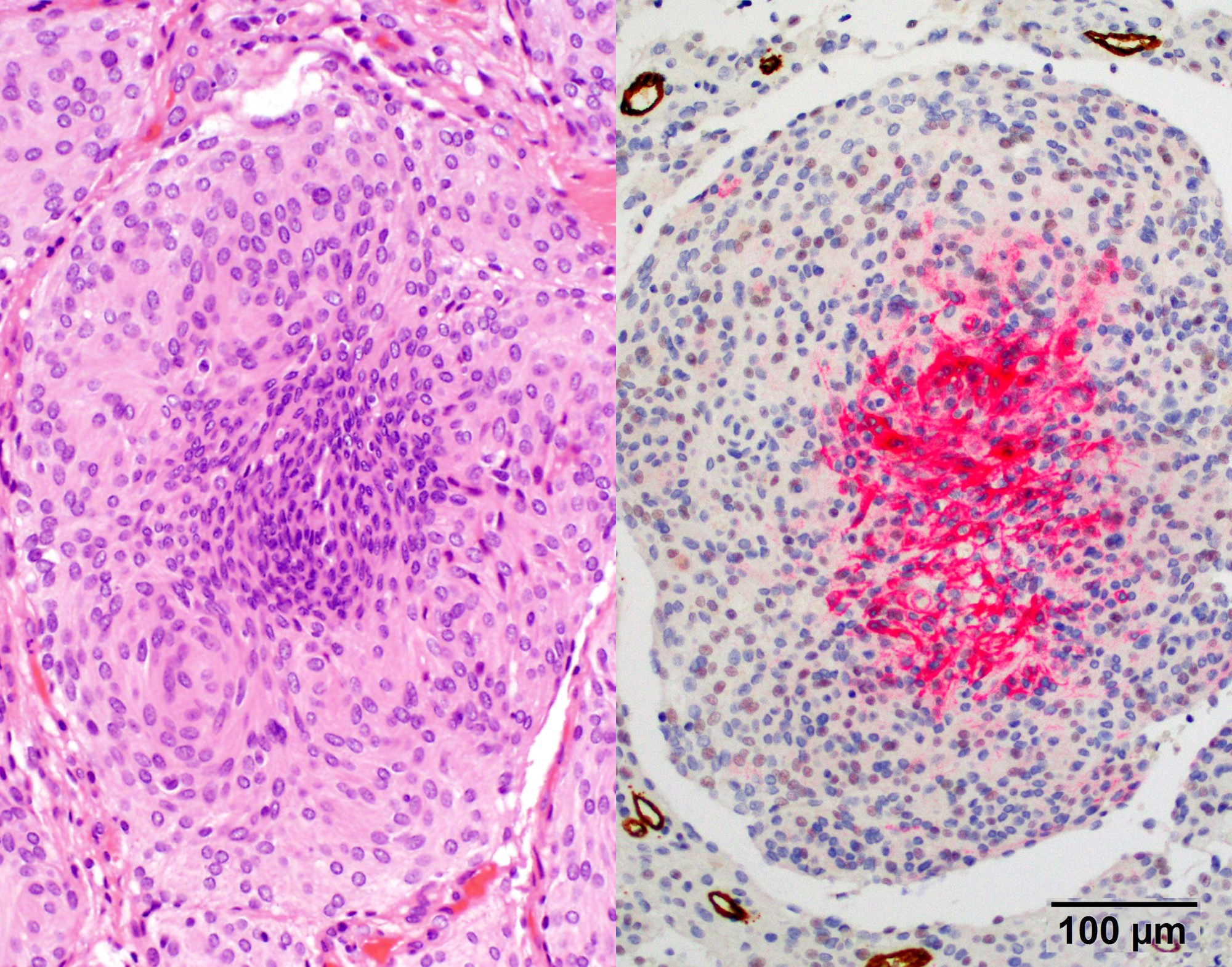
Small cell change - meningothelial
Small cell change - transitional
Spontaneous necrosis
Macronuclei
Sheeting
Brain invasion - nest
Brain invasion - protrusions
- Squash prep shows similar histology as standard meningioma but may also show occasional mitoses or macronucleoli
- GFAP (useful in highlighting tumor brain interphase in cases with brain invasion)
- STAT6 (useful to distinguish meningioma [negative] from solitary fibrous tumor / hemangiopericytoma [positive]) (Clin Neuropathol 2017;36:56)
- Majority of atypical meningiomas have loss of NF2 combined with either genome instability (large scale chromosomal alterations) or loss of SMARCB1 (Nat Commun 2018;9:16215)
- Recurrent losses of chromosome 1p, 6q, 14q,18q and gain of 1q are indicators of poor prognosis (Acta Neuropathol 2017;133:431)
- Non-NF2 meningiomas are enriched in mutations in TRAF2, KLF4, AKT1 and SMO, most of which are benign and preferentially locate in skull base (Science 2013;339:1077)
- DNA methylation profiling of meningioma distinguished 6 methylation classes (MCs), benign (ben) 1 - 3, intermediate (int) A and B and malignant (mal)
- DNA methylation based meningioma classification is reported to better predict tumor recurrence and prognosis than the WHO histological classification (Lancet Oncol 2017;18:682)
- NF2 mutant atypical meningiomas display increased H3K27me signal and a hypermethylated phenotype due to increased polycomb repressive complex 2 (PCR2) / EZH2 activity (Nat Commun 2018;9:16215)
- Brain, right frontal lobe mass, excision:
- Atypical meningioma (see comment)
- Comment: Section shows a meningioma with predominant meningothelial morphology and rare psammoma bodies. Multiple atypical features are present, including variably increased mitotic index up to 7 mitoses/10HPF (A7), multifocal microscopic necrosis, widespread small cell change, hypercellularity, and sheeted architecture. No macronucleoli or brain invasion is identified. Ki67 proliferation index is 12.7% per 1,000 nuclei count.
- Hemangiopericytoma:
- Anaplastic meningioma:
- With mitoses greater than 20/10 high power fields
- Meningioma:
- With atypical features insufficient for criteria above
- Necrosis:
- Due to prior radiation therapy or embolization (which are not considered spontaneous)
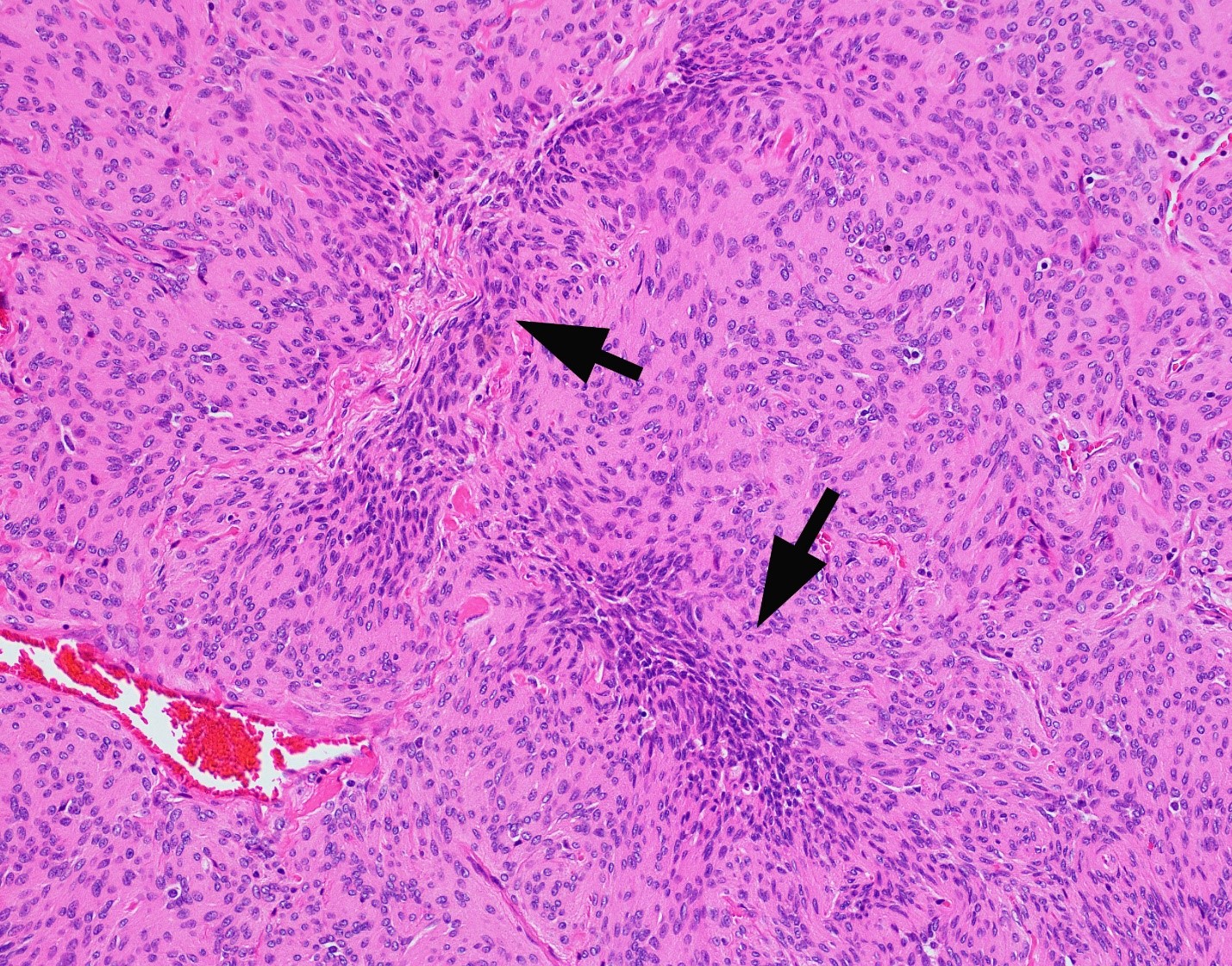
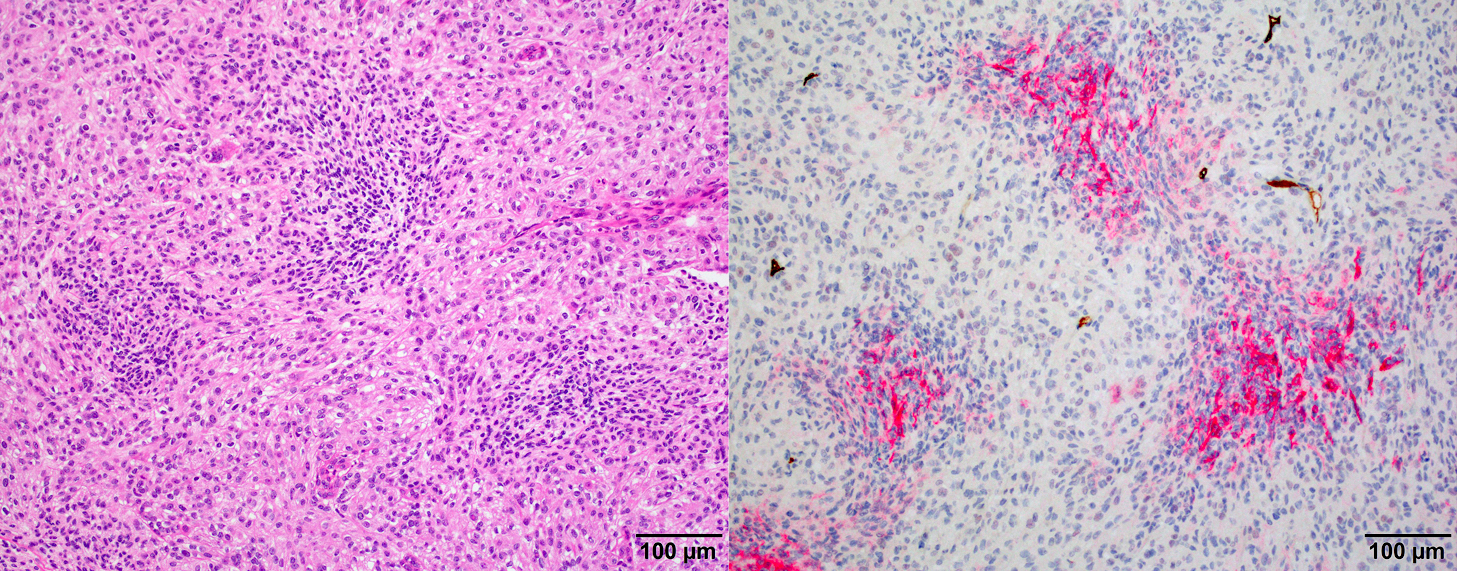
The arrows in the above image show which of the following?
- Blood vessels
- Lymphoplasmacytic inflammation
- Pseudopalisading necrosis
- Small cell change
The image shows an atypical meningioma with small cell change, characterized by reduced cytoplasm and increased N/C ratio in these regions. These regions may resemble lymphoplasmacytic inflammation on low power but on high power show nuclei that are similar to adjacent tumor cells.
Comment Here
Reference: Atypical meningioma
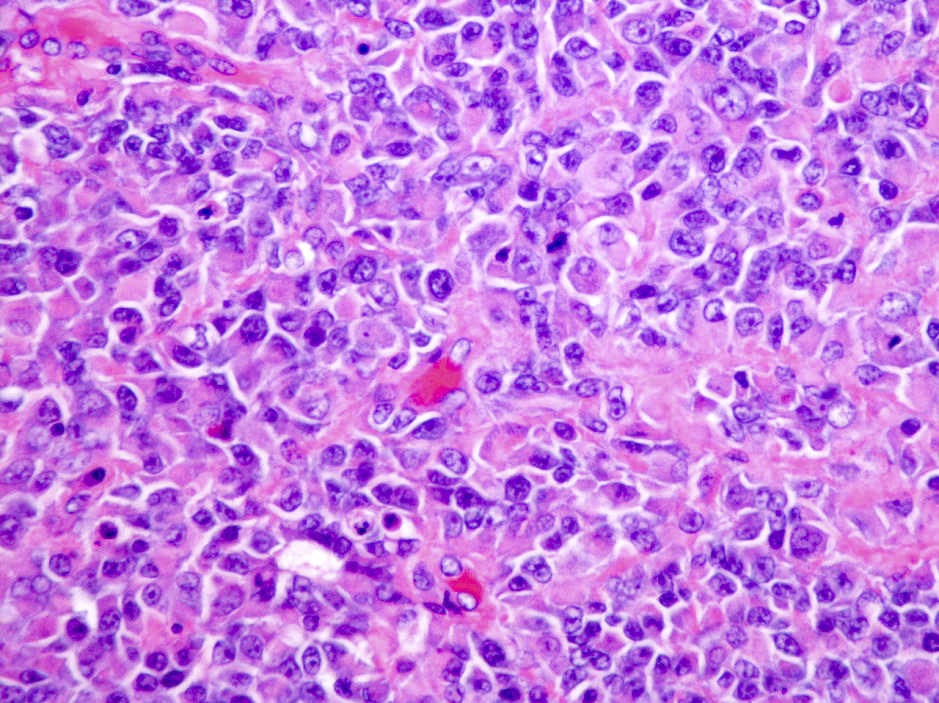
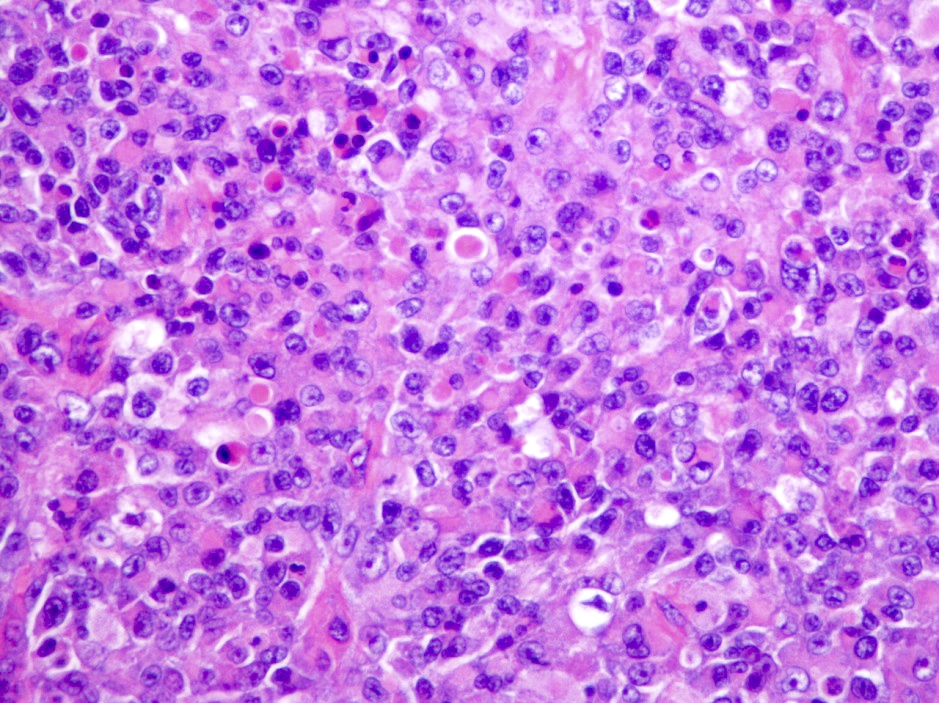
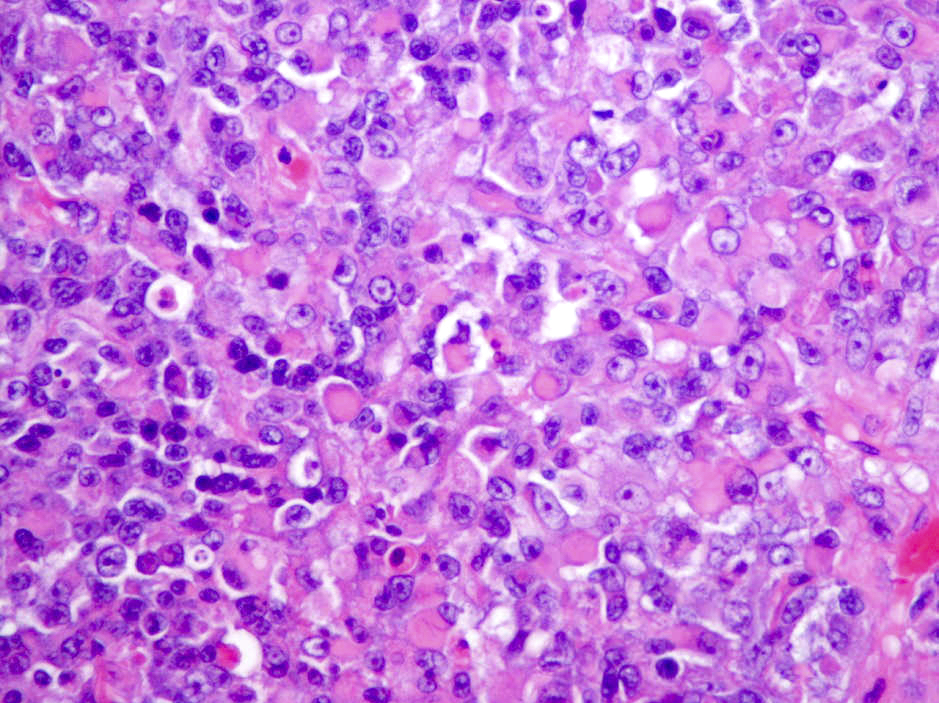
- High grade malignant CNS embryonal tumor composed of poorly differentiated cells with a variable number of rhabdoid cells
- Occurs predominantly in young children; diagnosis is based on demonstrating loss of SMARCB1 (INI1) or SMARCA4 (BRG1)
- Tumors with similar morphology but that lack characteristic molecular findings are classified as CNS embryonal tumors with rhabdoid features
- CNS embryonal tumor with a polyimmunophenotype and loss of nuclear SMARCB1 or SMARCA4 expression in tumor cells are required for the diagnosis of atypical teratoid / rhabdoid tumor (AT / RT)
- Tumors with similar morphology and immunophenotype but that lack a classifiable mutation are classified as CNS embryonal tumors (not elsewhere classified [NEC]) or not susceptible to further analysis (not otherwise specified [NOS])
- ICD-O: 9508/3 - atypical teratoid / rhabdoid tumor
- ICD-11: 2A00.1Y & XH7ZQ4 - other specified embryonal tumors of brain & atypical teratoid / rhabdoid tumor
- Accounts for 1 - 2% of all pediatric brain tumors and is very rare in adults (Front Oncol 2018;8:567)
- Age typically < 3 years; rare in children aged > 6 years (Neuro Oncol 2014;16:1392)
- Can occur in cerebral hemispheres, cerebellum or rarely in the spinal cord
- Cerebral hemispheres, cerebellar hemispheres, cerebellopontine angle, brainstem, spinal cord
- In adults, the most common sites are the cerebral hemispheres and sellar region
- Mutation or loss of the SMARCB1 locus at 22q11.2 is classic for this tumor (Nature 1998;394:203)
- Tumors with retained SMARCB1 can have biallelic inactivation and no expression of the SMARCA4 protein (Am J Hum Genet 2010;86:279)
- Function of SMARCB1 and SMARCA4 and their role in malignant transformation are unclear
- Familial cases in the setting of rhabdoid tumor predisposition syndrome 1 (SMARCB1 gene) or 2 (SMARCA4 gene) (Acta Neuropathol 2014;128:453)
- De novo germline mutations have accounted for 66% of germline mutations (Pediatr Blood Cancer 2011;56:7)
Images hosted on other servers:

Features of AT / RT subtypes
- Depends on age and location
- In infants, lethargy, vomiting and failure to thrive are common symptoms (J Neurosurg 1996;85:56)
- If > 3 years, headache and hemiplegia are reported (Childs Nerv Syst 2009;25:707)
- Cranial nerve palsy (mostly sixth and seventh nerve paresis) may also be present
- Adult sellar AT / RT often presents with headaches, visual impairment and endocrine disturbances (Mod Pathol 2022;35:1910)
- Diagnosis of AT / RT can be made on biopsy or cytology but staging requires cerebrospinal fluid (CSF) cytology (Radiology 1969;93:1351)
- No specific laboratory findings for this entity
- Isodense to hyperintense signal intensity on fluid attenuated inversion recovery (FLAIR) images with restricted diffusion
Contributed by Chunyu Cai, M.D., Ph.D. (Case #502)

MRI T1 postsagittal
Images hosted on other servers:
AT / RT in a 9 month old
Axial CT head, unenhanced
MRI brain, unenhanced and enhanced
- Poor; 5 year progression free and overall survival rate is only 60%, even for the favorable group
- Clinical staging (Radiology 1969;93:1351)
- 2 month old boy presented with rapidly increasing head circumference (J Pediatr Neurosci 2015;10:382)
- 2 year old boy presented with lethargy and vomiting (Asian J Neurosurg 2018;13:873)
- 23 year old man with an extra-axial dural based lesion (CNS Oncol 2020;9:CNS54)
- 55 year old woman presented with subarachnoid hemorrhage (Surg Neurol Int 2019;10:139)
- Surgery, high dose chemotherapy with radiation (Pediatr Blood Cancer 2017;64:e26663)
- Tan-pink to red soft tissue, appears demarcated from adjacent parenchyma
- Tumors with more mesenchymal tissue appear firm, tan-white
- Hemorrhage and necrosis can be seen (Autops Case Rep 2020;10:e2020205)
Images hosted on other servers:
Posterior fossa, cerebellum and brain stem tumors
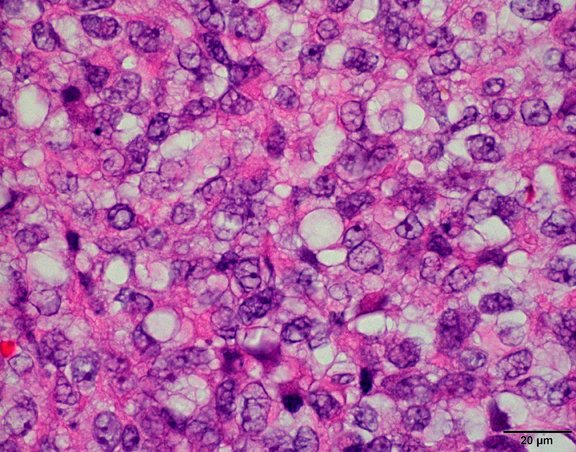
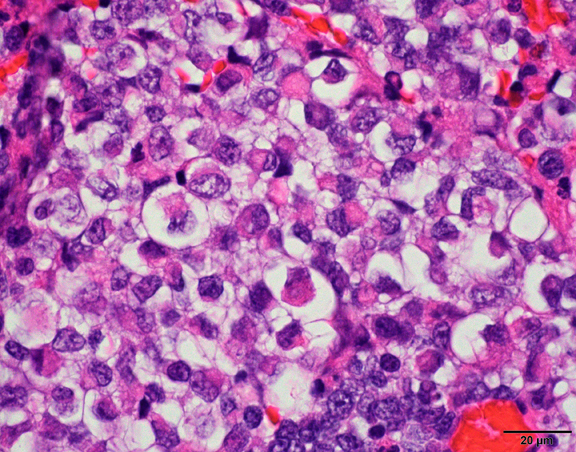
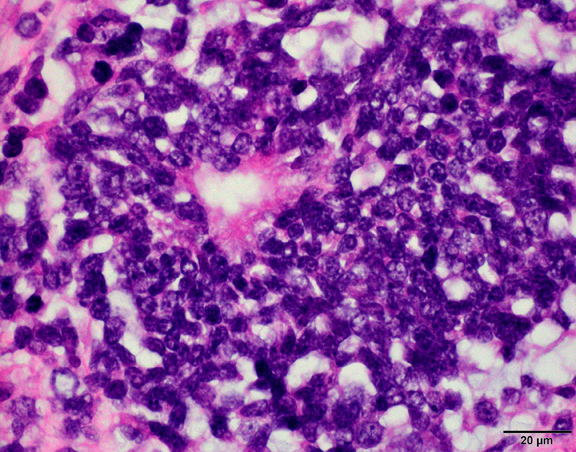
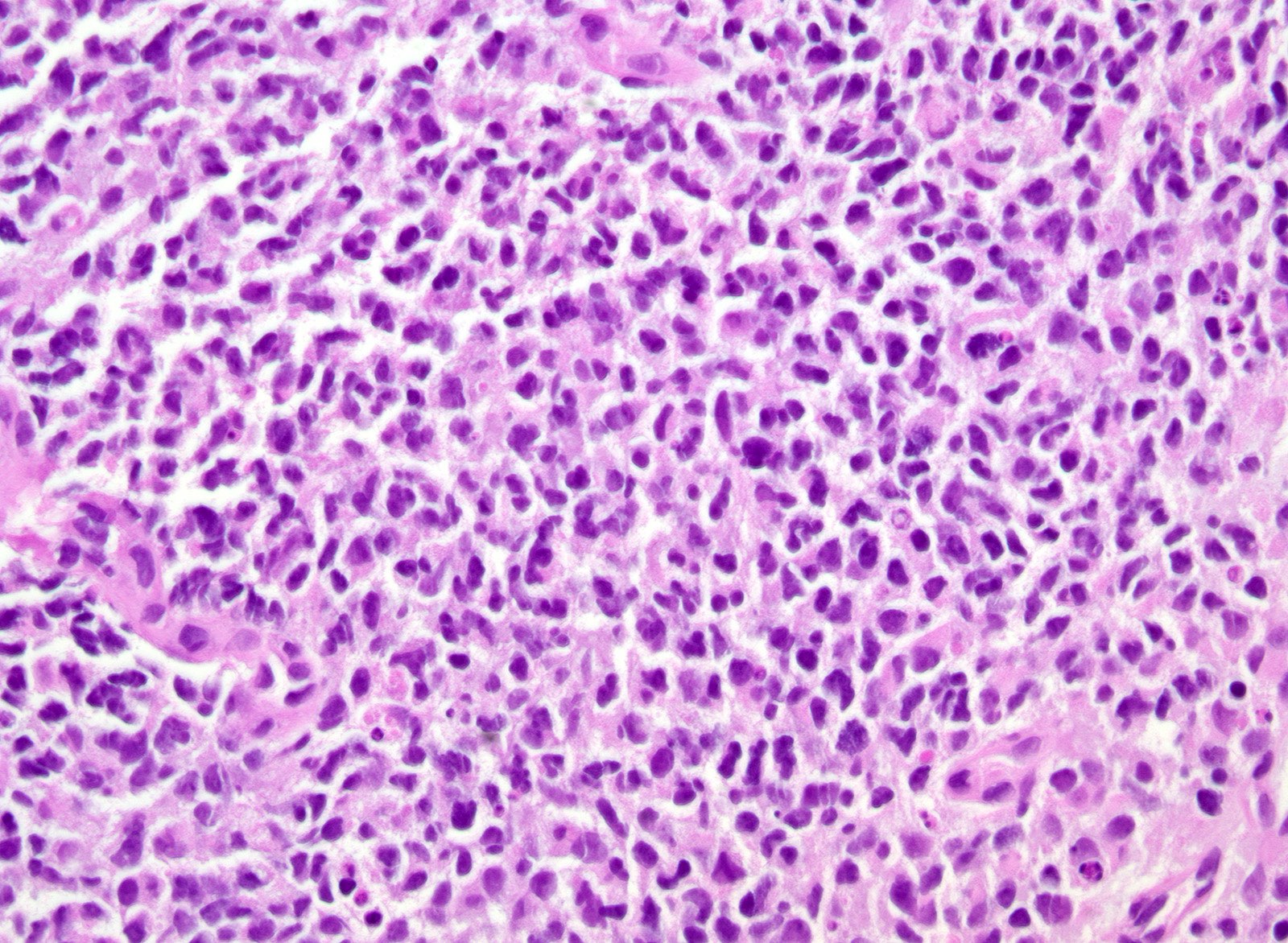
- Sheets of eosinophilic, embryonal cells with round to oval hyperchromatic nuclei and minimal cytoplasm
- Rhabdoid cells are larger cells with eccentrically located nuclei and eosinophilic cytoplasm
Images hosted on other servers:
Sheets of rhabdoid cells
Typical cytology
Touch preparation of rhabdoid cells
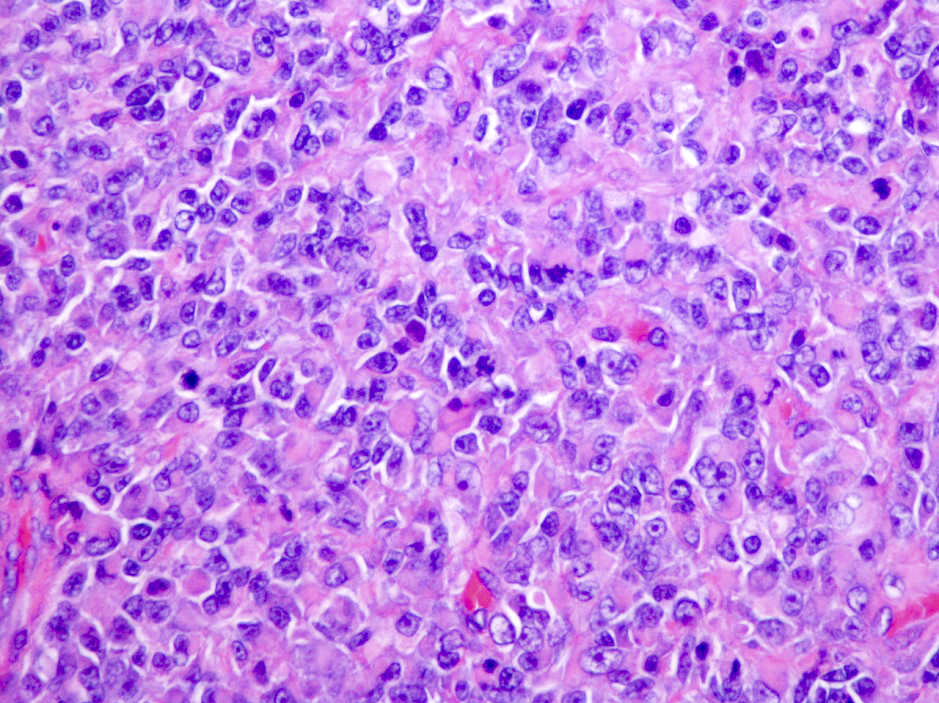
- Sheets of densely packed, immature cells with high N:C ratio
- Diagnostic feature on histology is the presence of cells with rhabdoid features, which includes (Neurooncol Pract 2019;6:163)
- Well defined cell borders
- Abundant cytoplasm with eosinophilic inclusions
- Eccentrically located nuclei containing vesicular chromatin
- Prominent eosinophilic nucleoli
- Mitotic figures can be numerous
- Geographic necrosis can be present
- Often diverse with a mixed histologic appearance (epithelioid, myxoid, spindled, chondroid) (Am J Surg Pathol 1998;22:1083)
- Primitive neuroectodermal component is most common (Am J Surg Pathol 2006;30:1462)
- Mesenchymal and epithelial features are less common
- Staghorn vasculature prominent in adult sellar AT / RT (Am J Surg Pathol 2017;41:932)
Contributed by Nirupama Singh, M.D., Ph.D., Chunyu Cai, M.D., Ph.D. (Case #502) and Geling Li, M.D., Ph.D.
Vesicular chromatin and prominent nucleoli
Eosinophilic globs
Mitotic figures
Myxoid area
Chondroid
Geographic necrosis
Gland
Large pale cell
Rhabdoid cell
Small blue cell
Densely packed immature cells
INI1
- Embryonal cells with round to oval hyperchromatic nuclei and minimal cytoplasm
- Rhabdoid cells are larger cells with eccentrically located nuclei and eosinophilic cytoplasm
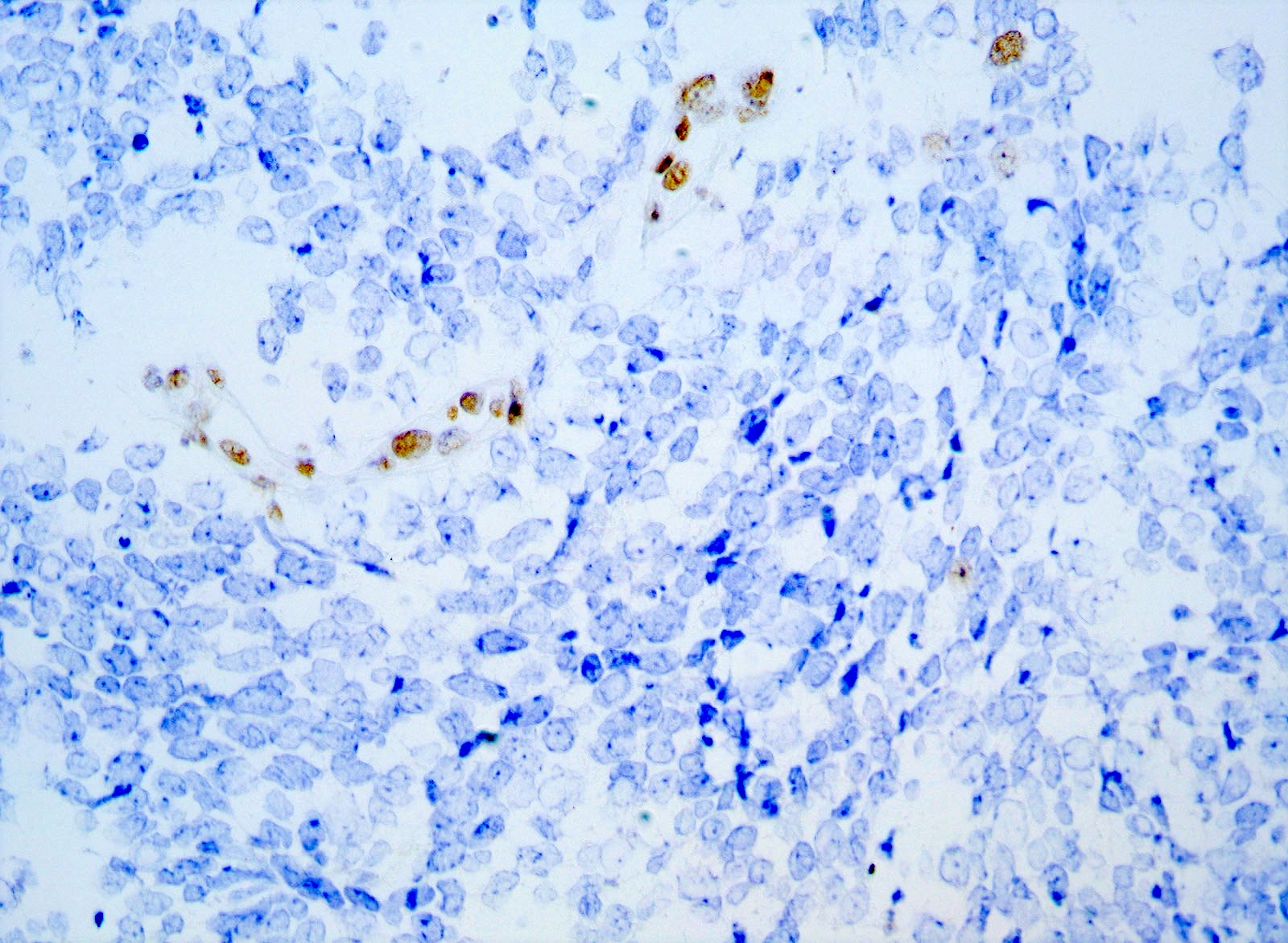
- Rhabdoid cells are positive for EMA, SMA and vimentin
- Cytokeratins, GFAP, NFP and synaptophysin staining are also observed
- Germ cell markers and skeletal muscle differentiation markers (MyoD1, myogenin)
- Nuclear loss of SMARCB1 (INI1) protein expression is highly sensitive
- Rhabdoid cell cytoplasm
- Spherical, paranuclear, cytoplasmic inclusions
Images hosted on other servers:
Rhabdoid cell
- Loss of SMARCB1 (INI1) expression at the protein level is seen in most AT / RTs (Cancer Biol Ther 2009;8:412, Pediatr Dev Pathol 2018;21:6, Neuropathology 2018;38:305)
- Mutations in SMARCA4 (BRG1) gene are rare
- Loss of all or part of chromosome 22 is frequent
- Gene expression and methylation profiling identified 3 potential subgroups that can potentially aid in subgroup based therapies (Neuro Oncol 2020;22:613)
- ATRT TYR: overexpression of tyrosinase
- Whole or partial loss of one copy of chromosome 22 accompanied by a point mutation in SMARCB1 on the other allele
- Majority with infratentorial location
- Younger patient age (median age at diagnosis: 12 months)
- ATRT SHH: overexpression of sonic hedgehog and Notch pathways
- Most display compound heterozygous point mutations in SMARCB1
- Further subtyped by supratentorial (ATRT SHH1) or infratentorial (ATRT SHH2) localization
- ATRT MYC: overexpression of MYC oncogene
- Homozygous, broad loss of SMARCB1 with lower overall DNA methylation levels
- Older patient age (median age at diagnosis: 27 months)
- Adult sellar AT / RT: clinically distinct entity, however, DNA methylation is similar to ATRT MYC subgroup (Am J Surg Pathol 2018;42:506)
- ATRT TYR: overexpression of tyrosinase
Images hosted on other servers:

Methylation array analysis

Cluster analysis
- Brain, suprasellar mass, biopsy:
- Atypical teratoid rhabdoid tumor (AT / RT), CNS WHO grade 4 (see comment)
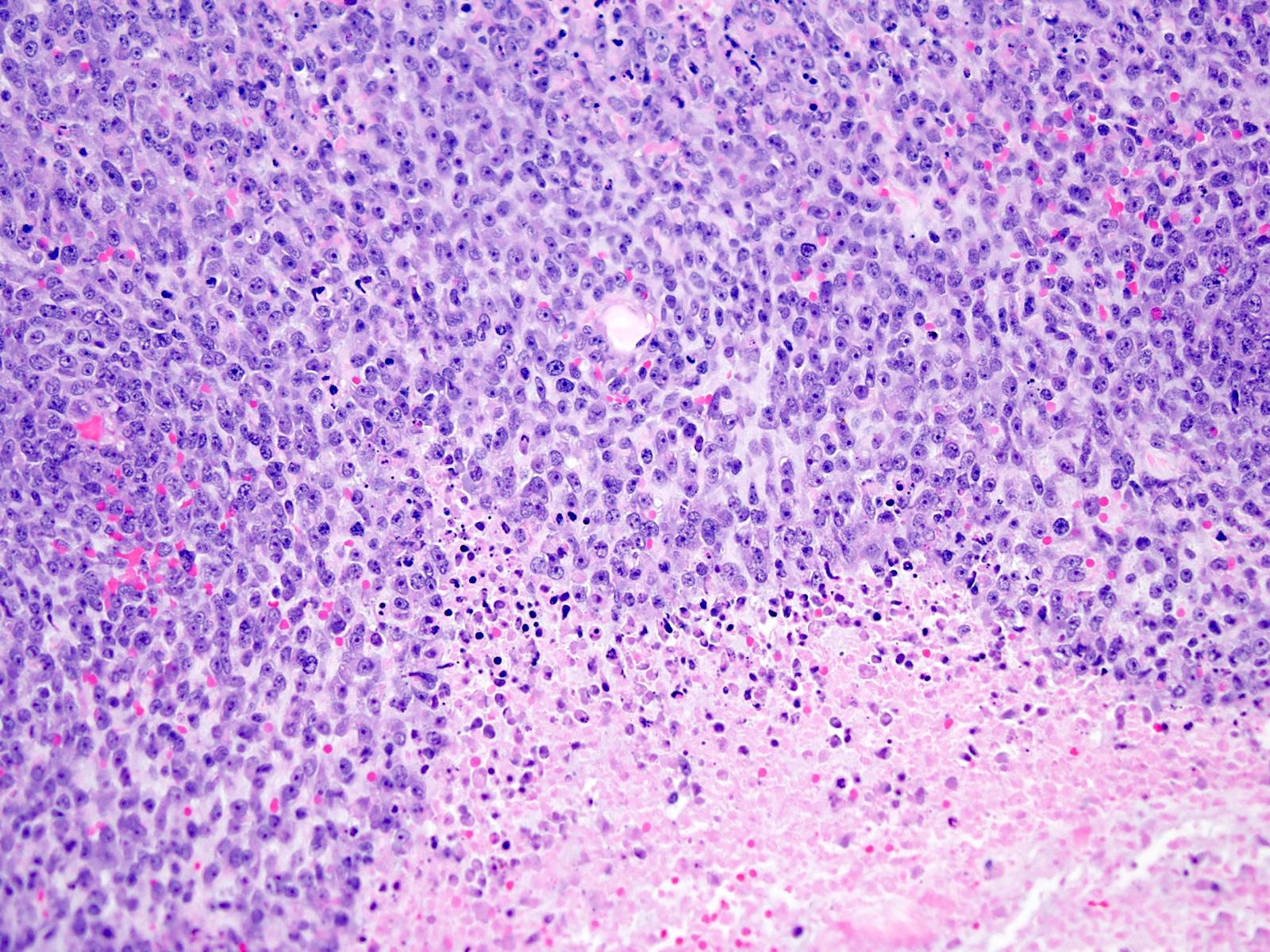
- Comment: Sections of the suprasellar tumor demonstrate patternless sheets of malignant blue cells with round to oval nuclei, vacuolated chromatin and scattered inconspicuous nucleoli. Nuclear profiles are focally irregular. Tumor cells have variable eosinophilic cytoplasm. Scattered mitotic figures and apoptotic cells are noted. Microvasculature is prominent. Necrosis is not seen. INI1 nuclear immunoreactivity is lost in tumor cells.
- Medulloblastoma:
- Larger, more atypical hyperchromatic nuclei
- Diffuse synaptophysin+
- Retained nuclear INI1
- Choroid plexus carcinoma:
- Rhabdoid meningioma:
- CNS embryonal tumors:
- Lacks rhabdoid inclusions
- Diffuse synaptophysin+
- Retained nuclear INI1
- Germinoma:
What is the key component of atypical teratoid / rhabdoid tumor (AT / RT)?
- Germline BAP1 mutation
- Loss of expression of SMARCB1 or SMARCA4
- N-MYC amplification
- TP53 mutation
Comment Here
Reference: Atypical teratoid / rhabdoid tumor
- 0 - 3 years
- 3 - 20 years
- 20 - 50 years
- 50 - 80 years
Comment Here
Reference: Atypical teratoid / rhabdoid tumor
- Undifferentiated round cell sarcoma with capicua-double homeobox 4 (CIC-DUX4) gene fusion (Am J Surg Pathol 2017;41:941)
- Aggressive sarcoma arising predominantly in soft tissues of children and young adults (Am J Surg Pathol 2017;41:941, Histopathology 2016;69:624)
- Unique clinical presentation, morphology, immunoprofile and genetic signature that are different from Ewing sarcoma
- Round to ovoid cytomorphology with a high nuclear to cytoplasmic ratio
- Expression of CD99, WT1, DUX4, ETV4 (Am J Surg Pathol 2016;40:313, Mod Pathol 2016;29:1324, Am J Surg Pathol 2017;41:423)
- CIC-DUX4 fusion from either a t(4;19)(q35;q13) or a t(10;19)(q26;q13) translocation (Am J Surg Pathol 2017;41:941)
- Undifferentiated round cell sarcoma with CIC-DUX4 fusion
- ICD-O: 8803/3 - Small cell sarcoma
- Wide age range (6 - 81 years, with a mean of 32 years) (Am J Surg Pathol 2017;41:941)
- Slight predominance in males
- Trunk (Am J Surg Pathol 2017;41:941)
- Distal extremities (Am J Surg Pathol 2017;41:941)
- Other sites include viscera, head and neck, upper extremities, bone (Histopathology 2016;69:624, Am J Surg Pathol 2016;40:1298)
- CIC-DUX4 fusion oncoprotein potentiates the transcriptional activity of CIC and activates the expression ETV1/4/5, which is a member of the E26 transformation specific (ETS) family of transcription factors (Sci Rep 2020;10:684)
- MYC amplification in majority of cases (Mod Pathol 2015;28:57)
- Most patients present with a rapidly growing, solitary mass in deep or superficial soft tissue (Am J Surg Pathol 2017;41:941)
- Metastatic disease may be present at initial presentation (Histopathology 2016;69:6247)
- Tissue sampling is the gold standard for a definitive diagnosis
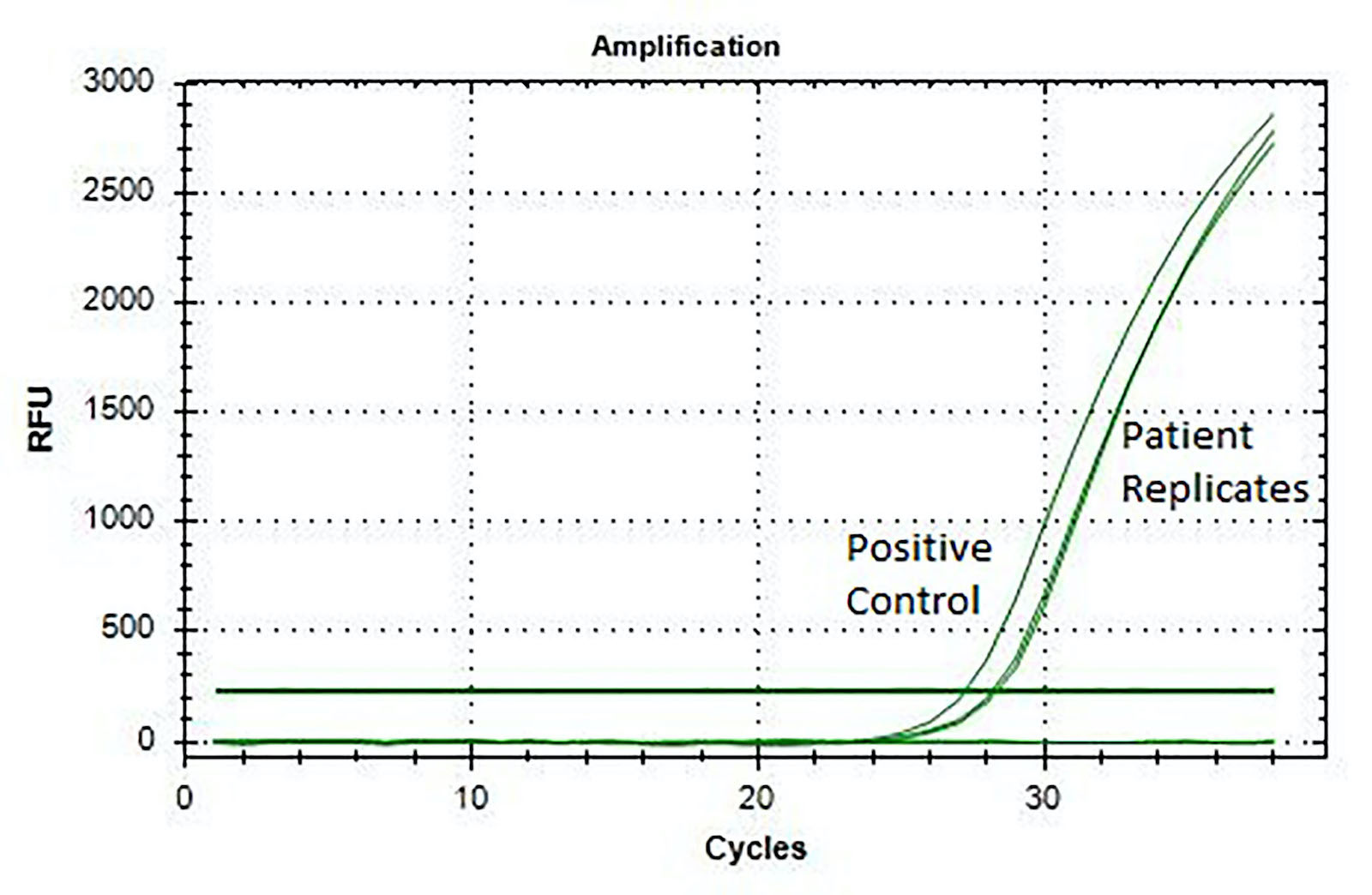
- Fluorescence in situ hybridization (FISH) (Histopathology 2017;71:461)
- Reverse transcriptase polymerase chain reaction (RT-PCR) (J Clin Pathol 2017;70:697, Histopathology 2017;71:461)
- Large heterogeneous appearing hypermetabolic mass on PET / CT
Contributed by Borislav Alexiev, M.D.
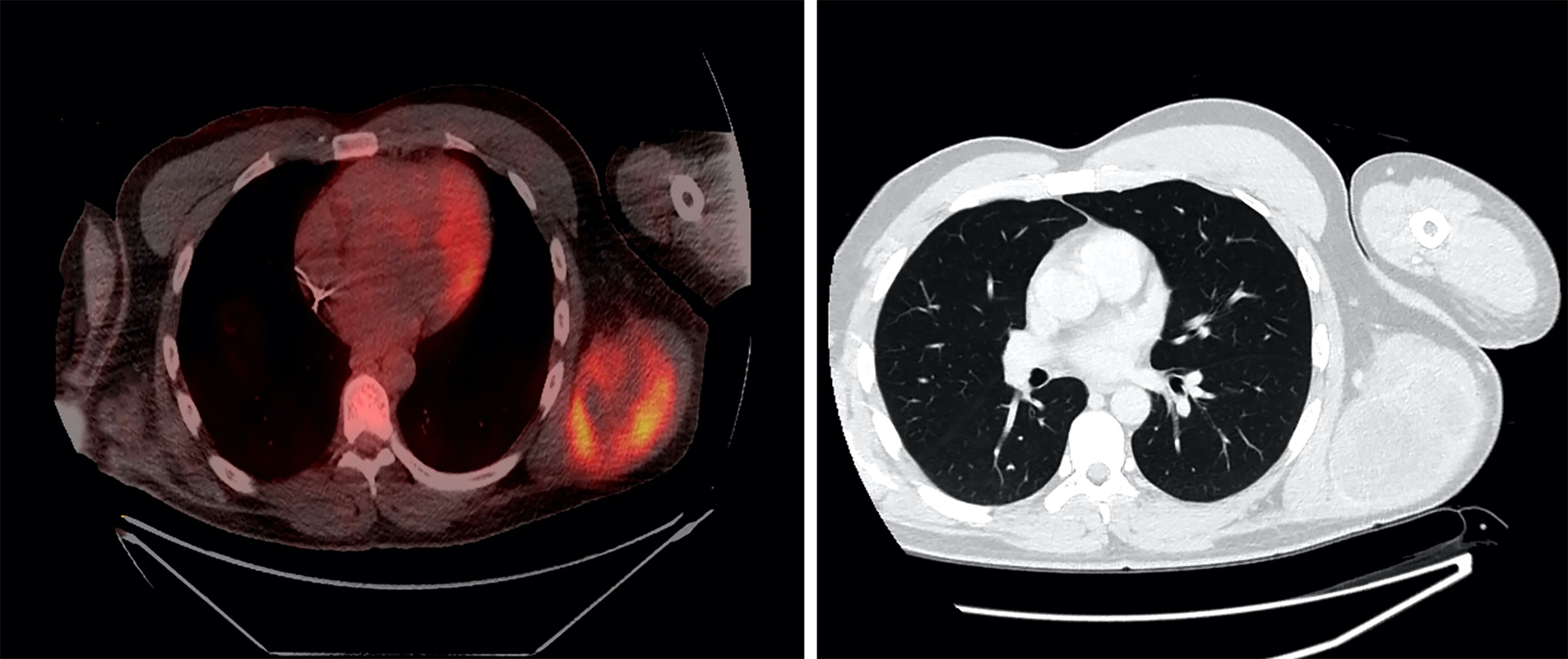
PET / CT
- CIC-DUX4 fusion positive sarcomas have a significantly unfavorable outcome compared to Ewing sarcoma (Hum Pathol 2019;86:57)
- High metastatic rate, mainly to the lung (Histopathology 2016;69:624)
- 43% 5 year overall survival (Am J Surg Pathol 2017;41:941)
- 12 year old boy with kidney mass (Pediatr Dev Pathol 2018;21:406)
- 13 year old girl with thigh mass (Diagn Cytopathol 2018;46:958)
- 24 year old man with a posterior mediastinal mass and a 69 year old woman with a gluteal mass (Cancer Cytopathol 2016;124:350)
- 38 year old man with deep abdominal wall mass (Pathol Res Pract 2017;213:1315)
- 40 year old man with mass in deep soft tissues in his thigh (Pathol Res Pract 2015;211:877)
- Patients treated with neoadjuvant chemotherapy showed an inferior survival compared to patients managed by surgery first (Am J Surg Pathol 2017;41:941)
- Further studies are needed to clarify the role of chemotherapy in CIC rearranged sarcomas (Genes Chromosomes Cancer 2012;51:207)
- Large mass with fleshy cut surface (Am J Surg Pathol 2004;28:523)
- Necrosis and hemorrhage
- Cystic changes
Contributed by Borislav Alexiev, M.D.
Soft tissue mass
- Diagnosis of CIC-DUX4 sarcoma on a small tissue fragment without molecular studies would be challenging
- Solid and often nodular growth pattern
- Less common architectural patterns include fascicular arrangement of neoplastic cells and reticular growth (Am J Surg Pathol 2004;28:523)
- Small round or ovoid cells with amphophilic or lightly eosinophilic cytoplasm (Genes Chromosomes Cancer 2012;51:207, Mod Pathol 2016;29:1523)
- Round or oval nuclei with variable chromatin patterns and small to medium sized nucleoli
- Higher degree of heterogeneity in nuclear shape and size, compared with the rather monomorphic appearance seen in Ewing sarcoma family of tumors (Genes Chromosomes Cancer 2012;51:207)
- Some cases may display a predominant spindle cell, epithelioid or plasmacytoid / rhabdoid morphology (Am J Surg Pathol 2017;41:941)
- Mitotic figures are common (Genes Chromosomes Cancer 2012;51:207)
- Most cases show geographic necrosis (Mod Pathol 2016;29:1523)
- Some degree of moderate nuclear pleomorphism may be observed
- Myxoid stroma in a third of cases (Am J Surg Pathol 2017;41:941)
Contributed by Borislav Alexiev, M.D.

Chest wall mass
Small round cell morphology
Solid growth pattern
Spindle cell morphology
Mitoses
Myxoid stroma
CD99 expression
WT1 expression
MYC expression
- Hypercellular smears, with tumor cells arranged in large groups and singly dispersed
- Individual cells with high nuclear to cytoplasmic ratio, eccentric round to ovoid nuclei, irregular nuclear contours and small nucleoli (Diagn Cytopathol 2018;46:958)
- Cytoplasmic vacuoles (Cancer Cytopathol 2016;124:350)
- Mitotic figures
- Necrosis
- Myxoid stromal component
- CD99 (Am J Surg Pathol 2017;41:941)
- WT1 (N terminal) (Genes Chromosomes Cancer 2012;51:207)
- WT1 (C terminal) (Mod Pathol 2016;29:1523)
- DUX4 (Am J Surg Pathol 2017;41:941)
- ETV4 (Mod Pathol 2016;29:1523)
- ERG (Virchows Arch 2017;470:373)
- FLI1 (Pathol Res Pract 2017;213:1315)
- Calretinin (Pathol Res Pract 2017;213:1315)
- MYC (Mod Pathol 2015;28:57)
- INI1, retained (Pathol Res Pract 2017;213:1315)
- Pancytokeratin AE1 / AE3 (Virchows Arch 2017;470:373)
- Myogenin (Am J Surg Pathol 2016;40:313)
- Desmin (Virchows Arch 2017;470:373)
- CCNB3 (Virchows Arch 2017;470:373)
- S100 (Am J Surg Pathol 2016;40:313)
- NY-ESO-1 (Virchows Arch 2017;470:373)
- NKX2.2 (Histopathology 2017;71:461)
- Heterogeneity: in cell density, from tightly packed to loosely unconnected areas (Ultrastruct Pathol 2020;44:237)
- Polygonal to pleomorphic cells with small processes
- Round, oval, polygonal or elongated nuclei
- Abundant glycogen in the cytoplasm and rare cell adhesions
- No neuroendocrine granules present
- CIC-DUX4 gene fusion, resulting from either a t(4;19) or t(10;19) translocation, is the most common genetic abnormality (Am J Surg Pathol 2017;41:941, J Clin Pathol 2017;70:697)
- Some tumors harbor CIC rearrangements with non-DUX4 partner genes including FOXO4, LEUTX, NUTM1 and NUTM2A (Sci Rep 2020;10:684)
- Trisomy for chromosome 8 (Mod Pathol 2015;28:57)
Contributed by Lawrence J. Jennings, M.D., Ph.D.
Real time RT-PCR
- Chest wall mass, excision:
- CIC-DUX4 associated undifferentiated small round cell sarcoma (see comment)
- Comment: There is a subcutaneous solid nodular proliferation of small to medium sized round / ovoid and spindle cells with scant amount of amphophilic or lightly eosinophilic cytoplasm. The cells contain medium sized round to oval vesicular nuclei with small nucleoli. High mitotic activity (21 mitoses/10 high power fields) and areas of necrosis are present. Patchy myxoid or edematous stromal changes are seen.
- Immunohistochemically the cells have strong expression of CD99, WT1 (N terminal) and MYC while are negative for pankeratin AE1 / AE3, EMA, myogenin, S100 and SOX10. INI1 is preserved. The tumor is positive for CIC-DUX4 fusion transcript.
- This constellation of morphological, immunohistochemical and molecular features strongly supports the diagnosis of CIC-DUX4 associated undifferentiated small round cell sarcoma. It is a sarcoma associated with an aggressive clinical course, with an inferior overall survival compared to Ewing sarcoma.
- Ewing sarcoma:
- Mean age at diagnosis of 15 years versus peak incidence in the fourth decade for CIC rearranged sarcomas (Am J Surg Pathol 2017;41:941)
- Most Ewing sarcomas occur in skeletal locations (Am J Surg Pathol 2017;41:941)
- Entirely uniform nuclei are more common in Ewing sarcomas (Am J Surg Pathol 2016;40:313)
- Prominence of the nucleoli, spindle cell elements and myxoid stroma are more common in the CIC rearranged sarcomas
- NKX2.2+ (Histopathology 2017;71:461)
- DUX4- (Am J Surg Pathol 2017;41:941)
- WT1-
- Pathognomonic translocations involving the EWSR1 gene (Am J Surg Pathol 2017;41:941)
- BCOR-CCNB3 (Ewing-like) sarcoma:
- Wide morphologic spectrum ranging from round to spindle cell phenotype (Am J Surg Pathol 2018;42:604)
- Monomorphic, ovoid nuclei with similar fine chromatin pattern
- Rich capillary network
- Myxoid or collagenous stroma
- Alternating hypercellular and hypocellular areas (Am J Surg Pathol 2018;42:604)
- BCOR+ (Am J Surg Pathol 2017;41:1713)
- CCNB3+ (Pathol Int 2015;65:410)
- SATB2+ (Am J Surg Pathol 2018;42:604)
- Cyclin D1+ (Am J Surg Pathol 2017;41:1713)
- NKX2.2- (Am J Surg Pathol 2018;42:604)
- WT1- (Am J Surg Pathol 2018;42:604)
- BCOR-CCNB3 fusions (Am J Surg Pathol 2018;42:604)
- Malignant peripheral nerve sheath tumor (MPNST):
- Arising (i) from a peripheral nerve (ii) from a preexisting benign nerve sheath tumor (usually neurofibroma) or (iii) in patients with NF1 disease (Am J Surg Pathol 2016;40:896)
- In the absence of these 3 associations, according to the WHO classification, the diagnosis was based on the histologic, IHC and ultrastructural features of Schwann cell differentiation (Am J Surg Pathol 2016;40:896)
- Typical histologic findings include fascicles of alternating hypercellular and hypocellular areas, branching hemangiopericytoma-like vascular pattern, whorls, palisades or rosette-like arrangements (Am J Surg Pathol 2016;40:896)
- S100 protein / SOX10 positive Schwann cells are reduced or absent (Hum Pathol 2017;67:1)
- Complete loss of trimethylated histone 3 lysine 27 expression is detectable in about half of MPNSTs (Hum Pathol 2017;67:1)
- DUX4-
- Synovial sarcoma:
- Biphasic: well formed glandular structures admixed with a monotonous atypical spindle cell proliferation in the stroma
- Monophasic: sheets of tumor cells with oval to spindle, vesicular nuclei surrounded by an indistinct rim of amphophilic to lightly eosinophilic cytoplasm
- Alternating zones of hypercellularity and hypocellularity
- Hemangiopericytoma-like vasculature
- Poorly differentiated morphology with rounded or spindle cells showing elevated nuclear grade, rhabdoid cells and high mitotic index
- Broad spectrum cytokeratins+ and EMA+ (Am J Surg Pathol 2005;29:569)
- CD99+ and BCL2+ (Am J Surg Pathol 2005;29:569)
- TLE1+ (Am J Clin Pathol 2011;135:839)
- NY-ESO-1+ and MAGE-A4+ (Oncol Lett 2019;17:3937)
- SS18-SSX fusion oncoprotein (Cancer Cell 2018;33:1128)
- Alveolar rhabdomyosarcoma:
- Alveolar, nested to solid growth pattern (Head Neck Pathol 2018;12:181)
- Primitive, undifferentiated or round blue cell pattern (Head Neck Pathol 2018;12:181)
- Classical rhabdomyoblastic differentiation with strap cells or cross striations is seldom seen
- Crush artifact
- Multinucleation
- Lymphovascular invasion
- Expression of muscle markers: desmin, myogenin, MyoD1 (Head Neck Pathol 2018;12:181)
- Aberrant expression of epithelial and neuroendocrine markers (Adv Anat Pathol 2013;20:387, Head Neck Pathol 2018;12:181)
- DUX4-
- Unique t(2;13)(q35;q14) or t(1;13)(p36;q14) chromosomal translocations, resulting in PAX3 / FOXO1 and PAX7 / FOXO1 fusion genes (Diagn Mol Pathol 2009;18:138, Adv Anat Pathol 2013;20:387)
- Desmoplastic small round cell tumor:
- Predominantly intraabdominal, pelvic or peritoneal based mass (Am J Clin Pathol 2000;114:345)
- Islands of monotonous small cells within prominent desmoplastic stroma (Int J Surg Pathol 2017;25:158)
- Solid variant, lacking evidence of desmoplastic stroma
- Polyphenotypic multidirectional differentiation, with expression of epithelial (pankeratin AE1 / AE3), muscle (desmin) and neural markers (NSE) (Int J Surg Pathol 2016;24:672)
- WT1+ (C terminal) ( Am J Surg Pathol 2000;24:830, Am J Clin Pathol 2000;114:345)
- DUX4-
- EWSR1-WT1 fusion gene (Int J Surg Pathol 2016;24:672, Am J Surg Pathol 2000;24:830)
- High metastatic rate, brain most common
- Diagnosis always requires clinicopathological and radiological correlation
- Prognosis is poor
- Tumor is always negative for ERG
- Tumor is characterized by cords of epithelioid cells distributed in a desmoplastic stroma
A 65 year old man presented with a left thigh mass. Hematoxylin eosin stains demonstrated proliferation of small to medium sized round / ovoid cells with medium sized round to oval vesicular nuclei and clear or pale eosinophilic cytoplasm. Increased mitotic activity, geographic necrosis and patchy myxoid stromal change were seen. Immunohistochemical stains for CD99, WT1 and DUX4 were positive in tumor cells while all of the following were negative: pankeratin AE1 / AE3, S100, SOX10, myogenin, NY-ESO-1, NKX2.2 and CCNB3. INI1 was retained. Which of the following is most likely the correct diagnosis?
- Synovial sarcoma, poorly differentiated
- Extraskeletal Ewing sarcoma
- Epithelioid sarcoma
- BCOR-CCNB3 (Ewing-like) sarcoma
- CIC-DUX4 associated undifferentiated round cell sarcoma
Comment Here
Reference: CIC-DUX4 fusion tumor
- CNS neuroblastoma, FOXR2 activated (CNS NB FOXR2) tumors are embryonal neoplasms characterized by poorly differentiated neuroblastic or neuronal differentiation with variable neuropil rich stroma and ganglion cells
- Characterized by the activation of FOXR2 by structural rearrangements
- Embryonal tumor with neuronal or neuroblastic differentiation and activation of FOXR2 rearrangement and fusion or a DNA methylation profile aligned with CNS neuroblastoma, FOXR2 activated
- CNS neuroblastoma, FOXR2 activated
- Formerly recognized as one of the primitive neuroectodermal tumors of CNS (CNS PNETs)
- ICD-O: 9500/3 - neuroblastoma, NOS
- ICD-10: C71 - malignant neoplasm of brain
- ICD-11: 2A00.1Y & XH85Z0 - other specified embryonal tumors of brain & neuroblastoma, NOS
- Supratentorial location in all tumors
- Slight female predilection (J Neuroimaging 2023;33:359)
- Range of ages at diagnosis, 1 - 20 years
- Median age at diagnosis is 5 years (Neuro Oncol 2021;23:1597)
- Supratentorial
- Thought to arise from neuroectodermal cells, although the exact cell of origin is not known (J Neuropathol Exp Neurol 2021;80:52)
- Most frequent genetic alterations are on the transcription factor gene FOXR2 (Cell 2016;164:1060)
- Although FOXR2 plays a causative role in the formation of CNS embryonal tumors, the exact underlying mechanisms have yet to be determined (Neuro Oncol 2019;21:993)
- Risk factors have not been reported
- Common symptoms: headache, vomiting, seizures and focal neurologic deficits (J Neuroimaging 2023;33:359)
- Embryonal tumor with neuronal or neuroblastic differentiation and activation of FOXR2 rearrangement and fusion or a DNA methylation profile aligned with CNS neuroblastoma, FOXR2 activated
- No specific laboratory findings for this entity
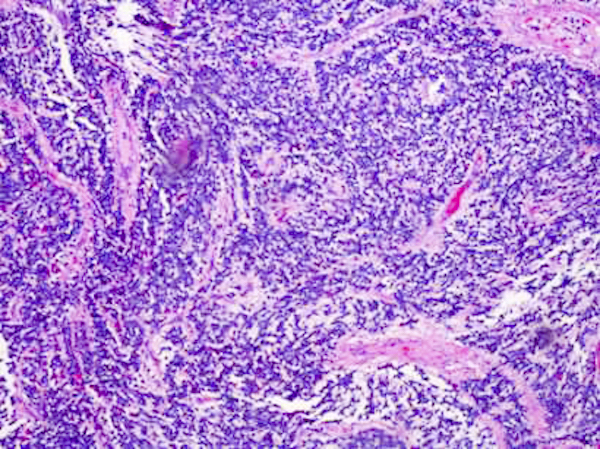
- Well circumscribed supratentorial mass with a mixed solid and cystic appearance (J Neuropathol Exp Neurol 2021;80:52)
- Solid component may show heterogeneous enhancement
Images hosted on other servers:
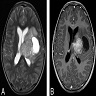
T2 weighted and postcontrast T1 weighted axial
- Favorable survival for patients with CNS NB FOXR2; higher rate of recurrences in nonirradiated and locally irradiated patients (Neuro Oncol 2021;23:1597)
- 3 year old girl with a cystic right frontal lobe tumor (Brain Tumor Pathol 2020;37:100)
- Case series of CNS neuroblastoma, FOXR2 activated (J Neuroimaging 2023;33:359)
- Maximal surgical resection followed by radiation to the brain and spinal cord; chemotherapy may also be given (NCI: Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment (PDQ®) - Patient Version [Accessed 5 September 2023])
- CNS embryonal tumors are often pink, soft, well circumscribed masses; if a prominent desmoplastic component is present, it is often tan-white and firm
- Poorly differentiated neuroepithelial cells and neurocytic cells (Cell 2016;164:1060)
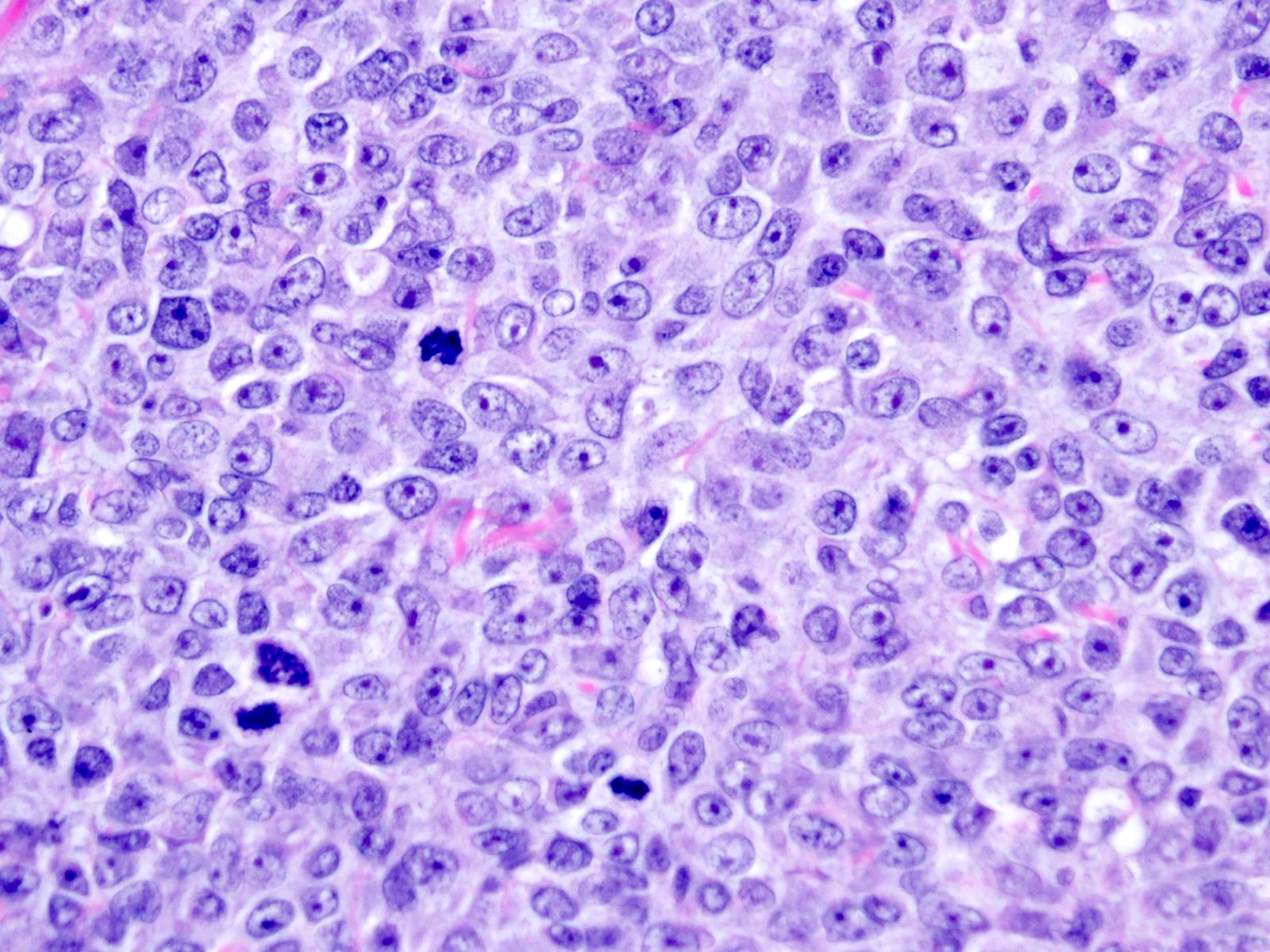
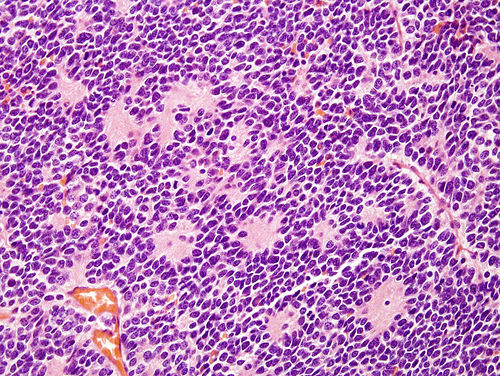
- Uniform small cells with high N:C ratio, oval to round with hyperchromatic nuclei
- Cells have little to no apparent cytoplasm in a background of neuropil with or without Homer Wright rosettes (J Neuropathol Exp Neurol 2021;80:52)
- Background of neuropil rich stroma
- Abundant mitotic activity
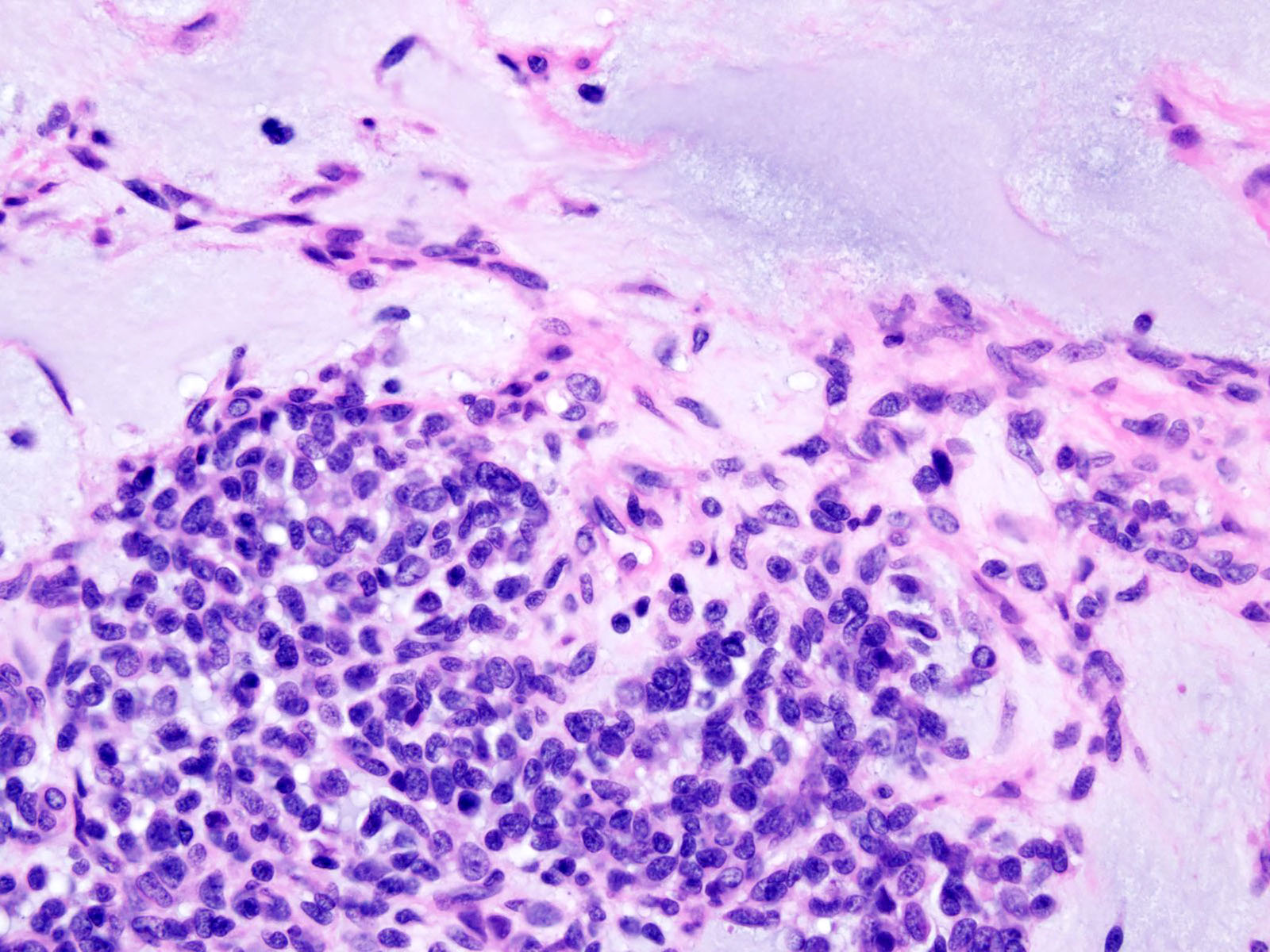
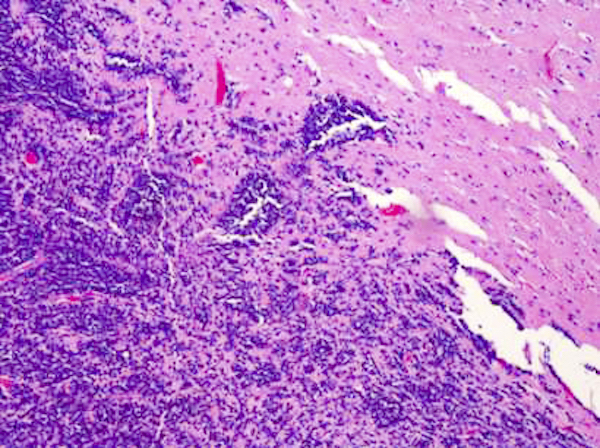
- Palisading necrosis can be present
- Infiltration of adjacent parenchyma is variable
Contributed by Nirupama Singh, M.D., Ph.D., Carmen Perrino, M.D. and P.J. Cimino, M.D., Ph.D.
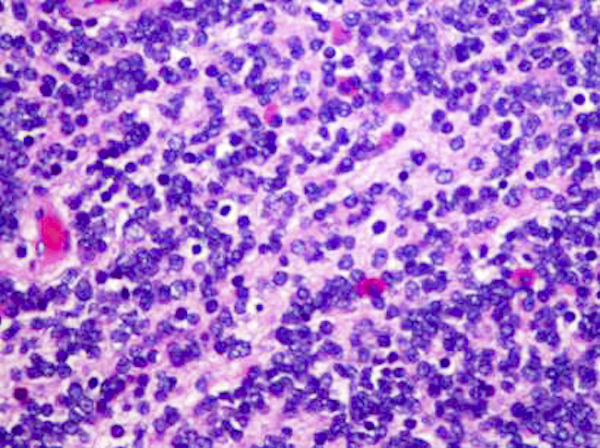
Neuroblasts with no cytoplasm
Homer Wright rosettes
Small tumor cells
Pseudorosettes
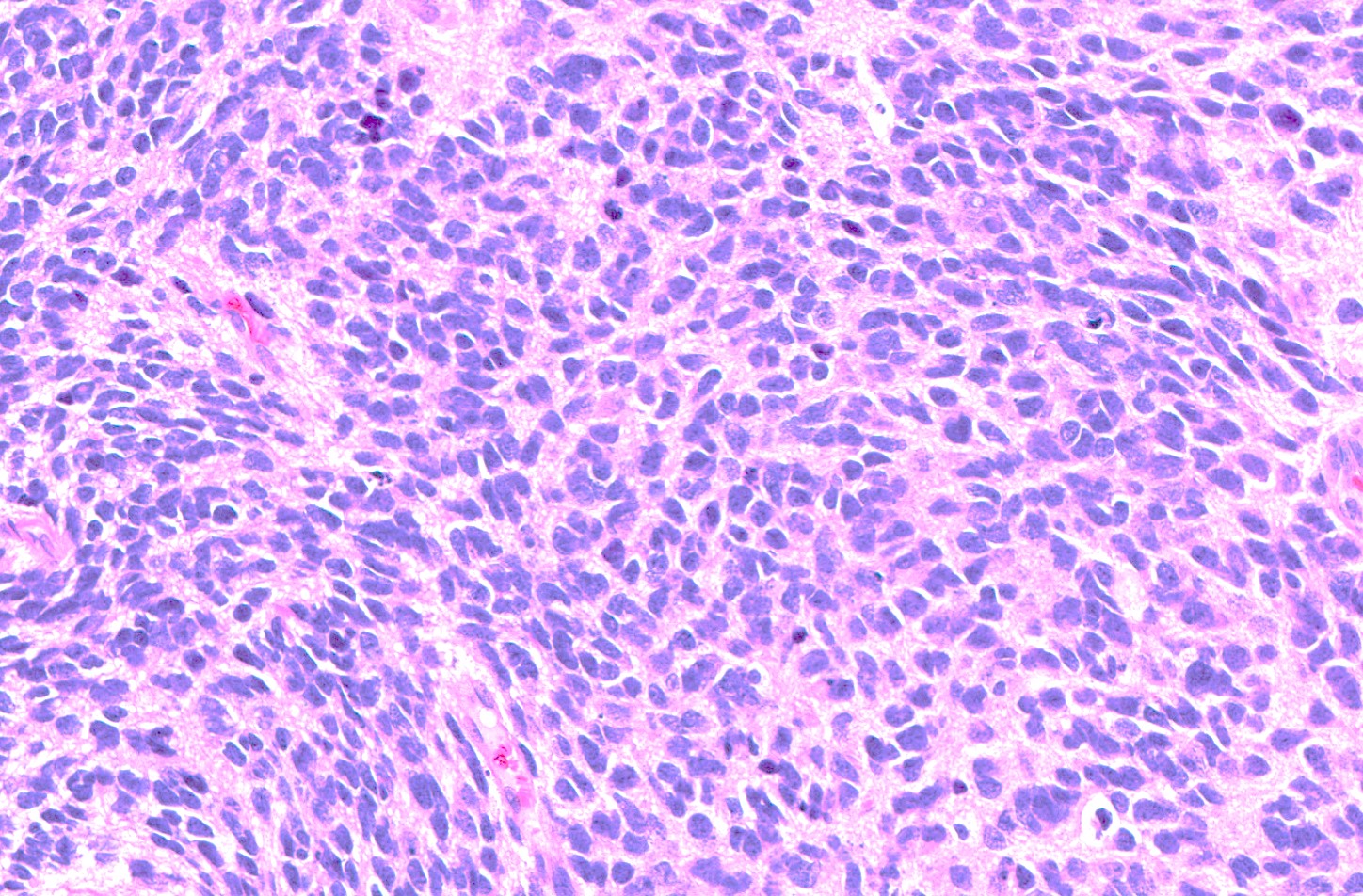
Embryonal cells

Neurocytic cells

Focal ganglion cells
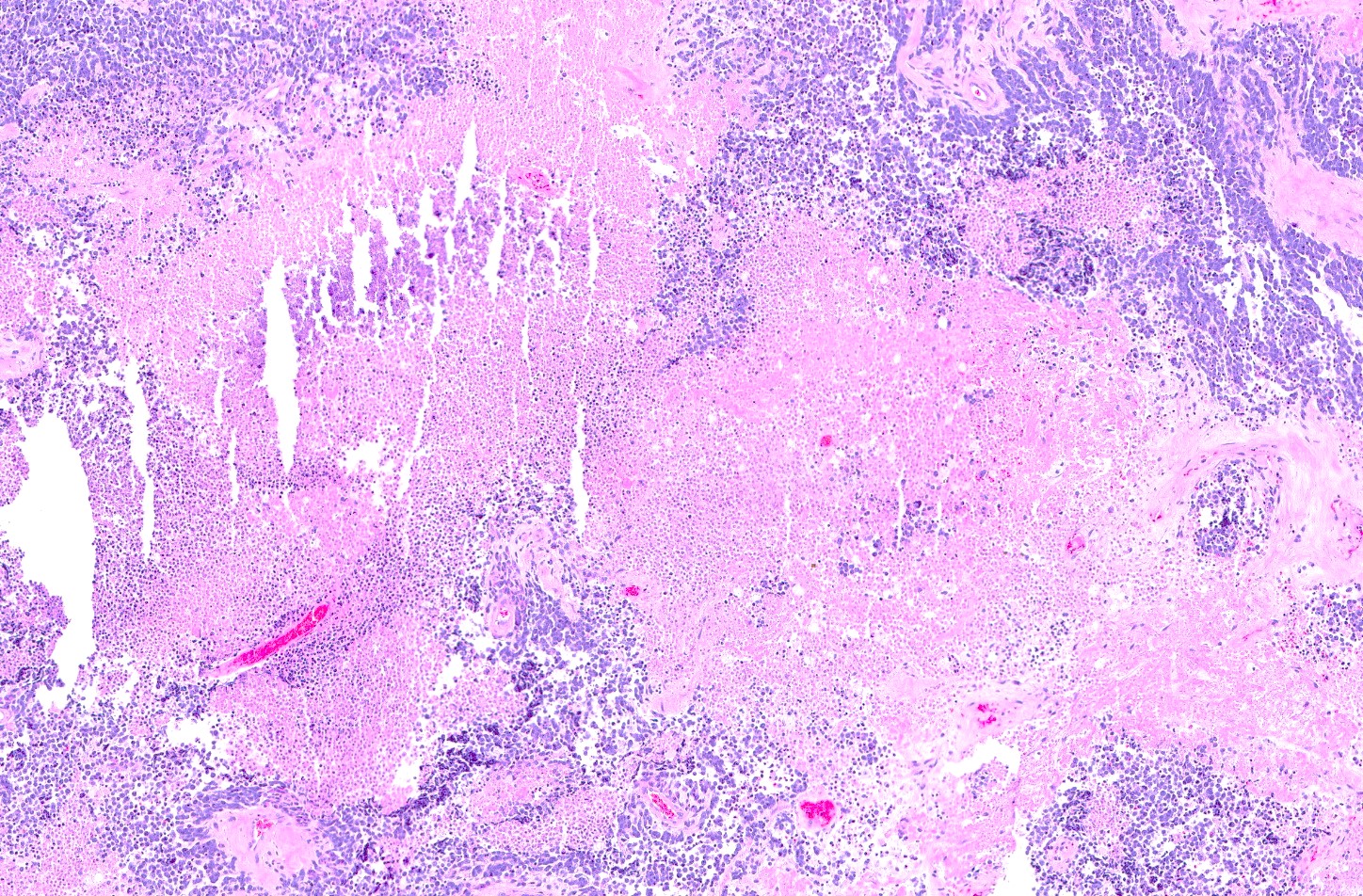
Necrosis
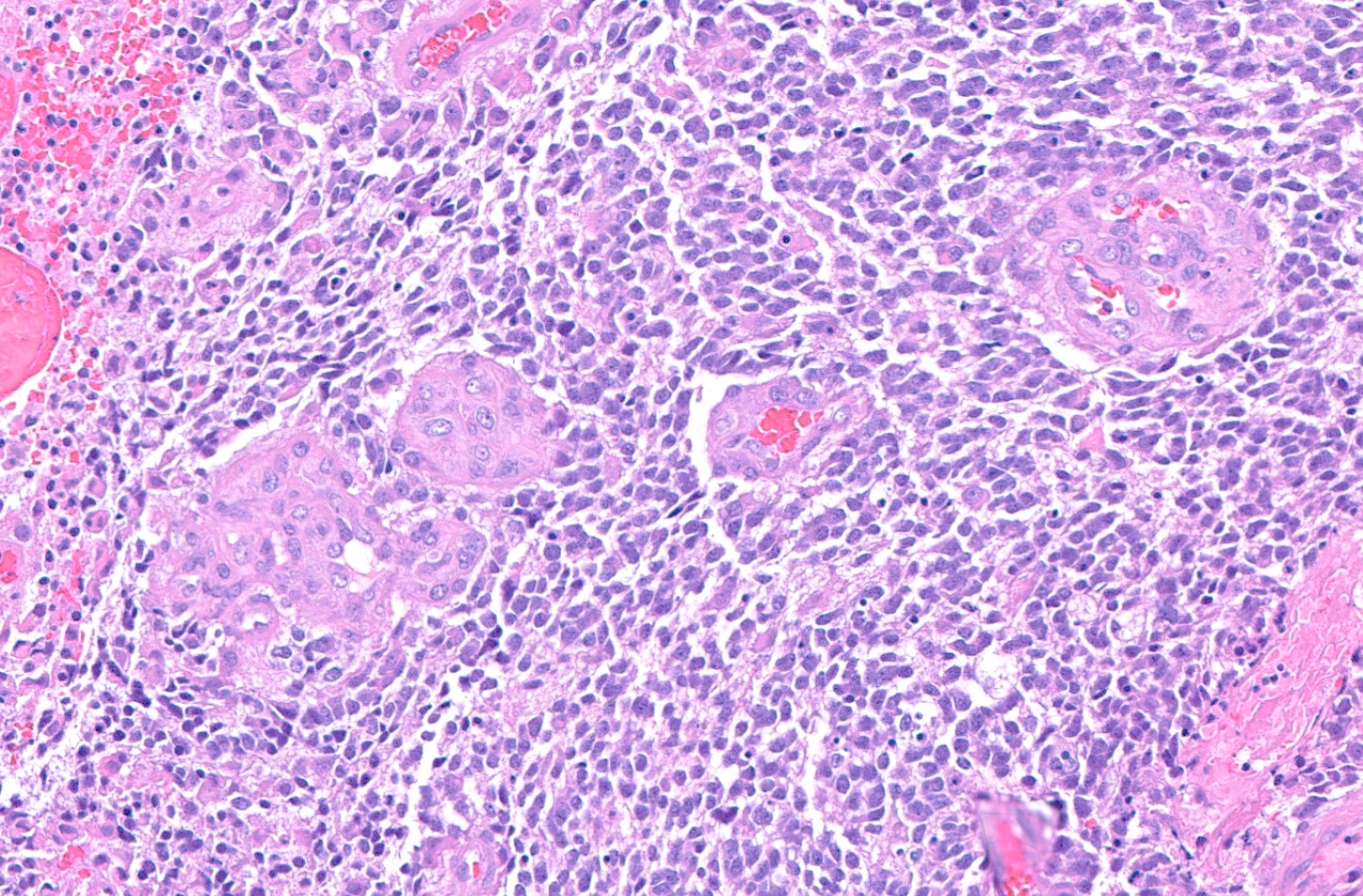
MVP
- Evaluation of cerebrospinal fluid (CSF) for the presence of tumor cells is required for staging (Radiology 1969;93:1351)
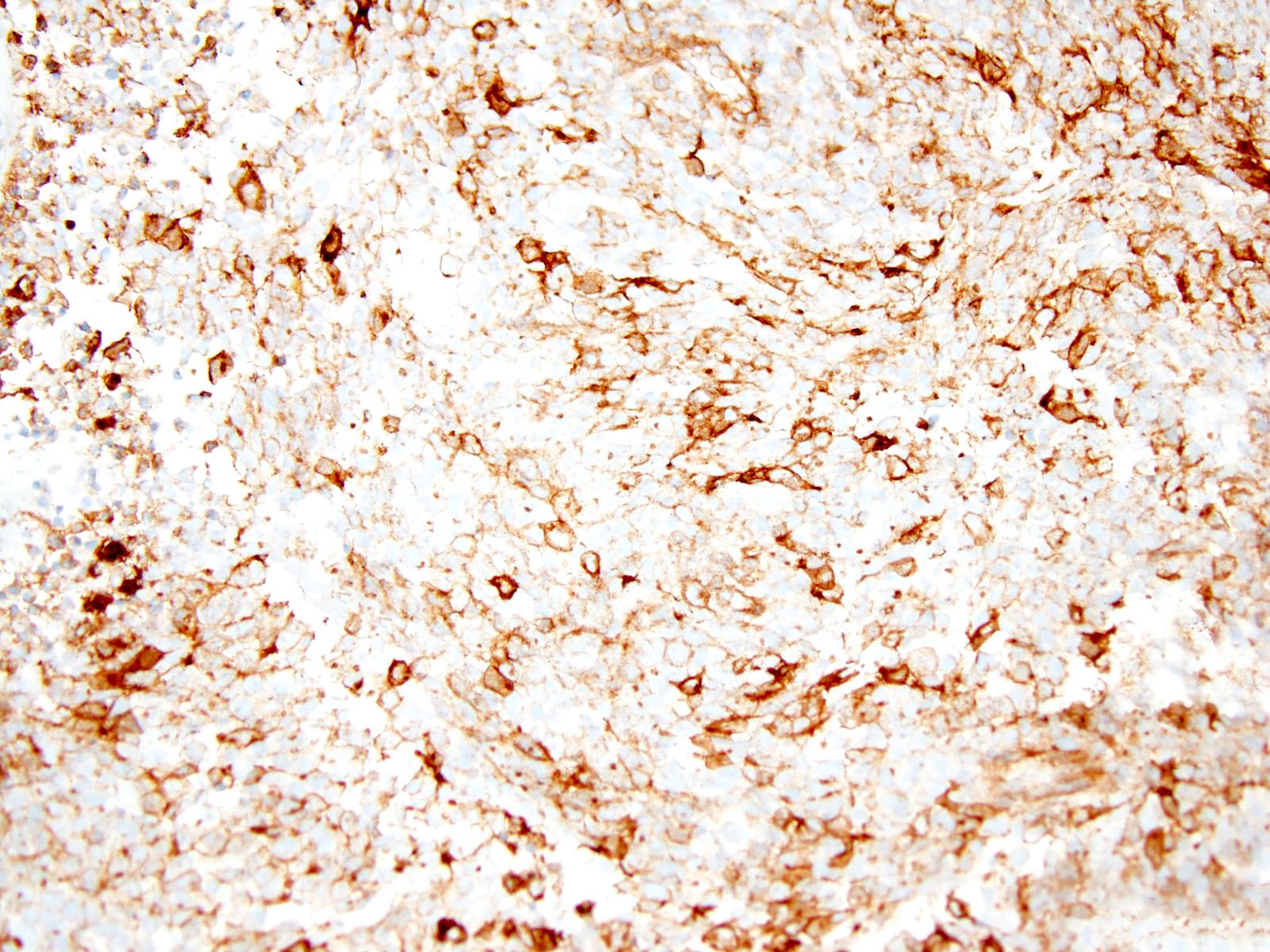
- Olig2
- Synaptophysin expression is accentuated in regions of neurocytic or ganglion cell differentiation (J Neuropathol Exp Neurol 2021;80:52)
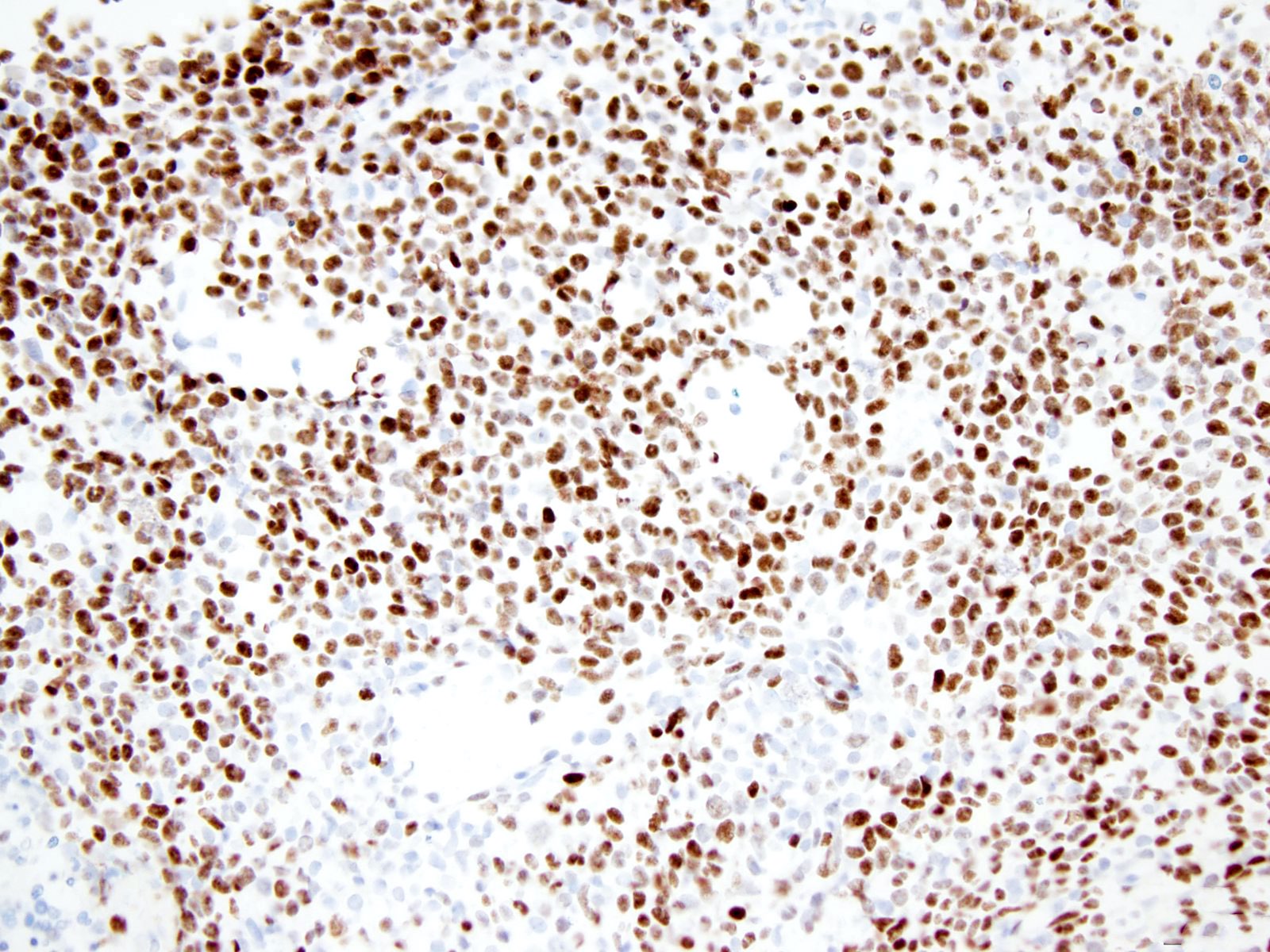
- Ki67 is high
- Tumor cells are often surrounded by cell processes
- Intermediate filaments and clear vesicles can be found (Brain Tumor Pathol 2020;37:100)
- Interchromosomal and intrachromosomal rearrangements in forkhead box R2, leading to increased FOXR2 gene expression
- FOXR2 locus on chromosome Xp11.21
- Common fusion partner is JMJD1C locus on chromosome 10q21.3
- Gain of chromosome 1q is present in most cases (Neuro Oncol 2021;23:1597)
- High FOXR2 expression can occur in a subset of high grade gliomas and medulloblastomas; however, DNA methylation profiling can distinguish these entities (Cell 2016;164:1060)
Images hosted on other servers:

Recurrent molecular alterations
Case presentation of CNS NB FOXR2
- Brain, suprasellar mass, resection:
- CNS neuroblastoma, FOXR2 activated, CNS WHO grade 4 (see comment)
- Comment: Sections of the suprasellar tumor demonstrate small, round, embryonic appearing cells with scant cytoplasm. Necrosis and abundant mitotic activity are appreciated. Immunohistochemical stains for synaptophysin and Ki67 were performed. Synaptophysin is patchy positive. Ki67 proliferation index is ~25%.
- Molecular pathology findings
- FOXR2 fusion
- Gain of chromosome 1q
- CNS embryonal tumors, NEC / NOS:
- Lacks FOXR2 fusion
- Histologically similar
- Embryonal tumor with multilayered rosettes (ETMR):
- Strong and diffuse cytoplasmic LIN28A+
- Cytokeratin, EMA, CD99+ in embryonal cells and rosettes
- Medulloblastoma:
- Larger, more atypical, hyperchromatic nuclei
- Diffuse synaptophysin+
- Atypical teratoid / rhabdoid tumor (AT / RT):
- Presence of rhabdoid cells
- Loss of INI1
- CNS tumor with BCOR internal tandem duplication (ITD):
- Requires ITD in exon 15 of BCOR
- Histologically similar
- Ewing sarcoma
- Lymphoma
- Neuroblastoma
- Testicular germ cell tumor
Comment Here
Reference: Neuroblastoma
This supratentorial mass is composed of small, round, embryonic appearing cells and Homer Wright rosettes. Whole genome analysis demonstrated a FOXR2 fusion. What is the most likely diagnosis?
- CNS neuroblastoma, FOXR2 activated
- Embryonal tumors with multilayered rosettes
- Ependymoma
- Medulloblastoma
Comment Here
Reference: Neuroblastoma
- Low grade, well circumscribed neuroendocrine neoplasm of cauda equina / filum terminale region
- CNS WHO grade 1
- Distinct from paragangliomas and pheochromocytomas outside the central nervous system (CNS) (Acta Neuropathol 2020;140:907)
- Grade 1, slow growing neuroendocrine neoplasm of cauda equina / filum terminale region
- Rare entity with a benign clinical course (J Neurooncol 2015;122:539)
- Generally affects adults
- Distinct from paragangliomas and pheochromocytomas outside the CNS (Acta Neuropathol 2020;140:907)
- Formerly, paraganglioma
- Acceptable: paraganglioma of the cauda equina, cauda equina paraganglioma (CEP)
- Also known as spinal paraganglioma, lumbar paraganglioma, paraganglioma of the filum terminale (Acta Neuropathol 2020;140:907, J Neurooncol 2015;122:539)
- ICD-O: 8693/3 - cauda equina neuroendocrine tumor (previously paraganglioma)
- ICD-11
- 2A02.0Y & XH1X68 - other specified gliomas of spinal cord, cranial nerves or other parts of the central nervous system & paraganglioma, benign
- 2A02.0Y & XH0EW6 - other specified gliomas of spinal cord, cranial nerves or other parts of the central nervous system & paraganglioma, NOS
- Rare: < 3% of spinal tumors, 3.5% of cauda equina / filum terminale tumors (Radiol Case Rep 2019;14:1185, J Neurooncol 2015;122:539, J Surg Tech Case Rep 2012;4:46)
- Generally affects adults (fourth to sixth decades); mean age: 49 years (range: 17 - 75 years) (Neurosurg Rev 2022;45:103, J Neurooncol 2015;122:539)
- Slight male predominance (M:F = 1.5:1) (Neurosurg Rev 2022;45:103)
- Majority are located in the cauda equina / filum terminale region (at the L1 level or below) (Neurosurg Rev 2022;45:103)
- Most are intradural extramedullary and attached either to filum terminale or (less often) to a caudal nerve root (J Neurooncol 2015;122:539, J Surg Tech Case Rep 2012;4:46)
- Rarely reported in thoracic and cervical regions (Histopathology 1997;31:167)
- Histogenetically, biologically and molecularly distinct from paragangliomas and pheochromocytomas outside the CNS (Acta Neuropathol 2020;140:907)
- Arises from specialized neural crest cells in the cauda equina / filum terminale region
- mRNA analyses revealed that cauda equina paragangliomas overexpress the transcription factor HOXB13 (which is developmentally expressed in the caudal extent of the spinal cord) as opposed to pheochromocytomas / paragangliomas of other regions of the body; this provides circumstantial evidence of their cellular origin (Neuropathol Appl Neurobiol 2021;47:889)
- Molecular alterations that drive tumorigenesis in cauda equina neuroendocrine tumors are unknown
- Almost all are sporadic (Acta Neuropathol 2020;140:893)
- Not associated with hereditary paraganglioma / pheochromocytoma syndrome due to mutations in SDH subunit genes (Acta Neuropathol 2020;140:907)
- No distinct clinical presentation; signs and symptoms are similar to other tumors in the region
- Most common: lower back pain (94%) with radiculopathy, sciatica (J Neurooncol 2015;122:539, World Neurosurg 2020:142:e66, Neurosurg Rev 2022;45:103)
- Less common: sensorimotor deficits and sphincter dysfunction (Radiol Case Rep 2019;14:1185, J Neurooncol 2015;122:539)
- Uncommon: fully developed cauda equina syndrome, signs of increased intracranial pressure, papilledema, subarachnoid hemorrhage (Neurosurg Rev 2022;45:103, World Neurosurg 2020:142:e66)
- Extremely rare: functionally active tumors with sympathetic secretory signs and symptoms of catecholamine hypersecretion (J Neurooncol 2015;122:539, J Surg Tech Case Rep 2012;4:46)
- Rarely a clinical or radiological diagnosis
- Neuroimaging: magnetic resonance imaging (MRI) (modality of choice) and computed tomography (CT) (Neurosurg Rev 2022;45:103)
- Biopsy
- WHO essential and desirable diagnostic criteria
- Essential
- Well demarcated tumor with zellballen architecture
- Synaptophysin or chromogranin immunoreactivity in chief cells
- Cauda equina location
- Methylation profile of cauda equina neuroendocrine tumor (for unresolved lesions)
- Desirable
- S100 positive sustentacular cells
- Cytokeratin positive chief cells
- Reticulin silver stain showing typical architecture
- Essential
- Cerebrospinal fluid (CSF) protein is usually markedly increased (Pract Neurol 2014;14:179)
- MRI
- Well circumscribed, intradural extramedullary, sausage shaped mass at cauda equina / filum terminale region (Radiol Case Rep 2019;14:1185)
- Solid, occasionally partly cystic
- Generally T1 hypointense to isointense and T2 isointense to hyperintense, with intense post contrast enhancement (Neurosurg Rev 2022;45:103)
- Although MRI findings are nonspecific and indistinguishable from other solid tumors in cauda equina region, certain characteristic MRI findings could suggest the diagnosis, including a salt and pepper appearance on T2 weighted images related to the hypervascular nature (42.1%), a peripheral hypointense rim (cap sign) on T2 weighted images caused by subcapsular hemosiderin (47.3%) and serpiginous flow voids on all sequences (78.9%) (J Neurooncol 2015;122:539)
- Perfusion weighted MR images show increased blood flow, consistent with hypervascular tumor (Radiol Case Rep 2019;14:1185)
- Polar sign may be seen in T1 contrast enhanced and T2 weighted MR images, representing subacute to chronic polar intratumoral hematomas (J Spine Neurosurg 2014;3:4)
- Spinal angiography reveals a well defined hypervascular lesion with intense early blush that persists well into the arterial and venous phases (silk cocoon appearance), which helps presurgical planning and differentiates CEPs from other spinal tumors (Radiol Case Rep 2019;14:1185, Neurosurg Focus 2015;39:E16)
- CT: may show scalloping of the vertebral bodies (J Surg Tech Case Rep 2012;4:46)
Images hosted on other servers:

MRI
Intradural enhancing lesion
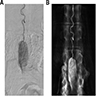
Spinal angiography

Scalloping of lumbar vertebrae
- Excellent prognosis; the vast majority are clinically benign and curable by total excision
- < 1% are locally invasive (Histopathology 1997;31:167)
- Local recurrence after excision (7%) (Neurosurg Rev 2022;45:103, Histopathology 1997;31:167)
- Occasional leptomeningeal dissemination (Acta Neurochir (Wien) 1996;138:475, J Neurosurg Spine 2017;26:501)
- Rare metastasis outside the CNS (reported only once) (Histopathology 1997;31:167)
- 42 year old man with gangliocytic paraganglioma of dorsolumbar spine (Asian J Neurosurg 2019;14:907)
- 50 year old man with functional conus cauda paraganglioma (J Surg Tech Case Rep 2012;4:46)
- 50 year old man with intradural paraganglioma of the thoracic spine (AJNR Am J Neuroradiol 1990;11:614)
- 61 year old woman with leptomeningeal dissemination of a low grade lumbar paraganglioma (J Neurosurg Spine 2017;26:501)
- 64 year old man with acute paraplegia due to hemorrhagic cauda equina paraganglioma (Asian J Neurosurg 2019;14:245)
- Complete microsurgical resection remains the first choice (Neurosurg Rev 2022;45:103)
- Preoperative embolization may not be necessary (J Neurooncol 2015;122:539)
- Postoperative radiotherapy may be helpful on an individualized basis (J Neurosurg Spine 2017;26:501, J Neurooncol 2015;122:539)
- Follow up: there is a low risk of local recurrence and leptomeningeal dissemination
Images hosted on other servers:

Intraoperative
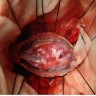
Lesion after durotomy
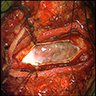
Hypervascular well marginated mass
- Well circumscribed, delicately encapsulated, oval to sausage shaped vascular mass
- Smooth surfaced, soft, red-brown and bleeds freely (Histopathology 1997;31:167)
- Size ranges from 10 mm to 112 mm in greatest dimension
- May show capsular calcification or cystic change
- Gross appearance resembles myxopapillary ependymoma
- If submitted with attached filum terminale, proximal and distal ends should be sampled as surgical margins
Images hosted on other servers:
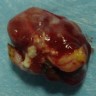
Lesion after en block removal
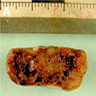
Hemorrhagic cystic change
- Discrete
- Lobular architecture
- Epithelioid cytologic features
- May show artifactual papillae mimicking myxopapillary ependymoma or ependymoma-like features with pseudorosettes (Folia Med (Plovdiv) 2022;64:1007, Turk Neurosurg 2012;22:353)
Images hosted on other servers:
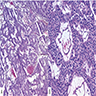
Ependymoma-like features
- Well differentiated, encapsulated and richly vascular tumor
- Organoid (zellballen) architecture: nests or lobules of chief (type I) cells surrounded by sustentacular (type II) cells with intervening delicate capillary and reticulin networks (J Neurooncol 2015;122:539)
- Chief cells
- Uniform round or polygonal epithelioid cells
- Round to oval nuclei with salt and pepper chromatin pattern and inconspicuous nucleoli (J Surg Tech Case Rep 2012;4:46)
- Finely granular eosinophilic cytoplasm, may be amphophilic or clear (Acta Neuropathol 2020;140:907)
- Sharp cell borders, particularly around vessels
- No or mild degenerative nuclear pleomorphism (endocrine atypia), of no prognostic significance
- Sustentacular cells
- Spindle shaped cells with attenuated long processes
- Perilobular; surround chief cell lobules
- Inconspicuous by routine light microscopy (visible by IHC for S100)
- Mitotic activity is highly variable (0 - 5/10 high power fields), of no prognostic significance (Acta Neuropathol 2020;140:893)
- Other common features
- Ganglion cell metaplasia (gangliocytic neuroendocrine tumors) and Schwann cell (ganglioneuromatous) differentiation in 25% of cases (Acta Neuropathol 2020;140:907)
- Ependymoma-like perivascular formations; similar but less pronounced and less fibrillar than in ependymoma (Acta Neuropathol 2020;140:907, Turk Neurosurg 2012;22:353)
- Papillary formations (Acta Neuropathol 2020;140:893)
- Less common features
- Carcinoid-like architectural features, including angiomatous, adenomatous and pseudorosette patterns
- Melanotic cells (melanotic neuroendocrine tumors) (Histopathology 1997;31:167)
- Oncocytic change (oncocytic neuroendocrine tumors) (Histopathology 1997;31:167)
- Predominant spindle cell proliferation arranged in an ill defined storiform pattern (Histopathology 1997;31:167)
- Ribboning architecture
- Extensive sclerosis (Histopathology 1997;31:167)
- Focal hemorrhagic necrosis (of no prognostic significance)
- Capsular calcification
- Bone invasion (Histopathology 1997;31:167)
Contributed by Eman Abdelzaher, M.D., Ph.D.


Encapsulated lesion
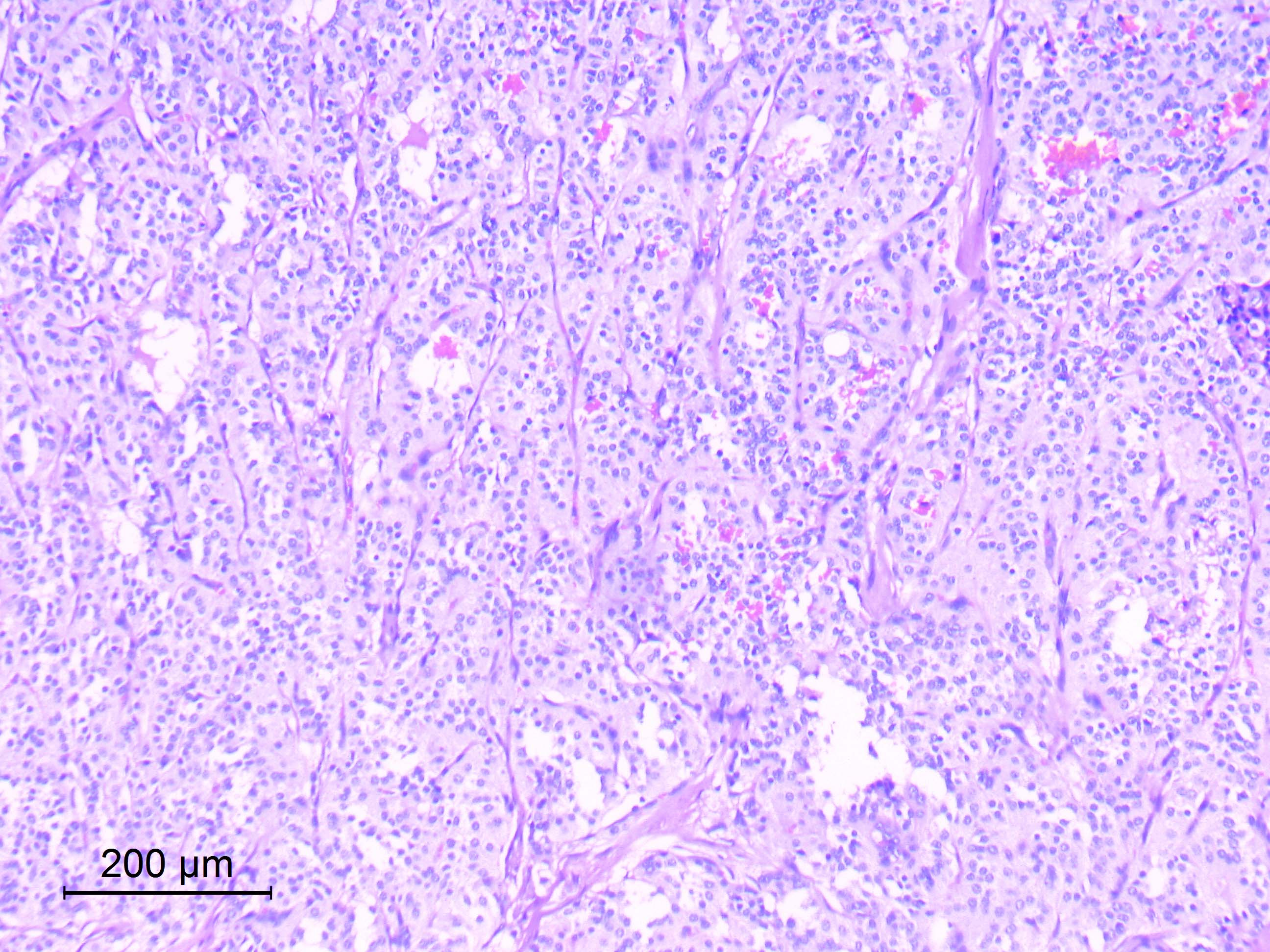
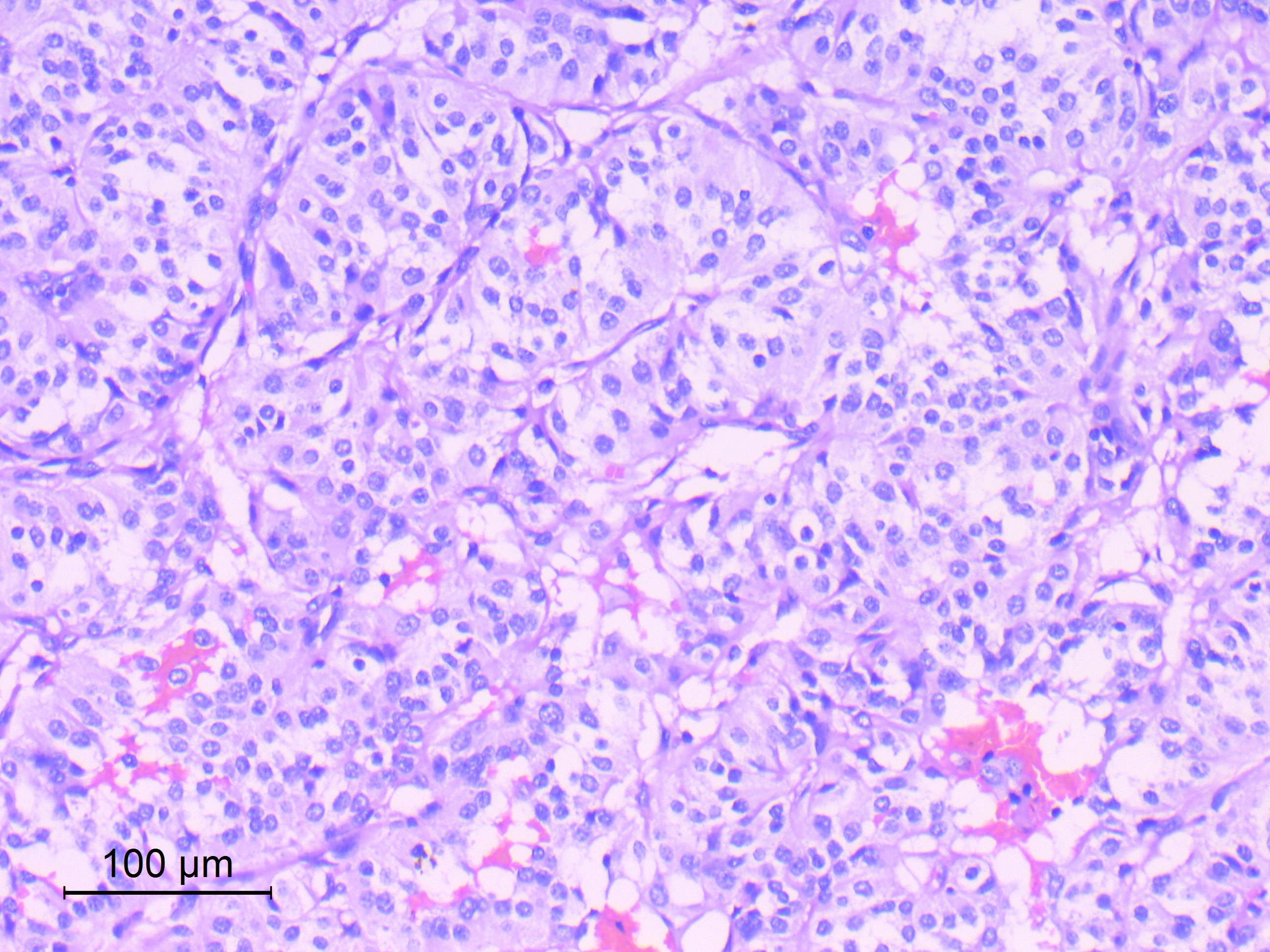
Zellballen architecture
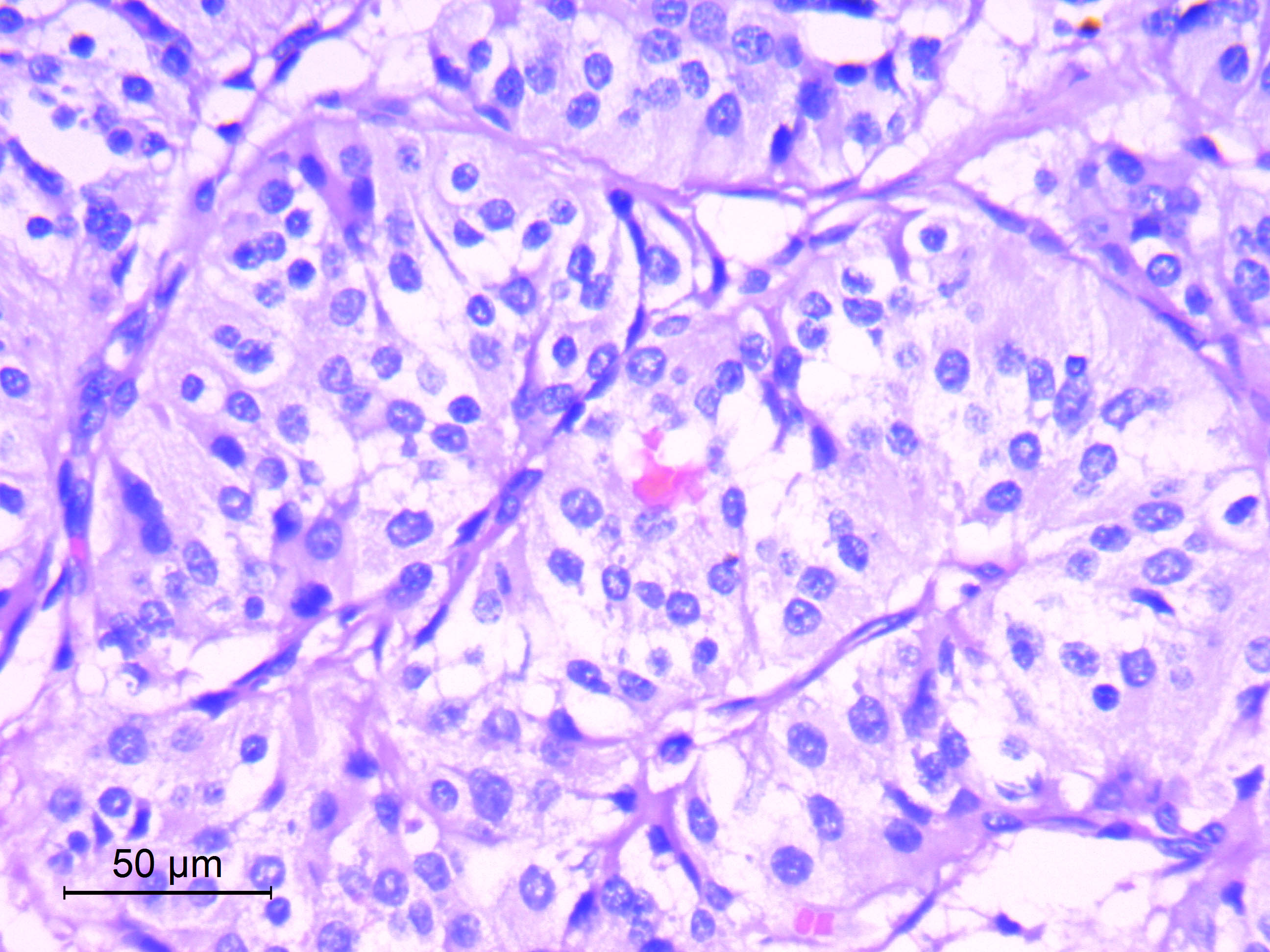
Zellballen architecture

Salt and pepper chromatin

Focal sclerosis
Reticulin

Synaptophysin

S100 positive sustentacular cells
S100 positive chief and sustentacular cells

GFAP negative

GFAP positive sustentacular cells
EMA
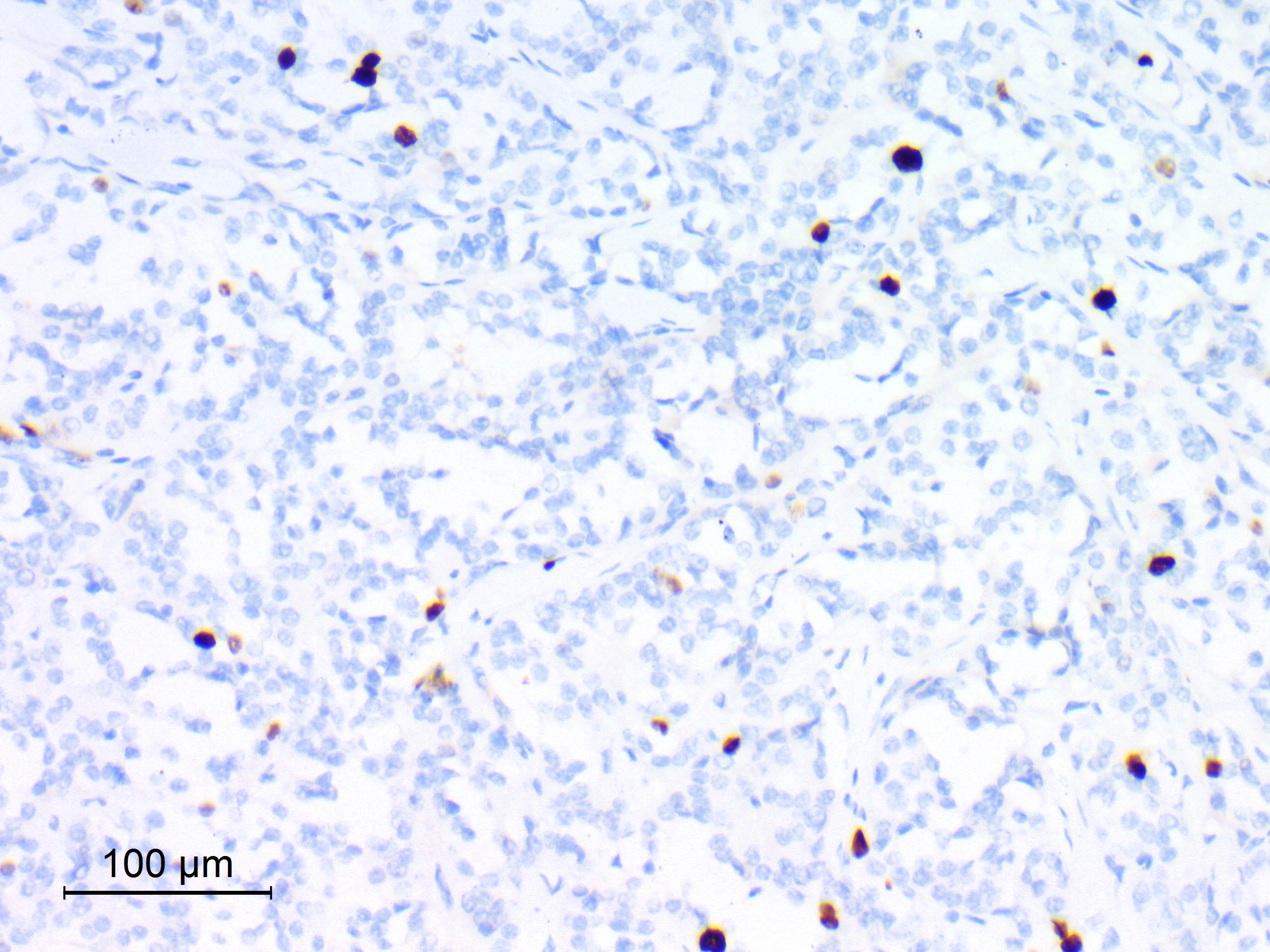
Low Ki67
- Largely discohesive smears that spread easily
- Uniform cells with plasmacytoid, triangular and polygonal shape and round to oval nuclei with salt and pepper chromatin (Lacruz: Central Nervous System Intraoperative Cytopathology, 2014 Edition, 2013)
- May show nuclear atypia
- Sharp cell borders (no fibrillar processes)
- May show ganglion cells
- Chief cells
- Synaptophysin (91 - 100%) (J Neurooncol 2015;122:539, Histopathology 1997;31:167, Ann Diagn Pathol 2022;57:151887)
- Chromogranin A (91 - 100% with variable intensity) (J Neurooncol 2015;122:539, Acta Neuropathol 2020;140:907, Histopathology 1997;31:167)
- Neuron specific enolase (NSE) (100%) (J Neurooncol 2015;122:539, Histopathology 1997;31:167)
- Cytokeratin (CAM5.2, AE1 / AE3, CK18 and MNF116) (21 - 100% with variable staining patterns): diffuse, strong with occasional prominent paranuclear staining (65%), focal or rare positive cells (35%) (Histopathology 1997;31:167, Acta Neuropathol 2020;140:907, Acta Neuropathol 2020;140:893, Ann Diagn Pathol 2022;57:151887)
- Vimentin (58%) (J Neurooncol 2015;122:539)
- SDHB: retained (Acta Neuropathol 2020;140:907)
- HOXB13 (Neuropathol Appl Neurobiol 2021;47:889)
- Sustentacular cells
- S100: inconsistent (84 - 100%) (J Neurooncol 2015;122:539, Histopathology 1997;31:167, Ann Diagn Pathol 2022;57:151887, Acta Neuropathol 2020;140:907)
- SOX10
- Ki67: heterogeneous proliferative activity; mean: 5.7% (range: 1 - 10%) (World Neurosurg 2020:142:e66, Acta Neuropathol 2020;140:893)
- Reticulin histochemical stain: emphasizes the lobular architecture
- Epithelial membrane antigen (EMA) (J Neurooncol 2015;122:539)
- GATA3 (Acta Neuropathol 2020;140:907)
- Creatine kinase (J Neurooncol 2015;122:539)
- GFAP: may be focally positive in chief cells (9 - 30%) and sustentacular cells (Ann Diagn Pathol 2022;57:151887, Histopathology 1997;31:167, Brain Pathol 2005;15:169)
- S100: may be focally positive in chief cells (Ann Diagn Pathol 2022;57:151887)
- Serotonin (5-HT) and various neuropeptides: may be positive in chief cells (somatostatin [34%], leuenkephalin [47%] and metenkephalin) (Histopathology 1997;31:167)
- Neurofilament proteins: may be positive in chief cells with paranuclear globular staining (13%) (Histopathology 1997;31:167)
- Chief cells
- Dense core (neurosecretory) granules with no evidence of catecholamine production (Neurosurg Rev 2022;45:103)
- Prominent, rough endoplasmic reticulum and Golgi apparatus (Neurosurg Rev 2022;45:103)
- Paranuclear intermediate filament whorls
- Sustentacular cells
- Electron dense with elongated processes
- Contain some intermediate filaments
- Lack neurosecretory granules
Images hosted on other servers:
Numerous perinuclear dense core granules
Prominent rough endoplasmic reticulum and Golgi apparatus
- Distinct DNA methylation and chromosomal copy number profiles as opposed to those of paragangliomas arising from other locations (Acta Neuropathol 2020;140:907)
- DNA methylation profiling and clustering analysis showed that CEPs are epigenetically distinct from extraspinal paragangliomas, pheochromocytomas, other neuroendocrine tumors and glial or ependymal neoplasms of the spinal cord (Acta Neuropathol 2020;140:907)
- Copy number analysis revealed diploid genomes in the vast majority of CEPs, whereas extraspinal paragangliomas were mostly aneuploid with recurrent trisomy 1q and monosomies of 1p, 3 and 11, none of which were present in the cohort of CEP (Acta Neuropathol 2020;140:907)
- RNA and DNA exome sequencing revealed that SDH mutations are absent in cauda equina paragangliomas (Acta Neuropathol 2020;140:893)
- CEPs are not driven by recurrent oncogenic gene fusions; fusion genes known to be relevant in paragangliomas / pheochromocytomas and associated with poor prognosis (e.g., MAML3 fusions) were not observed in CEPs (Acta Neuropathol 2020;140:893)
Images hosted on other servers:

DNA methylation cluster analysis of CEPs and other tumors

Chromosomal copy number analysis
Case 8: paraganglioma of the filum terminale
- Cauda equina mass, total resection:
- Cauda equina neuroendocrine tumor (cauda equina paraganglioma), CNS WHO grade 1 (see comment)
- Comment: Encapsulated tumor with zellballen architecture highlighted by reticulin histochemical stain. The tumor is composed of chief cells (synaptophysin positive) arranged in nests surrounded by S100 positive sustentacular cells.
- Molecular genetics: methylation profile of cauda equina neuroendocrine tumor
- Consider the possibility of paraganglioma for any cauda equina / filum terminale mass, as CEP is rarely diagnosed preoperatively
- Myxopapillary ependymoma:
- Perivascular myxoid change (Alcian blue+) (Histopathology 1997;31:167)
- Little reticulin
- Diffuse GFAP+ (Appl Immunohistochem Mol Morphol 2013;21:485)
- Synaptophysin- (Diagn Pathol 2008;3:40)
- Smears: tissue fragments, cells with fibrillar processes and smaller, darker nuclei
- Spinal ependymoma (including tanycytic pattern):
- Intramedullary, commonly cervical or cervicothoracic, demarcated tumors
- No capsule
- Little reticulin
- Tanycytic pattern characterized by spindle shaped cells with fascicular architecture
- GFAP+, EMA+ (dot-like microlumina) (Turk Neurosurg 2012;22:353, Ann Diagn Pathol 2022;57:151887)
- Chromogranin- and synaptophysin- (J Neurooncol 2005;74:1)
- Smears: tissue fragments, cells with fibrillar processes and smaller, darker nuclei
- Schwannoma:
- Biphasic with Antoni A and B areas
- Pericellular reticulin
- Strongly and diffusely S100+, SOX10+ (Arq Bras Neurocir 2018;37:105)
- Synaptophysin- and chromogranin- (Hum Pathol 2012;43:650)
- Smears: tissue fragments, spindle cells
- Hemangioblastoma:
- Vacuolated stromal cells
- Stromal cells variably lipid laden
- Oil red O+ staining
- Inhibin alpha+
- Chromogranin- and synaptophysin- (Vojnosanit Pregl 2004;61:273)
- Meningioma:
- Whorls, psammoma bodies
- EMA+ (J Neuropathol Exp Neurol 2017;76:289)
- Metastatic carcinoma:
- Pilocytic astrocytoma:
- Biphasic piloid and spongy areas (Acta Neuropathol 2015;129:775)
- GFAP+
- Little reticulin
- Melanocytoma:
- Most spinal melanocytomas arise at cervical and thoracic levels
- Positive for melanocytic markers (e.g., S100, SOX10, HMB45, MelanA)
- Chromogranin- and synaptophysin- (Radiol Case Rep 2015;10:1010)
- Cytokeratin- (Radiol Case Rep 2015;10:1010)
- WHO Classification of Tumours Editorial Board: Central Nervous System Tumours, 5th Edition, 2022, Burger: Diagnostic Pathology - Neuropathology, 2nd Edition, 2016, Burger: Tumors of the Central Nervous System (AFIP Atlas of Tumor Pathology), 1st Edition, 2007, Burger: Surgical Pathology of the Nervous System and its Coverings, 4th Edition, 2001
A 49 year old man presented with lower back pain and sciatica for a duration of 3 months. MRI showed cauda equina intradural extramedullary enhancing vascular mass. The received gross specimen was a delicately encapsulated oval mass with a red-brown cut section. The images shown above depict the histological features of the lesion and synaptophysin immunostain. What is the most likely diagnosis?
- Cauda equina paraganglioma
- Hemangioblastoma
- Myxopapillary ependymoma
- Schwannoma
Comment Here
Reference: Cauda equina neuroendocrine tumor
- Encapsulated tumors with zellballen architecture and synaptophysin immunoreactivity
- It is commonly functioning with clinical features of catecholamine hypersecretion
- It shares common genetic profile with extraspinal paragangliomas and pheochromocytomas
- Recurrence rate after total resection is high
Comment Here
Reference: Cauda equina neuroendocrine tumor
- Rare, well differentiated, intraventricular neoplasm with neuroepithelial differentiation, typically arising near the foramen of Monro
- Rare tumor, comprising 0.1 - 0.5% of all primary CNS neoplasms (J Neurooncol 2016;126:193)
- Intraventricular localization, usually involving the lateral or third ventricle(s) (Brain Pathol 1993;3:297)
- Clinical symptoms generally result from increased intracranial pressure due to obstructive hydrocephalus (Int J Radiat Oncol Biol Phys 2007;67:1145, J Clin Neurosci 2013;20:679)
- Microscopically appears as sheets of uniform, small - medium, round cells with fine chromatin stippling (salt and pepper) and occasional perinuclear clearing, interspersed with patches of fibrillary matrix (Brain Pathol 1993;3:297)
- CNS WHO grade 2 (Brain Tumor Res Treat 2016;4:49)
- Central neurocytoma
- ICD-O:
- ICD-10:
- ICD-11:
- ~0.1 - 0.5% of all primary brain tumors (J Neurooncol 2016;126:193)
- Overall incidence per 100,000 person years: 0.022, with individual rates varying by reported race (J Neurooncol 2019;143:123)
- Asian / Pacific Islander: 0.038
- Non-Hispanic White: 0.035
- Black: 0.026
- Hispanic White: 0.020
- Mean age at presentation: 20 - 34 years (J Neurooncol 2019;143:123)
- F:M = 1.02:1 (Brain Pathol 1993;3:297)
- Intraventricular mass, classically arising in the supratentorial ventricular system (Brain Pathol 1993;3:297)
- Anterior lateral ventricle (~50%)
- Lateral and third ventricle (~15%)
- Both lateral ventricles (~13%)
- Documented sites of origin include
- Foramen of Monro
- Septum pellucidum
- Corpus callosum
- Hypothalamus
- Large tumors may involve multiple sites
- Rarely reported in fourth ventricle and spinal cord (J Neurosurg 1994;81:288, Acta Neuropathol 2005;109:346, Mol Clin Oncol 2018;8:539)
- Neurocytomas occurring outside the ventricular system are termed extraventricular neurocytomas and are considered distinct entities (Acta Neurochir (Wien) 2014;156:349)
- Cell of origin
- Currently unknown, though favored to be a neuroglial progenitor cell due to dual differentiation potential (capable of forming both neurons and glial cells) (J Neurosci Res 1998;51:526, Lab Invest 1991;64:585)
- Frequent occurrence within lateral ventricle suggests origination from residual germinal matrix cells of subependymal plate (Lab Invest 1991;64:585, No Shinkei Geka 1995;23:1083)
- Infratentorial variants may arise from circumventricular organs (Acta Neuropathol 2005;109:346)
- Unknown at this time
Images hosted on other servers:
Coronal section diagram
- Most common presenting symptoms are related to increased intracranial pressure that is due to obstructive hydrocephalus (Int J Radiat Oncol Biol Phys 2007;67:1145, J Clin Neurosci 2013;20:679)
- Headache
- Vomiting
- Visual field changes
- Papilledema
- Presenting symptoms generally present for a short time prior to diagnosis (median: 1.7 - 3 months) (Int J Radiat Oncol Biol Phys 2007;67:1145)
- Based primarily on histologic and immunophenotypic features (Brain Pathol 1993;3:297)
- Requires intraventricular localization, oligodendroglioma-like cytology and synaptophysin expression
- Correlation with radiologic studies (e.g., MRI, CT) is essential; supportive findings include
- Intraventricular localization (required)
- Enhancing, multicystic mass
- MRI: T1 isointense, T2 heterogeneous, FLAIR hyperintense
- CT: may show calcifications
- Methylation profiling may aid diagnosis in unresolved cases (J Neurooncol 2022;159:725)
- Noncontrast computed tomography (J Clin Neurosci 2013;20:679, Neurosurg Clin N Am 2015;26:11)
- Highly variable appearance, often with mixed solid and cystic components that appear isodense and hypodense, respectively, to surrounding brain parenchyma
- Calcifications may be seen, typically partial or punctate
- Evidence of hydrocephalus or hemorrhage may be visible
- Magnetic resonance imaging (J Clin Neurosci 2012;19:681, J Clin Neurosci 2013;20:679, Neurosurg Clin N Am 2015;26:11)
- Classically an intraventricular mass with a multicystic, soap bubble appearance characterized by T1 and T2 isointense solid components and T2 hyperintense, fluid filled cysts
- Peripheral cyst walls may form spicules and cause undulation of the adjacent lateral ventricle wall (scalloping)
- Calcifications and flow voids may be seen on T1 sequences
- Heterogenous contrast enhancement
- No surrounding peritumoral edema on T2 / FLAIR
- Proton magnetic resonance spectroscopy (Eur Radiol 2009;19:2049, J Clin Neurosci 2012;19:681, Neurosurg Clin N Am 2015;26:11)
- Characteristic glycine peak at 3.55 ppm
- Prominent choline peak
- Inverted alanine peak
Images hosted on other servers:
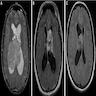
Intraventricular mass on MRI
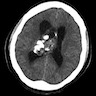
Calcifications on CT
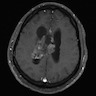
T1+C MRI
- Generally favorable prognosis (J Neurooncol 2016;126:193)
- 5 year overall survival rate: 96%
- 10 year overall survival rate: 82%
- Extent of resection is the only independent prognostic factor (Am J Surg Pathol 2012;36:220)
- Elevated proliferative index is associated with more aggressive behavior, with various thresholds proposed, but no single optimal cutoff established
- Proposed Ki67 thresholds: ~2 - 4% (J Neurooncol 2018;140:669, Neurology 2004;62:987, J Neuropathol Exp Neurol 1997;56:551, J Neurooncol 2016;126:193)
- e.g., Ki67 ≤ 4%: 90% 2 year progression free survival; Ki67 > 4%: 48% 2 year progression free survival (J Neurooncol 2016;126:193)
- Mitotic threshold of 3/10 HPF has been proposed (Am J Surg Pathol 2012;36:220, Int J Radiat Oncol Biol Phys 2007;67:1145)
- Proposed Ki67 thresholds: ~2 - 4% (J Neurooncol 2018;140:669, Neurology 2004;62:987, J Neuropathol Exp Neurol 1997;56:551, J Neurooncol 2016;126:193)
- 8 year old girl with atypical central neurocytoma arising in the posterior fossa (BMJ Case Rep 2019;12:e231626)
- 13 year old boy with periventricular atypical central neurocytoma harboring unique WSR1::ATF1 fusion and MUTYH mutation (BMJ Case Rep 2019;12:bcr-2018-226455)
- 17 year old boy with right lateral ventricular central neurocytoma and intraventricular hemorrhage (Case Rep Surg 2022;2022:9731987)
- 48 year old man with atypical central neurocytoma and craniospinal drop metastases as well as concomitant pituitary macroadenoma (Asian J Neurosurg 2020;15:140)
- 79 year old woman with third ventricular central neurocytoma treated with gamma knife radiation (Medicine (Baltimore) 2018;97:e13657)
- Surgical resection is standard of care (J Clin Neurosci 2013;20:1193)
- When complete resection is not possible, adjuvant radiotherapy improves survival (World Neurosurg 2020;137:e176)
- Adjuvant radiotherapy not recommended with complete resection of atypical central neurocytomas (Front Neurol 2020;11:834)
Images hosted on other servers:
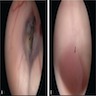
Endoscopic view of intraventricular tumor
- Gray, friable tissue (Brain Pathol 1993;3:297)
- May have macrocalcifications and hemorrhage (Brain Pathol 1993;3:297)
Images hosted on other servers:
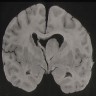
Intraventricular mass
- Sheets of isomorphous, round cells in a fibrillary background
- Minimal pleomorphism
- Necrosis or mitotic figures are typically absent
- Reference: Acta Cytol 2004;48:194
Contributed by Rebecca Yoda, M.D.

Cells with monotonous round nuclei
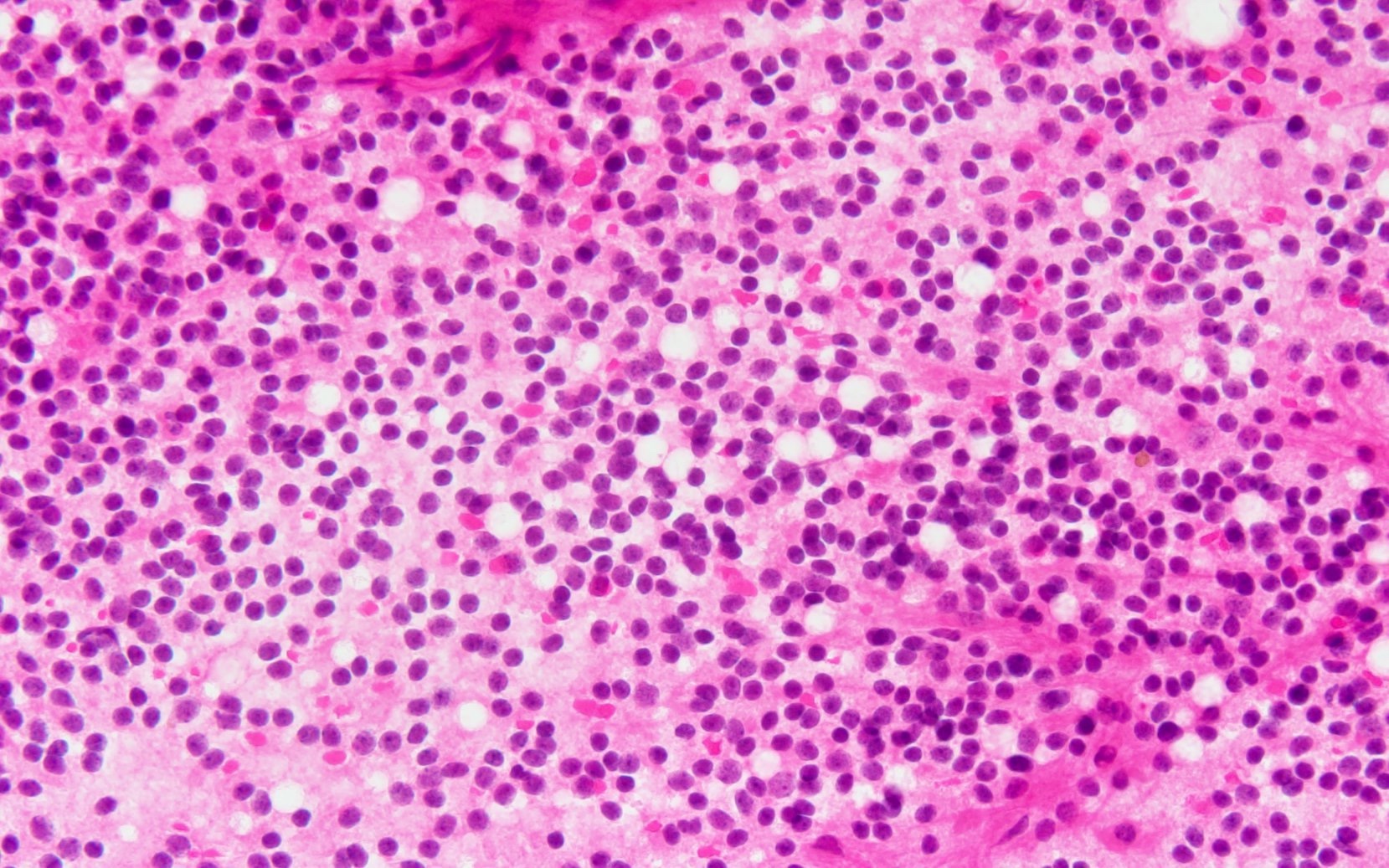
Intraoperative squash preparation
- Neuroepithelial neoplasm composed of uniform, small - medium cells growing in sheets with indistinct cytoplasm (Brain Pathol 1993;3:297)
- Nuclei are round with regular contours, finely stippled (salt and pepper) chromatin and micronucleoli
- Perinuclear clearing may be prominent (similar to oligodendrogliomas)
- Arborizing capillaries
- Large hyalinized blood vessels
- Other morphologic features may include
- Honeycomb-like architecture
- Patches of fibrillar, neuropil-like matrix mimicking pineocytomatous rosettes
- Perivascular pseudorosettes
- Homer-Wright rosettes
- Ganglioid cells
- Calcification is usually distributed throughout the tumor and may be prominent
- Lipomatous differentiation occurs rarely (World Neurosurg 2018;120:214)
- Hemorrhage is sometimes present; hemosiderin laden macrophages may be seen (Neurosurg Rev 2001;24:48)
- Designated as atypical central neurocytoma when anaplastic features are seen (J Neurosurg 1992;76:32, Brain Pathol 1993;3:297)
- Brisk mitotic activity
- Microvascular proliferation
- Necrosis
Contributed by Rebecca Yoda, M.D.
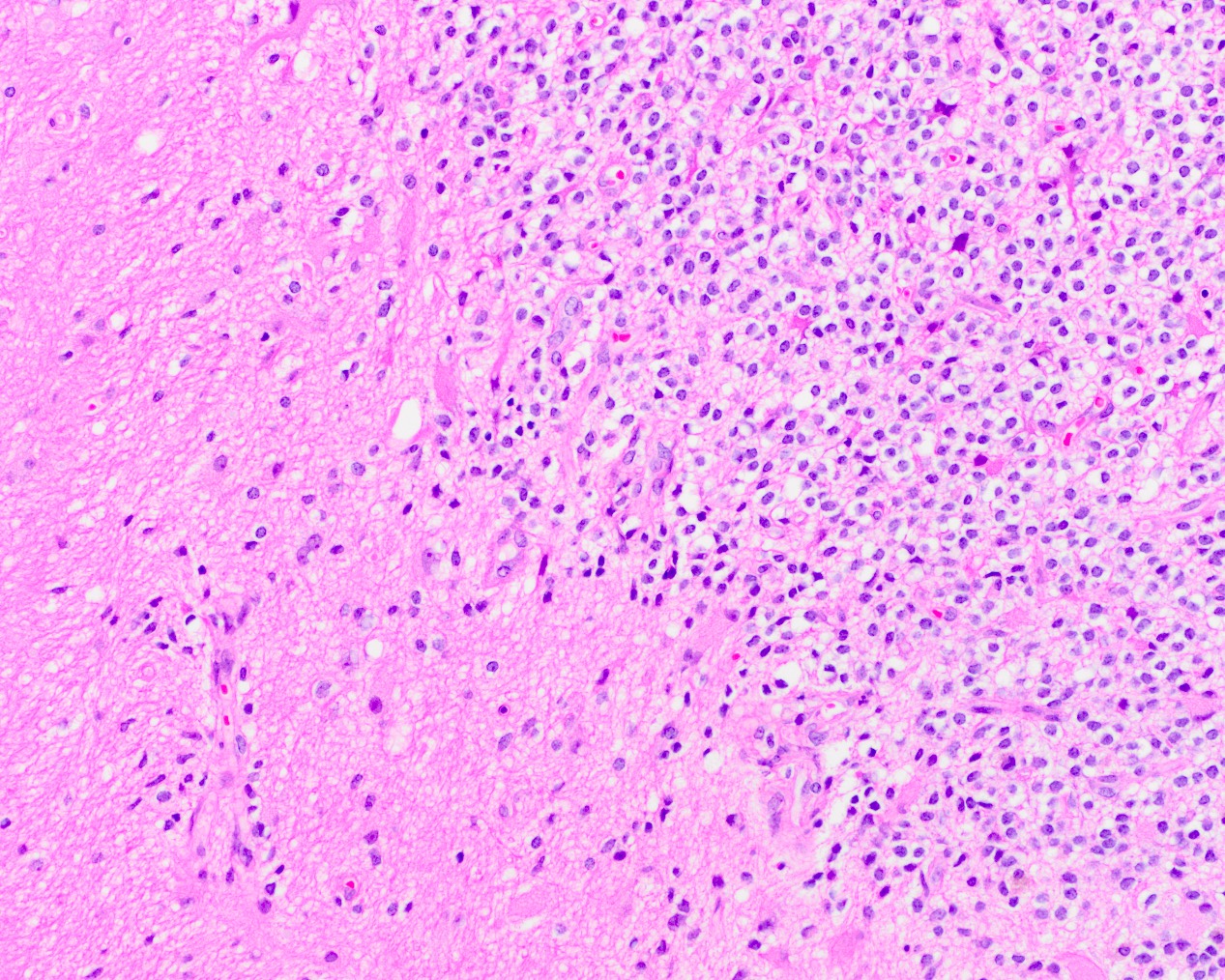
Circumscribed border
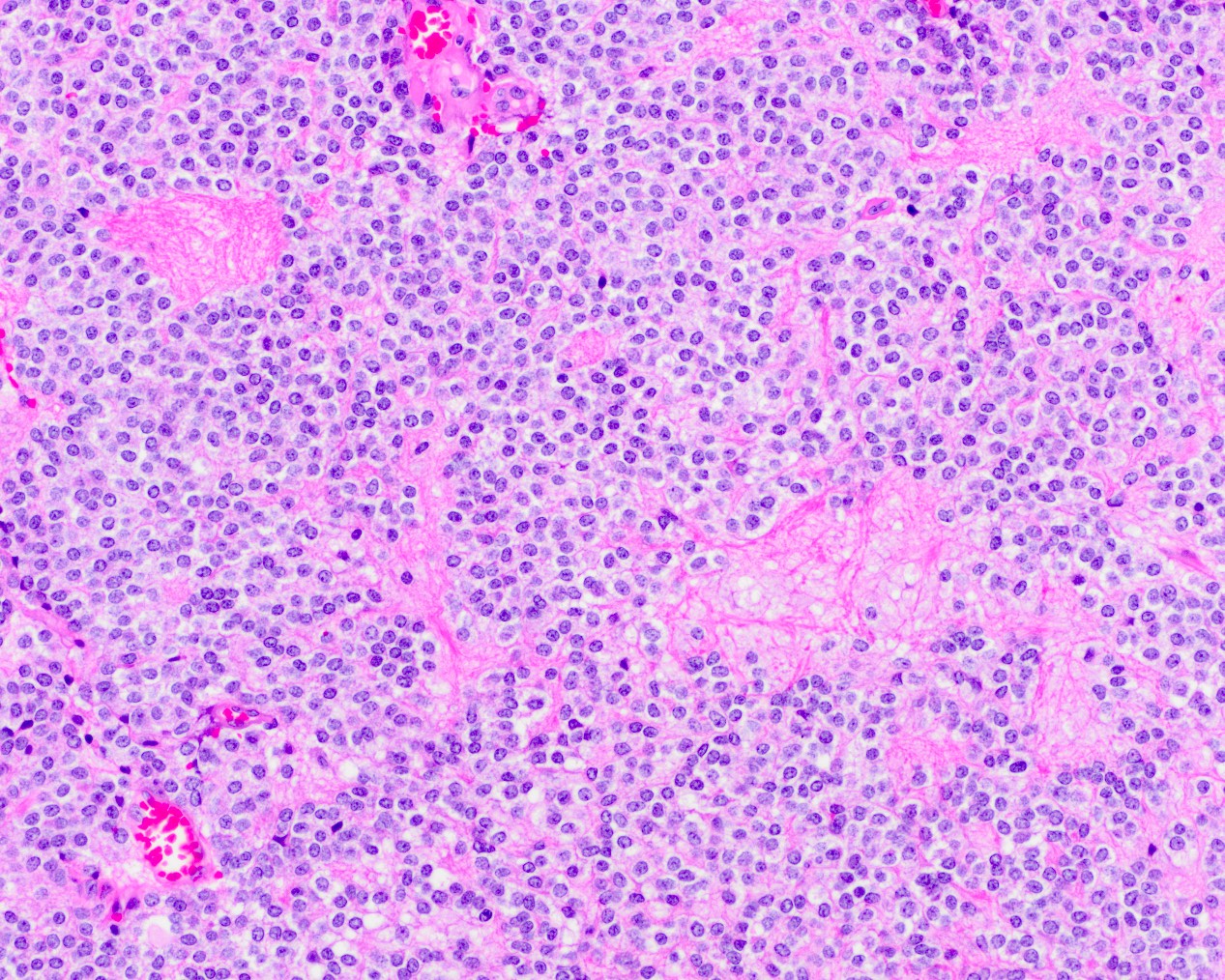
Fibrillary matrix
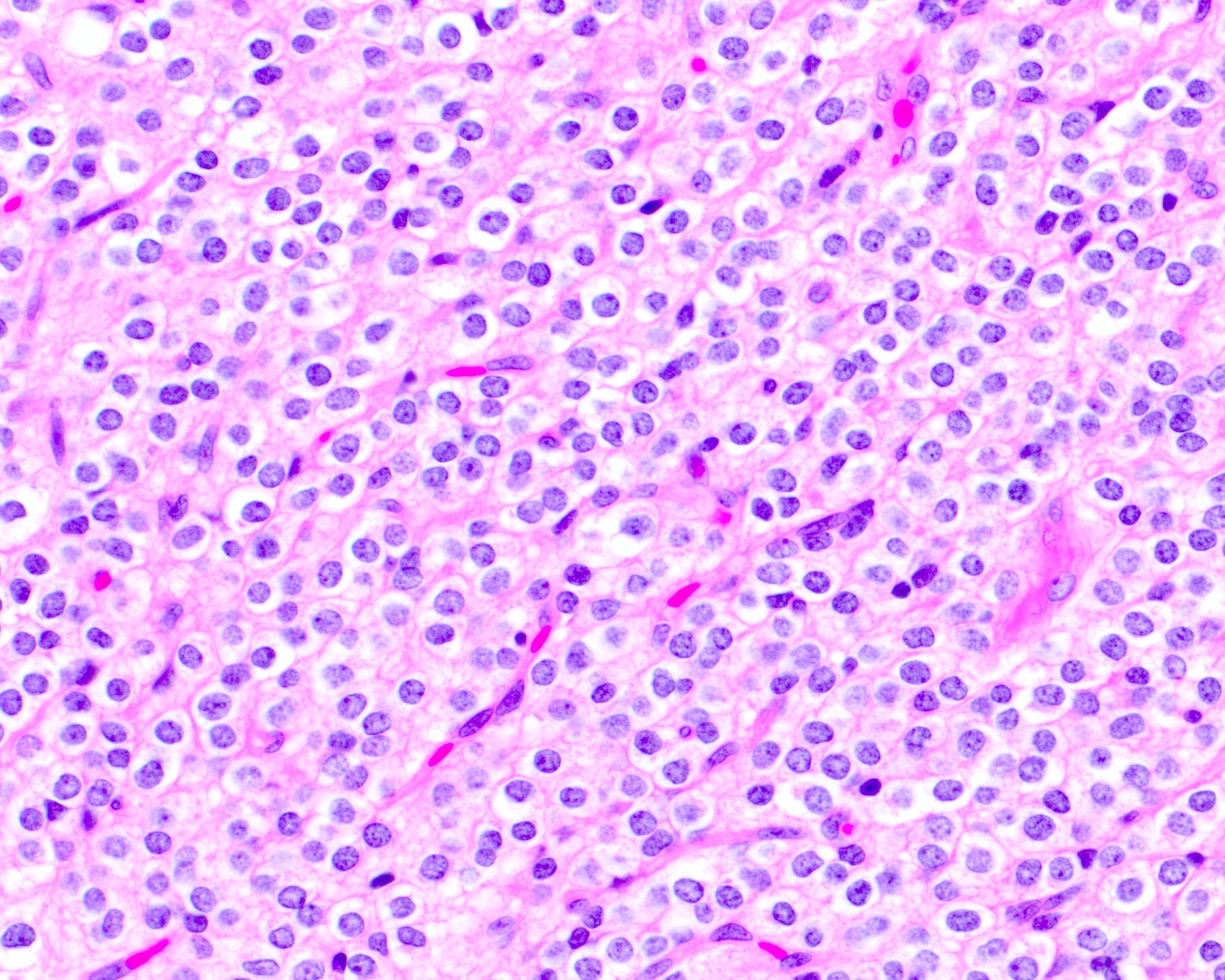
Perinuclear clearing
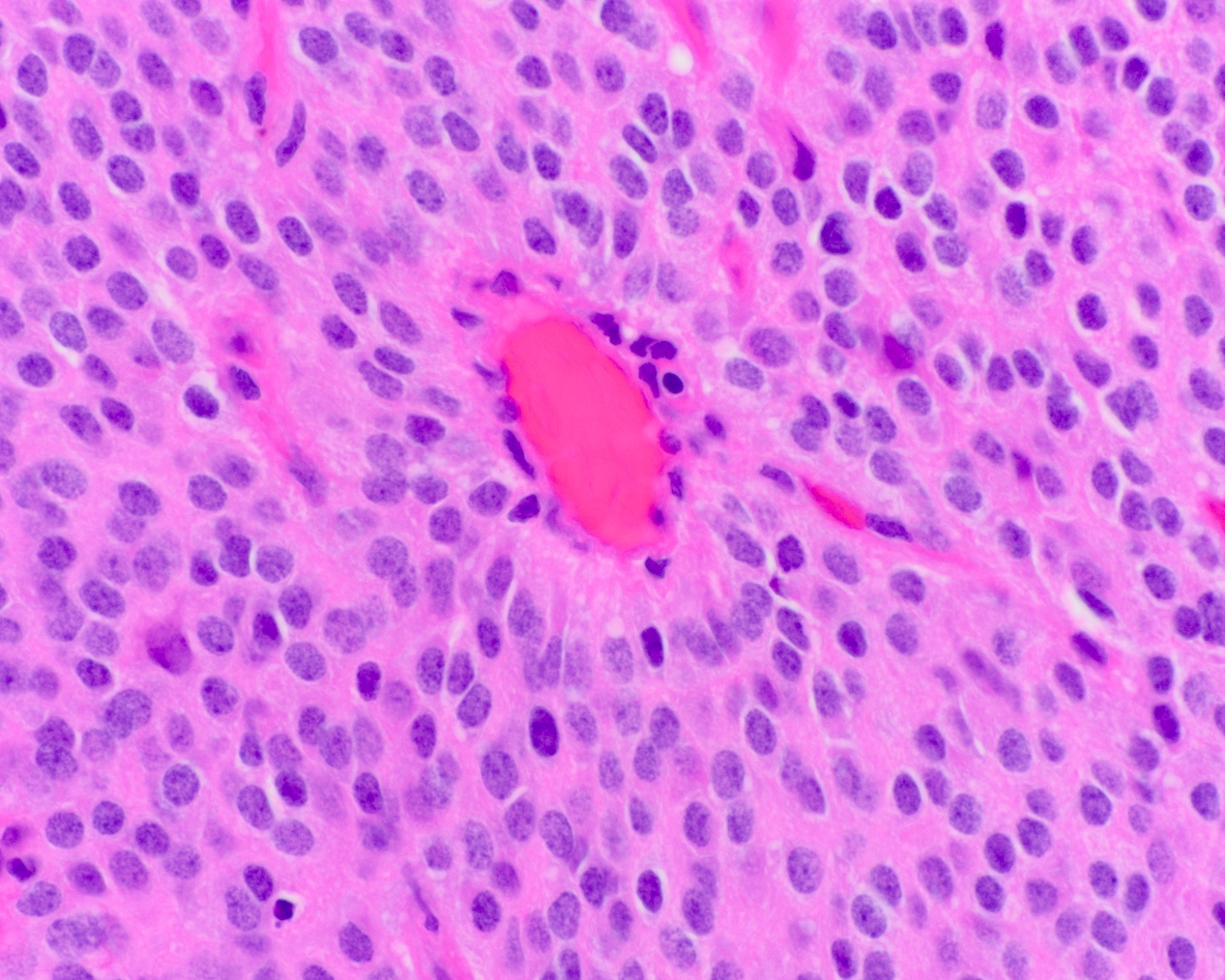
Perivascular pseudorosette
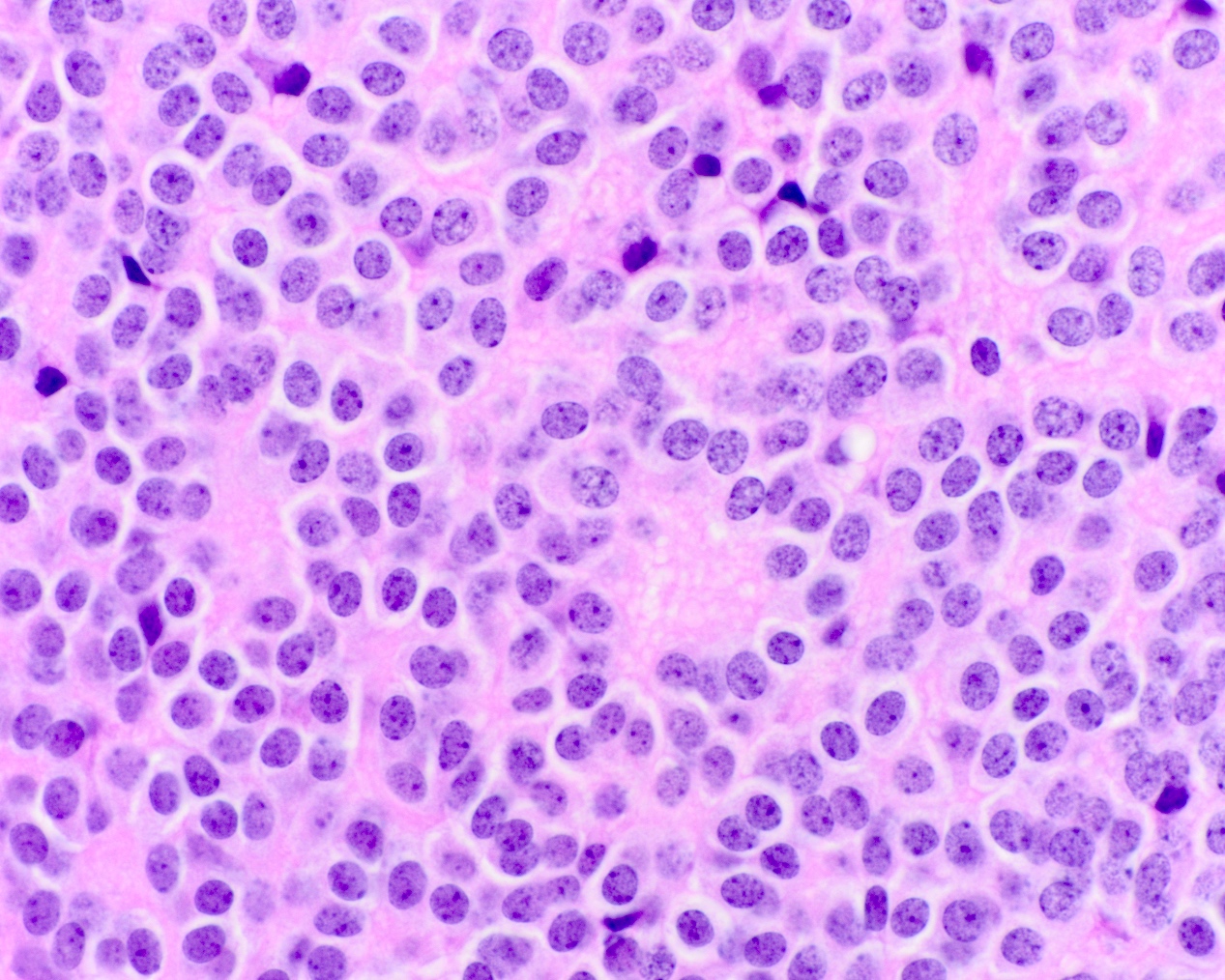
Neurocytic rosettes
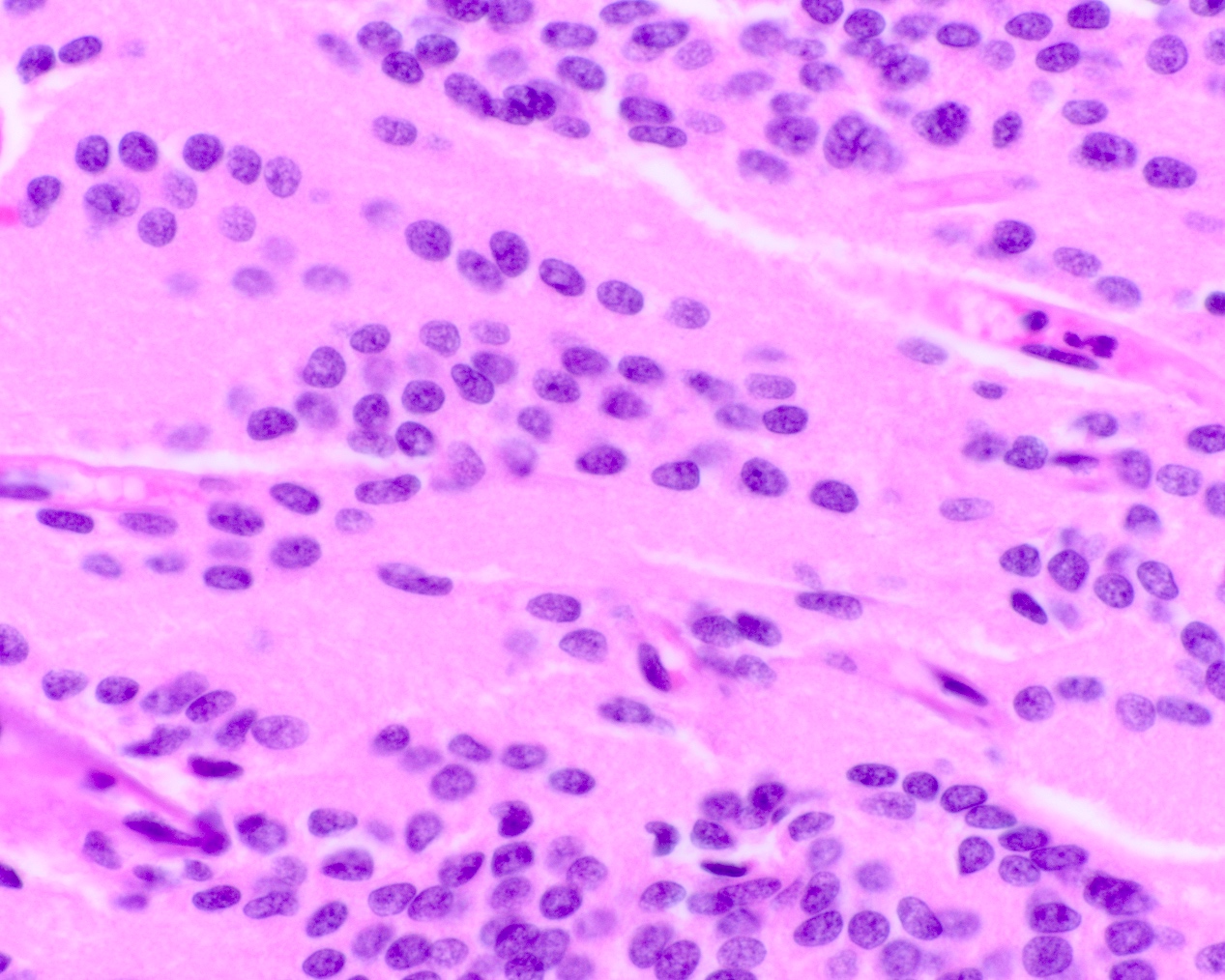
Linear arrangement

Calcifications
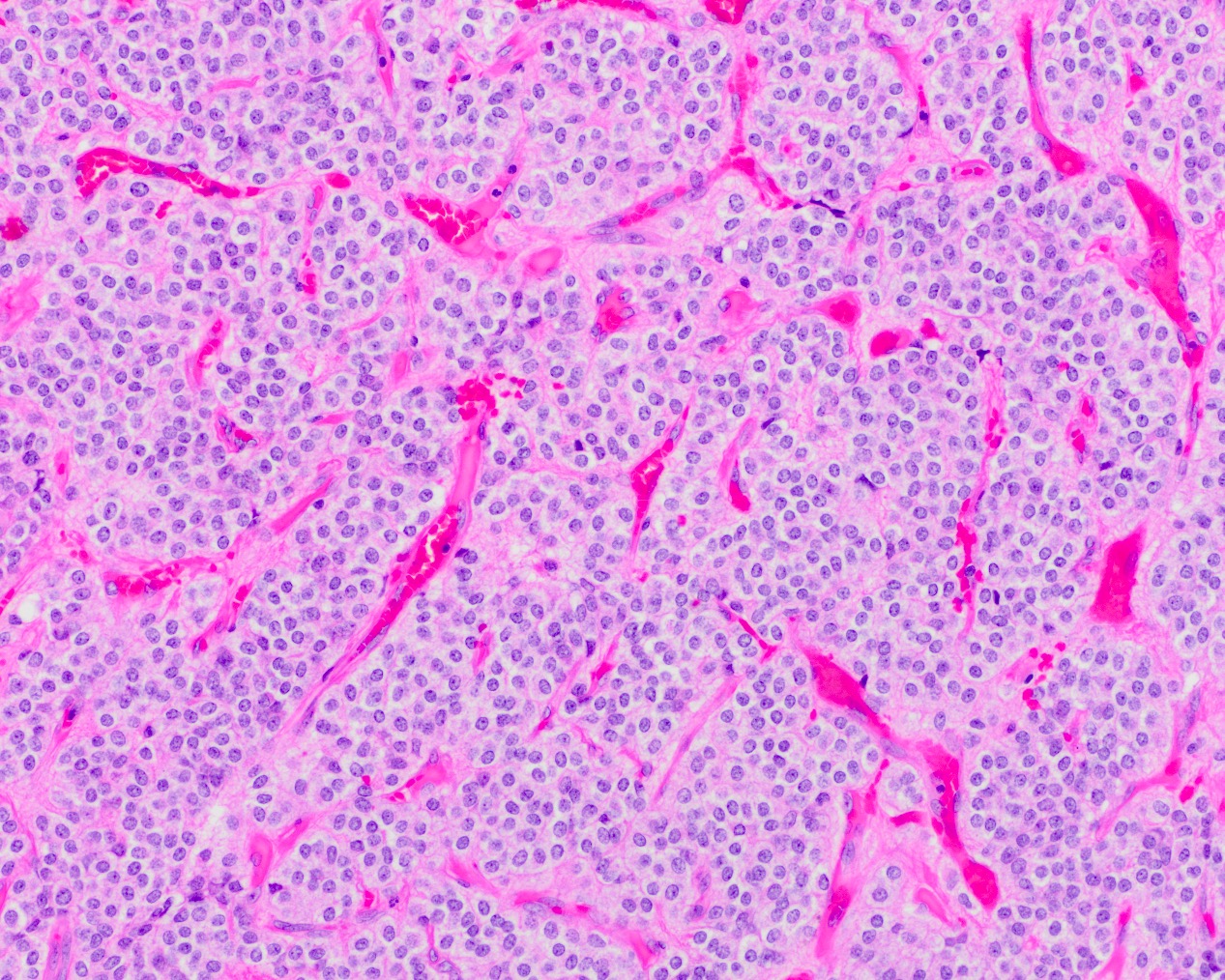
Branching capillaries
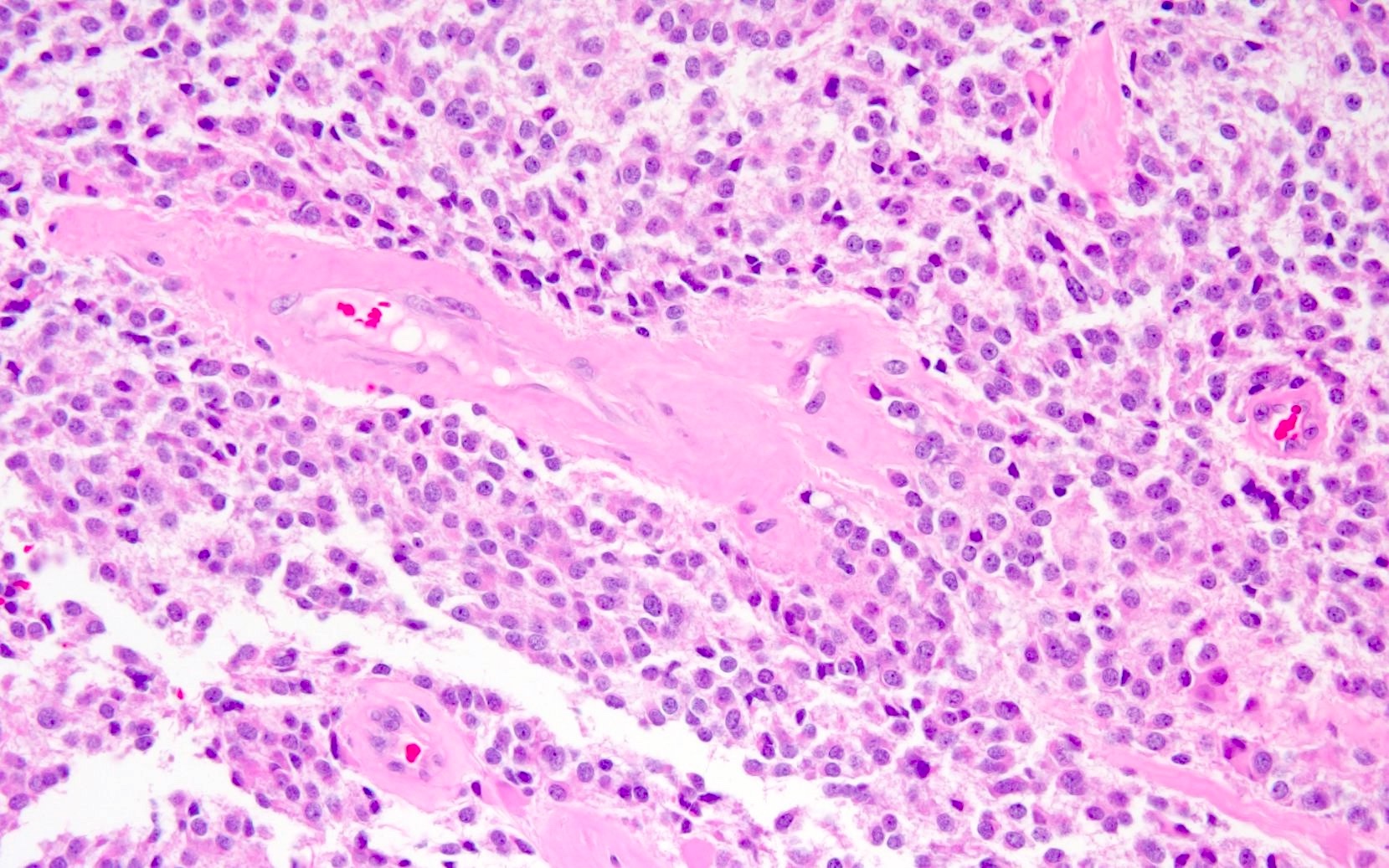
Hyalinized vessels
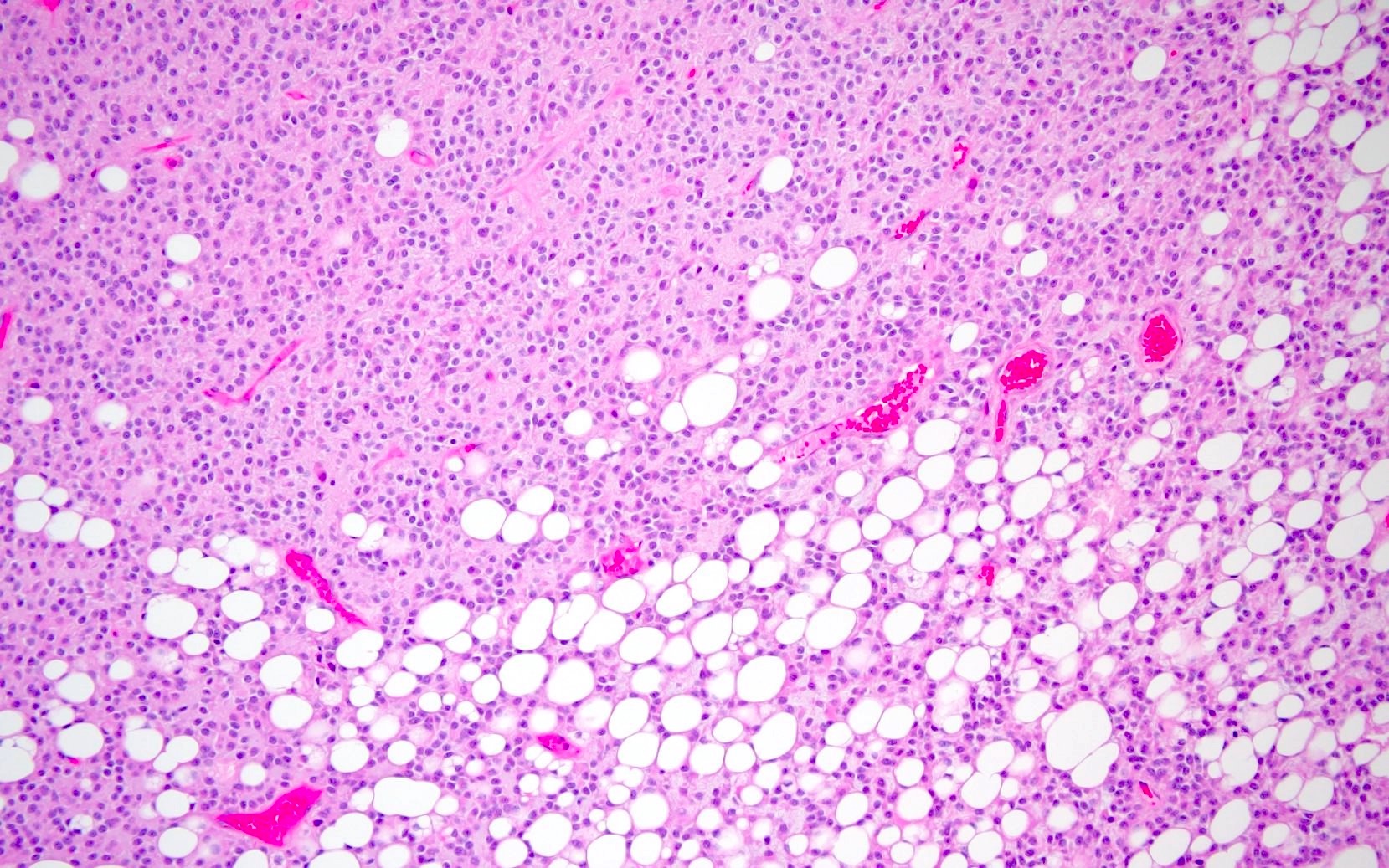
Lipomatous change
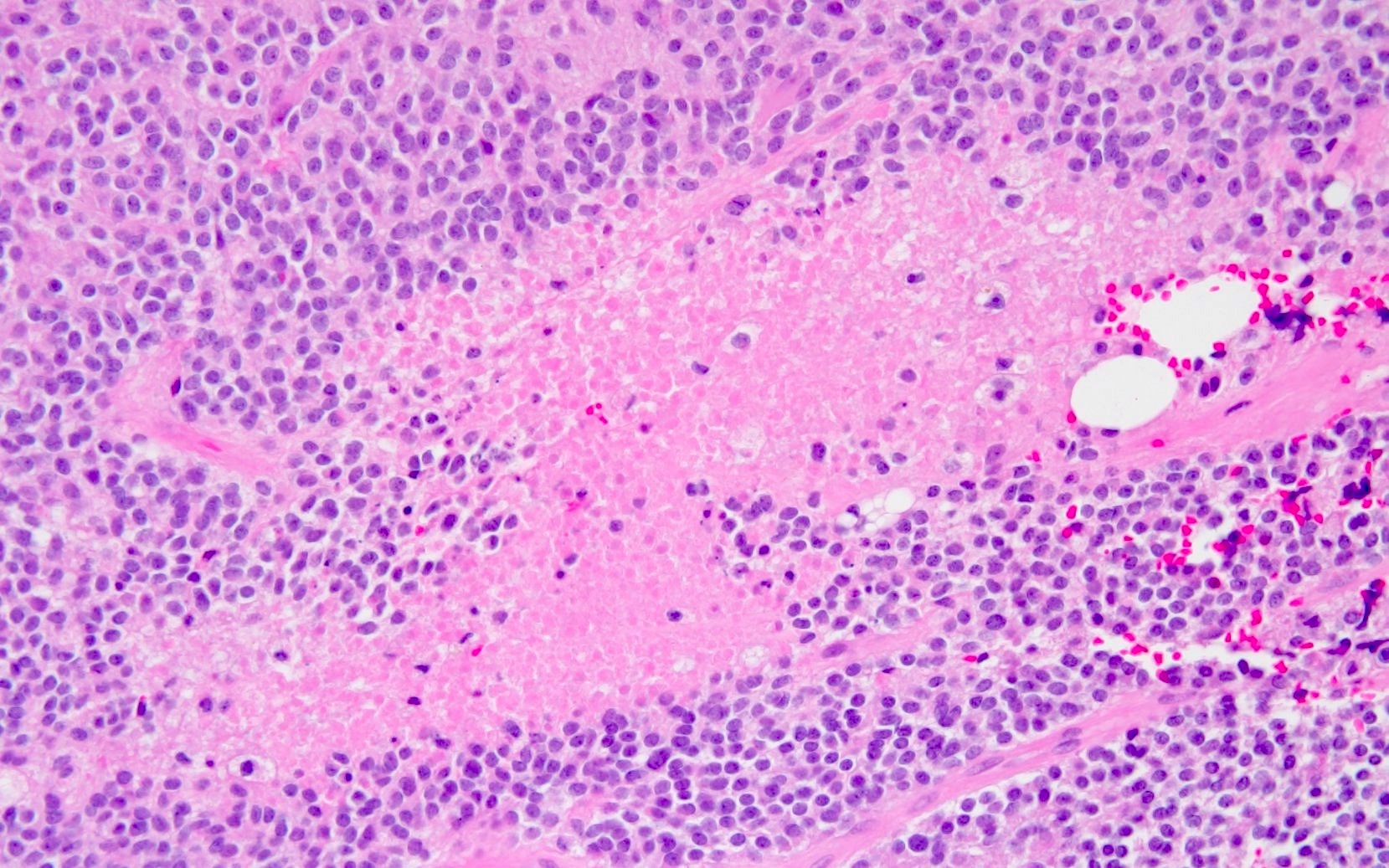
Necrosis
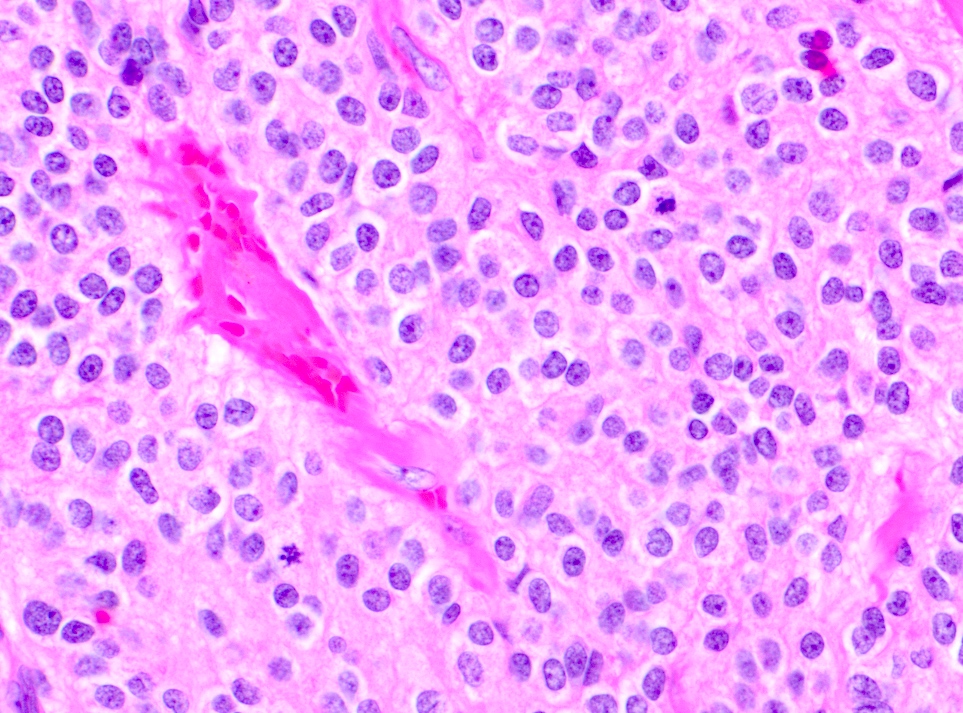
Mitotic activity
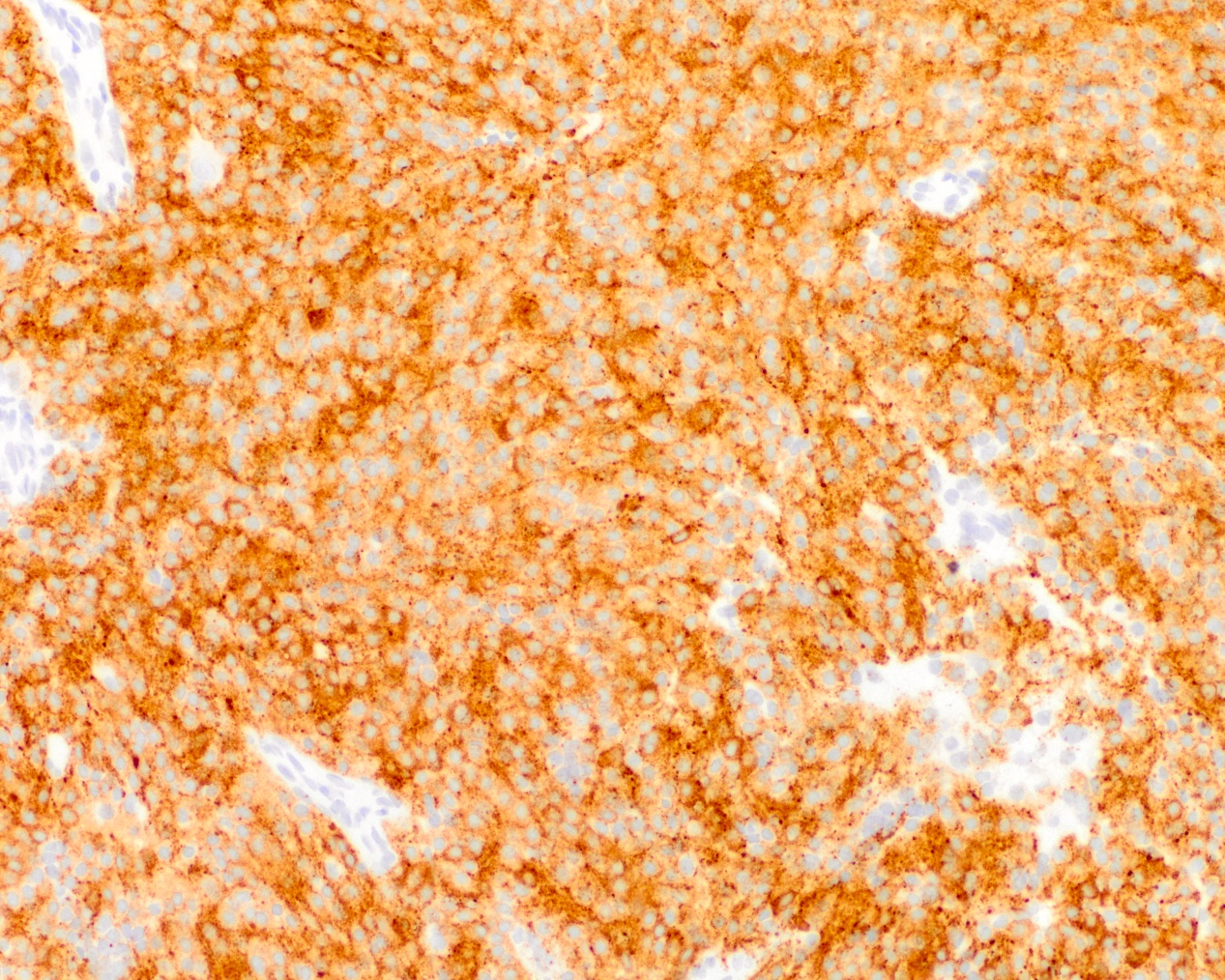
Synaptophysin IHC
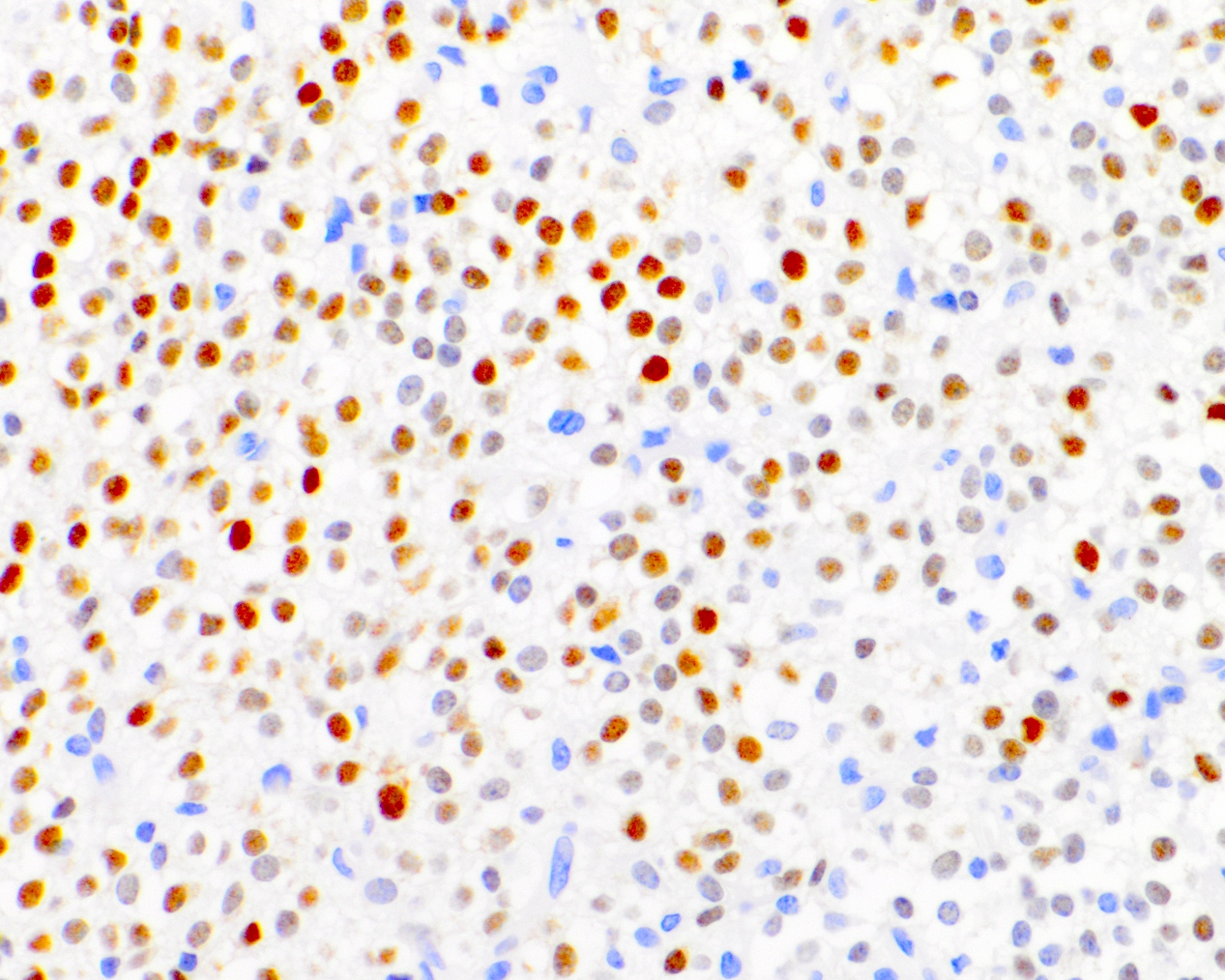
NeuN IHC
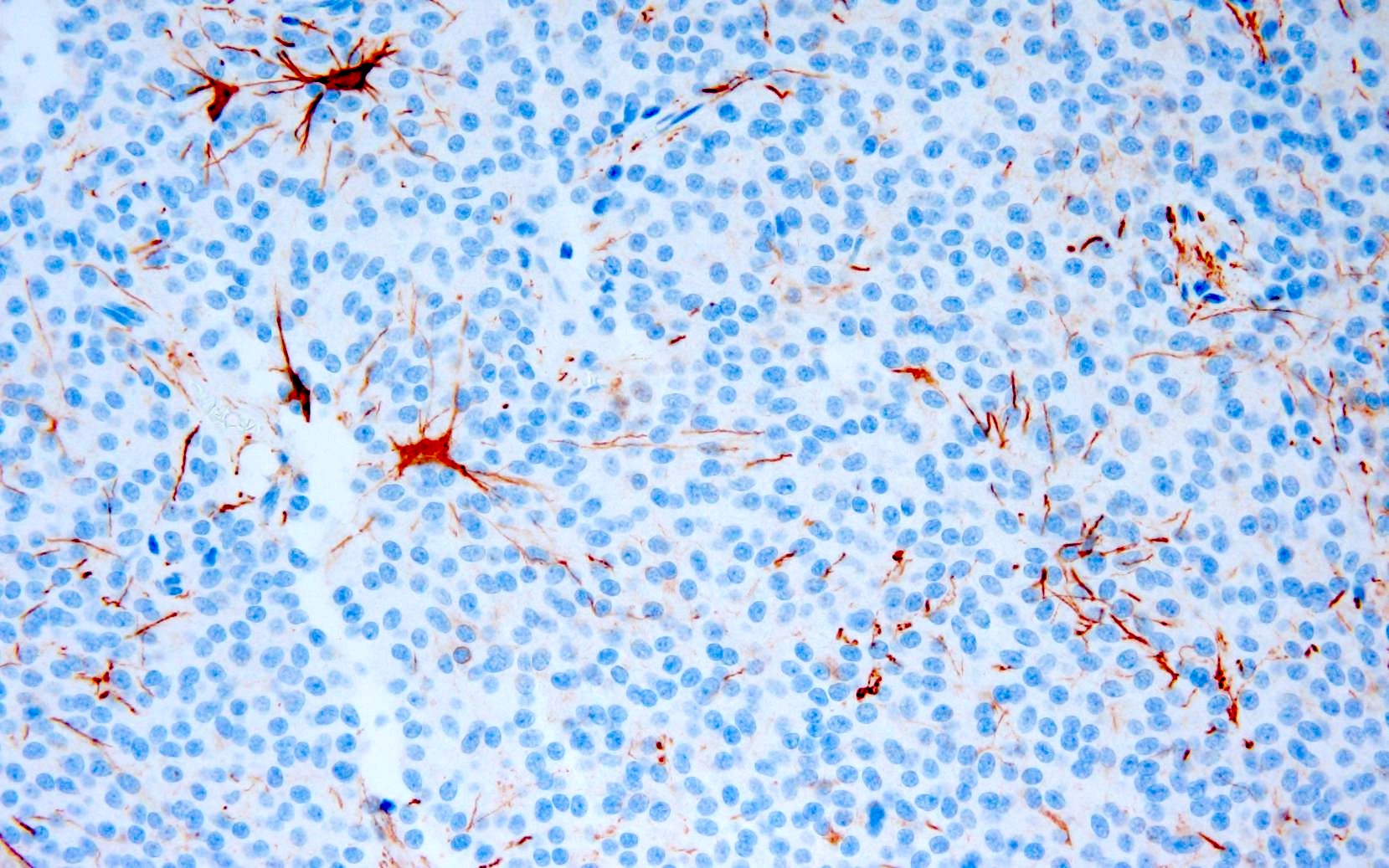
GFAP IHC
Contributed by Nazar M. T. Jawhar, M.D. (Case #119)
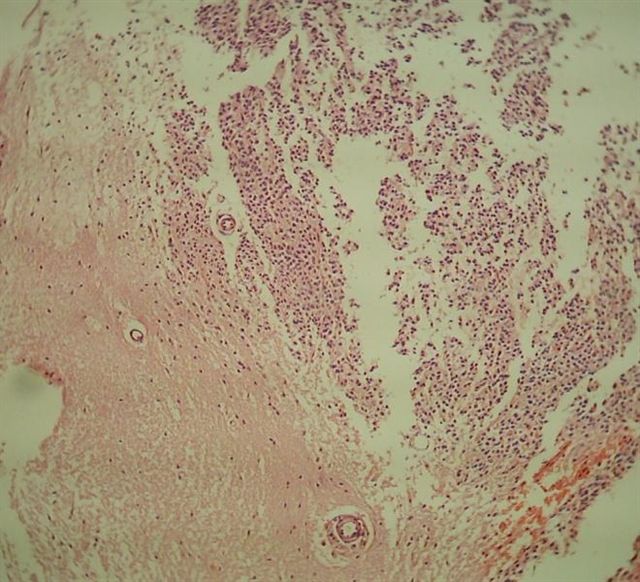
Circumscribed border
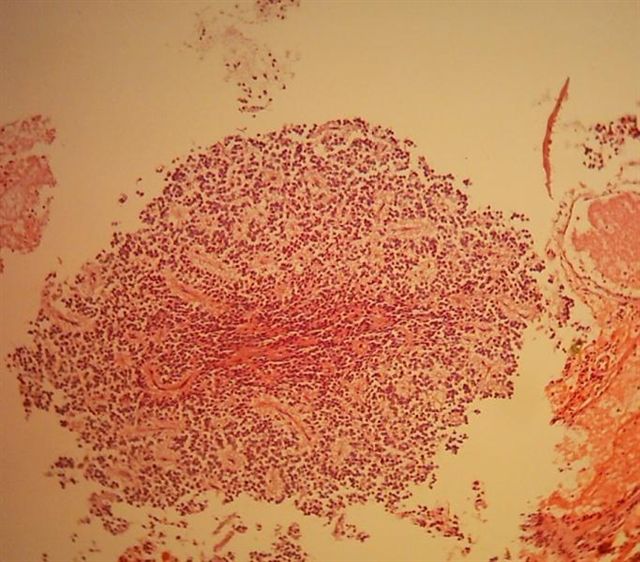
Perivascular pseudorosettes
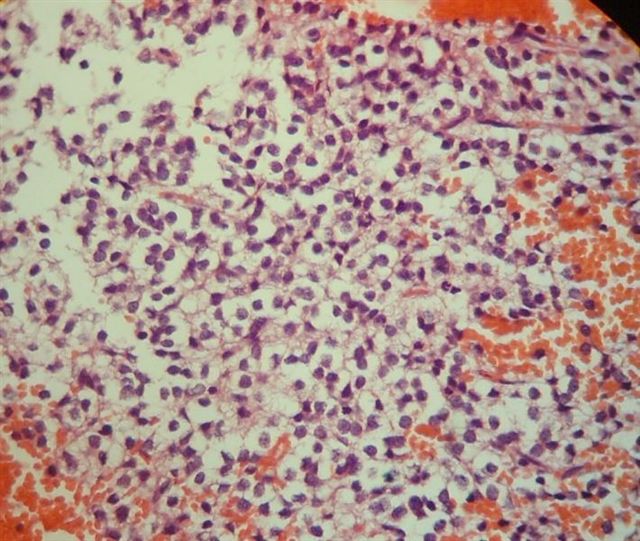
Perinuclear clearing
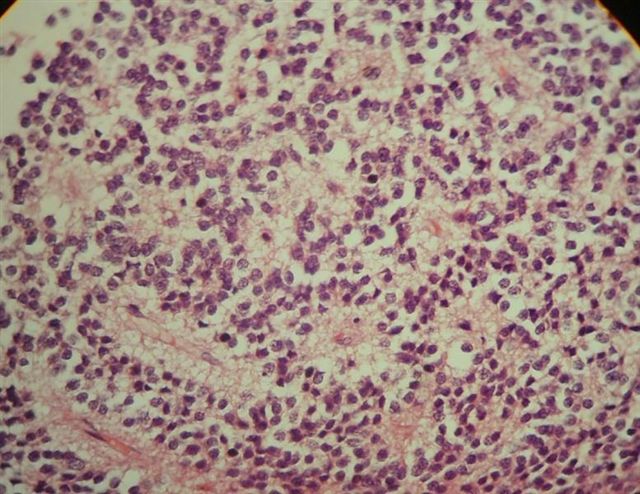
Rosettes and perivascular pseudorosettes
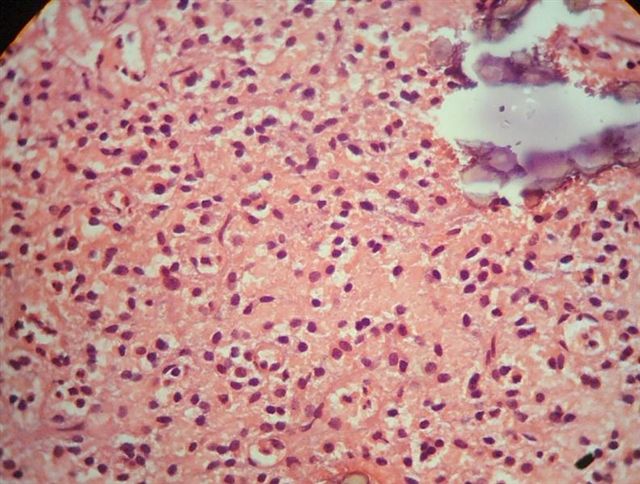
Calcification
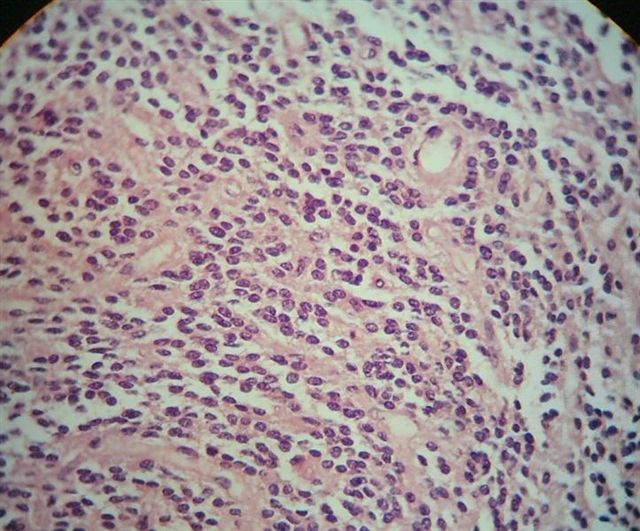
Linear cellular arrangement
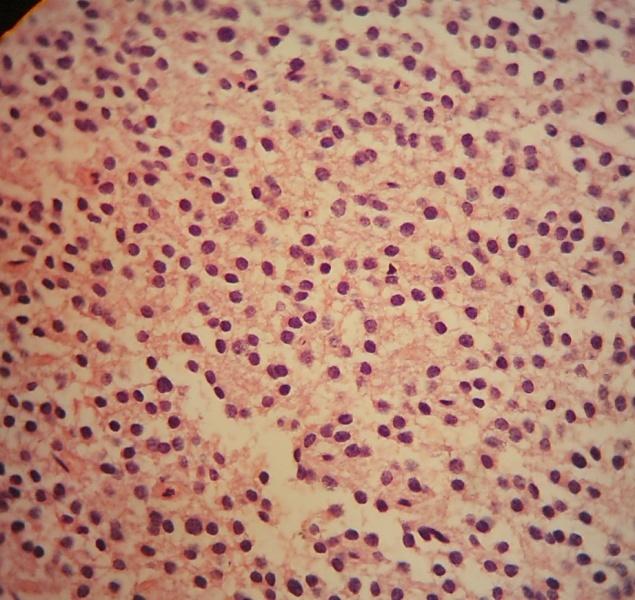
Uniform, round cells
Images hosted on other servers:

Left lateral ventricle tumor
- Cerebral spinal fluid cytology may be positive in the presence of disseminated tumor (J Neurosurg 1992;76:32)
- Crowded, cellular spheres composed of uniform small - medium cells
- Scant, cyanophilic cytoplasm on Papanicolau stain
- Neurocytic rosettes may be seen
- Squash preparations / direct smears (Acta Cytol 2004;48:194, Acta Cytol 2010;54:209)
- Monotonous, round cells with ill defined cytoplasm and without aggregation or clustering
- Nuclei tend to have finely granular chromatin and micronucleoli
- Hemosiderin laden macrophages or reactive astrocytes may be present
- Synaptophysin (Brain Pathol 1993;3:297)
- NeuN (Pathol Res Pract 2003;199:463)
- MAP2 (Brain Pathol 1993;3:297)
- Neuron specific enolase (Brain Pathol 1993;3:297)
- Ki67: cutoff for atypical central neurocytoma has not been established but suggested values range from ~2 - 4% (Neurology 2004;62:987, J Mol Biol 1989;210:709, J Neurooncol 2018;140:669, J Neuropathol Exp Neurol 1997;56:551)
- TTF1
- Clone 8G7G3/1 (7 - 21%) (World Neurosurg 2022;159:e62, Mod Pathol 2017;30:318)
- Clone SPT24 (47%) (Mod Pathol 2017;30:318)
- Chromogranin A (Brain Pathol 1993;3:297)
- Neurofilament (Am J Surg Pathol 2012;36:220)
- Olig2 (J Neurooncol 2017;135:57)
- GFAP: generally stains only entrapped astrocytes, though may be positive in rare cases (J Neuropathol Exp Neurol 1997;56:551, Acta Neuropathol 1996;91:573)
- Not utilized in routine diagnostics
- Cells typically appear more uniform than on H&E preparations (Brain Pathol 1993;3:297)
- Nuclei (Brain Pathol 1993;3:297, Pathol Res Pract 1995;191:100, Acta Neuropathol 1997;94:425)
- Regular and round
- Finely dispersed chromatin
- Small, well defined nucleoli
- Cytoplasm (Brain Pathol 1993;3:297, Pathol Res Pract 1995;191:100, Acta Neuropathol 1997;94:425)
- Prominent golgi apparatus
- Abundant mitochondria
- Dumbbell shaped lysosomal inclusions
- Parallel microtubule arrays
- Membrane bound, dense core neurosecretory granules
- Matrix (Brain Pathol 1993;3:297, Pathol Res Pract 1995;191:100, Acta Neuropathol 1997;94:425)
- Numerous cytoplasmic projections separating adjacent cell bodies
- Occasional synapses
Images hosted on other servers:
TEM showing neuronal features
Ultrastructure recapitulating neuropil and synapses
- No known recurrent mutations or chromosomal imbalances (Acta Neuropathol 2018;136:181)
- Microarray analysis (Acta Neuropathol 2007;113:303)
- One study reports frequent copy number aberrations including frequent MYCN gain
- No strong sensitivity or specificity for any region
- Transcriptomic analysis showed overexpression of genes related to (Neuropathology 2013;33:149)
- Wnt / beta catenin signaling pathway
- Sonic hedgehog signaling pathway
- Calcium function
- Maintenance of neural progenitors
- Methylation profile is unique to central neurocytoma but cannot distinguish between classic and atypical variants (Acta Neuropathol 2018;136:181, J Neurooncol 2022;159:725)
- Negative for 1p / 19q codeletion (J Neurosurg 2002;97:1350)
Images hosted on other servers:

DNA methylation based classification
- Brain, intraventricular mass, resection:
- Central neurocytoma, CNS WHO grade 2
- Oligodendroglioma:
- Infiltrative growth
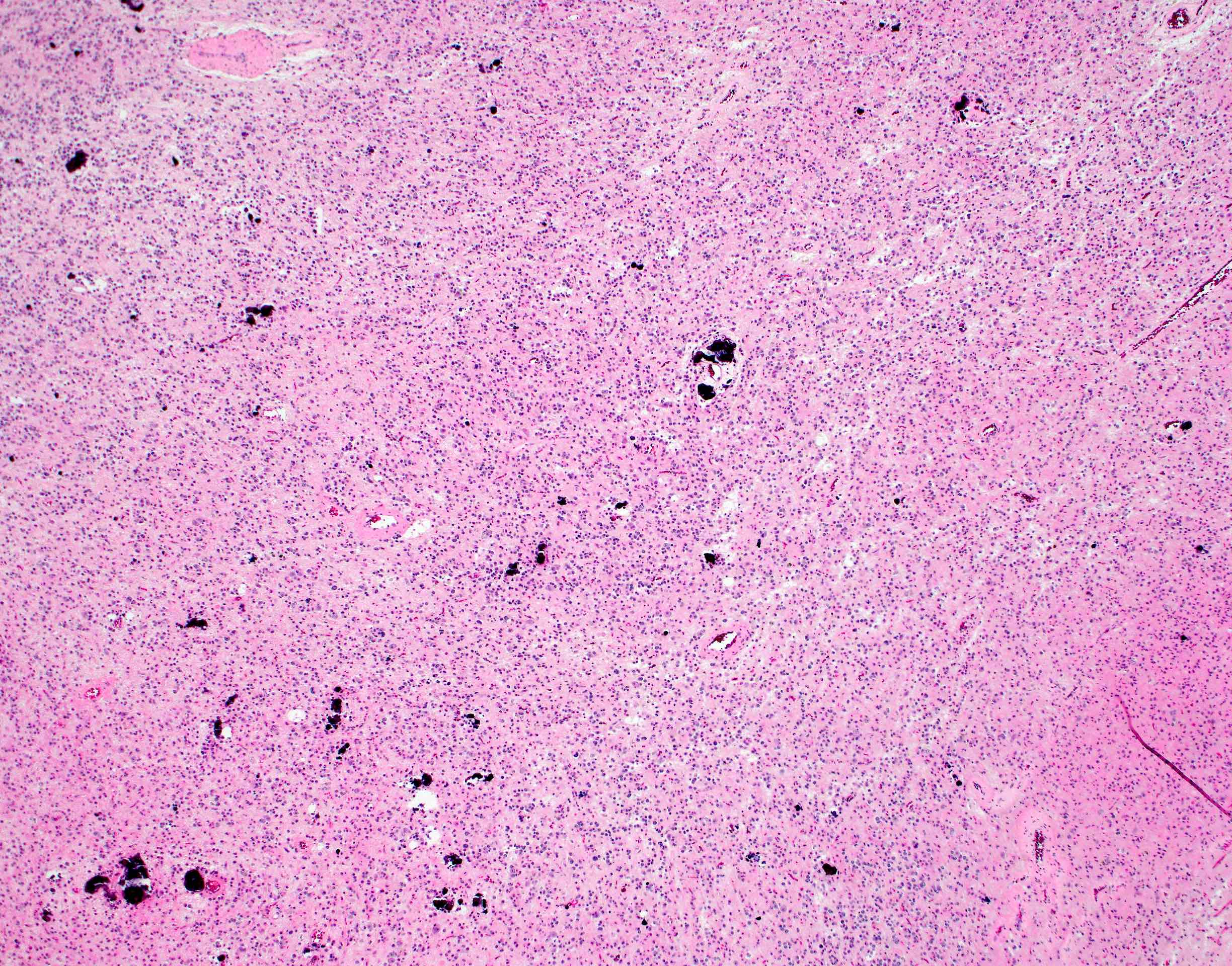
- IHC (J Neurooncol 2017;135:57)
- Strong and diffuse Olig2 positivity
- May be positive for IDH1 R132H
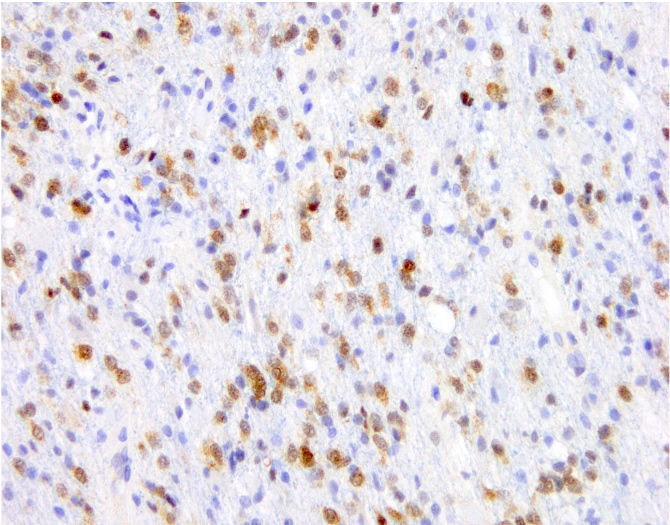
- Lacks diffuse neuronal differentiation (e.g., NeuN, synaptophysin)
- Molecular
- IDH1 / 2 mutation
- Chromosome 1p / 19q codeletion
- Infiltrative growth
- Ependymoma:
- Ganglioglioma:
- Mixed neuronal (ganglion cells) and glial neoplastic components
- IHC
- Positive for GFAP and CD56, frequently BRAF V600E positive (J Neurosci Rural Pract 2021;12:807, Pediatr Neurosurg 2019;54:36)
- Pineocytoma:
- Pineal region location
- IHC: positive for NFP (strong and diffuse)
- Distinct DNA methylation profile
- Choroid plexus papilloma
- Well developed papillary architecture
- IHC: positive for Kir7.1
- Subependymoma:
- Meningioma:
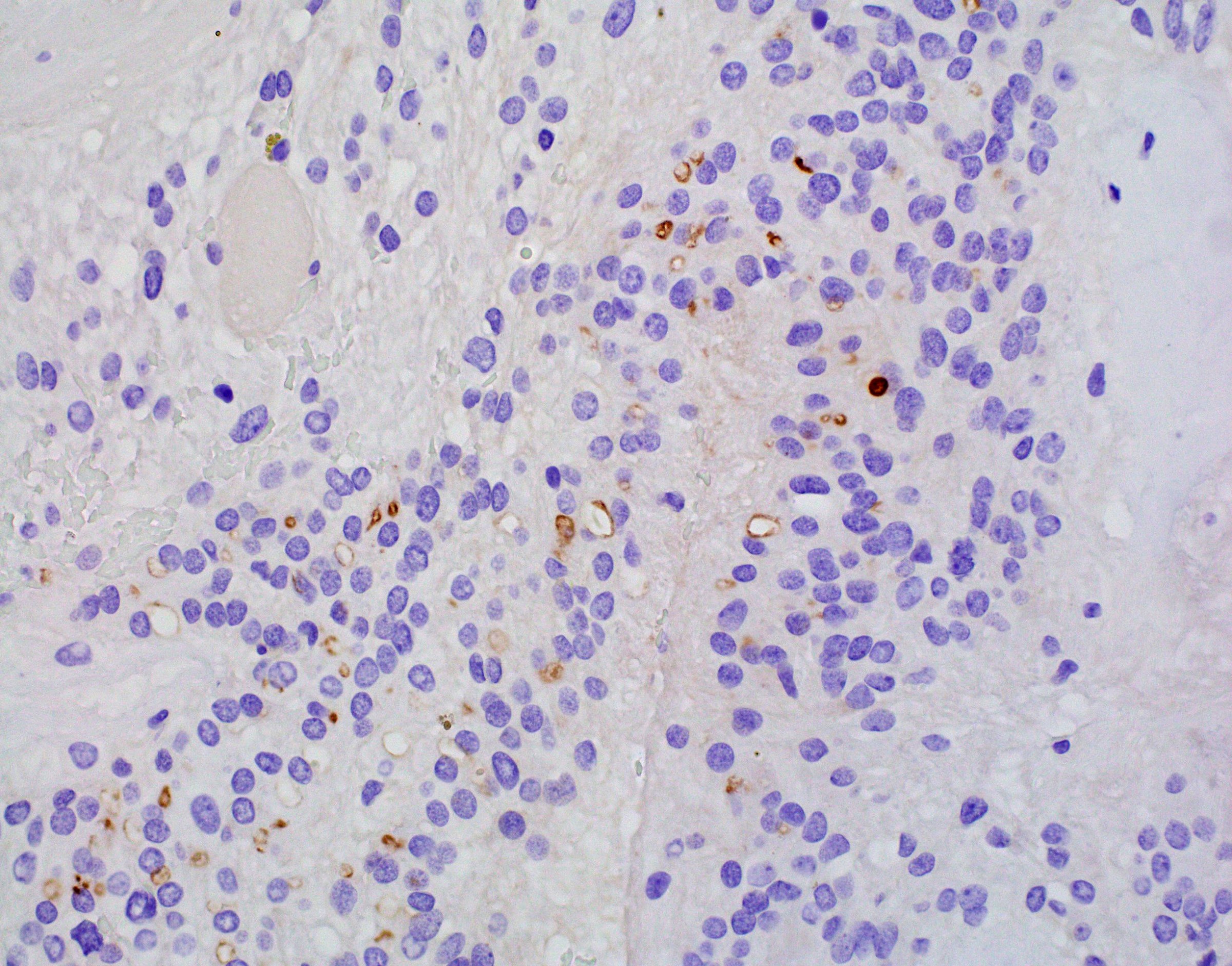
A 29 year old woman with an intraventricular brain mass is diagnosed with a central neurocytoma with histologic findings shown above. Which of the following immunohistochemical stains is most likely to be diffusely positive in this neoplasm?
- Chromogranin
- GFAP
- Olig2
- Synaptophysin
Comment Here
Reference: Central neurocytoma
- Hypercellularity
- Lipomatous differentiation
- Macronucleoli
- Microvascular proliferation
Comment Here
Reference: Central neurocytoma
- Cerebellar liponeurocytoma is a rare, slow growing tumor of the central nervous system with advanced neuronal or neurocytic differentiation, variable glial differentiation and lipoma-like changes
- Classified CNS WHO grade 2
- Typically occurs in the posterior cranial fossa
- To date, nearly 70 cases have been reported
- Rare, slow growing tumor with advanced neuronal or neurocytic differentiation and focal lipoma-like changes, typically occurring in the cerebellum
- Classified as CNS WHO grade 2
- Extent of surgical resection is the main prognostic factor
- Histologically composed of monotonous neurocytic cells with rare mitoses and intermingled lipidized cells
- Diffusely positive for neuronal markers syanptophysin, NeuN and MAP2
- Lipomatous medulloblastoma (not recommended)
- Lipidized medulloblastoma (not recommended)
- Medullocytoma (not recommended)
- Lipomatous glioneurocytoma (not recommended)
- Lipidized mature neuroectodermal tumor (not recommended)
- ICD-O: 9506/1 - cerebellar liponeurocytoma
- ICD-11: 2A00.3 & XH2GB0 - central neurocytoma of brain & cerebellar liponeurocytoma
- Rare tumor, first described in 1978 (Acta Neuropathol 1978;41:261)
- ~70 cases reported (World Neurosurg 2018;120:214)
- No significant sex predilection (World Neurosurg 2018;120:214)
- Mainly affects adults between the third and fifth decade, with a mean age of 46 years (World Neurosurg 2018;120:214, Neurosurg Rev 2022;45:1747)
- Rarely reported in children (Front Oncol 2021;11:759581, Acta Neuropathol 2005;109:346, Rare Tumors 2016;8:6240)
- Mostly located in the cerebellar hemispheres (World Neurosurg 2018;120:214)
- More rarely located in the vermis, paravermian region, fourth ventricle or cerebellopontine angle (World Neurosurg 2018;120:214, Neuropathology 2022;42:169, J Neurooncol 2012;108:513)
- Supposed to originate from GABAergic neurons in the cerebellar ventricular zone, which may aberrantly differentiate into adipocyte-like tumor cells (J Neurooncol 2012;108:513)
- Risk factors are unknown, though a possible inheritable predisposition was suggested (J Neurosurg 2016;125:57, J Clin Neurosci 2016;32:154)
- Headache or other symptoms and signs of raised intracranial pressure (Neuropathology 2022;42:169, Neurochirurgie 2021;67:579)
- Cerebellar signs, including ataxia or disturbed gait (Oncol Lett 2016;11:1061, Neurochirurgie 2021;67:579)
- Based on imaging (computerized tomography, magnetic resonance) / biopsy / resection specimen
- Computerized tomography (CT) scan: hypodense or isodense solid mass, with focal areas of hypoattenuation corresponding to high fat density (World Neurosurg 2018;120:214, J Neurooncol 2012;108:513)
- On magnetic resonance imaging (MRI), the tumor is (World Neurosurg 2018;120:214, Neuropathology 2022;42:169)
- Well circumscribed
- Hypointense or isointense in T1 weighted sequences, with focal hyperintensity, corresponding to lipidized foci
- Hyperintense in T2 weighted sequences
- Heterogeneously contrast enhanced in most cases
Contributed by Valeria Barresi, M.D., Ph.D.

T1 weighted MRI
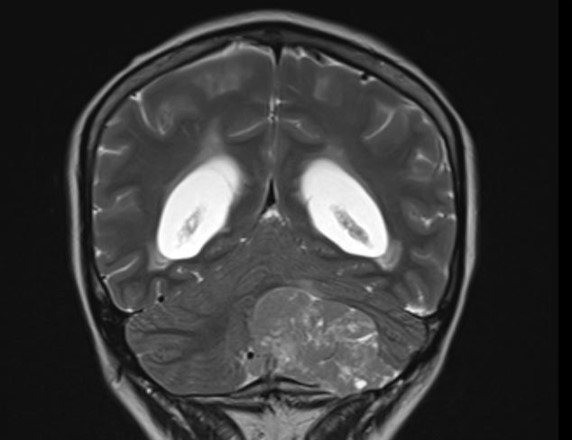
T2 weighted MRI
Images hosted on other servers:

Right cerebellopontine angle mass
- Progression free survival rates: 92.7%, 78.0% and 23.8% at 1, 5 and 10 years, respectively (Neurosurg Rev 2022;45:1747)
- Higher recurrence risk associated with incomplete surgical resection and high Ki67 (> 10%) labeling index (World Neurosurg 2018;120:214, World Neurosurg 2018;112:18, Neurosurg Rev 2022;45:1747)
- Adjuvant radiotherapy reduces the risk of recurrence after partial surgical resection (World Neurosurg 2018;120:214)
- No recurrences were observed in 6 patients receiving complete surgical resection of the tumor and adjuvant radiotherapy (World Neurosurg 2018;120:214)
- 5 year old boy with a mass located in the cerebellar vermis and fourth ventricle (Front Oncol 2021;11:759581)
- 39 year old man with a mass in the right cerebellar hemisphere (J Med Case Rep 2018;12:170)
- 50 year old woman with a tumor in the left cerebellar hemisphere (Int J Surg Case Rep 2021;82:105937)
- 55 year old man with a tumor in the cerebellar vermis (J Neurosci Rural Pract 2019;10:360)
- Surgery (Neurosurg Rev 2022;45:1747, World Neurosurg 2018;120:214)
- Adjuvant radiotherapy is controversial (Neurosurg Rev 2022;45:1747, World Neurosurg 2018;120:214)
- Well circumscribed, soft, gray-pink tumor (Neurosurg Rev 2022;45:1747)
- It may lack clear demarcation and be partly attached to the surrounding tissue in some cases (World Neurosurg 2018;112:18)
- Tumor composed of small, roundish, uniform cells (World Neurosurg 2018;112:18)
- Infrequent mitoses
- Microvascular proliferation and necrosis are typically absent
- Neuropil matrix around vessels may simulate perivascular pseudorosettes of ependymoma
- Lipid accumulation can be demonstrated using oil red O staining (Neuropathol Appl Neurobiol 1993;19:95)
Contributed by Valeria Barresi, M.D., Ph.D.

Neurocytic cells
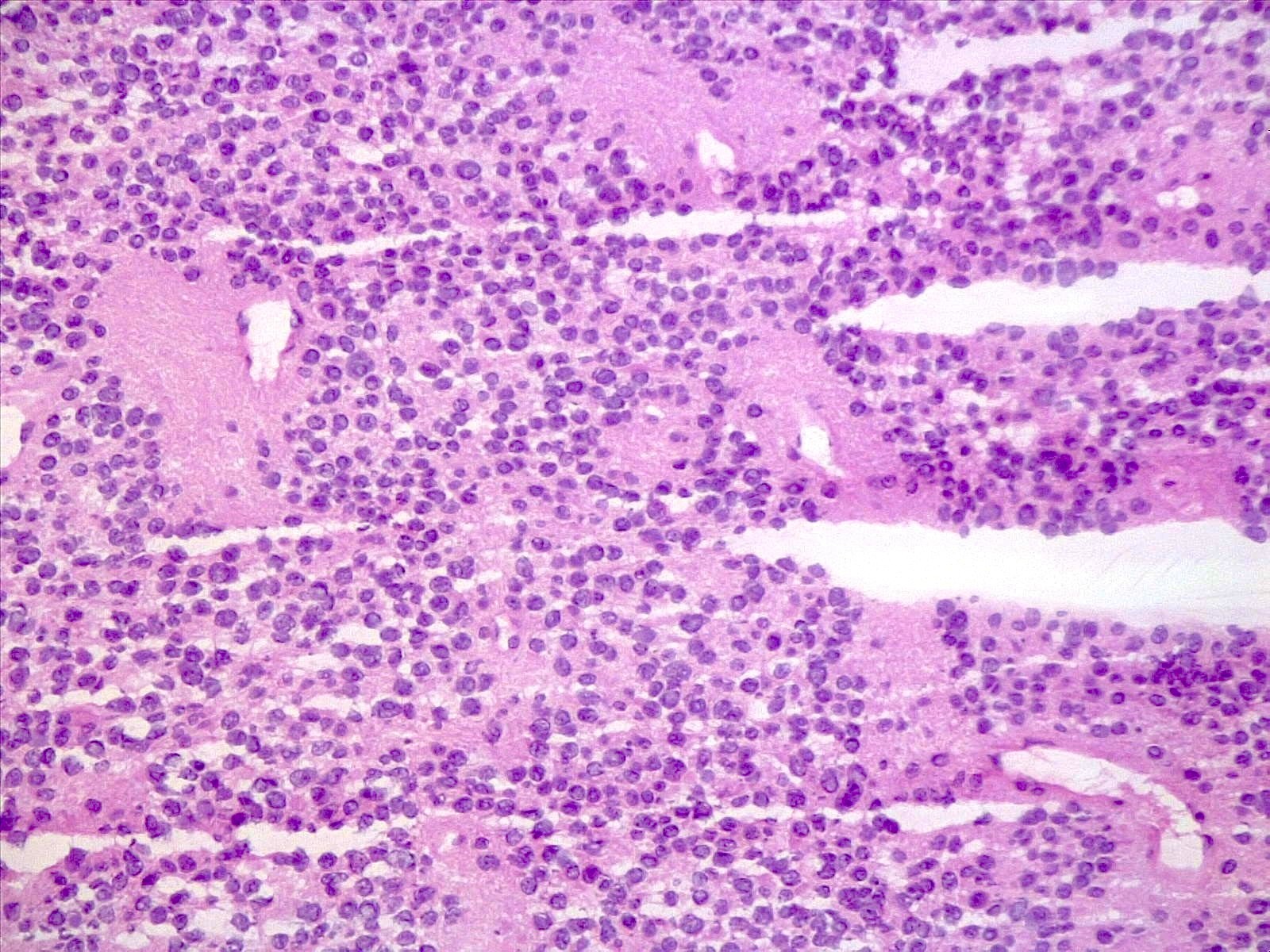
Perivascular fibrillary areas
- Tumor composed of small and uniform neurocytic cells with round to oval nuclei, clear cytoplasm and indistinct cell membrane, arranged in sheets or lobules (Front Oncol 2021;11:759581, Neuropathology 2022;42:169, Brain Tumor Pathol 2017;34:28)
- Presence of lipidized cells in groups or scattered throughout the tumor (Front Oncol 2021;11:759581, Neuropathology 2022;42:169, Brain Tumor Pathol 2017;34:28, Arch Pathol Lab Med 2012;136:965)
- Few mitoses
- Absent necrosis and microvascular proliferation
- Lipidized cells may be reduced or absent in recurrent tumors
Contributed by Valeria Barresi, M.D., Ph.D.
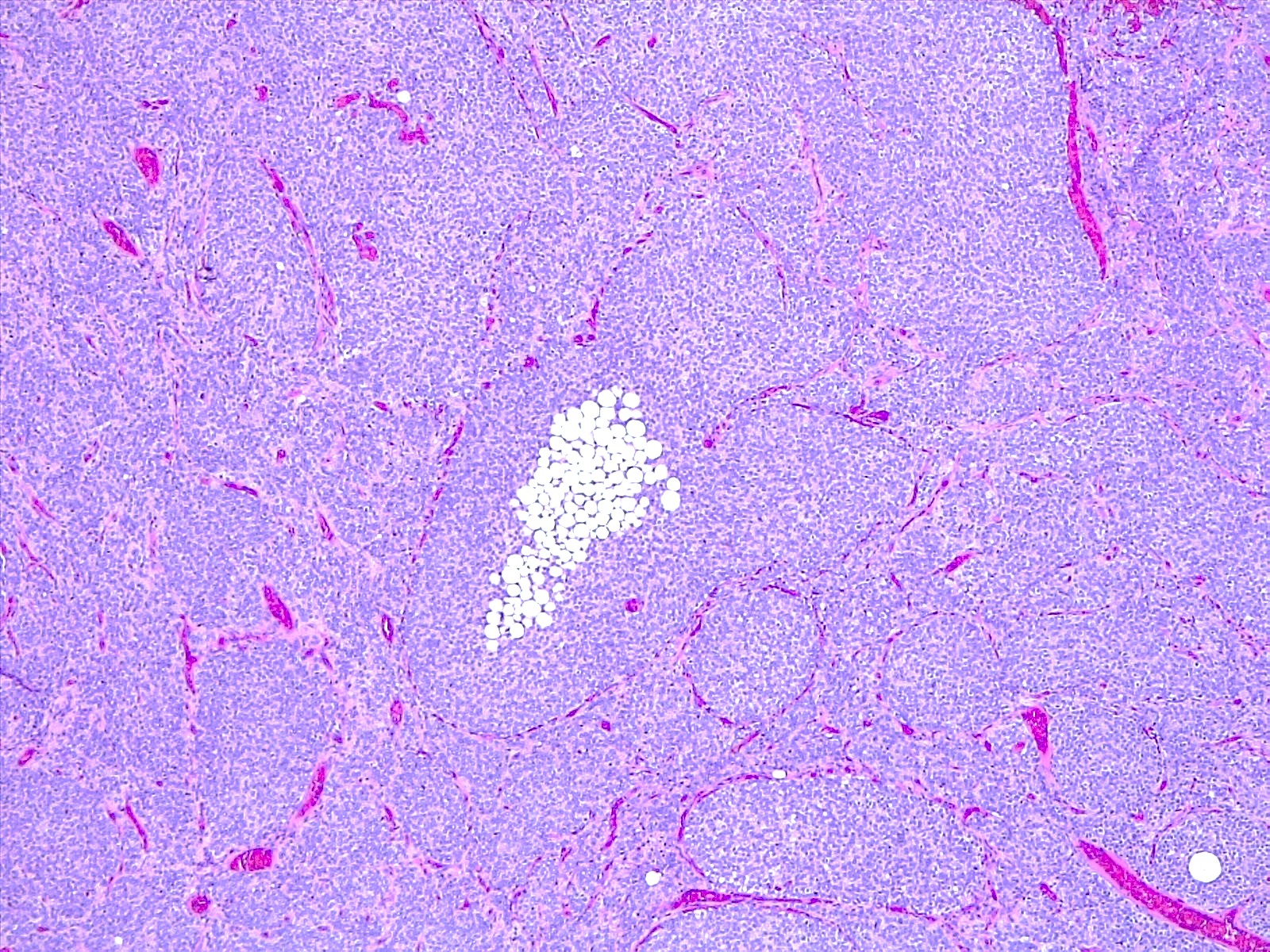
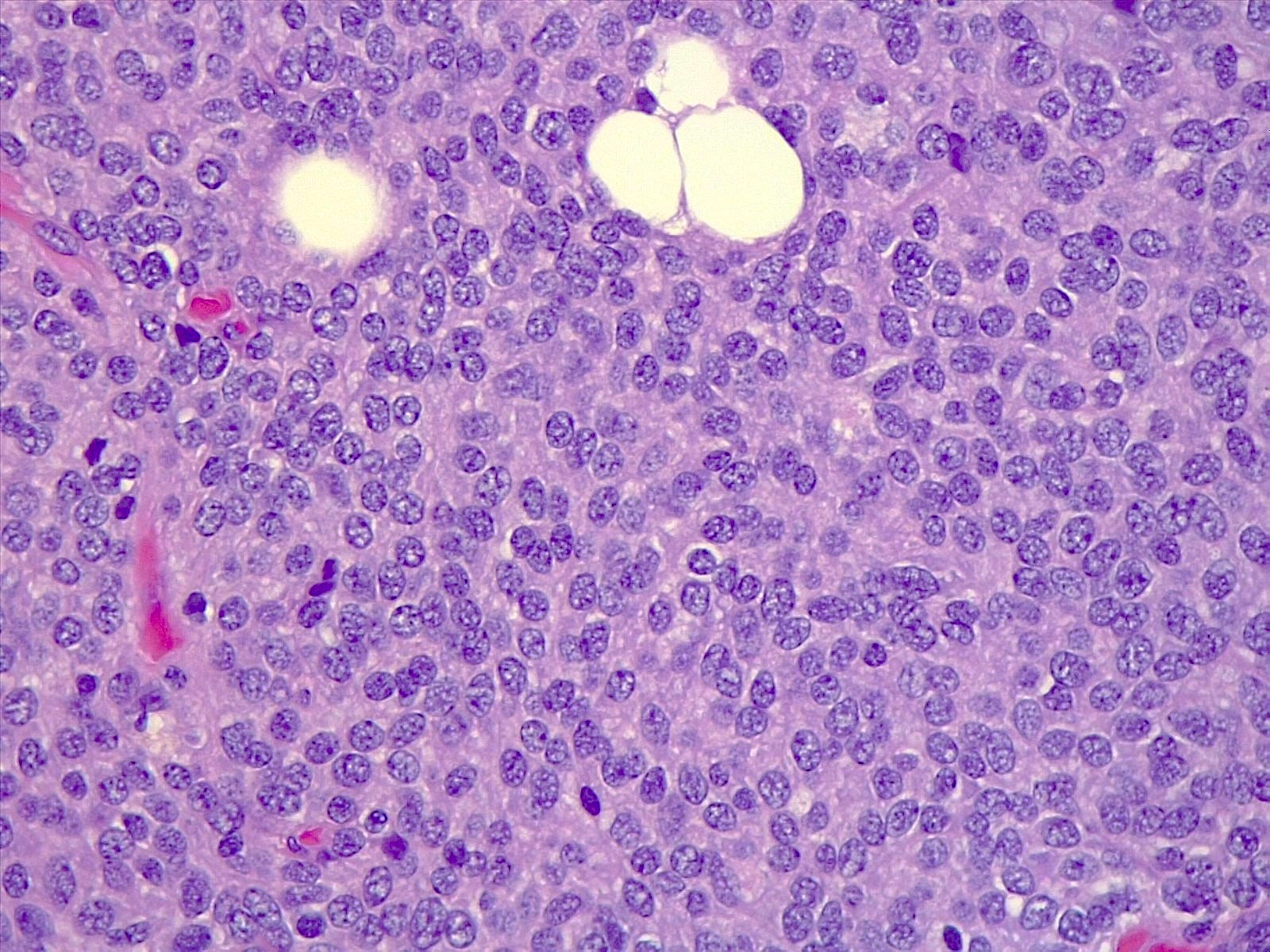
Lipidized cells
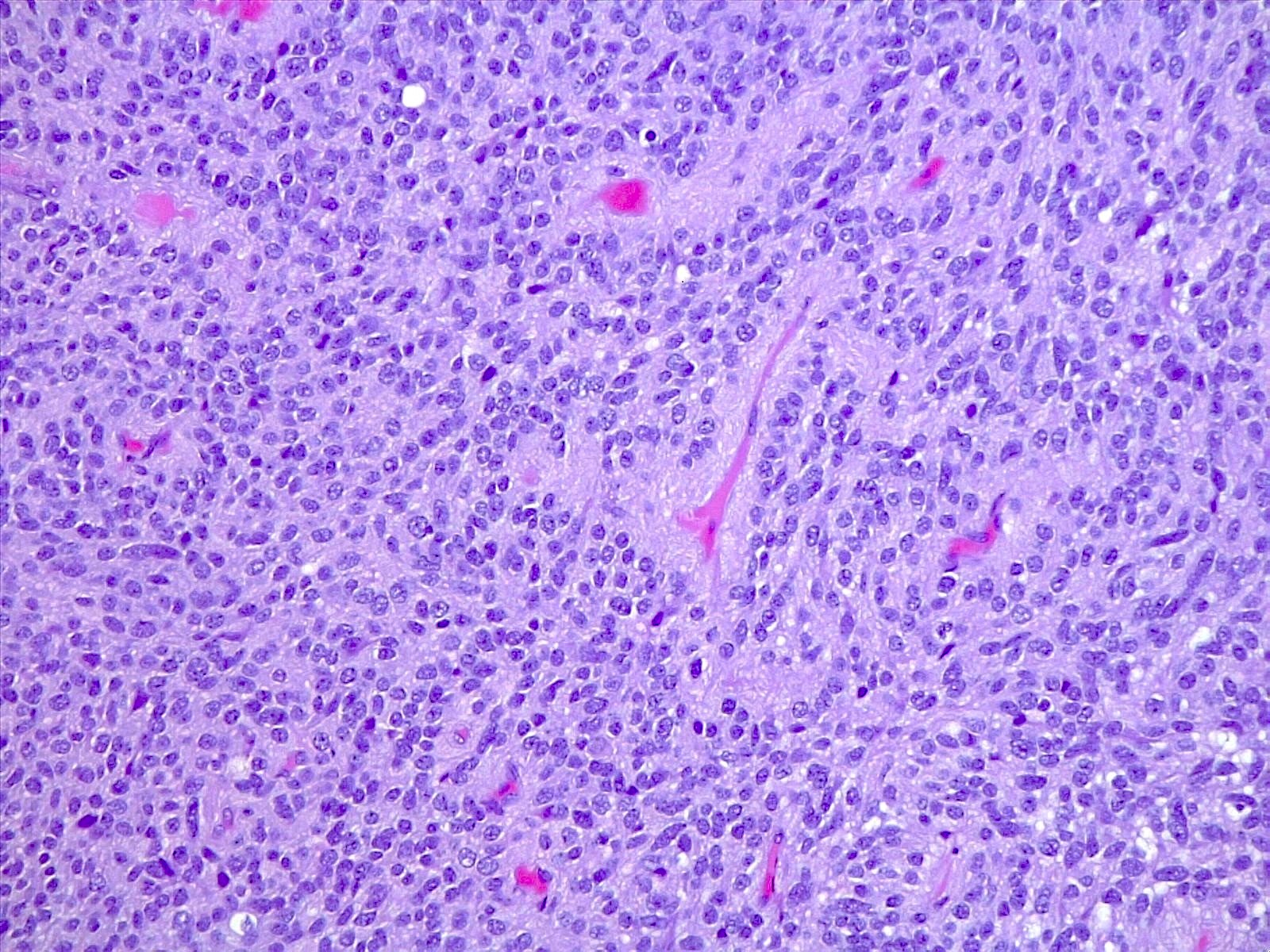
Perivascular matrix
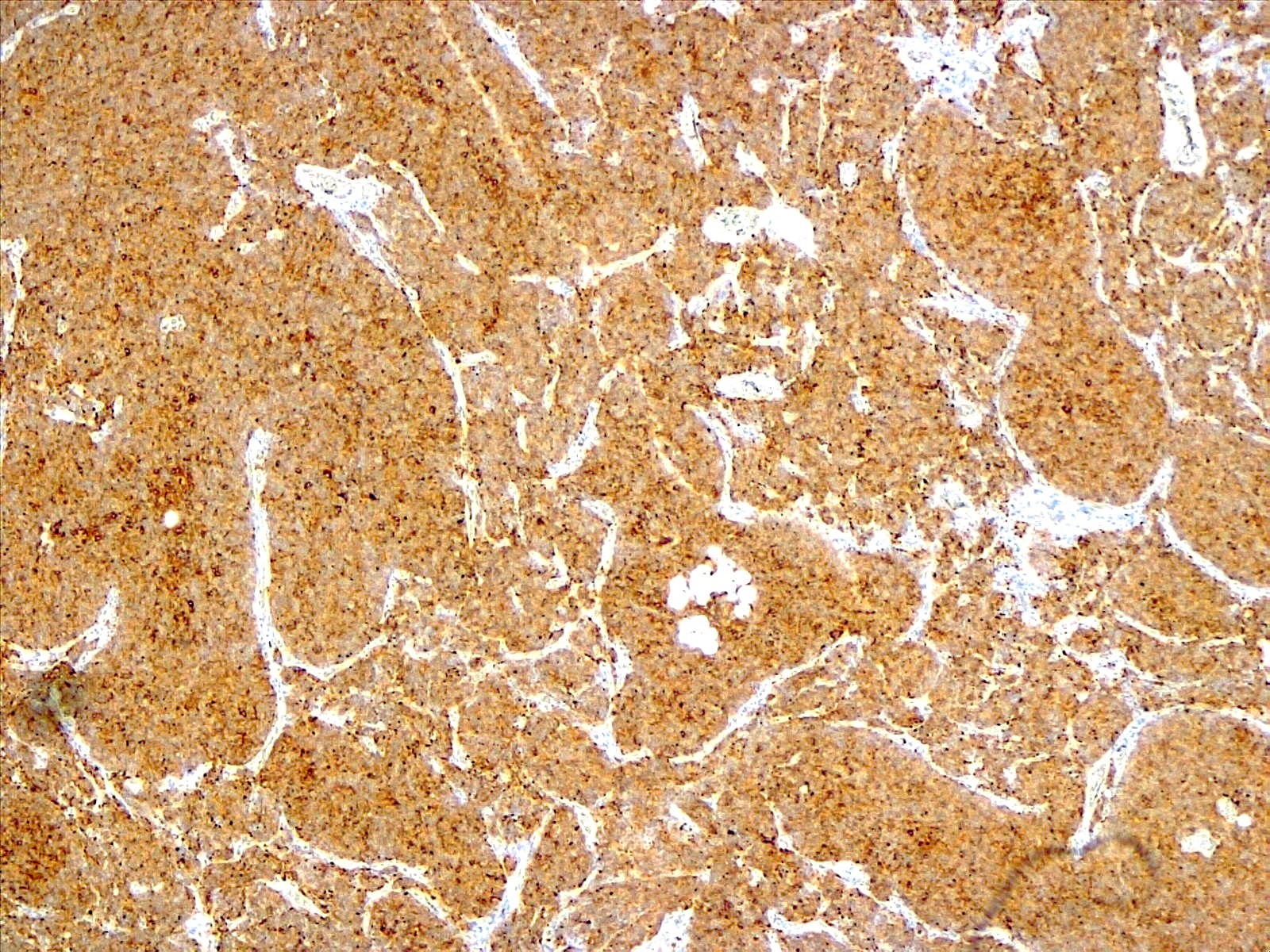
Synaptophysin immunostaining
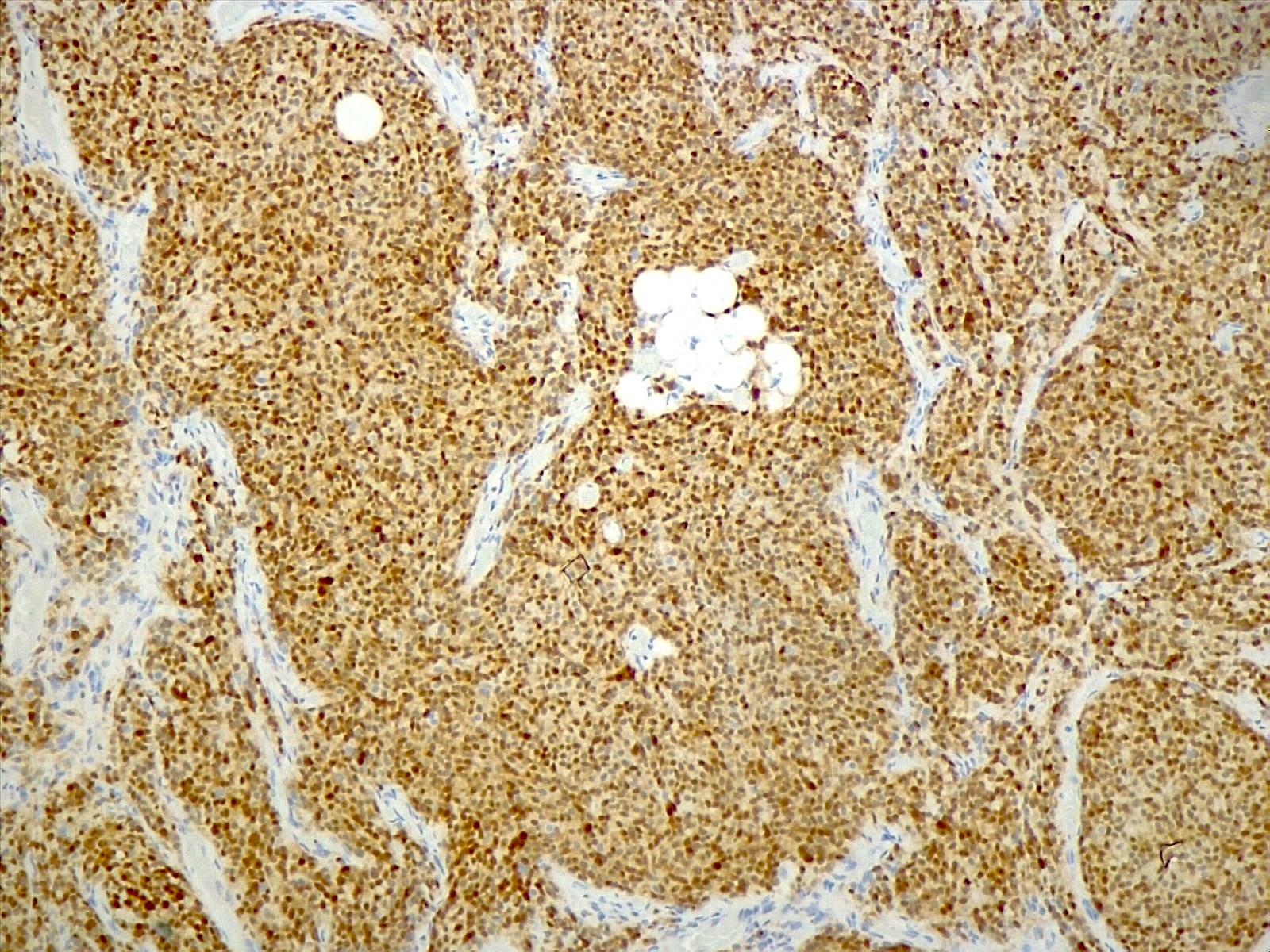
NeuN immunostaining
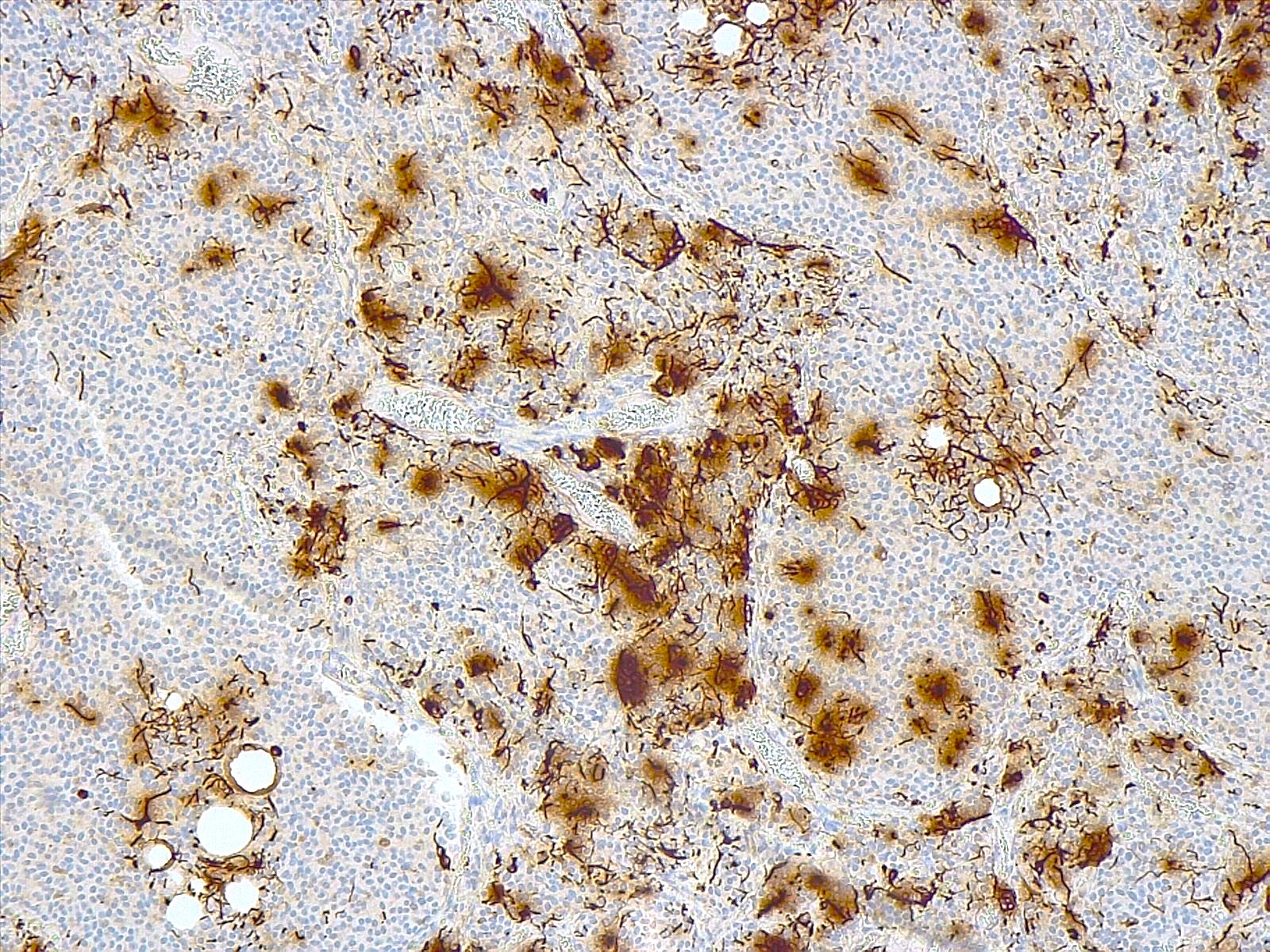
GFAP immunostaining
Olig2 immunostaining
EMA immunostaining
Ki67 immunostaining
Images hosted on other servers:
Cerebellar liponeurocytoma
- Small, uniform cells with rounded to oval nuclei showing salt and pepper chromatin and scant cytoplasm
- Some cells show a single, large cytoplasmic vacuole displacing the nucleus to the periphery of the cell, similar to adipocytes (Arch Pathol Lab Med 2012;136:965)
Contributed by Valeria Barresi, M.D., Ph.D.
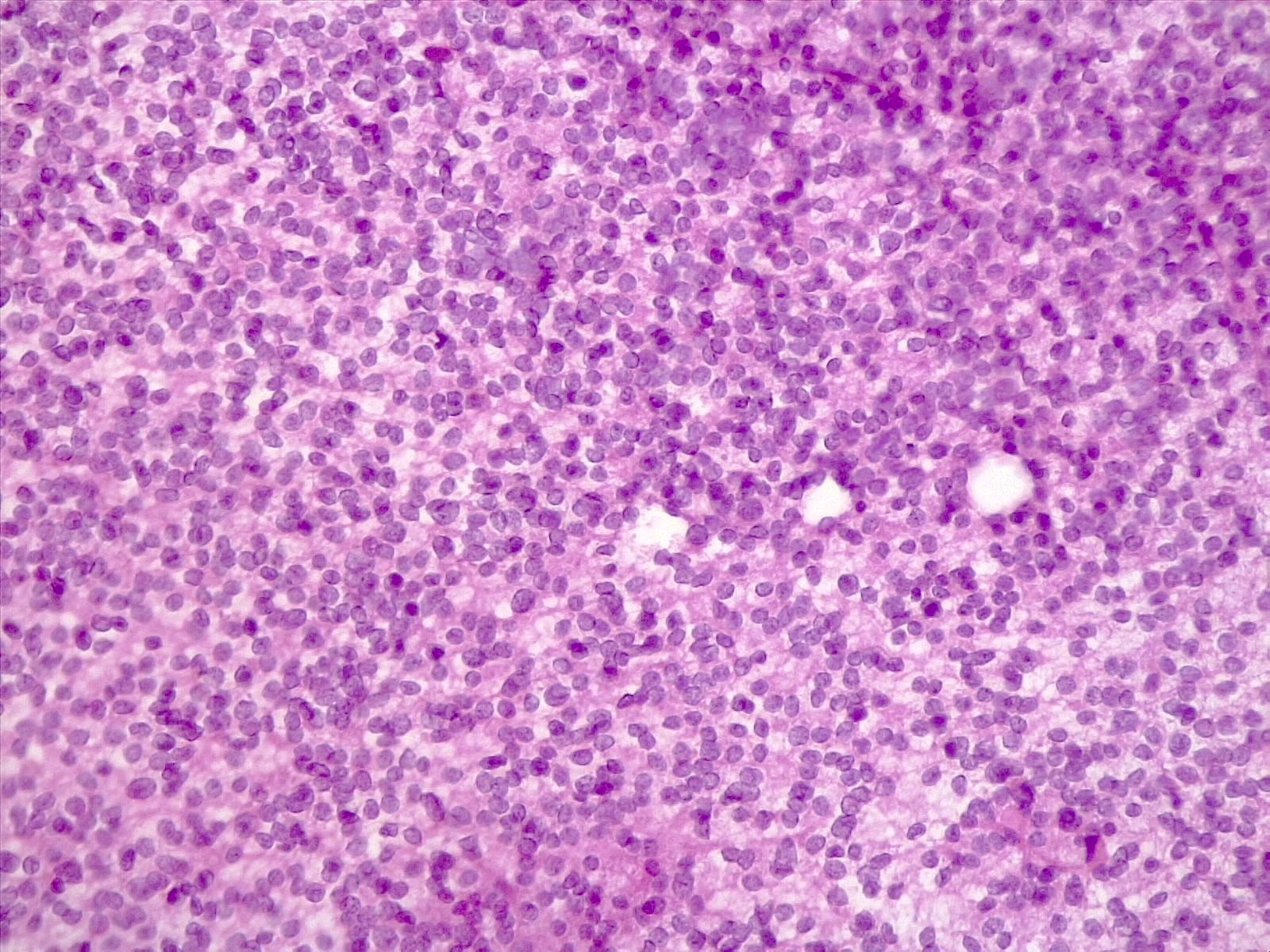
Sheets of uniform neurocytic cells
- Synaptophysin (~98%) (Arch Pathol Lab Med 2012;136:965, World Neurosurg 2018;120:214)
- MAP2 (100%) (Arch Pathol Lab Med 2012;136:965, World Neurosurg 2018;120:214)
- NSE (100%%) (Arch Pathol Lab Med 2012;136:965, World Neurosurg 2018;120:214)
- NeuN (100%) (Neuropathology 2022;42:169)
- GFAP (~85%) and S100 (~82%): expressed by reactive astrocytes and few tumor cells (Arch Pathol Lab Med 2012;136:965, World Neurosurg 2018;120:214)
- Chromogranin A (~55%) (Neuropathology 2022;42:169, World Neurosurg 2018;120:214)
- Ki67 (MIB1): usually 1 - 3% (Arch Pathol Lab Med 2012;136:965)
- Lipidized cells display large, nonmembrane bound lipid deposits within the cytoplasm and retain features of neuronal differentiation, including dense core neurosecretory granules (J Neurosurg 2001;95:700, Ultrastruct Pathol 2015;39:419)
- TP53 missense mutations in 20% of cases (Brain Pathol 2004;14:281)
- Lack of isochromosome 17 or mutations in APC, CTNNB and PTCH, which helps rule out medulloblastoma (Brain Pathol 2004;14:281)
- Recurrent focal losses of chromosomes 2p and 14 (Acta Neuropathol 2018;136:181)
- Unique DNA methylation profile (Clin Neuropathol 2022;41:12)
- Cerebellum, tumor resection:
- Cerebellar liponeurocytoma, CNS WHO grade 2 (see comment)
- Comment: The tumor is composed of monotonous neurocytic cells, with round nuclei, small nucleoli and scant cytoplasm, disposed in a diffuse growth pattern and showing foci of lipidized cells. Only scattered mitoses are present. Necrosis is absent. On immunohistochemistry, tumor cells are diffusely positive for synaptophysin and MAP2 and negative for Olig2 and EMA. Ki67 labeling index is < 5%.
- Medulloblastoma:
- Mainly affects children and young adults
- Lacks lipidized cells
- Shows high mitotic index and Ki67 labeling index
- May show necrosis
- May occasionally have foamy macrophages that mimic lipomatous differentiation of cerebellar liponeurocytoma
- Classic subtype is composed of small blue round cells with hyperchromatic nuclei of various shapes
- Desmoplastic / nodular subtype features reticulin free zones surrounded by densely packed, poorly differentiated, highly proliferative cells with moderately pleomorphic nuclei
- Large / anaplastic subtype is composed of markedly pleomorphic cells
- Harbors isochromosome 17q
- May harbor PTCH, CTNNB and APC mutations
- Clear cell ependymoma:
- Shows perivascular pseudorosettes and rosettes
- Lacks lipidized cells
- Positive for EMA (dot-like)
- Diffusely positive for GFAP
- Negative for synaptophysin
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted:
- Rare in the posterior fossa
- Lacks lipidized cells
- Positive for Olig2
- Negative for synaptophysin
- IDH1 / 2 mutated
- 1p / 19q codeleted
The tumor shown above was found in the left cerebellar hemisphere of a 45 year old woman. Only rare mitoses are present. It shows diffuse immunostaining for synaptophysin, focal immunostaining for GFAP and is negative for Olig2 and EMA. What is the most likely diagnosis?
- Cerebellar liponeurocytoma
- Ependymoma
- Medulloblastoma
- Oligodendroglioma
Comment Here
Reference: Cerebellar liponeurocytoma
- Rare, discrete, slow growing glial tumor of third ventricle of adults, characterized by chordoid architecture and myxoid background
- First described in 1998 (J Neuropathol Exp Neurol 1998;57:283)
- Cells may resemble ependyma of subcommissural organ, present in dorsocaudal third ventricle during embryonic life, which regresses after birth
- "Glioma" since GFAP+
- Uncommon ( < 50 cases reported)
- Usually middle aged women (median age 45 years, 63% female)
- Low grade neoplasm arising in third ventricle hypothalamic region
- Often attached to hypothalamic and suprasellar structures (63%)
- WHO grade 2
- WHO 2000 lists as "glial tumor of uncertain origin"
- Well circumscribed, solid, enhancing mass, 19% are cystic (AJNR Am J Neuroradiol 2001;22:464)
Images hosted on other servers:
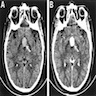
Axial CT scan reveals hyperdense mass
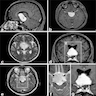
Various images
- May have poor outcome despite bland histology (Am J Surg Pathol 2002;26:1330, Neuropathol Appl Neurobiol 2005;31:354)
- 48 year old man with gradual diminution of vision (Neurol India 2011;59:469)
- 48 year old woman with suprasellar tumor coexisting with Rathke cleft cyst (Pathol Int 2003;53:780)
- 50 year old woman with chordoid glioma infiltrating optic chiasm (Surg Neurol Int 2011;2:53)
- 56 year old woman (Arch Pathol Lab Med 2004;128:e141)
- 60 year old woman (Hum Pathol 1999;30:723)
- Resection; rarely radiotherapy (Surg Neurol 2003;59:424), occasionally recurs due to incomplete excision (16%)
Images hosted on other servers:

Optic chiasm enlargement
- Discrete firm mass adherent to wall of third ventricle
- Chordoma-like features
- Clusters and cords of epithelioid cells in mucinous matrix
- Cells have abundant eosinophilic cytoplasm and round / oval nuclei with indistinct nucleoli
- Also lymphoplasmacytic infiltrates, Russell bodies, discrete border with peritumoral piloid gliosis
- May have chondroid metaplasia or papillary formations
- No / rare mitotic figures, no vascular proliferation, no necrosis
- GFAP, vimentin, CD34, EMA (focal), cytokeratin (focal) (Brain Pathol 1999;9:617)
- Low Ki67 index
- Variable S100
- Synaptophysin, neurofilament (usually), NSE (usually), desmin, p53
- Ependymal differentiation
- Apical pole has microvilli and basal pole has hemidesmosome-like structures connecting cell membranes to basal lamina
- Also submicroscopic cell body zonation and secretory granules (Am J Surg Pathol 2001;25:401)
- Chordoid meningioma: whorls, psammoma bodies and nuclear pseudoinclusions, EMA+, GFAP-, well formed desmosomes, 22q-
- Chordoma: infiltrates bone, physaliphorous cells, EMA+, diffusely keratin+, S100+, GFAP-, mitochondria rough ER complexes
- Chordoid meningioma is a histological subtype of meningioma characterized by cords or trabeculae of epithelioid (or spindle) cells embedded in a mucin rich stroma and resembling chordoma
- It is classified as CNS WHO grade 2, regardless of the presence of worrisome histological features (mitoses, spontaneous necrosis, brain invasion)
- Histological subtype of meningioma that is characterized by cords or trabeculae of epithelioid (or spindle) cells embedded in a mucin rich stroma and resembling chordoma
- CNS WHO grade 2
- Recurrence risk is associated with subtotal resection, high Ki67 labeling index and atypical histological features (mitoses, brain invasion, spontaneous necrosis, small cell with high N:C ratio, hypercellularity, macronucleoli, sheeting)
- Pure forms exist but it most commonly features intermingled areas of classical meningioma, with syncytial growth pattern, whorls and pseudoinclusions
- Immunohistochemistry: EMA+, podoplanin+, possible focal cytokeratin+, S100-, brachyury-, GFAP-
- Chordoid meningioma
- Rare meningioma subtype, accounting for 0.5 - 1% of intracranial meningiomas (Am J Surg Pathol 2000;24:899)
- M:F = 1:1
- Mainly affects adults (mean age: 47 years) (Am J Surg Pathol 2000;24:899)
- Mean age at presentation is nearly a decade younger than nonchordoid meningiomas (Acta Neuropathol Commun 2022;10:56)
- Mostly involves supratentorial meninges (88%) but may also occur at the (Am J Surg Pathol 2000;24:899)
- Skull base (World Neurosurg 2016;93:198)
- Cerebellar pontine angle (Am J Surg Pathol 2000;24:899)
- Spinal meninges (Am J Surg Pathol 2000;24:899)
- Cerebral ventricles (World Neurosurg 2016;93:198)
- Originates from arachnoid border cells or dura based cells (Acta Neurochir (Wien) 2021;163:57)
- Risk factors may be similar to general meningioma: ionizing radiation (Eur J Epidemiol 2020;35:591)
- Unspecific symptoms, including headache, limbs weakness or numbness, cranial nerve disorders, epilepsy (World Neurosurg 2016;93:198)
- It has been associated with Castleman disease (Cancer 1988;62:391)
- Based on imaging (CT, MRI) / biopsy / resection specimen
- At MRI, chordoid meningioma is hypoisointense on T1 weighted images and hyperintense on T2 and FLAIR, with contrast enhancement varying from homogeneous to heterogeneous (J Neurooncol 2018;137:575, Neuroradiology 2022;64:253)
- Contrast enhancement is more intense than in meningothelial, transitional or fibrous meningiomas (Neuroradiology 2022;64:253)
- Multicystic appearance is seen in some cases (Neuroradiology 2022;64:253)
Contributed by Valeria Barresi, M.D., Ph.D.
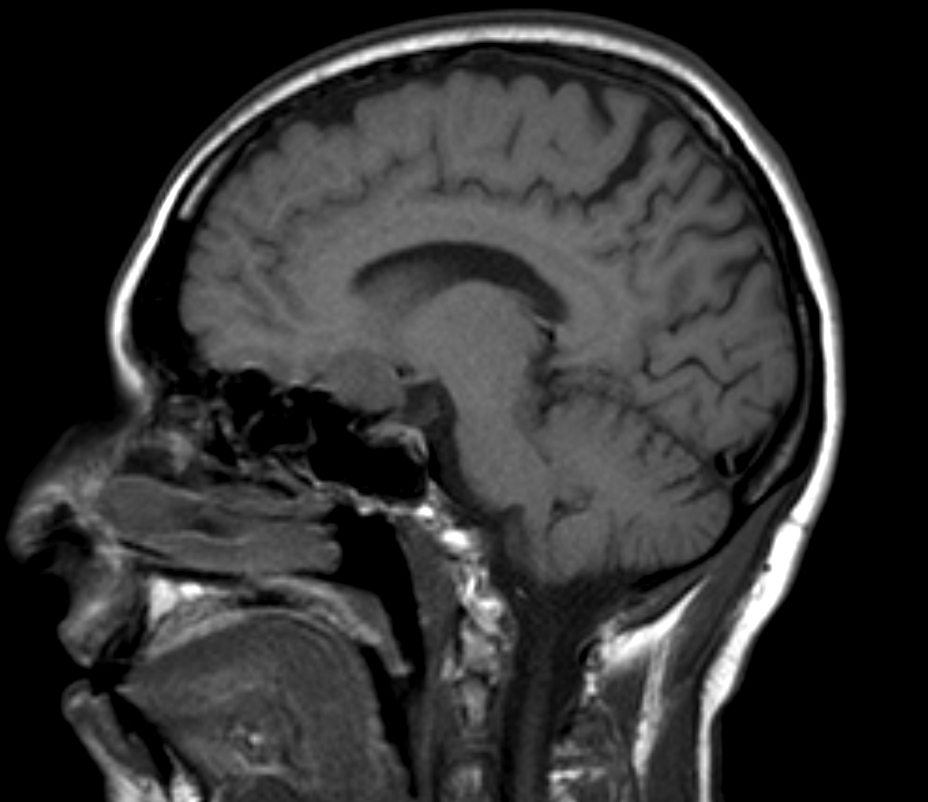
T1 weighted sagittal MRI

T1 sagittal contrast enhancement
T2 weighted axial MRI

FLAIR axial MRI
- In a meta analysis of 220 patients with chordoid meningiomas, 23% had tumor progression after surgery, with a median time to recurrence of ~131 months (J Neurooncol 2016;126:107)
- 3, 5 and 10 year progression free survival (PFS): 76.0%, 67.5% and 54.4%, respectively (J Neurooncol 2016;126:107)
- Significant risk factors for recurrence include:
- Subtotal resection (World Neurosurg 2016;93:198, J Neurooncol 2016;126:107)
- Ki67 labeling index ≥ 5% (J Neurooncol 2016;126:107)
- Histological atypical features (high mitotic index, spontaneous necrosis, brain invasion, macronucleoli) (Acta Neuropathol Commun 2022;10:56)
- NF2 mutation, 1p, 8p, 10, 14, 22q loss and homozygous deletion of CDKN2A / CDKN2B (Cancers (Basel) 2020;12:225, Acta Neuropathol 2018;136:975)
- Methylation class intermediate or malignant (Acta Neuropathol 2018;136:975, Acta Neuropathol Commun 2022;10:56)
- 22 year old woman presented with a foramen magnum lesion (Asian J Neurosurg 2018;13:834)
- 50 year old woman presented with progressive headaches (Arch Pathol Lab Med 2004;128:e115)
- 68 year old man presented with a tuberculum sellae lesion (NMC Case Rep J 2020;7:53)
- Surgery and adjuvant radiotherapy (World Neurosurg 2016;93:198)
- Soft / gelatinous consistency, smooth contour and translucent areas (NMC Case Rep J 2020;7:53, Diagn Cytopathol 2016;44:811)
- Differential diagnosis versus other tumor types: at least focal presence of psammoma bodies, meningothelial whorls or nuclear pseudoinclusions
- Differential diagnosis versus other meningioma subtypes: cords of cells with eosinophilic cytoplasm in a myxoid background (NMC Case Rep J 2020;7:53)
Images hosted on other servers:
Intraoperative histopathological findings

Vacuolated cells
- Histologically reminiscent of chordoma, consisting of epithelioid cells or spindle cells (often partly vacuolated) that are arranged in cords within a pale basophilic myxoid matrix (Am J Surg Pathol 2000;24:899, J Neurooncol 2010;100:465, Histopathology 2013;62:1002)
- Chordoid morphology must be the predominant pattern for diagnosis of chordoid meningioma and CNS WHO grade 2 designation
- Interspersed areas of more typical meningioma are frequent but pure chordoid cases may be seen (Am J Surg Pathol 2000;24:899, J Neurooncol 2010;100:465, Histopathology 2013;62:1002)
- Atypical features may be seen, including mitoses, brain invasion, spontaneous necrosis, macronucleoli, increased cellularity, sheeting or small cells with a high N:C ratio (Am J Surg Pathol 2000;24:899, Acta Neuropathol Commun 2022;10:56, Histopathology 2013;62:1002)
- Lymphoplasmacytic infiltrate in 59.5% (Am J Surg Pathol 2000;24:899)
Contributed by Valeria Barresi, M.D., Ph.D.
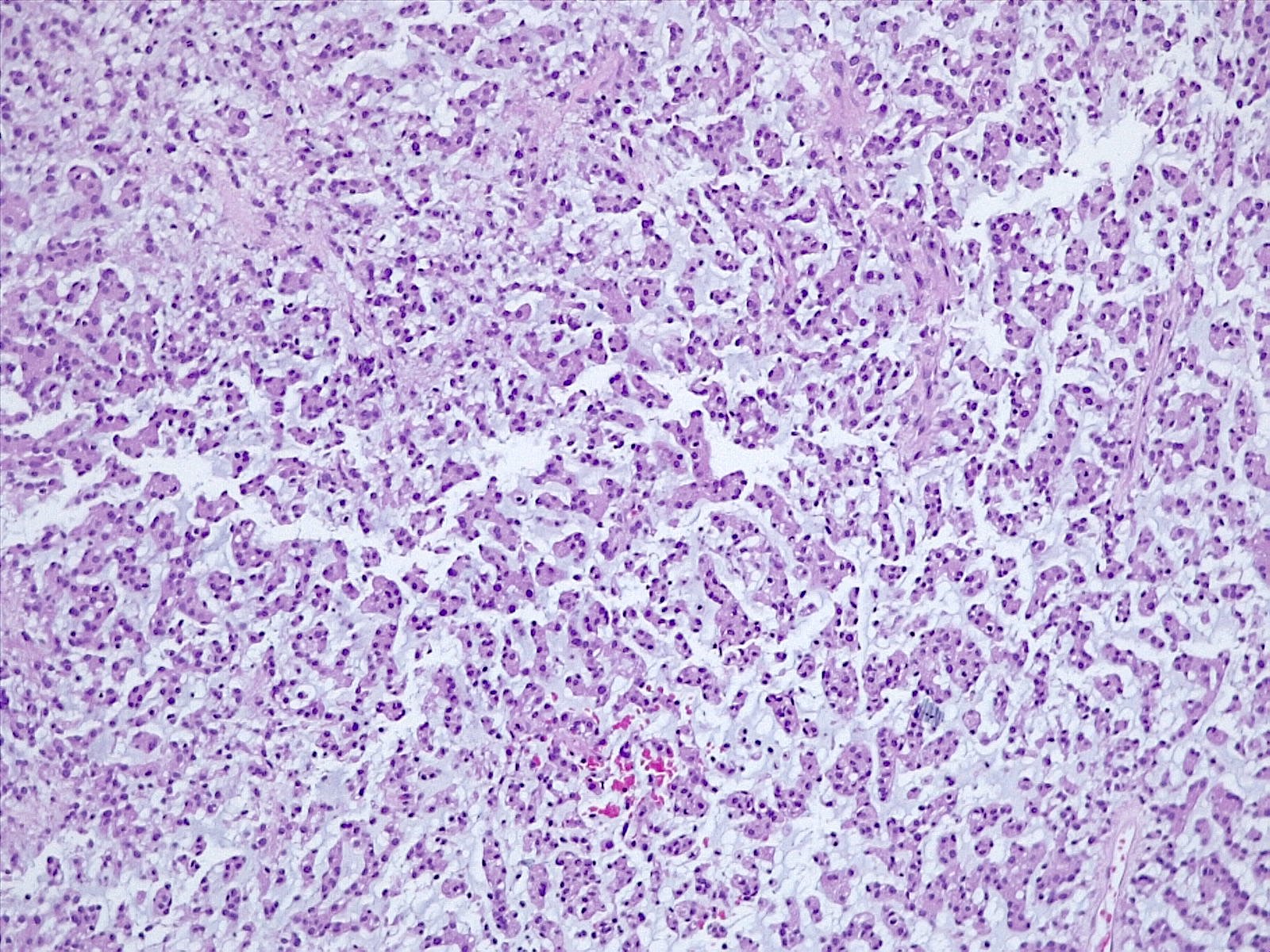
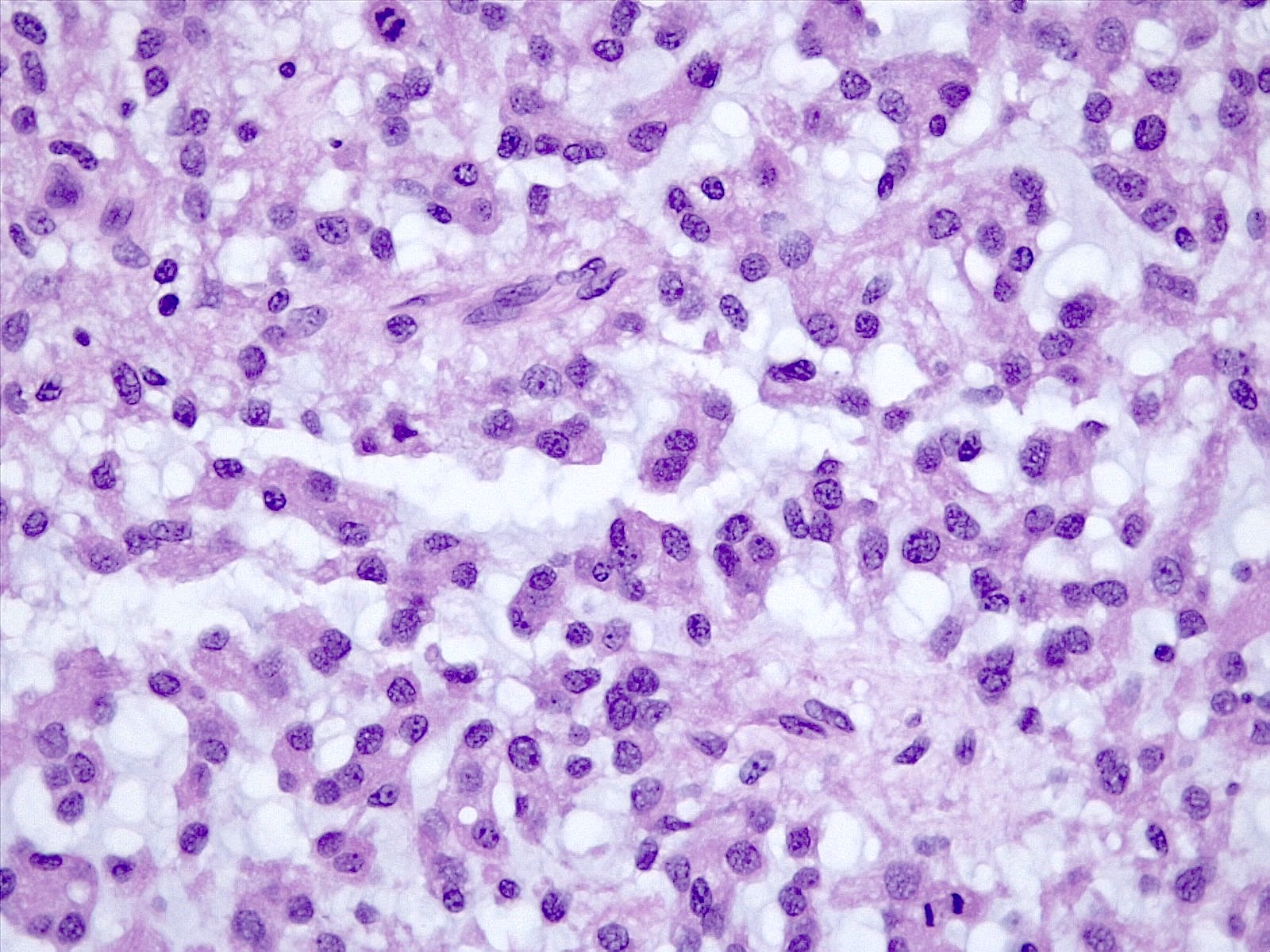
Epithelioid cells
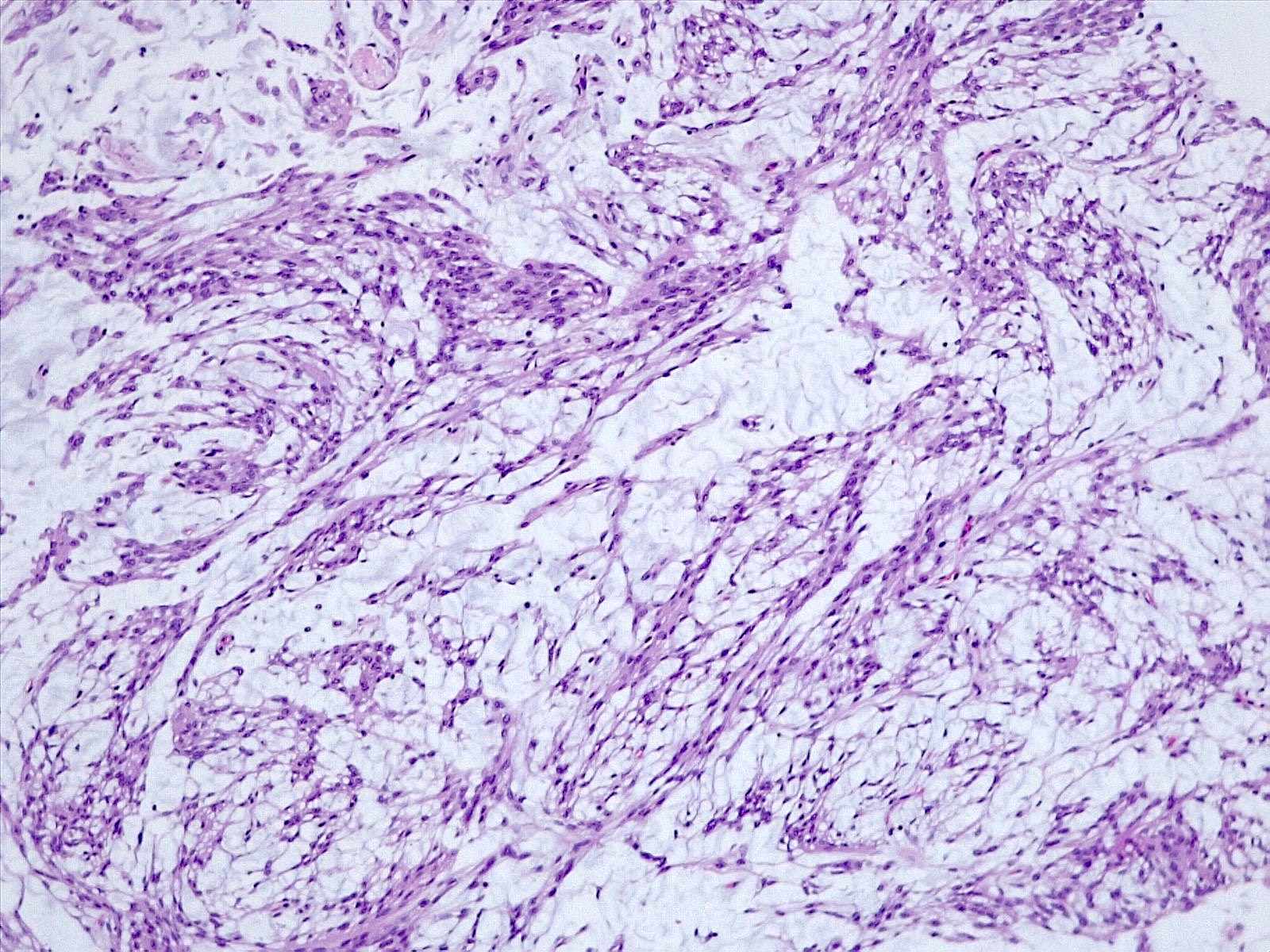
Spindle cells
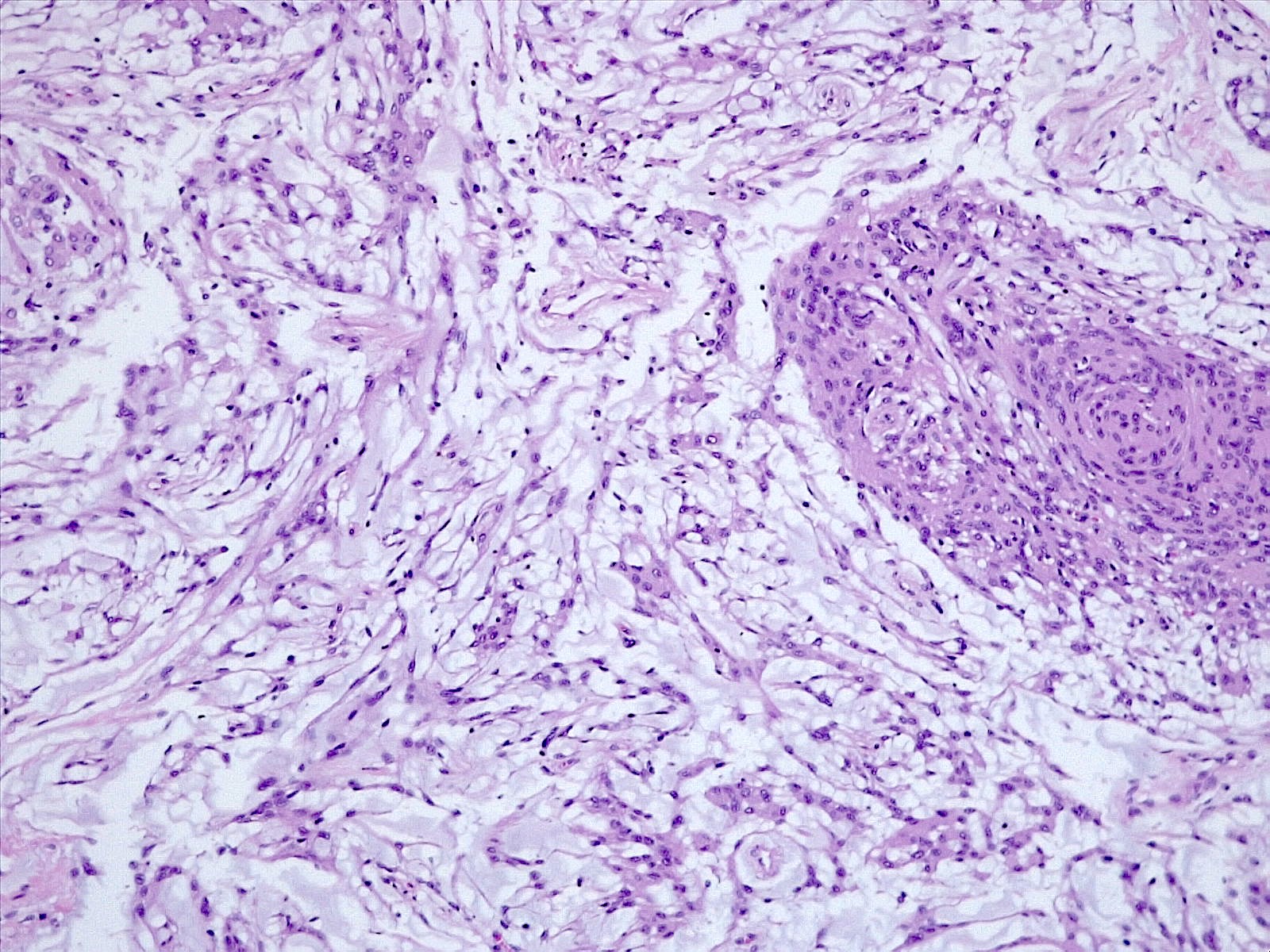
Intermingled typical meningioma
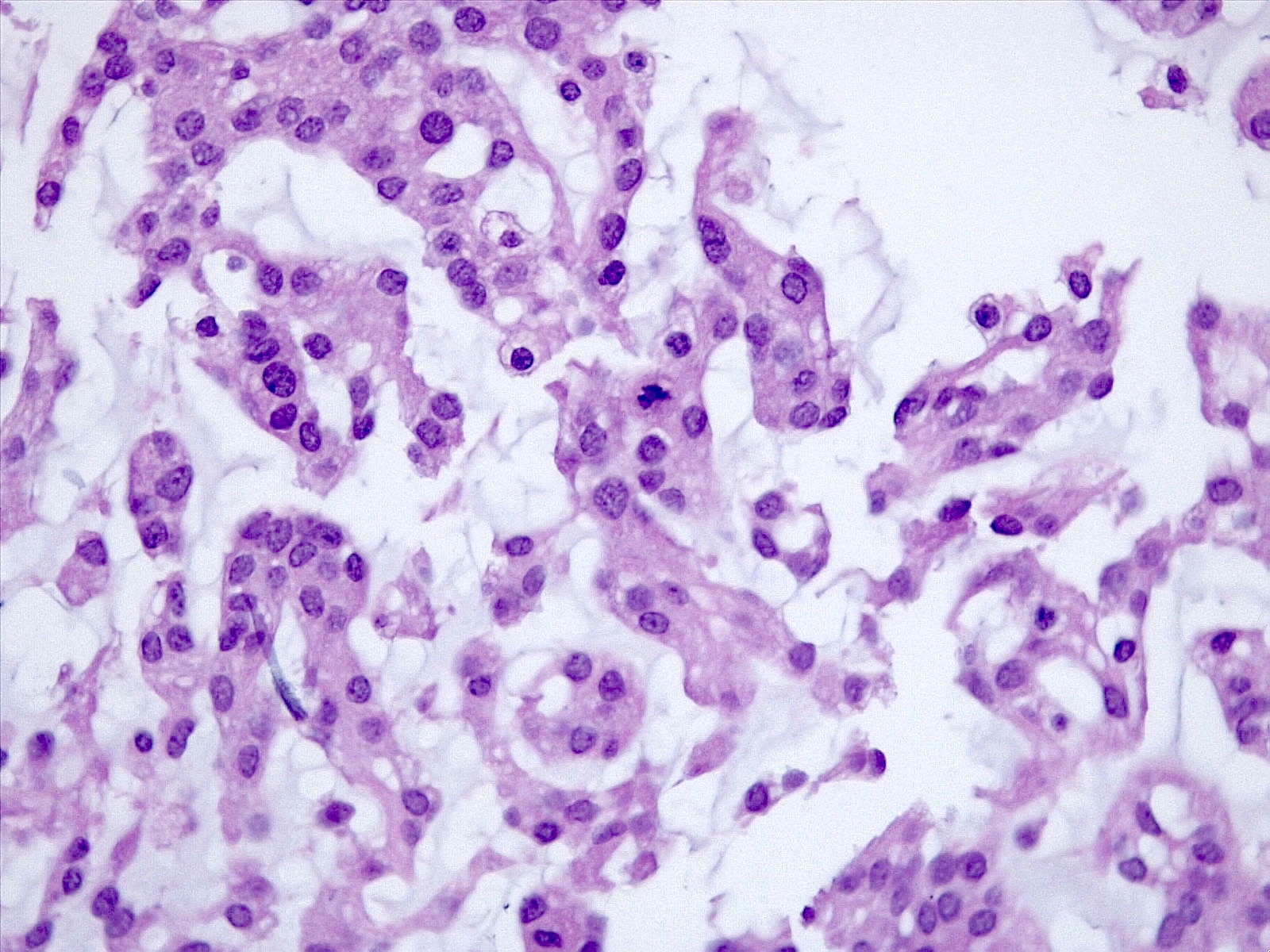
Mitoses

Alcian blue
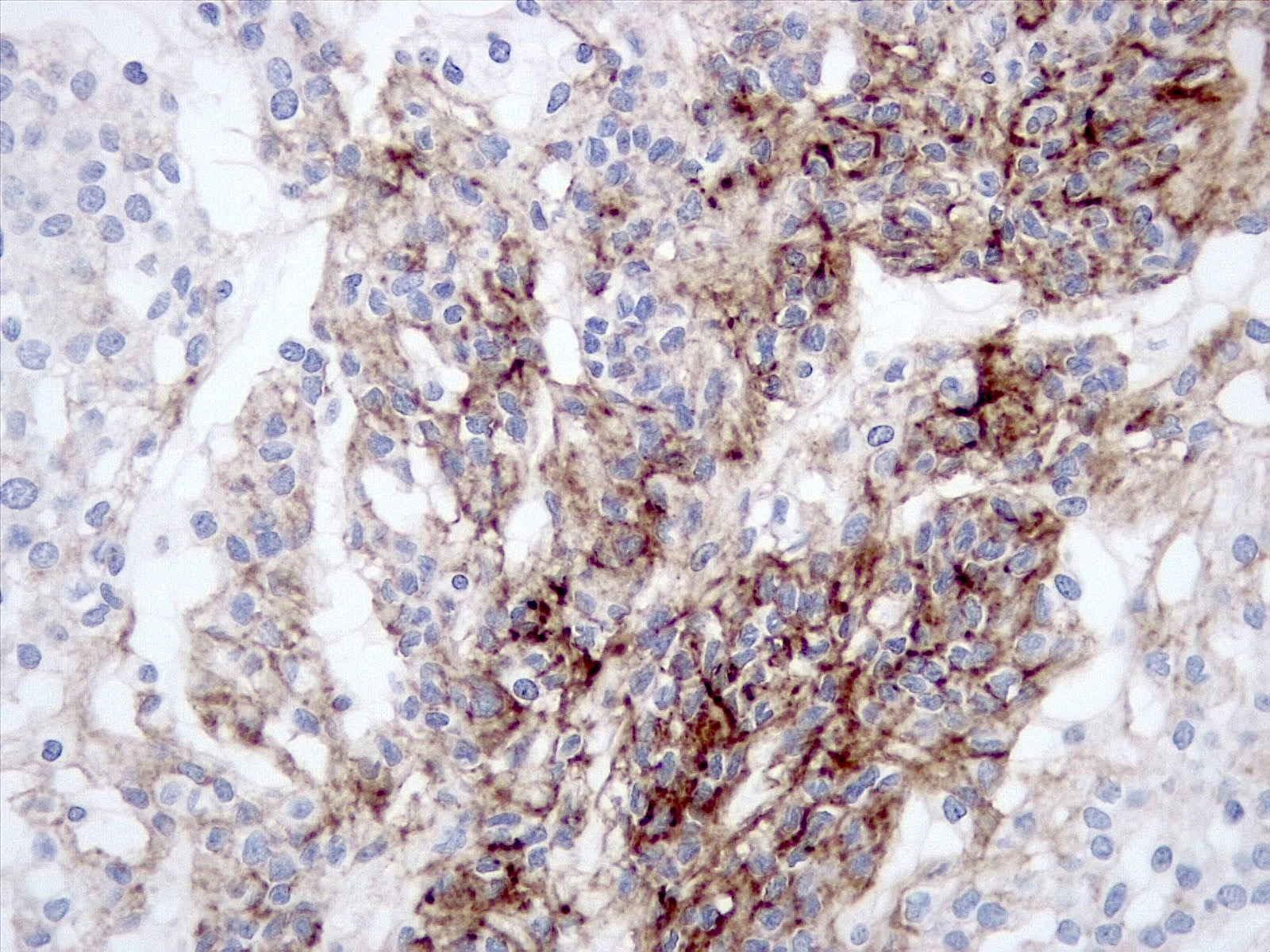
Podoplanin immunostaining
- Spindle shaped or epithelioid cells, with frequent intracytoplasmic vacuolization, that are arranged as loosely cohesive groups or have evident cord-like disposition, in a background of extracellular myxoid material (NMC Case Rep J 2020;7:53, Diagn Cytopathol 2016;44:811)
- Presence of meningothelial whorls and intranuclear pseudoinclusions (Diagn Cytopathol 2016;44:811)
- EMA (92.6%) (World Neurosurg 2016;93:198)
- Progesterone receptor (92.3%) (World Neurosurg 2016;93:198)
- SSTR2A (Neurosurg Rev 2022;45:467)
- Podoplanin (80%) (Am J Surg Pathol 2009;33:669)
- Mucoid matrix is red on mucicarmine stain, pink in the PASD reaction and bright blue on Alcian blue stain at pH 2 (Am J Surg Pathol 2000;24:899)
- S100 (weak extent with average intensity; 40%) (Am J Surg Pathol 2009;33:669)
- Pankeratin (weak and patch; 20%) (Am J Surg Pathol 2009;33:669)
- Brachyury (Am J Surg Pathol 2009;33:669)
- CK18 (Pathol Res Pract 2012;208:567)
- CK19 (Pathol Res Pract 2012;208:567)
- CD34 (Am J Surg Pathol 2009;33:669)
- Methylation profile is distinct from that of other meningioma subtypes, at whole genome DNA methylation analysis (Acta Neuropathol Commun 2022;10:56, Cancers (Basel) 2020;12:225)
- Frequent mutations in chromatin remodeling genes EP400 (73%), KMT2C (36%) and KMT2D (36%), at next generation sequencing (Acta Neuropathol Commun 2022;10:56, Cancers (Basel) 2020;12:225)
- Enrichment in chromosome 2p loss at copy number variation analysis (Acta Neuropathol Commun 2022;10:56, Cancers (Basel) 2020;12:225, Acta Neuropathol 2018;136:975)
- Lower frequency of AKT1, SMO, NF2, TRAF7, KLF4, SMO, AKT1 and SMARCB1 mutations compared with nonchordoid meningiomas (Acta Neuropathol Commun 2022;10:56)
- 1p, 8p, 10, 14, 22q loss and homozygous deletion of CDKN2A / CDKN2B detected in more aggressive cases (Cancers (Basel) 2020;12:225, Acta Neuropathol 2018;136:975, Neurol India 2018;66:156)
Images hosted on other servers:

FISH 18p, 22q, 14q, 1p
- Brain, extra-axial convexity mass:
- Meningioma, subtype chordoid, CNS WHO grade 2 (see comment)
- Comment: Meningothelial neoplasia showing epithelioid cells with vacuolated, eosinophilic cytoplasm or spindle cells, arranged in cords, within a myxoid matrix.
- Chordoma:
- Most commonly arises in the sacrococcygeal region and is usually extradural and bone invasive
- Can be intracranial at the clivus, have secondary invasion of the dura or rarely may be extradural
- Podoplanin negative, brachyury positive and showing extensive staining for CK AE1 / AE3, S100 and EMA
- Chondrosarcoma:
- Chordoid glioma:
- Rare glioma that is typically localized at the third ventricle
- GFAP positive
- Metastatic carcinoma:
- Metastases of carcinomas may be meningeal
- They show extensive cytokeratin staining, while chordoid meningioma features only focal staining for keratins
An extra-axial mass is found at the clivus without bone invasion. Histological examination shows a tumor with eosinophilic epithelioid cells in a myxoid stroma and focal areas with a syncytial growth pattern, whorls and nuclear pseudoinclusions. What is the most likely diagnosis?
- Chondrosarcoma
- Chordoid meningioma
- Chordoma
- Metastasis of carcinoma
Comment Here
Reference: Chordoid meningioma
- Chondrosarcoma
- Chordoid glioma
- Chordoid meningioma
- Chordoma
Comment Here
Reference: Chordoid meningioma
- Small cyst of choroid plexus containing CSF
- May be present throughout ventricular system but usually in glomus of lateral ventricles
- More prevalent in fetuses with chromosomal abnormalities (trisomy 18, trisomy 21, Aicardi syndrome)
- Common form affect fetuses in 1% of pregnancies; usually asymptomatic, resolves spontaneously by birth but large cysts can cause hydrocephalus
- Chromosomal abnormalities, specifically trisomy 18, should be considered if cysts are large ( > 1 cm), bilateral or irregular or if maternal age ≥ 32 years (AJR Am J Roentgenol 2009;192:32)
- In adults, usually asymptomatic, incidental postmortem finding
- On CT and MRI, usually show CSF density
Images hosted on other servers:
MR shows small cyst
- 20 week male fetus without any chromosomal abnormality (AJR Am J Roentgenol 2009;192:32)
- 53 year old woman with small lesion at foramen of Monro (Am J Neuroradiology 2002;23:841)
Images hosted on other servers:
Cyst at foramen of Monro
- Cyst wall lined by cuboidal to columnar epithelium with occasional cobblestone appearance typical of normal choroid plexus
- Some are devoid of epithelial lining
- Cyst lining has same immunohistochemical profile as normal choroid plexus: positive for vimentin, cytokeratin, S100, transthyretin, synaptophysin
- Choroid plexus tumors are rare neoplasms derived from the choroid plexus epithelium
- They occur within the ventricle system of the brain
- Rare intracranial tumor arising in the ventricle, mainly occurring in children
- 3 histological grades (WHO grade 1, 2, 3): choroid plexus papilloma (CPP), atypical choroid plexus papilloma (aCPP), choroid plexus carcinoma (CPC)
- Histological classification is based on architecture (preservation of papillary pattern), cellular density, cytology (nuclear pleomorphism), proliferation (mitoses) and necrosis / brain invasion
- Diagnostically, transthyretin (TTR), KIR7.1, cytokeratin and Ki67 immunohistochemistry are most helpful
- Based on methylation profiling, these tumors are now categorized in to 3 subtypes
- In line with other CNS tumors, integrated phenotype genotype diagnosis is preferred, which will guide appropriate management
- Conventionally, as per current WHO classification, these tumors are termed as choroid plexus papilloma (CPP, WHO grade 1), atypical choroid plexus papilloma (aCPP, WHO grade 2) and choroid plexus carcinoma (CPC, WHO grade 3) (Acta Neuropathol 2016;131:803)
- Based on recent DNA methylation profiling, they are being classified into 3 prognostically relevant epigenetic subgroups: supratentorial pediatric low risk choroid plexus tumors (CPP and aCPP), infratentorial adult low risk choroid plexus tumors (CPP and aCPP) and supratentorial pediatric high risk choroid plexus tumors (CPP and aCPP and CPC) (J Neurooncol 2020;148:39)
- Predominantly occurs in children (J Neurooncol 2015;121:151)
- Accounts for 2 - 5% of all pediatric brain tumors (J Neurosurg Pediatr 2012;10:398)
- Relatively higher incidence in infants (J Neurooncol 2015;121:151)
- M:F = 1.2:1 (J Neurooncol 2015;121:151)
- Genetic susceptibility:
- Aicardi syndrome (probable X chromosome abnormalities) (Brain Dev 2005;27:164)
- Li-Fraumeni syndrome (germ line TP53 mutation) (J Clin Oncol 2010;28:1995)
- Lateral ventricle (common in children)
- Fourth ventricle (distributed across all age groups)
- Rarely in spinal cord or ectopic sites
- Multifocal in exceptional circumstances
- So far, limited literature on pathogenesis or tumorigenesis
- Genetic or syndromic association present in a proportion of tumors
- Choroid plexus papilloma and atypical choroid plexus papilloma are genetically similar and are distinct from choroid plexus carcinoma (Clin Cancer Res 2015;21:184)
- Possible association with simian virus (SV40) reported (Virology 1995;212:710)
- Possible association with inflammatory pathway described (Acta Neuropathol Commun 2019;7:95)
- Hydrocephalus / raised intracranial pressure
- Headache, vomiting, papilledema
- Clinical features, imaging (CT or preferably MRI)
- Biopsy with histologic examination for definitive diagnosis
- Intraventricular papillary or lobulated lesions on MRI; hypo or isointense on T1, hyper or isointense on T2 and enhanced in postcontrast imaging (Cancer Imaging 2019;19:17)
Contributed by Kshitij Mankad, M.D.
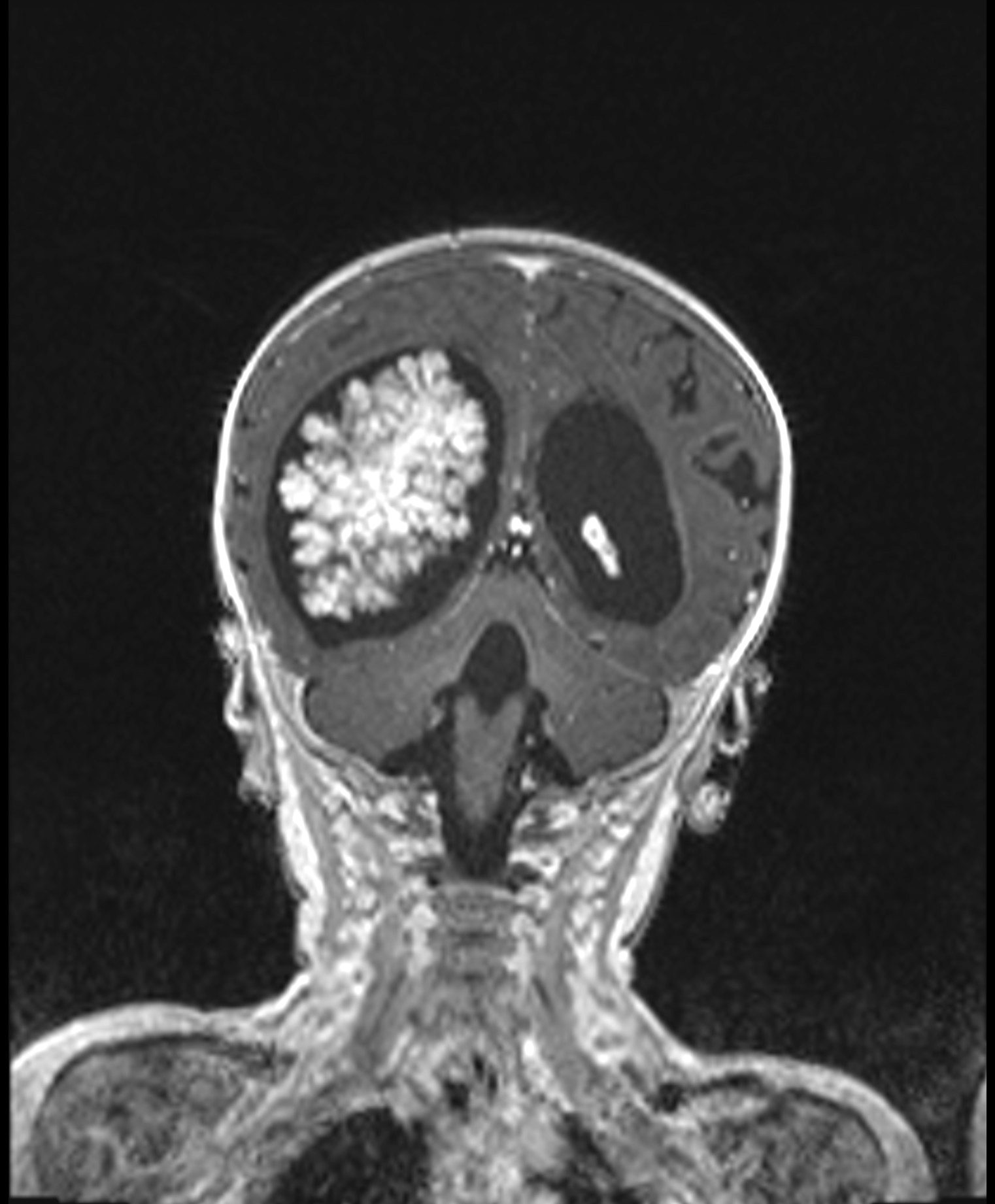
MRI coronal
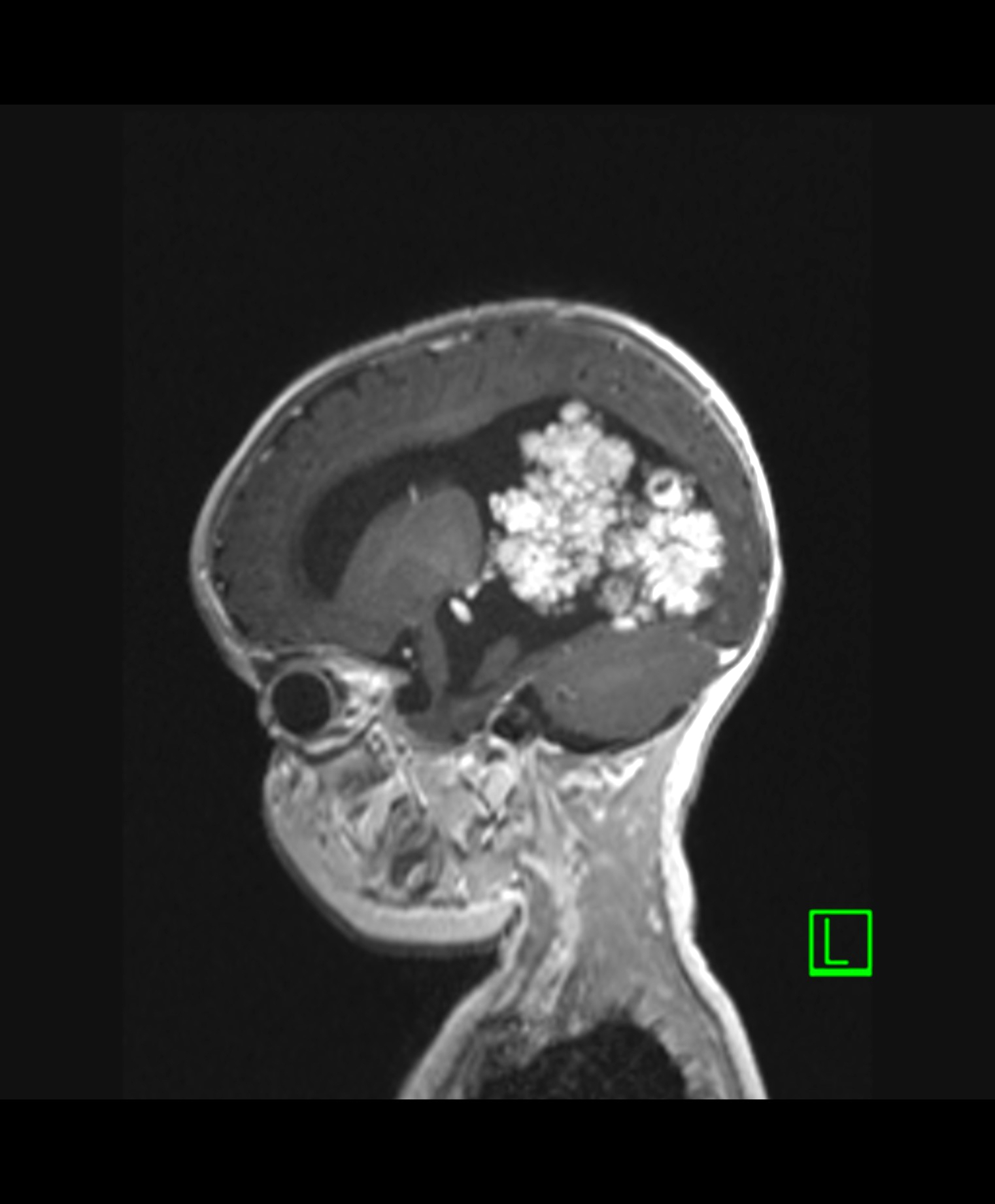
MRI sagittal
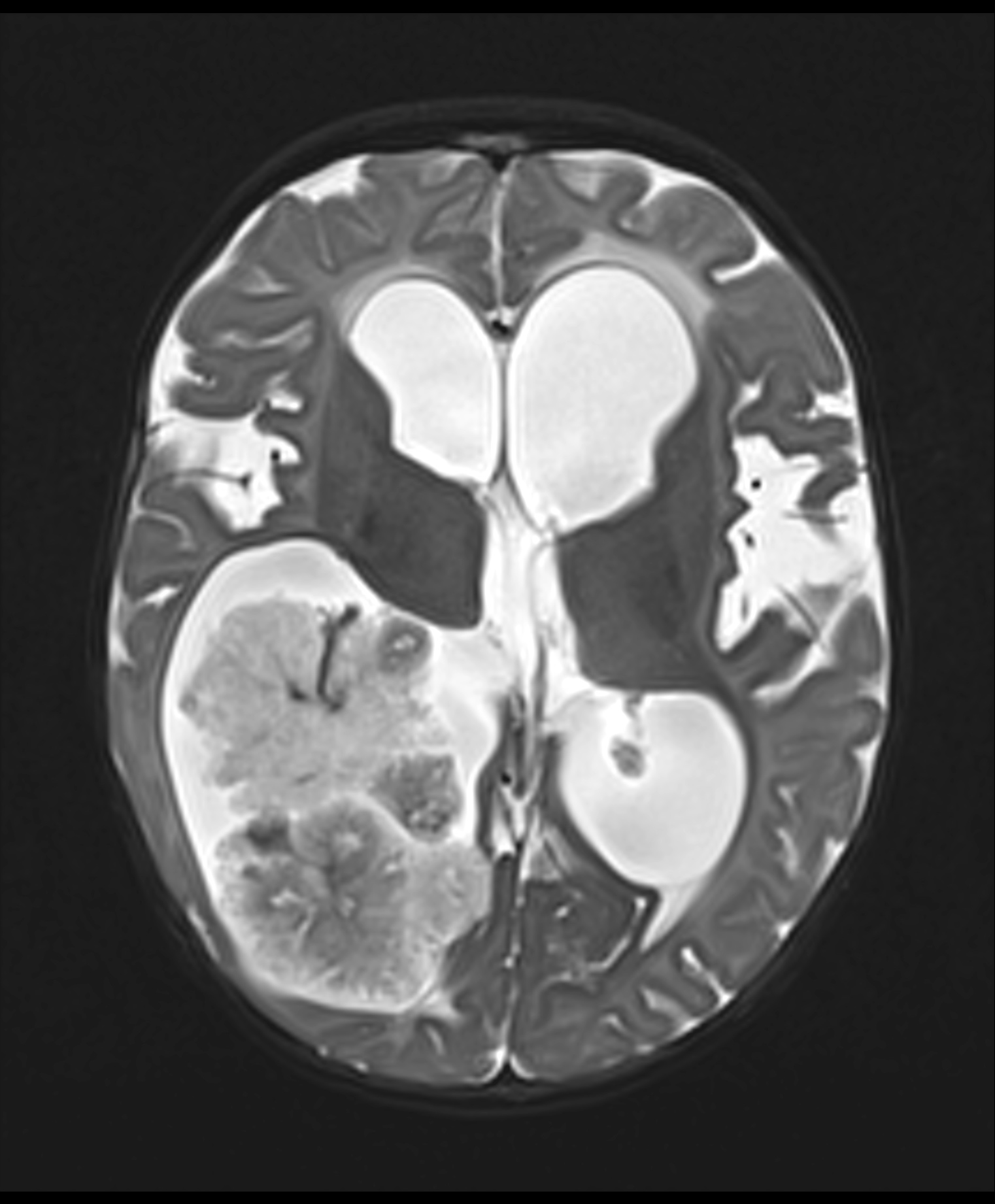
MRI transverse
- Historically, histological grade alone has been used for prediction of outcome
- 5 year survival rate for carcinoma reported approximately 62% (Pediatr Blood Cancer 2015;62:784)
- 5 year survival rate for papilloma and atypical papilloma is up to 100% and 89%, respectively (J Neurooncol 2009;95:383)
- Recent studies suggest 3 prognostic risk groups based on epigenetics, patient age and tumor location (Neuro Oncol 2016;18:790):
- Supratentorial pediatric low risk choroid plexus tumors
- Infratentorial adult low risk choroid plexus tumors
- Supratentorial pediatric high risk choroid plexus tumors
- Cerebrospinal fluid spread, drop metastasis and tumor recurrence after treatment are known and when present, carry poorer prognosis (BMJ Case Rep 2012;2012:bcr0120125681)
- 2 month old boy with recurrent choroid plexus carcinoma in the lateral ventricle (Childs Nerv Syst 2020;36:1601)
- 8 month old girl with choroid plexus papilloma and factor XIII deficiency (Pediatr Neurosurg 2018;53:413)
- 17 year old boy with an uncommon case of neck pain in choroid plexus papilloma (Medicine (Baltimore) 2018;97:e12466)
- 30 year old woman with cerebral intraparenchymal atypical choroid plexus papilloma (J Neurol Surg A Cent Eur Neurosurg 2019;80:53)
- 43 year old woman with a primary pigmented choroid plexus papilloma within the sella turcica (World Neurosurg 2017;105:1039.e13)
- Surgery to achieve gross total resection
- Adjuvant radiotherapy or chemotherapy, based on the clinical need
- Papillomas are well circumscribed cauliflower-like masses
- Cysts, hemorrhages and calcifications may be present
- Carcinomas are invasive, solid and necrotic
Images hosted on other servers:
Fourth ventricle tumor
- Choroid plexus papilloma (CPP, WHO grade 1):
- Papillary (finger-like) architecture, resembling normal choroid plexus
- Single layer of cuboidal to columnar monomorphic cells
- Loss of cobblestone surface
- Mild nuclear pleomorphism, mitotic activity rare (< 2/10 high power fields), lacks necrosis
- Atypical choroid plexus papilloma (aCPP, WHO grade 2):
- Higher cellularity relative to CPP
- Moderate nuclear pleomorphism, blurring of papillary pattern
- Occasional mitoses (> 2/10 high power fields), with or without necrosis
- Choroid plexus carcinoma (CPC, WHO grade 3):
- Frankly malignant
- High cellularity, hyperchromatic nuclei and nuclear pleomorphism
- Blurring of papillary pattern and solid arrangement
- Frequent mitoses (> 5/10 high power fields), necrosis, with or without brain invasion
Contributed by Ashirwad Merve, M.B.B.S., Ph.D.
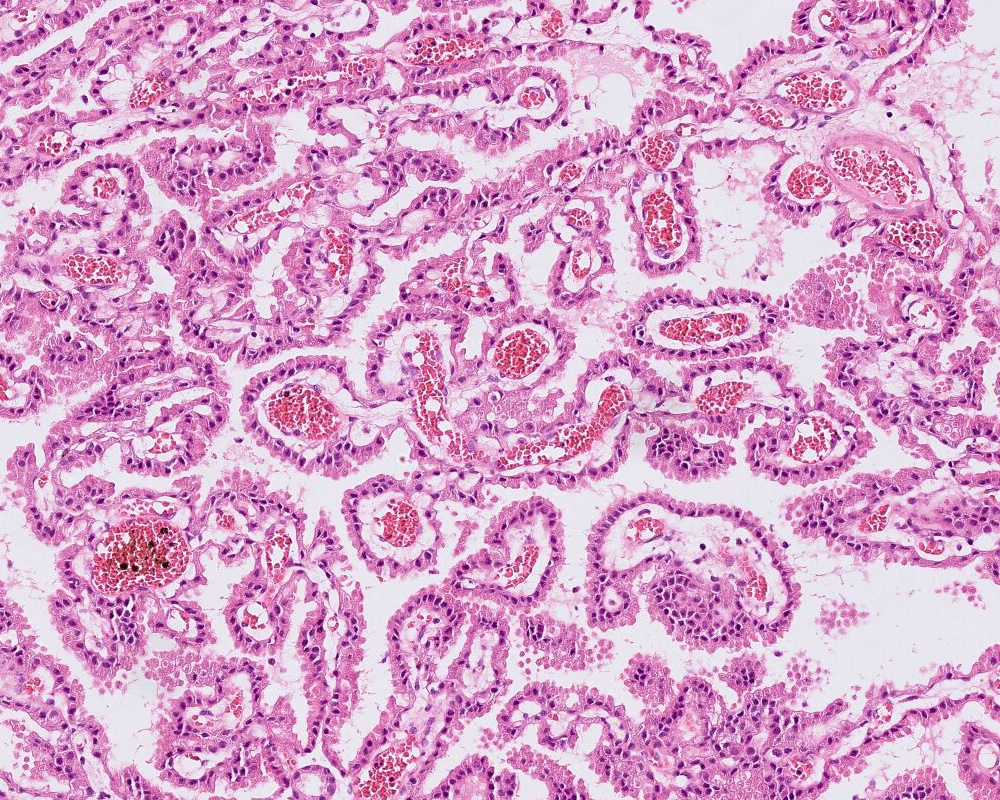

Choroid plexus papilloma
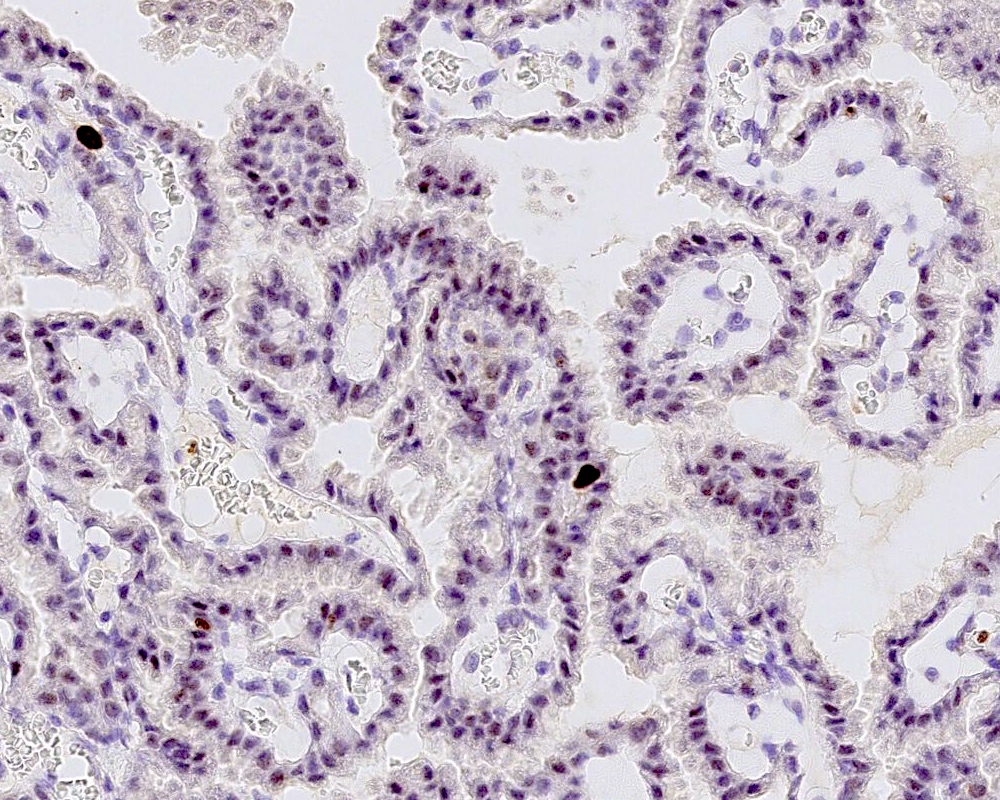
Ki67 in choroid plexus papilloma
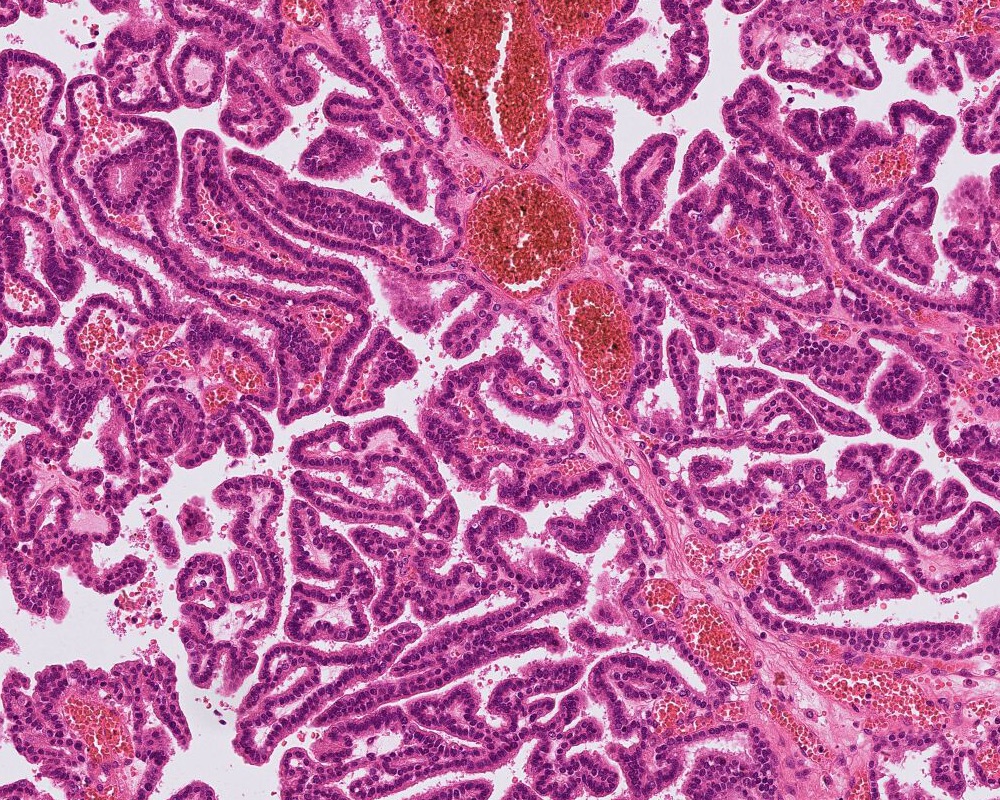
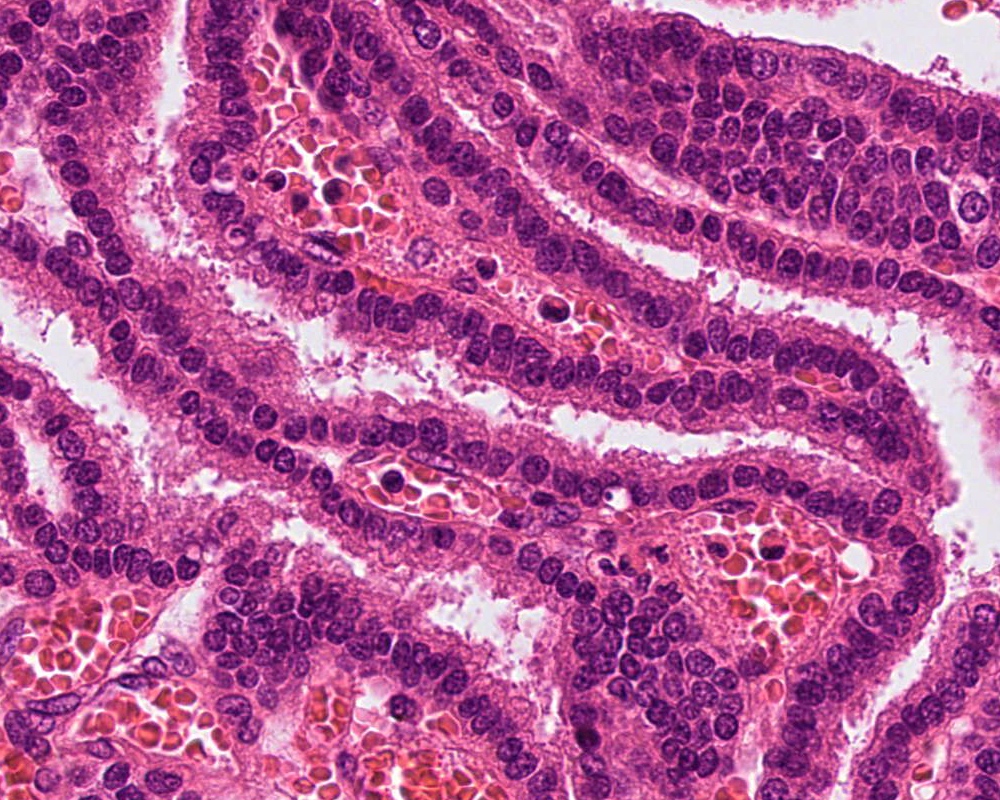
Atypical choroid plexus papilloma
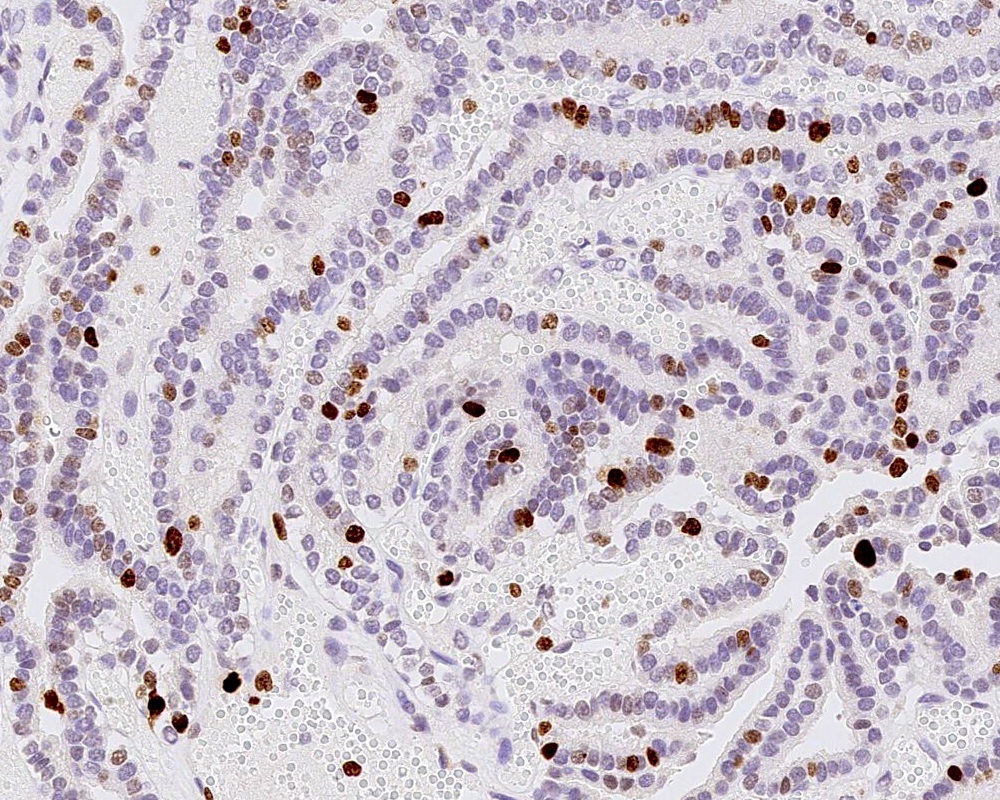
Ki67 in atypical choroid plexus papilloma
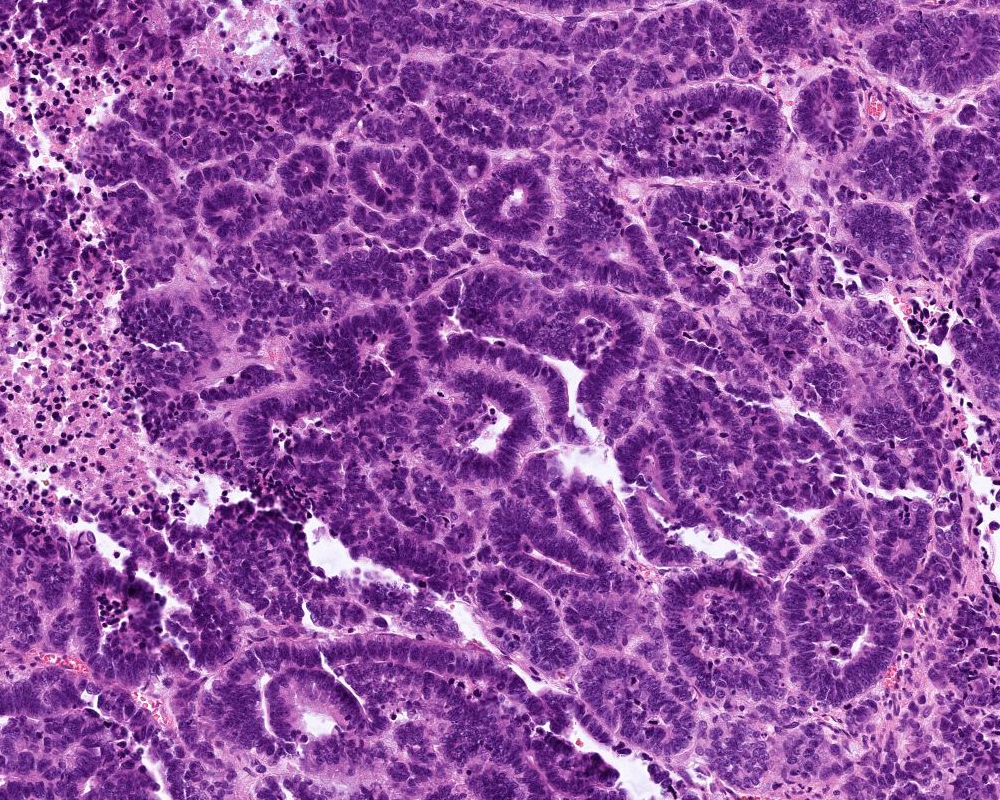
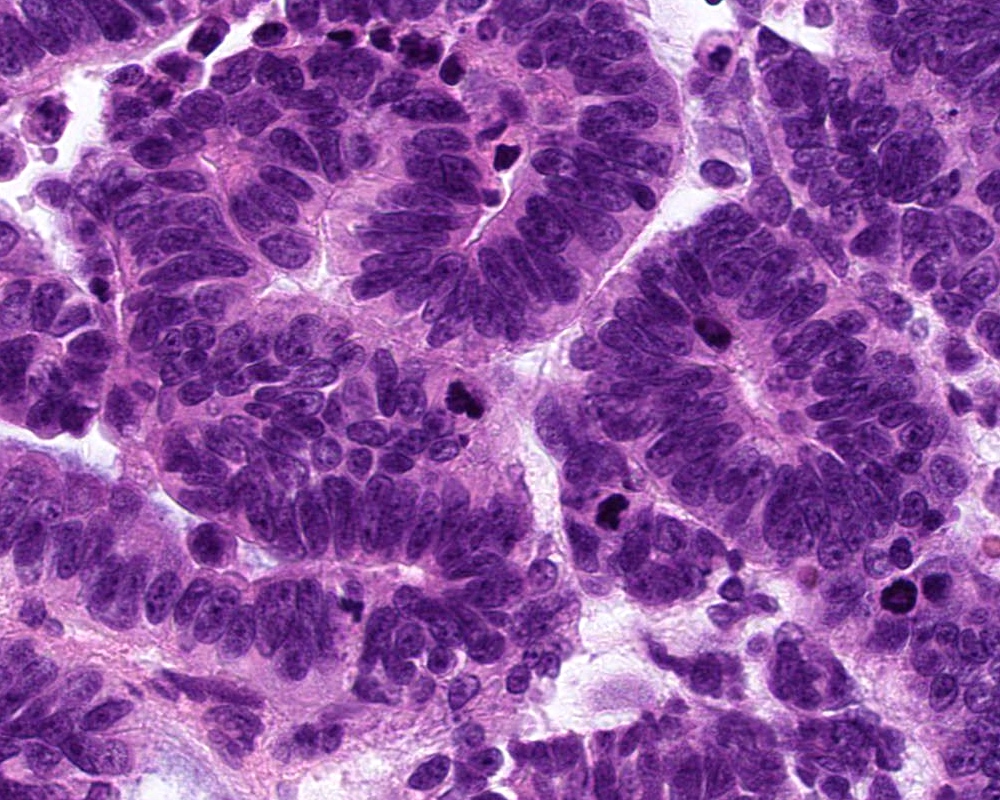
Choroid plexus carcinoma
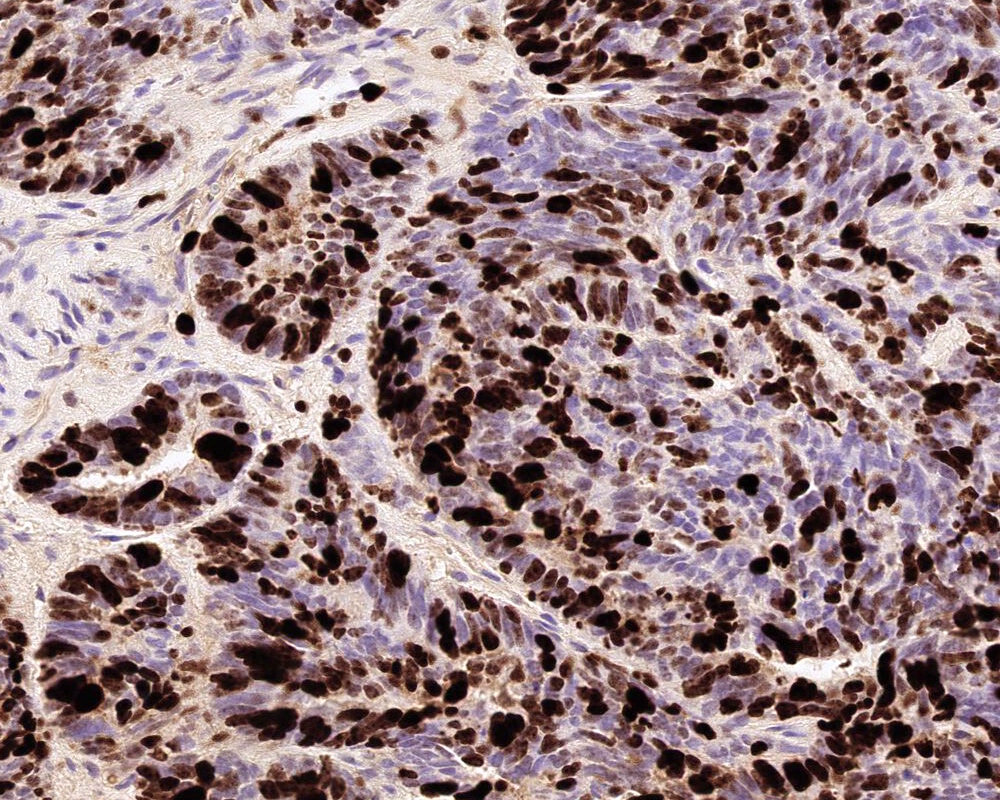
Ki67 in choroid plexus carcinoma
Transthyretin
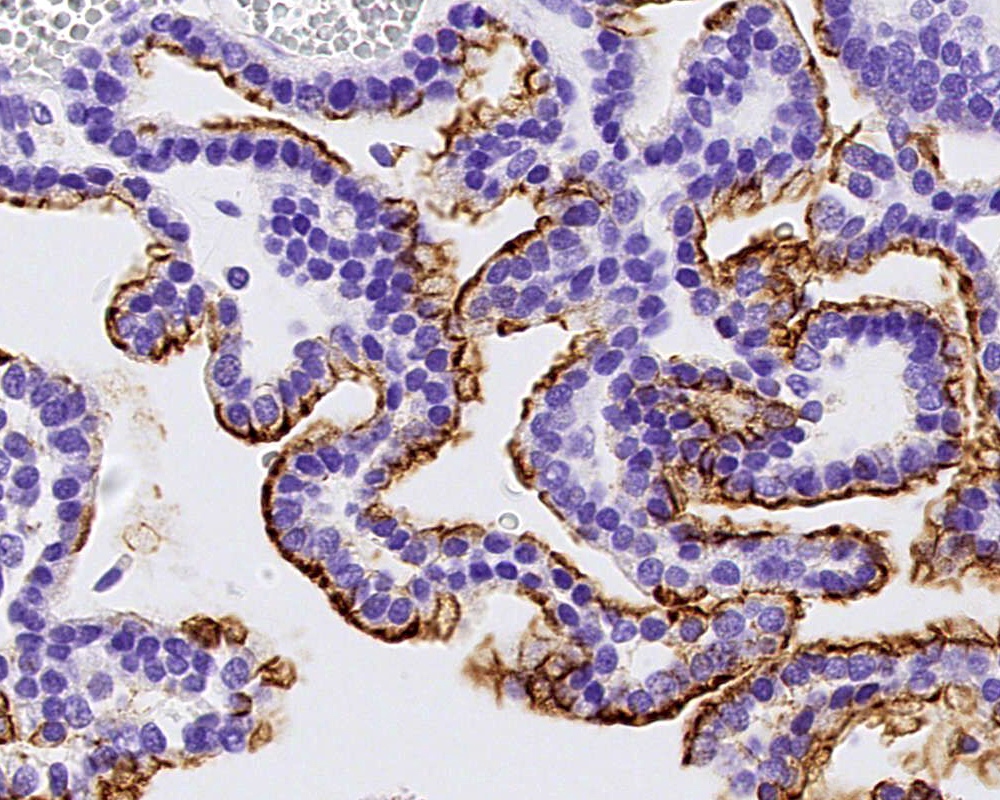
Cytokeratin
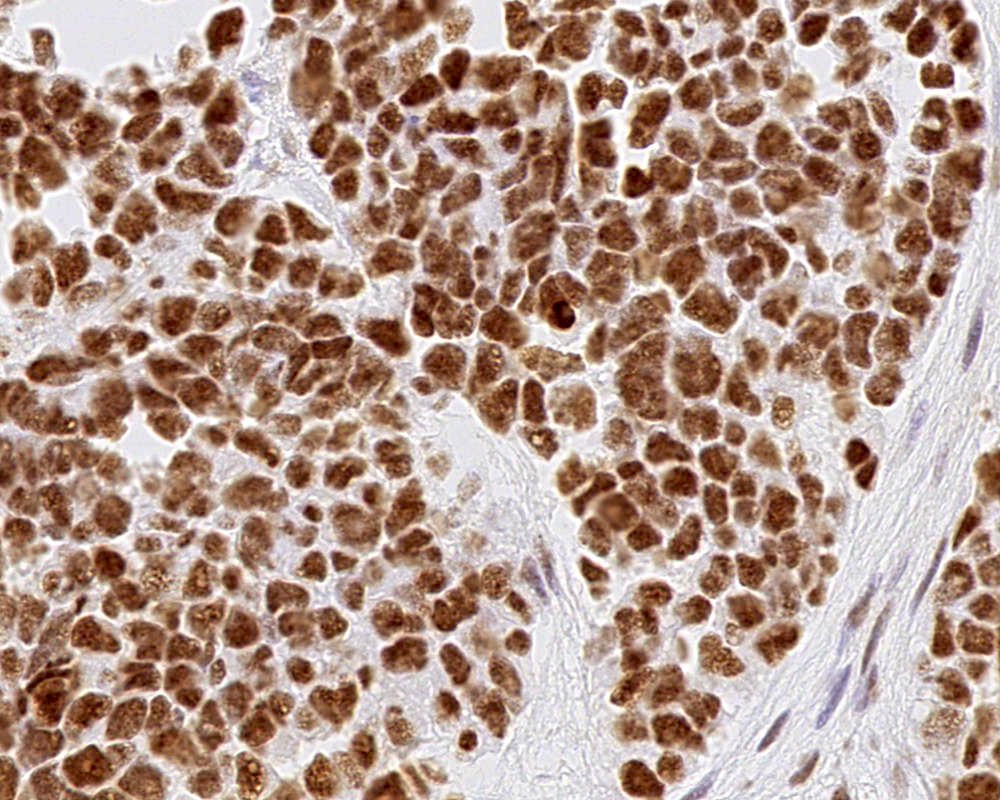
SMARCB1 (INI1)
- Intraoperative crush smear: papillary strands with fibrovascular cores
- Lesional cells exhibit relative cellular crowding and cytological atypia (in relation to normal choroid plexus epithelium)
Contributed by Ashirwad Merve, M.B.B.S., Ph.D.

Intraoperative smear
- Transthyretin, KIR7.1, EAAT1, S100, pancytokeratin (cytokeratin, MNF116, AE1 / AE3), CK7 > CK20
- Can be positive for GFAP, synaptophysin, S100
- Ki67 / MIB1 proliferation index may be used for grading (variable and subjective interpretation)
- Choroid plexus carcinoma: p53 variable, SMARCB1 / INI1 and SMARCA4 retained nuclear expression
- References: Neurosurgery 1988;23:384, Acta Neuropathol 1990;80:635, Am J Surg Pathol 2006;30:66, J Neuropathol Exp Neurol 1999;58:398
- CEA, EMA (weak or negative), LIN28a
- Reference: Acta Neuropathol 1990;80:635
- The following 3 methylation classes of choroid plexus tumors are recognized based on the German Cancer Research Center (DKFZ) Heidelberg classification:
- Plexus tumor, subclass adult
- Plexus tumor, subclass pediatric A
- Plexus tumor, subclass pediatric B (closely related to methylation cluster 3 described in Neuro Oncol 2016;18:790)
- TP53 sequencing and testing for germ line TP53 mutation is advisable in children with choroid plexus carcinoma
Contributed by Ashirwad Merve, M.B.B.S., Ph.D.
Methylation array copy number variation plot
- Lateral ventricle, tumor, excision:
- Atypical choroid plexus papilloma (WHO grade 2)
- Methylation class: Plexus tumor, subclass pediatric B, family class PLEX_T
- Molecular tests (sequencing): No TP53 mutation
- Comment: The tumor shows moderate atypia, histologically equivalent to WHO grade 2. The methylation array has classified as subclass B, which is closely related to methylation cluster 3 described in Neuro Oncol 2016;18:790.
- Metastatic adenocarcinoma with papillary structures:
- Variable morphology depending on the primary
- GATA3+ (breast carcinoma), TTF1+ (lung adenocarcinoma), PAX8 (renal, Müllerian thyroid origin carcinomas)
- Choroid plexus tumors are often diffusely synaptophysin+, which differs from metastatic papillary carcinoma (J Neuropathol Exp Neurol 1999;58:398)
- Ependymoma:
- Ependymal perivascular pseudo rosettes
- GFAP+, KIR7.1-, transthyretin-
- Intraventricular meningioma:
- Whorl formation, vesicular nuclei and syncytial cytoplasm
- SSTR2A+, KIR7.1-, transthyretin-
- Central neurocytoma:
- Uniform round cells with neuronal differentiation; no papillary structure
- NeuN+, MAP2+
- Germ cell tumor:
- Medulloblastoma:
- Small round blue cell tumor with or without Homer-Wright rosettes
- Synaptophysin+
- Embryonal tumor with multilayered rosettes (ETMR):
- Biphasic pattern with abundant neuropil and embryonal cells in multilayered rosettes
- LIN28a+
- Atypical teratoid / rhabdoid tumor (AT / RT):
- Poorly differentiated embryonal tumor
- INI1-
- Cribriform neuroepithelial tumor (CRINET) (J Neuropathol Exp Neurol 2009;68:1249):
- Intraventricular and papillary
- INI1-
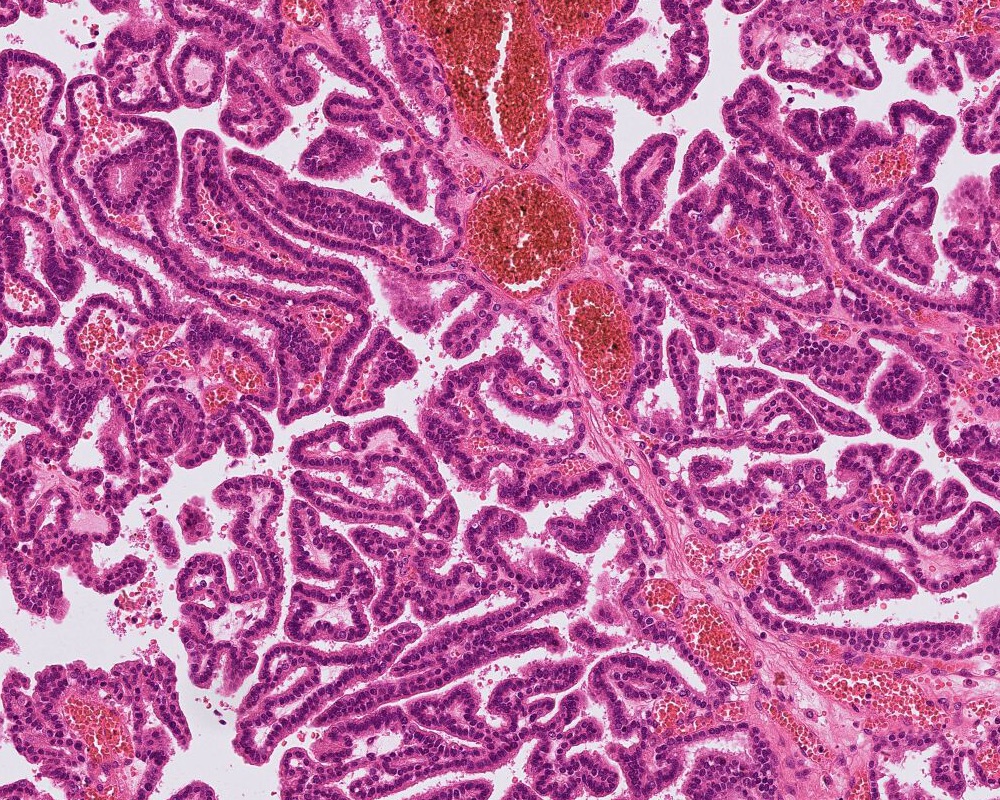
A 3 month old boy presented with restless crying and vomiting. His head size was larger than expected for his age. Imaging showed an 8 cm, lobulated, enhancing lesion in the right lateral ventricle. Histology is as above. Which of the following is true?
- Choroid plexus tumors are common in adults
- Recent studies suggest 3 distinct molecular entities based on methylation profiling, patient age and tumor location
- This is a choroid plexus carcinoma (WHO grade 3)
- TP53 mutation is strongly associated with choroid plexus papilloma
Choroid plexus tumors are rare intracranial tumors arising in the choroid plexus epithelium of the ventricles. They are more common in children. Histologically, they are classified into 3 categories: choroid plexus papilloma (CPP, WHO grade 1), atypical choroid plexus papilloma (aCPP, WHO grade 2) and choroid plexus carcinoma (CPC, WHO grade 3). Although any of these tumors can have CSF spread and recur following treatment, the latter is associated with poor outcome and higher recurrence rate. Recent studies have shown that CPP and aCPP are genetically similar but CPP and aCPP are genetically distinct from CPC. The CPCs are mostly driven by loss of function or mutations of tumor suppressor gene TP53 and may be associated germ line mutations (Li-Fraumeni syndrome). Furthermore, based on methylation profiling, tumor location and age, 3 distinct subgroups with prognostic significance have been described. The advances in understanding of genetics may help deliver appropriate personalized treatment.
Comment Here
Reference: Choroid plexus tumors (papilloma, atypical papilloma, carcinoma)
- Diffuse synaptophysin expression and Homer-Wright rosettes are features of choroid plexus carcinoma
- IDH1 is positive in choroid plexus carcinoma
- LIN28a is positive in choroid plexus carcinoma
- Nuclear INI1 (SMARB1) expression is retained in choroid plexus carcinoma but lost in atypical teratoid / rhabdoid tumor
Choroid plexus carcinomas can mimic other malignant tumors of the childhood, such as CNS embryonal tumors, and it is important to exclude them to guide appropriate treatment. INI1 (SMARCB1) and SMARCA4 immunohistochemistry expression is retained in CPC, which helps to differentiate from aggressive tumor atypical teratoid / rhabdoid tumor (AT / RT). A negative LIN28a differentiates from embryonal tumor with multilayered rosettes (ETMR, bearing C19MC alterations). Diffuse synaptophysin expression with patchy GFAP and presence of Homer-Wright rosettes should raise the possibility of medulloblastoma. Diffuse GFAP expression with possible IDH1 expression (in younger adults) with microvascular endothelial proliferation and necrosis would indicate a high grade glioma / glioblastoma. DNA methylation array and classification of CNS tumors will help in definitive classification. Nevertheless, it is important to have a solid histological diagnosis in case the array fails to confidently classify the tumor.
Comment Here
Reference: Choroid plexus tumors (papilloma, atypical papilloma, carcinoma)
- Rare variant, 0.2 - 0.8% of all meningiomas (J Neurooncol 2007;81:315)
- WHO grade 2 due to aggressiveness
- 20 - 60% chance of recurrence even with gross total resection
- More common in younger patients (mean age 29 years)
- Predilection for cauda equina and cerebellopontine areas
- Predilection for CP angle and cauda equina
- Indistinguishable from classic meningioma
- Dural based, homogenously enhancing
Contributed by Chunyu Cai, M.D., Ph.D. (Case #514)
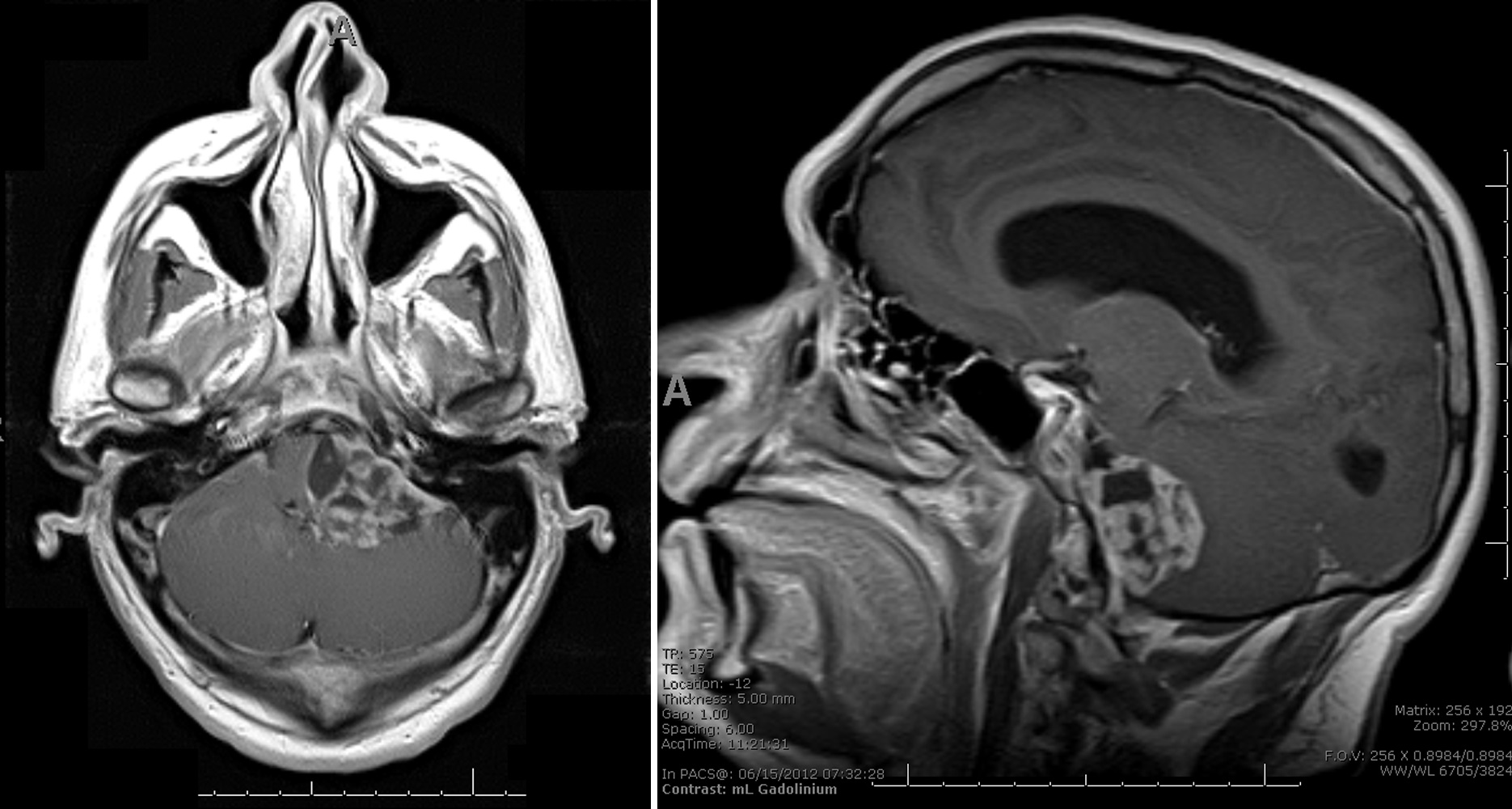
MRI
Images hosted on other servers:
Various images
- 13 year old girl with pediatric spinal clear cell meningioma (J Neurosurg Pediatr 2009;3:57)
- 14 year old boy with clear cell meningioma of the fourth ventricle (Pediatr Neurosurg 2010;46:462)
- 38 year old woman and two 60 year old men with clear cell meningiomas (Neurosurgery 2010;67:E870)
- 41 year old woman with clear cell meningioma of the cauda equina (J Spinal Disord Tech 2005;18:539)
- 51 year old man with progressive hearing loss in the left ear, intermittent dizziness, difficulty walking (Case of the Month #514)
- Clear cell meningioma of the fourth ventricle (Am J Surg Pathol 2003;27:131)
- Gross total resection
- Postsurgical radiation
- Patternless arrangement of clear cells with sometimes distinct cell borders
- Prominent perivascular and interstitial collagen
- Little to no mitotic activity
- May not display any characteristic meningioma features (whorls, psamomma bodies, intranuclear inclusions)
Contributed by Chunyu Cai, M.D., Ph.D. (Case #514)
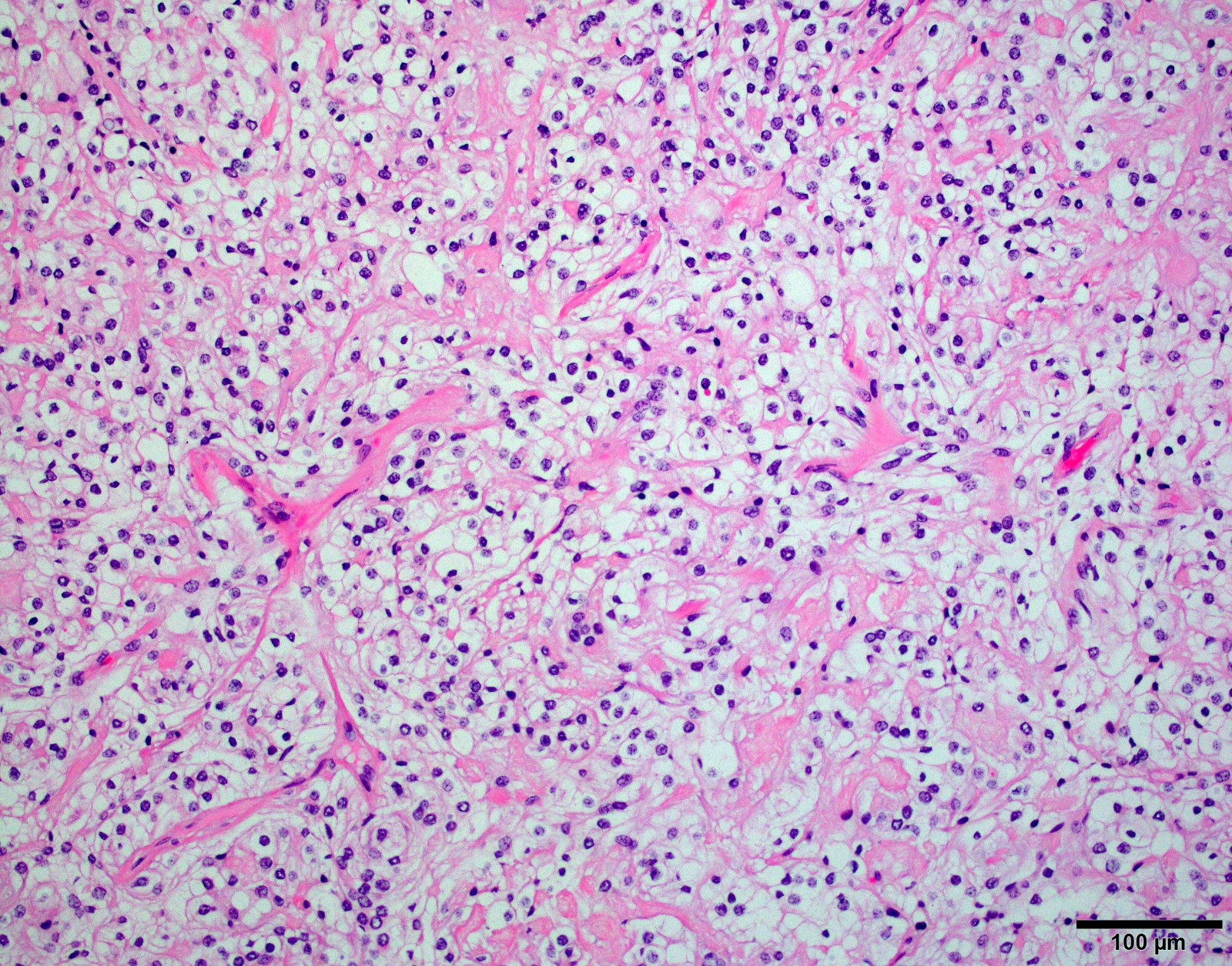
H&E clear cell

H&E blocky collagen
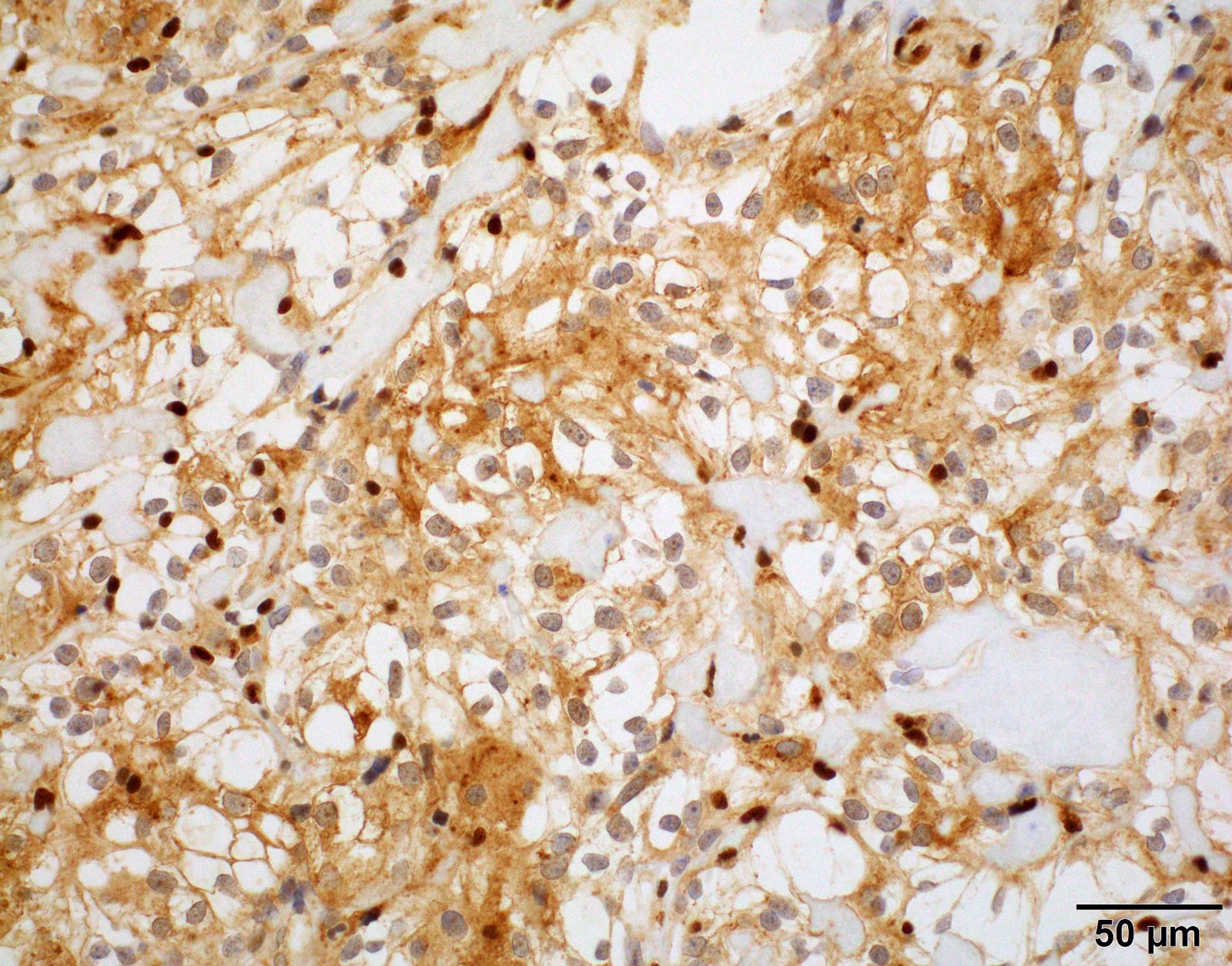
IHC SMARCE1
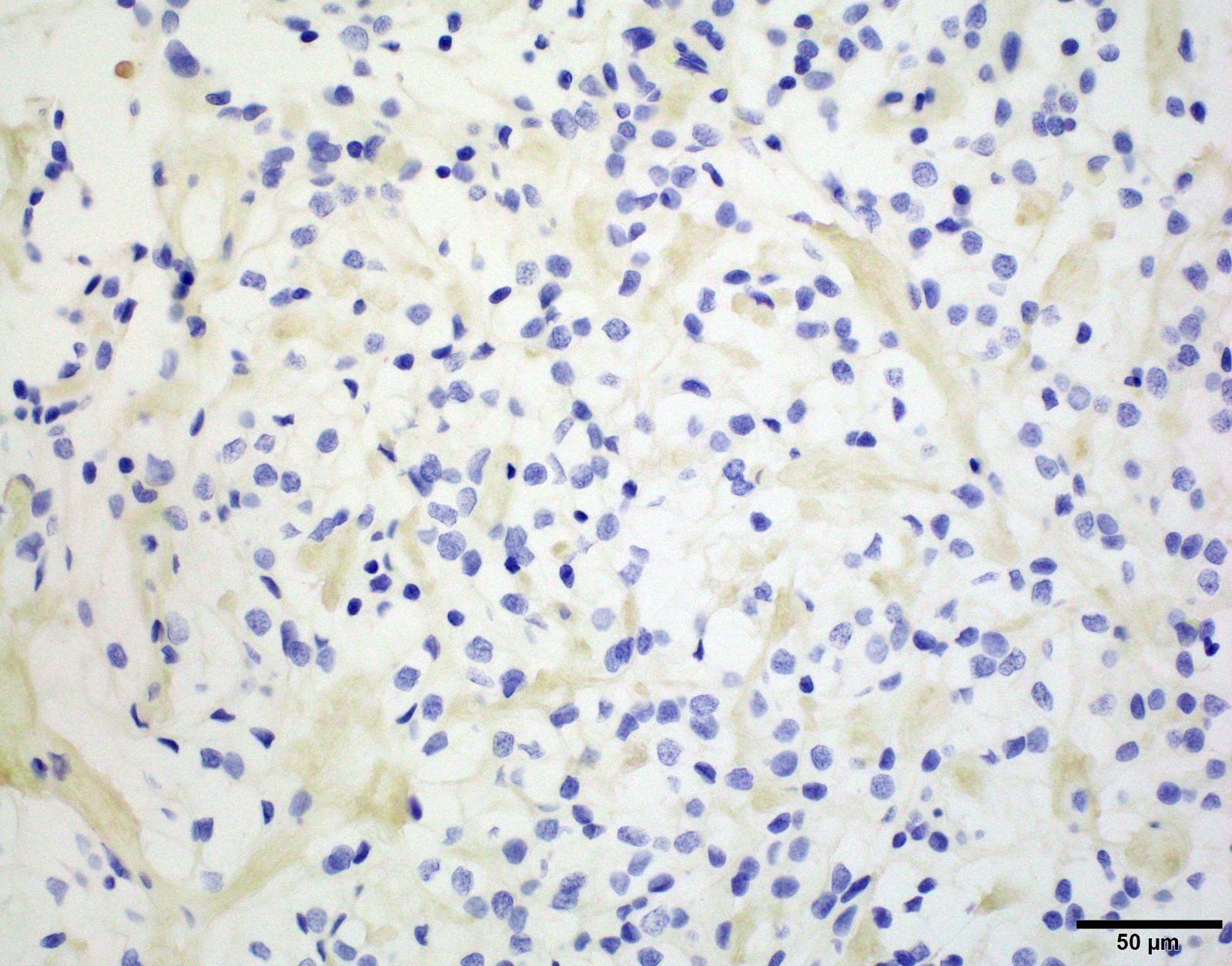
IHC CAIX
- Whorled, syncytial architecture composed of spindle to polygonal cells with vacuolated cytoplasm and bland nuclei (Diagn Cytopathol 1998;18:131)
- Abundant cytoplasmic glycogen
- Intermediate filaments
- Interdigitation of cell membranes, desmosomes
- Occasional cytoplasmic lumina (Ultrastruct Pathol 1999;23:51)
- Clear cell ependymoma: GFAP +
- Germinoma / seminoma: PLAP+, OCT3 / 4+, cKIT+
- Hemangioblastoma: inhibin+, NSE +
- Metastatic renal cell carcinoma: keratin+ (J Clin Neurosci 2005;12:685)
- Oligodendroglioma: GFAP +
- AKT1 mutation
- BAP1 mutation
- NF2 mutation
- SMARCE1 mutation
- TRAF7 and KLF4 co-mutations
Comment Here
Reference: Clear cell meningioma
- Benign, unilocular, epithelium lined, mucin filled cyst of third ventricle (eMedicine: Colloid Cysts [Accessed 31 October 2023])
- Uncertain histogenesis, mostly of endodermal derivation (Acta Neuropathol 1992;83:605)
- Site, radiologic and pathologic features are distinctive so differential diagnosis may be limited
- Benign, unilocular, epithelium lined, mucin filled cyst of third ventricle (eMedicine: Colloid Cysts [Accessed 31 October 2023])
- Usually adults (20 - 50 years); rare in children
- Located at anterosuperior third ventricle near foramen of Monro
- Excellent prognosis
- Colloid cyst of the third ventricle
- ICD-10: G93.0 - cerebral cysts
- Rare, 0.5 - 2.0% of all intracranial lesions and 10 - 20% of all intraventricular lesions (World Neurosurg 2019:123:351)
- Usually adults (20 - 50 years), rare in children, no sex predilection (World Neurosurg 2017:107:409)
- Anterosuperior third ventricle near foramen of Monro, attached to the third ventricular roof and choroid plexus
- Unusual sites: septum pellucidum, fourth ventricle (Acta Neurochir (Wien) 2004;146:397, Clin Neurol Neurosurg 2012;114:1095)
- Usually sporadic
- Genetic predisposition is suggested with limited reports of familial clusters (World Neurosurg 2017:107:409, Asian J Neurosurg 2020;15:414)
Images hosted on other servers:

Mechanisms of sudden death due to colloid cyst
Typical location of colloid cyst
- Due to its position, causes intermittent obstruction of cerebrospinal fluid (CSF) flow and obstructive hydrocephalus with manifestations of increased intracranial pressure
- Headache is the most common symptom
- May also cause nausea, vomiting, blurred vision, gait disturbance, urinary incontinence and personality changes (BMC Neurol 2022;22:397)
- Sudden impaction (ball valve effect on the foramen of Monro) causes abrupt, transient lower limb paralysis (drop attacks) and rarely, sudden death (Emerg (Tehran) 2015;3:162)
- May be asymptomatic
- Neuroimaging: computed tomography (CT) and magnetic resonance imaging (MRI)
- Biopsy
- Typical intraventricular location allows confident radiological diagnosis
- CT: unilocular hyperdense mass at or near the foramen of Monro (AJNR Am J Neuroradiol 2000;21:1470)
- MRI: spherical, usually nonenhancing, discrete cystic lesion at anterior third ventricle; most are intrinsically bright in precontrast T1 weighted MRI images (AJNR Am J Neuroradiol 2020;41:1833)
Contributed by Mohamed Kayed, M.D., Ph.D.
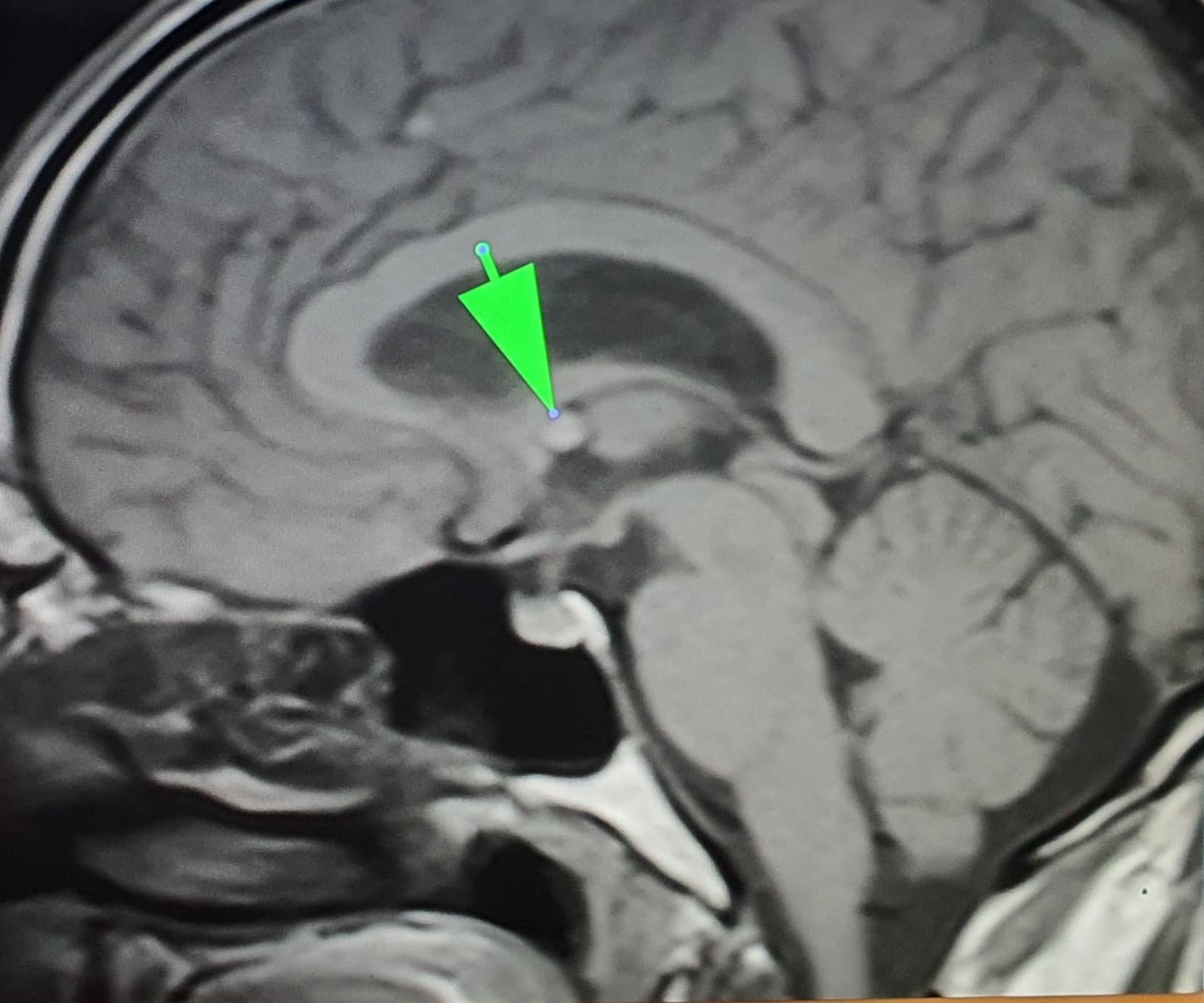
Small colloid cyst, T1 sagittal MRI
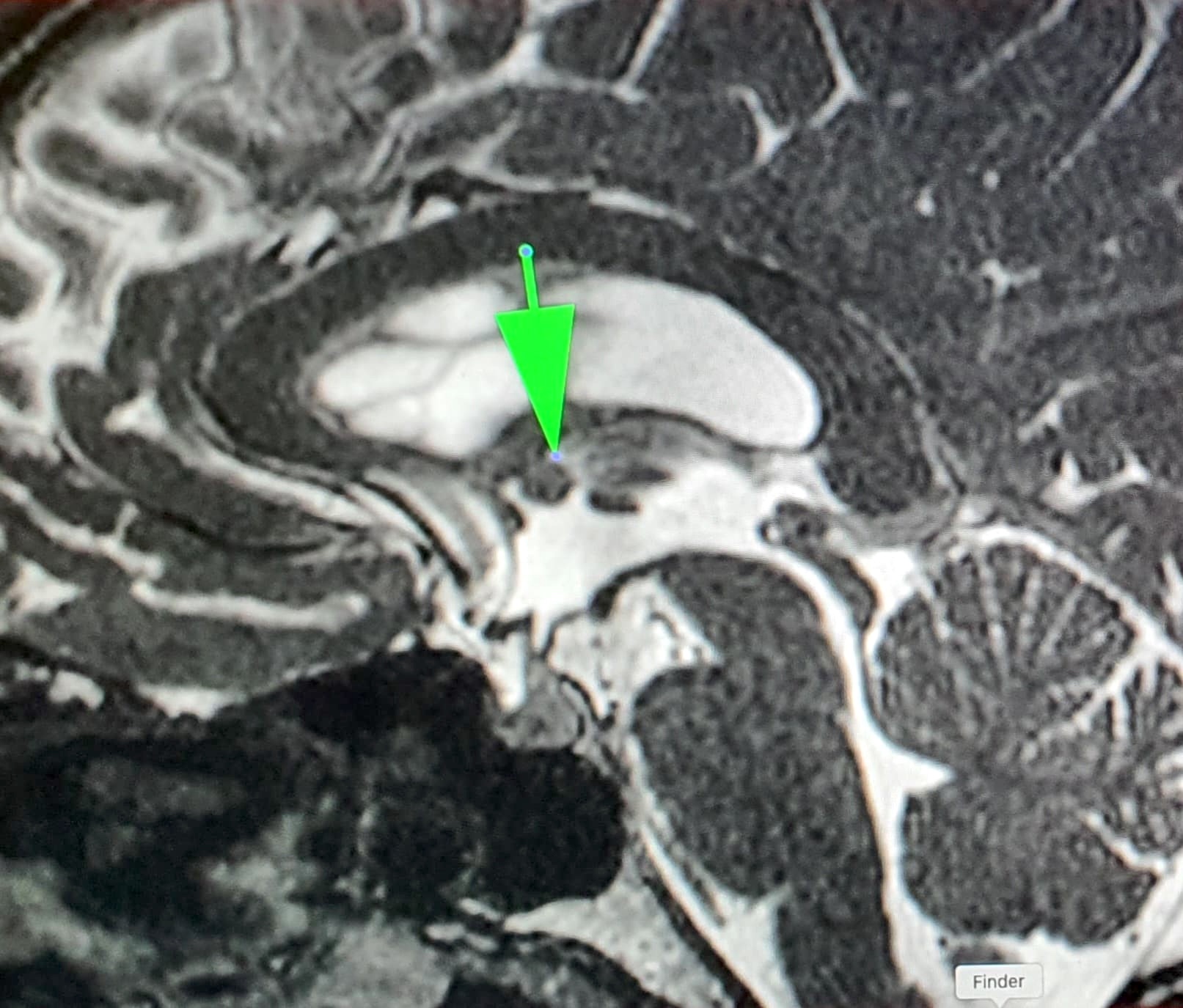
Small colloid cyst, T2 sagittal MRI

Large colloid cyst, MRI

Large colloid cyst, CT
Images hosted on other servers:
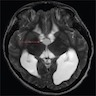
Extensive chronic hydrocephalus
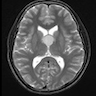
Large colloid cyst
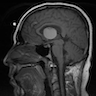
T1 hyperintense colloid cyst
- Excellent prognosis
- Rare cases associated with sudden death (World Neurosurg 2019:123:351)
- Spontaneous regression is very rare (BMC Neurol 2022;22:397)
- Malignant transformation has not been reported
- 10 year old girl with brainstem death due to a third ventricular colloid cyst (BJR Case Rep 2022;8:20220007)
- 31 year old woman with colloid cyst causing acute hydrocephalus during early pregnancy (Surg Neurol Int 2021:12:54)
- 49 year old man with spontaneous regression of colloid cyst (BMC Neurol 2022;22:397)
- 57 year old man with large colloid cyst obstructing the posterior third ventricle (J Neurosurg Case Lessons 2021;1:CASE2121)
- 77 year old woman with xanthogranulomatous colloid cyst (Surg Neurol Int 2019:10:169)
- Excision (microsurgical or endoscopic) is curative (World Neurosurg 2021:149:e298)
- Stereotactic aspiration (potential for cyst recurrence)
- Observation may be reasonable in some stable, asymptomatic cases
Images hosted on other servers:

Intraoperative view of a colloid cyst
- 1 - 2 cm; larger cysts have been reported
- Round, unilocular, translucent with thin, glistening wall
- Cyst filled with clear or turbid viscid mucin that solidifies after fixation (AJNR Am J Neuroradiol 2000;21:1470)
- Specimens received are often only a wrinkled membrane
Images hosted on other servers:
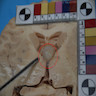
2 cm colloid cyst
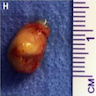
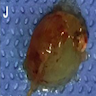
Endoscopically excised colloid cyst
- Hypocellular, fibrous wall lined by simple to pseudostratified columnar epithelium with variable cilia or goblet cells (resembles bronchial epithelium) (Acta Neuropathol 1997;93:271, Diagn Cytopathol 2002;27:27)
- Cyst lining may be modified by pressure atrophy (become low cuboidal or flattened) or degenerative changes
- Unlike Rathke cleft and endodermal (enterogenous) cysts, lining epithelium is not prone to squamous metaplasia
- Fragments of normal choroid plexus are frequently attached to cyst
- Cyst contents are amorphous and proteinaceous, may show ghosts of desquamated lining cells and eosinophilic filamentous material (degenerated nucleoprotein and phospholipid) resembling infectious organisms (Actinomyces)
- In chronic lesions, a xanthogranulomatous reaction may occur (Surg Neurol Int 2019:10:169)
Contributed by Eman Abdelzaher, M.D., Ph.D.
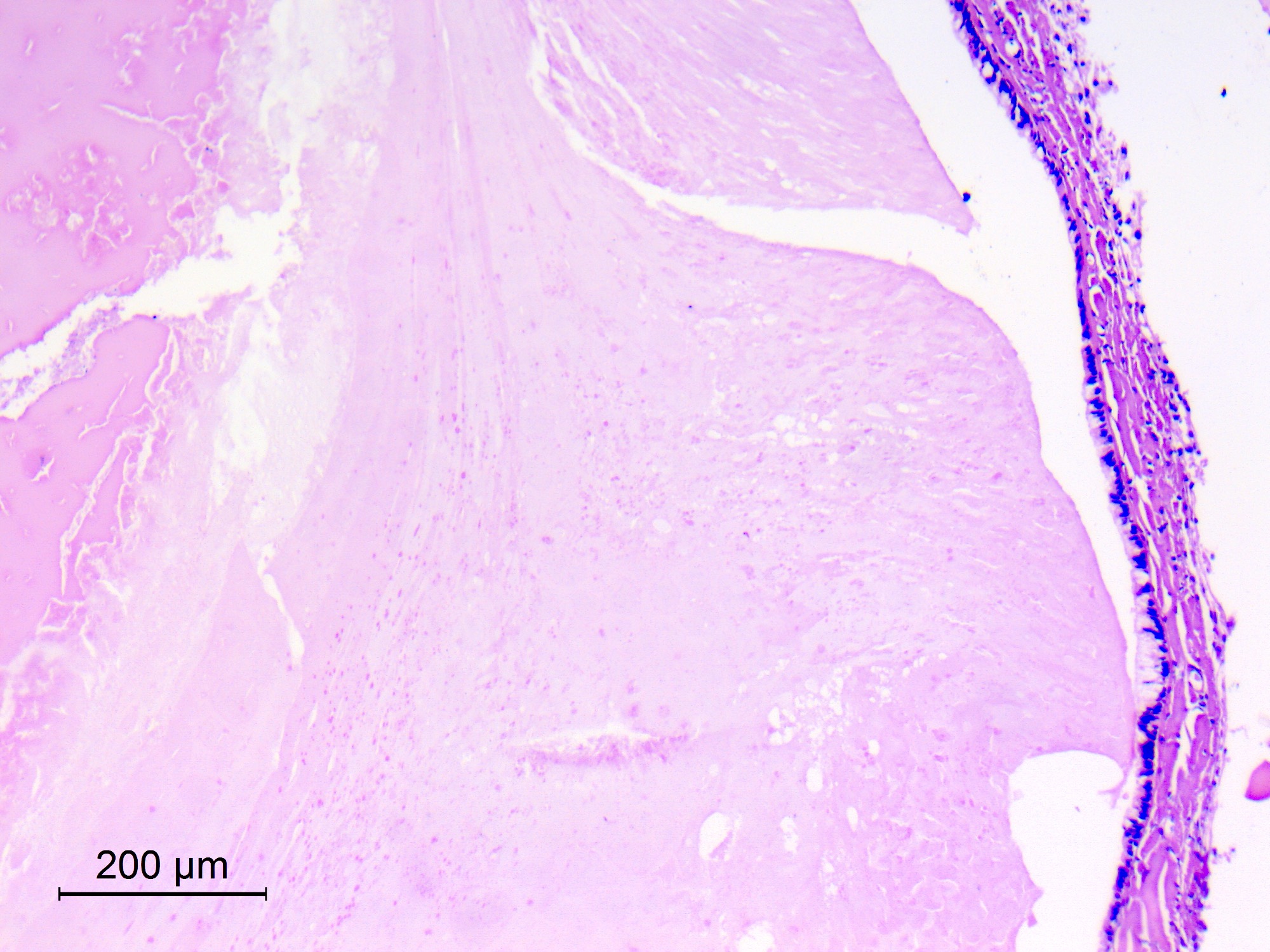
Fibrous wall and proteinaceous contents

Attached choroid plexus
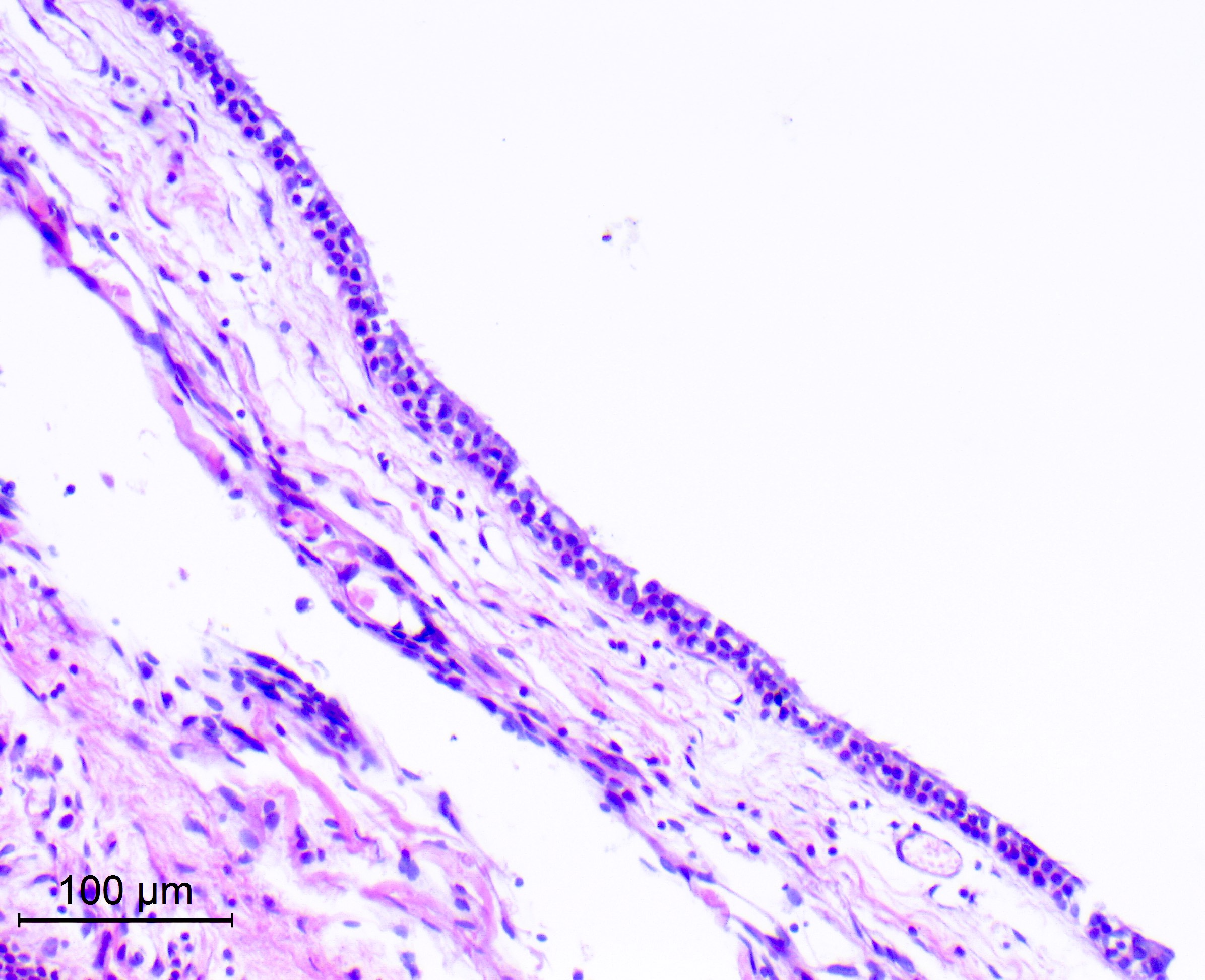
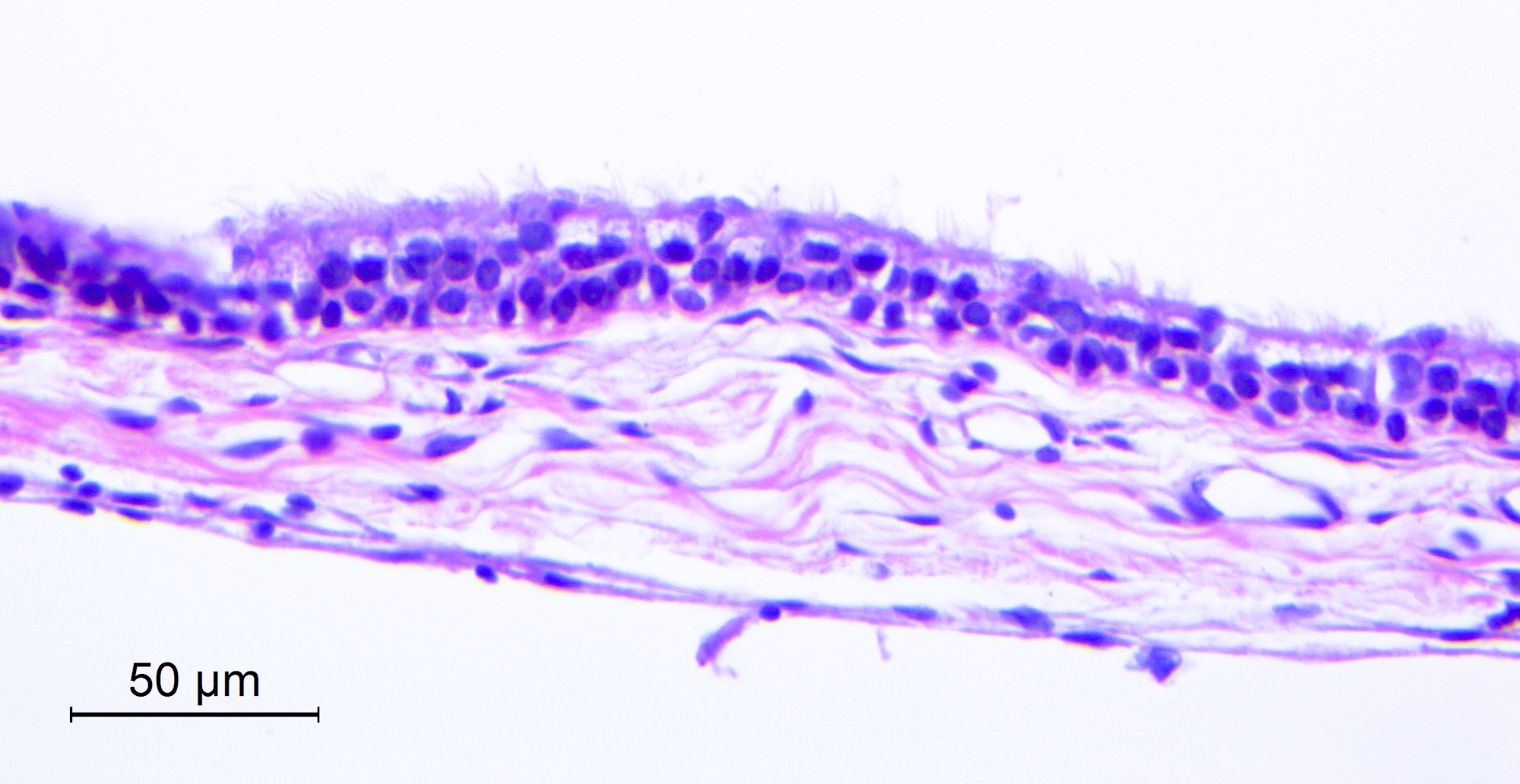
Pseudostratified ciliated cells
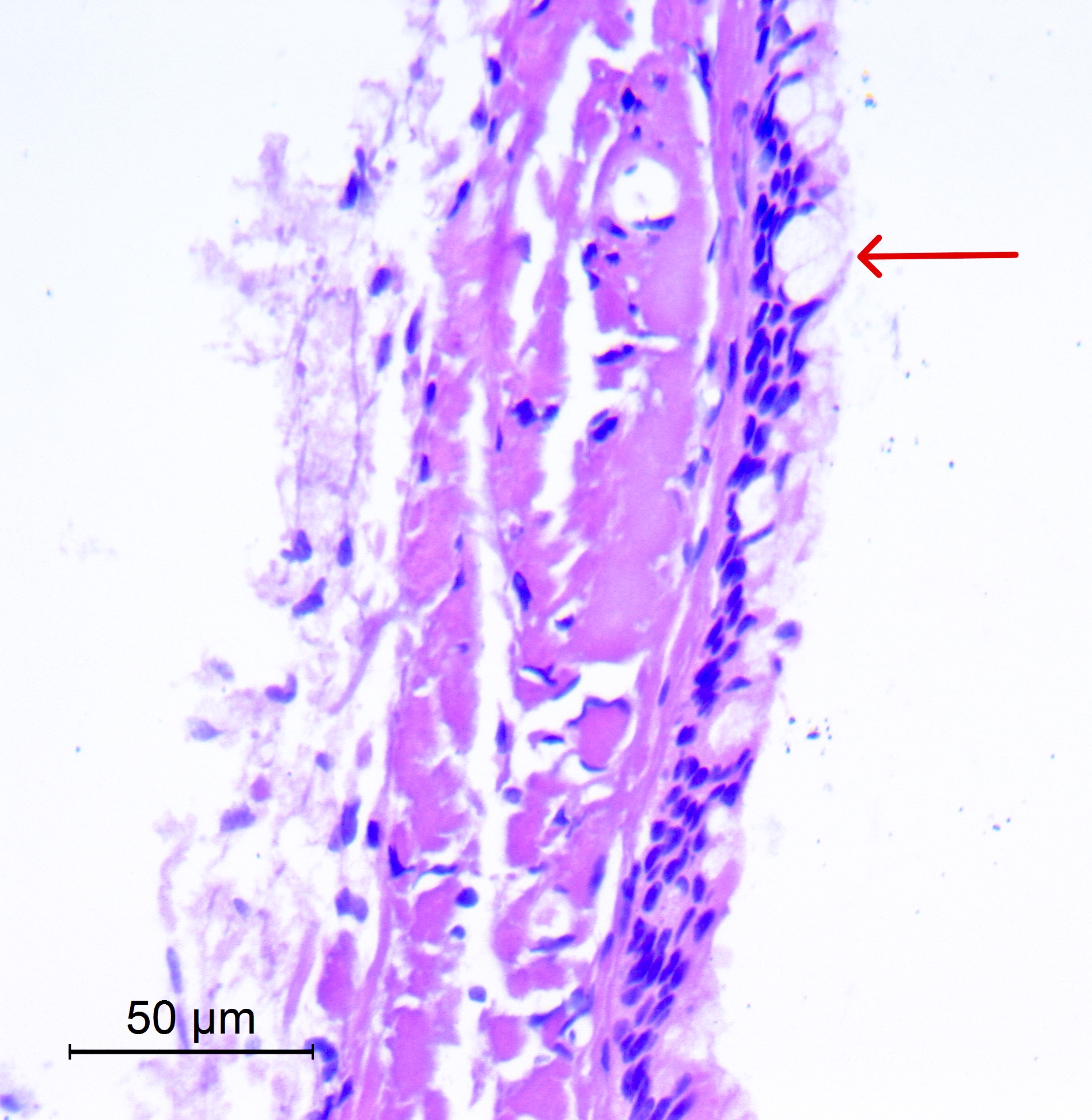
Goblet cells
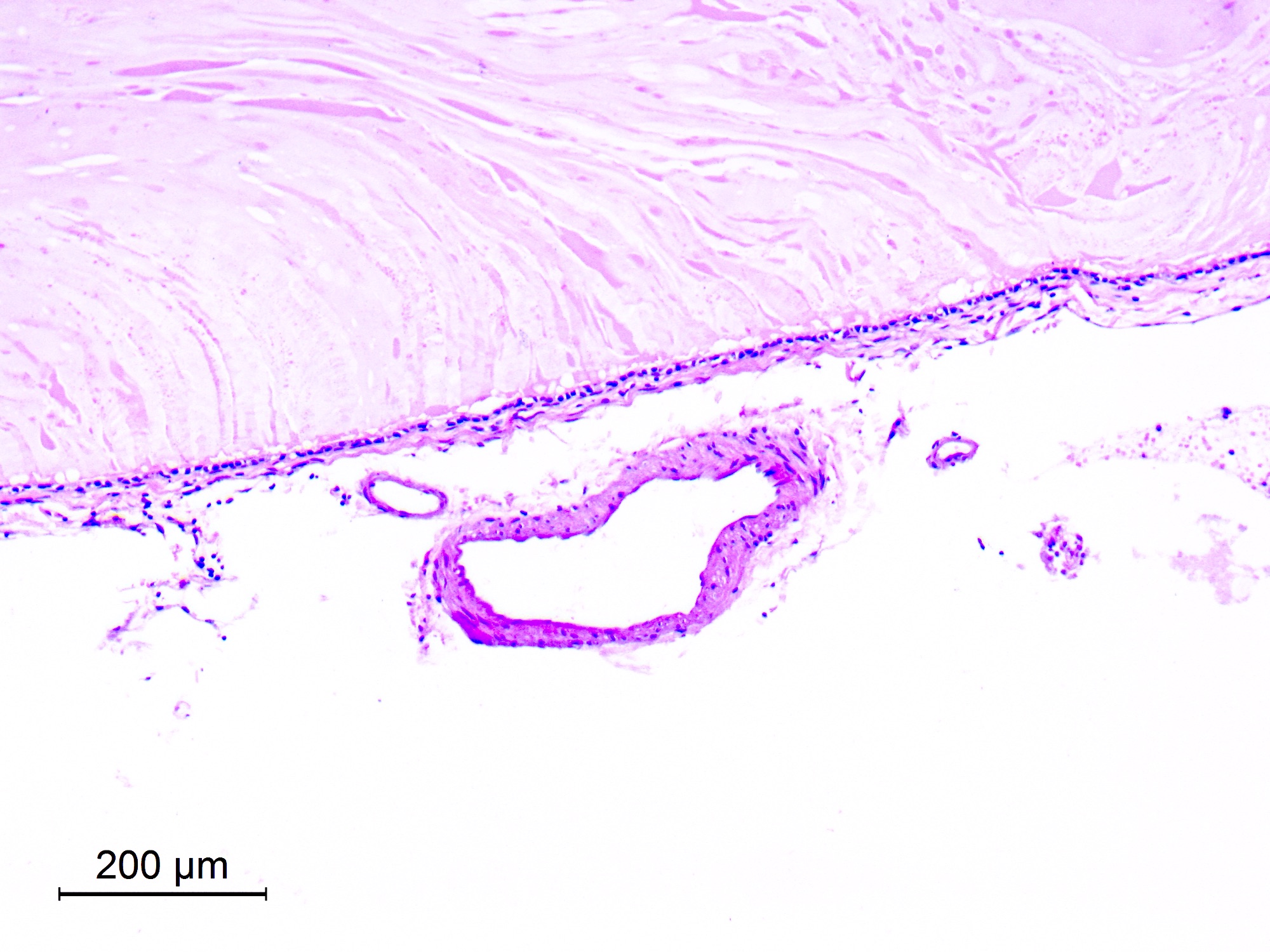
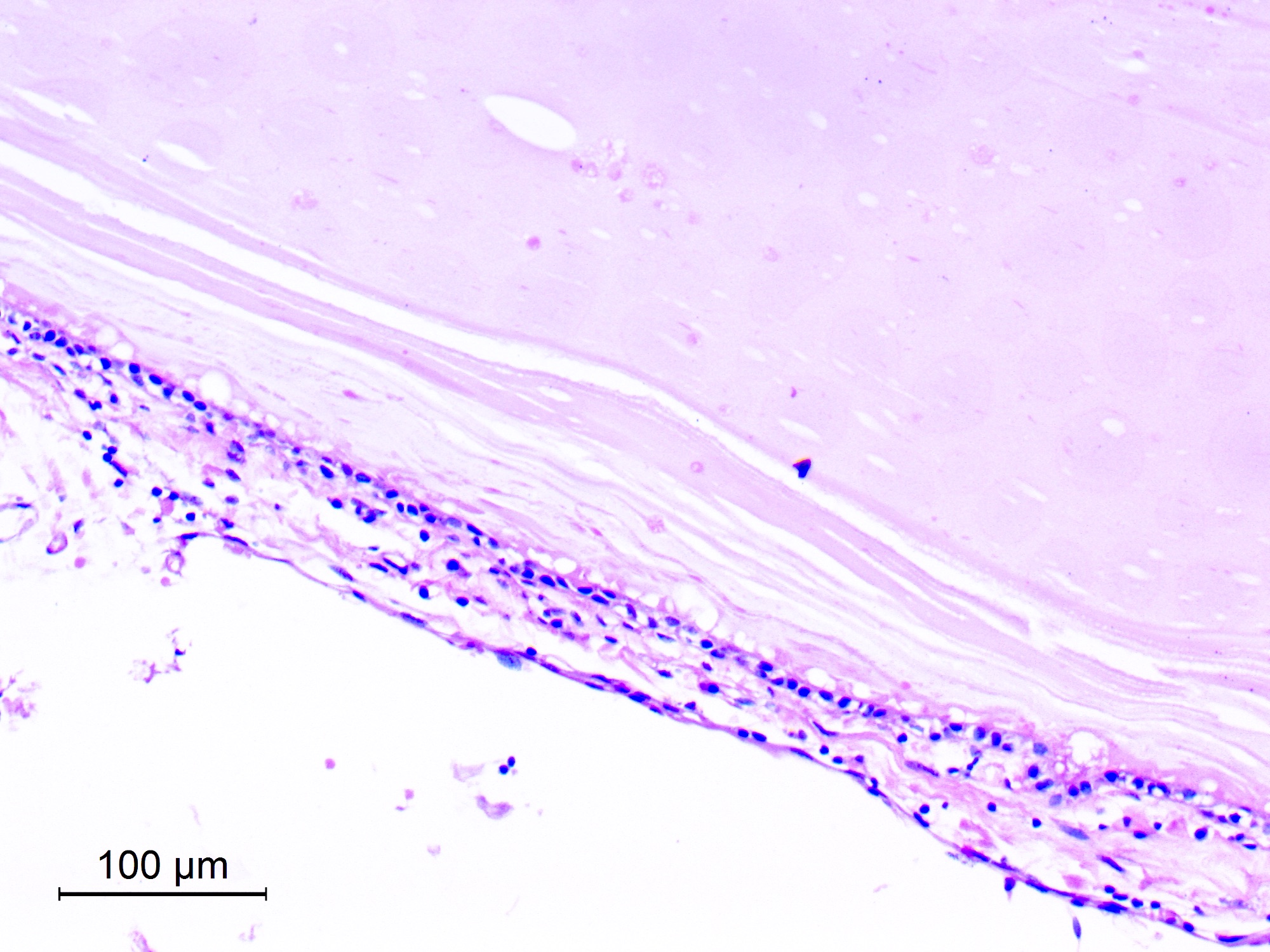
Cuboidal lining
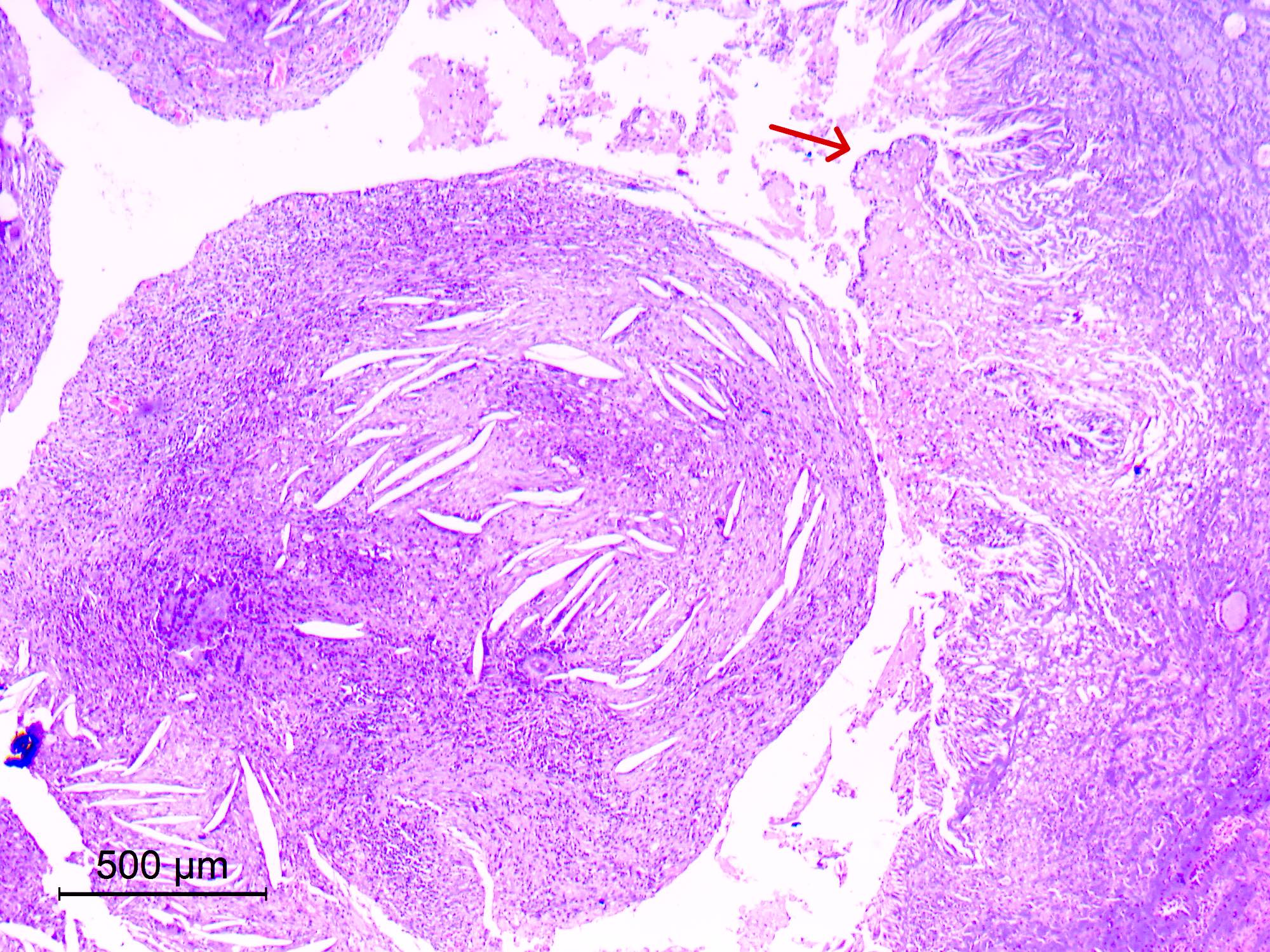
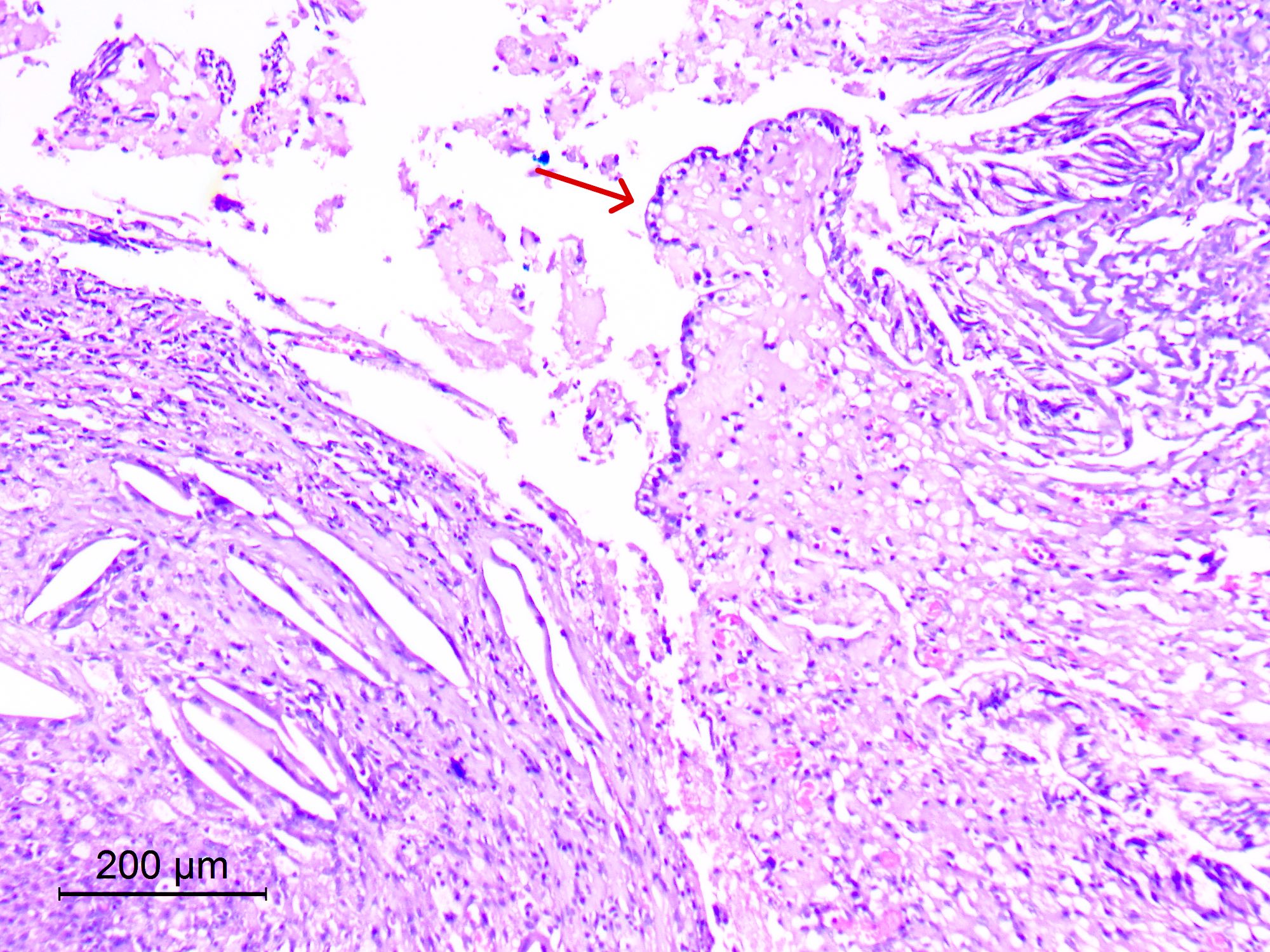
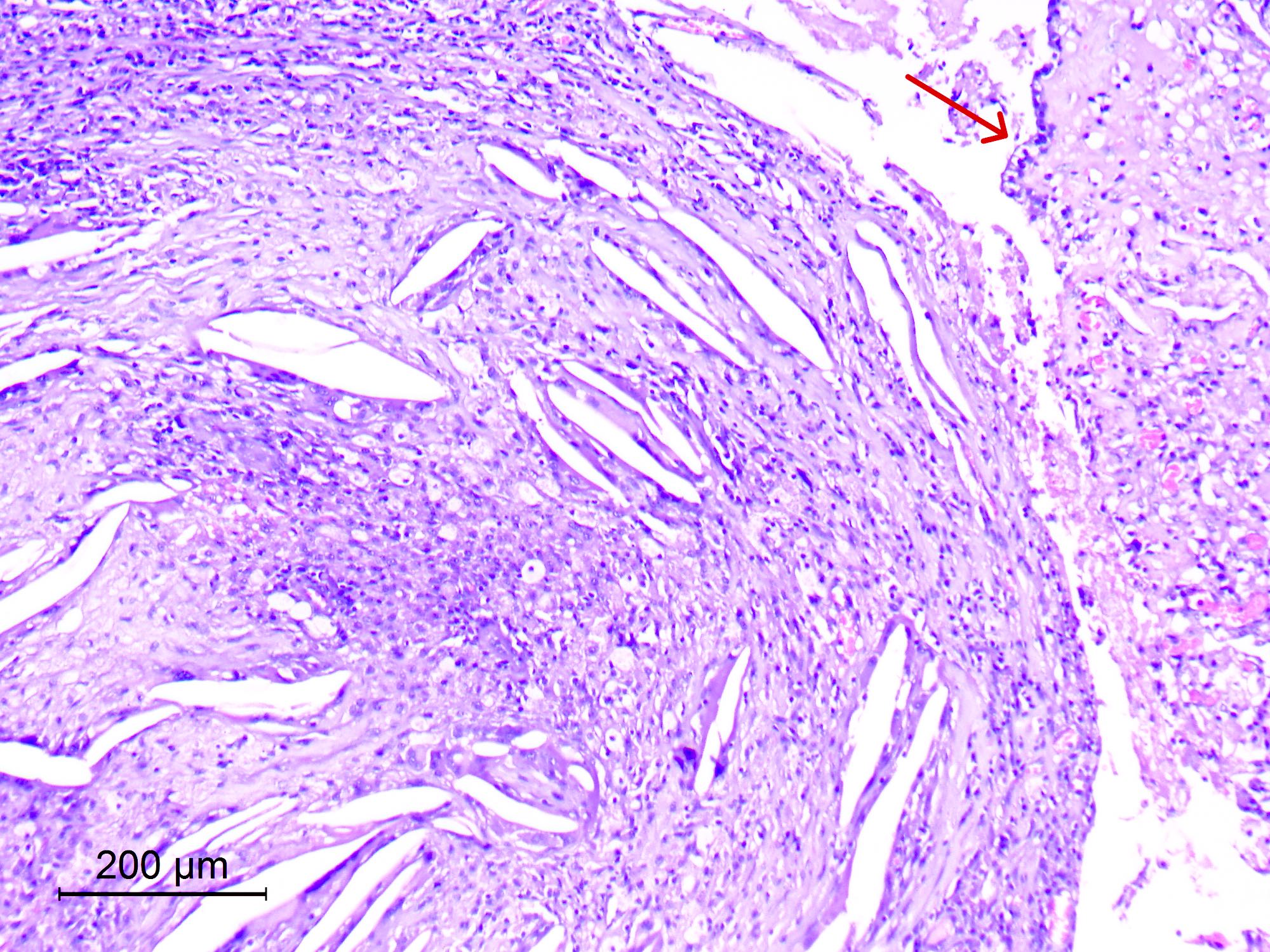
Xanthogranulomatous reaction
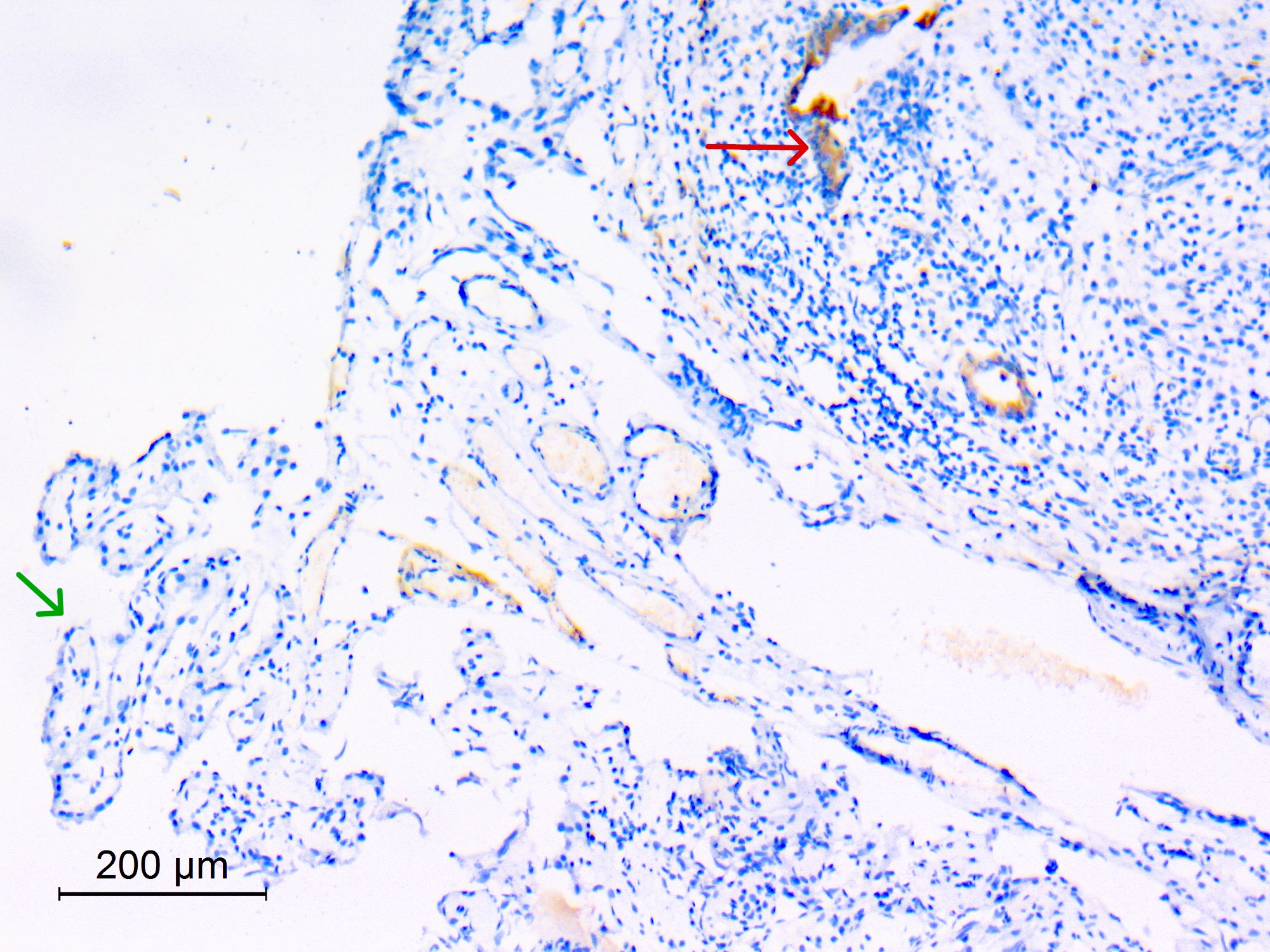

EMA

S100
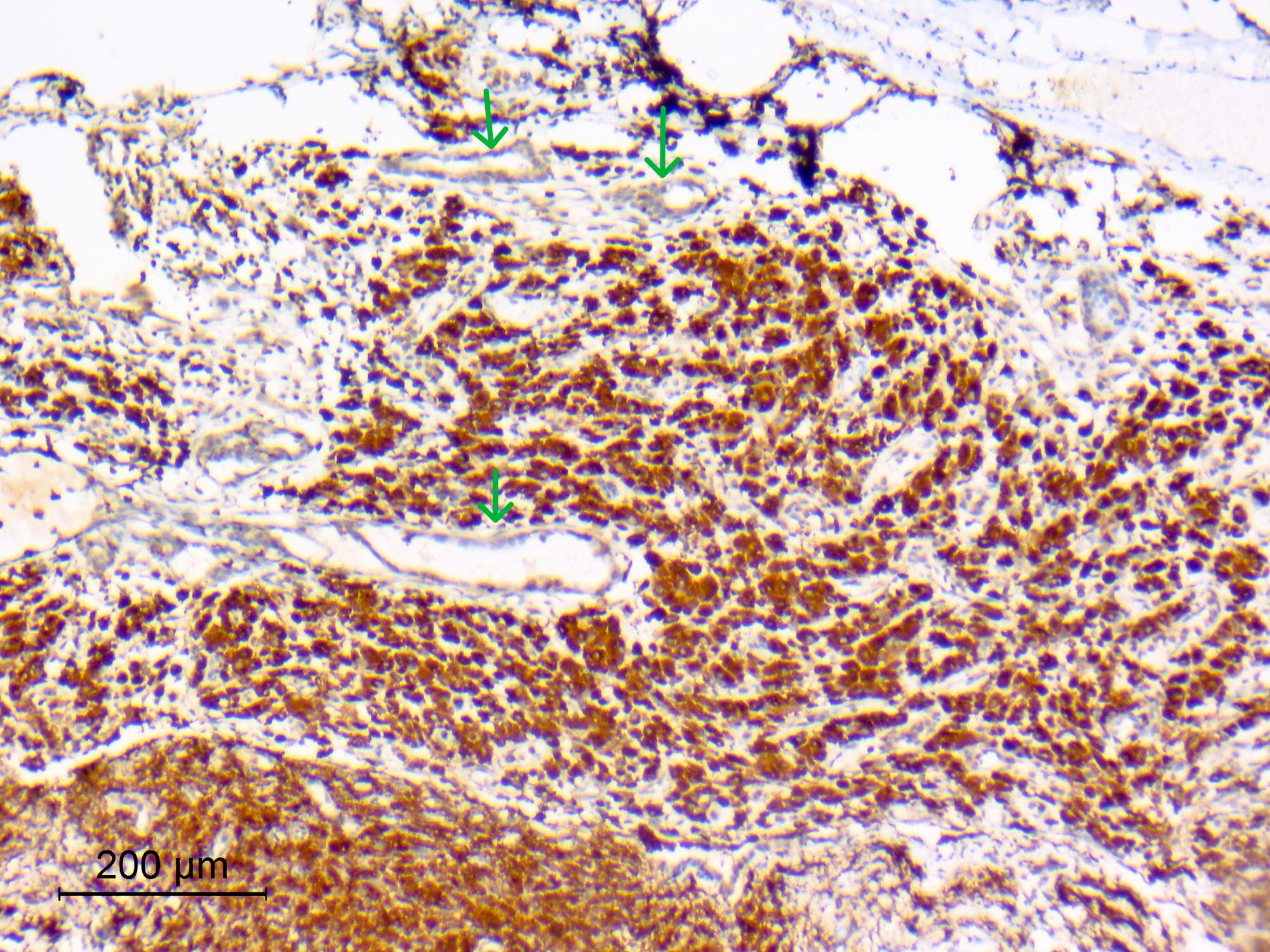
CD68
- Epithelial cells, cohesive sheets and individual ciliated cells and goblet cells (Diagn Cytopathol 2002;27:27)
- Abundant amorphous proteinaceous material with or without Actinomyces-like nucleoprotein arrays
- Presence of macrophages
Images hosted on other servers:
Thick proteinaceous material
- Epithelium: keratin (low and high molecular weight) (Hum Pathol 1992;23:811)
- Epithelium: EMA
- Mucin: PAS, mucicarmine (Diagn Pathol 2012:7:144)
- Epithelial nature of lining cells is evident by cytoplasmic tonofilaments and desmosomes
- 6 cell types: ciliated cells, nonciliated cells with surface microvilli, goblet cells, basal cells, nonspecific small cells and occasional neuroendocrine cells with neurosecretory granules (Acta Neuropathol 1992;83:605)
- Well formed basal lamina
Images hosted on other servers:
Ciliary pattern of colloid cyst
Colloid cyst
- Third ventricular cyst, endoscopic excision biopsy:
- Colloid cyst
- Rathke cleft cyst:
- Intrasellar or suprasellar location
- Prone to squamous metaplasia
- Normal choroid plexus:
- Sometimes dominant or only epithelial tissue
- Papillary with cobblestone lining epithelium
- No ciliated or goblet cells
- EMA generally negative
- Choroid plexus papilloma:
- Papillary with pseudostratified lining epithelium
- No ciliated or goblet cells
- Papillary craniopharyngioma with xanthogranulomatous change:
- Squamous lining
The brain cyst shown in the above image is strategically located near the foramen of Monro. Which of the following is the correct diagnosis?
- Arachnoid cyst
- Colloid cyst
- Endodermal (enterogenous) cyst
- Rathke cleft cyst
Comment Here
Reference: Colloid cyst
- Colloid cyst
- Endodermal (enterogenous) cyst
- Epidermoid cyst
- Rathke cleft cyst
Comment Here
Reference: Colloid cyst
- Genome wide DNA methylation based tumor classification is a valuable complementary method of CNS tumor classification that relies on computational analyses of tumor specific epigenetic modifications (Nature 2018;555:469, Acta Neuropathol 2018;136:181, Neuro Oncol 2022;24:571, Neuro Oncol 2021;23:S16)
- DNA methylation is a type of epigenetic modification in which a covalent bond in a nucleotide is enzymatically altered to produce a subtly different compound
- A common example and one of the best studied is cytosine converted to 5 methylcytosine (5mC); this is when a DNA methyl transferase enzyme replaces the hydrogen attached to the carbon atom in position 5 of the cytosine molecule with a methyl group
- In mammalian genomes, methylation typically occurs at the 5 prime cytosine of a cytosine guanine dinucleotide, which is connected by a phosphodiester bond (CpG) (Trends Genet 2022;38:676)
- DNA methylation classification
- Genome wide DNA methylation
- Whole genome DNA methylation
- DNA methylation
- Methylation classifier
- DNA methylation profiling
- Most contemporary technologies for DNA methylation profiling rely on bisulfite conversion of DNA extracted from formalin fixed paraffin embedded tissue
- Bisulfite treatment of DNA selectively deaminates cytosine and converts it to uracil but leaves 5 methylcytosine unaffected (Nucleic Acids Res 1980;8:4777, Proc Natl Acad Sci U S A 1992;89:1827)
- Resultant bisulfite treated DNA can then be amplified by polymerase chain reaction, which amplifies uracil and thymine as thymine and 5 methylcytosine as cytosine (Proc Natl Acad Sci U S A 1992;89:1827)
- Cytosine residues in the PCR products are therefore markers of 5 methylcytosine in the original input DNA (Proc Natl Acad Sci U S A 1992;89:1827)
- As an alternative to the amplification and sequencing approach, the similar hybridization behavior of uracil and thymine can be exploited to detect cytosine to uracil / thymine polymorphisms in bisulfite treated DNA using DNA hybridization technology (Genome Res 2006;16:383)
- Bisulfite sequencing
- Bisulfite treated and PCR amplified DNA can be sequenced by, for example, massively parallel short read sequencing
- All cytosines in the output sequences are interpreted as 5mC, so genome wide DNA methylation can be identified at nucleotide resolution (Proc Natl Acad Sci U S A 1992;89:1827, Nucleic Acids Res 2001;29:E65)
- Genome wide bisulfite sequencing is still prohibitively expensive for large scale implementation
- Early whole genome DNA methylation arrays
- Illumina GoldenGate DNA Methylation BeadArray; first generation genome wide DNA methylation array, introduced in 2006 (Genome Res 2006;16:383)
- Illumina Infinium HumanMethylation27K BeadChip; second generation genome wide DNA methylation array, introduced in 2009 (Epigenomics 2009;1:177)
- Illumina Infinium HumanMethylation450K BeadChip; third generation genome wide DNA methylation array, introduced in 2011 (Genomics 2011;98:288, Epigenetics 2011;6:692)
- Current whole genome DNA methylation array
- Illumina Infinium MethylationEPIC BeadChip; fourth generation genome wide DNA methylation array, introduced in 2015
- Version 1.0 includes 853,307 CpG sites, including 439,562 of the 482,421 CpGs (91%) included in the 450K microarray (Epigenomics 2016;8:389, Nucleic Acids Res 2017;45:e22)
- Adds 333,265 CpGs located in enhancer regions identified by ENCODE and FANTOM5 (Epigenomics 2016;8:389)
- Version 2.0 includes more than 935,000 CpG sites
- Over 186,000 new probes to target known enhancers, super enhancers, CTCF binding domains and open regions of chromatin associated with primary tumors identified by ATAC seq and ChIP seq experiments
- DNA input requirement: 250 ng
- Definitive classification of CNS tumor types, especially useful for diagnostically challenging cases or to confirm suspected diagnoses, as discussed above (Nature 2018;555:469, Acta Neuropathol 2018;136:181, Neuro Oncol 2022;24:571, Neuro Oncol 2021;23:S16, Clin Epigenetics 2019;11:185)
- DKFZ CNS tumor classifier: first comprehensive CNS tumor DNA methylation based classifier, published in 2018, based on random forest classifier trained on a reference cohort of 2801 samples from 91 methylation classes (Nature 2018;555:469)
- Emerging bioinformatic approaches have been introduced for clinical utility and validation (Neuro Oncol 2022;24:571, J Mol Diagn 2022;24:924)
- Tumor purity greatly influences methylation class prediction and interpretability (Acta Neuropathol 2023;145:365, Neuro Oncol 2022;24:571, PLoS One 2022;17:e0265557)
- Determining well established, clinically relevant medulloblastoma subtypes (Neuro Oncol 2019;21:214, Nature 2017;547:311)
- Determining proposed surrogate grading subtypes of meningioma (Lancet Oncol 2017;18:682, Nat Genet 2022;54:649, J Clin Oncol 2021;39:3839, Nature 2021;597:119)
- Discovery of new CNS tumor types
- Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC) (Neuropathol Appl Neurobiol 2020;46:422)
- High grade astrocytoma with piloid features (Acta Neuropathol 2018;136:273)
- Infant type hemispheric glioma (Cancer Discov 2020;10:942)
- Cribriform neuroepithelial tumor (Brain Pathol 2017;27:411)
- High grade glioma with pleomorphic and pseudopapillary features (HPAP) (Acta Neuropathol 2022;143:403)
- Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA) (Acta Neuropathol 2023;145:667)
- PATZ1 fusion positive neuroepithelial tumor (Acta Neuropathol 2021;142:841)
- PLAGL1 fusion positive neuroepithelial tumor (Acta Neuropathol 2021;142:827)
- Oligosarcoma, IDH mutant (Acta Neuropathol 2022;143:263)
- Resolving historical tumor types into molecularly coherent tumor classes
- Historical diffuse astrocytoma, IDH wild type; reclassified as glioblastoma, diffuse midline glioma and others (J Neuropathol Exp Neurol 2018;77:422, Acta Neuropathol 2015;130:407)
- Historical primitive neuroectodermal tumors of the central nervous system (PNET CNS); reclassified as several well defined entities (e.g., embryonal tumor with multilayered rosettes) and 4 newer tumor entities: CNS neuroblastoma, FOXR2 activated; CIC rearranged sarcoma; astroblastoma, MN1 altered; CNS tumor with BCOR internal tandem duplication (Cell 2016;164:1060)
- Historical embryonal tumor with abundant neuropil and true rosettes; reclassified as embryonal tumor with multilayered rosettes (ETMR) (Acta Neuropathol 2014;128:279)
- Historical ependymoblastoma; reclassified as ETMR (Acta Neuropathol 2014;128:279)
- Historical medulloepithelioma; reclassified as ETMR (Acta Neuropathol 2014;128:279)
- Defining prognostic tumor subtypes
- Diffuse pediatric type, high grade glioma (Acta Neuropathol 2017;134:507)
- Pineal parenchymal tumors (Acta Neuropathol 2020;139:243)
- Choroid plexus tumors (Neuro Oncol 2016;18:790)
- Intracranial mesenchymal tumor, FET::CREB fusion positive (Brain Pathol 2022;32:e13037)
- Glioblastoma (Acta Neuropathol 2022;144:155, Acta Neuropathol 2021;142:179, Brain Pathol 2018;28:656)
- Myxopapillary ependymoma (Neuro Oncol 2022;24:1689)
- Chordoma (Neuro Oncol 2022;24:442)
- Posterior fossa ependymoma (Acta Neuropathol 2022;144:373)
- IDH mutant astrocytoma (Acta Neuropathol 2021;141:85, Acta Neuropathol 2020;140:569)
- Diffuse leptomeningeal glioneuronal tumor (Acta Neuropathol 2022;144:1185)
- High grade astrocytoma with piloid features (Acta Neuropathol 2023;145:71)
- In addition to CNS tumor classification, the DNA methylation array allows for determination of alterations routinely assayed in surgical neuropathology on a single platform, including 1p / 19q codeletion status, somatic copy number alterations (chromosomal and gene level), MGMT promoter methylation status and chromosomal breakpoints inferring gene fusions (Neuro Oncol 2014;16:1630, Neuropathol Appl Neurobiol 2021;47:406)
- Some of these features may not be clinically validated in certain settings but their presence may suggest further orthogonal testing
- Like other studies, methylation profiling should be considered in conjunction with other histopathologic, genetic and clinical data in order to reach a diagnosis
- Misleading methylation profiles can occur (i.e., methylation matches that apparently conflict with histologic, genetic, radiographic or clinical features)
- Requires a relatively large amount of optimal input DNA (~250 ng)
- Some methylation classes do not necessarily correspond to a CNS WHO tumor type
- High confidence methylation classifier score may not be seen in up to 1/3 of cases (Neuro Oncol 2022;24:571, Clin Epigenetics 2019;11:185)
- Factors that may contribute to low classifier score (no methylation match)
- Low tumor cell content (low purity of sample)
- Failed DNA bisulfite conversion
- Poor quality DNA (e.g., old tissue blocks)
- Single bioinformatic pipelines are reliant on a single reference dataset
- Rare tumor types may not be present in reference dataset, so no class exists for them
Contributed by P.J. Cimino, M.D., Ph.D. and Chris Dampier, M.D.
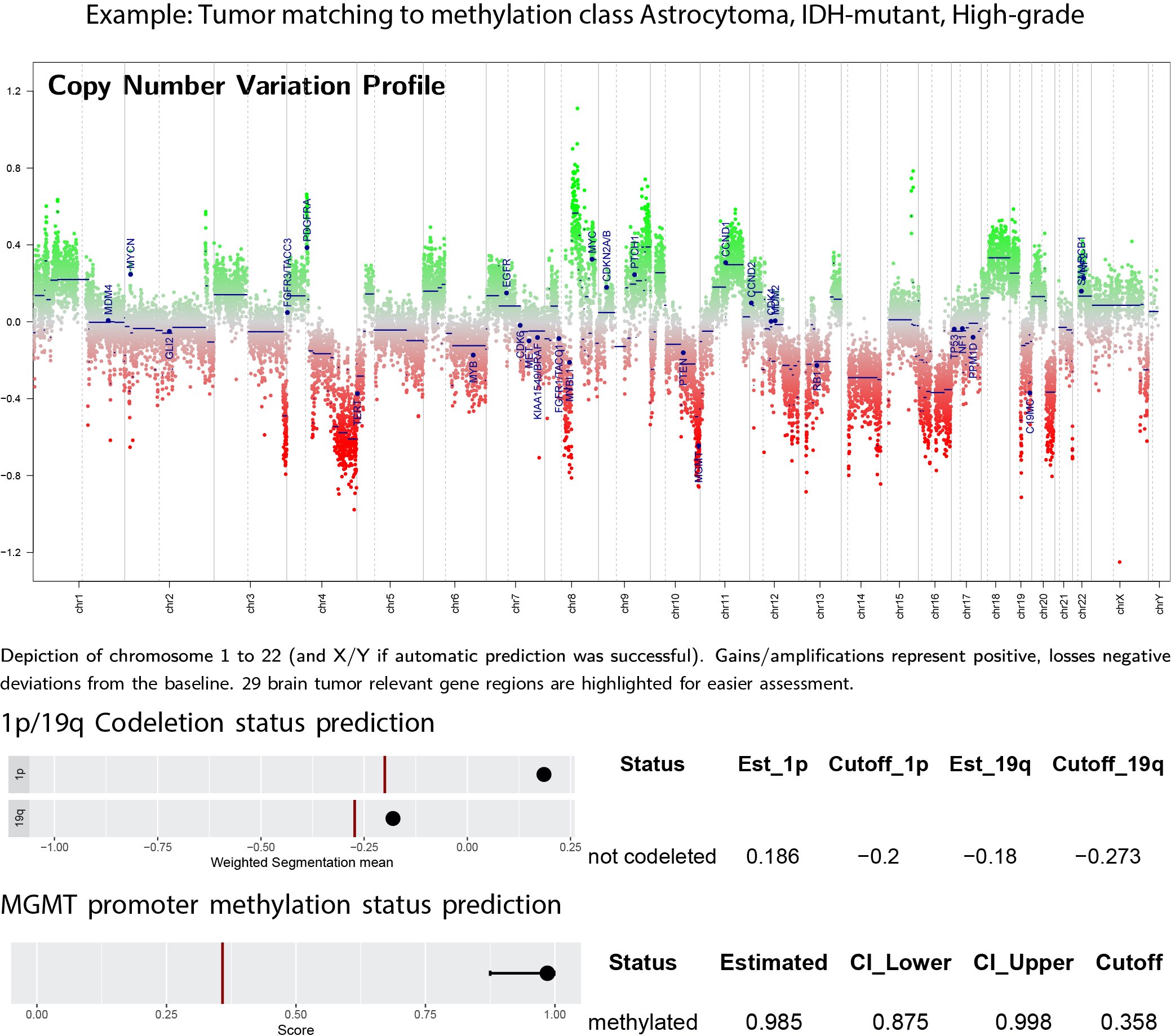
Methylation copy number plot
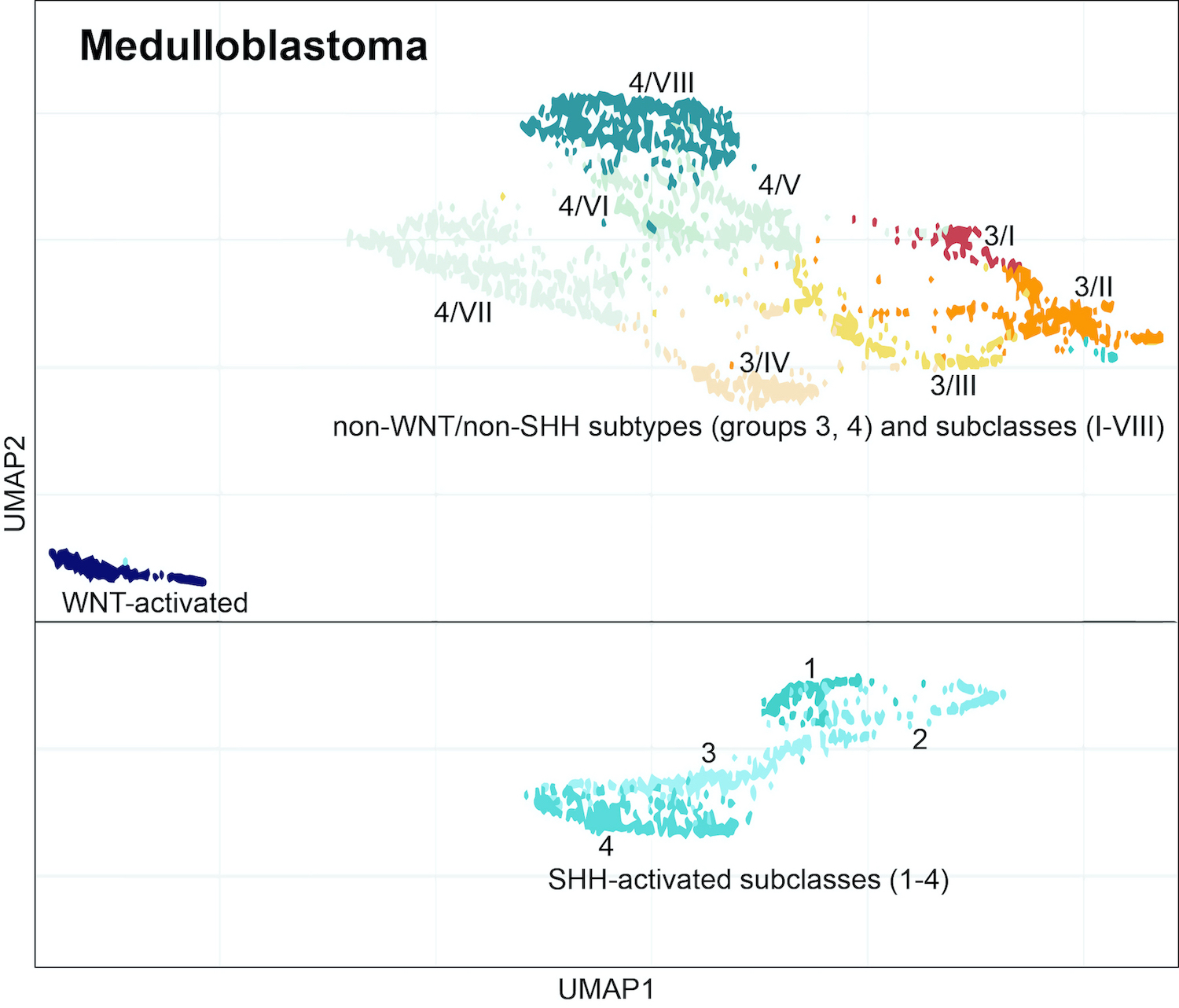
Dimensionality reduction of medulloblastoma
Images hosted on other servers:

Methylation based CNS tumor clustering

Impact of tumor purity

Workflow in surgical neuropathology

Heterogeneity of histologic diagnoses
- Brain, cervical spinal cord, biopsy:
- Histological classification: glial neoplasm with focal ependymal features
- CNS WHO grade 2
- Consensus methylation profiling class: spinal ependymoma
- Integrated diagnosis: spinal ependymoma, CNS WHO grade 2
A 7 year old girl undergoes stereotactic needle biopsy for diagnosis of an intracranial intrinsic right parietal lobe mass. There is scant tissue in the biopsy and nucleic acid extraction is expected to yield material for only a single genome wide assay (e.g., cytogenomic array, next generation sequencing, DNA methylation array). Which of these features are specifically detected by DNA methylation array profiling?
- Chromosomal copy number alterations
- Genomic breakpoints to suggest gene fusions
- Methylation class
- MGMT promoter methylation status
- Single gene mutations (e.g., specific H3 G34 mutation)
Comment Here
Reference: DNA methylation classification of CNS tumors
- Medulloblastoma histologic subtype
- Medulloblastoma molecular subtype
- Mitotic count
- Patient age
- TP53 mutational status in SHH activated medulloblastoma
Comment Here
Reference: DNA methylation classification of CNS tumors
- Developmental lesions arising from embryologic displacement of ectoderm into meninges, ventricles or rarely parenchyma (eMedicine: Imaging in CNS Dermoid Tumors)
- Benign cyst lined by keratinizing squamous epithelium and cutaneous adnexa
- Uncommon; much less common than epidermoid cyst
- Usually affects children
- Distribution more restricted than epidermoid cysts, usually affects midline (mostly fontanelle or posterior fossa as midline cerebellar lesions or within the 4th ventricle), may affect spinal canal
- May have sinus tract to skin surface in nasofrontal or occipital regions
- May rupture and cause chemical meningitis
- Thick walled with greasy pilosebaceous contents
- Fibrous wall lined by keratinizing squamous epithelium with skin adnexa, cyst contains squames, hair, sebum
- Hair shafts highlighted with polarized light
- May be hair matrix differentiation (shadow cells)
- Rupture may cause granulomatous inflammation with foreign body giant cell reaction
- Supratentorial superficially located cystic neuroepithelial tumors of infancy characterized by prominent desmoplasia with neoplastic glial component (desmoplastic infantile astrocytoma, DIA) or neoplastic glioneuronal component (desmoplastic infantile ganglioglioma, DIG)
- Corresponds to WHO grade 1
- DIA first described in 1982 by Taratuto et al. (J Neurosurg 1987;66:58)
- DIG first described in 1987 by VandenBerg et al.
- Since both lesions have similar radiological and clinical presentation, they are categorized together as desmoplastic infantile astrocytoma / ganglioglioma (DIA / DIG) in the WHO classification
- Incidence: rare ( < 0.1% of CNS tumors)
- Age and sex:
- Most < 1 year
- Rare in children and young adults (Neuropathology 2005;25:150)
- Male to female ratio of 1.5:1
- Supratentorial
- Usually frontoparietal
- Rapidly increasing head circumference, hydrocephalus, seizures
- Supratentorial
- Superficially located
- Large, involves more than one lobe
- Cystic with solid area / mural nodule
- Enhancing
- Desmoplastic area exhibits hypointense signal on T2
Images hosted on other servers:
Large right hemispheric mass
- Good prognosis
- Rarely, craniospinal seeding or metastases (Am J Surg Pathol 2002;26:1515, Mod Pathol 1997;10:945, Australas Radiol 2005;49:433, Pediatr Blood Cancer 2005;45:986)
- Tumor progression has been reported in incompletely resected tumors or tumors with anaplastic features
- 3 1/2 month old boy with mixed conventional and desmoplastic infantile ganglioglioma (Mod Pathol 2001;14:720)
- Gross total resection
- Chemotherapy if infiltrative or progressive
- Residual disease may not grow and may spontaneously regress (Neurosurgery 2003;53:979)
- Large (up to 13 cm), often involves multiple lobes
- Deep macrocystic portion
- Superficial solid leptomeningeal portion, firm to hard, focally attached to overlying dura
- Well delineated from normal brain
- Desmoplastic leptomeningeal component
- Involve the subarachnoid space and extends into Virchow-Robin spaces
- Neoplastic neuroepithelial cells in desmoplastic spindled stroma arranged in fascicular and storiform patterns with pericellular reticulin deposition lending a mesenchymal appearance
- Neoplastic neuroepithelial cells:
- Astrocytic cells:
- Only component in DIA
- Spindled or gemistocytic neoplastic astrocytes
- Neuronal component:
- Seen in DIG in addition to neoplastic astrocytes
- Small ganglion cells
- Uncommonly large ganglion cells or areas resembling ganglioglioma
- Astrocytic cells:
- Immature small cell component (unclear prognostic significance)
- Hypercellular poorly differentiated neuroepithelial cells
- No desmoplasia
- May show mitoses, vascular proliferation or necrosis
- Calcification common, chronic inflammatory cells uncommon
- Exceptionally, frank anaplastic features are encountered (high mitotic rate, vascular proliferation, palisading necrosis and high proliferation index)
- Low cellularity
- Dispersed or variably sized clusters of large neuronal cells with abundant granular cytoplasm, eccentric hyperchromatic nuclei with undulating nuclear membranes and occasional binucleation, prominent nucleoli
- Astroglial cells with smaller cytoplasmic rim, nuclear hyperplasia and more prominent irregularities in nuclear membranes
- May have prominent degenerative changes, foamy macrophages
- No vascular structures (Cytojournal 2005;2:1)
- Desmoplastic component: vimentin+, GFAP+, type IV collagen+, reticulin (pericellular pattern), trichrome (stroma)
- Astrocytic cells: GFAP+, S100+
- Neuronal cells: synaptophysin+, NeuN+, neuron specific enolase+
- Poorly differentiated small cells: vimentin+, GFAP+, synaptophysin+
- Proliferation index: < 2% (higher in the poorly differentiated small cell component)
- Astrocytic tumor cells are partly invested by pericellular basal lamina
- Pineal region neoplasm with ovoid or spindle shaped cells, containing inactivating alterations of the SMARCB1 / INI1 locus and variable amounts of desmoplasia and myxoid matrix
- Predominates in adults and adolescents
- Arises in the pineal region
- Small to medium sized cells with ovoid to spindle shaped nuclei
- Variable amounts of desmoplasia and myxoid matrix
- Inactivating SMARCB1 / INI1 alteration
- Desmoplastic myxoid tumor of the pineal region, SMARCB1 mutant
- ICD-11: 2A00.20 - tumors of the pineal gland or pineal region
- Incidence data not yet available
- M:F = 2:3 (Neuro Oncol 2022;24:847)
- Median patient age: 36.6 years old (Neuro Oncol 2022;24:847)
- Patient age range: 15 - 61 years old (Neuro Oncol 2022;24:847)
- Pineal region
- Precise pathophysiology is unclear
- Precise etiology is unclear
- Sequelae of obstructive hydrocephalus, such as blurred vision, headache, nausea, vomiting, dizziness
- Identification of SMARCB1 / INI1 inactivating alteration and protein loss
- Serum and cerebrospinal fluid alpha fetoprotein and beta human chorionic gonadotropin are normal
- MRI: T1 hypointense to slightly hyperintense, T2 / fluid attenuated inversion recovery (FLAIR) hyperintense, contrast enhancing (Neuropathology 2021;41:37, Virchows Arch 2021;479:835)
Contributed by Yue-E Wang, M.S., Ph.D., Jingjing Chen, Ph.D., Wei Wang, Ph.D., An-Li Zhang, M.D., Wenchao Zhou, Ph.D. and Hai-Bo Wu, M.D., Ph.D.

MRI - sagittal T1
MRI - sagittal T2
- Prognostic factors are not yet defined but reported prognosis much better than atypical teratoid / rhabdoid tumor
- 70% of reported patients are alive without recurrence, with a median follow up of 29.5 months (0 months - 7 years) (Neuro Oncol 2022;24:847)
- 24 year old woman with a myxoid tumor of the pineal region (Free Neuropathol 2021;2:14)
- 29 year old woman with hemorrhagic pineal region mass (Virchows Arch 2021;479:835)
- 33 year old man with pineal region mass (Neuropathology 2021;41:37)
- Surgical resection is the treatment of choice
- Chemotherapy or radiotherapy has been given to a subset of patients (Neuro Oncol 2022;24:847)
- Epithelioid to spindle shaped cells embedded in a desmoplastic stroma and loose myxoid matrix
- Rhabdoid cells may be rare
- No high grade / malignant features such as brisk mitotic activity and tumor necrosis
- Reference: Acta Neuropathol 2020;139:277
Contributed by Mariana Voudouri, M.D. and George Zanazzi, M.D., Ph.D.
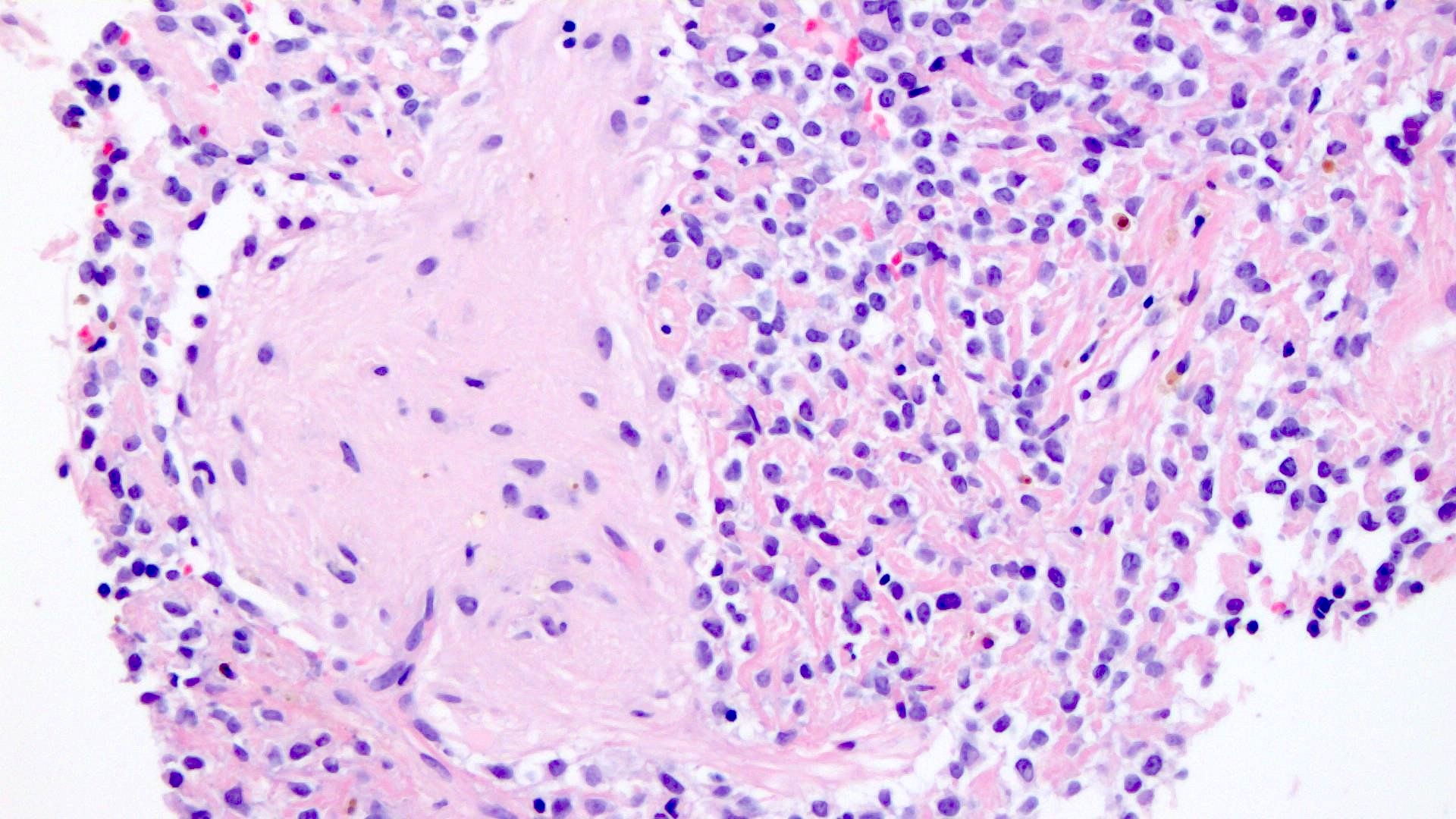
Epithelioid neoplasm with desmoplasia
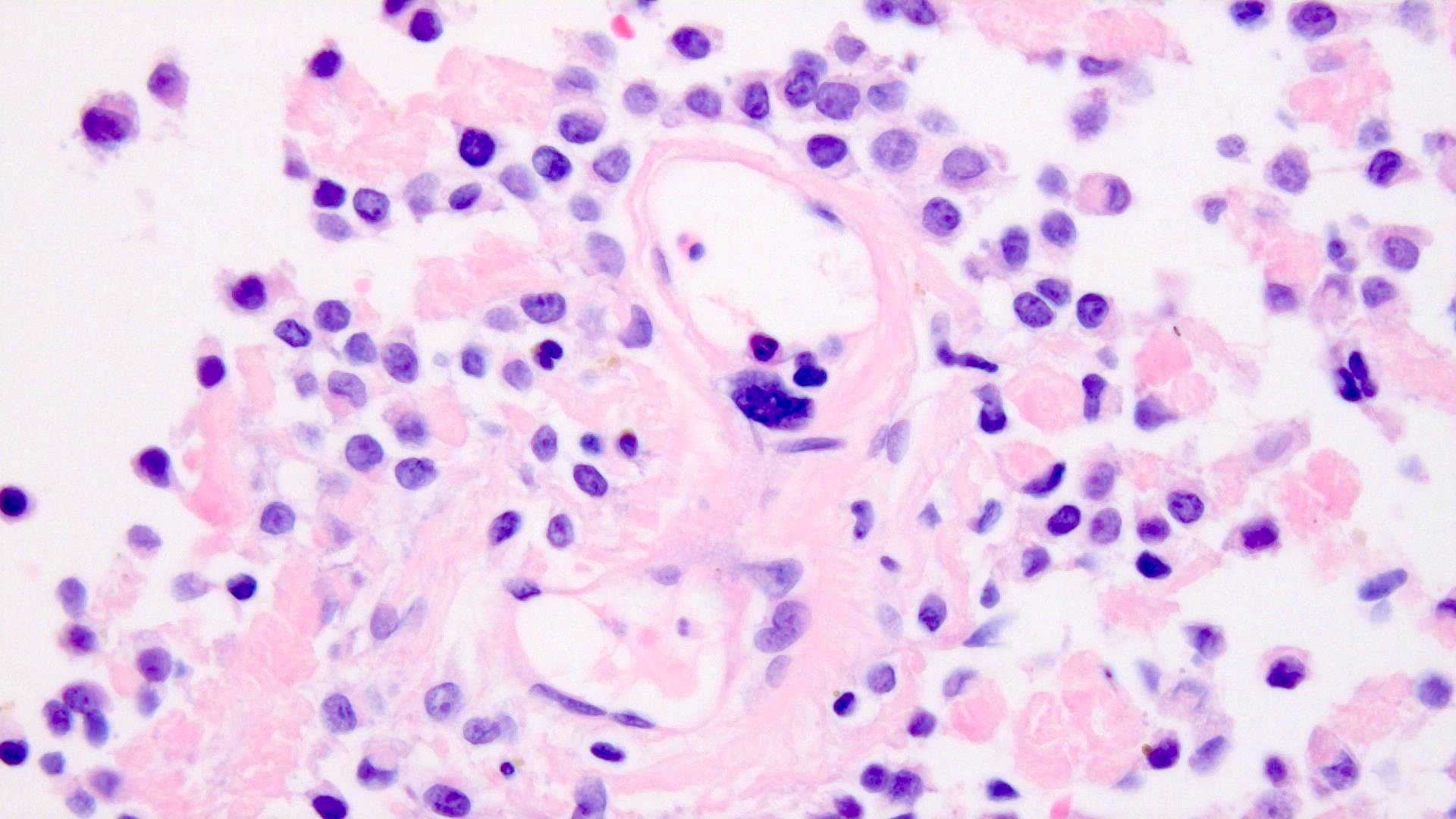
Cellular atypia
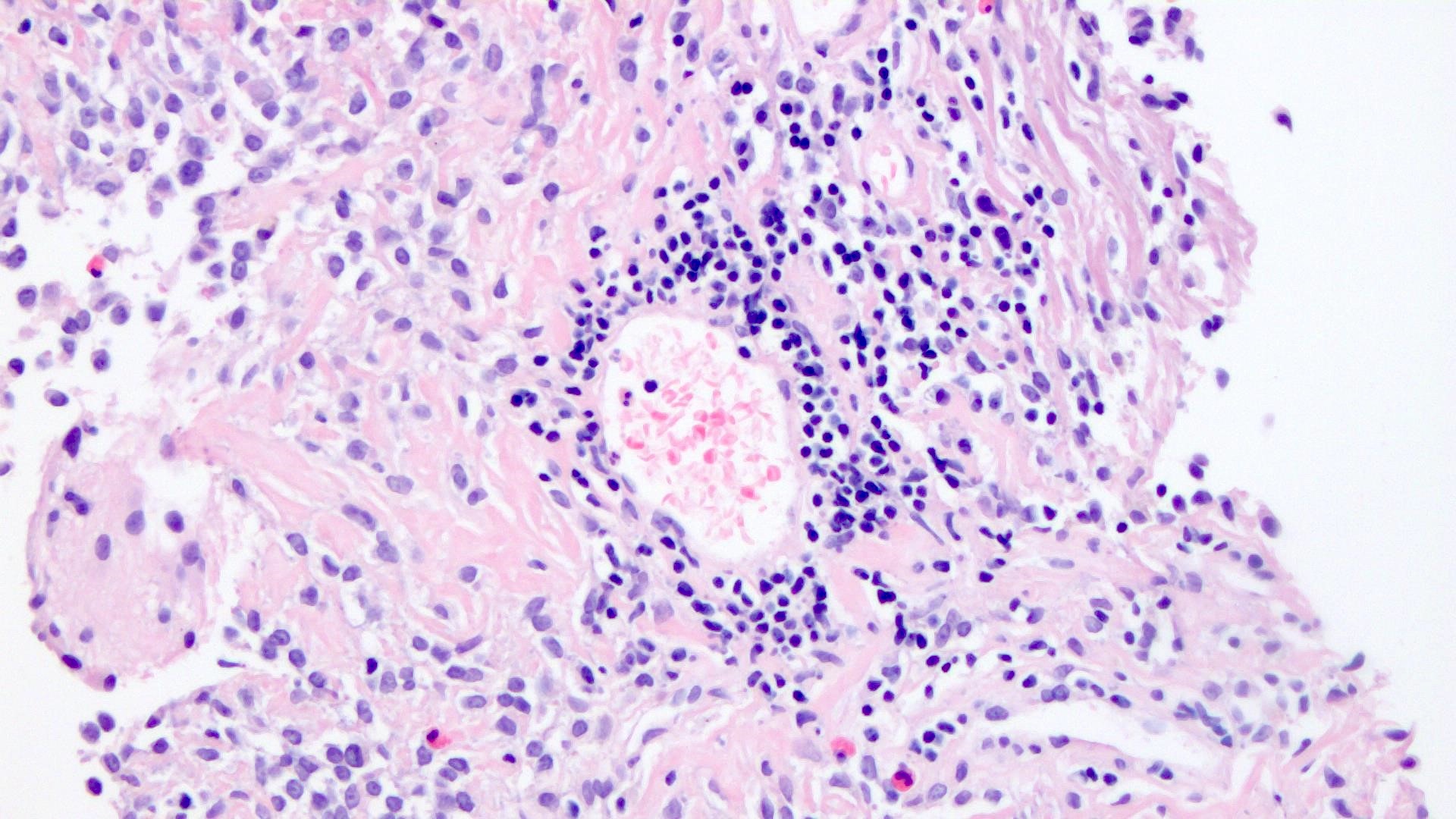
Perivascular tumor cell accumulation
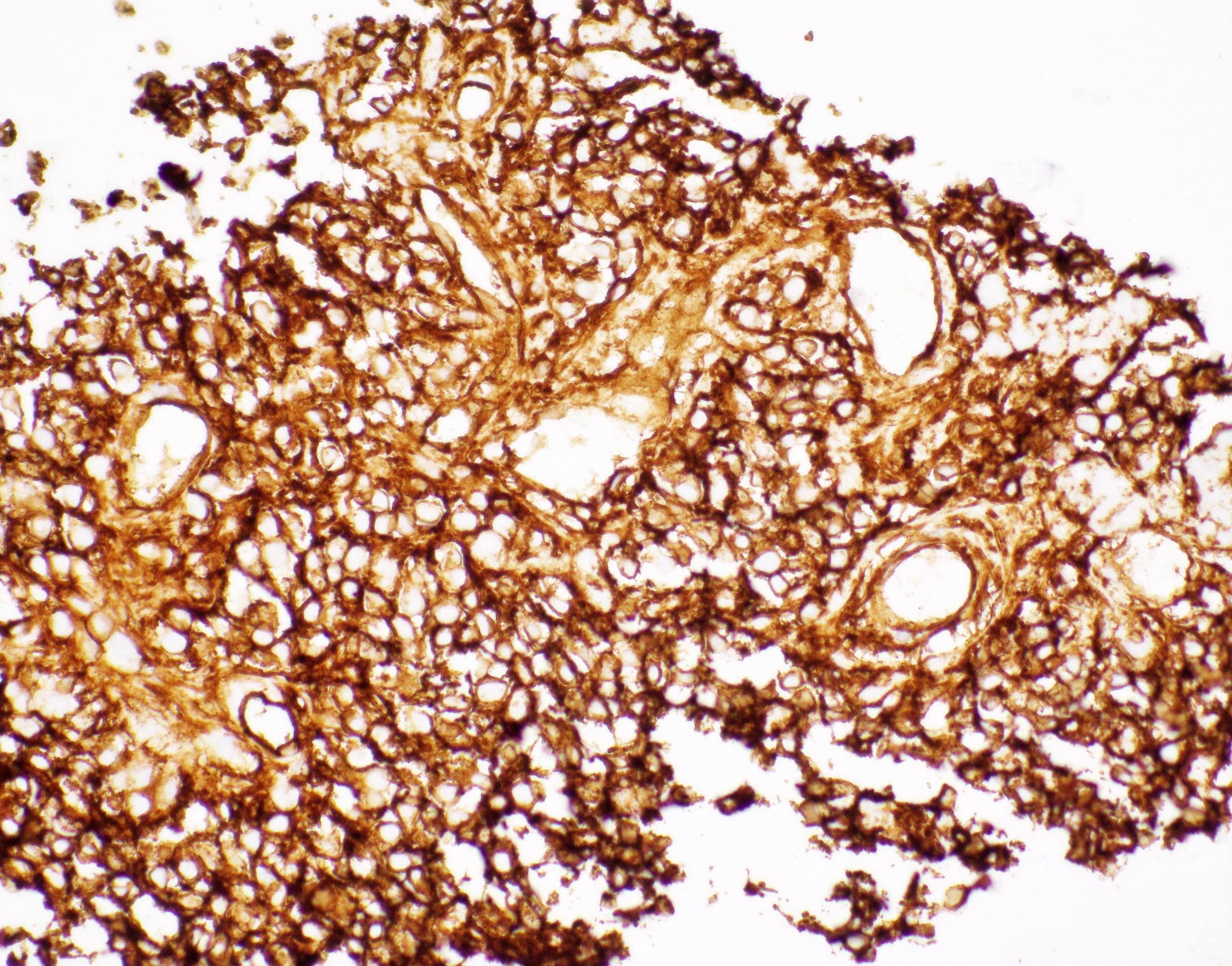
CD34
SMARCB1 / INI1
Contributed by Branavan Manoranjan, M.D., Ph.D., Abdelsimar T. Omar II, M.D., Hai-Bo Wu, M.D., Ph.D., Robert Nordal, M.D. and Yves Starreveld, M.D., Ph.D.
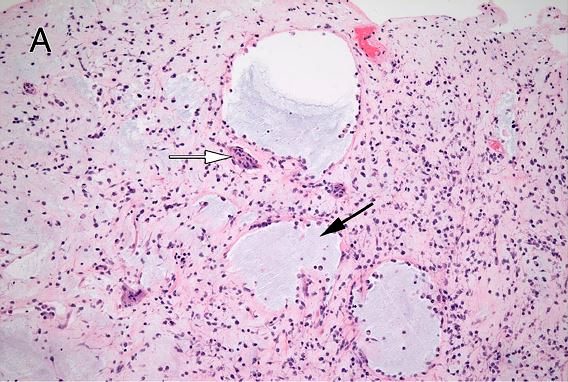
Microcysts and myxoid matrix
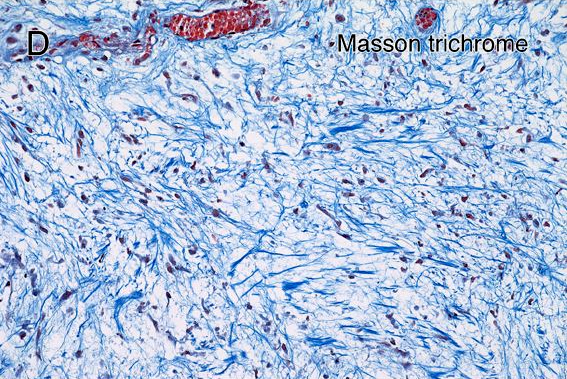
Trichrome
- CD34 (variably positive) (Acta Neuropathol 2020;139:277)
- EMA (variably positive) (Acta Neuropathol 2020;139:277)
- Vimentin (variably positive) (Acta Neuropathol 2020;139:277, Neuropathology 2021;41:37)
- Defined by SMARCB1 / INI1 inactivating alterations
- DNA methylation profile clusters adjacent to atypical teratoid / rhabdoid tumor - MYC and poorly differentiated chordomas
- Reference: Acta Neuropathol 2020;139:277
- Pineal region tumor, resection:
- Desmoplastic myxoid tumor of the pineal region, SMARCB1 mutant (see comment)
- Molecular information: SMARCB1 / INI1 loss of nuclear expression
- Immunohistochemistry: consistent with mutant
- Comment: A CNS WHO grade has not yet been assigned to this tumor entity.
- Meningioma:
- No SMARCB1 / INI1 inactivating alteration
- Solitary fibrous tumor:
- No SMARCB1 / INI1 inactivating alteration
- NAB2::STAT6 fusion with upregulated STAT6 expression
- Chordoma:
- Nuclear brachyury
- Cribriform neuroepithelial tumor:
- Cribriform architecture
- Predominantly in young children
- Tyrosinase staining
- Atypical teratoid / rhabdoid tumor, CNS WHO grade 4:
- Malignant with frequently increased mitotic activity and necrosis
- Predominantly in young children

A 35 year old woman presents with progressively worsening blurred vision, headache, nausea and vomiting. MRI shows a pineal region mass with areas of contrast enhancement. A representative hematoxylin and eosin stained image of the tumor is shown. The tumor cells are CD34 positive and exhibit loss of nuclear SMARCB1 / INI1. The diagnosis is consistent with which of the following?
- Desmoplastic myxoid tumor of the pineal region, SMARCB1 mutant
- Germinoma
- Meningioma, CNS WHO grade 1
- Pineocytoma, CNS WHO grade 1
Comment Here
Reference: Desmoplastic myxoid tumor, SMARCB1 mutant
- ATRX
- DICER1
- EWSR1
- SMARCB1
- Malignant, infiltrative, hemispheric, IDH wild type glioma with a G34R/V mutation in H3F3A
- Predominates in children and young adults
- Arises in the cerebral hemispheres
- Histopathologically heterogeneous, often resembling CNS embryonal tumor
- Histone H3 G34R/V mutations with concomitant TP53, ATRX and often PDGFRA mutations
- Poor prognosis: overall survival 12 - 24 months
- Other names used today or historically that pathologists may be more familiar with:
- Pediatric glioblastoma
- Pediatric high grade glioma
- CNS embryonal tumor
- Primitive neuroectodermal tumor (PNET)
- Incidence data not yet available
- M:F = 1.4:1 (Acta Neuropathol 2016;131:137)
- Median patient age: 19 years old (arises later than diffuse midline glioma, H3 K27 altered) (Acta Neuropathol 2016;131:137)
- Cerebral hemispheres
- Histone H3 G34R/V mutations impair SETD2 activity, reducing H3K36me3 to promote a unique gene expression profile that supports tumorigenesis (Proc Natl Acad Sci U S A 2020;117:27354)
- Histone H3 G34R/V mutations inhibit the neuronal differentiation of GSX2 / DLX expressing interneuron progenitors (Cell 2020;183:1617)
- PDGFRA signaling may promote gliomagenesis (Cell 2020;183:1617)
- Precise etiology is unclear, although recurrent histone H3 G34 R/V mutation is present
- Child with Li-Fraumeni syndrome and a diffuse hemispheric glioma, H3 G34 mutant has been reported (Acta Neuropathol 2021;142:591)
- Symptoms related to increased intracranial pressure, such as headache, nausea and vomiting
- May have site dependent neurological deficits
- Reference: Brain Tumor Pathol 2017;34:103
- Identification of a histone H3 G34R mutation (more commonly) or histone H3 G34V mutation that is confirmed by sequencing in a diffuse hemispheric glioma
- Large, hemispheric, bulky tumors that are T2 hyperintense on MRI (J Neuroradiol 2018;45:316)
- In one study, MRI showed multifocal lesions in 2 of 8 patients, contrast enhancement in 6 of 8, necrosis in 3 of 8, cysts in 3 of 8, hemorrhage in 1 of 8 and calcifications in 1 of 8 (Clin Nucl Med 2018;43:895)
Contributed by Mariana Voudouri, M.D. and George Zanazzi, M.D., Ph.D.
MRI of posterior left frontal tumor
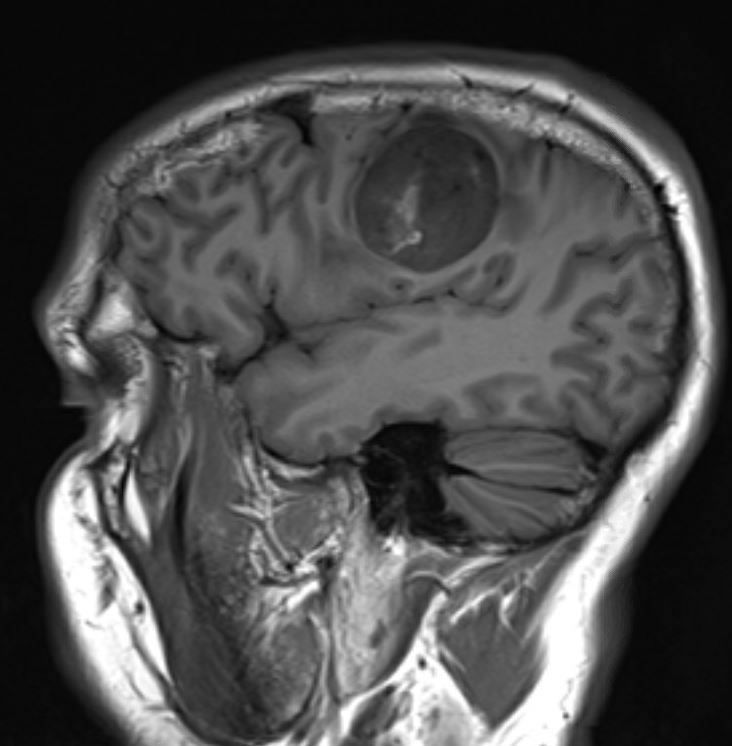
Sagittal T1
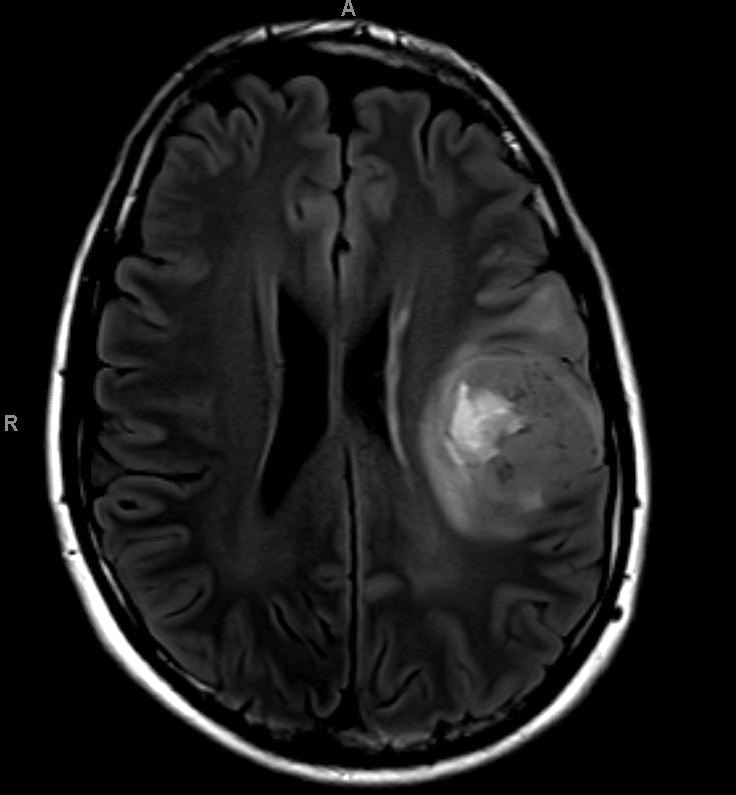
Axial T2 / FLAIR
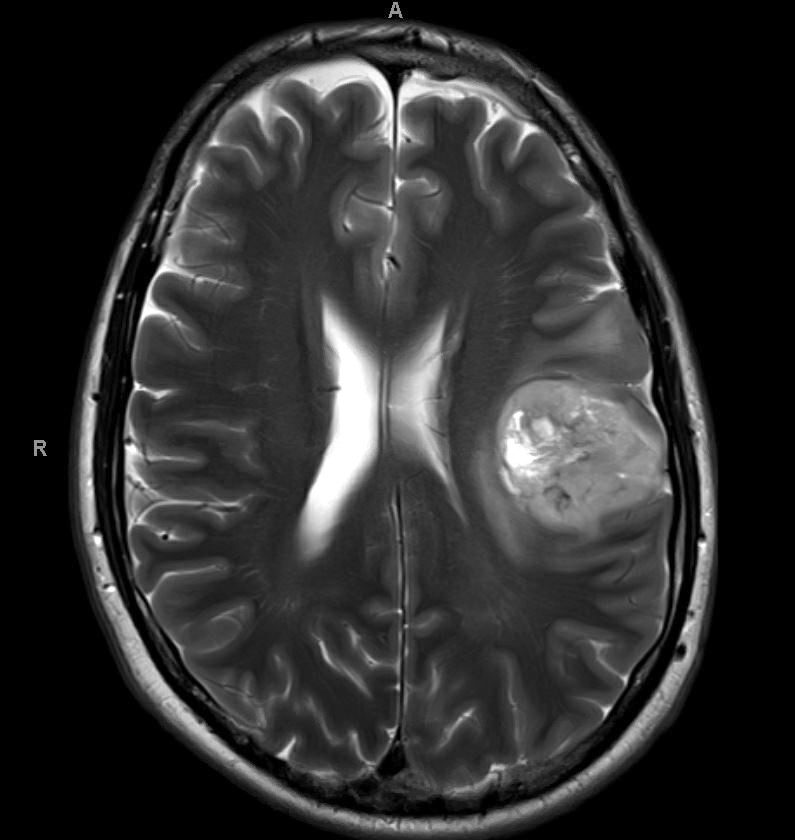
Axial postcontrast
- Generally poor prognosis
- Median survival is 12 - 24 months (Neurooncol Adv 2021;3:vdab061, Brain Tumor Pathol 2021;38:4)
- No significant difference in prognosis between high grade glioma and CNS embryonal tumor morphologies
- 13 year old boy with Li-Fraumeni syndrome and a left sided parietotemporal mass (Acta Neuropathol 2021;142:591)
- 16 year old boy with fronto-temporo-insular mass containing mixed glial and dysplastic neuronal components (Acta Neuropathol Commun 2019;7:78)
- 17 year old girl with a motor cortex mass and extraneural osseous metastases (Front Oncol 2019;9:373)
- 28 year old woman with a frontal lobe mass with embryonal histologic features and H3.3 G34V mutation (Indian J Pathol Microbiol 2020;63:262)
- 38 year old man with thalamus mass with histologically low grade gemistocytic astrocytoma and co-occurring K27M and G34R mutations in HIST1H3B (J Neuropathol Exp Neurol 2020;79:1038)
- Complete resection that is safely possible, followed by radiation and temozolomide
- Soft, tan-gray mass within the cortex and subcortical white matter
- Hemorrhage or necrosis may occur
- Tumor cells usually with either astrocytic morphology or CNS embryonal tumor morphology (Acta Neuropathol 2016;131:137)
- Diffusely infiltrating growth pattern, often with high grade features such as mitotic activity, microvascular proliferation or necrosis
- Even if high grade features are absent, the presence of an H3 G34R/V mutation confers a CNS WHO grade 4 (J Neuroradiol 2018;45:316)
Contributed by Mariana Voudouri, M.D. and George Zanazzi, M.D., Ph.D.
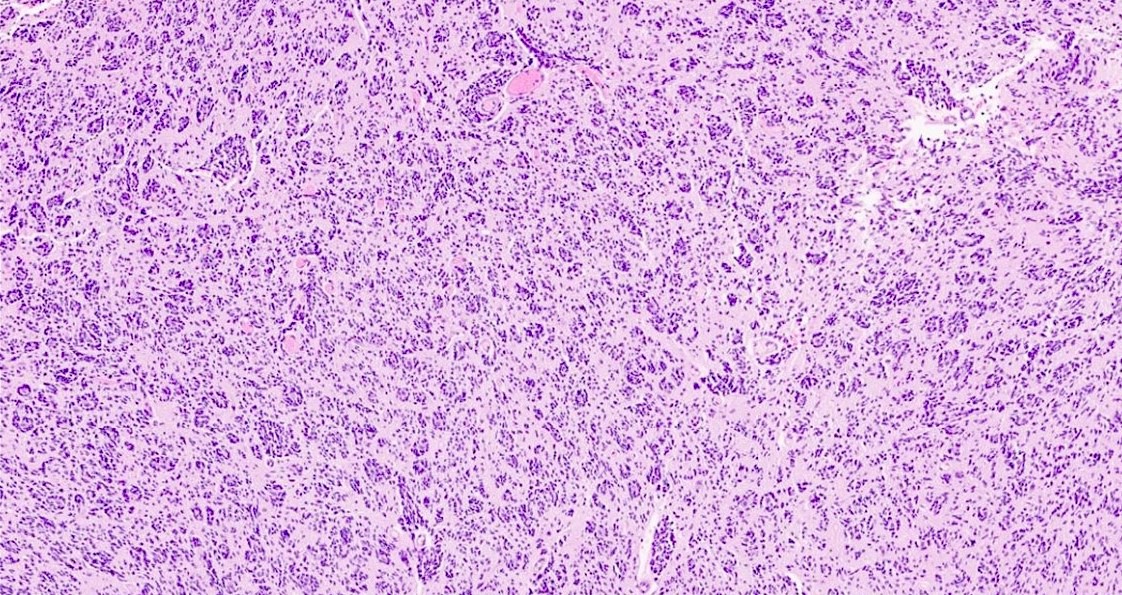
Hypercellular with clustering
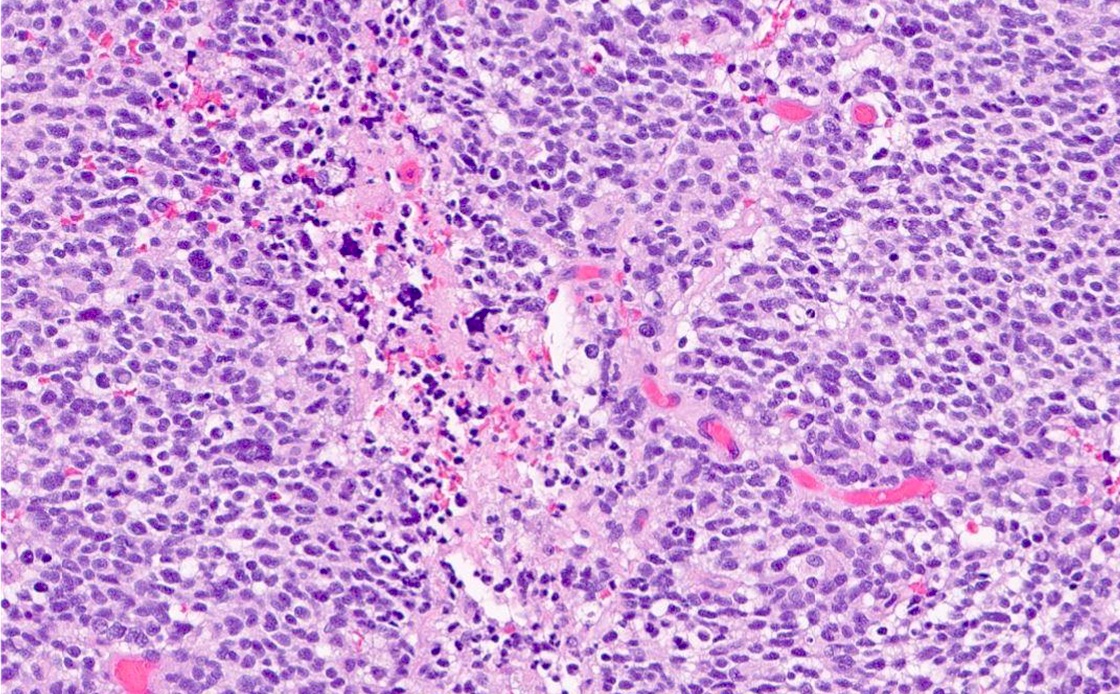
Area of necrosis
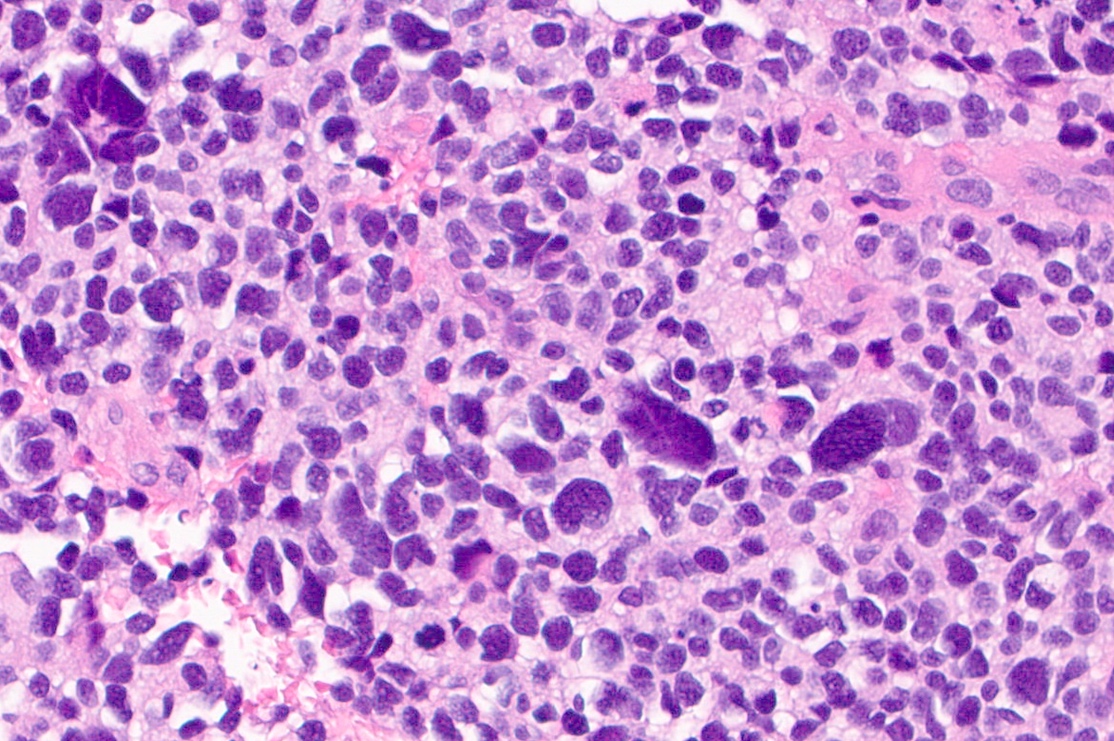
Marked pleomorphism

ATRX
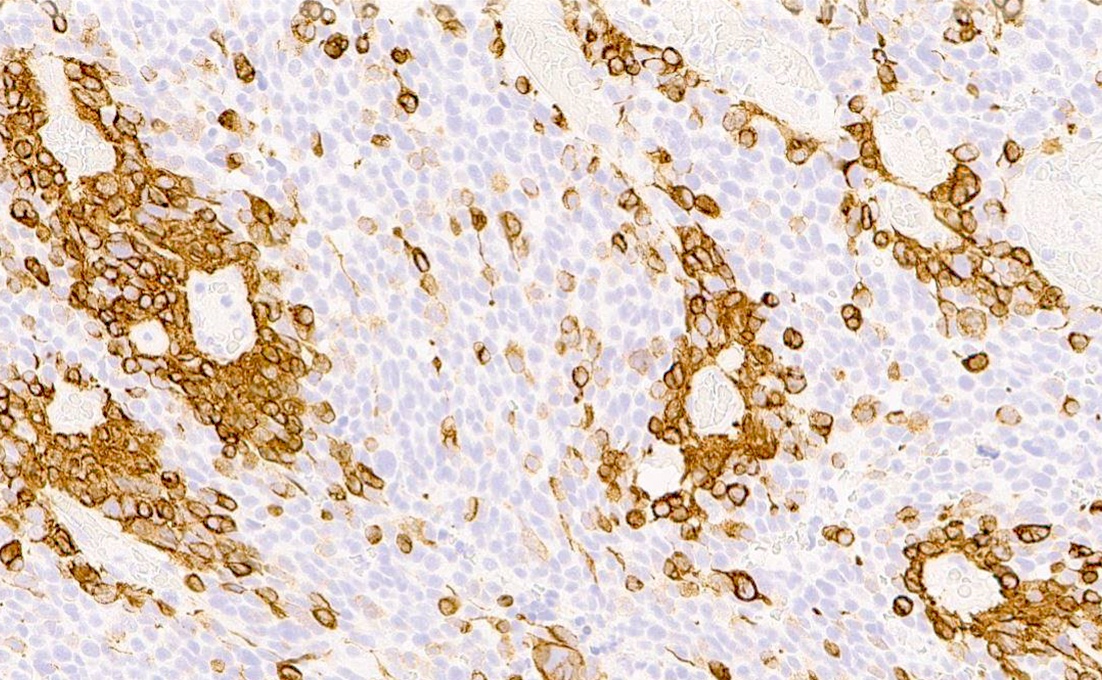
GFAP

Synaptophysin

p53
- H3F3A (G34R/V) (Am J Surg Pathol 2021;45:200)
- GFAP (variably positive) (J Neurooncol 2013;112:67)
- Synaptophysin (variably positive) (J Neurooncol 2013;112:67)
- p53 (strongly positive in most cases) (Nature 2012;482:226)
- Defined by recurrent G34R/V mutations in H3F3A
- Additional mutations:
- TP53
- ATRX
- PDGFRA
- Amplifications:
- NMYC
- PDGFRA (more common with astrocytic morphology) (Acta Neuropathol 2016;131:137)
- CCND2 (more common with CNS embryonal tumor morphology) (Acta Neuropathol 2016;131:137)
- Chromosomal alterations:
- Chromosome 3q and 4q loss (Acta Neuropathol 2016;131:137)
- Brain tumor, left frontal lobe, resection:
- Diffuse hemispheric glioma, H3 G34 mutant, WHO grade 4
- Histological diagnosis: CNS embryonal tumor, NEC, WHO grade 4
- Molecular information:
- H3 G34R: positive (immunohistochemistry; consistent with mutant)
- ATRX: nuclear expression loss (immunohistochemistry; consistent with mutation)
- p53: positive (immunohistochemistry; consistent with mutation)
- IDH: negative (R132H immunohistochemistry; consistent with wild type)
- CNS embryonal tumor, NOS, WHO grade 4:
- No H3 G34R/V mutation
- ATRX preserved
- Glioblastoma, IDH wild type, WHO grade 4:
- No H3 G34R/V mutation
- Olig2 positive
- Astrocytoma, IDH mutant, WHO grade 4:
- Astrocytoma, IDH mutant, WHO grade 3:
- Astrocytoma, IDH mutant, WHO grade 2:
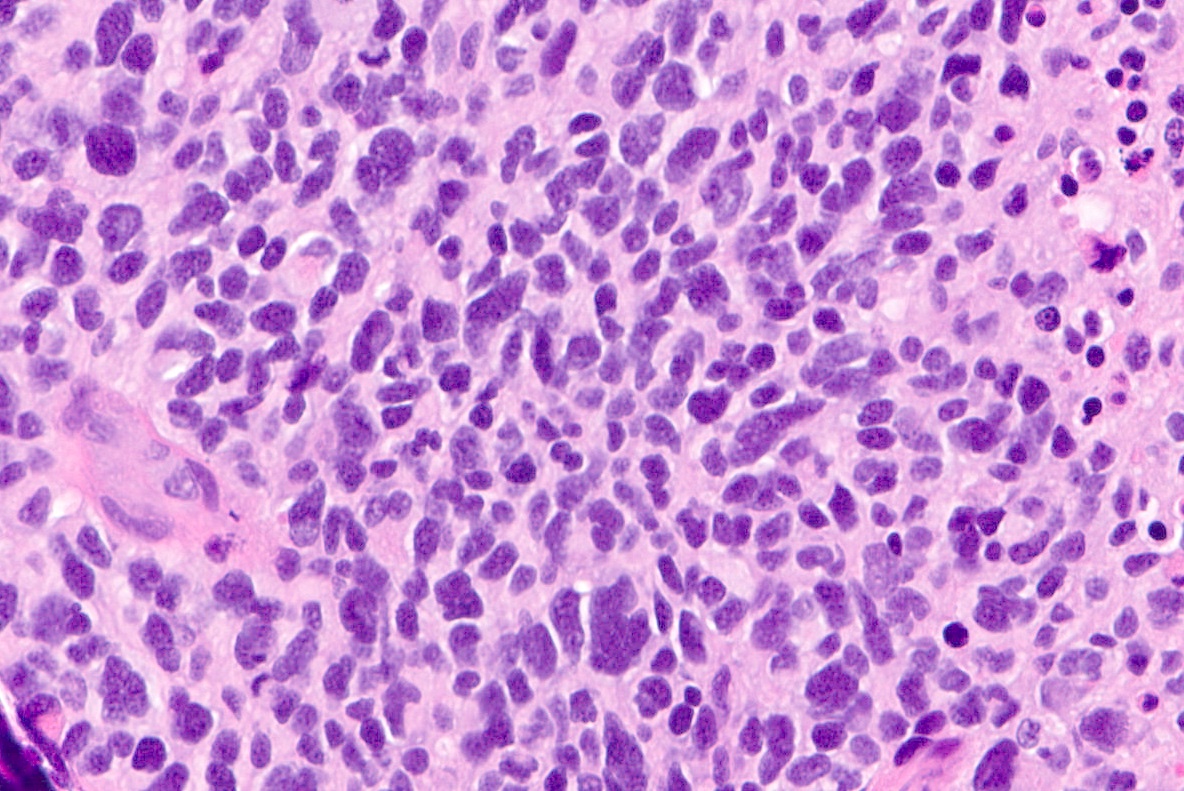
A 17 year old girl presents with word finding difficulties, right facial droop and right arm weakness. MRI shows a large, left frontal lobe mass in the brain with areas of contrast enhancement. A representative hematoxylin and eosin stained section of the resected tumor is shown. Some tumor cells are GFAP positive and there is abundant p53. There is loss of ATRX without an IDH mutation. The diagnosis is consistent with
- Diffuse hemispheric glioma, H3 G34 mutant, WHO grade 4
- Diffuse midline glioma, H3 K27 altered, WHO grade 4
- Glioblastoma, IDH wild type, WHO grade 4
- Pleomorphic xanthoastrocytoma, WHO grade 3
Comment Here
Reference: Diffuse hemispheric glioma, H3 G34 mutant
- Li-Fraumeni syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Tuberous sclerosis
- Indolent, low grade neuroepithelial neoplasm that typically shows widespread leptomeningeal and superficial parenchymal CNS dissemination and oligodendroglioma-like cytology. This tumor was added provisionally to the WHO classification scheme of CNS tumors in 2016, and is no longer a provisional entity in the 2021 WHO CNS5 classification
- Indolent, slow growing tumor with widespread and diffuse leptomeningeal involvement
- Typically affects children; rare in adults
- Presents with signs and symptoms of obstructive hydrocephalus
- Olig2 and S100 positive, monomorphic oligodendroglial-like cells in desmoplastic or myxoid matrix
- KIAA1549-BRAF fusion, 1p deletion common; 1p19q codeletion and BRAF V600E less common
- References: Acta Neuropathol 2018;136:239, Acta Neuropathol 2012;124:627
- Abbreviations: diffuse leptomeningeal glioneuronal tumor (DLGNT)
- Other historical terms (Acta Neuropathol 2016;131:803):
- Primary leptomeningeal oligodendrogliomatosis
- Disseminated oligodendroglioma-like leptomeningeal neoplasm (DOLN)
- Disseminated oligodendroglial-like neoplasm
- Diffuse leptomeningeal neuroepithelial tumor
- ICD-10: C70.1 - malignant neoplasm of spinal meninges
- Range: 5 months - 46 years
- Median age of diagnosis: 5 years
- M:F = 1.7:1
- Reference: Acta Neuropathol 2012;124:627
- Widespread, diffuse involvement of spinal and intracranial leptomeninges (Acta Neuropathol 2012;124:627)
- Within cranium, most common around posterior fossa, brainstem and base of brain
- May fill ventricles and spread along Virchow-Robin spaces (Acta Neuropathol 2012;124:627)
- May form cystic or solid tumor lesions in adjacent parenchyma; spinal > intracerebral
- Spread in subarachnoid space: plaque-like or multinodular pattern (Acta Neuropathol 2012;124:627)
- Unknown how tumor arises
- Precise etiology unknown, although recurrent genetic alterations are identified (see Molecular / cytogenetics description)
- Often acute, presents with signs and symptoms of increased intracranial pressure (Acta Neuropathol 2012;124:627)
- Due to obstructive hydrocephalus
- Nausea, vomiting and headache
- Opisthotonos
- Encasement or irritation of spinal or cranial nerves (Acta Neuropathol 2012;124:627)
- May lead to palsies related to motor, sensory or autonomic function
- Meningeal signs
- Ataxia
- Spinal cord compression
- Rarely, seizures
- Primarily made on radiologic features
- Confirmed by tissue biopsy with ancillary immunohistochemistry, cytogenetic (FISH) and molecular testing
- MRI shows widespread contrast enhancement and thickening along spinal cord; often extends intracranially to posterior fossa, brainstem and basal brain (J Belg Soc Radiol 2017;101:19)
- Small cystic or nodular T2 hyperintense lesions along subpial surface of spinal cord or brain
- Discrete intraparenchymal lesions, most common in spinal cord; sometimes without diffuse spread (Acta Neuropathol Commun 2020;8:95)
- Obstructive hydrocephalus with associated periventricular T2 hyperintensity
- See Case reports
- Mostly favorable prognosis in terms of mortality (Brain Tumor Pathol 2015;32:49, Acta Neuropathol 2012;124:627)
- Indolent tumor but with significant morbidity
- Poor prognosis (Brain Tumor Pathol 2015;32:49):
- Mitotic activity (of any degree)
- Glomeruloid microvascular proliferation
- MIB1 / Ki67 labeling index ≥ 4%
- 10 children, 2 - 14 years old; 9 patients received chemotherapy, 3 with progressive disease; 6 patient harbored genomic alterations affecting the MAPK pathway (J Neurooncol 2016;128:293)
- 4 year old boy with diffuse leptomeningeal thickening; review of imaging features (J Belg Soc Radiol 2017;101:19)
- 7 year old boy with low grade tumor versus 9 year old boy with high grade tumor (Hum Pathol 2017;70:105)
- 8 year old girl with imaging presentation mimicking tuberculous meningitis (Childs Nerv Syst 2011;27:187)
- 12 year old boy with ventriculoperitoneal shunt treated with temozolomide (Childs Nerv Syst 2020;36:459)
- 13 year old girl with predominant spinal infiltration (Childs Nerv Syst 2019;35:1609)
- 31 and 35 year old women with circumscribed intraparenchymal tumors (Acta Neuropathol Commun 2020;8:95)
- 36 year old woman with aggressive tumor (World Neurosurg 2018;114:53)
- 7 cases with typical and less common imaging features, showing the most characteristic imaging finding is diffuse intracranial and intraspinal nodular leptomeningeal thickening and enhancement (AJNR Am J Neuroradiol 2020;41:2155)
- Series of 36 patients; comprehensive description of presentation, imaging, histology, treatment and prognosis (Acta Neuropathol 2012;124:627)
- Surgical resection, particularly for parenchymal lesions (Childs Nerv Syst 2019;35:1609)
- Symptomatic treatment for hydrocephalus (Childs Nerv Syst 2020;36:459)
- Ventriculoperitoneal shunting
- Chemotherapy (Child Neurol Open 2015;2:2329048X14567531, Childs Nerv Syst 2018;34:329)
- Temozolomide, others
- Radiotherapy in disease progression (Child Neurol Open 2015;2:2329048X14567531, Childs Nerv Syst 2018;34:329)
- Targeted therapy (Pediatr Blood Cancer 2020;67:e28478)
- Postmortem examination confirms widespread, diffuse leptomeningeal spread in spinal and intracranial compartments (Brain Tumor Pathol 2018;35:209)
- Multifocal extension along Virchow-Robin spaces (Acta Neuropathol 2012;124:627)
- Growth along peripheral nerve roots and infiltration of basal cranial nerves and ganglia
- Low to moderate cellularity; diffuse growth or nests in leptomeninges (Brain Tumor Pathol 2015;32:49)
- Often with desmoplastic or myxoid changes
- Bland, monomorphous, oligodendroglial-like cells (Brain Tumor Pathol 2015;32:49)
- With or without perinuclear halo
- Uniform round nuclei with fine chromatin and inconspicuous nucleoli
- Some with ganglion or ganglioid cells; may be associated with neuropil
- Eosinophilic granular bodies (occasional), Rosenthal fibers (rare)
- Low mitotic activity
- Anaplasia is rare; associated with more aggressive clinical course (Brain Tumor Pathol 2015;32:49)
- Nuclear enlargement
- Atypia
- Increased mitotic activity
- Microvascular proliferation
- Necrosis
Contributed by Katherine Schwetye, M.D., Ph.D.
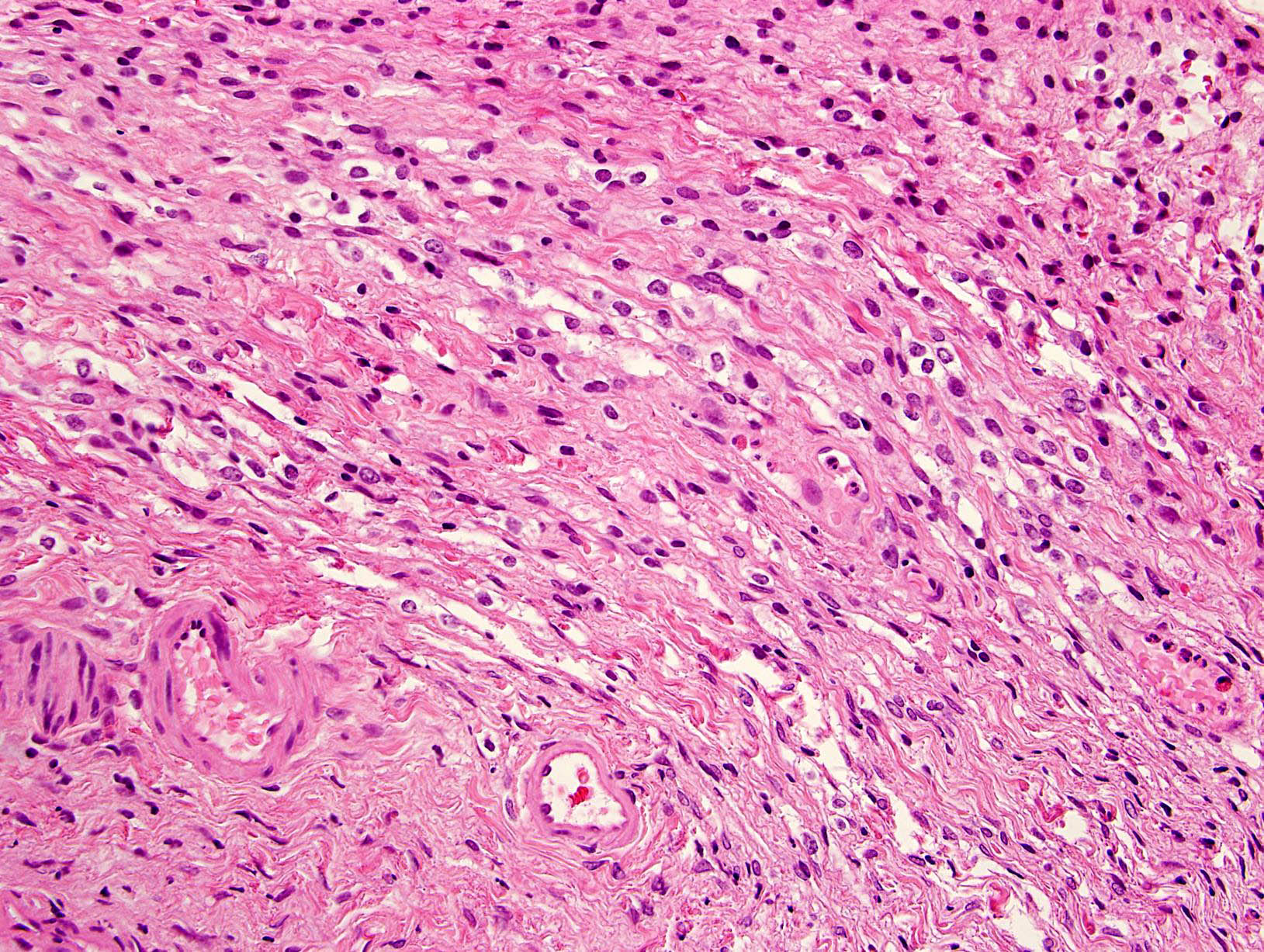
Oligodendroglial-like appearance
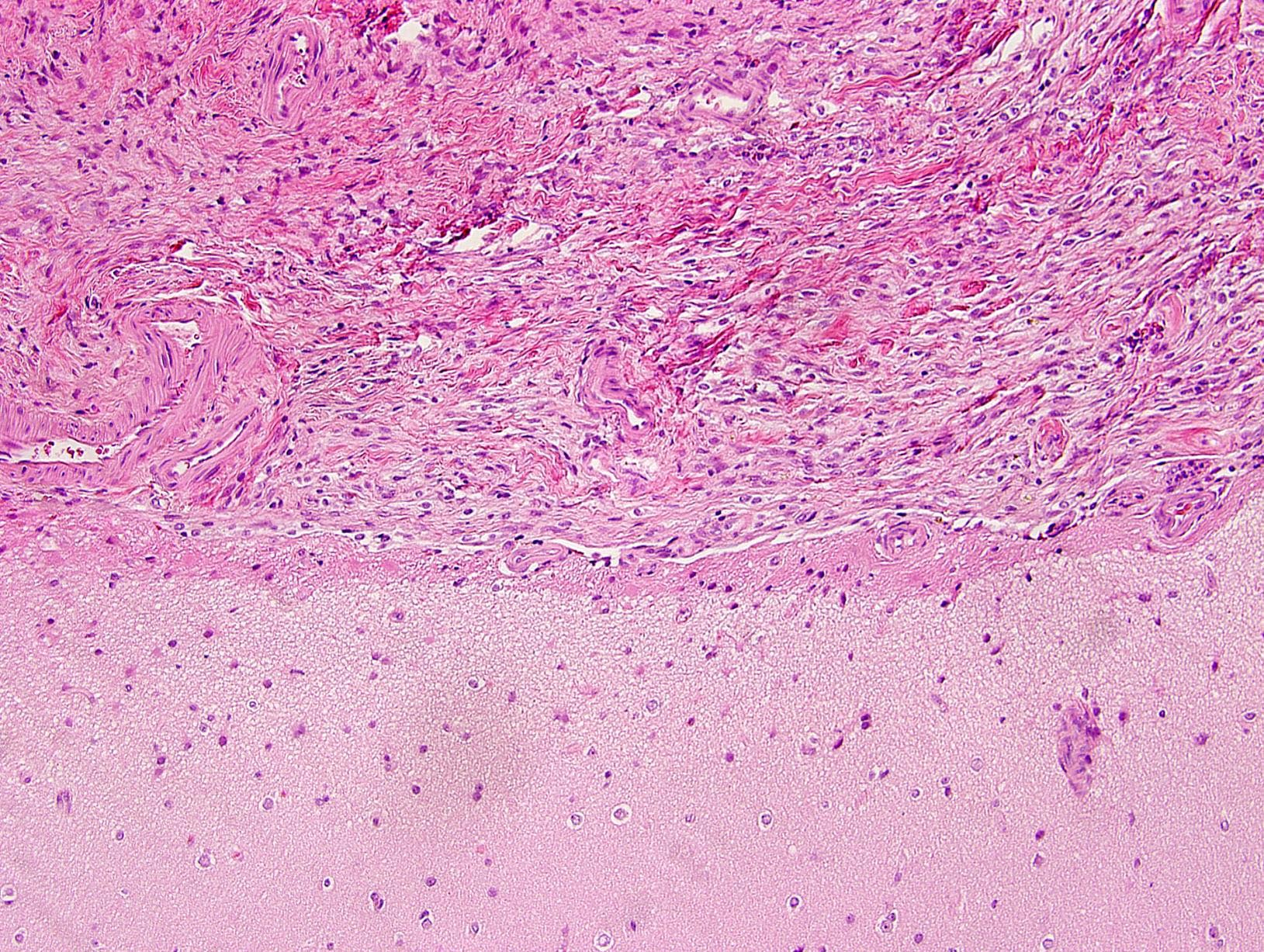
Tumor adjacent parenchyma
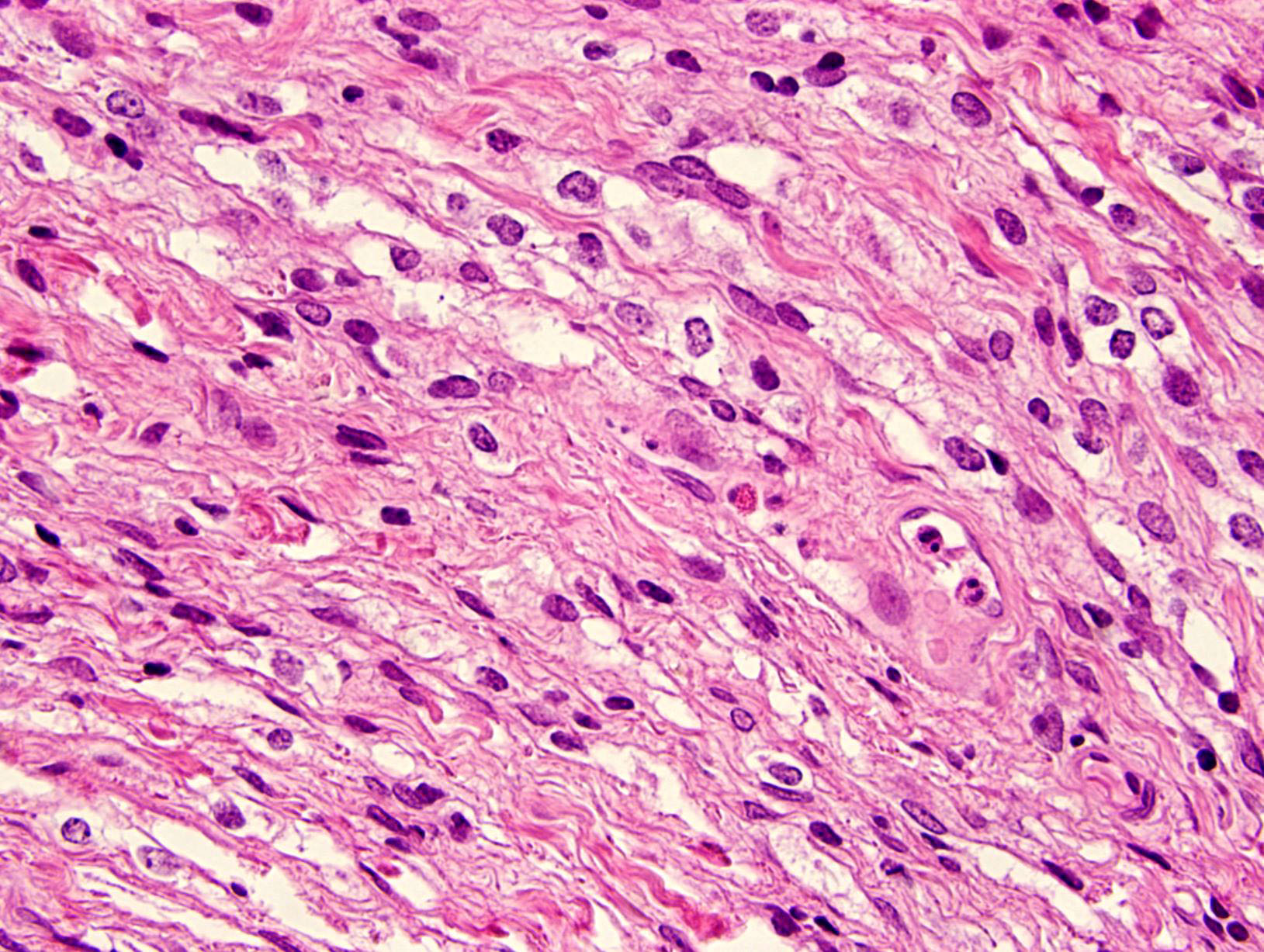
Bland cytomorphology and nuclear features
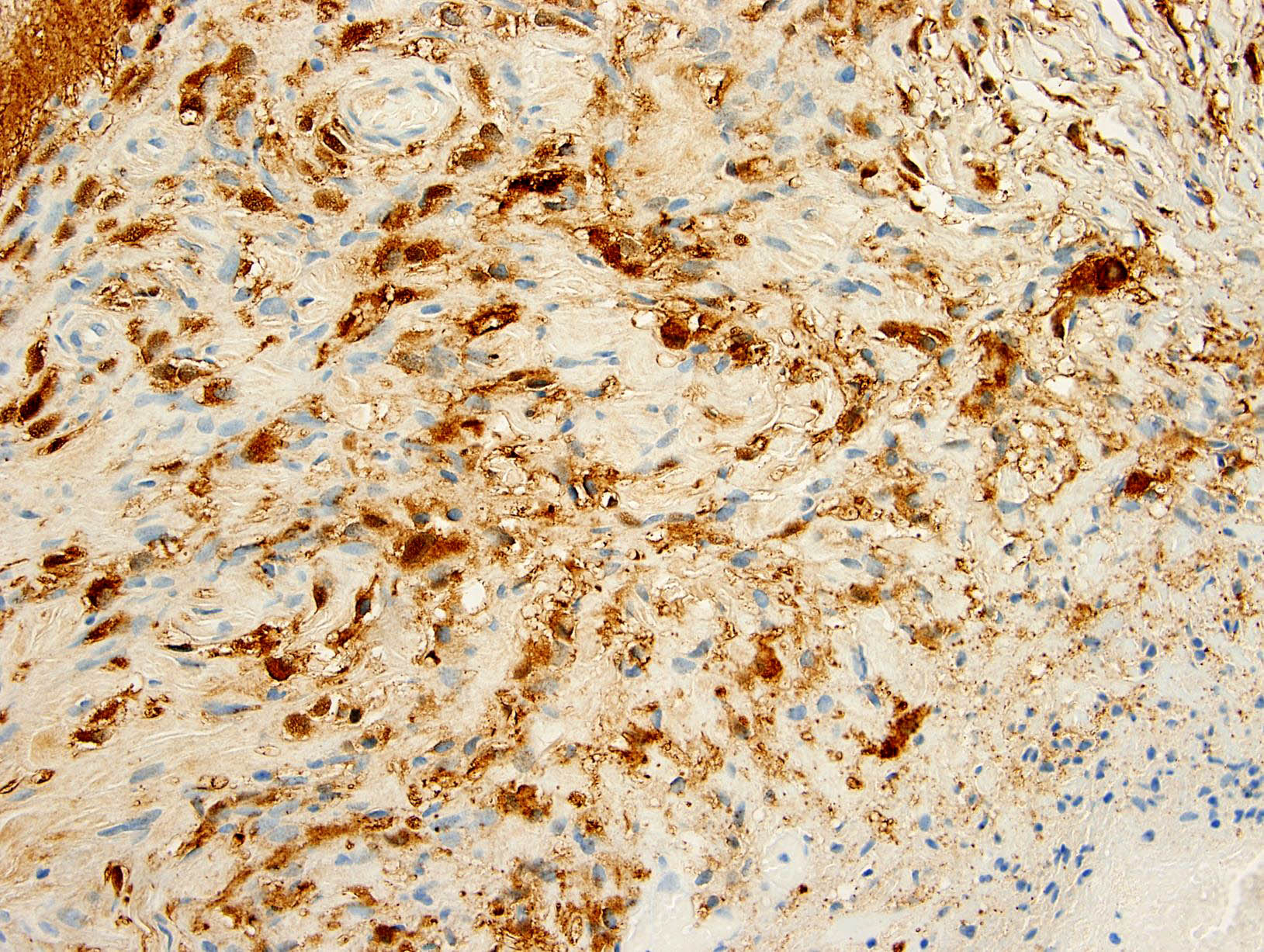
S100 positive
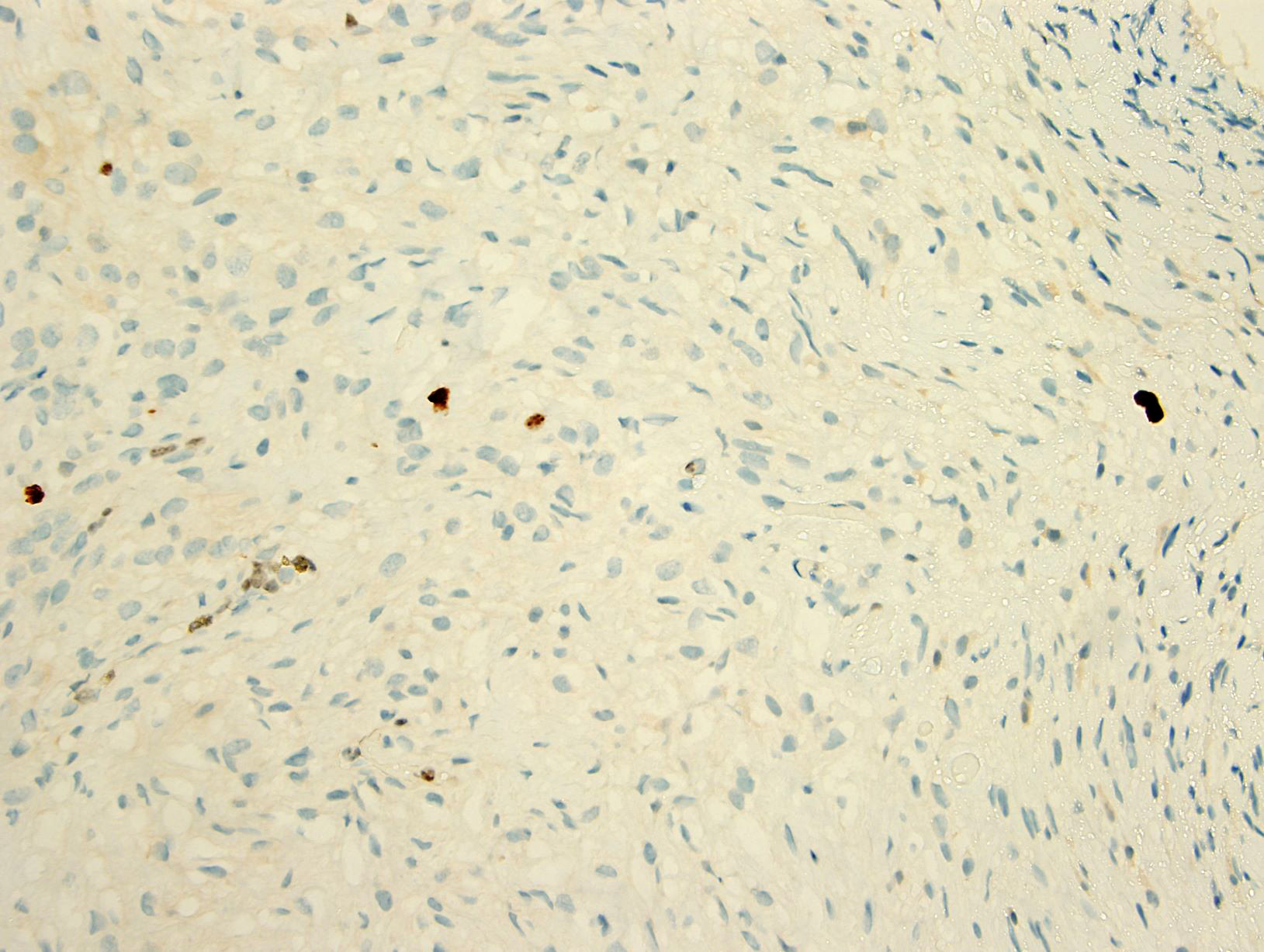
Ki67 low
These photographs were previously published in Human Pathology, Vol. 70, Schwetye KE et al., "Unusual high-grade features in
pediatric diffuse leptomeningeal glioneuronal tumor: comparison with a typical low-grade example", p.105-112 Copyright Elsevier (2017).
- Cerebrospinal fluid examination
- Increased protein levels
- Reactive lymphocytosis (rare feature)
- Typically negative for tumor cells (Childs Nerv Syst 2018;34:329)
- S100, Olig2, MAP2, synaptophysin (70%), GFAP (focal, in 50%)
- Ki67 ~1.5% (median)
- NeuN, neurofilaments, EMA and IDH1 R132H
- KIAA1549-BRAF fusion protein is most frequent (60 - 70%) (Acta Neuropathol 2012;124:627, Acta Neuropathol 2015;129:609)
- Chromosome 1p deletion (50 - 100%, varies by series) (Acta Neuropathol 2012;124:627, Acta Neuropathol 2015;129:609)
- DNA methylation profiling identifies 2 distinct classes of diffuse leptomeningeal glioneuronal tumor (Acta Neuropathol 2018;136:239)
- Chromosome 1p / 19q codeletion differs by methylation defined groups (47% versus 15%) (Acta Neuropathol 2018;136:239)
- Other reported alterations: TRIM33-RAF1, NTRK1/2/3 fusions, CDKN2A/B homozygous deletion, TERT C228T, C228A, ATRX point mutation, BCOR frameshift alteration, BRAF V600E (Acta Neuropathol 2018;136:239)
- Negative for IDH1 R132, IDH R172, H3F3A K27 alterations (Acta Neuropathol 2018;136:239)
- Spine, leptomeninges, biopsy:
- Diffuse leptomeningeal glioneuronal tumor (see comment)
- Comment: Hematoxylin and eosin examination of the biopsy material shows a moderately cellular, neoplastic proliferation within the leptomeninges with associated desmoplastic and myxoid change. The cells show relatively monomorphic, bland morphology, round nuclei with perinuclear haloes (reminiscent of an oligodendroglial-like appearance) and inconspicuous nucleoli. Mitotic activity is not identified. Necrosis is not identified. Immunohistochemical stains demonstrate reactivity for S100 and Olig2. Ki67 shows a low (< 2%) index of proliferation. FISH studies confirm the KIAA1549-BRAF rearrangement and deletion of chromosome 1p. Taken together, the radiologic, histomorphologic, immunohistochemical and cytogenetic features support a diagnosis of diffuse leptomeningeal glioneuronal tumor.
- Pilocytic astrocytoma:
- Typically intraparenchymal tumor without primary leptomeningeal spread
- More predominant Rosenthal fibers
- Glomeruloid microvascular proliferation does not alone confer worse prognosis
- Oligodendroglioma:
- Diffusely infiltrative glial tumor
- Typically intraparenchymal
- IDH1 / IDH2 mutations as a requisite feature for diagnosis
- Macrophage rich reactive / inflammatory process:
- CD68 positive macrophages
- Tumors metastatic to the leptomeninges:
- Clinical history, more likely adults
- H3F3 K27M
- IDH1 R132H
- KIAA1549-BRAF translocation and 1p deletion
- PTEN loss
Comment Here
Reference: Diffuse leptomeningeal glioneuronal tumor
Based on the image shown above in conjunction with MRI findings of an enhancing, spinal leptomeningeal process without a distinctive intraparenchymal lesion in a 5 year old boy, what is the expected pattern of immunoreactivity?
- S100 positive, Olig2 positive, H3F3a K27M positive
- S100 positive, Olig2 positive, IDH R132H positive
- S100 negative, Olig2 positive, IDH R132H positive
- S100 positive, Olig2 positive, IDH R132H negative
Comment Here
Reference: Diffuse leptomeningeal glioneuronal tumor
- Low grade, infiltrative, pediatric glioma with an alteration in a MAP kinase pathway gene such as FGFR1 or BRAF
- Tumor is IDH wild type, histone H3 wild type and does not have a homozygous deletion of CDKN2A
- Predominantly in children
- Arises anywhere in the central nervous system, often in the cerebral hemispheres or posterior fossa
- Histopathologically heterogeneous, often resembling low grade astrocytoma or oligodendroglioma
- MAP kinase pathway alteration without alterations in IDH1 / IDH2, histone H3 or homozygous deletion of CDKN2A
- Pediatric low grade glioma
- Formerly, pediatric oligodendroglioma or diffuse astrocytoma
- Predominantly in children
- Precise incidence data not yet available
- Cerebral hemispheres, diencephalon, brainstem, cerebellum and spinal cord, with possible site predilections depending on histology and genetic alterations (Cancer Cell 2020;37:569)
- Missense mutation, intragenic duplication or fusion may constitutively activate a receptor tyrosine kinase, such as FGFR or NTRK, leading to aberrant recruitment of the G protein RAS, which activates the RAF / MEK / ERK cascade (Acta Neuropathol Commun 2020;8:30)
- Missense mutations in RAS or RAF (especially BRAF V600E) can cause gliomagenesis in the absence of an upstream alteration (Acta Neuropathol Commun 2020;8:30)
- Precise etiology is unclear
- Symptoms related to increased intracranial pressure, such as headache, nausea and vomiting
- May have site dependent neurological deficits
- Longstanding epilepsy is common
- Identification of a MAPK pathway alteration (BRAF, FGFR1, FGFR2, NTRK1, NTRK2, NTRK3, MAP2K1, MET alterations) that is confirmed by sequencing in a diffuse low grade glioma (Nat Genet 2013;45:602, Acta Neuropathol 2016;131:833, Cancer Cell 2020;37:569)
- Absent IDH, histone H3 K27 or H3.3 G34R / V mutations
- Diffuse lesion, highlighted by T2 FLAIR on MRI, with possible cystic elements
- May be heterogeneously enhancing on MRI
Contributed by Mariana Voudouri, M.D. and George Zanazzi, M.D., Ph.D.
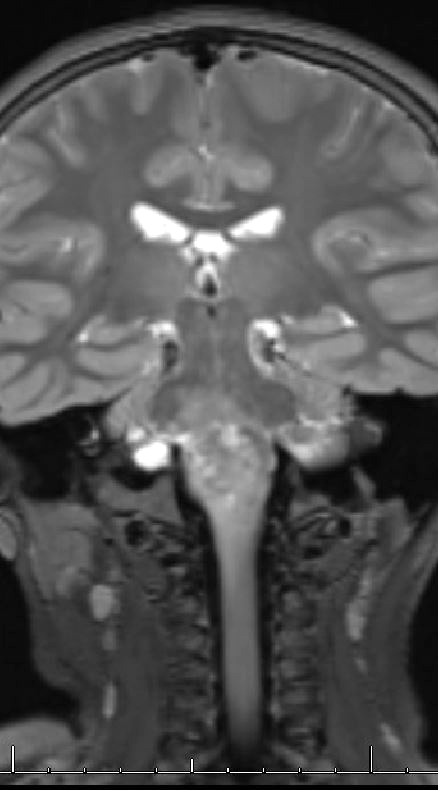
MRI coronal T2 brainstem tumor
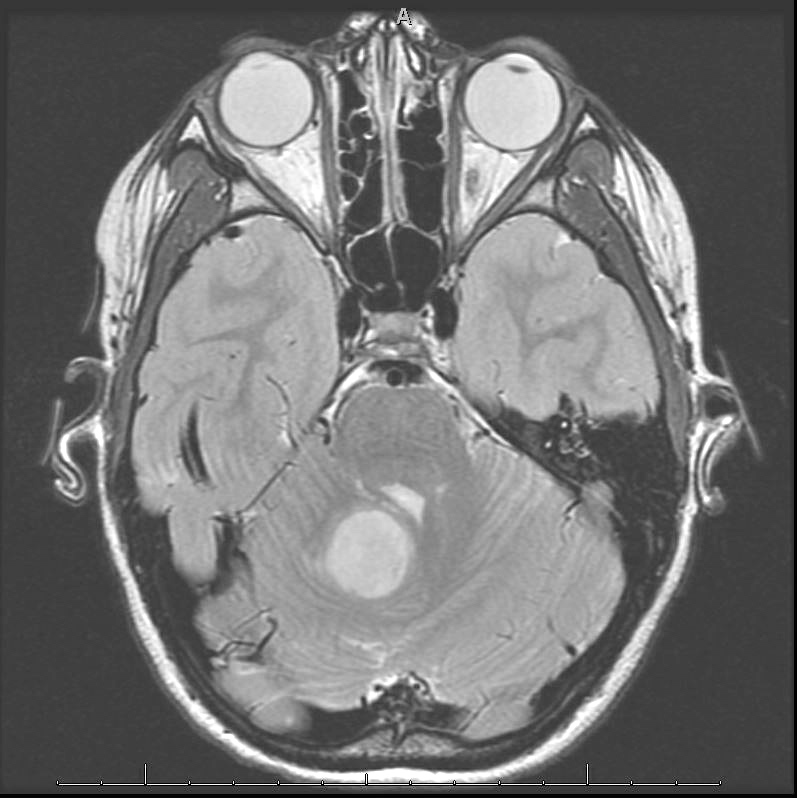
MRI axial T2 / FLAIR right cerebellar tumor
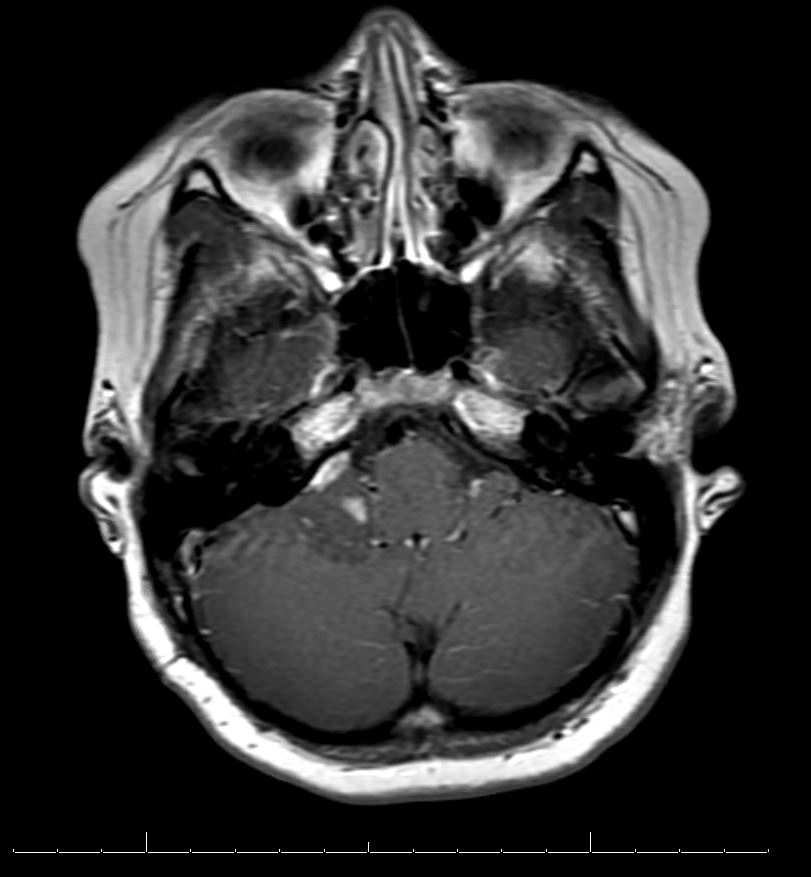
MRI axial T1 post contrast right cerebellar flocculus tumor
- Currently unclear but rarely undergo malignant transformation
- 9 year old boy with brainstem mass growing slowly over 8 years and low grade astrocytoma containing FGFR2-VPS35 fusion (Cold Spring Harb Mol Case Stud 2020;6:a005660)
- 15 year old with seizure and temporal lobe mass containing oligodendroglioma-like cells and BRAF V600E mutation (Brain Pathol 2020;30:515)
- 16 year old boy with developmental delay, germline 7q11.22 deletion and a low grade astrocytoma with BRAF V600E mutation in right temporal lobe (Clin Case Rep 2017;6:274)
- Surgical resection, although this may be limited by involvement of critical brain structures
- MEK inhibitors, such as selumetinib and trametinib, show promising effects in initial studies (Neuro Oncol 2017;19:1135, BMC Cancer 2019;19:1250)
- For BRAF V600E mutant tumors, dabrafenib has been used (Clin Cancer Res 2019;25:7303)
- Soft, tan-gray tissue
- Hemorrhage may occur (J Neurosurg 2020;134:733)
- General features: infiltrative, glial tumor cells without a neuronal component
- BRAF V600 altered tumors tend to have densely fibrillary areas and microcalcifications (Acta Neuropathol 2015;130:575)
- FGFR1 altered tumors tend to have oligodendrocyte-like cells, rare or no mitoses, no necrosis and no microvascular proliferation (Acta Neuropathol 2016;131:833)
- In a large study excluding NF1 patients, 5.9% of pediatric low grade gliomas exhibited astrocytic differentiation and 3.0% exhibited oligodendroglial differentiation (Cancer Cell 2020;37:569)
Contributed by Mariana Voudouri, M.D. and George Zanazzi, M.D., Ph.D.

Astrocytic morphology
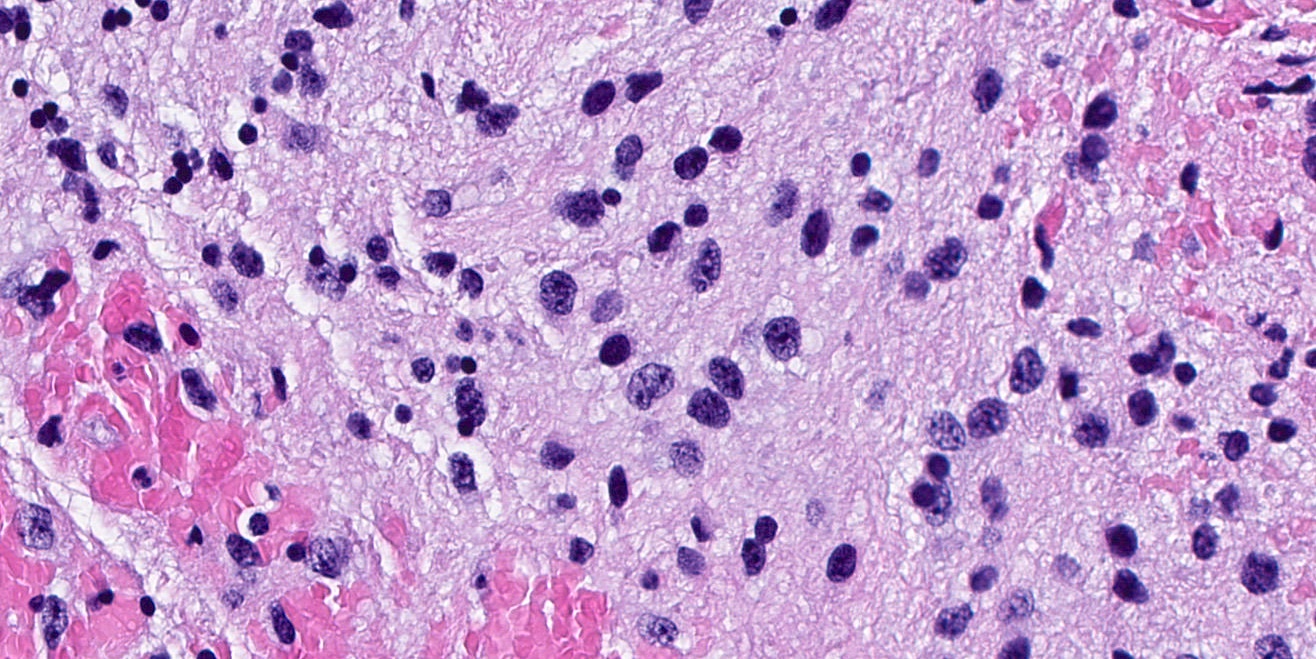
Pleomorphism
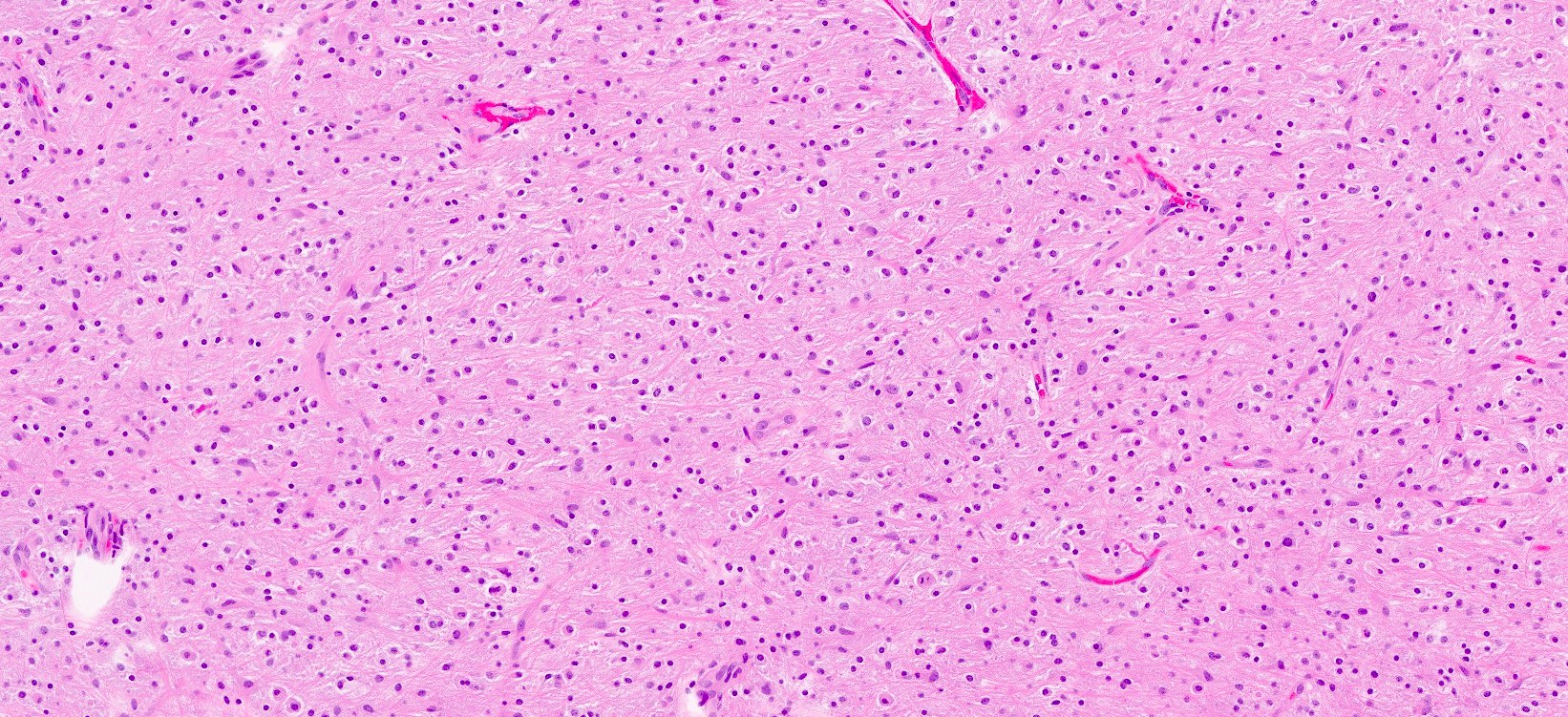
Oligodendroglial morphology
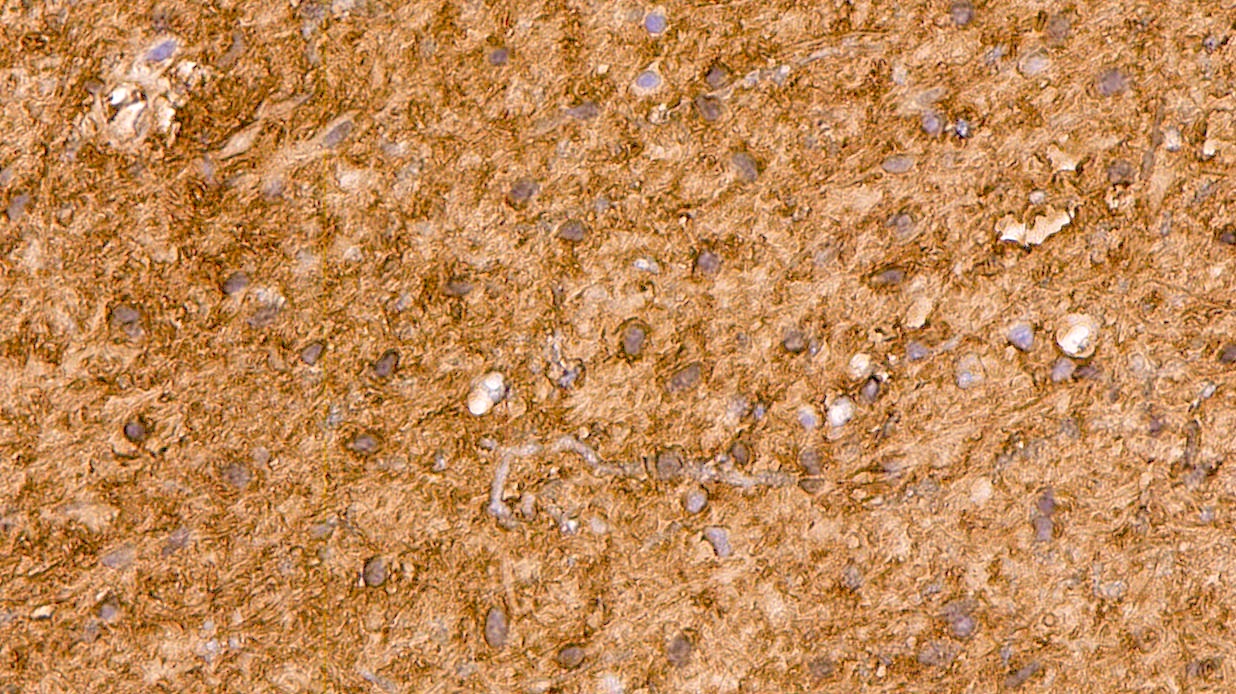
GFAP
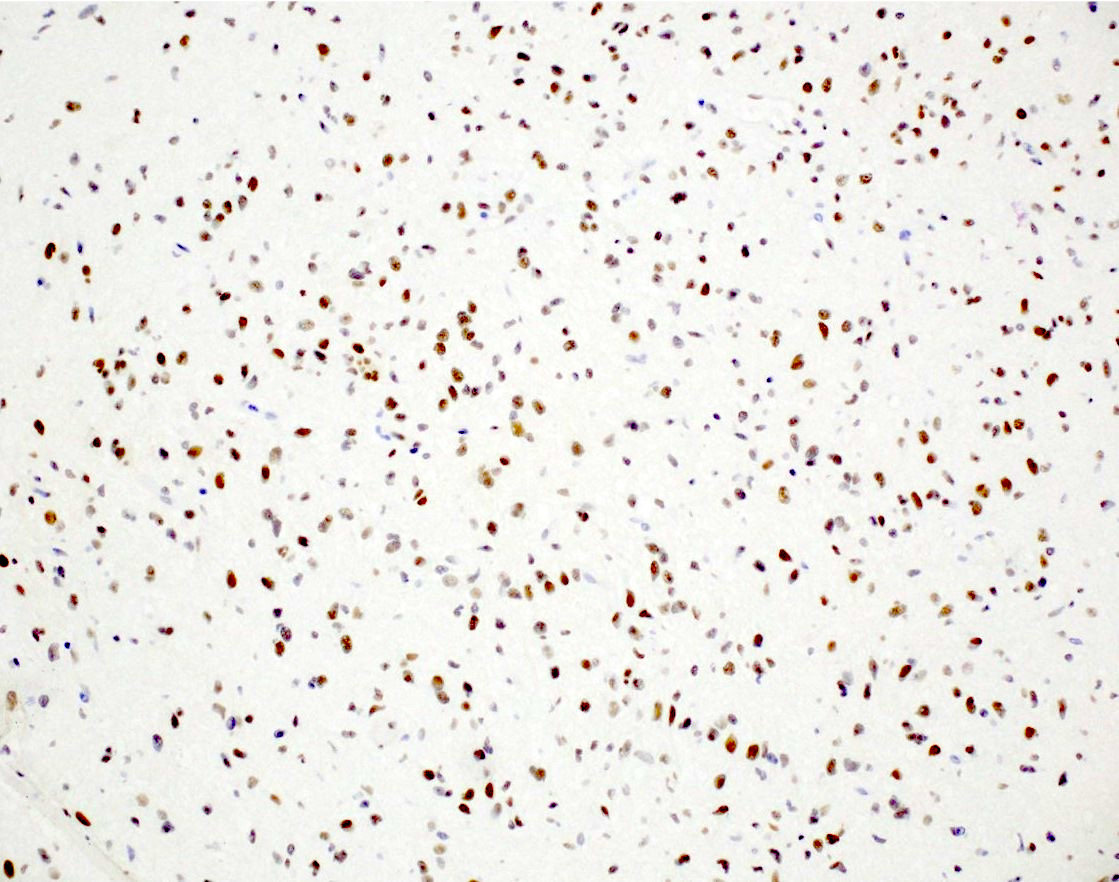
ATRX
Ki67
- GFAP (Nat Genet 2013;45:602)
- Olig2 (Am J Surg Pathol 2014;38:1058)
- BRAF V600E (if mutated)
- IDH1 (R132H)
- H3F3A K27M
- H3F3A G34R / V
- CD34 (although may be present in a few cells) (Acta Neuropathol 2015;130:575)
- FGFR alterations, such as FGFR1 tyrosine kinase domain duplications (Nat Genet 2013;45:602)
- BRAF alteration, usually V600E mutation (Nat Genet 2013;45:602)
- NTRK fusions (Acta Neuropathol Commun 2020;8:107)
- No CDKN2A / B homozygous deletion, MYB / MYBL1 rearrangement, IDH1 / IDH2 mutation or histone H3 K27 or G34 alterations (Neuro Oncol 2021;23:1231)
- Chromosomal alterations
- Chromosome 1p and 19q intact (Am J Surg Pathol 2014;38:1058)
- Brain tumor, right frontal lobe, resection:
- Integrated diagnosis: diffuse low grade glioma, MAPK pathway altered
- Histological diagnosis: diffuse astrocytoma, low grade
- Molecular information:
- IDH1 R132H: negative (immunohistochemistry; consistent with wild type)
- H3 G34R / V: negative (immunohistochemistry; consistent with wild type)
- CDKN2A: intact (fluorescence in situ hybridization)
- BRAF V600E: positive (immunohistochemistry; consistent with mutation)
- Comment: Sections demonstrate a diffusely infiltrating glioma with moderate cellularity. The tumor is composed of fibrillary astrocytic cells. No mitotic activity, microvascular proliferation or necrosis noted. No Rosenthal fibers or eosinophilic granular bodies are present. A CNS WHO grade has yet to be assigned.
- Diffuse astrocytoma, MYB or MYBL1 altered, CNS WHO grade 1:
- MYB or MYBL1 alterations
- Angiocentric glioma, CNS WHO grade 1:
- Angiocentric arrangements of tumor cells, including perivascular pseudorosettes, with EMA positivity
- Polymorphous low grade neuroepithelial tumor of the young, CNS WHO grade 1:
- Histologically variable with an oligodendroglioma-like component and widespread expression of CD34
- Pilocytic astrocytoma, CNS WHO grade 1:
- Well circumscribed, tumor cells with piloid processes, Rosenthal fibers, eosinophilic granular bodies
- Ganglioglioma, CNS WHO grade 1:
- Gangliocytic cells
- Dysembroplastic neuroepithelial tumor, CNS WHO grade 1:
- Glioneuronal elements and glial nodules (if complex DNET)
- Diffuse midline glioma, histone H3 K27 altered, CNS WHO grade 4:
- Presence of histone H3 K27 alteration, typically K27M
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted, CNS WHO grade 2:
- Presence of IDH mutation, chromosome 1p19q codeletion
- Astrocytoma, IDH mutant, CNS WHO grade 2:
- Presence of IDH mutation, ATRX loss
A nonenhancing tumor was discovered in the right frontal lobe of a 5 year old boy. Resection revealed fibrillary astrocytic tumor cells without mitotic activity, microvascular proliferation or necrosis. Tumor cells showed immunopositivity for Olig2 and GFAP. What molecular alteration would you expect?
- BRAF V600E
- Histone H3 G34R
- Histone H3 K27M
- IDH1 (R132H)
- EGFR
- MAP kinase
- TGF beta
- Wnt
- WHO 2021 definition: diffuse midline glioma, H3 K27-altered, is an infiltrative midline glioma with loss of H3 p.K28me3 (K27me3) and usually, either an H3 c.83A>T p.K28M (K27M) substitution in one of the histone H3 isoforms (H3.1, H3.2 or H3.3), aberrant overexpression of EZHIP or an EGFR mutation (CNS WHO grade 4)
- Predominant in children but also occurs in adults
- Arises in the midline; the brain stem, thalamus and spinal cord are the most common locations
- High grade features, such as mitotic activity, microvascular proliferation and necrosis, may be seen but are not necessary for the diagnosis
- Diffusely infiltrative of both adjacent and distant brain structures
- Poor prognosis with 2 year survival rate of < 10%
- Brain stem and pontine lesions previously termed brain stem glioma and diffuse intrinsic pontine glioma (DIPG)
- ICD-10: C71.9 - malignant neoplasm of brain, unspecified
- Official incidence data are not available; this is because, historically, infiltrative gliomas arising in the midline have not been distinguished from other infiltrative gliomas in large registries
- M = F
- Median age at diagnosis is 5 - 11 years, with tumors that arise in the pons occurring at a younger age (~7 years) than those that arise in the thalamus (~11 years) (Neuro Oncol 2014;16:iv1)
- Those harboring EGFR amplifications, containing mutations in isoform histone H3.3 or resulting from EZHIP overexpression, typically occur at 7 to 8 years of age, while those with K27M mutations in isoform histone H3.1 or H3.2 tend to occur in younger patients (~5 years) (Cancer Cell 2017;32:520)
- Most commonly located in the spinal cord, pons and thalamus
- Occasionally arises in the cerebellum
- No genetic susceptibility is known; however, rare cases of H3 K27-altered tumors have been seen in patients with genetic tumor syndromes such as NF1, Li-Fraumeni syndrome or mismatch repair deficiency
- Tumorigenesis is thought to be driven by disruption of polycomb repressive complex 2 (PRC2) mediated differentiation and self renewal (Cancer Cell 2019;35:140, Genes Dev 2014;28:1758)
- Common genetic and epigenetic characteristics of midline gliomas are suggestive of a distinct developmental origin (Nat Rev Cancer 2014;14:651, Nat Rev Cancer 2014;14:92)
- Nestin- / Olig2- expressing neural precursor-like cell has been suggested as the possible cell of origin in one study (Proc Natl Acad Sci USA 2011;108:4453)
- Another found the majority of tumor cells resembled an oligodendrocyte precursor cell (Science 2018;360:331)
- Patients typically present with evidence of cerebrospinal fluid obstruction or brain stem dysfunction, such as cranial nerve abnormalities, ataxia and long tract signs that develop over a short period of time (Front Oncol 2012;2:205)
- Tumors that arise in the thalamus are often associated with motor weakness and gait disturbance (Neuro Oncol 2011;13:680)
- Typically diagnosed by imaging (MRI) and stereotactic biopsy; resection is often not possible given the involvement of critical brain stem structures
- Grade 4 of 4
- Presence of H3 K27-alteration in an infiltrative glioma arising in the midline is sufficient for a grade 4 designation, even in the absence of necrosis or microvascular proliferation (Acta Neuropathol 2014;128:573)
- On MRI, typically T1 hypointense and T2 hypertintense
- Contrast enhancement (typically < 25% of tumor volume), hemorrhage or necrosis may be seen (J Neurooncol 2011;105:119)
- Tumors arising in the pons typically present as large expansile masses that occupy > 67% of the pons (Front Oncol 2012;2:205)
- Exophytic component may be present
- Infiltration into adjacent structures, such as the cerebellar peduncles, midbrain or medulla is frequent
Contributed by Rawia Mubarak Mohamed, M.D. and Najla Saleh Ben Gashir, M.D. (Case #477)
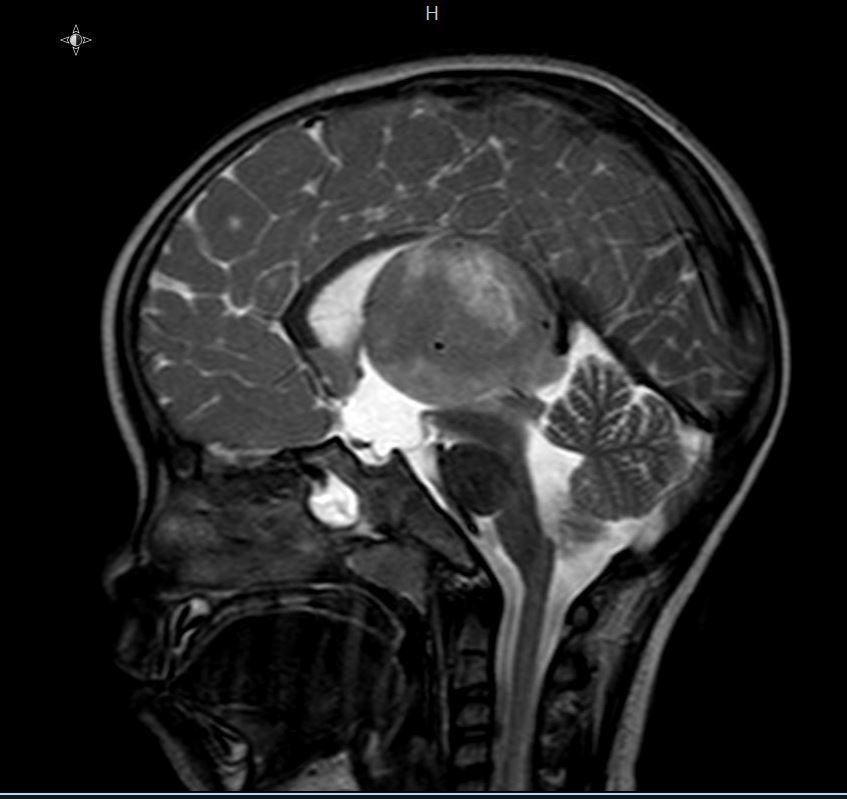
Tumor in thalamic region
Images hosted on other servers:
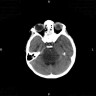
CT - axial noncontrast: pontine tumor
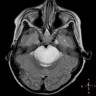
MRI - axial T2: pontine tumor
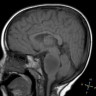
MRI - sagittal T1: pontine tumor
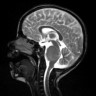
MRI - sagittal T2: pontine tumor
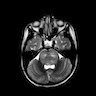
MRI - axial T2: pontine tumor
- Prognosis is almost universally poor
- Presence of an H3 K27M alteration confers a worse prognosis over wild type diffuse midline gliomas (Acta Neuropathol 2014;128:573, Acta Neuropathol 2012;124:439)
- 5 year old girl with headache and vomiting for 1 week (Case #477)
- 30 year old woman and 69 year old man with mosaic H3.3 K27M protein expression (Acta Neuropathol 2017;134:961)
- 45 year old man with an H3.1 K27M mutant diffuse cerebellar astrocytoma (Brain Tumor Pathol 2018;35:29)
- 51 year old woman with a diffuse midline glioma with primitive neuroectodermal tumor-like appearance and neuropil-like islands (Neuropathology 2018;38:165)
- Surgical resection is often highly limited, due to the involvement of critical brain structures (PDQ Cancer Information Summaries: Childhood Astrocytomas Treatment [Accessed 25 February 2019])
- Radiation or chemotherapy is standard
- Infiltrative nature causes enlargement and distortion of involved anatomical structures
- Focal discoloration and softening, indicating hemorrhage or necrosis, may be present (Neurol Med Chir (Tokyo) 2003;43:375)
- Tumor cells typically have an astrocytic morphology and may be small and monomorphic to (occasionally) large and pleomorphic (Neurol Med Chir (Tokyo) 2003;43:375)
- Show an infiltrative growth pattern with tumor cells diffusely growing among native neurons and invading into adjacent structures
- Occasionally, an oligodendroglial-like pattern with halos may be seen
- Some cases will not show mitoses, necrosis or microvascular proliferation consistent with a WHO grade 2 histologic appearance; however, in the presence of H3 K27-alteration, WHO grade 4 is warranted given the aggressive nature of these tumors (Acta Neuropathol 2014;128:573)
Contributed by John DeWitt, M.D., Ph.D.
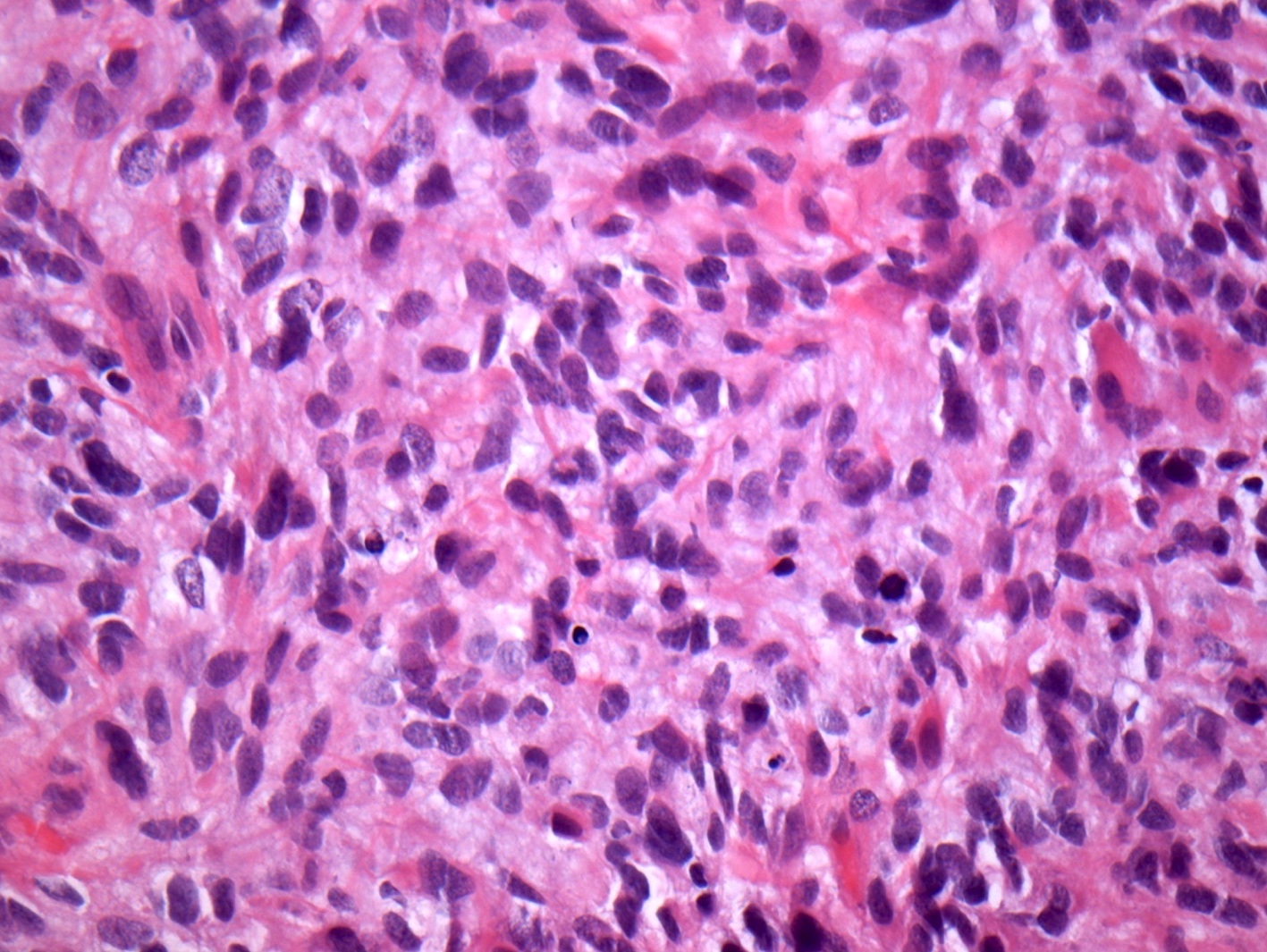
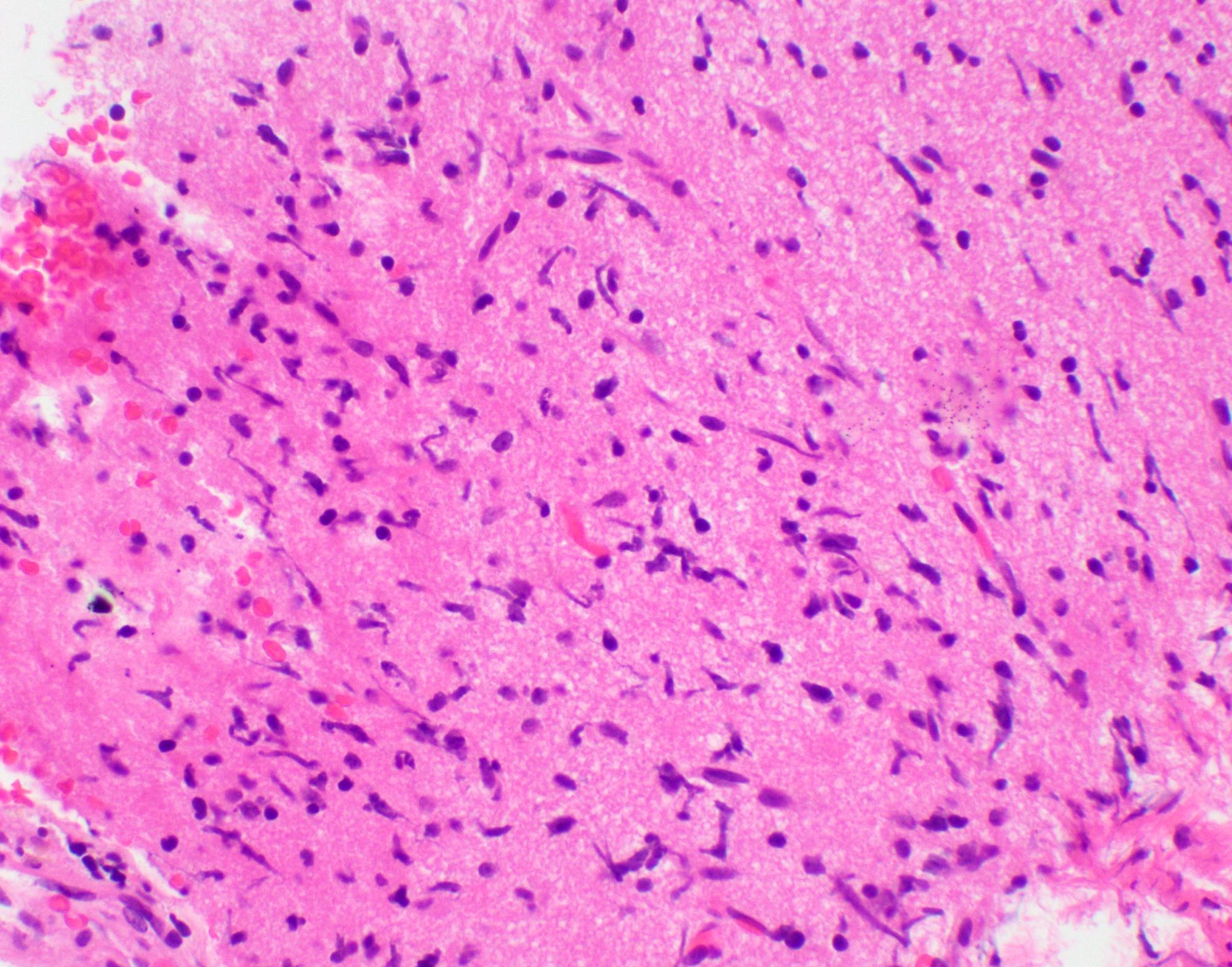
Diffusely infiltrating astrocytic tumor cells
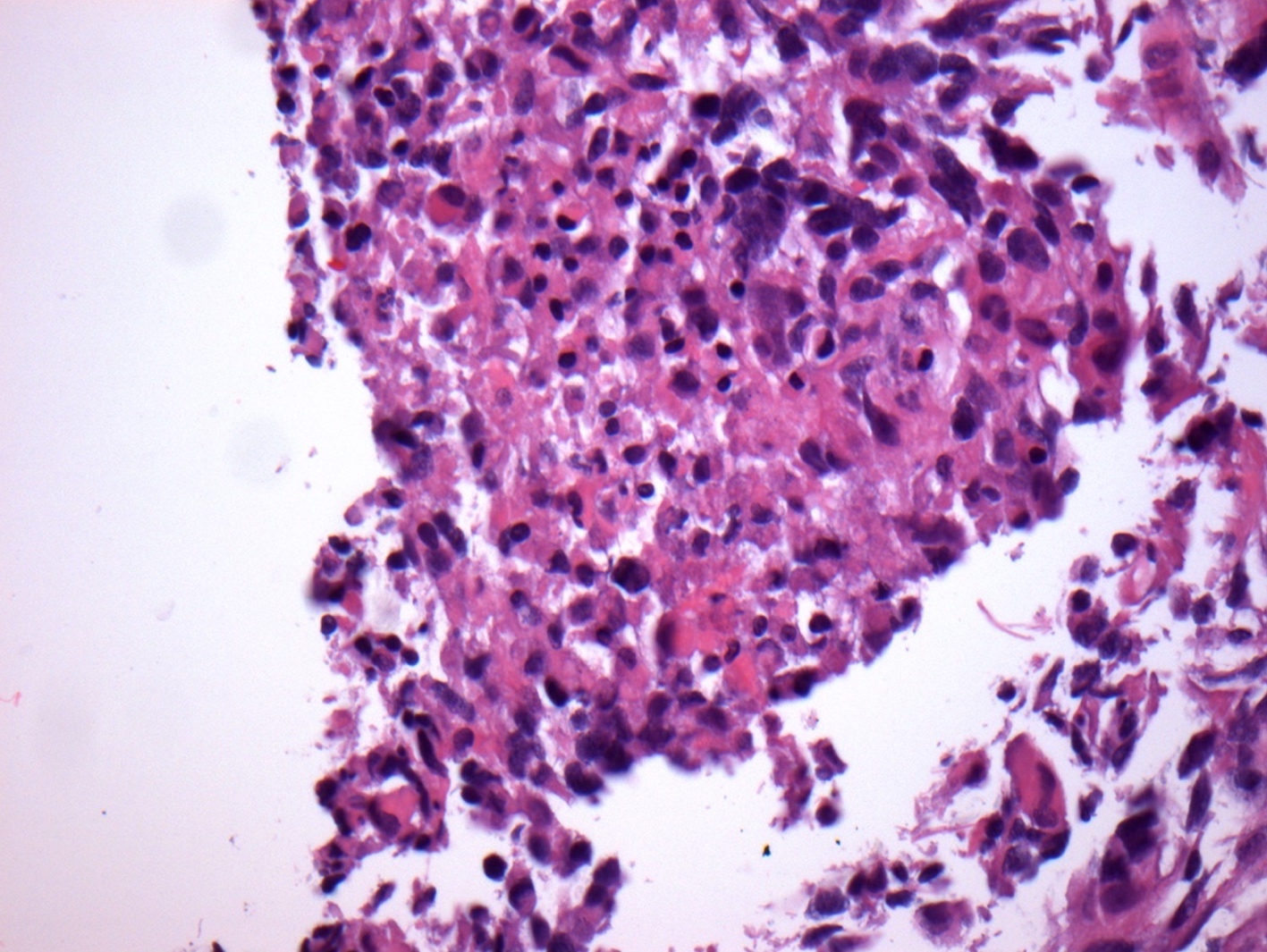
Necrosis
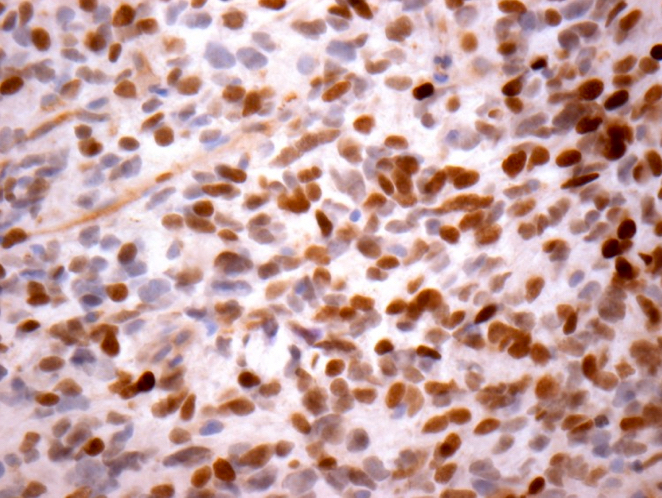
H3 K27M
R132H-IDH1
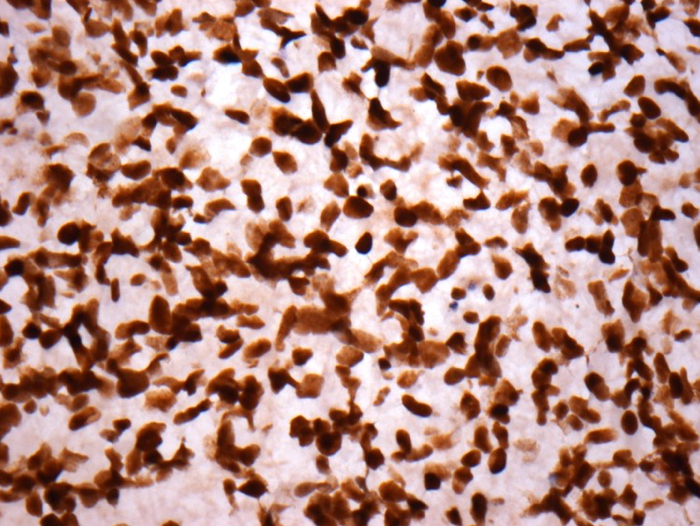
ATRX
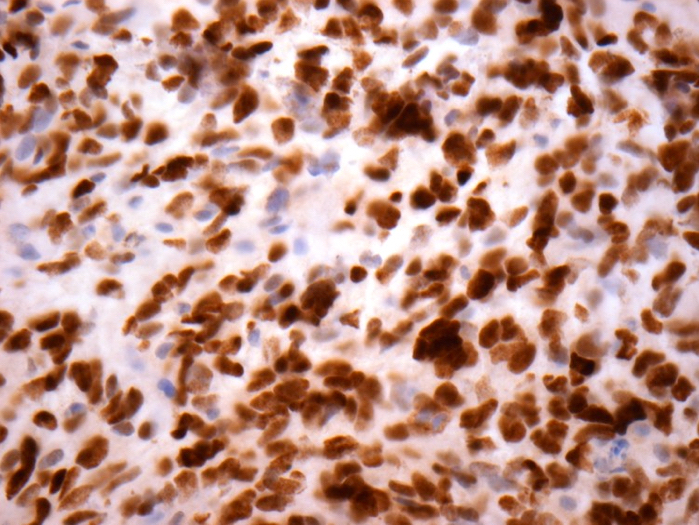
p53
Contributed by Chunyu Cai, M.D., Ph.D.
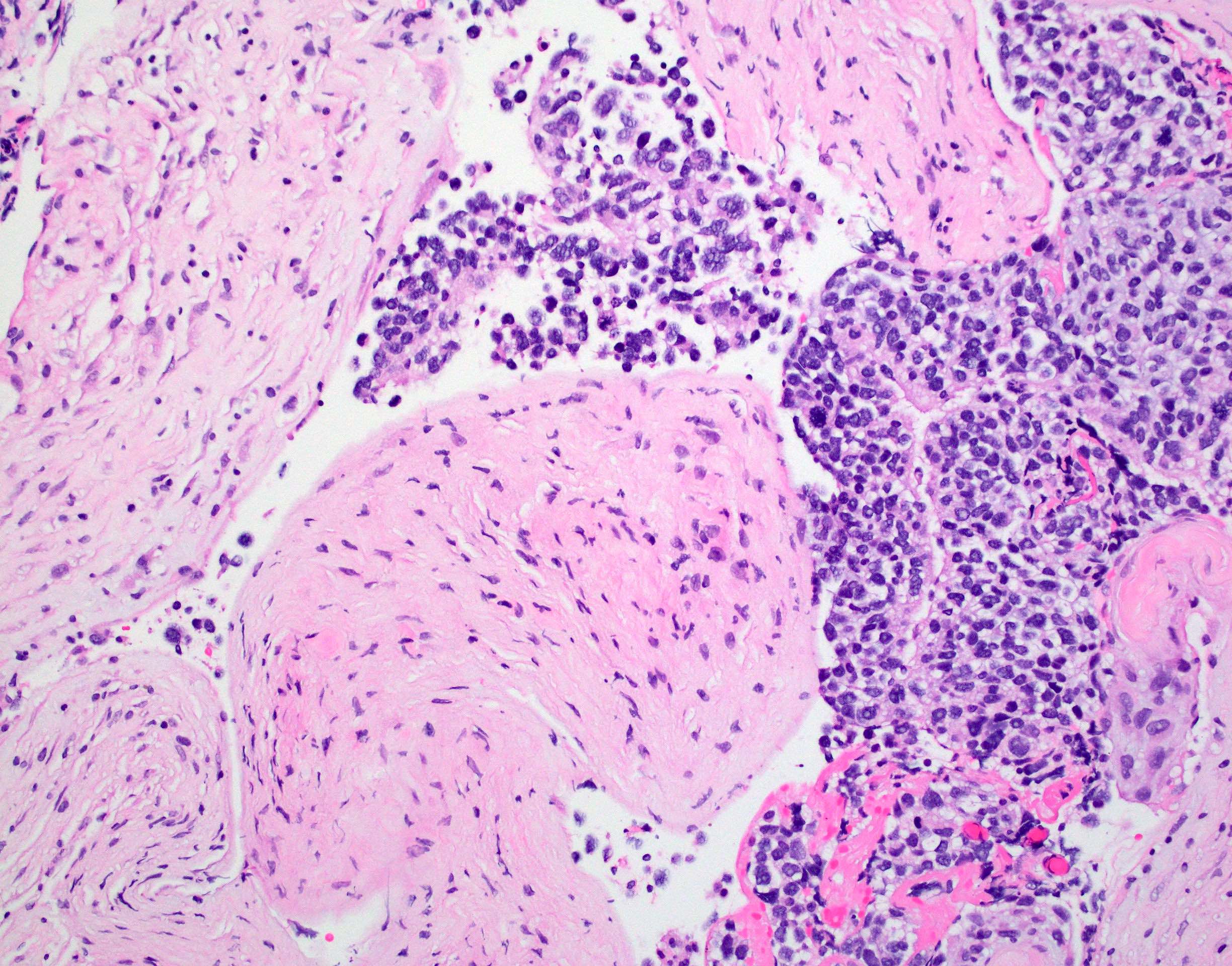
Clusters of tumor cell seeding
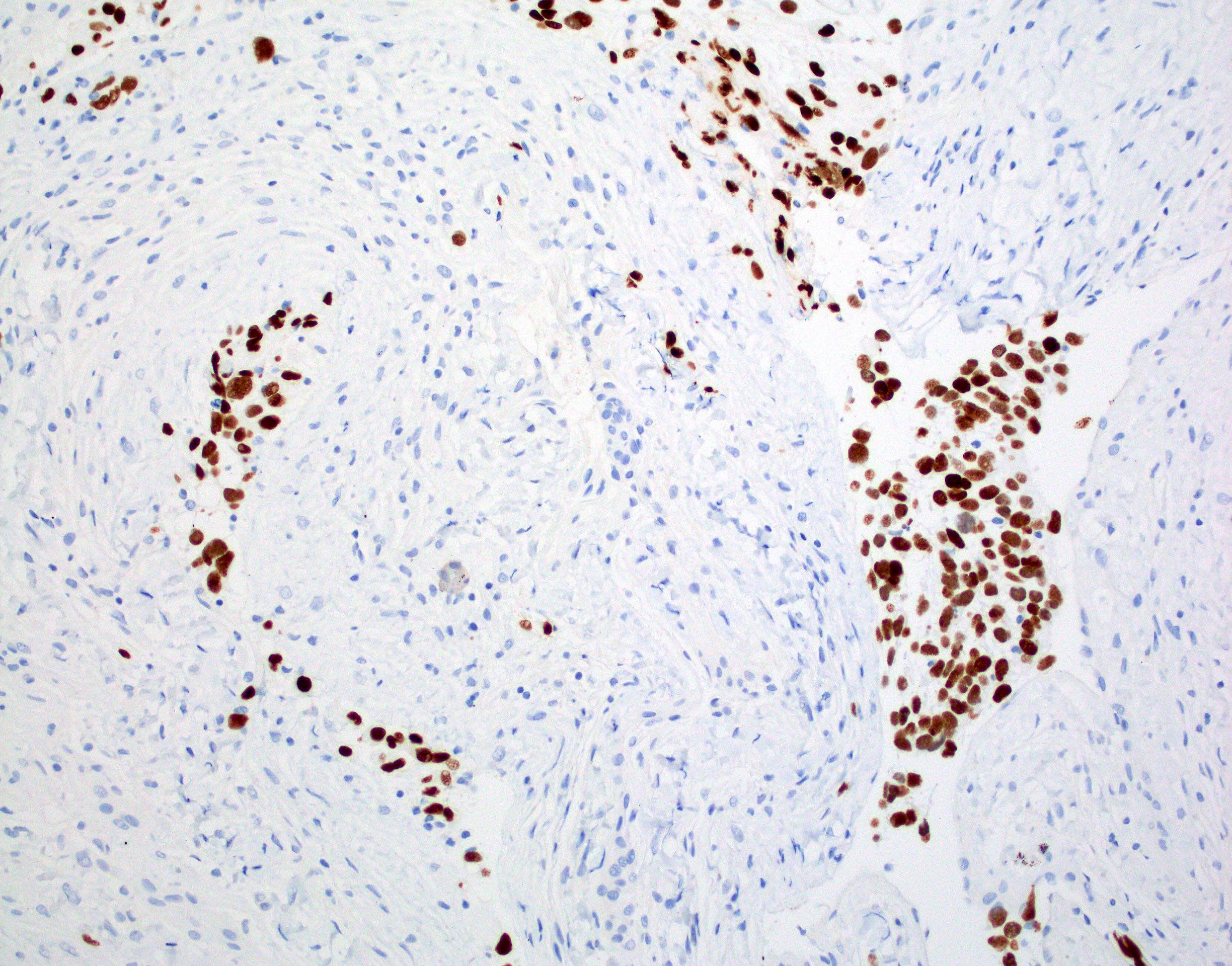
H3 K27M
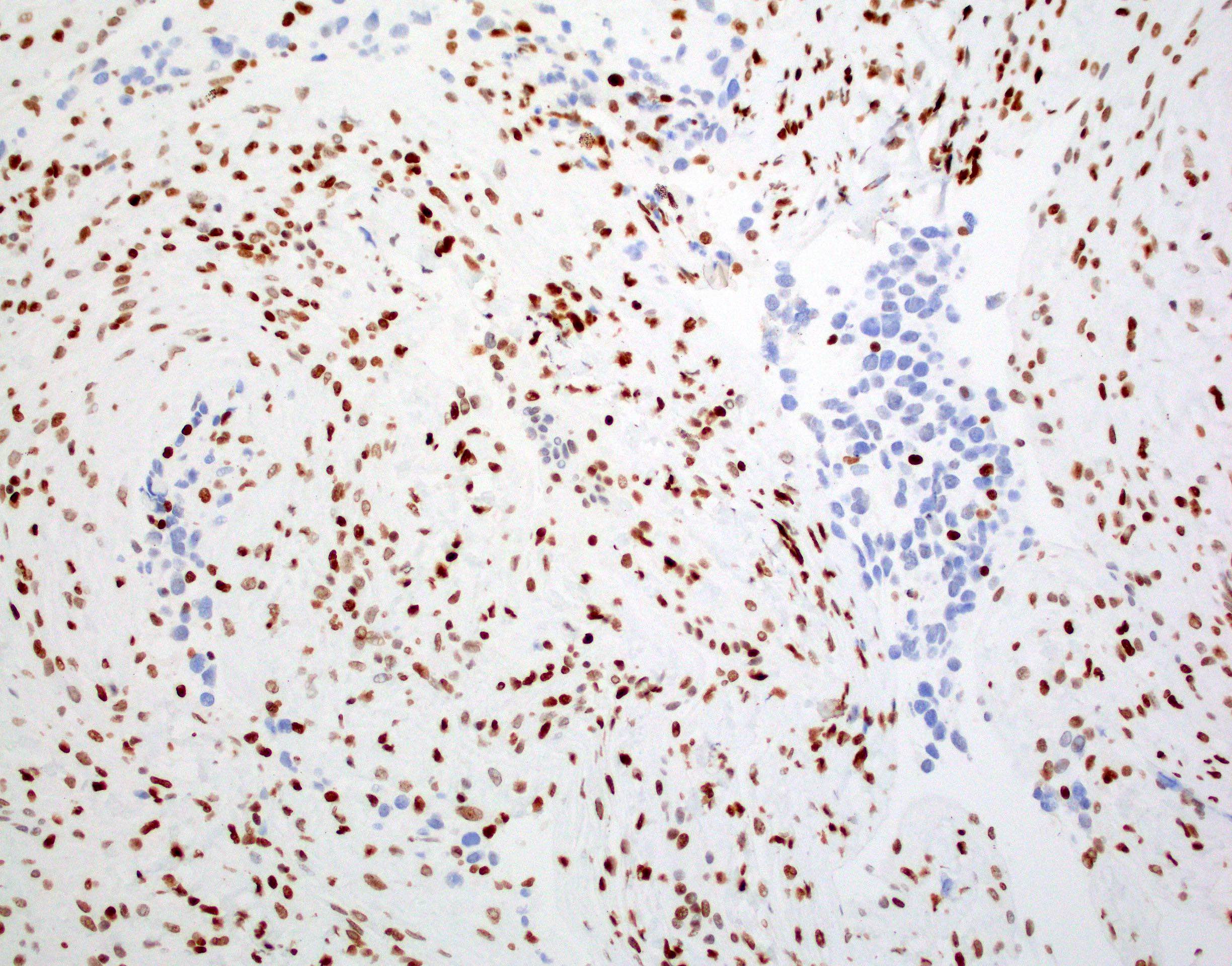
H3K27Me3
Contributed by Rawia Mubarak Mohamed, M.D. and Najla Saleh Ben Gashir, M.D. (Case #477)
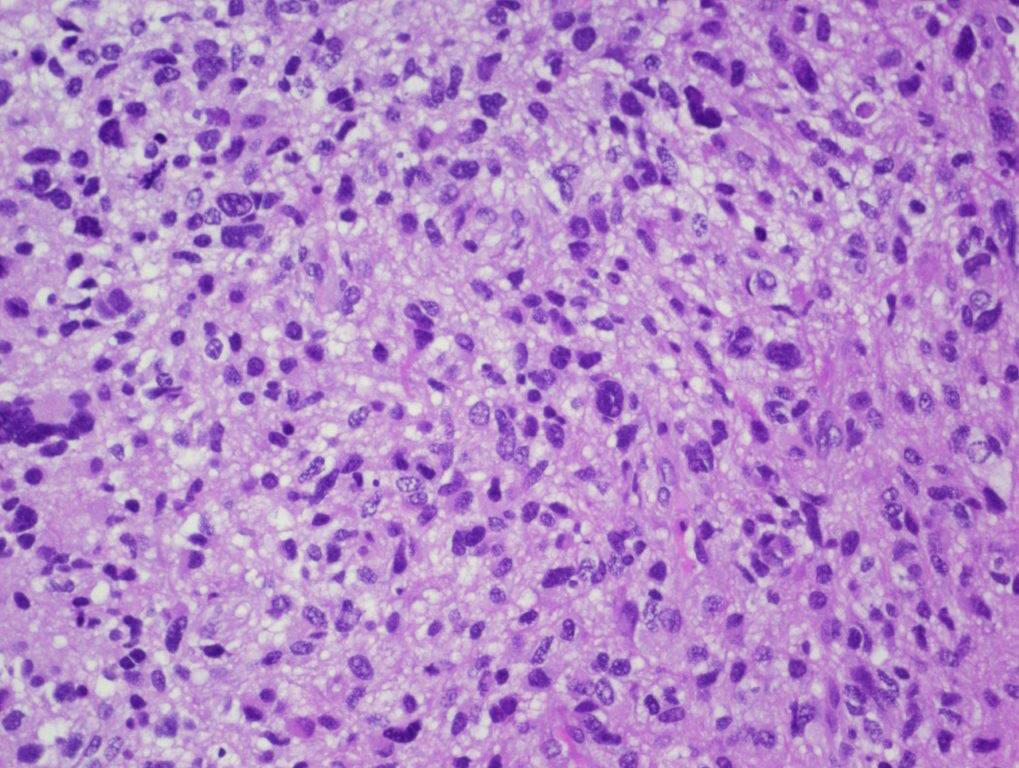
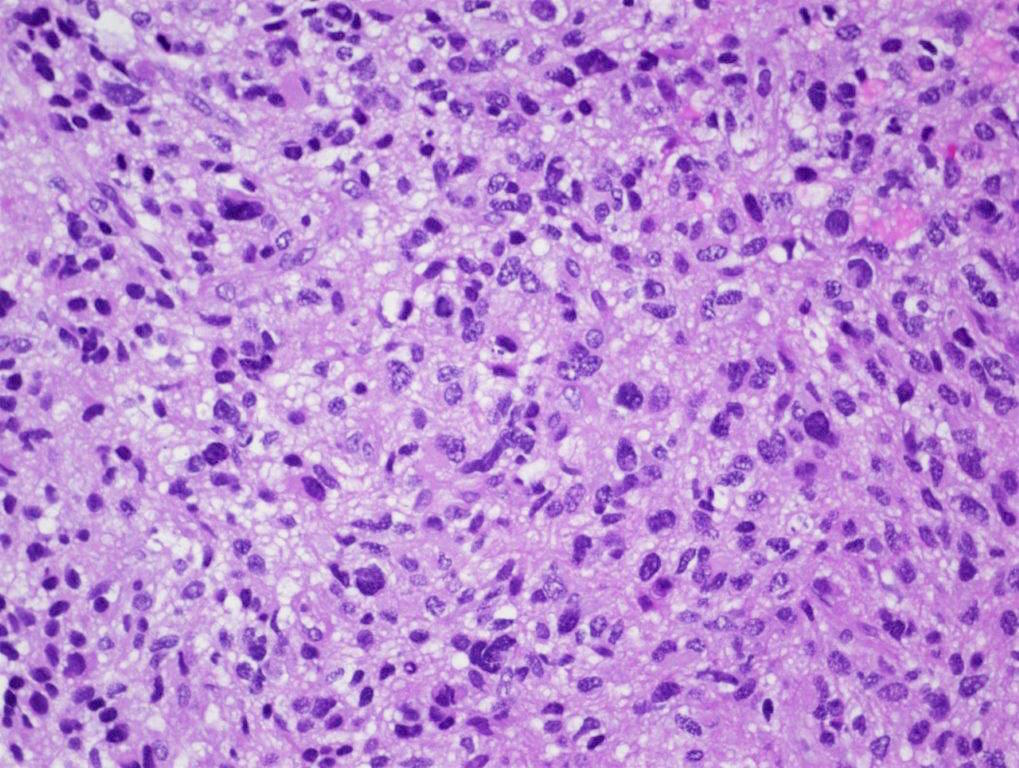
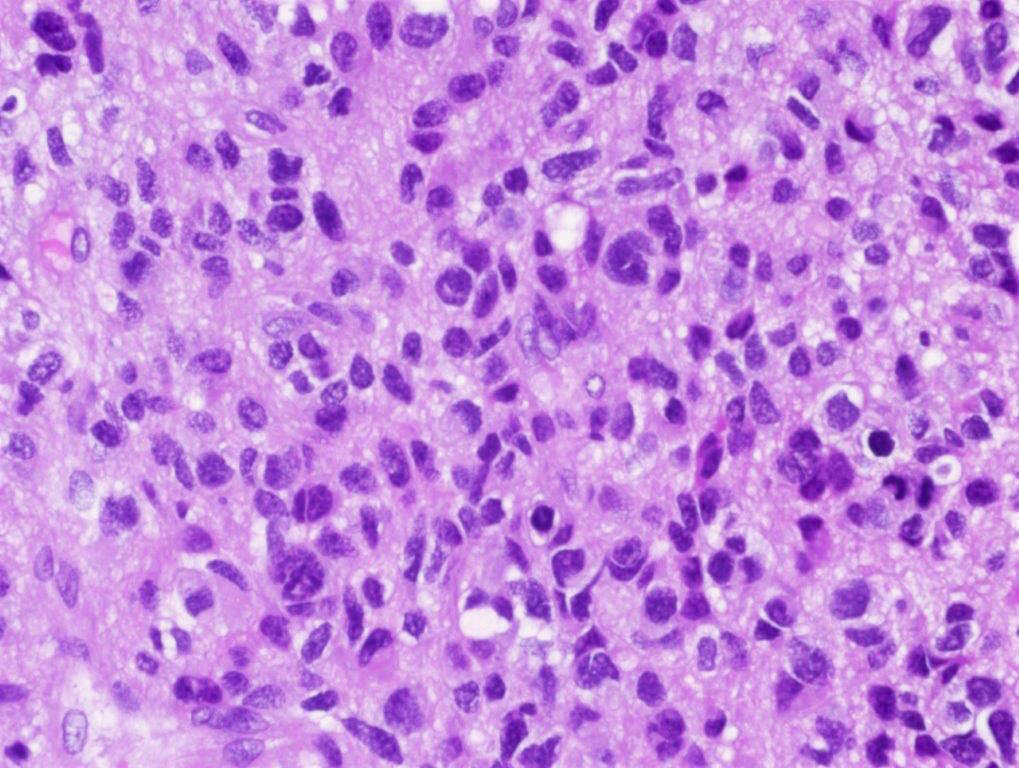

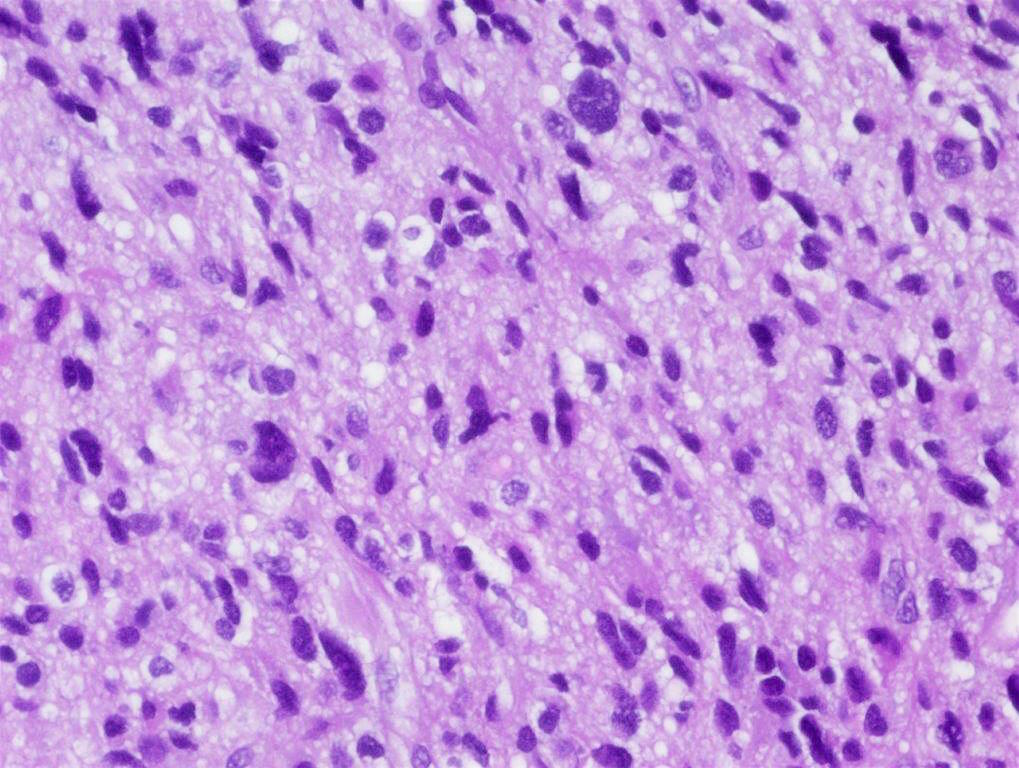
High grade infiltrating glial cells
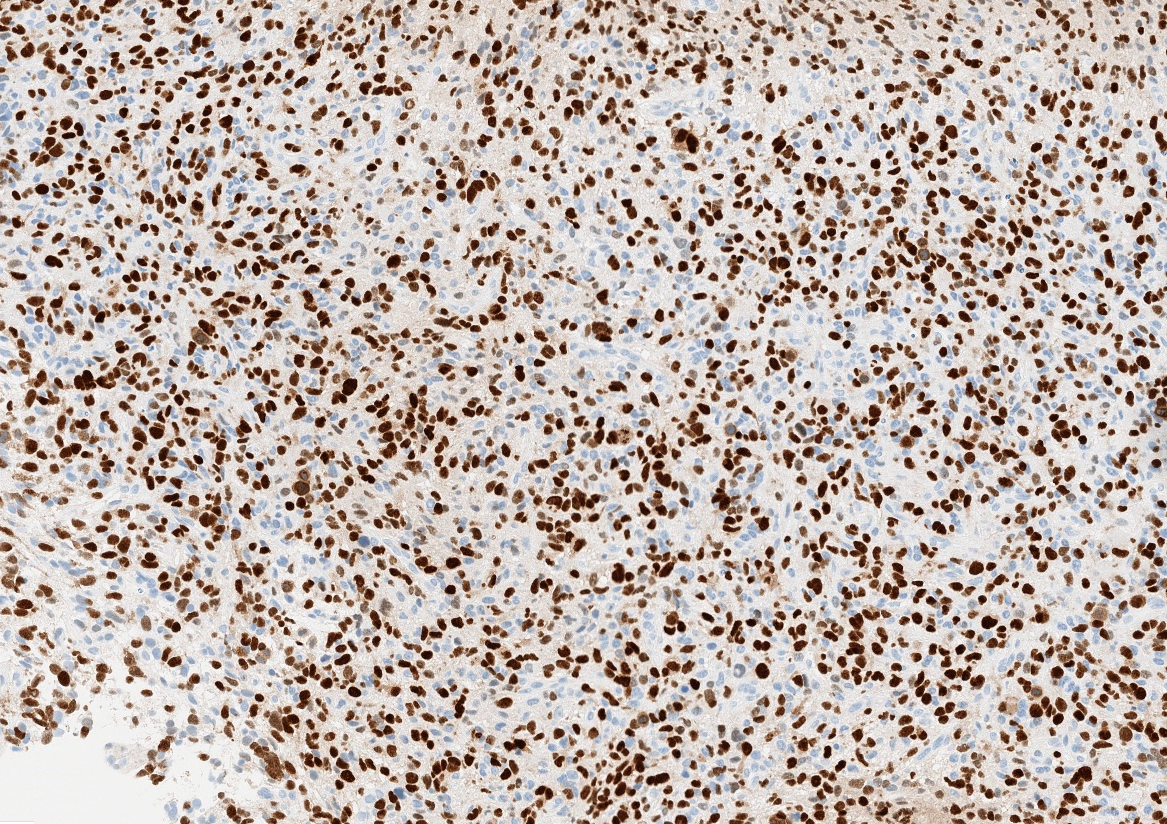
Olig2
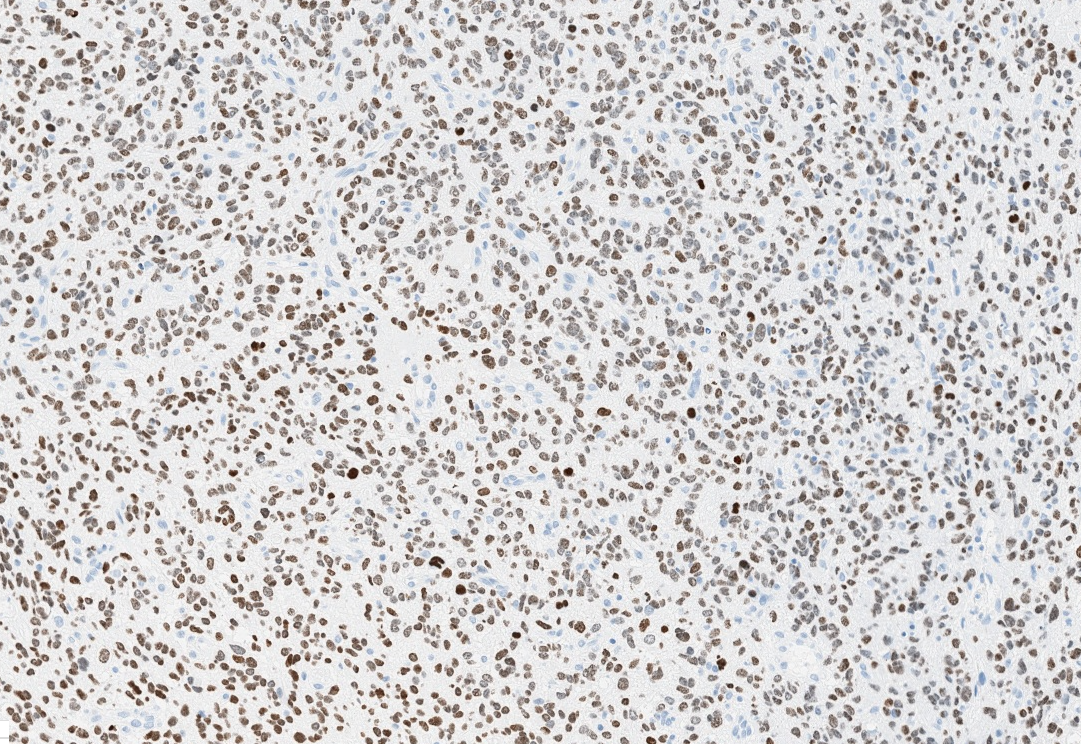
H3 K27M
- H3F3A K27M (specific immunostain)
- S100
- ATRX nuclear loss varies by tumor location, very low in pontine gliomas (1/13) but high in thalamus (9/12) or spinal cord gliomas (4/7) (Brain Pathol 2016;26:569)
- GFAP (variably positive)
- Olig2, MAP2
- p53 (strong positivity in ~50% of cases)
- Reference: Brain Pathol 2016;26:569
- R132H-IDH1
- Keratins (although cocktails may show cross reactivity with GFAP)
- H3K27me3: H3 K27-altered tumors show loss of H3K27me3 staining, a finding that by itself is not specific, however, may be helpful in identifying diffuse midline gliomas with H3 p.K28I (K27I) mutation or EZHIP overexpression, which are negative for H3K27M IHC but show loss of nuclear H3K27me3 expression
- Reference: Brain Pathol 2016;26:569
- Various molecular techniques including sequencing, expression arrays and methylation patterns are consistent with 4 distinct subtypes of diffuse midline gliomas that are defined by oncohistone alterations:
- H3.3 c.83A>T p.K28M (K27M) mutant
- H3.1 or 3.2 c.83A>T p.K28M (K27M) mutant
- H3 wild type with aberrant overexpression of EZHIP
- EGFR mutation or amplification
- Additional mutations seen in diffuse midline glioma, H3 K27-altered (Nat Genet 2014;46:451, Nat Genet 2014;46:457, Nat Genet 2014;46:444, J Neurosurg 1999;90:833):
- TP53 (~50%)
- PDGFRA amplification (~30%)
- CDK4 / 6 or CCND1 - 3 amplification (~20%)
- ACVR1 mutation (~20%)
- PPM1D mutation (~15%)
- MYC / PVT1 amplification (~15%)
- ATRX mutation (~15%)
- CDKN2A / B homozygous deletion (< 5%)
- Brain, pons, biopsy:
- Integrated diagnosis: diffuse midline glioma, H3 K27-altered, WHO grade 4 of 4 (see comment)
- Histological diagnosis: astrocytoma with proliferative activity
- CNS WHO grade 4 of 4
- Molecular information:
- IDH: negative (R132H immunohistochemistry; consistent with wild type)
- ATRX: nuclear expression retained (immunohistochemistry; consistent with wild type)
- p53: negative (immunohistochemistry; consistent with wild type)
- H3K27M: positive (immunohistochemistry; consistent with mutant)
- Comment: The specimen consists of core biopsy specimens of white matter with moderately atypical infiltrating astrocytic tumor cells. There is no evidence of vascular proliferation or necrosis. Scattered mitoses are seen. Although the histologic grade is that of a grade 3 astrocytoma, the positivity for H3K27M is consistent with a diagnosis of diffuse midline glioma, H3 K27-altered, which are considered grade 4 lesions due to their historically aggressive clinical behavior
- Integrated diagnosis: diffuse midline glioma, H3 K27-altered, WHO grade 4 of 4 (see comment)
- Astrocytoma, IDH mutant, CNS WHO grade 2:
- Grade 2 histology with IDH mutation present
- Astrocytoma, IDH mutant, CNS WHO grade 3:
- Grade 3 histology with IDH mutation present
- Astrocytoma, IDH mutant, CNS WHO grade 4:
- Grade 4 histology with IDH mutation present or lower grade histology with IDH mutation and homozygous deletion of CDKN2A / CDKN2B
- Glioblastoma, IDH wild type:
- Astrocytoma lacking IDH or H3 K27M mutations with either grade 4 histology or any of the following molecular alterations: EGFR amplification, TERT promoter mutation, gain of chromosome 7 / loss of chromosome 10
- Tumors do not show loss of H3 K27 trimethylation
- Posterior fossa (PFA) ependymoma (Childs Nerv Syst 2017;33:1047, Nat Commun 2019;10:2146):
- Will show loss of H3K27Me3 (trimethyl mark) as in diffuse midline glioma, H3 K27-altered and rare cases also contain H3 K27 mutations
- Found in the posterior fossa where diffuse midline glioma, H3 K27-altered may also occur
- Differentiated by ependymal differentiation (round to oval glial tumor cells with perivascular pseudorosettes) as opposed to astrocytic differentiation of diffuse midline glioma, H3 K27-altered
- Molecular studies including sequencing and methylation profiling could be considered in histologically ambiguous cases

This tumor was found arising in the pons of a 7 year old boy. The presence of which molecular alteration would warrant a WHO grade 4 designation?
- 1p / 19q codeletion
- H3 K27M mutation
- IDH1 mutation
- KIA1549-BRAF fusion
- TP53 mutation
- Cerebellar hemisphere
- Frontal lobe
- Lateral ventricle
- Temporal lobe
- Thalamus
- Despite its name, it is typically not found in the midline
- It may lack high grade histologic features but is still considered grade 4
- The prognosis varies based on the histologic features
- This diagnosis includes midline gliomas that are diffusely infiltrating but have not been tested for the H3 K27M mutation
Comment Here
Reference: Diffuse midline glioma, H3 K27-altered
- Cortex based glioneuronal neoplasm that is often located in the mesial temporal lobe of adolescents and young adults and associated with medically refractory epilepsy, usually with activating mutations of FGFR1, CNS WHO grade 1 (Neurosurgery 1988;23:545)
- Presents clinically with intractable seizures, usually in children and young adults (Neurosurgery 1988;23:545, Brain Pathol 2012;22:350)
- Radiographically is sharply demarcated, nodular, cortical lesion(s) without edema or enhancement (AJNR Am J Neuroradiol 2003;24:829, Neuroradiology 2009;51:433, J Neuropathol Exp Neurol 2011;70:859)
- Composed of astrocytes, oligodendrocytes (or oligodendrocyte-like cells) and neurons with neurons often appearing to float in a myxoid matrix between columns of oligodendroglial cells (Neurosurgery 1988;23:545, Brain Pathol 2012;22:350)
- Activating genetic alterations in FGFR1 (Acta Neuropathol 2016;131:847)
- CNS WHO grade 1 (J Neuropathol Exp Neurol 2011;70:859)
- Dysembryoplastic neuroepithelial tumor
- Simple dysembryoplastic neuroepithelial tumor
- Complex dysembryoplastic neuroepithelial tumor
- 1.2:1 = M:F (N Engl J Med 2017;377:1648)
- 5.9% of epilepsy surgery cases (N Engl J Med 2017;377:1648)
- Cerebral cortex
- Most common sites: temporal lobe, especially medial (67%), frontal lobe (16%), other cortex (16%) (J Neuropathol Exp Neurol 2011;70:859)
- Activating alteration in FGFR1 (receptor tyrosine kinase) > activation of MAPK pathway > dysregulated cell growth, proliferation and differentiation (Acta Neuropathol 2016;131:847)
- Most cases are sporadic (Acta Neuropathol 2016;131:833)
- Germline mutations in MAPK pathway genes, including FGFR1, NF1 and PTPN11 (Acta Neuropathol 2016;131:847, Epilepsy Res 2013;105:384, Am J Med Genet A 2017;173:1061)
Images hosted on other servers:

Relative distribution of LEATs
Proposed modification of LEAT classification
Nodular appearing neoplasm
- Intractable seizures
- Absence of focal neurologic deficits (Neurosurgery 1988;23:545)
- Cortical glioneuronal tumor with presence of specific glioneuronal component
- FGFR1 alteration or DNA methylation profile can help in challenging cases
- Preferable to make diagnosis in context of early onset focal epilepsy
- Sharply demarcated, nodular, cortical lesion without edema or enhancement (AJNR Am J Neuroradiol 2003;24:829)
Images hosted on other servers:
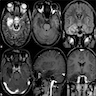
Representative imaging features in adolescent
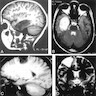
Typical imaging features

10 year old girl with seizure
- Benign lesion with low rate of recurrence after resection (J Neuroradiol 2001;28:230)
- Rare case reports of malignant transformation (Ann Diagn Pathol 2003;7:240, J Neurooncol 2009;94:283, Radiol Case Rep 2022;17:939)
- 18 year old woman with left parietal mass (Radiol Case Rep 2022;17:939)
- 26 year old woman with superficial right frontal mass (J Radiol Case Rep 2013;7:7)
- 27 year old man with right temporal mass (BMJ Case Rep 2013;2013:bcr2013010469)
- Surgical resection (Childs Nerv Syst 2015;31:847)
- Radiation or chemotherapy is generally not applicable
Images hosted on other servers:
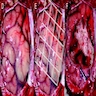
Surgical resection of epileptogenic tumor
- Usually poorly demarcated (Epileptic Disord 2002;4:99)
- Located predominantly in gray matter and subcortical white matter
- May contain solid, mucoid or cystic components
Images hosted on other servers:
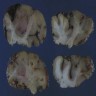
Frontobasal surgical specimen
- Growth pattern
- Intracortical and multinodular (Neurosurgery 1988;23:545)
- Pathognomonic appearance
- Bundles of axons lined by small oligodendroglia-like cells form columns oriented perpendicularly to the cortical surface with intervening cytologically normal neurons floating in a myxoid matrix (Neurosurgery 1988;23:545)
- Absence of the following:
- Dysplastic ganglion-like cells
- High cellularity
- Necrosis
- Perivascular lymphoid infiltrates
- Eosinophilic granular bodies
- Simple form
- Pathognomonic component alone
- Complex form
- Pathognomonic component along with glial nodules, resembling other glioma types (J Neurooncol 1999;41:267)
Contributed by P.J. Cimino, M.D., Ph.D. and Chris Dampier, M.D.
Nodular growth pattern
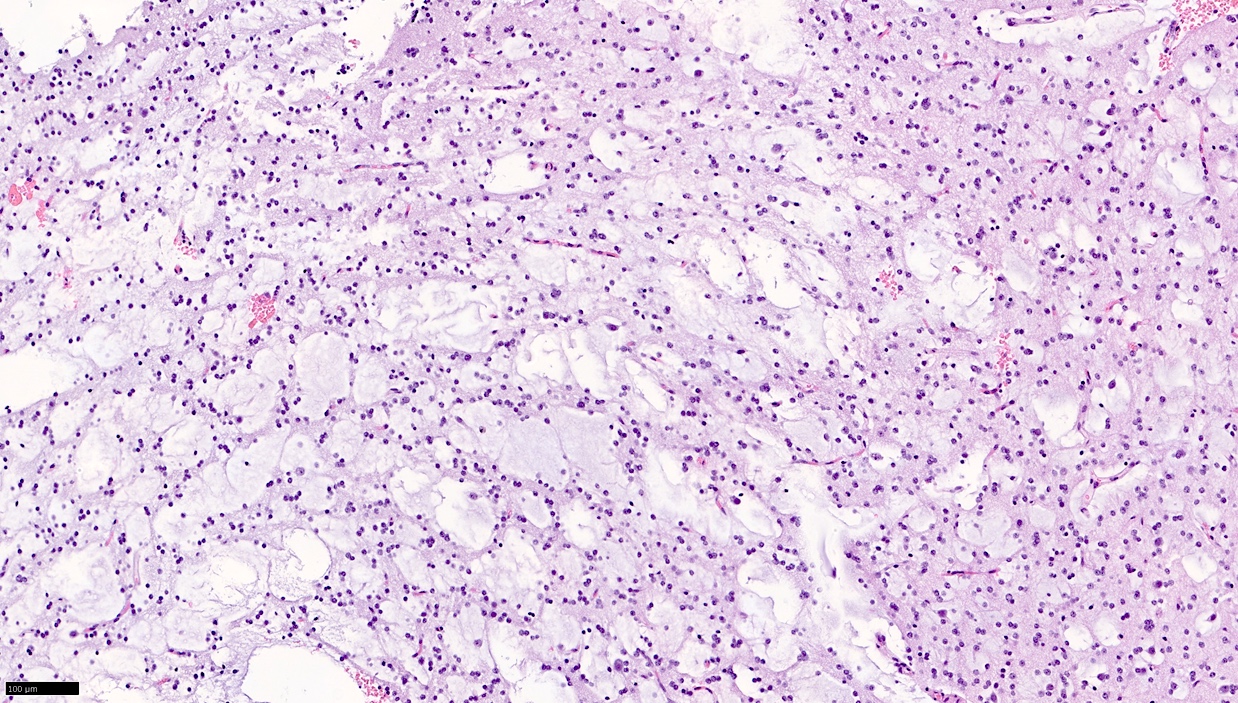
Specific glioneuronal (pathognomonic) component
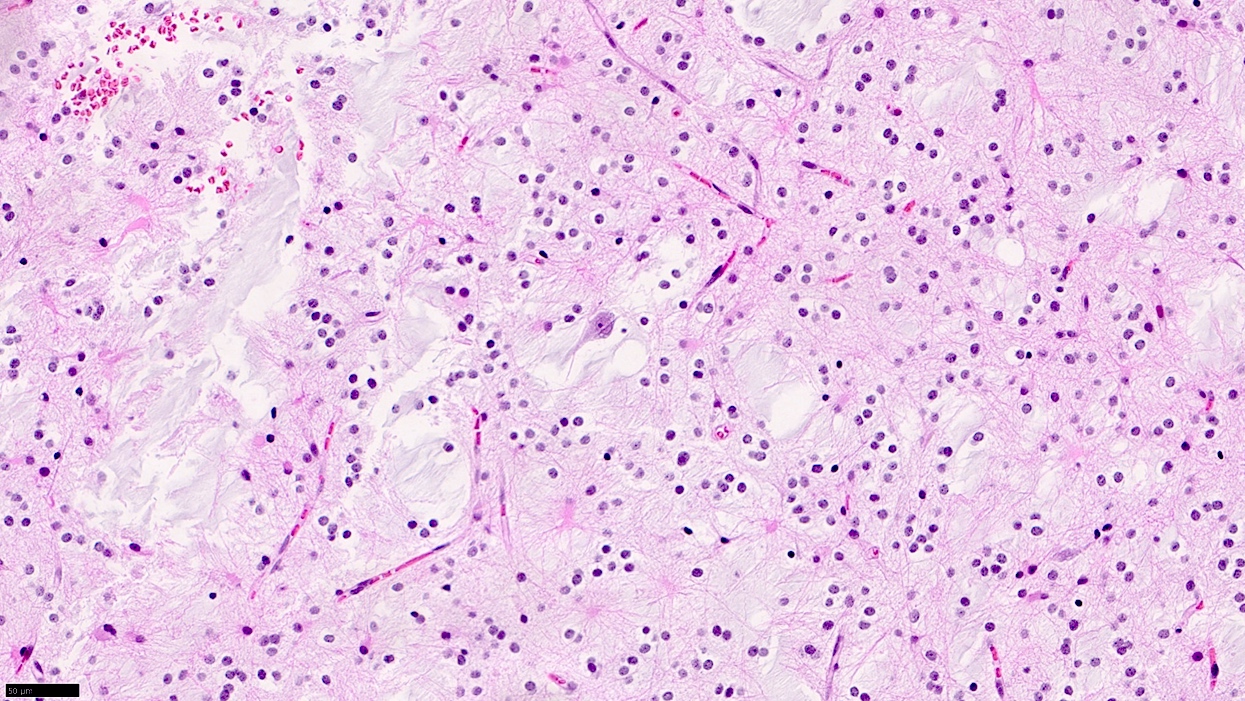
Floating neuron
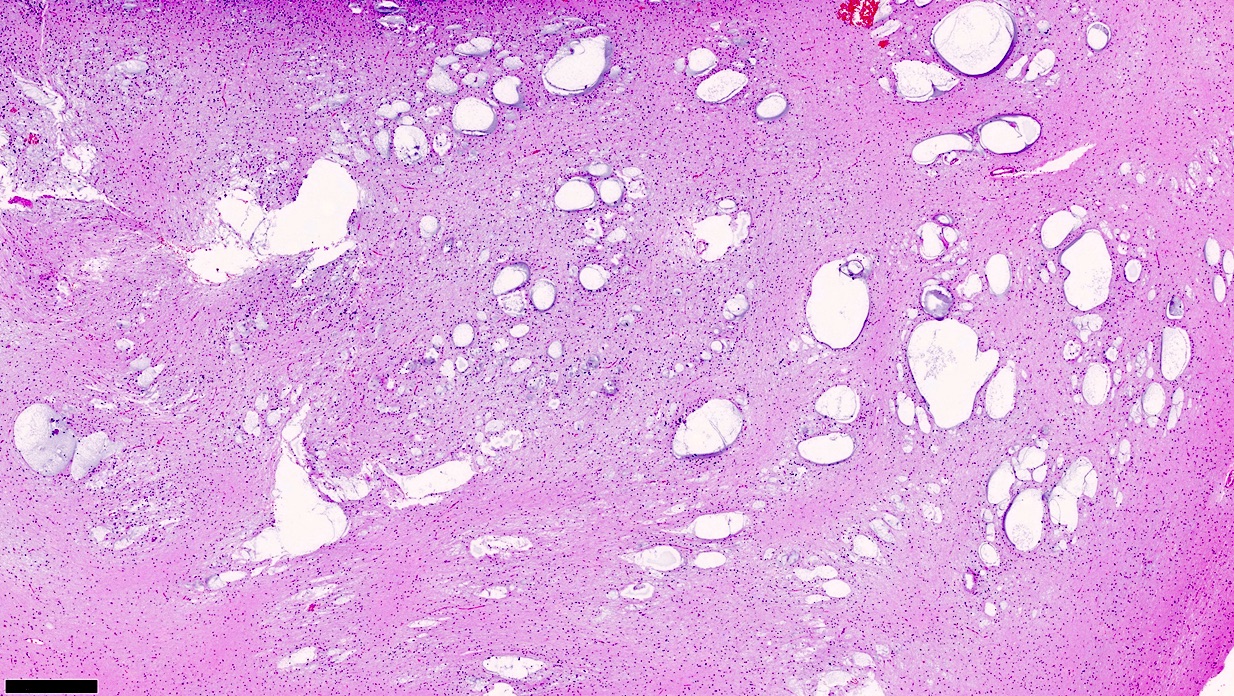
Cystic component
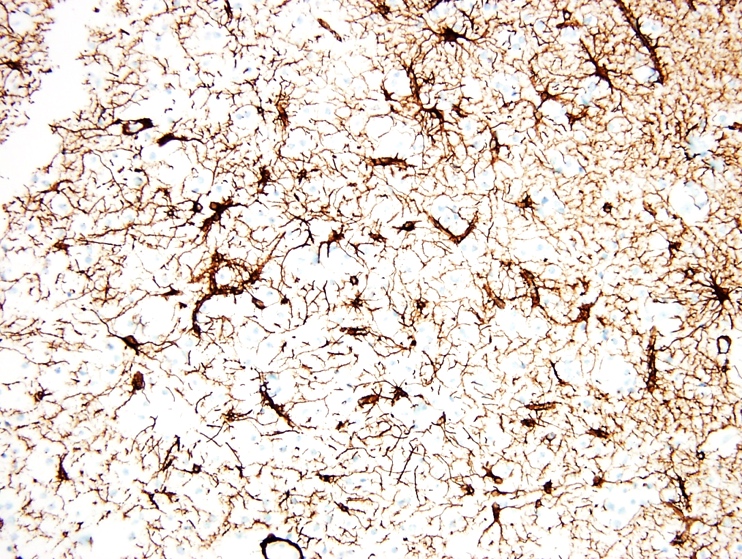
GFAP
IDH1 p.R132H
- Smear preparation (alcohol fixed, H&E stained) (Acta Cytol 2022;66:142)
- Uniform round cells
- Mucinous and fibrillary background
Images hosted on other servers:
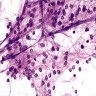
Smear preparations
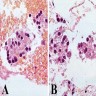
Partially arranged as columnar structures
Fine neuritic processes
- S100 (in the oligodendroglial-like cells) (J Pathol Transl Med 2015;49:438)
- Olig2 (in the oligodendroglial-like cells) (Neuropathology 2013;33:246)
- PDGFRA (in the oligodendroglial-like cells)
- NeuN (in the floating neurons) (J Pathol Transl Med 2015;49:438)
- Alcian blue (extracellular)
- GFAP (in the oligodendroglial-like cells) (J Pathol Transl Med 2015;49:438)
- MAP2 (in the oligodendroglial-like cells) (Acta Neuropathol 2004;108:89)
- CD34 (76.5% of cases are negative) (World Neurosurg 2019;121:e761)
- BRAF p.V600E
- IDH1 p.R132H (Acta Neuropathol 2011;121:241)
- Activating alterations in FGFR1 (Acta Neuropathol 2016;131:847)
- Internal tandem duplication
- Missense mutations
- FGFR1::TACC1 fusion (rare)
- Complete duplication of FGFR1 (rare)
- PTPN11 alteration (rare) (Am J Med Genet A 2017;173:1061)
- NF1 alteration (rare) (Epilepsy Res 2013;105:384)
- Chromosomal polysomies (gains of chromosome 5, chromosome 6, chromosome 7; loss of chromosome 22) unusual but reported (Neuropathol Appl Neurobiol 2015;41:743, Acta Neuropathol 2016;131:847)
Contributed by P.J. Cimino, M.D., Ph.D. and Chris Dampier, M.D.

SCNA plot
Dysembryoplastic neuroepithelial tumor (DNET)
- Brain, temporal lobe, biopsy:
- Dysembryoplastic neuroepithelial tumor, CNS WHO grade 1
- Positive for FGFR1 alteration
- Neoplastic
- Oligodendroglioma:
- IDH1/2 mutation
- Codeletion of whole chromosome arms 1p and 19q
- Ganglioglioma:
- Dysplastic ganglion-like cells
- Perivascular lymphocytic infiltrate
- Eosinophilic granular bodies
- BRAF p.V600E alteration (Acta Neuropathol 2011;121:397)
- Myxoid glioneuronal tumor:
- Located primarily in the septum pellucidum
- PDGFRA p.K385 alteration (Brain Pathol 2020;30:479)
- Multinodular and vacuolating neuronal tumor:
- Predominant neuronal component (Brain Pathol 2013;23:515)
- MAP2K1 or BRAF alteration
- Polymorphous low grade neuroepithelial tumor of the young:
- Diffuse growth pattern (Acta Neuropathol 2017;133:417)
- Extensive CD34 immunopositivity
- BRAF p.V600E or FGFR2/3 fusion
- Diffuse astrocytoma, MYB or MYBL1 altered:
- Monomorphic glial neoplasm (Acta Neuropathol 2020;139:193)
- MYB or MYBL1 alteration
- Angiocentric glioma:
- Perivascular orientation of tapered cells
- MYB::QKI fusion or other MYB alteration (Nat Genet 2016;48:273)
- Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters:
- Specific methylation profile (Neuropathol Appl Neurobiol 2020;46:422)
- Monosomy chromosome 14
- Oligodendroglioma:
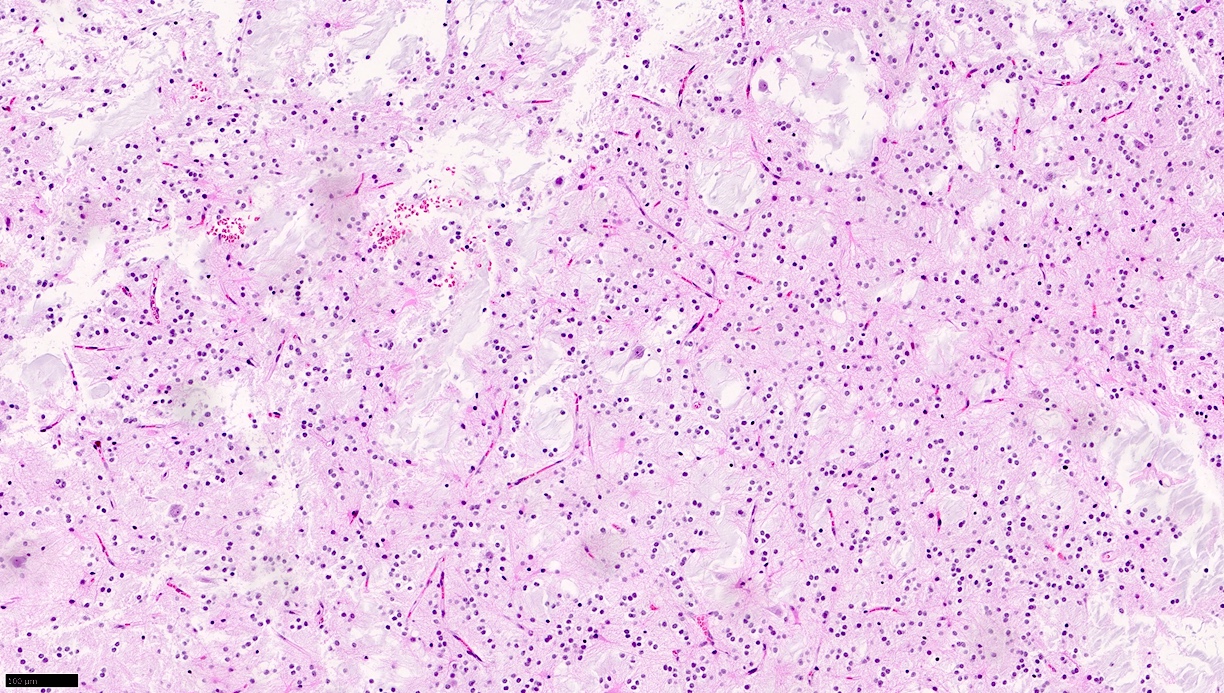
The tumor shown above is from the mesial temporal lobe of a child. What is the most common underlying genetic alteration in this entity?
- BRAF V600E mutation
- FGFR1 alteration
- IDH1 R132H
- NF1 loss
- TP53 mutation
Comment Here
Reference: Dysembryoplastic neuroepithelial tumor
- Associated with NF2
- Associated with seizures
- Harbor several cytogenetic alterations
- Most frequently occur in adults
- Show diffuse CD34 immunoreactivity
Comment Here
Reference: Dysembryoplastic neuroepithelial tumor
- Hamartomatous cerebellar lesion characterized by enlarged cerebellar folia and replacement of internal granular layer by dysplastic ganglion cells (Clin Neurol Neurosurg 2001;103:105, Acta Neurol Scand 2002;105:137)
- Due to or associated with germline PTEN mutation (Am J Hum Genet 2003;73:1191)
- PTEN mutations are identified in all adult onset cases but not in childhood onset cases
- Associated with Cowden disease (OMIM: Cowden Syndrome 1; CWS1), an autosomal dominant disorder with trichilemmomas, hamartomas, intestinal polyposis, palmoplantar keratoses, oral papillomas; increased incidence of breast, GU, CNS and thyroid tumors; due to abnormalities in 10q23
- Rare, < 200 cases described
- Usually young adults, may present in childhood
- Presents with dysmetria, headache, ataxia, mass effect
- Unilateral cerebellar affection with distorted architecture and enlarged folia
- Characteristic striped appearance on FLAIR and T2 W MRI images
- 16 year old girl with headaches and gait disturbance (University of Pittsburgh: A Cerebellar Mass)
- 44 year old woman with Cowden disease (Can J Neurol Sci 2004;31:542)
- Surgical resection if symptomatic
- Good prognosis; recurs in 25% of cases but no malignant potential
- Discrete region of hypertrophy and coarse gyral pattern
- Relative preservation of cerebellar architecture
- Diffuse enlargement of internal granular layer and molecular layers, with replacement of internal granular layer by dysplastic ganglion cells of different sizes and axonal hypermyelination of molecular layer
- Clear vacuoles in white matter and molecular layer
- Common: calcification and ectatic vessels
- No mitotic figures, no necrosis, no endothelial proliferation
- Dysplastic ganglion cells are synaptophysin+ with loss of PTEN protein expression; also are phosphorylated AKT+ and phosphorylated S6+ (indicates AKT / mTOR pathway activation)
- Ganglion cell tumor
- Infiltrating glioma with entrapped neurons
- CNS embryonal tumor with biphasic architecture
- Aggressive CNS embryonal tumor with multilayered rosettes (ETMR), amplicon C19MC altered and C19MC unaltered or not tested (Acta Neuropathol 2016;131:803)
Essential features
- Belongs to ETMR, a new subclassification of embryonal tumors
- Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity
- Overexpression of LIN28A protein in C19MC altered embryonal tumors
- Poor prognosis
ICD coding
- ICD-10: C71.9 - malignant neoplasm of brain, unspecified
Epidemiology
- M = F
- Mostly < 2 years of age
Sites
- Supratentorial in both hemispheres (70%)
- Infratentorial in cerebellum and brain stem (30%)
Clinical features
- Signs and symptoms of increased intracranial pressure (headache, nausea and visual disturbances)
- Focal neurologic signs (ataxia and weakness)
Grading
- WHO grade 4
Diagnosis
- Overexpression of LIN28A protein is highly sensitive and specific (Acta Neuropathol 2012;124:875)
Radiology description
- Contrast enhancing large tumor masses on CT and MRI
Case reports
- 33 month old boy with brainstem tumor (J Neurosurg Pediatr 2015;16:291)
Treatment
- Complete surgical removal, radiotherapy and high dose chemotherapy (J Neurooncol 2016;126:99, Childs Nerv Syst 2016;32:299)
Microscopic (histologic) description
- Biphasic architecture
- Dense clusters of small cells with round / polygonal nuclei, scanty cytoplasm and indistinct cell bodies along with large, paucicellular, fibrillary / neuropil like areas along with neoplastic neurocytic and ganglion cells
- Mitosis and apoptotic bodies in hypercellular area
- Multilayered rosettes
Positive stains
- Nestin and vimentin in neuroepithelium, multilayered rosettes and tubular structures
- Focal cytokeratins, EMA and CD99 in small cell areas
- INI1
- Ki67: 20% - 80%
Negative stains
Molecular / cytogenetics description
- Amplicon at 19q13.42 is sensitive and specific diagnostic marker for medulloepithelioma with C19MC alteration (Acta Neuropathol 2014;128:279)
- Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity
- Aggressive CNS embryonal tumor with multilayered rosettes (ETMR) and amplicon C19MC upregulation
- Rare tumor of children aged < 4 years
- Other tumors in this group are
- Embryonal tumor with abundant neuropil and true rosettes
- Medulloepithelioma
- References: Acta Neuropathol 2016;131:803, Acta Neuropathol 2014;128:305
Essential features
- Belongs to ETMR with or without alteration of C19MC, a new subclassification of embryonal tumors
- Rosettes are frequent and characteristic of embryonal tumors
- Ependymoblastoma lacks a neuropil-like matrix and ganglion cell elements
- Overexpression of LIN28A protein in C19MC altered embryonal tumors
- Poor prognosis
ICD coding
- ICD-O: 9478/3 - embryonal tumor with multilayered rosettes C19MC altered
Epidemiology
- M = F
- Mostly < 2 years of age
Sites
- Supratentorial in both hemispheres (70%)
- Infratentorial in cerebellum and brain stem (30%)
Clinical features
- Signs and symptoms of increased intracranial pressure (headache, nausea and visual disturbances)
- Focal neurologic signs (ataxia and weakness)
Diagnosis
- Overexpression of LIN28A protein (Acta Neuropathol 2012;124:875)
Radiology description
- Contrast enhancing large tumor masses on CT and MRI
Case reports
- 12 month old boy with embryonal tumor with multilayered rosettes of the fourth ventricle (J Neurosurg Pediatr 2015;16:579)
Treatment
- Complete surgical removal, radiotherapy and high dose chemotherapy (Childs Nerv Syst 2016;32:299)
Gross description
- Well circumscribed
- Grayish pink with areas of necrosis and hemorrhage
Microscopic (histologic) description
- Rosettes are frequent and characteristic of ETMR
- Sheets and clusters of poorly differentiated cells
- Lacks neuropil-like matrix and ganglion cells
- Rosettes are intermixed with small to medium sized embryonal cells with high N:C ratio
Positive stains
Negative stains
Molecular / cytogenetics description
- Amplicon at 19q13.42 is a sensitive and specific diagnostic marker for ETMR (Acta Neuropathol 2014;128:279)
- CNS embryonal tumor with prominent pseudostratified neuroepithelium that resembles the embryonic neural tube and poorly differentiated neuroepithelial cells
- Aggressive CNS embryonal tumor with multilayered rosettes (ETMR) and amplicon C19MC altered, C19MC unaltered or C19MC not tested
- Significant proportions of medulloepithelioma have not shown C19MC alterations (Acta Neuropathol 2016;131:803)
Essential features
- Belongs to ETMR, a new subclassification of embryonal tumors, some of which have C19MC alterations
- C19MC altered embryonal tumors overexpress LIN28A protein
- Poor prognosis
ICD coding
- ICD-10: C71.9 - malignant neoplasm of brain, unspecified
Epidemiology
- M = F
- Mostly < 2 years of age
Sites
- Supratentorial in both hemispheres (70%)
- Infratentorial in cerebellum and brain stem (30%)
Clinical features
- Signs and symptoms of increased intracranial pressure (headache, nausea and visual disturbances)
- Focal neurologic signs (ataxia and weakness)
Grading
- WHO grade 4
Diagnosis
- Overexpression of LIN28A protein is highly sensitive and specific (Acta Neuropathol 2012;124:875)
Radiology description
- Contrast enhancing large tumor masses on CT and MRI
Case reports
- 17 year old girl with unusual occurrence of supratentorial medulloepithelioma (J Neurosci Rural Pract 2014;5:261)
Treatment
- Complete surgical removal, radiotherapy and high dose chemotherapy (Strahlenther Onkol 2011;187:757, Childs Nerv Syst 2016;32:299)
Microscopic (histologic) description
- Neoplastic pseudostratified neuroepithelium resembling embryonic neural tube, with papillary, tubular and trabecular arrangements
- Sheets of poorly differentiated cells with hyperchromatic nuclei and high N:C ratio
- Multilayered rosettes may be seen
- Periodic acid-Schiff positive external limiting membrane
- No cilia or blepharoplasts on luminal surface of tubules
- Abundant mitotic figures
- Rarely mesenchymal differentiation and melanin pigment
Positive stains
- Patchy expression of cytokeratins and EMA in neuroepithelium
- Ki67 is high in embryonal cells
- Rare GFAP
- LIN28A
Negative stains
Molecular / cytogenetics description
- Amplicon at 19q13.42 is sensitive and specific diagnostic marker for medulloepithelioma with C19MC alteration (Acta Neuropathol 2014;128:279)
- Cyst lined by simple columnar epithelium (with or without cilia or mucus globules)
- Rare, benign; probably of malformative nature
- Also called neurenteric, enteric, enterogenic, bronchogenic, respiratory cyst
- Usually intraspinal, rarely intracranial
- Favored sites: posterior fossa and spinal cord (usually anterior to cord, intradural, extramedullary)
- All ages are affected: infants, children and adults
- Like the closely related Rathke cleft cyst and colloid cyst, cyst is often bright on precontrast images
- Prenatally diagnosed large intracranial cyst (J Neurosurg 2007;106:506)
- 35 year old woman with intramedullary cyst of spinal cord during pregnancy (J Neurol Neurosurg Psychiatry 2001;71:528)
- 43 year old man with cystic mass at cerebellopontine angle (Arch Pathol Lab Med 2003;127:e45)
- 78 year old man with cyst in front of medulla (AJNR Am J Neuroradiology 2001;22:496)
- Complete excision; occasionally is adherent to adjacent structures
- Recurrence may occur
- Usually 1 cm or less, simple, thin walled, filled with opalescent mucinous material
- Columnar epithelium resting on collagen layer
- Goblet cells often present
- Variable cilia, may have squamous metaplasia
- More complex lesions replicate gastrointestinal or respiratory epithelium (Appl Immunohistochem Mol Morphol 2004;12:230)
- Well developed stereocilia, distinct basal cells, thin basement membrane
- Choroid plexus cyst: positive for vimentin, S100, synaptophysin, transthyretin
- Colloid cyst: in third ventricle
- Cystic tumors
- Glioependymal cyst: abuts onto gliotic neuropil, S100+, GFAP+
- Papillary endolymphatic sac tumor: at cerebellopontine angle, arises within bone, more complex architecture
- Circumscribed neuroepithelial tumor with histological and molecular evidence of ependymal differentiation that may arise in the supratentorial region, posterior fossa or spinal cord
- 2021 CNS WHO classification has reclassified ependymal tumors according to their location and methylation profiles
- Some traditional histological subtypes, including the papillary, clear cell, tanycytic and anaplastic subtypes, have been discontinued
- Supratentorial ependymal tumors include supratentorial ependymoma ZFTA fusion positive (ST-ZFTA), supratentorial ependymoma, YAP1 fusion positive (ST-YAP1) or supratentorial subependymoma (ST-SE)
- Posterior fossa ependymal tumors include posterior fossa ependymoma group A (PFA), group B (PFB) and posterior fossa subependymoma (PF-SE)
- Spinal ependymal tumors include spinal ependymoma (SP-EP), spinal ependymoma, MYCN amplified (SP-MYCN), spinal subependymoma (SP-SE) and myxopapillary ependymoma (SP-MP)
- Common pathology features across all ependymoma subtypes include perivascular pseudorosettes, ependymal rosettes, positive GFAP and S100 cytoplasmic positivity, dot-like EMA perinuclear reactivity and negative Olig2 nuclear reactivity
- ICD-10
- C71.0 - malignant neoplasm of cerebrum, except lobes and ventricles
- C71.1 - malignant neoplasm of frontal lobe
- C71.2 - malignant neoplasm of temporal lobe
- C71.3 - malignant neoplasm of parietal lobe
- C71.4 - malignant neoplasm of occipital lobe
- C71.5 - malignant neoplasm of cerebral ventricle
- C71.7 - malignant neoplasm of brain stem
- C71.9 - malignant neoplasm of brain, unspecified
- C72.9 - malignant neoplasm of central nervous system, unspecified
- D33.2 - benign neoplasm of brain, unspecified
- Ependymal tumors comprise 1.9% of all primary CNS tumors and 6.9% of primary glial neoplasms, with incidence rates of ~0.4/100,000 in the U.S. (Neuro Oncol 2015;17:iv1)
- ST-ZFTA, ST-YAP1 and PFA predominantly occur in children, while SP-MYCN predominantly occurs in middle aged adults; other subtypes (ST-SE, PF-SE, PFB, SP-EP, SP-MP, SP-SE) can occur in all ages (Acta Neuropathol 2024;147:24)
- M:F = 1.38:1 (Acta Neuropathol 2024;147:24)
- Typically associated with ventricular lining in the spinal cord or cerebrum but can occur intraparenchymally / paraventricularly, particularly in supratentorial tumors
- Spinal ependymomas commonly in cervical and thoracic spine
- Spinal myxopapillary ependymoma predominantly in conus medullaris and filum terminale
- Posterior fossa ependymomas around fourth ventricle
- References: Childs Nerv Syst 2003;19:270, Crit Rev Oncol Hematol 2007;63:81
- Single cell RNA sequencing across all major molecular ependymoma groups revealed hierarchical cellular populations, including undifferentiated neural stem cells, radial glia cells and more differentiated cells towards ependymal, astrocytic and neuronal lineages (Cancer Cell 2020;38:44)
- Proportion of undifferentiated or less differentiated cells correlates with poor prognosis and increased recurrence
- Aberrant radial glia-like cells are potential cells of origin for supratentorial ependymoma, with ZFTA::RELA fusions and neural stem cell-like cells the origin of posterior fossa ependymoma (Cancer Cell 2020;38:44)
- Spinal ependymoma likely arises from mature adult ependymal cells due to their highly similar transcriptomic profiles through single cell RNA sequencing (Acta Neuropathol 2024;147:22)
- Unknown
- Spinal cord ependymomas present with pain, weakness, sensory loss or radiculopathy, depending on level affected
- Intracerebral ependymomas present with obstructive hydrocephalic symptoms as well as features of mass effect, neurologic deficits or seizures, depending on location
- Reference: Pediatr Neurosurg 2019;54:98
- Diagnosis is made by integrating histologic features, tumor location and molecular findings (Neuro Oncol 2021;23:1231)
- Commonly discrete, well circumscribed mass with avid uniform contrast enhancement
- Hyperdense on computed tomography and T2 hyperintense on magnetic resonance imaging
- Reference: Childs Nerv Syst 2009;25:1203
Contributed by Chunyu Cai, M.D., Ph.D.
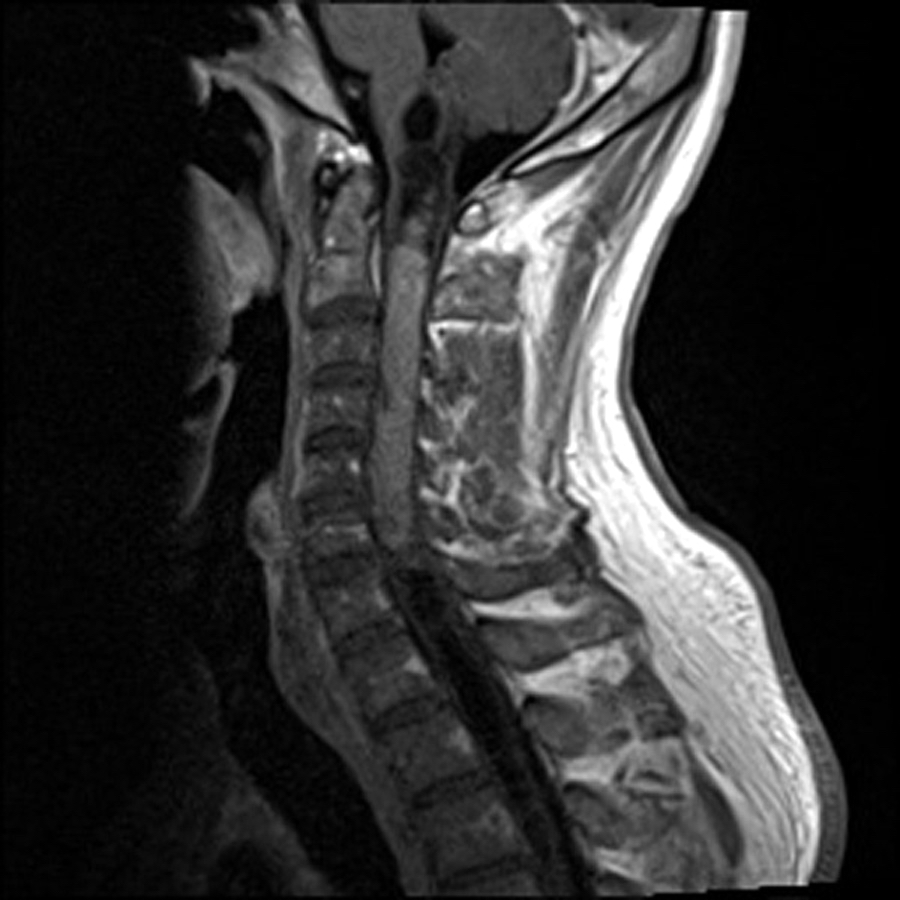
MRI cervical spine ependymoma
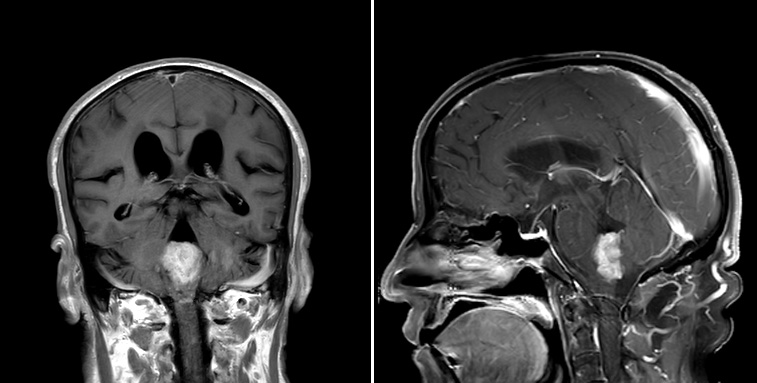
MRI posterior fossa ependymoma
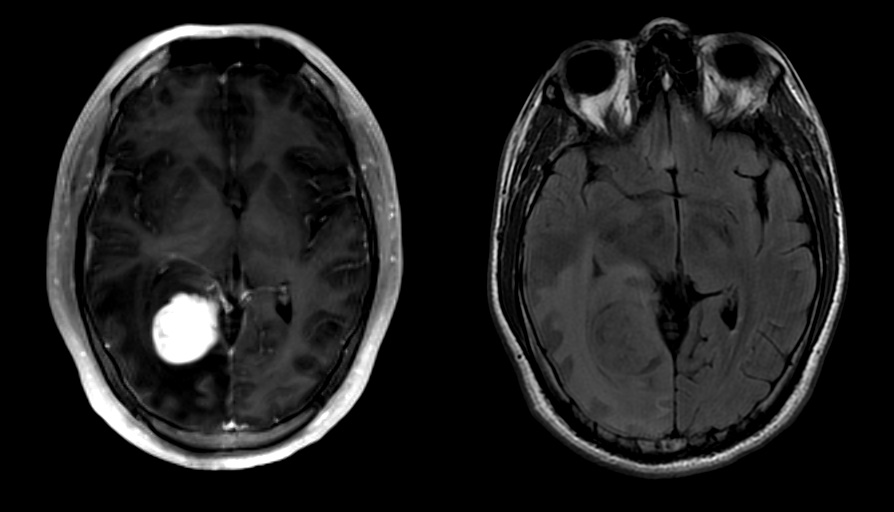
MRI supratentorial ependymoma
Images hosted on other servers:


MRI: ependymoma of fourth ventricle
- Spinal ependymoma (SPEP), generally indolent
- Recent studies suggest the presence of an NF2 mutation may confer a higher risk of recurrence
- Spinal ependymoma, MYCN amplified (SPMYCN) has poor prognosis
- Ependymomas with ZFTA fusions
- 6% of ependymomas occur in posterior fossa and those occur exclusively in children and have worse prognosis
- Supratentorial ependymoma with ZFTA fusion and combined homozygous CDKN2A / CDKN2B losses have worse prognosis
- Supratentorial ependymomas with YAP1 fusions have good prognosis
- Of posterior fossa ependymomas
- PFA is characterized by H3K27 trimethyl loss on IHC and has worse prognosis than PFB
- Gain of 1q and loss of 6q are additional risk factors that predict even worse outcomes in PFA
- Complete resection is a predictor of good prognosis in PFA, PFB, PF-SE and SP-MP but not in ST-ZFTA
- References: Acta Neuropathol 2024;147:24, Brain Pathol 2020;30:863, Acta Neuropathol 2024;147:22
- 26 year old woman with neck pain (Cureus 2020;12:e7981)
- 28 year old man with incidental cerebellopontine angle mass (Medicine (Baltimore) 2019;98:e15019)
- 37 year old man with cervicomedullary mass (Asian J Neurosurg 2020;15:190)
- 44 year old woman with extramedullary lumbar spine mass (Yeungnam Univ J Med 2020;37:128)
- Gross total resection if possible, with or without adjuvant radiotherapy
- Multiple pathway inhibitors (ALK, EGFR, ErbB2, FAK, MEK, mTOR, TEAD, VEGF) are in clinical trials for NF2 associated tumors (Acta Neuropathol 2024;147:22)
- Gray-red tumors with or without cystic degeneration, hemorrhage or necrosis
- Intracranial tumors are well circumscribed
- Ependymomas of fourth ventricle are typically exophytic
Images hosted on other servers:
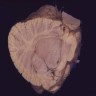
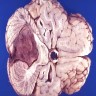
Arising from floor of fourth ventricle
- Cellular tumor with sharp border with brain parenchyma
- Perivascular pseudorosettes, true ependymal rosettes and lumina (J Neurosurg Spine 2018;30:133)
Contributed by Chunyu Cai, M.D., Ph.D.
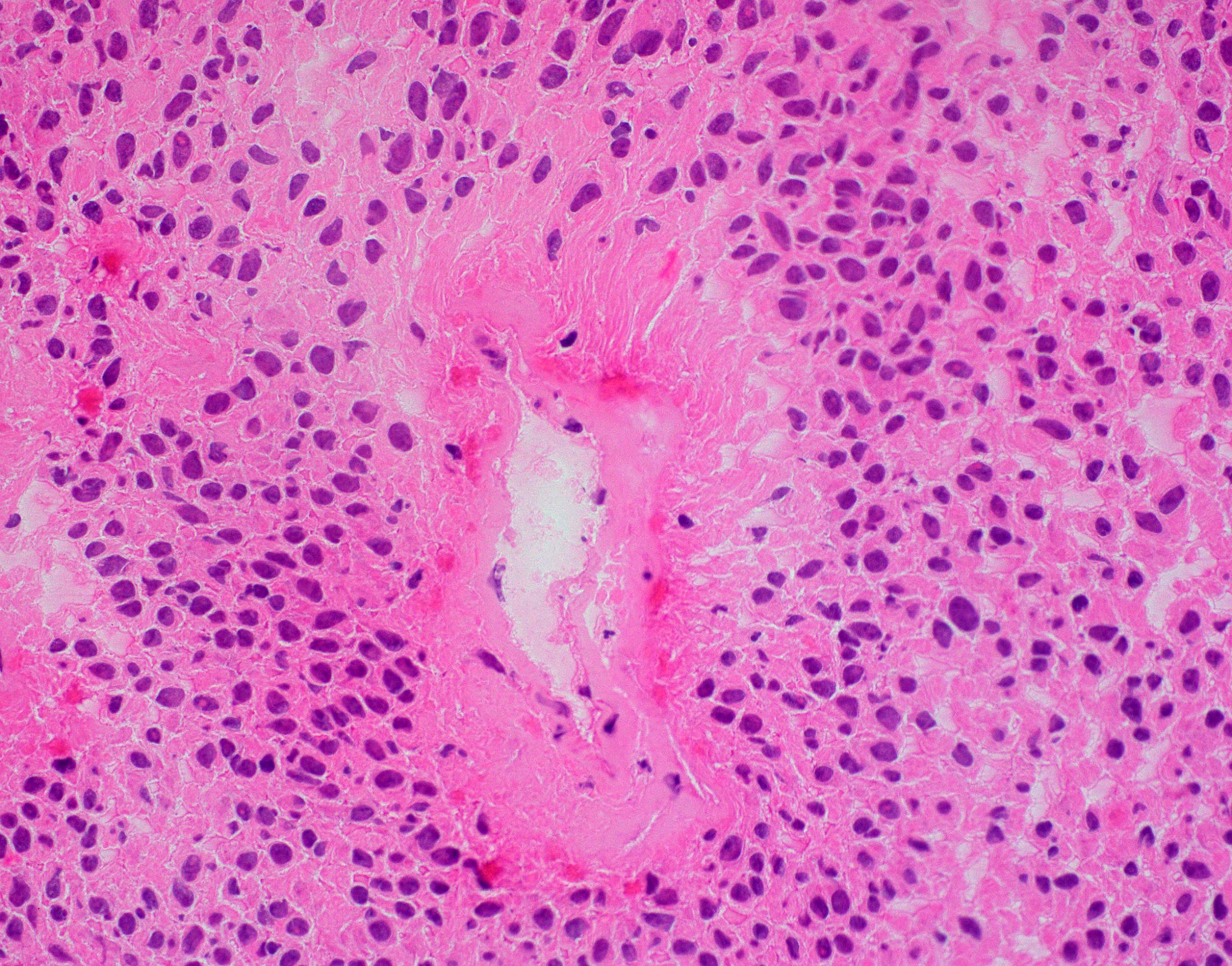
Fibrillary perivascular pseudorosette

Perivascular pseudorosette
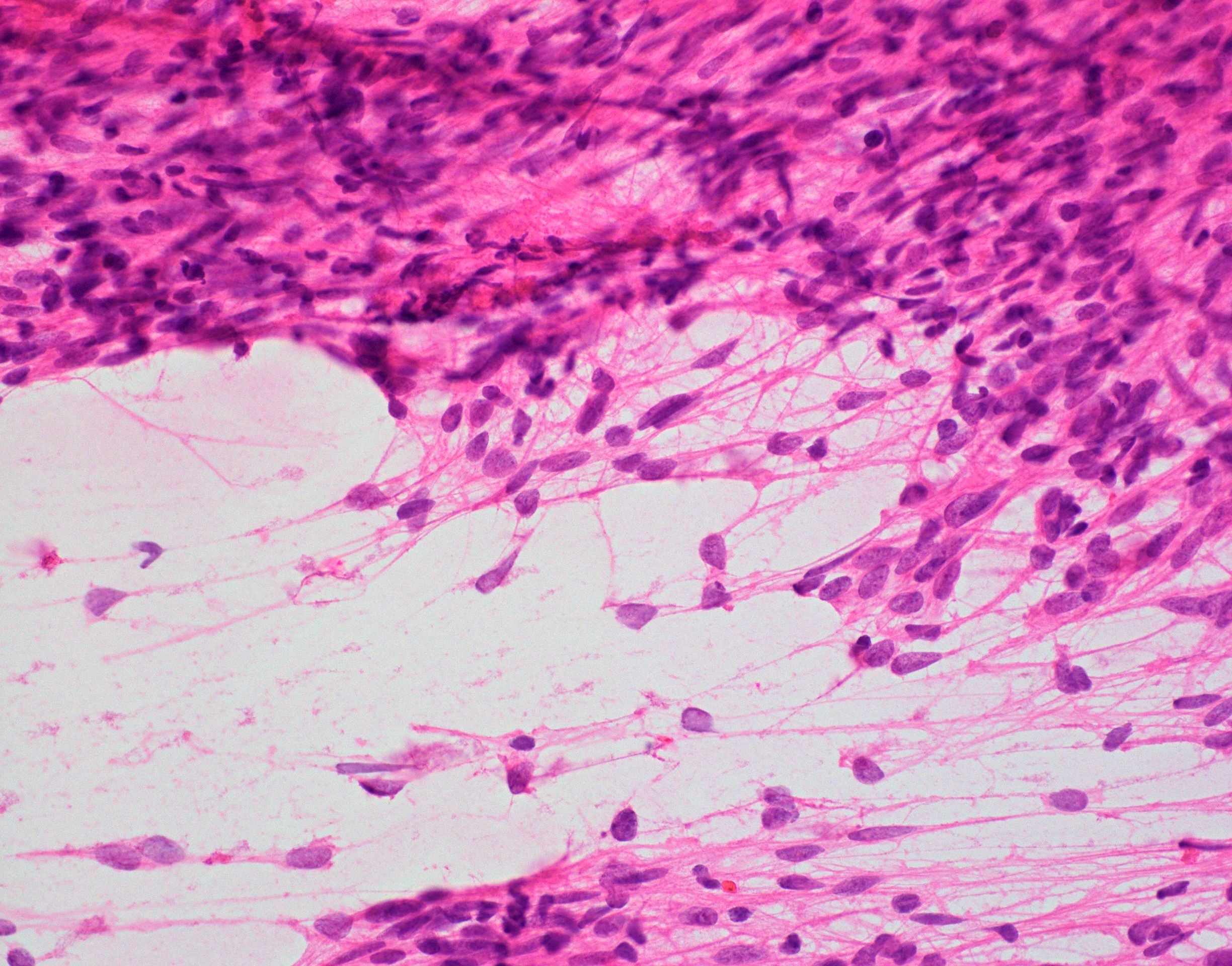
Fibrillary cytoplasm
- Cellular tumor with typically sharply circumscribed borders; may be infiltrative
- Monomorphic round to oval cells with speckled chromatin
- Perivascular pseudorosettes, true ependymal rosettes, lumina and fibrillar areas
- May see gemistocyte-like cells and hypercellular nodules, particularly in posterior fossa tumors
- Can have nonpalisading necrosis, areas of cystic or myxoid degeneration, calcifications, degenerative atypia, neuronal differentiation and rarely metaplastic elements
- Morphologic subtypes have no clinicopathological significance and include papillary, clear cell and tanycytic
- Utility of histological grading is debated; the 2021 WHO still recommend assigning either WHO grade 2 or grade 3 to an ependymoma, according to its histopathological features as part of the integrated diagnosis
- Myxopapillary ependymoma are now assigned WHO grade 2 in the 2021 WHO CNS tumor classification
- Subependymoma remains a WHO grade 1 (Neuro Oncol 2021;23:1231)
Contributed by Chunyu Cai, M.D., Ph.D., Maria Martinez-Lage, M.D. and Eman Abdelzaher, M.D., Ph.D.
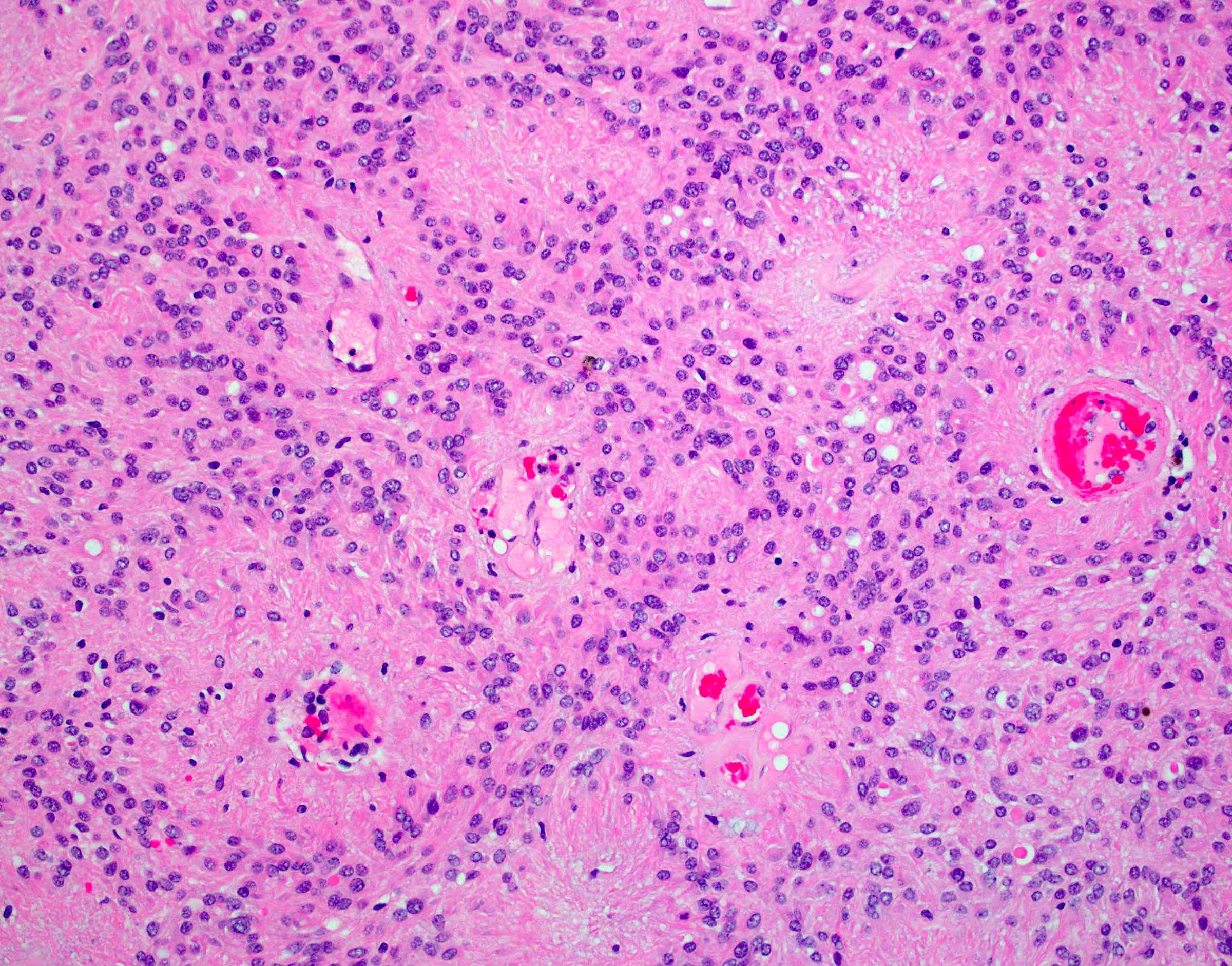
Perivascular pseudorosettes
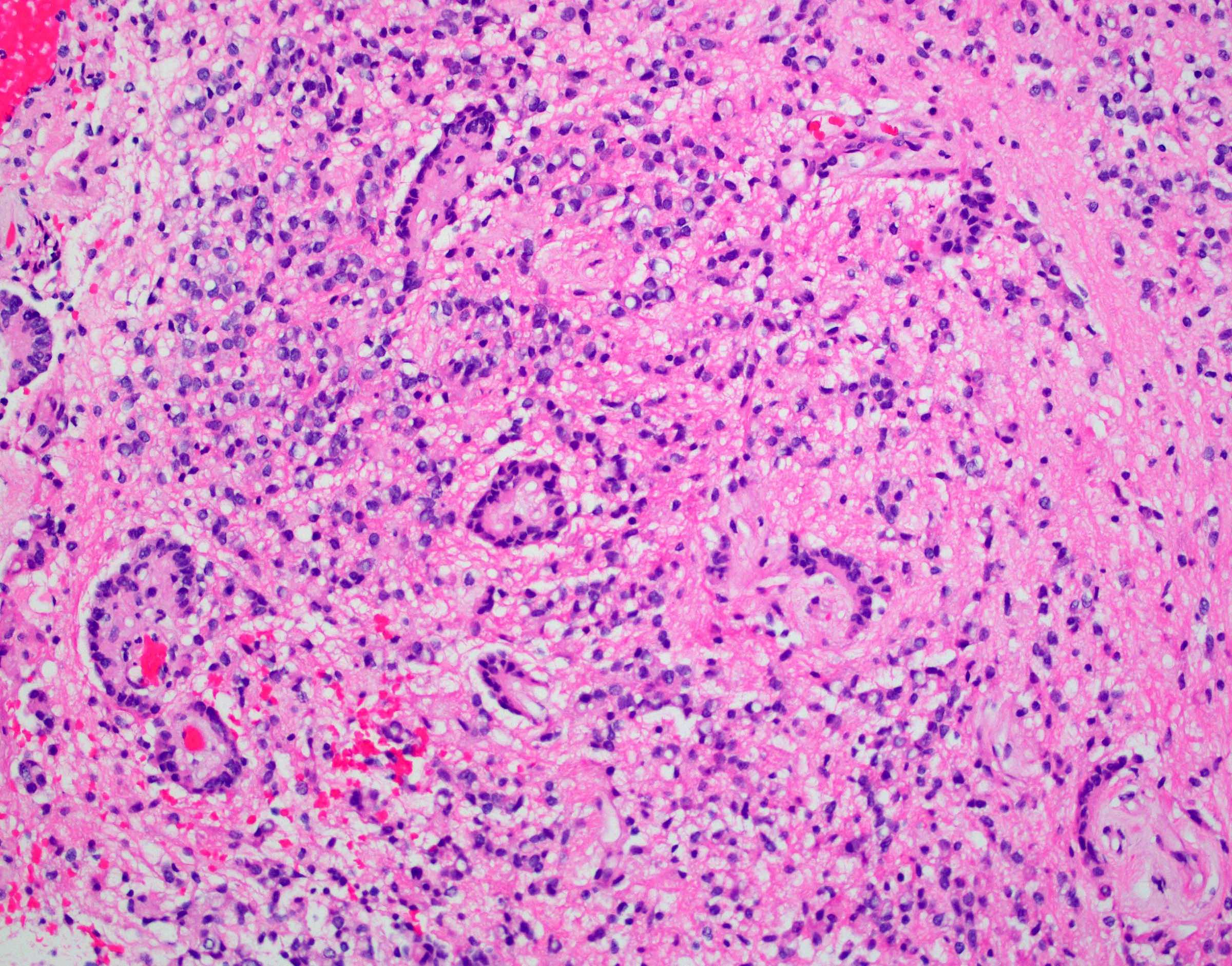
Ependymal rosettes
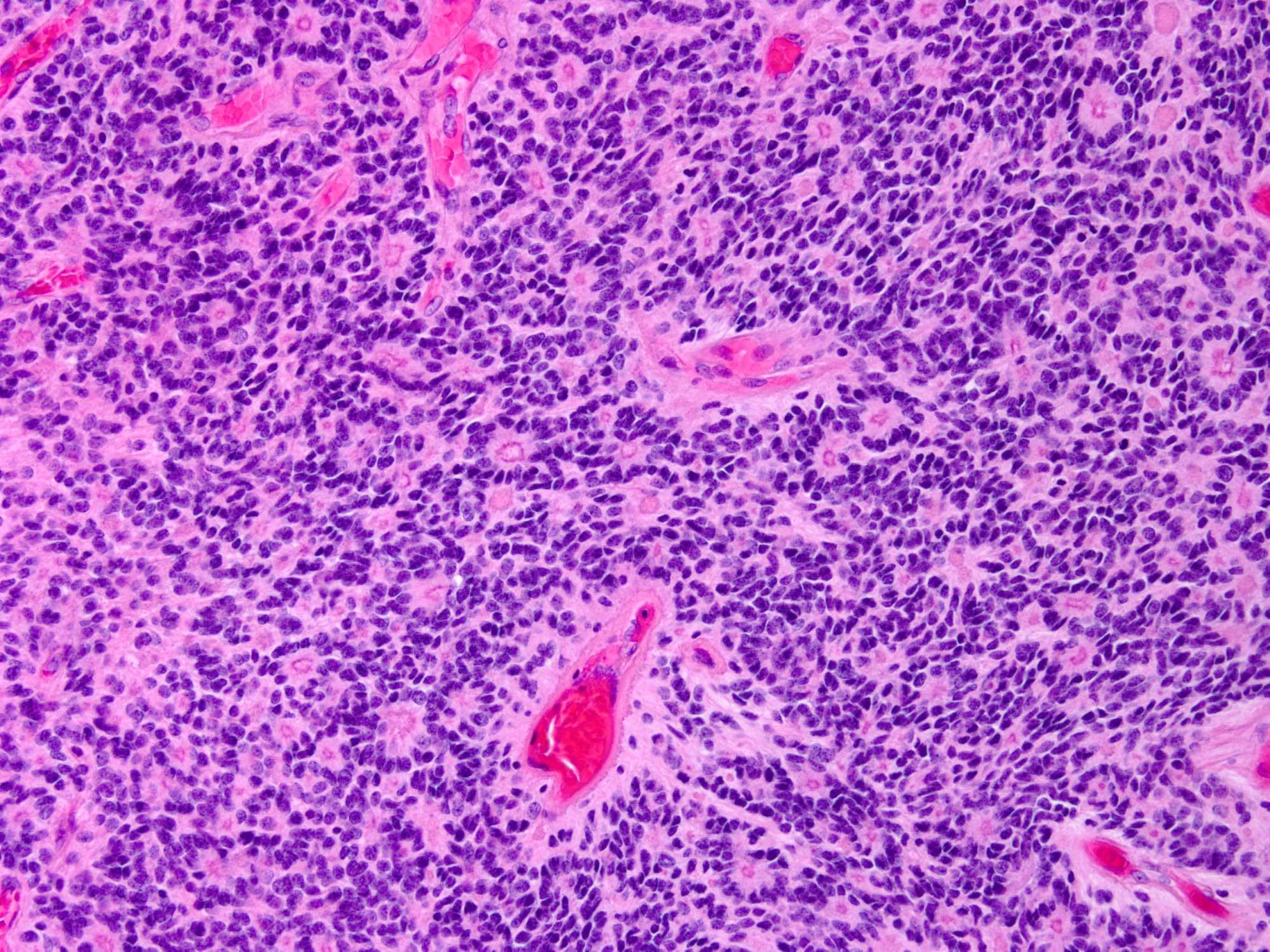
True ependymal rosettes
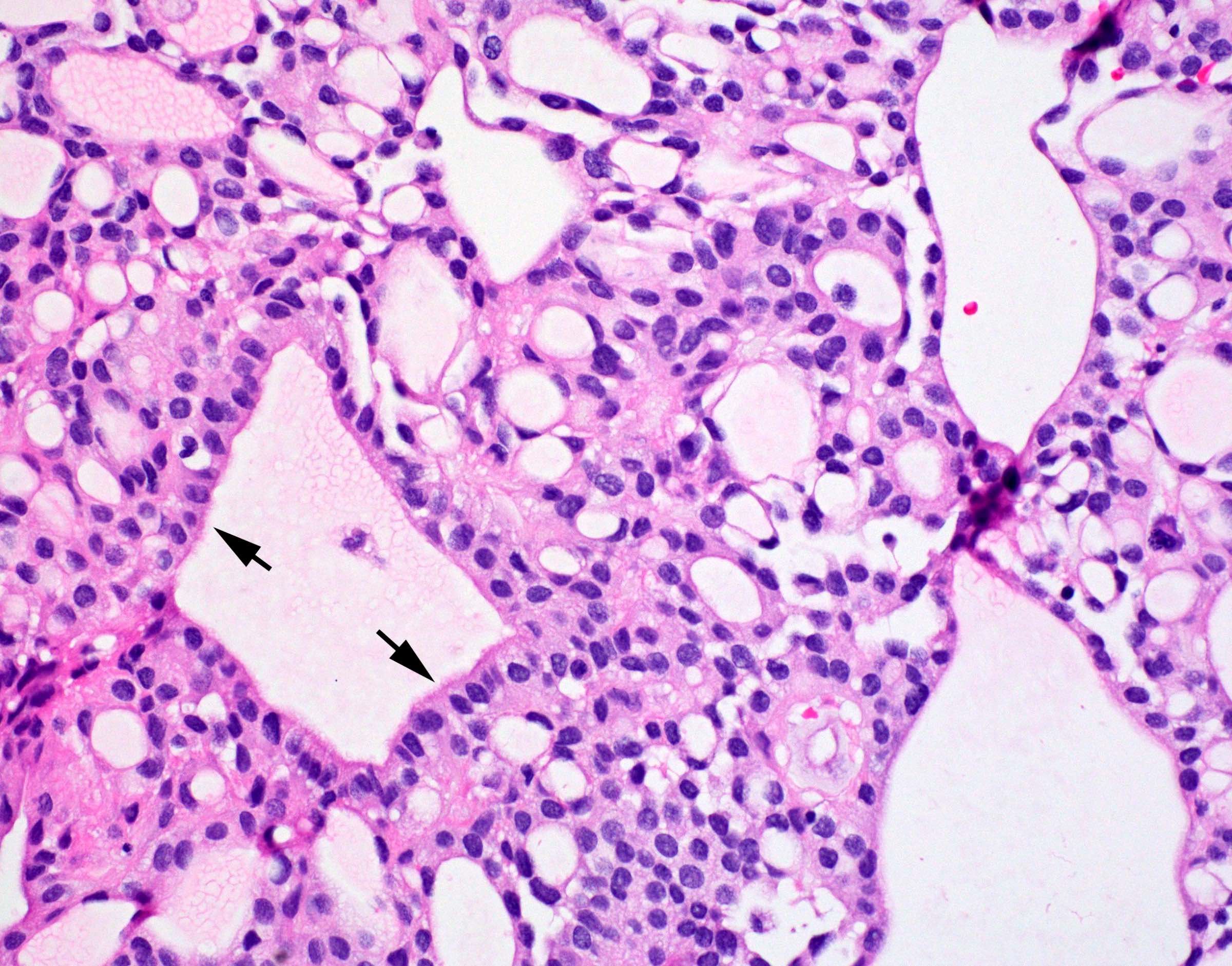
Ependymoma canal
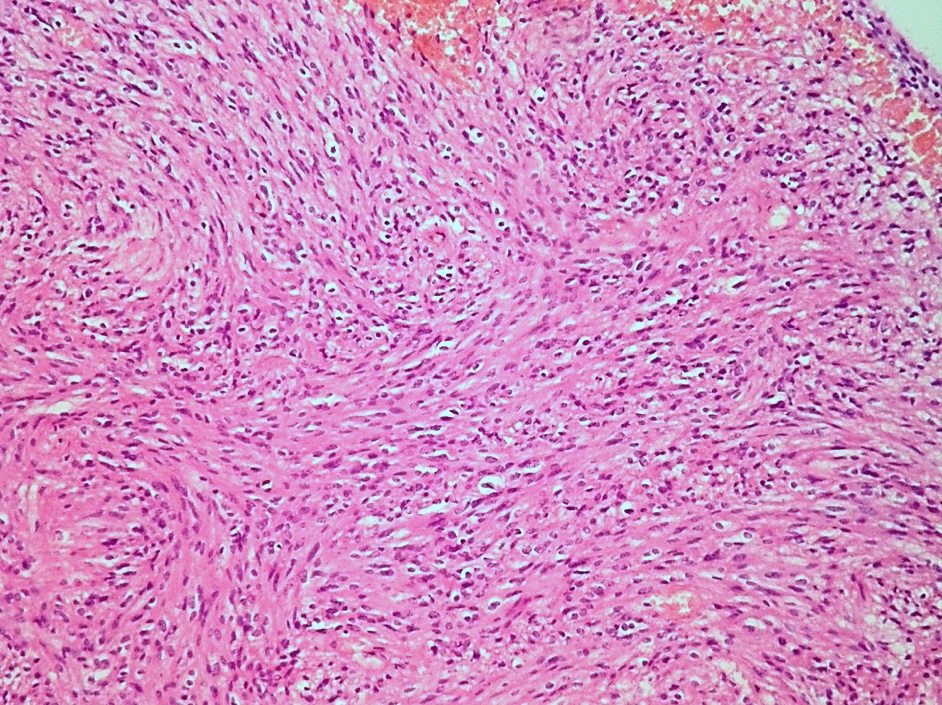
Tanycytic ependymoma
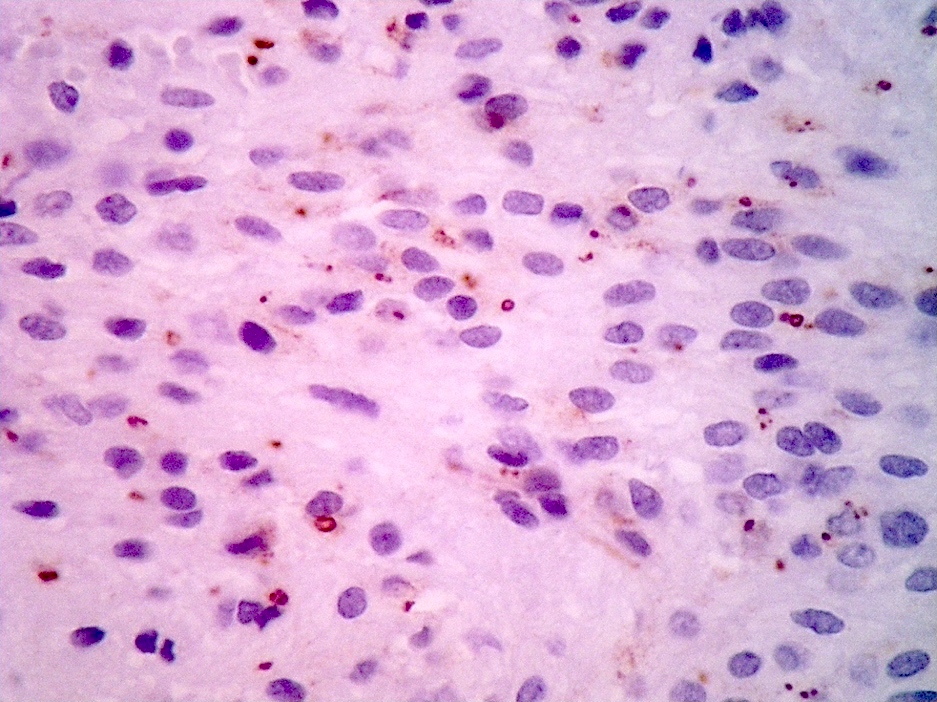
EMA showing dot-like and ring-like positivity
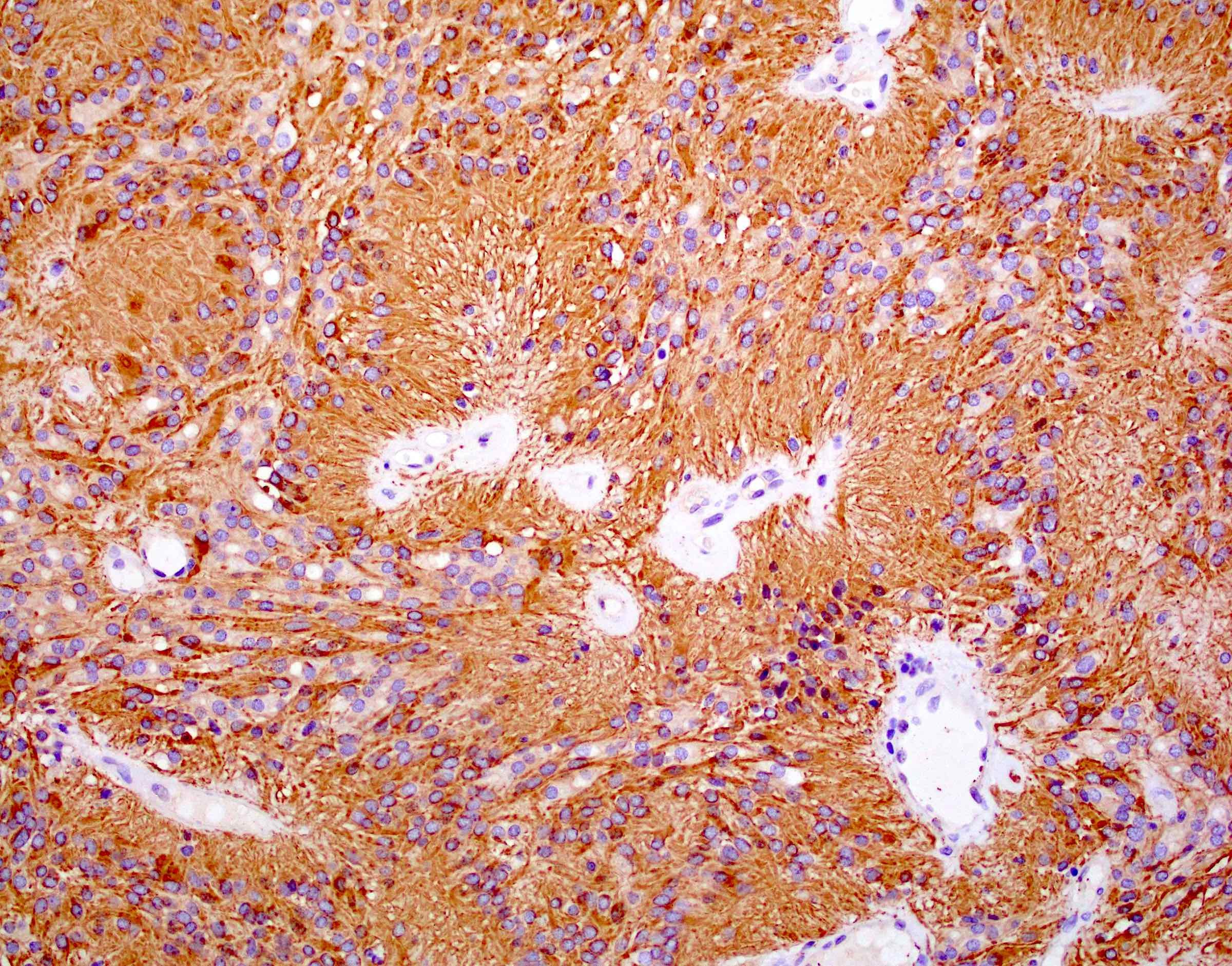
GFAP
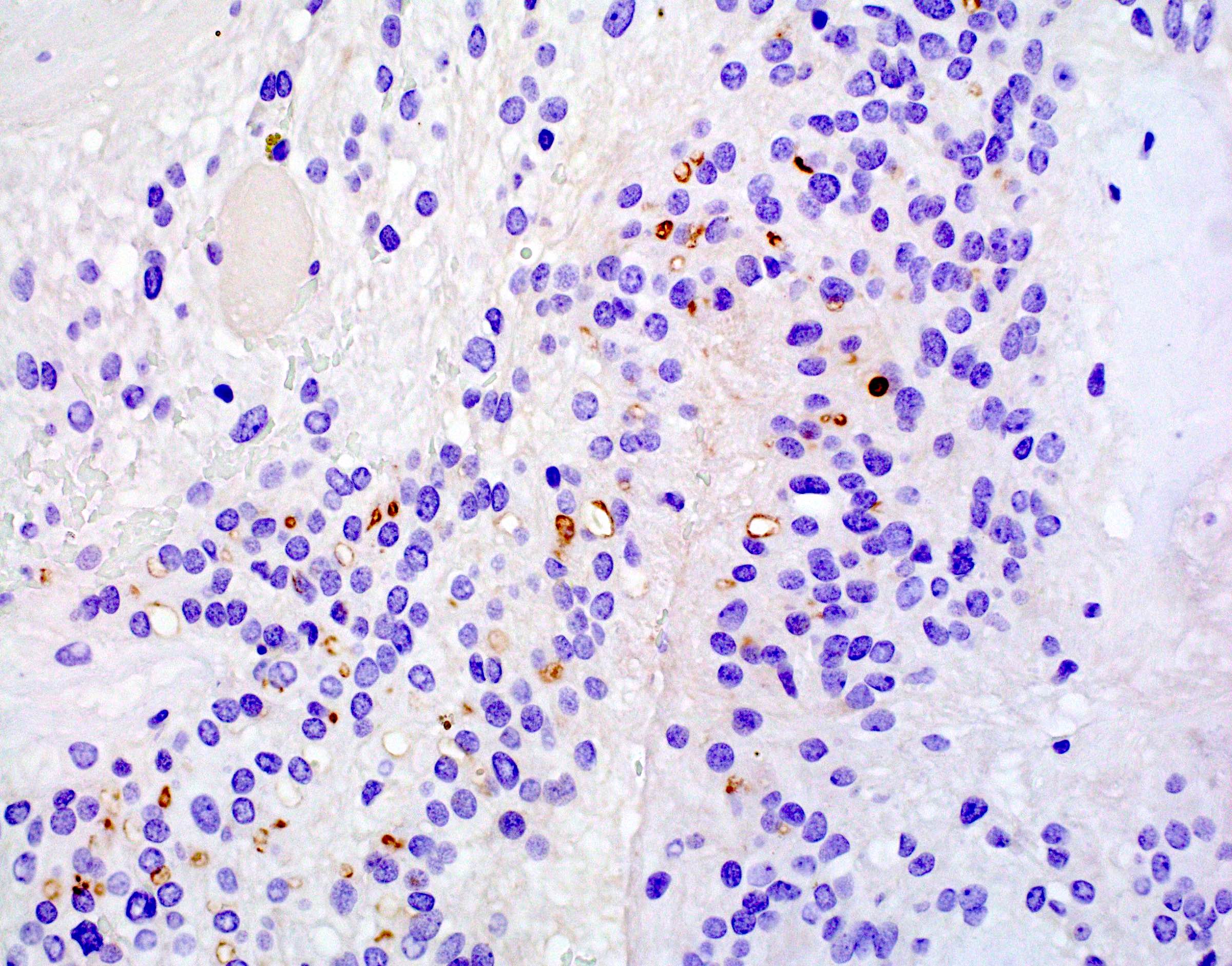
EMA
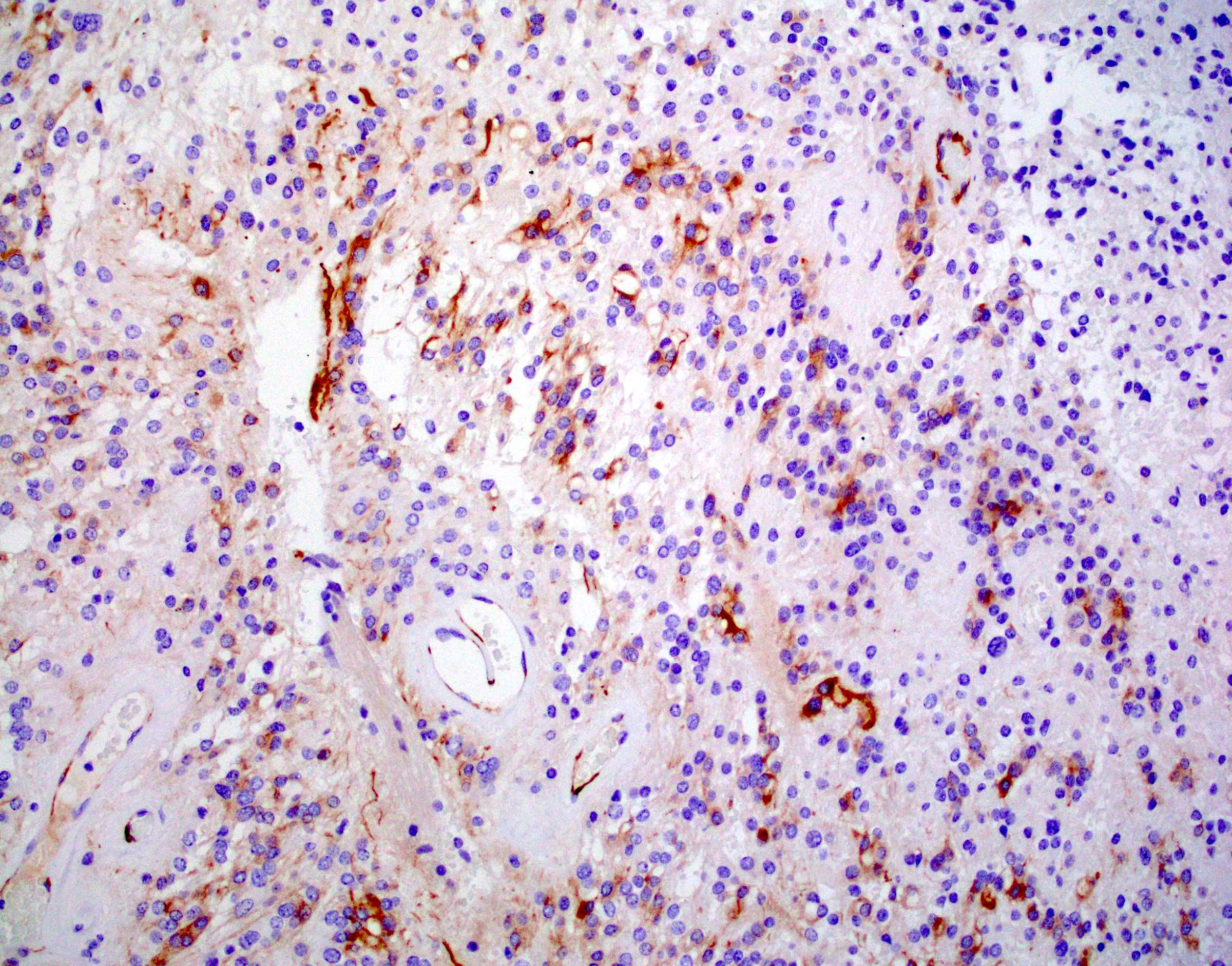
MAP2
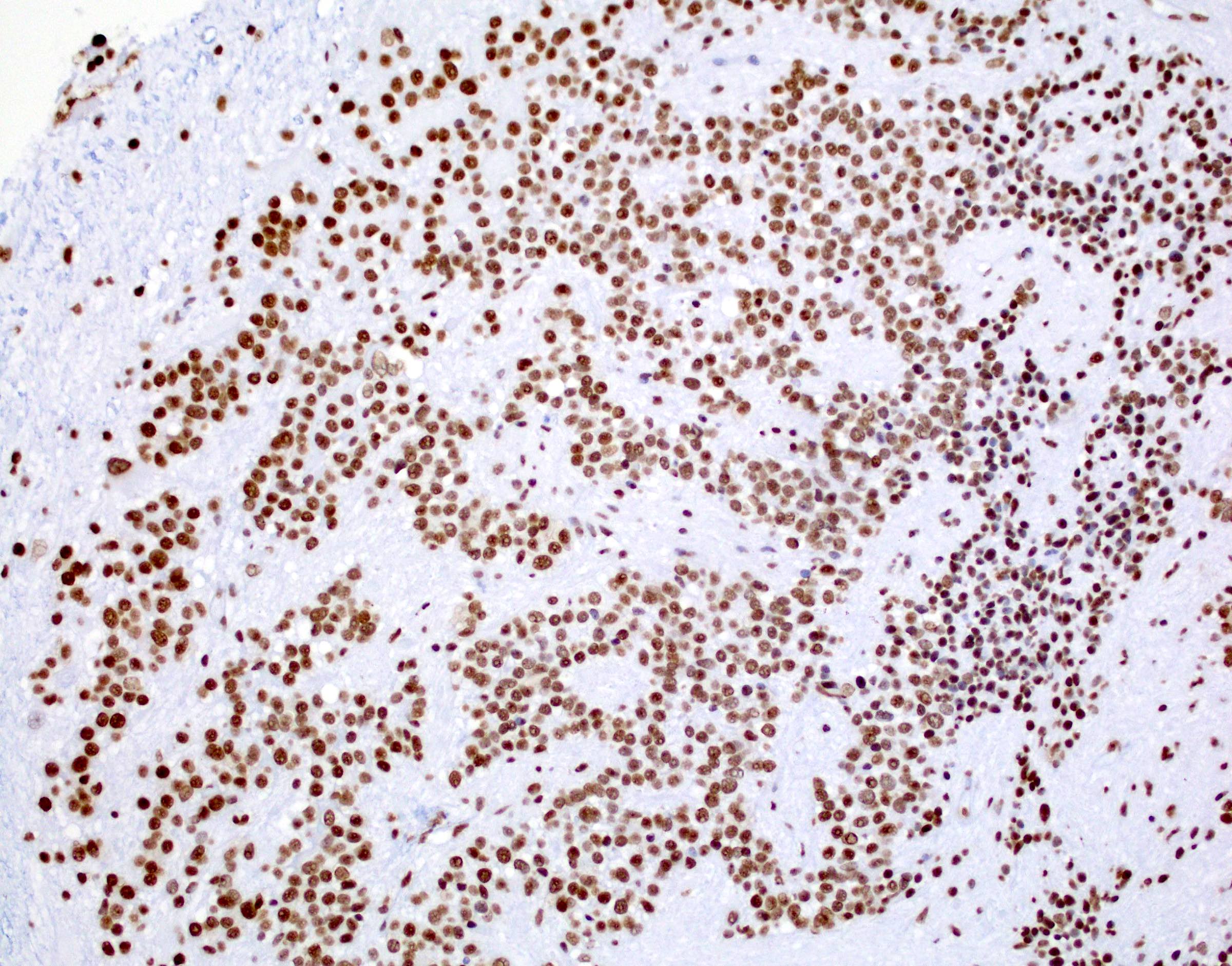
H3K27me IHC PFB
Images hosted on other servers:
Ependymoma
- Spindle shaped cells with oval to elongated nuclei and delicate fibrillary cytoplasm with occasional intracytoplasmic lumina, which can be arranged around blood vessels
- Occasional nuclear grooves and inclusions can be seen (J Pathol Transl Med 2019;53:104)
- Positive for S100, GFAP, vimentin
- Perinuclear dot-like pattern of EMA and D2-40 staining
- CD56 staining in lumina and tumor cells
- Variable membranous or dot-like staining for CD99
- Can have focal staining for keratin (CAM 5.2) and synaptophysin
- L1CAM can be positive in some supratentorial ependymomas but mostly is seen in RELA fusion tumors (Am J Surg Pathol 2019;43:56)
- In posterior fossa ependymomas, decreased expression of H3K27me3 is seen in posterior fossa group A, which has a worse prognosis (Acta Neuropathol 2017;134:705)
- Negative for neuronal markers (NeuN, chromogranin), Olig2 and IDH1
- Negative for reticulin
- Features of ependymal cells
- Cilia with a 9+2 microtubular pattern
- Blepharoblasts and microvilli of the lumina
- Zipper-like junctional complexes of lateral aspect
- Lack of basement membrane
- Reference: CNS Oncol 2014;3:49
- Molecular groups based on fusion and methylation profiling (Cancer Cell 2015;27:728)
- Supratentorial ependymoma, ZFTA fusion positive (formerly C11orf95::RELA fusion tumors), comprises 60% of supratentoral ependymomas and portends a poor prognosis
- Supratentorial ependymoma, YAP1 fusion positive, has a better prognosis
- Posterior fossa ependymoma, group A
- Posterior fossa ependymoma, group B
- Spinal ependymomas with NF2 mutations
- Spinal ependymoma, MYCN amplified
- Molecular characterization of histopathological ependymoma variants (Acta Neuropathol 2020;139:305)
- Tanycytic ependymomas were mostly located in spinal cord; DNA methylation match to spinal or myxopapillary ependymoma
- Clear cell ependymomas were mostly supratentorial; DNA methylation match to RELA fusion positive ependymomas
- Papillary ependymomas match to posterior fossa group B or myxopapillary ependymoma
Ependymal tumors by Dr. Rodriguez
- Brain, fourth ventricular mass, excision:
- Posterior fossa group B ependymoma, WHO grade 2 (see comment)
- Comment: Sections show an ependymal neoplasm with perivascular pseudorosettes and multiple foci of well formed ependymal canals. Solid, papillary and clear cell components are variably present. No necrosis, microvascular proliferation or mitotic figures are identified. Tumor cell nuclei are oval and uniform. Focal areas of hemosiderin laden macrophages are present.
- Immunohistochemically, a subset of the tumor cells express GFAP. EMA is negative. MIB1 proliferation index is < 1%. H3K27me nuclear expression is retained in over 95% of tumor cells.
- The collective findings of floor of fourth ventricle location, adult age and retained H3K27me nuclear expression in tumor cells are most consistent with posterior fossa group B (PFB) ependymoma. Correlation with NGS result is recommended.
- Infiltrating glioma:
- Astroblastoma:
- Astroblastic pseudorosette and IHC profile that resembles ependymoma
- Located almost exclusively in cerebral hemispheres
- Stout broad process that is strongly GFAP+
- MN1 rearrangement detectable by FISH (Cell 2016;164:1060, Brain Tumor Pathol 2019;36:112)
- Medulloblastoma:
- Posterior fossa ependymoma can radiographically and histologically resemble medulloblastoma, with monotonous cells and true rosettes
- Medulloblastoma is diffusely synaptophysin+
- Subtype specific markers: GAB1, YAP1, filamin A, beta catenin (nuclear)
- Schwannoma:
- Meningioma:
- Astrocytoma, IDH mutant
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted
- Posterior fossa ependymoma
- Supratentorial ependymoma, YAP1 fusion positive
- Supratentorial ependymoma, ZFTA fusion positive
Comment Here
Reference: Ependymoma
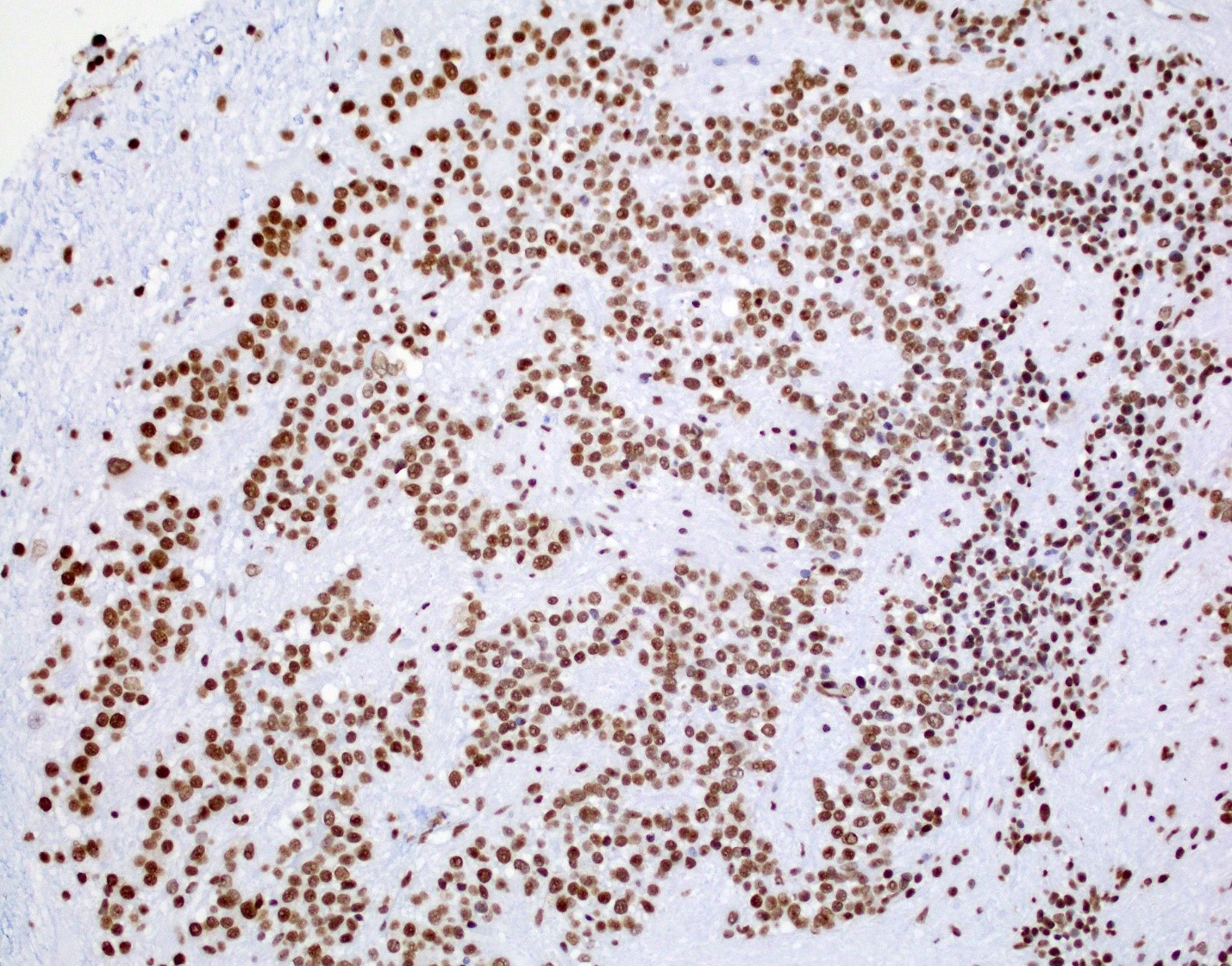
Which immunohistochemical stain can help differentiate posterior fossa group A (PFA) ependymomas from group B (PFB)?
- EMA
- H3K27me3
- Ki67
- L1CAM
- S100
Comment Here
Reference: Ependymoma
- Benign cyst lined by keratinized stratified squamous epithelium and lacking skin adnexa (eMedicine: Brain Epidermoid Imaging)
- More common than dermoid cyst (1% of intracranial masses)
- Also called epidermoid or epidermoid tumor
- Occurs throughout neuriaxis
- Favored site: cerebellopontine angle
- Other sites: brainstem, cranial diploe, intraspinal, pineal gland, sella, temporal lobe
- Rarely undergoes malignant degeneration
- Discrete, extra-axial with signal characteristics reflecting keratinous contents
- Diffusion MRI helps distinguish from arachnoid cysts
Images hosted on other servers:
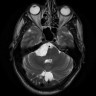
Cerebellopontine angle
- 68 year old man with cyst and sudden death (Am J Forensic Med Path 2002;23:368)
- Woman with cerebellopontine angle cyst that degenerated into squamous cell carcinoma (Neurosurg 2002;97:1237)
- Well defined round mass, thin walled, pearly white
- Fibrous wall lined by keratinizing squamous epithelium; contains anucleate squames but no skin adnexae and no hair
- Craniopharyngioma, adamantinous: wet keratin, adamantinomatous epithelium
- Craniopharyngioma, papillary: no anucleate squames
- Dermoid cyst: skin adnexa
- Rathke cleft cyst or endodermal cyst with extensive sqaumous metaplasia: have residual columnar epithelium but no anucleate squames
- Neurocytic tumor within brain parenchyma
- Median age 34 years, range 5 - 76 years
- Involves cerebrum
- References: Am J Surg Pathol 2001;25:1252
- Circumscribed, solitary and 57% cystic
- Poor prognostic factors: incomplete excision, atypical features, high cell proliferation rates; also older patient age
- 54 year old woman with atypical neurocytoma with oligodendroglia-like spread (Hum Pathol 2004;35:1156)
- Total excision prevents recurrence
- Sheets, clusters, ribbons or rosettes of monotonous tumor cells with round and regular vesicular nuclei and distinct nucleoli embedded in matrix of fine neuropil
- Ganglion cells in 66%
- Atypical if necrosis, vascular proliferation and 3+ mitotic figures/10 HPF
- Synaptophysin (strong), GFAP (46%; also is staining of entrapped astrocytes at periphery)
- Neuritic type processes with microtubules, dense core granules
- Ependymoma
- Oligodendroglioma: no ganglion cell differentiation, no salt and pepper chromatin and no neuropil islands; usually more infiltrative, synaptophysin-
- Uncommon, well differentiated, slow growing glioneuronal tumors composed predominantly of neoplastic mature ganglion-like cells (gangliocytoma) or a mixture of neoplastic ganglion-like cells and atypical glial cells (ganglioglioma)
- Ganglioglioma and gangliocytoma are considered discreet tumor entities by the 2021 WHO classification of CNS tumors
- Neoplastic ganglion cells often display dysplastic / dysmorphic features
- Characterized by molecular alterations resulting in activation of the MAPK pathway (BRAF V600E is most common)
- Commonly associated with epilepsy
- ICD-O:
- ICD-11:
- 2A00.21 & XH5FJ3 - mixed neuronal - glial tumors & ganglioglioma, NOS
- 2A00.21 & XH6KA6 - mixed neuronal - glial tumors & gangliocytoma
- Ganglioglioma:
- Incidence: 0.186 cases per 100,000 population worldwide (J Neurooncol 2017;131:525)
- ~2% of all primary intracranial tumors (Childs Nerv Syst 2016;32:1839, Cancer 2004;101:146)
- Predilection for children and young adults (majority are < 20 years old; median age is 12 years) but can occasionally be seen in older patients (N Engl J Med 2017;377:1648)
- Gangliocytoma:
- Generally similar to that of ganglioglioma
- Relative incidence in epilepsy related surgical resections is 1 - 3% (Brain Pathol 2012;22:350, N Engl J Med 2017;377:1648)
- Can occur throughout the CNS; temporal lobe is most common (~70%) (Acta Neuropathol 1994;88:166, J Neuropathol Exp Neurol 2002;61:575)
- Ganglioglioma:
- Results from genetic alterations that lead to activation of the MAPK pathway and therefore cause increased cell proliferation
- Gangliocytoma:
- Data not available but likely closely resembles that of ganglioglioma
- References: Acta Neuropathol Commun 2020;8:30, Folia Neuropathol 2013;51:283
- No known risk factors or environmental exposures for ganglioglioma or gangliocytoma
- Symptoms are related to the location of the tumor (N Engl J Med 2017;377:1648)
- Tumors in the cerebrum, especially temporal lobe, are associated with seizures / epilepsy (J Neuropathol Exp Neurol 2002;61:575)
- Vast majority are sporadic; small proportion are associated with neurofibromatosis type 1 (NF1) (J Neuropathol Exp Neurol 2008;67:240, Acta Neuropathol Commun 2018;6:47)
- Magnetic resonance imaging (MRI) of brain
- Stereotactic biopsy can be performed, especially in sites unamenable to surgical excision
- Circumscribed, cystic mass with enhancing mural nodule
- Classically, well delineated, T1 hypointense, T2 hyperintense cyst with enhancing mural nodule (Radiographics 2002;22:1177)
- Variable calcifications and enhancement
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
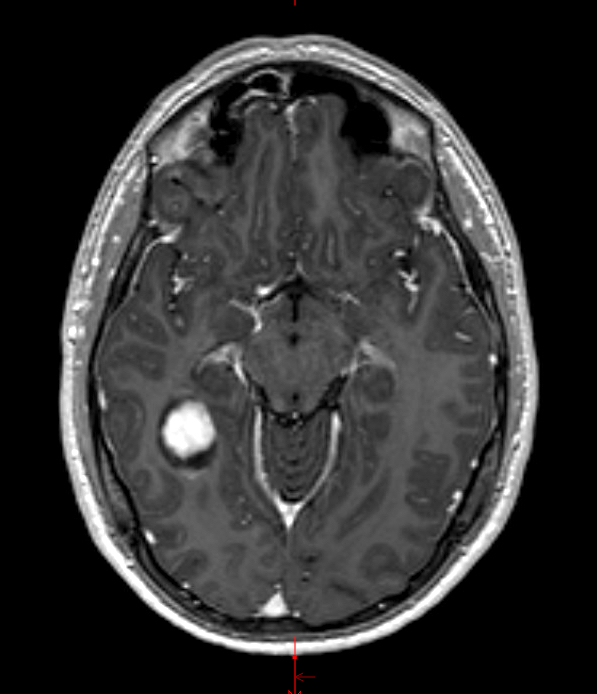

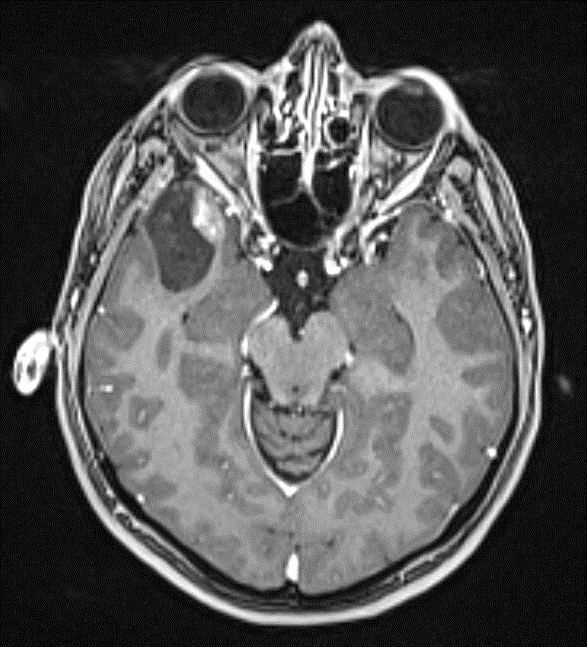
Temporal lobe mass
- Ganglioglioma:
- Low grade tumor with excellent overall prognosis (15 year overall survival is 83 - 94%) (J Neurosurg 2012;117:825)
- Complete surgical excision is best prognostic factor (Neuro Oncol 2014;16:409)
- Potential clinical benefit of RAF and MEK inhibitors is still under investigation (Neurology 2021;97:e673, Cancer Control 2021;28:10732748211040013)
- Although rare, the co-occurrence of BRAF V600E and CDKN2A / CDKN2B homozygous deletion or midline tumors with concurrent BRAF V600E and H3 K27M mutation seem to have worse outcomes (J Clin Oncol 2017;35:2934, Acta Neuropathol Commun 2018;6:47, Cancer Cell 2020;37:569, Brain Pathol 2018;28:103)
- See Microscopic (histologic) description for discussion about anaplastic ganglioglioma
- Gangliocytoma:
- Benign tumors with excellent long term survival
- Specific prognostic factors have not been identified
- 3 year old boy with progressive quadriparesis, gait disturbance and sphincter deficit (Prague Med Rep 2018;119:122)
- 16 year old girl with worsening vision (Surg Neurol Int 2020;11:392)
- 22 year old man with spinal cord lesion resected 9 years prior (Front Oncol 2019;9:177)
- 43 year old woman with chronic headaches (Medicine (Baltimore) 2018;97:e11583)
- Complete surgical resection, where possible (J Neurosurg 2012;117:825)
- Progressive, recurrent or refractory cases may benefit from RAF or MEK inhibitor treatment (J Clin Oncol 2013;31:e159, J Transl Med 2014;12:356)
- Well demarcated, variably solid and cystic mass forming lesion
- Lacks areas of hemorrhage and necrosis
- Well demarcated mass but can show brain infiltration at the tumor edge
- Admixture of neuron-like cells and atypical glial cells (ganglioglioma) or predominantly neuron-like cells (gangliocytoma)
- Variable microcalcifications, perivascular lymphocytes, eosinophilic granular bodies
- Reference: Acta Cytol 2022;66:142
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
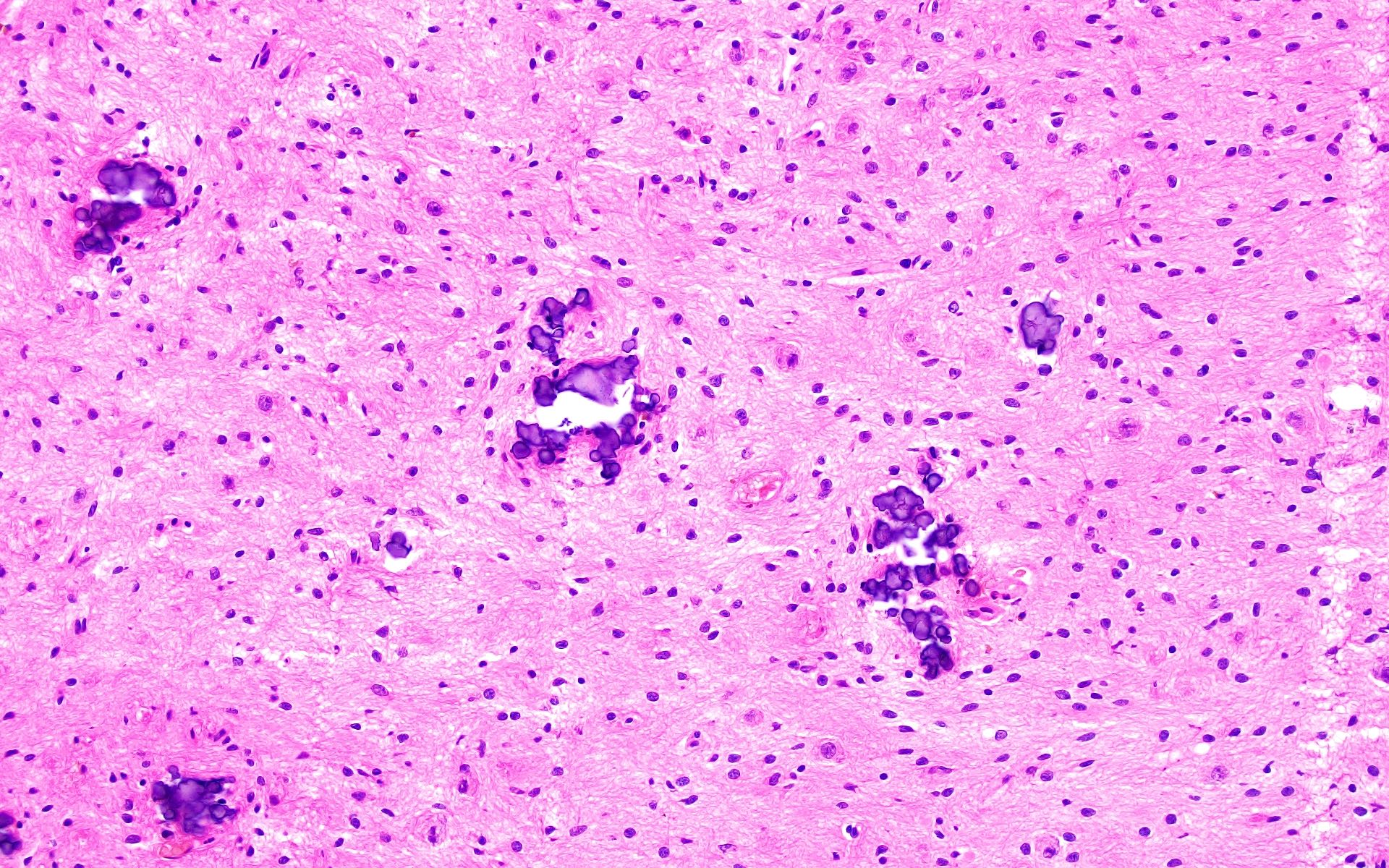
Low grade glioneuronal tumor
Admixture of mildly atypical glial cells
- Ganglioglioma:
- Biphasic tumor with variable mixture of mature appearing ganglion-like cells and atypical glial cells
- Ganglion-like cells demonstrate dysmorphic features:
- Binucleation
- Neuron clustering (kissing neurons)
- Cytomegaly with ballooning cytoplasm
- Peripheral accumulation of Nissl substance
- Lack of organized cytoarchitecture (Acta Neuropathol 1994;88:166)
- Atypical glial cells are hyperchromatic, moderately enlarged and resemble those of fibrillary astrocytomas, pilocytic astrocytoma or oligodendroglioma (Hum Pathol 2016;49:107, Clin Interv Aging 2014;9:1981)
- Eosinophilic granular bodies more common than Rosenthal fibers (Acta Neuropathol 1994;88:166)
- Other common findings:
- Dystrophic calcifications
- Perivascular lymphoid infiltrates
- Prominent capillary network
- Low to absent mitotic activity (J Neuropathol Exp Neurol 2002;61:575)
- Compact growth but microscopic infiltration can be seen along the tumor edge (Acta Neuropathol 2014;128:39, Nat Rev Neurol 2016;12:732)
- Rare cases with anaplastic features (anaplastic ganglioglioma) have been reported:
- Tumors with ganglioglioma-like histology and increased mitoses, microvascular proliferation, necrosis
- Many reported cases lack molecular analyses to exclude other high grade gliomas (J Neuropathol Exp Neurol 2016;75:971, Neuro Oncol 2017;19:678)
- Anaplastic ganglioglioma is a term that should be used with caution, especially if complete molecular characterization is not available (Neuropathol Appl Neurobiol 2022;48:e12847)
- Gangliocytoma:
- Generally similar to that of ganglioglioma except lacking atypical neoplastic glial cells
- Composed almost entirely of large, dysmorphic ganglion cells (as described above), often arranged in irregular clusters
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
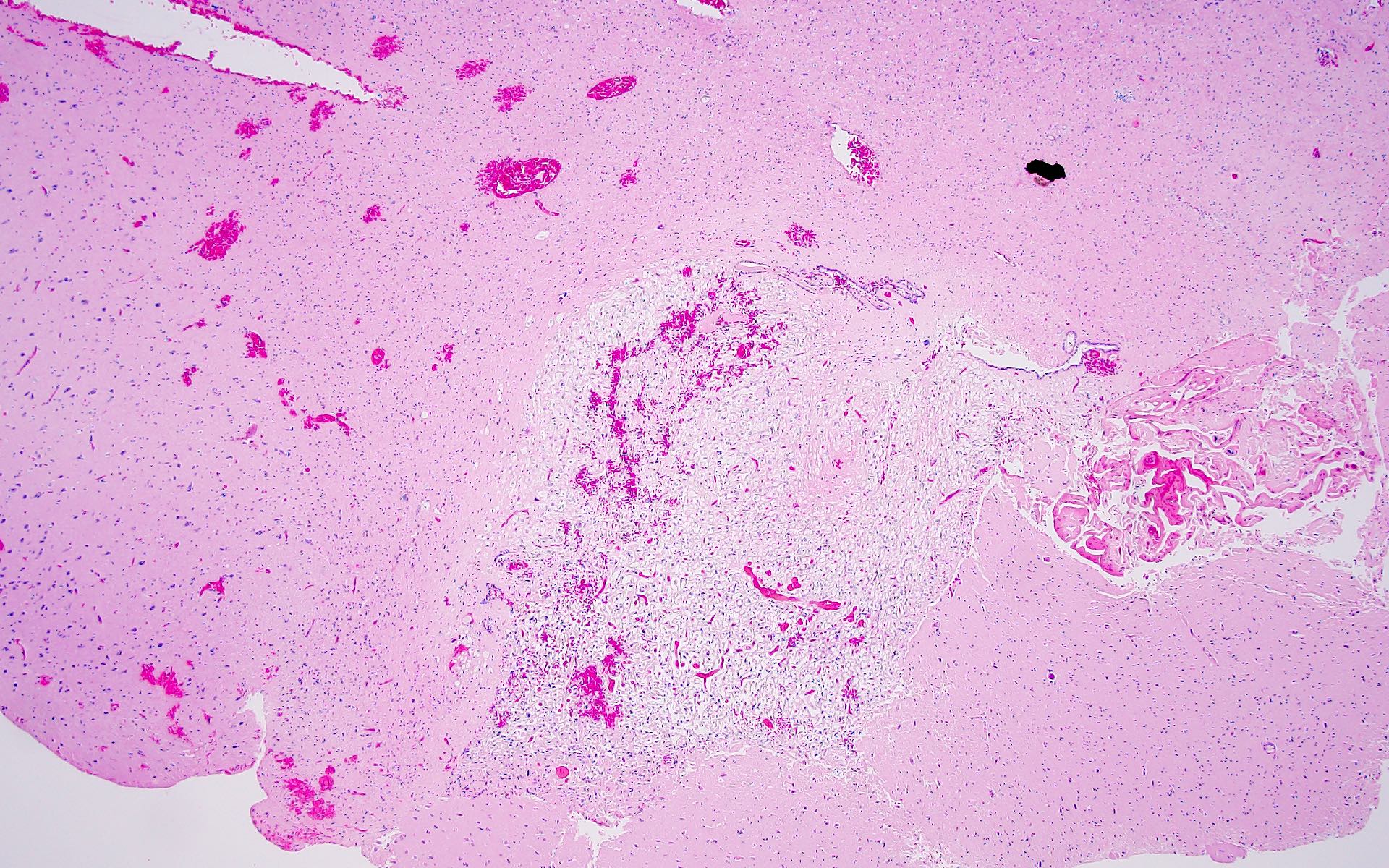
Well circumscribed brain lesion
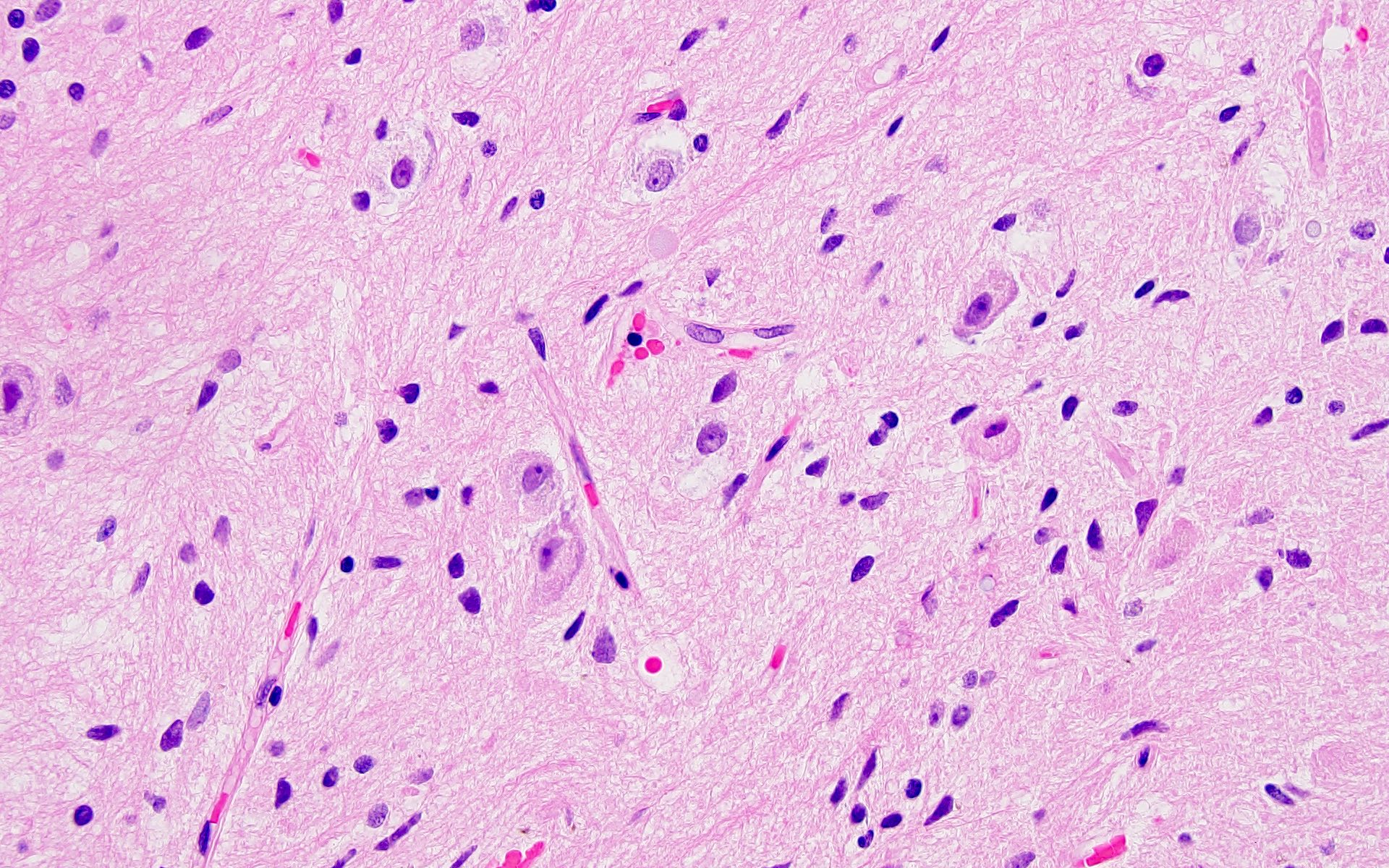
Mixed glioneuronal tumor
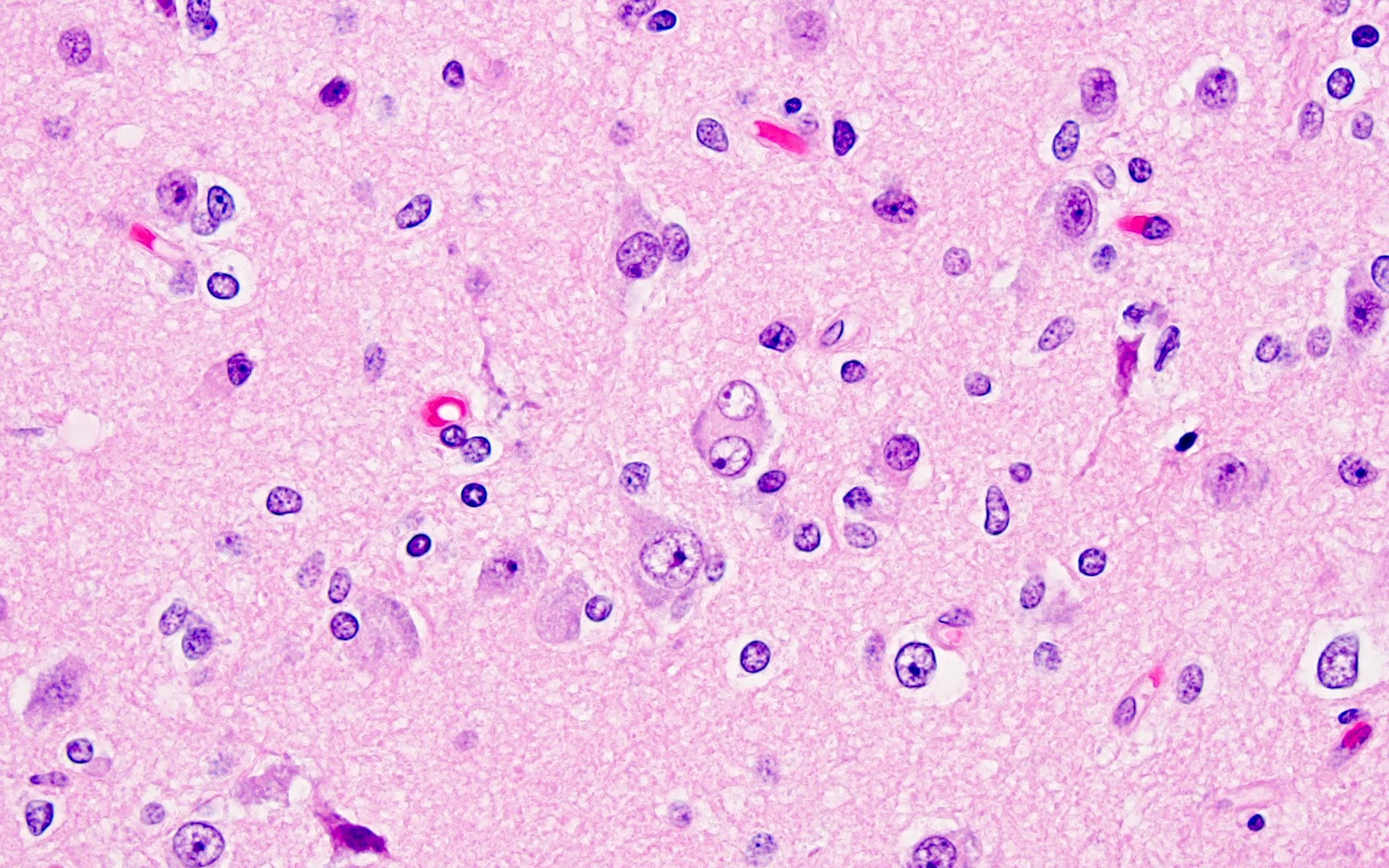
Ganglioglioma with binucleate neurons
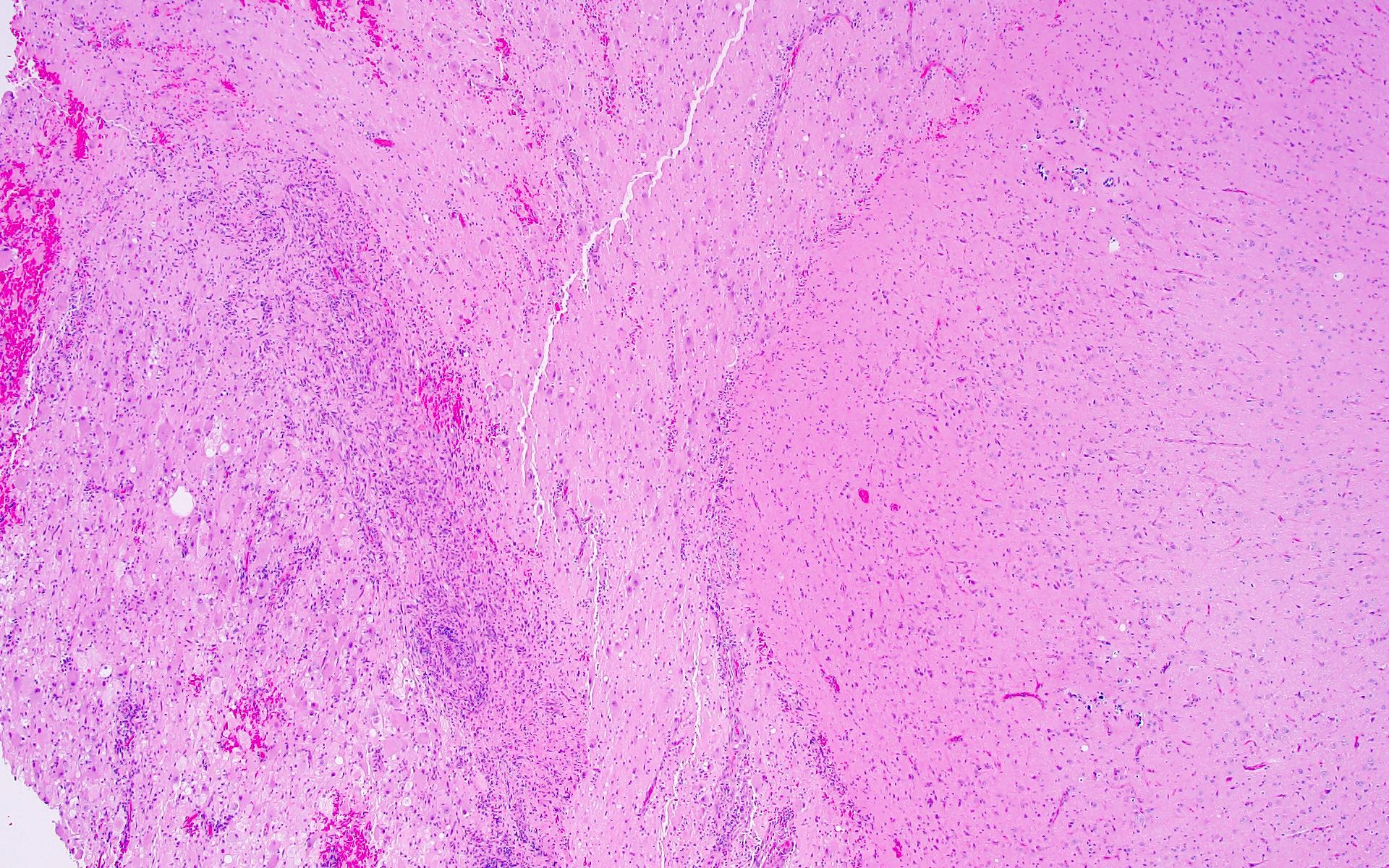
Well demarcated mass
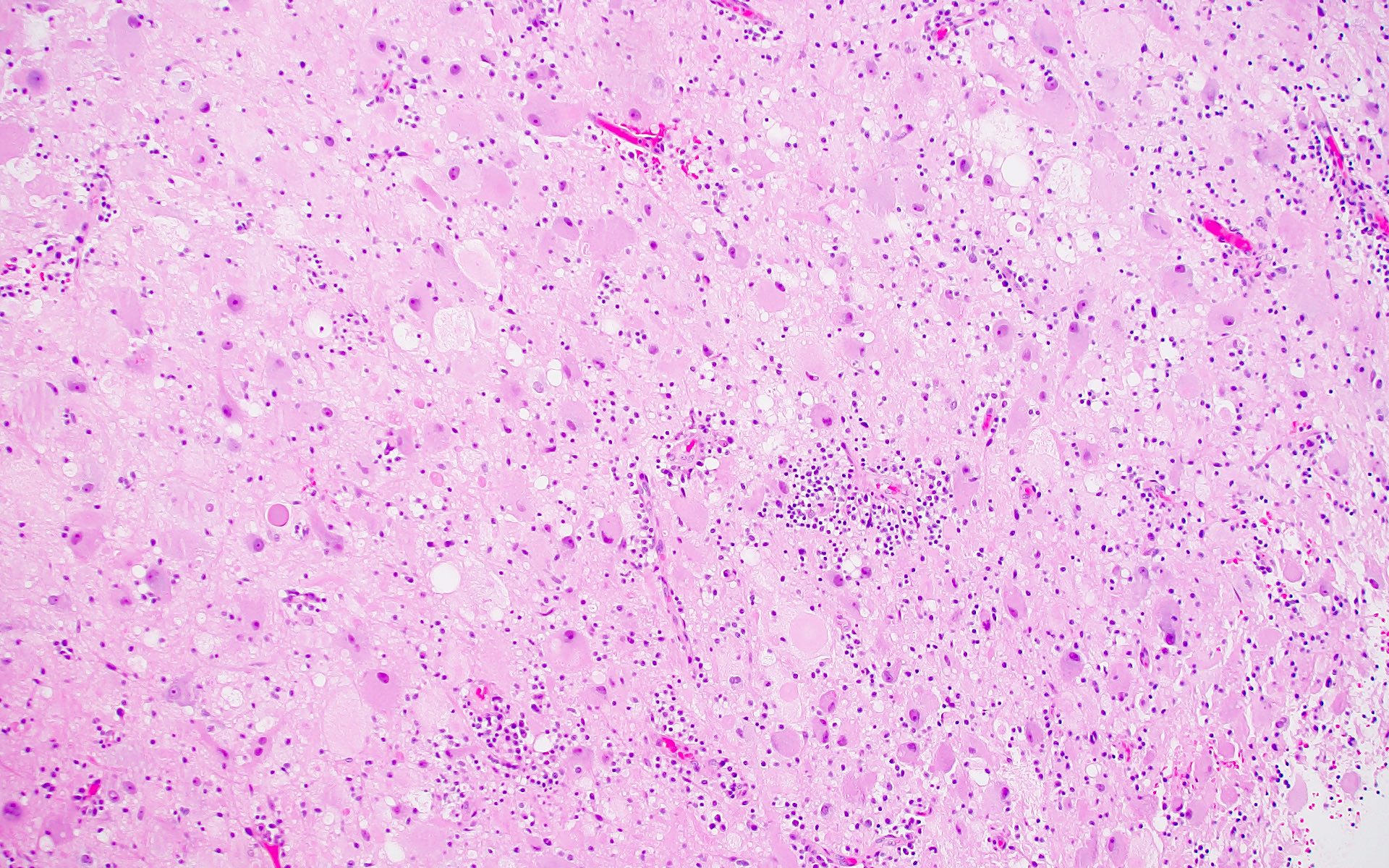

Dysplastic ganglion-like cells in gangliocytoma
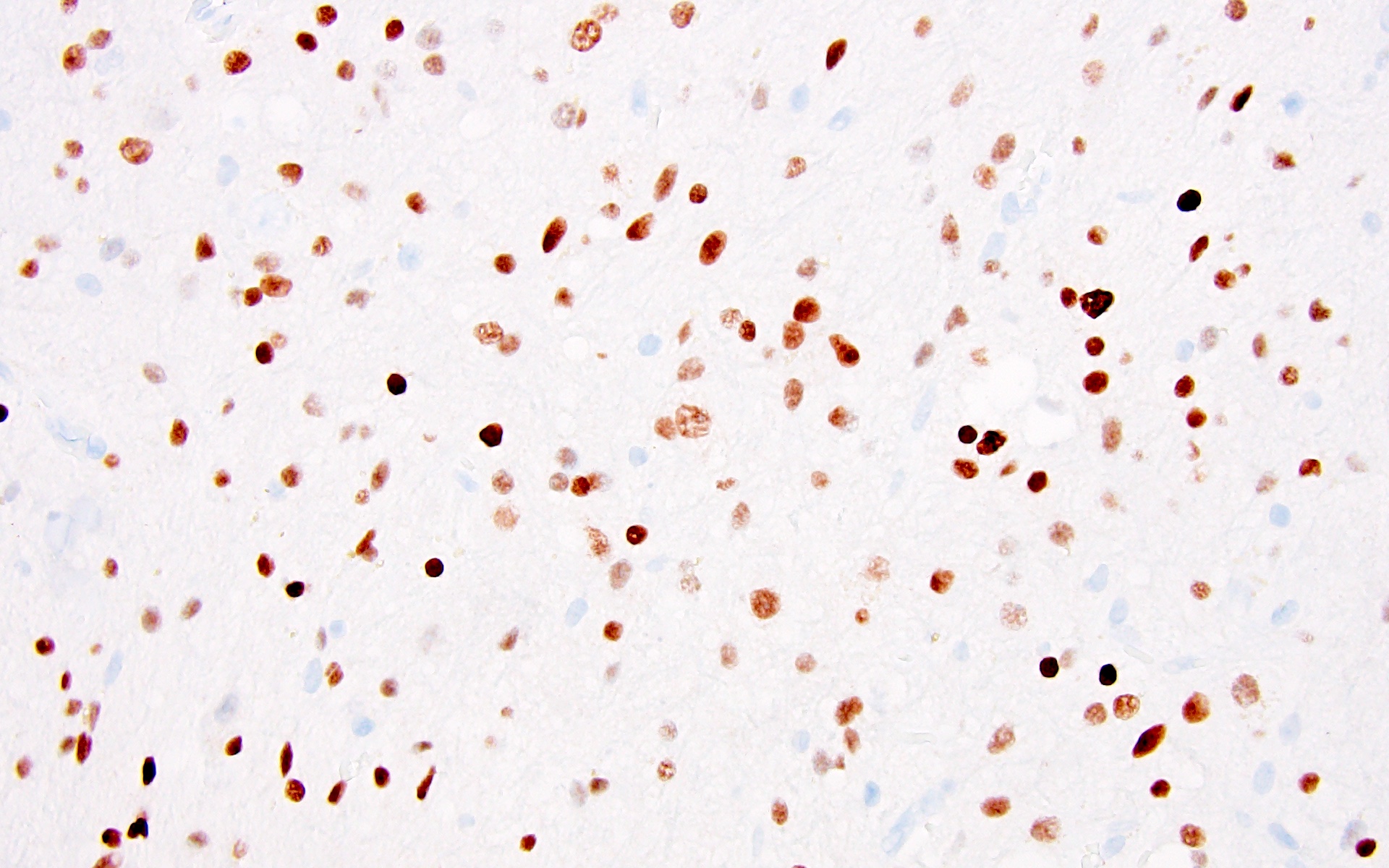
Olig2 IHC
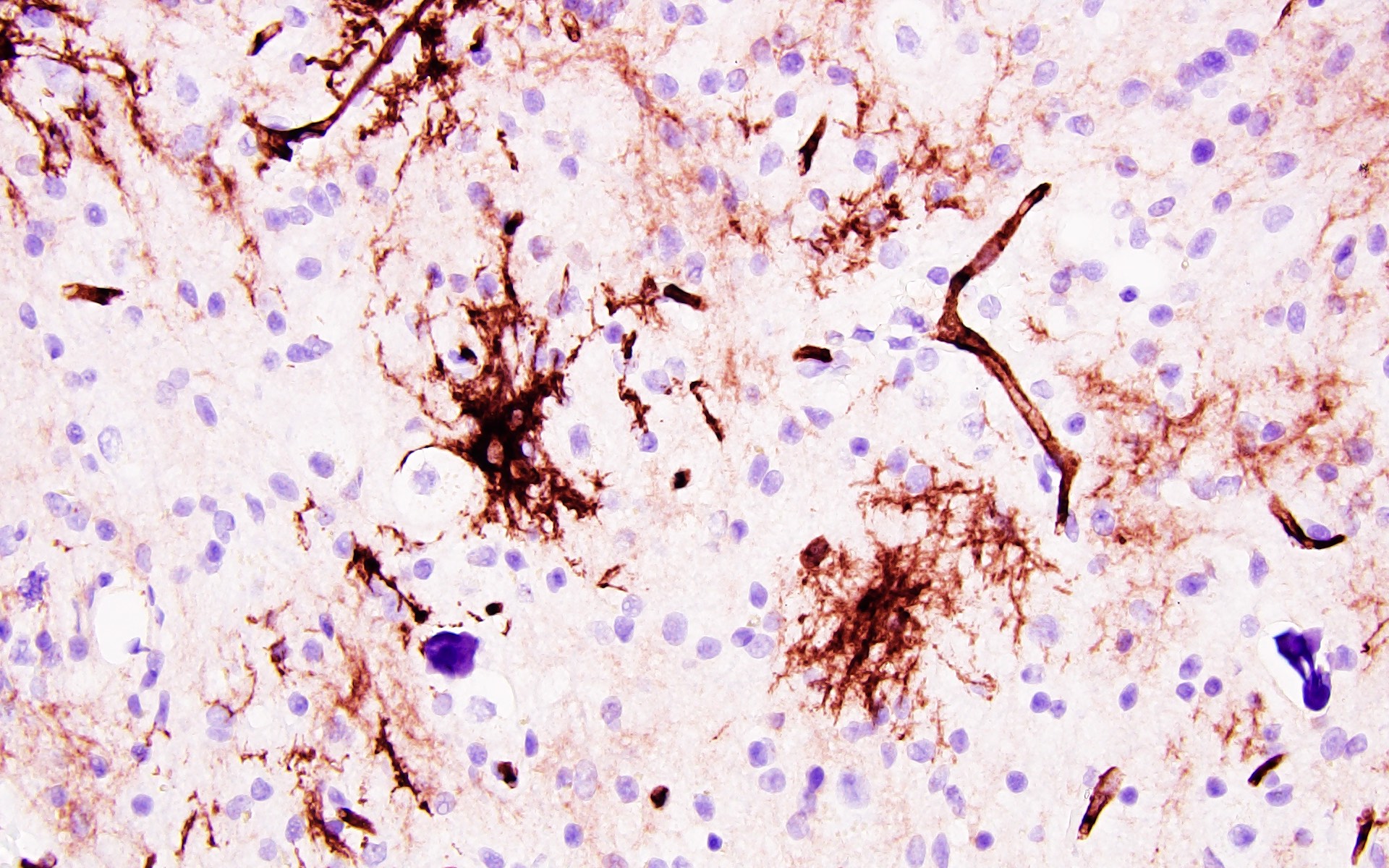
CD34 IHC

Synaptophysin IHC
Images hosted on other servers:

Circumscribed glioneuronal tumor
- Ganglion-like cells: MAP2, neurofilament, synaptophysin (neural markers)
- No reliable marker to distinguish nonneoplastic neurons from neoplastic ganglion-like cells
- Neoplastic glial cells: GFAP, Olig2 (Cancer 1997;79:989)
- CD34, either diffuse or patchy (arborescent pattern), within the tumor or adjacent cortex (Acta Neuropathol 1999;97:481)
- Ki67 low (< 5%) (J Neuropathol Exp Neurol 1995;54:513)
- BRAF V600E mutation specific antibody is available and generally positive in dysplastic ganglion-like cells (Acta Neuropathol 2013;125:891)
- Ganglion-like cells often lack NeuN immunoreactivity (which is positive in nonneoplastic neurons)
- Negative for IDH1 R132H staining
- Not routinely used for clinical / diagnostic purposes
- Ultrastructural characteristics of ganglioglioma include:
- Ganglion-like cells with numerous dense core granules within the perikarya and cellular processes and abundant rough endoplasmic reticulum (Cancer 1997;79:989)
- Ganglioglioma:
- BRAF V600E is most common molecular alteration, seen in 10 - 60% of tumors (highest occurrence in cortical tumors, lowest in spinal cord tumors) (Acta Neuropathol 2011;121:397, Acta Neuropathol 2016;131:833, Hum Pathol 2016;49:107)
- Other alterations include other BRAF mutations, BRAF gene fusions (most commonly with KIAA1549), RAF1 gene fusions, KRAS activating mutations and NF1 inactivating mutations / deletions (Acta Neuropathol Commun 2018;6:47)
- BRAF V600E mutation can be detected by immunohistochemistry or gene sequencing (including droplet digital PCR, ddPCR) (Acta Neuropathol 2013;125:891)
- If negative for BRAF V600E, other MAPK activating alterations should be considered utilizing a large next generation sequencing panel or gene fusion panel (Acta Neuropathol Commun 2018;6:47)
- Ganglioglioma demonstrates a unique genome wide methylation pattern, which can be helpful in diagnostically challenging cases (Nature 2018;555:469)
- Gangliocytoma:
- No signature molecular profile published to date
Neuronal and glioneuronal tumors
by Dr. Fausto Rodriguez (PathCast)
- Brain mass, temporal lobe, resection:
- Ganglioglioma, BRAF V600E mutant, WHO grade 1
- Histologic classification: ganglioglioma
- WHO grade: 1
- Molecular information: BRAF V600E
- Focal cortical dysplasia (FCD):
- Presence of MAPK activating alteration (such as BRAF V600E) can be helpful to exclude FCD
- If any genetic alterations are identified, FCD generally harbors alterations in the PI3K / AKT / mTOR pathway (Epilepsia 2011;52:158, Neuropathol Appl Neurobiol 2018;44:6)
- Dysplastic neurons typically retain NeuN immunoreactivity and shows poorly delineated cortical thickening, rather than a well circumscribed tumor mass
- Infiltrating low grade glioma:
- NeuN positive neurons with a neoplastic glial population are more in keeping with an infiltrating low grade glioma with entrapped neurons
- Will often see IDH1 / IDH2 mutations in low grade infiltrating gliomas
- Low grade infiltrating gliomas in children can have BRAF V600E mutations or MAPK alterations but will not show dysplastic ganglion cells
- Polymorphous low grade neuroepithelial tumor of youth (PLNTY):
- FGFR2 / FGFR3 gene fusions or BRAF V600E mutation
- Numerous calcifications and diffuse CD34 immunoreactivity (Acta Neuropathol 2017;133:417)
- At least focally infiltrative growth and oligodendroglial-like component
- Dysembryoplastic neuroepithelial tumor (DNT):
- Multinodular growth
- Floating neurons in mucoid matrix
- Frequently FGFR1 alterations
- Typically lack of CD34 staining (Neuropathol Appl Neurobiol 2019;45:95)
- Multinodular vacuolating neural tumor (MVNT):
- Multinodular growth typically without dysmorphic or binucleate neurons
- Lack of CD34 staining, Olig2 positive neuronal cells and prominent vacuolization (Acta Neuropathol 2018;135:485)
- Pleomorphic xanthoastrocytoma (PXA):
- Presence of xanthomatous cells, nuclear pleomorphism, reticulin rich network surrounding individual tumor cells and co-occurrence of both BRAF V600E and CDKN2A / CDKN2B homozygous deletion (Brain Pathol 2019;29:85)
- Desmoplastic infantile ganglioglioma (DIG):
- Usually occurs in infants and present with much larger tumors
- Histology demonstrates markedly desmoplastic stroma
- Hypothalamic hamartoma (Pediatr Dev Pathol 2018;21:324, Eur J Hum Genet 2015;23:92):
- Congenital lesion located along the floor of the hypothalamus
- Patients often present with gelastic seizures
- Pathognomonic lesion for Pallister-Hall syndrome (autosomal dominant, associated with mutations of GLI3)
- Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease):
- Unique cerebellar lesion in patients with Cowden syndrome (PTEN hamartoma syndrome) (Hered Cancer Clin Pract 2010;8:6)
- Loss of PTEN, though MAPK alterations typically seen in ganglioglioma are absent
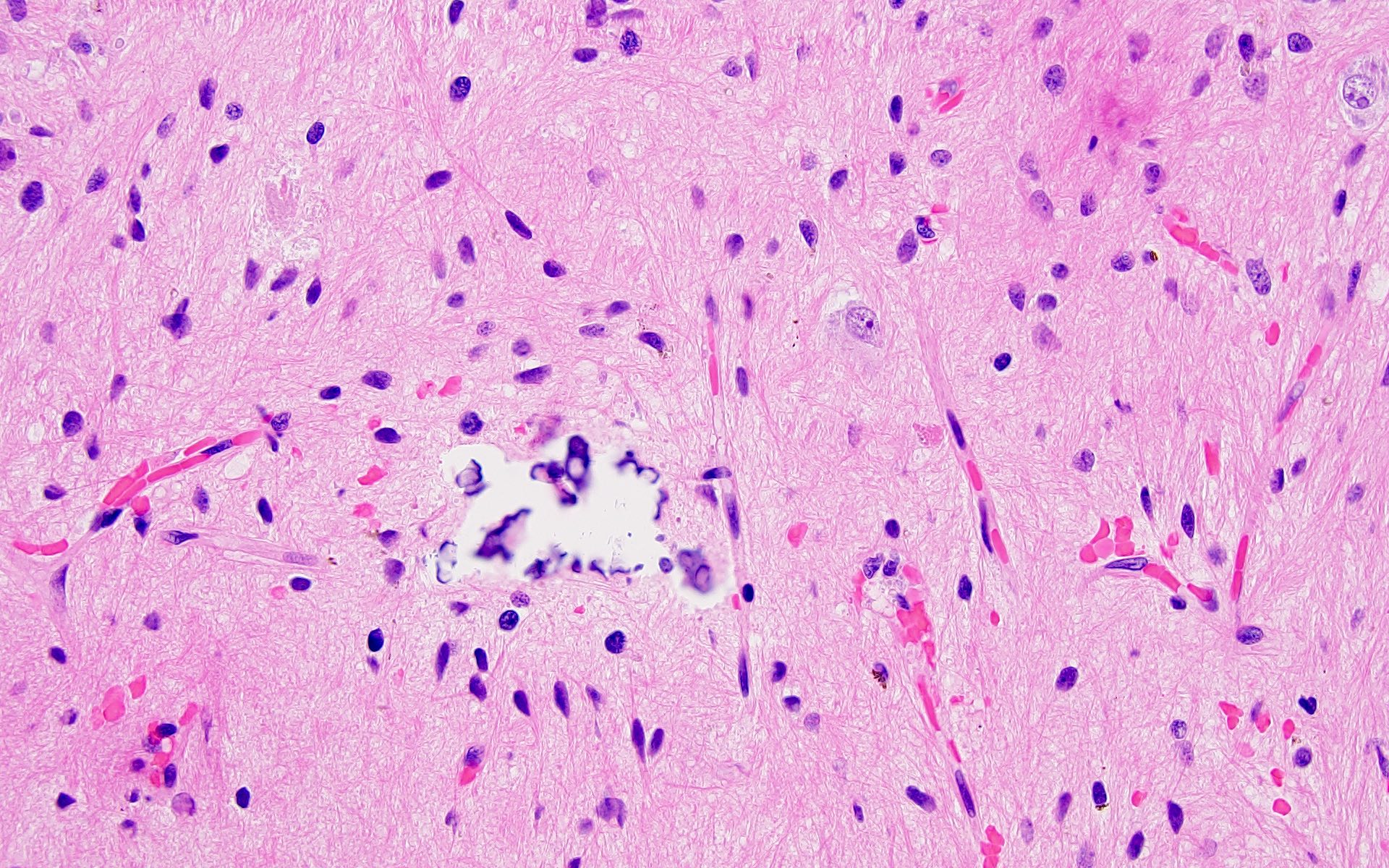
A 15 year old patient presents with chronic seizures that are refractory to medical management. Magnetic resonance imaging (MRI) of the brain reveals a cystic lesion with an area of solid enhancement located in the medial temporal lobe. Surgical resection was performed, revealing the histology shown above. What is the most likely diagnosis?
- Ganglioglioma
- Glioblastoma
- Oligodendroglioma
- Pilocytic astrocytoma
- Subependymal giant cell astrocytoma
Comment Here
Reference: Gangliocytoma & ganglioglioma
- BRAF V600E mutation
- H3 K27M mutation
- IDH1 R132H mutation
- NF1 mutation
- NF2 mutation
Comment Here
Reference: Gangliocytoma & ganglioglioma
- Prognosis poorer than germinoma - median survival 22 months in cases with high hCG levels (J Neurooncol 2004;66:225)
- Serum levels of hCG are helpful
Case reports
- 1 month old infant with intracranial choriocarcinoma (Arch Pathol Lab Med 1990;114:1079)
- 8 year old boy with intracranial choriocarcinoma causing precocious puberty and cured with combined modality therapy (J Paediatr Child Health 1993;29:464)
Microscopic (histologic) description
- Syncytiotrophoblasts (large multinucleated cells) and cytotrophoblasts
Positive stains
Differential diagnosis
- Metastatic choriocarcinoma:
- From gonads or placenta
- Prognosis poorer than germinoma
- May be associated with precocious puberty
- Characterized by rapid and bulky growth and spread to liver and lungs
- 60% have metastases at presentation
Treatment
- Chemotherapy
- Radiation therapy
- Surgery
Positive stains
- Most common intracranial germ cell neoplasm
- Often teenagers and young adults
- Two - thirds male
- May be mixed with other germ cell tumors
- May derive from ectopic rests, transformation of resident germ cells or migration of germ cells late in development
- Most common site is pineal region
- Also anterior or posterior third ventricle, rarely fourth ventricle
- Rarely associated with dysgenetic syndromes
- Immunostains useful because biopsy is often small
- Relatively good prognosis (5 - 10 year survival is 75 - 95%) versus 25 - 40% for nongerminoma germ cell tumors (Pediatr Neurol 2002;26:369)
- Very sensitive to radiotherapy and chemotherapy; nongerminomatous germ cell tumors are less radiosensitive
- Metastases may be due to surgical displacement of tumor
- Spinal cord metastases occur in 10 - 15% of patients
Staging / staging classifications
- T1: smaller than 5 cm in diameter and located in the suprasellar, intrasellar or pineal region
- T2: larger than 5 cm in diameter and located in the perisellar region
- T3: may be smaller than 5 cm in diameter but invades and encroaches on the third ventricle
- T4: extends into the anterior, middle or posterior fossa
- N: not indicated for CNS tumors
- M0: no evidence of gross subarachnoid or hematogenous metastasis
- M1: microscopic tumor cells found in the CSF
- M2: gross nodular seeding in the ventricular system or cranial subarachnoid spaces
- M3: gross nodular seeding in the spinal subarachnoid spaces
- M4: metastasis outside the cerebrospinal axis
Case reports
- 23 year old man with headaches and visual difficulties (Arch Pathol Lab Med 2003;127:497)
Treatment
- Resection difficult due to high collagen content
- Radiation therapy helpful
Gross description
- Soft, gray-pink, homogenous
- Variable encapsulation
- Usually poorly circumscribed and infiltrative
Microscopic (histologic) description
- Resembles seminoma / dysgerminoma
- Large, epithelioid cells with abundant PAS+ cytoplasm, large, round nuclei and irregular and pleomorphic nuclei
- May have prominent nests of lymphocytes with occasional granulomatous inflammation that may obscure tumor cells (Neurol Med Chir (Tokyo) 2005;45:415)
- Lymphocytes may smear in small biopsies
- Frequent mitotic activity and necrosis
- Syncytiotrophoblasts in 14%
- Less anaplasia than embryonal carcinoma
- No cells intermediate in size between lymphocytes and large germinoma cells
Cytology description
- Loose fragments or single large pleomorphic and polygonal cells with vacuolated cytoplasm, enlarged oval nuclei and prominent nucleoli
- Frequent mitotic figures, naked nuclei, foamy background
- Also smashed lymphoid cells with streaking
Positive stains
- PLAP, PAS+ cytoplasm, CD117 / c-kit, OCT4 (strong, often diffuse)
Negative stains
Electron microscopy description
- Glycogen in cytoplasm, sparse cytoskeletal elements, prominent nucleoli
Differential diagnosis
- Carcinoma:
- Usually metastatic, keratin+, EMA+, 13% are PLAP+
- Embryonal carcinoma:
- 25% are PLAP+
Additional references
- Tissue derived from ectoderm, endoderm and mesoderm (at least 2 of 3 germinal layers)
- Usually well differentiated / grade I of IV
- Incidence varies with age: 30 - 50% of congenital brain tumors, 2% of brain tumors in infants and children, 0.5% at all ages
- Congenital cases are usually fatal; may replace cerebral hemispheres (Pediatr Pathol 1987;7:333)
- Mature teratomas:
- Have well differentiated tissue from all three germinal layers, including neuroectoderm, epithelium (solid, cystic, glandular or tubular), cartilage or other mesenchymal elements, glial and neuronal tissue
- Immature teratomas:
- Have less differentiated tissue from any of the three germinal layers
- 50% with intracranial tumors die within one year
- Pineal teratomas are more common in males but saccrococcygeal teratomas are more common in females
- Must sample thoroughly for correct diagnosis
Terminology
- Growing teratoma syndrome
- Enlarging teratoma after tumor treatment (Neurosurgery 2005;56:188)
Prognostic factors
- Poor prognostic factors: tissue resembling medulloepithelioma, neuroblastoma, retinoblastoma or ependymoblastoma
Case reports
- 27 week old fetus with massive macrocephaly and generalized anasarca (Arch Pathol Lab Med 2004;128:102)
- Congenital intracranial teratoma of the lateral ventricle (Neurol India 2001;49:170)
- 3 day old girl with congenital intracranial immature teratoma of the lateral ventricle (Neurol Res 2005;27:53)
Treatment
- Newborns: complete surgical excision (difficult)
Additional references
- Rare intracranial tumor, usually in pineal or suprasellar regions
- Also called endodermal sinus tumor
- Prognosis poorer than germinoma (median survival 2 years or less)
Case reports
- 15 year old girl with an unusual presentation of an intraparenchymatous frontal yolk sac tumor (Neurol India 2001;49:395)
- 22 year old man with Down syndrome and pineal yolk sac tumor with a solid pattern (J Clin Pathol 2004;57:882)
Gross description
- Usually large
Microscopic (histologic) description
- Tubulopapillary structures with vacuolated cuboidal cells, cystic spaces with eosinophilic hyaline bodies and Schiller-Duval bodies
Positive stains
Additional references
- An aggressive, infiltrating, astrocytic glioma that lacks mutations in IDH1, IDH2 and histone H3 genes and is:
- Histologically defined by brisk mitotic activity and microvascular proliferation or necrosis
- Or molecularly defined by the presence of TERT promoter mutation, EGFR gene amplification or copy number changes in the form of combined gain of chromosome 7 and loss of chromosome 10 (Acta Neuropathol 2018;136:793, Acta Neuropathol 2018;136:805)
- Characterized by diffusely infiltrative growth pattern with nuclear atypia and either:
- Mitotic activity, necrosis or microvascular proliferation or
- Molecularly defined by the present of TERT promoter mutation, EGFR amplification or combined gain of chromosome 7 and loss of chromosome 10
- Lacks mutations in IDH1 / IDH2 and histone H3 genes
- Various morphologic subtypes have been recognized (giant cell, small cell, epithelioid, sarcomatous / gliosarcoma) with similar prognosis
- Primitive neuronal component has increased rate of cerebrospinal fluid dissemination (Brain Pathol 2009;19:81)
- Glioblastoma, IDH wild type
- Glioblastoma multiforme (not recommended)
- Diffuse astrocytoma with molecular features of glioblastoma (no longer recommended)
- ICD-O: 9440/3 - glioblastoma, NOS
- ICD-11: 2A00.00 & XH5571 - glioblastoma of brain & glioblastoma, IDH wild type
- Most common and most malignant astrocytic glioma in adults (Pharmacol Res 2021;171:105780, Cancer Epidemiol Biomarkers Prev 2014;23:1985, StatPearls: Glioblastoma Multiforme [Accessed 5 July 2022])
- Accounts for 14.3% of all primary CNS tumors and 49.1% of all malignant CNS tumors in adults and up to 2.2% of all CNS tumors in children (Neuro Oncol 2021;23:iii1)
- More common in males than females (1.6:1)
- More common in older adults above the age of 55
- Highest incidence between the ages of 75 - 84 years
- Incidence rate by race: white to black = 1.98:1; white to Asian or Pacific Islander = 2.44:1
- Most commonly in supratentorial regions (frontal, temporal, parietal and occipital lobes), with highest incidence in the frontal lobes
- Most often centered in subcortical white matter
- Many cases show infiltration into cortex and across the corpus callosum with spread to contralateral hemisphere
- Rare cases reported in the cerebellum and spinal cord (World Neurosurg 2013;80:e237, J Neurosurg Spine 2010;13:67, Neurooncol Adv 2021;3:vdab154)
- Cell of origin undetermined
- Some studies suggest a variety of CNS cell types can undergo malignant transformation with features of glioblastoma (GBM) (oligodendrocyte precursor cells, neural precursor cells, astrocytes and neurons)
- Sequencing of human glioblastomas suggests that a neural precursor cell in the subventricular zone may be the cell of origin (Nature 2018;560:243)
- Etiology of most GBMs remains unknown
- Rare cases associated with genetic tumor syndromes: Lynch syndrome, Li-Fraumeni syndrome, tuberous sclerosis and neurofibromatosis type 1 (Neuro Oncol 2014;16:896)
- Only validated risk factor is ionizing radiation to the head and neck (Neurooncol Adv 2021;3:vdab109, Neuro Oncol 2002;4:278, UpToDate: Risk Factors for Brain Tumors [Accessed 5 July 2022])
- Some radiation associated gliomas with histologic features of glioblastoma may be molecularly more consistent with diffuse pediatric type high grade glioma, H3 wild type and IDH wild type (Acta Neuropathol 2019;137:139)
- Conflicting evidence over a possible association with traumatic brain injury (TBI) (Surg Neurol Int 2016;7:78, J Int Med Res 2018;46:2170, Cureus 2020;12:e8019)
- Symptoms are dependent on tumor location
- May present with signs of increased intracranial pressure (i.e., headaches, nausea, emesis), seizures or focal neurologic deficits (visual field defects, hemiparesis, aphasias, etc.) (BMJ 2021;374:n1560)
- Computed tomography (CT) and magnetic resonance imaging (MRI) for radiologic assessment
- Biopsy or surgical resection required for definitive diagnosis
- May be able to detect glioblastoma by circulating tumor DNA (ctDNA) in the blood and cerebrospinal fluid (CSF) of some patients, though this is under investigation (J Neurosurg 2018;129:334, Cancers (Basel) 2019;11:950, Mol Oncol 2022;16:2098, Cancers (Basel) 2020;12:1219)
- MRI: T2 / fluid attenuated inversion recovery (FLAIR) bright infiltrative lesion(s) with postcontrast T1 showing irregular peripheral rim enhancement with central necrosis
- May cross the corpus callosum
- May be multifocal
- Lack of contrast enhancement may be observed in molecularly defined glioblastoma
- Certain subtypes (i.e., gliosarcoma, epithelioid, giant cell) may appear well circumscribed (Neurosurg Rev 2021;44:3335, Eur Radiol 2019;29:429, Surg Pathol Clin 2020;13:249)
Contributed by Bharat Ramlal, M.D.
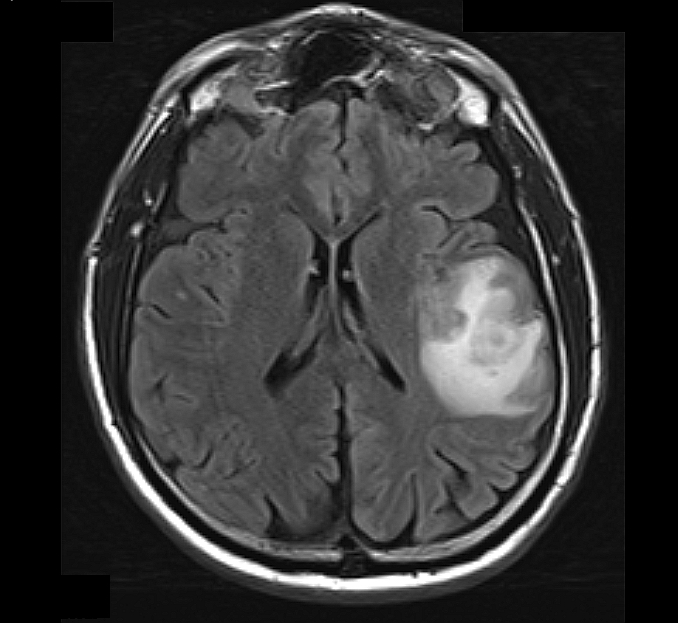
T2 FLAIR MRI
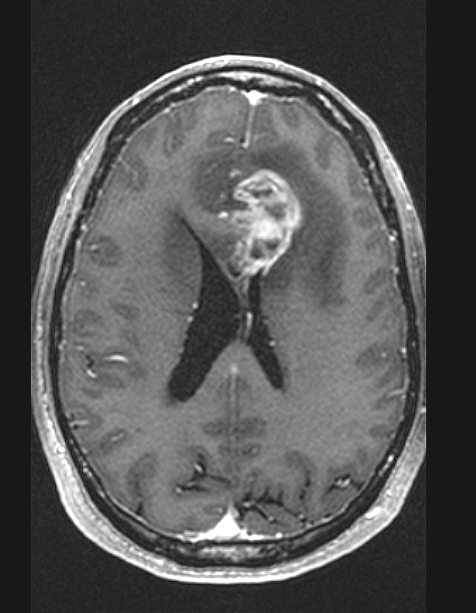

T1 postcontrast MRI
- Poor prognosis, with a median survival of 8 months and 5 year survival rate of only 6.8% (Neuro Oncol 2021;23:iii1)
- Most patients die within 15 - 18 months after therapy with chemoradiation
- Longer survival is observed in patients who are diagnosed at a younger age (< 50 years), have high performance status and gross total resection (often difficult) (J Clin Oncol 2009;27:5743)
- MGMT promoter methylation is a positive predictor of response to alkylating and methylating chemotherapy and may be an independent prognostic factor for longer overall survival (J Cell Physiol 2018;233:378, J Clin Oncol 2009;27:5743)
- Brisk cytotoxic T cell infiltrates may be associated with longer survival (J Clin Neurosci 2010;17:1381)
- FGFR3::TACC3 fusion GBMs may be associated with better outcomes and may be a target for therapy (Neuro Oncol 2020;22:1614, Clin Cancer Res 2015;21:3307)
- Shorter survival times than patients with IDH mutant grade 4 astrocytomas with similar histological findings (Clin Cancer Res 2013;19:5146, Acta Neuropathol 2018;136:153)
- 46 year old man with glioblastoma and subsequent scalp and pulmonary metastases (J Int Med Res 2020;48:300060520930459)
- 47 year old woman with primary intraventricular epithelioid glioblastoma (World Neurosurg 2018;112:257)
- 47 year old man with traumatic brain injury secondary to a fall and subsequent development of GBM (Cureus 2020;12:e8019)
- 76 year old woman with primary glioblastoma of the cauda equina (Neurooncol Adv 2021;3:vdab154)
- Surgical resection where possible in younger patients (≤ 70 years old) and patients with good performance status, followed by radiotherapy with concurrent and adjuvant temozolomide (TMZ)
- Unmethylated tumors, standard brain radiotherapy alone may be attempted (NCNN: NCNN Guidelines - Central Nervous System Cancers [Accessed 5 July 2022])
- Tumor treating fields (TTFields / Optune) under investigation - alternating electric field therapy using low intensity energy to stop glioma proliferation; relatively recent treatment option with rare reports showing favorable outcomes (Front Oncol 2020;10:477)
- Ill defined whitish gray mass with areas of hemorrhage and necrosis
- Can expand gyri and cross the corpus callosum
- Hypercellular infiltrative lesion with variable morphology
- Infiltration often difficult to assess on frozen sections but entrapped neurons may be useful
- Nuclear hyperchromasia and nuclear elongation, possible giant cells
- Mitotic activity
- Necrosis; geographic or pseudopalisading
- Microvascular proliferation
Contributed by Bharat Ramlal, M.D.
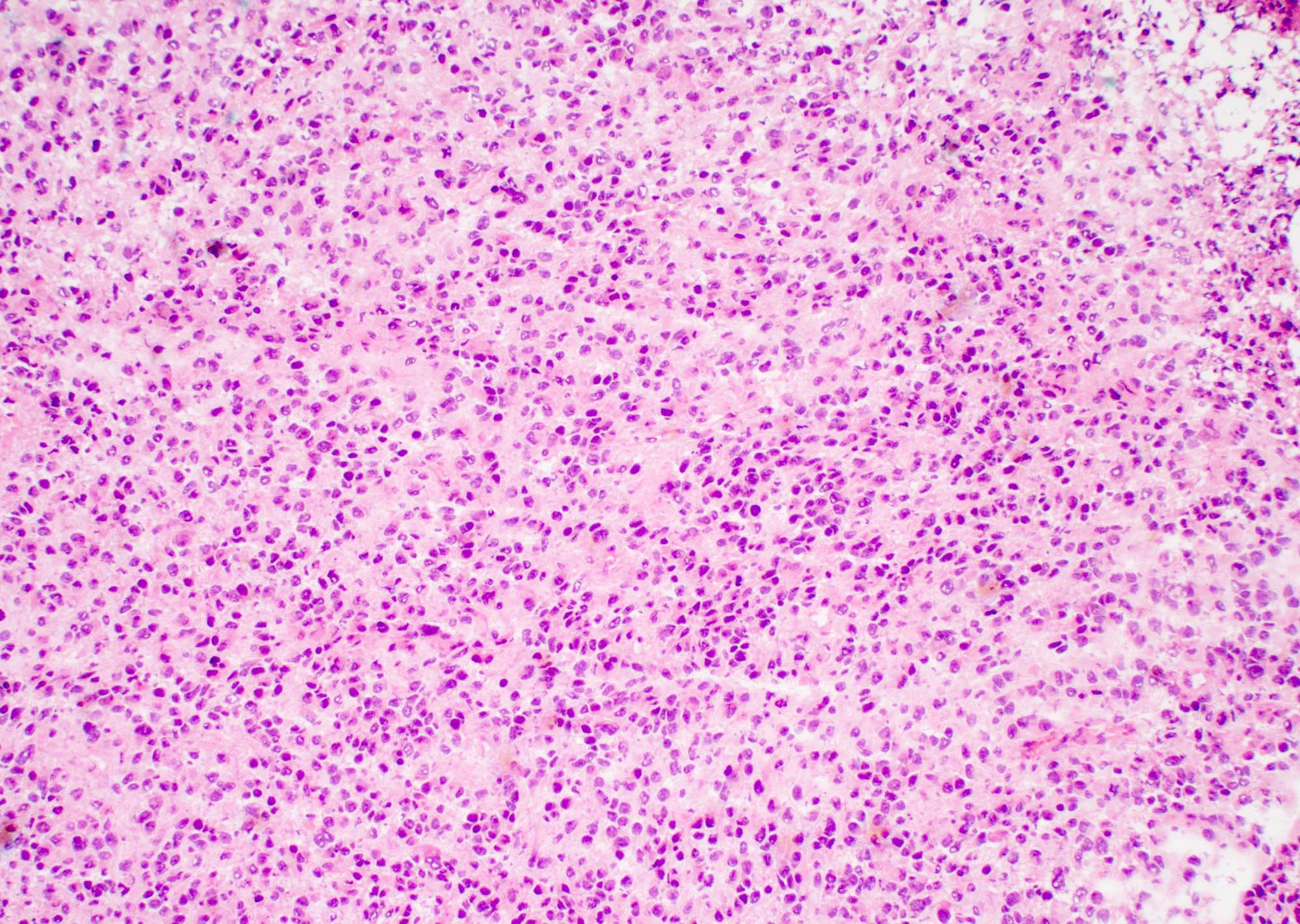
Markedly hypercellular neoplasm
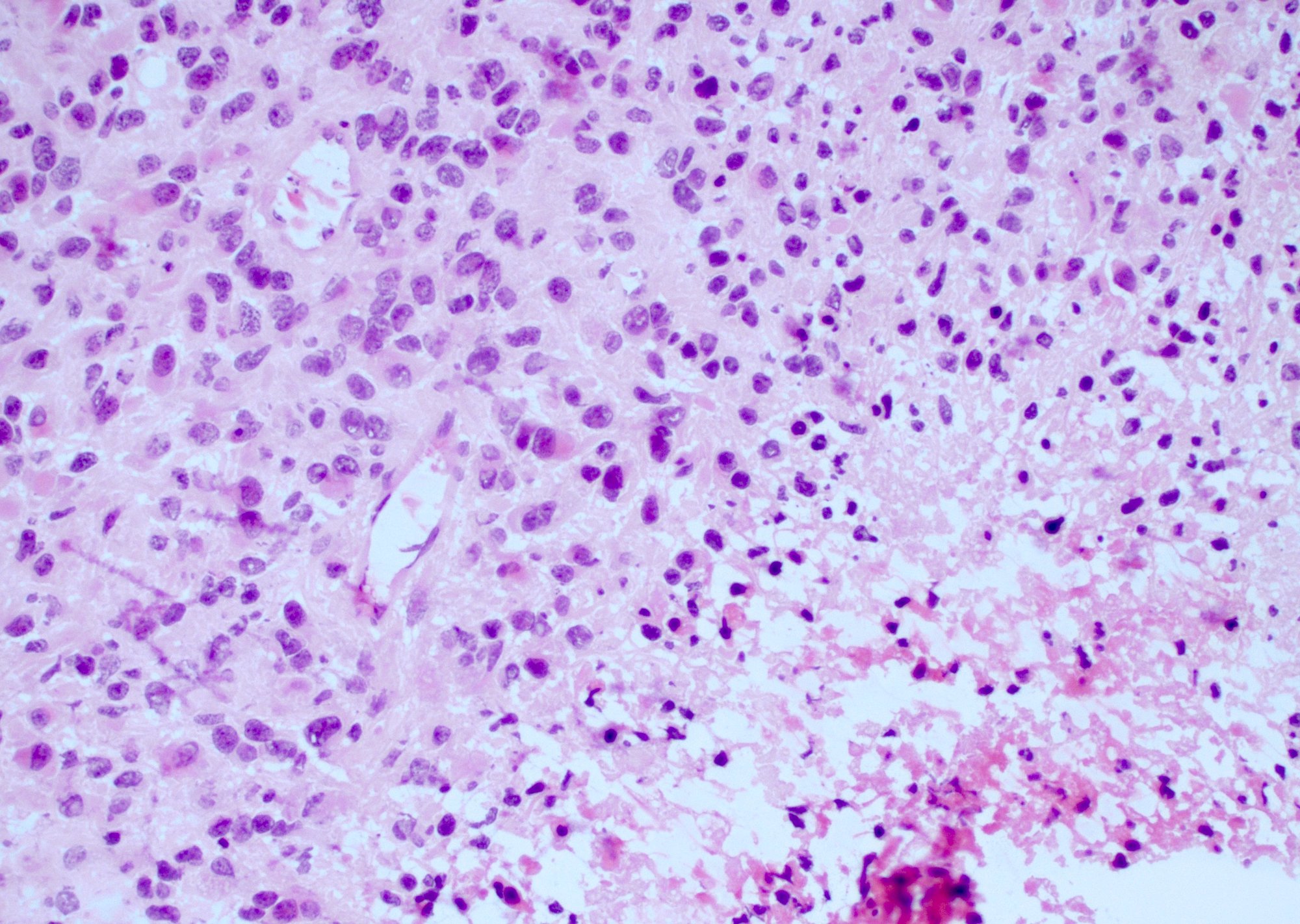
Nuclear atypia and palisading tumor cells
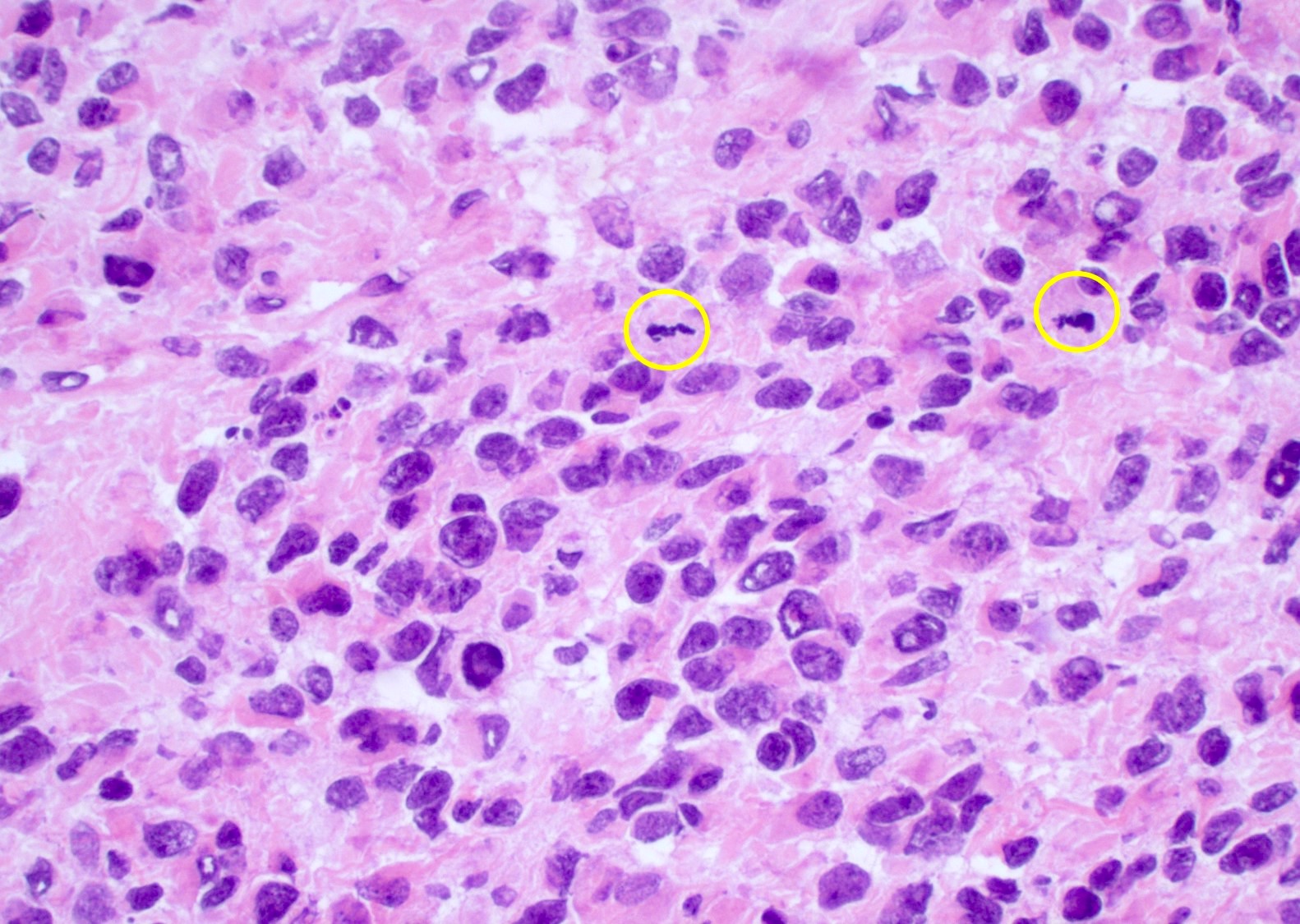
Pleomorphic, astrocytic tumor cells
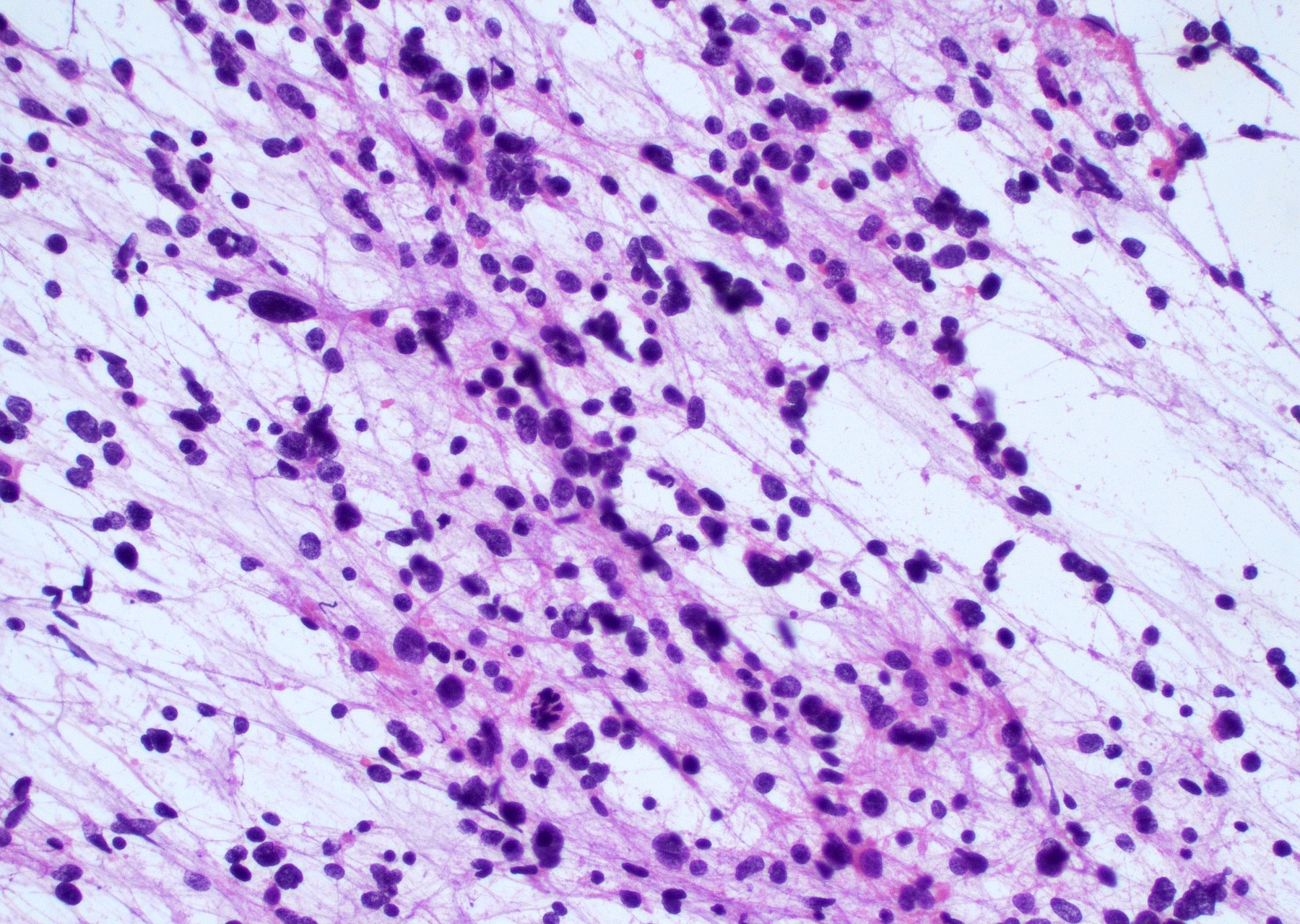
Intraoperative smear preparation
- Infiltrating, hypercellular astrocytic neoplasm often with hyperchromatic, elongated nuclei and irregular nuclear membranes
- Typically mitotically active, though not required if molecular criteria are met
- Microvascular proliferation or necrosis is required for a histologic diagnosis of GBM
- Microvascular proliferation: multilayered, small caliber vessels with glomeruloid appearance (J Neuropathol Exp Neurol 1992;51:488)
- Necrosis: can be geographic or pseudopalisading with neoplastic cells surrounding central necrosis
- Greater association of thrombosis and necrosis in IDH wild type GBM than in IDH mutant grade 4 astrocytomas (Acta Neuropathol 2016;132:917)
- Variable cell morphology: undifferentiated / primitive neuronal cells, astrocytic, gemistocytic, oligodendroglial-like, small cell, lipidized, granular, epithelioid, giant cells, mesenchymal metaplasia and epithelial metaplasia
- Primitive neuronal cells (embryonal): markedly increased cellularity composed of cells with high N/C ratio, brisk mitotic activity with apoptotic bodies, nuclear molding, sometimes with neuroblastic rosettes
- Typically has conventional infiltrating astrocytic component, which is morphologically distinct
- Loss of glial markers, expression of neuronal markers (synaptophysin)
- Higher risk of CSF dissemination but similar survivals as classic GBM
- Associated with MYC amplifications
- Astrocytic: fibrillary, elongated processes
- Gemistocytic: abundant eosinophilic cytoplasm with eccentric nuclei
- Oligodendroglial-like: cells with small, round nuclei with perinuclear clearing in a vascular background
- Can be associated with FGFR3::TACC3 fusion positive GBM (Brain Pathol 2018;28:674)
- Small cell change: monomorphic cells with small, round or angulated, hyperchromatic nuclei and brisk mitotic activity
- Associated with EGFR amplification (Clin Neuropathol 2005;24:163, J Neuropathol Exp Neurol 2001;60:1099)
- Lipidized / xanthomatous cells: cells with abundant foamy cytoplasm
- Be sure to exclude pleomorphic xanthoastrocytoma
- Granular cells: large cells with small nuclei and abundant granular cytoplasm
- PAS positive
- May be CD68 positive but negative for CD163
- Epithelioid: large eosinophilic cells with prominent nucleoli
- May resemble rhabdoid cells with more eccentric nuclei
- May be immunoreactive to cytokeratins but negative for CAM5.2
- May be more sharply demarcated with less infiltration
- Often associated with BRAF V600E mutations, usually in younger individuals (Discov Med 2018;26:51, Int J Clin Exp Pathol 2020;13:1529)
- Giant cell: well circumscribed tumors composed of markedly pleomorphic and bizarre cells, including multinucleated tumor cells
- Often have a rich reticulin network
- Associated with mutations in TP53, RB1, NF1 and POLE (Neurooncol Adv 2020;2:vdz059, Acta Neuropathol Commun 2021;9:200, Neuro Oncol 2015;17:1356)
- POLE mutated tumors associated with ultramutated phenotype and may have a better prognosis
- Mesenchymal / sarcomatous: may be well circumscribed; corresponds to cellular differentiation along various lineage; sarcomatous (spindled and fibroblastic), osseous, chondroid or myogenic differentiation (see Gliosarcoma)
- Sarcomatous component usually comprised of GFAP negative spindled cells with reticulin deposition rich
- Associated with mutations in TP53, PTEN, TERT and CDKN2A deletion and MDM2 and CDK4 coamplification (J Neuropathol Exp Neurol 1995;54:651, Am J Pathol 2000;156:425, Acta Neuropathol 2001;101:321)
- Epithelial metaplasia: rare but may include squamous or adenomatous differentiation
- Keratin pearls, epithelial whorls: CK5/6 positive
- Glandular structures
- Primitive neuronal cells (embryonal): markedly increased cellularity composed of cells with high N/C ratio, brisk mitotic activity with apoptotic bodies, nuclear molding, sometimes with neuroblastic rosettes
Contributed by Bharat Ramlal, M.D. and Meaghan Morris, M.D., Ph.D.
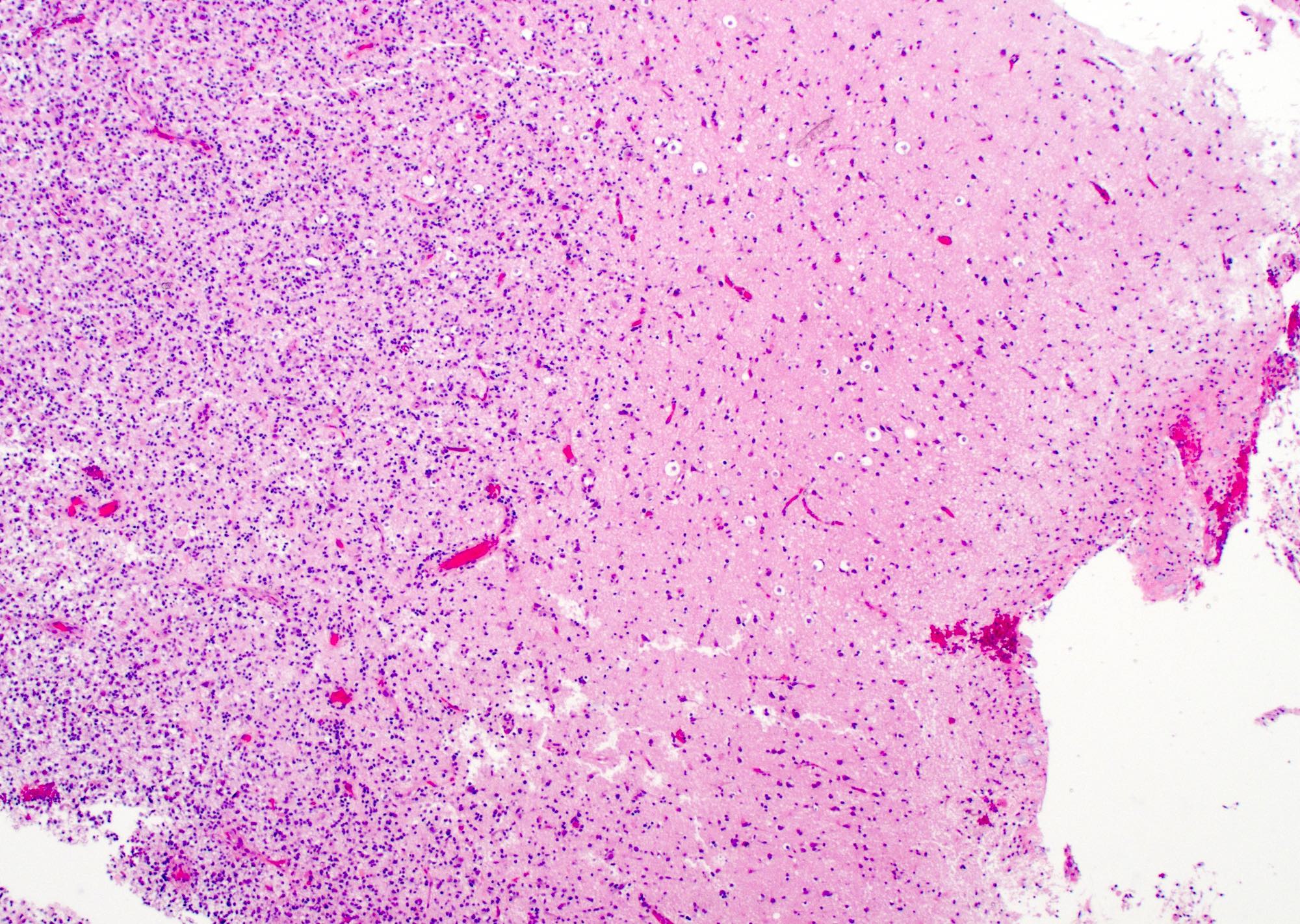
Hypercellular neoplasm
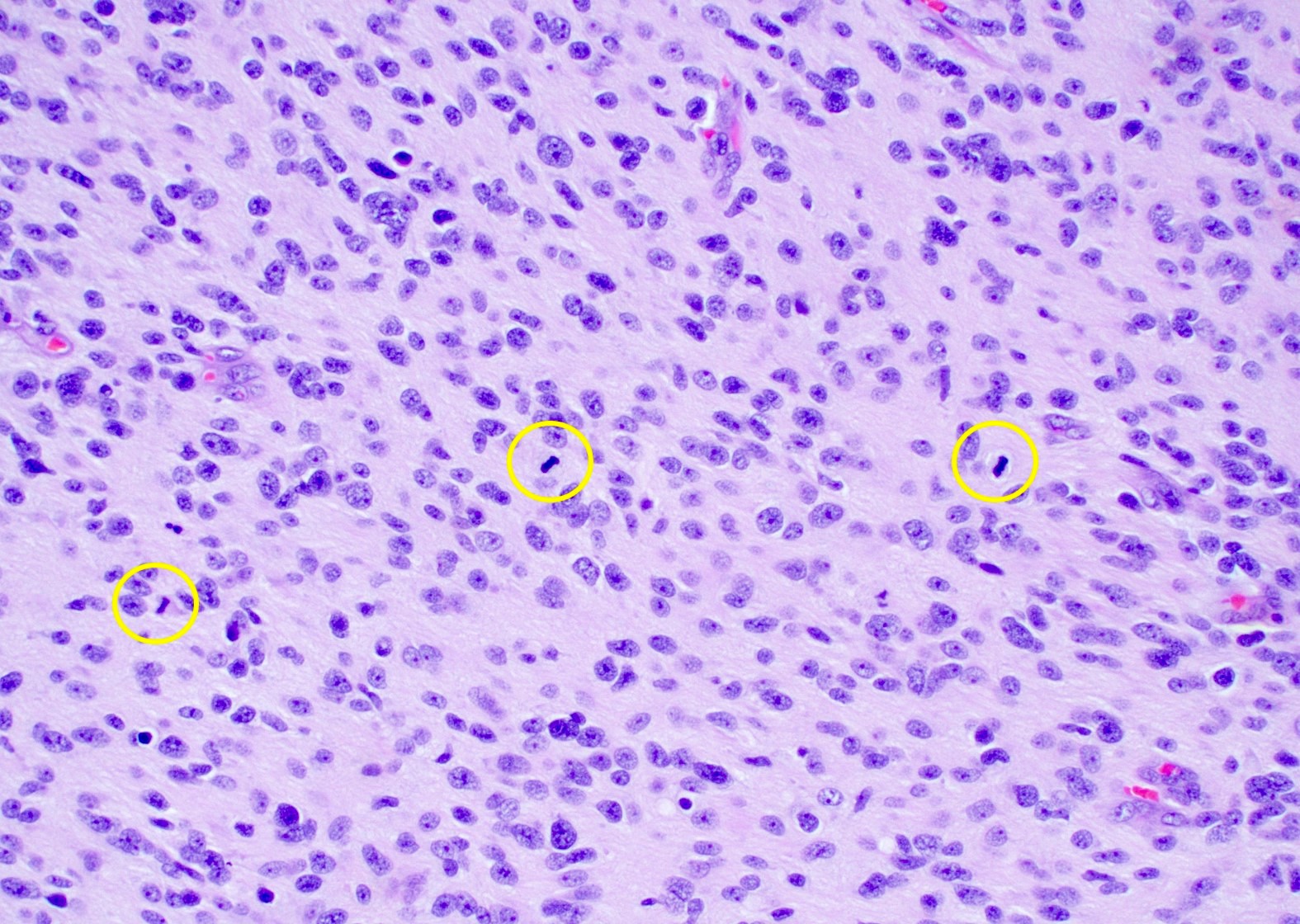
Nuclear atypia and brisk mitotic activity
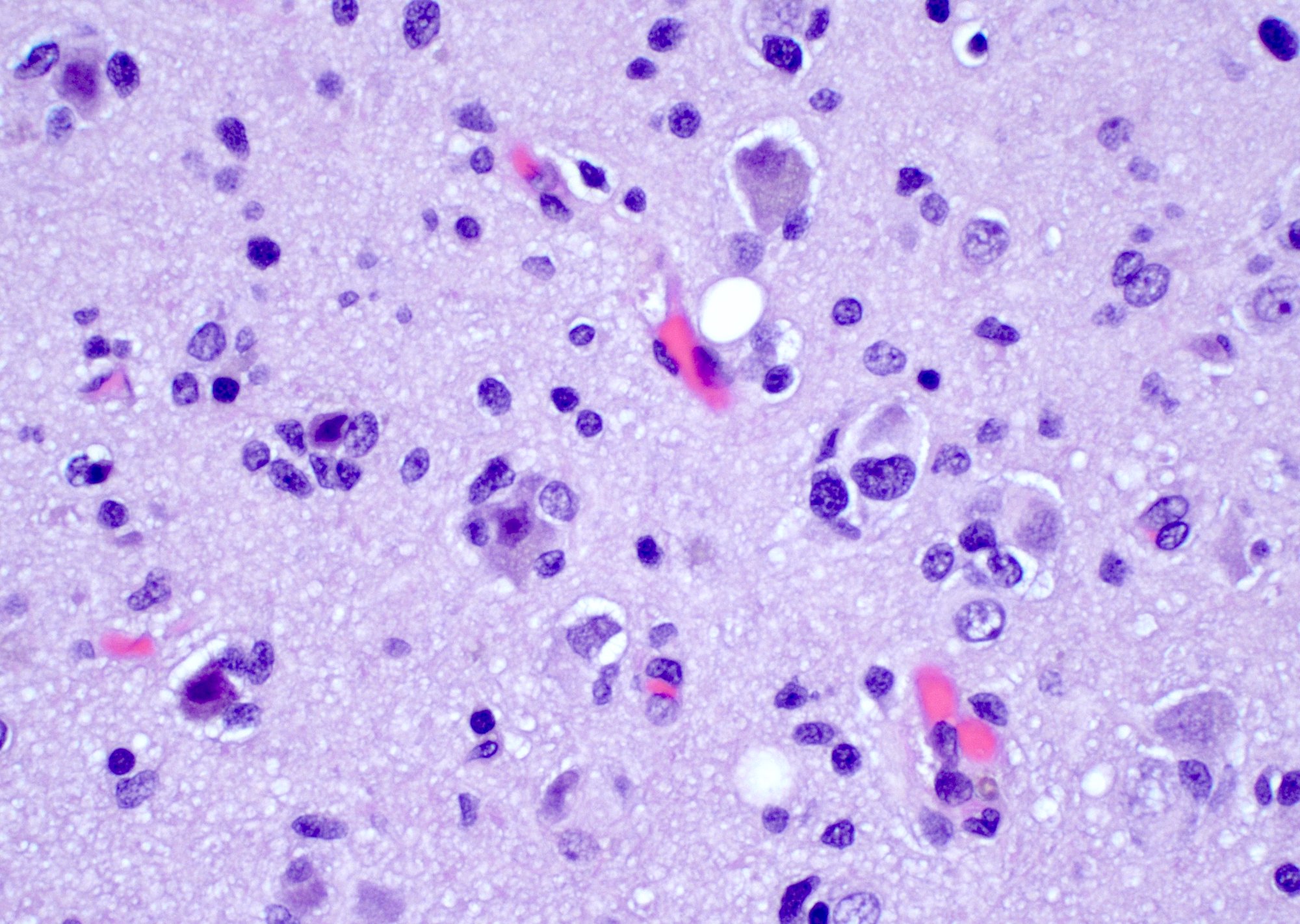
Atypical astrocytic tumor cells
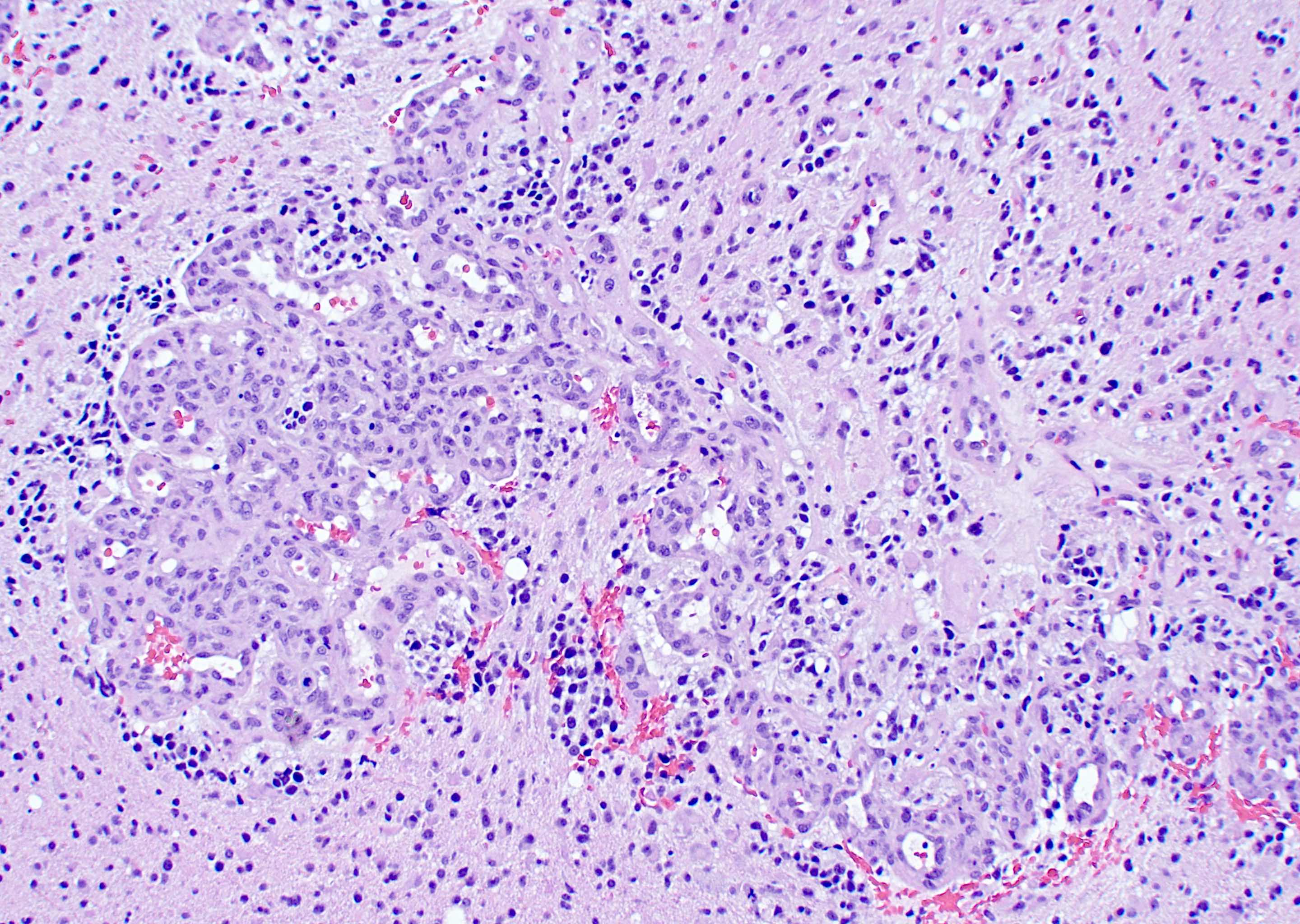
Glomeruloid microvascular proliferation

Pseudopalisading necrosis
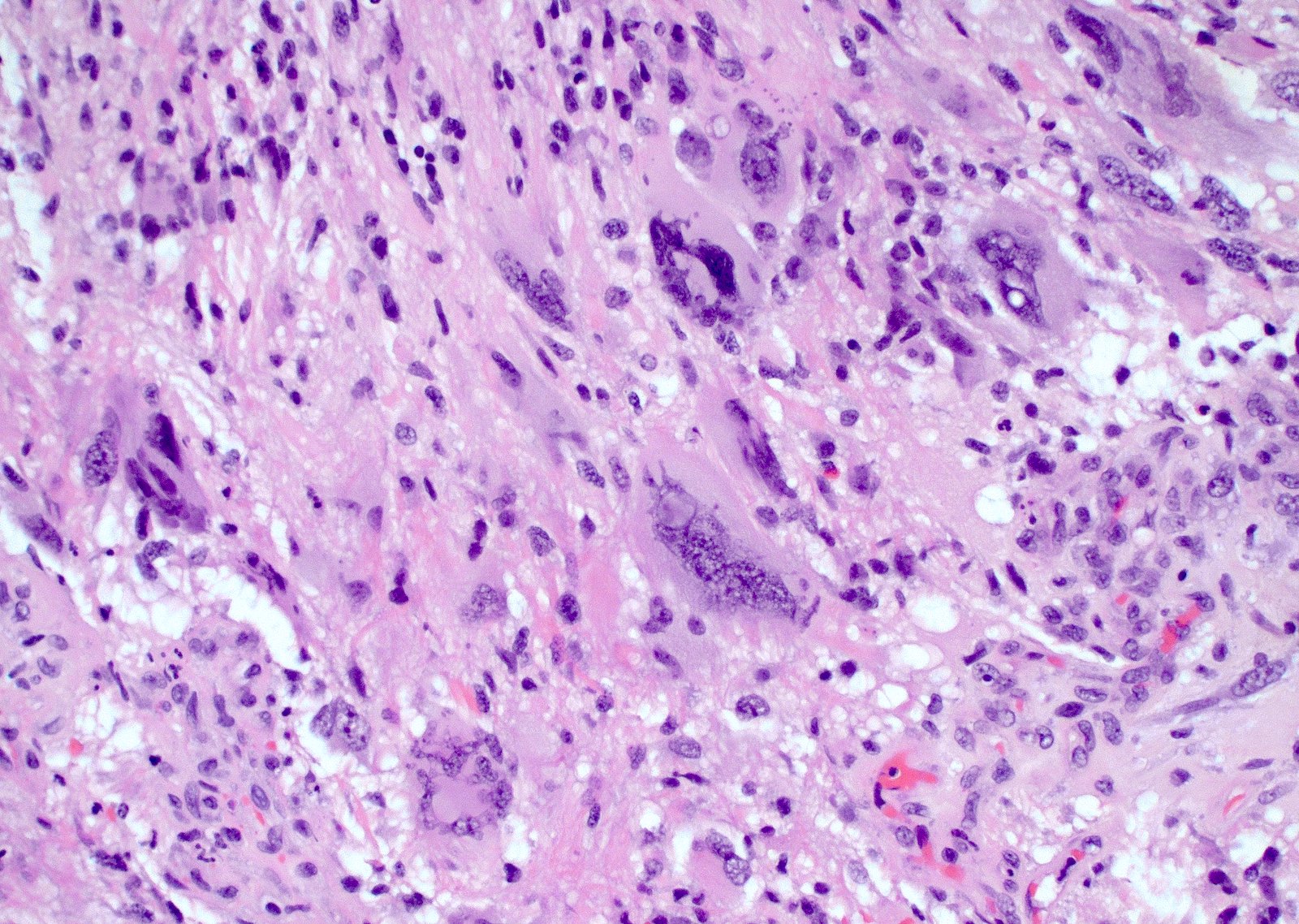
Marked nuclear pleomorphism
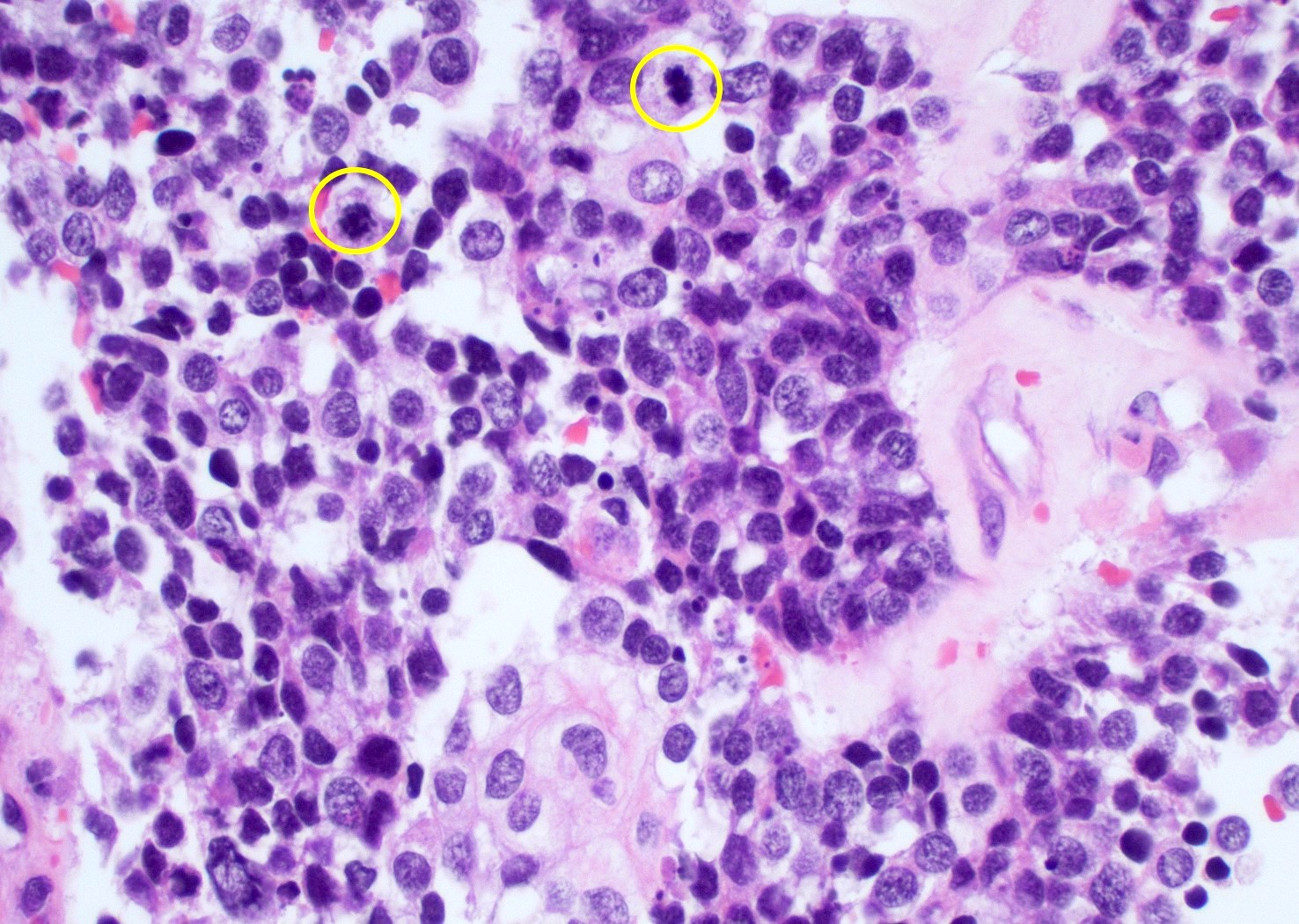
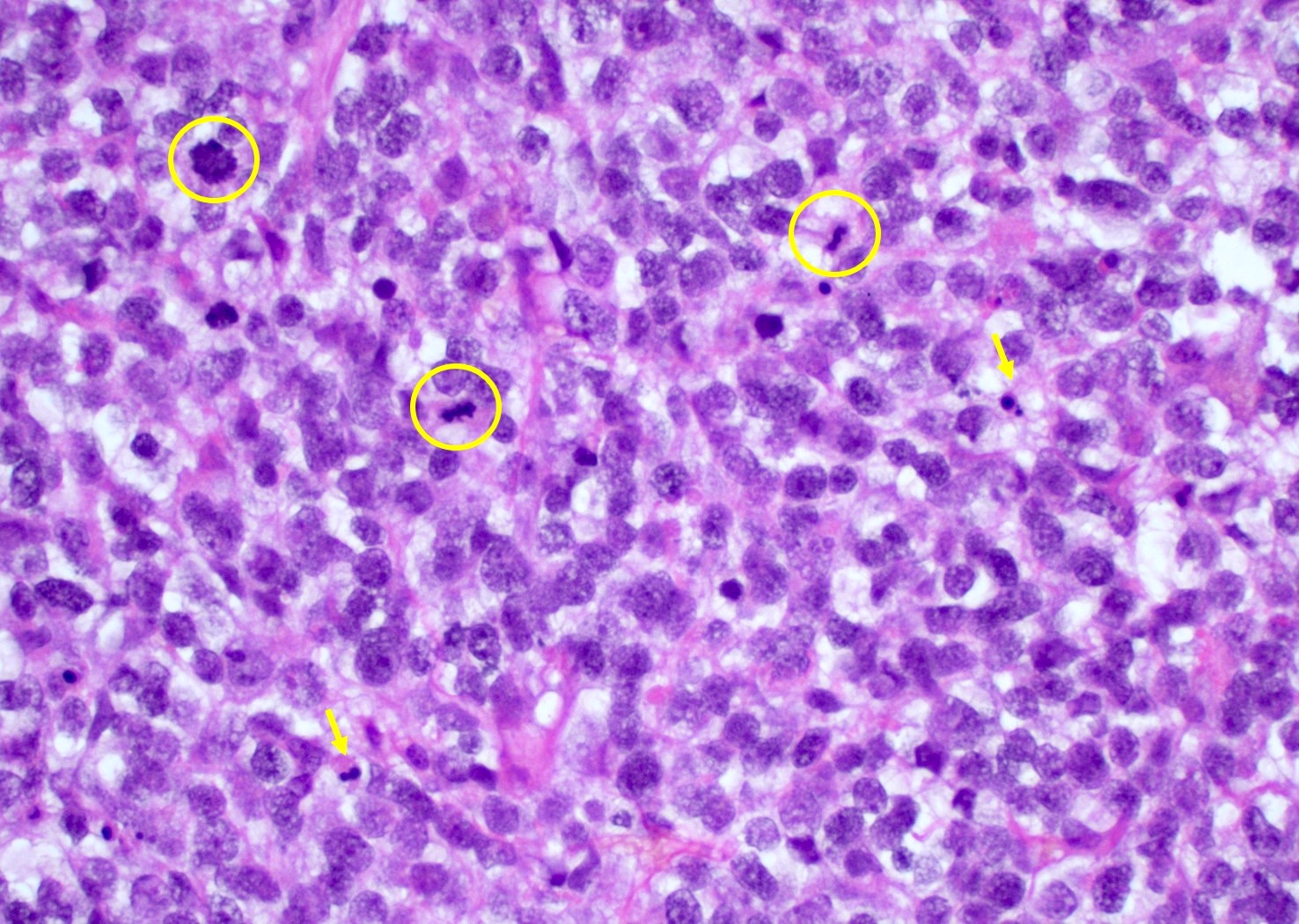
Primitive neuronal component
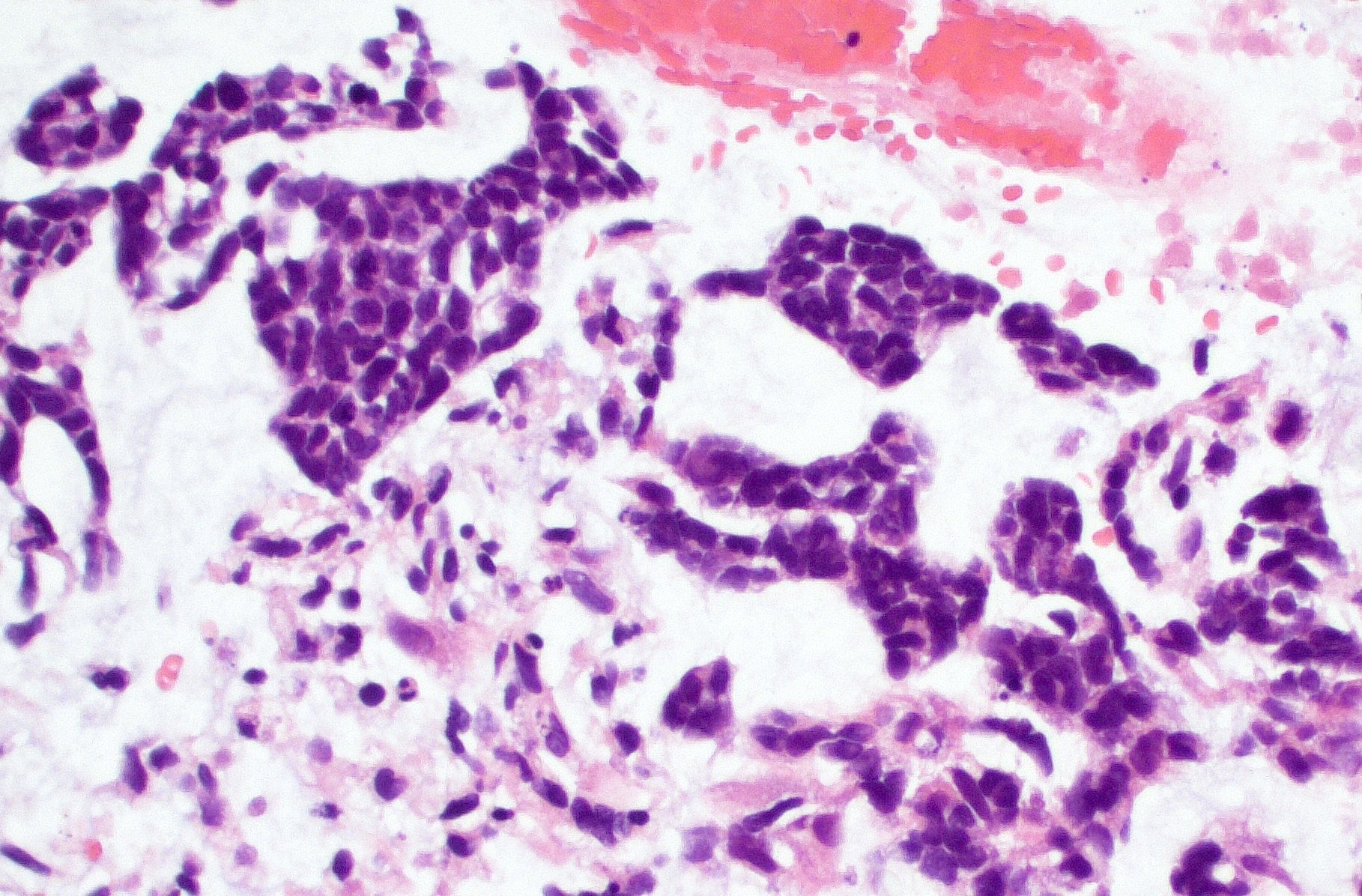
Adenoid morphology

Epithelioid glioblastoma
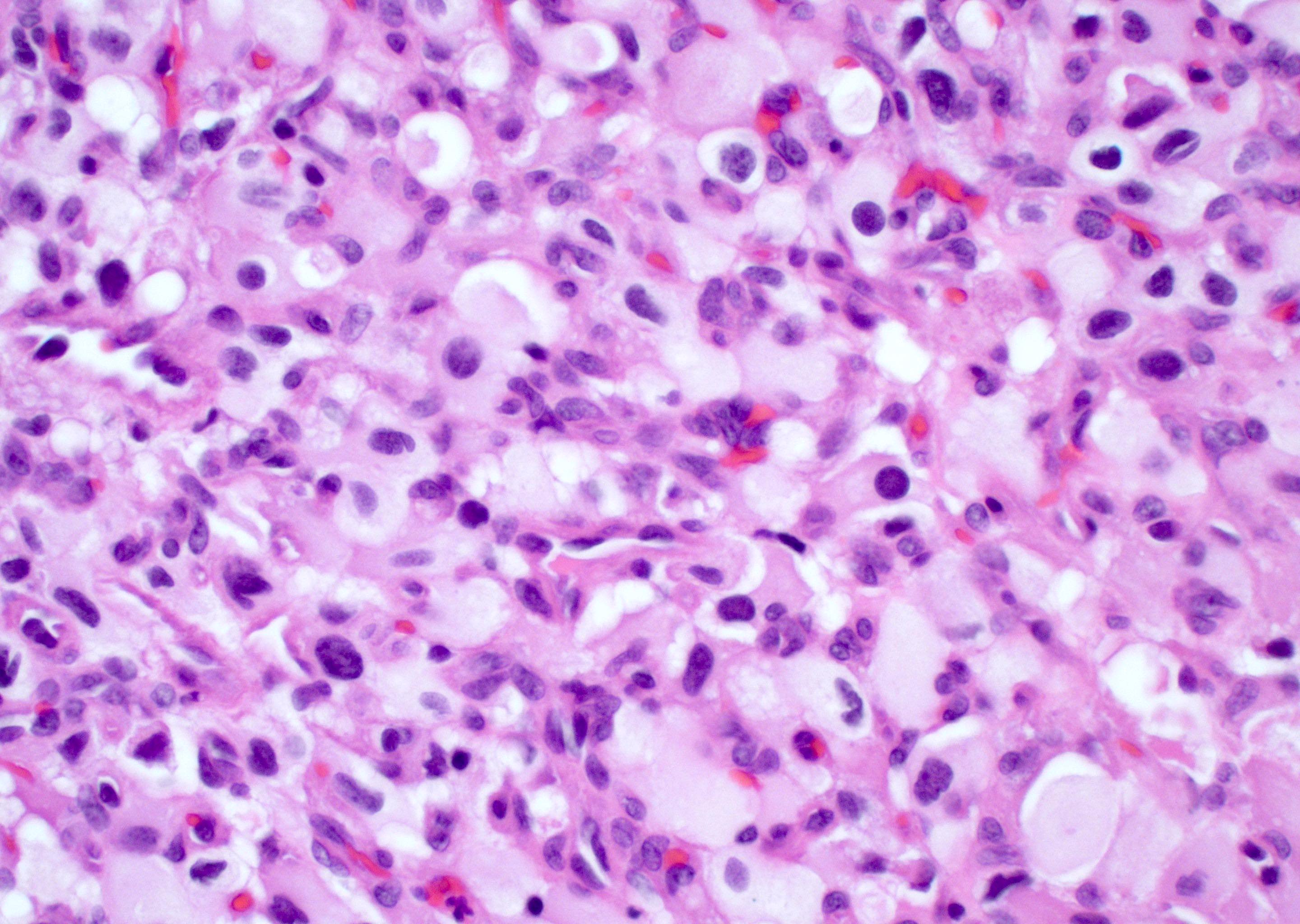

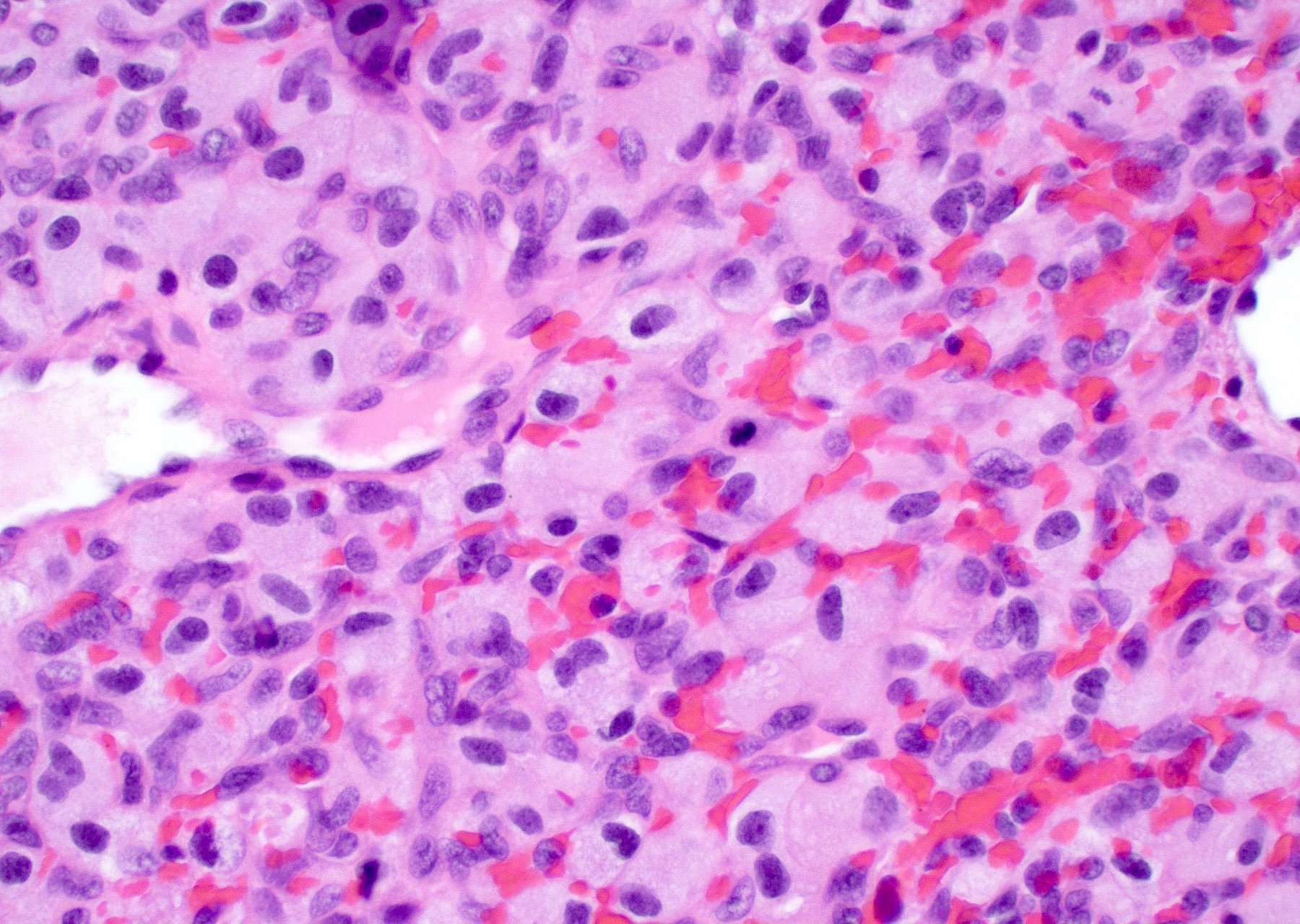
Granular cell morphology
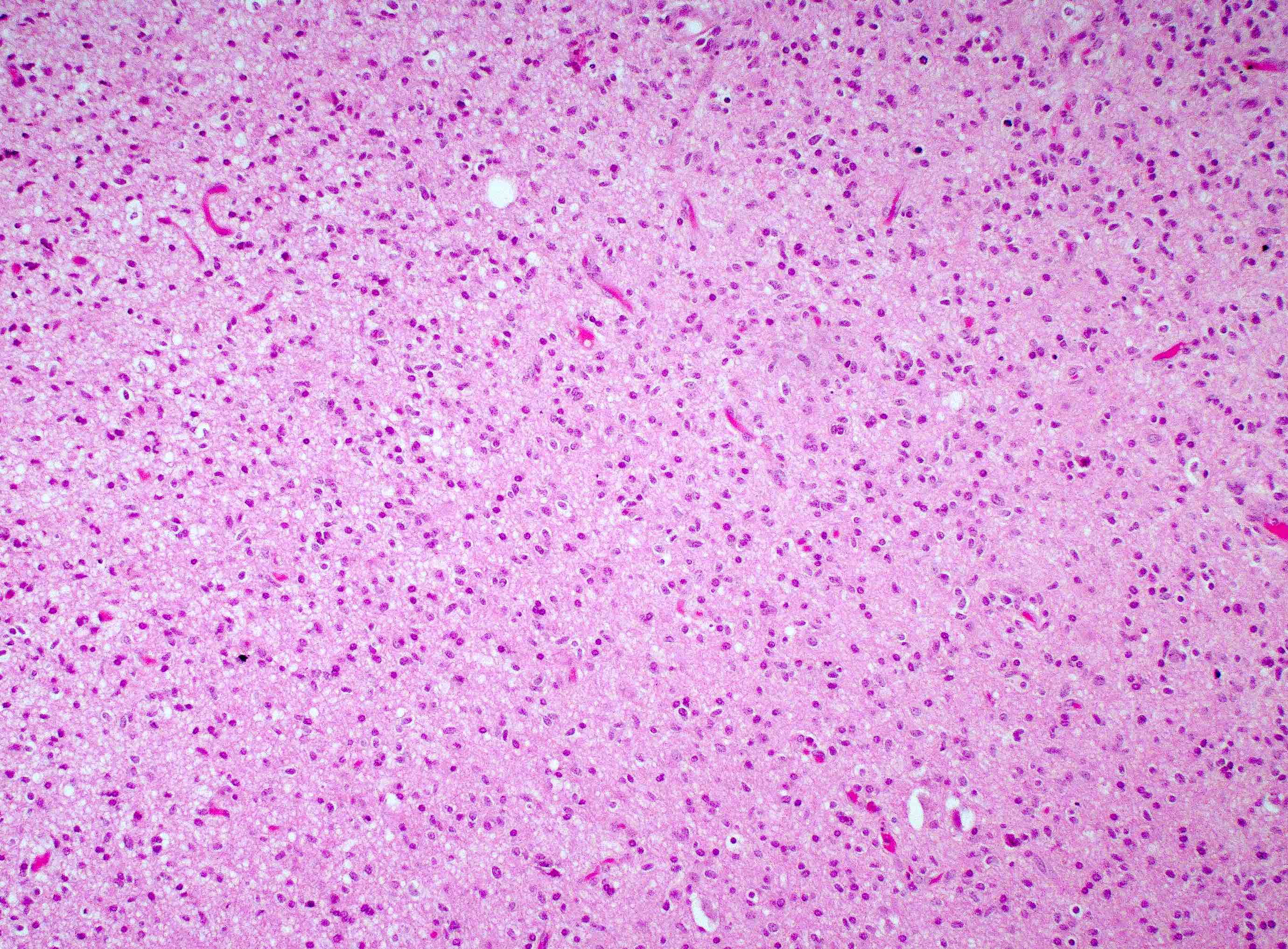
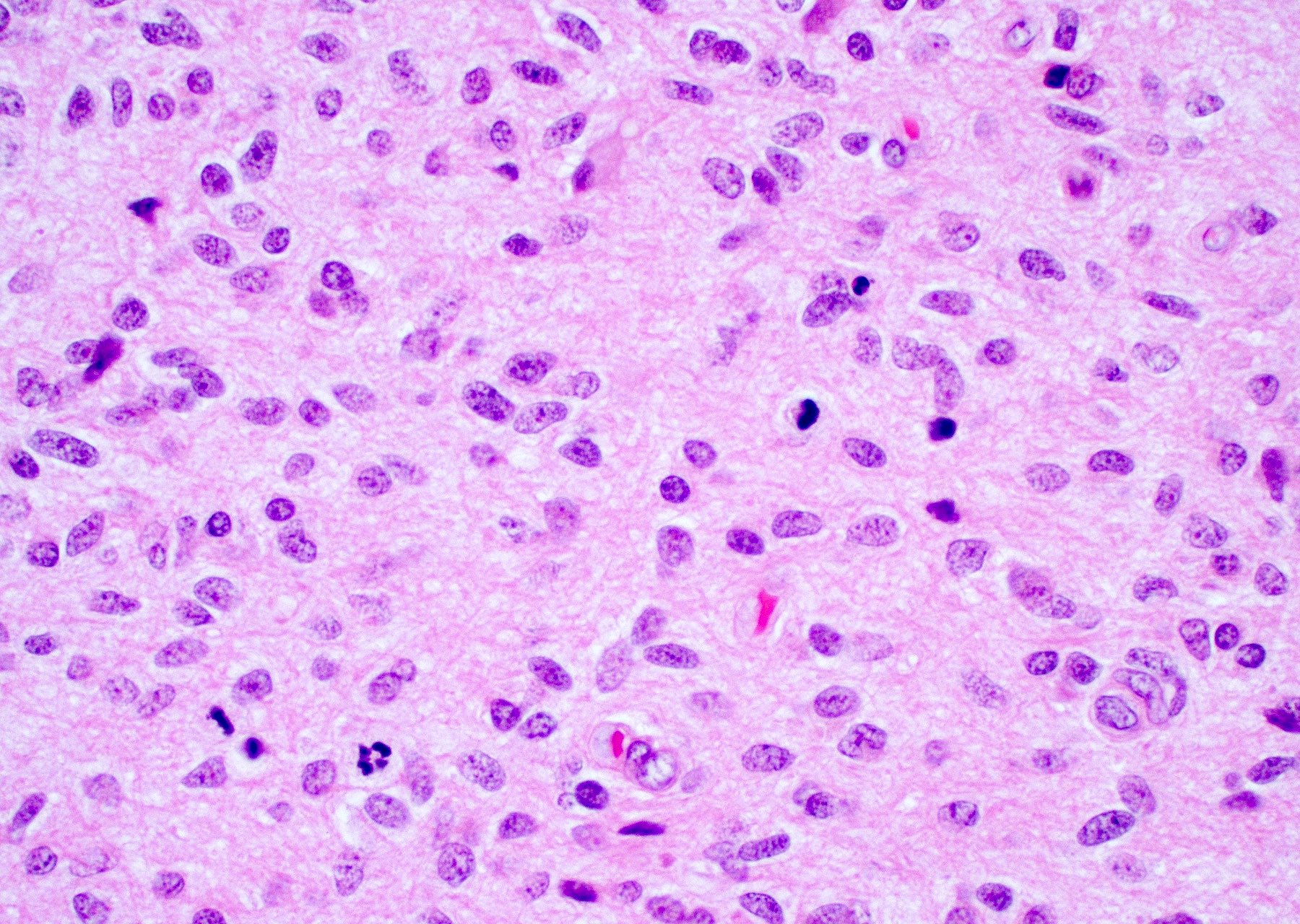
Small cell glioblastoma

IDH1 R132H
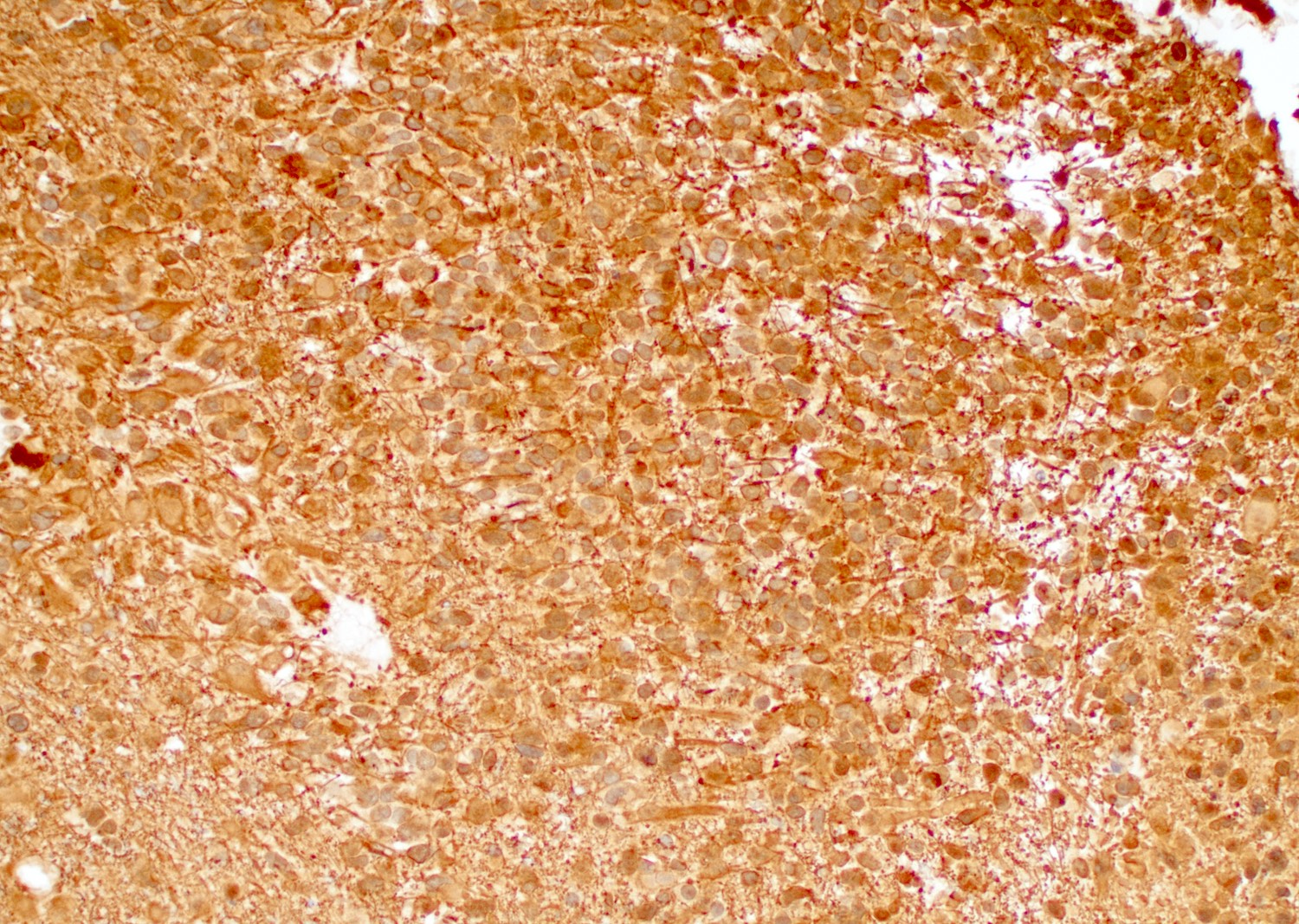
GFAP
Olig2

Ki67

Synaptophysin
- Intraoperative smears may show marked cellularity, with moderate to markedly pleomorphic astrocytic / gemistocytic cells with fine fibrillar glial processes (Diagn Cytopathol 1986;2:312)
- May show necrosis
- Mitotic figures can be observed
- GFAP, Olig2, S100
- GFAP may be lost in GBMs with sarcomatoid or primitive neuronal components
- Cytokeratin AE1 / AE3 may cross react with glial filaments (Pathol Oncol Res 2015;21:817)
- ATRX and H3K27 trimethylation usually retained
- EGFR (Indian J Med Res 2017;146:738)
- Nuclear p53 in up to 30% of GBM; more common in giant cell GBM
- BRAF V600E in epithelioid GBMs (Am J Surg Pathol 2015;39:528)
- Synaptophysin, chromogranin, INSM1 in primitive neuronal component; usually negative in glioblastoma lacking a primitive neuronal component
- Ki67 usually high; can be low in molecularly defined GBMs
- IDH1 (R132H mutant protein) (Acta Neuropathol 2016;131:803)
- H3 (K27M mutant protein) (Acta Neuropathol 2016;131:803, Ann Pathol 2021;41:137)
- H3 (G34R mutant protein) (Acta Neuropathol 2016;131:803, Brain Tumor Pathol 2021;38:4)
- Bundles of cytoplasmic filaments 80 - 90 angstroms in diameter (Am J Pathol 1973;73:589)
- Pleomorphic nuclei and prominent nucleoli with nuclear infoldings and cytoplasmic invaginations (intranuclear pseudoinclusions)
- Lacks mutations in IDH1 / IDH2 and histone H3 genes
- Lack of IDH1 immunohistochemistry sufficient in patients ≥ 55 years of age meeting histologic criteria for glioblastoma with nonmidline tumors (Acta Neuropathol 2016;131:803, Oncotarget 2016;7:31393)
- Molecularly defined GBM: even in low grade appearing tumors and tumors lacking necrosis or microvascular proliferation (Acta Neuropathol 2018;136:805, Acta Neuropathol 2018;136:793):
- TERT promoter mutation
- EGFR gene amplification or combined whole chromosome 7 gain and whole chromosome 10 loss (+7 / -10)
- Alterations of RTK genes, PI3K pathway genes and PTEN in up to 90% of GBM (Cell 2013;155:462, Nature 2008;455:1061)
- CDKN2A / CDKN2B deletion can be present
- BRAF V600E in up to 50% of GBMs with epithelioid features (Am J Surg Pathol 2013;37:685)
- MYC or MYCN amplification associated with primitive neuronal component
- Radiation induced gliomas: PDGFRA, TP53, CDKN2A deletion (Neurooncol Adv 2021;3:vdab109, Nat Commun 2021;12:5530, Acta Neuropathol 2019;137:139)
- Some radiation associated gliomas with histologic features of glioblastoma may be molecularly more consistent with diffuse pediatric type high grade glioma, H3 wild type and IDH wild type (Acta Neuropathol 2019;137:139)
- If present, gene fusions most commonly involve the receptor tyrosine kinase (RTK) family (Cell 2013;155:462, Cancers (Basel) 2020;12:1219, Front Oncol 2020;10:593578):
- EGFR
- FGFR3
- MET
- NTRK1 / NTRK2 / NTRK3
- PDGFRA
- Brain, right frontal lobe, biopsy:
- Glioblastoma, IDH wild type, CNS WHO grade 4
- Histologic diagnosis: glioblastoma
- Grade: 4
- Molecular information: EGFR amplified, negative for IDH1 / IDH2 and histone H3 alterations
- Astrocytoma, IDH mutant:
- Younger individuals, presence of IDH1 / IDH2 gene mutations by immunohistochemistry or sequencing
- Often have ATRX mutation (loss of expression) and p53 mutations (nuclear overexpression)
- Diffuse hemispheric glioma, H3 G34 mutant:
- Older adolescents and young adults (age 11 - 30) with hemispheric mass
- May have classic GBM morphology or primitive neuronal / embryonal morphology
- Characterized by histone H3 G34 mutation on sequencing
- Classically lack Olig2 expression, show ATRX loss of expression (mutant) and have p53 mutations (nuclear overexpression)
- Diffuse midline glioma, H3 K27 altered:
- Midline tumor (brainstem, thalamus, spinal cord, less often basal ganglia or cerebellum)
- Most positive for histone H3K27M mutant protein (nuclear)
- All show loss of histone H3K27 trimethylation (H3K27me3)
- Diffuse pediatric type high grade glioma, H3 wild type and IDH wild type:
- Typically in a child or young adult
- Frequent PDGFRA or EGFR alterations or MYCN amplification
- Methylation profiling may be helpful in difficult cases
- Pleomorphic xanthoastrocytoma:
- Prominent xanthomatous cells
- Often well circumscribed
- Lower grade lesions have no necrosis and low mitotic activity
- Eosinophilic granular bodies (EGBs), Rosenthal fibers and perivascular lymphocytic cuffing
- Often BRAF V600E positive IHC / sequencing and CD34 positive
- Pericellular reticulin deposition
- Lymphoma:
- Tumefactive multiple sclerosis / demyelinating diseases:
- Macrophage rich lesions (CD163 positive) with perivascular lymphoplasmacytic inflammation
- Loss of myelin (LFB) with axonal preservation (neurofilament)
- Creutzfeldt cells: astrocytic cells with nuclear fragmentation may mimic mitotic figures
- Astrocytes have a reactive (fibrillary) appearance, which can be highlighted by GFAP IHC
- Abscess:
- Abundant necrosis with mixed acute and chronic inflammation
- Peripheral granulation tissue and fibrosis
- Neutrophilic inflammation predominates
- Microorganisms may be identified
Which histologic subtype of glioblastoma, IDH wild type (pictured above) is important to diagnose due to increased risk of cerebrospinal fluid (CSF) dissemination?
- Epithelioid
- Gemistocytic
- Primitive neuronal component
- Sarcomatoid
Comment Here
Reference: Glioblastoma, IDH wild type
- Epithelioid
- Gemistocytic
- Giant cell
- Oligodendrolial-like features
Comment Here
Reference: Glioblastoma, IDH wild type
- Rare, benign intraparenchymal and often paraventricular cyst lined by simple epithelium or glial tissue, S100+ or GFAP+, resting on neuroglia (J Neuroradiol 1995;22:48)
- Usually intracranial, not midline but may affect spinal cord; may affect adult cerebellum or represent burned out pilocytic astrocytoma
- Not in communication with ventricle or CSF spaces
- Rarely ruptures and causes meningitis
- Cyst lined by glial tissue
Images hosted on other servers:

MR: cystic lesion of right frontal lobe
- 15 year old boy with recurrent intramedullary cyst at C2-C3 (Neurol India 2003;51:111)
- 19 year old woman with thalamic glial cyst causing hydrocephalus due to hemorrhage (Neurol Med Chir (Tokyo) 1997;37:284)
- 42 year old man with clinical symptoms of expansive cerebellar lesion (J Neuroradiol 2001;28:209)
- Resembles arachnoid cyst
Images hosted on other servers:

Ependymal cyst
- Simple columnar or cuboidal cells, often ciliated, resting on neuroglia; no fibrous capsule
- Alternatively, wall lined by gliosis, Rosenthal fibers present, variable hemosiderin; no epithelial lining
- Neuroepithelial origin
- Endodermal cyst: CK+
- Most common form of intra-axial primary tumors of the central nervous system (CNS)
- Includes adult and pediatric type diffuse gliomas, circumscribed gliomas and ependymal tumors
- Note: pediatric type gliomas occur more frequently in children and adolescents; however, they can also occur in young adults (and vice versa)
- Glioneuronal and neuronal tumors will not be discussed here
- Increasing importance of molecular diagnostics for glioma classification (IDH1 / IDH2, histone H3, etc.)
- Group of primary CNS neoplasms with glial differentiation
- Range in biologic aggressiveness from WHO grade 1 (benign) to WHO grade 4 (malignant)
- Changes with the 2021 CNS WHO (5th edition) (Neuro Oncol 2021;23:1231)
- Arabic numbers are now used instead of Roman numerals for grading (i.e., WHO grade 1, 2, 3, 4 instead of WHO grade I, II, III, IV)
- Simplified classification of adult type diffuse gliomas into 3 groups
- Glioblastoma, IDH wildtype, WHO grade 4
- Astrocytoma, IDH mutant, WHO grade 2 - 4
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted, WHO grade 2 - 3
- Additional molecular surrogates for WHO grade 4 designation (i.e., grade 4 designation should be applied even without the presence of grade 4 histology) (Neuro Oncol 2021;23:1231)
- Glioblastoma, IDH wildtype
- Polysomy 7 and monosomy 10 (+7 / -10)
- EGFR amplification
- TERT promoter mutation
- Astrocytoma, IDH mutant
- CDKN2A / CDKN2B homozygous deletion
- Glioblastoma, IDH wildtype
- Intra-axial: within the brain or spinal cord parenchyma
- Extra-axial: not within the brain or spinal cord parenchyma (e.g., meningioma)
- Intramedullary: within the spinal cord parenchyma
- Extramedullary: not within the spinal cord parenchyma (e.g., schwannoma)
- Supratentorial: above tentorial membrane - cerebrum
- Infratentorial: below tentorial dura - brainstem, cerebellum, spinal cord
- ICD-O
- 9400/3 - astrocytoma, IDH mutant, grade 2
- 9401/3 - astrocytoma, IDH mutant, grade 3
- 9445/3 - astrocytoma, IDH mutant, grade 4
- 9450/3 - oligodendroglioma, IDH mutant and 1p / 19q codeleted, grade 2
- 9451/3 - oligodendroglioma, IDH mutant and 1p / 19q codeleted, grade 3
- 9440/3 - glioblastoma, IDH wildtype
- 9421/1 - diffuse astrocytoma, MYB or MYBL1 altered
- 9431/1 - angiocentric glioma
- 9413/0 - polymorphous low grade neuroepithelial tumor of the young
- 9421/1 - diffuse low grade glioma, MAPK pathway altered
- 9385/3 - diffuse midline glioma, H3 K27 altered
- 9385/3 - diffuse hemispheric glioma, H3 G34 mutant
- 9385/3 - diffuse pediatric type high grade glioma, H3 wildtype and IDH wildtype
- 9385/3 - infant type hemispheric glioma
- 9421/1 - pilocytic astrocytoma
- 9421/3 - high grade astrocytoma with piloid features
- 9424/3 - pleomorphic xanthoastrocytoma
- 9384/1 - subependymal giant cell astrocytoma
- 9444/1 - chordoid glioma
- 9430/3 - astroblastoma, MN1 altered
- 9391/3 - supratentorial ependymoma, NOS
- 9396/3 - supratentorial ependymoma, ZFTA fusion positive
- 9396/3 - supratentorial ependymoma, YAP1 fusion positive
- 9391/3 - infratentorial ependymoma, NOS
- 9396/3 - posterior fossa group A (PFA) ependymoma
- 9396/3 - posterior fossa group B (PFB) ependymoma
- 9391/3 - spinal ependymoma, NOS
- 9396/3 - spinal ependymoma, MYCN amplified
- 9394/1 - myxopapillary ependymoma
- 9383/1 - subependymoma
- ICD-11
- 2A00.0Y& XH6PH6 - other specified gliomas of brain & astrocytoma, NOS
- 2A00.0Y& XH2HK4 - other specified gliomas of brain & diffuse astrocytoma, IDH mutant
- 2A00.0Y& XH7K31 - other specified gliomas of brain & oligodendroglioma, IDH mutant and 1p / 19q codeleted
- 2A00.0Y& XH5571 - glioblastoma of brain & glioblastoma, IDH wildtype
- 2A00.0Y& XH41C5 - other specified gliomas of brain & angiocentric glioma
- 2A00.2Y - other specified tumors of neuroepithelial tissue of brain
- 2A00.0Y - other specified gliomas of brain
- 2A00.0Y& XH7692 - other specified gliomas of brain & diffuse midline glioma, H3 K27M mutant
- 2A00.0Y& XH29Q5 - other specified gliomas of brain & pilomyxoid astrocytoma
- 2A00.0Y& XH99U2 - other specified gliomas of brain & pleomorphic xanthoastrocytoma
- 2A00.0Y& XH1L48 - other specified gliomas of brain & subependymal giant cell astrocytoma
- 2A00.0Y& XH9HV1 - other specified gliomas of brain & chordoid glioma
- 2A00.0Y& XH1DC5 - other specified gliomas of brain & astroblastoma
- 2A00.0Y & XH1511 - other specified gliomas of brain & ependymoma, NOS
- 2A00.0Z & XH1511 - other and unspecified neoplasms of brain or central nervous system & ependymoma, NOS
- 2A00.0Y& XH15U1 - other specified gliomas of brain & myxopapillary ependymoma
- 2A00.0Y& XH8FZ9 - other specified gliomas of brain & subependymoma
- CNS tumors account for only 2% of all malignant neoplasms (Neuro Oncol 2021;23:iii1)
- 40% with neuroepithelial origin
- In children, CNS tumors are the leading cause of cancer related death (NCHS Data Brief 2016;257:1, Neuro Oncol 2018;20:iv1)
- Children and adolescents are more likely to acquire pediatric type diffuse gliomas and certain circumscribed gliomas, whereas adults are more likely to acquire adult type diffuse gliomas, though there is considerable overlap (Neuro Oncol 2021;23:iii1)
- Brain
- Spinal cord
- Generally, not well understood
- Most glial tumors are likely derived from populations of CNS progenitor cells, including neural progenitor cells, glial progenitor cells, oligodendrocyte precursor cells, etc. (Cells 2021;10:621)
- In most cases, there is not a strong causal connection
- History of prior craniospinal radiation exposure is a known risk factor for glioma development (J Natl Cancer Inst 2006;98:1528, Neurooncol Adv 2021;3:vdab109)
- Gliomas in hereditary cancer disorders (Neuro Oncol 2010;12:104, Curr Neurol Neurosci Rep 2021;21:64, Neurooncol Pract 2021;8:375)
- Neurofibromatosis type 1: pilocytic astrocytoma (including optic pathway glioma), high and low grade gliomas, which can be difficult to classify
- Neurofibromatosis type 2: ependymomas, most commonly spinal
- Tuberous sclerosis: subependymal giant cell astrocytoma (SEGA)
- Noonan syndrome: pilocytic astrocytoma
- Enchondromatosis (Ollier disease, Maffucci syndrome): diffuse gliomas (IDH mutant)
- Li Fraumeni syndrome: grade 4 gliomas in younger patients, including diffuse pediatric type high grade glioma, H3 wildtype and IDH wildtype; IDH mutant astrocytomas in young adults (Acta Neuropathol Commun 2023;11:3)
- Lynch syndrome (Turcot syndrome): glioblastoma
- Clinical presentation highly variable, depending on patient age, tumor location and tumor type (Acta Neurol Scand 2015;131:88)
- Common presenting symptoms include seizure, cognitive disorder, headache, nausea / vomiting, focal neurologic deficit
- Combination of clinical findings, imaging (especially magnetic resonance imaging [MRI]) and tissue based biopsy / resection (Lancet Oncol 2017;18:e315)
- Highly variable, depending on location and tumor type (Radiology 2022;304:494, Childs Nerv Syst 2009;25:1203)
- Circumscribed gliomas
- Well circumscribed, variably enhancing, occasionally seen as a cystic mass with enhancing mural nodule
- Diffuse low grade gliomas
- Expansile but usually without enhancement
- T2 FLAIR mismatch sign is a specific (but not sensitive) indicator in adults for astrocytoma, IDH mutant
- Calcifications and cystic components can occasionally be seen in oligodendroglioma, IDH mutant and 1p / 19q codeleted
- Diffuse high grade gliomas
- Expansile and generally with variable enhancement
- Glioblastoma often displays a characteristic ring enhancement with surrounding vasogenic edema and diffusion restriction
- Ependymoma
- Often conspicuous on T2 weighted FLAIR sequences and usually visible on unenhanced T1 weighted images
- Circumscribed gliomas
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
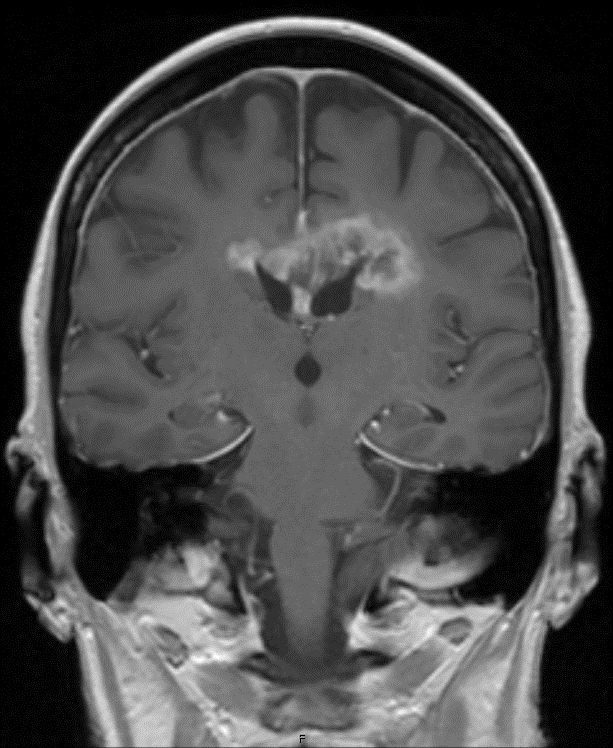
Glioblastoma MRI
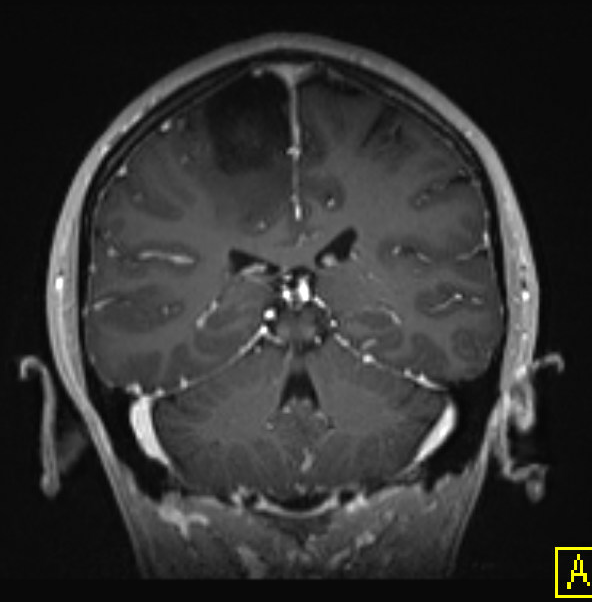
Glioma MRI
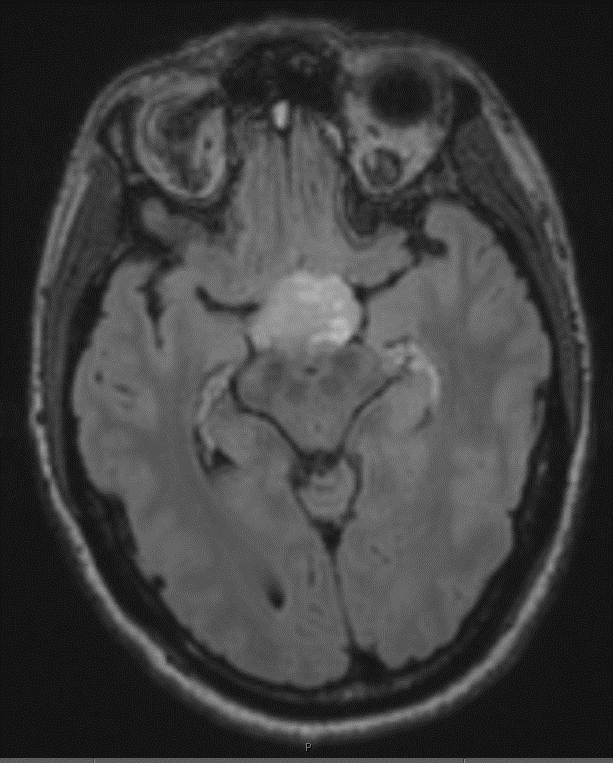
Optic pathway glioma MRI
Spinal ependymoma MRI
- CNS WHO grade (BMC Cancer 2020;20:35)
- WHO grade 1: generally benign; potentially curable with resection
- WHO grade 2 - 3: intermediate prognosis; survival generally many years with eventual tumor progression
- WHO grade 4: malignant; survival limited to months
- Even low grade tumors can cause significant morbidity and mortality if located in delicate parts of the brain, such as the brainstem
- Other factors that influence prognosis include patient age, comorbid conditions, location of tumor, presence of hydrocephalus, extent of surgical resection, response to adjuvant therapy and molecular features of the tumor (BMC Cancer 2020;20:35, Ann Palliat Med 2021;10:863)
- 3 month old girl presents with altered mental status (BMJ Case Rep 2019;12:e228248)
- 39 year old woman with progressive unilateral vision loss (Brain Tumor Pathol 2021;38:59)
- 53 year old woman with unsteady gait (Acta Radiol Open 2020;9:2058460120980143)
- 69 year old woman with radiation induced spinal glioblastoma (BMJ Case Rep 2020;13:e238372)
- 73 year old woman with spinal cord mass (Folia Neuropathol 2020;58:377)
- Depends on the location, tumor type and grade (Neurosurg Rev 2017;40:1)
- Generally, gross total resection is preferred for most tumors; however, watch and wait approach with close clinical monitoring can be considered for some low grade tumors, especially those in locations unamenable to surgery (World Neurosurg 2013;79:537, World Neurosurg 2014;82:e105)
- Adjuvant radiation or chemotherapy (temozolomide) is often performed for high grade gliomas (N Engl J Med 2005;352:987)
- Targeted therapies available for tumors with certain molecular alterations (Mol Cancer 2022;21:39)
- Several experimental approaches under investigation, including immunotherapeutic approaches (Cancer Lett 2020;476:1, Cell Commun Signal 2023;21:74)
- Can be useful in some cases to determine circumscribed versus infiltrative growth patterns
- Helpful to identify high grade features, such as mitotic activity, microvascular proliferation and necrosis
- Due to freezing artifact, nuclear atypia is difficult to assess on frozen section (see Cytology description)
- References: Surg Pathol Clin 2015;8:27, Semin Diagn Pathol 2002;19:192
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
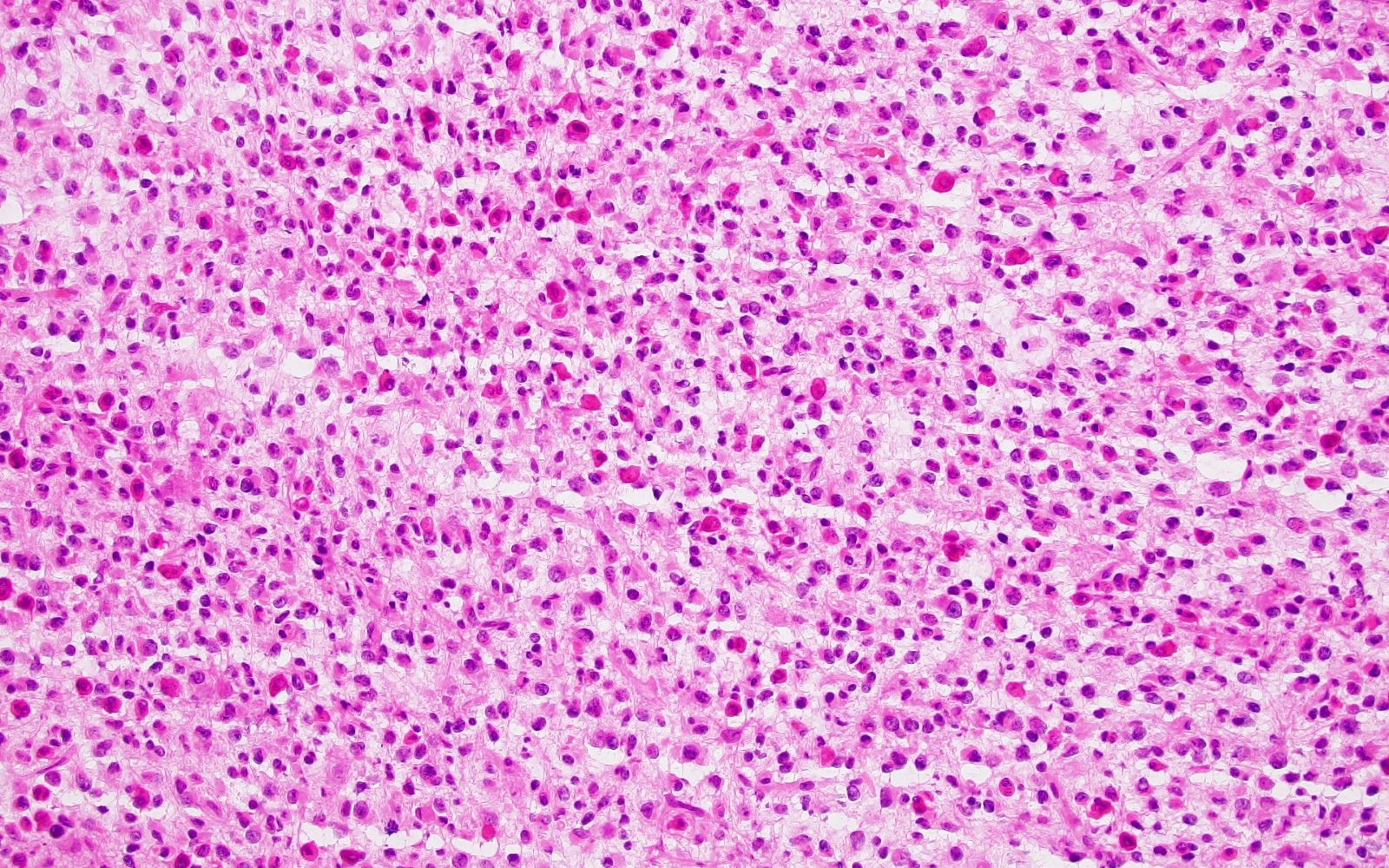
Glioma with astrocytic differentiation
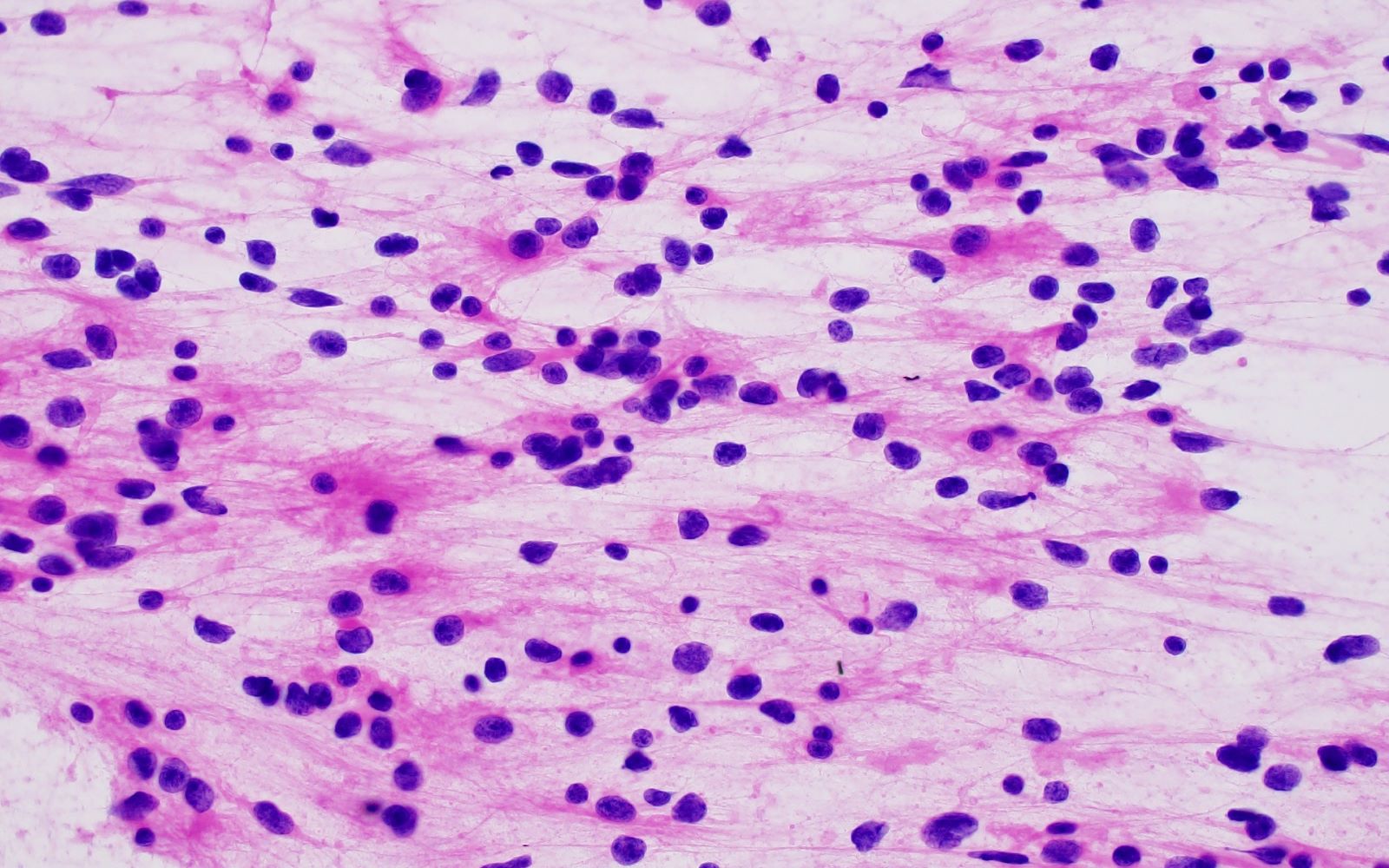
Glioma smear preparation
- Diffuse gliomas
- Variably hypercellular with individual tumor cells infiltrating the brain parenchyma
- Diffuse infiltration results in ill defined margin with adjacent brain tissue
- Secondary structures of Scherer, indicative of infiltrative growth
- Perineuronal satellitosis by tumor cells
- Perivascular condensation by tumor cells
- Subpial accumulation of tumor cells
- Spread of tumor cells along white matter tracks
- Variable nuclear atypia, including nuclear enlargement, irregular nuclear contours, hyperchromasia, multinucleation, etc.
- Some histologic features are suggestive of particular diagnostic entities (see individual chapters for more complete descriptions)
- Astrocytoma, IDH mutant: astrocytic differentiation often in a densely fibrillary background with microcysts
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted: round, monotonous nuclei with characteristic perinuclear halos (fried egg appearance), microcalcifications and chicken wire blood vessels are common
- Glioblastoma, IDH wildtype: densely cellular with brisk mitotic activity and often with microvascular proliferation and pseudopalisading necrosis; tumor microthrombi are characteristic (Acta Neuropathol 2016;132:917)
- Diffuse astrocytoma, MYB or MYBL1 altered & diffuse low grade glioma, MAPK altered: diffuse growth pattern, mild nuclear atypia, minimal to no mitotic activity, absence of anaplastic features (vascular proliferation, necrosis)
- Angiocentric glioma: diffuse glioma with uniform bipolar spindle cells characteristically arranged radially around blood vessels
- Polymorphous low grade neuroepithelial tumor of the young (PLNTY): diffuse growth pattern and tumor cells with oligo-like features (monotonous nuclei, perinuclear halos), minimal mitotic activity, calcifications are common
- Pediatric type diffuse high grade gliomas: distinguishing histologic characteristics are not generally seen; requires immunohistochemistry and molecular testing
- Infant type hemispheric glioma: highly cellular, infiltrative and frequently well demarcated from adjacent brain; often with leptomeningeal involvement
- Circumscribed astrocytic neoplasms
- Pilocytic astrocytoma
- Circumscribed, biphasic (compact and loose) astrocytic neoplasm with piloid cellular processes, bipolar tumor cells, low proliferation and often Rosenthal fibers or eosinophilic granular bodies
- High grade astrocytoma with piloid features
- Histopathologic features vary considerably and molecular testing is generally required for diagnosis
- Pleomorphic xanthoastrocytoma
- Predominantly circumscribed astrocytoma with pleomorphic nuclei, occasional cells with bubbly (xanthomatous) cytoplasm, eosinophilic granular bodies, perivascular lymphocytes and reticulin rich background
- Subependymal giant cell astrocytoma (SEGA)
- Circumscribed, moderately cellular astrocytic neoplasm composed of polygonal cells with abundant eosinophilic cytoplasm and often multinucleated cells
- Chordoid glioma
- Glial neoplasm composed of chords / ribbons of epithelioid cells in a variably myxoid background
- Astroblastoma, MN1 altered
- Glial neoplasm characterized by the presence of astroblastic perivascular pseudorosettes: radially oriented tumor cells with thick cellular processes oriented towards a central blood vessel
- Pilocytic astrocytoma
- Ependymal tumors
- Ependymoma
- Circumscribed neoplasm characterized by monomorphic round tumor cells, often arranged around blood vessels to form perivascular pseudorosettes
- Subependymoma
- Round, uniform tumor cells arranged in clusters / nests within a dense fibrillary background
- Microcystic changes and calcifications are common
- Myxopapillary ependymoma
- Glioma with papillary structures and often prominent myxoid background
- Ependymoma
- Reference: Neuro Oncol 2021;23:1231
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
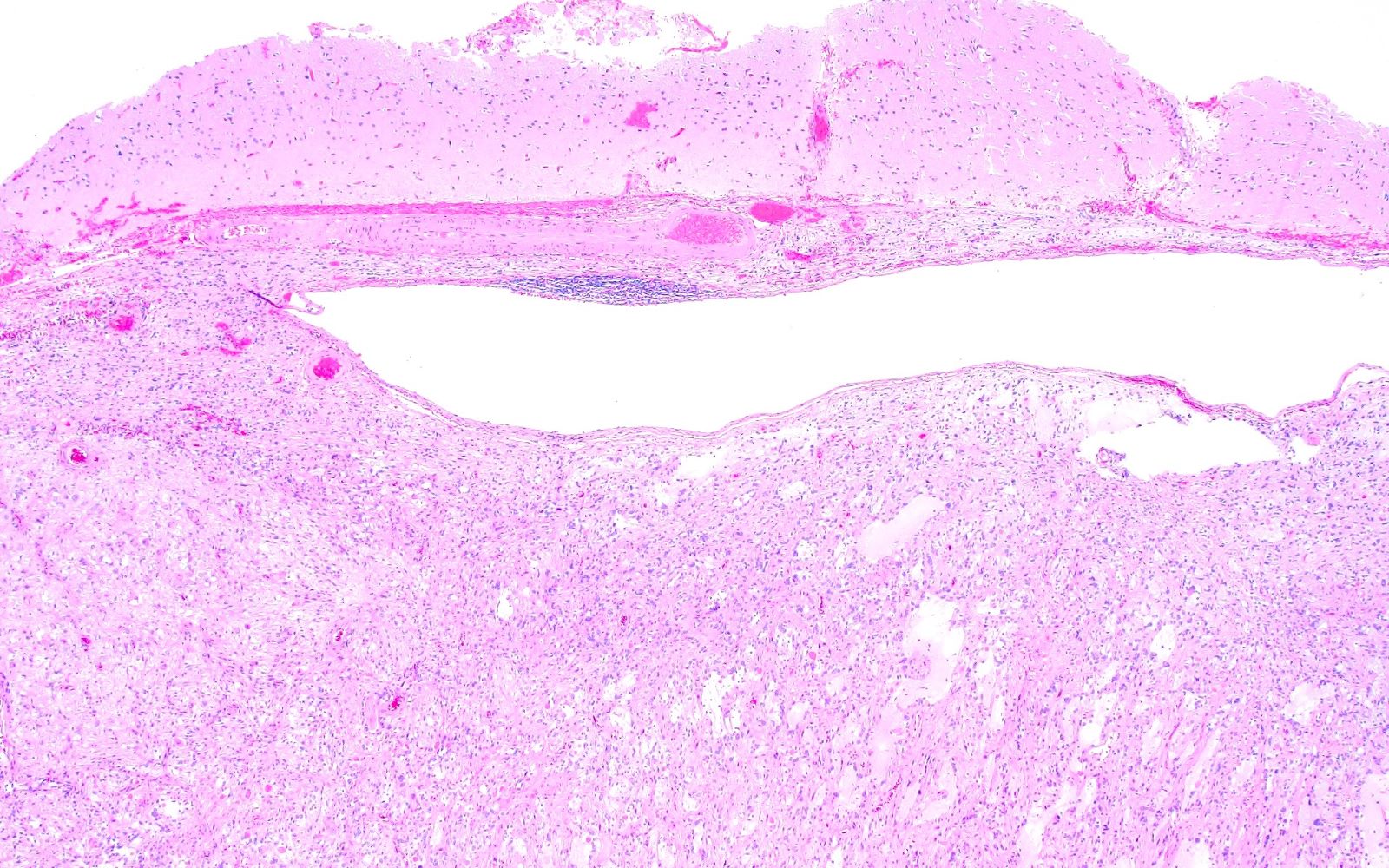
Circumscribed glioma
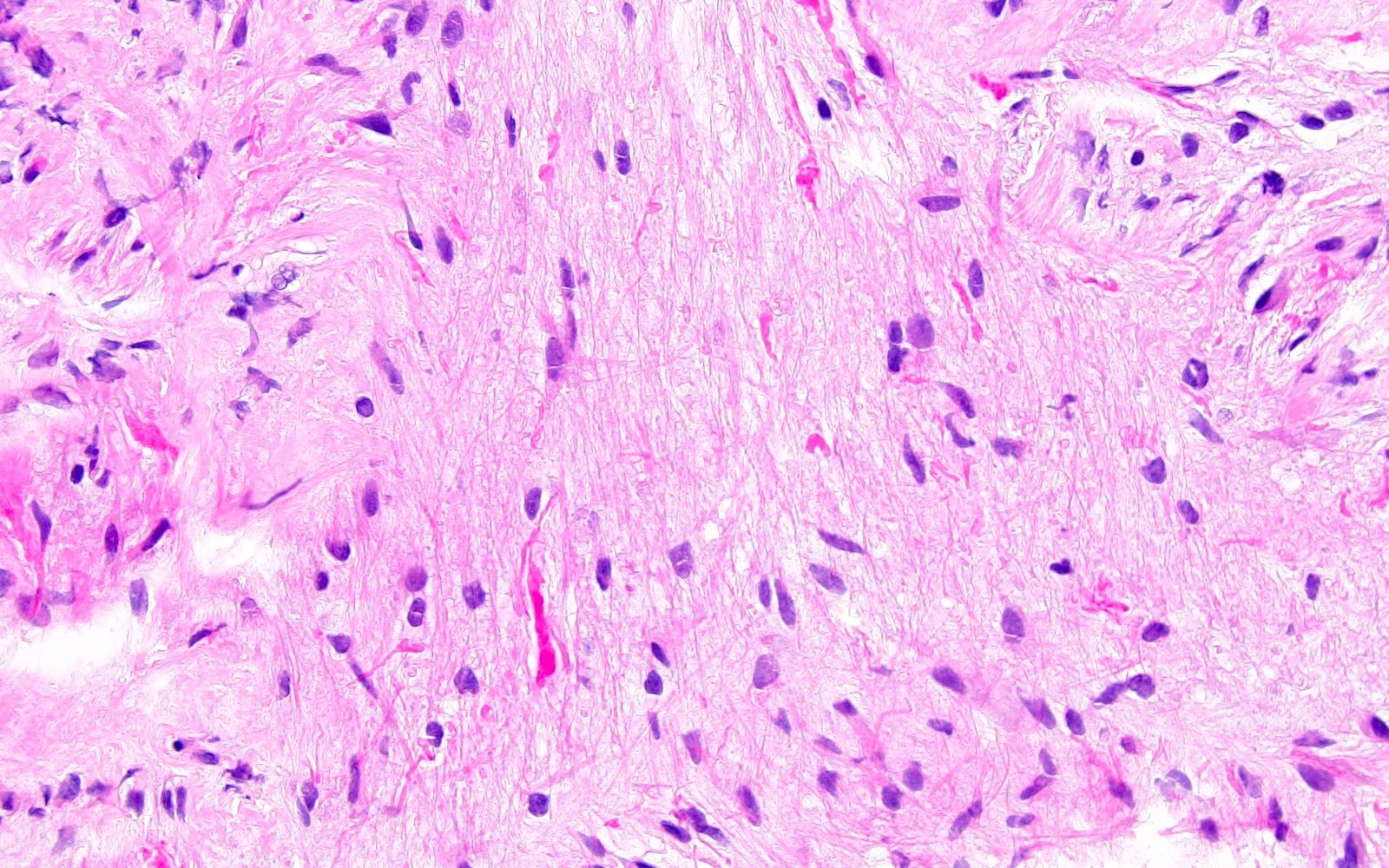
Pilocytic astrocytoma
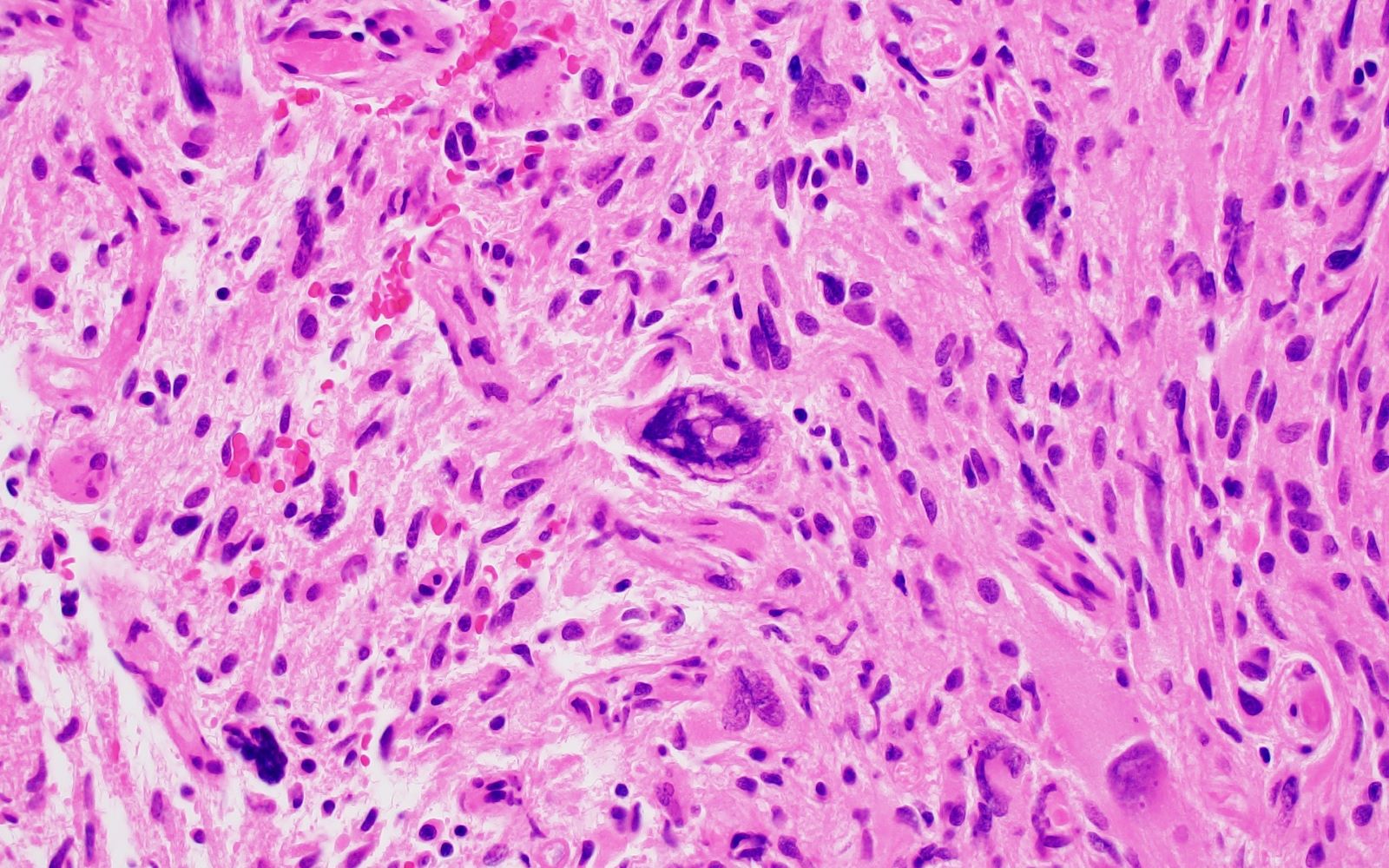
Pleomorphic xanthoastrocytoma (PXA)
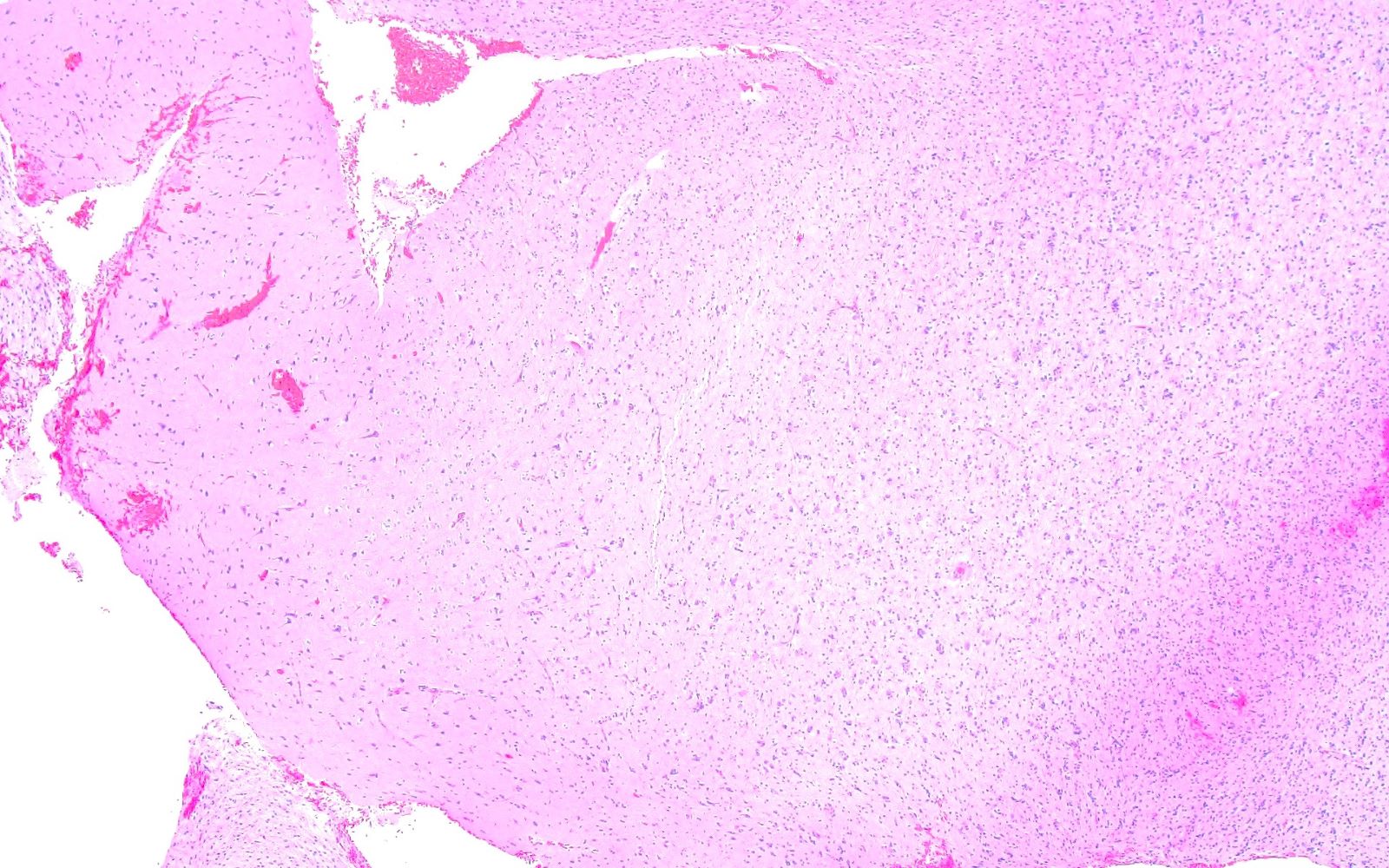
Glioma with infiltrating edge
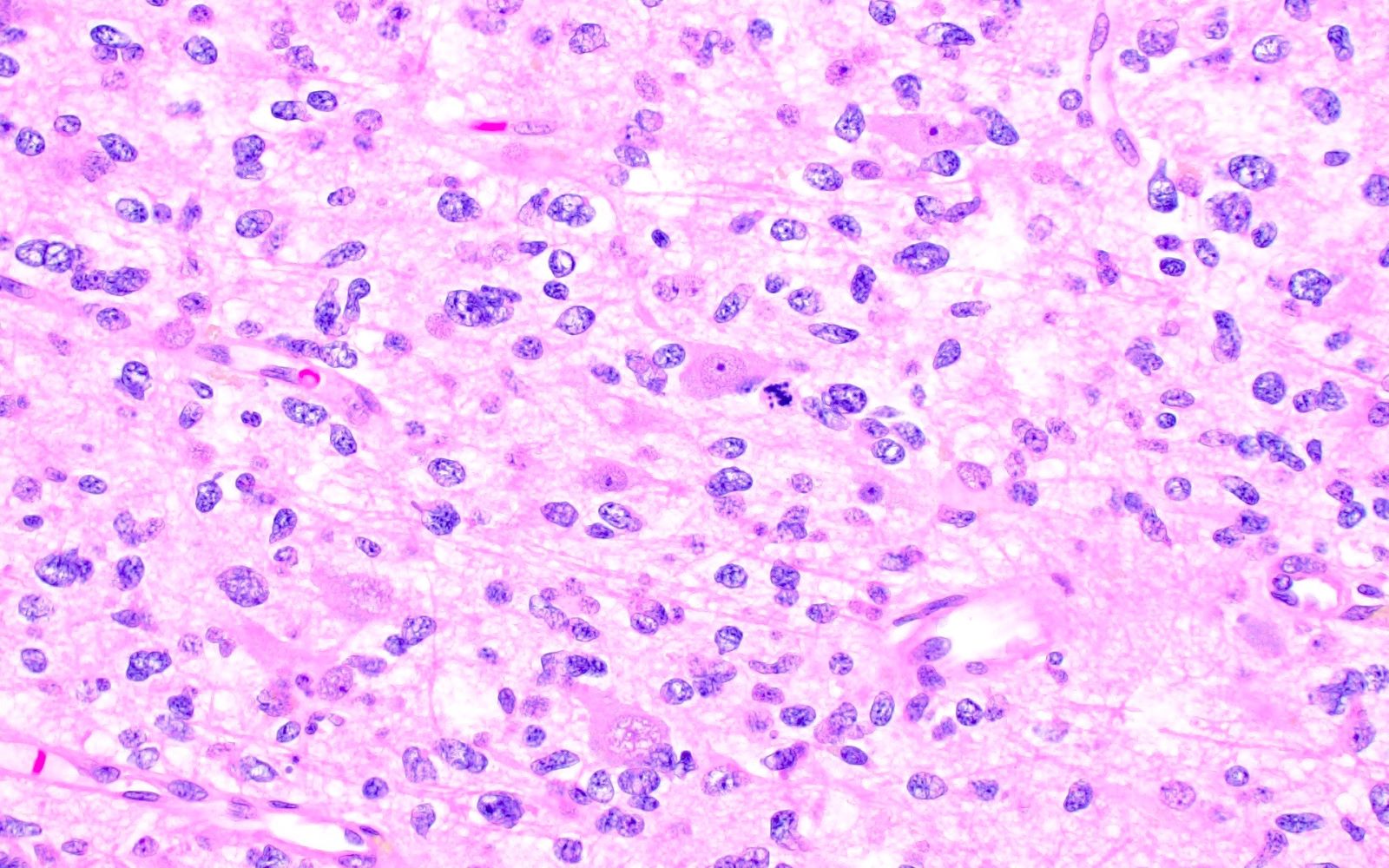
Diffuse glioma entrapped neurons
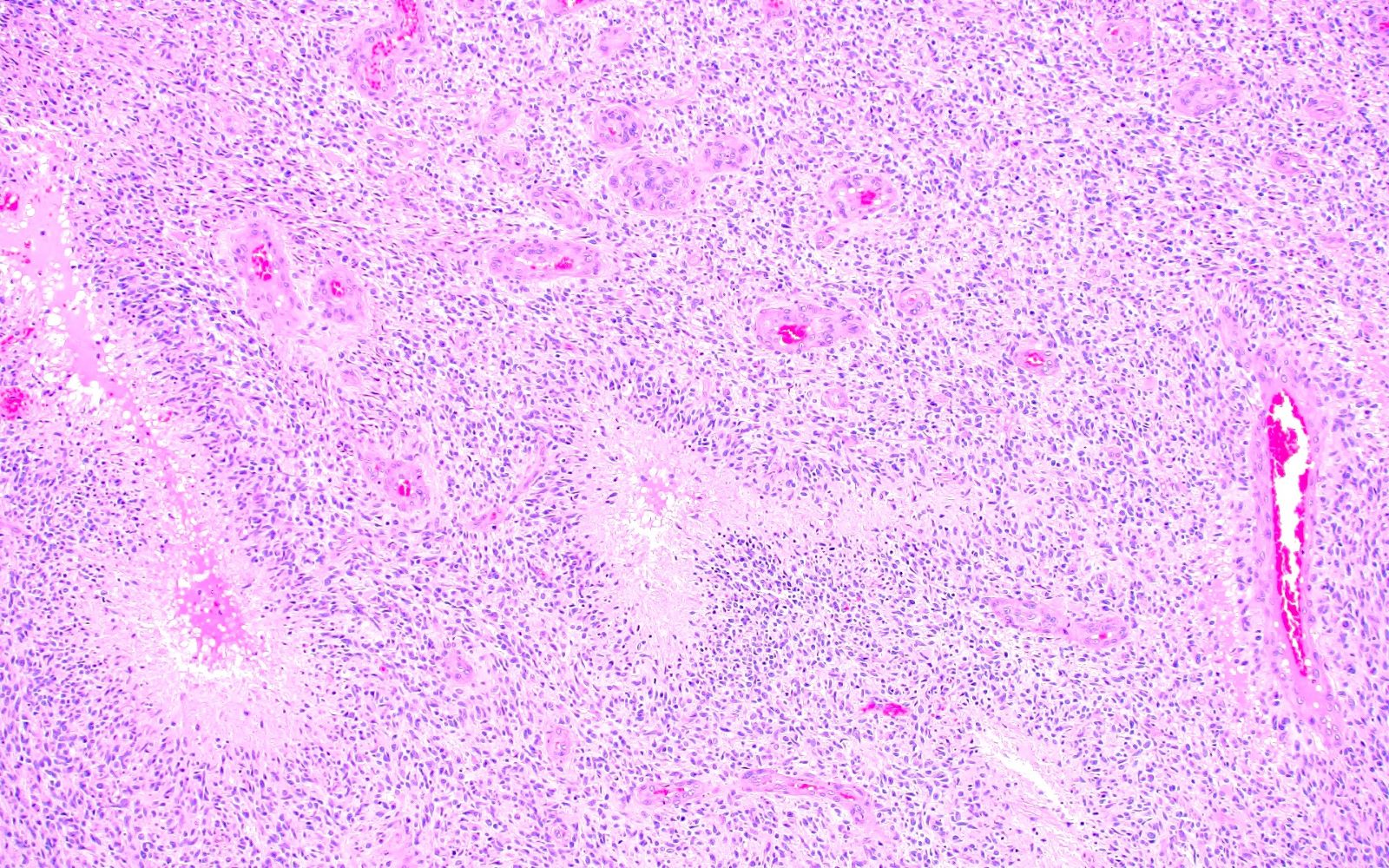
High grade infiltrating glioma
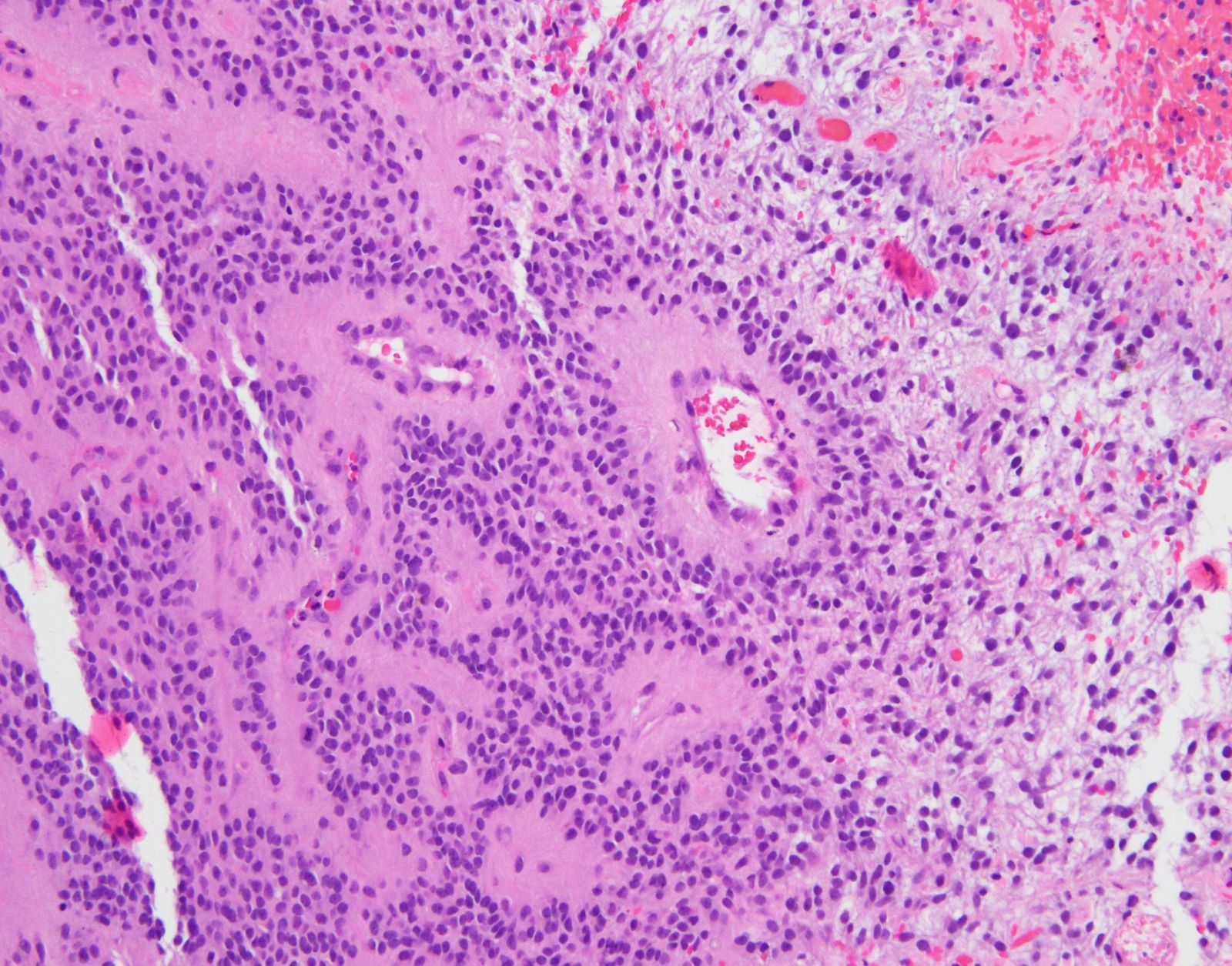
Perivascular pseudorosettes
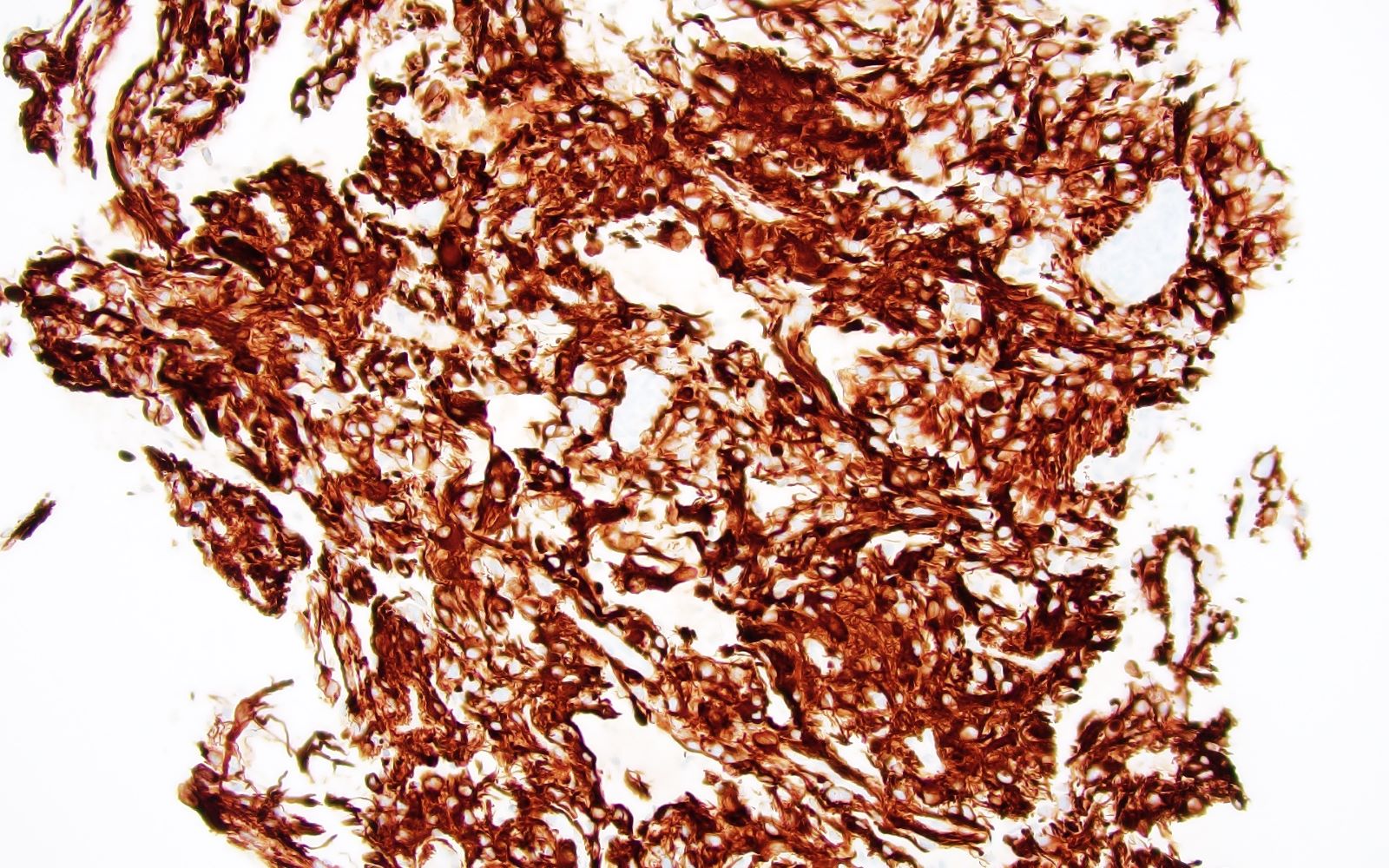
GFAP positive glioma
Images hosted on other servers:

Glioblastoma, not otherwise specified
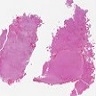
Diffuse astrocytoma, not otherwise specified
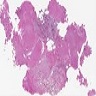
Oligodendroglioma, not otherwise specified
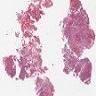
Ependymoma, not otherwise specified
Subependymoma
- Adult type diffuse gliomas
- Astrocytoma, IDH mutant (J Neuropathol Exp Neurol 2016;75:4, N Engl J Med 2015;372:2481, Acta Neuropathol 2018;136:153)
- By definition, presence of IDH1 / IDH2 mutation and intact chromosomes 1p and 19q
- Co-occurring ATRX and TP53 mutations are common
- Presence of CDKN2A / CDKN2B homozygous deletion associated with poor outcome and sufficient for WHO grade 4 designation, irrespective of histologic grade
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted (N Engl J Med 2015;372:2481, CNS Oncol 2015;4:287)
- By definition, presence of IDH1 / IDH2 mutation and whole arm deletion of chromosomes 1p and 19q
- Co-occurring CIC and TERT promoter mutations are common
- Glioblastoma, IDH wildtype (Acta Neuropathol 2018;136:793, N Engl J Med 2005;352:997)
- By definition, IDH1 / IDH2 wildtype
- Characteristic molecular findings
- TERT promoter mutation
- EGFR amplification
- Polysomy 7 and monosomy 10 (+7 / -10)
- Presence of any of these 3 molecular alterations is sufficient for WHO grade 4 designation in an adult type, IDH wildtype diffuse glioma, irrespective of histologic grade
- MGMT promoter methylation associated with better response to alkylating chemotherapy (temozolomide)
- Many other co-occurring molecular and cytogenetic alterations can be encountered
- Astrocytoma, IDH mutant (J Neuropathol Exp Neurol 2016;75:4, N Engl J Med 2015;372:2481, Acta Neuropathol 2018;136:153)
- Pediatric type diffuse low grade gliomas
- Diffuse astrocytoma, MYB or MYBL1 altered (Acta Neuropathol 2019;137:683)
- Defined by the presence of MYB or MYBL1 fusion, most common fusion partners include PCDHGA1, MMP16 and MAML2
- Typically not MYB::QKI fusion, which characterizes angiocentric glioma (see below)
- Angiocentric glioma (Acta Neuropathol 2019;137:683, Nat Genet 2016;48:273)
- Characterized by alteration of MYB on chromosome 6q, most commonly MYB::QKI fusion
- Polymorphous low grade neuroepithelial tumor of the young (PLNTY) (J Neuropathol Exp Neurol 2021;80:821)
- Characterized by MAPK pathway activating alterations, including BRAF V600E and fusions involving FGFR2 and FGFR3
- Diffuse low grade glioma, MAPK altered (Nat Genet 2013;45:602)
- Defined by the presence of a MAPK pathway activating alteration, in addition to wildtype IDH and H3 as well as absence of CDKN2A / CDKN2B homozygous deletion
- Diffuse astrocytoma, MYB or MYBL1 altered (Acta Neuropathol 2019;137:683)
- Pediatric type diffuse high grade gliomas
- Diffuse midline glioma, H3 K27 altered (Acta Neuropathol 2015;130:815)
- Defined by loss of H3 K27 trimethylation with presence of H3 K27 mutation, EGFR amplification or EZHIP overexpression in a diffusely infiltrating glioma located along the midline
- Can be alternately defined by loss of H3 K27 trimethylation with a methylation profile of diffuse midline glioma in a diffusely infiltrating glioma located along the midline
- Diffuse hemispheric glioma, H3 G34 mutant (Acta Neuropathol 2016;131:137)
- Defined by the presence of H3 G34 mutation in a diffusely infiltrating glioma located in a cerebral hemisphere
- Diffuse pediatric type high grade glioma, H3 wildtype and IDH wildtype (Acta Neuropathol 2017;134:507)
- Diffuse glioma lacking IDH and H3 alterations; often with PDGFRA alteration, EGFR alteration or MYCN amplification
- Infant type hemispheric glioma (Nat Commun 2019;10:4343)
- Characterized by the presence of a receptor tyrosine kinase abnormality, especially fusions involving ROS1, MET1, ALK or NTRK family gene
- Diffuse midline glioma, H3 K27 altered (Acta Neuropathol 2015;130:815)
- Circumscribed astrocytic neoplasms
- Pilocytic astrocytoma (Cancer Res 2008;68:8673, Nat Genet 2013;45:602)
- Cerebellar tumors commonly harbor KIAA1549::BRAF fusion (7q34 duplication); other MAPK pathway alterations can also be encountered
- High grade astrocytoma with piloid features (Acta Neuropathol 2018;136:273)
- Presence of a MAPK pathway alteration along with CDKN2A / CDKN2B homozygous deletion, amplification of CDK4 or ATRX mutation
- DNA methylation profiling is only definitive method for diagnosis
- Pleomorphic xanthoastrocytoma (Hum Pathol 2019;86:38, Brain Pathol 2019;29:85)
- Characterized by the presence of both BRAF V600E and CDKN2A / CDKN2B homozygous deletion
- Subependymal giant cell astrocytoma (SEGA) (Oncotarget 2017;8:95516)
- Seen almost exclusively in patients with tuberous sclerosis (germline mutations in TSC1 or TSC2)
- Chordoid glioma (Nat Commun 2018;9:810)
- Characterized by PRKCA D463 mutation
- Astroblastoma, MN1 altered (Brain Tumor Pathol 2019;36:112)
- Defined by presence of MN1 alteration, most commonly MN1::BEND2 fusion
- Pilocytic astrocytoma (Cancer Res 2008;68:8673, Nat Genet 2013;45:602)
- Ependymal tumors
- Ependymoma (Cancer Cell 2015;27:728, Brain Pathol 2020;30:863)
- Supratentorial ependymoma characterized by ZFTA fusion or YAP1 fusion
- Posterior fossa ependymoma characterized by unique methylation patterns
- Spinal ependymoma characterized by NF2 mutation or MYCN amplification
- Subependymoma (Neuro Oncol 2018;20:1616)
- Recurrent losses of chromosome 19 and partial losses of chromosome 6
- Myxopapillary ependymoma (Neuro Oncol 2018;20:1616)
- Recurrent gains of chromosome 16 and losses of chromosome 10
- Note: ependymomas are defined by both anatomic location as well as molecular features
- Ependymoma (Cancer Cell 2015;27:728, Brain Pathol 2020;30:863)
- Many of these tumors form distinct clusters on DNA methylation profiling (Nature 2018;555:469)
- Brain, left frontal lobe mass, resection:
- Integrated diagnosis: glioblastoma, IDH wildtype, WHO grade 4
- Histopathologic classification: glioblastoma
- CNS WHO grade: 4
- Molecular information: TERT promoter mutation, EGFR amplification
- Brain, right parietal mass, resection:
- Integrated diagnosis: supratentorial ependymoma, not otherwise specified (NOS)
- Histopathologic classification: ependymoma
- CNS WHO grade: 3
- Molecular information: genetic material extracted from FFPE tissue was of insufficient quality or quantity for molecular testing
- Reactive gliosis (ischemic, inflammatory, demyelination, infectious, etc.):
- Glioneuronal tumors, especially in the case of a circumscribed astrocytic neoplasm:
- Generally display some degree of neuronal differentiation, including dysmorphic ganglioid cells or expression of neural antigens (such as synaptophysin)
- See individual topics for additional differential considerations for each tumor type
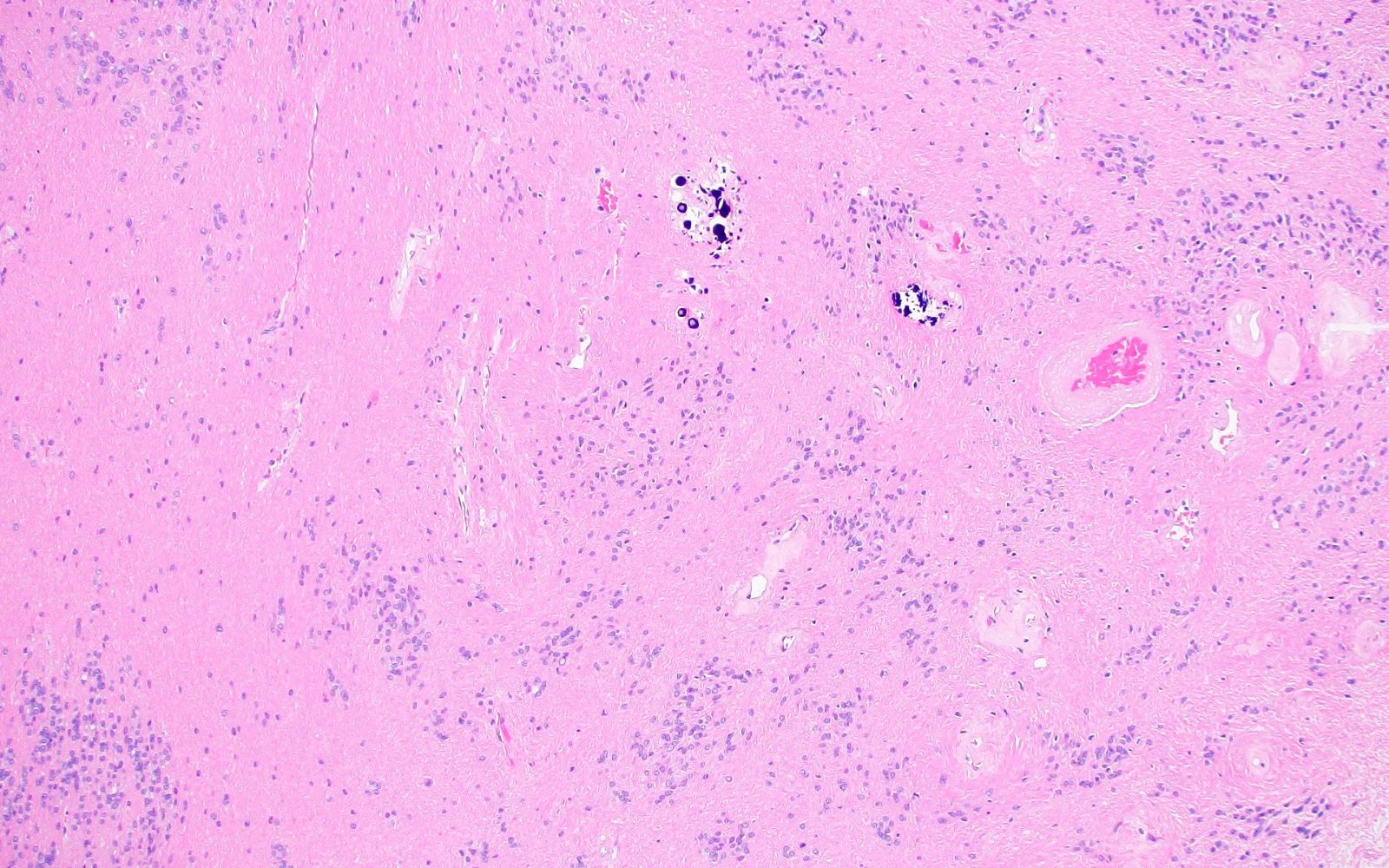
During autopsy, an incidental mass is discovered in the fourth ventricle. The histopathology is shown in the photomicrograph above. What is the most likely diagnosis?
- Astrocytoma, IDH mutant, WHO grade 2
- Pilocytic astrocytoma
- Reactive astrogliosis
- Subependymoma
Comment Here
Reference: Glioma overview
- 1
- 2
- 3
- 4
Comment Here
Reference: Glioma overview
- Rare, classic variant of glioblastoma (GBM), WHO grade 4
- Biphasic glial and mesenchymal differentiation
- De novo: primary gliosarcoma (GS); secondary GS develops in previously resected or irradiated glioblastoma
- Biphasic glial and mesenchymal differentiation
- Clinical and genetic similarities to glioblastoma; likewise, treatment and management is similar to that of glioblastoma
- Overall, an aggressive tumor with slightly worse prognosis than glioblastoma (Neuro Oncol 2009;11:183)
- Gliosarcoma (GS)
- Classic variant of glioblastoma (GBM)
- ICD-10: C71.9 - malignant neoplasm of brain, unspecified
- Comprises ~2% of all glioblastoma cases (Neuro Oncol 2009;11:183)
- Typically affects adults in the fourth to seventh decades of life (Neuro Oncol 2009;11:183)
- Slight male predominance, similar to glioblastoma (Neuro Oncol 2009;11:183)
- Pediatric incidence is rare but up to ~13% of all gliosarcoma; age range of 3 - 19 years (BMC Pediatr 2021;21:101)
- Majority are supratentorial, with tendency towards peripheral localization and dural attachment (Eur Radiol 2019;29:429)
- Temporal > parietal > frontal > occipital lobes; rarely involve occipital region or posterior fossa (Neuro Oncol 2009;11:183)
- Can involve and invade into overlying meninges, bone and extracranial soft tissue (Case Rep Radiol 2016;2016:1762195)
- Mesenchymal / sarcomatous component is associated with propensity to invade and metastasize
- Gliosarcoma has greater propensity for extracranial metastasis than other CNS tumors, including glioblastoma (J Neurooncol 2007;83:39)
- Most common sites of extracranial metastases include lung, liver and lymph node but are also reported in adrenal gland, spleen, kidney, oral mucosa, skin, bone marrow and bone
- Metastatic tumor deposits contain sarcomatous with or without glial components
- Similar genetic alterations (p53 mutations, p16 deletions and PTEN mutations) both in glial and sarcomatous components support monoclonal origin (Am J Pathol 2000;156:425)
- Prior history of radiation for CNS tumor is a known risk factor
- Presents similarly to glioblastoma
- Symptoms and signs depend on tumor location, increased intracranial pressure
- Common symptoms include headache, nausea, vomiting, seizures, dysphasia, dizziness, cognitive dysfunction, altered mental status and personality changes
- Common signs include focal neurological deficits, visual field defects and papilledema
- Reference: Cancer 2010;116:1358
- Based on clinical history, imaging, histology, immunohistochemical and molecular features
- Usually infiltrative, supratentorial, peripheral, cortical based with dural base / invasion or skull involvement (Eur Radiol 2019;29:429)
- Rarely infratentorial or intraventricular
- CT imaging:
- Large necrotic areas and heterogenous contrast enhancement, similar to glioblastoma (AJNR Am J Neuroradiol 1985;6:527, Neurosurgery 1990;26:261)
- Hyperdense with well defined margins homogenous enhancement, mimicking meningioma (AJNR Am J Neuroradiol 1985;6:527, Neurosurgery 1990;26:261)
- MRI:
- Well demarcated mass with a heterogeneous appearance (Eur Radiol 2019;29:429)
- Heterogeneously isointense to hypointense on the T1 weighted images (Eur Radiol 2019;29:429)
- Mixed hypointense / homointense / hyperintense on the T2 weighted images (Eur Radiol 2019;29:429)
- Unevenly thickened walls with strong rim and a ring-like or paliform enhancement patterns in 78.26% (36/46) of cases (Eur Radiol 2019;29:429)
- Hemorrhage at different stages; salt and pepper sign on T2 or T1 weighted images in 67.39% (31/46) of cases (Eur Radiol 2019;29:429)
Contributed by Nicolas Kostelecky, M.D.
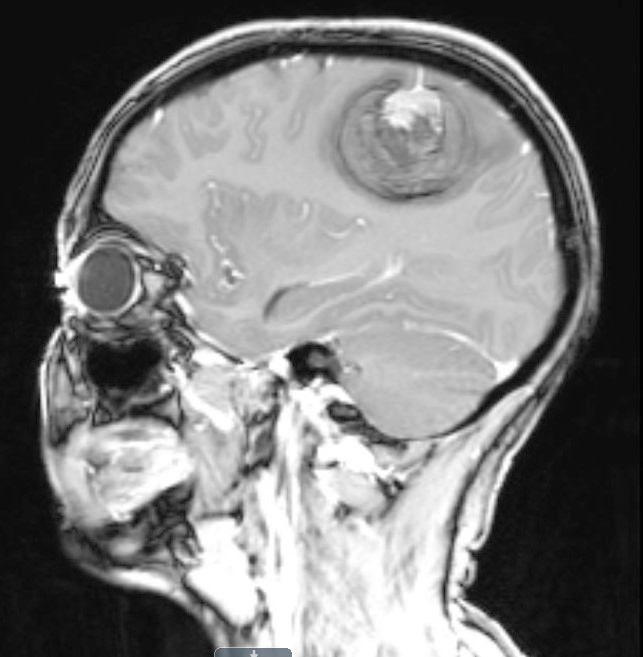
Sagittal T1 postcontrast
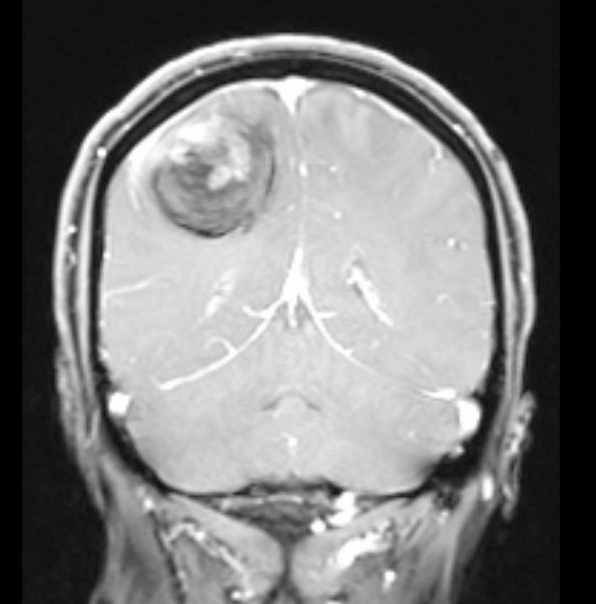
Coronal T1 postcontrast
Images hosted on other servers:

Spectrum of
radiologic
phenotypes
(n = 4)
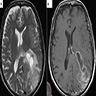
Heterogeneous, solid / cystic, with rim enhancement
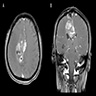
Solid / cystic with leptomeningeal involvement

Cystic / solid; temporoparietal region
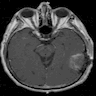
Before surgery
- Median overall survival with treatment (both primary and secondary gliosarcoma): 17.5 months (J Neurooncol 2015;125:401)
- Median overall survival with treatment (primary gliosarcoma alone): 24.7 months (J Neurooncol 2015;125:401)
- Most important prognostic factors of overall survival: age, extent of resection and adjuvant radiotherapy (Neuro Oncol 2009;11:183)
- 8 year old boy with giant parieto-occipital gliosarcoma (Surg Neurol Int 2018;9:111)
- 16 year old boy, 23 year old woman, 35 year old man and 35 year old woman with gliosarcoma (World J Oncol 2017;8:53)
- 37 year old woman with primary gliosarcoma; 2 recurrences and extracranial metastasis, including molecular evidence for clonal evolution (Cold Spring Harb Mol Case Stud 2020;6:a004671)
- 69 year old man with primary gliosarcoma and multiple extracranial metastases (Brain Tumor Res Treat 2020;8:53)
- Multimodal therapy, similar to glioblastoma (J Clin Neurosci 2014;21:478, Clin Neurol Neurosurg 2019;182:98)
- Treatment includes maximal surgical resection, followed by radiotherapy and chemotherapy (J Clin Neurosci 2014;21:478, Clin Neurol Neurosurg 2019;182:98)
- Given the higher propensity to metastasize, screening for extracranial metastases is recommended (J Neurooncol 2007;83:39)
- Usually well circumscribed, lobular mass in superficial location, weakly attached to dura (Neurosurgery 1990;26:261)
- Glial component is soft with foci of yellowish necrosis or red-brown recent and remote hemorrhage
- Sarcomatous component is firm and well circumscribed, facilitating separation from adjacent brain tissue
- Mixed tumor of biphasic differentiation with glial and sarcomatous components
- Proportions of glial and sarcomatous components vary; may show mosaic pattern
- Glial component typically shows typical features of glioblastoma, including pleomorphic astrocytic cells with nuclear atypia, mitosis, pseudopalisading necrosis and microvascular proliferation
- Sarcomatous component shows densely packed, spindle shaped cells with nuclear atypia; can show a variety of morphologies: fibrosarcoma, osteosarcoma, chondrosarcoma, angiosarcoma, rhabdosarcoma or undifferentiated pleomorphic sarcoma
Contributed by Irfan Yasin, M.B.B.S., Katherine Schwetye, M.D., Ph.D. and Miguel Guzman, M.D.
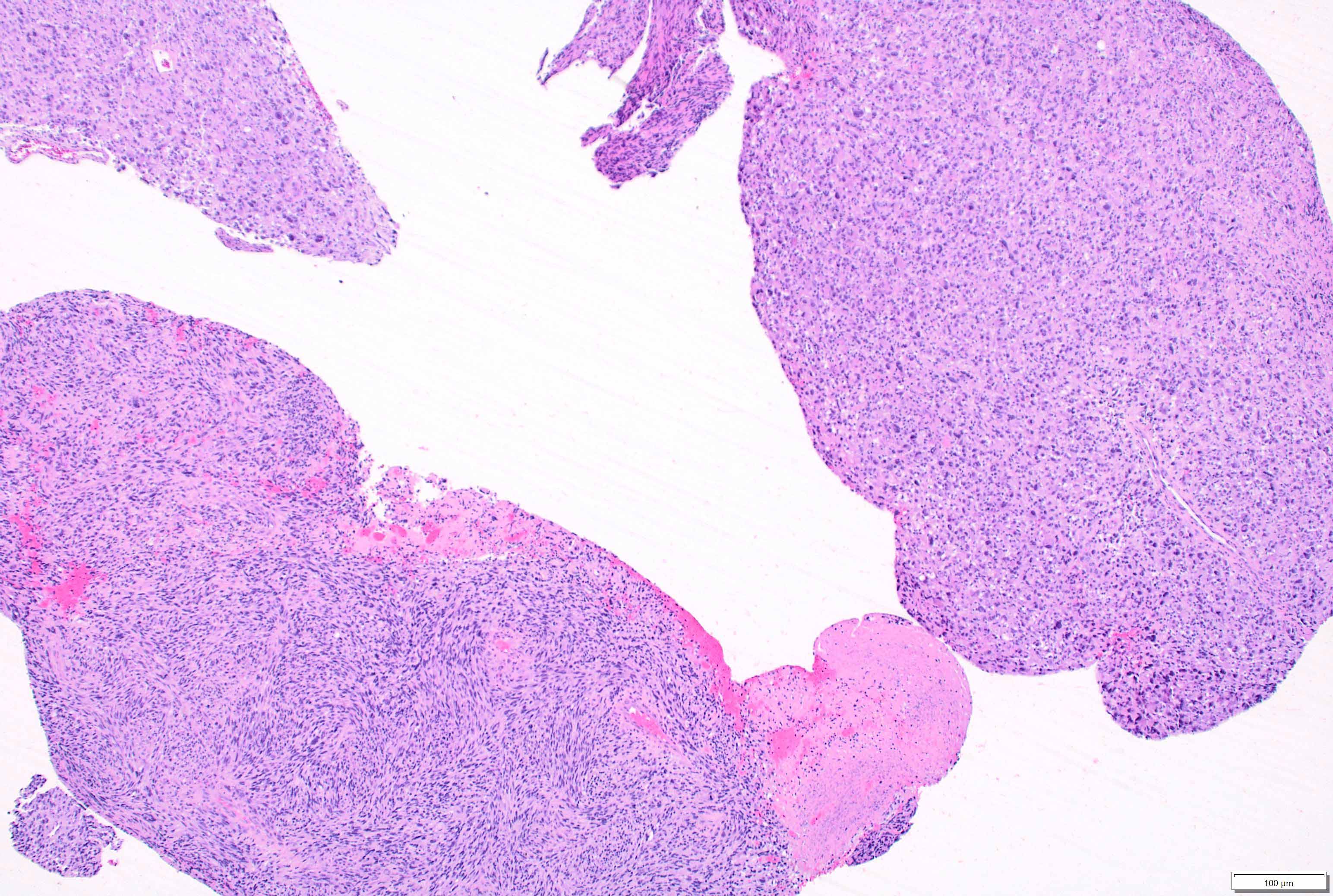
Glial and sarcomatous component
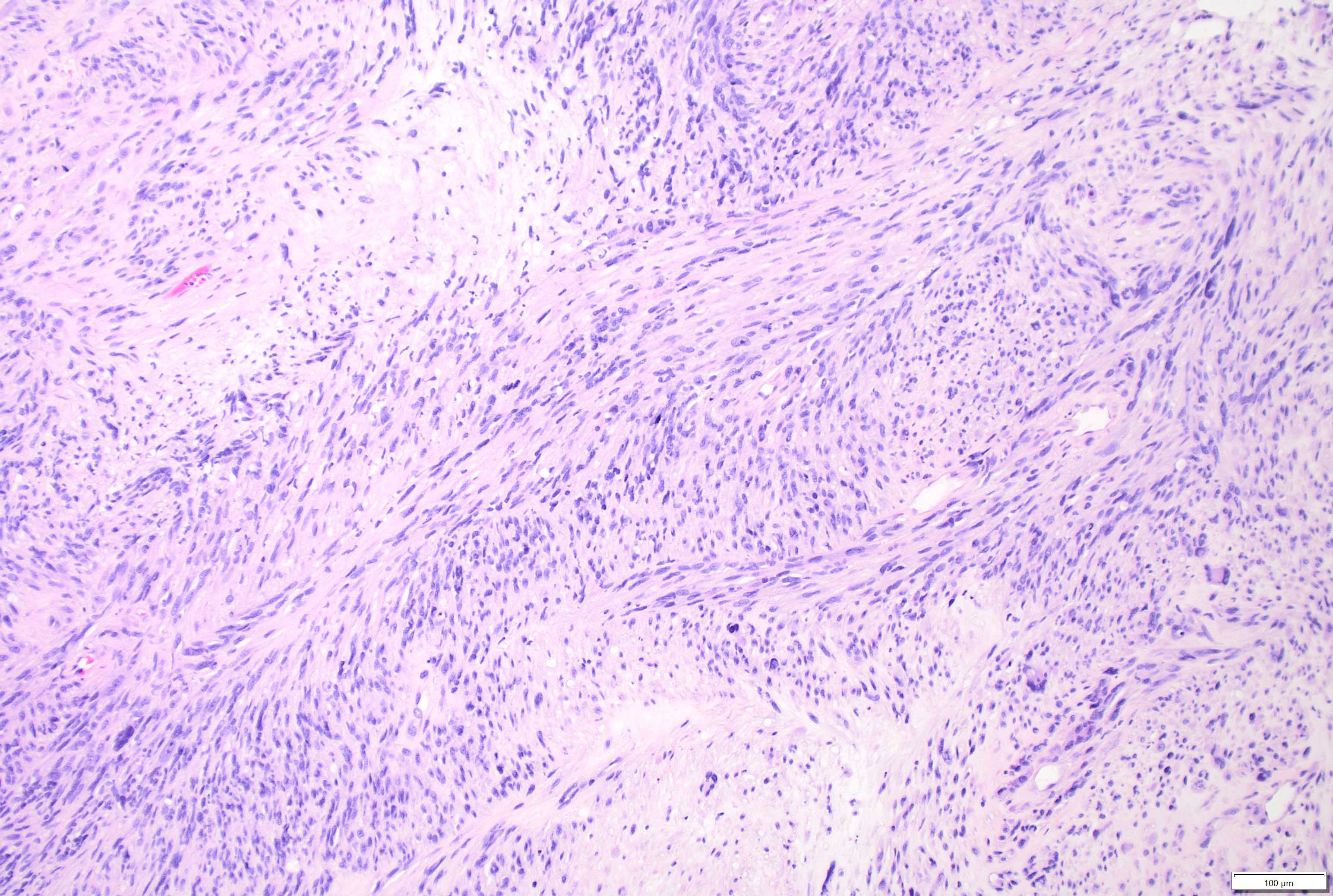
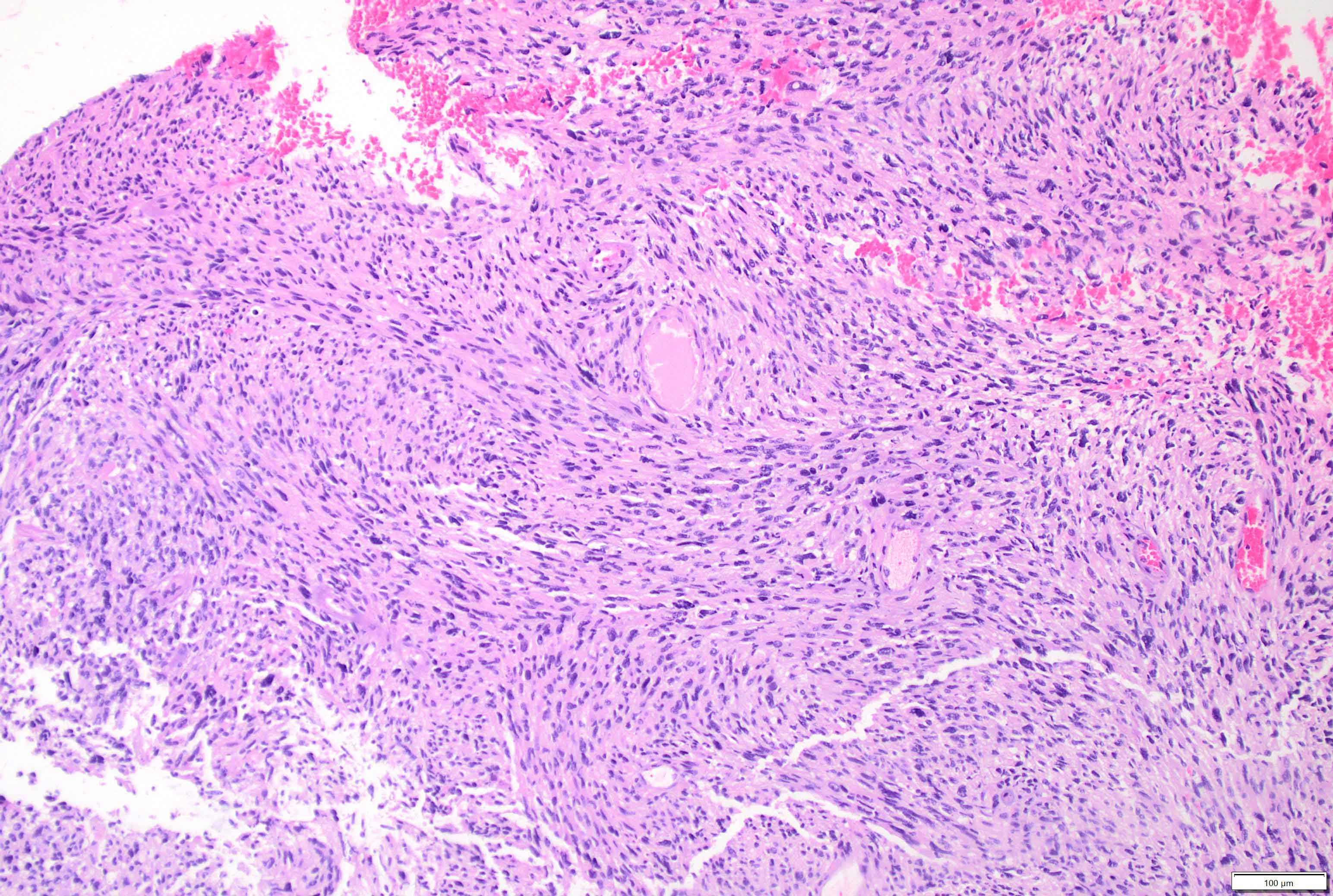
Sarcomatous component
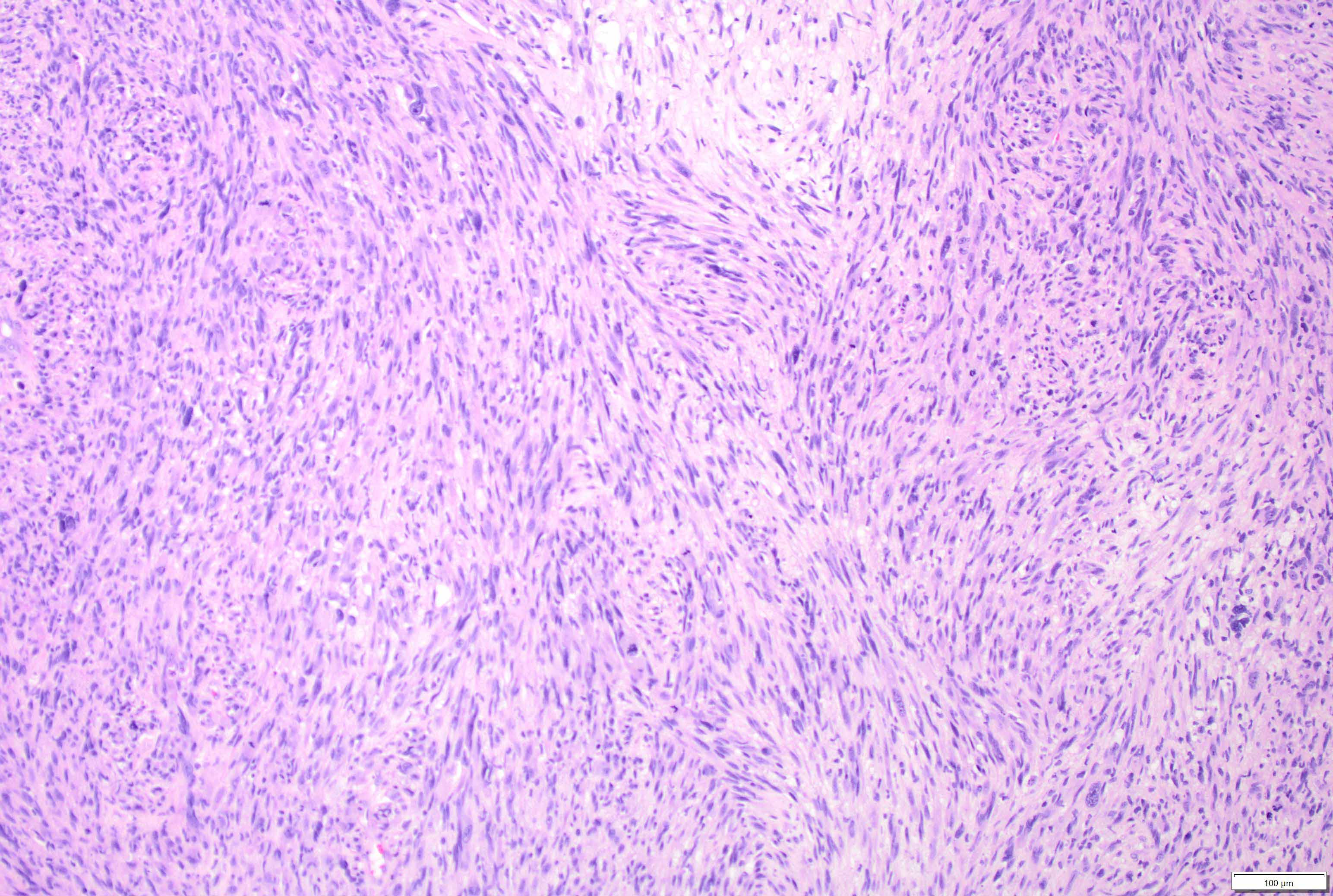
Herringbone architecture
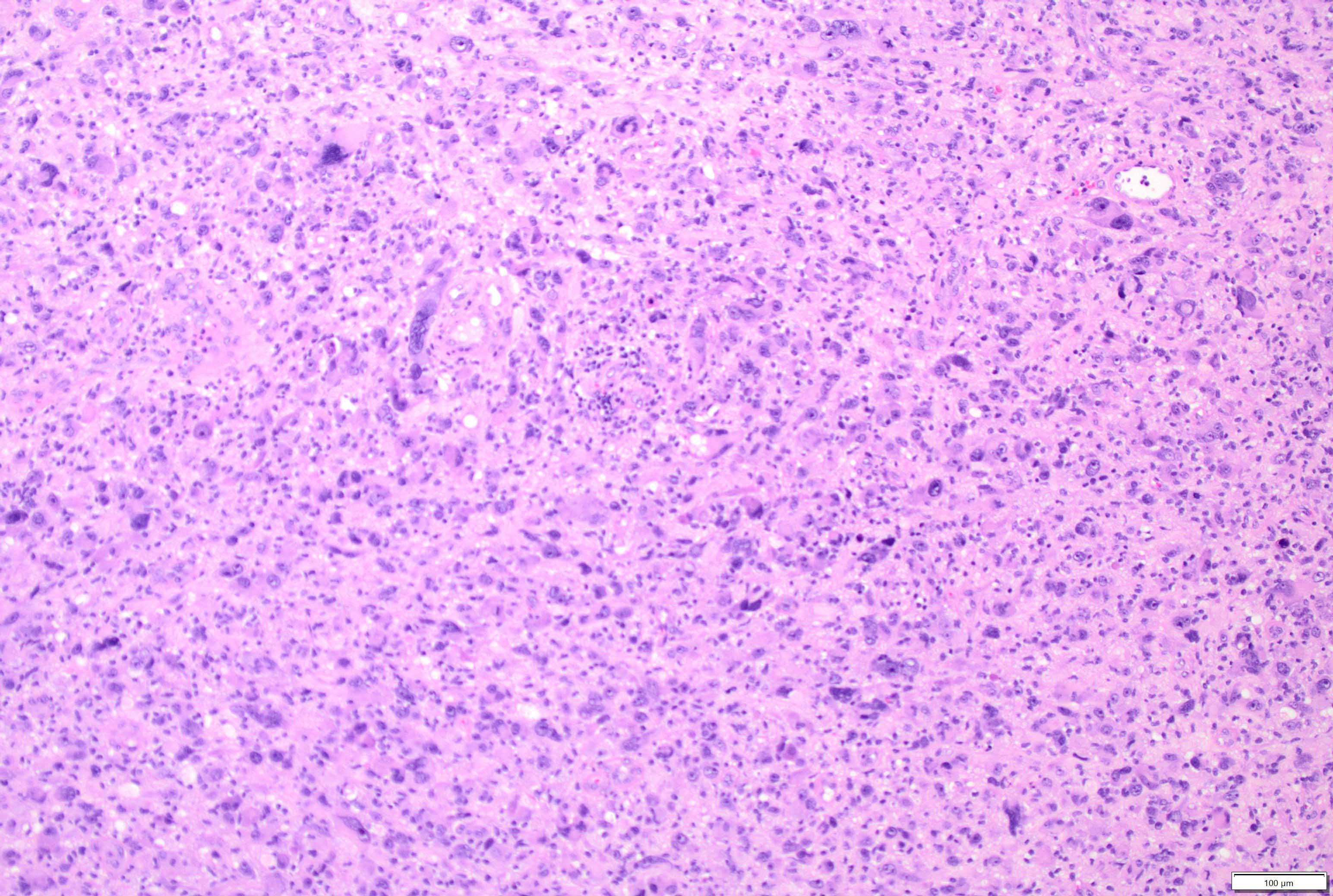
Severe atypia
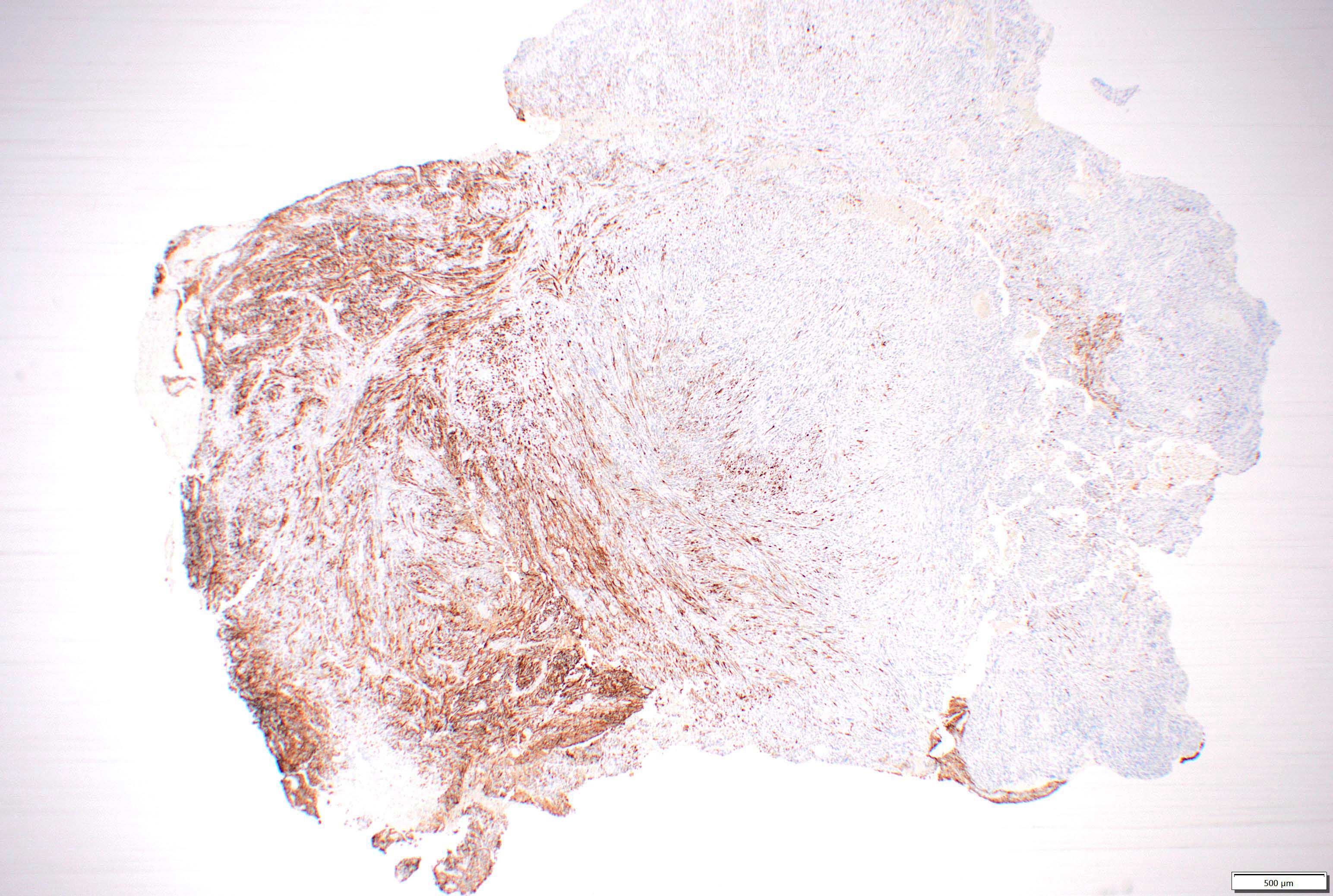
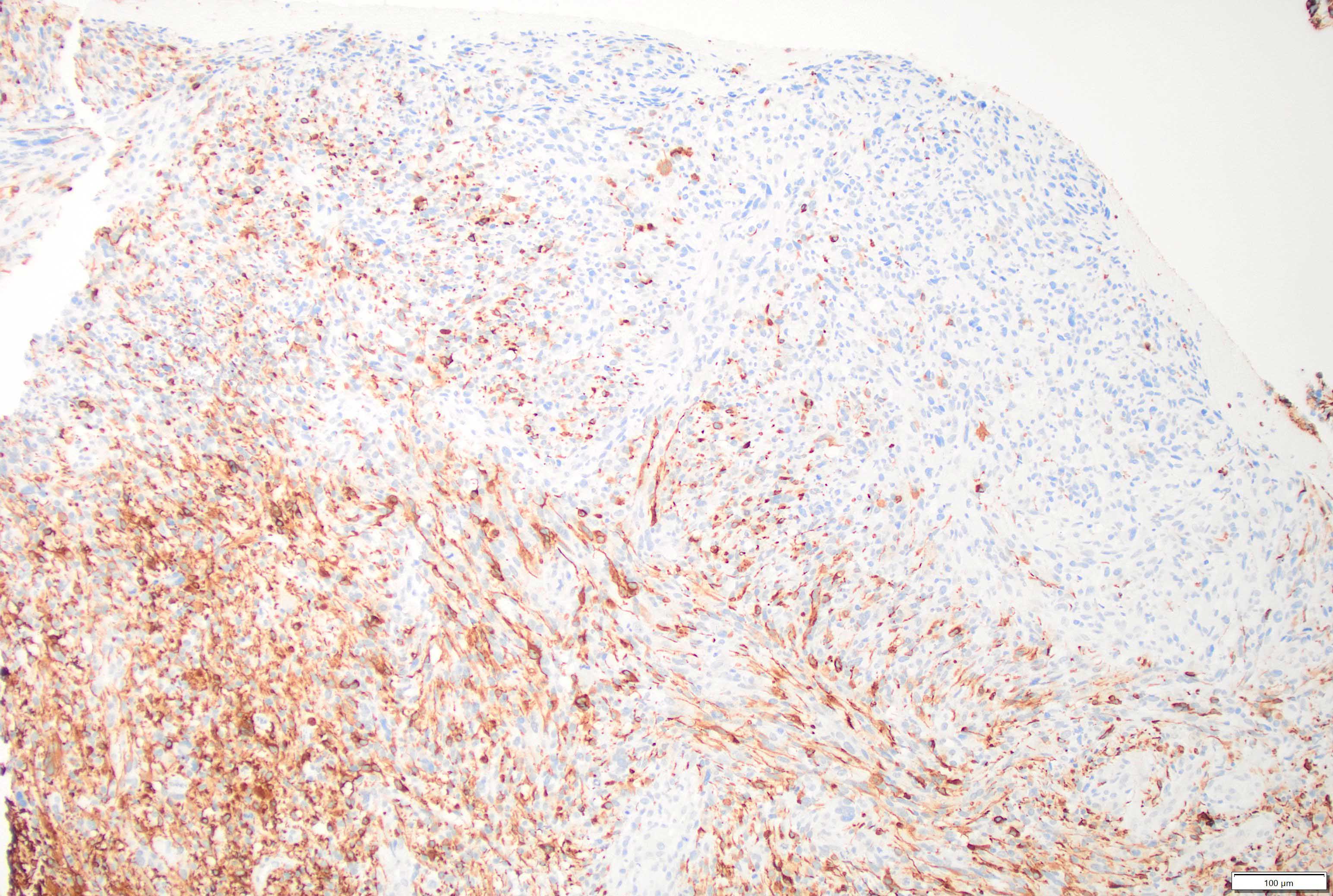
GFAP
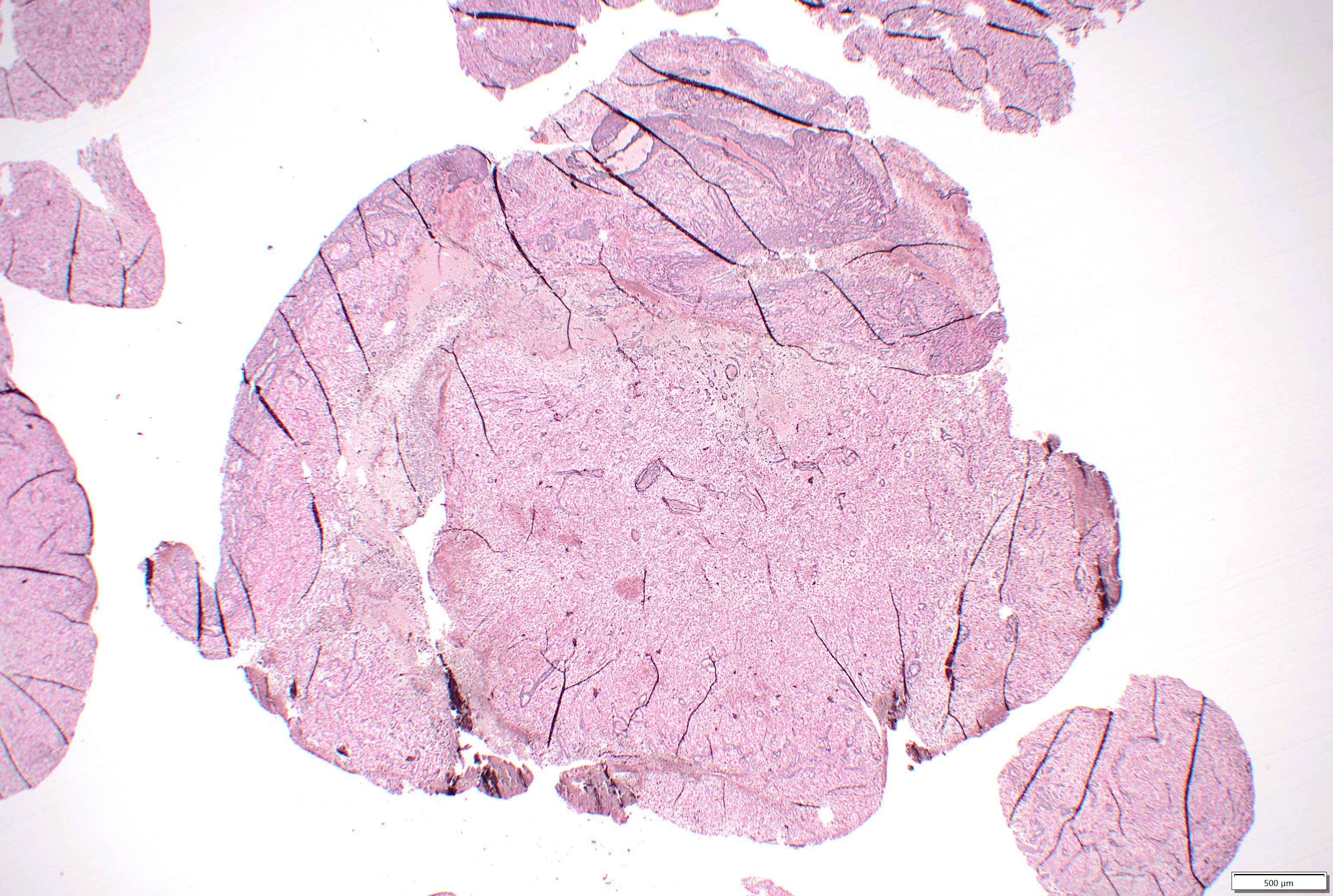
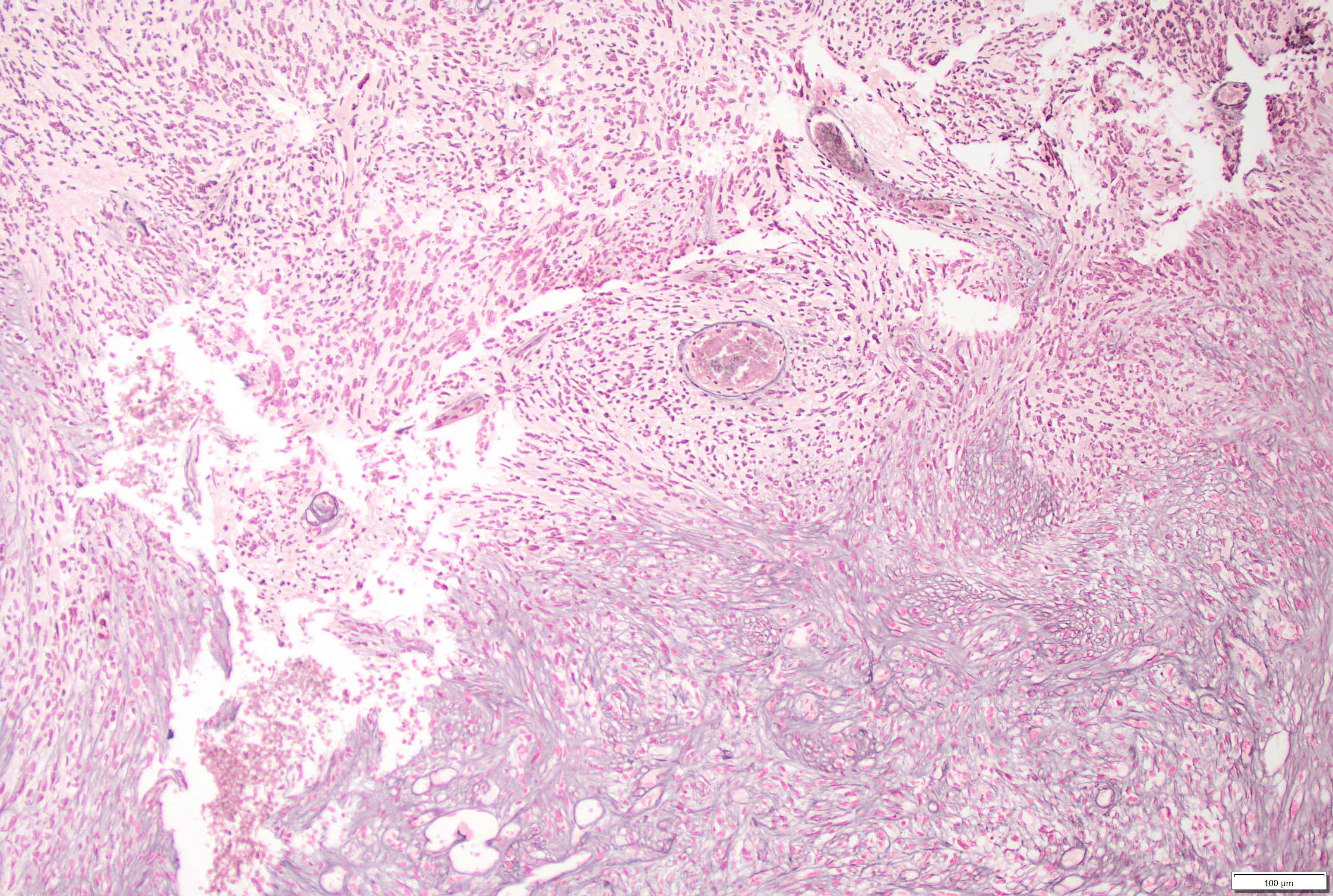
Reticulin
Images hosted on other servers:
H&E; intermixed glial and sarcomatous elements
- Smears demonstrate biphasic neoplastic cell populations with mesenchymal and glial components
- Highly cellular, rich arborizing vessels, high mitotic rate and necrosis
- Glial component has pleomorphic round / oval nuclei
- References: Diagn Cytopathol 2009;37:906, Acta Cytol 2016;60:490, Diagn Cytopathol 2004;30:77
- GFAP in glial region but mostly negative in sarcomatous region
- Olig2 is positive in the glial region (93% of cases) (Mod Pathol 2013;26:315)
- Reticulin in sarcomatous region and blood vessels in glial region
- Vimentin in sarcomatous region
- Other stains in sarcomatous region depending on differentiation: CD34, desmin, etc.
- p53 may be positive in both glial and sarcomatous region
- References: Neuroradiol J 2013;26:639, Neuropathol Appl Neurobiol 1991;17:177
- Olig2, unlike other glioblastomas
- Most common genetic aberrations in gliosarcoma similar to those of primary glioblastoma: PTEN and PIK3 mutations, p53 mutation and p16 deletion (Cancers (Basel) 2019;11:284)
- Among 19 commonly altered genes in gliosarcoma: most frequent are TERT promoter (92%), PTEN (66%), TP53 (60%) and NF1 (41%) (Sci Rep 2021;11:18009)
- Altered genes that are potentially targetable by FDA approved therapies: BRAF, EGFR, CDKN2A, NF1 and PTEN (Sci Rep 2021;11:18009)
- Less frequent pathologic EGFR alteration in gliosarcoma as compared to glioblastoma (BMC Neurol 2021;21:231)
- MGMT methylation and IDH1 mutation are rare events in primary gliosarcoma (J Neurooncol 2018;137:303)
- Gliosarcomas overexpress mesenchymal proteins, indicating epithelial mesenchymal transition (EMT) (Brain Pathol 2012;22:670)
- Brain, cerebrum, right, mass, excision:
- Gliosarcoma, IDH wildtype, WHO grade 4 (see microscopic description and comment)
- Microscopic examination shows highly pleomorphic, glial and spindled tumor cells with pseudopalisading necrosis and microvascular proliferation. The spindle cell component has herringbone architecture. The more pleomorphic glial elements show high cellularity. Tumor is remarkable for abundant atypical mitosis. There are areas of geographical necrosis.
- IHC: The glial elements are positive for GFAP. Reticulin is markedly increased in the spindle cell areas with concomitant negativity for GFAP. This dual immunostaining pattern with strong reactivity of glial component for GFAP and reticulin positivity in mesenchymal component supports a diagnosis of glioblastoma.
- Molecular: PCR based analysis of IDH1 and IDH2 genes (exon 4) shows wildtype phenotype. Analysis for MGMT gene promoter hypermethylation shows no hypermethylation.
- Glioblastoma with meningeal invasion:
- Lacks alternating areas of glioma and sarcoma
- Oligosarcoma (Acta Neuropathol 2022;143:263):
- Recurrence after oligodendroglioma or de novo
- Belongs to IDH mutant and 1p19q codeleted category in 2021 WHO
- Smooth muscle differentiation by proteomic profiling
- Embedded in a dense network of reticulin fibers
- Frequent p53 accumulation, positivity for SMA and CALD1, loss of Olig2, gain of H3K27me3, CDKN2A / CDKN2B deletions, loss of NF1 and gain of YAP1
- Frequent mutations in IDH, TERT promoter, FUBP1, CIC, NF1 and TP53
- Primary sarcomas of the CNS:
- Lack glial differentiation
- Metastatic sarcomas:
- Clinical history of a primary sarcoma elsewhere
- Lack glial differentiation
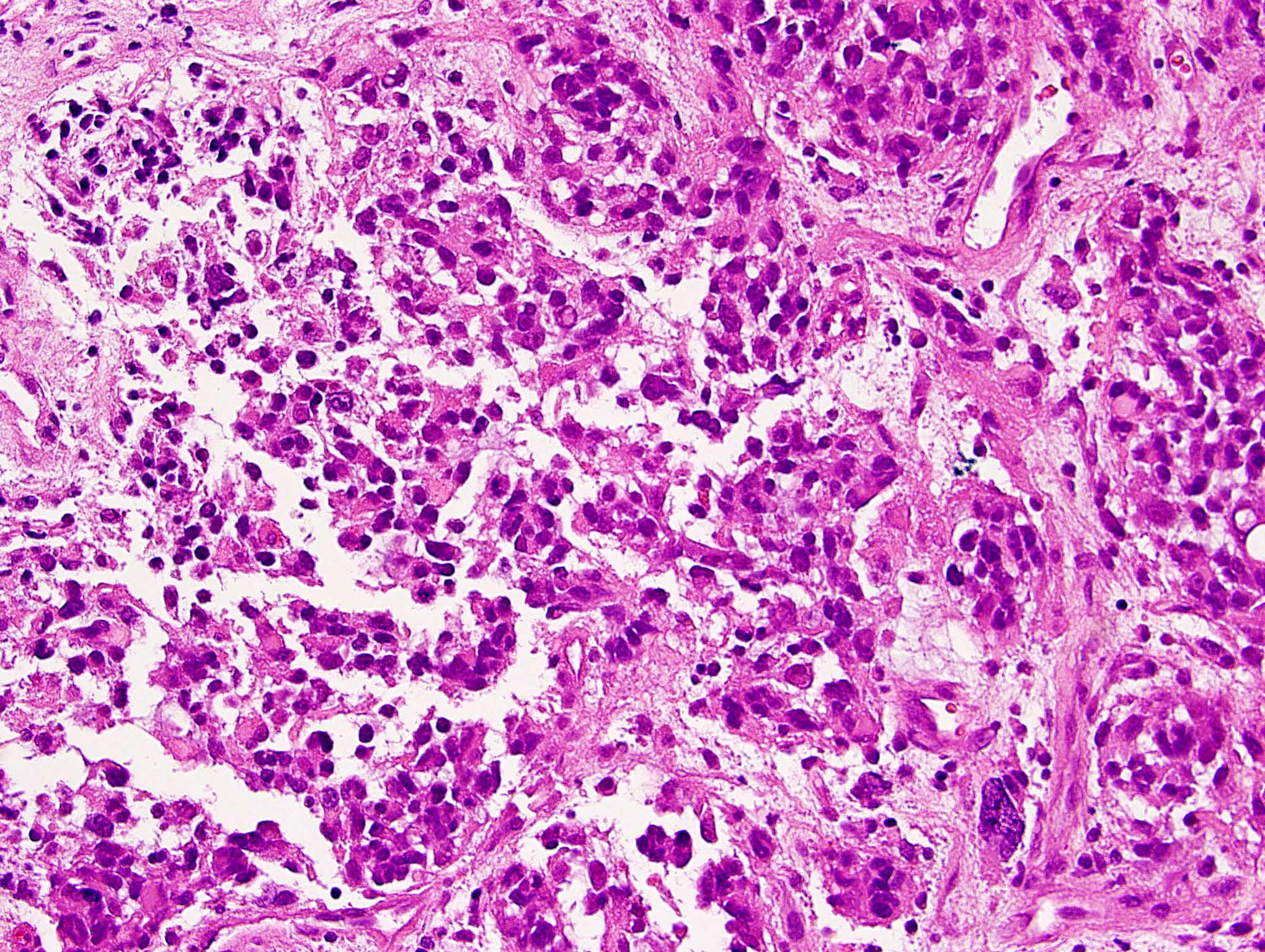
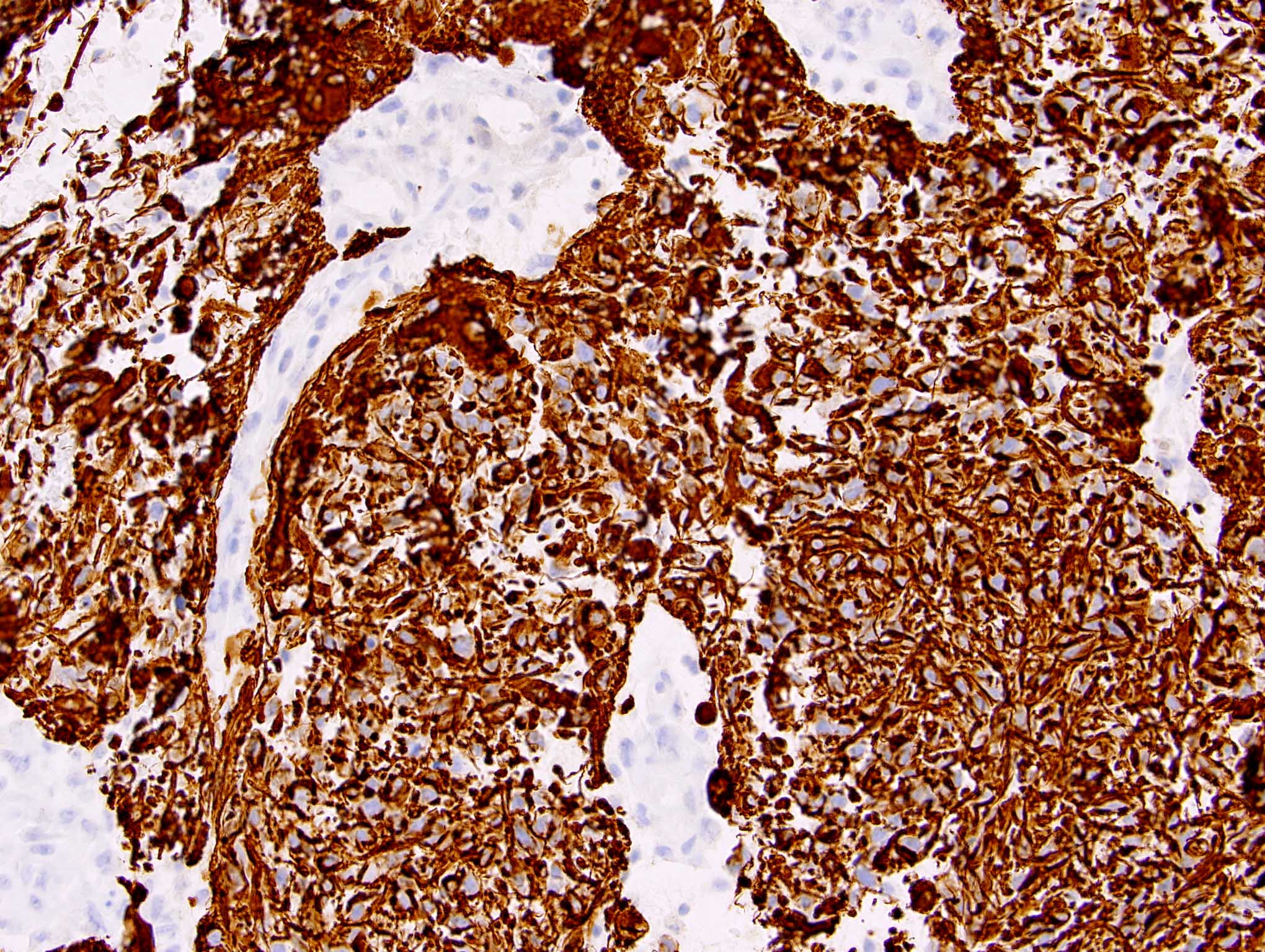
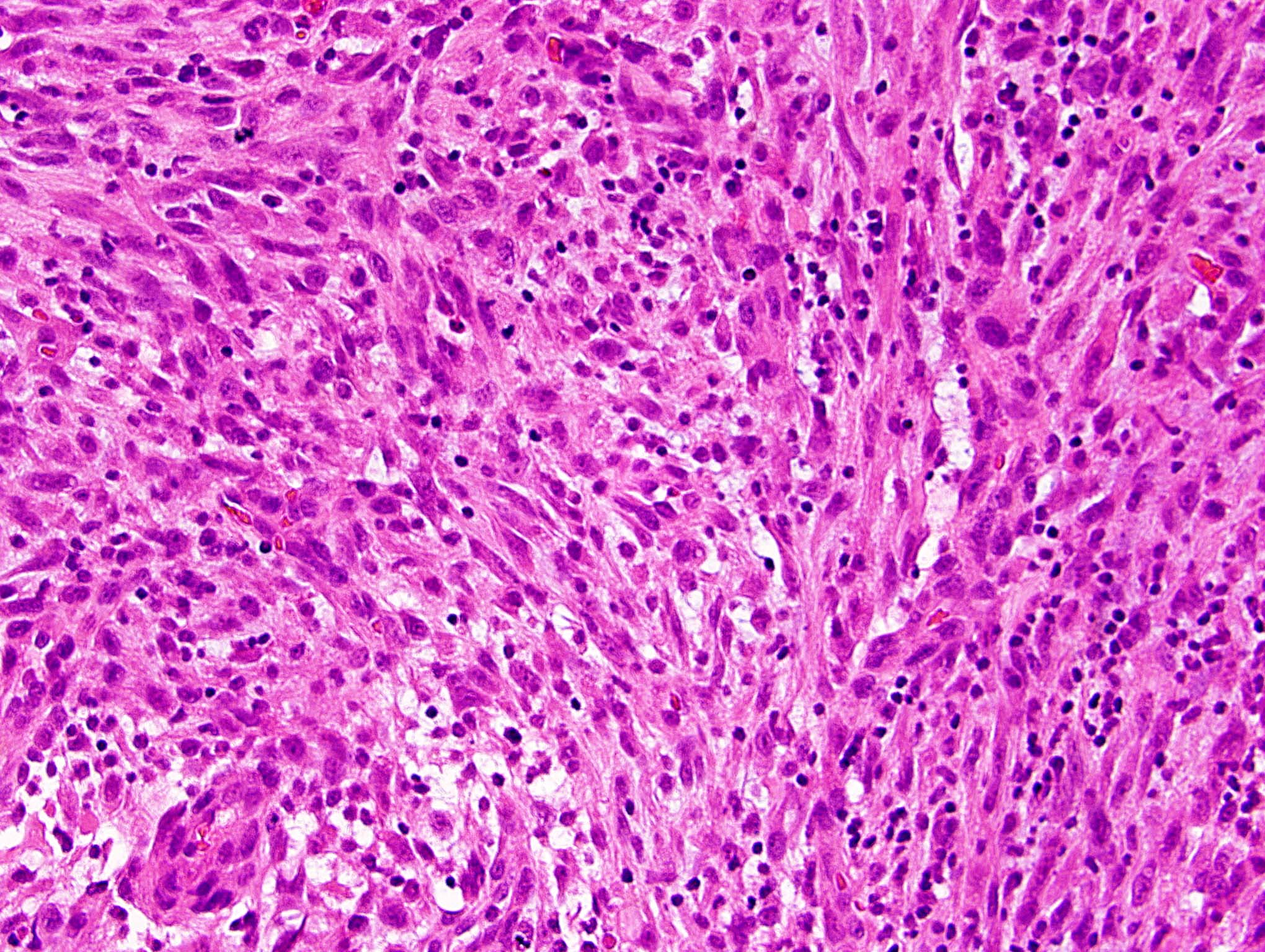
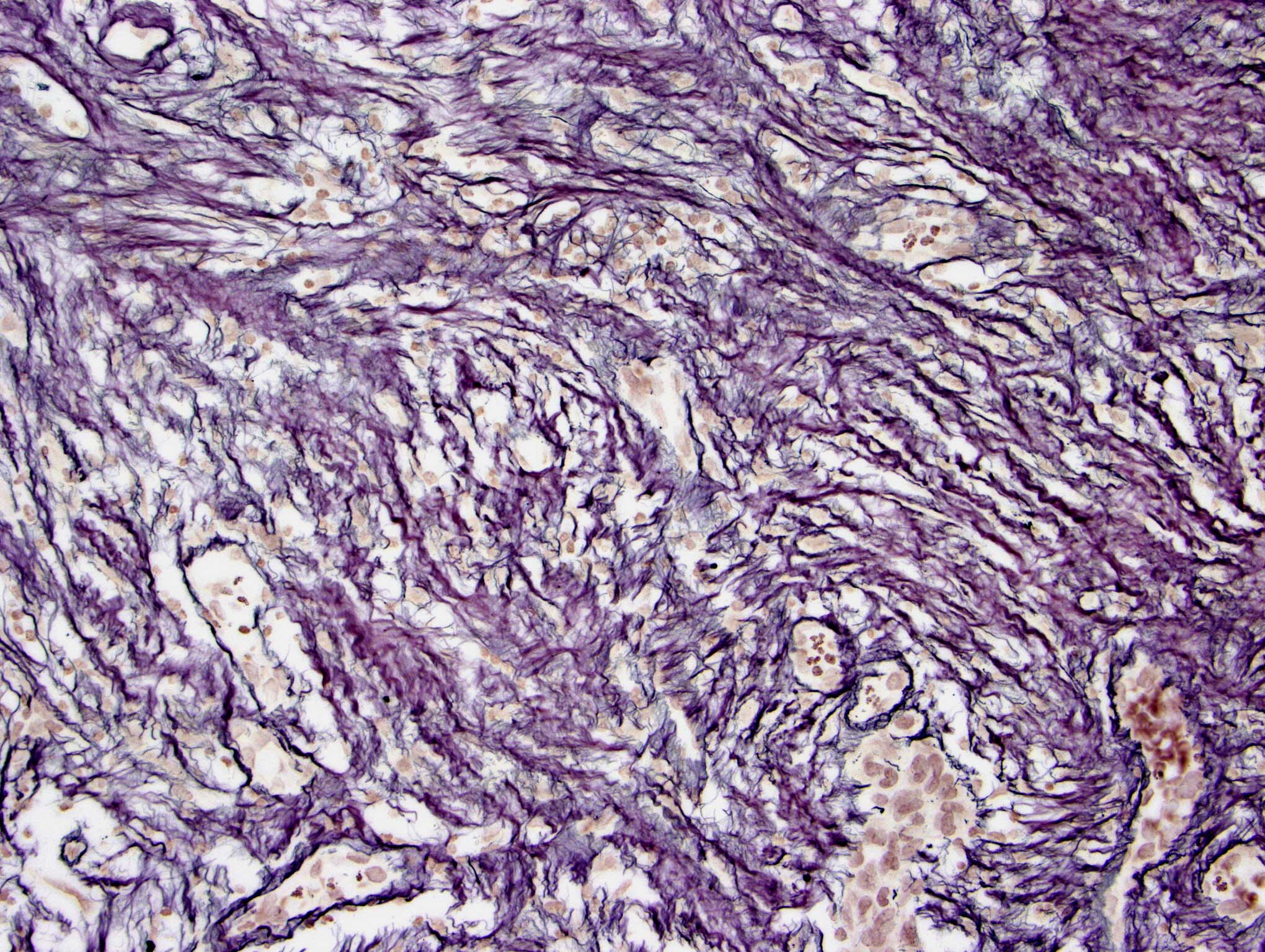
A 58 year old man with a medical history that is significant for resection and treatment (including radiotherapy) of a glioblastoma (IDH wildtype) 2 years prior, presents with a headache. MRI shows a heterogeneously enhancing mass associated with the dura near his prior resection site. The tumor is resected and shows the above on H&E and immunostains. Which of the following is most likely true of this tumor?
- It is a metastatic sarcoma
- It is a recurrent glial malignancy, this time with sarcomatous differentiation: secondary gliosarcoma
- Most likely hypermethylated in the MGMT gene promoter region
- WHO grade is 3, since we do not see necrosis
Comment Here
Reference: Gliosarcoma
- Frequent EGFR alteration and MGMT hypermethylation
- Frequent IDH mutations and loss of ATRX expression
- Frequent infratentorial and spinal cord localization
- Slightly increased incidence in males
- Evaluate the arachnoid, gray and white matter
- Section, if possible, from the arachnoid through the gray to the white matter
- Sample interface between the lesion and normal appearing brain
- EM helpful for poorly differentiated or unusual tumors (clear cell ependymoma), toxic - metabolic disease, infection; recommended to save tissue for EM in challenging cases
- Infarcts: section perpendicular to surface of brain, submit sections of arachnoid, gray and white matter; save gray matter for EM if considering CADASIL or mitochondrial disease
- Hematopoietic lesions: also B5, cell culture media (for flow cytometry), touch preparations, flash frozen tissue for molecular analysis
- Infectious disorders: recommended that surgeon obtain cultures from sterile operating field
- Toxic - metabolic disorders: recommend fixation of gray and white matter in formalin, glutaraldehyde, frozen tissue bank
- Margins: usually impossible to assess due to piecemeal nature of resection or infiltrative growth pattern; may be able to determine for childhood pilocytic astrocytoma of cerebellum, cerebral dysembryoplastic neuroepithelial tumor and meningioma
- Safety: recommended that surgeon contact lab if patient is HIV+ (use cytology, not frozen section) or may have Creutzfeldt-Jakob disease (decline frozen sections since cannot decontaminate cryostat); decontaminate cryostat used for HIV+ tissue by replacing blade, removing frozen debris and wiping surfaces with 10% bleach in water; for CJD specimens that are sectioned, recommended to place cassettes in formalin for 24 hours, then transfer to 100% formic acid for one hour, then return to fresh formalin for 48 hours, then process; label cassettes with pencil (formic acid dissolves ink), wrap needle biopsies in lens paper, handled embedded tissue as if infectious, dispose of blade, section waste, wipe microtome and cutting station with bleach, incinerate all towels, gloves and waste
- Abnormal mass: solid tissue not identifiable as gray or white matter
- Cyst wall: flat tissue from 0 to 3 mm thick - compare to surgeon's impression or radiographs
- Gliosis: often yellow or gray and firm
- Necrosis: soft, friable, often cavitates
- Semiliquid or gelatinous masses: usually tumors, including oligodendroglioma, lymphoma and pituitary adenoma
- Vascular malformation: vessels larger than usual aggregate of arachnoid vessels, often associated with hemorrhage, may involve meninges or parenchyma
- Indications: (a) determine if lesional tissue is present, (b) determine if adequate sampling, (c) provide preliminary information to assist neurosurgeon (see below) and (d) perform special techniques (culture, B5 for lymphoma, touch preparations)
- Need not give definitive diagnosis
- Should not grade astrocytic glial neoplasm unless is clearly a glioblastoma (since tumors often have variable grades)
- For some tumors, attempts at total resection are made (meningioma, schwannoma, solitary metastases, cysts, ependymoma, hemangioblastoma, cerebellar pilocytic astrocytoma, craniopharyngioma) so intraoperative consultation may be helpful
- Recommended to assess undefined lesions by both frozen section and cytologic preparation
- Cytologic preparation: adds fine nuclear detail, reveals glial type processes or epithelioid features of carcinomas; shows discohesiveness associated with pituitary adenoma, oligodendroglioma, medulloblastoma or lymphoma; may be more accurate than frozen section (Stereotact Funct Neurosurg 1995;65:187)
- Recommended to obtain touch preparations (touch glass slide to wet tissue, fix before it dries, then stain)
- Artifacts: long empty cavities in parenchyma (due to cryostat) vs. microcysts which contain cells and eosinophilic proteinaceous material)
- References: Arch Pathol Lab Med 2005;129:1635, Arch Pathol Lab Med 1997;121:481, Mod Pathol 1988;1:378, J Neurol Neurosurg Psychiatry 1988;51:332, Arch Pathol Lab Med 2005;129:1653
- Specimen type
- Specimen size (greatest dimension, additional dimensions are optional)
- Tumor site
- Tumor size (greatest dimension, additional dimensions are optional)
- Histologic type
- Histologic grade (WHO I - IV, other, cannot be determined, not applicable)
- Margins (involved, uninvolved, cannot be assessed, not applicable)
- Results of additional studies performed (electron microscopy, cytogenetics, molecular testing, immunohistochemistry)
- References: Arch Pathol Lab Med 2001;125:1162
- No TNM classification exists for CNS tumors (Edge: AJCC Cancer Staging Manual, 8th Edition, 2017) for these reasons:
- For T component, histology and location are more important than tumor size
- For N component, brain and spinal cord tumors do not propagate in lymphatics, so lymph nodes are not staged
- For M component, metastatic disease is rarely seen (except for CSF seeding in some pediatric tumors) and extremely rare outside of the CNS
- WHO classification and WHO grading systems should be used
- Generally favorable prognostic factors: younger age, total resection versus subtotal resection; higher Karnofsky performance scale
- Benign, slowly growing, highly vascular neoplasm containing neoplastic stromal cells which usually involves the cerebellum, brainstem or spinal cord
- Generally associated with loss or inactivation of the VHL gene, with frequent occurrence in von Hippel-Lindau (VHL) disease (Am J Clin Pathol 1997;107:459)
- Circumscribed, enhancing solid cystic mass, most frequently located in the cerebellum and observed as a cyst with enhancing mural nodule (AJNR Am J Neuroradiol 1992;13:1343)
- Composed of neoplastic stromal cells with foamy cytoplasm within a background network of numerous small vessels
- Associated with loss of function of VHL, with frequent occurrence in von Hippel-Lindau (VHL) disease (Am J Clin Pathol 1997;107:459)
- Other neoplasms associated with VHL may be histologic mimics (e.g. clear cell renal cell carcinoma, paraganglioma, pheochromocytoma)
- WHO grade 1
- Hemangioblastoma
- Capillary hemangioblastoma (not recommended)
- Lindau tumor (not recommended)
- Angioblastoma (not recommended)
- ICD-O: 9161/1 - hemangioblastoma
- ICD-10:
- D33.0 - benign neoplasm of brain, supratentorial
- D33.1 - benign neoplasm of brain, infratentorial
- D33.2 - benign neoplasm of brain, unspecified
- D33.3 - benign neoplasm of cranial nerves
- D33.4 - benign neoplasm of spinal cord
- D33.7 - benign neoplasm of other specified parts of central nervous system
- D33.9 - benign neoplasm of central nervous system, unspecified
- ICD-11:
- 0.141 per 100,000 person years (Front Oncol 2020;10:570103)
- Cerebellar
- ~1 - 2% of primary CNS tumors (Surg Neurol 1977;7:79)
- ~7 - 12% of primary posterior fossa tumors (Surg Neurol 1977;7:79)
- Spinal
- ~2% of spinal tumors (Neuroepidemiology 2016;46:14)
- Genetics
- ~70% sporadic (J Neurosurg 1989;70:24, Neurosurgery 2001;48:55)
- ~30% familial with VHL syndrome (J Neurosurg 1989;70:24, Neurosurgery 2001;48:55)
- Mean age at presentation (J Neurosurg 1989;70:24)
- Sporadic ~47 yrs
- VHL ~29 yrs
- Sex: M:F = 1 - 1.25:1 (Neuroepidemiology 2016;46:14, Front Oncol 2020;10:570103)
- Occur throughout the central nervous system
- Common: cerebellum > brainstem > spinal cord (Front Oncol 2020;10:570103)
- Rare: cerebrum, peripheral nervous system (J Neurosurg 2014;120:1055)
- VHL syndrome - multiple hemangioblastomas (Neurosurgery 2001;48:55, J Neurosurg 2014;120:1055)
- VHL loss of function implicated in most cases
- Normal VHL protein (pVHL)
- Regulates cell cycle (Proc Natl Acad Sci U S A 1998;95:993)
- Regulates cellular hypoxia signaling via the HIF (hypoxia inducible factor) complex, which under hypoxic conditions leads to enhanced levels of growth factors (e.g. vascular endothelial factor, platelet derived growth factor, transforming growth factor alpha) (Nat Rev Cancer 2015;15:55)
- Pseudohypoxia hypothesis
- Loss of function of VHL → loss of regulation of HIF complex → increased expression of growth factors in absence of hypoxia → increased angiogenesis and tumor growth (Best Pract Res Clin Endocrinol Metab 2010;24:957)
- VHL syndrome (Eur J Hum Genet 2011;19:617)
- Autosomal dominant tumor predisposition syndrome resulting from inactivating germline mutation in the VHL tumor suppressor gene
- Biallelic VHL inactivation
- Normal VHL protein (pVHL)
- Sporadic: up to 78% of sporadic cases with VHL loss of function (J Neurol Neurosurg Psychiatry 2001;70:644, Cancer Res 1998;58:504, Acta Neuropathol Commun 2014;2:167)
- Familial (VHL syndrome): ~60% of VHL disease patients develop hemangioblastoma (J Neurol Neurosurg Psychiatry 1999;67:758)
Images hosted on other servers:
VHL regulation of the HIF pathway
- Symptoms
- General
- Increased intracranial pressure / hydrocephalus (J Neurosurg 2008;108:210)
- Secondary polycythemia due to erythropoietin production (Neurosurgery 2013;72:930, Blood 1998;92:3388, AJNR Am J Neuroradiol 1992;13:1343)
- Hemorrhage (World Neurosurg 2015;83:1180.e13, Neurosurgery 2005;57:71, Neurosurg Rev 2010;33:11)
- Cerebellar
- Dysmetria, ataxia (J Neurosurg 2008;108:210)
- Spinal
- Pain, hypesthesia, incontinence (Neurosurgery 2001;49:321, AJNR Am J Neuroradiol 2001;22:206)
- VHL syndrome sites of involvement (Eur J Hum Genet 2011;19:617)
- CNS: hemangioblastoma
- Retina: hemangioblastoma
- Head / neck: paraganglioma
- Kidney: clear cell renal cell carcinoma, cysts
- Adrenal: pheochromocytoma
- Pancreas: neuroendocrine islet cell tumor, cysts
- Inner ear: endolymphatic sac tumor
- Epididymis: papillary cystadenoma
- Broad ligament: cystadenoma
- General
- Based on histologic and immunophenotypic features (Acta Neuropathol 2013;125:333)
- Clinical history of VHL syndrome or evidence of genetic alterations leading to VHL inactivation is supportive
- Secondary polycythemia (~5 - 40% of cases) (Neurosurgery 2013;72:930, Blood 1998;92:3388, AJNR Am J Neuroradiol 1992;13:1343)
- General
- Well demarcated enhancing mass ranging from solid to cystic, frequently presenting as a cyst with an enhancing mural nodule (AJNR Am J Neuroradiol 1992;13:1343)
- Spinal cord tumors often associated with syrinx (AJR Am J Roentgenol 1989;152:1087)
- Magnetic resonance imaging (MRI)
- Nodule: T1 hypointense to isointense, T2 hyperintense (AJNR Am J Neuroradiol 1992;13:1343)
- Serpentine flow voids in the nodular portion (AJNR Am J Neuroradiol 1992;13:1343)
- Often abuts the pia (Cancer Imaging 2012;12:237)
- Cyst wall rarely enhances (Cancer Imaging 2012;12:237)
- Computed tomography
- Mural nodule is isodense to the brain on noncontrast scans (J Comput Assist Tomogr 1982;6:912)
- Calcification usually absent (J Comput Assist Tomogr 1982;6:912)
Images hosted on other servers:
T1+C MRI posterior fossa
- Excellent prognosis with complete surgical resection (Childs Nerv Syst 2020;36:2537, JAMA Neurol 2018;75:620, J Neurosurg 2008;108:210)
- Outcome better in sporadic cases than in VHL syndrome cases (Brain Tumor Pathol 2000;17:111)
- 41 year old man with a family history of VHL syndrome and a synonymous VHL mutation (BMC Med Genet 2020;21:42)
- 42 year old man with cerebellopontine angle tumor (Surg Neurol Int 2017;8:264)
- 54 year old woman with a past medical history of VHL syndrome and bilateral clear cell renal cell carcinoma found to have an enhancing intramedullary mass at C6 / C7 (J Clin Neurosci 2015;22:215)
- 63 year old man with suspected posterior inferior cerebellar artery aneurysm (World Neurosurg 2019;122:165)
- Surgical resection - standard of care (JAMA Neurol 2018;75:620)
- Stereotactic radiosurgery may provide some control for unresectable tumors but data on safety and long term efficacy is limited (J Neurosurg 2015;122:1469, Neuro Oncol 2010;12:80)
Images hosted on other servers:
Cystic mass in medulla
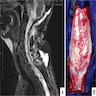
Multiple spinal masses
- Solid or cystic with mural nodule (J Neurosurg 2003;98:82)
- Well circumscribed and pseudoencapsulated
- Highly vascular
- Red with yellow / orange cut surface regions attributed to lipid content
Contributed by Rebecca Yoda, M.D.

Hemangioblastoma cut surfaces
Images hosted on other servers:

Variegated red-yellow tumor
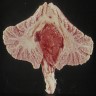
Hemangioblastoma in situ
Contributed by P.J. Cimino, M.D., Ph.D.
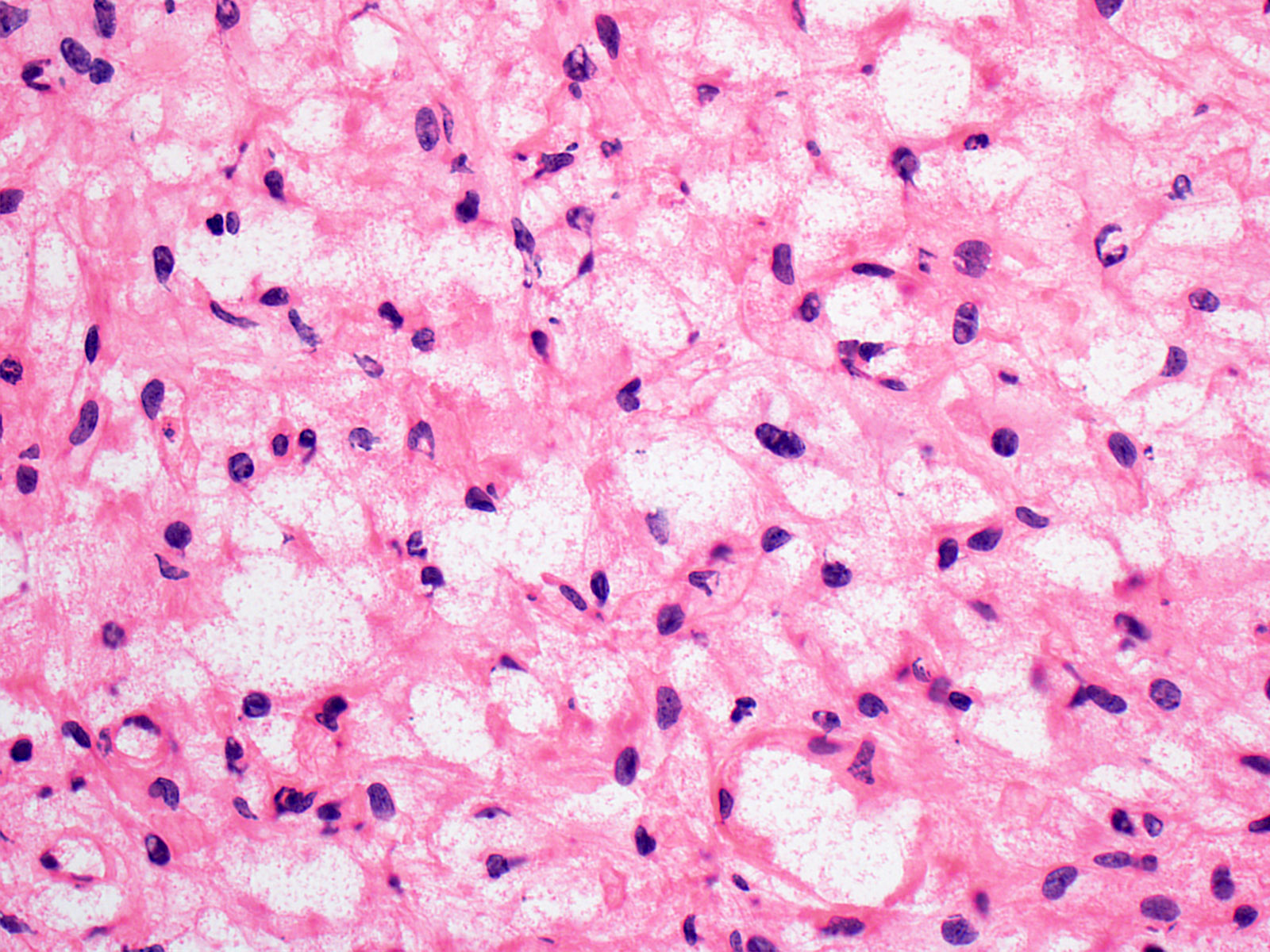
Moderate hypercellularity

Smear preparation
- Architecture (Acta Neuropathol 2013;125:333)
- Composed of neoplastic stromal cells arranged between numerous small vessels
- Compact, generally noninfiltrative growth with variable lobularity
- Stromal component
- Mild nuclear pleomorphism with degenerative atypia
- Clear, foamy cytoplasm with lipid containing vacuoles
- Rare hyaline globules
- Vascular component
- Numerous thin walled vessels
- Well demarcated border with neoplasm
- Other findings
- Mitotic figures rare to absent
- Intratumoral hemorrhage
- Mast cells
- Cyst-like spaces
- Adjacent parenchyma may have piloid gliosis with Rosenthal fibers
- Extramedullary hematopoiesis (~15%) (Neurosurgery 1991;29:34)
- Histologic variants
- Reticular (Neuropathol Appl Neurobiol 2005;31:618)
- Capillaries > stromal cells
- More common
- Cellular (Neuropathol Appl Neurobiol 2005;31:618)
- Stroma cells > capillaries
- Less common
- Reticular (Neuropathol Appl Neurobiol 2005;31:618)
Contributed by P.J. Cimino, M.D., Ph.D.

Circumscribed border

Vascular and stromal cells
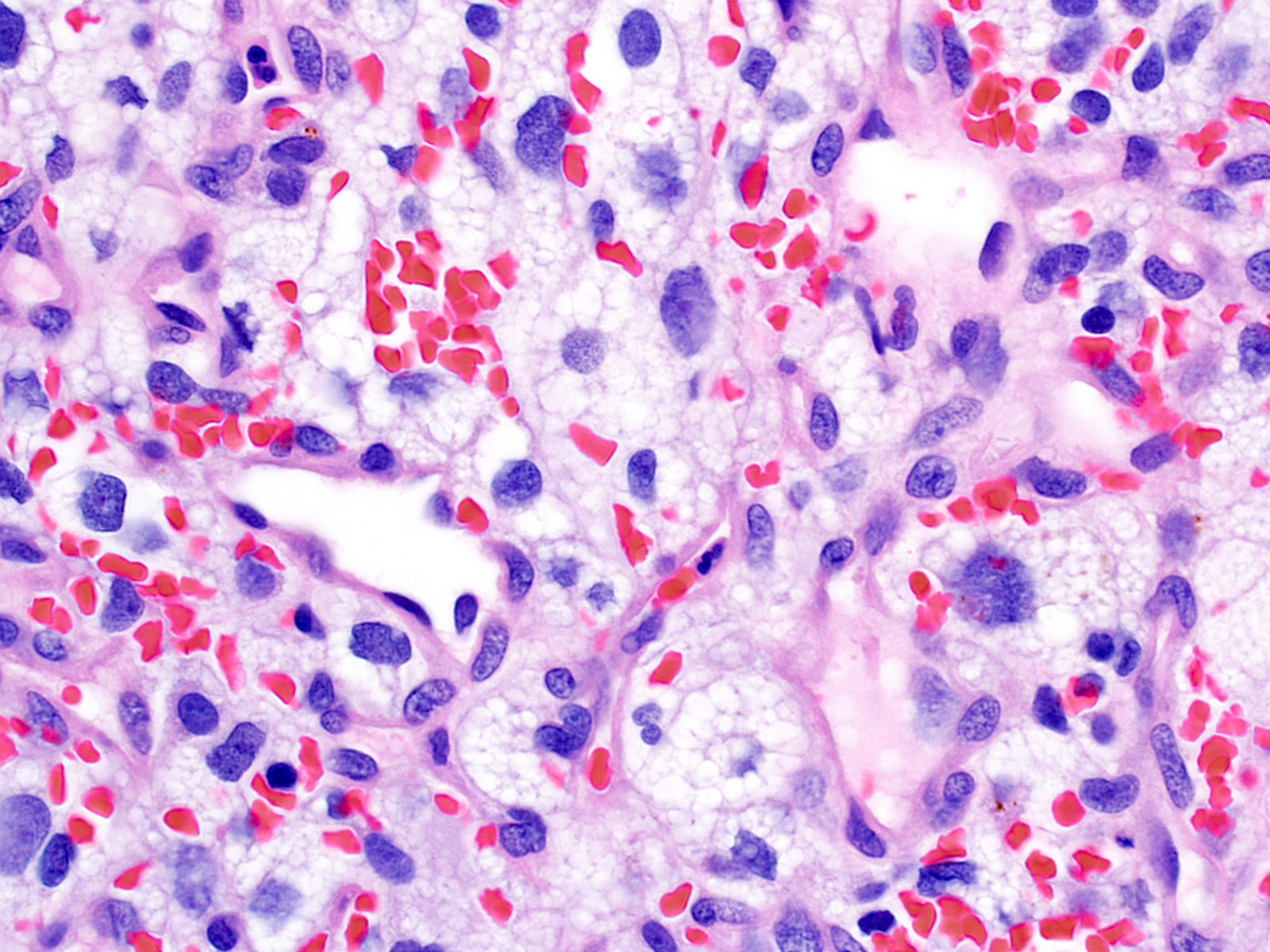
Foamy cytoplasm

Large branching vessels
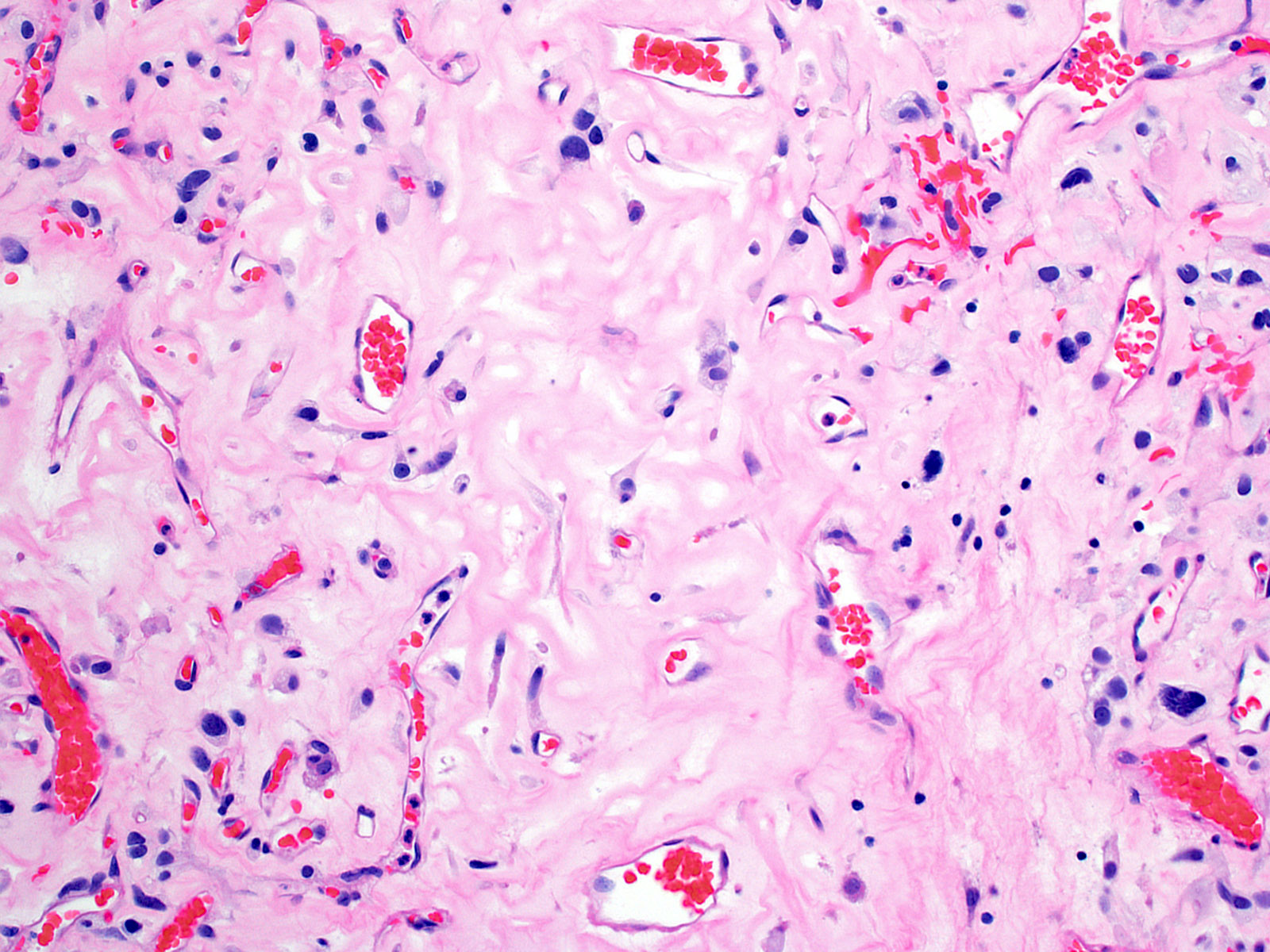
Hyalinized stroma
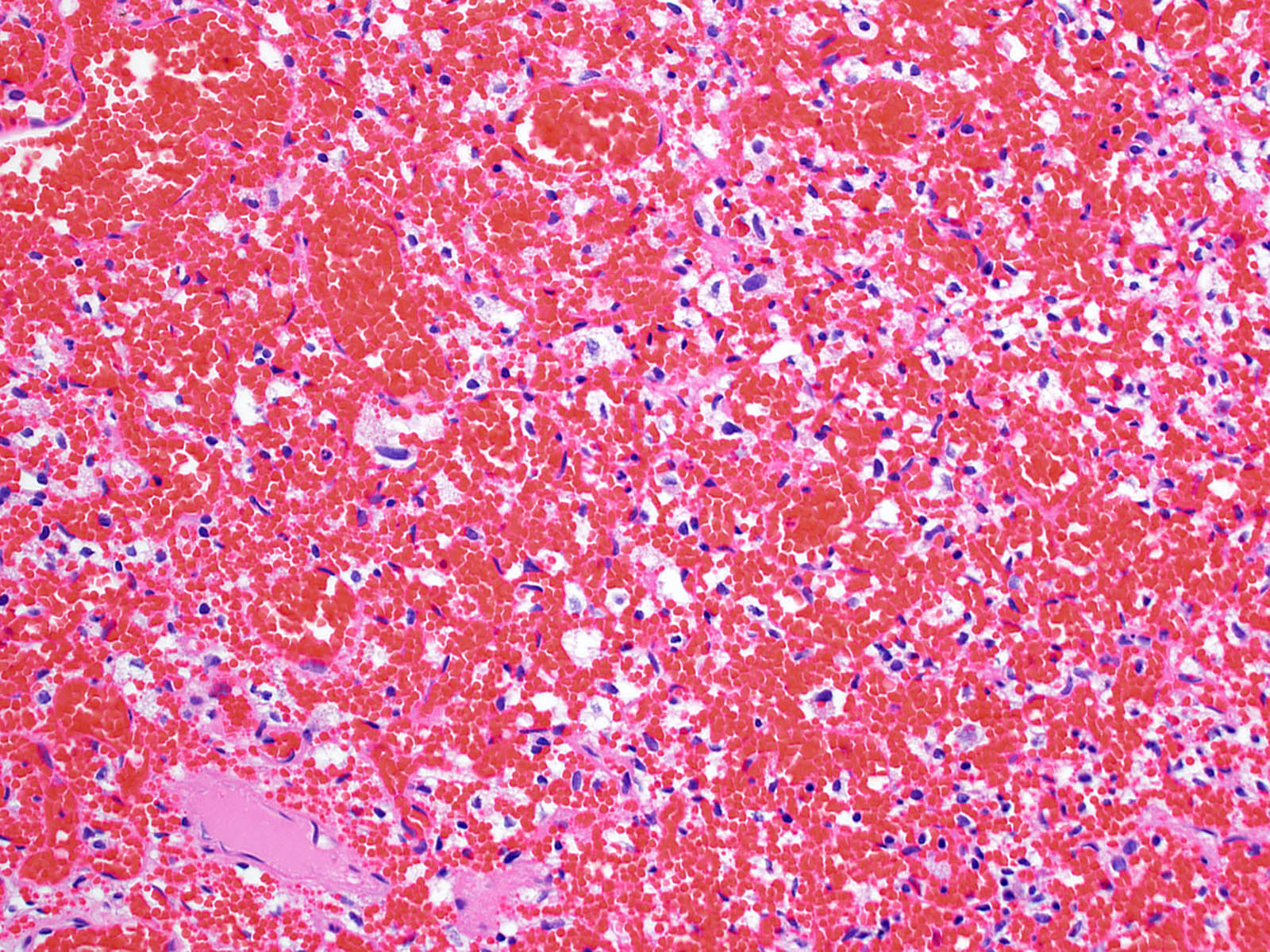
Extensive hemorrhage
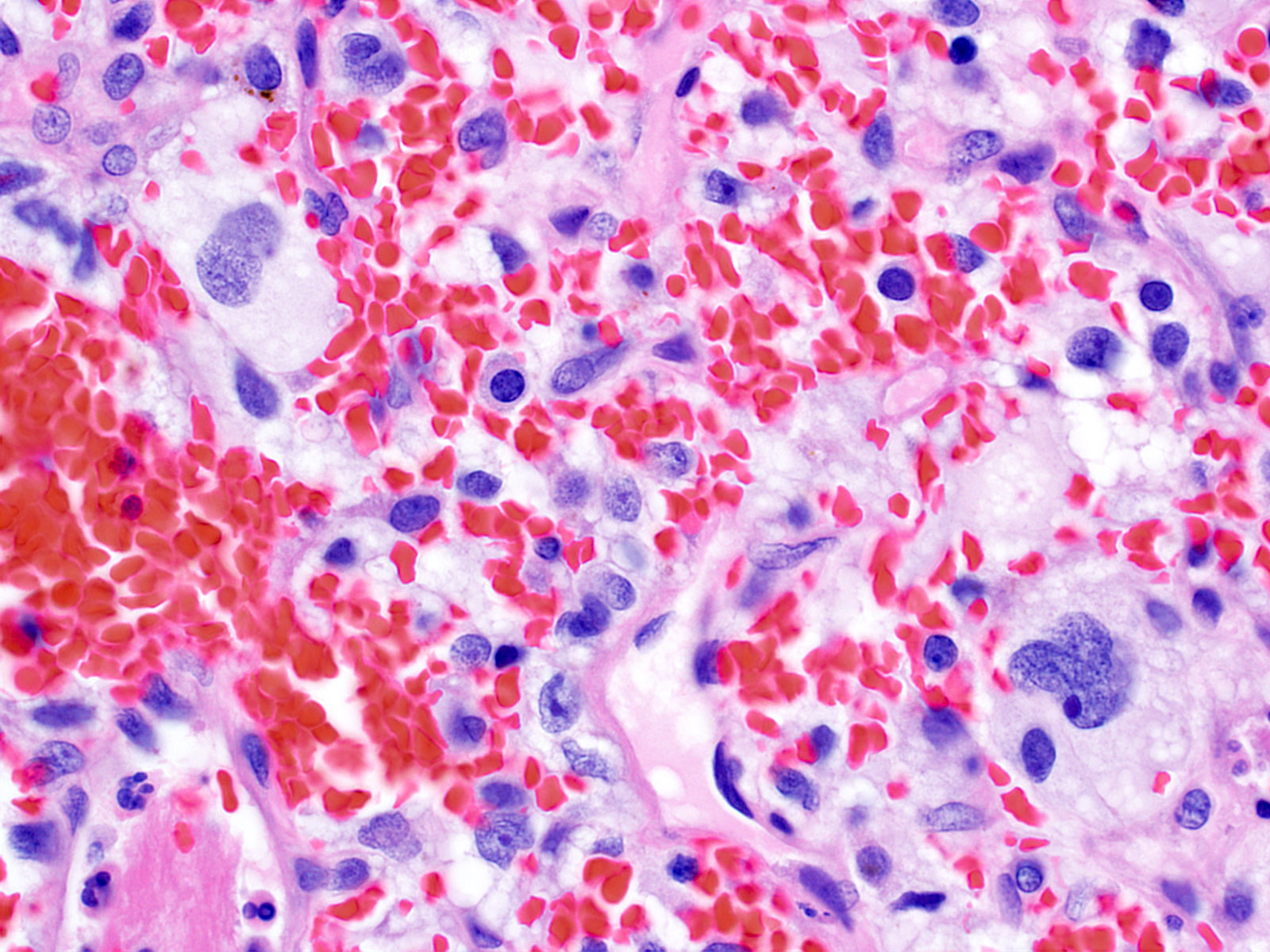
Nuclear atypia
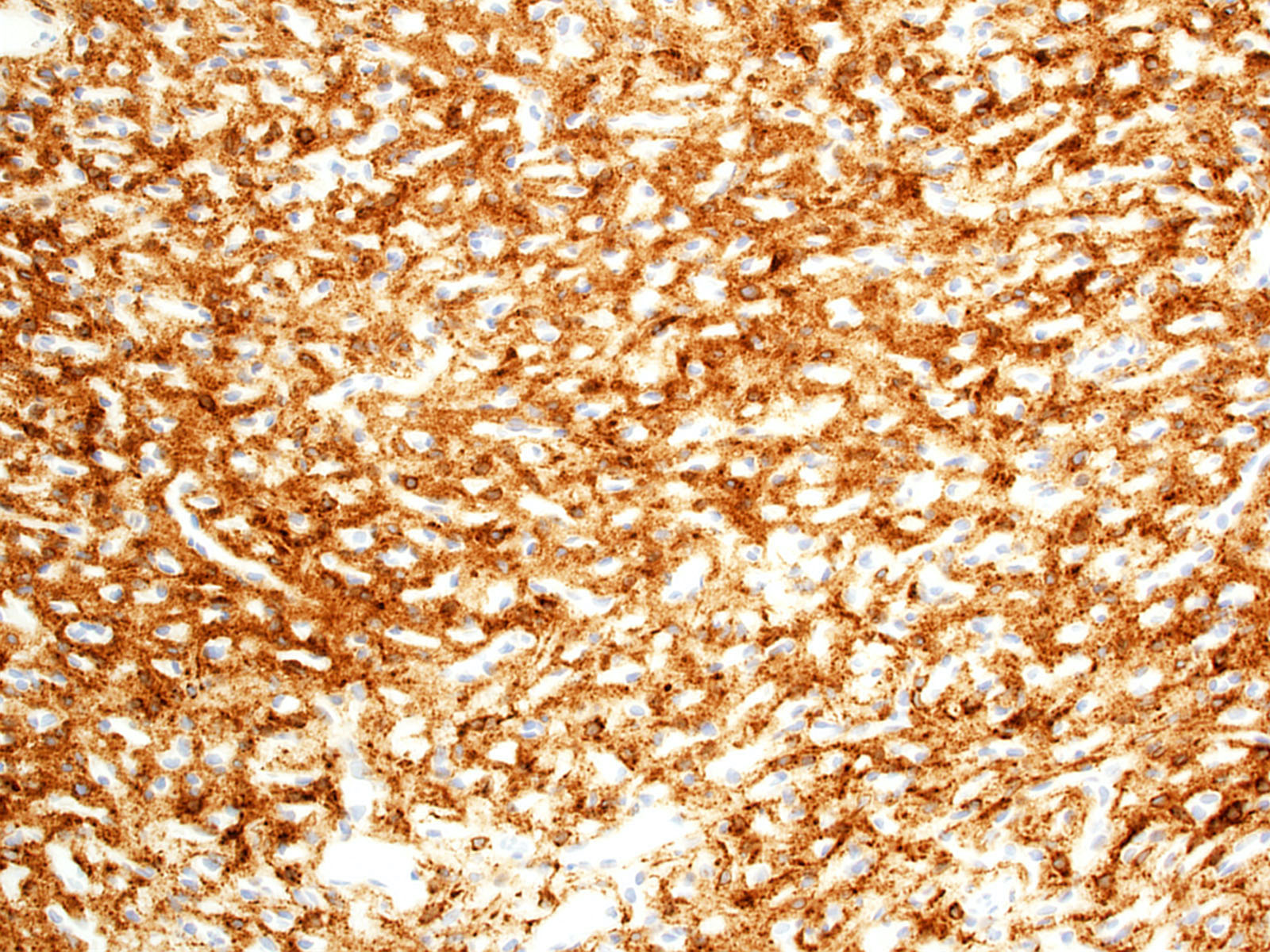
Inhibin

CAIX
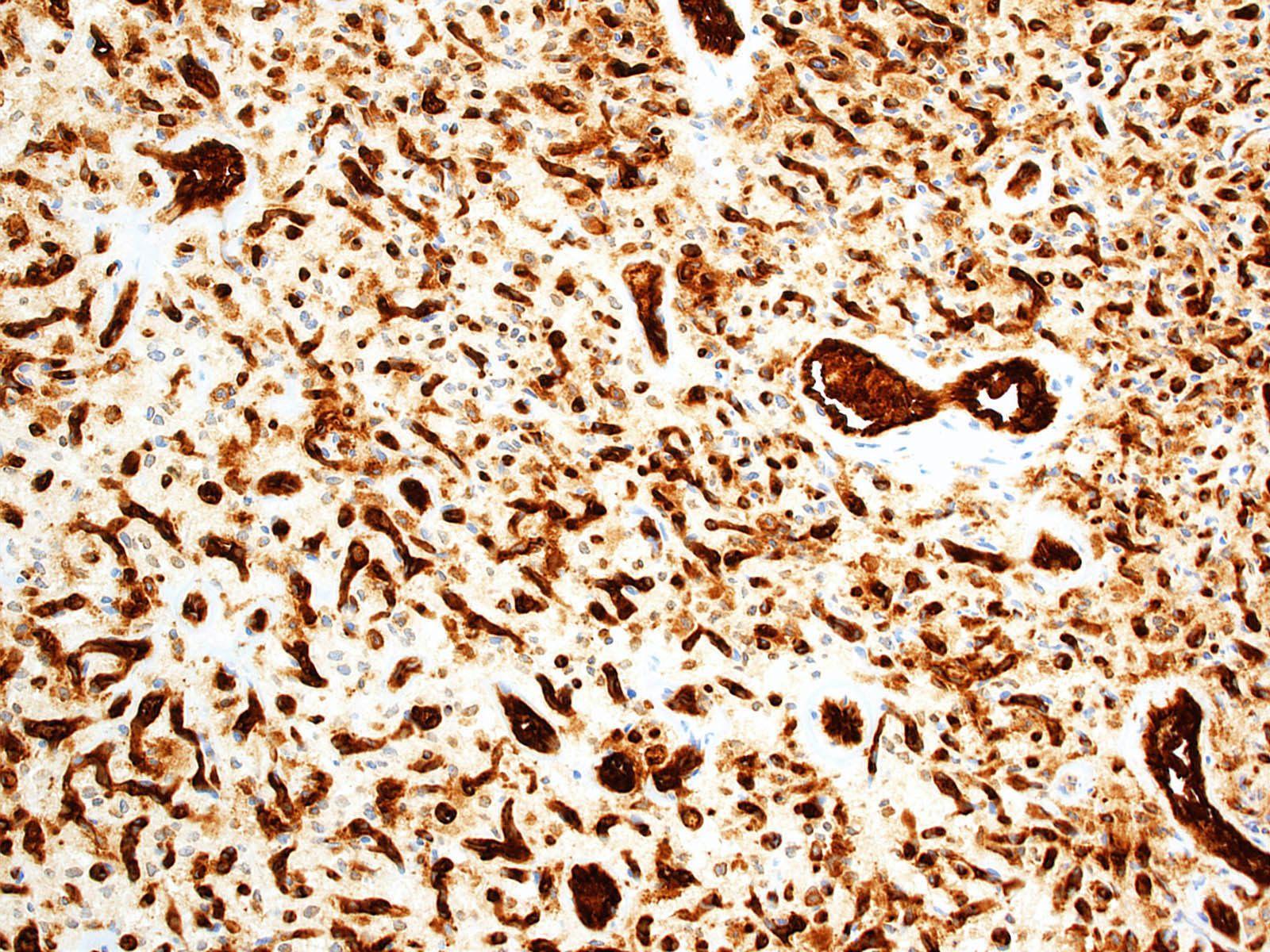
CD31
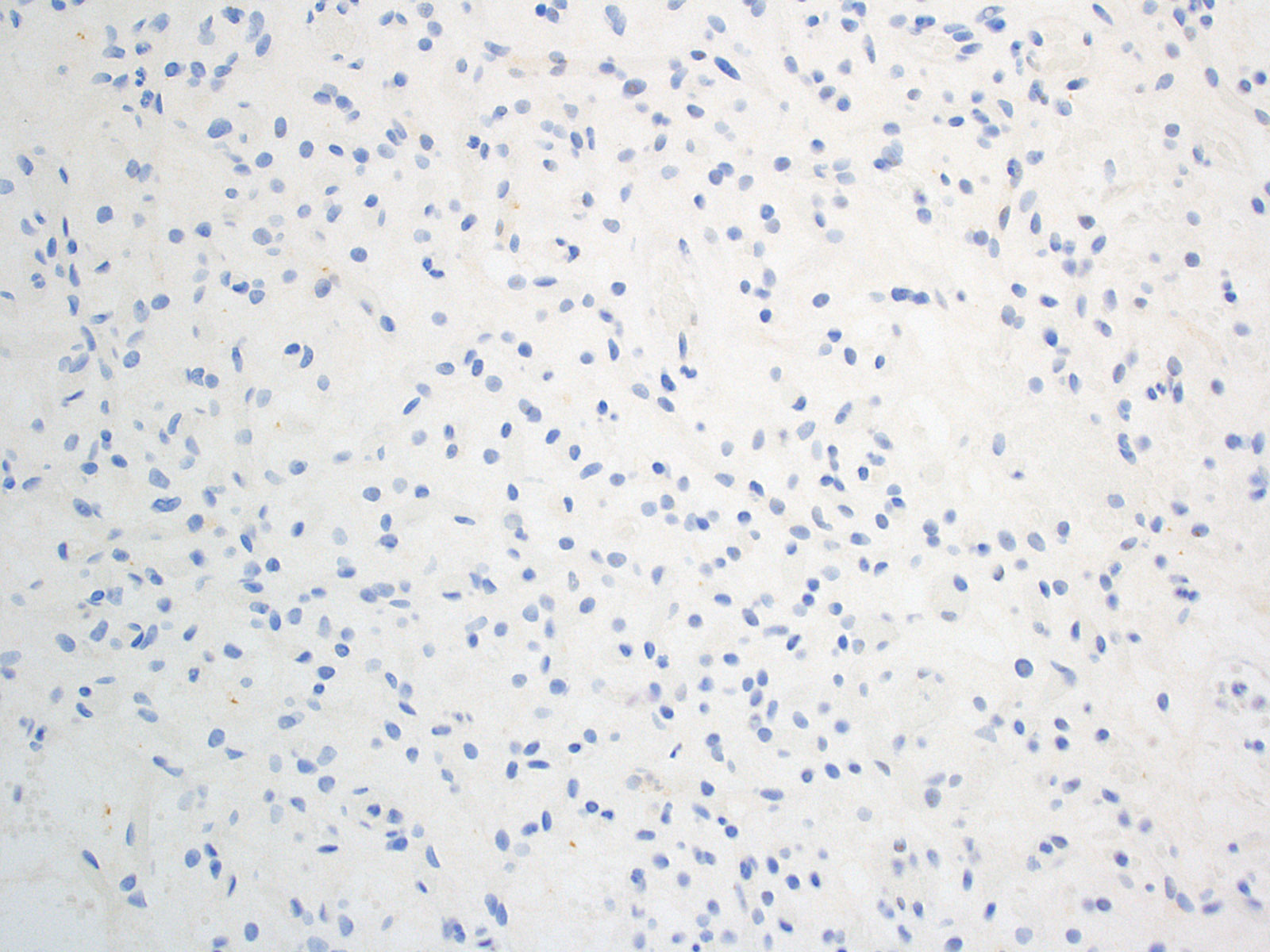
PAX8
Images hosted on other servers:
Hemorrhagic tumor
Highly vascular tumor
- Generally resistant to smear (squash preparation), resulting in larger tissue clumps
- Successful cytologic smear preparations include the following (Acta Cytol 1998;42:1104):
- Cytoplasm
- Foamy
- Indistinct cytoplasmic borders
- Nuclei
- Mildly pleomorphic
- Hyperchromatic, speckled chromatin
- Nuclear grooves
- Hemosiderin
- Clumping of cohesive cells often obscures cytologic features
- Cytoplasm
- Stromal cells
- Inhibin alpha (Am J Surg Pathol 2011;35:262, Am J Surg Pathol 2003;27:1152, Neuropathology 2010;30:580, Am J Surg Pathol 2008;32:1051)
- N-CAM1 (CD56) (Arch Pathol Lab Med 2007;131:641)
- S100 (100%; scattered positive cells) (Am J Surg Pathol 1989;13:207)
- Carbonic anhydrase IX & XII (Neuro Oncol 2005;7:465)
- Brachyury (91% cytoplasmic) (Am J Surg Pathol 2012;36:1052)
- Vimentin (86 - 100%) (Mod Pathol 1989;2:638)
- Aquaporin 1 (Am J Surg Pathol 2008;32:1051)
- TFE3 (Am J Transl Res 2020;12:4498)
- Oil red O (Arch Pathol Lab Med 2007;131:641)
- Vascular cells
- Endothelial cell markers (e.g. CD31, CD34) (Arch Pathol Lab Med 2007;131:641)
- Reticulin (highlights vessel walls)
- AE1 / AE3 (Am J Surg Pathol 2008;32:1051)
- CAM5.2 (Arch Pathol Lab Med 2007;131:641)
- RCC marker (Neuropathology 2010;30:580)
- PAX8 (Am J Surg Pathol 2011;35:262)
- PAX2 (5%) (Am J Surg Pathol 2011;35:262)
- CD10 (0 - 12%) (Am J Surg Pathol 2008;32:1051, Neuropathology 2010;30:580)
- EMA (0 - 36%; membranous or diffuse cytoplasmic) (Am J Surg Pathol 1989;13:207, Am J Surg Pathol 2008;32:1051)
- NSE (33%) (Am J Surg Pathol 1989;13:207)
- GFAP (18%; scattered individual cells) (Histol Histopathol 1996;11:1049)
- Lipid droplet containing stromal cells seen between capillaries (J Neuropathol Exp Neurol 2002;61:1027)
Images hosted on other servers:
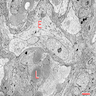
EM lipid droplets
- VHL syndrome
- Biallelic inactivation of VHL (J Neurol Neurosurg Psychiatry 2001;70:644)
- Sporadic
- Up to 78% of cases show loss / inactivation of VHL (J Neurol Neurosurg Psychiatry 2001;70:644, Cancer Res 1998;58:504, Acta Neuropathol Commun 2014;2:167, Neuro Oncol 2017;19:1228)
- Distinctive epigenetic classifying signature can be determined by genome wide DNA methylation array profiling (Acta Neuropathol 2018;136:181)
- MicroRNA (miRNA) patterns include elevated miRNA-9 and decreased miRNA-200a (Brain Pathol 2012;22:522)
Images hosted on other servers:

VHL next generation sequencing

Recurrent loss of chromosome 3

DNA methylation based classification

Idiogram of VHL mutation
Hemangioblastoma MRI
Hemangioblastoma histopathology
- Brain, cerebellum, excision:
- Hemangioblastoma, WHO grade 1
- Metastatic clear cell renal cell carcinoma:
- Anaplasia, prominent nucleoli
- Mitotic activity
- Necrosis
- Usually lacks foamy / vacuolated cytoplasm
- Positive for AE1 / AE3, CAM5.2, EMA, CD10, PAX2, PAX8, RCC marker (Am J Surg Pathol 2008;32:1051, Arch Pathol Lab Med 2007;131:641, Mod Pathol 2005;18:788, Neuropathology 2010;30:580, Adv Anat Pathol 2010;17:377)
- Negative for inhibin alpha, brachyury, N-CAM1 (J Neuropathol Exp Neurol 2017;76:289, Mod Pathol 2005;18:788, Am J Surg Pathol 2003;27:1152, Arch Pathol Lab Med 2007;131:641, Am J Surg Pathol 2012;36:1052)
- Meningioma (clear cell, microcystic, angiomatous subtypes):
- Usually positive for EMA (weak, patchy cytoplasmic), PR, SSTR2A (J Neuropathol Exp Neurol 2017;76:289)
- Negative for inhibin alpha, brachyury (J Neuropathol Exp Neurol 2017;76:289, Am J Surg Pathol 2012;36:1052)
- Solitary fibrous tumor:
- Positive for nuclear STAT6 (J Neuropathol Exp Neurol 2017;76:289)
- Negative for inhibin alpha (J Neuropathol Exp Neurol 2017;76:289)
- Glioma:
- Smear shows fibrillary processes, less cohesion
- Ependymoma:
- Perivascular pseudorosettes, ependymal rosettes and canals
- Positive for GFAP (Appl Immunohistochem Mol Morphol 2000;8:25)
- EMA may show paranuclear dot-like or ring-like staining (Appl Immunohistochem Mol Morphol 2000;8:25)
- Pilocytic astrocytoma:
- Biphasic appearance
- Compact fibrillar portions with Rosenthal fibers
- Loose microcystic portions with eosinophilic granular bodies
- Generally positive for GFAP, Olig2 (Am J Surg Pathol 2012;36:43, J Neuropathol Exp Neurol 2004;63:499)
- MAPK pathway activating genetic alterations (e.g. KIAA1549-BRAF fusion) (Nat Genet 2013;45:602)
- Biphasic appearance
- Diffuse glioma:
- Neoplastic glioma cells infiltrate background brain parenchyma (single cell infiltration, entrapped axons and neurons)
- Generally positive for GFAP, Olig2 (Acta Neuropathol 1986;72:15, J Neuropathol Exp Neurol 2004;63:499)
- Paraganglioma:
- Smear demonstrates individual cells
- Neuroendocrine cytologic features with uniform, round nuclei
- Zellballen architecture
- Diffusely positive for synaptophysin, chromogranin (J Clin Med 2018;7:280)
- Sustentacular cells positive for S100 (J Clin Med 2018;7:280)
- Capillary angioma / hemangioma:
- Lobular architecture
- Absence of extravascular stromal cells

A 47 year old man presents with ataxia. MRI reveals a solitary cerebellar cyst with an enhancing mural nodule. A representative histologic photo of the surgical specimen is shown. On immunohistochemical stains, stromal cells are negative for AE1 / AE3 and PAX8. Which of the following IHC stains is most likely to be positive in stromal cells?
- CAM5.2
- CD31
- Inhibin alpha
- PAX2
- RCC marker
Comment Here
Reference: Hemangioblastoma
A 32 year old woman is found to have multiple enhancing intra-axial mixed solid cystic masses in the cerebellum and spinal cord as well as numerous bilateral renal cysts and a renal mass. A biopsy of a cerebellar mass is shown. A germline mutation is most likely to be present in which gene?
- FLCN
- NF2
- PKD1
- TSC1
- VHL
- FLCN - Birt-Hogg-Dubé syndrome (increased risk of renal cysts and tumors; no CNS involvement)
- NF2 - neurofibromatosis type 2 (increased risk of various lesions primarily affecting the nervous system, including schwannoma, meningioma, ependymoma; no renal involvement)
- PKD1 - autosomal dominant polycystic kidney disease (associated with bilateral renal cysts and berry aneurysms; no other CNS involvement)
- TSC1 - tuberous sclerosis (renal manifestations may include angiomyolipoma and cysts; CNS manifestations may include cortical tubers and subependymal giant cell astrocytoma; no known association with hemangioblastoma)
Comment Here
Reference: Hemangioblastoma
- Circumscribed astrocytic glioma with high grade piloid or glioblastoma-like histological features and distinct DNA methylation profile
- Recently recognized entity, included in the 2021 WHO classification of CNS tumors
- Circumscribed astrocytic glioma occurring throughout the entire CNS, mostly in the posterior fossa
- High grade histological features associated with piloid or glioblastoma-like morphology
- Distinct DNA methylation profile and MAPK pathway gene alterations, often associated with homozygous deletion (HD) of CDKN2A / 2B or ATRX alterations
- CNS WHO grade currently not assigned (clinical behavior roughly similar to CNS WHO grade 3)
- Anaplastic astrocytoma with piloid features (not recommended by CNS WHO 2021)
- ICD-O: 9421/3 - high grade astrocytoma with piloid features
- ICD-11: 2A00.0Y & XH6PH6 - other specified gliomas of brain & astrocytoma, NOS
- Comprehensive epidemiological data currently not available
- Rare, ~1 - 3% of CNS tumors (Neuropathol Appl Neurobiol 2020;46:478, Acta Neuropathol Commun 2019;7:24, Nature 2018;555:469)
- Very rare in pediatric population (Lancet Child Adolesc Health 2020;4:121, Acta Neuropathol 2020;139:287)
- Median age: 40 years; range: 4 - 88 years (Acta Neuropathol 2018;136:273, Acta Neuropathol Commun 2019;7:24, Acta Neuropathol 2020;139:287)
- No sex predilection (Acta Neuropathol 2018;136:273, Acta Neuropathol Commun 2019;7:24, Acta Neuropathol 2020;139:287)
- Can occur anywhere in the CNS
- Posterior fossa, particularly cerebellum, is most common (74%) (Acta Neuropathol 2018;136:273)
- More rarely supratentorial (17%) or spinal (7%) (Acta Neuropathol 2018;136:273)
- Cell of origin is currently unknown
- 3 pathways are probably involved in the pathogenesis (Acta Neuropathol 2018;136:273)
- MAPK pathway gene alterations are likely the initiating event (driver mutation)
- Deregulation of retinoblastoma tumor suppressor protein cell cycle pathway by CDKN2A / 2B inactivation (homozygous deletion) or occasionally CDK4 amplification
- Activation of telomere maintenance by ATRX alterations and rarely TERT promoter mutations
- Numerous chromosomal alterations and frequent structural aberrations (Acta Neuropathol 2018;136:273)
- Partial gains of 12q and 17q
- Losses of 1p and 8p
- Partial losses of 14 and 19q
- Generally sporadic without significant known risk factors
- In some cases, may occur from pre-existing low grade astrocytic tumors (Acta Neuropathol 2018;136:273)
- Uncommon association with irradiation but no established etiological role (Acta Neuropathol 2018;136:273)
- Rarely reported in patients with NF1 gene alterations (Acta Neuropathol 2018;136:273, Acta Neuropathol 2023;145:71)
- Clinical signs and symptoms depend on tumor location
- Magnetic resonance imaging (MRI), followed by stereotactic brain biopsy or surgical resection
- CNS WHO 2021 diagnostic criteria
- Essential
- Histological aspect of an astrocytic glioma and
- DNA methylation profile of HGAP
- Desirable
- MAPK pathway gene alteration
- CDKN2A / 2B HD or CDK4 amplification
- ATRX mutation or loss of nuclear expression
- Anaplastic histological features
- Essential
- MRI
- T1 hypointense / isointense, T2 hyperintense (J Neurooncol 2021;153:109)
- Mostly no diffusion restriction; peripheral irregular enhancement with central necrosis or mixed heterogeneous enhancement (AJNR Am J Neuroradiol 2024;45:468)
- Some tumors may show glioblastoma-like radiological features (i.e., ring enhancing mass) (J Neurooncol 2021;153:109)
Contributed by Antonio d’Amati, M.D.

T2 MRI of brain

T1 MRI of brain

Coronal FLAIR MRI of brain
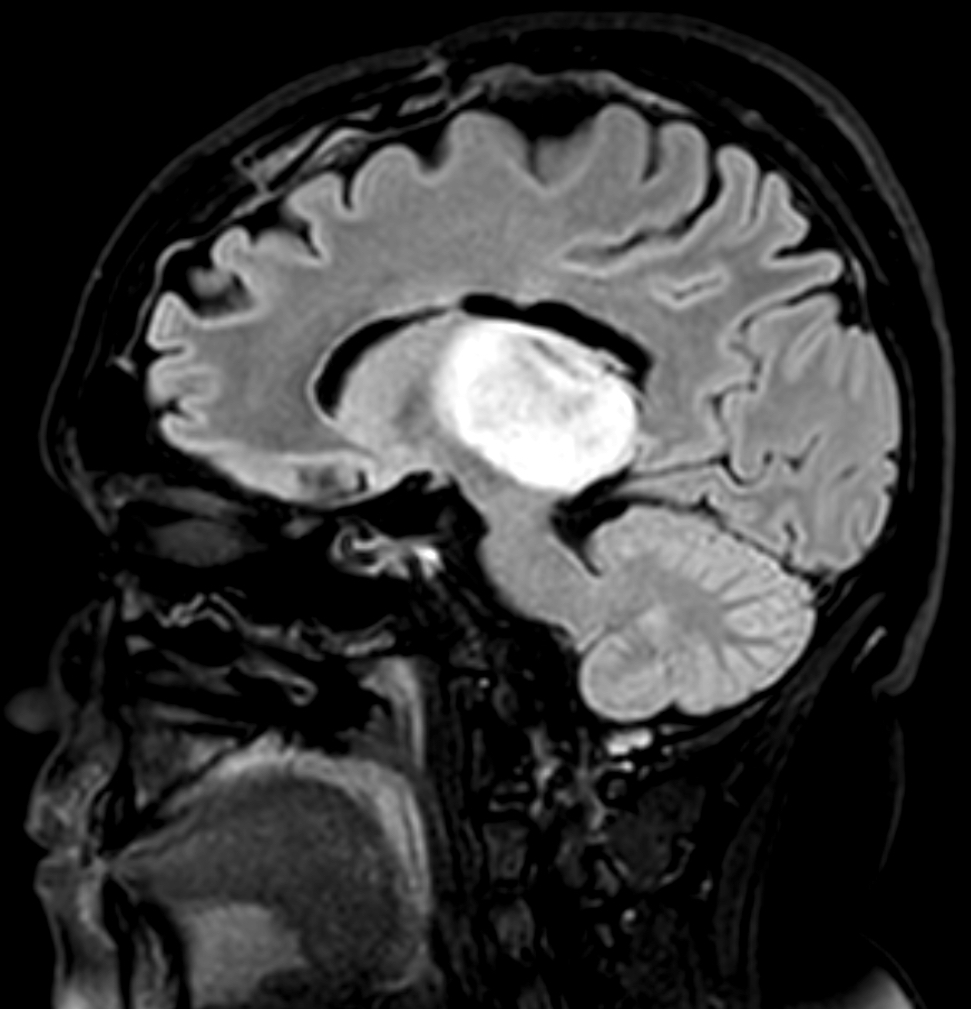
Sagittal FLAIR MRI of brain
- Prognostic data are currently limited to a single retrospective study (Acta Neuropathol 2018;136:273)
- 5 year overall survival: 50%
- No prognostic association with high grade histological features (e.g., mitoses or necrosis)
- No prognostic association with MGMT promoter methylation status
- CNS WHO grade currently not assigned (clinical behavior roughly similar to CNS WHO grade 3)
- 68 year old man presented with dizziness, double vision and an infratentorial cystic mass (Clin Neuroradiol 2024;34:279)
- 8 cases, mean age 45.5 years, with different locations and clinical presentations (AJNR Am J Neuroradiol 2024;45:468)
- 6 cases, mean age 55.2 years, with different locations and clinical presentations (J Neurooncol 2021;153:109)
- Complete surgical resection, if possible, followed by adjuvant radiation and chemotherapy (J Neurooncol 2021;153:109, AJNR Am J Neuroradiol 2024;45:468)
- Mostly well demarcated solid lesion
- Areas of cystic degeneration, necrosis or hemorrhage may be variably present
Contributed by Antonio d’Amati, M.D.
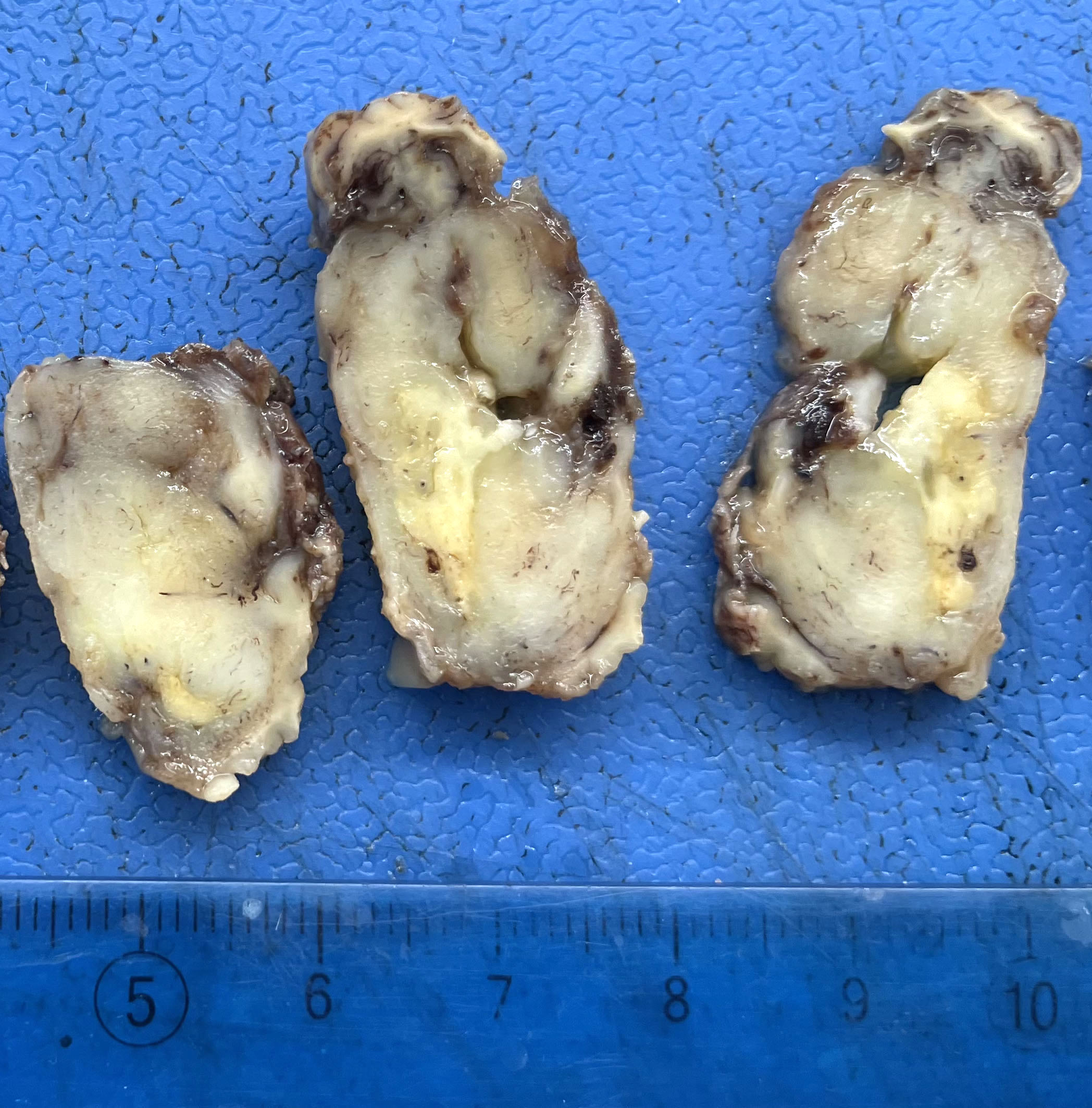
Cerebellar surgical resection
- Histological features are very variable and not sufficient for diagnosis, without additional molecular testing
- Astrocytic glioma, with moderate cell density
- Predominantly solid / circumscribed tumor growth but possible invasion into the adjacent parenchyma
- Growth patterns (Acta Neuropathol 2018;136:273)
- Glioblastoma-like
- Pleomorphic xanthoastrocytoma-like
- Pilocytic astrocytoma-like (cells with hair-like piloid cytoplasmic processes)
- Eosinophilic granular bodies or Rosenthal fibers (~30% of cases) (Acta Neuropathol 2018;136:273)
- High grade histological features
- Mitoses (usually ≥ 1 mitoses/10 high power fields [HPF] of 0.55 mm in diameter) (Acta Neuropathol 2018;136:273)
- Microvascular proliferations (hypertrophy, multilayering or glomeruloid proliferations)
- Necrosis (~30% of cases) (Acta Neuropathol 2018;136:273)
Contributed by Francesca Gianno, M.D., Ph.D.

Piloid features

Spindle cells

Sheets of cells

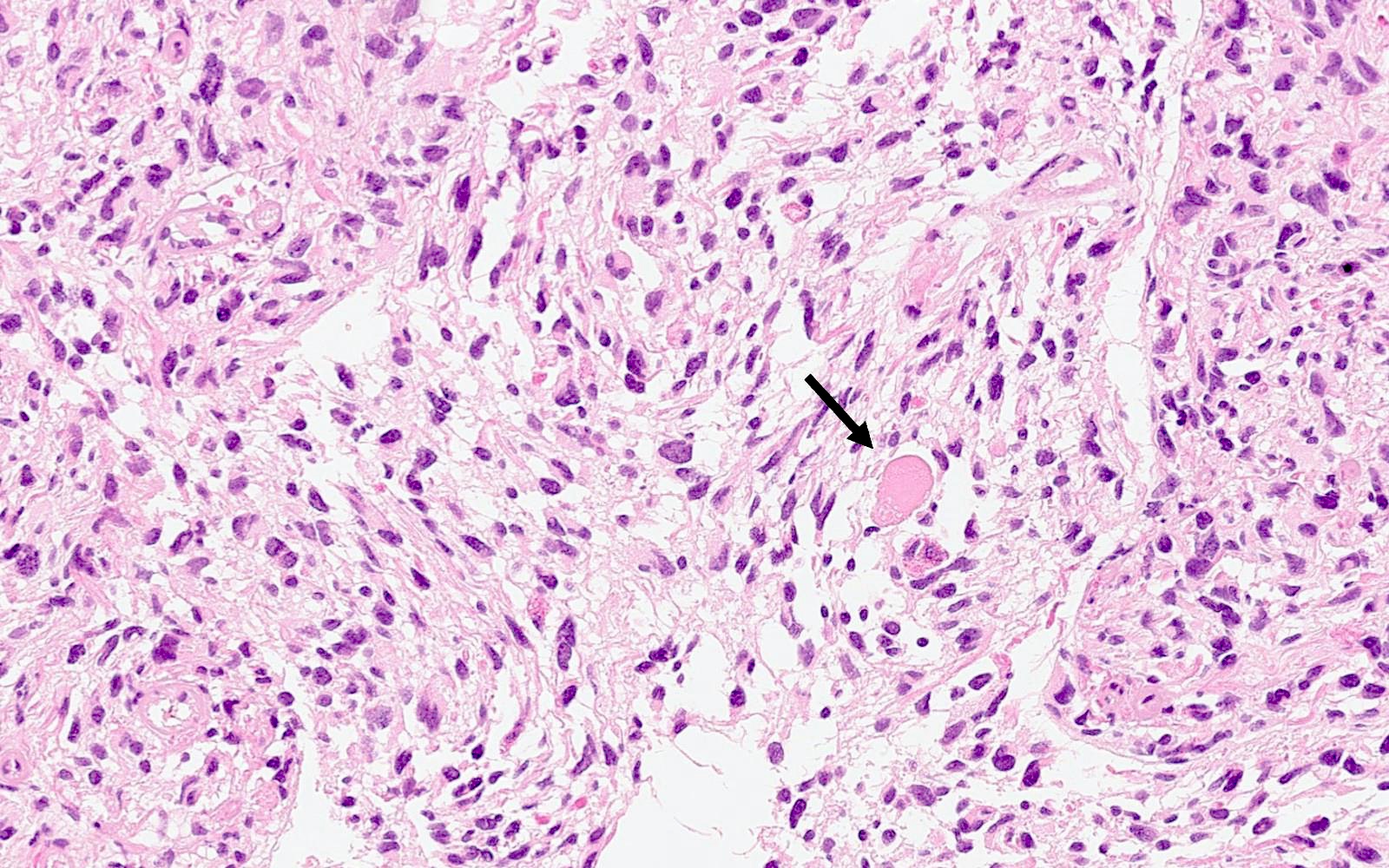
Rosenthal fibers

Eosinophilic granular bodies

Pleomorphic cells
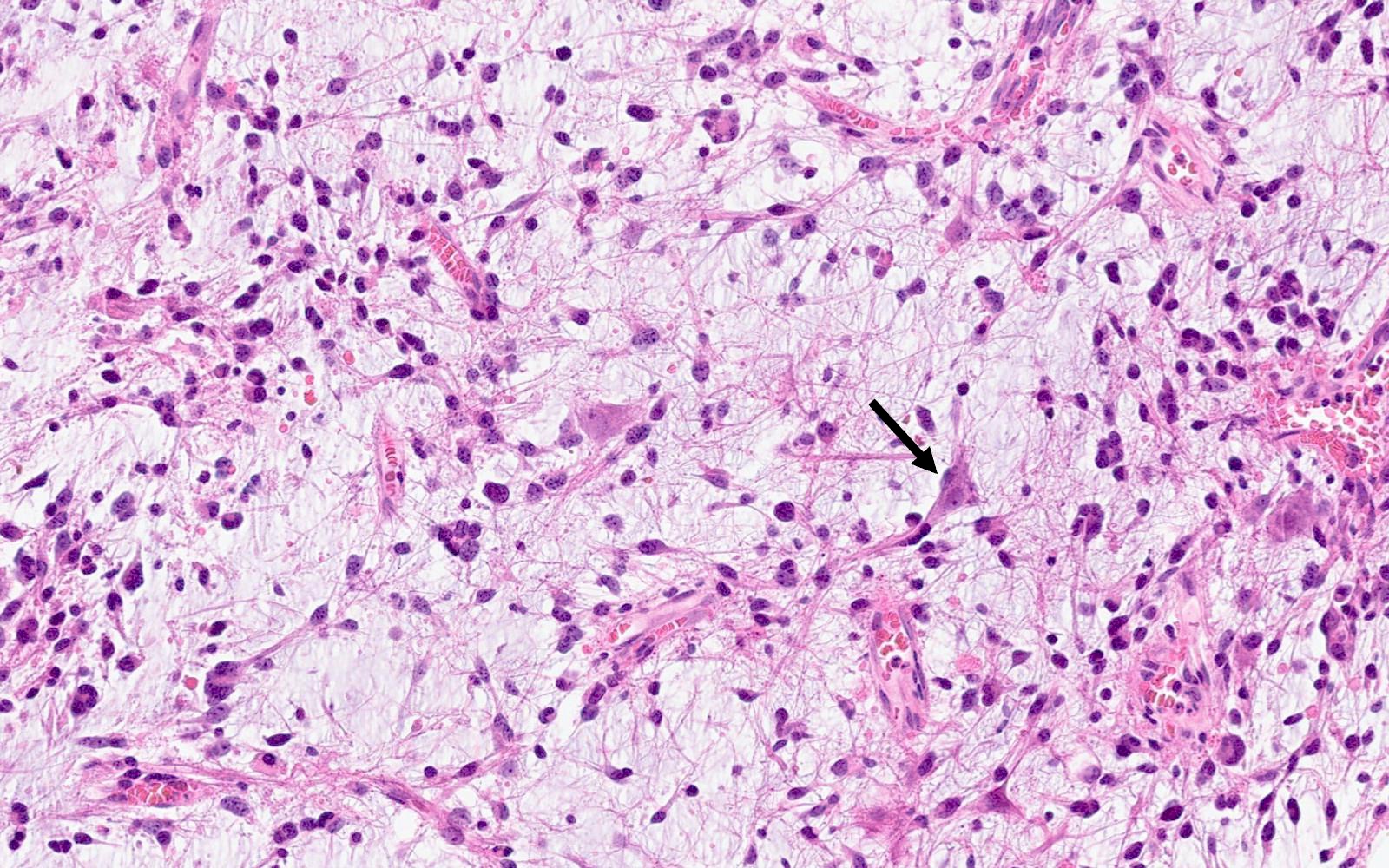

Myxoid background

GFAP

Olig2

ATRX

Low Ki67
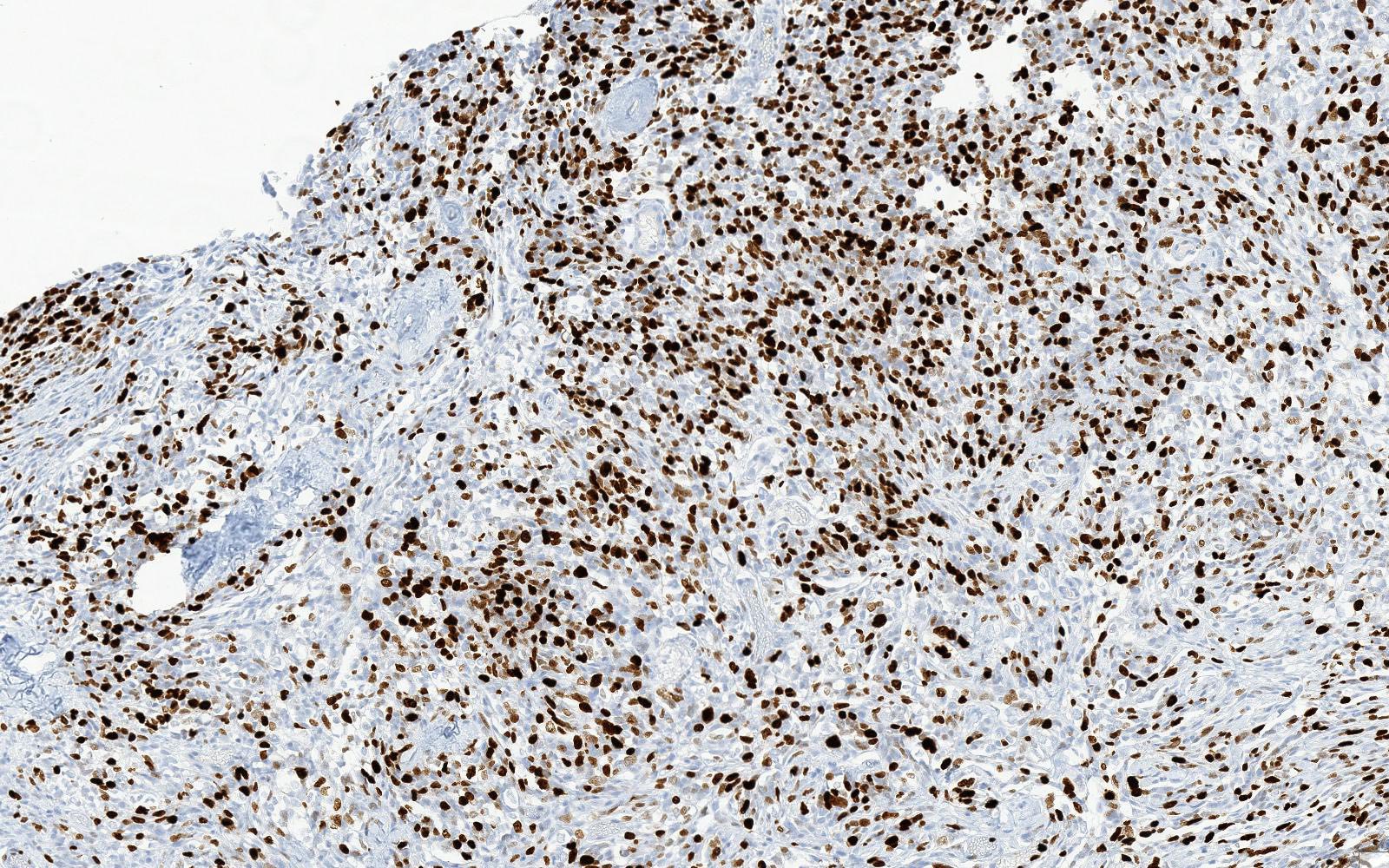
High Ki67
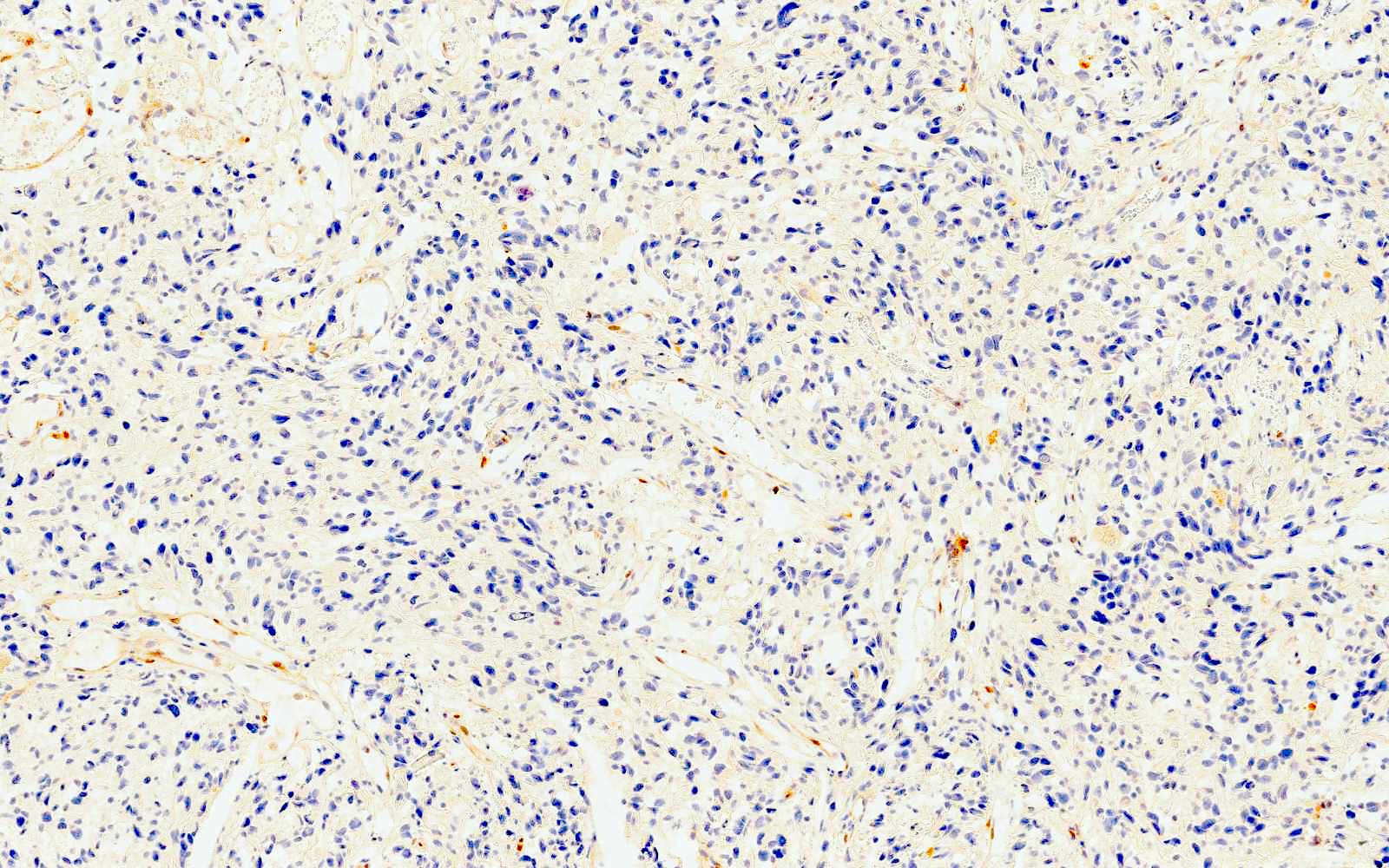
p16
- GFAP
- Olig2
- Ki67 variable (5 - 15%) (J Neurooncol 2021;153:109)
- IDH1 R132H
- ATRX (loss of nuclear expression in ~40% of cases) (Acta Neuropathol 2018;136:273)
- H3 K27M (very rare positive cases) (Acta Neuropathol 2018;136:273)
- Not routinely used for diagnostic purposes
- Currently, DNA methylation profiling is the only method for establishing a definitive diagnosis of HGAP (Acta Neuropathol 2018;136:273)
- Various MAPK pathway gene alterations reported, in order of frequency (Acta Neuropathol 2020;139:287, Acta Neuropathol 2018;136:273)
- NF1 alterations
- KIAA1549::BRAF fusions
- FGFR1 alterations
- KRAS mutations
- BRAF mutations
- CDKN2A or CDKN2B homozygous deletion (80% of cases) (Acta Neuropathol 2018;136:273)
- Alternatively, CDK4 amplification (rarely)
- ATRX alterations (45% of cases) (Acta Neuropathol 2018;136:273)
- Alternatively, TERT promoter mutations (rarely)
- H3 K27M mutations (3% of cases); definitive classification of these cases yet to be established (Acta Neuropathol 2018;136:273)
Contributed by Simone Minasi, Ph.D. and Francesca Romana Buttarelli, Ph.D.

CDKN2A homozygous deletion
KIAA1549::BRAF fusion
Images hosted on other servers:

DNA methylation data

Copy number plots
- Brain, cerebellum, resection:
- Integrated diagnosis: high grade astrocytoma with piloid features (HGAP)
- Histologic diagnosis: astrocytic glioma, with high grade histologic features
- Immunohistochemical results
- GFAP+
- Olig2+
- IDH1 R132H-
- Loss of ATRX nuclear expression
- Ki67: 10%
- CNS WHO grade: not assigned
- Molecular information
- DNA methylation class: HGAP (score 0.99)
- NF1 mutation
- CDKN2A homozygous deletion
- Diffuse gliomas (including glioblastoma, IDH wild type; astrocytoma, IDH mutant; diffuse midline glioma, H3 K27 altered):
- Diffusely infiltrative
- Piloid features, eosinophilic granular bodies or Rosenthal fibers uncommon
- Distinct molecular features
- Pleomorphic xanthoastrocytoma:
- Most commonly localized in superficial cerebral cortex
- Reticulin deposition
- Expression of CD34 and neuronal antigens
- Frequent BRAF p.V600E mutations (very rare in HGAP)
- Pilocytic astrocytoma:
- High grade histologic features usually absent (if present: in the pediatric population, favor anaplastic pilocytic astrocytoma; in adult population, favor HGAP)
- Preserved ATRX expression (loss in 45% of HGAP cases)
- Distinct molecular alterations (some HGAP cases may have KIAA1549::BRAF fusions)
- Distinct DNA methylation profile
A 43 year man has a cerebellar mass. Histologic features are shown in the image above. Immunohistochemistry revealed IDH1 R132H negativity and loss of ATRX nuclear expression. Molecular analyses only revealed NF1 mutation and CDKN2A homozygous deletion. What is the most likely diagnosis?
- Ganglioglioma
- Glioblastoma, IDH wild type
- High grade astrocytoma with piloid features (HGAP)
- Pilocytic astrocytoma
- Pleomorphic xanthoastrocytoma
Comment Here
Reference: High grade astrocytoma with piloid features (HGAP)
- ATRX alterations
- CDK4 amplification
- KIAA1549::BRAF fusions
- Specific DNA methylation profile
- TP53 mutations
Comment Here
Reference: High grade astrocytoma with piloid features (HGAP)
- See Bone topic
- See Lymph node topic
- See Skin nonmelanocytic tumor topic
-
Definition / general
- See also Lymph node topic
- Rare lytic skull lesion or parasellar mass
- Children and young adults
- May arise from primary histiocytic proliferation with secondary atrophy or from demyelination and gliosis of unknown origin (J Child Neurol 2000;15:150)
-
Definition / general
- See also Lymph node topic
- Also called sinus histiocytosis with massive lymphadenopathy
- Often (25 - 43%) has extranodal involvement
- Rare (100 cases/year in US) and isolated CNS findings are extremely rare
- Benign but may be associated with systemic disease
- Children and young adults (mean age 41 years, range 22 - 63 years)
- CNS tumors usually associated with dura
- References: Mod Pathol 2001;14:172, Clin Neuropathol 2005;24:112 (isolated CNS disease)
Case #488
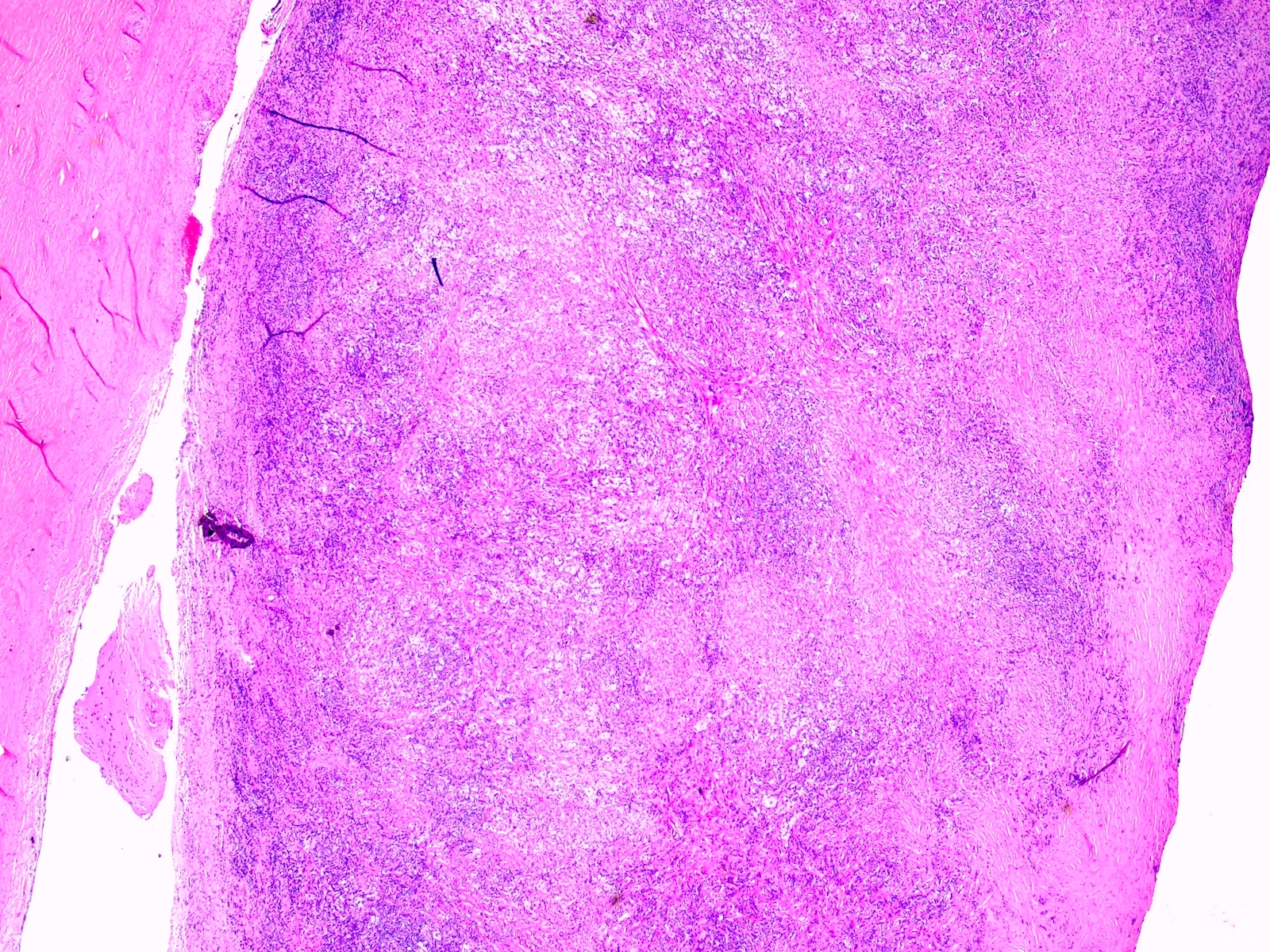
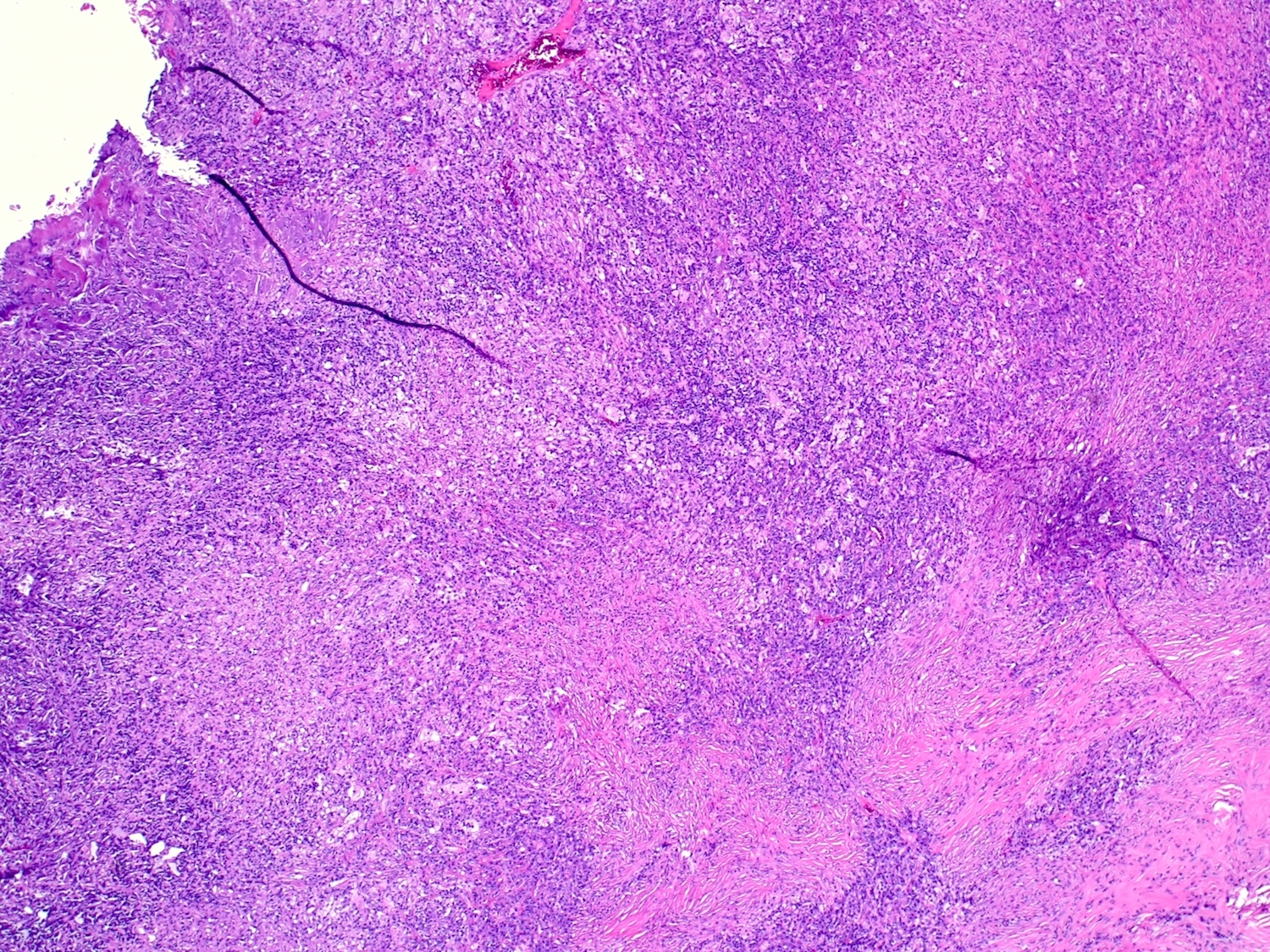
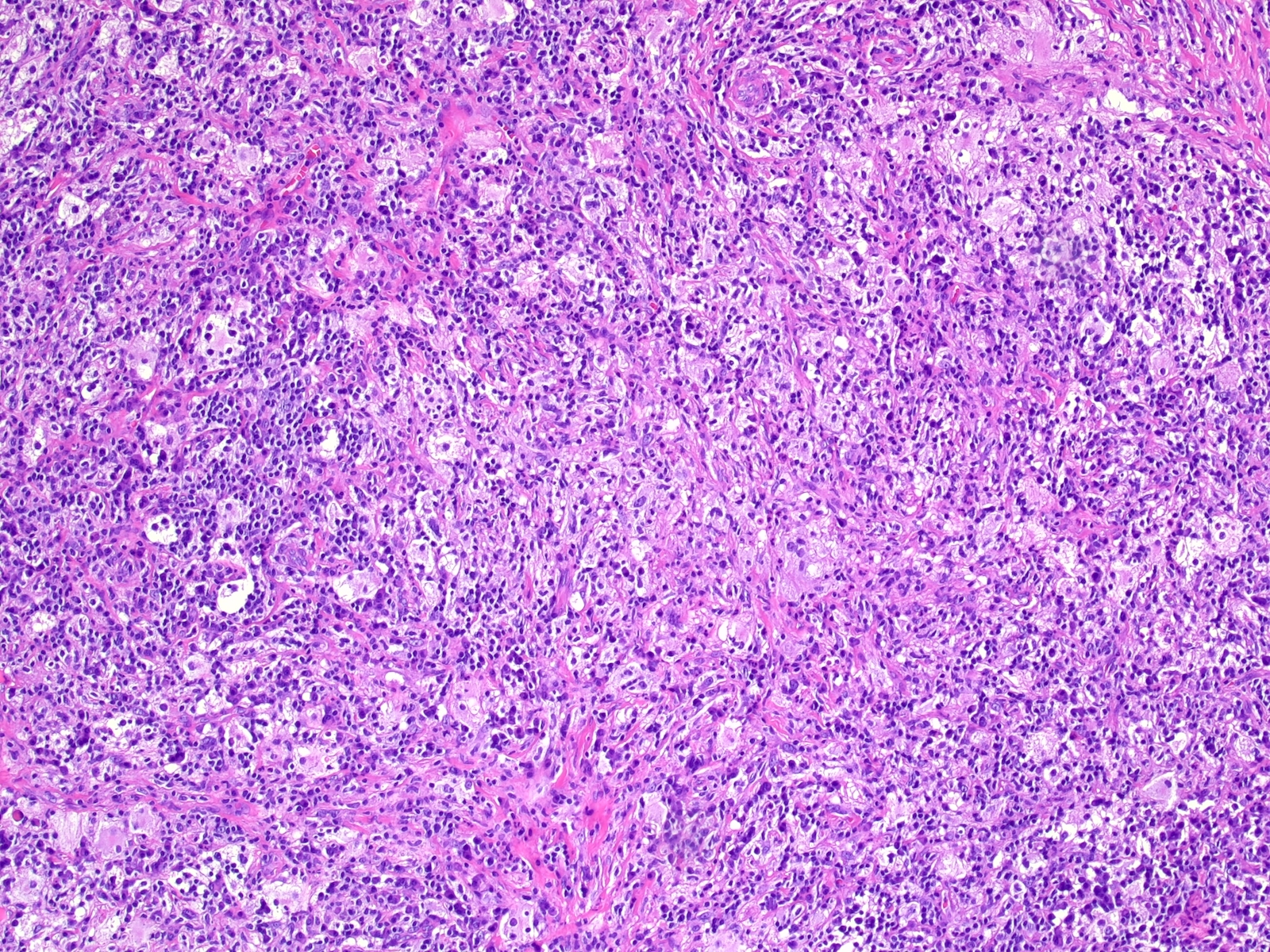
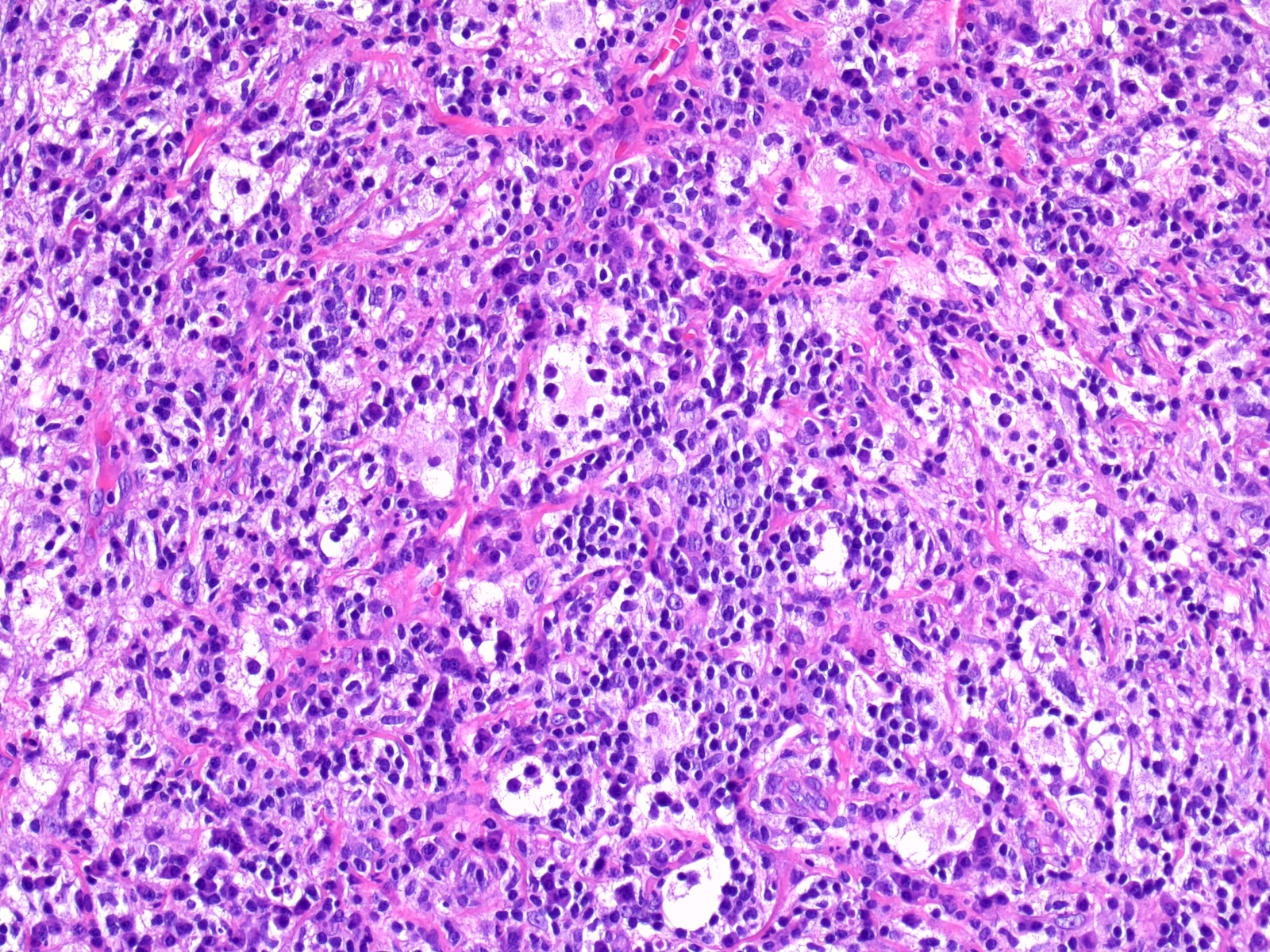
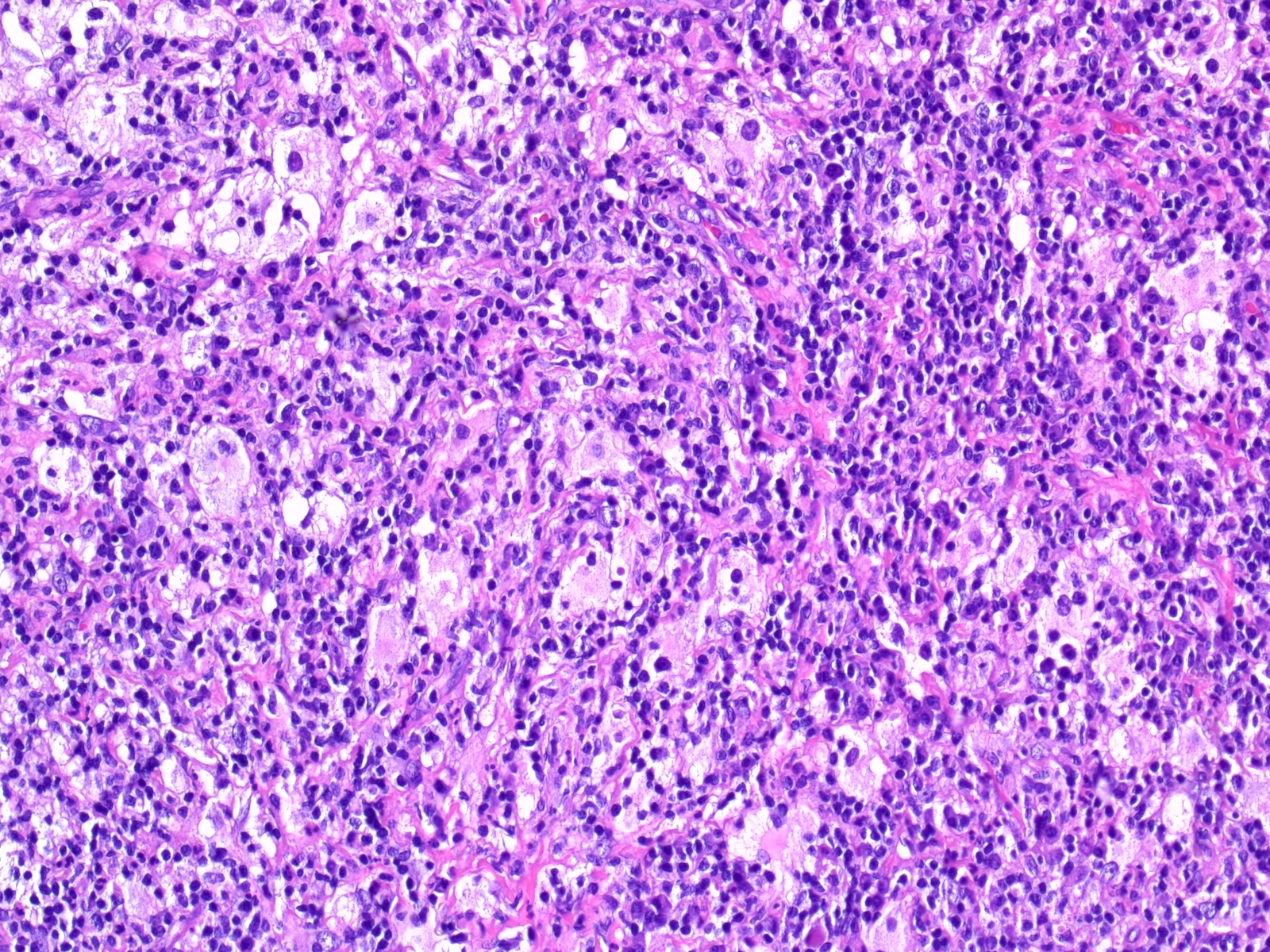
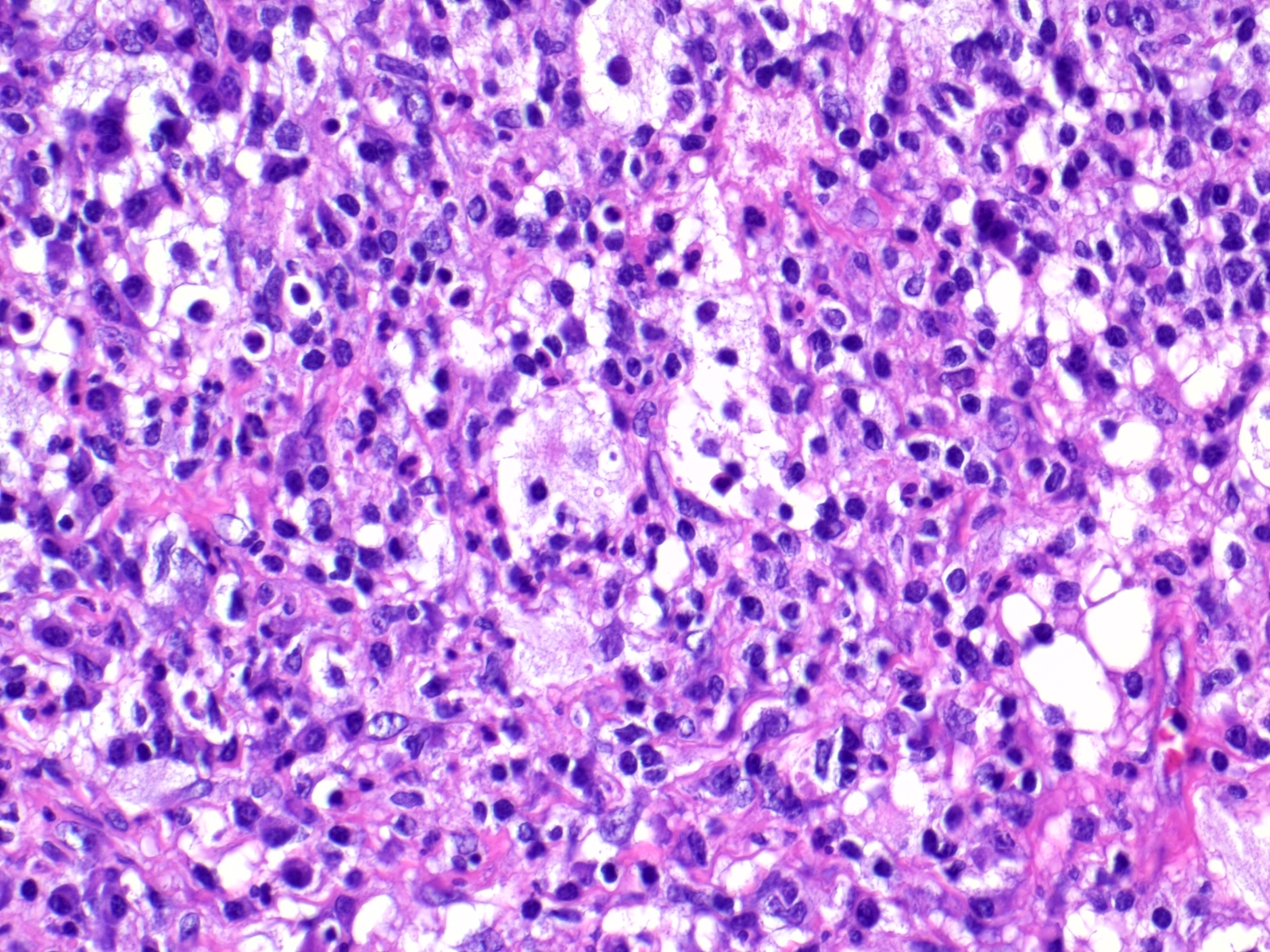
Rosai-Dorfman disease of CNS
- In distinguishing CNS Rosai-Dorfman disease (RDD) from other histiocytoses, what is the correct immunophenotype for RDD?
- S100-, CD1a-
- S100+, CD1a+
- S100+, CD1a-
- S100-, CD1a+
Explanation: CNS Rosai-Dorfman disease forms lymphohistiocytic masses with prominent pale S100+, CD1a- histiocytes. Answer A (S100-, CD1a-) is the staining pattern of histiocytes in Erdheim-Chester disease. Answer B (S100+, CD1a+) is the staining pattern of histiocytes in Langerhans cell histiocytosis.
Comment Here
Reference: Histiocytic tumors
- Extranodal large B cell lymphoma characterized by lymphoma cells, predominantly within lumina of blood vessels, especially capillaries, with the exclusion of larger arteries and veins
- Few to no circulating lymphoma cells in peripheral blood
- Rare, mature B cell lymphoma limited to intravascular spaces
- Most common in skin and central nervous system
- Classical variant most common clinical presentation (neurological and cutaneous manifestations)
- Neoplastic cells are located in lumina of small to intermediate sized vessels
- Lymphoma cells are large with prominent nucleoli and vesicular chromatin
- Pan B cell markers positive, as well as BCL2 and MUM1
- Aggressive behavior with short overall survival
- Intravascular large B cell lymphoma (ILBCL)
- Malignant angioendotheliomatosis, angioendotheliomatosis proliferans syndrome, intravascular lymphomatosis, angioendothelilotropic lymphoma (all obsolete)
- ICD-O: 9712/3 - intravascular large B cell lymphoma
- Rare; accounts for 1% of B cell lymphomas (Cancer Sci 2021;112:3953)
- Older patients; median age 70 years (Blood 2018;132:1561)
- Slightly more common in males
- Selective growth within lumina of small blood vessels, particularly capillaries
- Extranodal sites, including bone marrow, with sinusoidal or perivascular involvement
- Most common sites of involvement: skin, nervous system, kidneys, lungs, endocrine glands
- Lymph node involvement rare (Blood 2018;132:1561)
- Localization to blood vessel lumens partially explained by lack of CD29 (β1 integrin) and CD54 (ICAM1), both of which are important for transvascular lymphocyte migration (Blood 2018;132:1561)
- Unknown
- Classic / western form:
- Clinical presentation ranges from a few mild symptoms (fever of unknown origin, pain, organ specific local symptoms) to severe symptoms (B symptoms and signs of multiorgan failure) (Cancer Sci 2021;112:3953)
- CNS involvement (present in 35%) with heterogeneous symptoms, including sensory and motor deficits or neuropathies, meningoradiculitis, paresthesias, hemiparesis, seizures and altered mental state
- Skin lesions (present in 40%) are heterogeneous, including painful indurated erythematous eruptions, cellulitis, peau d’orange, small red palpable spots, nodules with or without ulceration, tumors and erythematous and desquamated plaques (Blood 2018;132:1561)
- Lymphadenopathy typically absent
- May present with disseminated intravascular coagulation leading to critical bleeding after biopsy (Cancer Sci 2021;112:3953)
- Cutaneous variant:
- 25% of patients
- More frequent in western countries
- Patients usually younger, with a median age of 59
- Single or multiple skin lesions; no other sites involved
- Disease progression is less aggressive and associated with better overall survival (Blood 2018;132:1561)
- Hemophagocytic syndrome associated form:
- Presents in Asian populations with multiorgan failure, hepatosplenomegaly and pancytopenia
- Bone marrow infiltration common
- Skin and CNS involvement are rare
- Rapid aggressive onset and progression with median survival of 2 - 8 months (Blood 2018;132:1561)
- Biopsy of affected or enlarged organ (Blood 2018;132:1561)
- Most commonly random skin biopsy in conjunction with bone marrow biopsy (Br J Haematol 2019;187:328)
- Anemia, leukopenia, thrombocytopenia and unexplained hypoxemia are most frequently observed
- Low levels of serum albumin
- High LDH and ferritin (Br J Haematol 2019;187:328)
- Lung involvement presents with ground glass appearance and nodules (Blood 2018;132:1561)
- Organ enlargement (liver, spleen, kidney and adrenal gland) is common (Cancer Sci 2021;112:3953)
- Brain MRI shows hyperintense lesions in pons, nonspecific white matter lesions, infarct-like lesions or meningeal enhancement
- PET CT can detect bone marrow involvement (Br J Haematol 2019;187:328)
- Median overall survival of 105 months, with chemotherapy 135 months
- 5 year survival of approximately 50 - 60% (Br J Haematol 2019;187:328)
- 52 year old woman with diffusely PET avid kidneys (Intern Med 2017;56:827)
- 63 year old woman with multiple strokes (Medicine (Baltimore) 2018;97:e12793)
- 72 year old man with known right kidney tumor (Int J Surg Pathol 2021;29:653)
- 73 year old man with progressive exertional dyspnea, fever and calcified mediastinal lymphadenopathy (Respir Med Case Rep 2019;29:100989)
- 83 year old man with diffuse gallbladder wall thickening (Case Rep Radiol 2018;2018:2494207)
- Chemoimmunotherapy usually with addition of CNS oriented therapy (Cancer Sci 2021;112:3953)
- Lymphoma cells can be found in any organ vessel lumina (Br J Haematol 2019;187:328)
- Lymphoma cells are large with high nuclear to cytoplasmic ratio with single or multiple prominent nucleoli and scant cytoplasm
- Morphologic spectrum from centroblasts to immunoblasts / plasmablasts, including rare forms with anaplastic morphology
- Different growth patterns:
- Discohesive - lymphoma cells are preferentially within the central portion of the blood vessels with a free floating appearance
- Cohesive pattern - lymphoma cells almost completely fill the lumen to the point that assessment of vascular structure tends to be difficult
- Marginating pattern - less frequent, lymphoma cells preferentially adhere to endothelium, leaving the central portion of the lumen free (Blood 2018;132:1561)
- Infiltration patterns in bone marrow:
- Pure intrasinusoidal infiltration - neoplastic cells are confined within the intrasinusoidal spaces
- Intrasinusoidal infiltration with extravasation - neoplastic cells proliferate within intrasinusoidal space but with extravasation
- Diffuse interstitial infiltration - neoplastic cells proliferate diffusely within bone marrow (Br J Haematol 2019;187:328)
Contributed by Kathryn Gibbons, M.D.
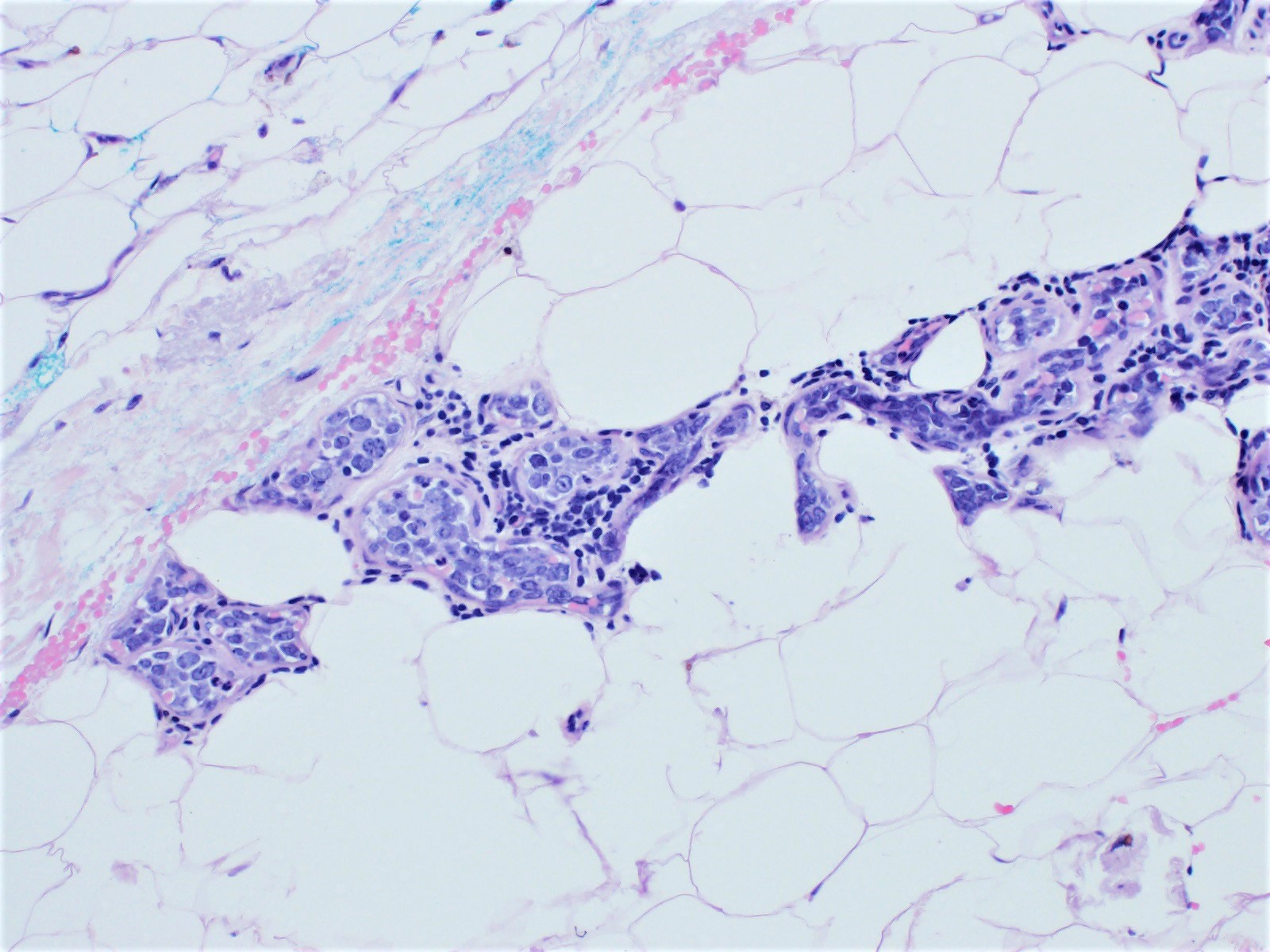
ILBCL involving skin and subcutaneous tissue
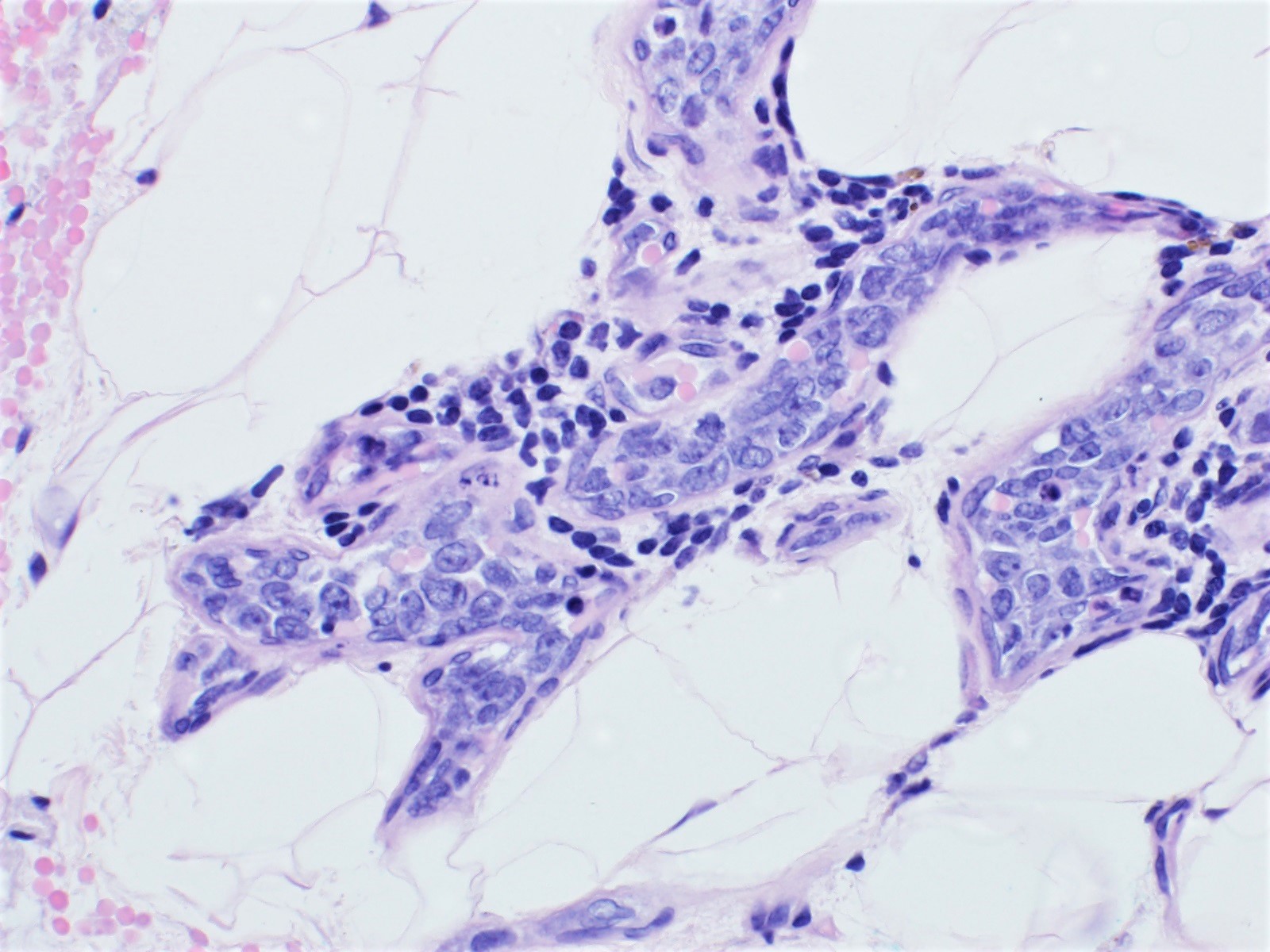
Large lymphoma cells in small vessels
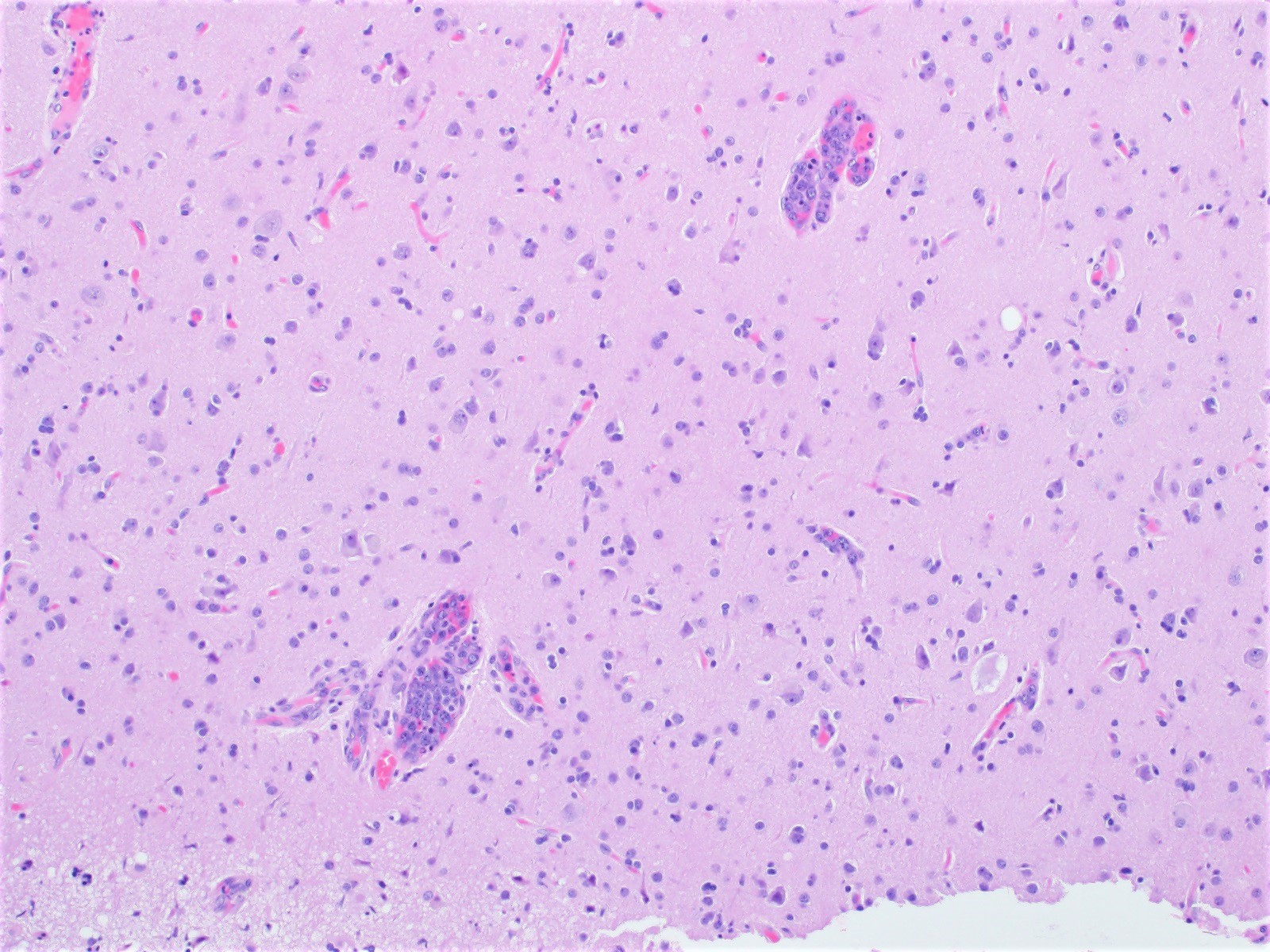
ILBCL involving the brain parenchyma
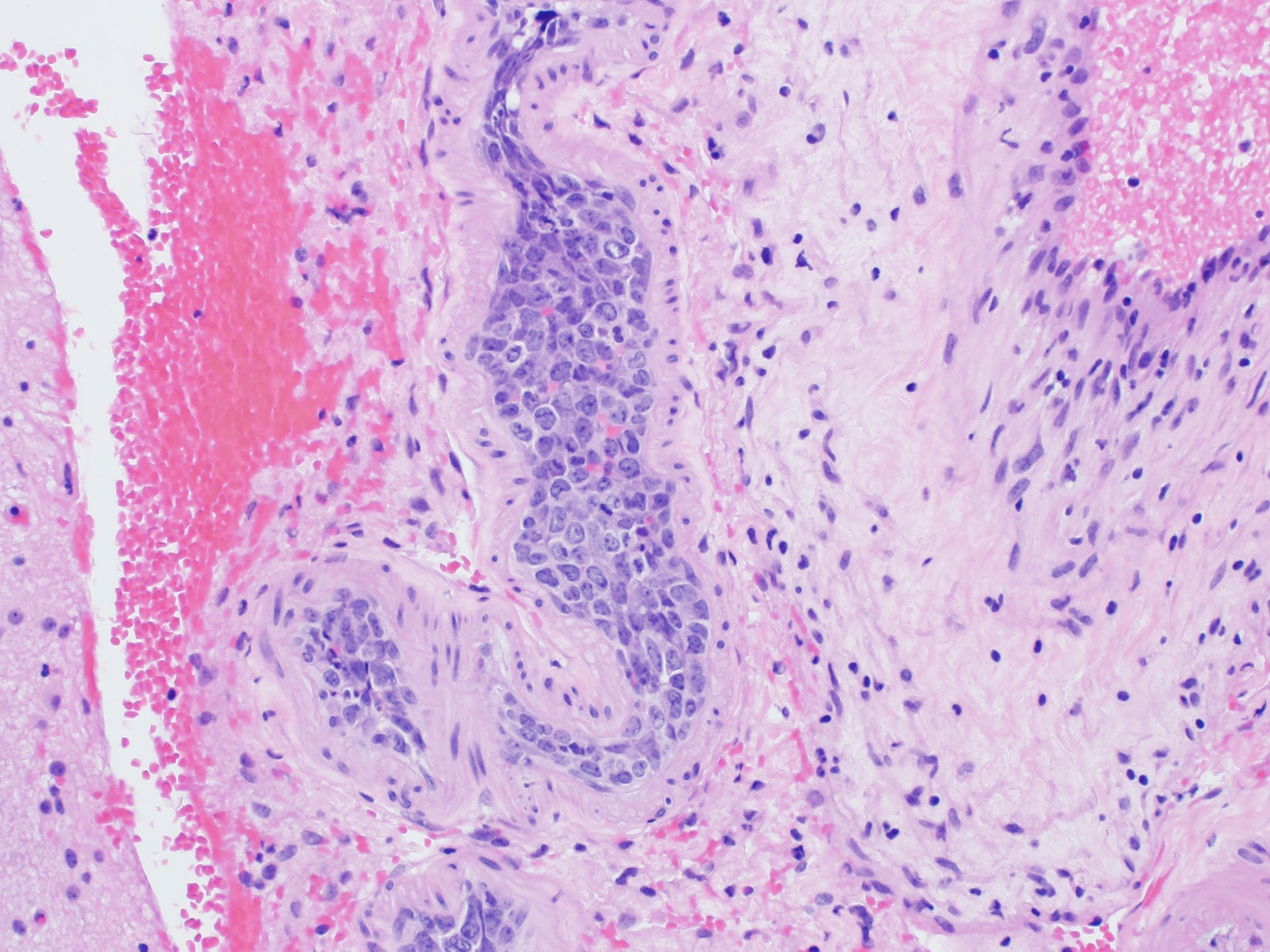
Vessel with involvement by ILBCL
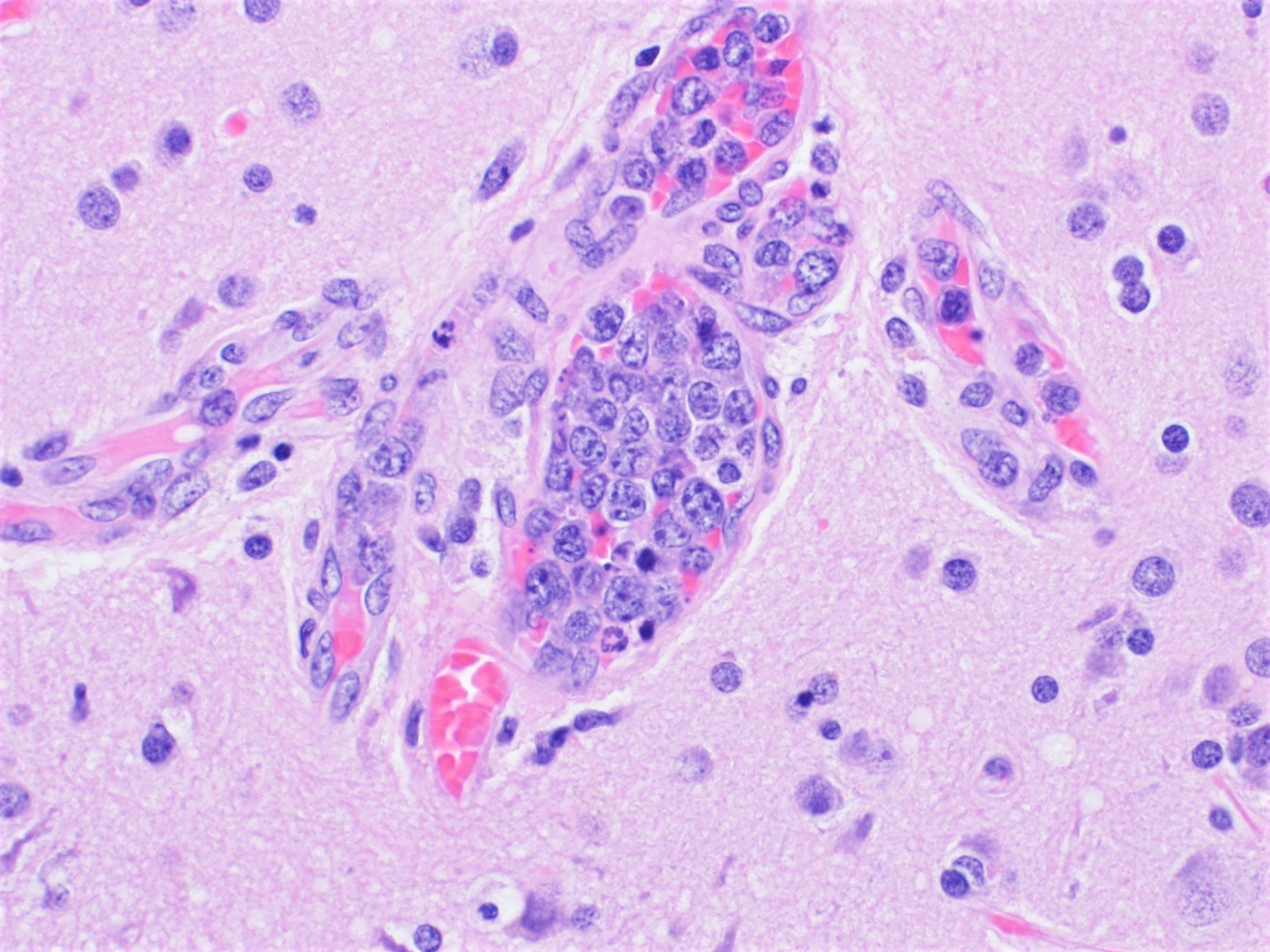
Large lymphoma cells in the vessels
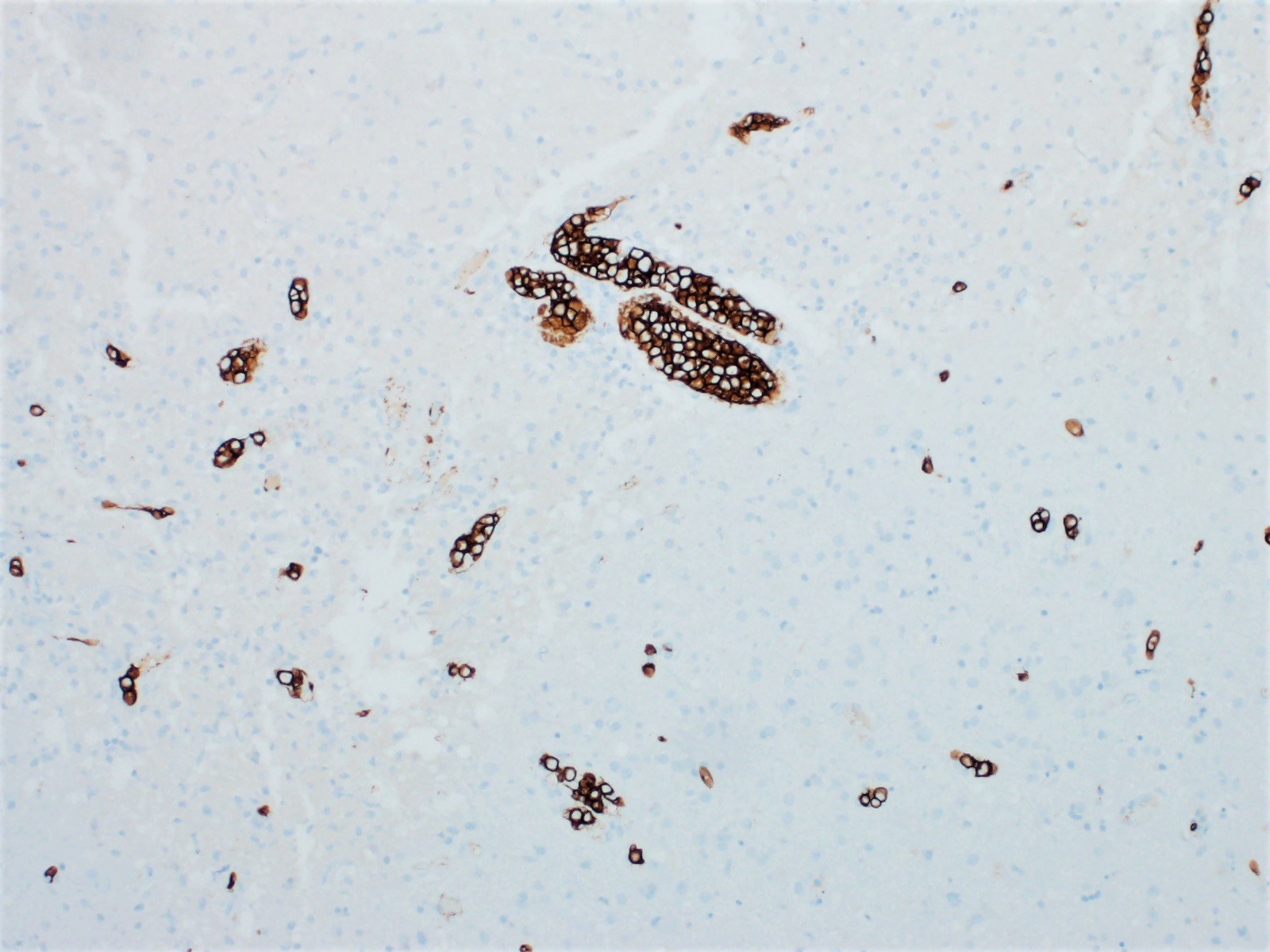
CD20
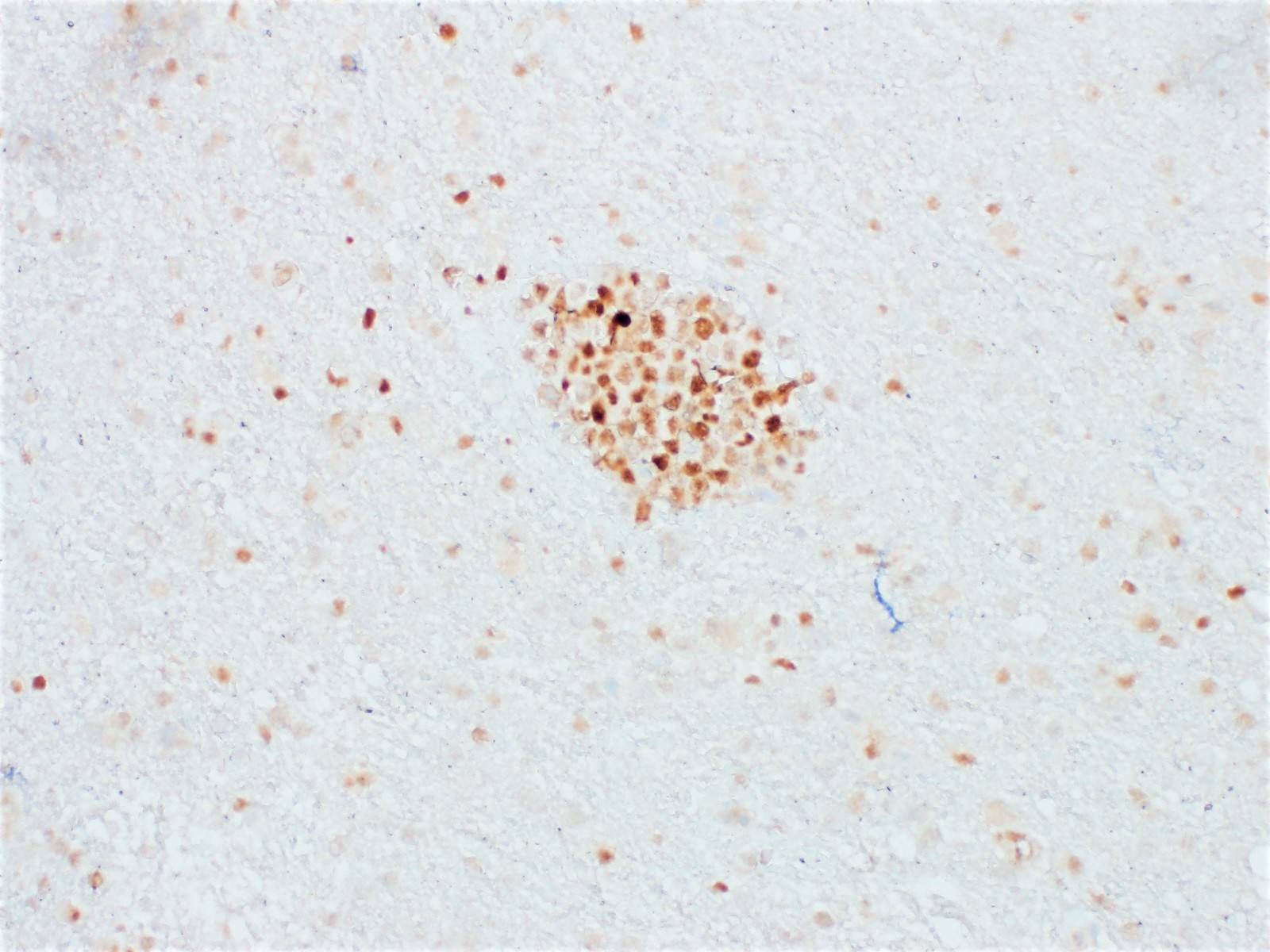
BCL6
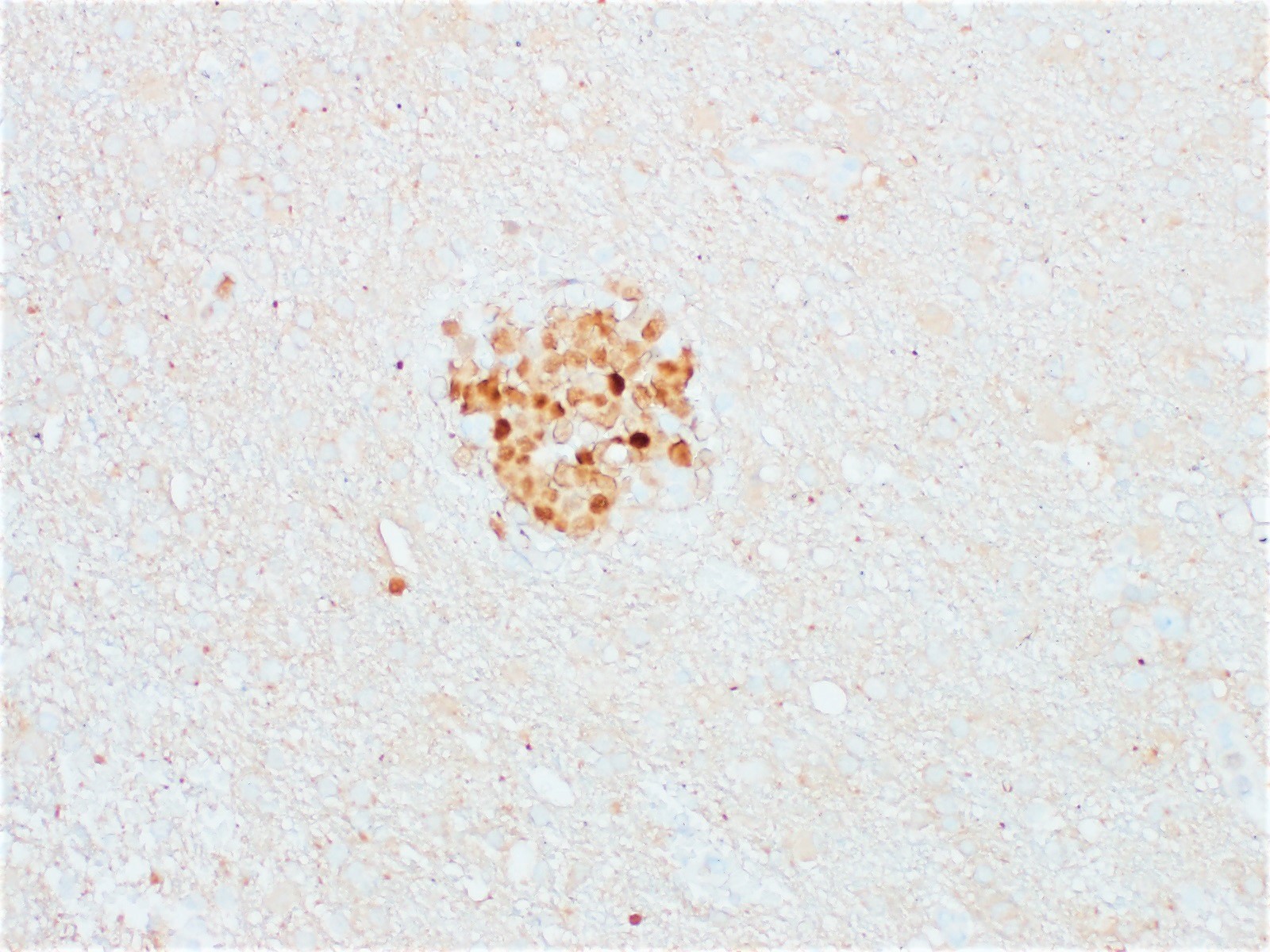
MUM1
Images hosted on other servers:
Lymphoma cells in blood vessels of subcutaneous adipose tissue
Lymphoma cells limited to lumina of blood vessels
- Pan B cell markers: CD20, CD79a, PAX5
- BCL2 positive (Blood 2018;132:1561)
- Based on the Hans algorithm, 95% show nongerminal center phenotype with MUM1 / IRF4 expression (Cancer Sci 2021;112:3953)
- CD5 positive in 22 - 38% (Blood 2018;132:1561)
- Substantial proportion demonstrates PDL1 expression
- CD34 useful to highlight endothelium (Cancer Sci 2021;112:3953)
- Rare cases show positivity for EBV or HHV8 (Mod Pathol 2009;22:618, Diagn Pathol 2020;15:72, J Investig Med High Impact Case Rep 2014;2:2324709614526702)
- CD10 and BCL6 mostly negative (positive in approximately 20% of cases)
- Cyclin D1, CD30 (Blood 2018;131:2086)
- Clonal rearrangements of immunoglobulin genes (Blood 2018;132:1561)
- MYD88 L265P in 44% (Blood 2018;131:2086)
- CD79b Y196 mutations in 26%
- Similar mutations to DLBCL, activated B cell type (Cancer Sci 2021;112:3953)
- Skin, left thigh, punch biopsy:
- Intravascular large B cell lymphoma (see comment)
- Comment: Biopsy shows large atypical CD20 positive B cells present within vascular spaces in the subcutaneous fat. These cells coexpress CD5, BCL6 and MUM1 and are negative for CD10 and CD3. Overall, these findings are consistent with intravascular large B cell lymphoma.
- Lymphomatoid granulomatosis:
- Angiodestructive and angiocentric lesions showing a mixture of B cells, T cells, immunoblasts and histiocytes
- B cells are EBV positive
- Primary CNS lymphoma:
- Lymphoma cells infiltrate brain parenchyma
- Diffuse large B cell lymphoma:
- Cytologically and immunophenotypically identical cells but typically presents with lymphadenopathy or mass
- CNS vasculitis:
- Cells are less atypical and involve vessel walls in addition to vessel lumina (Arch Pathol Lab Med 2012;136:333)
- Other lymphomas presenting as intravascular lymphoma:
- Extracavitary primary effusion lymphoma as well as T and NK/T cell lymphomas, in rare cases, have been reported to show an intravascular pattern (Am J Surg Pathol 2014;38:426, BMC Cancer 2018;18:1115, Am J Surg Pathol 2013;37:617)
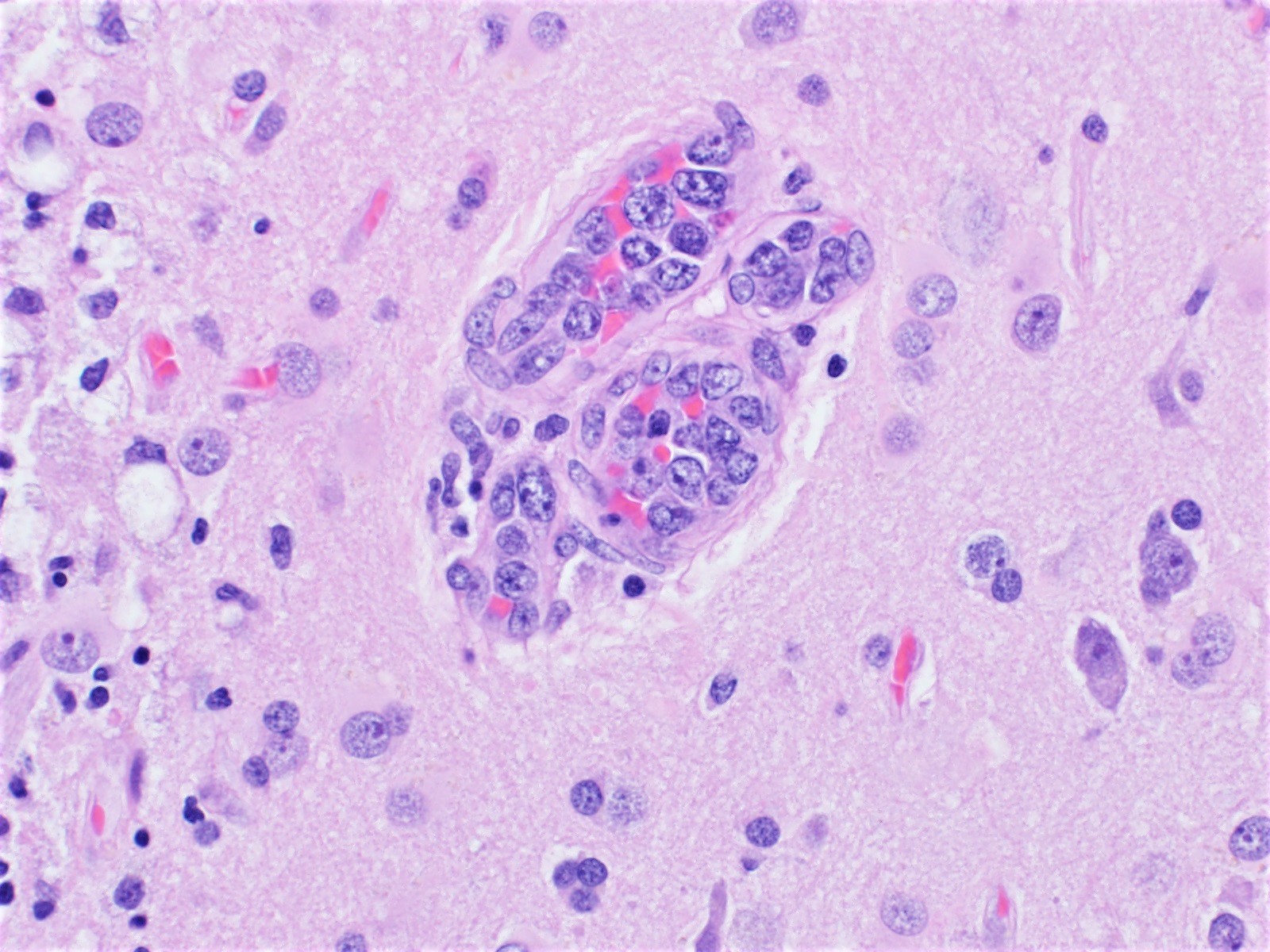
The image shown above is from a brain biopsy in a patient with fever and seizures. Large cells are strongly positive for CD20. What is the correct diagnosis?
- CNS vasculitis
- Intravascular large B cell lymphoma
- Marginal zone lymphoma
- Plasmablastic lymphoma
- CD20+, CD3-, CD5-, CD10+, BCL6+, BCL2+, c-MYC+
- CD20+, CD5+, Cyclin D1+
- CD20+, PAX5+, MUM1+, BCL2+
- CD20-, CD2+, CD3+, CD5-, CD7+, CD56+
- Lymphomatoid granulomatosis is an angiocentric and angiodestructive lymphoproliferative disease involving extranodal sites composed of Epstein–Barr virus (EBV) positive B cell admixed with reactive T cells, which usually predominate (Blood 2016;127:2375)
- Angiocentric and angiodestructive lymphoproliferative disease involving extranodal sites composed of EBV positive B cell admixed with reactive T cells
- Angiocentric immunoproliferative lesion
- Very rare
- Usually occurs in adults; sometimes seen in children with immunodeficiency disorders (Cancer 1979;43:360)
- M:F ≥ 2:1
- More common in western countries than in Asia (Am J Surg Pathol 2015;39:141)
- Pulmunary involvement > 90% patients (Cancer 1979;43:360)
- Other common sites include: brain, kidney, liver and skin (Am J Surg Pathol 2010;34:e35, Arch Dermatol 1996;132:1464)
- Uncommon sites include: upper respiratory tract and gastrointestinal tract (Am J Surg Pathol 1999;23:1356)
- Rare sites: lymph nodes and spleen (Am J Surg Pathol 2015;39:141, Indian J Pathol Microbiol 2018;61:228)
- Lymphomatoid granulomatosis due to the predominance of T cells on pathologic examination, was initially thought to be a T cell disorder (Am J Med 1982;72:467)
- Subsequently, lymphomatoid granulomatosis was determined to be a B cell lymphoproliferative disorder secondary to EBV, with a prominent angiocentric T cell infiltrate (Mod Pathol 1990;3:435)
- Lymphomatoid granulomatosis has a complex relationship with the host’s underlying immune function and defective immune surveillance of EBV infected B cells, particularly a functional defect in CD8+ cytotoxic T cells, is hypothesized to lead to disease development (Blood 1996;87:4531)
- Particular study suggests that immunologic deficits are likely preexistent and that a quantitative or qualitative defect in mainly CD8+ cytotoxic T cells may be prerequisite for disease (Cancer 1982;49:2070)
- Resembles an EBV driven lymphoproliferative disorder (Blood 1996;87:4531)
- Also common in many immunodeficiency states, such as AIDS, allogenic organ transplant, Wiskott-Aldrich syndrome and X linked lymphoproliferative syndrome (Cancer Surv 1997;30:233)
- Normal counterpart: mature B cell, transformed by EBV (Blood 2016;127:2375)
- Most commonly: cough, dyspnea and chest pain (Am J Surg Pathol 2010;34:e35, Am J Surg Pathol 2015;39:141)
- Other common symptoms: fever, malaise, weight loss, neurological symptoms, arthralgias, myalgia and gastrointestinal symptoms (Am J Surg Pathol 1999;23:1356)
- Patients with CNS disease; may show diplopia, hearing loss, dysarthria, ataxia, altered mental status or be asymptomatic (Radiology 2005;237:265)
- Skin manifestations can be painful and include subcutaneous nodules, dermal nodules, maculopapular eruptions, macular erythema and ulcerations (Dermatol Clin 2015;33:489)
- Few patients (2.8%) are asymptomatic (Cancer 1979;43:360)
- Combination of light microscopy and immunohistochemical markers or in situ hybridization for EBV coded small RNA (EBER)
- Chest Xray may show single or multiple nodules with poorly defined margins
- CT scans show well defined and poorly defined nodules throughout both lungs (AJR Am J Roentgenol 2000;175:1335)
Contributed by Nicholas Joseph Dcunha, M.B.B.S., M.D. and Elanthenral Sigamani, M.B.B.S., M.D.
Mass in lung (CT scan)
- Prognostic features are difficult to determine due the rarity of the disease
- 14 year old boy with trisomy 21 and history of B lymphoblastic leukemia / lymphoma diagnosed 1.5 years previously on maintenance chemotherapy, with right lower lobe consolidation (Fetal Pediatr Pathol 2018;37:7)
- 22 year old man with lymphomatoid granulomatosis of the CNS (Neuropathology 2019;39:479)
- 56 year old renal allograft recipient; 11 years after transplantation (Transpl Infect Dis 2008;10:52)
- 72 year old woman with asymptomatic right lung mass (Arch Bronconeumol 2018;54:108)
- 79 year old man with bulky lung mass (Intern Med 2018;57:3163)
- In a prospective NCI study, patients with low grade lymphomatoid granulomatosis received primary therapy with IFN-α while patients with high grade lymphomatoid granulomatosis received primary therapy with DA-EPOCH-R (dose adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin and rituximab) every 3 weeks for up to 6 cycles of therapy (Blood 2018;132:785)
- Lesions in the lung are mainly nodular and involve middle and lower lung fields; may exhibit central necrosis and cavitate (Am J Surg Pathol 2010;34:e35, Arch Dermatol 1996;132:1464)
- Kidney and brain lesions are also nodular and associated with central necrosis (Am J Surg Pathol 2015;39:141)
- Skin lesions range from nodules in subcutaneous tissue to dermal ulcers with necrosis; occasionally plaques or maculopapular rash-like presentation (Am J Surg Pathol 2001;25:1111)
- Angiocentric and angiodestructive polymorphous lymphoid infiltrate (Cancer 1979;43:360)
- Lymphocytic vasculitis with infiltration of the vessel wall; may also show infarct like tissue necrosis or fibrinoid necrosis of vessel wall (Blood 1997;90:4099)
- Infiltrate is predominantly composed of lymphocytes with admixed plasma cells, immunoblasts and histiocytes; background lymphocytes; may show atypia but are not overtly neoplastic
- Lymphomatoid granulomatosis is composed of variable number of EBV positive B cells admixed with inflammatory background (Am J Surg Pathol 2010;34:e35)
- EBV positive cells resemble immunoblasts or are multinucleated; may show atypia
- Classic Reed-Sternberg cells are absent
- Well formed granulomas are typically not seen in lungs and most other extranodal sites except skin where granulomatous reaction might be seen in subcutaneous tissue (Am J Surg Pathol 2001;25:1111)
- Grading is based on the relative number of EBV positive B cells to the reactive lymphocyte background (Am J Surg Pathol 2015;39:141)
- Grade I: EBV positive cells are < 5 per high power field; absent or rare large transformed cells on light microscopy; necrosis if present is focal
- Grade II: EBV positive cells are 5 - 50 per high power field; few large transformed cells on light microscopy; necrosis is more commonly seen
- Grade III: EBV positive cells are > 50 per high power field; large transformed cells are easily seen on light microscopy; large areas of necrosis are common
Contributed by Nicholas Joseph Dcunha, M.B.B.S., M.D. and Elanthenral Sigamani, M.B.B.S., M.D.
Lymphocytic vasculitis with angioinvasion (lung)
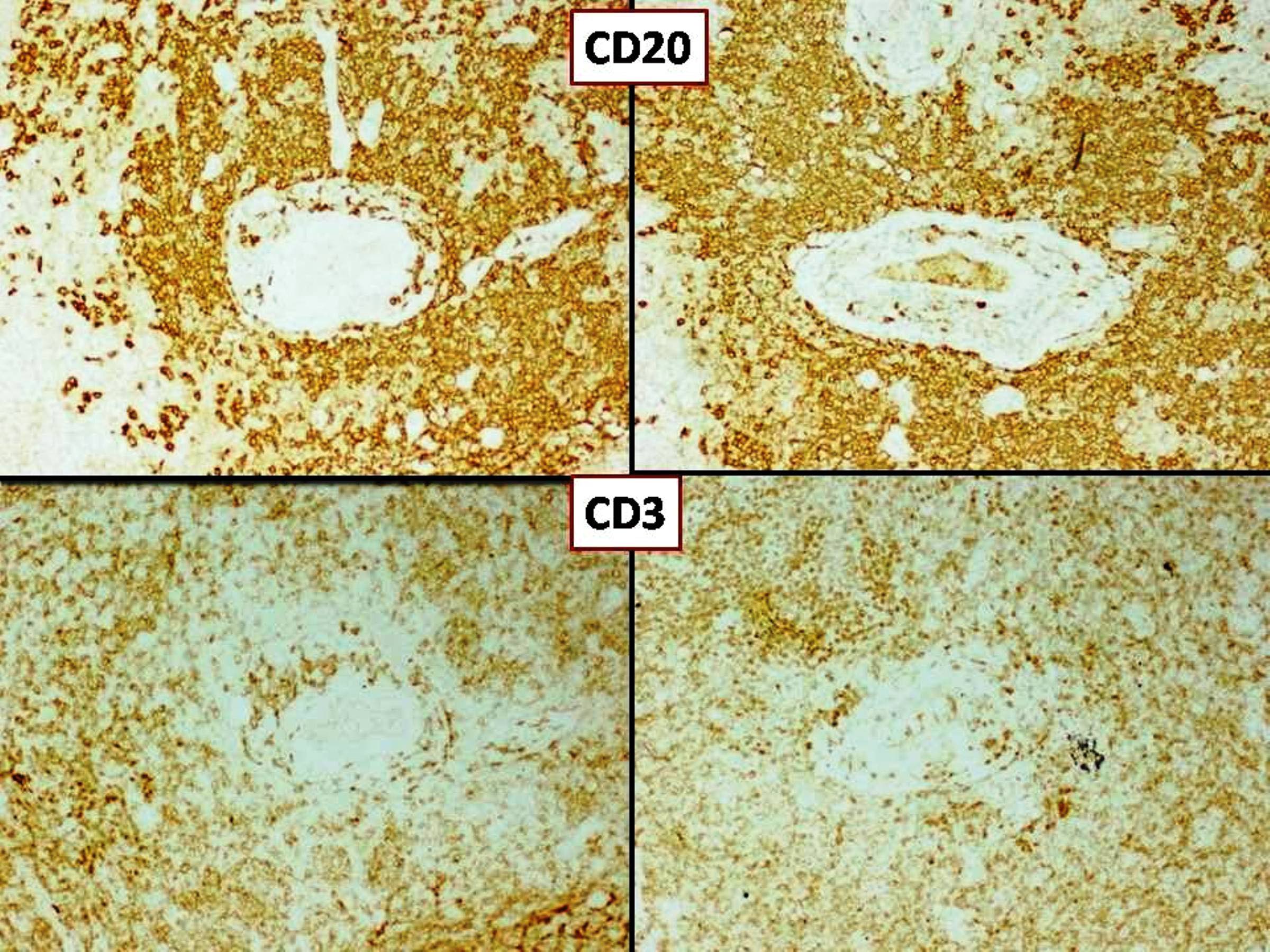
CD20 in tumor cells and CD3

CD30 and EBV LMP1 IHC
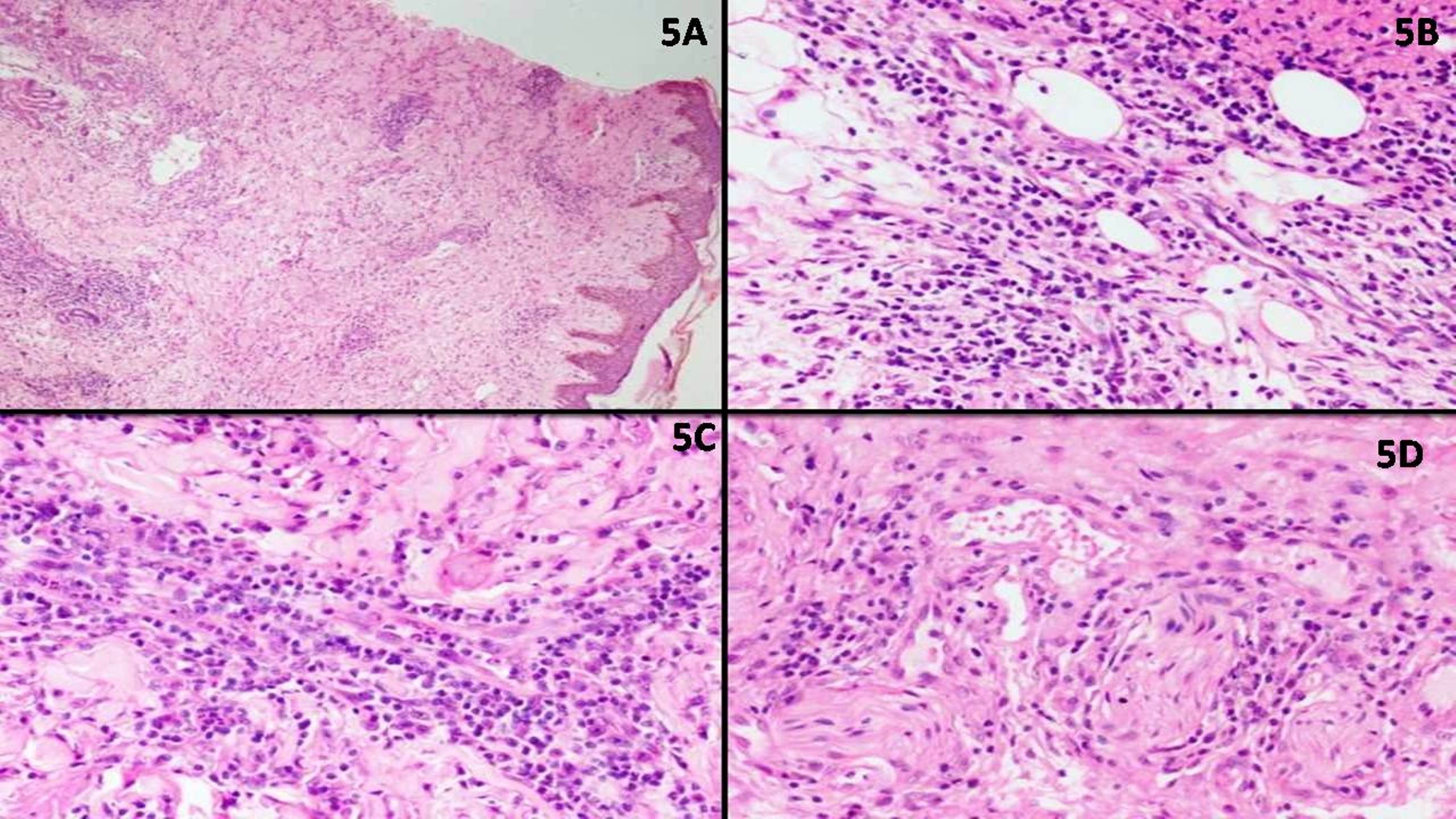
Lymphocytic vasculitis with angioinvasion (skin)
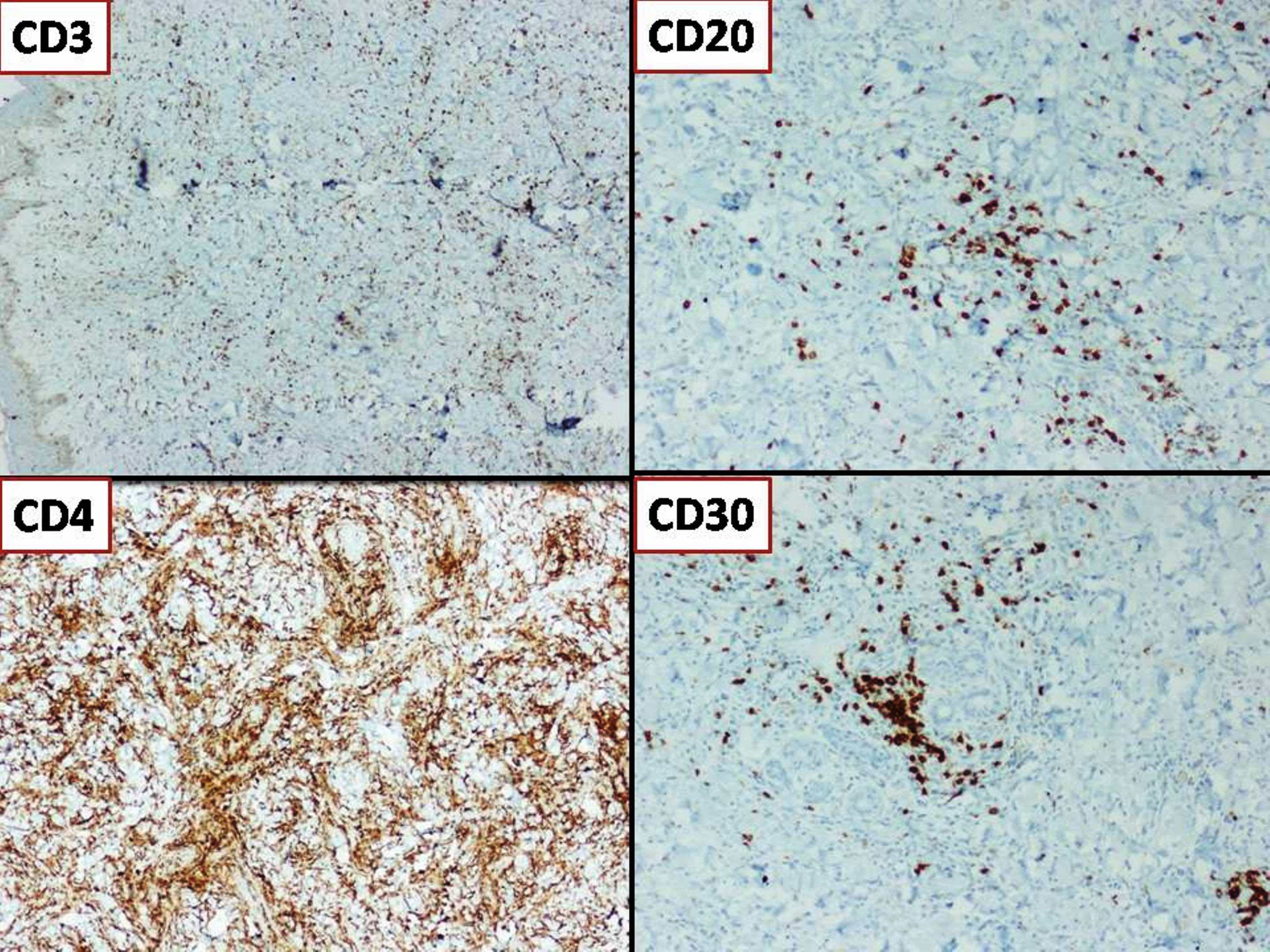
CD20 and CD30 in tumor cells; CD3 and CD4 in reactive T cells
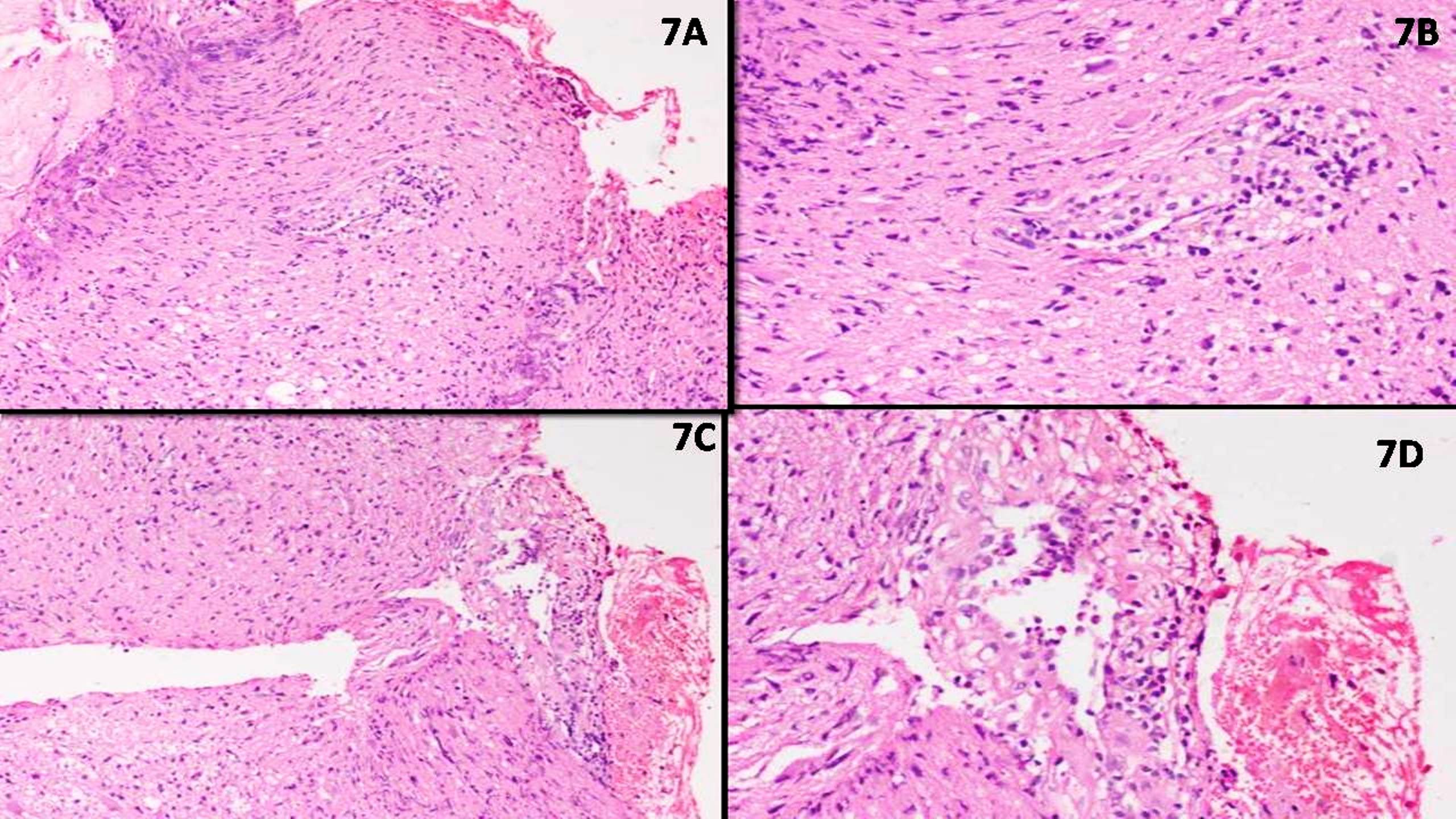
Lymphocytic vasculitis with angioinvasion (CNS)
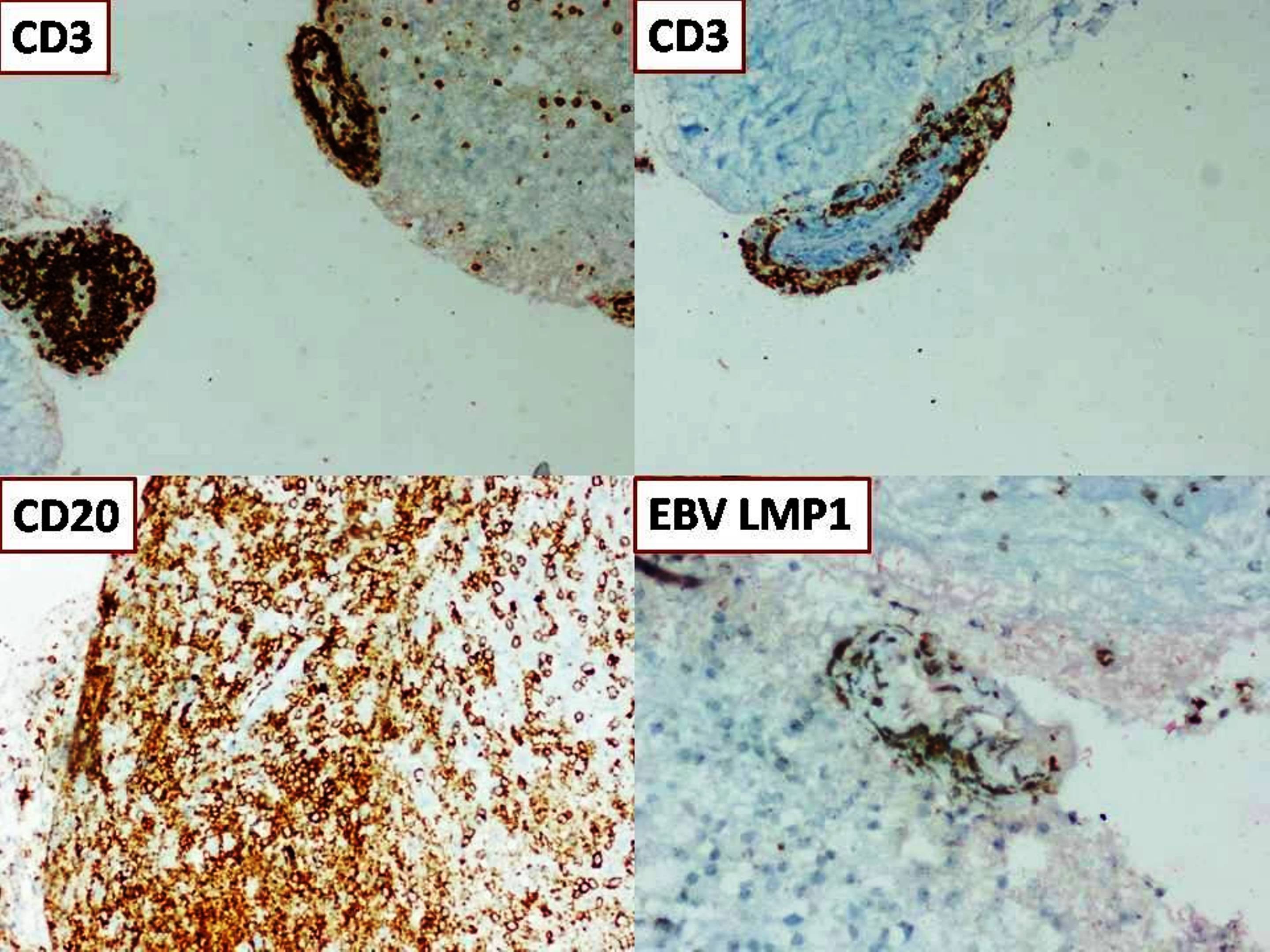
CD20 and EBV LMP1 in tumor cells; CD3 in reactive T cells
- B cell component: CD20, EBV LMP1, EBER, CD30 (variably) (Am J Surg Pathol 1998;22:1093)
- EBNA2 is frequently positive, consistent with latency type III (Am J Surg Pathol 2015;39:141)
- Background T cells: CD3 with CD4 more often than CD8 (Am J Surg Pathol 2015;39:141)
- Most cases of grade II and III lymphomatoid granulomatosis show clonality for IG genes (Am J Surg Pathol 1994;18:753)
- Clonality of grade I lymphomatoid granulomatosis is less consistent; some cases of lymphomatoid granulomatosis might be polyclonal (Am J Surg Pathol 2015;39:141)
- T cell receptor (TR) genes do not show clonality (Hum Pathol 1991;22:1150)
- Lung tissue, bronchoscopic biopsy:
- Lymphomatoid granulomatosis, grade I, biopsies from left lung, upper and lower lobes (see comment)
- Comment: The small reactive lymphoid cells are CD3 positive. The scattered large cells are positive for CD20 and CD79a and show faint membrane positivity for EBV LMP1. The MIB1 proliferation index is low (approximately 10 - 20%). Special stains for microorganisms (AFB TB, PASD, GMS) are negative.
- Microscopic description: Dense interstitial and diffuse infiltrates of small lymphoid cells with round nuclei, clumped chromatin and scattered large lymphoid cells with round nuclei, visible nucleoli and moderate amounts of cytoplasm. Admixed with these are a few aggregates of histiocytes and occasional eosinophils. The lymphoid cells display angioinvasion with transmural infiltration by lymphocytes which adhere to the endothelium with focal luminal occlusion. There are extensive areas of necrosis with necrotic outlines of blood vessels. The adjacent lung tissue shows extensive type II pneumocyte hyperplasia.
- EBV positive diffuse large B cell lymphoma (polymorphous variant):
- Does not show angiocentric and angiodestructive infiltrate
- Extranodal NK / T cell lymphoma:
- Shows angiocentric and angiodestructive infiltrate; atypical cells are T cells / NK cells and not B cells like in lymphomatoid granulomatosis
- Classic Hodgkin lymphoma (CHL):
- Shows polymorphous background; does not show angiocentric and angiodestructive infiltrate
- Tumor cells in classic Hodgkin lymphoma are CD30 and CD15 positive, CD20 and CD45 negative, while tumor cells in lymphomatoid granulomatosis are CD20 and CD45 positive and are negative for CD15
- Classic Reed-Sternberg-like cells are rarely seen in lymphomatoid granulomatosis
- Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL):
- May show background of reactive T cells but nodular lymphocyte predominant Hodgkin lymphoma is a tumor predominantly of the lymph nodes, while lymphomatoid granulomatosis rarely involves lymph nodes
- EBV positivity is a must for lymphomatoid granulomatosis but rare in nodular lymphocyte predominant Hodgkin lymphoma
- Polymorphous lymphoid infiltration associated with immunodeficiency / transplant:
- May show polymorphous background with large atypical cells
- Does not show angiocentric or angioinvasive lymphoid infiltrate
- B lymphocytes
- Histiocytes
- NK cells
- T lymphocytes
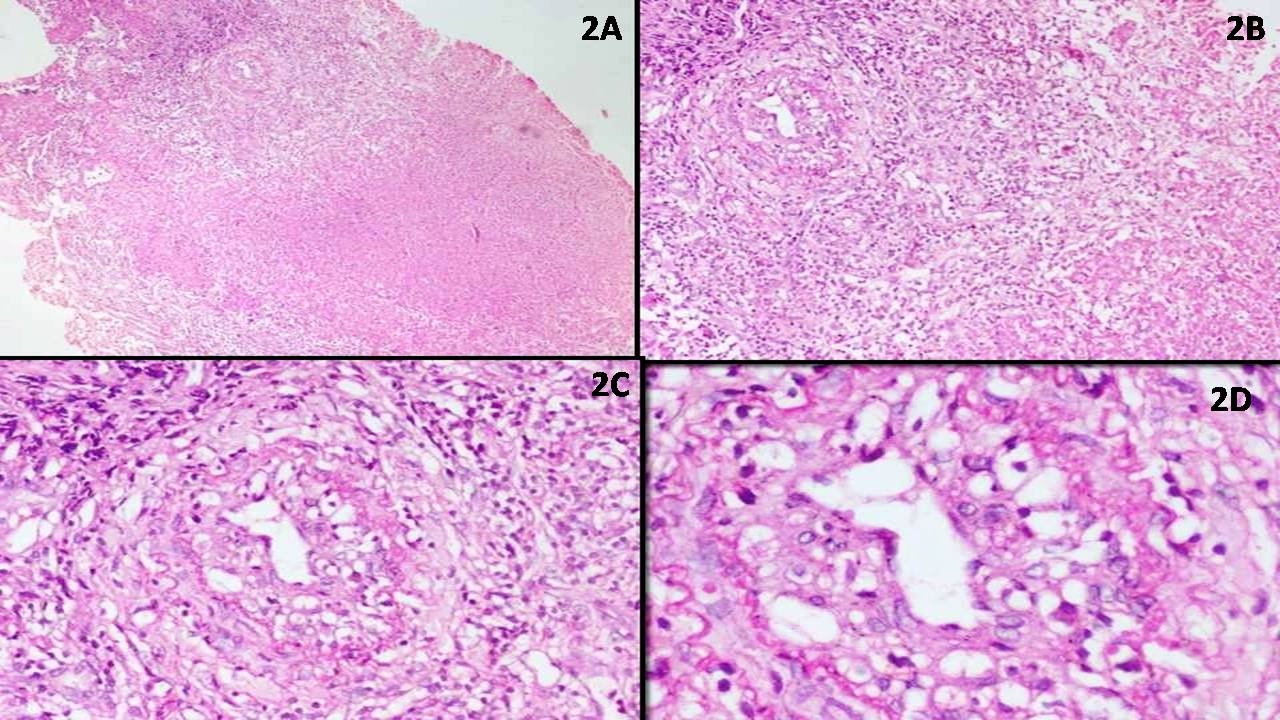
Which of these findings is essential for the diagnosis of lymphomatoid granulomatosis?
- Angioinvasion by tumor cells
- Granulomatous reaction
- Necrosis
- Reed-Sternberg-like cells
- 4 genetically defined groups and 4 histologically defined groups (Nature 2017;547:311)
- Genetically defined: (a) WNT (for wingless integration site) activated, (b) SHH (sonic hedgehog) activated (either TP53 mutant or TP53 wild type), non-WNT / non-SHH, either (c) medulloblastoma group 3 or (d) medulloblastoma group 4 (Nature 2017;547:311)
- Histologically defined: classic, desmoplastic / nodular, medulloblastoma with extensive nodularity and large cell / anaplastic medulloblastoma (Neuropathol Appl Neurobiol 2002;28:257)
- Classic is more common in childhood and desmoplastic / nodular in infants and adults (Neuropathol Appl Neurobiol 2002;28:257)
- Small, round undifferentiated cells with mild to moderate nuclear pleomorphism (Neuropathol Appl Neurobiol 2002;28:257)
- High mitotic count (Neuropathol Appl Neurobiol 2002;28:257)
- Cerebellum and paratentorial brain, fourth ventricle (Neuropathol Appl Neurobiol 2002;28:257)
- Medulloblastoma is the most common CNS embryonal tumor of childhood and is second only to pilocytic astrocytoma for all intracranial neoplasms (Acta Neuropathol 2012;123:473)
- Classic medulloblastoma (Acta Neuropathol 2012;123:473)
- Non-WNT / non-SHH tumors or WNT activated
- Midline location
- Desmoplastic / nodular medulloblastoma (Acta Neuropathol 2012;123:473)
- Cerebellar hemispheres and midline
- Bimodal age distribution
- Gorlin syndrome: nevoid basal cell carcinoma syndrome
- SHH activated
- Favorable outcome in young children compared to nondesmoplastic
- Medulloblastoma with extensive nodularity (Acta Neuropathol 2012;123:473)
- Closely related to desmoplastic medulloblastoma
- SHH activated
- Predominantly occurs in infants
- Excellent outcome
- Large cell / anaplastic medulloblastoma (Acta Neuropathol 2012;123:473)
- Undifferentiated cells with marked nuclear pleomorphism, prominent nucleoli, cell wrapping and high mitotic count and apoptotic counts
- Associated with all genetic variants but more common in SHH activated and in group 3
- Fourth ventricle or cerebellar parenchyma
- Signs and symptoms of increased intracranial pressure (headache, nausea, vomiting)
- Diagnosis of medulloblastoma requires histologic examination of tumor tissue and molecular genetic analyses
- Integrated pathology diagnosis that includes both morphologic subtype and molecular subgroup along with any additional molecular data is recommended (Pediatr Dev Pathol 2022;25:23)
- Solid, intensely contrast enhancing masses on CT and MRI (Neurochirurgie 2021;67:6)
- 3 prognostic subgroups (high risk, low risk and standard risk) depending on (Neuro Oncol 2021;23:1163)
- Clinical findings: age at diagnosis, presence or absence of metastases at diagnosis, extent of resection of the tumor (Neuro Oncol 2021;23:1163)
- Histopathological findings: histological group (large cell / anaplastic = poor prognosis; desmoplastic / nodular = good prognosis), presence of tumor cells in the cerebrospinal fluid (Neuro Oncol 2021;23:1163)
- Biological / molecular findings: immunohistochemical / genetic subgroup (non-WNT / non-SHH = groups 3 and 4 having a poor prognosis; WNT activated = good prognosis), SHH activated TP53 mutation (associated with a poorer prognosis), MYC / MYCN amplification (associated with a poorer prognosis) (Neuro Oncol 2021;23:1163)
- 2 year old boy who presented with a midline cerebellar mass (Neuropathology 2016;36:372)
- 4 year old boy with a cerebellar hemispheric mass (Pediatr Blood Cancer 2016;63:1128)
- 6 year old boy with a mass lesion located in the upper middle cerebellum (J Neuropathol Exp Neurol 2021;80:205)
- Depends on risk stratification
- Total resection
- Chemotherapy and radiation therapy
- Reference: J Neurooncol 2022;157:37
Images hosted on other servers:

Cerebellar tumor extending into fourth ventricle
- Classic medulloblastoma (Neuropathol Appl Neurobiol 2002;28:257)
- Small, blue, round cell tumor
- Syncytial arrangement of densely packed undifferentiated cells (embryonal cells)
- Mitosis with apoptotic bodies
- Homer Wright rosettes
- Desmoplastic / nodular medulloblastoma (Neuropathol Appl Neurobiol 2002;28:257)
- Densely packed, undifferentiated cells with hyperchromatic and pleomorphic nuclei that produce dense intercellular reticulin fiber network with nodular reticulin free zones
- Medulloblastoma with extensive nodularity (Neuropathol Appl Neurobiol 2002;28:257)
- Expanded lobular architecture due to reticulin free nodular zones becoming enlarged and rich in neuropil-like tissue
- Large cell / anaplastic medulloblastoma (Neuropathol Appl Neurobiol 2002;28:257)
- Anaplasia with marked nuclear pleomorphism, high mitotic count and high apoptotic count
- Nuclear molding and cell wrapping
- Medulloblastomas with melanotic or myogenic differentiations (Neuropathol Appl Neurobiol 2002;28:257)
Contributed by Arnault Tauziede-Espariat, M.D., John DeWitt, M.D., Ph.D., Meaghan Morris, M.D., Ph.D.,
Nirupama Singh, M.D., Ph.D. and Eman Abdelzaher, M.D., Ph.D.
Genetically defined
WNT (for wingless integration site) activated
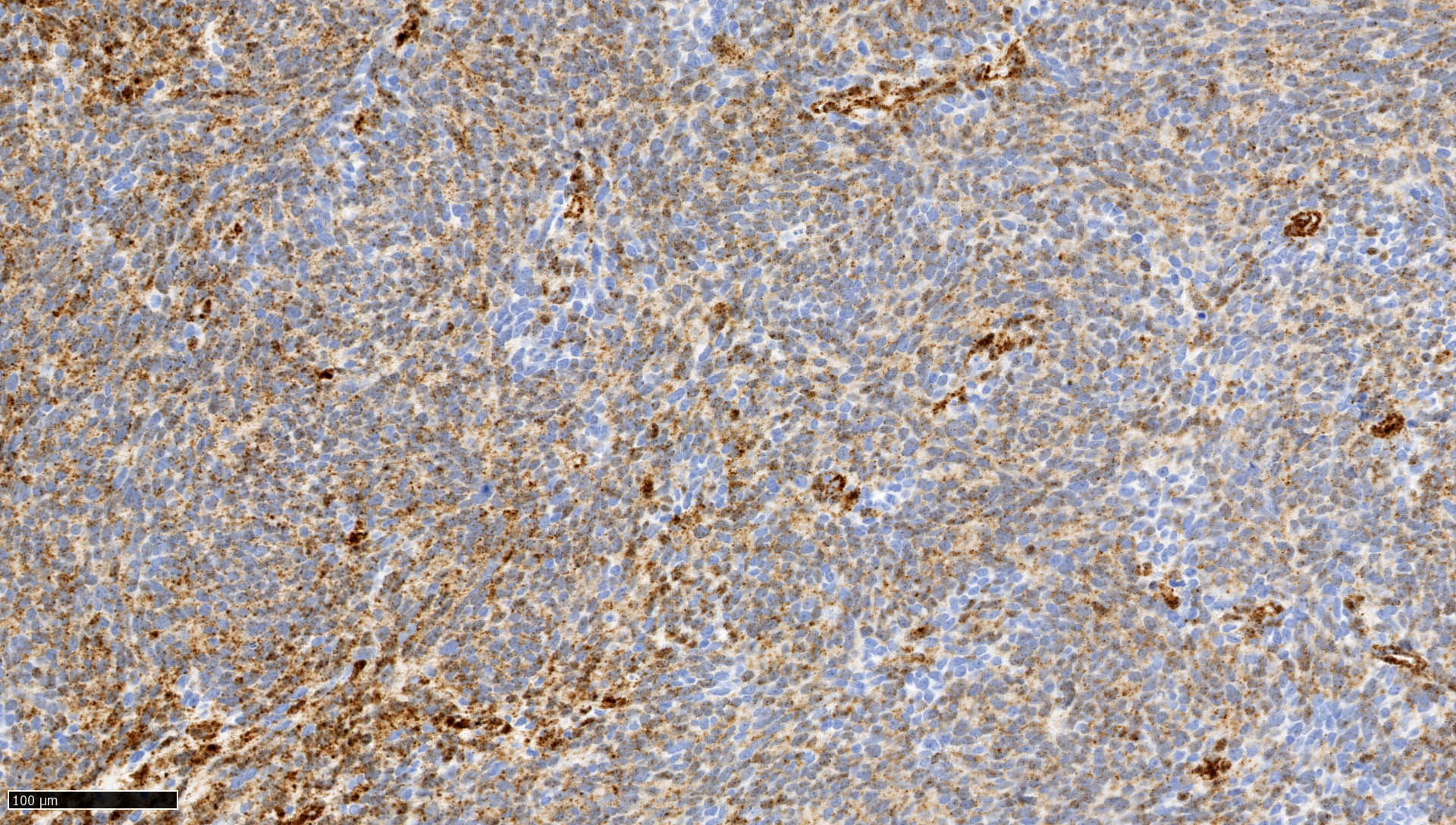
YAP1 immunoreactivity
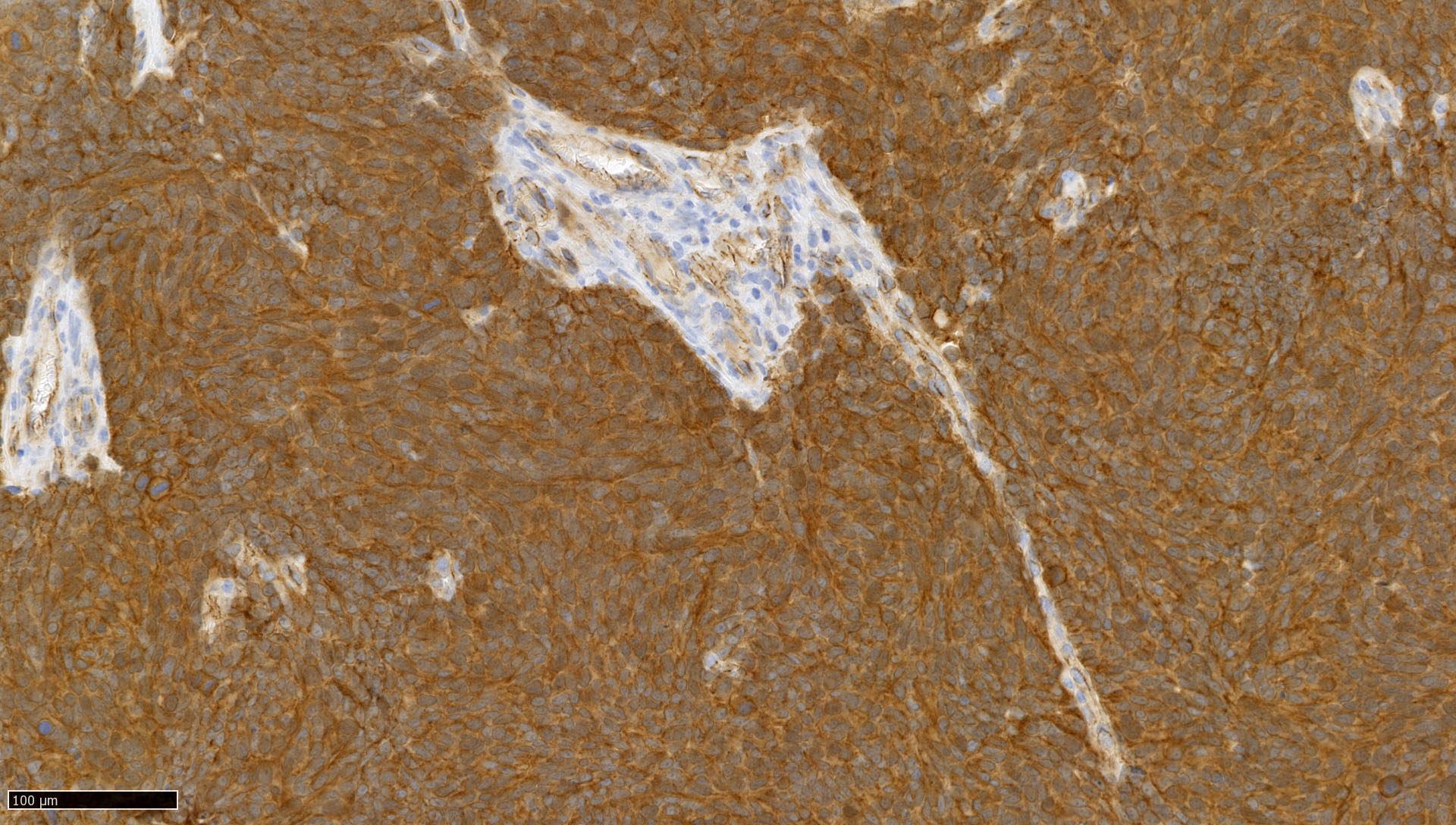
Beta catenin immunoreactivity
SHH (sonic hedgehog) activated (either TP53 mutant or TP53 wild type)
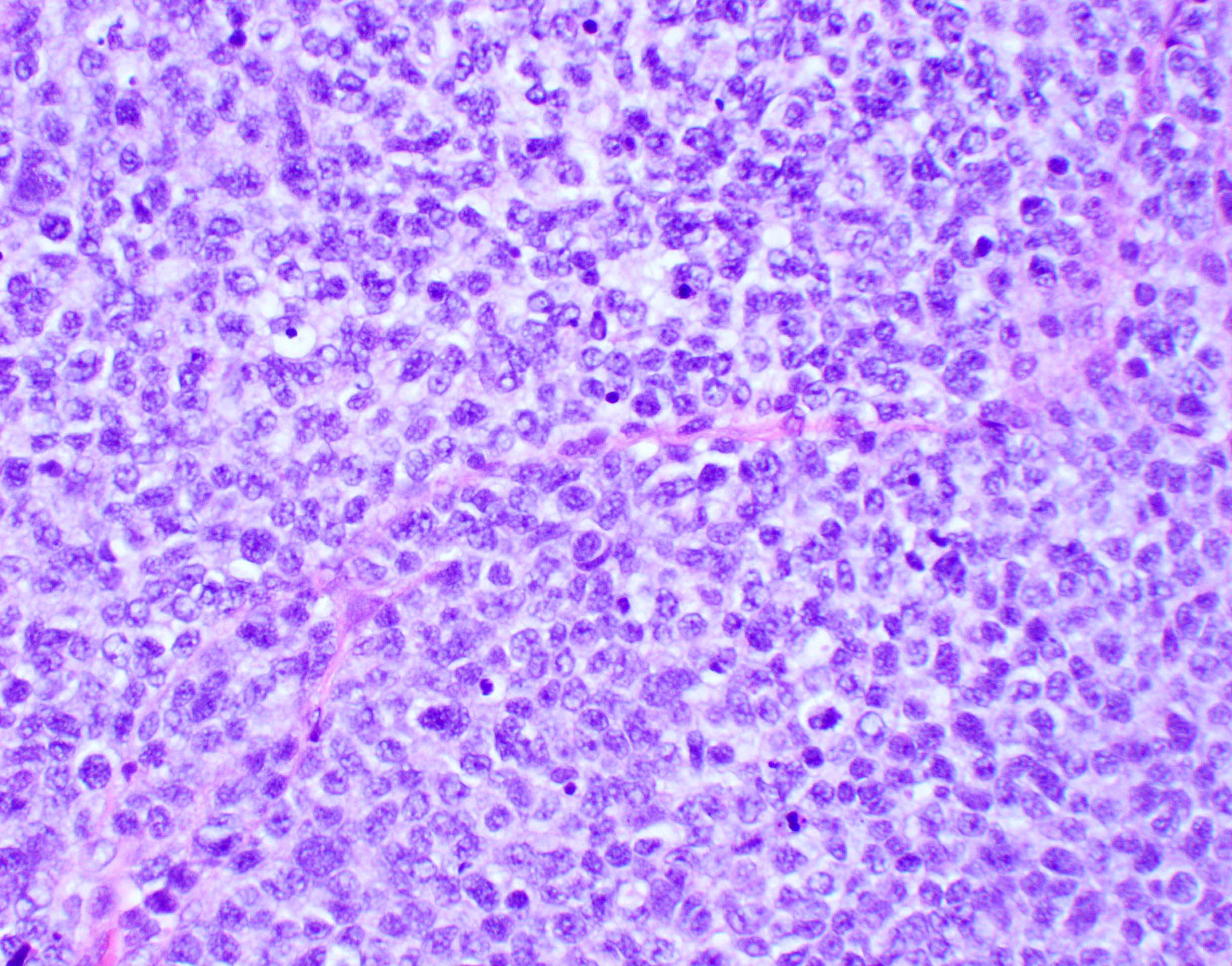
Sheets of undifferentiated cells
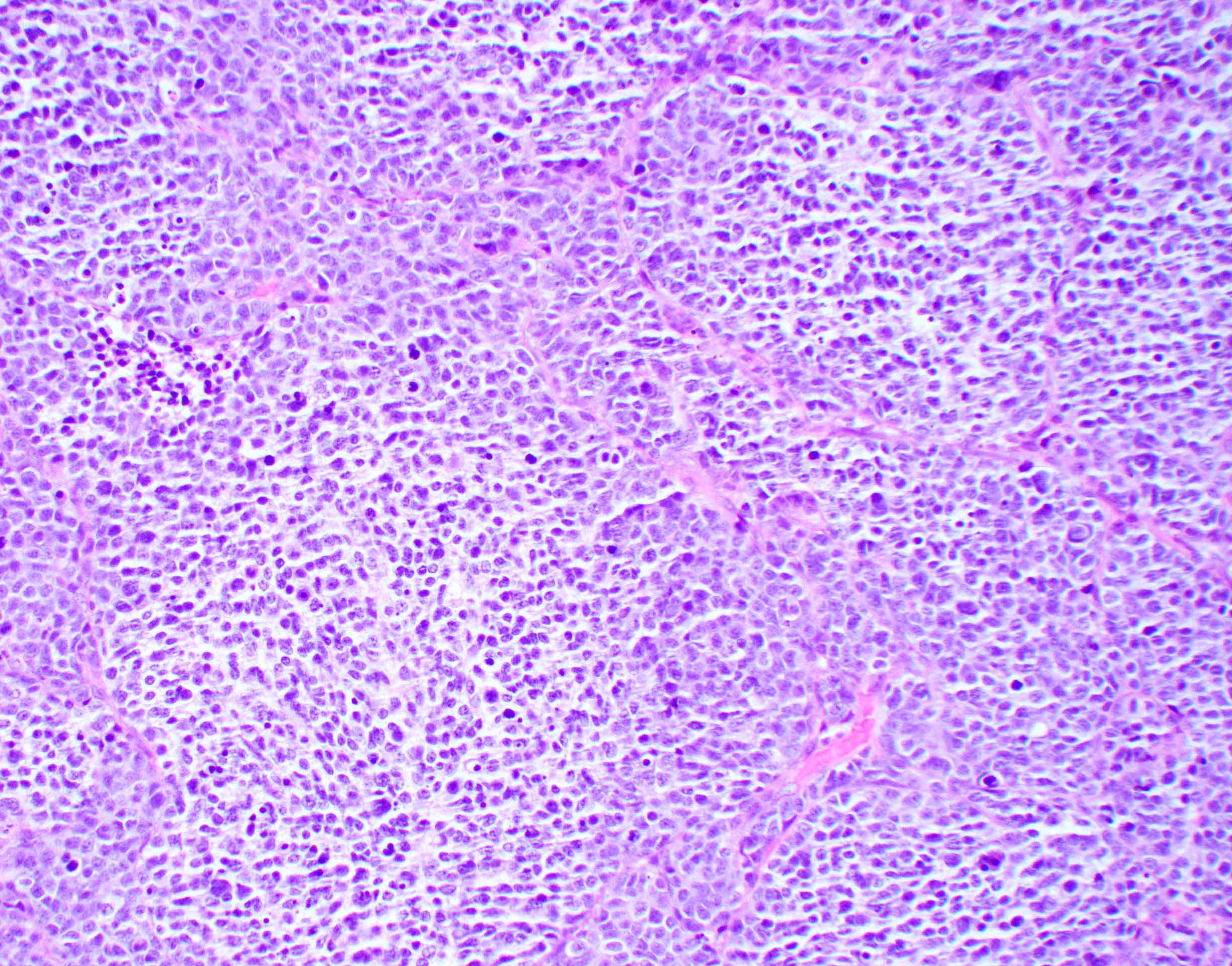
Vague nodular formation
Vague nodular formation with reticulin
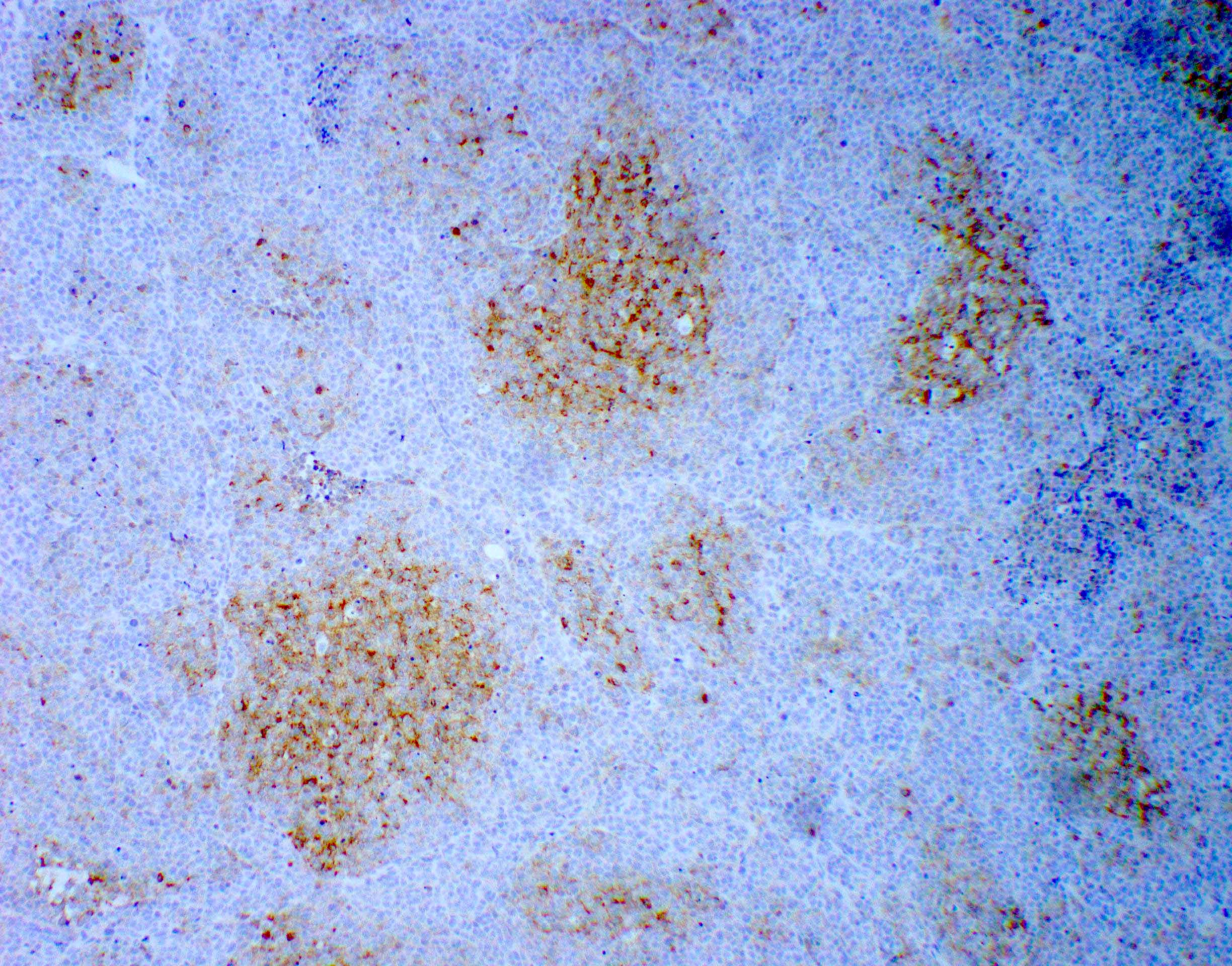
Neuron specific enolase (NSE)
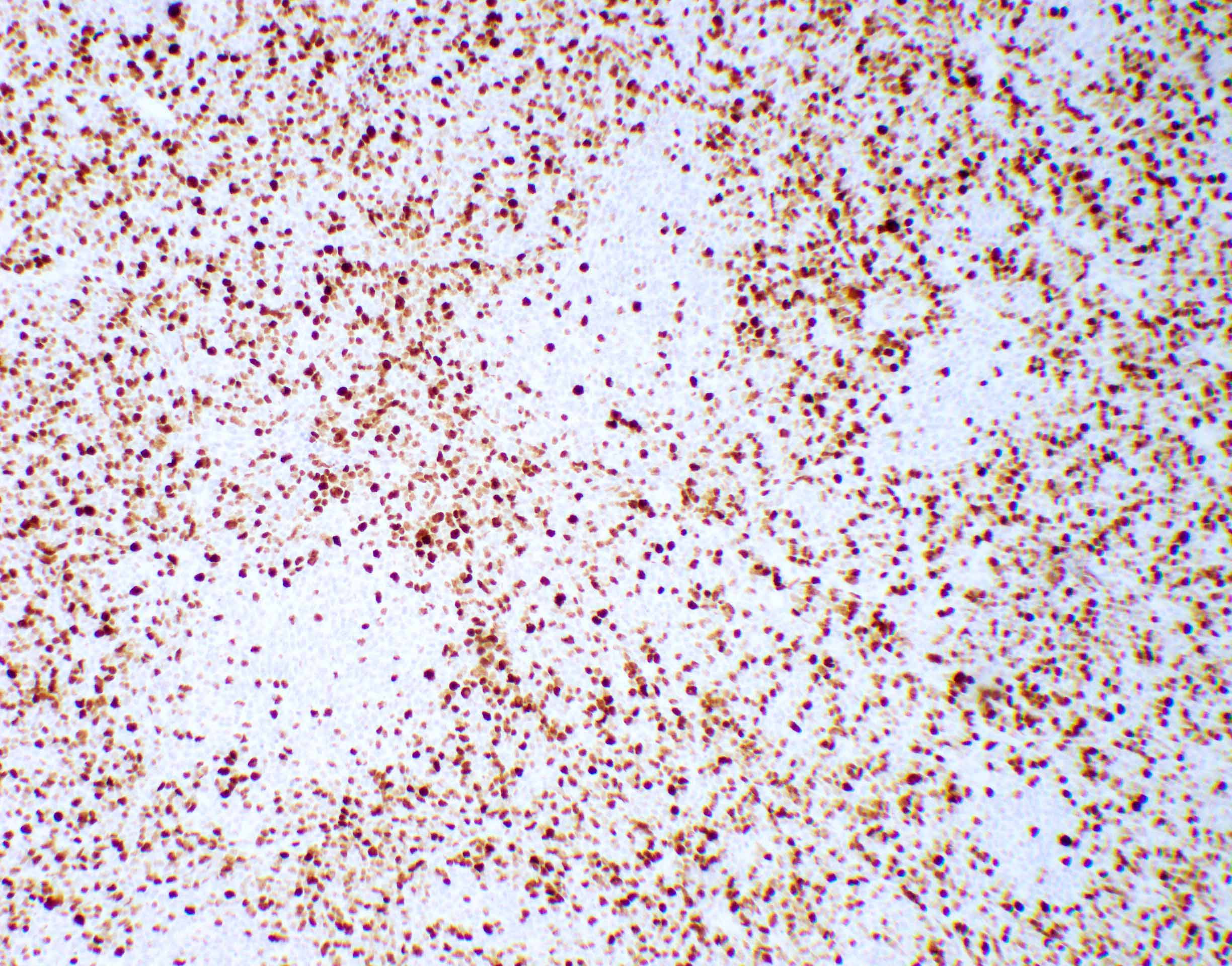
Ki67
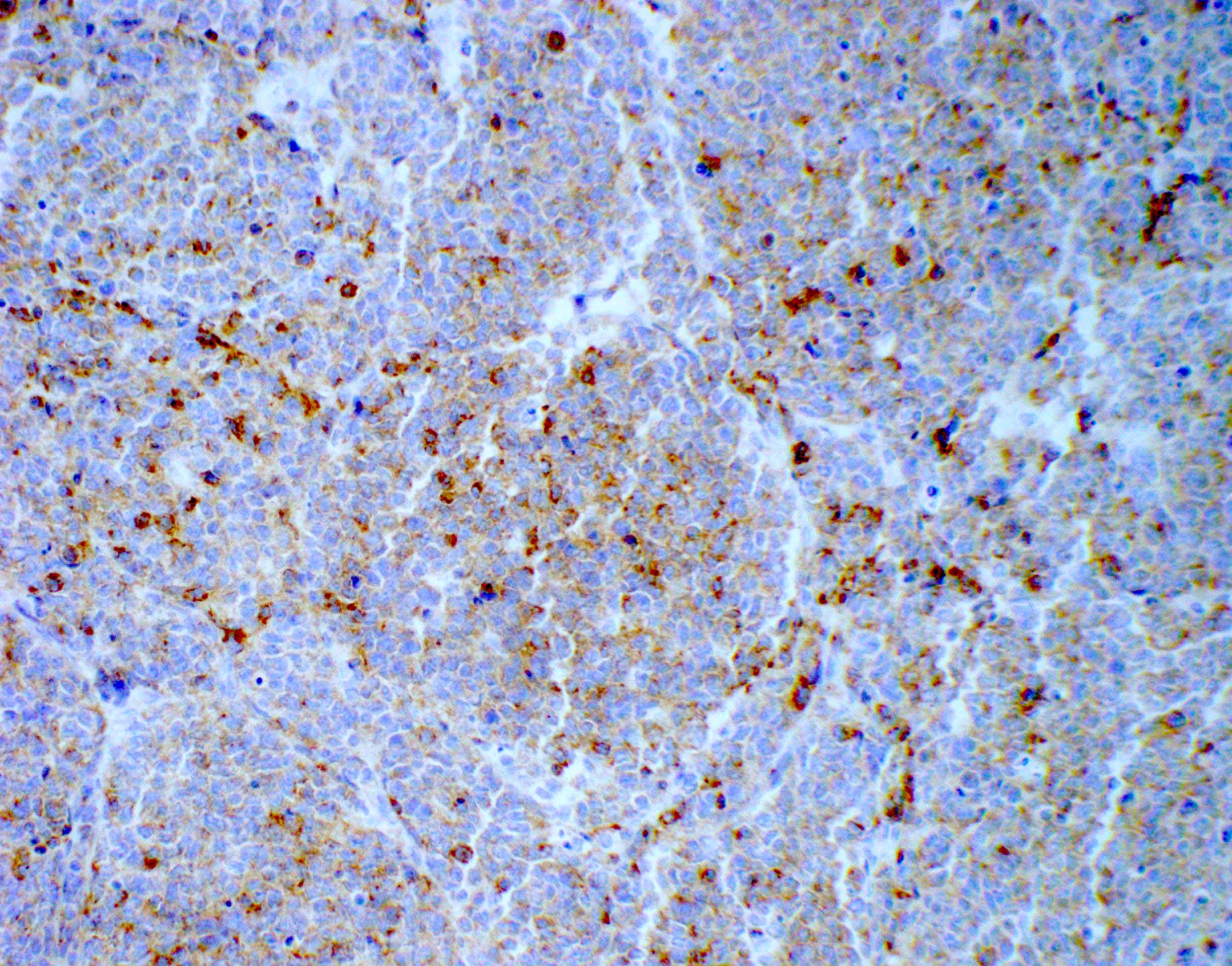
Synaptophysin
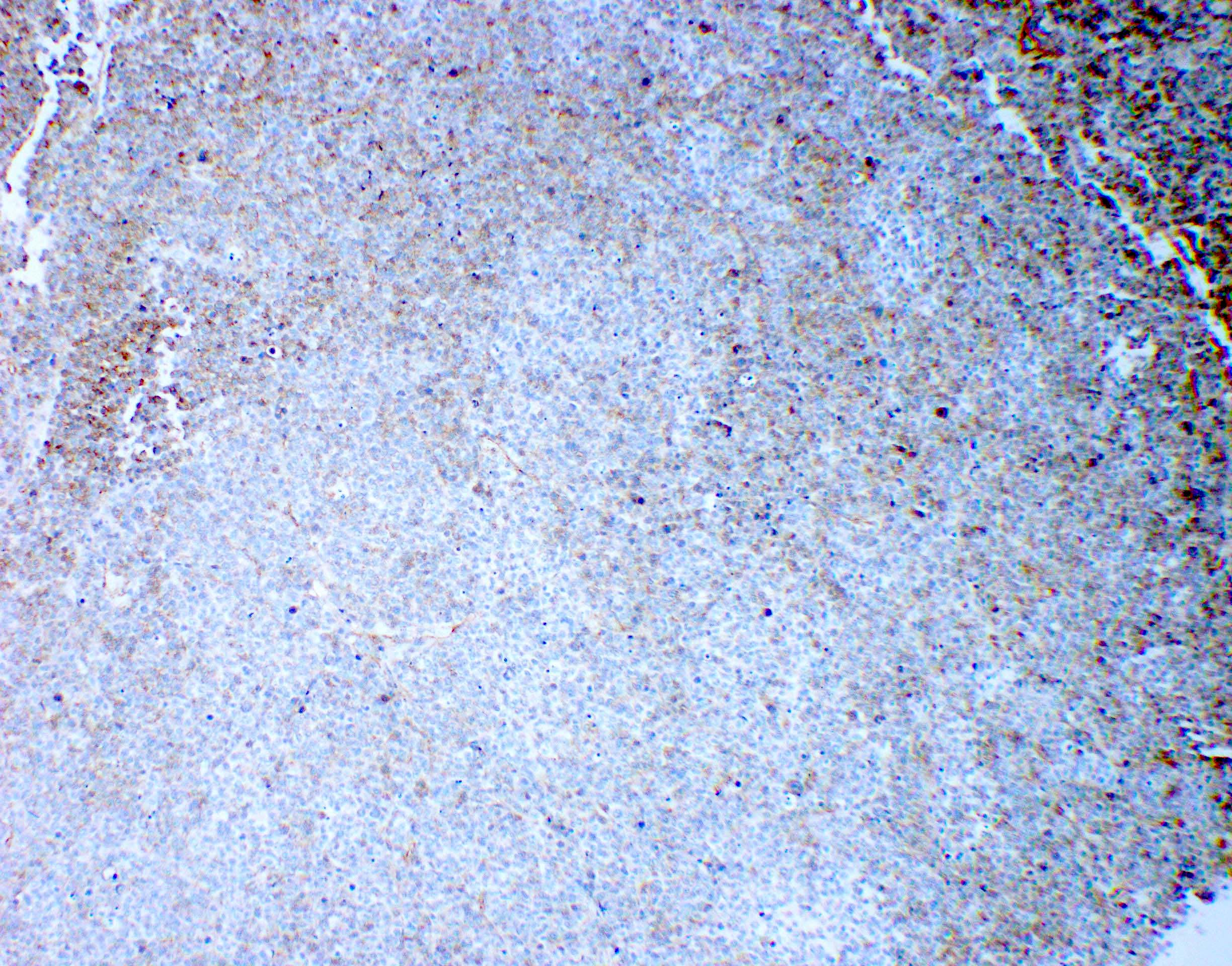
Beta catenin
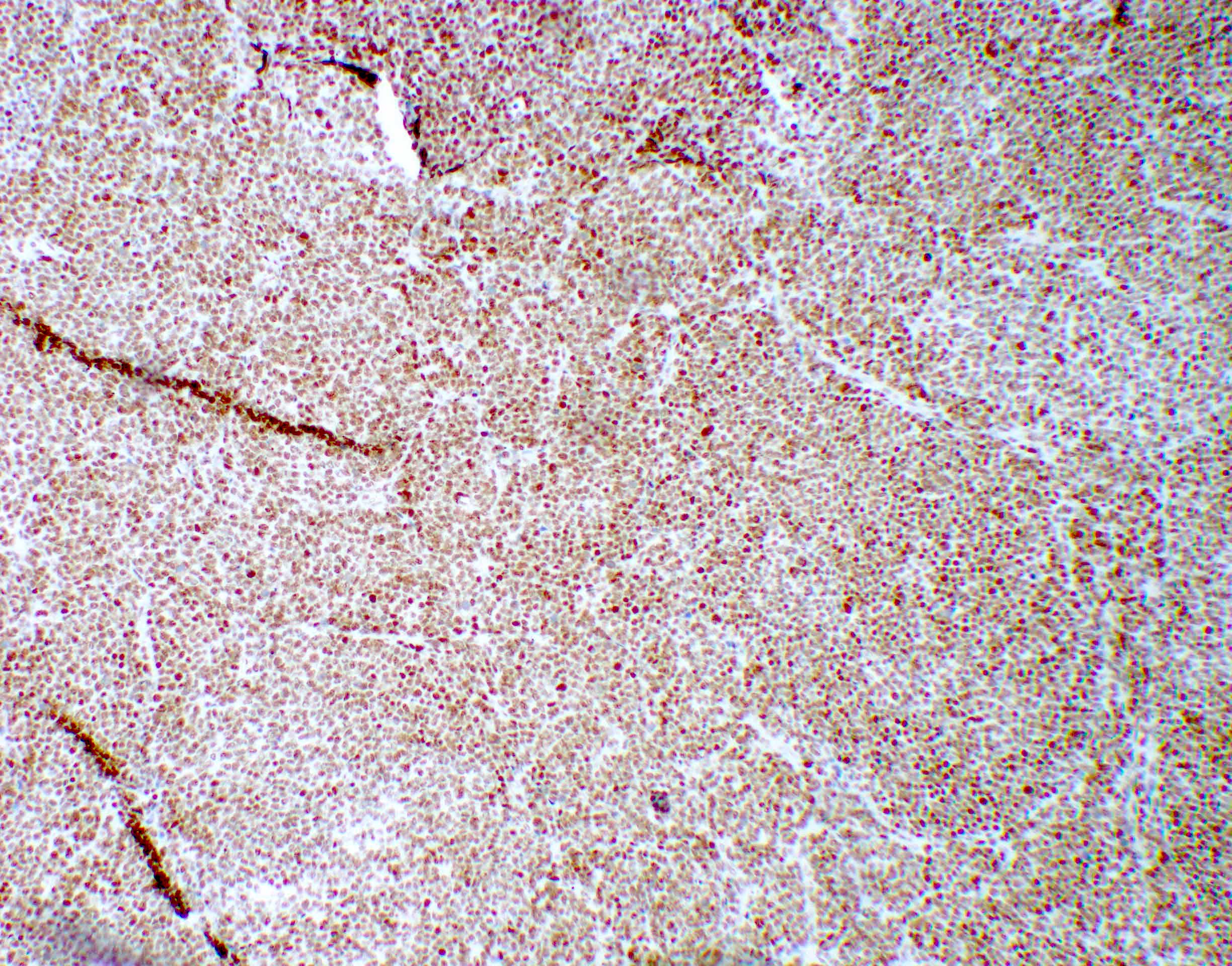
INI1
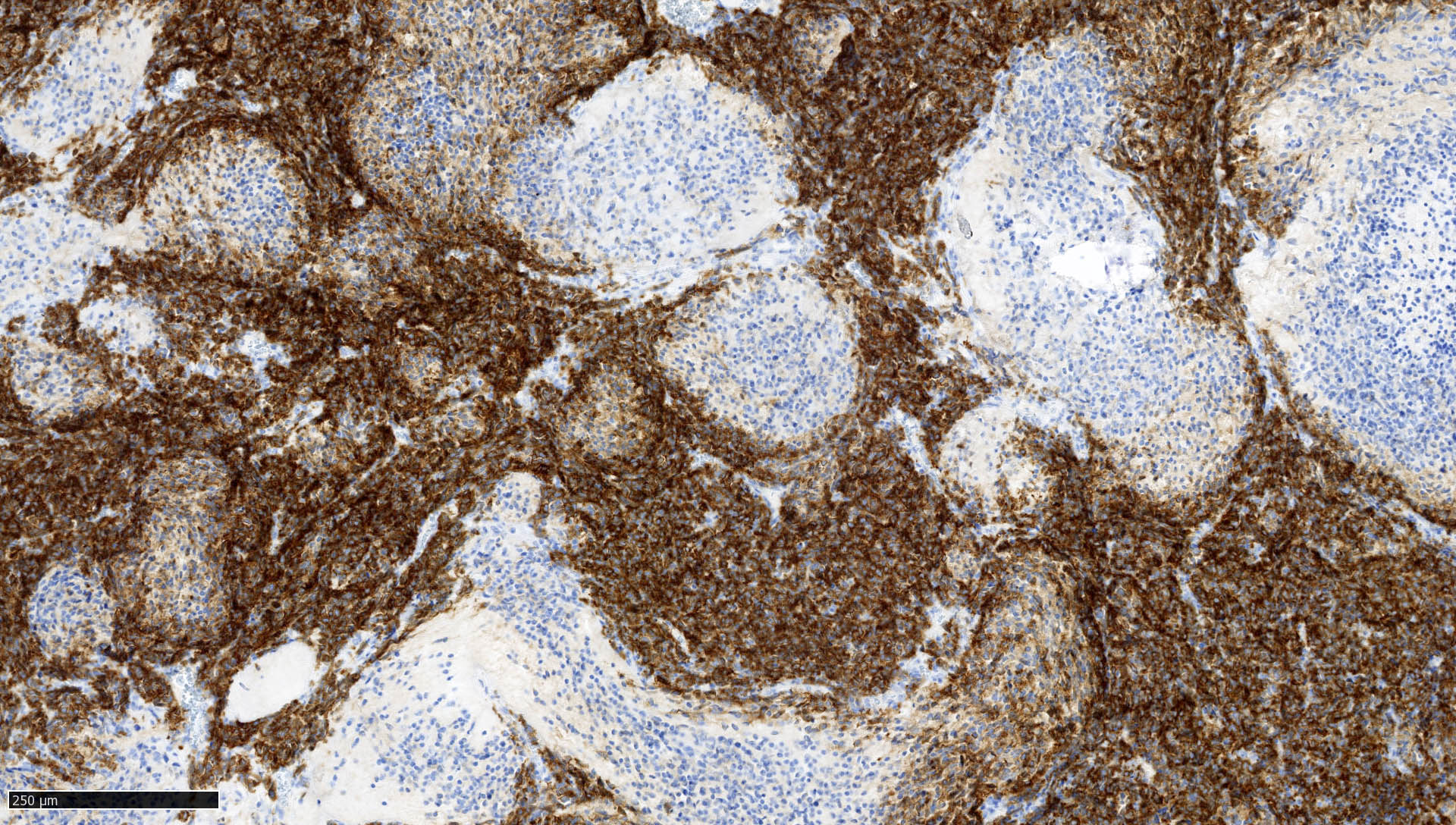
YAP1 immunoreactivity
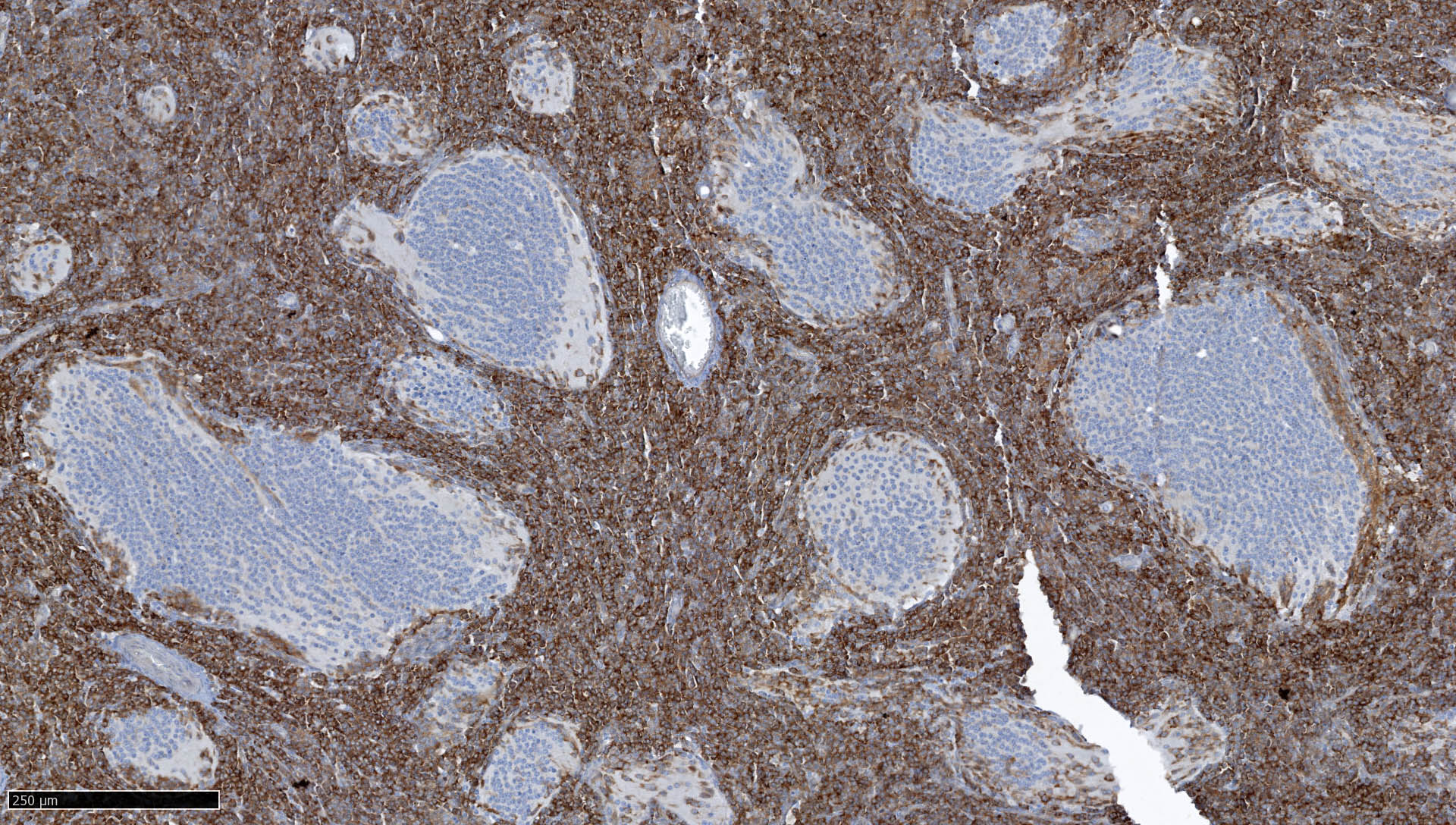
GAB1 immunoreactivity
OTX2 immunoreactivity
Histologically defined
Classic
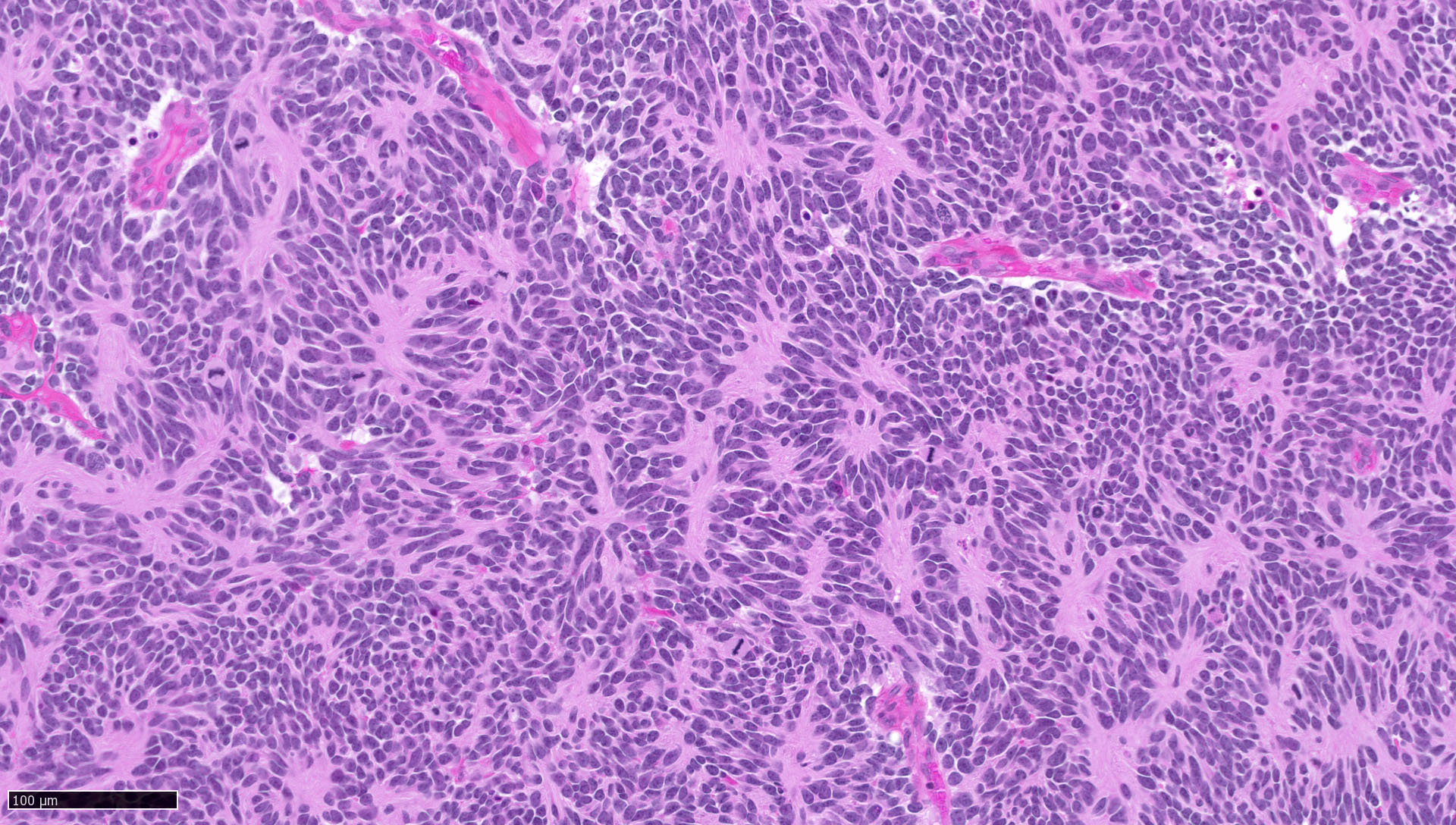
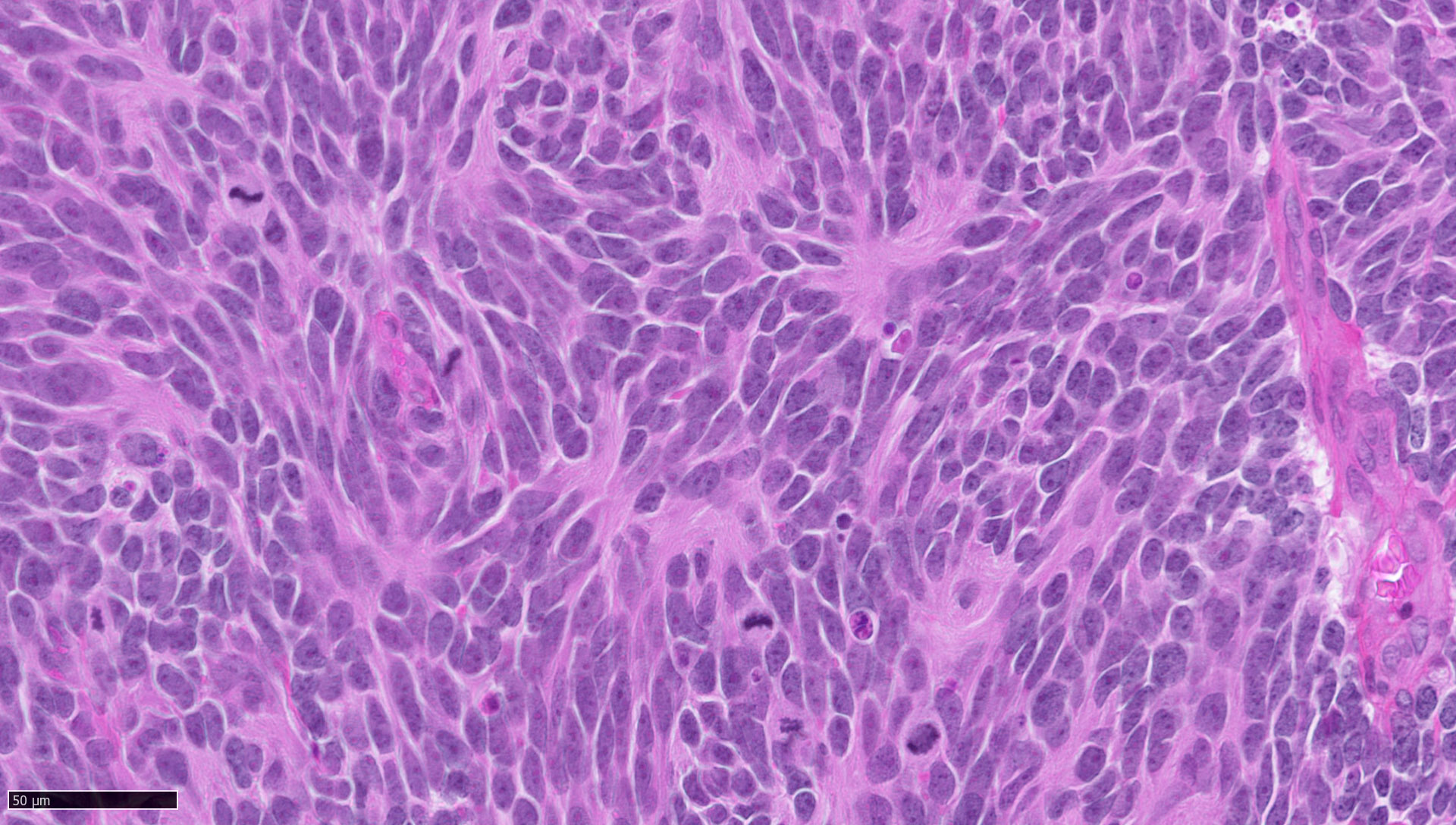
Homer Wright rosettes
Desmoplastic / nodular medulloblastoma
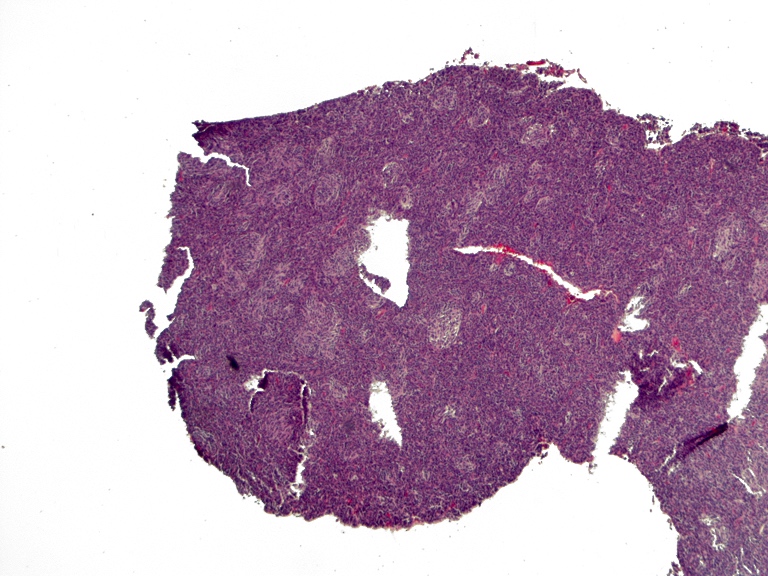
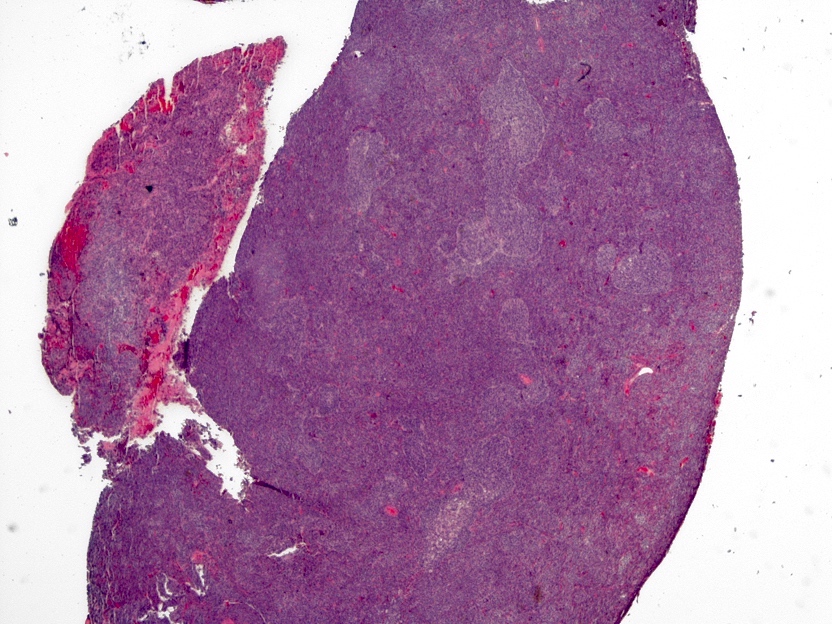
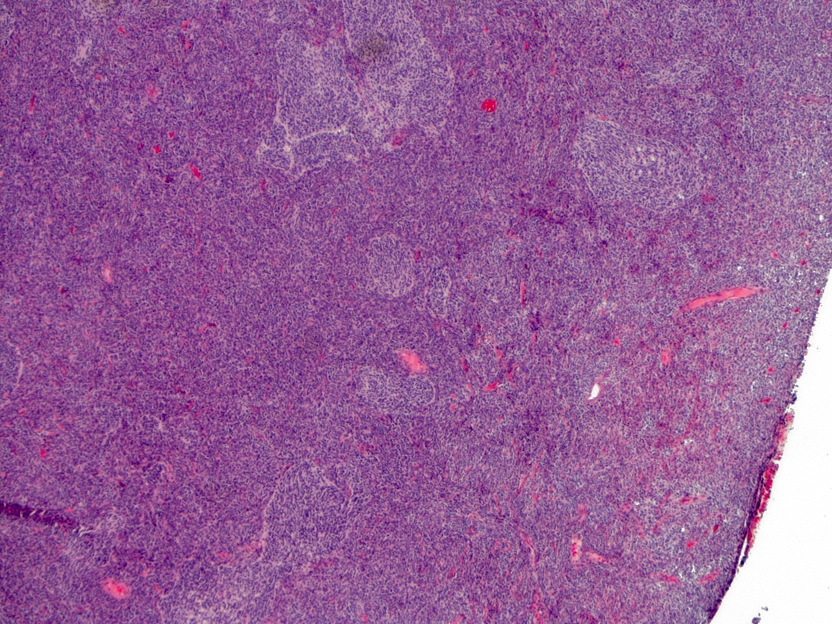
Desmoplastic medulloblastoma
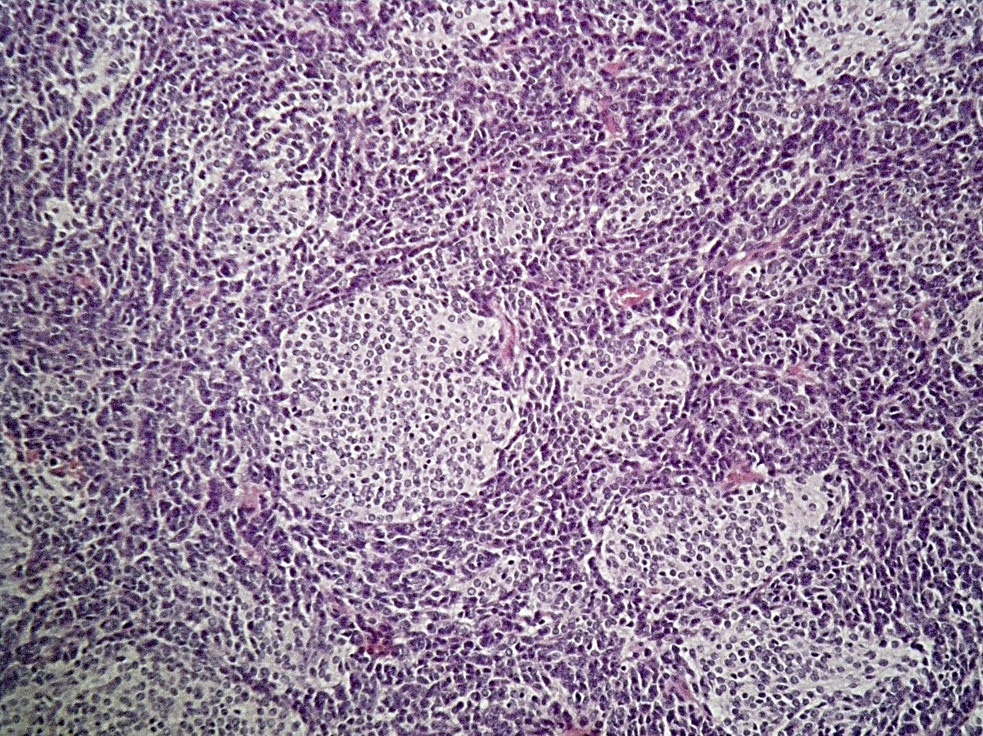
Pale nodular islands
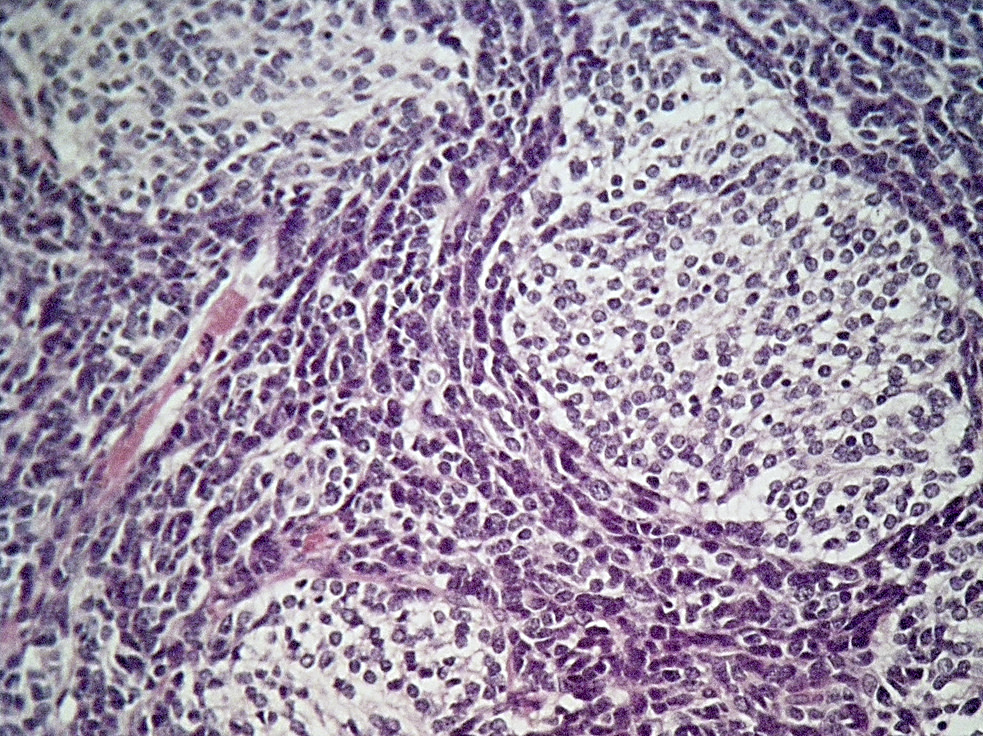
Pale islands formed of uniform cells
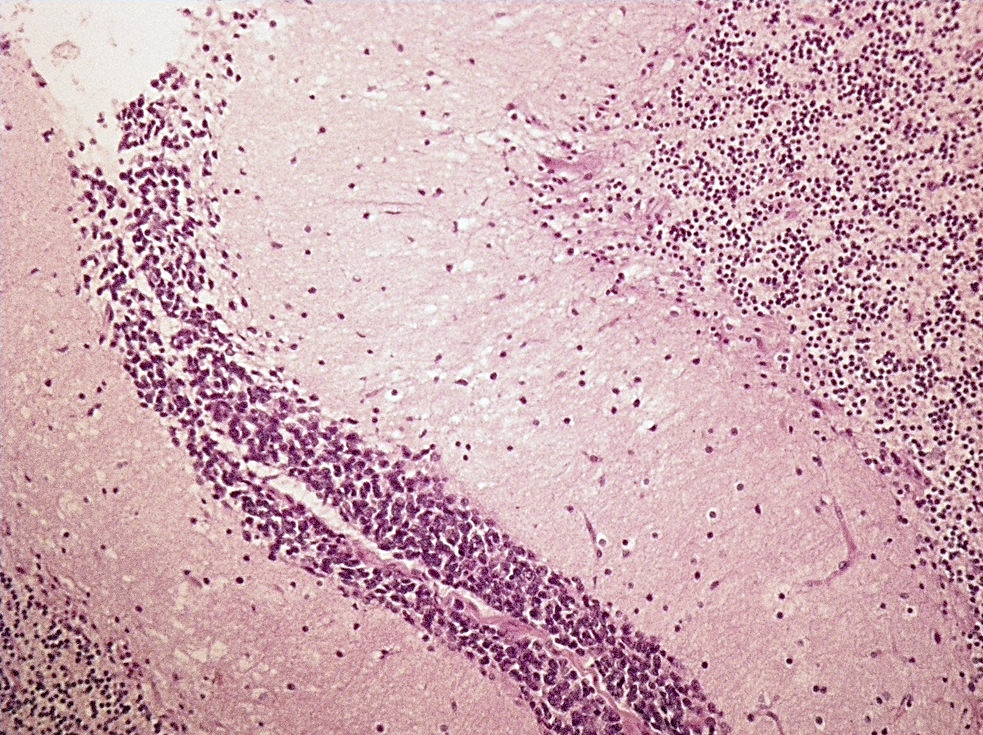
Tumor in the subarachnoid space
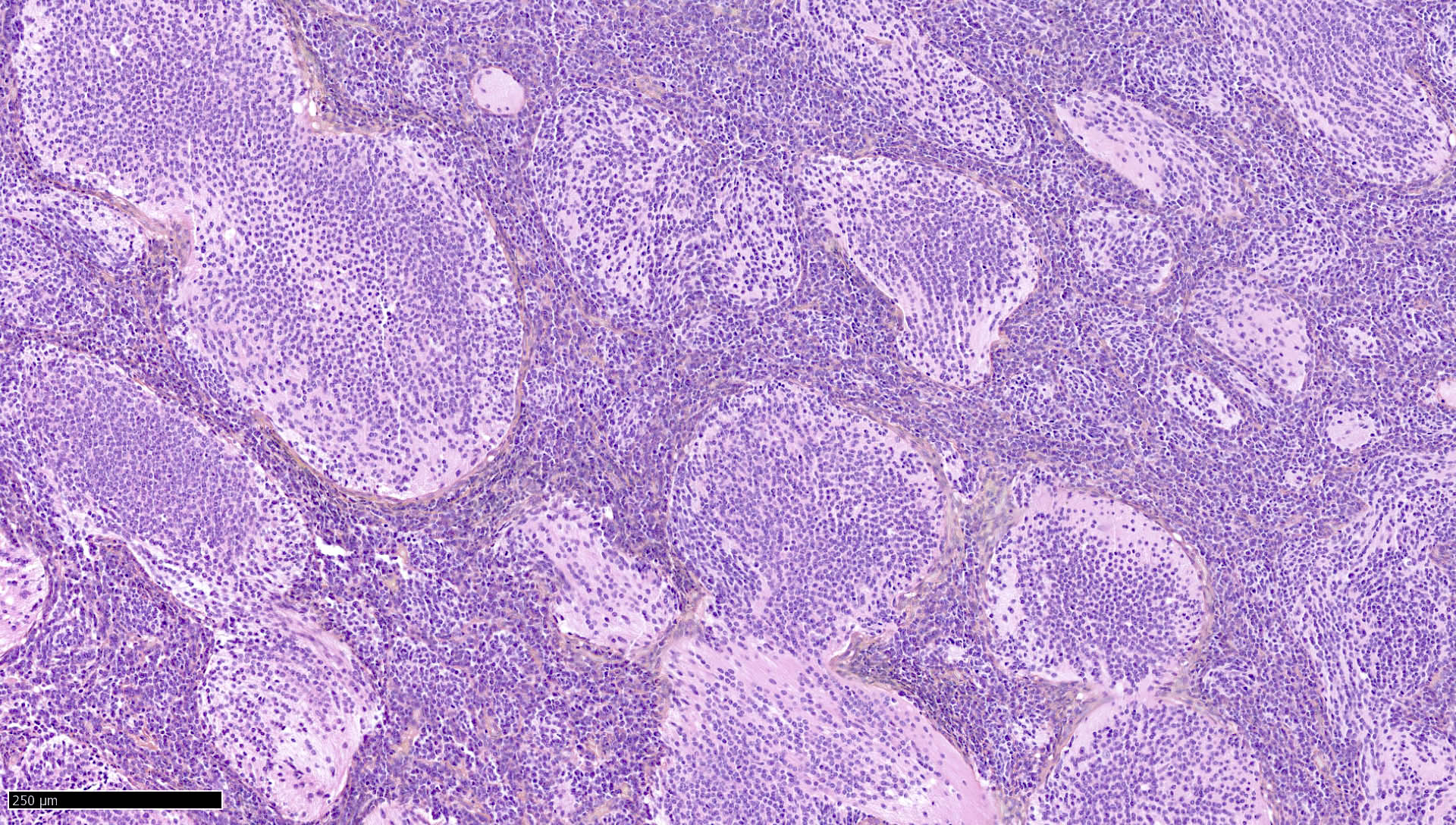
Desmoplastic / nodular medulloblastoma
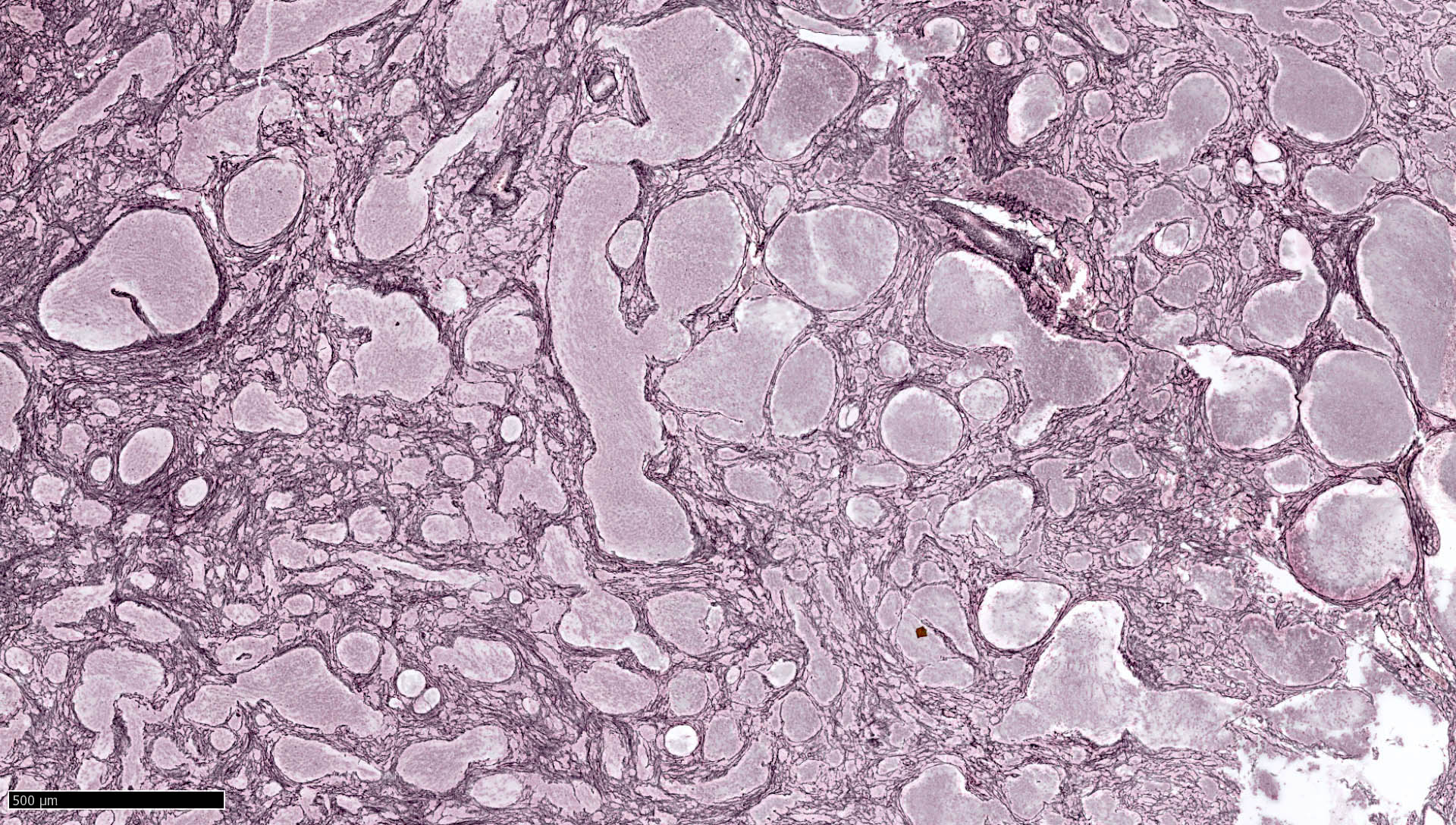
Reticulin fiber network
Medulloblastoma with extensive nodularity
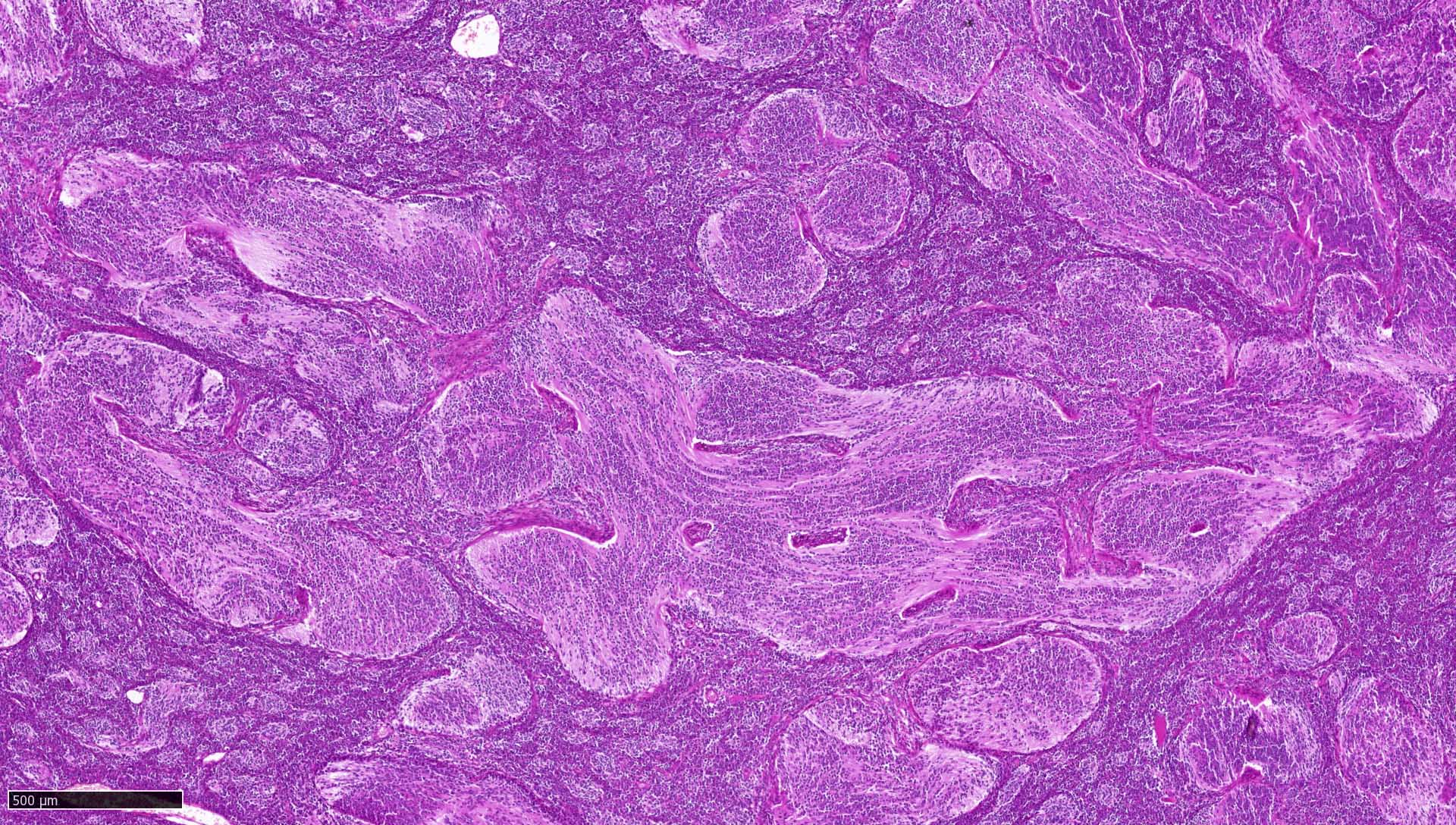
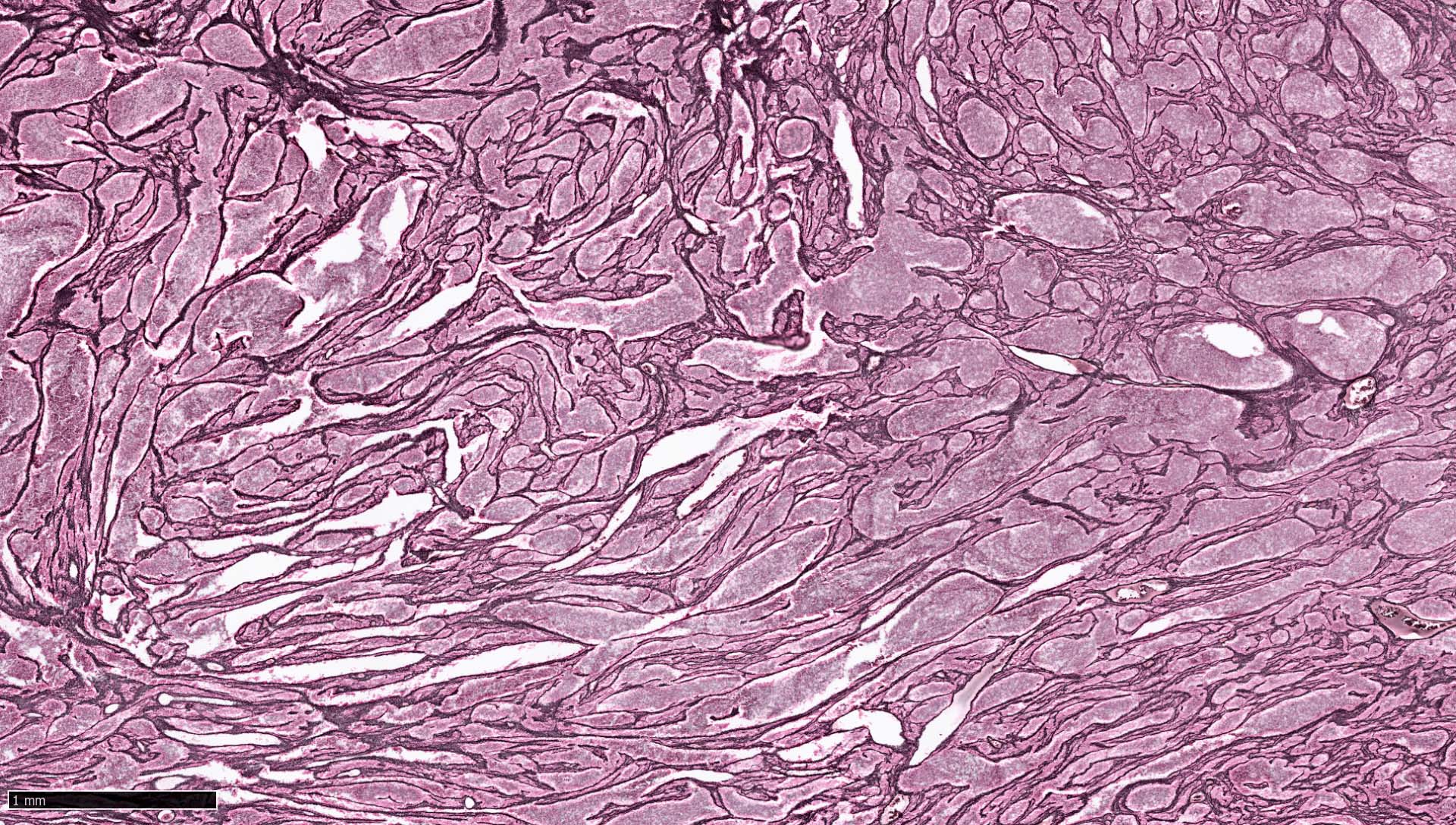
Extensive nodularity
Large cell / anaplastic medulloblastoma
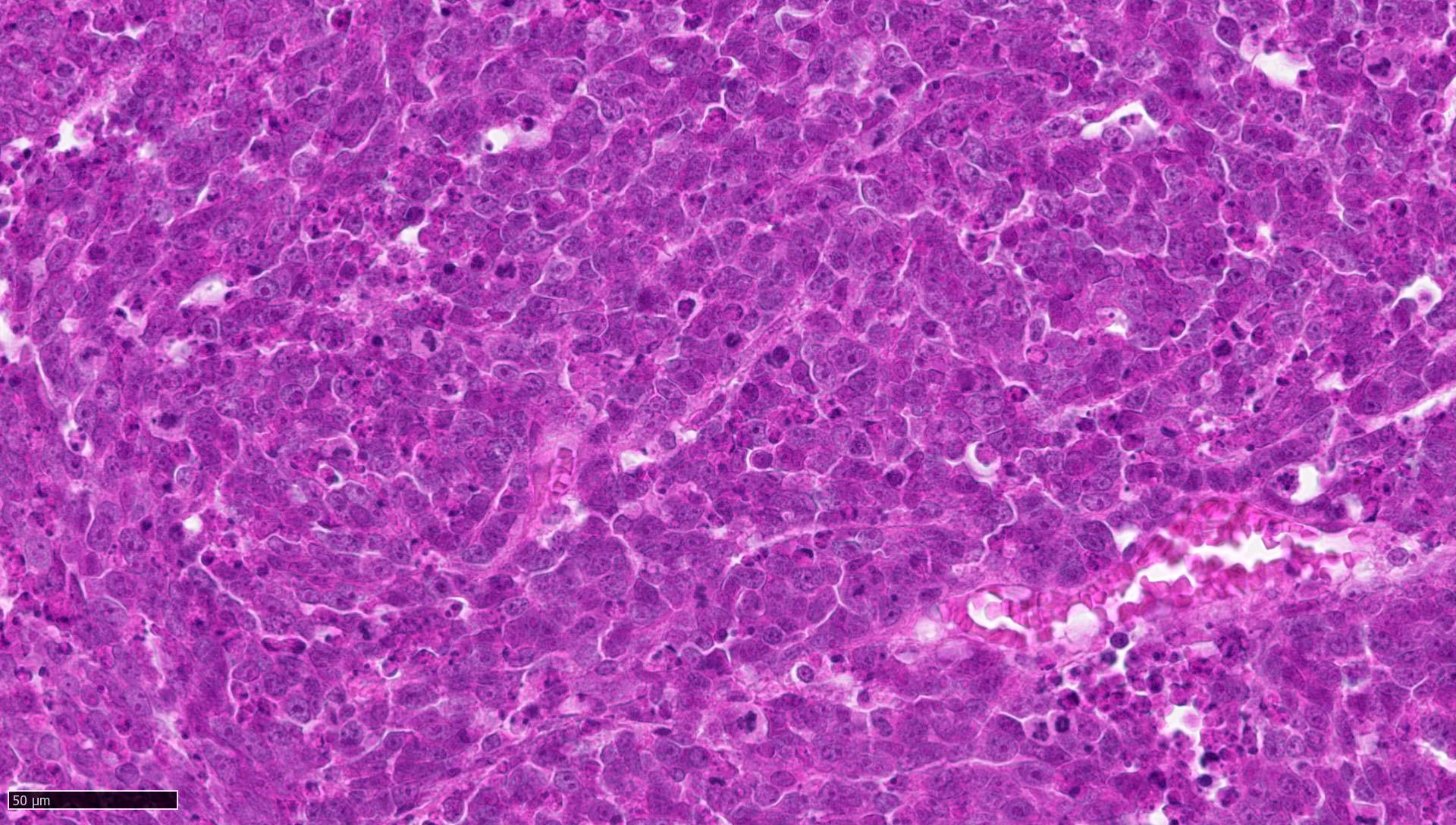
Apoptotic bodies, prominent nucleoli
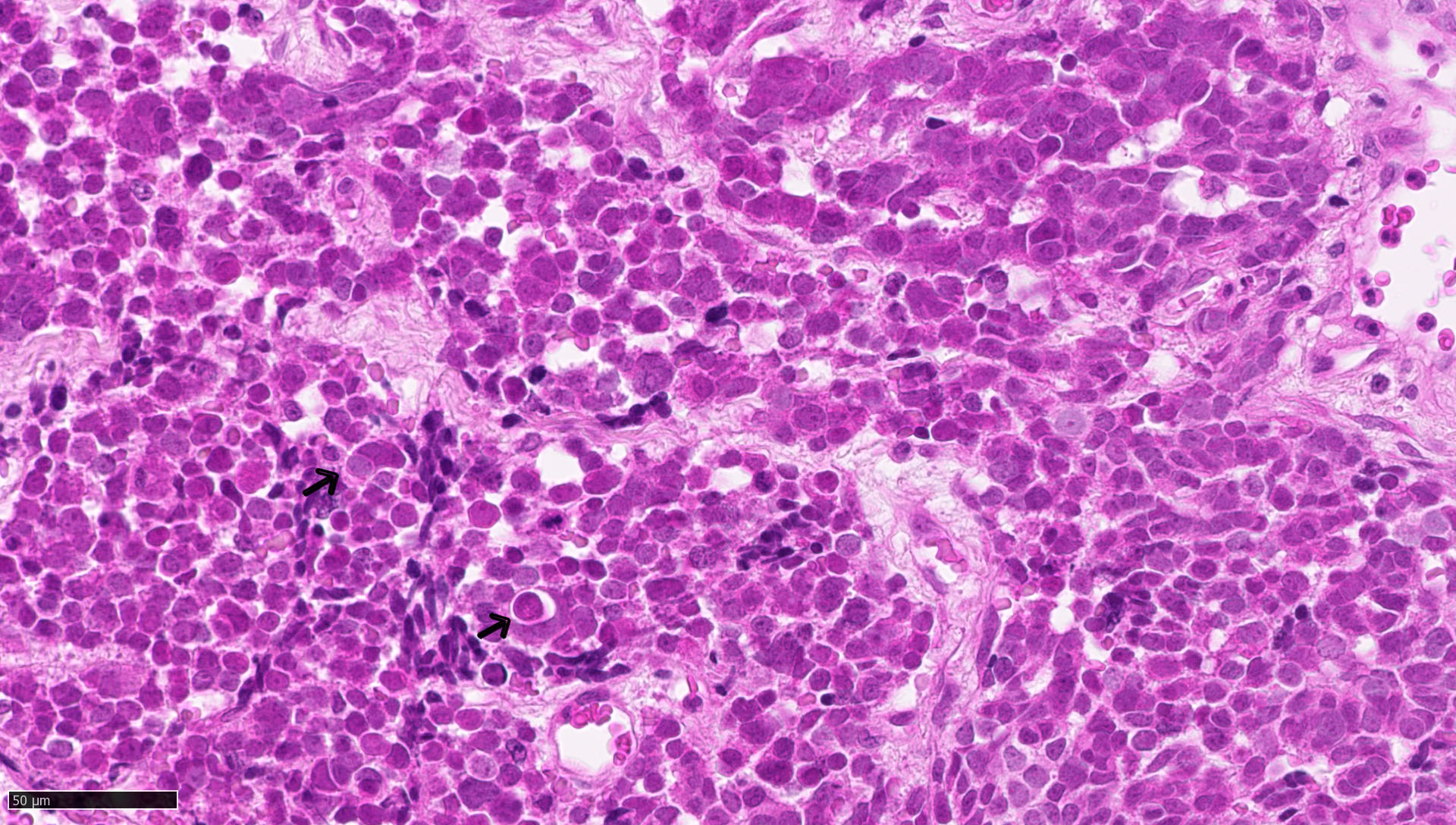
Cell wrapping
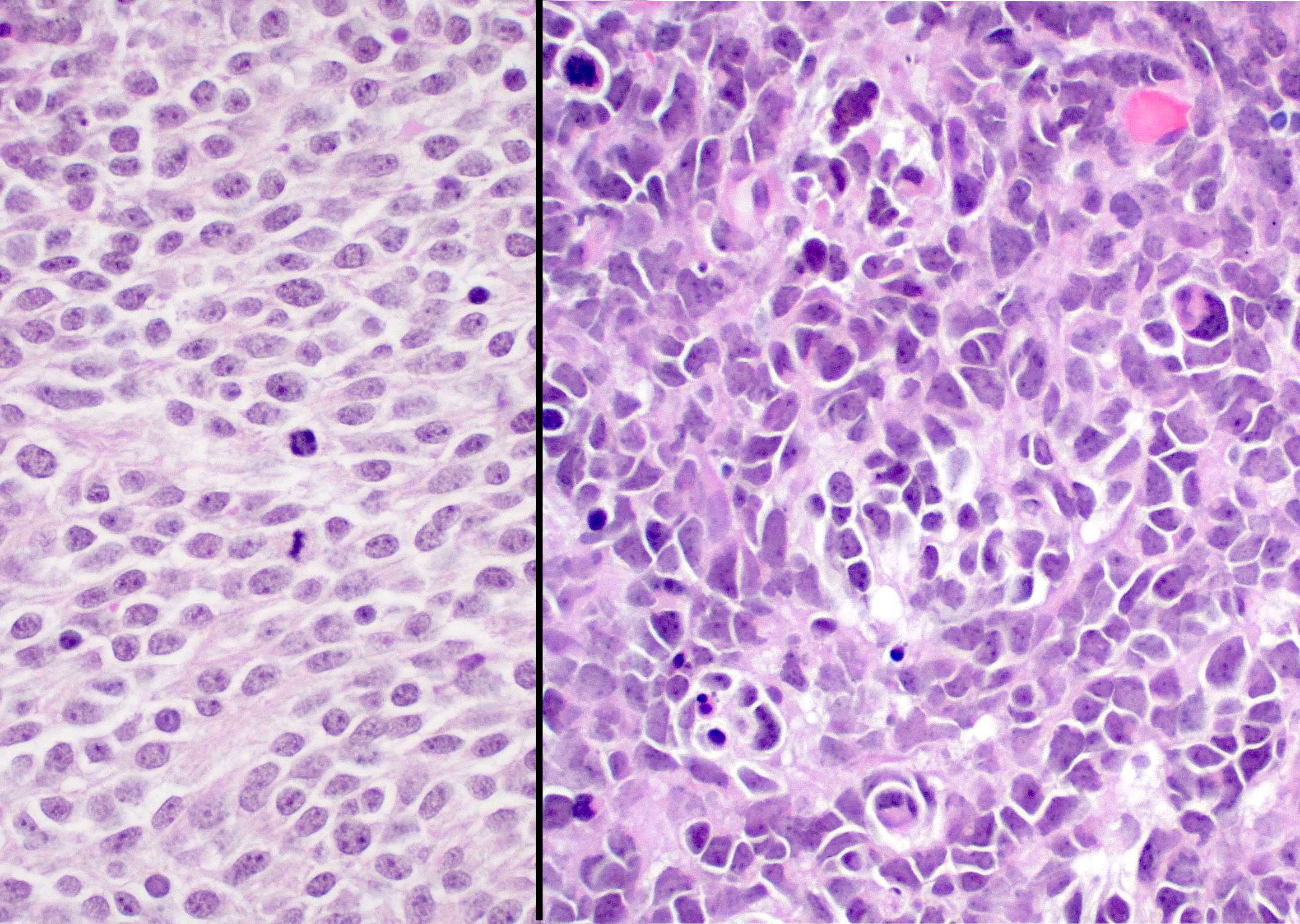
Anaplasia in medulloblastoma
Special morphology
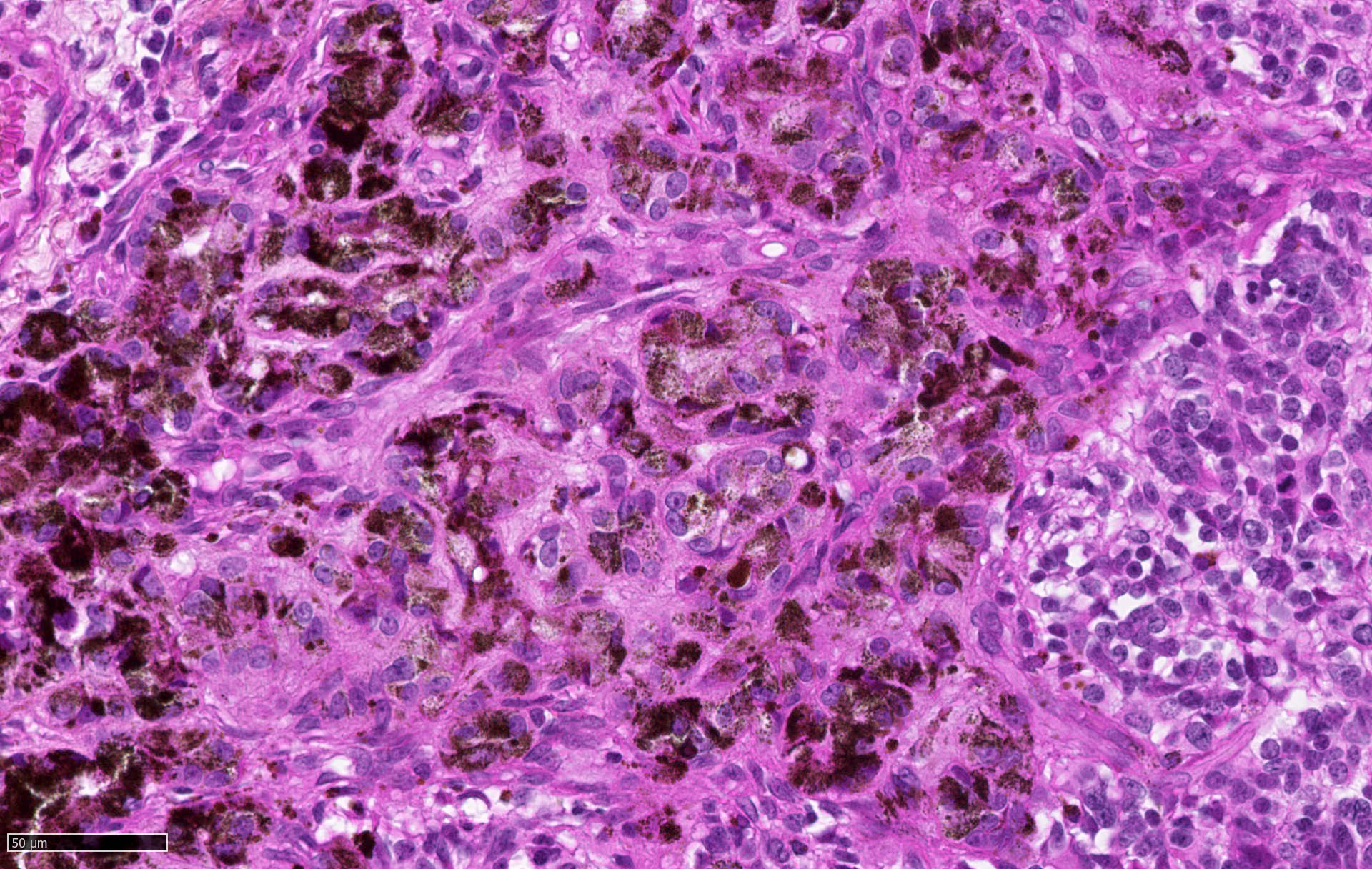
Melanocytic differentiation
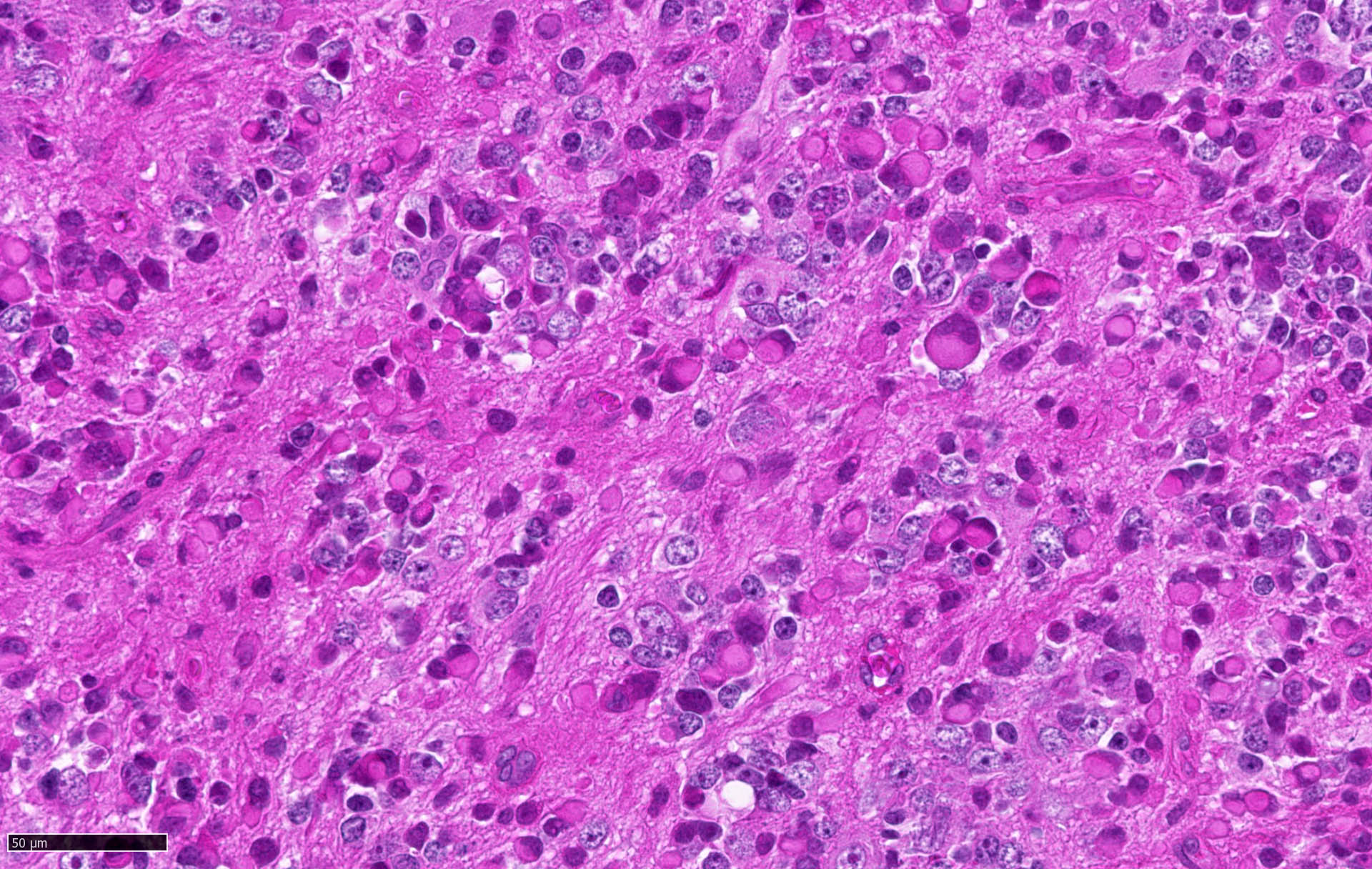
Myogenic differentiation
- Clusters of tumor cells with hyperchromatic nuclei
Contributed by Arnault Tauziede-Espariat, M.D.
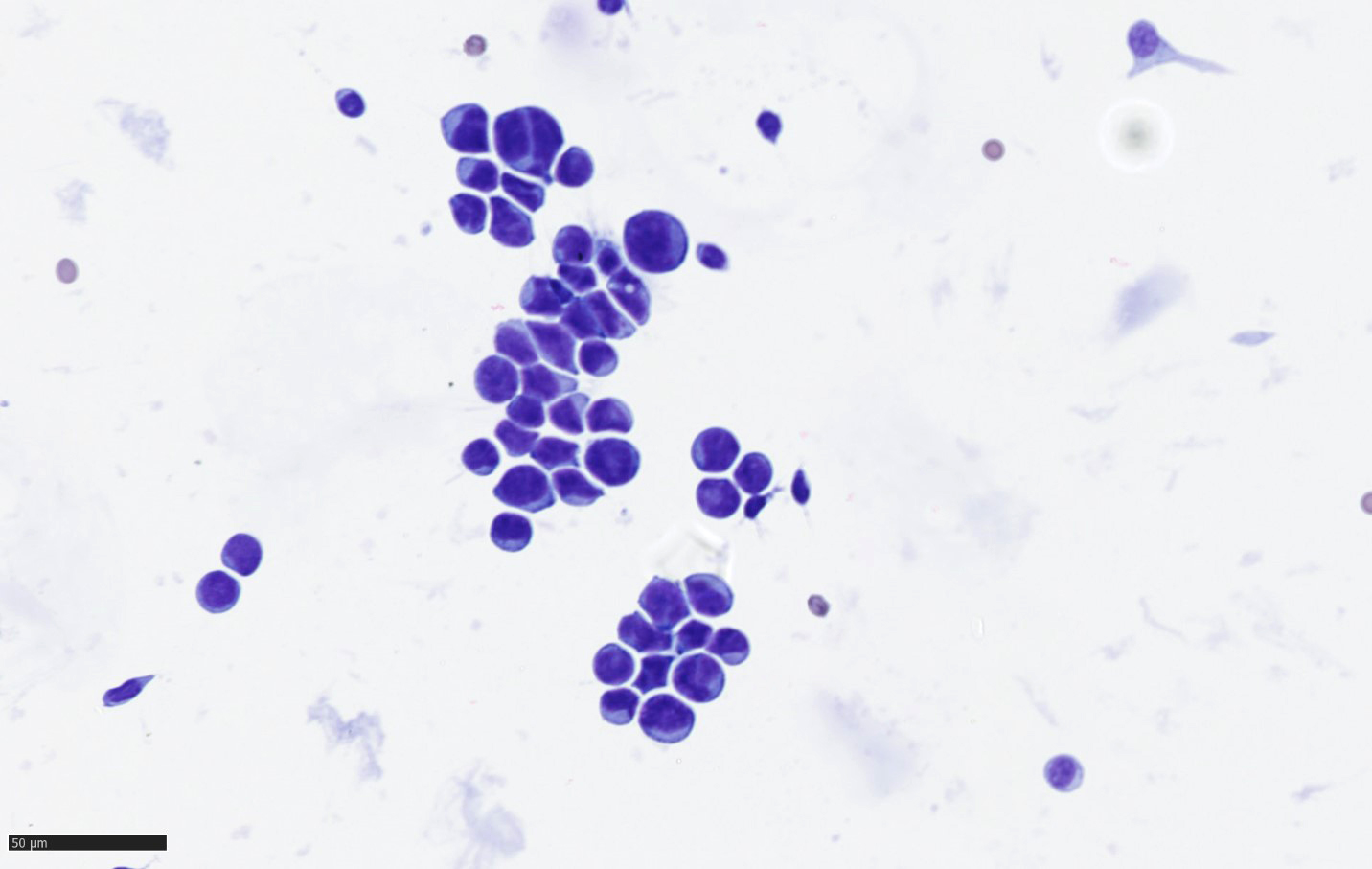
MGG staining
- Synaptophysin, NeuN
- Nuclear SMARCB1 / INI1 and SMARCA4 are retained
- Medulloblastoma with melanocytic differentiation: HMB45, MelanA
- Medulloblastoma with myogenic differentiation: desmin and myogenin
- Reference: Neuropathol Appl Neurobiol 2002;28:257
- GFAP rarely positive in tumor cells
- LIN28A
- References: Acta Neuropathol 2012;124:875, Acta Neuropathol 1985;67:1
- Genetic subgroups may be presumed due to immunohistochemical markers (Am J Surg Pathol 2021;45:558)
| Genetic subgroups | Immunohistochemical markers | |||||
| GAB1 | OTX2 | p75 NGFR | Filamin A | |||
| WNT activated | + n | - | + n and c / - | + n | - | + c |
| Non-WNT / non-SHH (groups 3 and 4) | - | - | - | + n | - | - |
| SHH activated | - (only c and m) | + c | + n and c | - | + c | + c |
Contributed by Arnault Tauziede-Espariat, M.D.
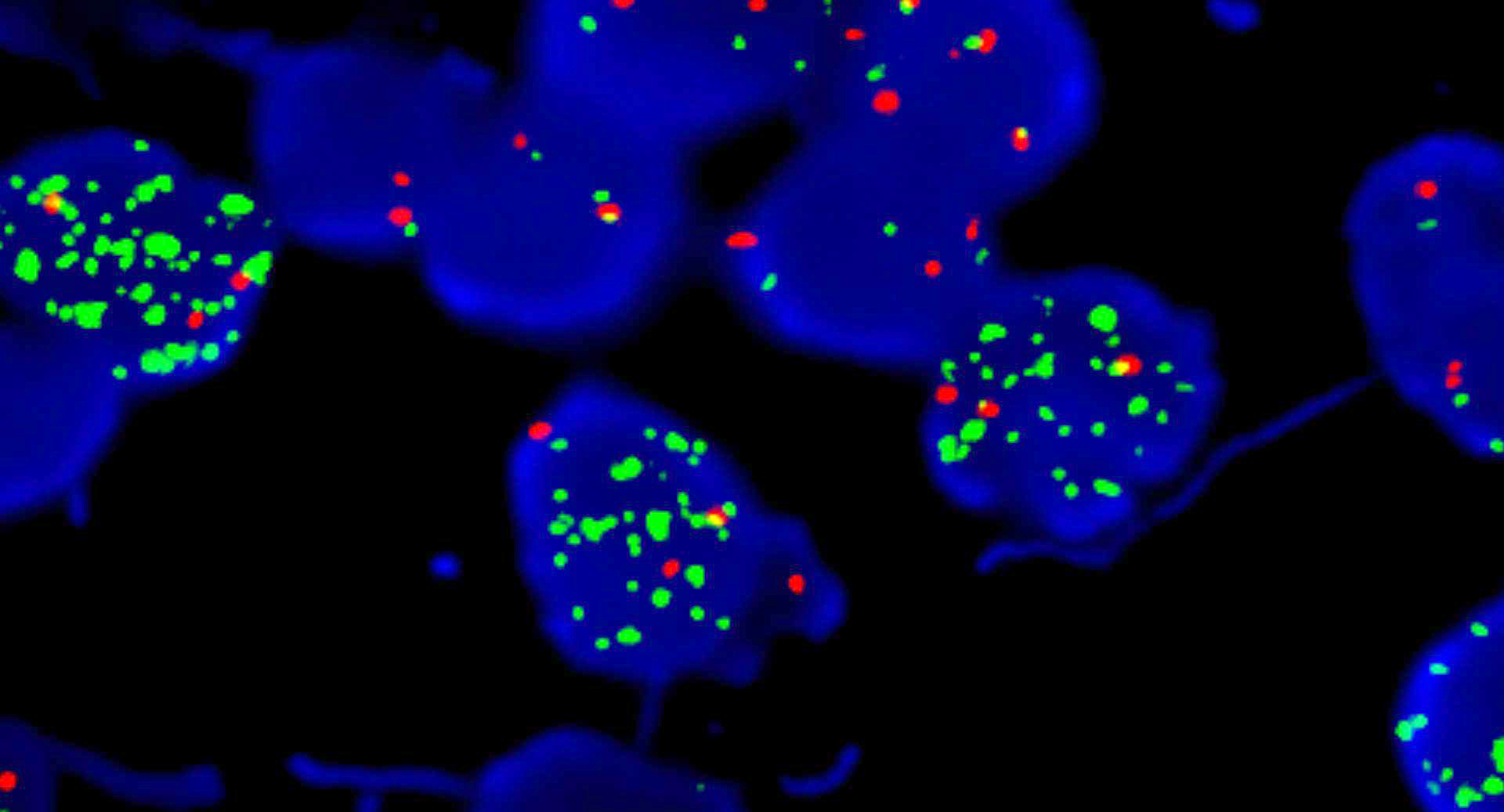
MYCN amplification
- Brain, cerebellar tumor, resection:
- Classic medulloblastoma (see comment)
- Comment: Medulloblastoma, WNT activated, CNS WHO Grade 4
- Molecular pathology findings
- CTTNB1 p.Ser33Cys mutation
- Monosomy 6
- Ependymoma:
- GFAP positive, synaptophysin negative
- Embryonal tumors with multilayered rosettes:
- LIN28A positive
- Atypical teratoid / rhabdoid tumor:
- INI1 loss
- CNS tumor with BCOR internal tandem duplication (ITD):
- BCOR positive
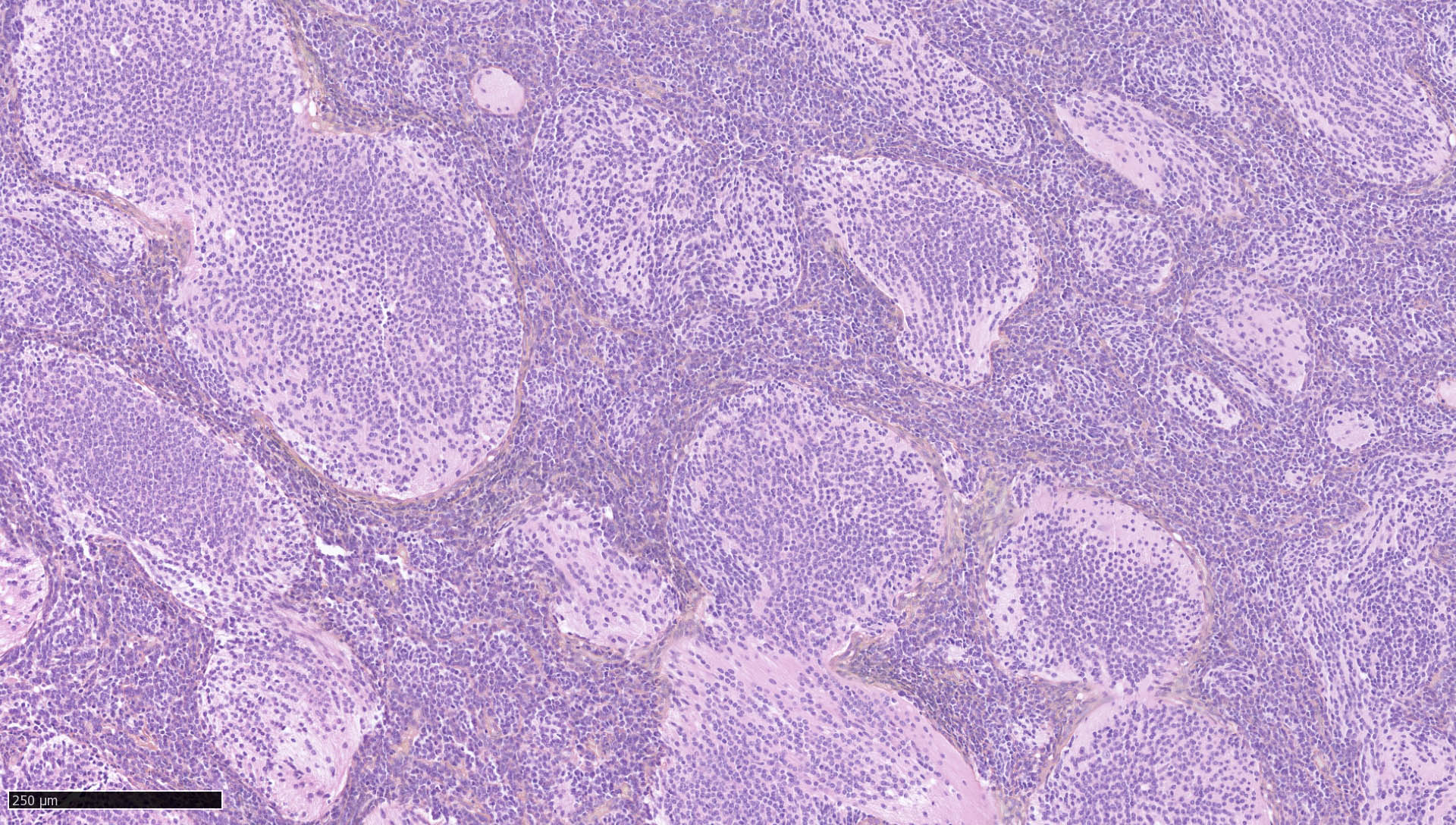
The tumor shown above is found in the cerebellum in a child. What is the diagnosis?
- Anaplastic / large cell medulloblastoma
- Classic medulloblastoma
- Desmoplastic / nodular medulloblastoma
- Myomedulloblastoma
- Medulloblastomas, SHH activated show YAP1 and OTX2 immunoreactivities
- Medulloblastomas, WNT activated show YAP1 and GAB1 immunoreactivities
- Medulloblastomas, non-WNT / non-SHH show OTX2 expression
- Beta catenin cytoplasmic expression is specific of medulloblastomas, WNT activated
- p53 overexpression is specific of medulloblastomas, SHH activated
Comment Here
Reference: Medulloblastoma
- Melanocytes are a normal yet sparse cell of the leptomeninges, most often seen over the anterior / lateral cord, brainstem, base of brain
- Can give rise to rare primary intracranial melanocytic tumors
- WHO recognizes three categories of primary CNS melanocytic lesions: diffuse melanocytosis, melanocytoma, malignant melanoma
- Diffuse melanocytosis: strongly associated with neurocutaneous melanosis, a rare congenital syndrome with giant congenital pigmented skin nevi and high rate of CNS melanoma usually presenting before age 2 (Semin Cutan Med Surg 2004;23:138)
- Melanocytoma: less than 0.1% of brain tumors, arises at any age
- Melanoma: incidence of 0.005 cases per 100,000; reported in ages 15 - 71 with a peak in the fourth and fifth decade
- Diffuse melanocytosis can involve infra or supratentorial leptomeninges but has highest frequency in cerebellum, brain stem, temporal lobes
- Involves subarachnoid space and superficial cortex
- Melanocytomas can occur in any area of meninges; however, have a predilection for cervical and thoracic spinal cord (intradural, extramedullary) and Meckel cave
- Melanomas can also occur anywhere within meninges but have predilection for spinal cord, posterior fossa, Meckel cave
- Diffuse melanocytosis presents most commonly with features of neurocutaneous melanosis - congenital nevi, hydrocephalus, mass effect and neuropsychiatric symptoms
- Melanocytoma and melanoma present with mass effect / cord compression symptoms
Images hosted on other servers:
Diffuse melanocytosis
MRI of thoracic area
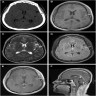
Abnormal signal in the left temporal lobe
- Diffuse melanocytosis: poor prognosis even when histologically benign
- Melanocytoma: good prognosis with resection, rarely transforms into malignant melanoma
- Can be called intermediate grade when Ki67 and mitoses are present but no obvious melanoma
- Malignant melanoma: poor prognosis (6 years in spine) yet better than metastatic melanoma to CNS (6 months) (J Neurosurg 1987;66:47)
- 11 year old girl with malignant blue nevus of ear associated with two intracranial melanocytic tumors (Hum Pathol 2004;35:1292)
- 29 year old man with primary cerebellopontine angle melanoma (Turk Neurosurg 2012;22:469)
- 34 year old woman with primary spinal cord melanoma (J Korean Neurosurg Soc 2010;48:157)
- 35 year old woman with intermediate grade melanocytoma (University of Pittsburgh: A 35 Year Old Woman with Progressive Bilateral Leg Weakness)
- 37 year old woman with leptomeningeal metastases from a primary CNS melanoma (World J Surg Oncol 2014;12:265)
- 48 year old woman with intraventricular melanoma (AJNR Am J Neuroradiol 1999;20:691)
- 57 year old man with leptomeningeal and liver tumors consistent with melanocytoma (University of Pittsburgh: A 57 Year Old Male with Leptomeningeal and Liver Tumors)
- Spectrum of CNS abnormalities in neurocutaneous melanocytosis (Dev Med Child Neurol 2012;54:563)
- Gross total resection
- Adjuvant chemoradiation therapy for malignant melanoma
- Usually solitary, well demarcated, dural based with black or reddish brown discoloration
- Melanocytoma:
- Solitary, circumscribed lesions - do not invade adjacent structures
- Nests (reminiscent of whorls) of relatively uniform cells with variable melanin pigment
- Bland, oval nuclei with eosinophilic nucleoli
- Mitoses no more than 1/10 HPF
- Malignant melanoma:
- Hypercellular sheets or nests of spindled or epithelioid cells
- May have significant pleomorphism
- Atypical mitoses (5/10 HPF)
- Invasion of adjacent structures or necrosis may be seen
- Prominent nucleoli
Contributed by Rana Al-Zaidi, M.B.B.S.
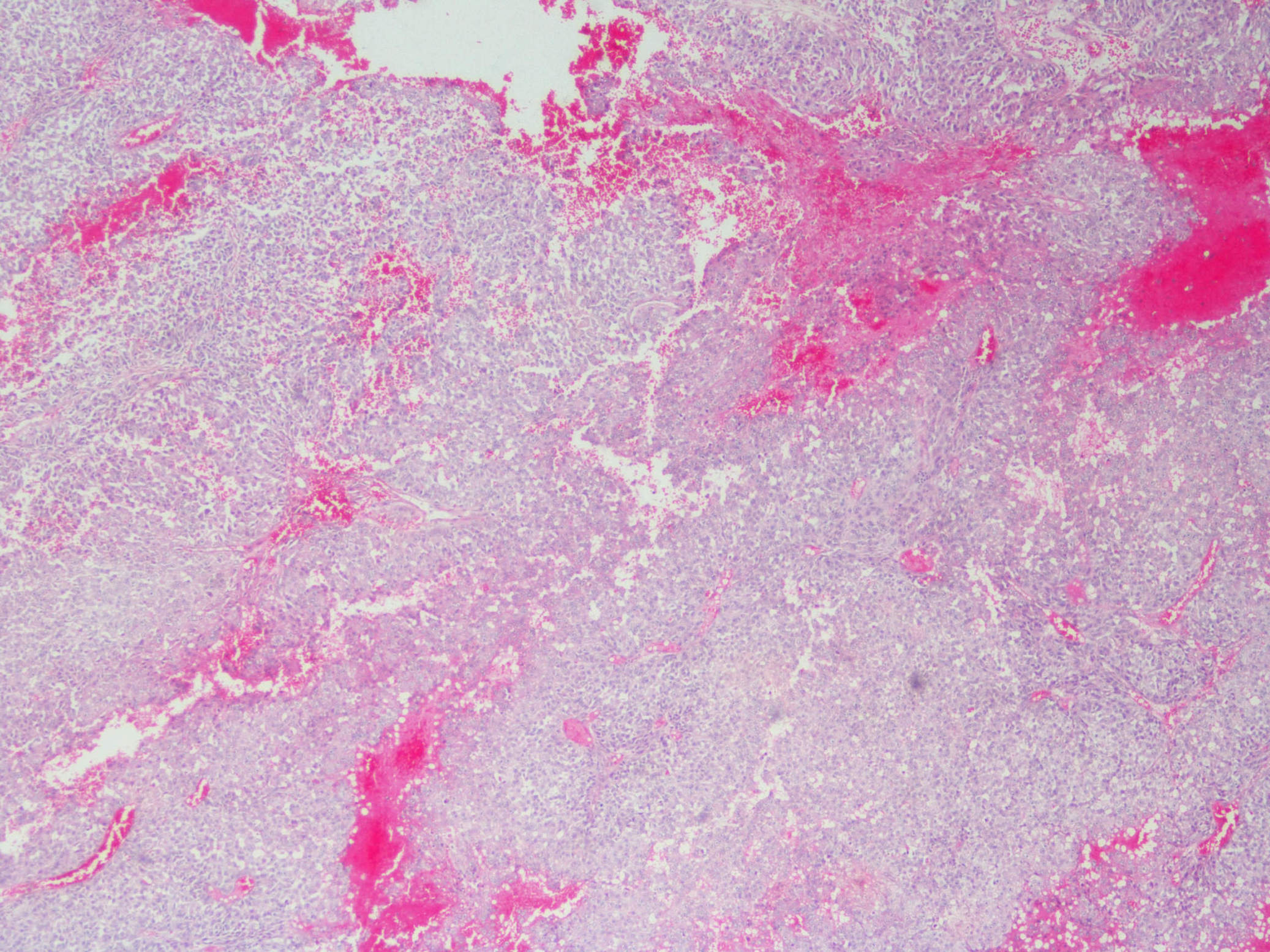
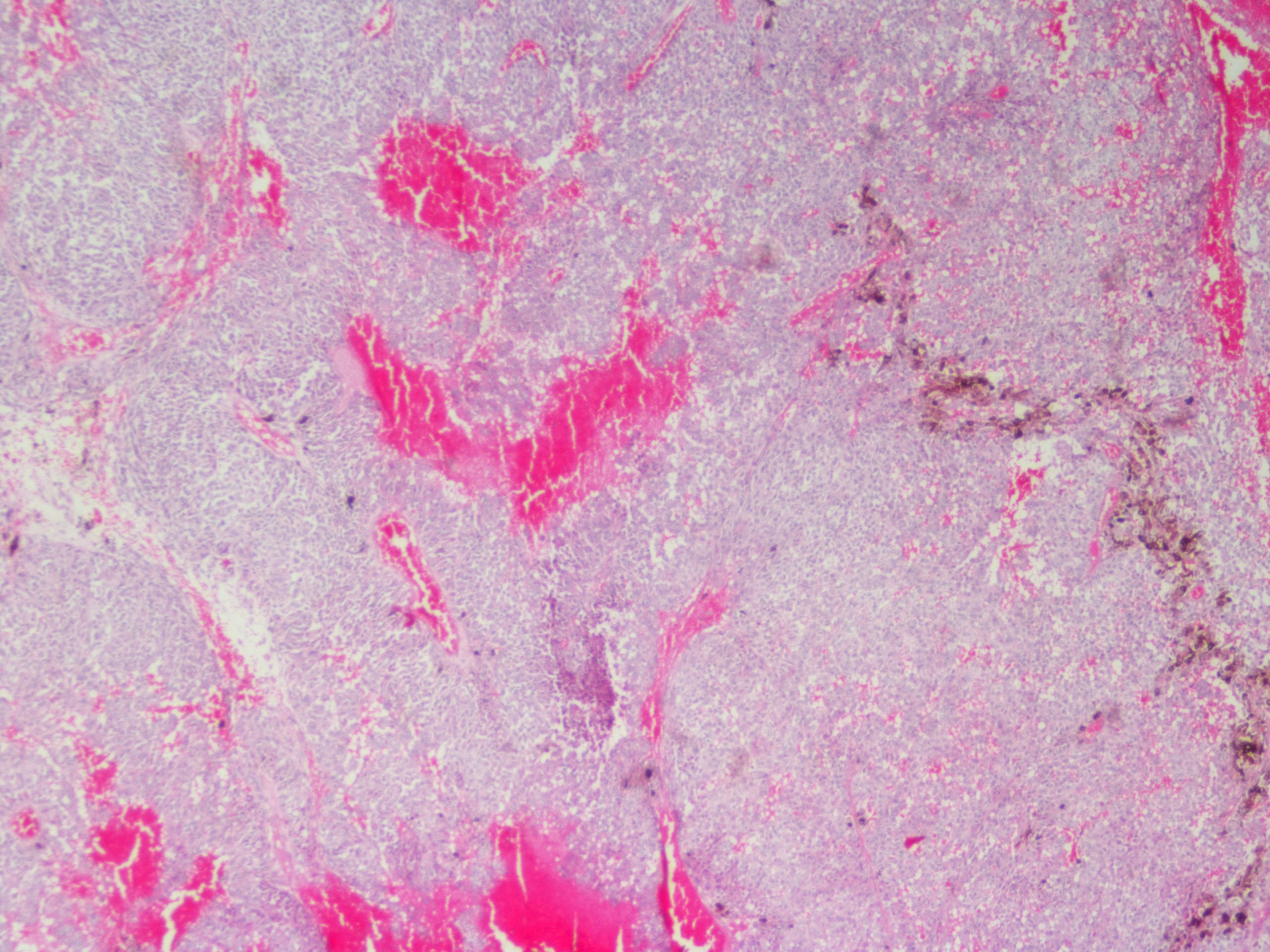

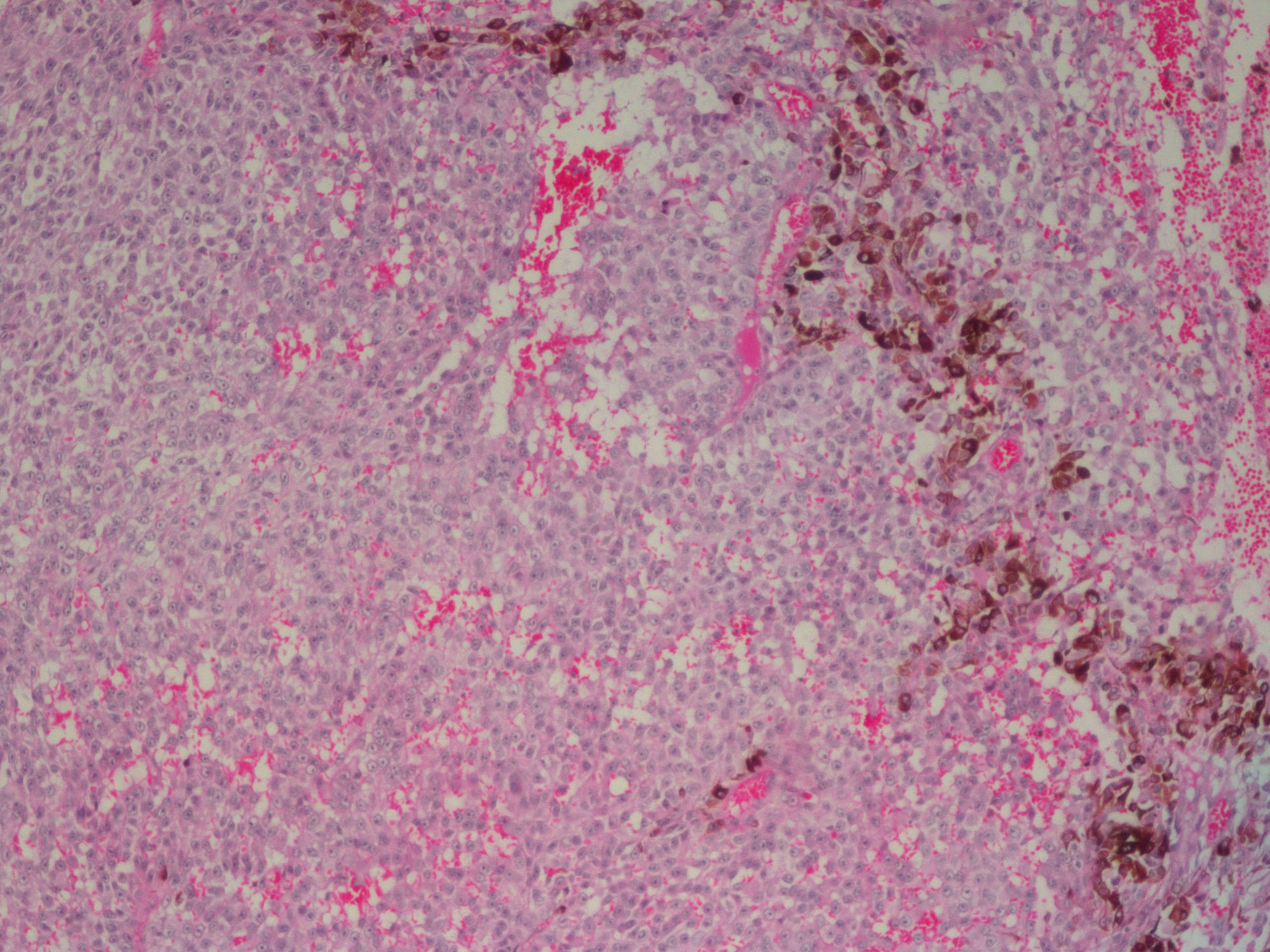
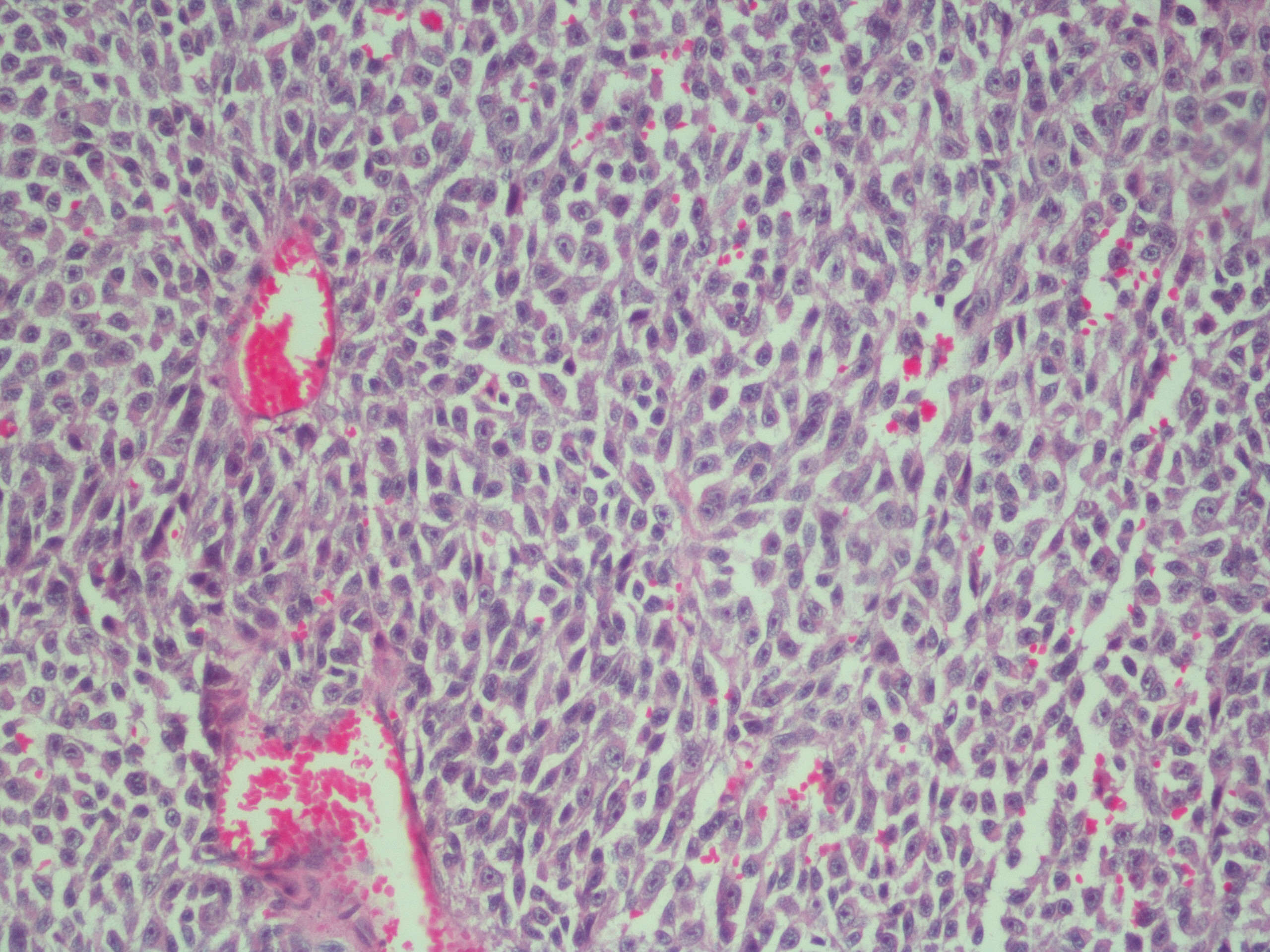
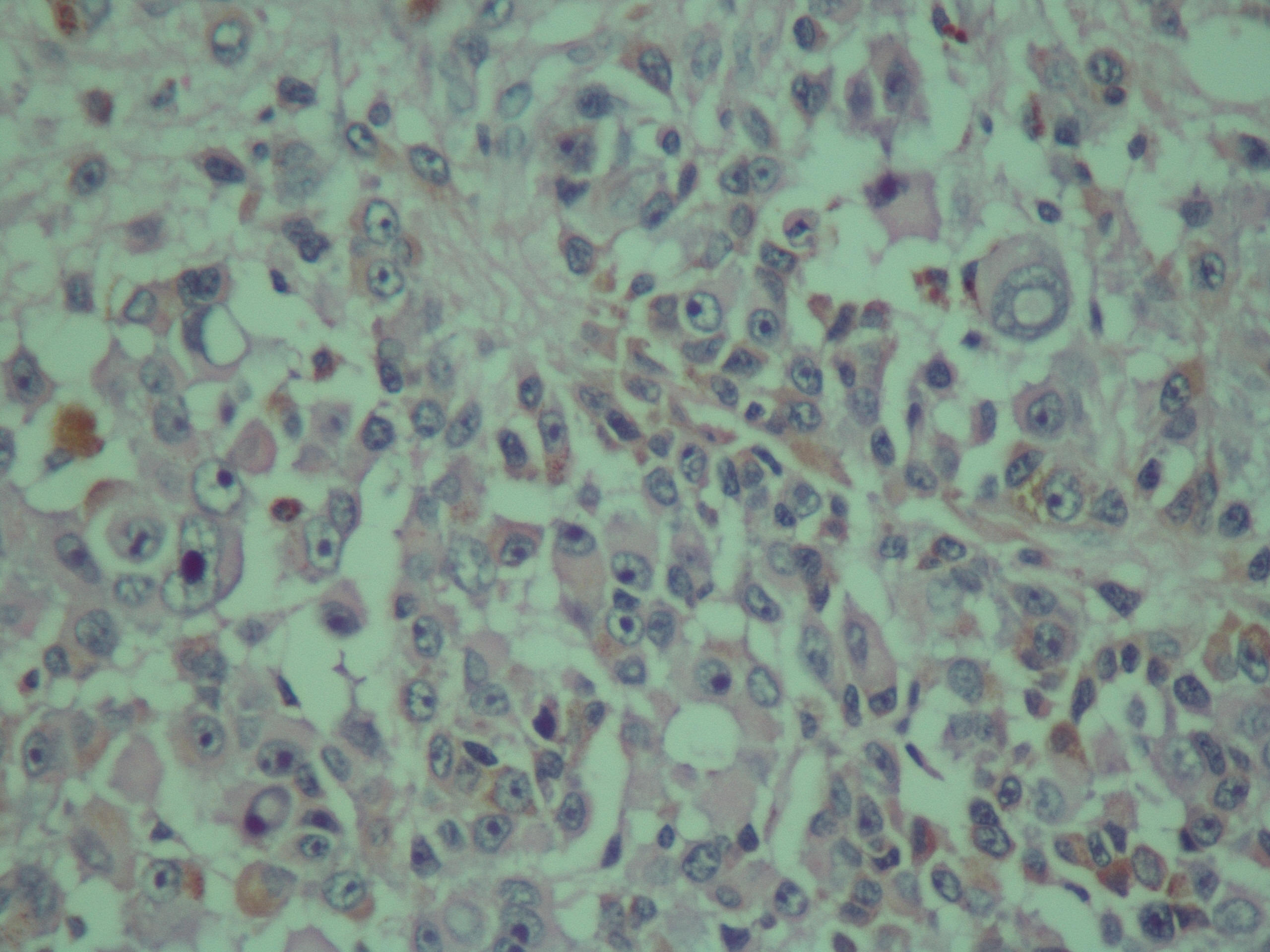
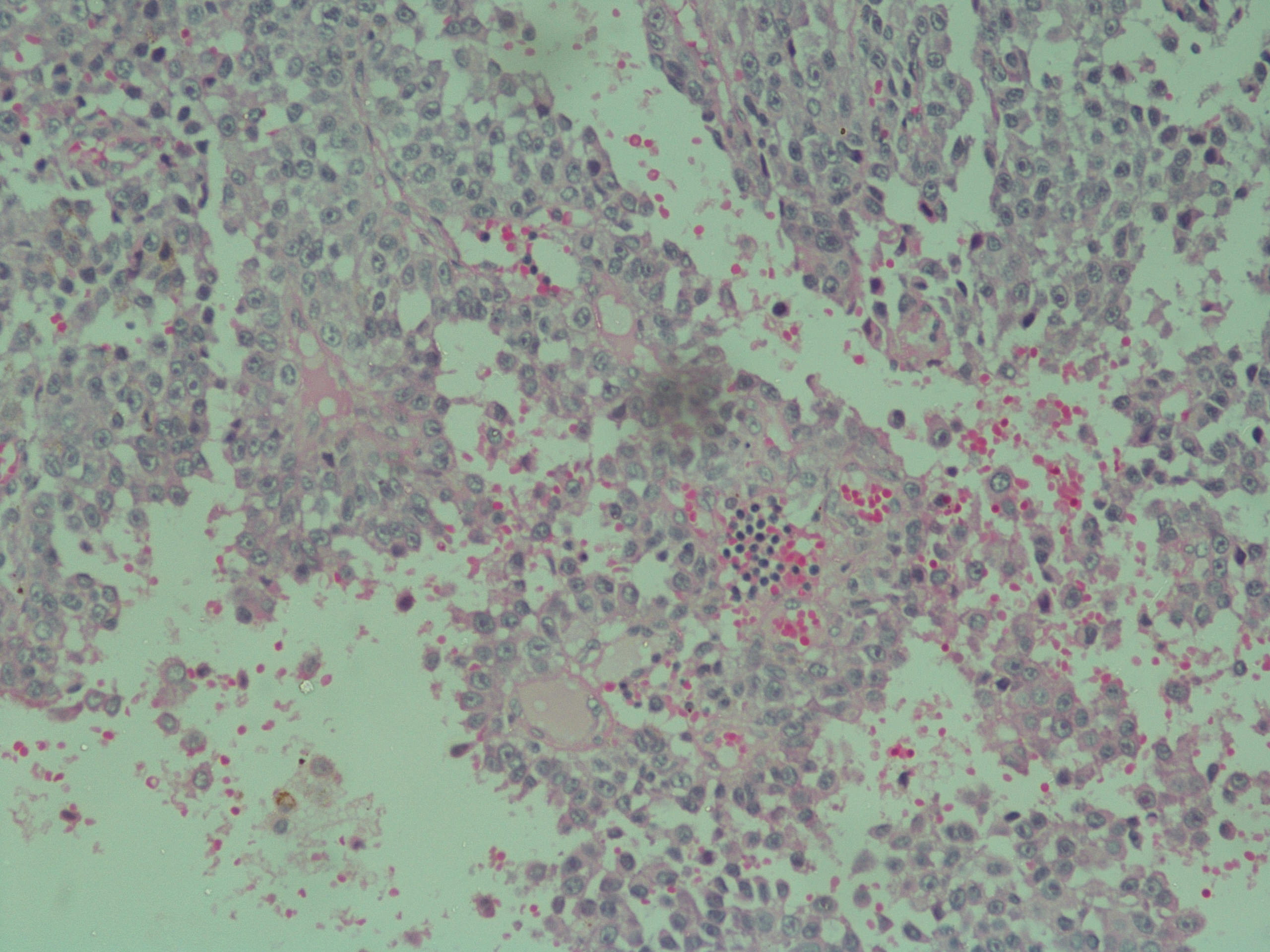
55 year old man with nausea, vomiting and sudden loss of consciousness and 4 cm temporal lesion - primary CNS melanoma
Contributed by Jesse Kresak, M.D.
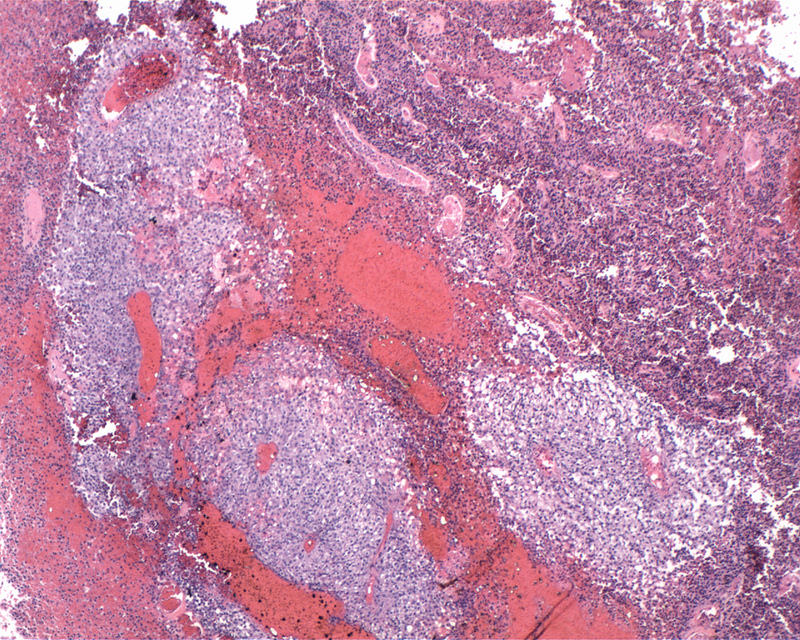
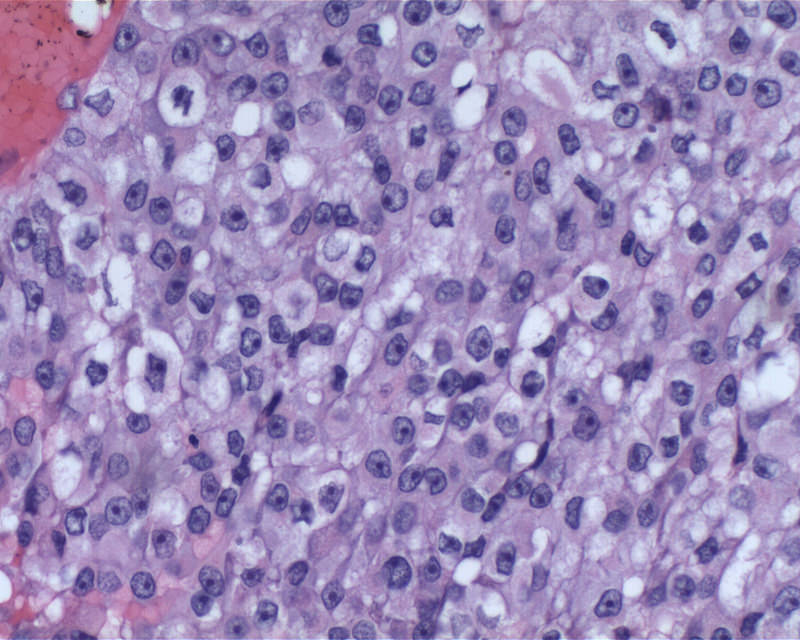
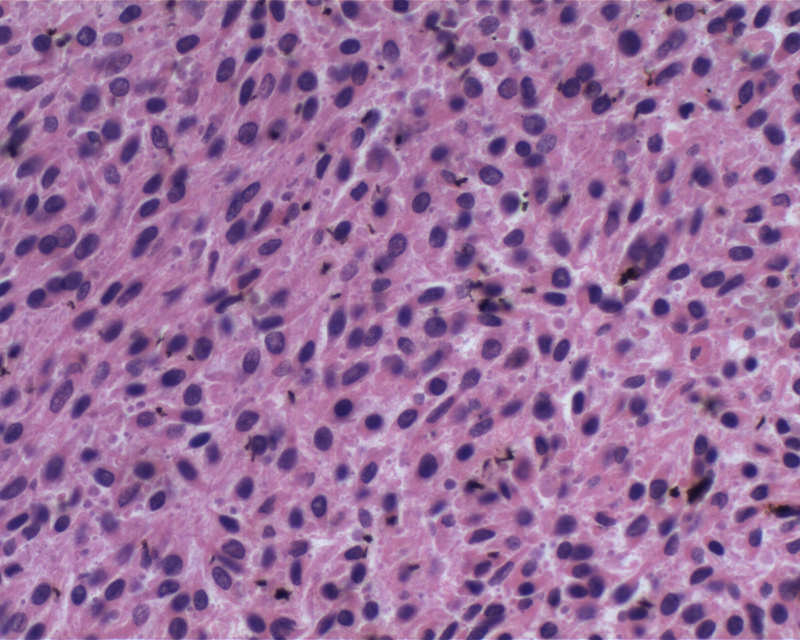
Melanocytoma
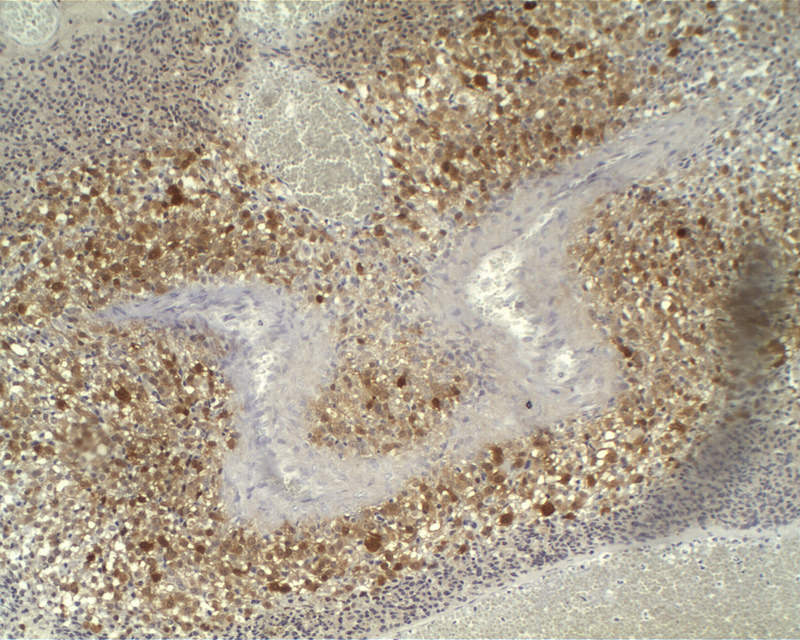
Melanocytoma: S100
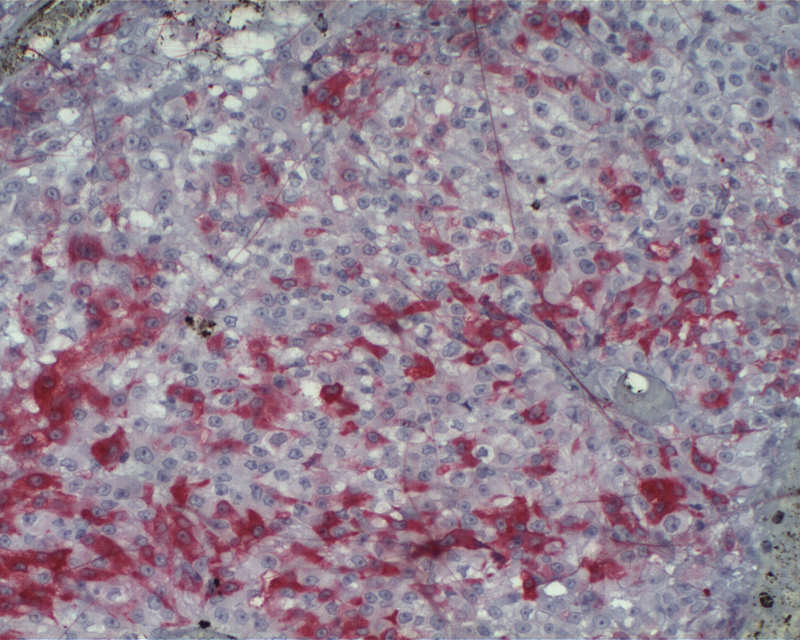
Melanocytoma: MelanA

Melanocytoma: HMB45
Images hosted on other servers:
Melanoma cells in CSF
- Melanosomes present, no junctions, no desmosomes
- Melanocytic schwannoma
- Metastatic melanoma
- Also called diverticulum
- Usually spinal
Images hosted on other servers:
Various images (figures 1A - 4C)
- 20 year old woman post motor vehicle accident (J Surg Tech Case Rep 2011;3:40)
- Lined by fibrous tissue resembling dura, with or without arachnoid inner membrane
- Rare, benign, hamartomatous lesion of children and young adults characterized by leptomeningeal and meningovascular proliferation
- Presents with seizures and headaches (or asymptomatic)
- Mean age 21 - 28 years, range 1 - 70 years
- 25% associated with neurofibromatosis II
- May be associated with other developmental anomalies (Clin Neuropathol 2013;32:37, Pediatr Dev Pathol 2014;17:292)
- Two cases, 9 months to 33 years (Am J Surg Pathol 1999;23:872)
- 7 year old girl with a 6 month history of partial complex seizures of increasing frequency (University of Pittsburgh: Seven Year Old Girl with Seizures)
- 11 year old boy with intractable seizures (Arch Pathol Lab Med 2003;127:e349)
- 13 year old boy with sudden, unexpected death (Pediatr Dev Pathol 2005;8:240)
- 15 year old boy with meningioangiomatosis and oligodendroglioma (Arch Pathol Lab Med 1996;120:587)
- 16 year old boy with a genetic alteration (Am J Surg Pathol 2002;26:125)
- 30 year old pregnant woman with fetal malformations (Pediatr Dev Pathol 2014;17:292)
- 30 year old man with a frontal lobe mass (University of Pittsburgh: A Male in His 30s with a Frontal Lobe Mass)
- 33 year old man (Case Rep Oncol Med 2012;2012:296286)
- Cortical blindness (Neurology 2013;81:511)
- Surgical excision cures most seizures (Clin Neurol Neurosurg 2013;115:1407)
- Thick and opaque leptomeninges with abnormal vessels but no evidence of neoplasia
- Often with plaque or rind-like meningothelial proliferation at cortical surface with tongue-like projections into underlying gray matter, predominantly in a perivascular pattern
- Also increased cortical vascularity
- Often leptomeningeal calcification
- Variable psammoma bodies and osteoid
- May have entrapped reactive glial tissue or neurons
- Also variable neurofibrillary tangles
- No / rare mitotic activity, no necrosis, no marked pleomorphism
- Numerous thin walled capillaries, bland spindle cells, occasional large cells with prominent nucleoli, variable meningothelial whorls and neurons (Acta Cytol 2001;45:1069)
- EMA
- Progesterone receptor: variable
- CD34: will highlight vascular elements
- Atypical meningioma: increased mitotic activity and either increased cellularity, high N/C, prominent nucleoli, sheet-like growth or necrosis
- Desmoplastic infantile astrocytoma: prominent desmoplastic stroma with neuroepithelial cells
- Invasive meningioma: infiltrative growth into brain parenchyma with destruction, no prominent vascular component
- Schwannoma: encapsulated, Schwann cells in palisading or myxoid patterns, strong S100+, EMA-
- Vascular malformation: Sturge-Weber syndrome or arteriovenous malformation, no perivascular meningothelial elements
- Most common primary CNS tumor, arising from arachnoid cap cells associated with dura mater or choroid plexus, accounting for 36% all CNS tumors (Neuro Oncol 2015;17:iv1)
- Growing along external surface of brain, spinal cord or rarely, within the ventricular system
- 3 grades exist based on WHO criteria:
- Most are slow growing WHO grade 1 (benign)
- 20 - 25% are WHO grade 2 (increased likelihood of recurrence)
- 1 - 6% are WHO grade 3 (malignant with metastatic potential)
- While there are 15 WHO recognized morphological meningioma variants, this topic focuses more on WHO grade 1 variants
- WHO grade 1 meningiomas have low mitotic index (< 4/10 high power field), no brain invasion and < 3 atypical features (necrosis, small cell change, sheeted architecture, macronuclei and hypercellularity)
- ICD-10:
- D32.9 - benign neoplasm of meninges, unspecified
- D32.0 - benign neoplasm of cerebral meninges
- D32.1 - benign neoplasm of spinal meninges
- C70 - malignant neoplasm of meninges
- C70.0 - malignant neoplasm of cerebral meninges
- C70.1 - malignant neoplasm of spinal meninges
- C70.9 - malignant neoplasm of meninges, unspecified
- D42 - neoplasm of uncertain behavior of meninges
- D42.0 - neoplasm of uncertain behavior of cerebral meninges
- D42.1 - neoplasm of uncertain behavior of spinal meninges
- D42.9 - neoplasm of uncertain behavior of meninges, unspecified
- Mainly adults, mean age of 65
- F > M
- 66% of cerebral meningiomas occur in women; 90% of spinal cord meningiomas occur in women (Neuro Oncol 2012;14:v1)
- Rare cases may occur in pediatric population, which are typically associated with genetic syndromes or childhood radiation (Acta Neuropathol 2021;142:873)
- Extra-axial mass growing along external surface of brain, spinal cord, rarely within the ventricular system or outside of the CNS (ectopic)
- Common CNS sites:
- Convexity
- Skull base
- Falx and tentorium
- Spinal cord
- Common CNS sites:
- Ectopic:
- Head and neck region is the most common ectopic site, including scalp skin, intraosseous, orbit, paranasal sinuses, ear and temporal bone (Head Neck Pathol 2009;3:116)
- Rare cases in lung, salivary glands and mediastinum have also been reported (Cancer 1996;78:2328, J Med Case Rep 2011;5:271, World J Surg Oncol 2012;10:17)
- Meningiomas driven by chromosome 22q alterations (e.g. NF2, SMARCB1) arise in neural crest cell derived meninges, including convexities, falx, tentorium and spinal cord
- Meningiomas driven by hedgehog signaling pathway, PI3K signaling, TRAF7, KLF4 and POLR2A arise in the mesodermal derived meninges of the midline and paramedian anterior, central and ventral posterior skull base (Oncogene 2021;40:875)
- Risk factors:
- Radiation, either high dose or low dose (Nat Commun 2017;8:186, Acta Neuropathol 2017;134:155)
- Hormone replacement therapy or oral contraceptives (J Clin Oncol 2008;26:279, J Neurosurg 2013;118:649)
- Germline mutations in NF2 or SMARCB1 and SMARCE1 predispose to familial and multiple meningiomas (Neurogenetics 2012;13:1, J Med Genet 2011;48:93, Nat Genet 2013;45:295)
- Common symptoms include headaches, seizures and focal neurological deficit due to tumor compression (Neurosurg Clin N Am 2016;27:239)
- Grade 1 variants
- Angiomatous:
- Vascular component should exceed 50% of total tumor area
- Can further divide into microvascular and macrovascular variants
- Mean Ki67 index is ~2%
- Does not recur if entirely resected
- Differential diagnosis includes hemangioblastoma for the microvascular variant and vascular malformations for the macrovascular variant (Am J Surg Pathol 2004;28:390)
- Genetically characterized by polysomy of chromosome 5, 13 and 20 (Oncotarget 2014;5:10596)
- Fibroblastic:
- Spindle cell neoplasm with few or no meningothelial nests or whorls; often has thick bundles of collagen
- Resembles schwannoma or solitary fibrous tumor but is positive for meningothelial markers SSTR2A and EMA and negative for STAT6
- NF2 mutated meningiomas are predominantly fibroblastic or transitional and more commonly located in convexity, falx or tentorium (Nat Genet 2013;45:285, Oncogene 2021;40:875)
- Lymphoplasmacyte rich:
- Rare, comprising < 1% of meningiomas
- Extensive lymophoplasmacyte infiltrates that overshadow an inconspicuous meningothelial component
- May be associated with Castleman disease or other hematopoietic neoplasm (Int J Clin Exp Med 2013;6:504, World Neurosurg 2017;106:152)
- Meningothelial:
- Most common variant
- Lobulated architecture, often contains meningothelial whorls
- Syncytial cells with indistinct cell membranes, eosinophilic cytoplasm
- Round uniform nuclei, intranuclear pseudoinclusions common
- May have sparse psammoma bodies
- Meningiomas with SMO or AKT1 mutations are predominantly meningothelial and more commonly located in the midline or paramedian skull base (Nat Genet 2013;45:285, Oncogene 2021;40:875)
- Metaplastic:
- Rare, < 1% of all meningiomas
- May contain foci of bone, cartilage, fat or xanthomatous morphology (J Korean Neurosurg Soc 2020;63:657, Chin J Cancer Res 2013;25:112)
- Microcystic:
- Cells have elongated processes and vacuolated cytoplasm that resembles microcysts
- May have prominent nuclear pleomorphism but usually low mitotic index
- Often rich in vasculature and overlaps with angiomatous meningioma
- Small areas of classic meningothelial nests may be present
- Microcystic areas are weakly but diffusely positive for hypoxic marker carbonic anhydrase IX (J Neuropathol Exp Neurol 2019;78:1081)
- Psammomatous:
- Found in spinal region
- Numerous psammoma bodies, intervening meningothelial cells hard to find
- Secretory:
- Eosinophilic round secretions (pseudopsammoma bodies) positive for CEA and PAS
- Genetically characterized by combined KLF4 and TRAF7 mutations (Acta Neuropathol 2013;125:351)
- Transitional:
- Mixed meningothelial and fibroblastic features
- Usually prominent whorls, psammoma bodies and clusters of syncytial cells
- NF2 mutated meningiomas are predominantly fibroblastic or transitional (Nat Genet 2013;45:285)
- Angiomatous:
- Grade 2 variants
- Grade 3 variants
- Upcoming 2021 WHO CNS tumor classification emphasizes that the criteria defining atypical or anaplastic (i.e. grade 2 and 3) meningioma should be applied regardless of the underlying subtype (Neuro Oncol 2021;23:1231)
- Diagnose by imaging and pathology of biopsy / resection specimen
- Extra-axial mass with dural tail
- Uniformly contrast enhancing
- Extensive peritumoral edema is usually associated with brain invasion (Neuro Oncol 2021;23:324)
- With the caveat that several special grade I variants (angiomatous, microcystic, secretory and lymphoplasmacyte rich) can have prominent peritumoral edema (J Neurooncol 2013;111:49)
Contributed by Chunyu Cai, M.D., Ph.D.
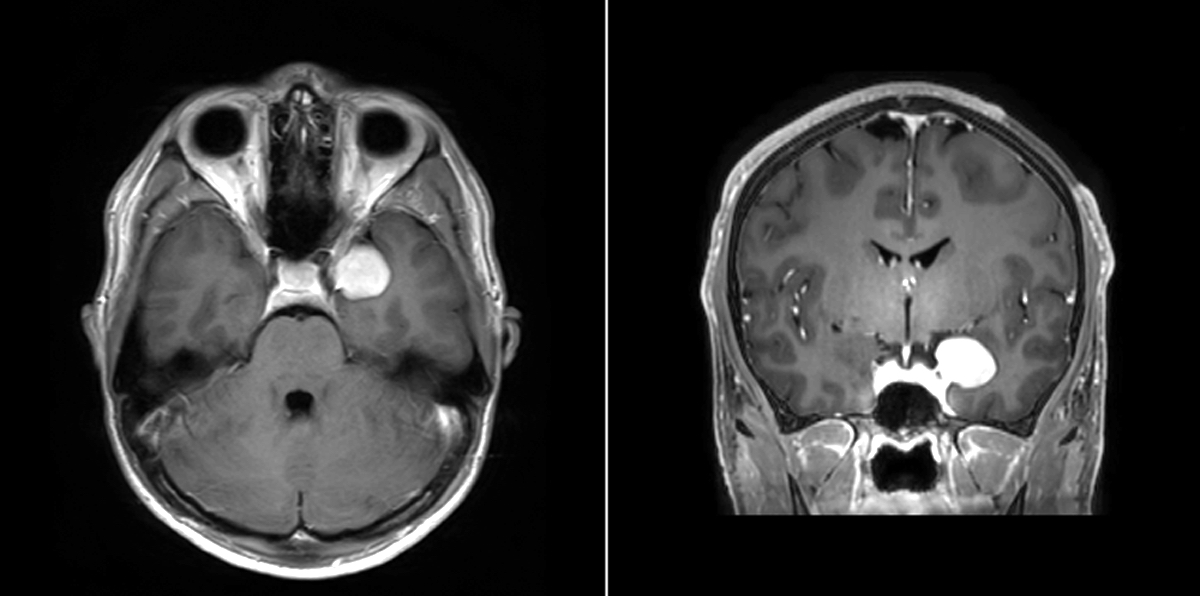
MRI benign meningioma
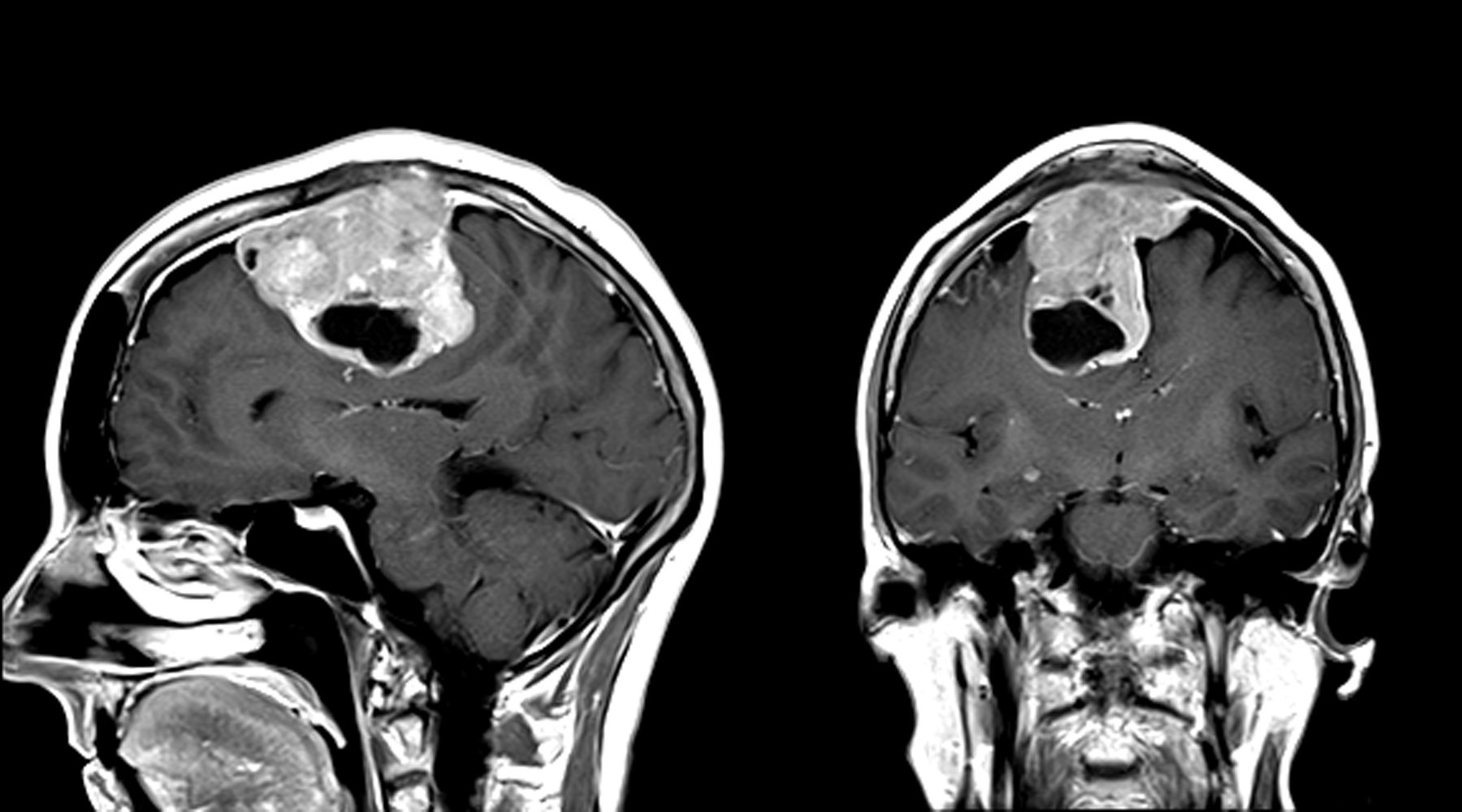
MRI atypical meningioma
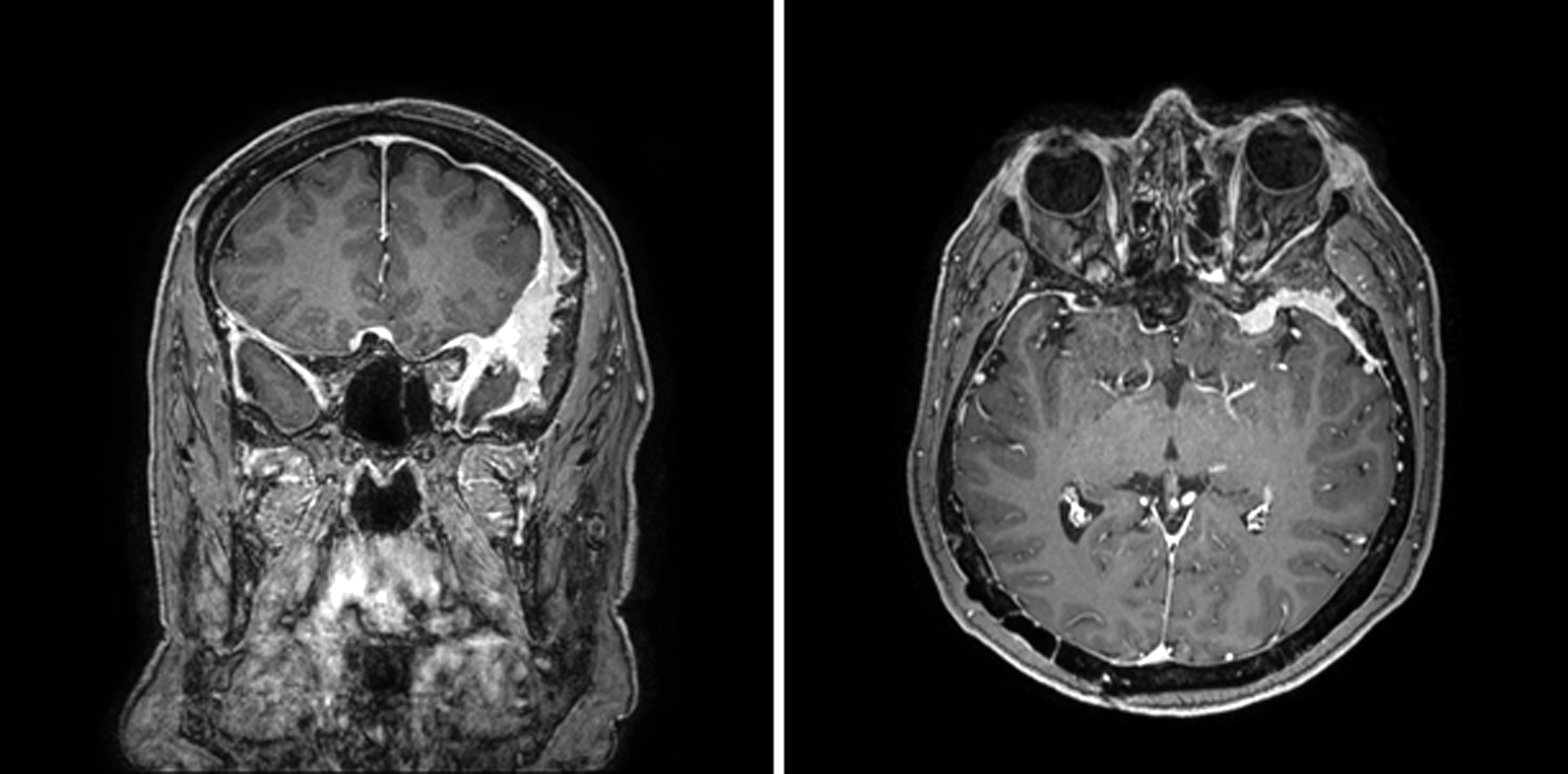
MRI en plaque meningioma
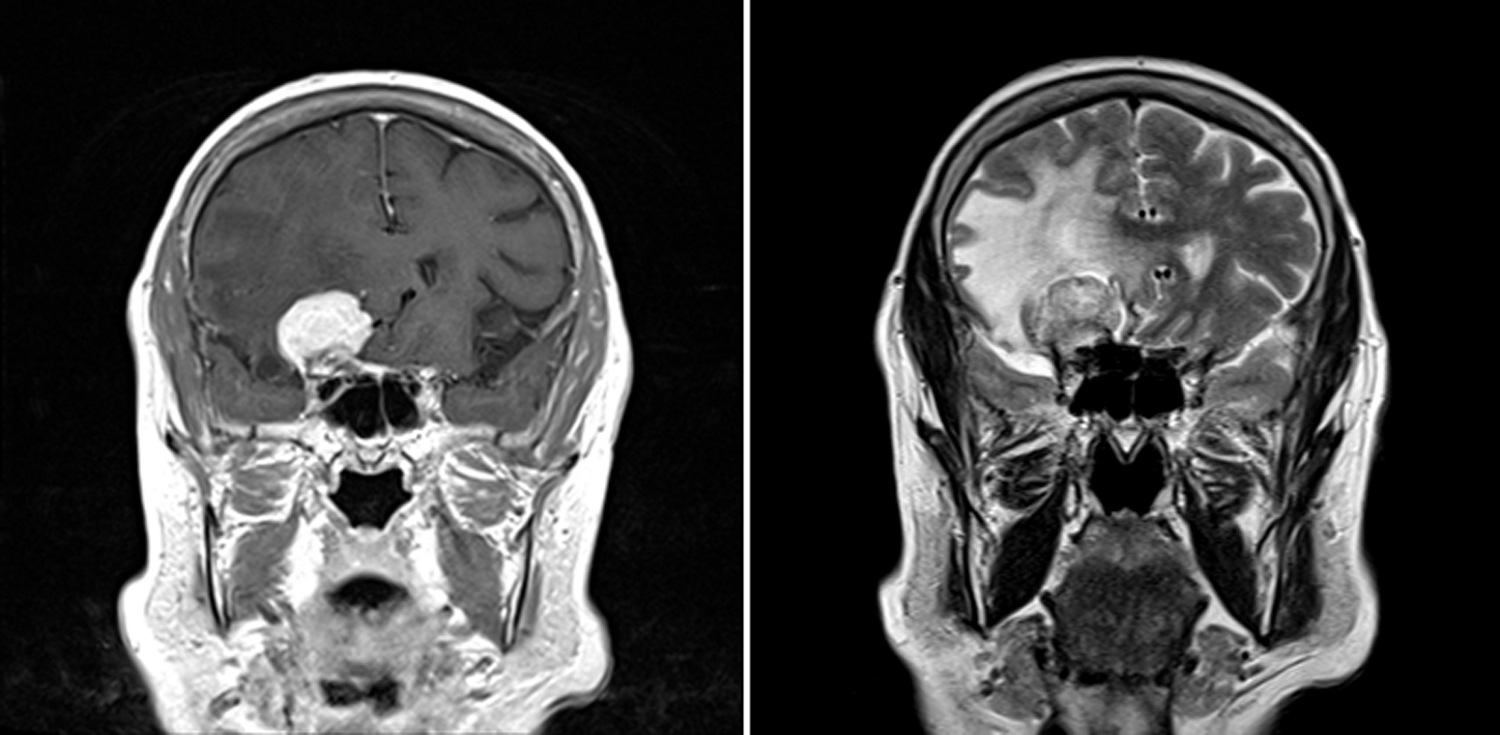
MRI secretory meningioma
- Extent of surgery and WHO grading (J Neurosurg 2015;122:4, Neuro Oncol 2016;18:863)
- Most recently, DNA methylation profiling is reported to better predict tumor recurrence and prognosis than the WHO histological classification (Lancet Oncol 2017;18:682)
- Recurrent losses of chromosome 1p, 6q, 14q, 18q and gain of 1q are indicators of poor prognosis (Acta Neuropathol 2017;133:431)
- Loss of H3K27 trimethylation (H3K27me3) by IHC predicts poor prognosis in grade 1 and 2 meningiomas but not grade 3 (Acta Neuropathol 2018;135:955)
- TERT promoter mutation is seen in high grade meningioma progressed from low grade, not in primary atypical meningioma (Brain Pathol 2014;24:184)
- 22 year old man with microcystic meningioma, WHO grade I (Case #104)
- 32 year old man with xanthomatous meningioma (Turk Neurosurg 2019;29:141)
- 36 year old man with lymphoplasmacyte rich meningioma (World J Clin Cases 2020;8:4272)
- 49 year old woman with dumbbell meningioma of the upper cervical spinal cord (J Orthop Sci 2013;18:1042)
- 52 year old woman with thoracic psammomatous meningioma with osseous metaplasia (World J Surg Oncol 2019;17:150)
- 56 year old man with angiomatous meningioma of orbit mimicking as malignant neoplasm (Orbit 2011;30:183)
- 61 year old man with left brain mass (Case #373)
- Observation, if asymptomatic
- Gross total resection is usually curative
- Postoperative radiation if incompletely excised or WHO grade 2 or 3 (Neurosurg Focus 2015;38:E3)
- Rounded and well circumscribed
- Attached to dura
- Tumor separates readily from brain
- May grow en plaque (along dural surface) and cause reactive (hyperostotic) bone changes
- Reference: Perry: Practical Surgical Neuropathology - A Diagnostic Approach, 1st Edition, 2010
Images hosted on other servers:
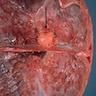
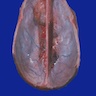
Tumor displaces brain without invading
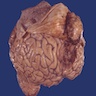
Compression of underlying cortex
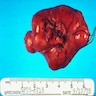
Resected tumor
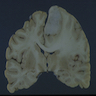
Adherent to falx cerebri
- Arachnoid plane exists between meningioma and CNS parenchyma
- Histology varies by variant (see Grading)
Contributed by Chunyu Cai, M.D., Ph.D.
Meningothelial meningioma:
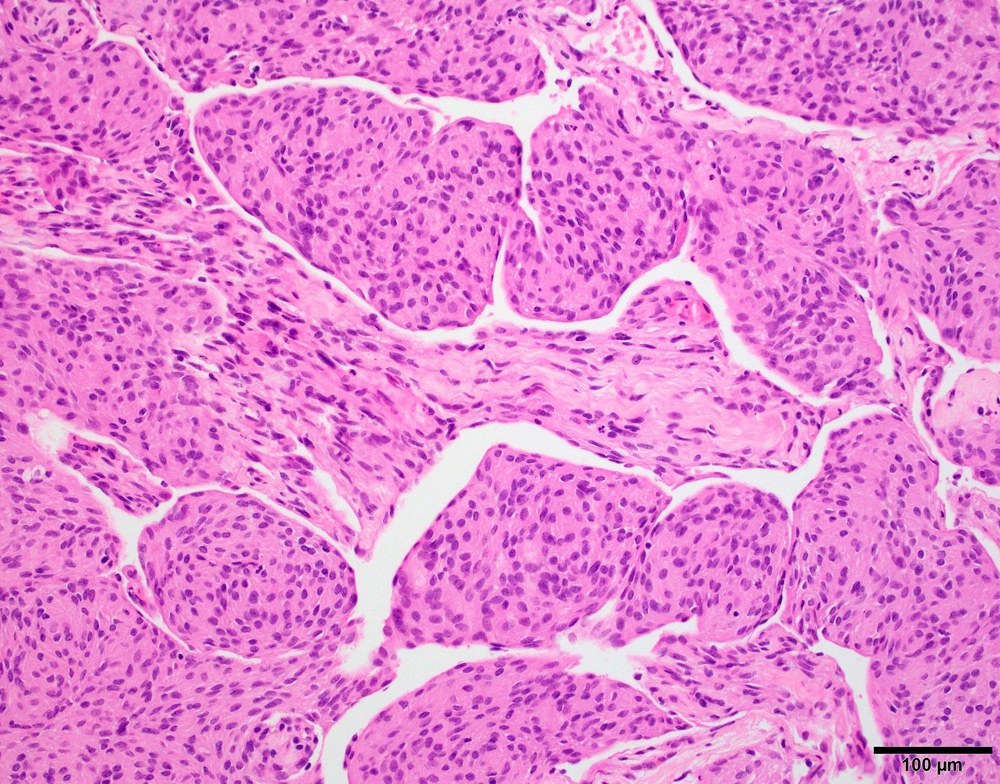
Meningothelial meningioma
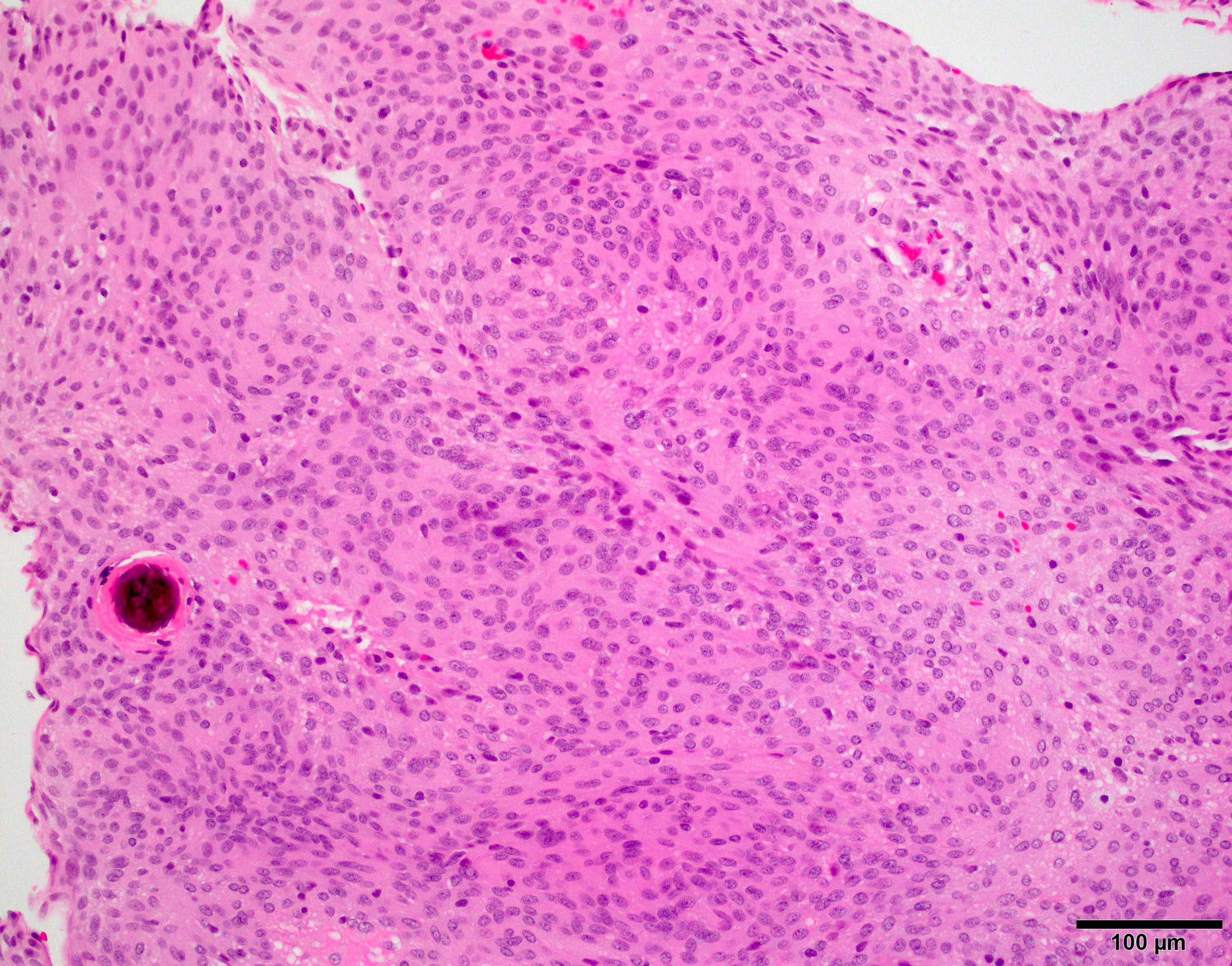
Meningothelial meningioma syncycial

Meningothelial meningioma SSTR2
Fibrous meningioma:
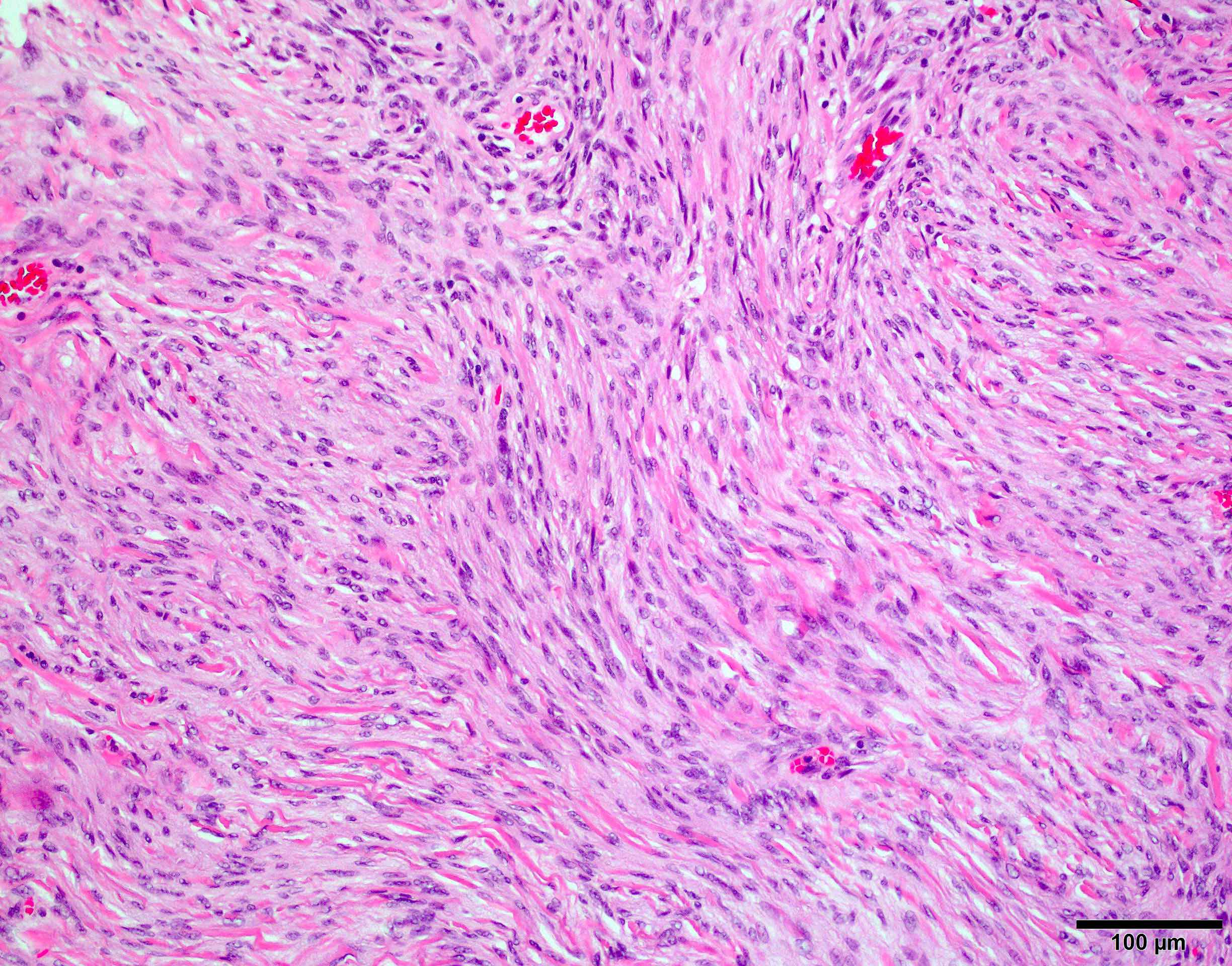
Fibrous meningioma
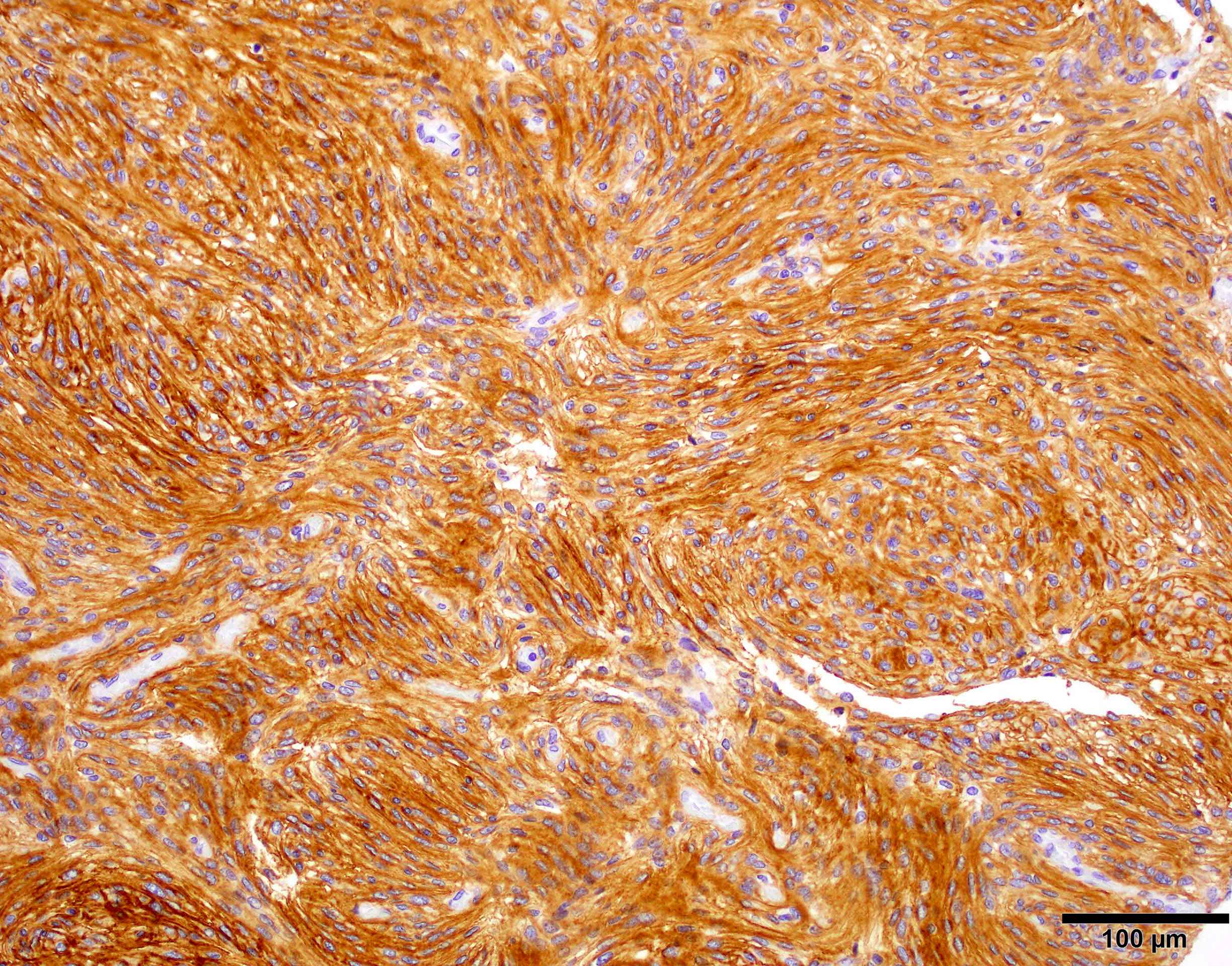
Fibrous meningioma SSTR2a
Transitional meningioma:
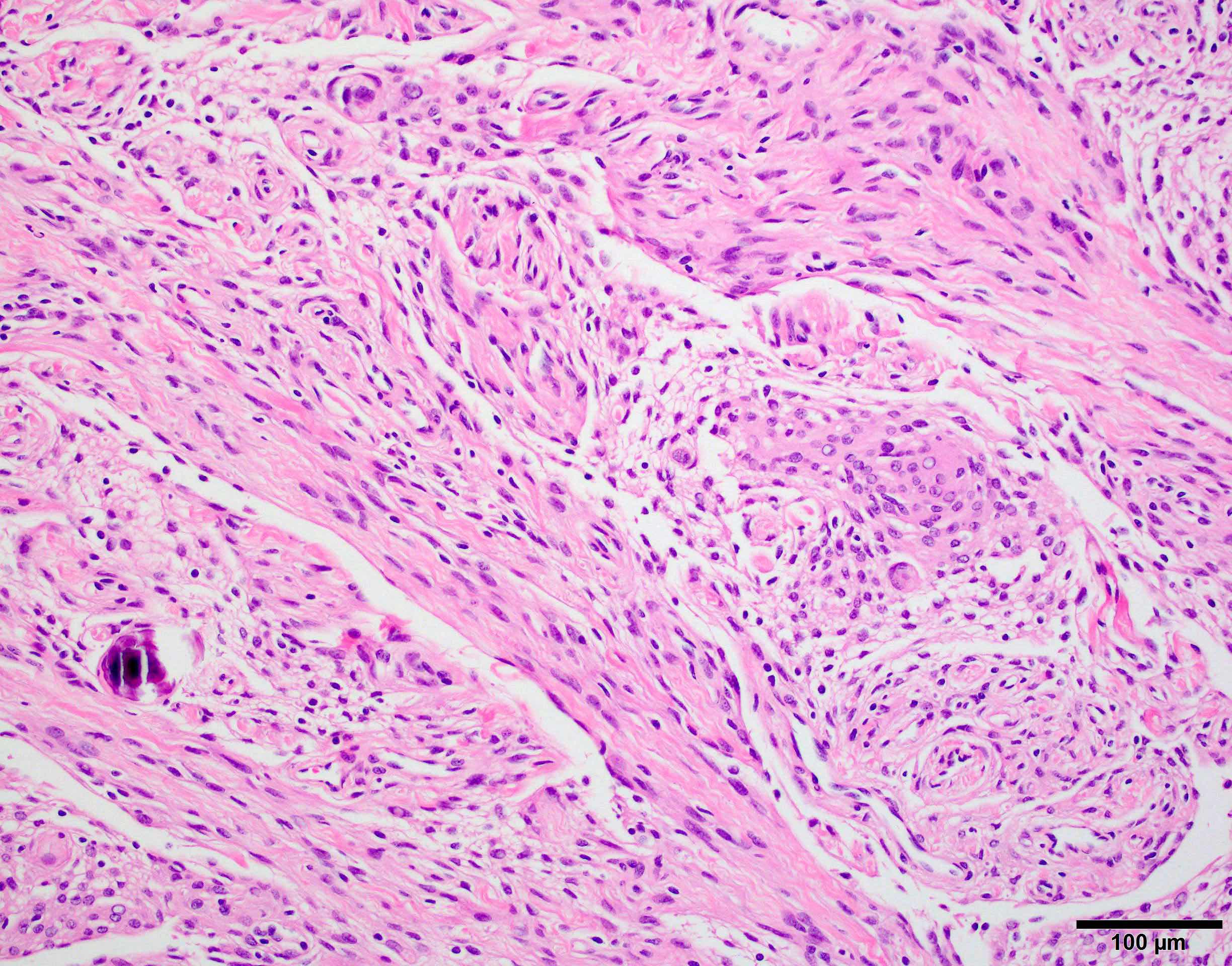
Transitional meningioma
Angiomatous meningioma:

Angiomatous meningioma macrovascular
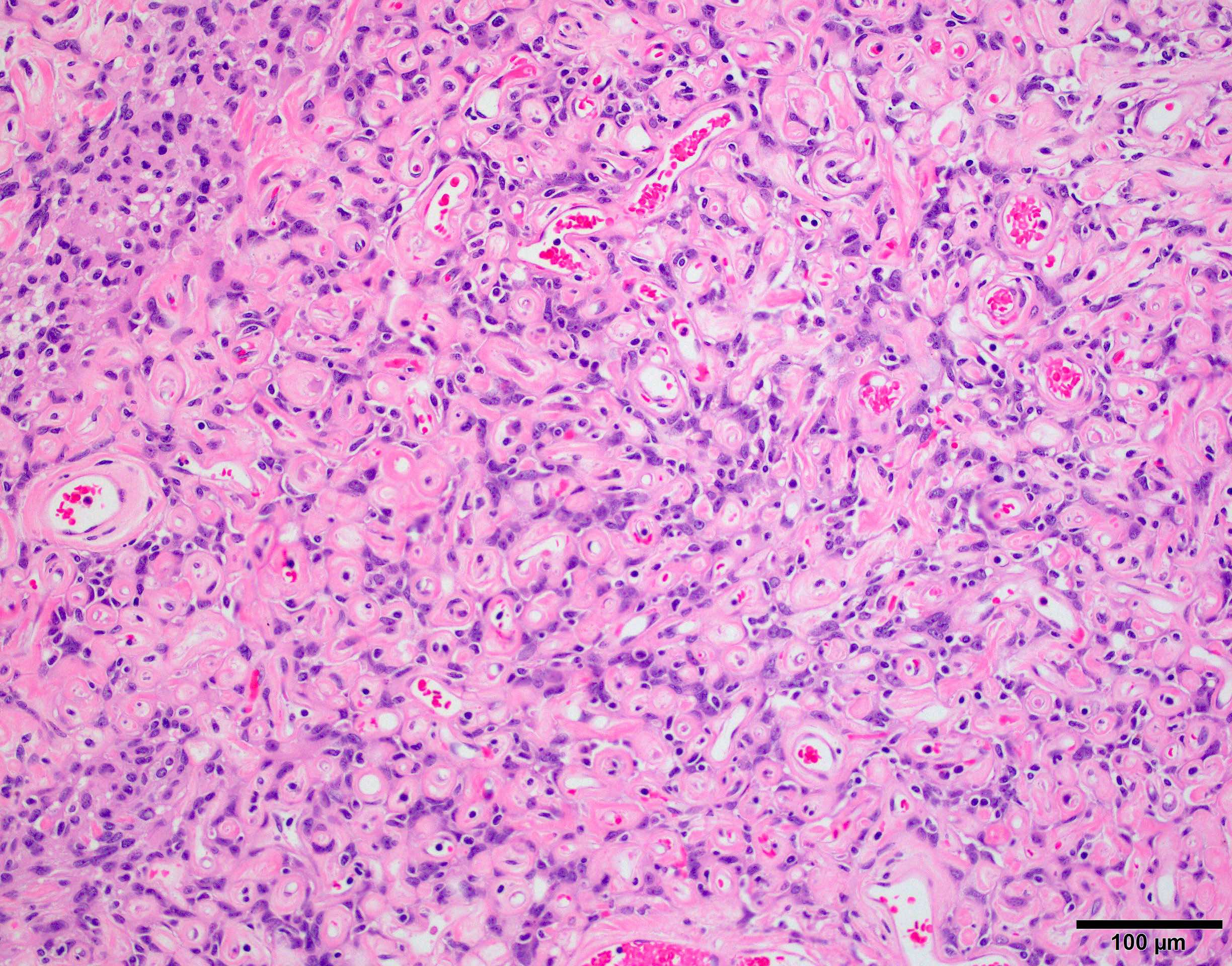
Angiomatous meningioma microvascular
Microcystic meningioma:
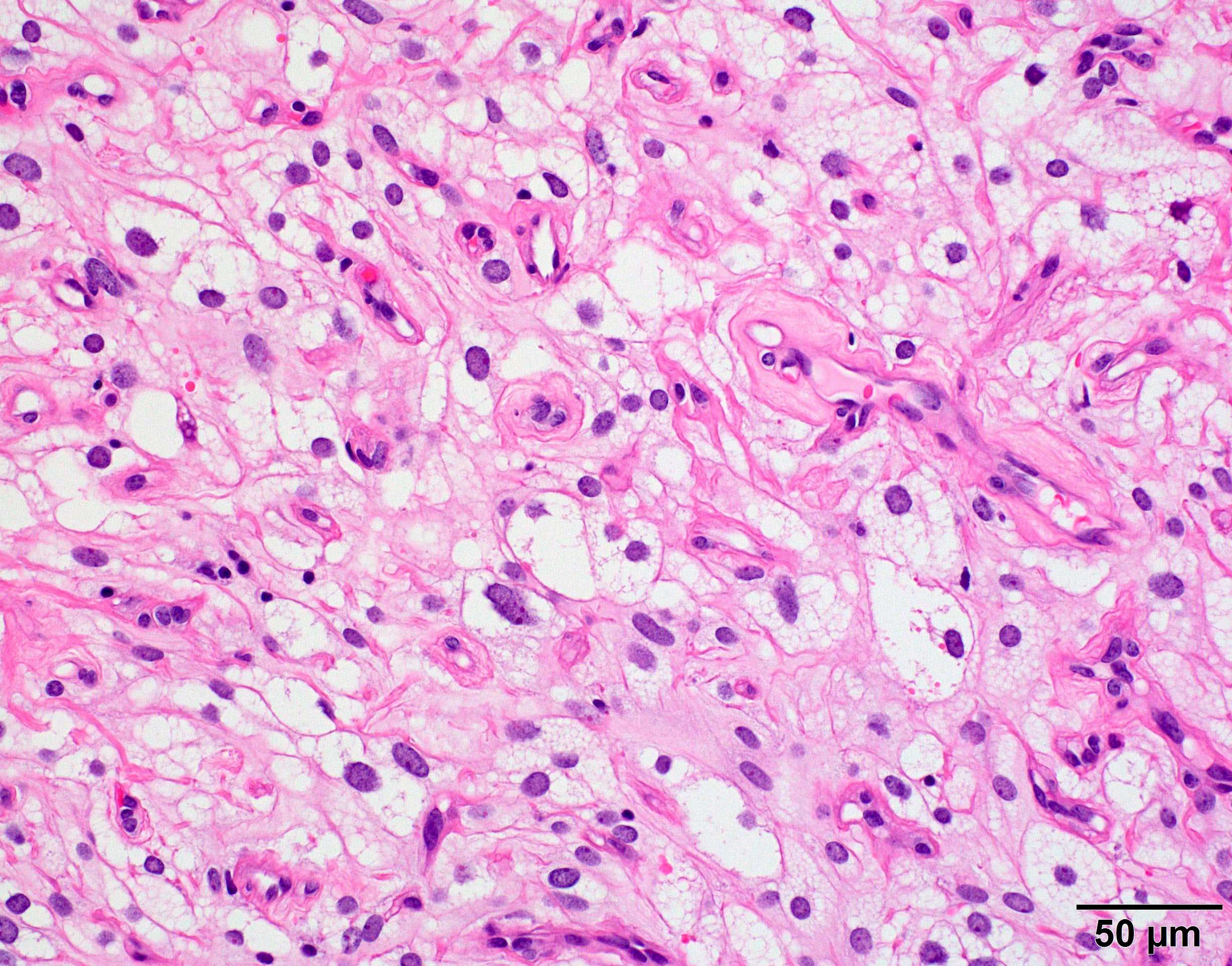
Microcystic meningioma vacuolated cytoplasm
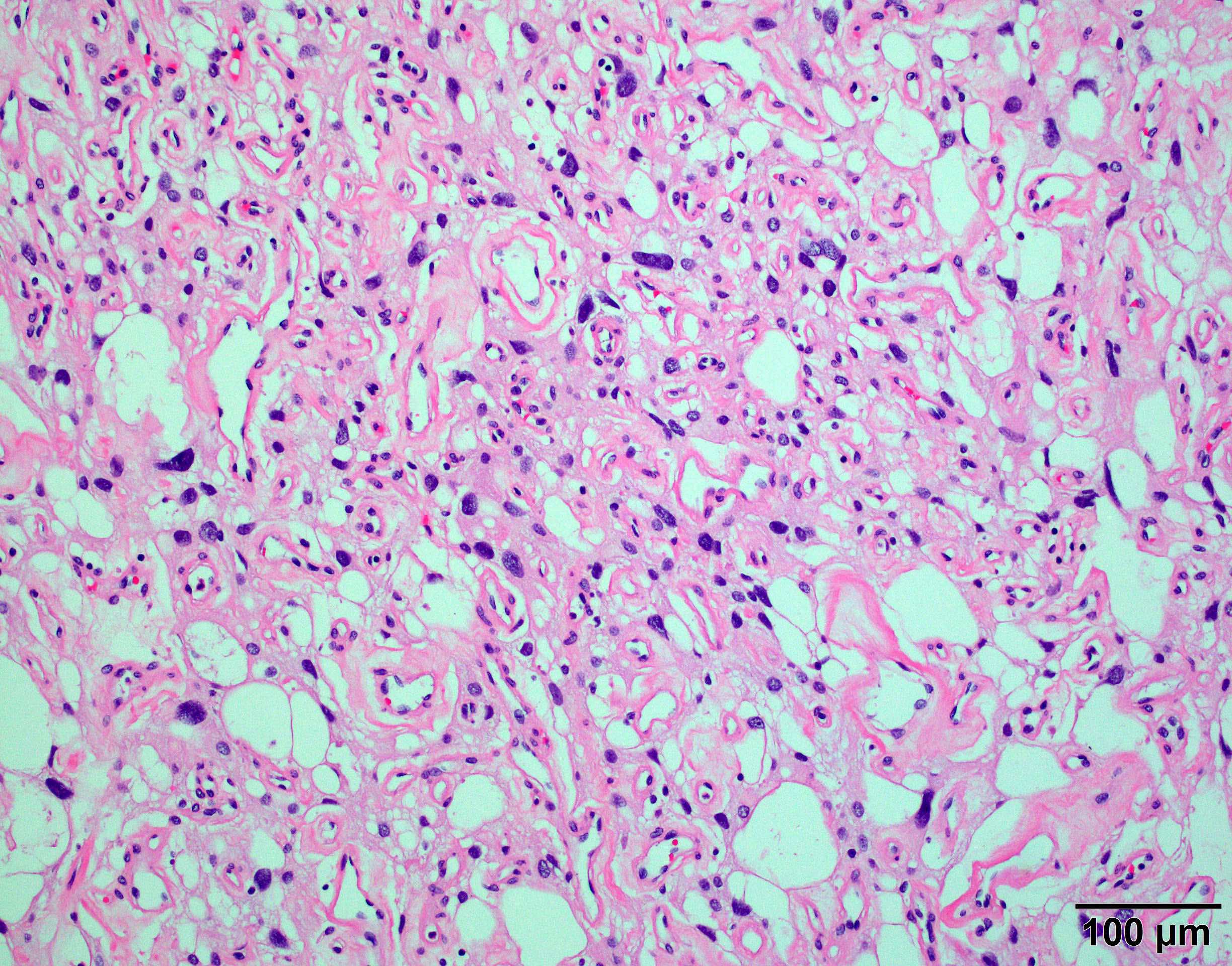
Microcystic meningioma nuclear pleomorphism
Clear cell meningioma:
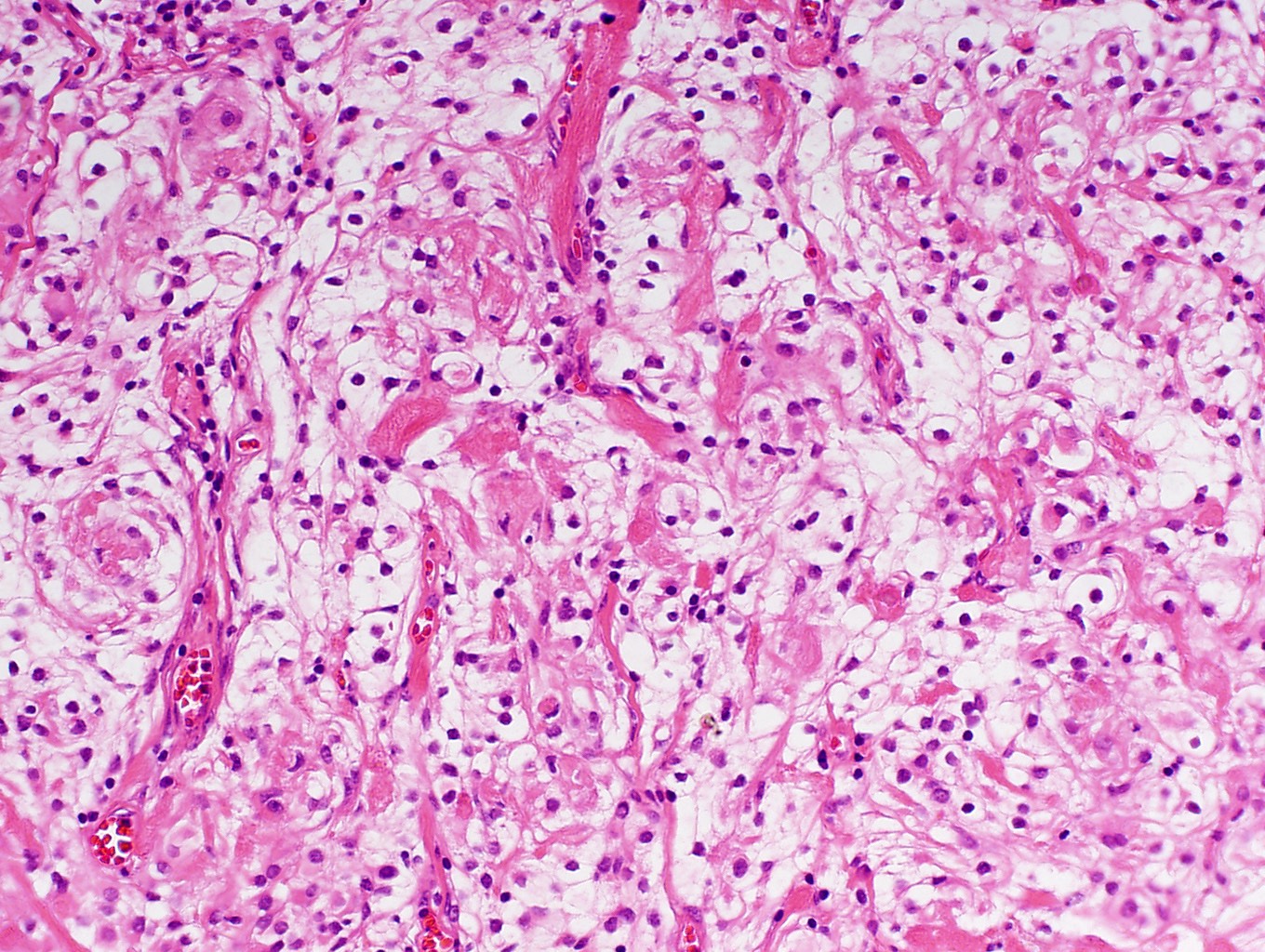
Clear cell meningioma
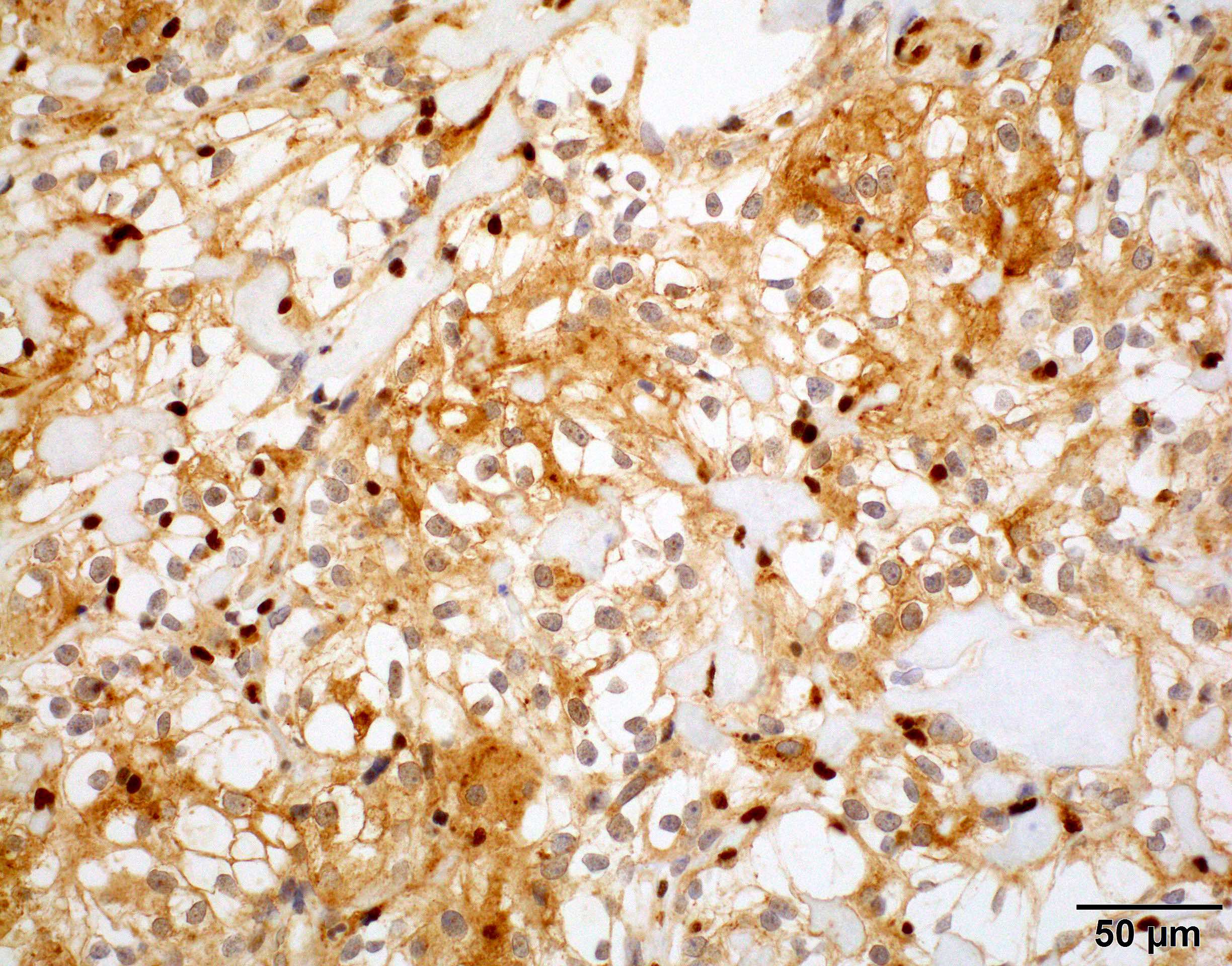
Clear cell meningioma SMARCE1
Lymphoplasmacyte rich meningioma:
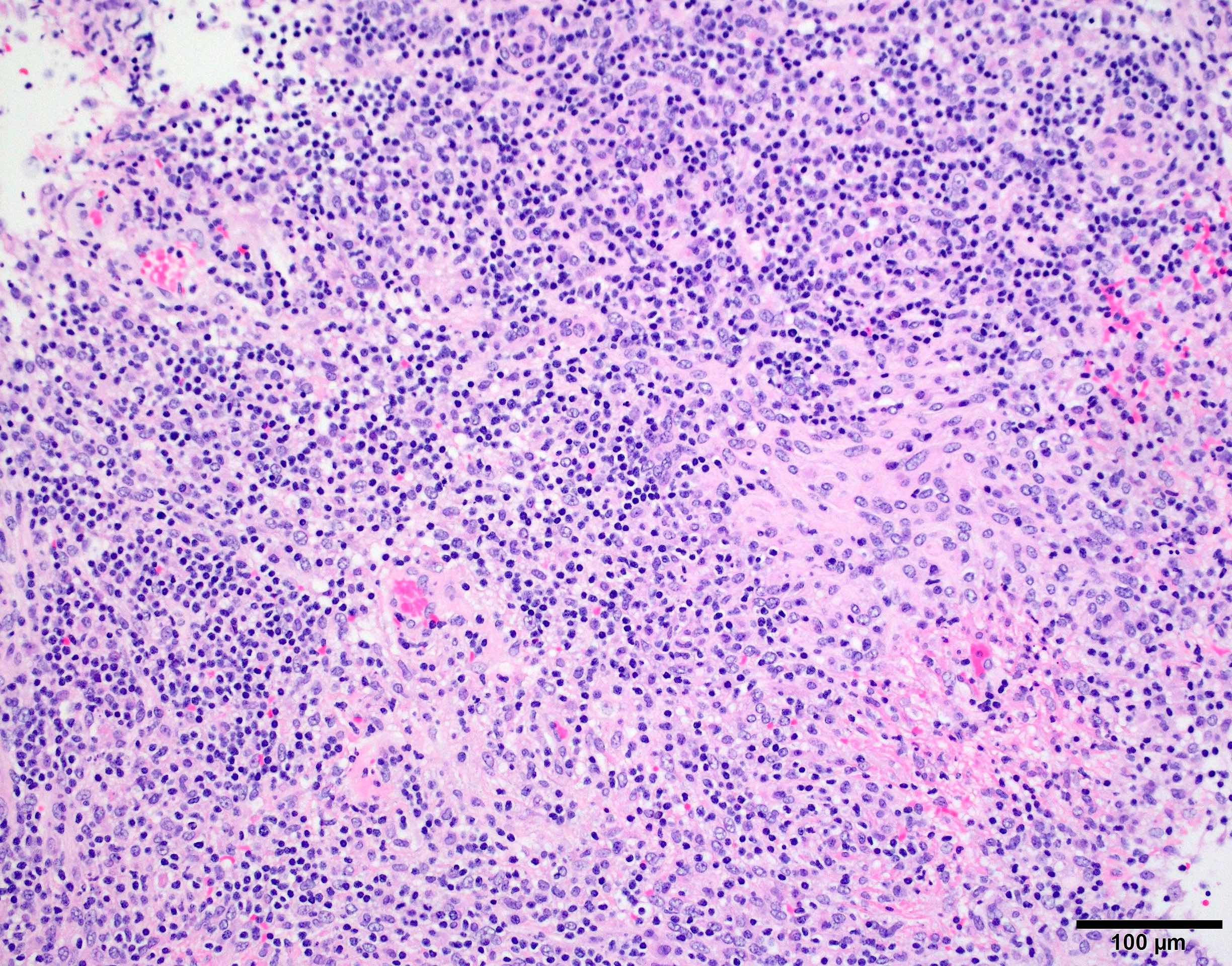
Lymphoplasmacyte
rich meningioma
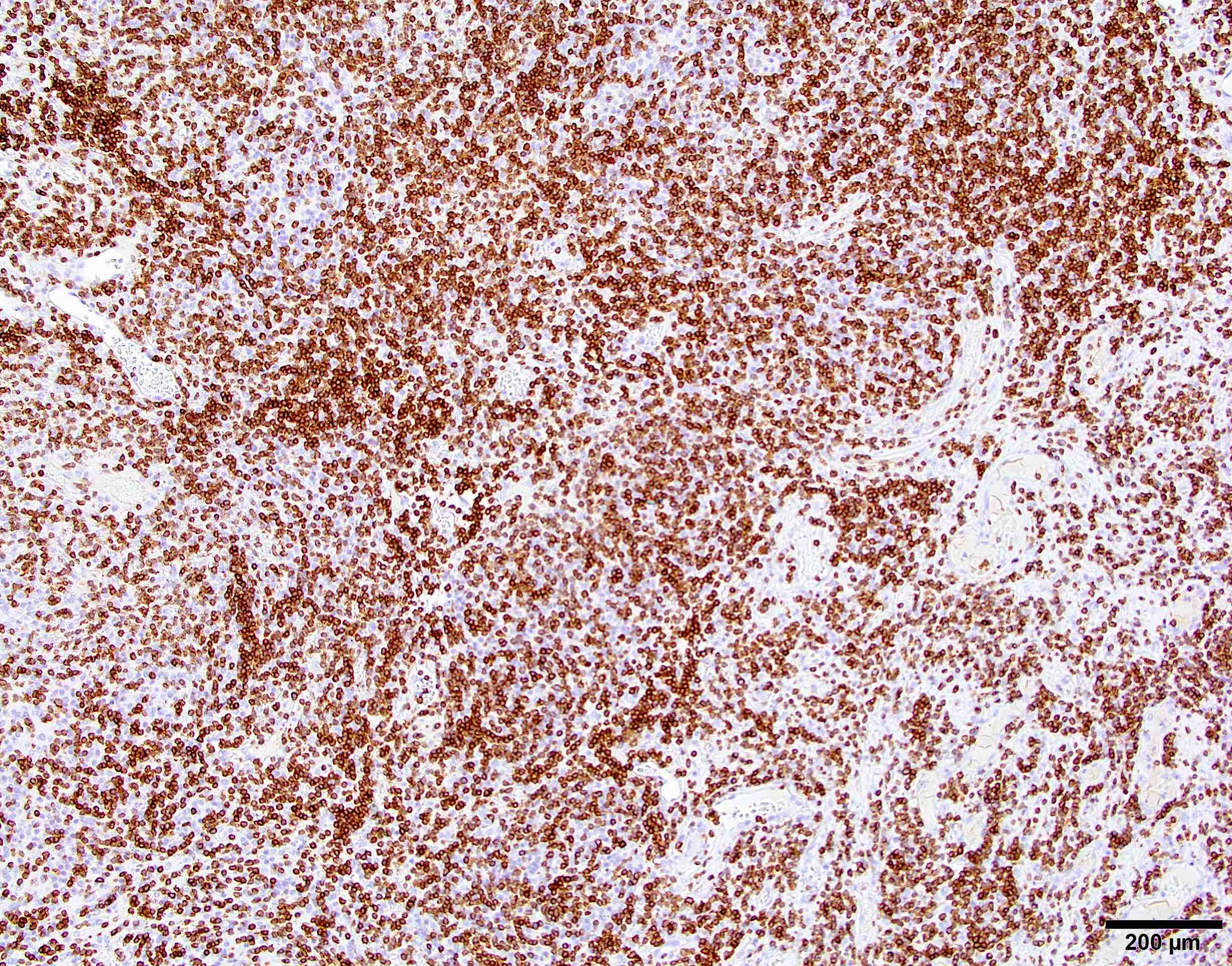
Lymphoplasmacyte
rich meningioma
CD3
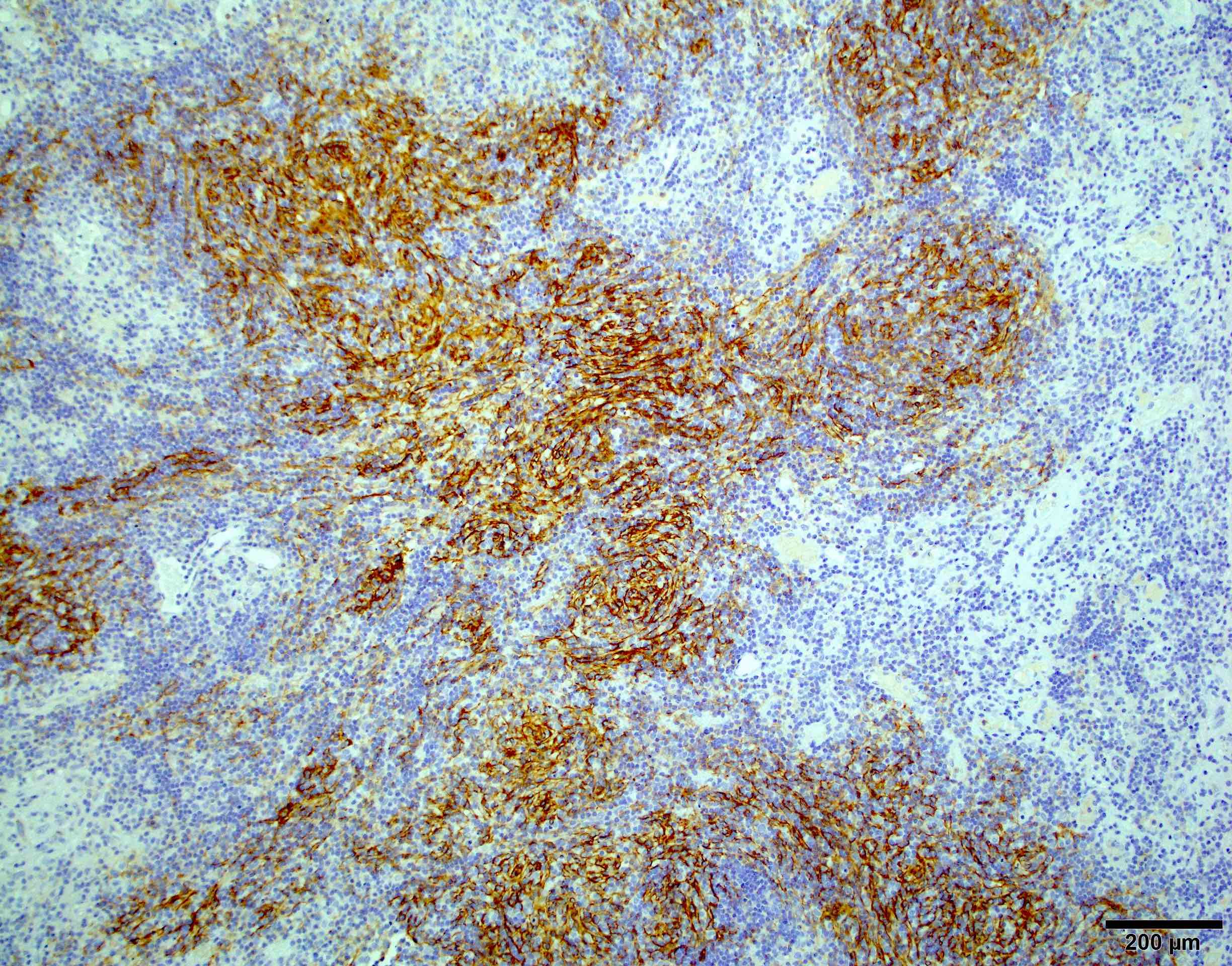
Lymphoplasmacyte
rich meningioma
SSTR2
Secretory meningioma:
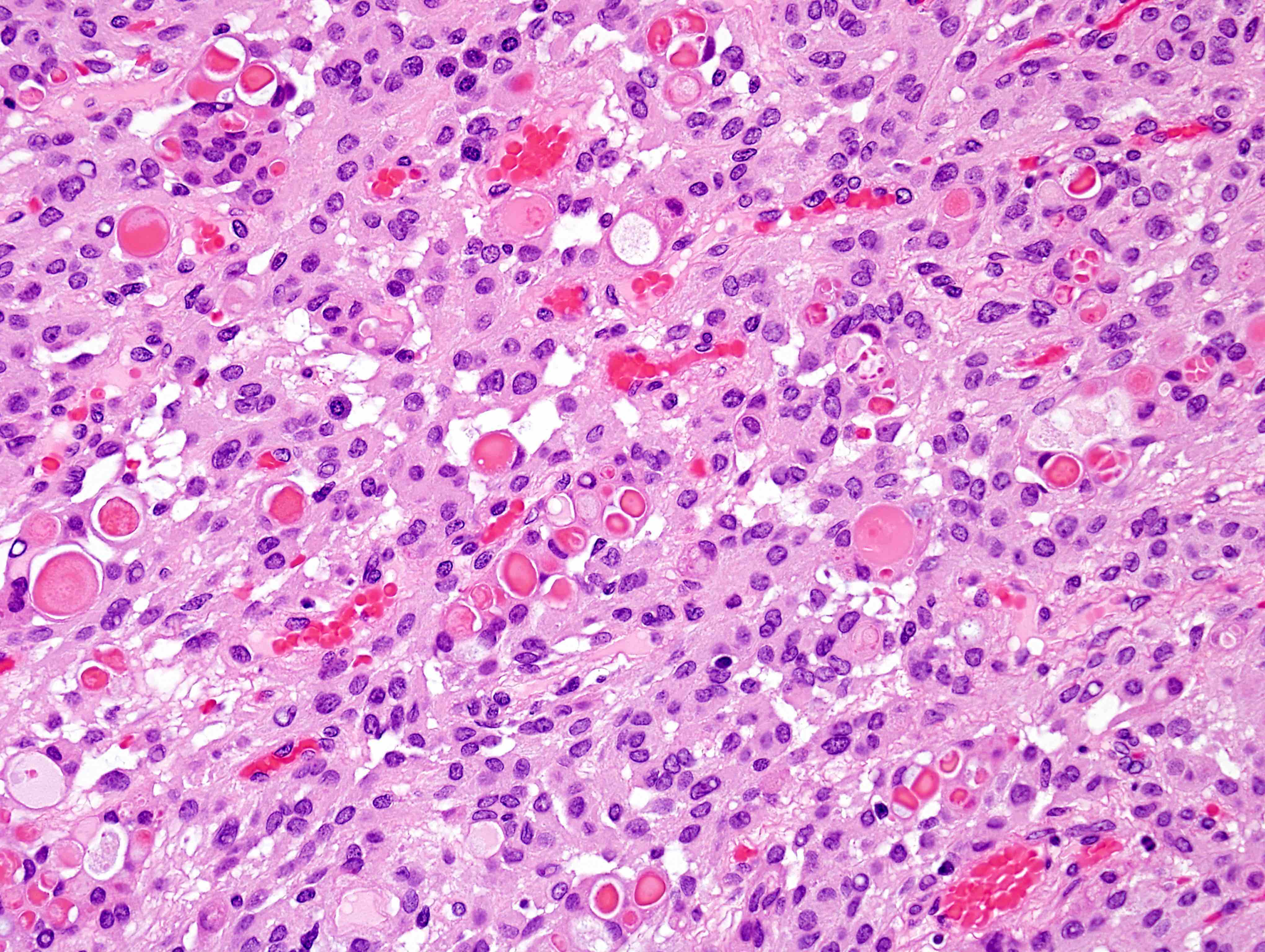
Secretory meningioma
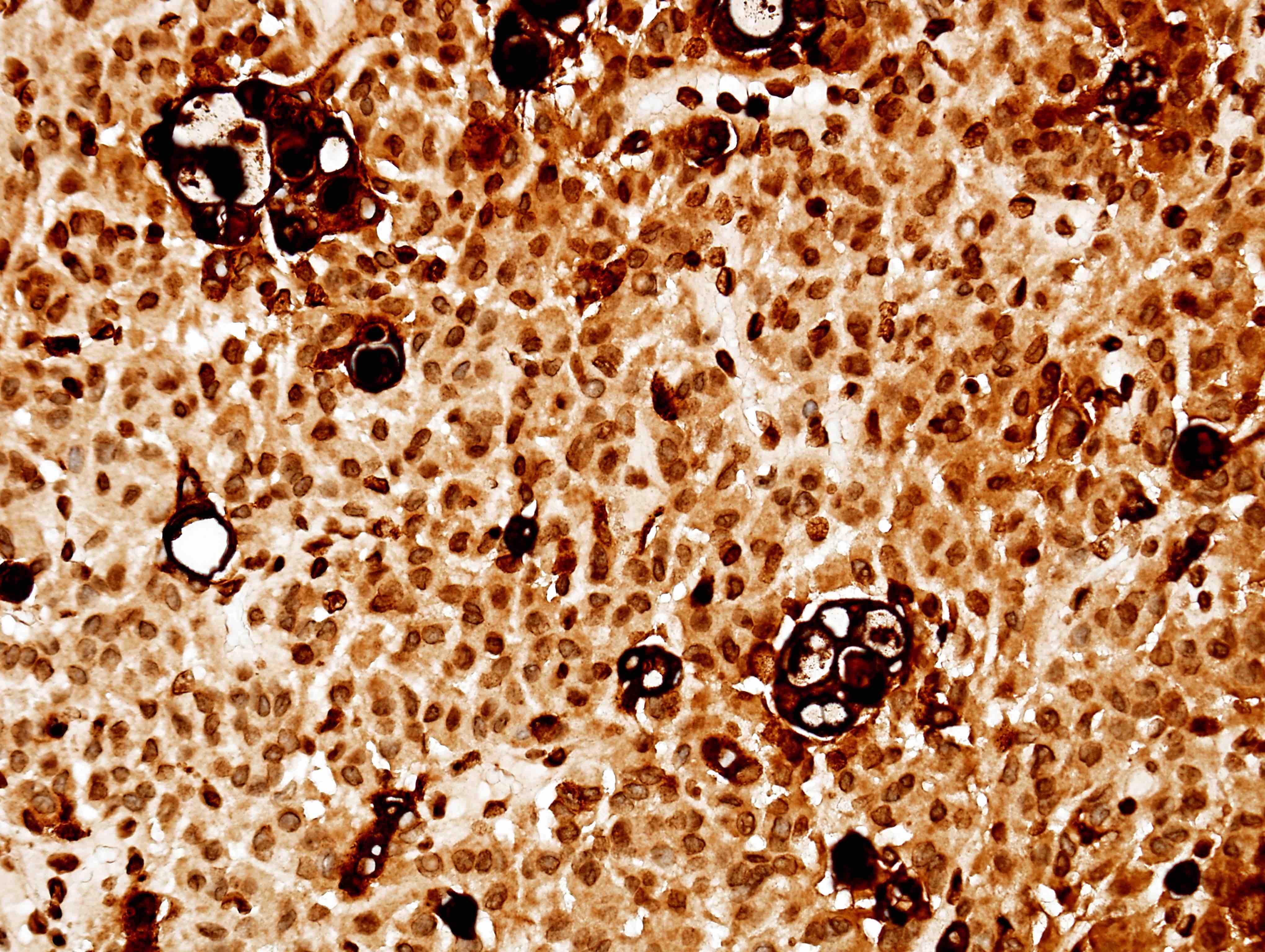
Secretory meningioma pCEA

Secretory meningioma EMA
Other meningioma:
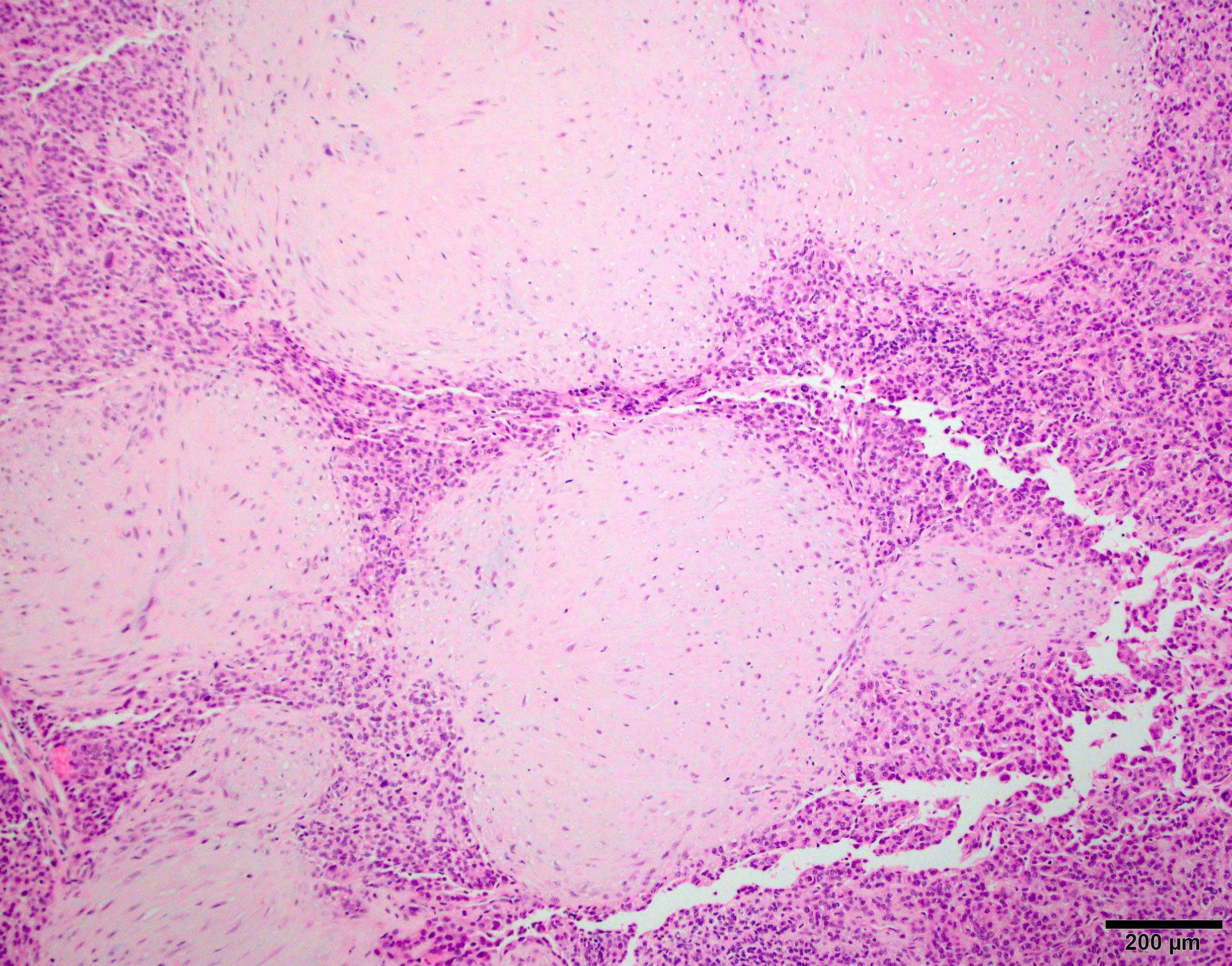
Metaplastic meningioma

Chordoid meningioma
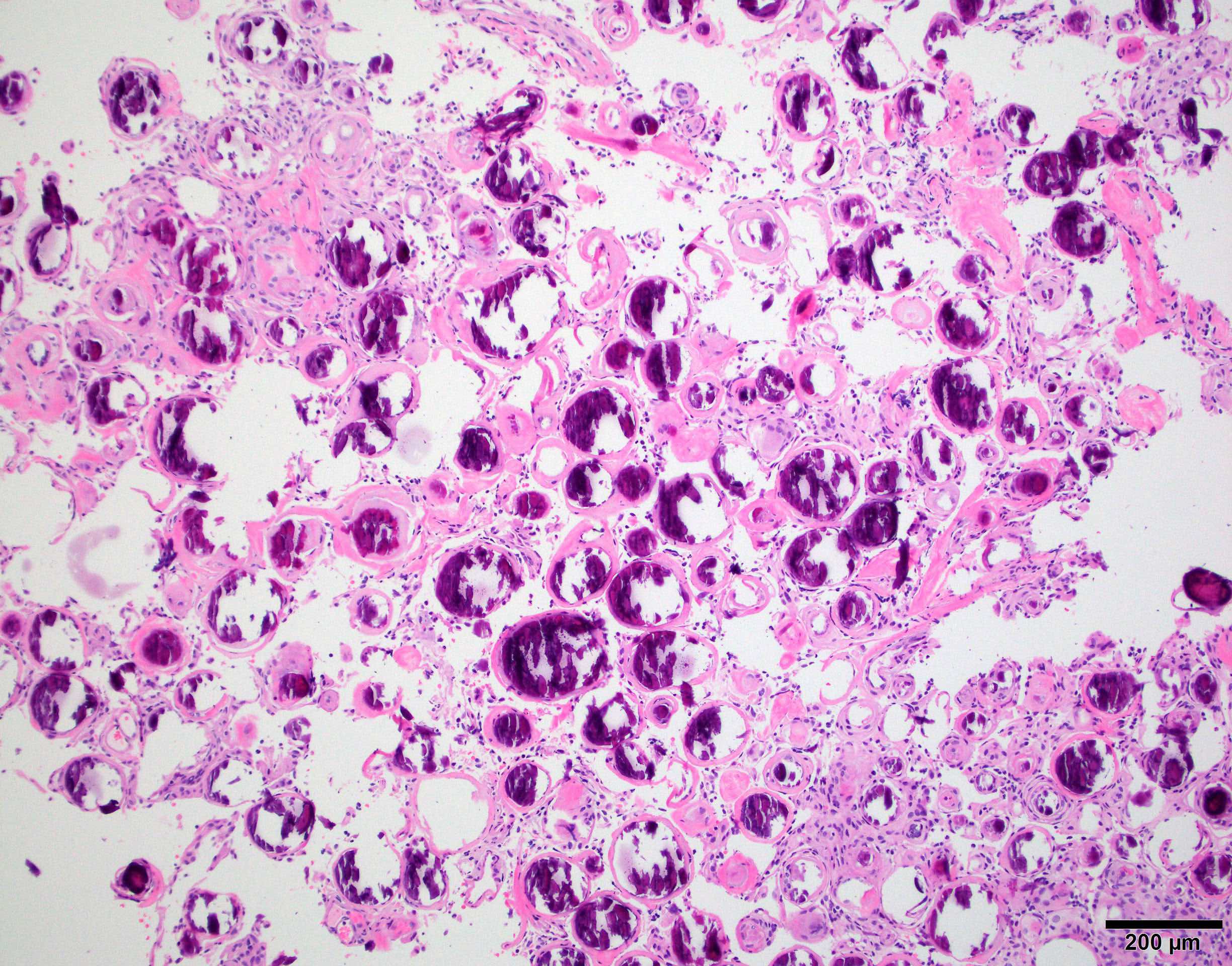
Psammomatous meningioma
Contributed by Andrey Bychkov, M.D., Ph.D.

Meningioma
- Epithelioid cells with round to oval nuclei and streaked cytoplasm; may contain intranuclear pseudoinclusions
- Meningothelial nests or whorls and psammoma bodies are highly characteristic for meningioma on brain tumor crush preps (Burger: Smears and Frozen Sections in Surgical Neuropathology 1st Edition, 2009)
Contributed by Chunyu Cai, M.D., Ph.D.
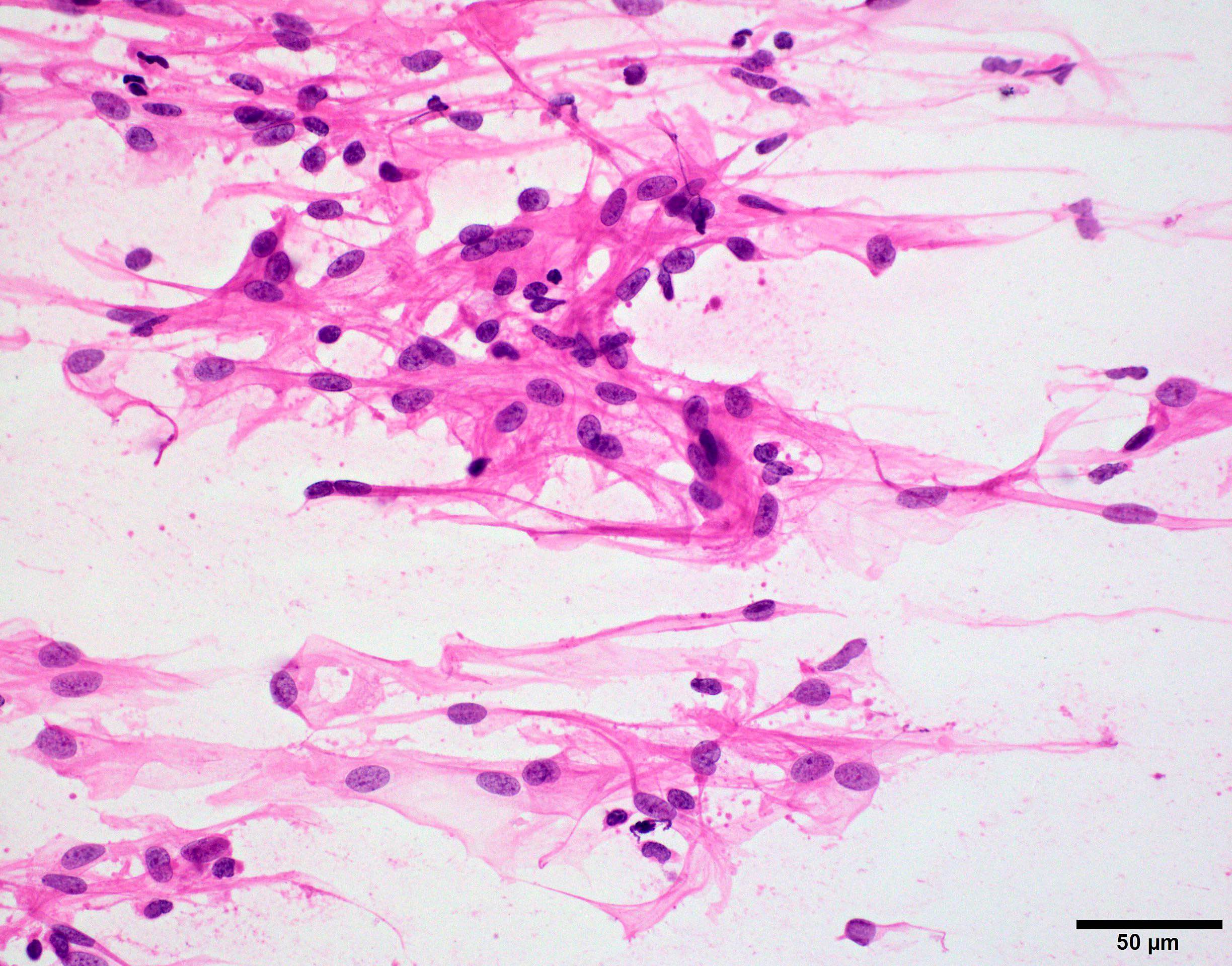
Streaked cytoplasm
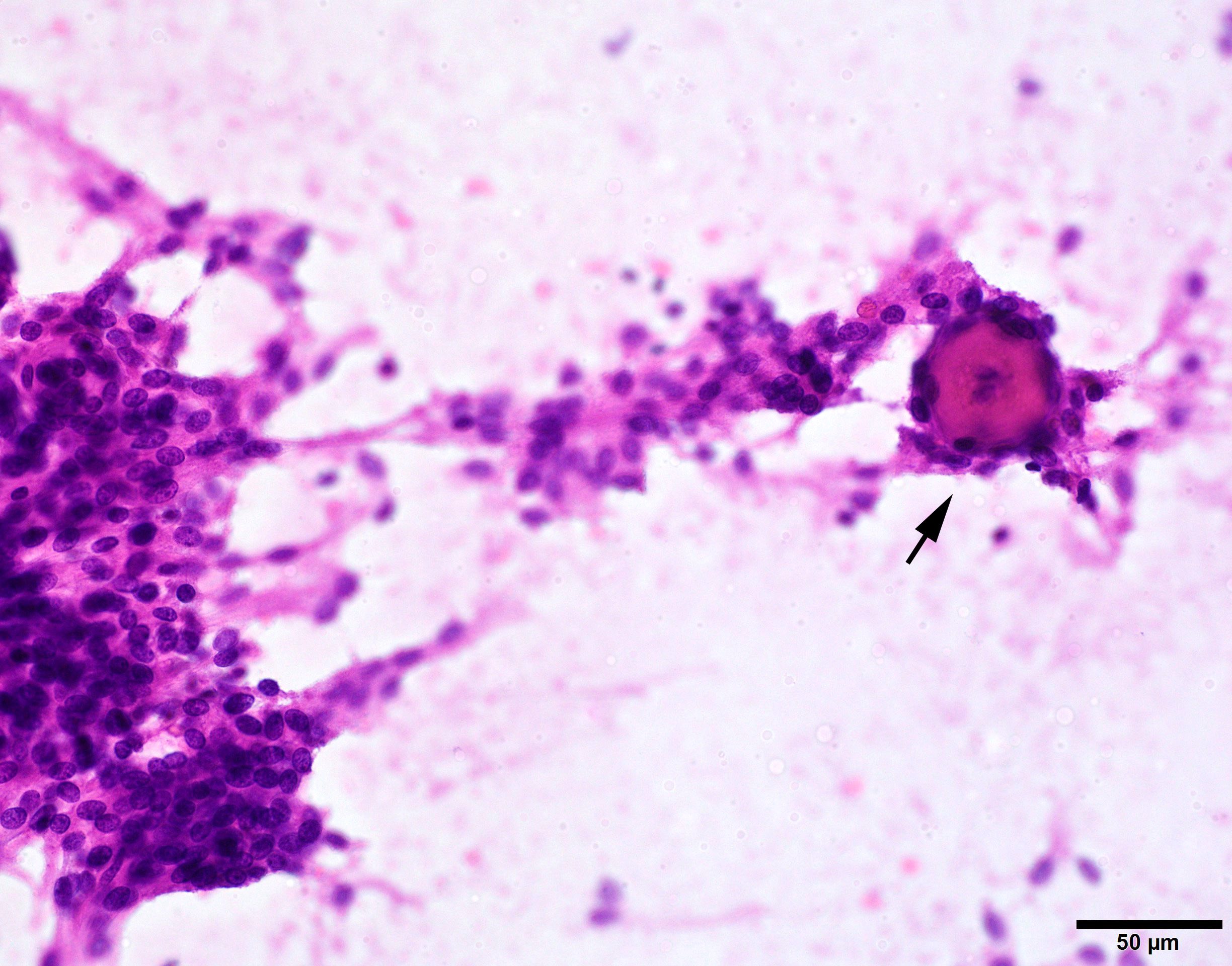
Psammoma body

Cellular whorls
Images hosted on other servers:
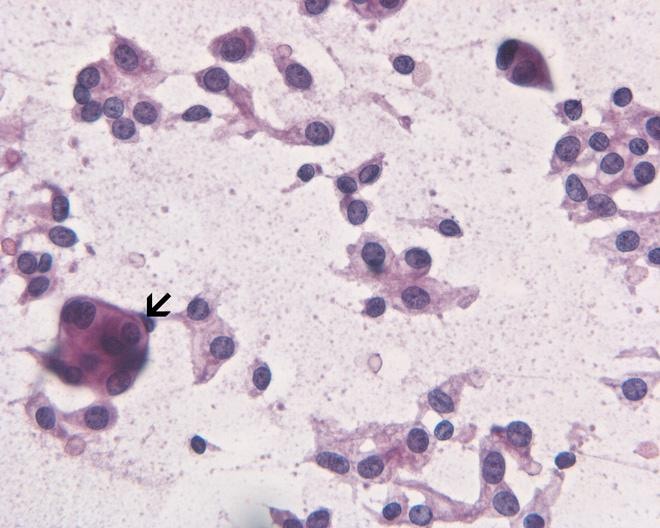
Meningothelial tumor
- Somatostatin receptor 2a (SSTR2a) is a specific meningioma marker in CNS tumors (Acta Neuropathol 2015;130:441, J Neuropathol Exp Neurol 2017;76:289)
- EMA: usually patchy, not diffuse
- Progesterone receptor: diffuse strong nuclear in low grade meningiomas and diminished in high grade meningiomas (J Clin Neurosci 2014;21:421)
- Glial fibrillary acidic protein (GFAP):
- Useful in highlighting entrapped brain in cases with brain invasion
- Differentiates chordoid meningioma (negative) from chordoid glioma (positive) (Am J Surg Pathol 2009;33:669)
- Brachyury: differentiates chordoid meningioma (negative) from chordoma (positive) (Am J Surg Pathol 2009;33:669)
- STAT6: useful to distinguish meningioma (negative) from solitary fibrous tumor / hemangiopericytoma (positive) (J Thorac Cardiovasc Surg 1989;98:291)
- Inhibin: differentiates angiomatous or microcystic meningiomas (negative) from hemangioblastoma (positive) (J Neuropathol Exp Neurol 2017;76:289)
- SOX10: differentiates fibrous meningioma (negative) from schwannomas (positive)
- S100 expression can be seen in ~33% of all meningiomas, particularly fibrous meningiomas (J Neuropathol Exp Neurol 2017;76:289)
- Fibrous, transitional and psammomatous morphologies are associated with NF2 mutation, while meningothelial, secretory and microcystic variants are non-NF2 (Oncogene 2021;40:875)
- Majority of atypical meningiomas have loss of NF2 combined with either genome instability (large scale chromosomal alterations) or loss of SMARCB1 (Nat Commun 2017;8:14433)
- Non-NF2 meningiomas are enriched in mutations in TRAF2, KLF4, AKT1 and SMO, most of which are benign and preferentially located in the skull base (Science 2013;339:1077)
- Clear cell meningioma has been linked to SMARCE1 mutation (J Pathol 2014;234:436)
- Nearly 100% of secretory meningioma contain TRAF7 / KLF4 comutations, mutually exclusive to NF2 (Science 2013;339:1077)
- Angiomatous meningiomas contain multiple chromosome alterations, particularly gains of 5 and 20 (100% and 89%); blood vessels are nonneoplastic in origin (Oncotarget 2014;5:10596)
- DNA methylation profiling of meningioma distinguished 6 methylation classes (MCs) in adults, benign (ben) 1 - 3, intermediate (int) A and B and malignant (mal)
- DNA methylation based meningioma classification is reported to better predict tumor recurrence and prognosis than the WHO histological classification (Lancet Oncol 2017;18:682)
- Pediatric meningiomas are mostly associated with genetic syndromes or childhood radiation
- Global DNA methylation profile grouped separately from adult meningiomas and forms 3 groups: group 1 (clear cell and papillary meningiomas), 2A (enriched in NF2 driven high grade meningiomas) and 2B (enriched for rhabdoid, chordoid and other non-NF2 driven high grade meningiomas)
Molecular classification of meningioma
- Brain, inferior skull base mass (resection):
- Meningioma, transitional (WHO grade 1) (see comment)
- Comment: The tumor is a transitional meningioma. There is no evidence of necrosis, brain invasion, sheet-like growth or increased cellularity. There are no prominent nucleoli or small cells with a high nuclear to cytoplasmic ratio. Mitotic figures are not identified (0/10 high power fields). No brain is present for evaluation of brain invasion.
- Meningothelial meningioma versus meningothelial hyperplasia:
- Meningothelial hyperplasia can be up to 100 cell layers thick and several millimeters in greatest dimension but has a discontinuous growth pattern and never invades dura (Brain Pathol 2005;15:109)
- Meningothelial meningioma is usually a single mass
- Fibrous meningioma versus solitary fibrous tumor (SFT) / hemangiopericytoma:
- Fibrous meningioma versus schwannoma:
- Intracranial myxoid neoplasms (Am J Surg Pathol 2009;33:669, Brain Pathol 2021;31:e12918):
- Chordoid meningioma: SSTR2a+, EMA+, S100-, GFAP-, brachyury-
- Chordoma: brachyury+, EMA+, S100-, SSTR2a-, GFAP-
- Chordoid glioma: intra-axial, near third ventricle, GFAP+, S100+, EMA+, SSTR2a-
- Chondrosarcoma: S100+, EMA-, brachyury-, GFAP-, SSTR2a-
- Intracranial myxoid mesenchymal tumors: EWS or FUS fusions+, S100+, EMA variable, GFAP-, SSTR2a-, brachyury-
- Clear cell meningioma versus microcystic meningioma / metastatic clear cell renal cell carcinoma:
- Clear cell meningioma: SSTR2a+, SMARCE1 loss, CA9-
- Microcystic meningioma: nuclear pleomorphism, SSTR2a+, CA9+, SMARCE1 retained (J Neuropathol Exp Neurol 2019;78:1081)
- Metastatic clear cell renal cell carcinoma: SSTR2a-, PAX8+, RCC+, CA9+ (Appl Immunohistochem Mol Morphol 2010;18:422)
- Angiomatous meningioma and microcystic meningioma versus hemangioblastoma:
A 57 year old man presented with an intracranial convexity based tumor with histology shown in the above image. What is the best set of immunostains to determine the diagnosis?
- p53 and EMA
- S100 and CD34
- SMA and SOX10
- SSTR2a and STAT6
Comment Here
Reference: Meningioma
- Angiomatous, microcystic, meningothelial
- Psammomatous, secretory, fibrous
- Secretory, angiomatous, metaplastic
- Secretory, microcystic, lymphoplasmacyte rich
- Transitional, microcystic, meningothelial
Comment Here
Reference: Meningioma
- Most common brain tumors in community hospitals are metastases (50% of intracranial tumors in hospitalized patients)
- Primary is usually known: melanoma or carcinoma (lung, breast, kidney and GU), occasionally germ cell tumor
- Common with choriocarcinoma
- Common presentation is adult with seizures or ataxia
- Dural metastases are often from breast and prostate, but also unusual primaries; may have survival of 2+ years (Arch Pathol Lab Med 2001;125:880)
- Metastases to vertebral column: often prostate, breast or hematopoietic neoplasm
- Metastases with unknown primary: lung, colon, kidney
- Multiple lesions are suggestive of metastasis vs. primary CNS tumor
- Usually to cerebrum, but no distinct patterns
Meningeal carcinomatosis:
- Represents 4-8% of metastatic brain tumors
- Diffuse spread of tumor in subarachnoid space
- Associated with carcinoma of lung and breast and ALL
- Poor prognosis
- Repeat lumbar punctures and immunocytochemistry may be helpful in differentiating from aseptic meningitis (Arch Pathol Lab Med 2000;124:759)
- Favorable prognostic factors: single metastasis, younger age, surgical resection of metastasis, primary in lung (non-small cell), breast, melanoma, renal cell or ovary
- 18 year old woman with primary alveolar soft part sarcoma in tongue when 8 years old and multiple pulmonary metastases (Case #222)
- 26 year old man with metastatic renal collecting duct carcinoma (Arch Pathol Lab Med 1999;123:638)
- 37 year old man with metastatic salivary gland pleomorphic adenoma to spinal cord (Arch Pathol Lab Med 2003;127:887)
- 38 year old man with contralateral adenoid cystic carcinoma of external ear canal (Arch Pathol Lab Med 2002;126:87)
- A middle aged woman with a history of lung carcinoma presented with a single cerebellar mass (Case #425)
- 77 year old woman with obstructive hydrocephalus due to gallbladder carcinoma (Arch Pathol Lab Med 2001;125:1120)
- Leptomeningeal melanomatosis due to shoulder melanoma (Hum Pathol 2003;34:625)
- Sharply demarcated lesion at gray-white matter junction, surrounded by edema
Images hosted on other servers:
Lung metastasis
- Epithelial cells with discrete cell boundaries, pushing margin except for small cell carcinoma (infiltrative)
Case #222

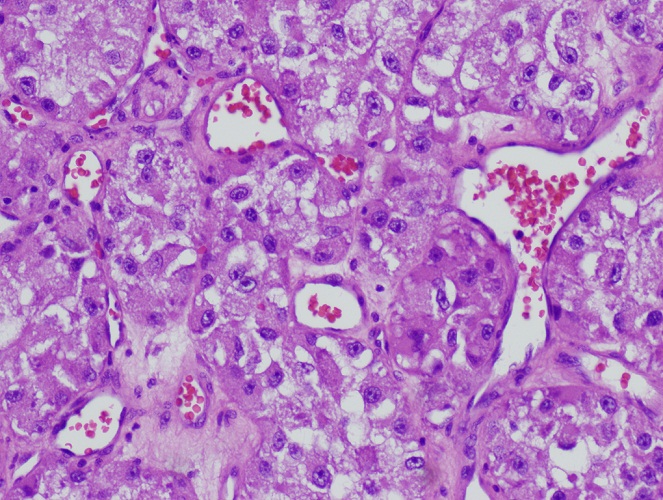
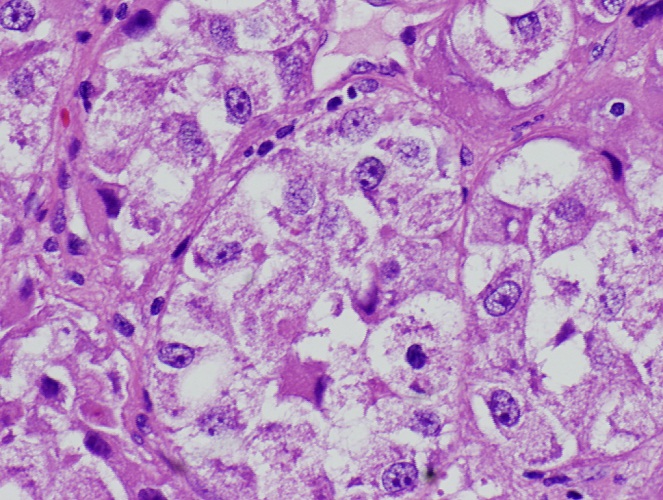
Alveolar soft part sarcoma (H&E)
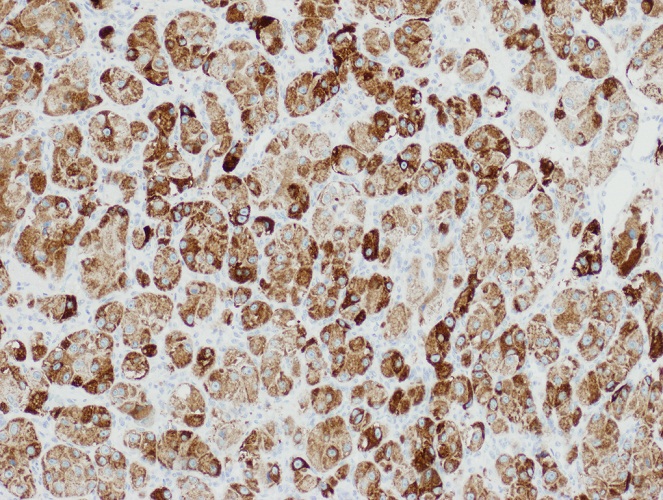
MyoD1

PASD
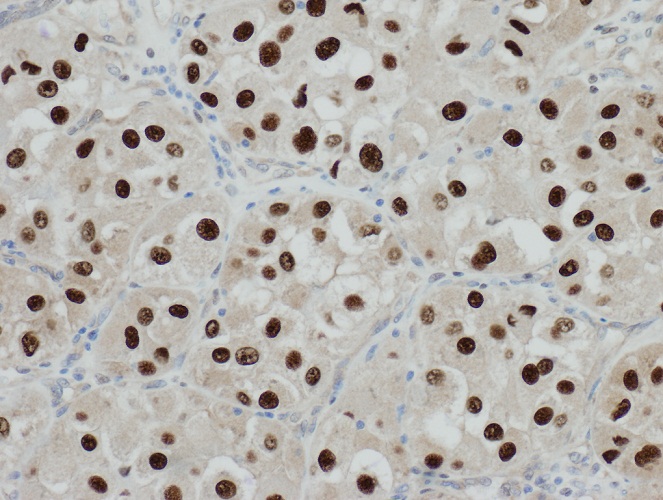
TFE3
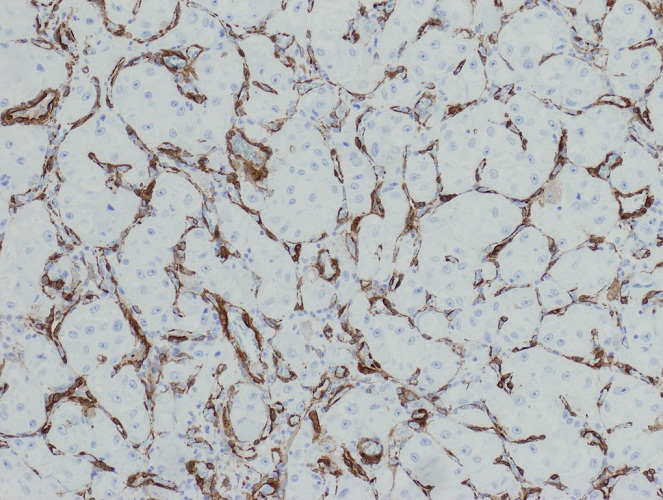
Vimentin
Contributed by Ankur Sangoi, M.D. (Case #425)
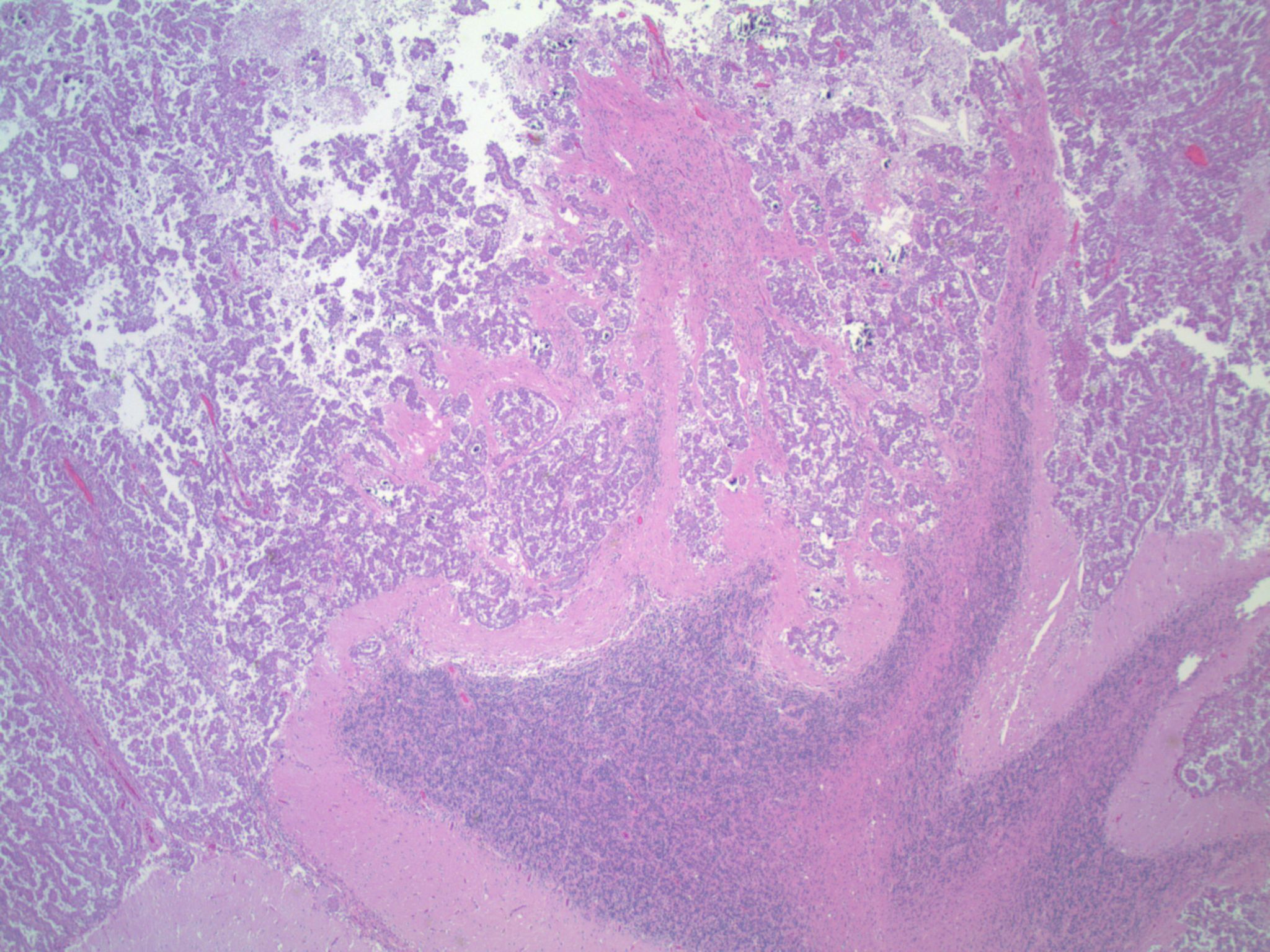
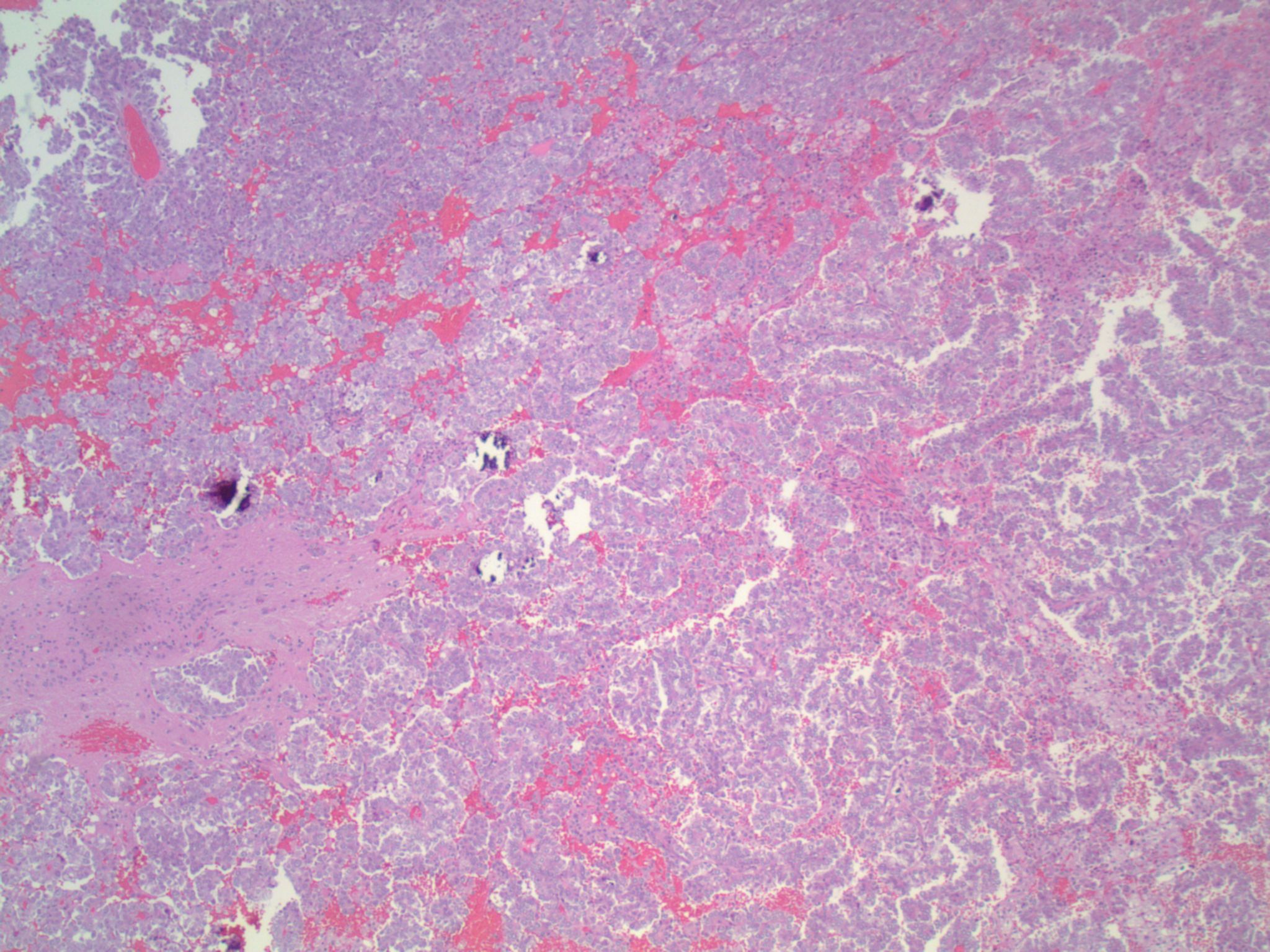
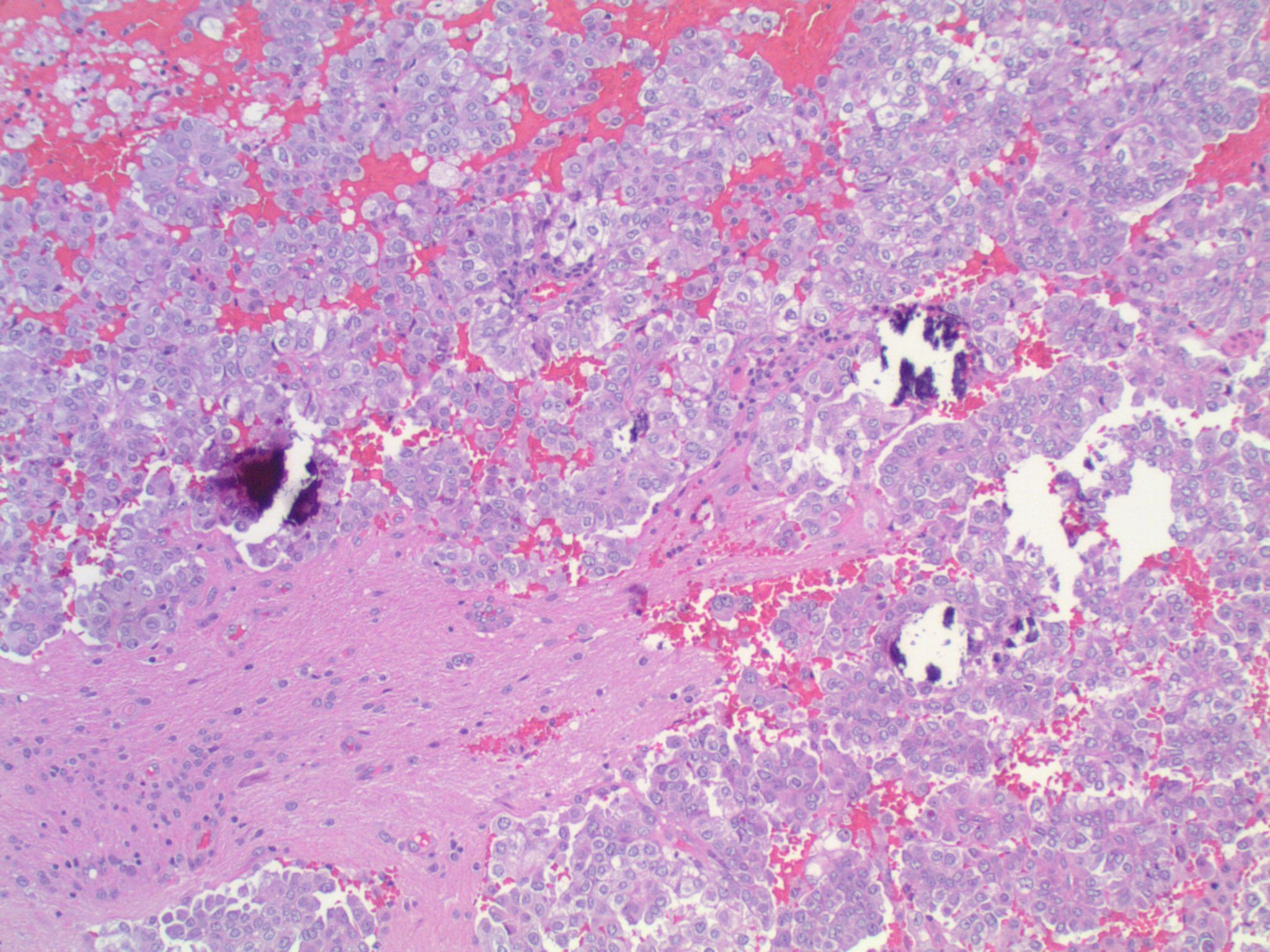
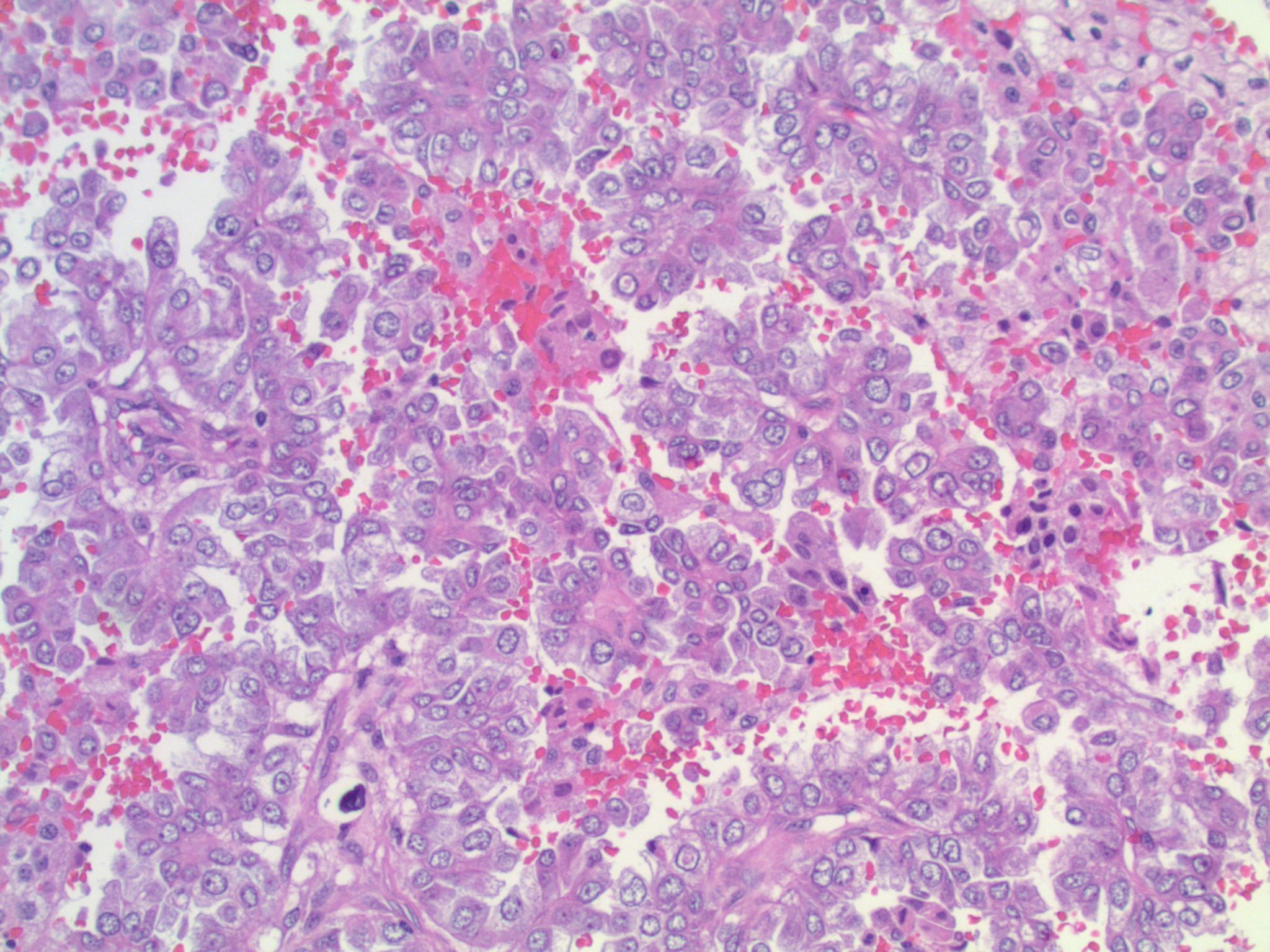
Metastatic lung adenocarcinoma, ALK rearranged
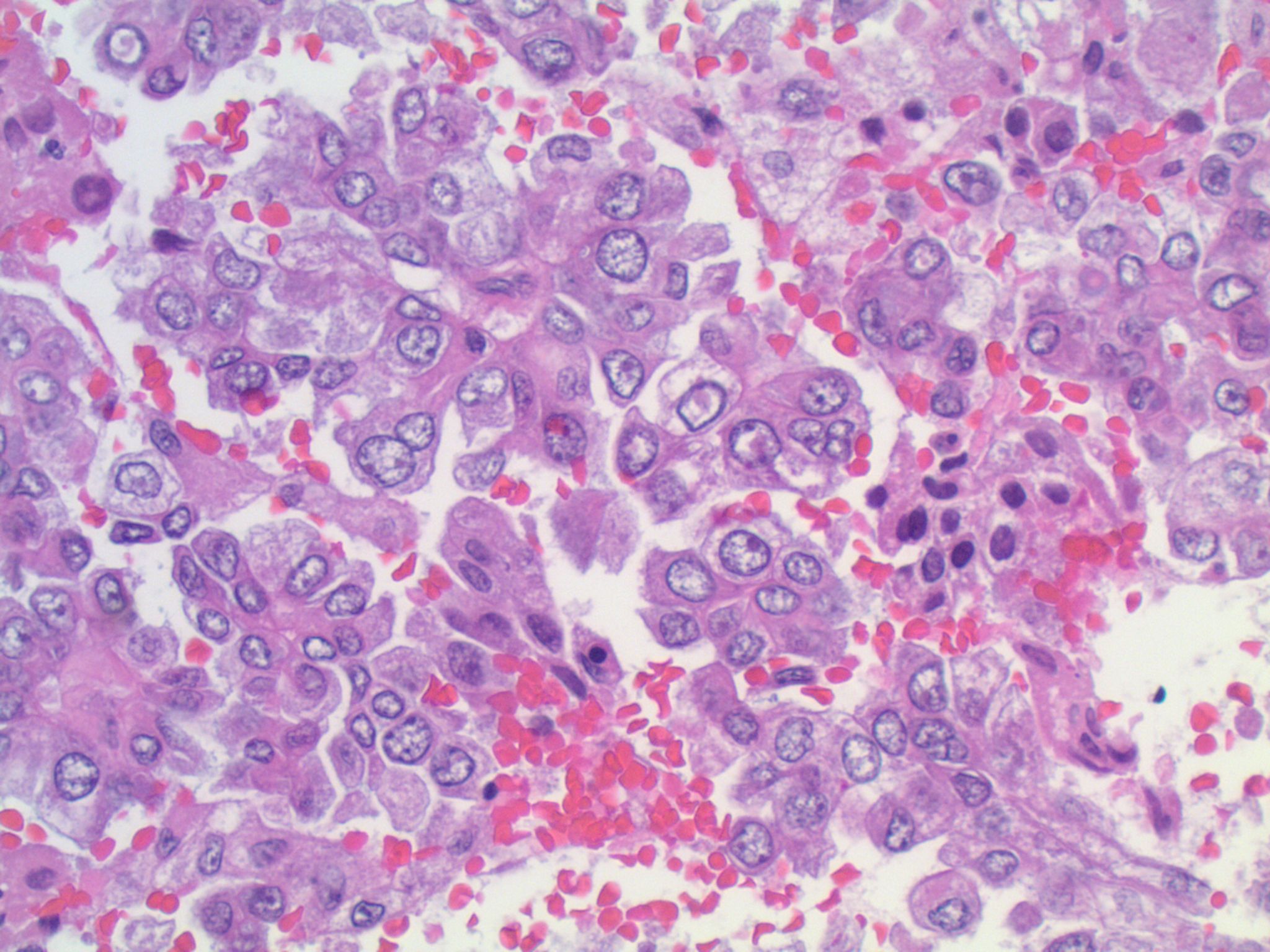
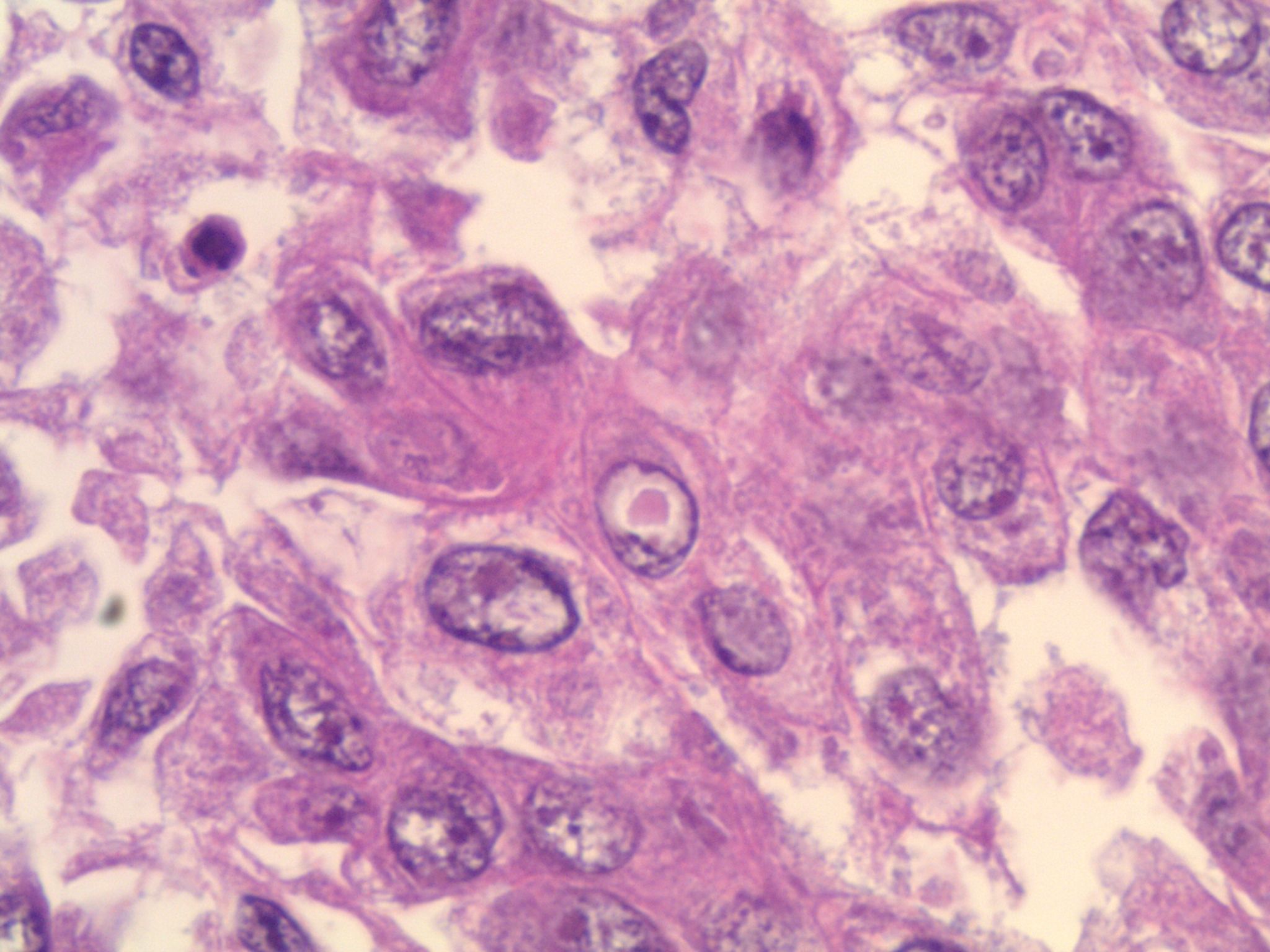

Metastatic lung adenocarcinoma, ALK rearranged

Napsin A
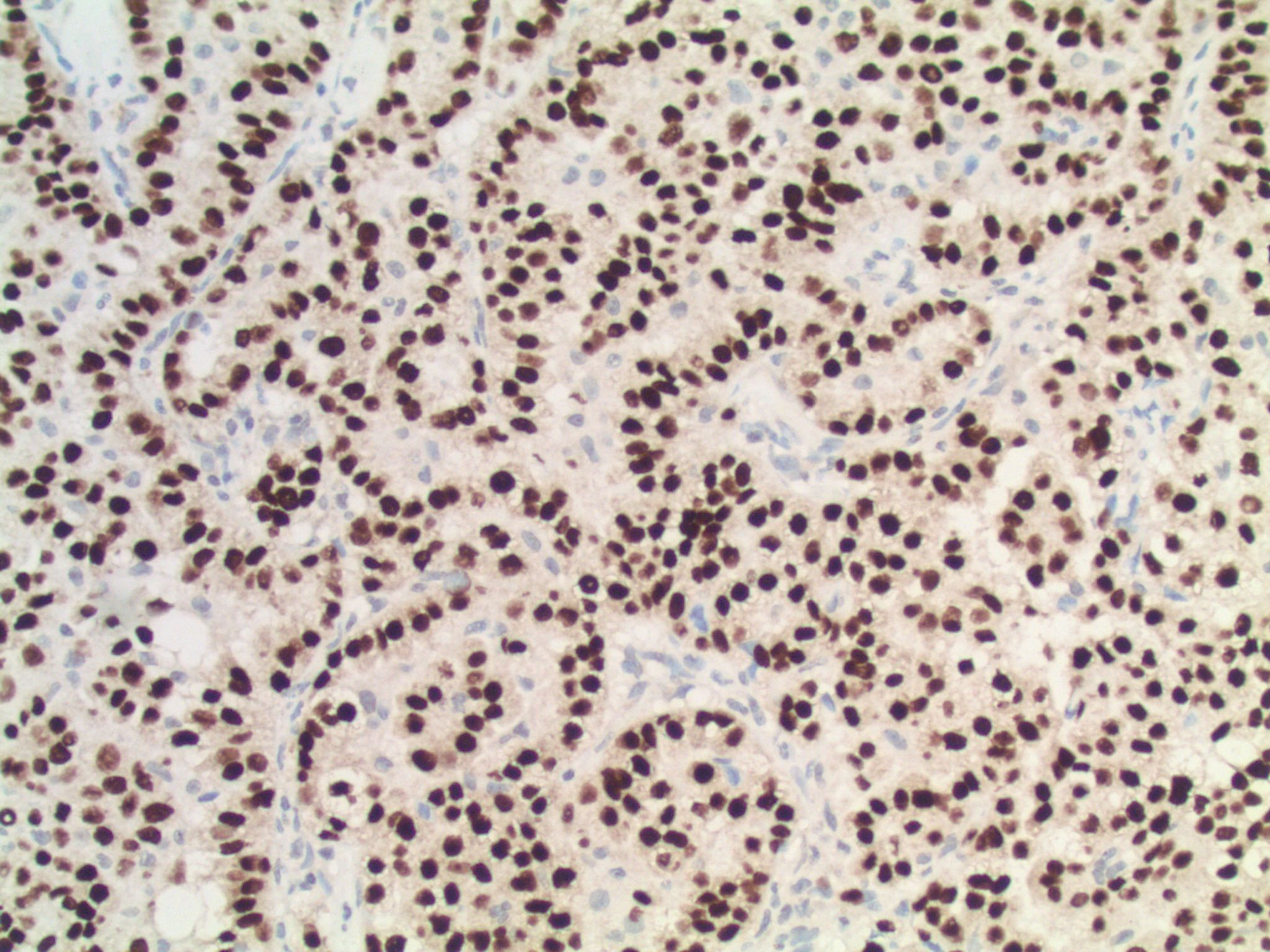
TTF1
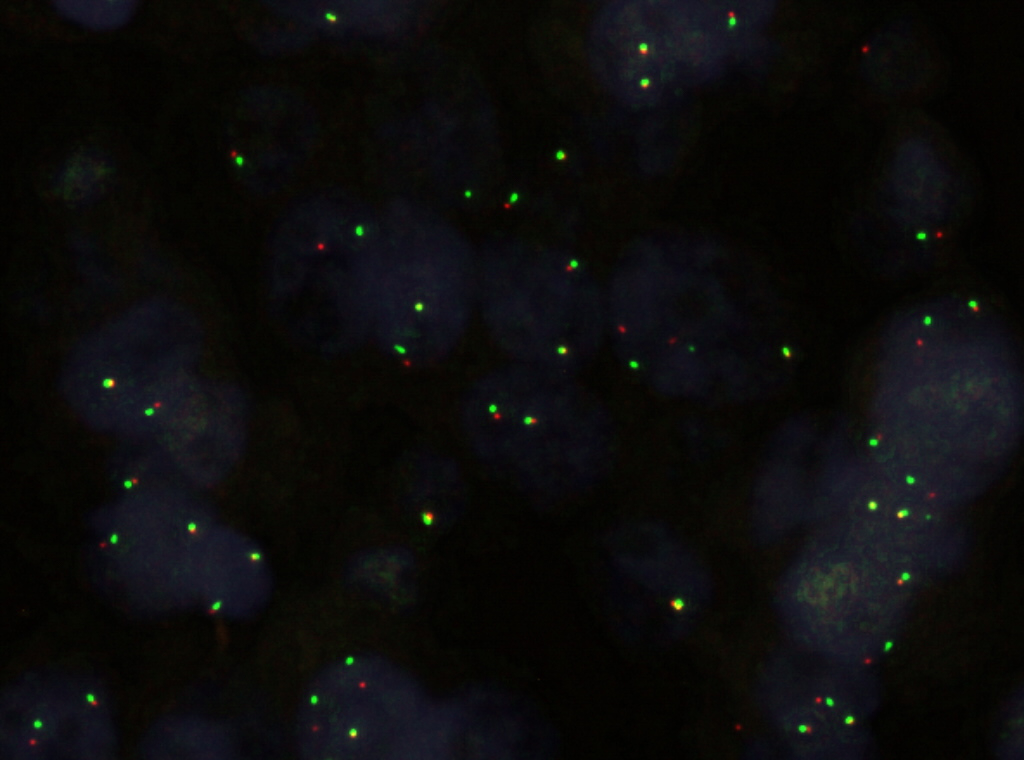

ALK break-apart FISH images (ALK gene in RED)
Images hosted on other servers:
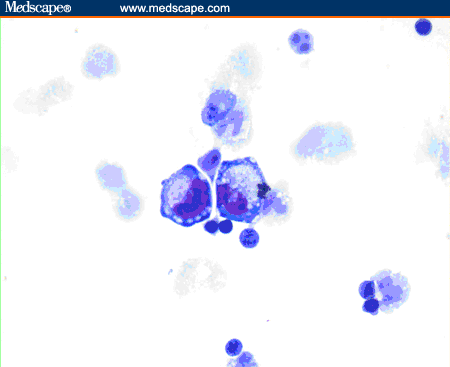
Metastatic GI carcinoma (signet ring cells)
- General: TTF1 (lung and thyroid, Hum Pathol 2002;33:642), cytokeratin and CAM 5.2, CK7, CK20 and PSA
- Breast carcinoma: CK7+, CAM5.2+, CK20-, TTF1-
- Lung adenocarcinoma: CK7,+ CAM5.2+, TTF1+, CK20-
- Colon carcinoma: CK20+, CK7-, TTF1-
- Renal cell carcinoma: RCC+, CAM5.2+, vimentin+
- Anaplastic glioma: no distinct borders, have fibrillary cytoplasmic processes
- Glioblastoma
- Hemangioblastoma vs. metastatic renal cell carcinoma
- Melanoma
- Meningioma: syncytial borders
- PNET vs. metastatic small cell carcinoma: nonfibrillar cells
- CNS WHO grade 2 glial neoplasm with a variably papillary architecture characterized by a radial arrangement of spindled or epithelioid to cuboidal cells around blood vessels with perivascular myxoid change (intermediate layer of Alcian blue positive myxoid stroma) and mucin rich microcyst formation (Neuro Oncol 2021;23:1231)
- Glioma with papillary architecture and perivascular myxoid change or at least focal myxoid microcysts
- Immunoreactive for glial fibrillary acidic protein (GFAP)
- For unresolved tumors, DNA methylation profile is aligned with myxopapillary ependymoma
- ICD-O: 9394/1 - myxopapillary ependymoma
- ICD-11: 2A00.0Y & XH15U1 - other specified gliomas of brain & myxopapillary ependymoma
- Incidence rates are 0.6 - 1.0 cases per 1 million person years
- M:F = 1.4 - 2.1:1
- Seen in all ages but most common in adults
- 2 age peaks: 25 - 29 years and 45 - 49 years (J Neurooncol 2016;129:251, Neuro Oncol 2015;17:588)
- Almost exclusively occurs in the conus medullaris and filum terminale of the spinal cord (Mod Pathol 1997;10:304, Minerva Chir 1997;52:629)
- Occasional examples in lateral and fourth ventricles, inside or outside the brain or in cervicothoracic spinal cord but most often is in a sacrococcygeal or presacral location; rarely reported in mediastinum, lung, uterine adnexae, etc. (Brain Tumor Pathol 2012;29:25, Clin Neuropathol 2004;23:53, Cancer 1985;56:883, J Neurooncol 1993;15:251, J Clin Neurosci 2018;47:202)
- When this tumor occurs at higher levels of neuroaxis, origin from conus / filum terminale should be excluded (Pathology 1996;28:373)
- Pathophysiology is unknown; various recurring chromosomal copy number abnormalities have been described
- No consistent structural variants or other driving mutations have been demonstrated (J Clin Neurosci 2018;50:144, Neuro Oncol 2018;20:1616)
- Unknown
- Chronic lower backache
- Sciatica
- Sensory or motor deficits leading to impotence, urinary and fecal incontinence, myelopathy
- Tissue biopsy with immunohistochemistry
- Morphology (see Microscopic (histologic) description)
- Immunophenotype (see Positive stains)
- Diagnostic molecular pathology: unique DNA methylation profile (Acta Neuropathol 2020;139:305, Cancer Cell 2015;27:728, Neuro Oncol 2018;20:1616)
- Well circumscribed, sharply demarcated, ovoid, contrast enhancing mass in the cauda equina / conus / filum terminale region
- Hyperintense on T1 and T2 weighted MRI with intense contrast enhancement
Contributed by Ahmed Ijaz Gilani, M.B.B.S., M.D., Ph.D.
MRI T1 with contrast
MRI T2
- Relatively favorable prognosis with 10 year survival rates > 90.6% (J Neurooncol 2016;126:165, Neuro Oncol 2015;17:588)
- Often resistant to complete removal due to locally advanced growth or cerebrospinal fluid borne seeding of the thecal sac or more rostral neuroaxis; this may be seen in > 50% of pediatric cases (World Neurosurg 2020;136:e245, J Neurooncol 2016;126:165)
- Many patients live with this disease but require repeated operations and adjuvant therapy
- Tumors arising in the conus have a poorer prognosis than tumors in the cauda equina as the former adhere strongly to the spinal cord and are less amenable to resection
- Radiotherapy improves progression free survival (Neuro Oncol 2015;17:588)
- Anaplastic histology may increase risk of aggressive behavior (Brain Pathol 2019;29:75)
- 6 year old girl with 1 week history of progressive upper and lower extremity weakness and bilateral upper extremity dysesthesia (Childs Nerv Syst 2022;38:223)
- 9 year old girl with slow growing painless mass in the sacrococcygeal region (Am J Dermatopathol 2021;43:e273)
- 22 year old man with acute exacerbation of chronic back pain shooting down both thighs secondary to preoperative intracranial dissemination of spinal myxopapillary ependymoma (World Neurosurg 2021;145:13)
- 32 year old man with primary seeding of myxopapillary ependymoma into multiple lumbosacral areas (World Neurosurg 2017;99:812.e21)
- 34 year old man with multifocal lumbosacral mass presenting with drop metastasis (Spinal Cord Ser Cases 2022;8:43)
- 36 year old woman with spinal cord myxopapillary ependymoma who refused surgery and presented 5 years later with paraparesis (World Neurosurg 2019;121:239)
- Surgical excision is the mainstay of treatment
- Often lifted cleanly from the operative bed; others dissected piecemeal but still in toto
- Low risk of recurrence in either of the above scenarios
- Radiotherapy reserved for subtotally resected tumors
- Some authors recommend radiotherapy for tumors excised completely but piecemeal (Mod Pathol 1997;10:304, Neurosurgery 1992;30:202, Cancer 1985;56:883)
- Often encapsulated, soft in consistency, pink to tan-gray
- May be grossly gelatinous or show hemorrhage and cystic changes
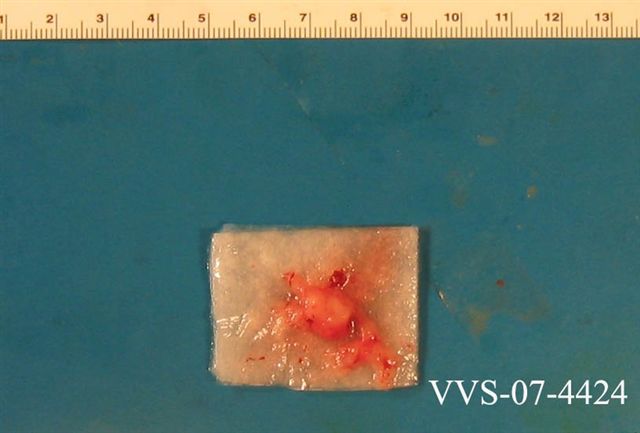
- Intraoperative squash / smear preparation and frozen section reveals epithelioid to spindled tumor cells arranged around microcysts and forming papillary structures with perivascular myxoid change; these features are diagnostic in the appropriate clinical setting
- Most common pattern is radial arrangement of cuboidal to epithelioid elongated glial tumor cells around hyalinized fibrovascular (central, often hyalinized blood vessels) cores in a papillary configuration
- Accumulation of basophilic myxoid material around blood vessels (myxoid stroma) and in microcysts
- Myxoid material is highlighted by PAS and Alcian blue positive staining
- In cases composed of confluent sheets of epithelioid cells with little or no papillary structures, PAS and Alcian blue positivity is useful in reaching a correct diagnosis
- Fascicular growth and spindle cells are common
- Pleomorphic tumor giant cells can be seen
- Occasionally tumor cells show distinctive eosinophilic balloons; these are PAS positive spherules that demonstrate spiculated reticulin staining (Am J Surg Pathol 1996;20:1091)
- Uncommon examples reported as anaplastic myxopapillary ependymomas show hypercellularity and reduced mucin in association with at least 2 of the following features: ≥ 5 mitoses / 10 high power field, Ki67 labeling index ≥ 10%, microvascular proliferation, spontaneous necrosis (Brain Pathol 2019;29:75)
Contributed by Ahmed Ijaz Gilani, M.B.B.S., M.D., Ph.D. and Aisha Hassan Memon, M.B.B.S.
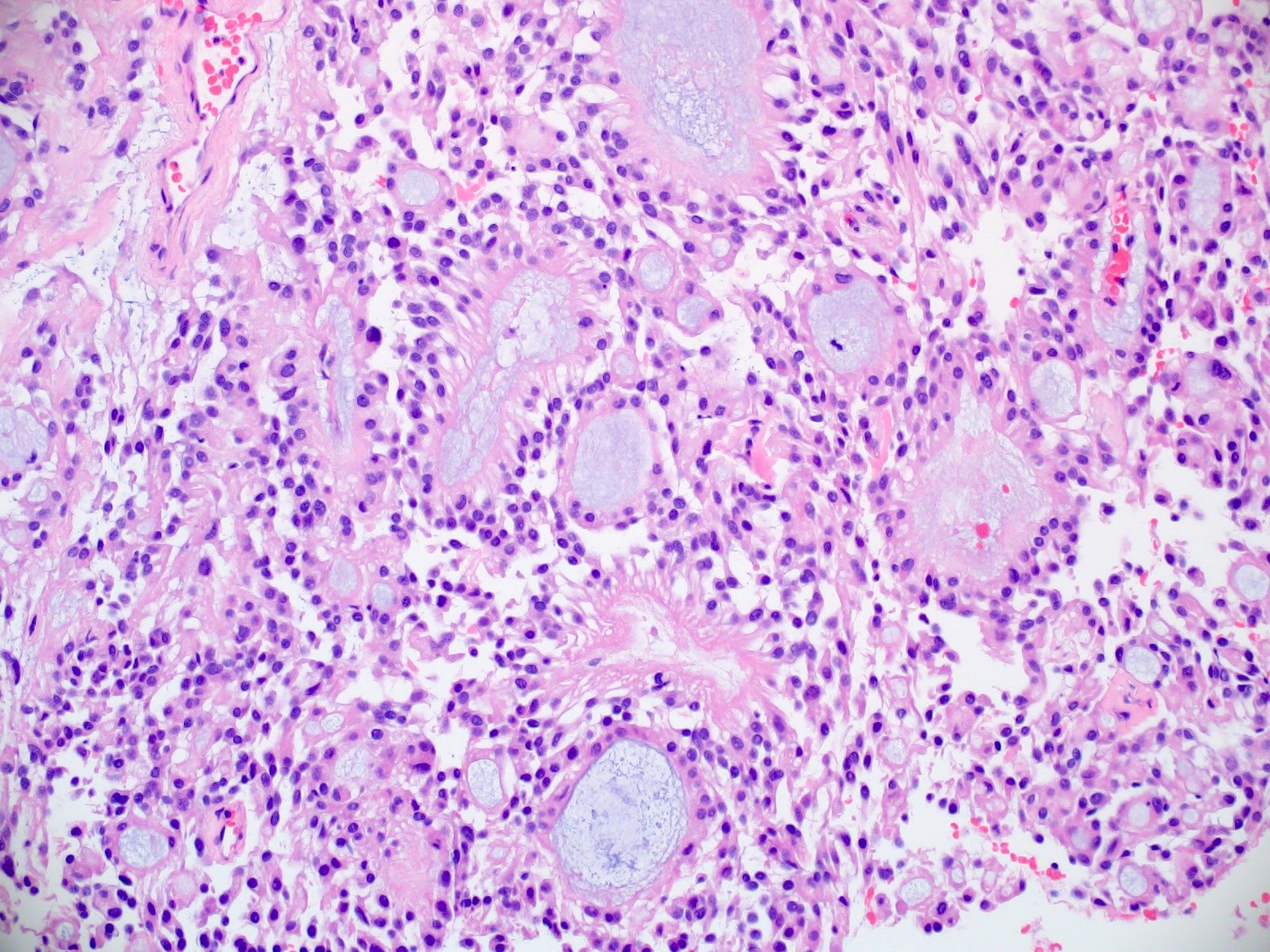
Radial arrangement
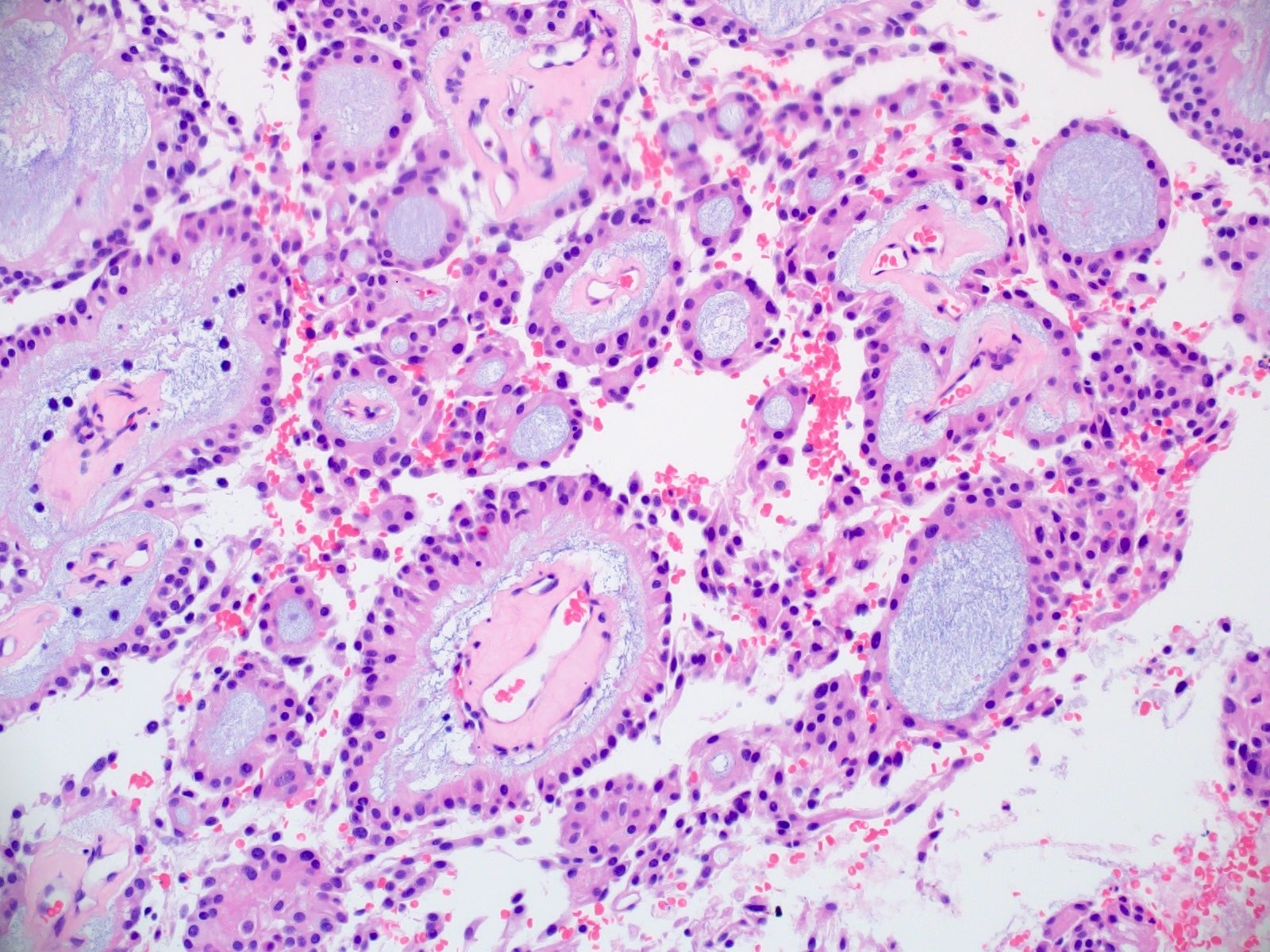
Epithelioid tumor cells
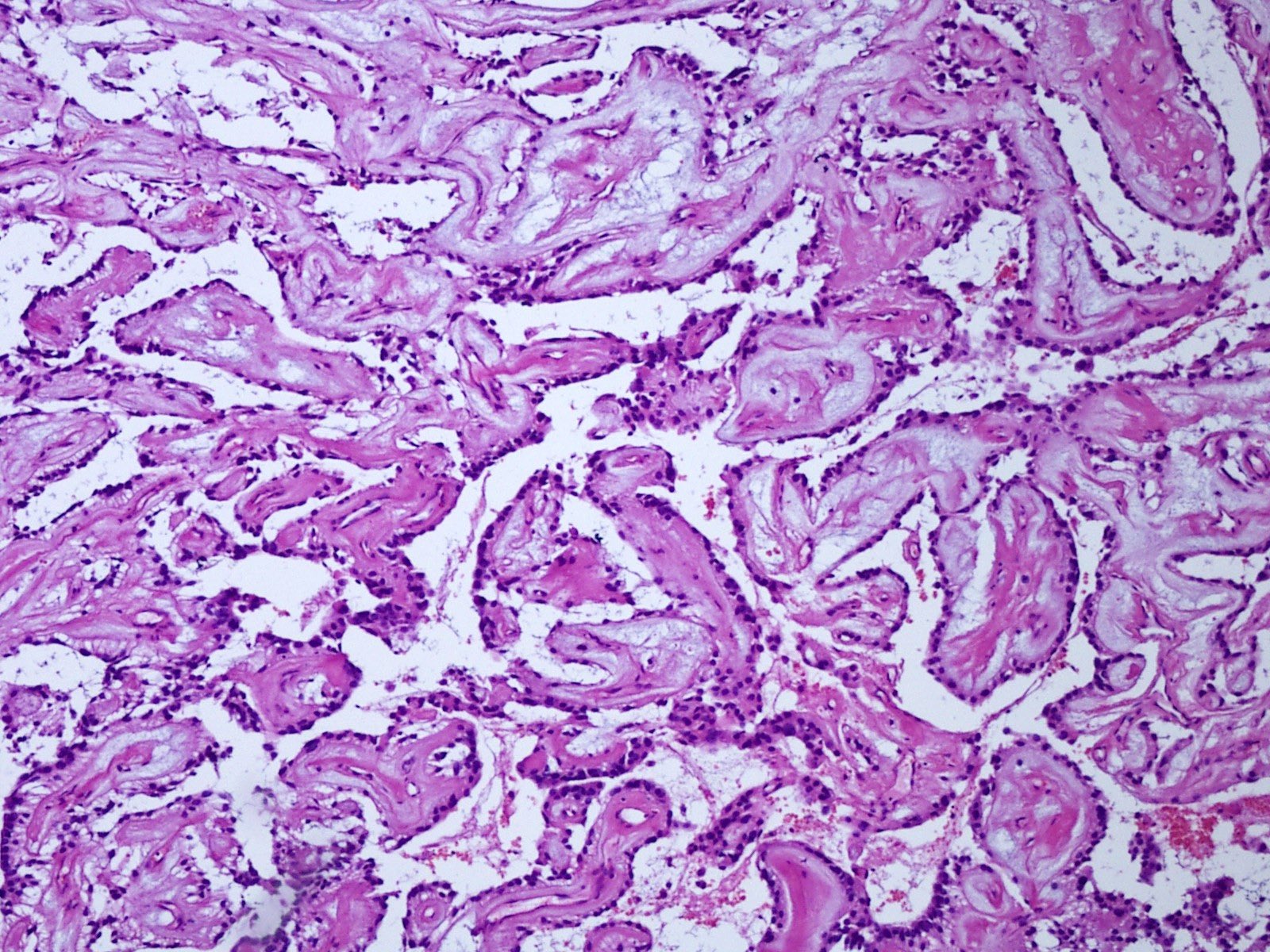
Papillary structures
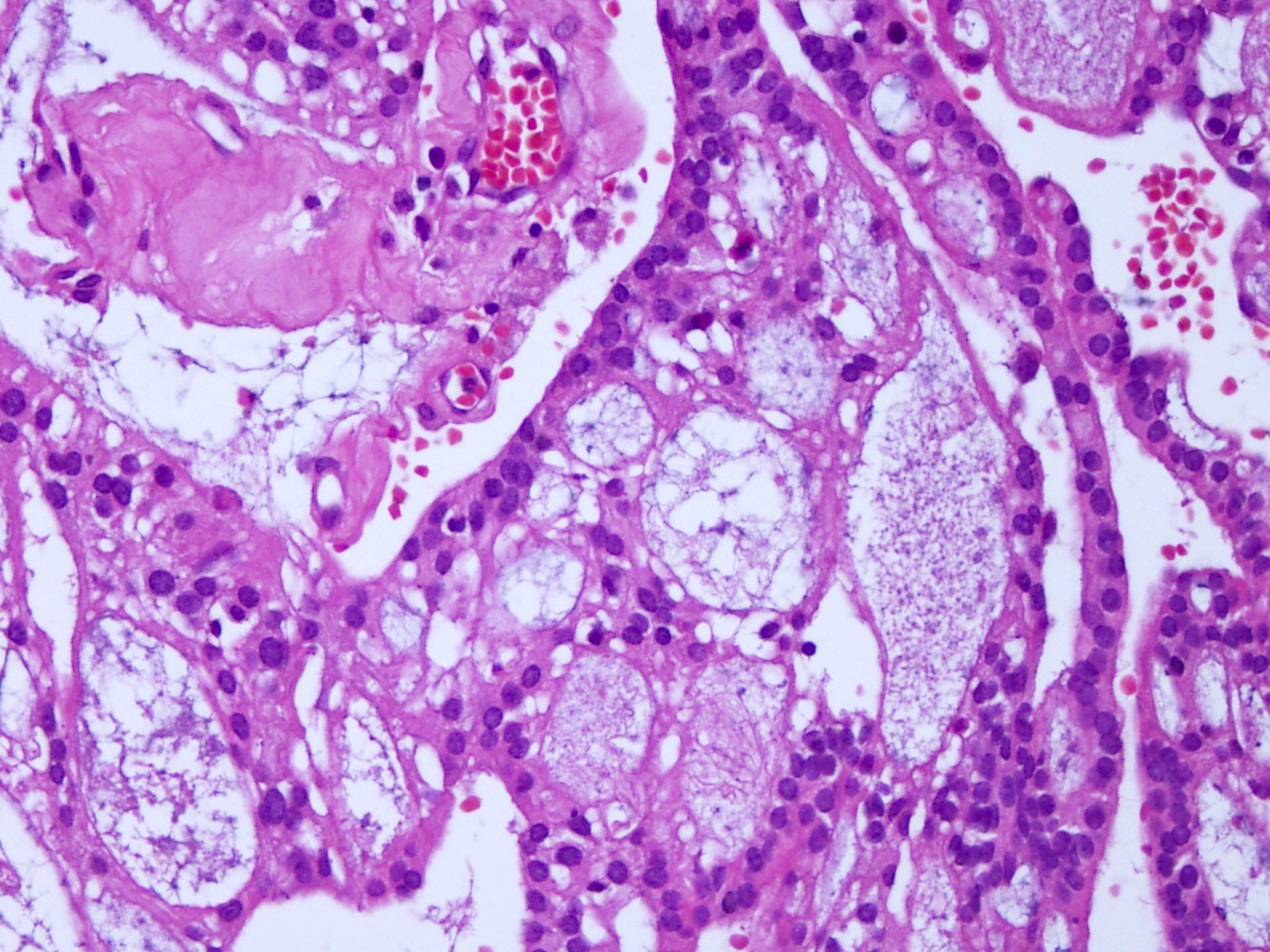
Microcystic pattern
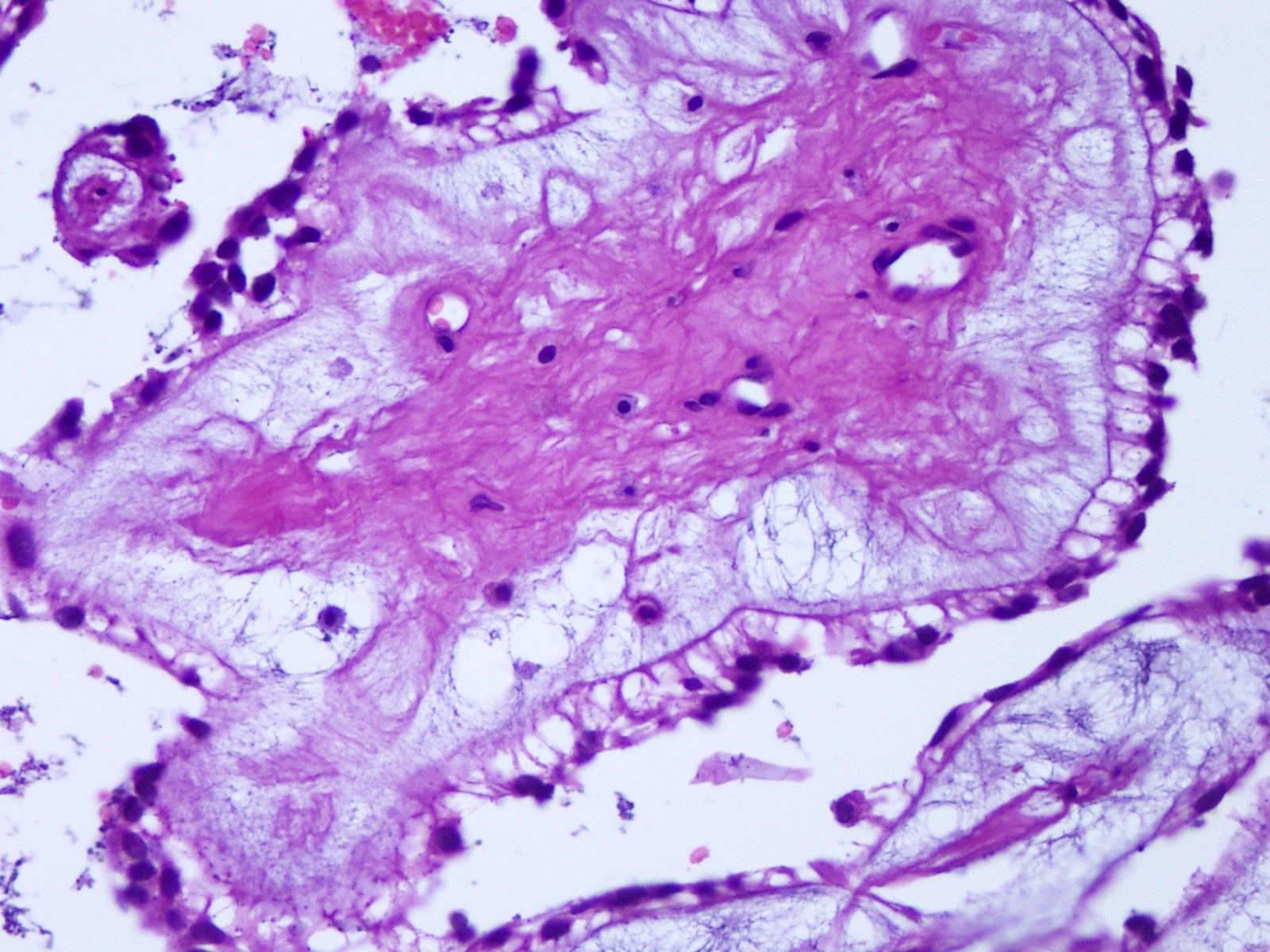
Hyalinized fibrovascular core with myxoid matrix
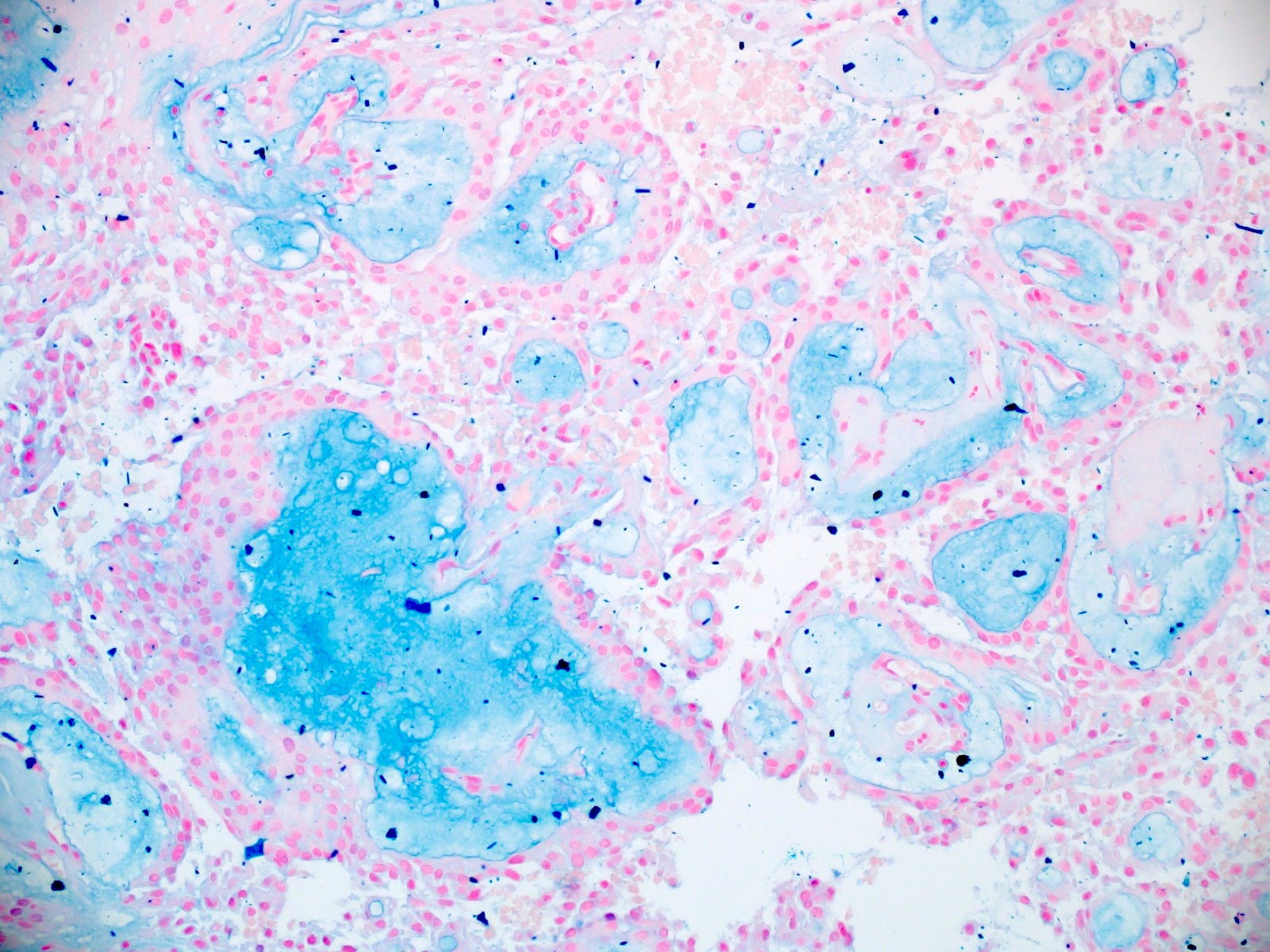
Alcian blue
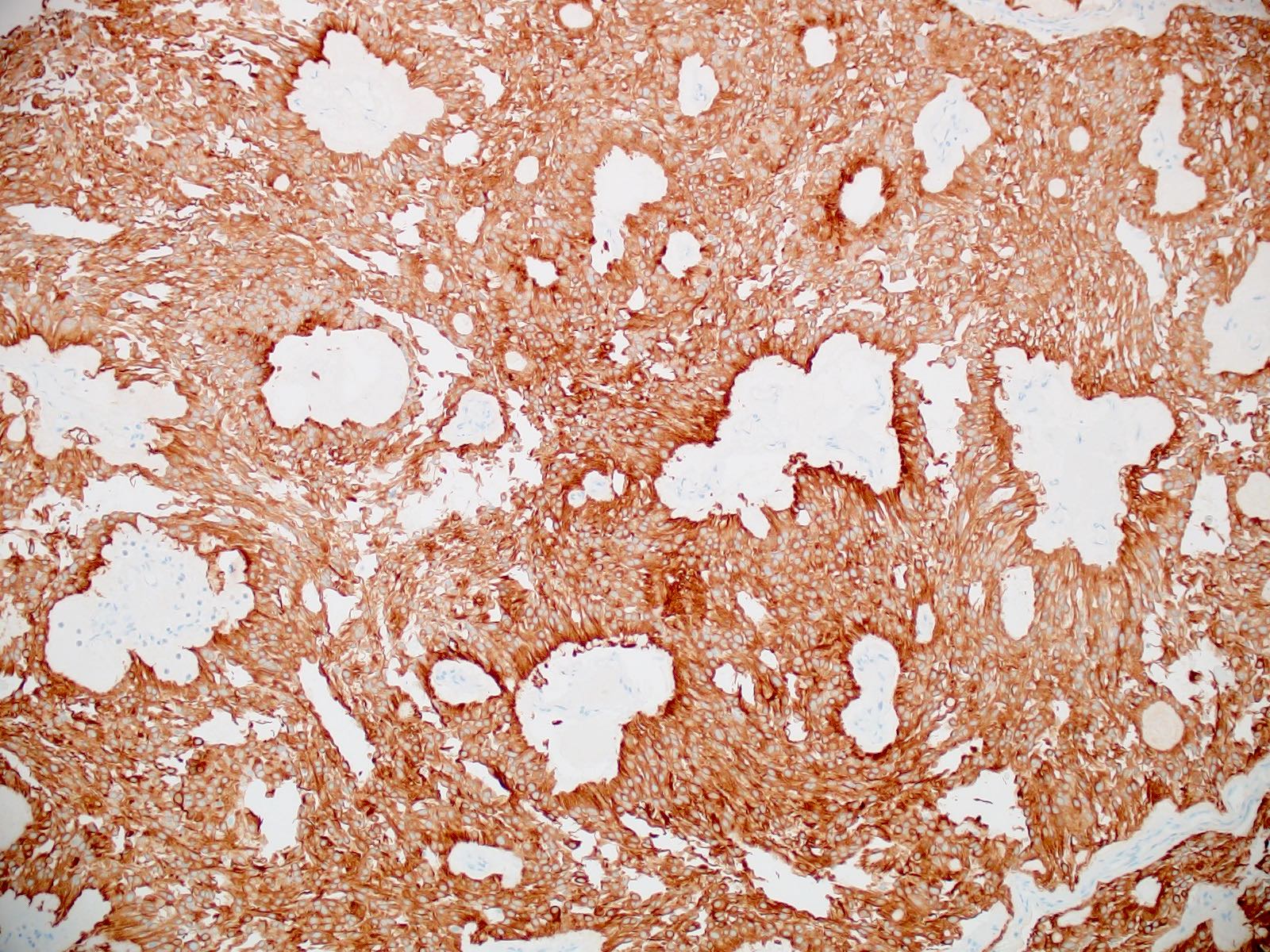
GFAP
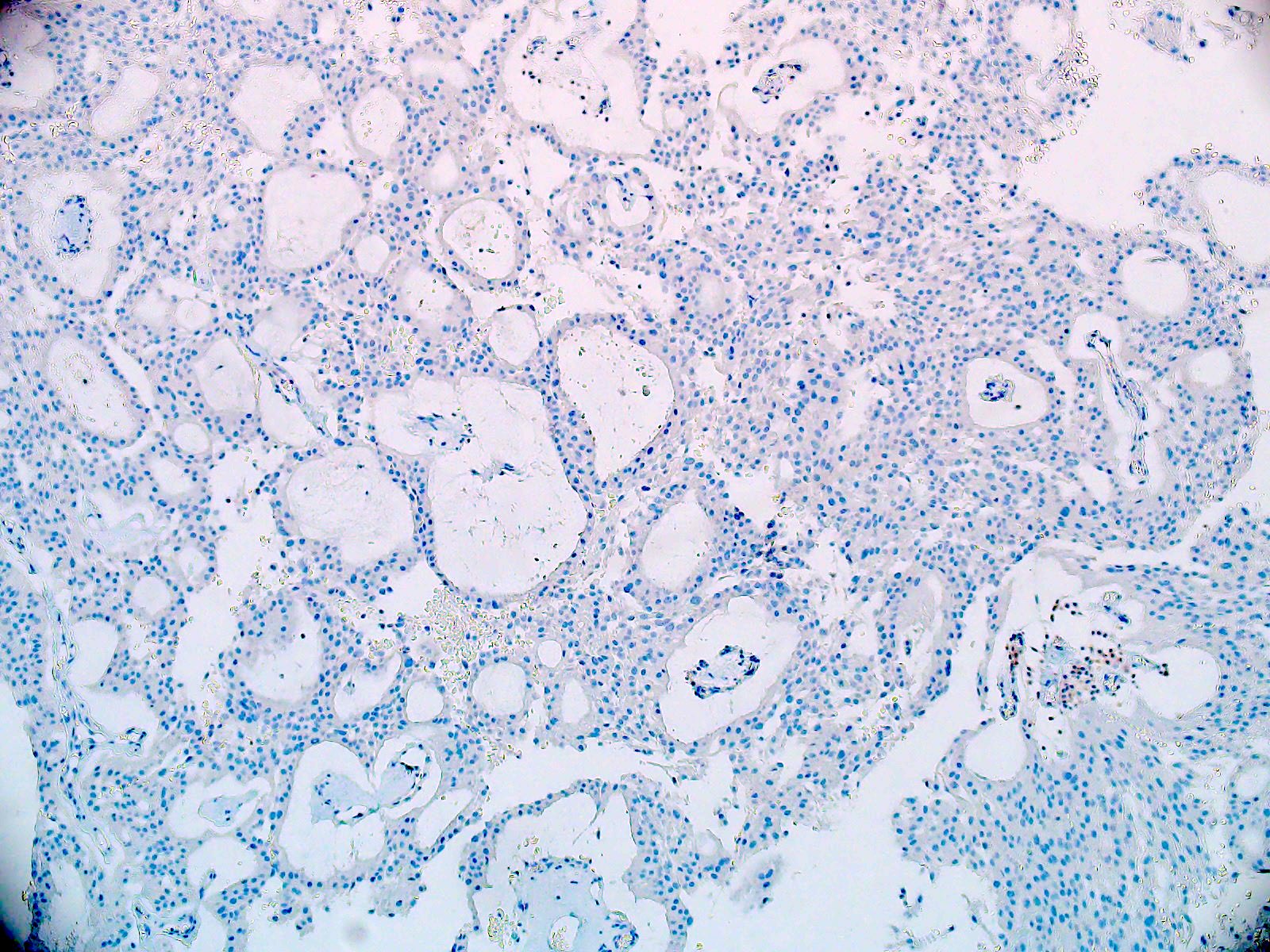
Olig2
EMA
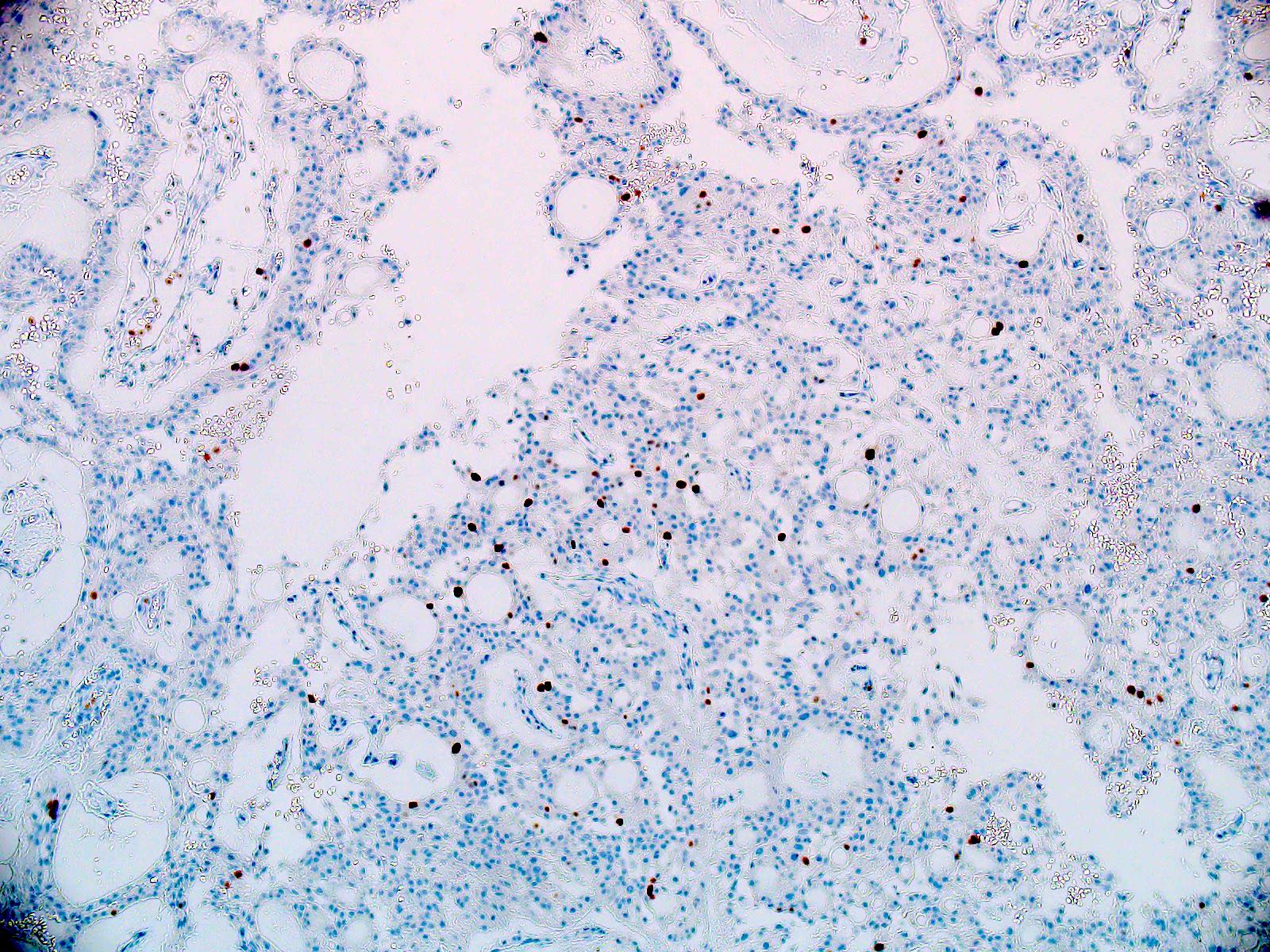
Ki67
- Cytology usually recapitulates the histologic findings; myxoid matrix and cells show nuclear uniformity and process formation
- Epithelioid to spindle cells
- Arrangement of tumor cells around blood vessels forms papillary structures with perivascular myxoid change
- Tumor cells are arranged around myxoid microcysts
Contributed by Ahmed Ijaz Gilani, M.B.B.S., M.D., Ph.D. and Brittany Pakalniskis, M.D. (Case #399)
Stromal globules
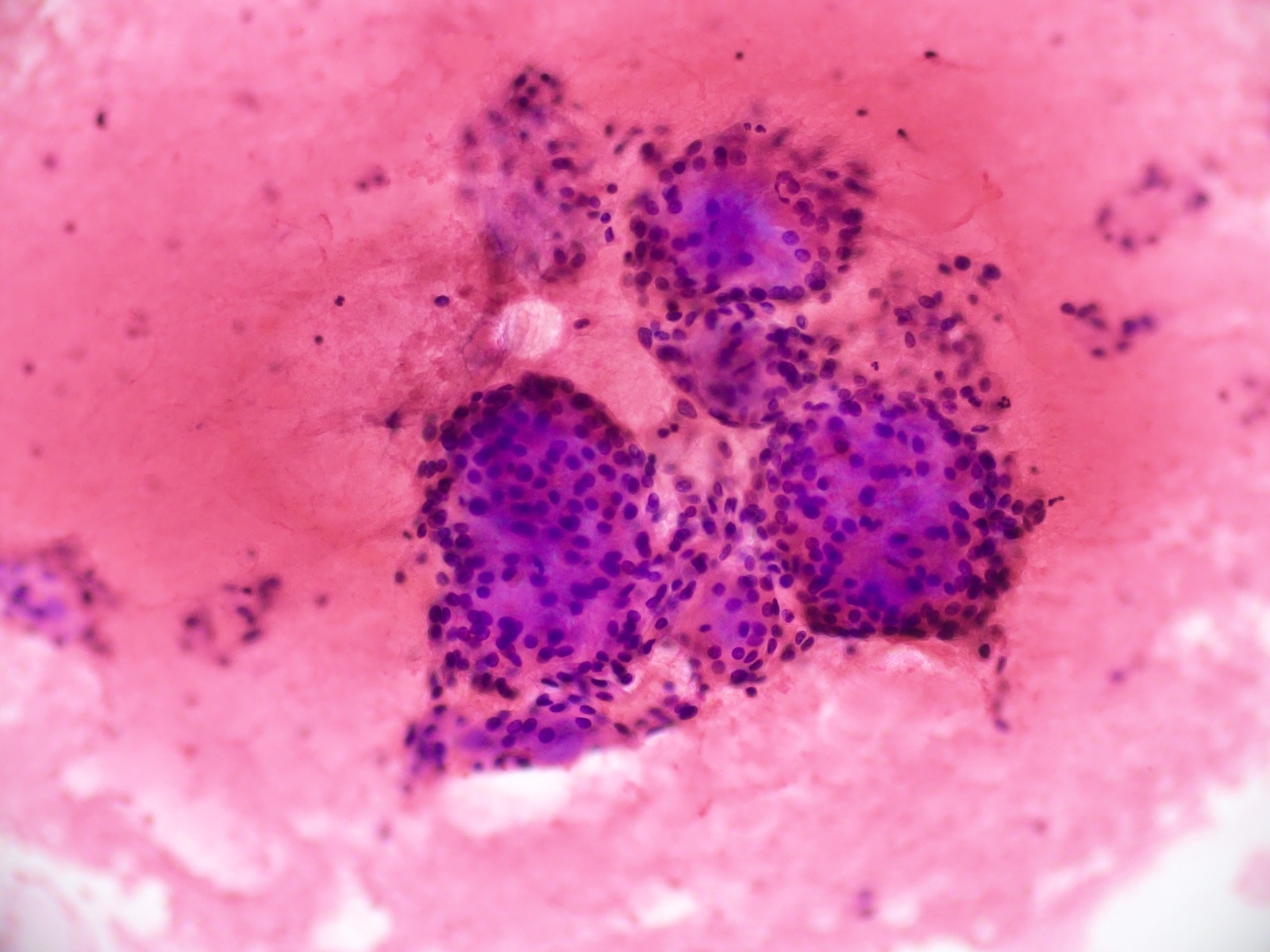
Papillary structures and rosettes
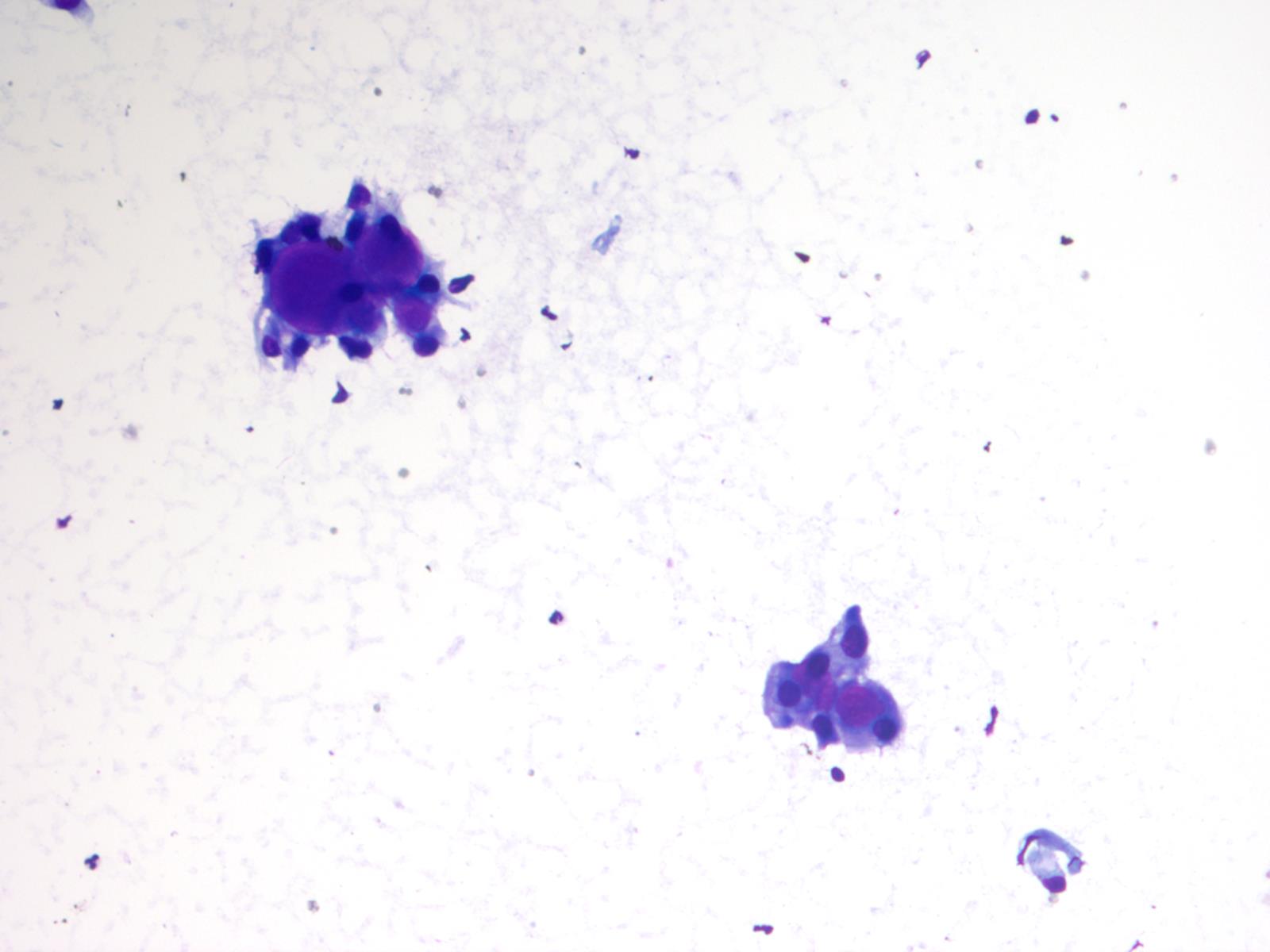
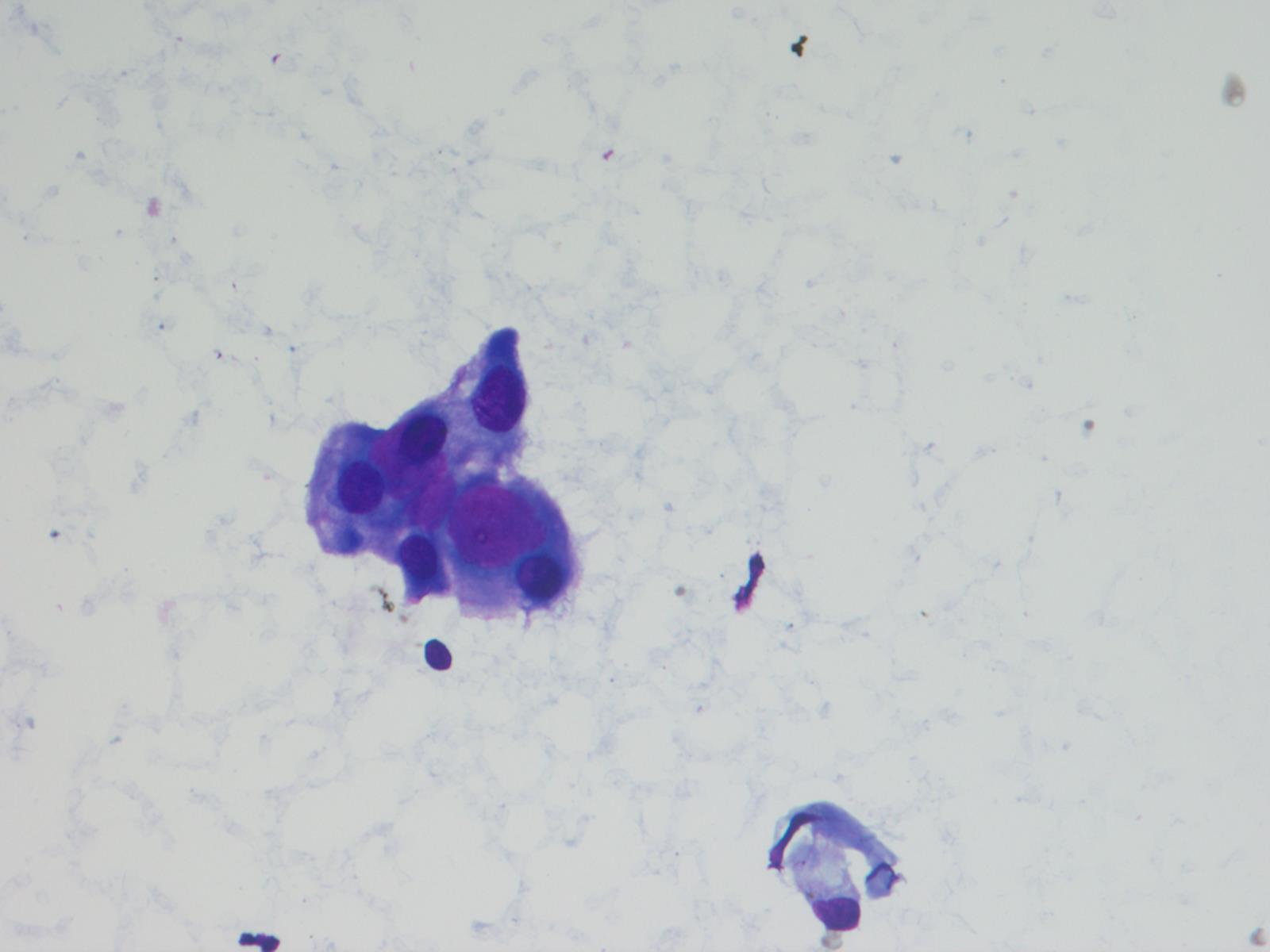
Stromal globules (Diff-Quik stain)
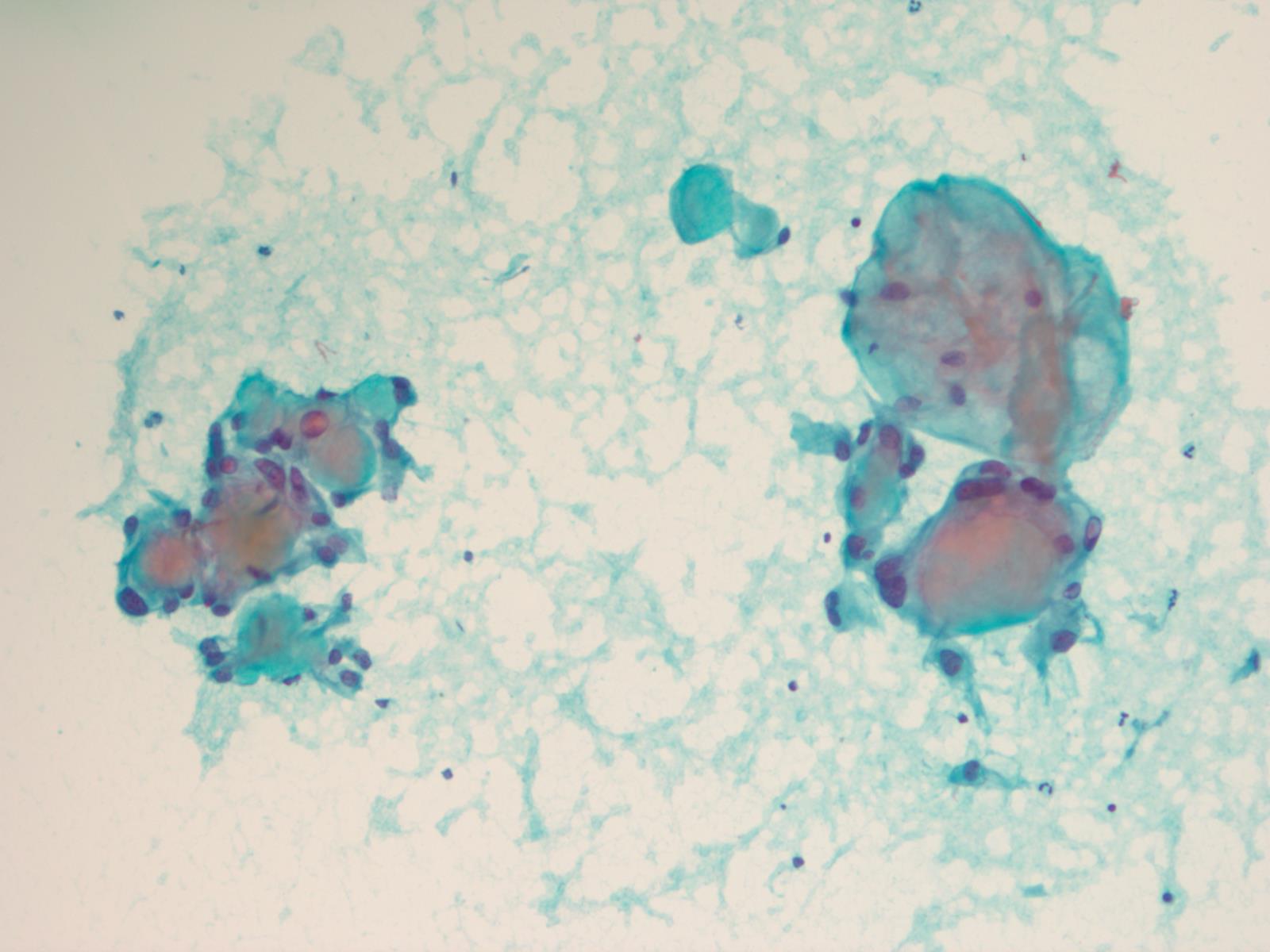
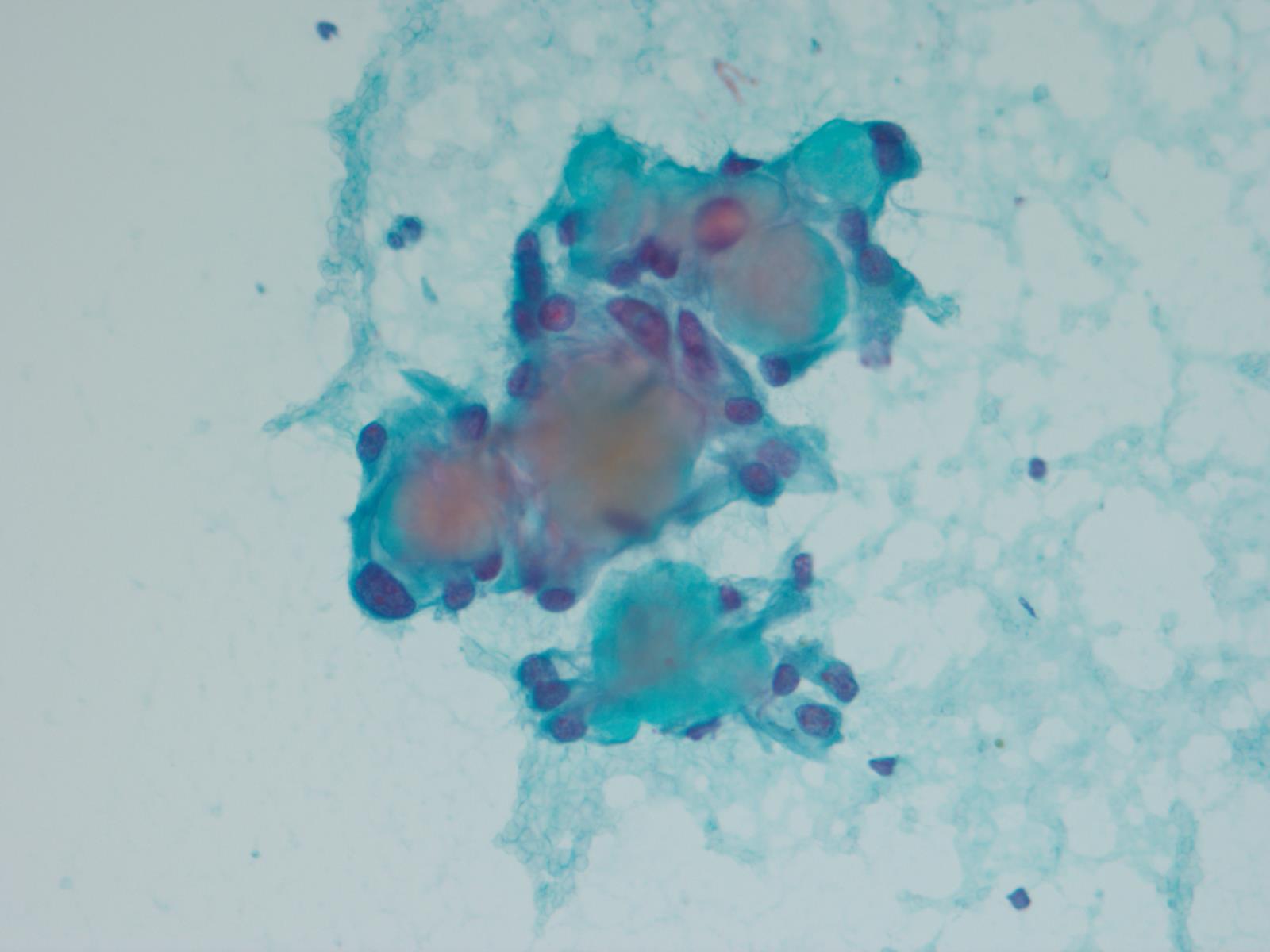
Stromal globules (Pap stain)
- Positive for GFAP (diffuse expression of GFAP distinguishes myxopapillary ependymomas from other tumors in the differential diagnosis, such as metastatic carcinomas, chordomas, paragangliomas, myxoid chondrosarcomas, schwannomas, etc.)
- Positivity for S100 is typical; CD99, vimentin and CD56 positivity is seen frequently
- Positivity for cyclooxygenase 2 (COX2) is seen in the majority of cases (60%) (Neuropathology 2006;26:422)
- Often positive for keratin AE1 / AE3 cocktail (Appl Immunohistochem Mol Morphol 2013;21:485, Appl Immunohistochem Mol Morphol 2000;8:25)
- Olig2 (tumor nuclei are negative), EMA (dot-like cytokeratin expression seen in other ependymoma variants is often absent)
- Negative for cytokeratin CAM 5.2, CK5/6, CK7 and CK20 (Appl Immunohistochem Mol Morphol 2013;21:485, Appl Immunohistochem Mol Morphol 2000;8:25)
- Zipper-like intercellular junctional complexes, occasional cilia, surface and intraluminal microvilli are typical features of ependymal differentiation
- In addition, interdigitating cell processes and microtubular aggregates bond by endoplasmic reticulum
- Unique DNA methylation profile (however, histologically classic ependymomas may also cluster on DNA methylation with myxopapillary ependymomas)
- Prognostic significance of DNA methylation profile of myxopapillary ependymomas in context of uncharacteristic histological features (little myxoid change, pseudorosetting, spindle cells or tanycytic features) is not known
- Recurrent gain of chromosome 16 and losses of chromosome 10 have been noted (Acta Neuropathol 2020;139:305, Cancer Cell 2015;27:728, Neuro Oncol 2018;20:1616)
- Lower spinal mass, resection specimen:
- Myxopapillary ependymoma, CNS WHO grade 2
- Schwannoma:
- Arises from nerve roots (myxopapillary ependymomas usually arise in the filum terminale)
- Usually, distinctive histological appearance from ependymoma; however, a paucicellular ependymoma with fascicular architecture may simulate schwannoma
- No Antoni A or B areas are present in ependymomas
- There are no nuclear or cytoplasmic inclusions in ependymomas
- Myxopapillary ependymomas have a delicate rather than densely collagenized capsule seen in schwannomas
- Schwannomas show pericellular reticulin and extensive immunostaining for laminin and type IV collagen
- Both tumors are S100 and GFAP positive
- Schwannomas show more intense staining for S100 compared with GFAP; the reverse is true for myxopapillary ependymomas
- Paraganglioma:
- More often arises from nerve roots than ependymomas
- Presence of zellballen pattern or ganglion cells confirms the diagnosis of paraganglioma
- Chromogranin positive (myxopapillary ependymomas are negative)
- Chordoma:
- Mostly extradural
- Mucinous matrix of myxopapillary ependymoma may simulate chordoma; however, mucin distribution in chordoma is diffuse and tends not to be confined in perivascular spaces
- Chordomas are strongly immunoreactive for low molecular weight keratins and EMA; negative for GFAP
- Metastatic adenocarcinoma:
- Myxoid chondrosarcoma:
- Myxoid or chondroid matrix with lacunar-like spaces supports the diagnosis of myxoid chondrosarcoma
- Myxoid chondrosarcoma cells would be negative for GFAP
- CD99
- GFAP
- Keratin
- Olig2
- S100
Comment Here
Reference: Myxopapillary ependymoma
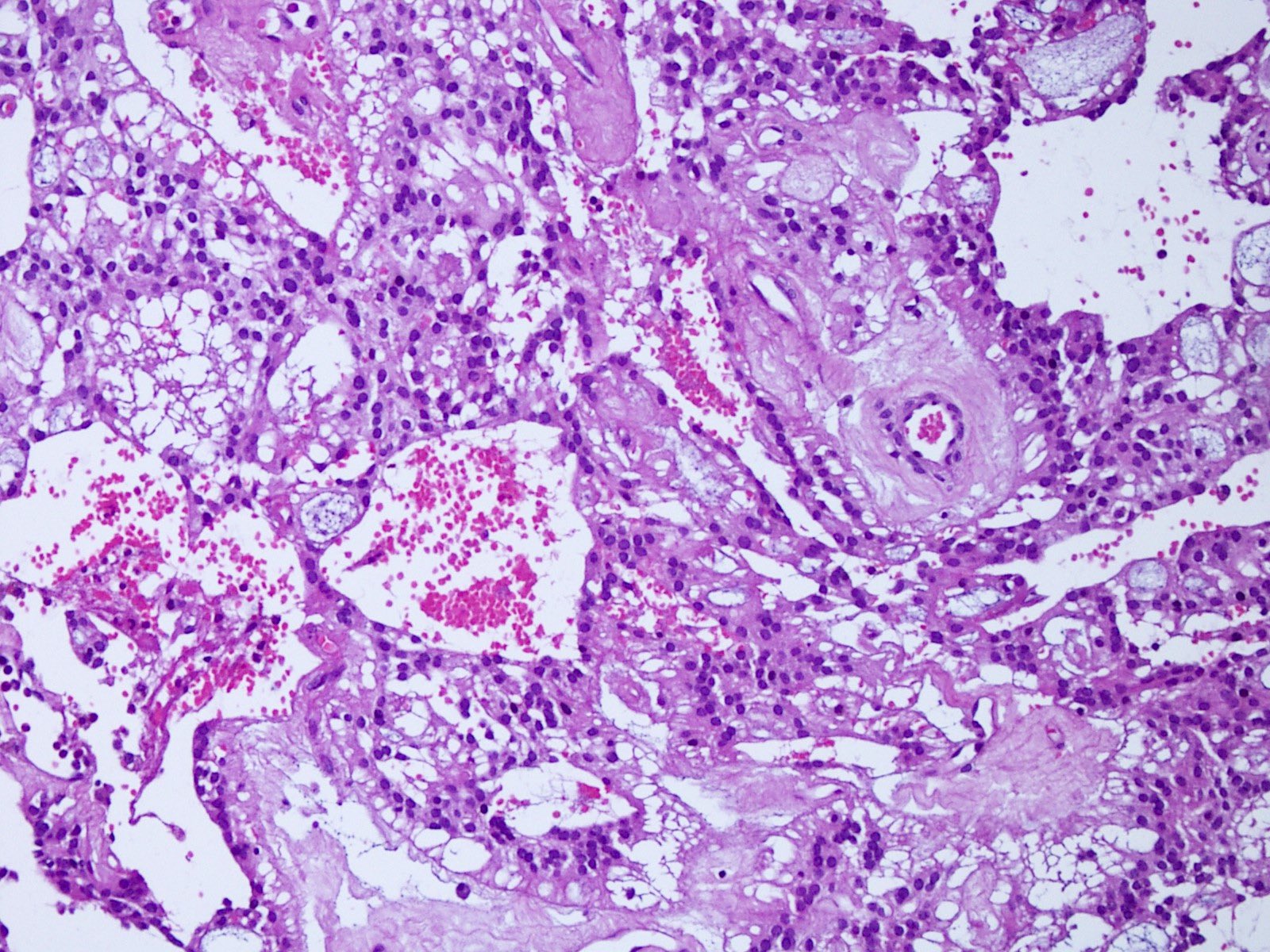
In the 5th edition of the 2021 WHO classification for CNS, which histological grade does myxopapillary ependymoma correspond to?
- Grade 1
- Grade 2
- Grade 3
- Grade 4
- Unassigned
Comment Here
Reference: Myxopapillary ependymoma
- CNS WHO 2021 definition: diffusely infiltrating glioma with IDH1 or IDH2 mutation and codeletion of chromosome arms 1p and 19q (CNS WHO grade 2 or 3)
- Diffusely infiltrating glial neoplasm with IDH1 or IDH2 mutation and 1p / 19q whole arm codeletion (both features are required for diagnosis)
- Morphology resembles nonneoplastic oligodendrocytes with round monotonous nuclei and perinuclear halos
- Chicken wire vasculature, microcalcifications and microcysts are characteristic (Neuro Oncol 2014;16:1244)
- Astrocytic differentiation does not preclude diagnosis if molecular features are present
- Small gemistocytes (mini gemistocytes) with rounded bellies of eosinophilic, eccentrically placed cytoplasm are occasionally seen, especially in grade 3 tumors (Acta Neuropathol 1984;64:265)
- Presence of other atypical features (including multinucleated giant cells, sarcomatous features, neurocytic differentiation or ganglion-like cells) does not preclude a diagnosis of oligodendroglioma if the requisite molecular features are present (Acta Neuropathol 2010;120:237, J Neuropathol Exp Neurol 2002;61:947, Neuropathology 2014;34:323)
- Most epidemiologic data is based on histologic, rather than molecular, classification of oligodendroglioma
- Incidence of 0.23 cases per 100,000 population in the United States (Neuro Oncol 2019;21:v1)
- Incidence of CNS WHO grade 3 oligodendroglioma is 0.11
- Of all brain tumors in the United States:
- 0.9% are oligodendroglioma WHO grade 2
- 0.4% are oligodendroglioma WHO grade 3
- Peak incidence in fourth and fifth decades of life (Neuro Oncol 2020;22:iv1)
- Rare in infants and children (Am J Surg Pathol 2014;38:1058)
- Slight male predominance (Neuro Oncol 2019;21:v1)
- Infiltrative neoplasm involving the white and gray matter
- Can occur anywhere in the neuraxis; most common locations (Neuro Oncol 2020;22:iv1):
- Frontal lobes: 59%
- Temporal lobes: 14%
- Parietal lobes: 10%
- Occipital lobes: 1%
- Rarely observed in midline structures, brainstem, cerebellum or spinal cord
- Leptomeningeal spread occasionally observed, particularly at recurrence (Neurology 2019;92:e2483)
- Cell (or cells) of origin for oligodendroglioma remains unknown
- IDH mutation is likely the initiating event (driver mutation), which precedes 1p / 19q codeletion (Adv Anat Pathol 2015;22:50, Biomed Res Int 2014;2014:540236)
- IDH mutations give rise to metabolic alterations, with increased production of 2-hydroxyglutarate (2HG)
- Increased 2HG inhibits histone demethylation, causing a hypermethylation phenotype in neoplastic cells: glioma CpG island methylated phenotype (G CIMP) (Nature 2012;483:479, Acta Neuropathol 2013;125:621)
- Generally sporadic without significant known risk factors
- Rare instances of familial oligodendroglioma and genetic alterations with associated increased risk of developing oligodendroglioma (Neuro Oncol 2018;20:1625, Cancer 2005;103:2363, J Natl Cancer Inst 2014;107:384)
- About 67% of patients present with seizure (PLoS One 2017;12:e0188419, Nat Rev Neurol 2017;13:340)
- Other common presenting symptoms: headache, focal neurologic deficits or cognitive / mental status change, depending on anatomic location
- WHO grade 2:
- Well differentiated tumor lacking anaplastic features (brisk mitotic activity, microvascular proliferation, necrosis)
- WHO grade 3:
- Prominent anaplastic features (necrosis, microvascular proliferation or brisk mitotic activity) are compatible with anaplastic oligodendroglioma, IDH mutant and 1p / 19q codeleted, WHO grade 3
- Strict mitotic activity criteria do not currently exist
- Some authors suggest ≥ 6 mitotic figures per 10 high power fields in resection specimens for grade 3 designation (J Neuropathol Exp Neurol 2001;60:248)
- Fewer mitotic figures might be sufficient for grade 3 designation in small biopsy specimens if other anaplastic features (vascular proliferation or necrosis) or significant nuclear atypia are present
- CDKN2A homozygous deletion may serve as a molecular marker of CNS WHO grade 3 in IDH mutant and 1p / 19q codeleted oligodendrogliomas (Neuro Oncol 2019;21:1519)
- Magnetic resonance imaging (MRI), followed by stereotactic brain biopsy or surgical resection
- Methods to detect IDH gene mutation:
- Immunohistochemistry for IDH1 R132H (positive in > 90% of tumors) (Acta Neuropathol 2009;118:599)
- IDH2 mutations overrepresented in oligodendrogliomas compared with astrocytomas (Biomed Res Int 2014;2014:540236)
- Sanger sequencing
- Droplet digital polymerase chain reaction (ddPCR)
- Next generation sequencing
- MRI techniques to detect 2-hydroxyglutarate and therefore IDH mutation are under investigation (J Clin Invest 2013;123:3659)
- Immunohistochemistry for IDH1 R132H (positive in > 90% of tumors) (Acta Neuropathol 2009;118:599)
- Methods to detect 1p / 19q codeletion:
- Fluorescent in situ hybridization (FISH)
- Array comparative genomic hybridization
- Polymerase chain reaction (PCR)
- Computed topography (CT):
- Mixed density (hypodense and isodense) located in cortex or subcortical white matter (Radiology 2017;284:316)
- High attenuation areas, likely from calcifications
- MRI:
- Heterogeneous on T1 and T2 weighted imaging
- Typically no diffusion restriction
- Poorly circumscribed borders (AJNR Am J Neuroradiol 2017;38:678)
- Cystic changes are relatively common (Radiology 2017;284:316)
- Contrast enhancement present in < 20% of WHO grade 2 tumors and > 70% of WHO grade 3 tumors (AJNR Am J Neuroradiol 2012;33:852, Eur J Cancer 2019;107:15)
- Elevated 2HG by magnetic resonance spectroscopy could serve as radiologic surrogate of IDH mutation status (Nat Med 2012;18:624)
Contributed by Jared T. Ahrendsen, M.D., Ph.D. and John DeWitt, M.D., Ph.D.
MRI, brain: T1 post
MRI, brain: T2
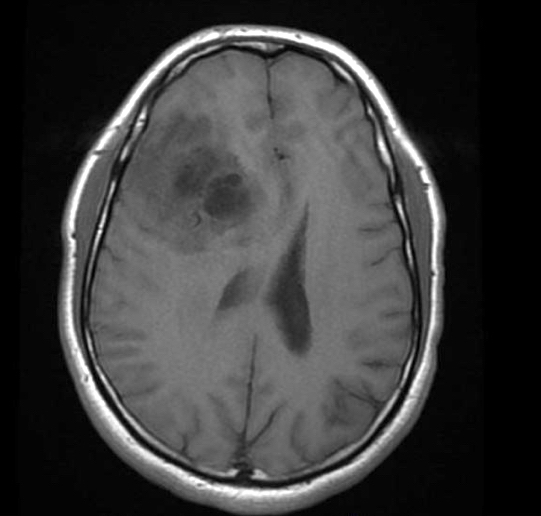
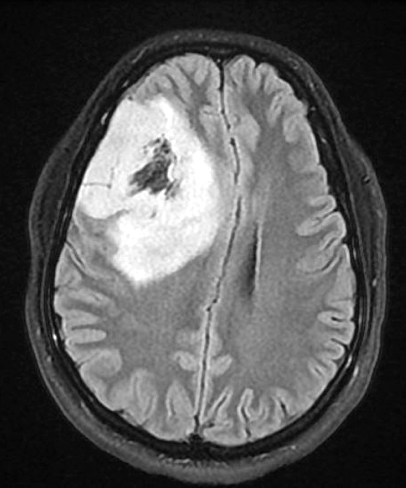
MRI: frontal lobe tumor with cystic change
Images hosted on other servers:
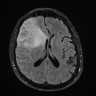
Axial flair frontal lobe tumor

Axial flair large temporal lobe tumor
- Slow growing and relatively long overall survival
- Median overall survival: 11.6 years; 10 year overall survival rate: 51 - 63% (J Neuropathol Exp Neurol 2005;64:479, Neuro Oncol 2020;22:iv1)
- Longer median survival compared with grade 2 IDH mutant astrocytoma (median overall survival: 10.9 years) (Acta Neuropathol 2015;129:867)
- Favorable features (Crit Rev Oncol Hematol 2008;66:262):
- Younger age at diagnosis
- Tumor location in frontal lobe
- Presentation with seizures
- Macroscopically complete surgical resection
- CNS WHO grade 2 histology
- Higher postoperative Karnofsky score
- Unfavorable features:
- Contrast enhancement on MRI
- CNS WHO grade 3 histology (Eur J Cancer 2020;137:10, Oncotarget 2014;5:1515)
- CDKN2A / CDKN2B homozygous deletion (J Neuropathol Exp Neurol 2004;63:314, Neuro Oncol 2019;21:1519)
- Local recurrence and malignant transformation are common
- 26 year old man presents with nausea, headache and rash (JAAD Case Rep 2019;6:1)
- 43 year old woman with headaches, blurry vision and a right parietal mass (Front Neurol 2018;9:700)
- 44 year old man with sudden right sided optic neuritis (BMJ Case Rep 2018;2018:bcr2018225318)
- 48 year old man presents with seizures (Front Oncol 2021;10:601452)
- 55 year old man with mass lesion in the superior left temporal gyrus (Brain Pathol 2019;29:693)
- Gross total resection, if possible
- Adjuvant chemotherapy (temozolomide) and radiotherapy
- Given to patients with symptomatic or progressive tumors, tumors with CNS WHO grade 3 histology or those with large postoperative residual tumor
- References: Crit Rev Oncol Hematol 2008;66:262, Lancet 2005;366:985
- Variably well defined, gray-pink mass
- Mucoid change can give a gelatinous consistency
- Areas of cystic degeneration, calcifications, hemorrhage or necrosis can be seen
- Moderately cellular, diffusely infiltrating neoplasm
- Glia with mild to moderate nuclear atypia
- Round nuclei with speckled chromatin
- Calcifications, perineuronal satellitosis or perivascular accumulation of tumor cells may be seen
- Will not see perinuclear halos on frozen section or smear preparations
- Anaplastic features (necrosis, vascular proliferation, mitoses) may be seen in WHO grade 3 tumors
- Reference: J Cytol 2011;28:147
Contributed by Jared T. Ahrendsen, M.D., Ph.D.
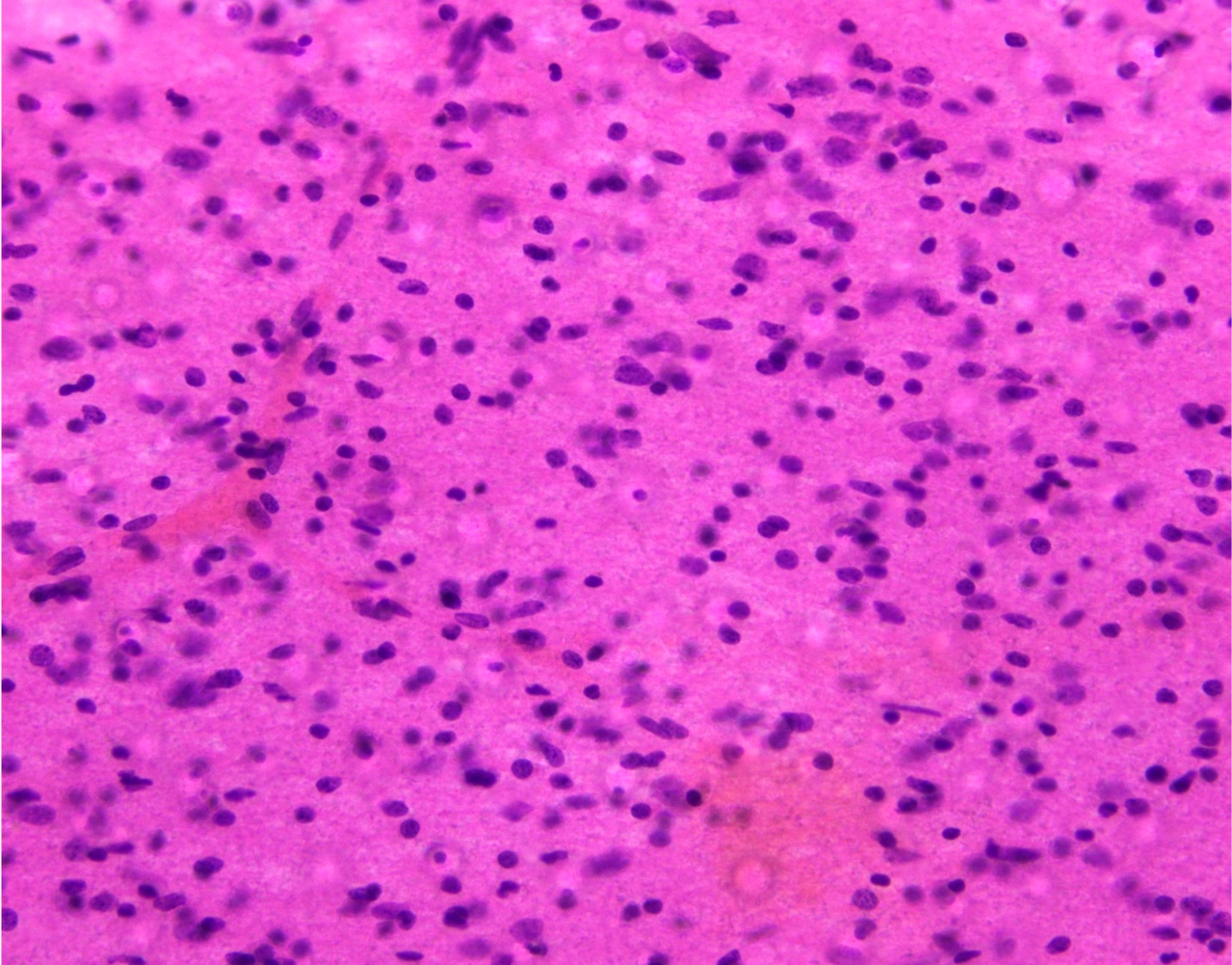
Intraoperative smear preparation
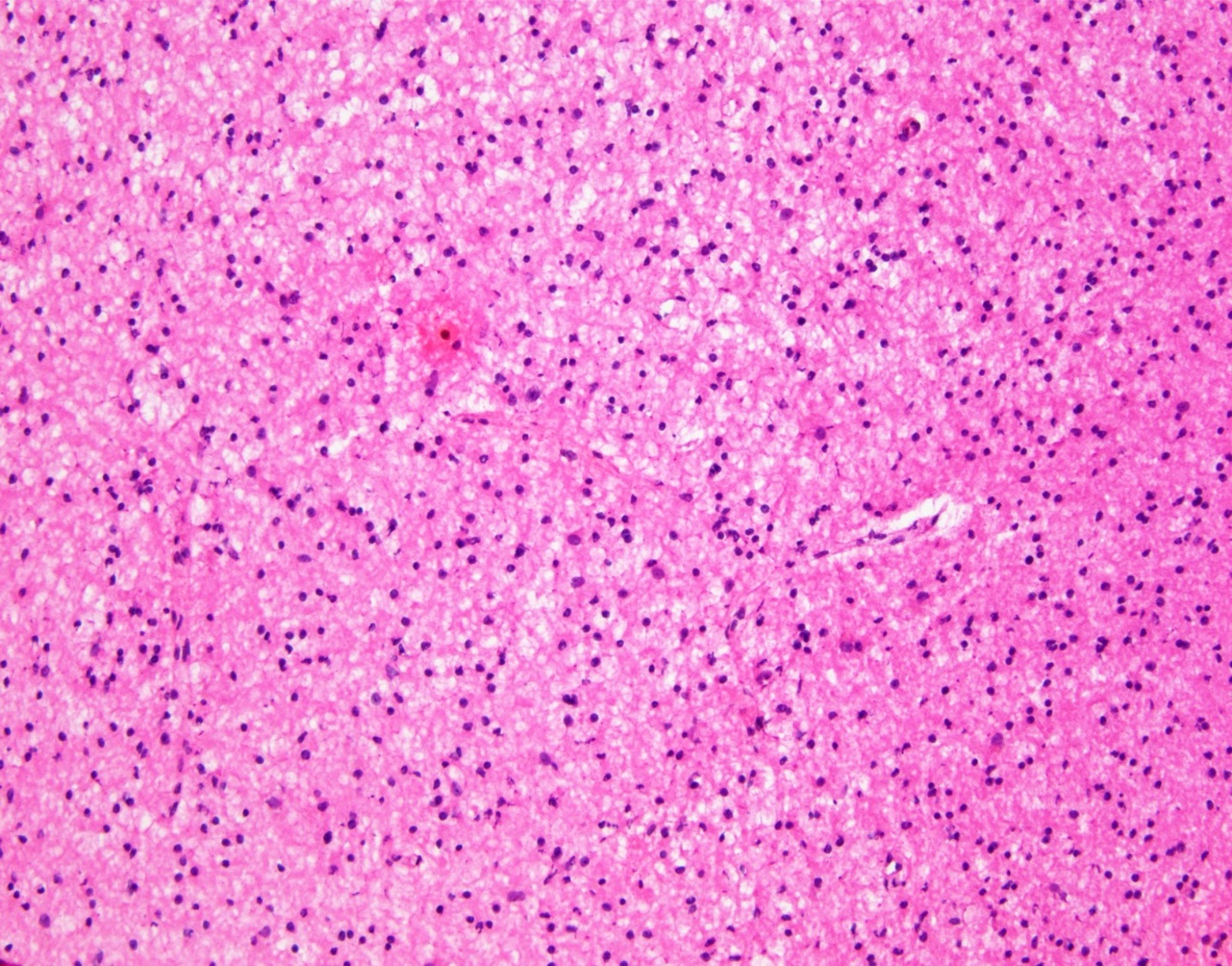
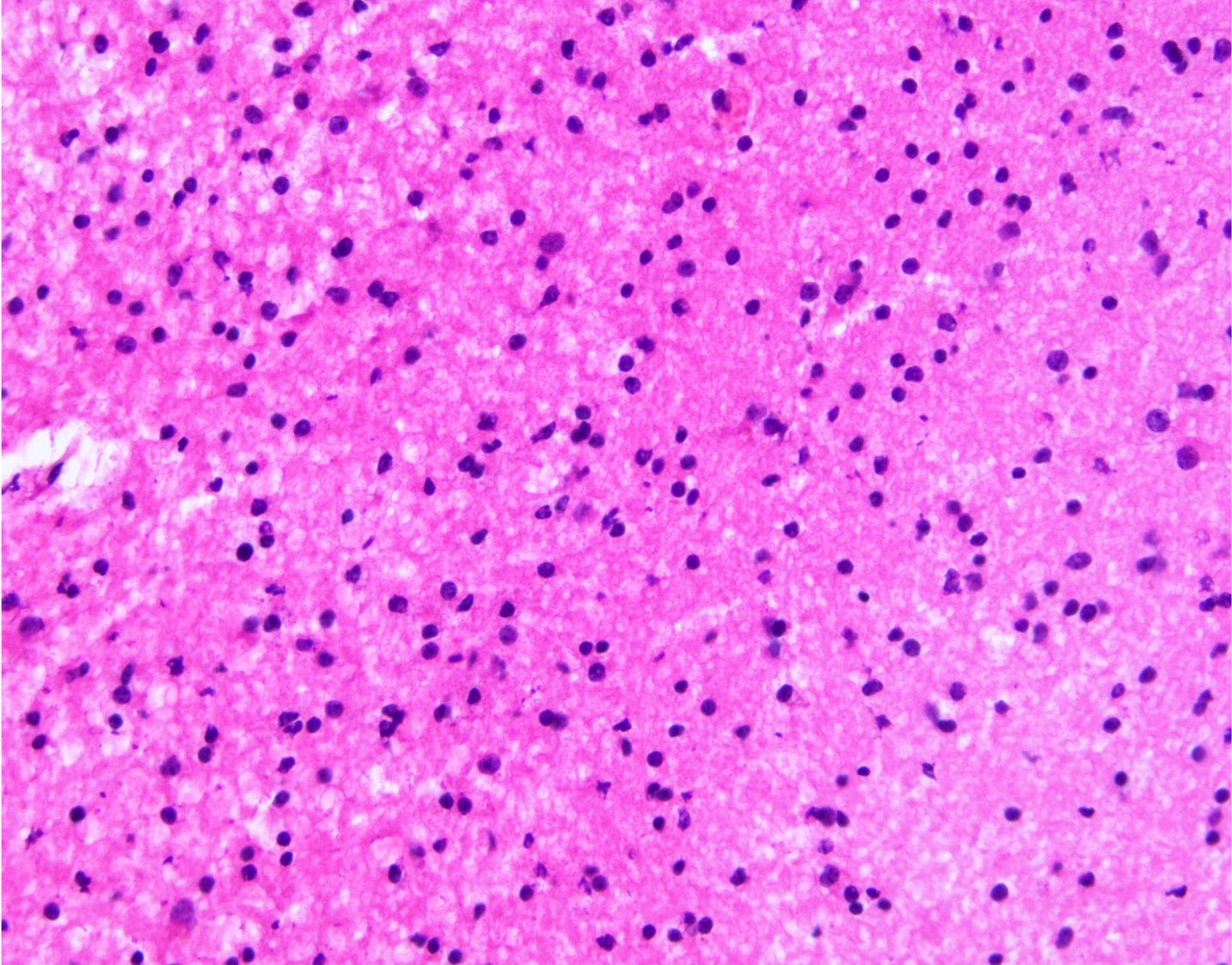
Intraoperative frozen section
- Closely packed cells with small, round, monotonous nuclei (slightly larger than a normal oligodendrocyte)
- Perinuclear clearing (fried egg appearance)
- Formalin fixation artifact
- Will not be seen on frozen sections or smear preparations
- Network of thin walled, branching blood vessels (chicken wire vasculature)
- Microcalcifications (calcospherites) are characteristic
- Presence of perineural, perivascular or subpial aggregates of tumor cells (secondary structures of Scherer)
- Occasional mitoses and moderate nuclear atypia are still consistent with grade 2 designation (J Neuropathol Exp Neurol 2001;60:248)
- Not uncommon to find well differentiated / fibrillary astrocytic morphology (Acta Neuropathol 1984;64:265)
- Features of CNS WHO grade 3 oligodendroglioma:
- Presence of microvascular proliferation
- Presence of necrosis
- Presence of brisk mitotic activity
- Strict mitotic figure cutoffs do not currently exist; some authors suggest ≥ 6 mitoses per 10 high power fields for WHO grade 3 designation in tumors without necrosis or vascular proliferation (Neuro Oncol 2014;16:1244, Neuro Oncol 2016;18:888)
Contributed by Jared T. Ahrendsen, M.D., Ph.D. and John DeWitt, M.D., Ph.D.
Diffusely infiltrating glial neoplasm
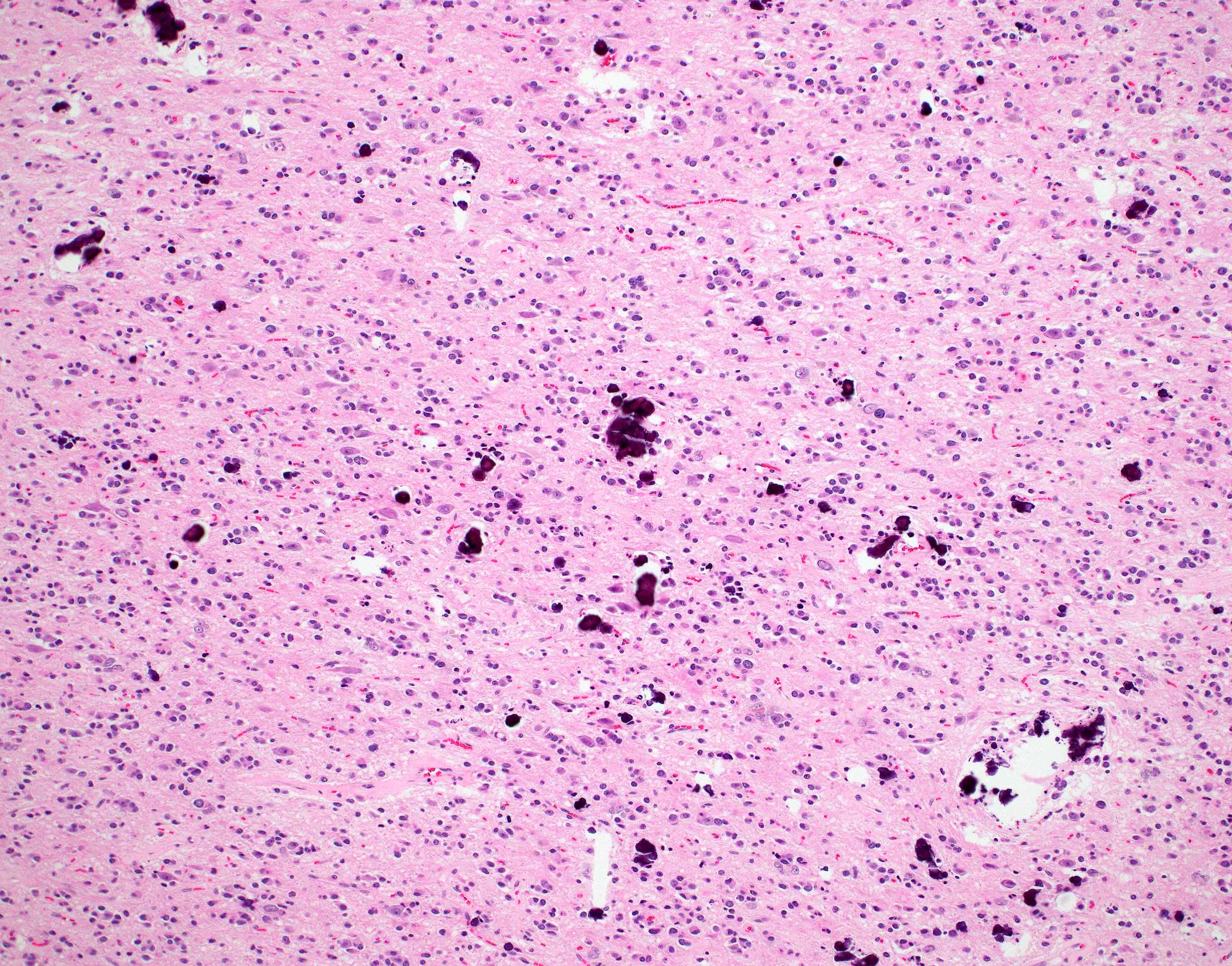
Microcalcifications
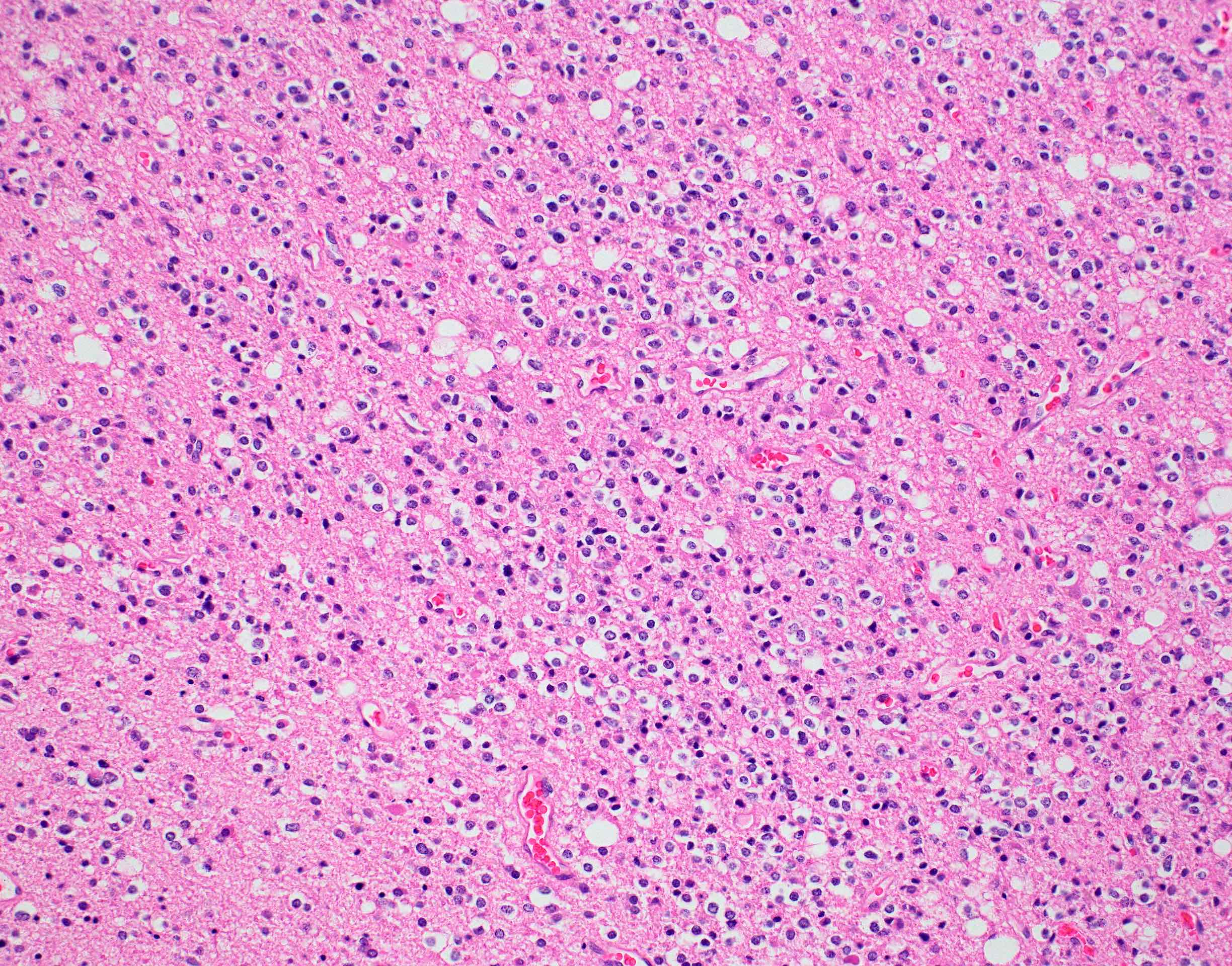
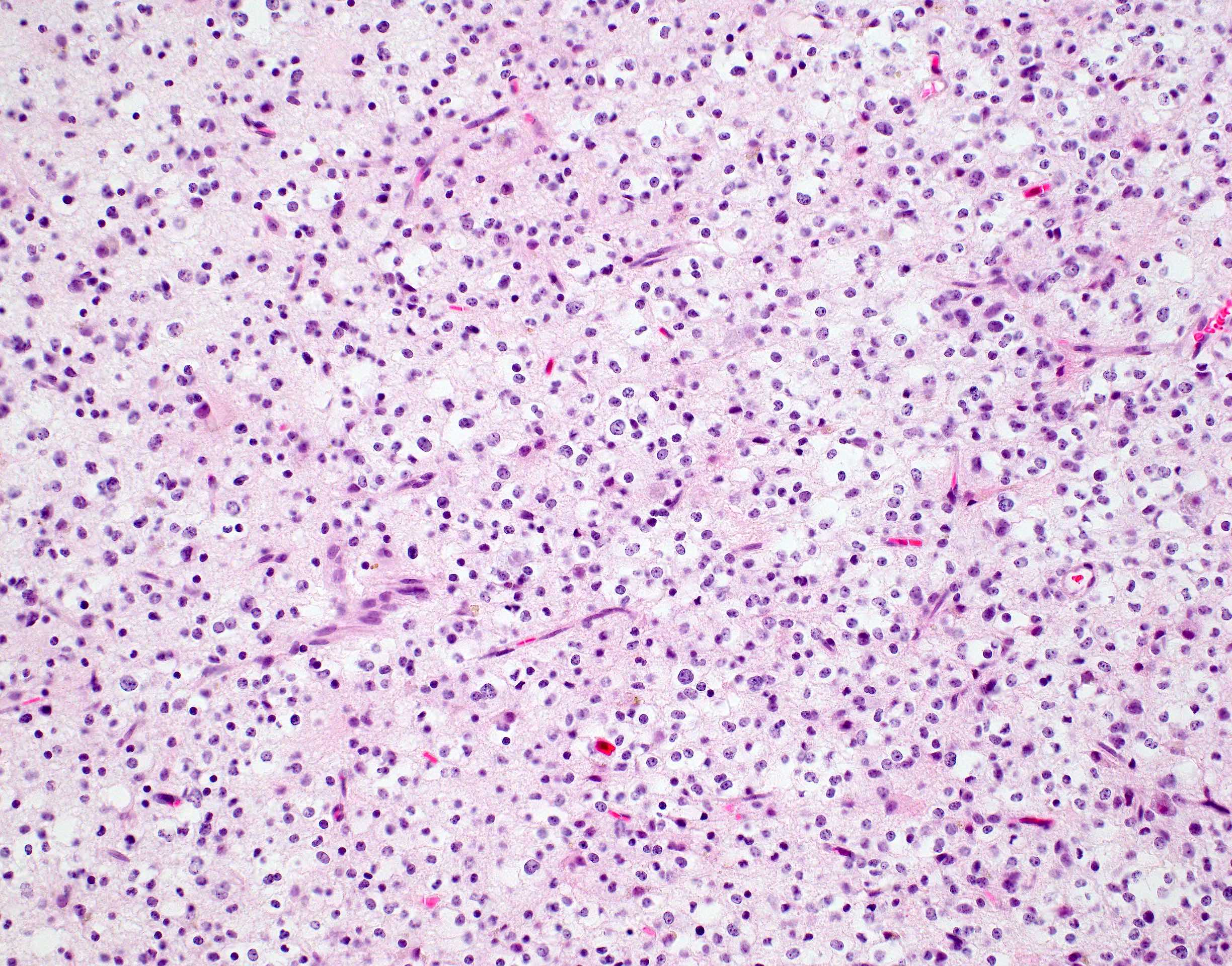
Perinuclear halos
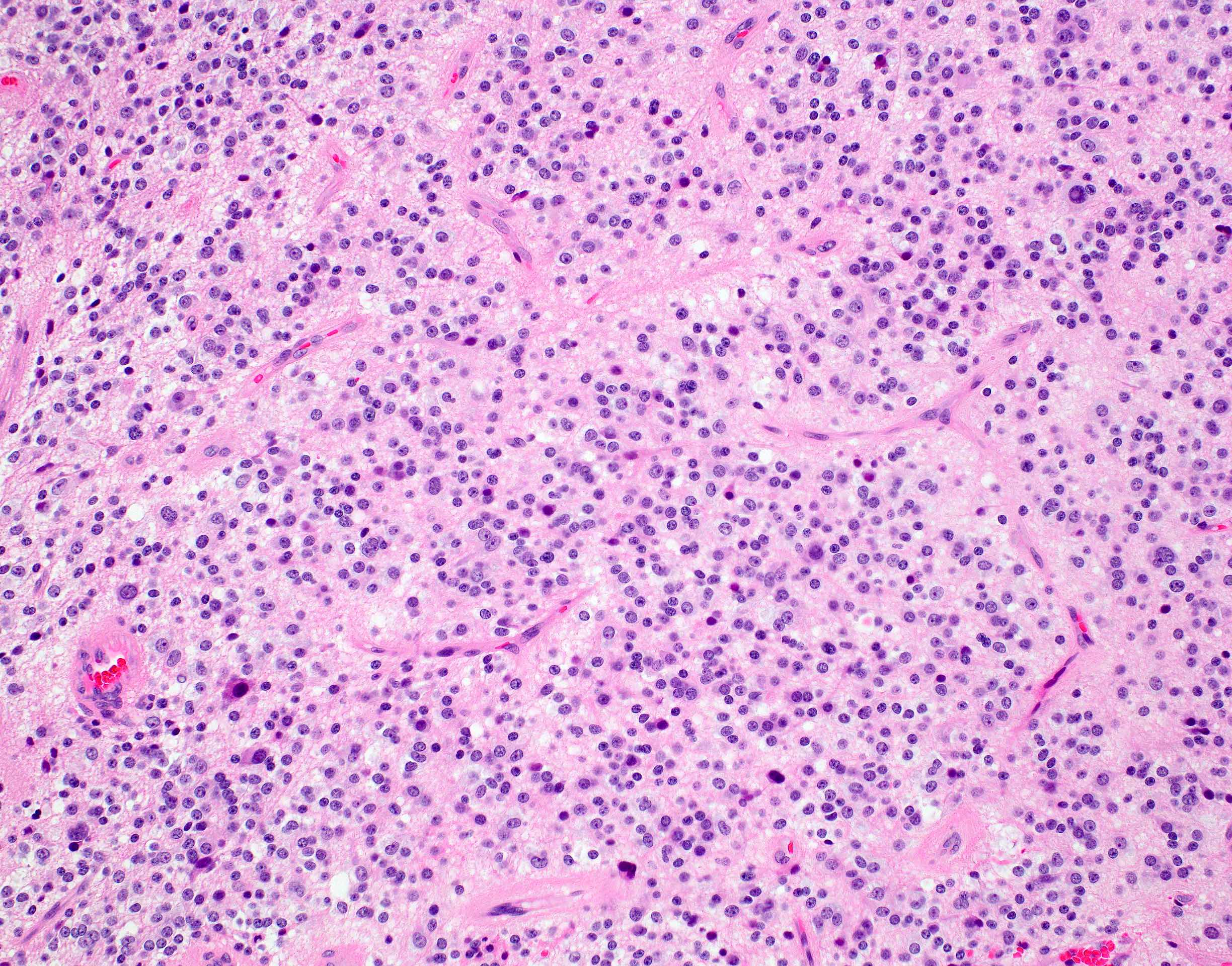
Chicken wire vasculature
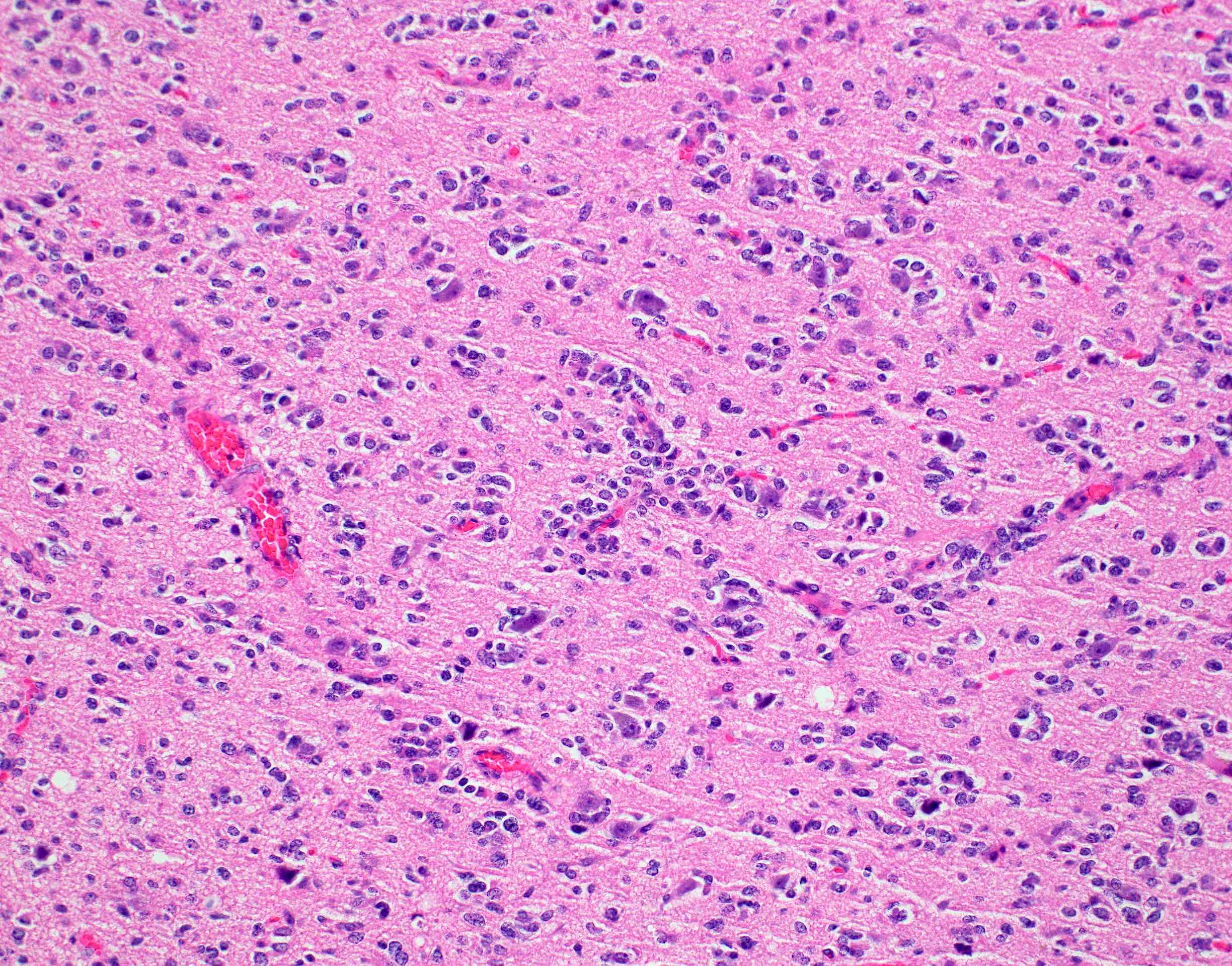
Secondary structures of Scherer
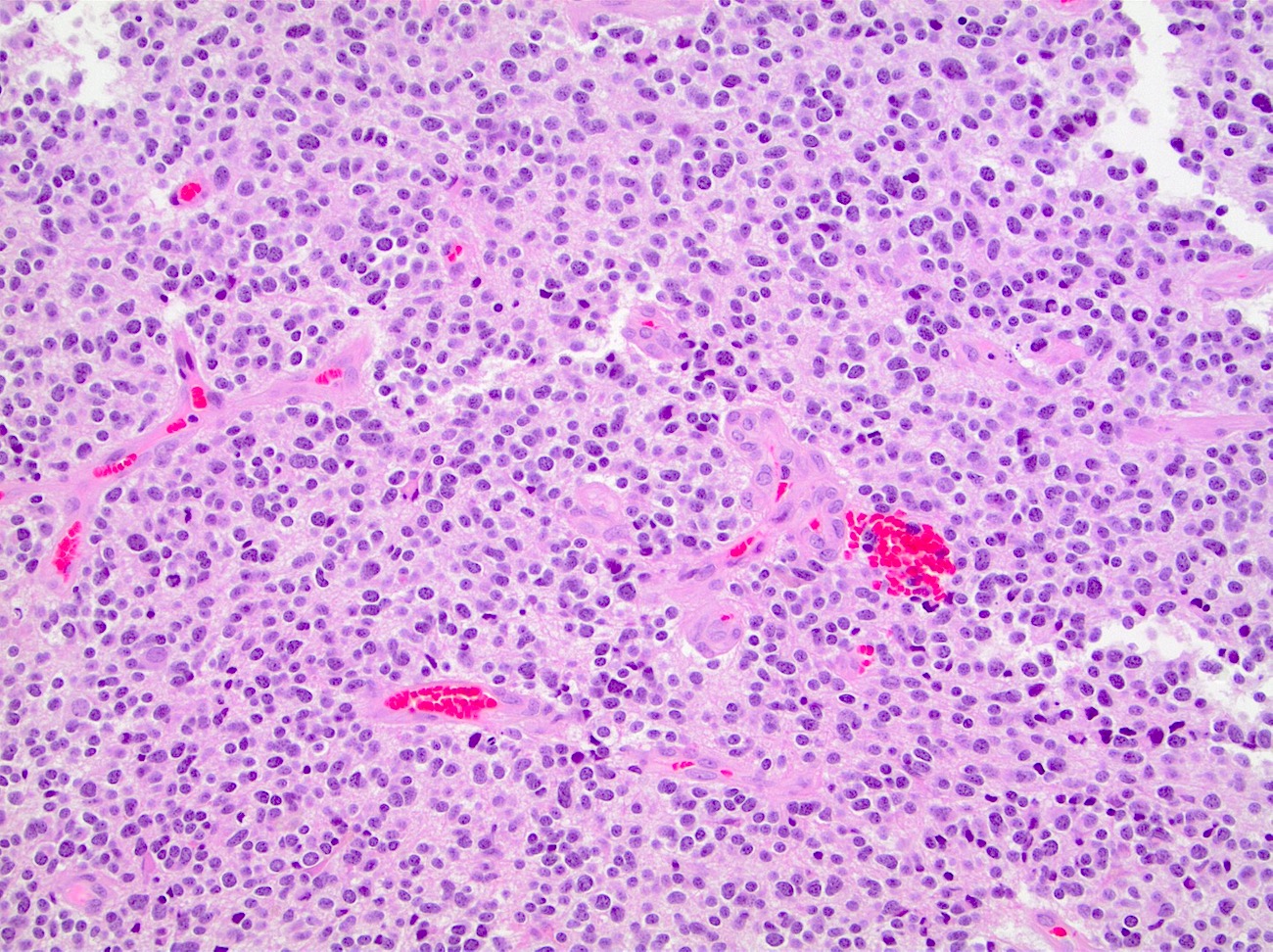
Cellularity and vascular proliferation
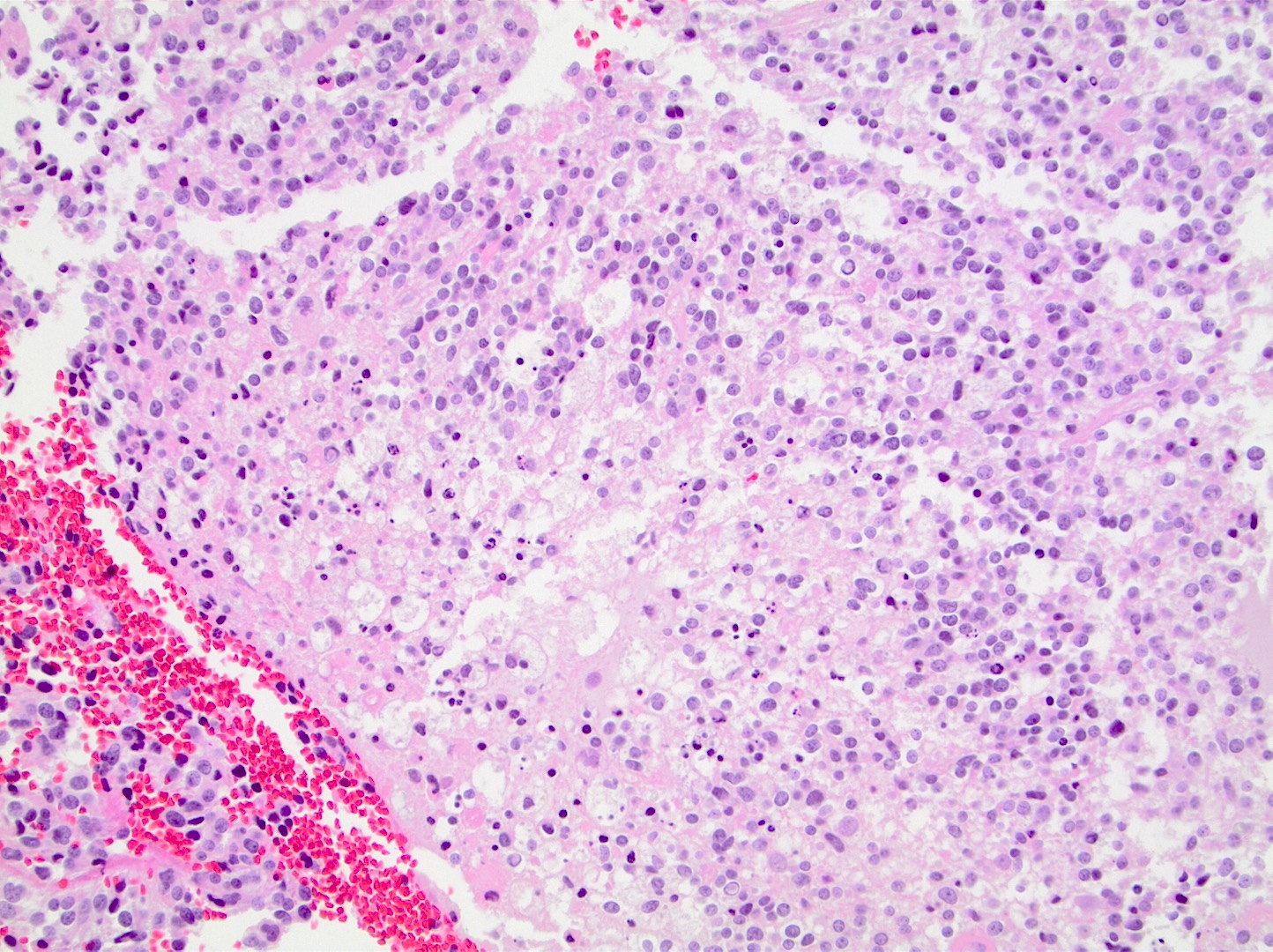
Focal necrosis
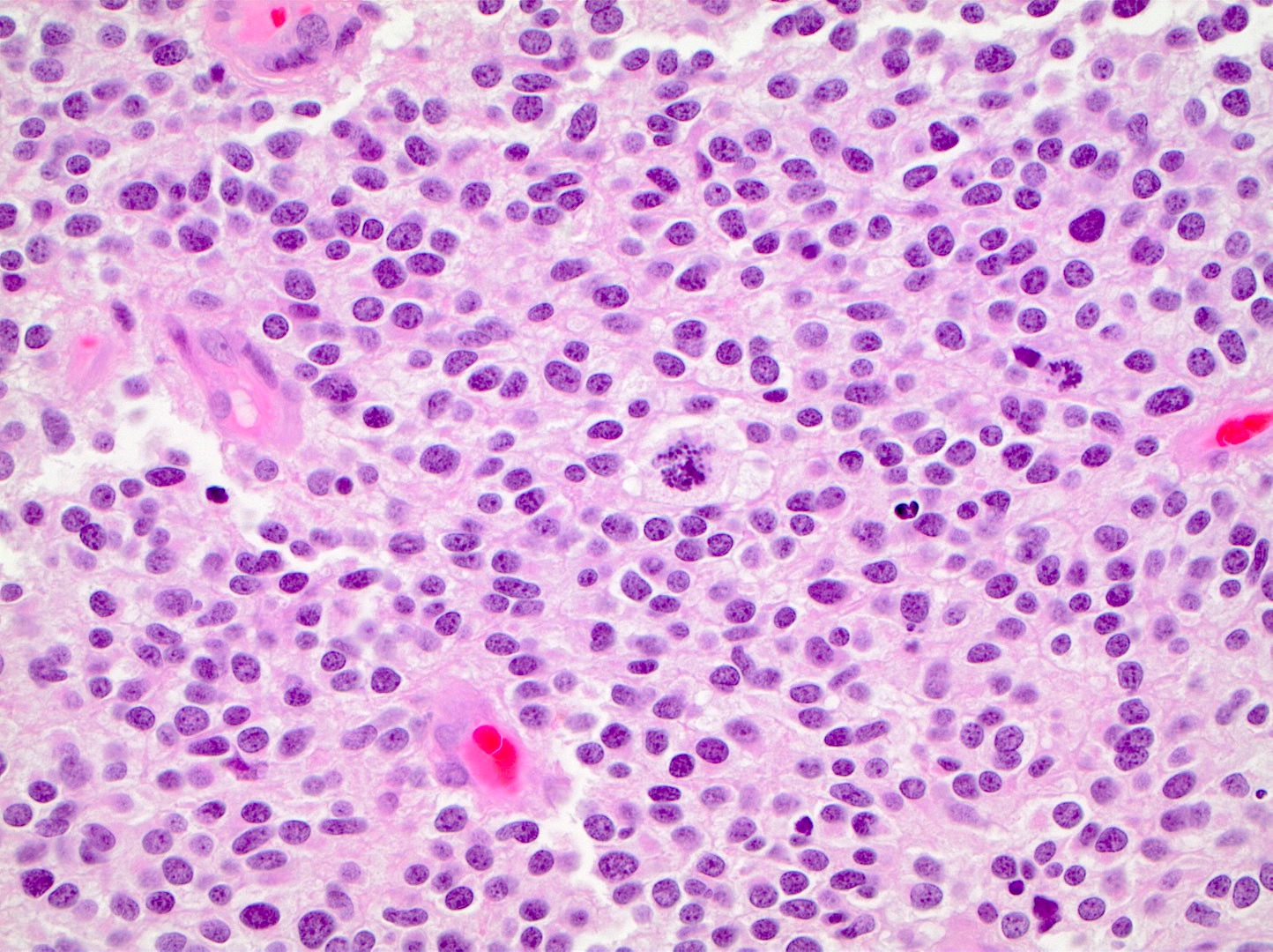
Brisk mitotic activity
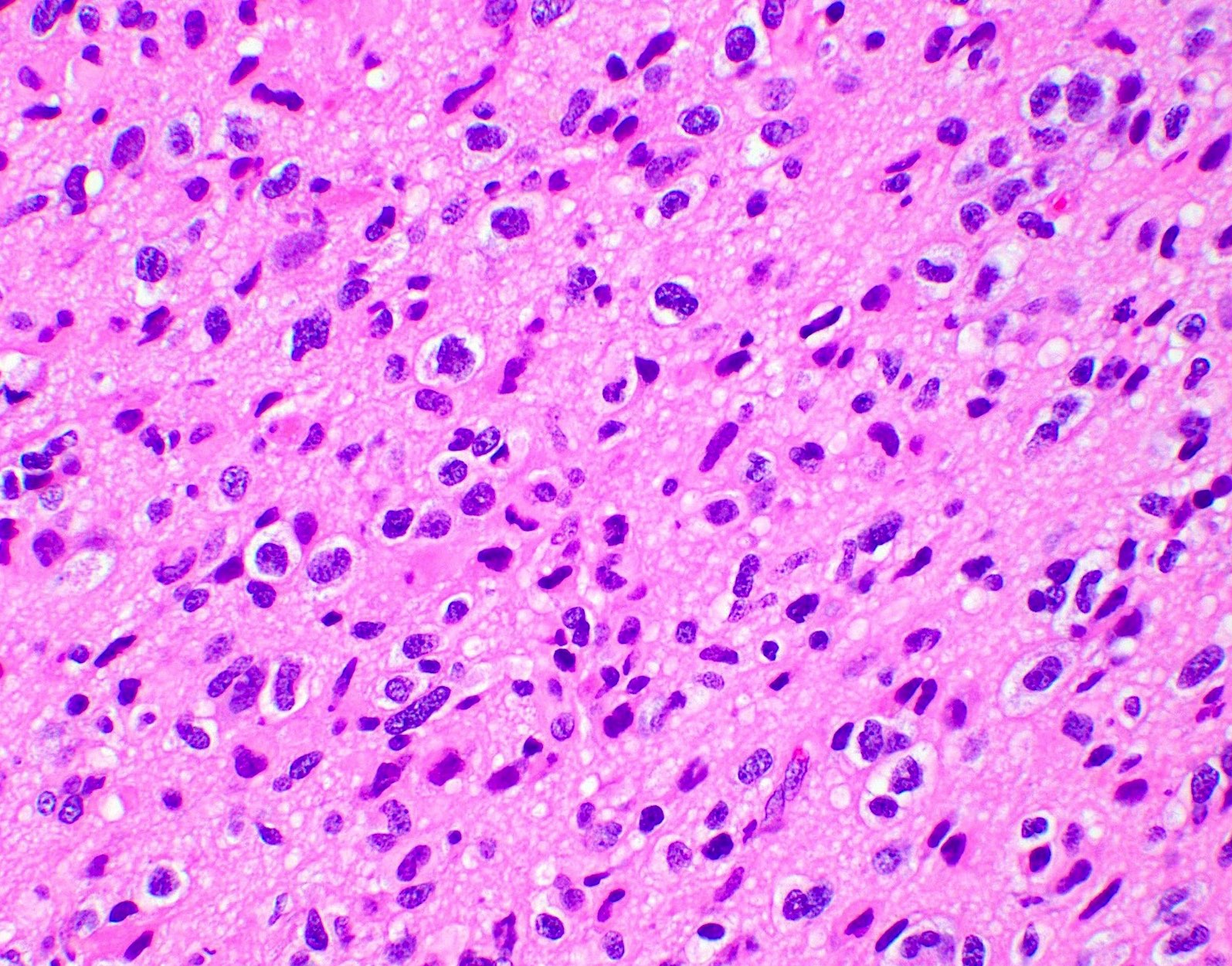
Mixed astrocytic histology
IDH1 R132H

p53
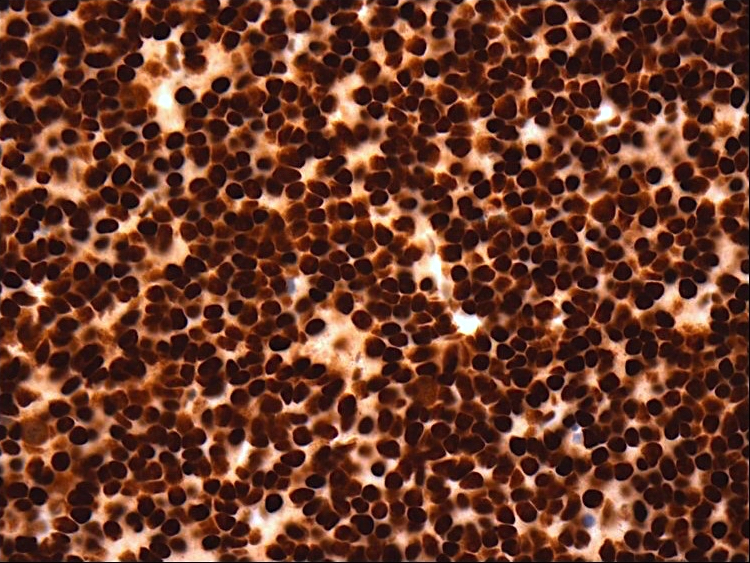
ATRX
Images hosted on other servers:

Oligodendroglioma,
resection

Oligodendroglioma,
IDH1 R132H,
resection
- IDH1 (R132H)
- Positive in > 90% of oligodendrogliomas (Acta Neuropathol 2009;118:599)
- Negative staining is not incompatible with oligodendroglioma if 1p / 19q codeletion is present
- Olig2, GFAP
- ATRX (retained; wildtype pattern)
- p53 (weak staining in rare cells; wildtype staining pattern)
- Ki67:
- Grade 2 tumors: usually < 5% of tumor nuclei
- Grade 3 tumors: generally > 10% of tumor nuclei (Neuro Oncol 2014;16:1244, Neuro Oncol 2016;18:888)
- Trio of IDH1 (R132H), ATRX and p53 is useful to distinguish oligodendroglioma from IDH mutant astrocytoma (Acta Neuropathol 2012;124:615)
- Loss of H3K27 trimethylation (Acta Neuropathol 2020;139:597)
- Keratins (although certain cytokeratin cocktails may demonstrate crossreactivity)
- Not routinely used for diagnostic purposes
- Oligodendroglioma is a molecularly defined diagnosis requiring demonstration of both:
- IDH1 or IDH2 mutation
- Unbalanced translocation between chromosome 1 and 19, resulting in whole arm loss of 1p and 19q chromosomal material (1p / 19q codeletion)
- Incomplete or partial deletions are not compatible with oligodendroglioma diagnosis
- Other common molecular alterations:
- TERT promotor mutation in vast majority (Nat Genet 2015;47:458)
- Early (clonal) event in tumorogenesis (Oncotarget 2019;10:3641)
- Often absent in teenagers with oligodendroglioma (Acta Neuropathol Commun 2018;6:95)
- CIC mutation in 70% (N Engl J Med 2015;372:2499)
- FUBP1 mutations in 20 - 30% (Science 2011;333:1453)
- NOTCH1 mutation in 15% (Nat Genet 2015;47:458)
- Loss of H3K27 trimethylation by immunohistochemistry (Acta Neuropathol 2020;139:597)
- TERT promotor mutation in vast majority (Nat Genet 2015;47:458)
- Molecular alterations associated with tumor progression:
- Increased copy number alterations (Neuro Oncol 2018;20:66)
- CDKN2A / CDKN2B deletions on chromosome 9p21 (PLoS One 2016;11:e0168728)
- PIK3CA mutation (Clin Cancer Res 2019;25:4375)
- TCF12 mutation (Nat Commun 2015;6:7207)
- MYC amplification (Nat Commun 2016;7:11263)
- Epigenetic changes:
- Glioma CpG island methylator phenotype (G CIMP) (Cancer Cell 2010;17:510)
- MGMT promotor methylation is detectable in the majority of tumors (Int J Cancer 2005;113:379)
- Brain, frontal lobe, left, tumor, resection:
- Integrated diagnosis: oligodendroglioma, IDH1 R132H mutant and 1p / 19q codeleted
- Histologic diagnosis: oligodendroglioma
- CNS WHO grade: 3
- Molecular information:
- IDH1 R132H mutation
- 1p / 19q codeletion
- TERT promotor mutation
- Astrocytoma, IDH mutant:
- Other tumors with oligo-like morphology:
- Dysembryoplastic neuroepithelial tumor (DNET)
- Central neurocytoma
- Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
- Clear cell ependymoma
- Metastatic clear cell carcinomas
- These tumors lack IDH mutations and 1p / 19q codeletion and are generally circumscribed instead of infiltrative
- Macrophage rich lesions:
- Stain positive with macrophage markers

A 42 year old man presents to the emergency room with new onset seizures. Brain magnetic resonance imaging (MRI) reveals a nonenhancing infiltrative mass lesion in the right frontal lobe. A biopsy is performed, shown in the image above. What molecular features are most likely present?
- BRAF V600E mutation
- EGFR amplification
- IDH mutation and 1p / 19q codeletion
- IDH, p53 and ATRX mutations
- Polysomy 7 and monosomy 10 (+7 / -10)
Comment Here
Reference: Oligodendroglioma, IDH mutant and 1p / 19q codeleted
- ATRX mutation
- BRAF V600E
- EGFR amplification
- p53 mutation
- TERT promotor mutation
Comment Here
Reference: Oligodendroglioma, IDH mutant and 1p / 19q codeleted
- Suprasellar (usually) epithelial encapsulated neoplasm with well differentiated nonkeratinizing squamous epithelium and papillary fibrovascular stroma
- BRAF V600E mutation in almost all cases
- WHO grade 1
- Encapsulated tumor with well differentiated nonkeratinizing squamous epithelium and papillary fibrovascular stroma
- Suprasellar papillary squamous epithelioma
- Ciliated craniopharyngioma
- Ciliated and goblet cell craniopharyngioma
- Incidence: 10% of all craniopharyngiomas
- Age: almost always adults
- No sex predilection
- Reference: J Neuropathol Exp Neurol 2017;76:126
- Suprasellar or intraventricular (third ventricle)
- Possible transition from Rathke cleft cyst
- Visual disturbances (Orphanet J Rare Dis 2007;2:18)
- Obstructive hydrocephalus
- Mental / personality changes
- Hyperprolactinemia (pituitary stalk effect)
- Diencephalic syndrome (rare)
- Lesions usually solid (J Neurosurg 1995;83:206)
- If cystic, may have mural nodule
- No calcifications on CT scan
- MRI: contrast enhancing solid or cystic mass
- Full pituitary endocrine workup is usually mandatory
- Visual acuity and visual field assessment is also performed to show any deficits and rule out papilledema
- More spherical in outline and usually lacks the prominent cystic component (Childs Nerv Syst 2019;35:169, J Neurosurg 1995;83:206)
- Solid or contains a few smaller cysts
- May have cyst and mural nodule configuration
- Tends to displace adjacent structures
- MRI:
- T1 weighted images: 85% of cysts are hypointense
- T1 weighted images: solid component iso to hypointense
- Vividly contrast enhancing
- CT:
- Cysts small and insignificant
- Near cerebrospinal fluid (CSF) density
- Solid component near soft tissue density
- Vivid enhancement
- Calcifications very rare
Contributed by Chunyu "Hunter" Cai, M.D., Ph.D.
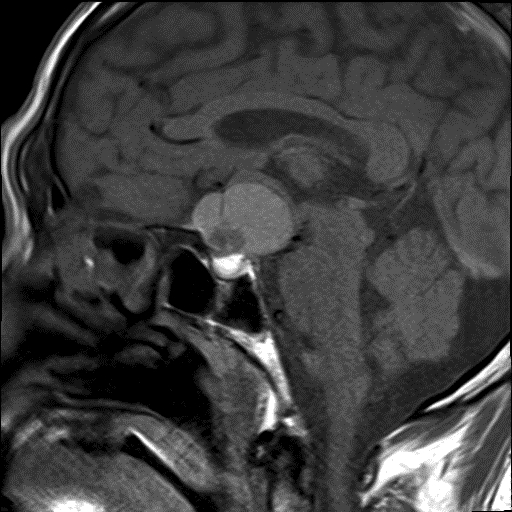
T1 precontrast sagittal
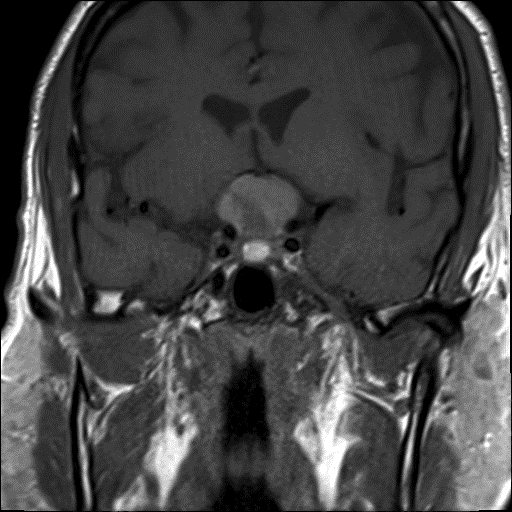
T1 precontrast coronal
Images hosted on other servers:
Suprasellar mass
Solid suprasellar mass with small cystic component
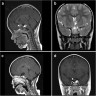
Preoperative MRI
- Prognosis slightly better than adamantinomatous variant (Neurosurgery 1994;35:1001)
- 4 year old girl with BRAF V600E mutation (Childs Nerv Syst 2019;35:169)
- 65 year old man with recurrent papillary craniopharyngioma with BRAF V600E mutation (Acta Neurochir (Wien) 2017;159:2217)
- 80 year old man with mixed type craniopharyngioma (Neuropathology 2012;32:171)
- Gross total resection is optimal; smoother surface than adamantinomatous tumors, which facilitates excision (Acta Neurochir (Wien) 2017;159:2217)
- Some reports show dramatic shrinkage of papillary craniopharyngioma using anti-BRAF agents (dabrafenib and trametinib) (Pituitary 2016;19:544, Eur J Endocrinol 2016;174:R139, Neurosurg Focus 2016;41:E3)
- Discrete, encapsulated mass (Acta Neurochir (Wien) 2017;159:2217, J Neurosurg 1995;83:206)
- Not densely adherent to adjacent brain
- No cholesterol rich, thick, oily cyst contents
- If cystic, contains clear liquid
- Low power highlights papillary configuration with cauliflower-like morphology
- Solid sheets of well differentiated nonkeratinizing squamous epithelium (J Neuropathol Exp Neurol 2017;76:126, J Neurosurg 1995;83:206)
- Crude papillae around fibrovascular cores
- Small collagenous whorls
Contributed by Nelli S. Lakis, M.D., M.Sc. and Chunyu "Hunter" Cai, M.D., Ph.D.
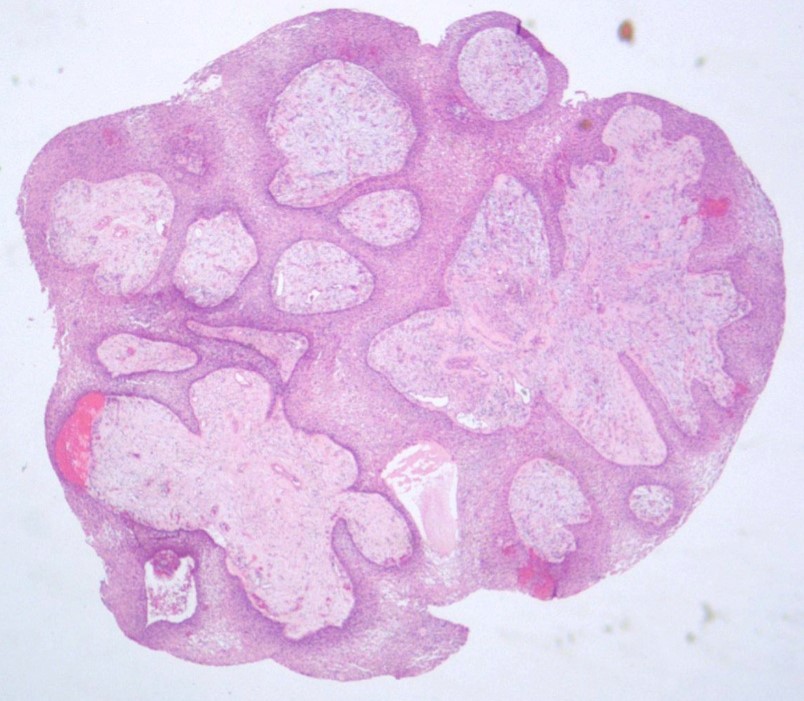
Papillary architecture
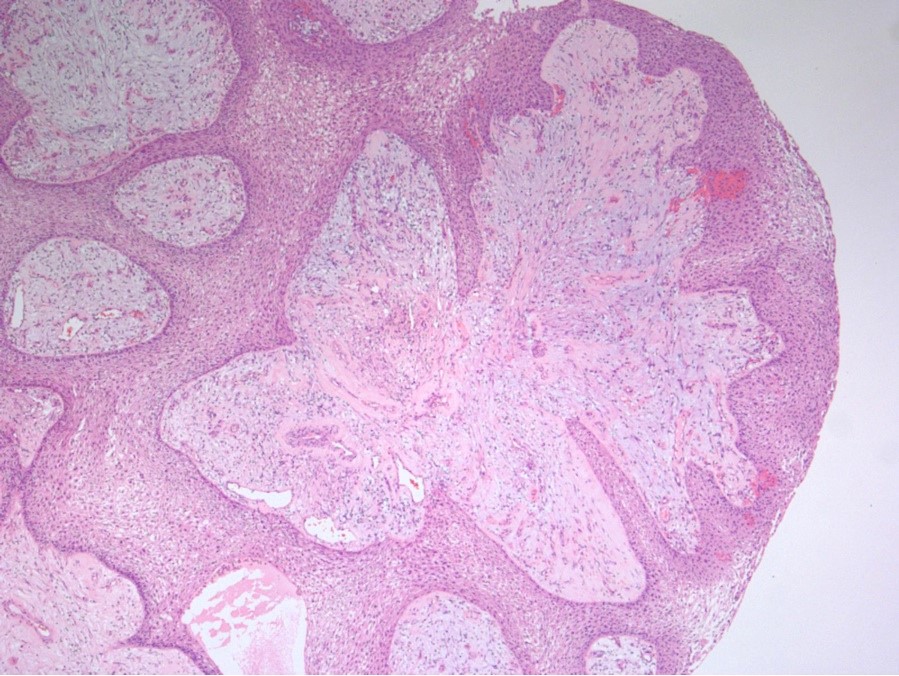
Fibrovascular cores
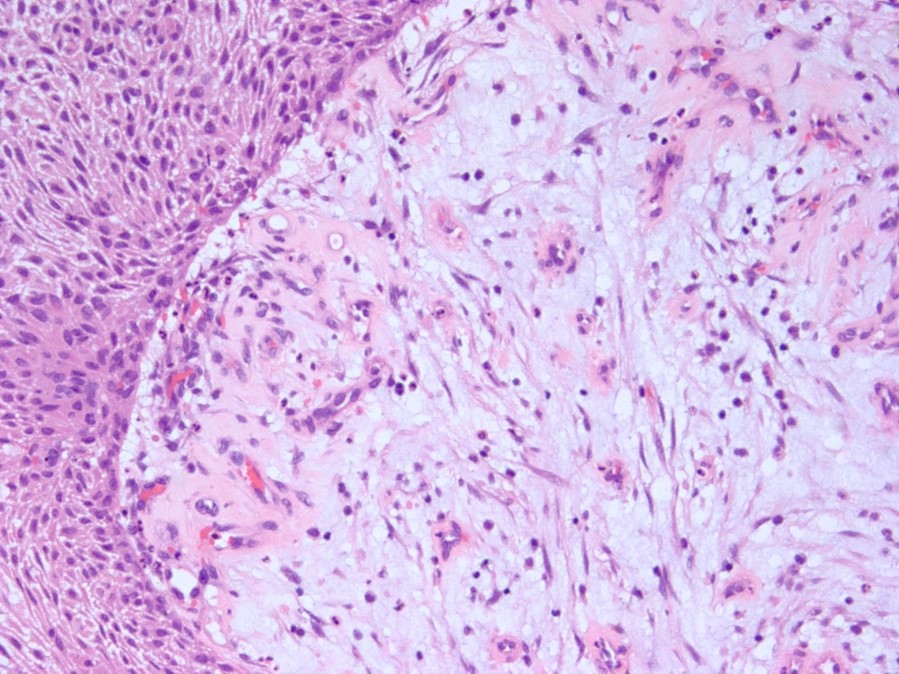
Stroma
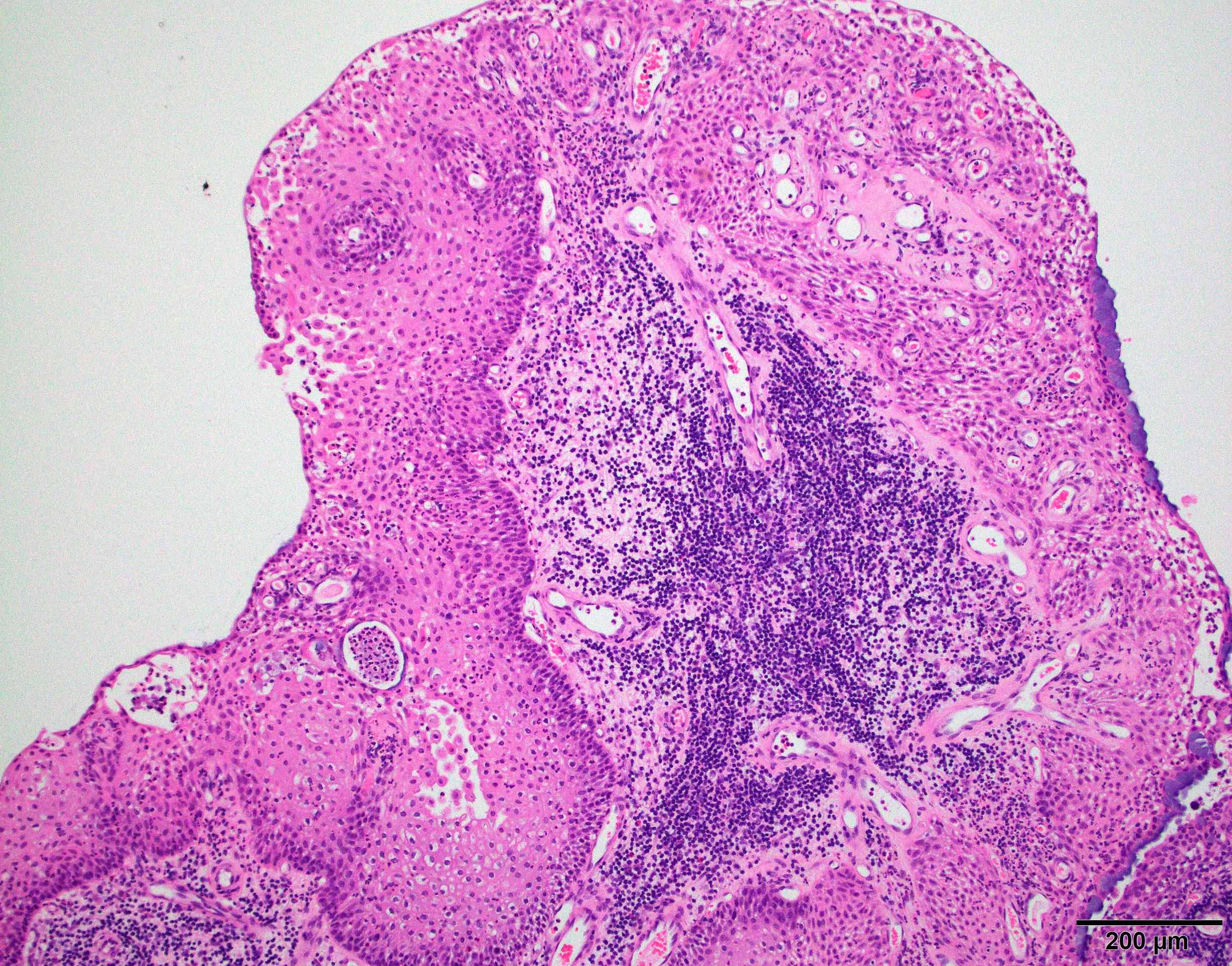
Lymphocytic inflammation
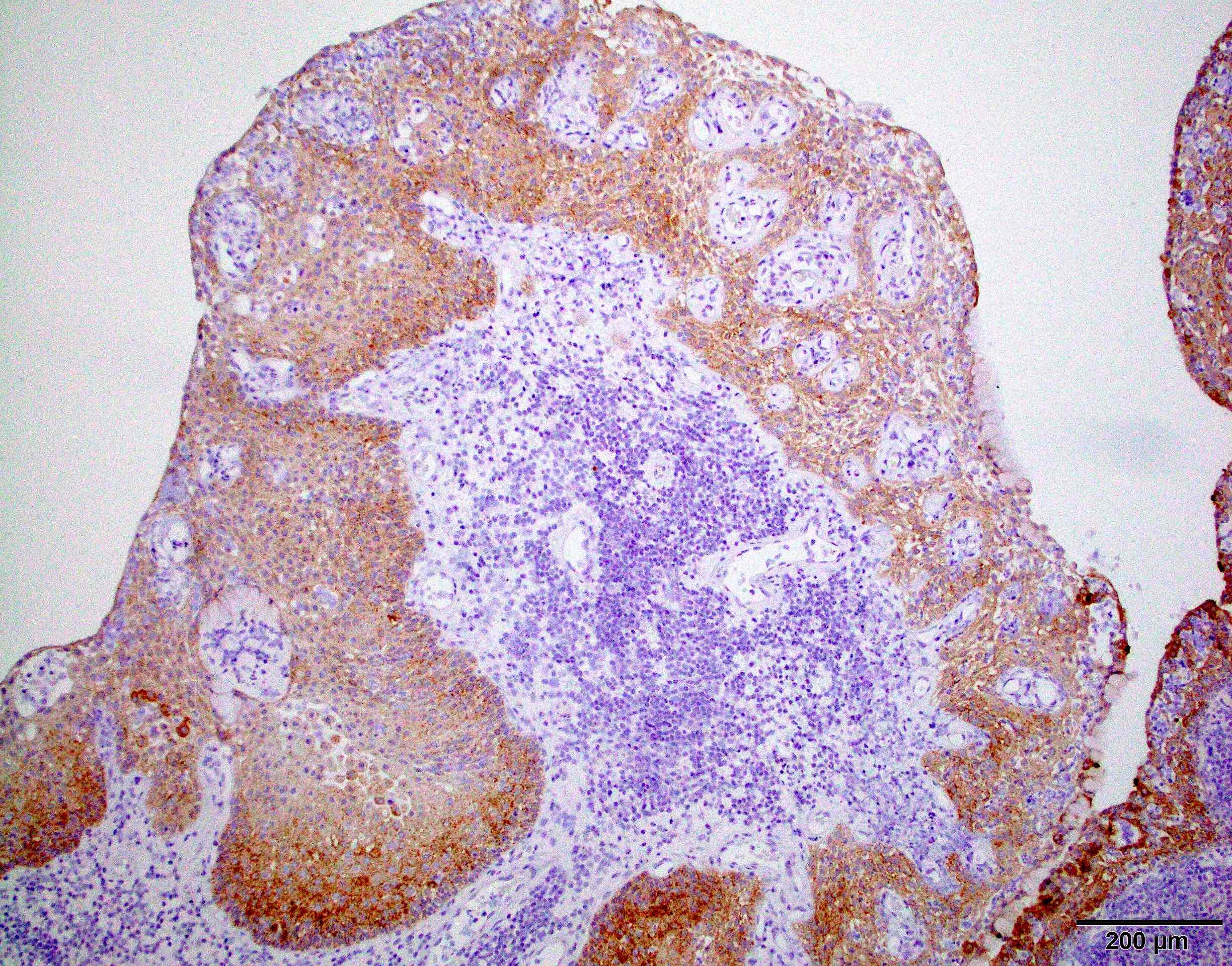
BRAF V600E immunostain
Images hosted on other servers:
54 year old man with papillary craniopharyngioma
- Sheets of epithelial cells
- Individual nucleated squamous cells
- CK7, EMA (J Neuropathol Exp Neurol 2017;76:126)
- BRAF VE1 parallels presence of BRAF V600E mutation in 95% of cases
- Can have scant PAS positive goblet cells
- Only membranous beta catenin; nuclei and cytoplasm negative
- Reference: Neuropathol Appl Neurobiol 2015;41:733
- BRAF V600E mutation in 95% of cases
- Sellar / suprasellar region, suprasellar mass, endoscopic resection:
- Papillary craniopharyngioma (see comment)
- Comment: BRAF V600E immunostain shows positive cytoplasmic reactivity, supporting / confirming the diagnosis of papillary craniopharyngioma.
- Adamantinomatous craniopharyngioma:
- Irregular, infiltrative borders
- Complex architecture
- Wet keratin
- Calcifications
- Peripheral palisading
- Loose stellate reticulum
- Positive nuclear beta catenin in keratin whorls (may be focal)
- Epidermoid cyst:
- Uniloculate with thin layer of keratinizing squamous epithelium and keratohyalin granules
- Rathke cleft cyst with squamous metaplasia:
- Usually cystic without solid component, squamous epithelium with ciliated or mucus containing cells
- Clin Neuropathol 2003;22:229, J Egypt Natl Canc Inst 2015;27:139, J Korean Neurosurg Soc 2017;60:108, Acta Neuropathol Commun 2016;4:20, AJNR Am J Neuroradiol 2015;36:E55, eMedicine: Surgery for Craniopharyngiomas Treatment & Management [Accessed 30 April 2021], Radiopaedia: Craniopharyngioma [Accessed 30 April 2021], Radiopaedia: Craniopharyngioma (papillary) [Accessed 30 April 2021], Kleinschmidt-DeMasters: Diagnostic Pathology - Neuropathology, 2nd Edition, 2016, Louis: WHO Classification of Tumours of the Central Nervous System, 4th Edition, 2016
A 56 year old man is being evaluated due to headaches and visual disturbances. A brain MRI shows a 5 cm solid and cystic suprasellar lesion. The lesion is resected and a histopathologic examination reveals fibrovascular cores and abundant well differentiated squamous epithelium. Which of the following is a feature of the most likely diagnosis?
- BRAF V600E mutation
- Most commonly occurs in children
- Nuclear palisading
- Nuclear staining with beta catenin
- Adamantinomatous craniopharyngioma
- Dermoid cyst
- Papillary craniopharyngioma
- Rathke cleft cyst
- Rare, low grade biphasic neoplasm characterized by pseudopapillae lined by glial cells with interpapillary neuronal elements
- Favorable outcome suggests that it corresponds to WHO grade 1
- First described by Komori et al. (Am J Surg Pathol 1998;22:1171)
- Age: young adults
- Sex: no gender predilection
- Cerebral hemisphere
- > Temporal lobe
- Supratentorial, discrete, contrast enhancing
- Often cystic with occasional cyst mural nodule pattern
Images hosted on other servers:
Heterogeneously enhancing mass in pineal gland
- 14 year old girl with papillary glioneuronal tumor (Arq Neuropsiquiatr 2004;62:869)
- 18 year old man with papillary glioneuronal tumor (Arch Pathol Lab Med 2000;124:1820)
- 58 year old man with malignant papillary glioneuronal tumor of the pineal gland (Am J Case Rep 2013;14:164)
- Complete surgical resection
- Favorable clinical outcome
- Biphasic pattern with glial and neuronal components
- Glial component: small uniform glial cells line pseudopapillae with hyalinized vascular cores; no atypia or mitosis
- Neuronal component: interpapillary zone is occupied by neurocytes with uniform nuclei and occasional perinuclear halos; occasionally ganglion and ganglioid cells are admixed
- Vascular proliferation and necrosis are exceptional
- The surrounding brain tissue shows gliosis
- Nonspecific, uniform cells
- Glial elements: GFAP+
- Neuronal elements: synaptophysin+
- Low proliferation index (Ki67 is 1 - 2%)
- Three cell types: astrocytic, neuronal, glioneuronal progenitor cells
- Astroblastoma: dot-like EMA reactivity, synaptophysin negative
- Clear cell ependymoma: synaptophysin negative
- DNET: intracortical, specific glioneuronal elements with intervening floating neurons
- Extraventriular neurocytoma: no pseudopapillae lined by glial cells
- Oligodendroglioma: nonenhancing, diffusely infiltrative
- Pilocytic astrocytoma: biphasic piloid and spongy areas
- RGNT: 4th ventricle, neurocytic rosettes with synaptophysin positive cores
- Low grade glioneuronal tumor with a biphasic pattern composed of pseudopapillary glial structures and interpapillary neuronal components; also with PRKCA gene fusion (mainly SLC44A1::PRKCA fusion)
- Corresponds to WHO grade 1
- Rare, low grade glioneuronal neoplasm with biphasic histological and immunophenotypic pattern characterized by pseudopapillae lined by GFAP positive glial cells with interpapillary synaptophysin positive neuronal elements (PMID: 26899971, PMID: 35512827, PMID: 26671581)
- First described by Komori et al. (Am J Surg Pathol 1998;22:1171)
- PGNT is characterized by PRKCA gene fusion (mainly SLC44A1::PRKCA fusion) and a distinctive methylation profile
- Supratentorial, well delineated and often cystic tumors with predilection to young adults
- Favorable prognosis with gross total resection
- Not recommended by WHO: pseudopapillary ganglioneurocytoma, pseudopapillary neurocytoma with glial differentiation
- ICD-O: 9509/1 - papillary glioneuronal tumor
- ICD-11: 2A00.21 & XH3XU4 - mixed neuronal glial tumors & papillary glioneuronal tumor
- Rare (PMID: 37787321)
- Age: predilection for young adults (median age at diagnosis is 16 years (range: 6 - 54 years) (PMID:30759284, PMID: 34038055 PMID: 28823671)
- No gender predilection (PMID:30759284, PMID: 35512827)
- Supratentorial, with predilection for temporal and frontal lobes (PMID:30759284, PMID: 28823671, PMID: 35512827, PMID:26671581)
- Propensity for the ventricular system, with > 60% of tumors in a periventricular or intraventricular location (PMID:30759284, PMID: 28823671 PMID: 26671581)
- It can occur anywhere in the cerebrum, cerebellum, or brainstem (PMID: 28823671)
- PRKCA gene fusion is the hallmark of PGNT, mainly SLC44A1::PRKCA fusion; less frequently NOTCH1::PRKCA fusion (PMID:30759284, PMID:26671581)
- A reciprocal translocation t(9;17)(q31 ;q24) results in the canonical SLC44A1::PRKCA fusion with consequent generation of a constitutively expressed oncoprotein (PMID:22725730)
- PRKCA encodes protein kinase C alpha (PKCA) involved in MAPK signaling pathway (PMID:26671581)
- Unknown
- Principal manifestations include headache and seizures and mass effects (PMID: 26899971, PMID: 34038055)
- Occasionally asymptomatic (PMID: 35512827)
- Hemorrhagic onset has been reported (PMID:16771176, PMID:22717600)
- Neuroimaging: MRI and CT
- Biopsy: diagnosis should be heavily weighted towards molecular findings because morphological analyses frequently result in mistyping (PMID:30759284)
- WHO diagnostic criteria
- WHO essential diagnostic criteria
- Biphasic histological and immunophenotypic pattern with pseudopapillary glial lining and interpapillary neuronal components
- PRKCA gene fusion (mostly SLC44A1::PRKCA)
- For unresolved lesions: methylation profile of papillary glioneuronal tumor
- WHO desirable diagnostic criteria
- Well delineated, solid and cystic tumor
- WHO essential diagnostic criteria
- MRI
- Supratentorial, well delineated
- Cystic or solid, some cases are cystic with a mural nodule (PMID: 26899971, PMID: 34038055, PMID: 35512827, PMID: 22725730)
- Solid component is hypo- to isointense on T1 weighted images and iso- to hyperintense on T2 weighted images (PMID: 35512827)
- Contrast enhancing (PMID: 26899971)
- No or only minimal peritumoral edema and little mass effect (PMID: 35512827)
- No diffusion restriction (PMID: 35512827)
Contributed by Eman Abdelzaher, M.D., Ph.D.
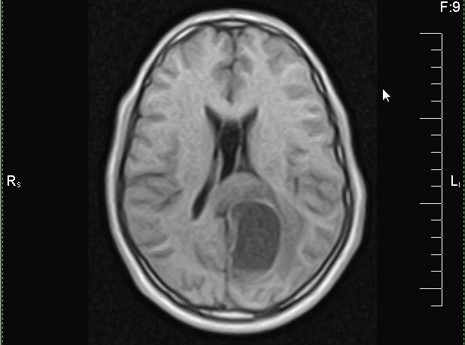
MRI, axial T1
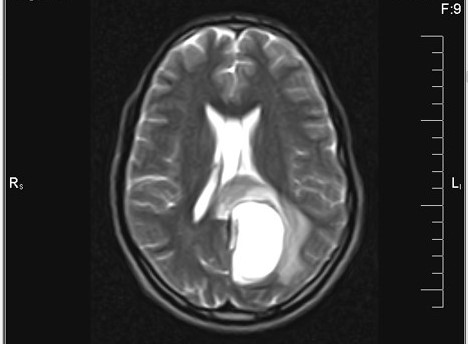
MRI, axial T2
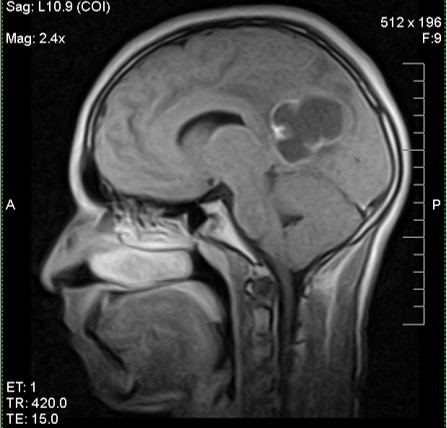
MRI, sagittal T1 with contrast

Axial T1 with contrast
Images hosted on other servers:
Left temporal lobe mass, MRI
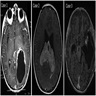
Contrast enhancement and ventricular association
- Outcome is usually favorable, with a reported median 5 year progression free survival > 80% (PMID: 30759284)
- Gross total resection constitutes the main prognostic factor; male sex and low Ki67 proliferation index were also reported as positive prognostic indicators (PMID: 28823671, PMID: 35512827)
- Recurrence was reported in one molecularly confirmed case, while it was documented in 11.8 % of morphologically diagnosed cases, with higher rates associated with incomplete resection (PMID:30759284, PMID: 24553727)
- Increased Ki67 proliferation index (> 20%) or anaplastic features with tumoral progression or dissemination have been reported in morphologically diagnosed cases but have not been described in PRKCA fused cases (PMID:28823671, PMID:19119904, PMID: 24553727)
- 14 year old boy with intraventricular papillary glioneuronal tumor (PMID: 37787321)
- 16 year old girl with suprasellar papillary glioneuronal tumor mimicking craniopharyngioma (PMID: 32695553)
- 38 year old man with papillary glioneuronal tumor growing slowly for 26 years (PMID: 35854961)
- 48 year old woman with papillary glioneuronal tumor masquerading as malignant brain tumor (PMID: 37621982)
- 64 year old man with papillary glioneuronal tumor with an excessive angiomatous component (PMID: 29336376)
- Gross total resection is optimal (PMID: 34038055)
- Radiation therapy and chemotherapy are used rarely, on a case by case basis (PMID: 28823671)
Images hosted on other servers:
Right occipital lobe tumor
Left frontal lobe tumor
- Well circumscribed, solid or cystic lesions
- Calcification and hemorrhage may be observed
- Characterized by a biphasic pattern created by a glial pseudopapillary component and an interpapillary neuronal component with variable proportions of both components (PMID: 26899971, PMID:9777979)
- Glial component is composed of a single or pseudostratified layer of small cuboidal glial cells with round nuclei and scant cytoplasm, lining pseudopapillary structures (present in 93% of molecularly confirmed PGNTs) with hyalinized vascular cores (PMID:30759284)
- Neuronal component is composed of uniform neurocytes or medium sized neurons disposed in a neuropil background (PMID:9777979)
- Ganglion cells (21.4%), microcalcifications (35.7%) may be seen (PMID:30759284)
- Rarely, eosinophilic granular bodies may be encountered (PMID:9777979, PMID:26671581)
- Occasional mitoses may be seen (PMID: 30759284)
- Microvascular proliferation and necrosis are absent (PMID:30759284, PMID:9777979)
- Anaplastic features were rarely reported in non-molecularly confirmed cases (PMID: 19119904, PMID:19157003)
- Peritumoral reactive gliosis
- Nonspecific
- May exhibit vague papillary structures with hyalinized cores lined by bland tumor cells (PMID: 37787321, PMID: 22020040)
- Loosely cohesive clusters and singly scattered tumor cells with mildly pleomorphic round to oval nuclei with stippled chromatin, small nucleoli and scant to moderate cytoplasm (PMID: 33991783, PMID: 37787321, PMID: 22020040)
- Biphenotypic immunophenotype: glial in pseudopapillary structures and neuronal in interpapillary areas
- GFAP: positive in glial cells ensheathing pseudopapillae and in scattered cells in the interpapillary areas (PMID: 26899971, PMID: 22725730)
- S100: positive in glial cells (PMID: 35512827, PMID: 22725730)
- Olig2: positive in glial cells; in some cases, oligodendrocyte-like cells (Olig2+ and GFAP-) may be seen (PMID:15906048)
- Synaptophysin: positive in neuronal component (PMID: 26899971)
- a href="https://www.pathologyoutlines.com/topic/stainsneun.html">NeuN: positive in neuronal component (PMID: 26899971)
- NFP / neurofilament: mostly confined to ganglioid / ganglion cells
- Ki67: generally low (does not exceed 2%) but elevated proliferative activity (ranging from 10% to > 50%) has been reported in non-molecularly confirmed cases (PMID:24709681, PMID:19157003 PMID: 34038055)
- Chromogranin A: variable (PMID:9777979, PMID:26671581)
- CD34: may be focally positive (PMID:26671581)
- p53 (PMID:26671581)
- H3-K27 (PMID:26671581)
- BRAF V600E (PMID:26671581)
- EMA (PMID: 32695553)
- IDH1 R132H (PMID: 32695553, https://doi.org/10.3171/CASE21266)
- Three cell types: astrocytic, neuronal, glioneuronal progenitor cells
- Astroblastoma: dot-like EMA reactivity, synaptophysin negative
- Clear cell ependymoma: synaptophysin negative
- DNET: intracortical, specific glioneuronal elements with intervening floating neurons
- Extraventriular neurocytoma: no pseudopapillae lined by glial cells
- Oligodendroglioma: nonenhancing, diffusely infiltrative
- Pilocytic astrocytoma: biphasic piloid and spongy areas
- RGNT: 4th ventricle, neurocytic rosettes with synaptophysin positive cores
- WHO grade 3
- Aggressive, high rate of recurrence, may metastasize
- Often occurs in younger patients
- May be difficult to recognize as meningothelial
- Helps to refer to imaging or surgeon to verify a dural based lesion
- Most often supratentorial
- However, can occur in posterior fossa and spinal cord
- MRI may show irregular tumor borders, heterogenous tumoral enhancement and peritumoral edema suggesting a high grade meningioma
Images hosted on other servers:
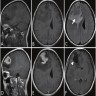
Right frontal papillary meningioma
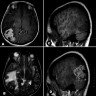
Large, lobulated, irregular mass
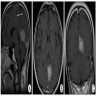
Mass with a broad base
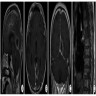
Postoperative shows severe leptomeningeal seeding
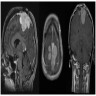
Avidly enhancing meningioma
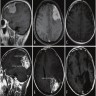
Preoperative sagittal and axial adolinium enhanced MR images
- Complete surgical resection is the most favorable prognostic indicator (Neurosurgery 2013;73:777)
- Low MIB1 index and retention of progesterone receptor (PR) have also been reported as markers of improved prognosis
- 3 year old boy with cerebellar papillary meningioma (Am J Surg Pathol 1999;23:844)
- 13 year old boy with papillary meningioma with pleural metastasis (Acta Neurol Scand 2000;102:200)
- 15 year old girl with cystic papillary meningioma (Childs Nerv Syst 2005;21:322)
- 19 year old man with spinal papillary meningioma (Acta Neurochir (Wien) 2000;142:703)
- 25 year old woman with jugular foramen papillary meningioma (Brain Tumor Pathol 2004;21:143)
- 29 year old woman with meningioma with rhabdoid, papillary and clear cell features (Clin Neuropathol 2011;30:291)
- 43 year old man with papillary meningioma with leptomeningeal seeding (J Korean Neurosurg Soc 2011;49:124)
- Gross total resection
- Postsurgical radiotherapy (Front Oncol 2013;3:227)
- Radiosurgery (Radiat Oncol 2014;9:38)
- Dural based
- May be well circumscribed or readily adherent to brain parenchyma
- Size can vary widely
- Papillary structures are best appreciated at edges of tumor
- Papillary features may be focal but at least 50% of tumor should have papillary features to call papillary meningioma, WHO grade 3
- Cross sections through papillae may give an ependymoma-like histology with perivascular pseudorosettes
- May have areas of classic meningioma or may be difficult to appreciate any meningothelial features (whorls, psammoma bodies, nuclear pseudoinclusions)
- Often has features of atypical meningioma: hypercellularity, brain invasion, increased mitoses, prominent nucleoli
- Not distinct from a classic meningioma smear
- However, may appreciate papillary clusters from low power
Images hosted on other servers:
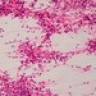
Meningioma smear
- Meningioma features of interdigitating cell processes, desmosomes and intermediate filaments (Histopathology 2001;38:318)
Images hosted on other servers:
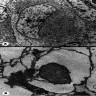
Bundles of intermediate filaments
- First described in 2003 (Am J Surg Pathol 2003;27:505)
- 4 of 6 were women, ages 19 - 53 years
- May derive from ependymal cells of subcommissural organ (also chordoid glioma)
- Well circumscribed pineal mass
- Epithelial-like growth with vessels covered by a layer of tumor cells
- Cells are large, columnar / cuboidal with clear cytoplasm, round or infolded nuclei at basal pole of tumor cells
- May have rosettes
- Abundant rough endoplasmic reticulum with distended cisternae filled with secretory product
- Also microvilli and perinuclear intermediate filaments
- Pineal parenchymal tumor: synaptophysin+, keratin-, vimentin-
- Circumscribed, well differentiated astrocytic neoplasm with piloid (hair-like) processes, most commonly occurring in children and young adults, WHO grade 1 (Acta Neuropathol 2016;131:803)
- Most common glioma in children (Neuro Oncol 2020;22:iv1)
- Radiologic appearance often cystic with an enhancing mural nodule (Radiographics 2004;24:1693)
- Generally circumscribed, biphasic, astrocytic neoplasm with piloid processes, Rosenthal fibers and eosinophilic granular bodies (EGBs)
- Activating genetic alterations in components of MAPK pathway (most frequently BRAF fusions) (Nat Genet 2013;45:927, Nat Genet 2013;45:602)
- WHO grade 1 (Acta Neuropathol 2016;131:803)
- Pilocytic astrocytoma
- Juvenile pilocytic astrocytoma (not recommended)
- Spongioblastoma (not recommended)
- Pilomyxoid astrocytoma (subtype)
- Optic nerve glioma
- M = F (Neuro Oncol 2014;16:iv1)
- 5.4% of all gliomas (J Neuropathol Exp Neurol 2005;64:479)
- Most common glioma in children (Neuro Oncol 2014;16:iv1)
- Most common glioma during first 2 decades of life (Neuro Oncol 2014;16:iv1)
- Found throughout neuraxis
- Most common sites: cerebellum (42%), supratentorial (36%), optic pathway / hypothalamus (9%), brainstem (9%), spinal cord (2%) (J Neurosurg 2003;98:1170)
- Infratentorial location most common in childhood (J Neurooncol 2017;131:163)
- Mutation in MAPK pathway component → pathway activation → transcription factor activation → increased cell growth and proliferation
- Biallelic inactivation of NF1 in the setting of neurofibromatosis type 1 (Genome Res 2013;23:431)
- Most cases are sporadic
- Germline mutations in MAPK pathway genes, including FGFR1, NF1, PTPN11 and RAF1
Images hosted on other servers:
MAPK pathway mutations
- Neurological deficits depending on tumor location
- Nonlocalizing signs
- Macrocephaly
- Headache
- Endocrinopathy
- Increased intracranial pressure
- Optic pathway tumors in 15% of patients with neurofibromatosis type 1 (Ophthalmology 1984;91:929)
- Diagnosis may be made on histologic features alone (Acta Neuropathol 2015;129:775)
- When microscopic findings are limited / ambiguous, molecular testing may be necessary to assess for gene fusions and other alterations
- Well demarcated enhancing mass (Insights Imaging 2014;5:387)
- Cystic mass with enhancing mural nodule (~66% of cases), solid or heterogeneous mixed solid - cystic
- Fusiform mass in optic pathway
Contributed by P.J. Cimino, M.D., Ph.D.
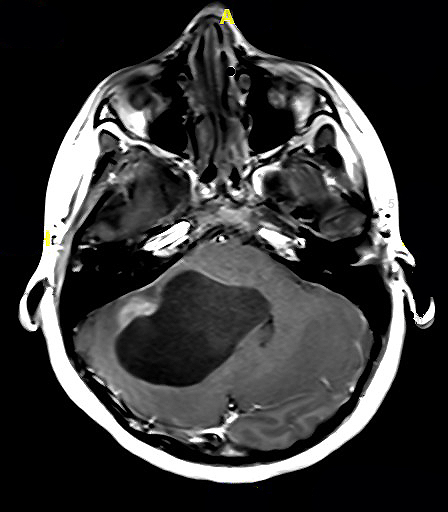
T1 postcontrast MRI
- 10 year survival > 95% after surgical intervention alone (J Neurosurg 2003;98:1170)
- Optic nerve tumors in NF1 patients: favorable (Ann Neurol 2007;61:189)
- Deep, midline location: high rates of incomplete resection, recurrence, shorter survival (Neuropathol Appl Neurobiol 2013;39:693)
- Supratentorial: poor (Cancer 1993;72:1335)
- Partial resection: poor (Neurosurgery 2003;53:544)
- Age > 40: poor (J Neurooncol 2020;148:187)
- Anaplastic features in adults: poor, no definitive WHO grade (Am J Surg Pathol 2010;34:147, Acta Neuropathol 2020;139:287, Acta Neuropathol 2016;131:803)
- Pilomyxoid astrocytoma variant: unclear prognosis, no definitive WHO grade (Acta Neuropathol 2016;131:803)
- 13 year old girl with left cerebellar cystic mass (NMC Case Rep J 2019;6:95)
- 33 year old woman with mesial temporal lobe mass (Case Rep Pathol 2020;2020:5903863)
- 39 year old woman with optic nerve glioma (Brain Tumor Pathol 2021;38:59)
- Gross total surgical resection
- Radiation or chemotherapy may be considered for subtotally resected or recurrent cases
- Variable success reported for MAPK pathway targeted therapies (Proc Natl Acad Sci U S A 2013;110:5957, Nat Med 2013;19:1401)
Images hosted on other servers:

Intraoperative surgical image
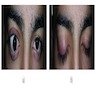
Exophthalmos - optic nerve glioma

Orbital pilocytic astrocytoma
- Usually well circumscribed (Acta Neuropathol 2015;129:775)
- Commonly cystic
- May have myxoid or mucoid appearance
- May contain calcifications or hemosiderin staining
Images hosted on other servers:

Intraocular pilocytic astrocytoma
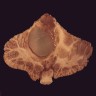
Cerebellar tumor
Contributed by P.J. Cimino, M.D., Ph.D.
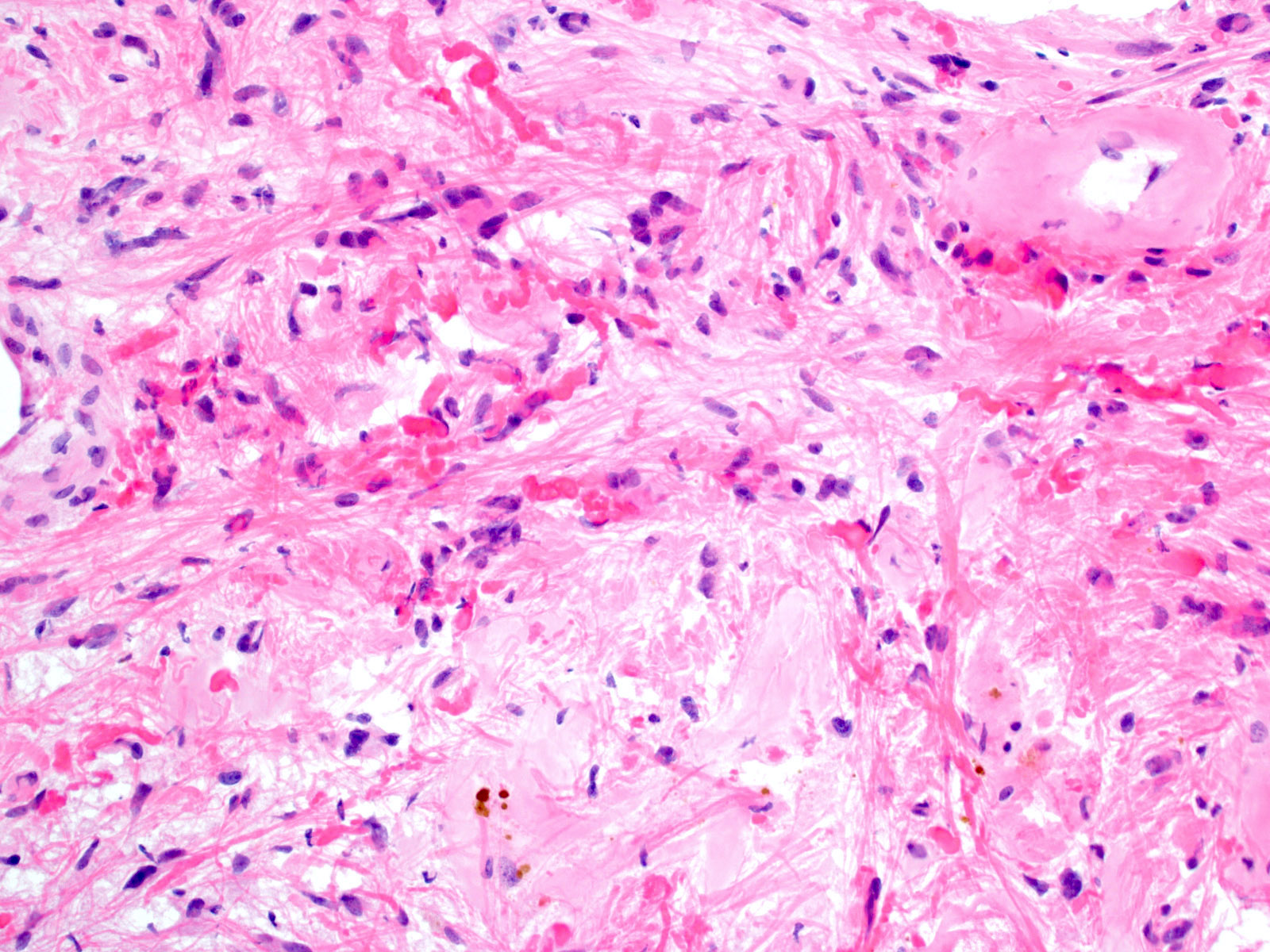
Atypical piloid glial cells
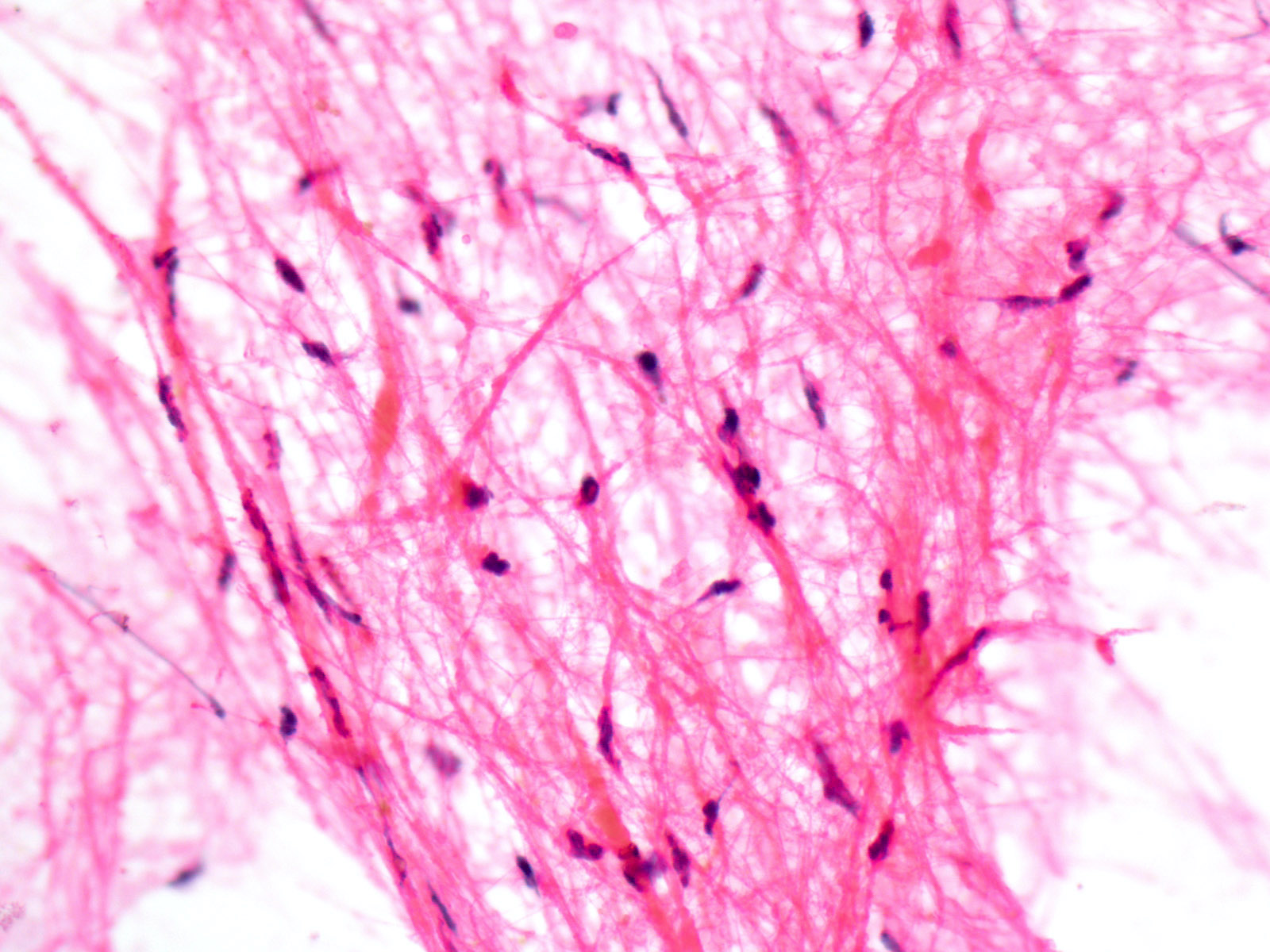
Smear (squash) preparation
- Growth pattern
- Predominantly solid / circumscribed; often limited peripheral infiltration
- Frequent extension into subarachnoid space
- Biphasic appearance
- Compact fibrillar portions: elongated nuclei, bipolar piloid processes, Rosenthal fibers
- Loose microcystic portions: round to oval nuclei, cobweb-like processes, eosinophilic granular bodies
- Occasional "pennies on a plate" multinucleated cells
- Oligodendroglioma-like areas may be present
- Glomeruloid vessels
- Regressive / degenerative changes
- Degenerative atypia
- Vascular hyalinization
- Infarct-like necrosis
- Calcification
- Perivascular lymphocytes
- Anaplasia in a minority of cases
- Increased mitotic activity (> 4 mitoses/10 high power fields) with or without necrosis (Am J Surg Pathol 2010;34:147)
- Pilomyxoid astrocytoma subtype
- Variant with angiocentric arrangement of monophasic bipolar tumor cells in a myxoid background
Contributed by P.J. Cimino, M.D., Ph.D.
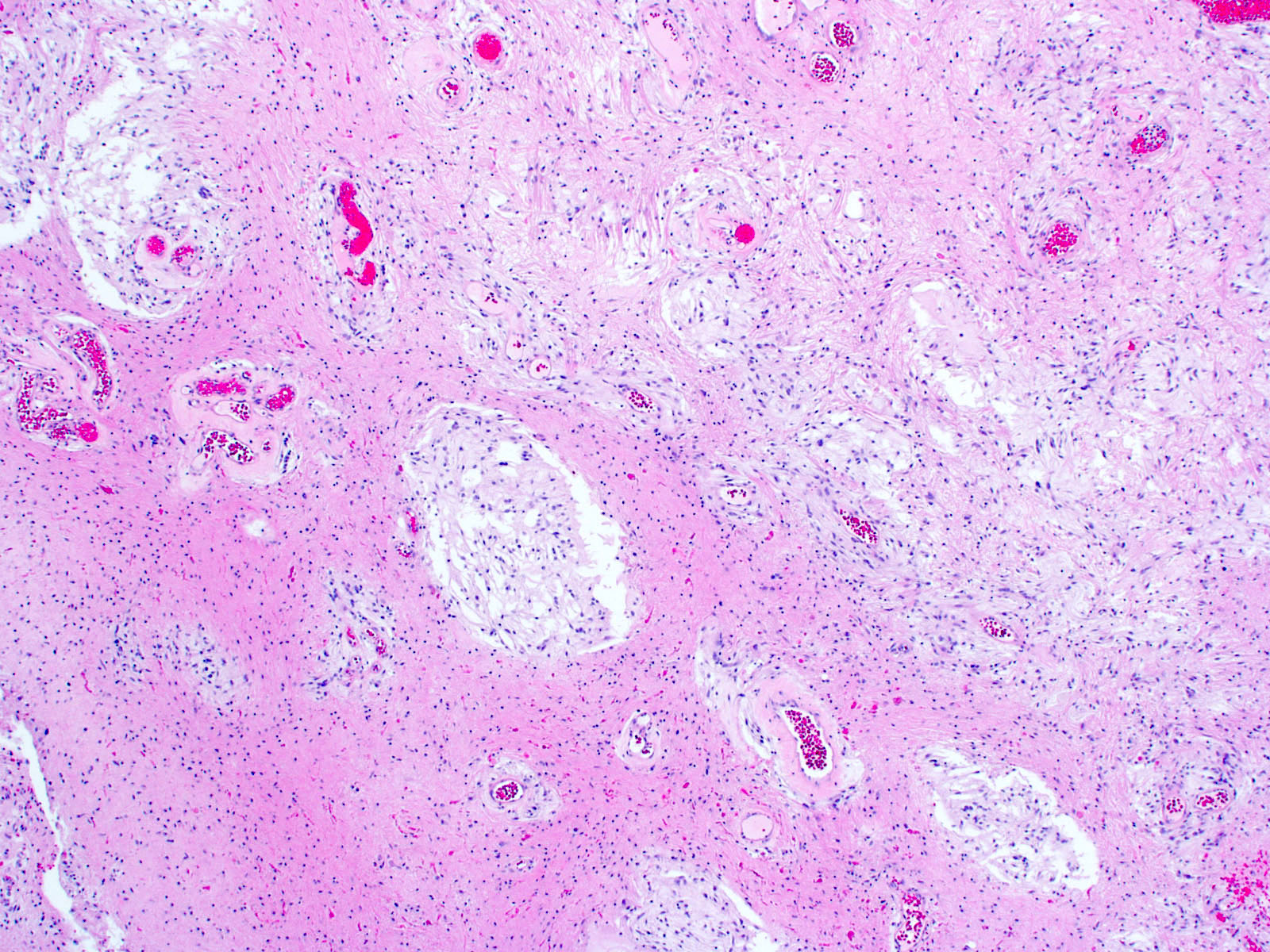
Biphasic appearance
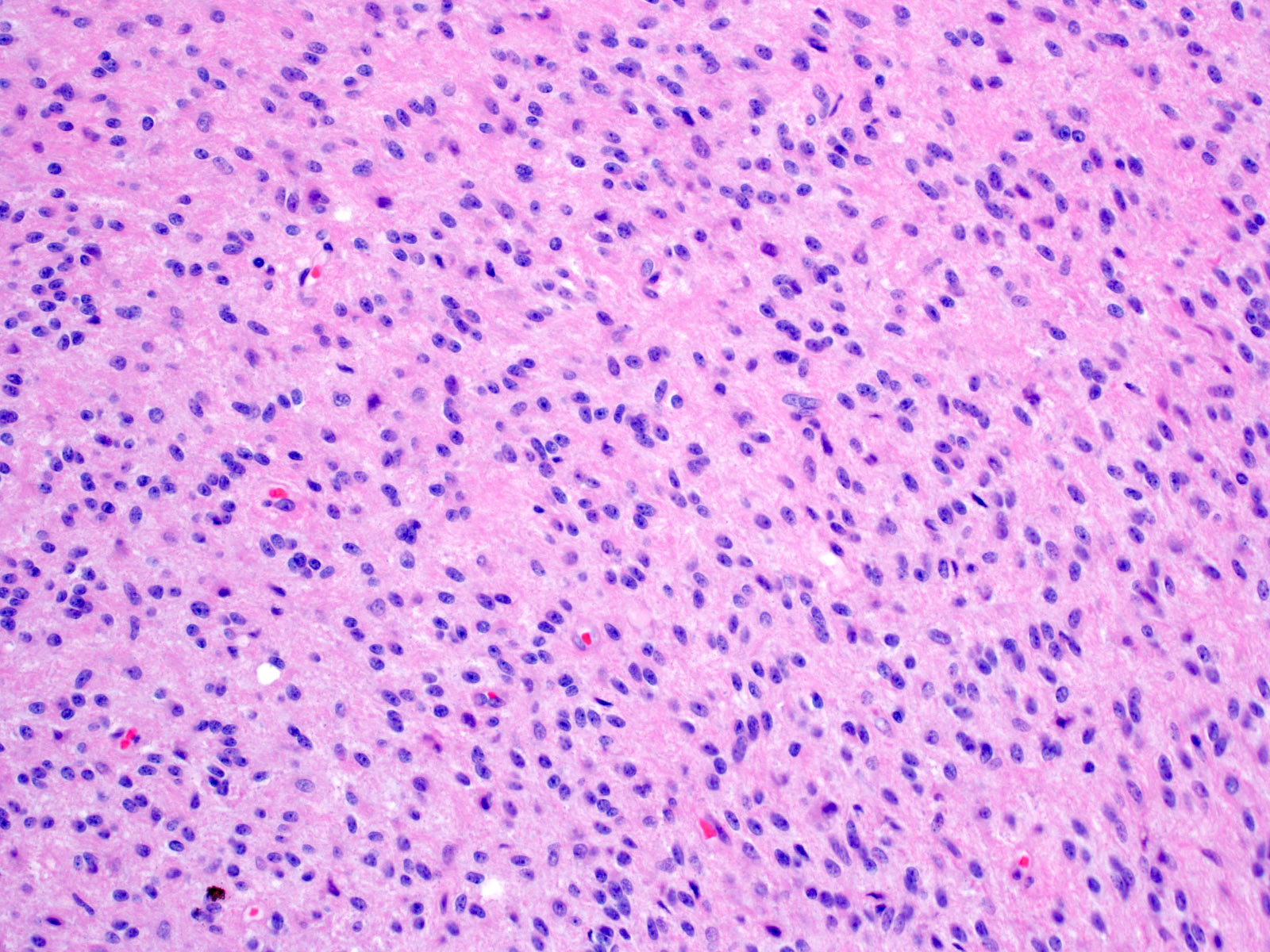
Compact fibrillary area
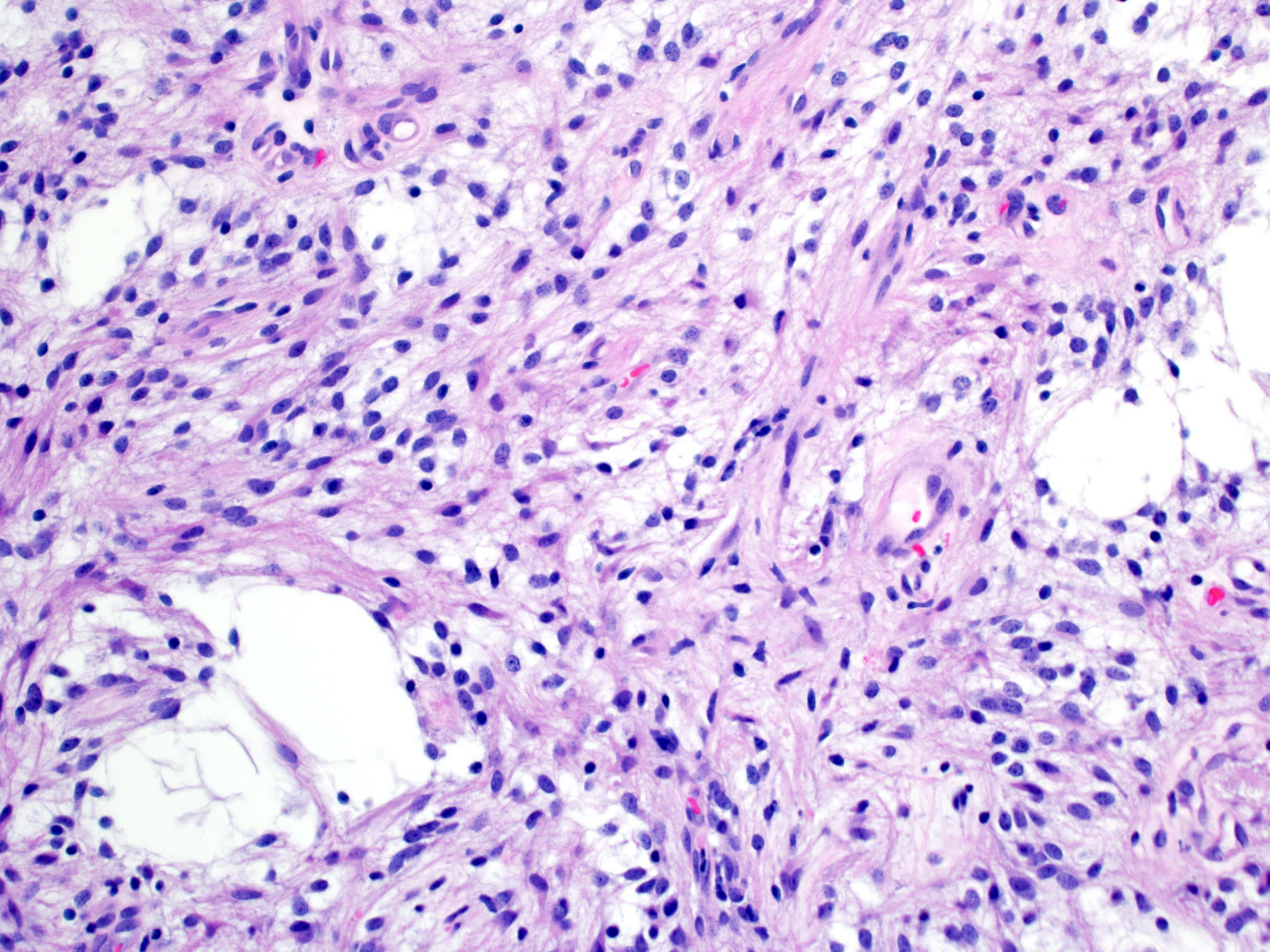
Cystic area
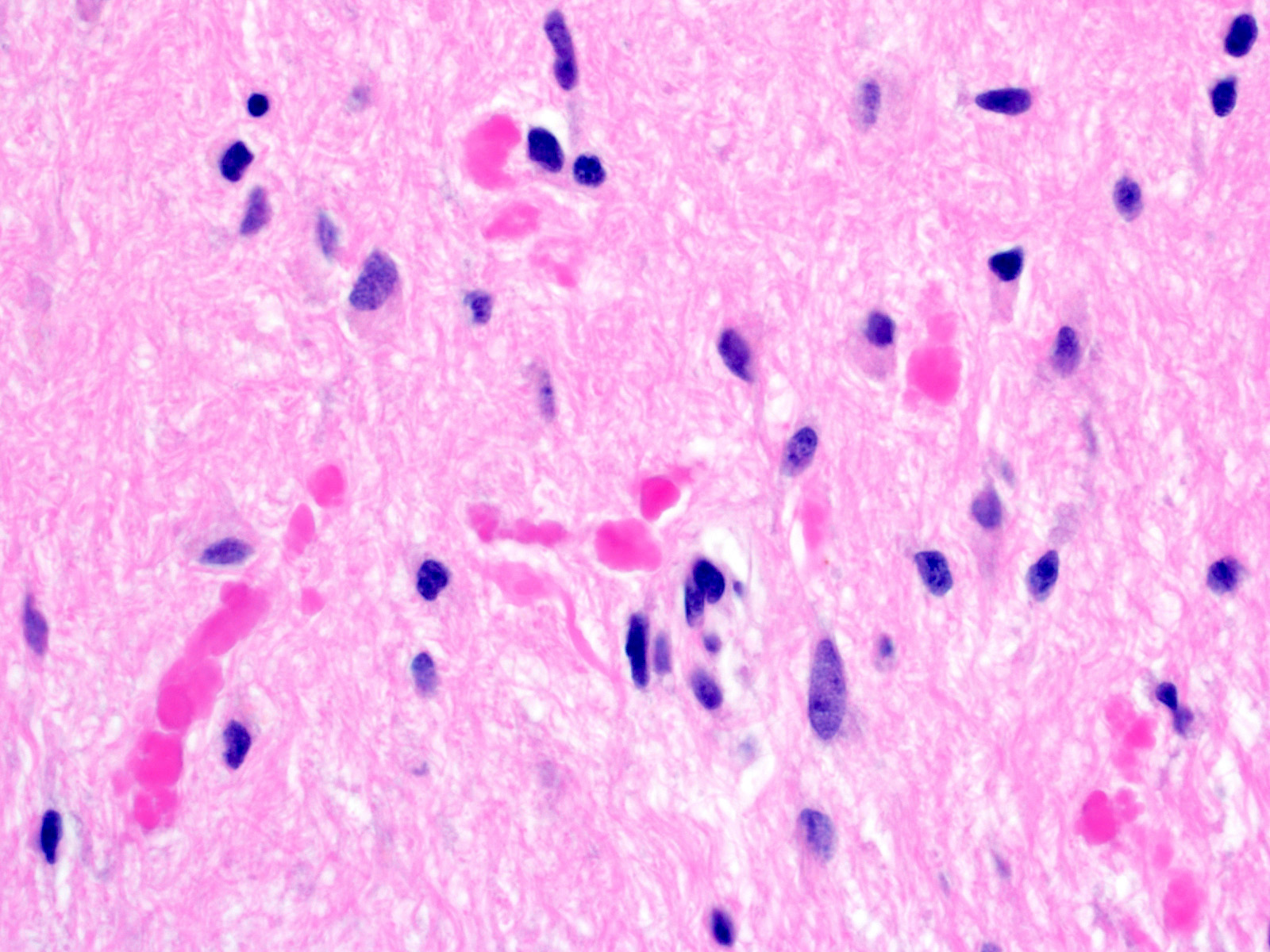
Rosenthal fibers
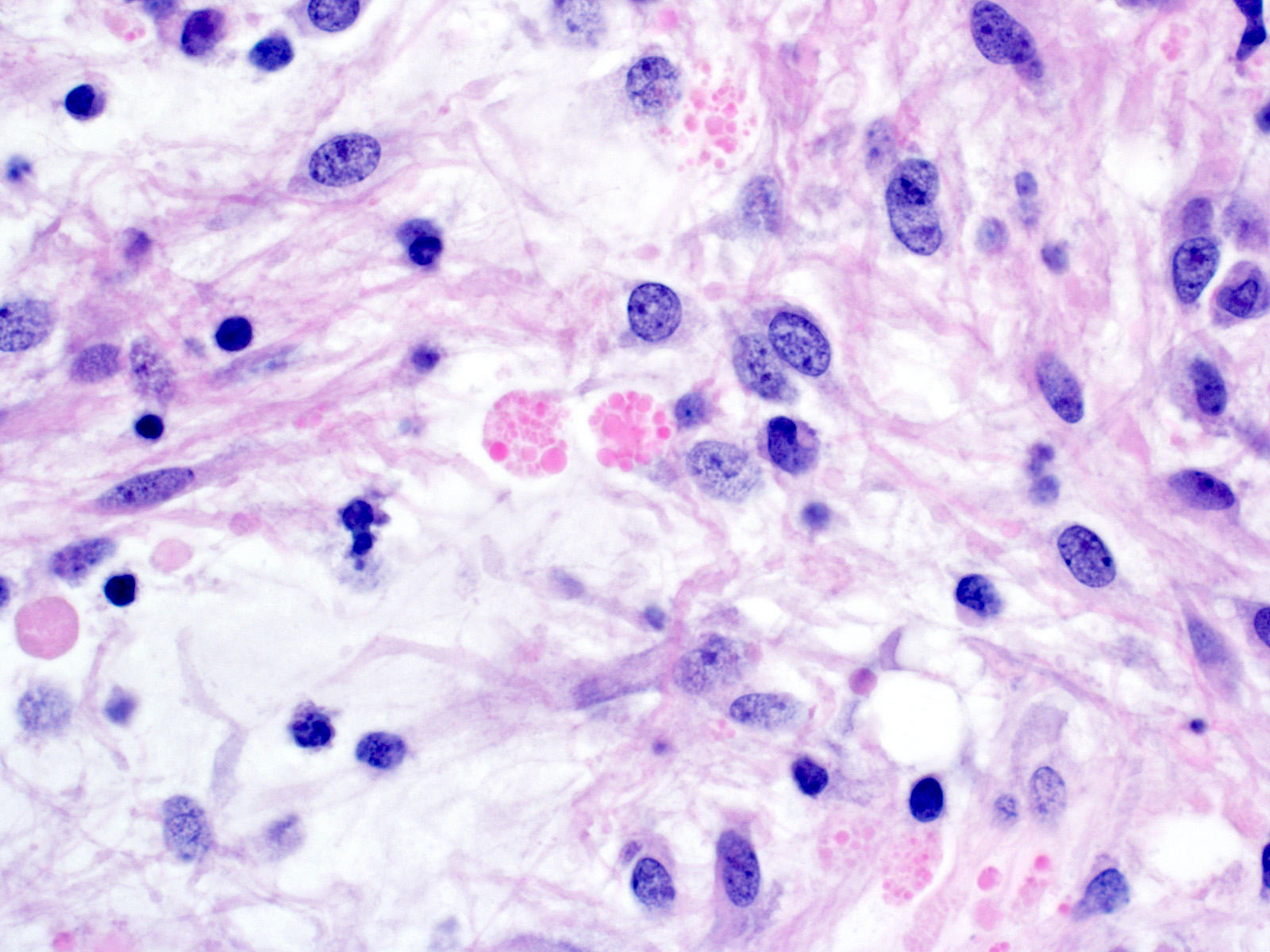
Eosinophilic granular bodies
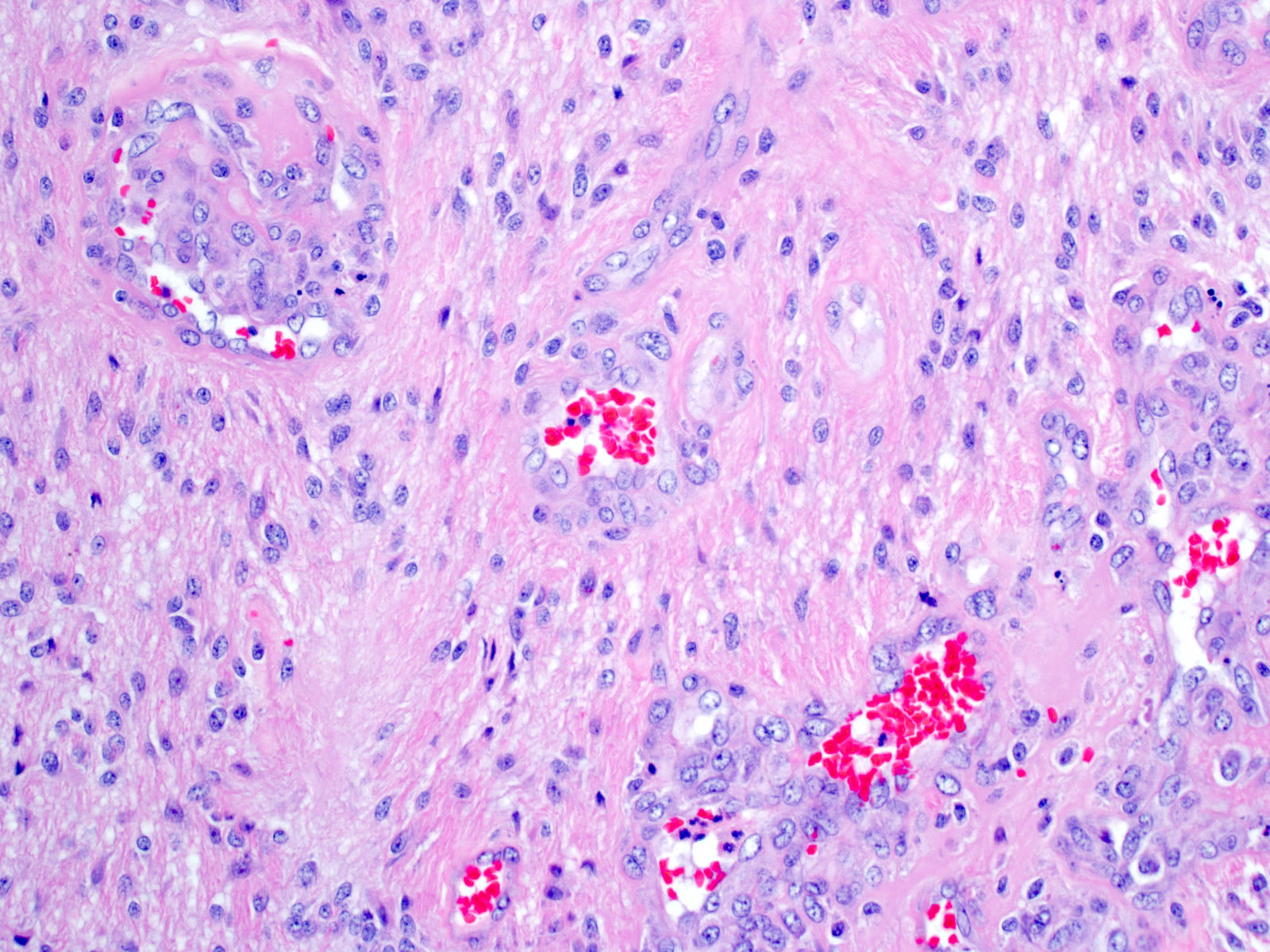
Microvascular proliferation
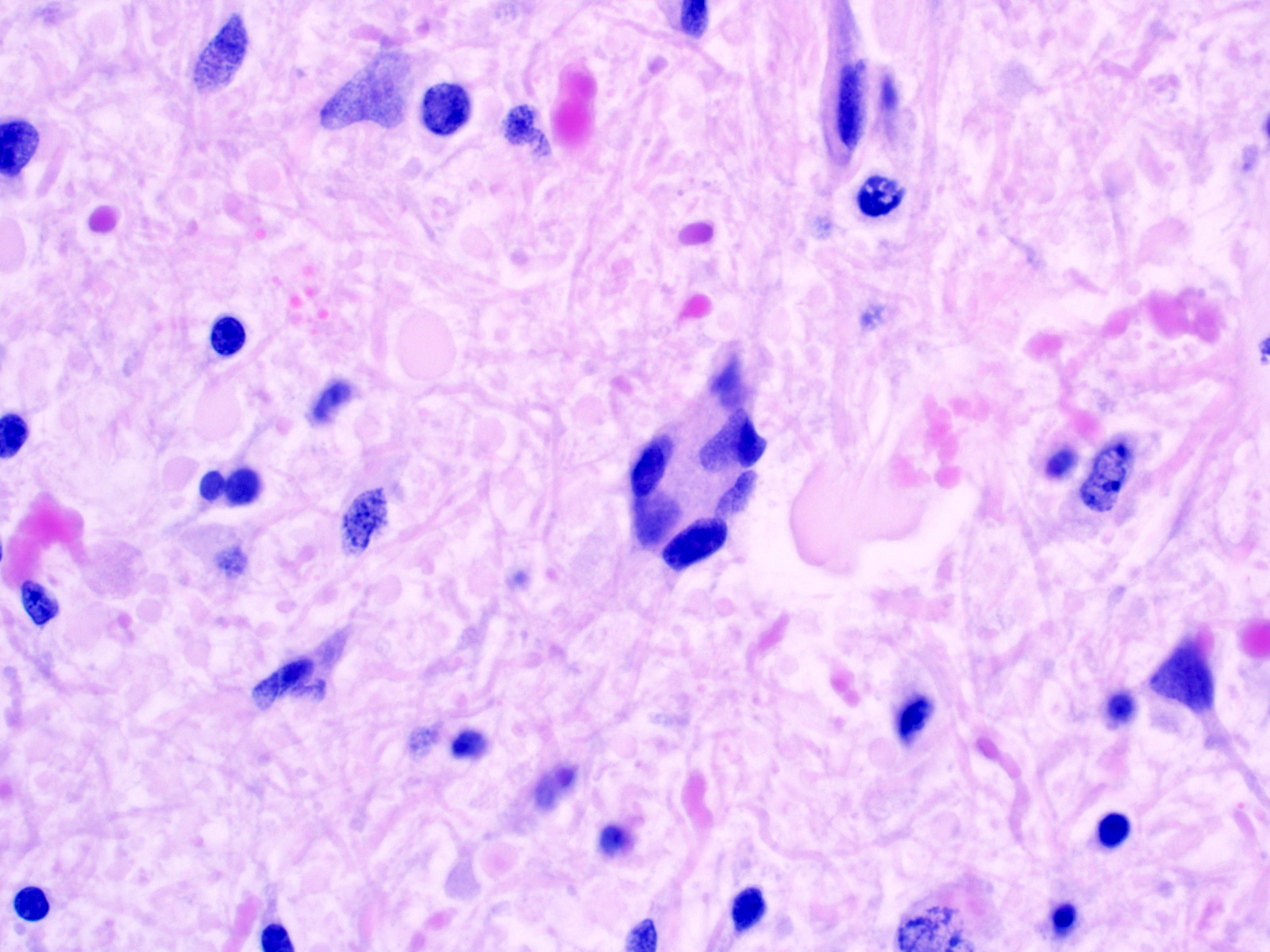
Multinucleated cells
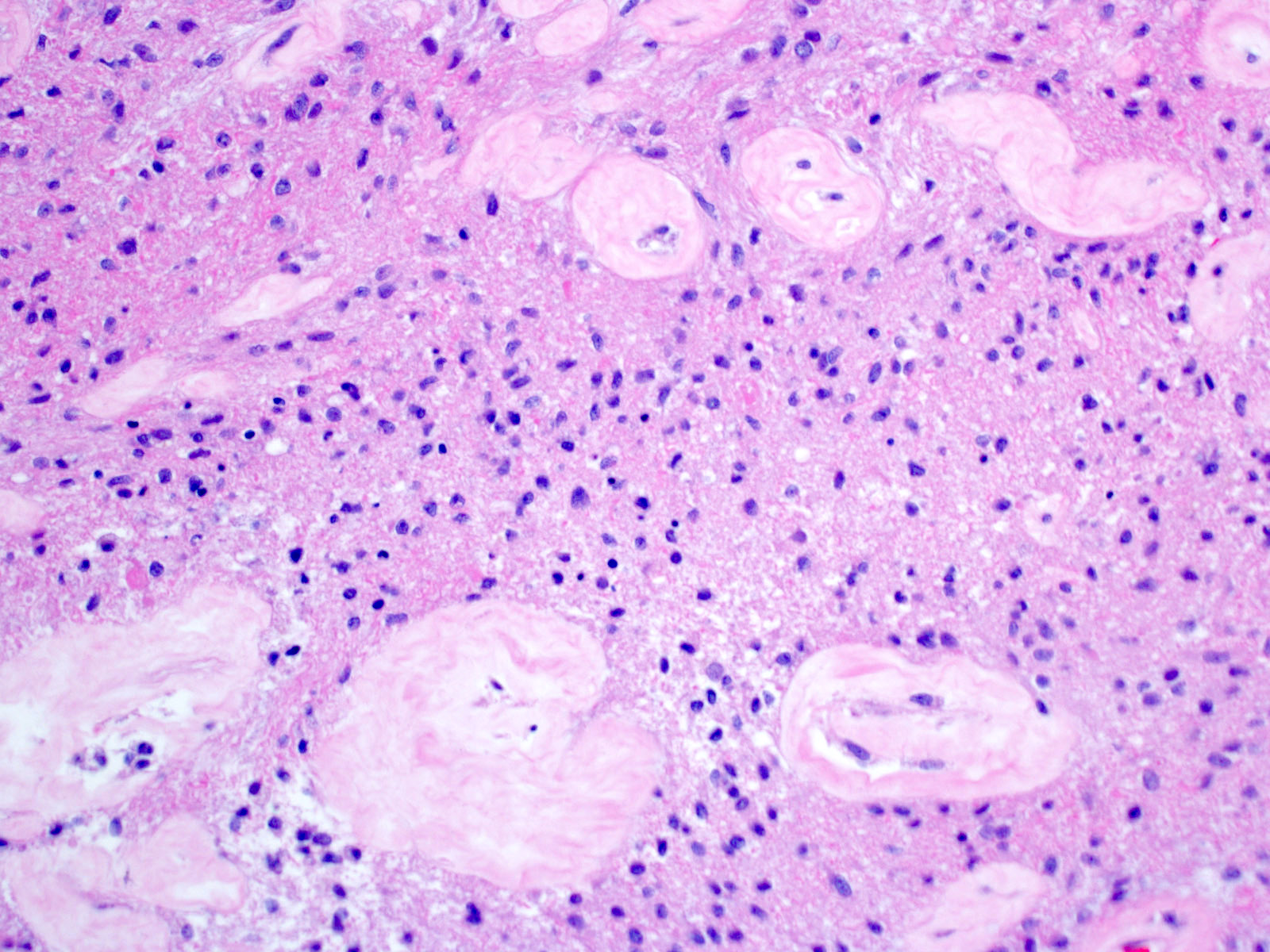
Hyalinized vessels
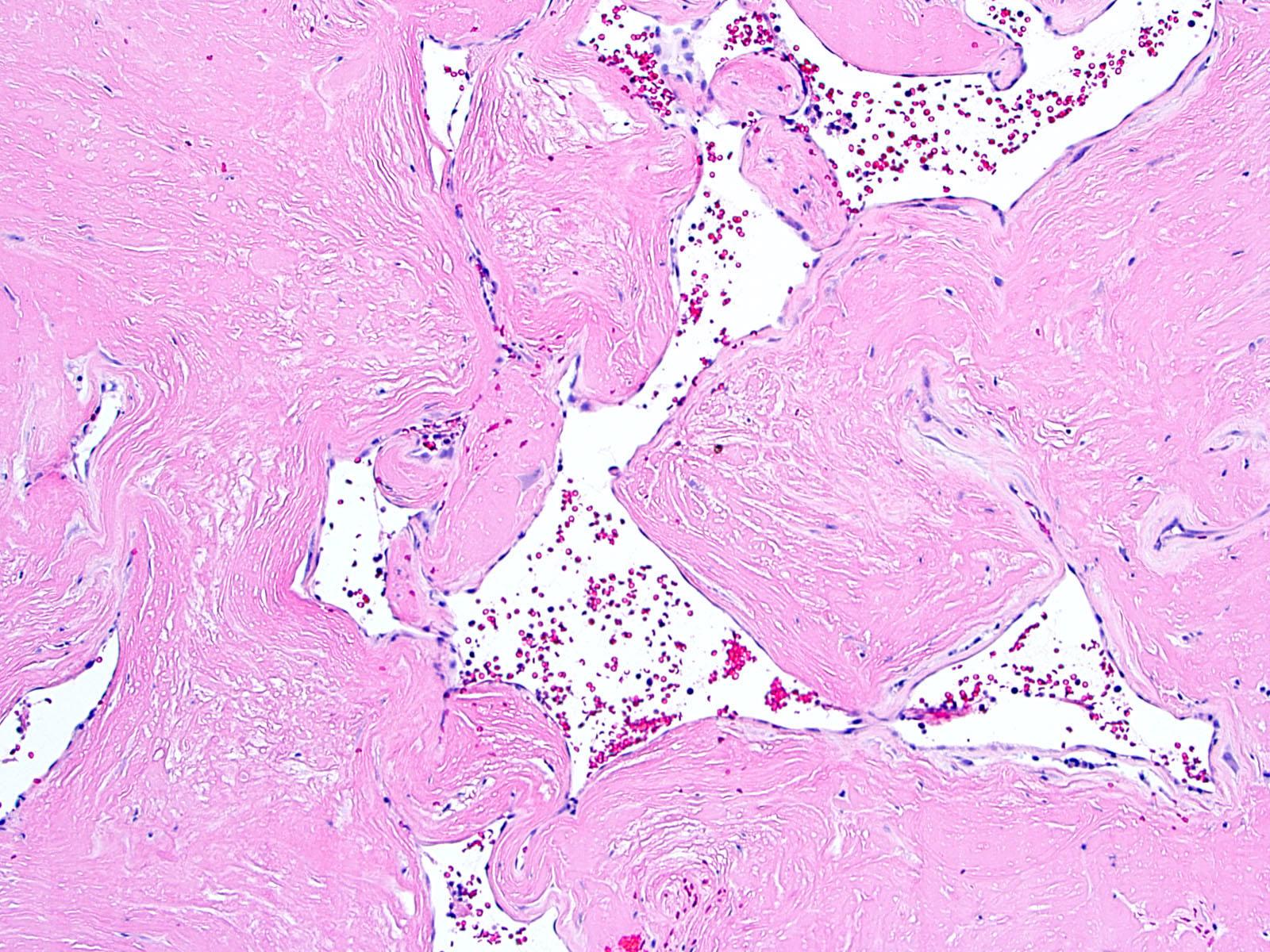
Extensive regressive features
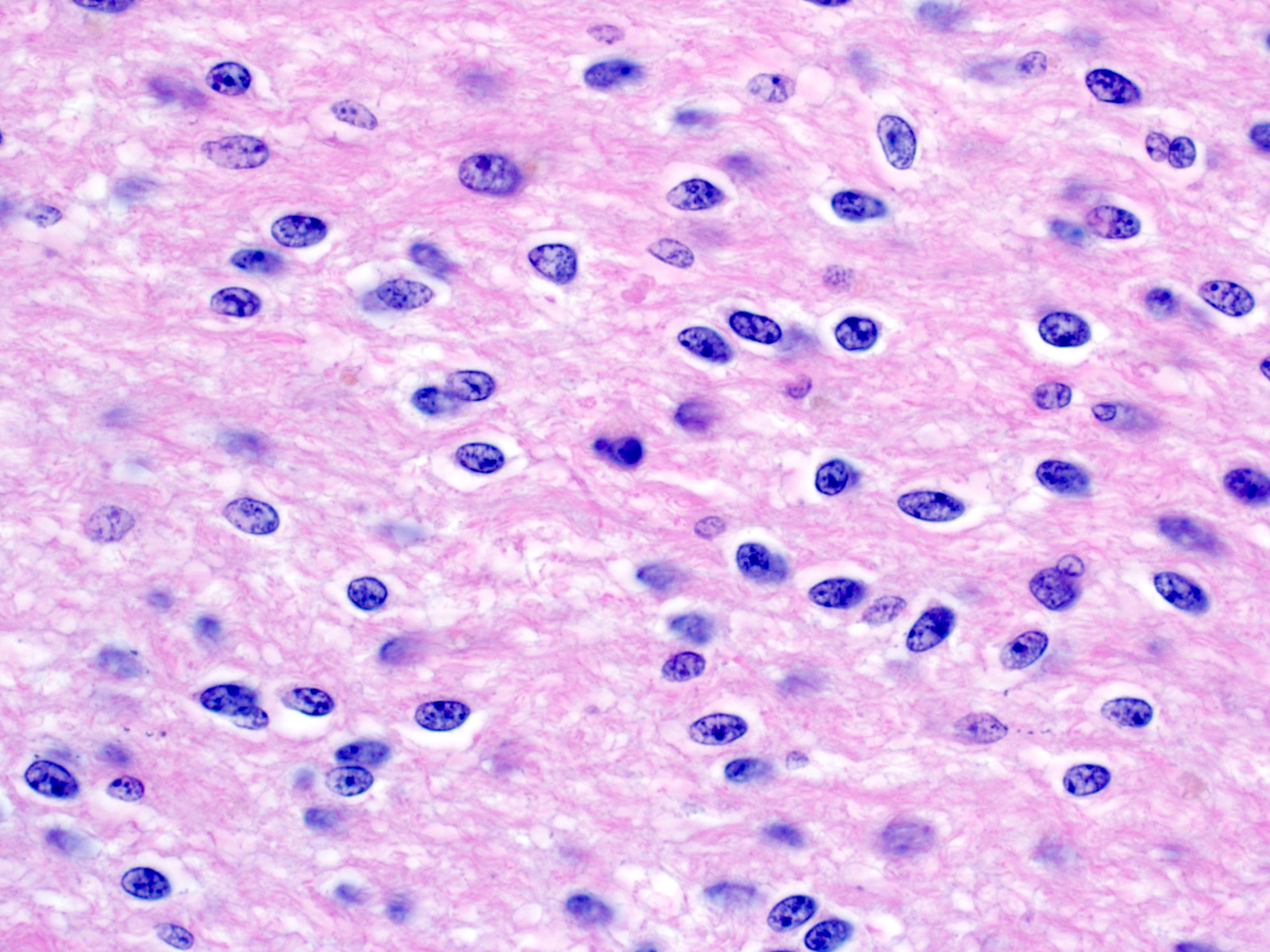
Oligodendroglial-like
cytomorphology
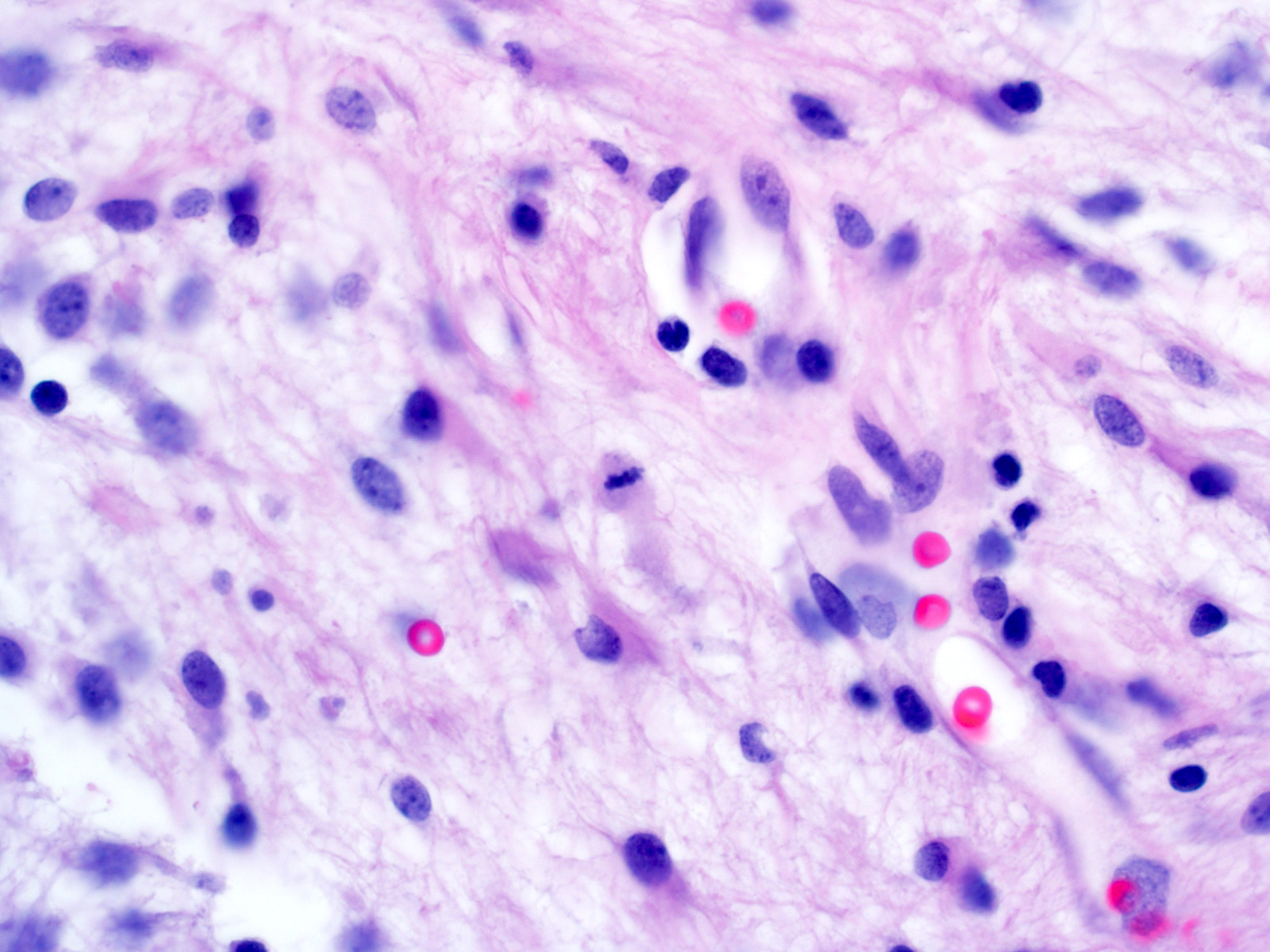
Mitosis

GFAP piloid processes
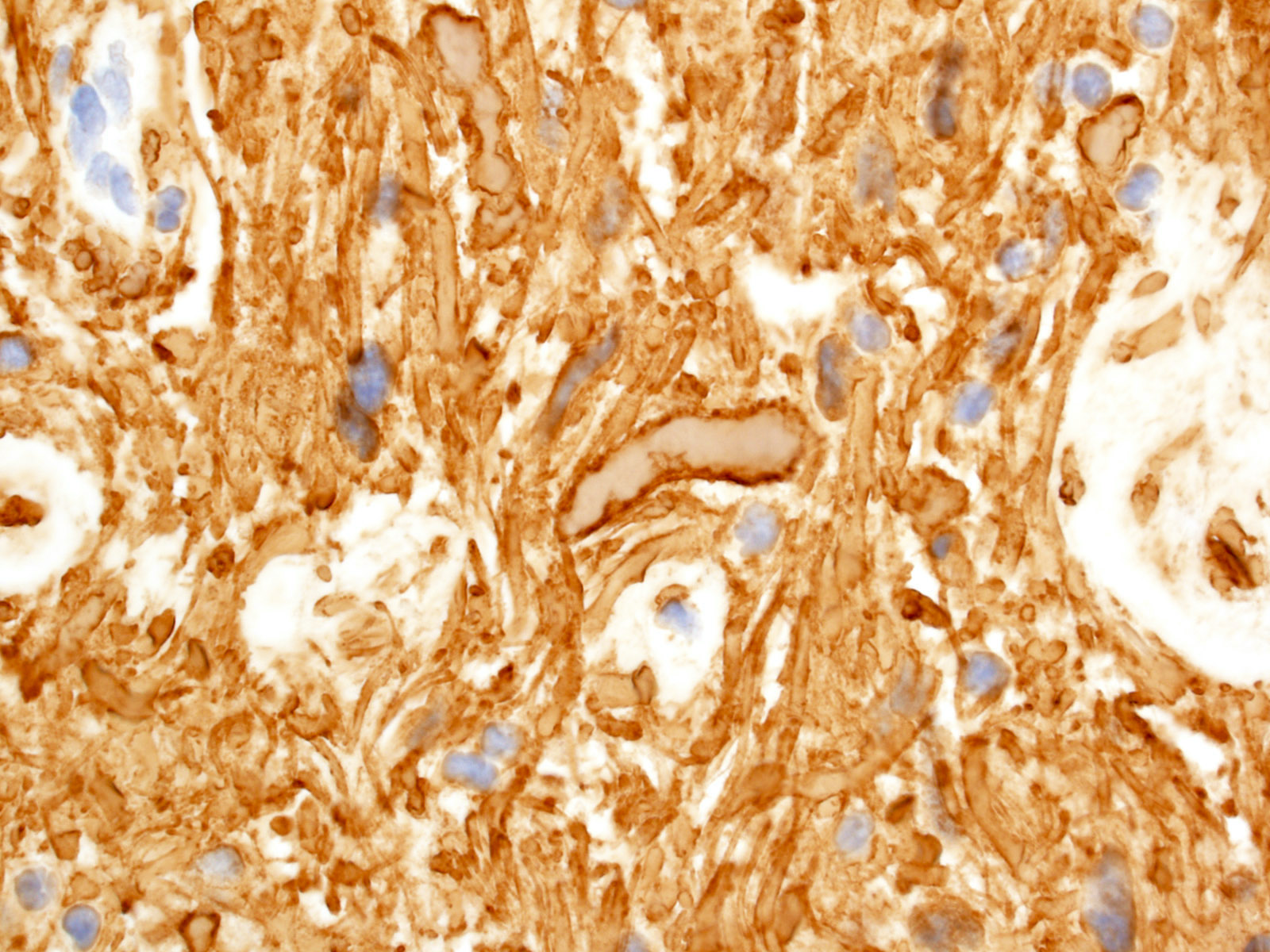
Rosenthal fiber - GFAP
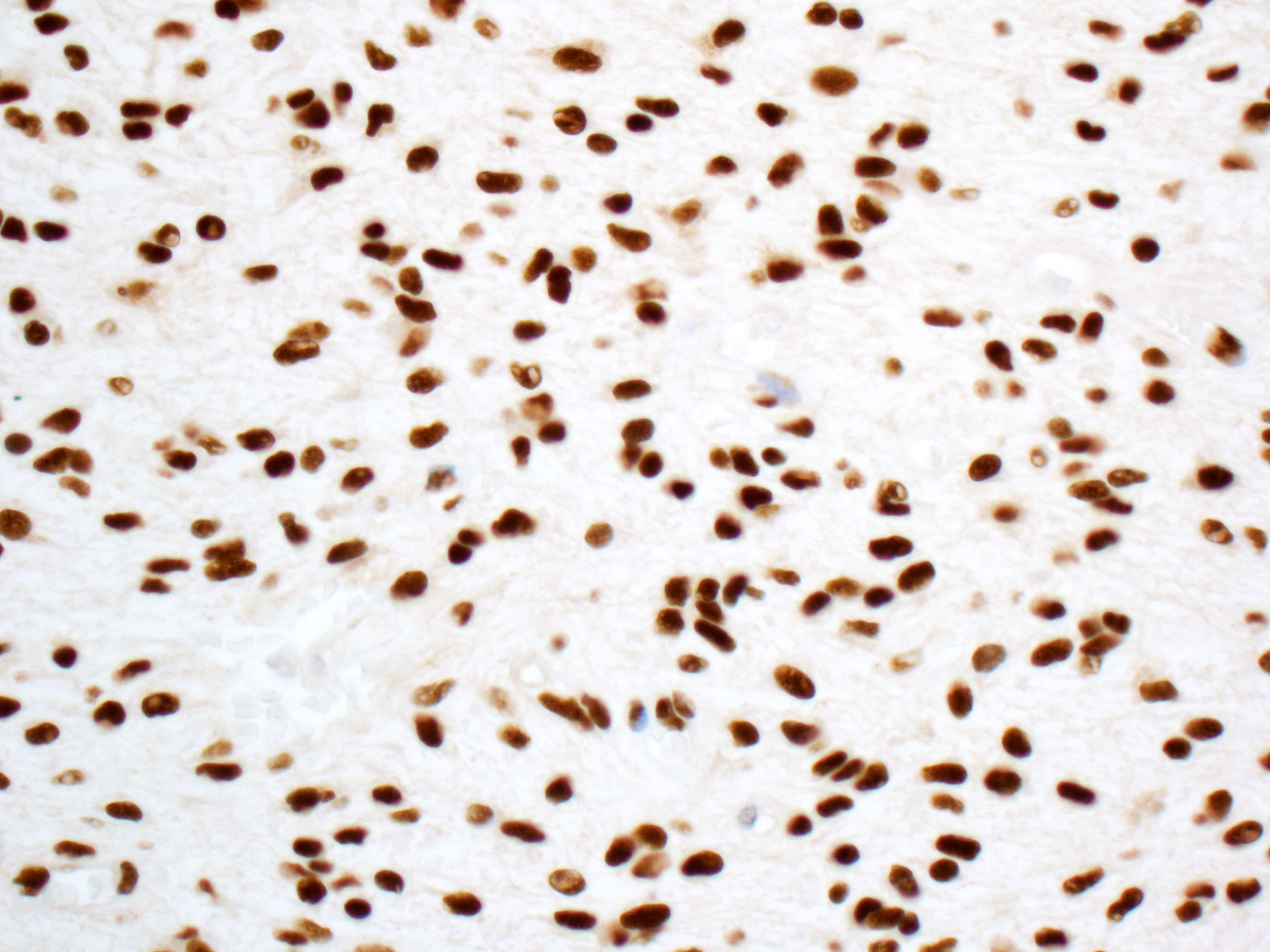
ATRX retained nuclear immunoreactivity
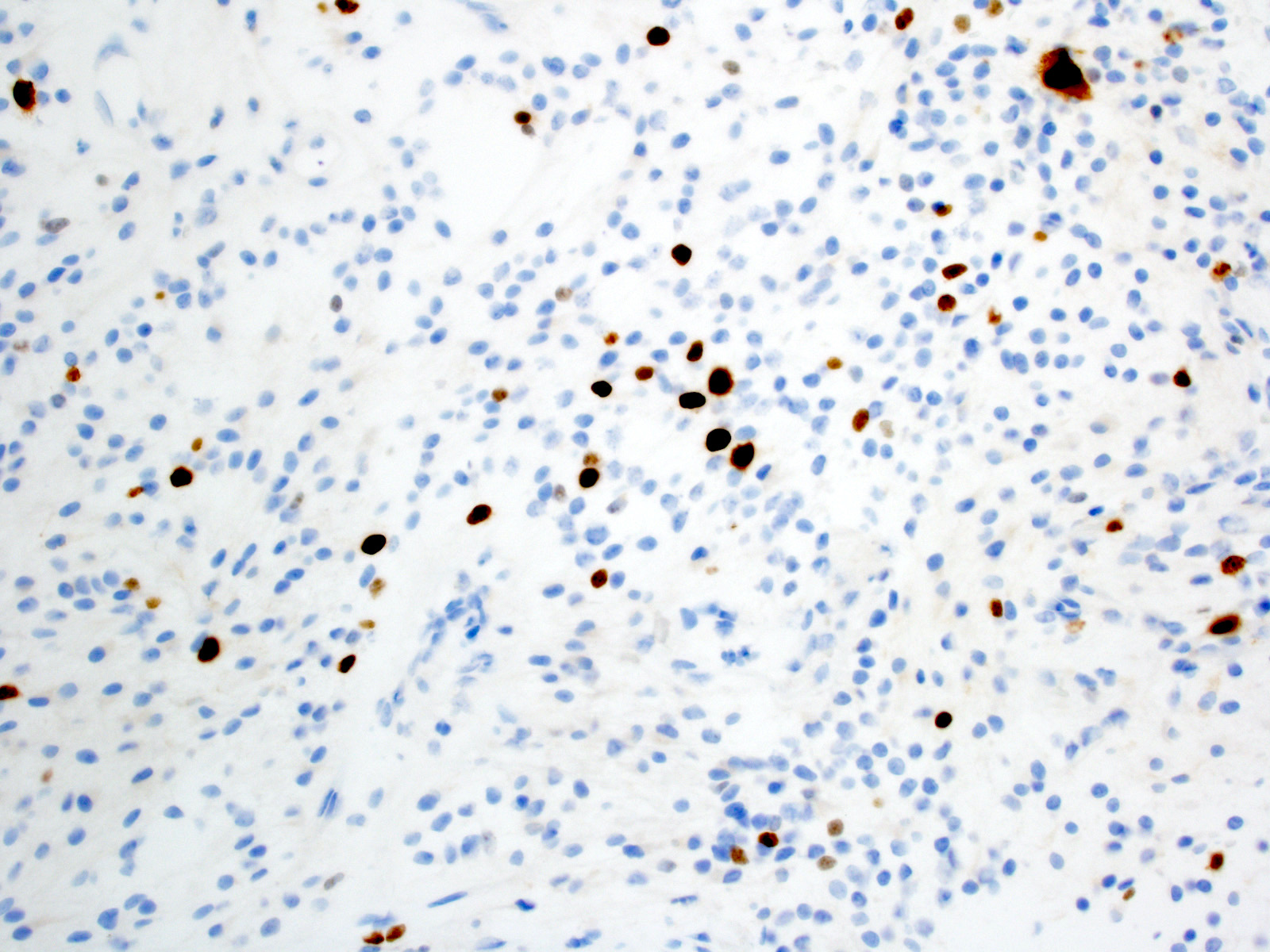
Increased Ki67 with anaplasia
Masson trichrome - EGBs
Images hosted on other servers:
Pilocytic astrocytoma (grade I)
- Smear preparation (alcohol fixed, H&E stained) (Cancer Cytopathol 2015;123:331)
- Mildly atypical glial cells with long piloid processes
- Ovoid nuclei with delicate chromatin
- Rosenthal fibers
- Eosinophilic granular bodies
- Tumor cells:
- GFAP (Am J Surg Pathol 2012;36:43)
- Olig2 (J Neurooncol 2011;104:423)
- S100 (Am J Surg Pathol 2012;36:43)
- SOX10 (J Neuropathol Exp Neurol 2016;75:295)
- ATRX (retained nuclear staining) (Surg Neurol Int 2018;9:29, Acta Neuropathol 2020;139:287)
- Ki67 (generally low but increased with anaplasia) (J Neuropathol Exp Neurol 1999;58:46, Hum Pathol 1998;29:1511, J Clin Oncol 2003;21:2968, Am J Surg Pathol 2010;34:147)
- Eosinophilic granular bodies (EGBs) (Hum Pathol 1998;29:1511):
- Rosenthal fibers (Hum Pathol 1998;29:1511, Acta Neuropathol 1985;66:155):
- IDH1 R132H (Brain Pathol 2010;20:245)
- p53 (weak / low to absent) (Histopathology 1998;33:446)
- BRAF V600E (~5% of cases) (Brain Behav 2017;7:e00641)
- Rosenthal fibers: amorphous, electron dense elements surrounded by intermediate filaments within astrocytic processes (J Pathol Transl Med 2015;49:427)
- Eosinophilic granular bodies: round body, electron dense homogeneous material
Images hosted on other servers:

Eosinophilic granular bodies
- Activating alterations in components of MAPK pathway (Nat Genet 2013;45:927, Nat Genet 2013;45:602)
- > 70% KIAA1549-BRAF fusion due to 7q34 tandem duplication
- ~90% cerebellar cases
- ~50% supratentorial cases
- ~5% other BRAF fusion
- ~5% BRAF V600E
- ~8% NF1 loss
- < 5% FGFR1 mutation
- < 5% FGFR1 fusions / internal tandem duplication
- ~2% NTRK fusions
- PTPN1 mutation (rare)
- KRAS mutation (rare)
- RAF1 fusions (rare)
- > 70% KIAA1549-BRAF fusion due to 7q34 tandem duplication
- Chromosomal polysomies (5, 6, 7, 11, 15) detected by comparative genomic hybridization (CGH) (J Neuropathol Exp Neurol 2006;65:1049, Br J Cancer 2000;82:1218)
- Infratentorial / supratentorial tumors show distinct gene expression and DNA methylation signatures (Acta Neuropathol 2013;126:291)
- H3 K27M mutation (rare) (Brain Pathol 2019;29:126)
- Negative for IDH1 / IDH2 exon 4 mutations
- Usually negative for TP53 mutations
Images hosted on other servers:

KIAA1549-BRAF fusion by FISH

Copy number variation plot

NTRK2 fusion by next generation sequencing

NF1 biallelic inactivation

Pilocytic astrocytoma methylation classifier
Pilocytic astrocytoma
Rosenthal fibers in pilocytic astrocytoma
- Brain, cerebellum, biopsy:
- Pilocytic astrocytoma, WHO grade 1 (see comment)
- Comment: Positive for KIAA1549-BRAF fusion.
- Neoplastic
- Diffuse glioma (including oligodendroglioma, IDH mutant astrocytoma, IDH wildtype astrocytic gliomas, diffuse midline glioma, H3 K27M mutant):
- Diffusely infiltrative
- Eosinophilic granular bodies and Rosenthal fibers uncommon
- Distinct molecular features
- High grade astrocytoma with piloid features:
- High grade histological features
- Distinct molecular features
- Frequent loss of ATRX nuclear staining (Acta Neuropathol 2018;136:273)
- Diffuse leptomeningeal glioneuronal tumor:
- Primary leptomeningeal location
- Commonly not reactive for GFAP
- Distinct molecular features
- Pleomorphic xanthoastrocytoma:
- Most frequently in superficial cerebral cortex
- Pleomorphic, occasionally xanthomatous cells
- Reticulin rich foci
- Ganglioglioma:
- Neoplastic dysmorphic ganglion cells
- Dysembryoplastic neuroepithelial tumor:
- Predominantly cortical
- Mucin rich nodules
- Floating neurons in microcysts
- Diffuse glioma (including oligodendroglioma, IDH mutant astrocytoma, IDH wildtype astrocytic gliomas, diffuse midline glioma, H3 K27M mutant):
- Nonneoplastic
- Reactive piloid gliosis:
- Hypocellular
- Lack of genetic alterations
- Vascular malformation
- Reactive piloid gliosis:
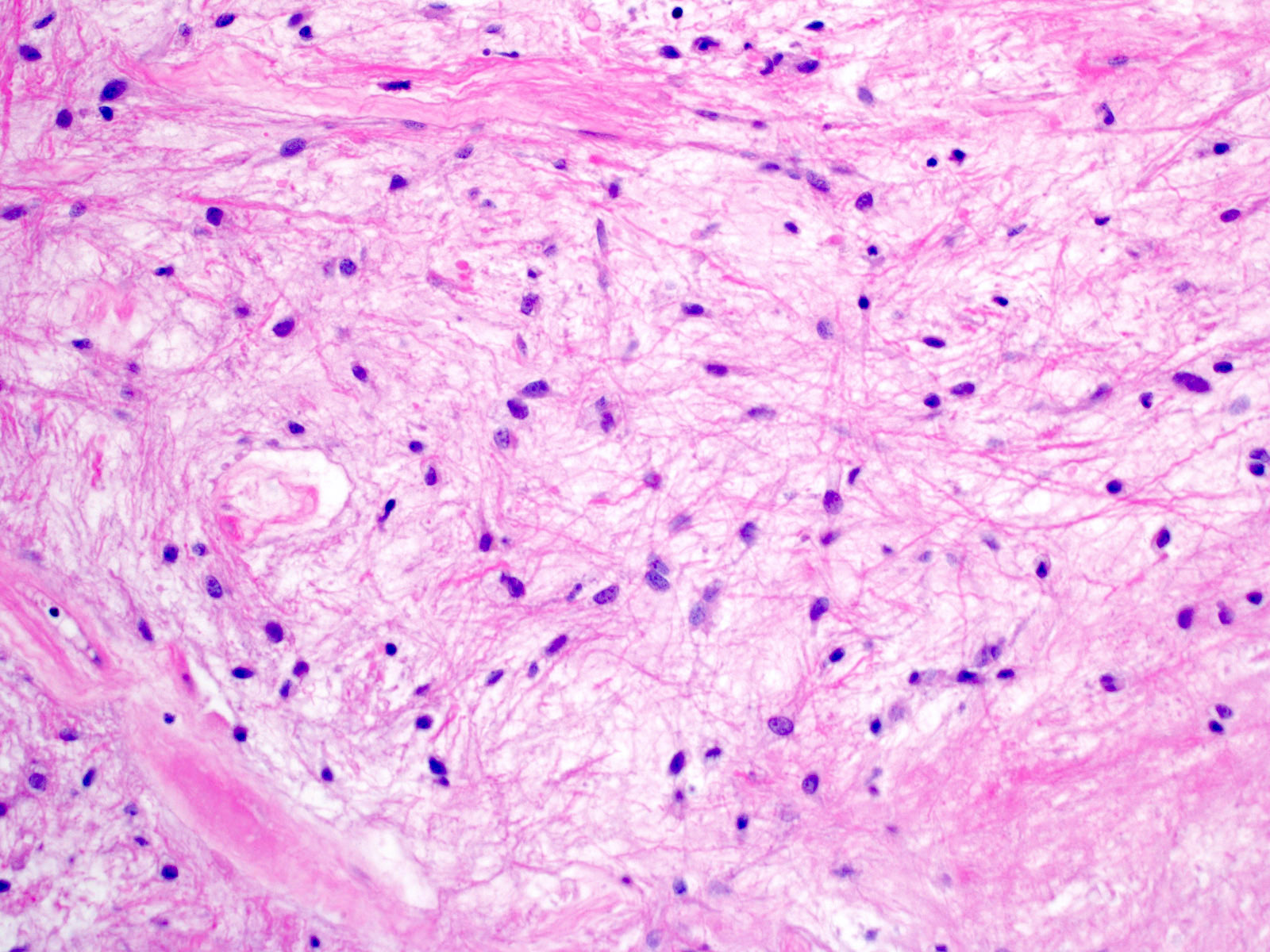
The above tumor is from the cerebellum of a child. What is the most common underlying genetic alteration in this entity?
- TP53 mutation
- IDH1 R132H
- BRAF V600E mutation
- BRAF fusion involving KIAA1549
- NF1 loss
Comment Here
Reference: Pilocytic astrocytoma
- Show diffuse p53 immunoreactivity
- Most frequently occur in adults
- Demonstrate reticulin rich foci
- Associated with neurofibromatosis 2
- Potentially curable with resection
Comment Here
Reference: Pilocytic astrocytoma
- A variant of pilocytic astrocytoma composed of monomorphous bipolar piloid cells with an angiocentric pattern in a myxoid background (MedGenMed 2004;6:42)
- A variant of pilocytic astrocytoma with distinct histologic and clinical features
- Predominantly affects infants and young children
- Preferentially located in the hypothalamic / chiasmatic region
- Histologically characterized by small monomorphous bipolar cells, a myxoid background and perivascular arrangement
- Generally has more aggressive behavior than pilocytic astrocytoma due to local recurrence and cerebrospinal fluid dissemination
- Pilomyxoid astrocytoma (PMA)
- ICD-O: 9425/3 - pilomyxoid astrocytoma
- Rare tumor (Brain Tumor Pathol 2019;36:52)
- Tends to occur during infancy and early childhood (median 10 months) (Einstein (Sao Paulo) 2012;10:236)
- Male predominance (Am J Surg Pathol 2010;34:1783)
- Most common: hypothalamic / chiasmatic region
- Other less common sites: thalamus, cerebellum, brain stem, cerebral cortex and spinal cord (Brain Tumor Pathol 2019;36:52, Brain Pathol 2015;25:429, Am J Surg Pathol 2010;34:1783)
- Cell of origin is unclear
- Pilocytic and pilomyxoid astrocytoma may be part of a single disease spectrum
- They share common genetic alterations (BRAF duplication / fusion); some pilomyxoid astrocytomas mature into classic pilocytic astrocytomas over time and intermediate forms exist (Annu Rev Pathol 2013;8:361, J Neurooncol 2007;81:191, Am J Surg Pathol 2010;34:1783)
- Associated with neurofibromatosis type 1 in a minority of cases (Pediatr Blood Cancer 2006;46:377)


- Most common: manifestations of increased intracranial pressure or parenchymal compression (MedGenMed 2004;6:42)
- Some present with diencephalic syndrome (Einstein (Sao Paulo) 2012;10:236)
- MRI with contrast
- Definitive diagnosis requires tissue biopsy
- MRI findings (Neurol Res 2008;30:945, MedGenMed 2004;6:42):
- Well circumscribed
- The majority (84.6%) are solid
- T1: isointense or hypointense signal
- T2 / FLAIR: hyperintense signal
- No diffusion restriction
- Homogeneous or heterogeneous enhancement pattern
- No peritumoral edema
- Craniospinal imaging is advisable to detect dissemination
Images hosted on other servers:

Chiasmatic / hypothalamic pilomyxoid astrocytoma in a 3 month old infant:
DWI: no sign of restricted diffusion
T2w: homogeneous hyperintense signal
T1w: hypointense signal
T1w+ contrast: solid mass with marked enhancement
- Displays a more aggressive behavior with a higher rate of local recurrence, a substantial rate of cerebrospinal fluid dissemination and shorter survival than pilocytic astrocytoma (Neurosurgery 2004;54:72, Brain Tumor Pathol 2019;36:52)
- Some have a favorable prognosis, especially if completely resected
- Better prognosis than diffuse astrocytomas
- 3 month old girl with disseminated pilomyxoid astrocytoma with novel MUTYH mutation (BMJ Case Rep 2016 Oct 26;2016)
- 4 month old child with pilomyxoid astrocytoma of the right temporal / suprasellar region (Cytopathology 2017;28:74)
- 13 month old girl with giant sellar pilomyxoid astrocytoma (Int J Clin Exp Med 2018;11:1038)
- 11 year old girl with pilomyxoid astrocytoma of the brainstem (Rare Tumors 2013;5:65)
- 22 year old woman with a left frontal lobe pilomyxoid astrocytoma (J Cancer Res Ther 2015;11:667)
- Surgery
- Chemotherapy and radiotherapy for recurrent tumors
- Targeted therapy is promising (Pediatr Blood Cancer 2014;61:2099)
- Well circumscribed, solid gelatinous mass (Scheinemann: Pediatric Neuro-oncology, 2015)
- May be infiltrative
- Perivascular bipolar cells in a mucoid background (J Ayub Med Coll Abbottabad 2014;26:611)
Images hosted on other servers:
Perivascular bipolar cells in a mucoid background
- Well demarcated, may be infiltrative (Brain Tumor Pathol 2019;36:52, Am J Surg Pathol 2010;34:1783)
- Solid and compact (i.e. the tumor is devoid of axons with negative staining for neurofilaments) (Acta Neuropathol 2015;129:775)
- Highly monomorphous, small bipolar piloid cells
- Angiocentric arrangement of tumor cells (perivascular pseudorosettes) with formation of pseudopapillary structures is typical (Acta Neuropathol 2015;129:775)
- Markedly myxoid matrix
- Low mitotic activity (Am J Surg Pathol 2010;34:1783)
- Uncommon: glomeruloid vascular proliferation, necrosis, eosinophilic granular bodies (Brain Tumor Pathol 2019;36:52, Am J Surg Pathol 2010;34:1783)
- Absence of classic pilocytic astrocytoma features such as biphasic (solid / microcystic) pattern or Rosenthal fibers (Brain Pathol 2015;25:429)
- See table above summarizing the histologic features of pilocytic and pilomyxoid astrocytoma (Neurosurgery 2004;54:72)
- Classic pilocytic astrocytomas with focal pilomyxoid changes should not be diagnosed as pilomyxoid astrocytomas
- Intermediate tumors with hybrid features of pilocytic and pilomyxoid astrocytoma were reported and tend to occur at older age than pilomyxoid astrocytomas (median = 36 months)
- This might represent a maturational effect as some pilomyxoid astrocytomas and intermediate tumors mature into pilocytic astrocytoma-like neoplasms over time (Am J Surg Pathol 2010;34:1783)
- According to WHO 2016 classification, a definite grade assignment for pilomyxoid astrocytoma is not recommended (Brain Tumor Pathol 2019;36:52)
Contributed by Eman Abdelzaher, M.D., Ph.D.
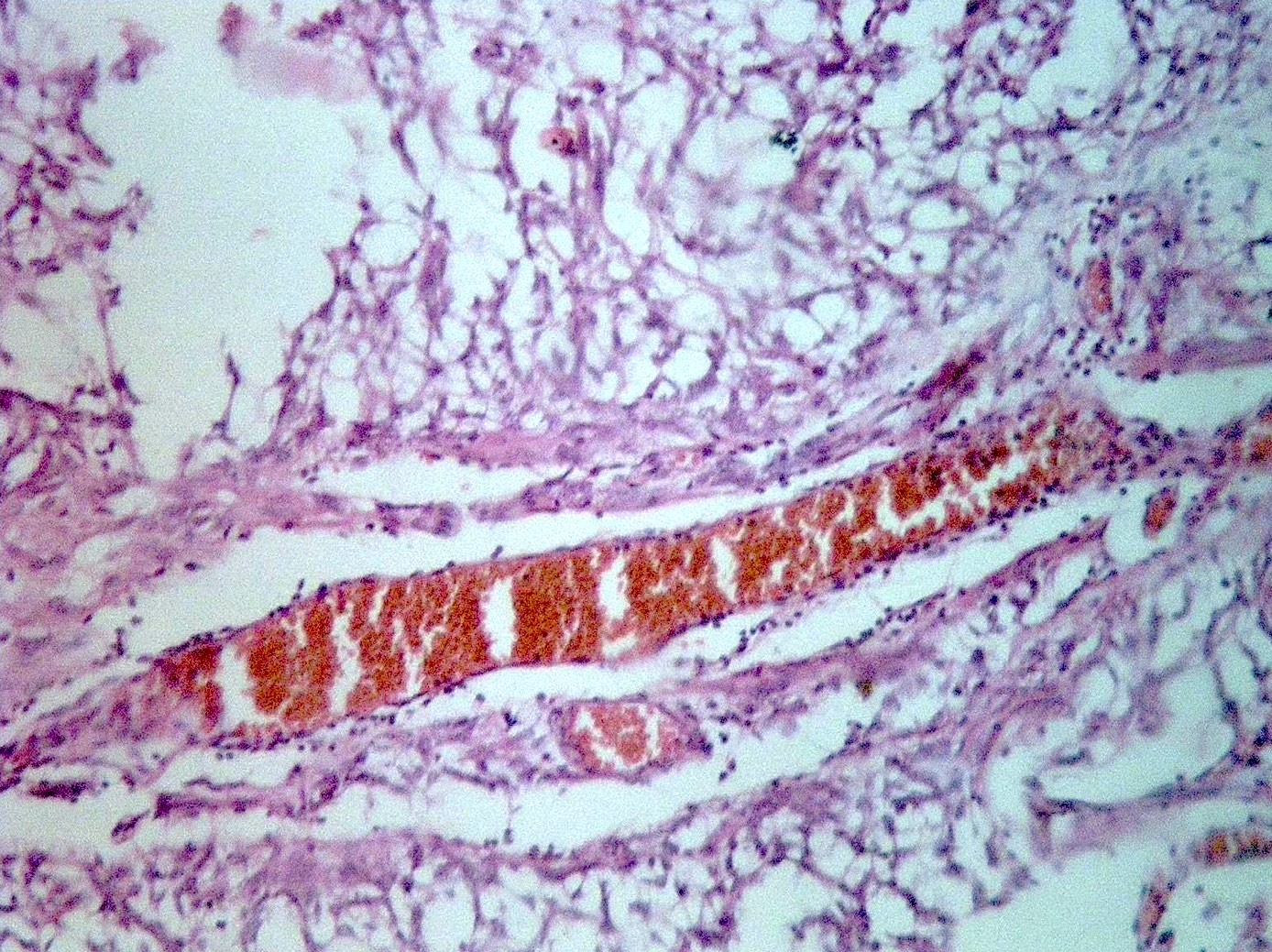
Angiocentric pattern
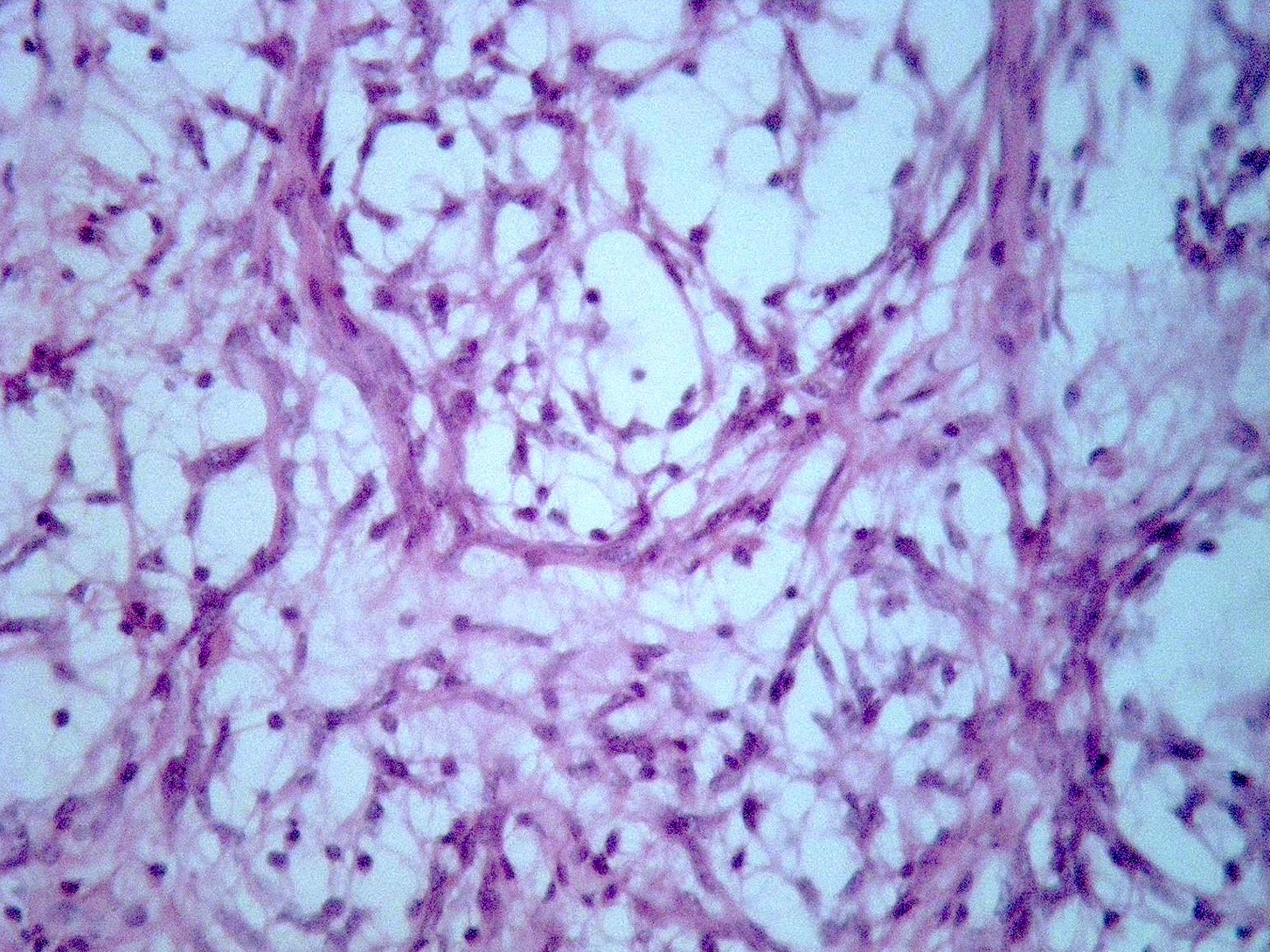
Monomorphous cells in a myxoid background
- Similar to pilocytic astrocytoma but no Rosenthal fibers (Brain Tumor Pathol 2019;36:52)
- Monomorphous piloid cells with bipolar fibrillary processes and hyperchromatic, elongated nuclei (Brain Tumor Pathol 2019;36:52)
- Angiocentric arrangement
- Myxoid background
- GFAP, Olig2, S100, vimentin
- Synaptophysin (of variable extent, in > 50%) (Am J Surg Pathol 2010;34:1783)
- Ki67: variable (2 - 20%) (Neurosurgery 2003;53:544)
- Bipolar tumor cells with elongated thin processes extending to rest upon the basal lamina of blood vessels
- Abundant intermediate (glial) filaments can be seen in some cells
- Apical surfaces may show microvilli, blebs and occasional cilia
- Vesicles and coated pits, as well as dense core granules and synaptoid complexes may be seen (Adesina: Atlas of Pediatric Brain Tumors, 2010)
Images hosted on other servers:

Bipolar, thin processes
extending to basal
lamina of vessels;
dense core granules
- Similar genetic alterations as in pilocytic astrocytomas (J Neuropathol Exp Neurol 2013;72:2, Neuropathol Appl Neurobiol 2013;39:693)
- Most common alteration is the BRAF duplication / fusion resulting in activation of the MAPK pathway (Brain Tumor Pathol 2019;36:52, Annu Rev Pathol 2013;8:361)
- KIAA1549-BRAF fusion in half of cases by FISH (J Neuropathol Exp Neurol 2013;72:2, Neuropathol Appl Neurobiol 2013;39:693)
- The majority show no genetic susceptibility; rare cases occur in the context of neurofibromatosis type 1 and Noonan syndrome (Pediatr Blood Cancer 2006;46:377, Pediatr Blood Cancer 2015;62:1084)
- Brain, suprasellar region, biopsy:
- Pilomyxoid astrocytoma
- Ancillary tests:
- Strong positive GFAP staining
- Focally positive for synaptophysin
- Ki67 proliferation index: 3%
- Pilocytic astrocytoma:
- Biphasic pattern, Rosenthal fibers, more often cystic, myxoid areas and angiocentric pattern only focal
- Ependymoma:
- More cellular and less myxoid, true rosettes, well developed perivascular pseudorosettes
- Olig2-, synaptophysin-
- Angiocentric glioma:
- Corticocentric, minimal or no myxoid background
- EMA+

A 10 month old boy presents with a solid enhancing hypothalamic mass on MRI. Biopsy shows a highly myxoid tumor with perivascular arrangement of monomorphous cells. What is the most likely genetic alteration in this tumor?
- BRAF duplication / fusion
- H3F3A K27M mutation
- IDH1 and IDH2 mutations
- SMARCB1 / INI1 mutations
Comment Here
Reference: Pilomyxoid astrocytoma
- Has a favorable prognosis with no reported risk of recurrence
- Preferentially located in the cerebellum with rare cases involving other sites
- Shares common genetic alterations with pilocytic astrocytoma
- Tends to affect adults
Comment Here
Reference: Pilomyxoid astrocytoma
- Glial cyst within the pineal gland
- Present in 2 - 3% of adults
- May resemble a cystic tumor
- Usually incidental finding, affect all age groups
- Minority of cases, usually in young adults, are large enough to compress aqueduct and cause symptoms
- Fine needle aspiration may provide rapid diagnosis (Cancer 2005;105:80)
- Midline, well circumscribed, 1 - 3 cm
- 22 year old man with sudden death (J Clin Path 1996;49:267)
- Smooth external surface and inner lining
Images hosted on other servers:

Pineal cyst
- Inner lining is glial tissue which is hypocellular, highly fibrillated with many Rosenthal fibers; hemosiderin is common
- Glial layer is surrounded by compressed, often calcified, pineal parenchyma
- Outer layer is leptomeningeal connective tissue (Ann Diagn Path 1997;1:11)
- Small, uniform polygonal cells
- Other cysts
- Pilocytic astrocytoma
- Pineocytoma
- Features intermediate between pineocytoma and pineoblastoma
- 10% of pineal parenchymal tumors
- Occasionally associated with CNS or extraneural metastasis but otherwise difficult to predict prognosis
- Better survival than pineoblastoma
- Marked hypercellularity but relatively bland nuclear features
- No / mild nuclear pleomorphism
- No / rare pinocytomatous rosettes
- Minimal mitotic activity
- Often +4q, +12q, -22 (Genes Chromosomes Cancer 2001;30:99)
- Grade 4 of 4
- Second most common pineal gland tumor after germ cell tumor
- Usually age 20 years or less
- Frequent CNS metastases or spinal seeding, which is the main cause of death
- 5 year survival is 58%
- Poor prognostic factors:
- 7+ mitotic figures/10 HPF
- Presence of necrosis
- No neurofilament staining
- 15 year old girl with intracranial midline mass (Arch Pathol Lab Med 2004;128:707)
- 18 and 26 year old men with vertebral metastases from pineoblastoma (Arch Pathol Lab Med 2001;125:939)
- Osseous metastasis of pineoblastoma (J Neurooncol 2005;74:53)
- Surgery, variable radiation therapy
- Prognosis is usually poor
- Located in pineal gland
- May appear well demarcated from surrounding brain tissue but usually infiltrates into surrounding structures
- Sheets of densely packed cells with high grade (anaplastic / undifferentiated) features including high N/C ratio with minimal cytoplasm and large hyperchromatic nuclei
- Also necrosis, frequent mitotic figures
- Focal nuclear molding
- Homer Wright or Flexner-Wintersteiner rosettes
- May have lower grade features of pineocytoma and pineal parenchymal tumor of intermediate differentiation
- Often infiltrates into surrounding structures
Images hosted on other servers:
Small blue cell neuroepithelial malignant tumor
- NSE, synaptophysin, retinal S antigen
- Occasional cytoplasmic dense core granules
- Short immature cell processes
- Occasional junctional complexes
- No definite synapses
Images hosted on other servers:


Presence of abundant filopodia and rare intercellular junctions but no definitive synapses
- Glial neoplasms: GFAP+
- Medulloblastoma
- Pineocytoma: better differentiated cells with more cytoplasm, smaller cells, no / rare mitotic figures
- Malignant embryonal neoplasm of the pineal gland (CNS WHO grade 4)
- Malignant embryonal neoplasm (CNS WHO grade 4)
- High mitotic index
- Located in pineal region
- Pineoblastoma
- ICD-O: 9362/3 - pineoblastoma
- ICD-10: C75.3 - malignant neoplasm of pineal gland
- ICD-11: 2A00.20 & XH1ZH1 - tumors of the pineal gland or pineal region & pineoblastoma
- Pineal tumors are rare, representing < 1% of all CNS neoplasms (Neuro Oncol 2023;25:iv1)
- Pineoblastoma represents ~30 - 35% of all pineal parenchymal tumors
- Pineoblastoma typically arise in children / adolescents, with the overall median age of 6 years, varying depending on the molecular subtype (Acta Neuropathol 2021;141:771)
- miRNA processing altered 1: 8.5 years old
- miRNA processing altered 2: 11.6 years old
- RB1 altered: 2.1 years old
- MYC / FOXR2 activated: 1.3 years old
- Pineal gland but may infiltrate the surrounding brain parenchyma
- Cerebrospinal fluid (CSF) dissemination in 25 - 30% of patients and can show leptomeningeal disease as well as distant spinal metastases (BMJ Case Rep 2015;2015:bcr2015210343, Arch Pathol Lab Med 2001;125:939)
- Can be associated with DICER1 tumor predisposition syndrome (Acta Neuropathol 2021;141:771)
- RB1 altered subgroup can be seen in association with germline RB1 mutations (Acta Neuropathol 2020;139:243)
- Rare cases in patients with familial adenomatous polyposis (Dis Colon Rectum 2005;48:2343)
- Patients typically present with features of increased intracranial pressure and obstructive hydrocephalus (headache, nausea, vomiting, papilledema) (Childs Nerv Syst 1998;14:53, Coll Antropol 2013;37:35)
- If the tumor compresses the superior colliculus, patients may present with Parinaud syndrome (upward gaze palsy) (Curr Med Imaging 2020;16:1044)
- Brain imaging, usually magnetic resonance imaging (MRI)
- Requires tissue for definitive diagnosis, with molecular studies performed to further subclassify
- Large, irregular, lobulated mass with varying degrees of calcification and ill defined borders with adjacent structures (Neuroradiology 2000;42:509)
- Weak / heterogeneous contrast enhancement on MRI and CT (Neuroradiology 2000;42:509)
- Frequently associated with obstructive hydrocephalus (J Comput Assist Tomogr 1995;19:509)
Contributed by Jared T. Ahrendsen, M.D., Ph.D.

Axial MRI T1 postcontrast

Axial MRI T2
- Young patient age, disseminated disease, extent of surgical resection and extent of adjuvant therapy are all important prognostic factors (Cancer 2012;118:173, Clin Transl Oncol 2012;14:827)
- Staging (based on the Chang system for medulloblastoma) (Radiology 1969;93:1351)
- M0: no evidence of gross subarachnoid or hematogenous metastases
- M1: microscopic tumor cells in CSF
- M2: gross nodular disease seen in cerebral / cerebellar subarachnoid space or in third / lateral ventricles
- M3: gross nodular disease seen in spinal subarachnoid space
- M4: metastases outside of the CNS
- Clinical outcomes vary by molecular subgroup (Acta Neuropathol 2021;141:771)
- miRNA processing altered 1: intermediate prognosis
- miRNA processing altered 2: favorable prognosis
- RB1 altered and MYC / FOXR2 activated: poor prognosis
- 2 year old boy who received a multivisceral transplant (Pediatr Transplant 2012;16:E110)
- 3 year old girl with pineal anlage tumor (Childs Nerv Syst 2022;38:1625)
- 21 year old pregnant woman with pineoblastoma (Anticancer Res 2019;39:2627)
- Pineoblastoma without retinoblastoma in a patient with germline RB1 mosaicism (JAMA Ophthalmol 2022;140:437)
- Following surgical resection, patients typically receive adjuvant craniospinal irradiation (CSI) and chemotherapy (Acta Neuropathol 2020;139:259)
- Soft, friable, tan-pink to tan-gray mass
- May see areas of hemorrhage or necrosis (Cancer 1980;45:1408)
- Densely cellular, poorly differentiated neoplasm with numerous mitoses, necrosis, hyperchromasia and high N:C ratio
Contributed by Jared T. Ahrendsen, M.D., Ph.D., Nitin R. Wadhwani, M.D. and Rachel A. Multz, M.D.

Small round blue cells

Malignant embryonal cells
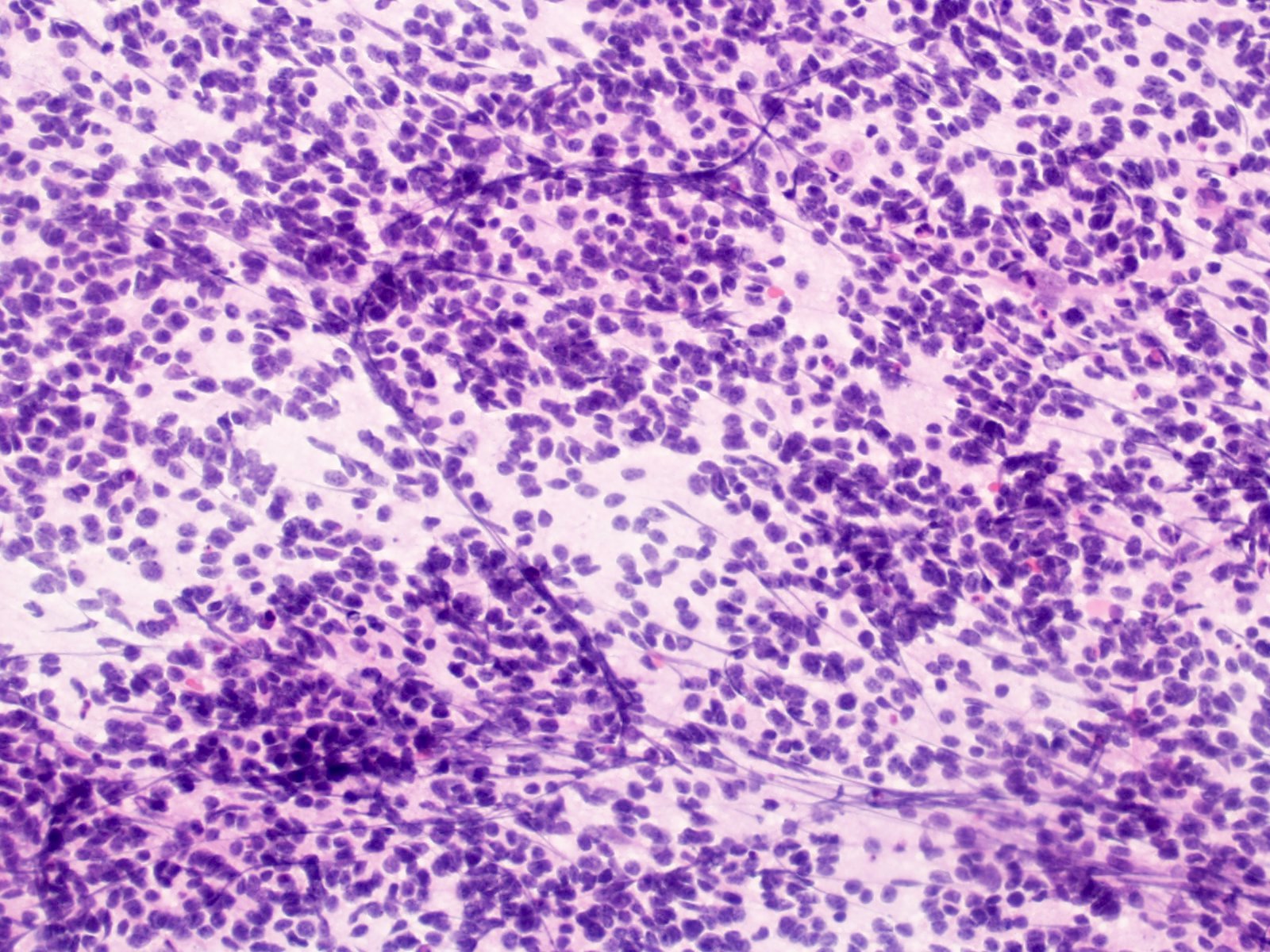
Smear preparation
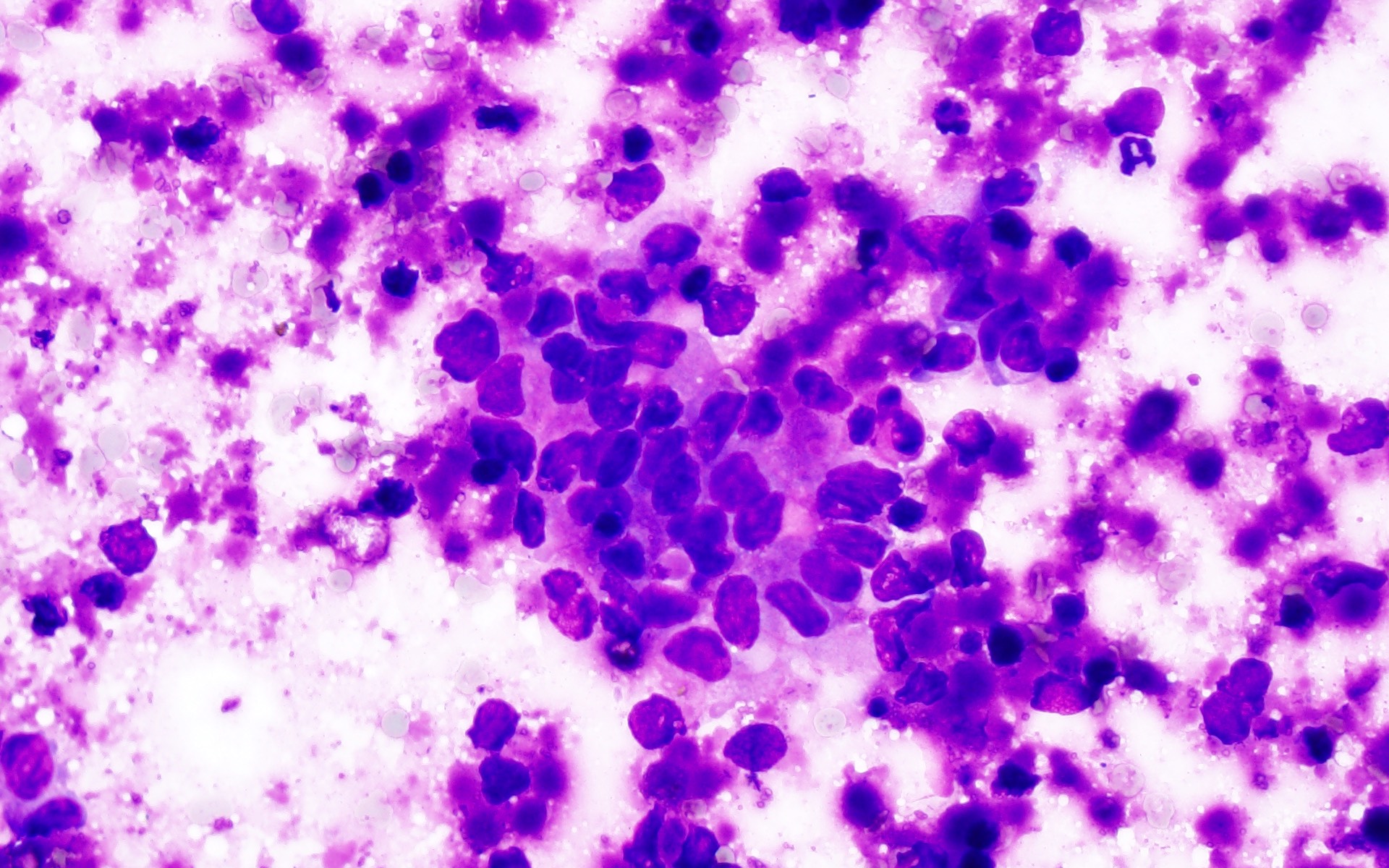
Diff-Quik smear preparation
Images hosted on other servers:
Diff-Quik
- Hypercellular, poorly differentiated, small round blue cell tumor (embryonal morphology)
- High N:C ratio, nuclear molding, brisk mitotic activity, necrosis
- Homer-Wright rosettes (eosinophilic material as false lumen) and less commonly Flexner-Wintersteiner rosettes (true lumen formation)
- May or may not see invasion of adjacent brain structures
- Reference: Brain Pathol 2000;10:49
Contributed by Jared T. Ahrendsen, M.D., Ph.D.

With necrosis


Histology
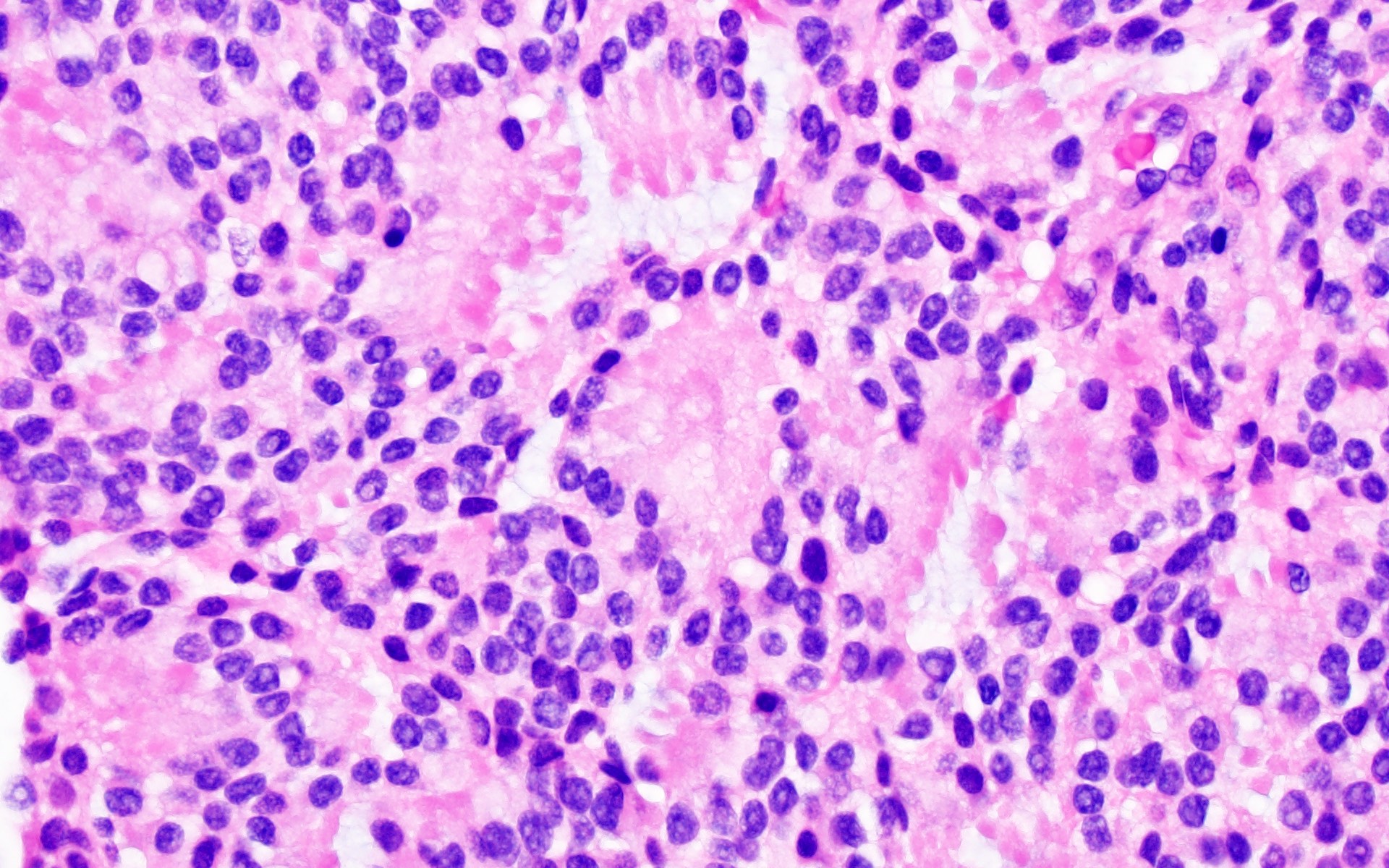
Homer-Wright pseudorosettes

Mitotic activity
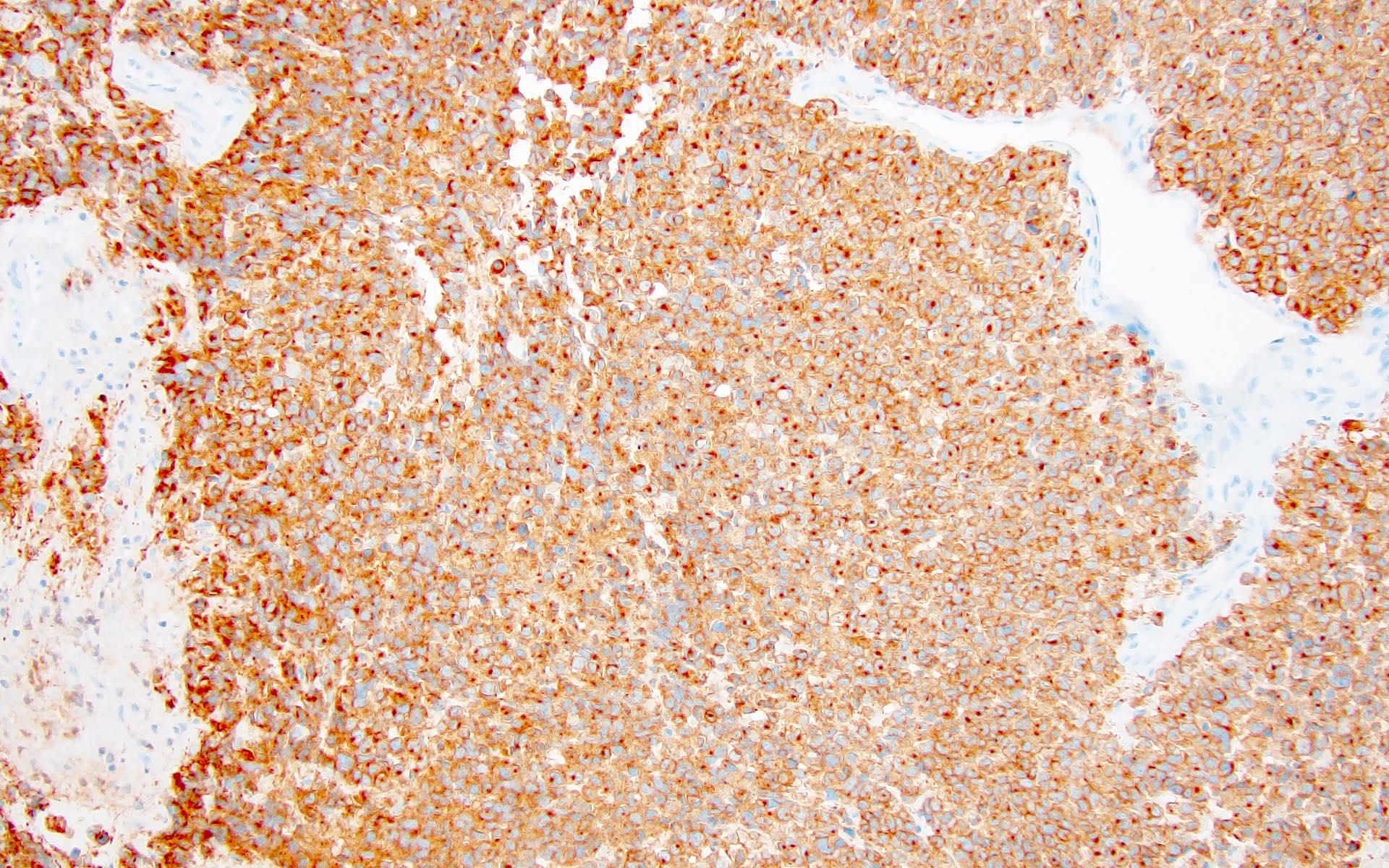
Synaptophysin positive

Variably chromogranin positive
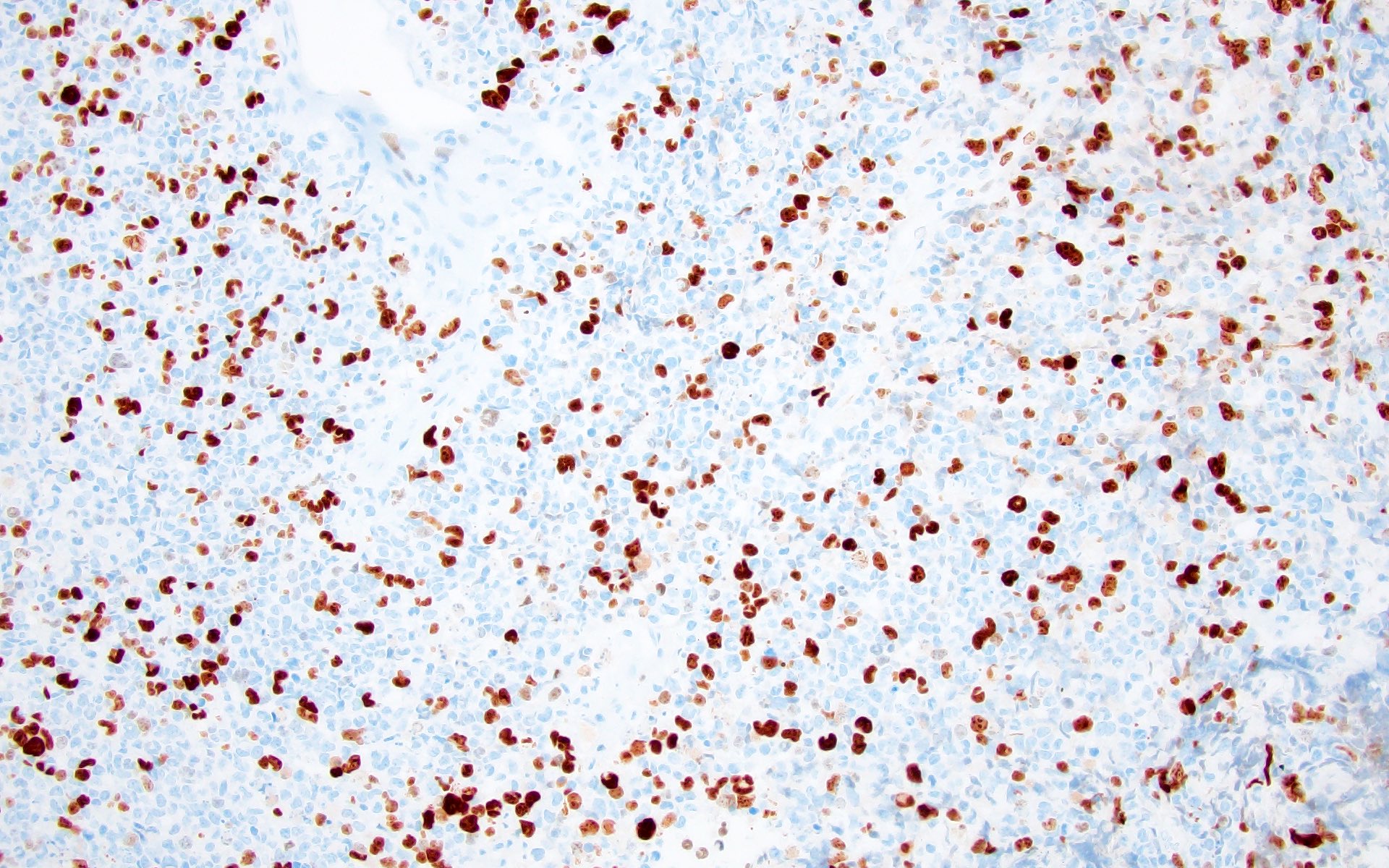
High Ki67
- As CSF cytology is used as part of the staging criteria for patients with pineoblastoma, recognizing tumor cells on cytology specimens is crucial
- Tumor cells will show high N:C ratio, brisk mitotic activity and necrosis (Cytopathology 2023 Dec 15 [Epub ahead of print])
- Sheet-like architecture and the presence of Homer-Wright rosettes may or may not be appreciated by cytology
- Synaptophysin, neuron specific enolase (NSE)
- Weakly positive for neurofilament and chromogranin A (Neuropathology 2002;22:66)
- Diffusely positive for CRX, a retinal / pineal lineage specific transcription factor (Am J Surg Pathol 2017;41:1410)
- Retained INI1 (SMARCB1) and BRG1 (SMARCA4) expression (Brain Pathol 2013;23:19)
- High Ki67 proliferation index (Clin Neuropathol 2008;27:325)
- GFAP, Olig2, cytokeratins, LIN28A (Neuropathology 2002;22:66)
- No longer used in routine clinical practice
- 4 distinct subgroups of pineoblastoma have been identified from DNA methylation profiling (Acta Neuropathol 2020;139:223, Acta Neuropathol 2020;139:243, Acta Neuropathol 2021;141:771)
- miRNA processing altered 1
- Germline DICER1 variant or alterations in DICER1, DROSHA and DGCR8 (loss of function) by molecular testing
- Gains of chromosomes 7 and 12, loss of chromosomes 16 and 22q seen on cytogenetics
- miRNA processing altered 2
- Germline DICER1 variant or alterations in DICER1 and DROSHA (loss of function) by molecular testing
- Loss of chromosomes 8, 14q, 16 and 20 seen on cytogenetics
- RB1 altered
- Germline RB1 variant, somatic loss of function RB1 mutation or gain of miR17 / 92 seen on molecular testing
- Gains of chromosomes 1q and 6p and loss of chromosome 16 seen on cytogenetics
- MYC / FOXR2 activated
- FOXR2 overexpression or MYC amplification seen on molecular testing
- Gains of 8q and loss of 16q seen by cytogenetics
- miRNA processing altered 1
Images hosted on other servers:

Overview of pineoblastoma subtypes
- Brain, pineal tumor, resection:
- Pineoblastoma, miRNA processing altered 1, CNS WHO grade 4
- Pineal parenchymal tumor of indeterminate differentiation (PPTID):
- Can be hypercellular but will have less cytologic atypia and mitotic activity, minimal to no rosette formation and no necrosis
- KBTBD4 mutation, which is absent in pineoblastoma (Acta Neuropathol 2020;139:243)
- Medulloblastoma:
- Can see alterations in SHH and WNT by molecular studies
- Unique DNA methylation profiles
- Negative for CRX pineal lineage transcription factor
- Atypical teratoid / rhabdoid tumor (AT / RT):
- Loss of INI1 (SMARCB1) expression
- Variably positive for GFAP and cytokeratins
- Embryonal tumor with multilayered rosettes (ETMR):
- LIN28A positive
- C19MC alteration
A 5 year old boy presents with an intracranial mass that appears to arise adjacent to the splenium of the corpus callosum. A resection is performed and shows the tumor morphology above. The tumor cells are positive for synaptophysin, NSE and INI1 and negative for GFAP and Olig2. What is the most likely molecular alteration in this tumor?
- Biallelic loss of SMARCB1
- DICER1 mutation
- H3 K27M mutation
- IDH1 R132H mutation
- SHH pathway activation
Comment Here
Reference: Pineoblastoma
- Beta catenin
- CRX
- H3 K27M
- INI1
- LIN28A
Comment Here
Reference: Pineoblastoma
- Grade 1 of 4
- Mostly adults age 25 - 35 years
- Slow growing
- Average survival 7 years
- Well circumscribed, gray, hemorrhagic
- Similar to normal pineal glands well differentiated cells but hypercellular
- Fibrovascular stroma highlights expansive lobules of tumor cells with uniform round nuclei
- Pinocytomatous rosettes (large, loose, Homer Wright-like rosettes with central fibrillar zones surrounded by neoplastic cells with round nuclei)
- May have features of neuronal differentiation (ganglion cells)
- Noninfiltrative, no / rare mitotic figures, no necrosis, no / minimal atypia
- Pituicytomas are tumors that arise from the pituicytes of the neurohypophysis and pituitary infundibulum
- 4 subtypes
- Pituicytoma
- Oncocytic pituicytoma (first described as spindle cell oncocytoma in 2002) (Am J Surg Pathol 2002;26:1048)
- Granular cell pituicytoma
- Ependymal pituicytoma
- Pituicytoma derives from light and dark normal pituicytes, oncocytic pituicytoma derives from oncocytic pituicytes, the granular cell subtype derives from granular pituicytes and the ependymal subtype derives from the elongated ependymal pituicytes (Am J Surg Pathol 2013;37:1694)
- These tumors and cells are characterized by the expression of the transcription factor TTF1
- Pituicytoma belongs to the group of TTF1 expressing tumors of the neurohypophysis and infundibulum
- Histologic features include fascicles of spindle cells or epithelioid cells expressing TTF1, S100 protein and GFAP
- Surgical resection is curative; excellent outcome
- Not recommended
- Infundibuloma
- Low grade glioma of sellar and suprasellar region
- Pilocytic astrocytoma
- Spindle cell oncocytoma of adenohypophysis
- Granular cell myoblastoma / schwannoma / neuroma
- Pituitary ependymoma / sellar ependymoma
- Classic pituicytoma represents ~0.07% of all primary sellar neoplasms (Eur J Endocrinol 2007;156:203)
- ~50% of posterior pituitary tumors (World Neurosurg 2021;145:148)
- Usually in adults: 40 - 60 years
- No obvious male or female predominance
- Exceptional in children (3% of posterior pituitary tumors)
- Sella and suprasellar region
- TTF1 expression indicates an origin from neurohypophysis and infundibulum (Am J Surg Pathol 2013;37:1694)
- Given the low incidence of diabetes insipidus, an origin from the lower infundibulum was proposed (Pituitary 2012;15:227)
- Unknown
- Main signs and symptoms at onset (World Neurosurg 2021;145:148)
- Headache
- Visual field defects
- Hypopituitarism
- Occurrence of polydipsia and polyuria (diabetes insipidus) is rare
- Apoplexy is exceptional
- Pituicytoma can also present in association with primary tumors of adenohypophyseal cells (collision tumors)
- Evidence of a sellar and suprasellar (or suprasellar only) lesion involving the infundibulum
- Usually no excess of pituitary hormones
- Diagnosis is made on histology
- Reference: World Neurosurg 2021;145:148
- Low serum levels of pituitary hormones
- Increase in prolactin due to pituitary stalk compression
- Occasional increase in growth hormone or adrenocorticotrophic hormone (ACTH) / peripheral cortisol due to coexistence of a pituitary neuroendocrine tumor
- References: World Neurosurg 2021;145:148, Endocrine 2020;70:15
- Can be suspected when exclusively suprasellar and the anterior hypophysis is spared
- Indistinguishable from other clinically nonfunctioning primary sellar tumors when it involves the sellar and suprasellar region
- MRI features: isointense on T1 weighted sequences and often inhomogeneous intensity on T2 weighted sequences; homogenous or heterogeneous contrast enhancement
- Reference: World Neurosurg 2021;145:148
Contributed by Federico Roncaroli, M.D.
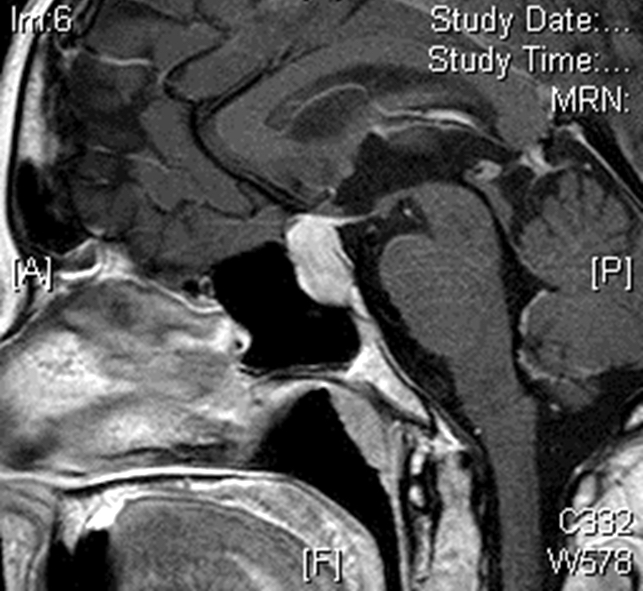
Sagittal postcontrast MRI
- Behaves as a benign tumor when completely resected
- Recurrence can occur when incompletely excised
- Oncocytic tumors have higher frequency of recurrence, possibly due to rich vascularity and adhesion
- Radiation therapy has been reported for recurrent / residual disease
- Metastasis has not been reported
- 1 recurrent case showed nuclear atypia and mitoses (Pathology 2011;43:389)
- Unclear relevance of atypia and increased proliferation as a prognostic factor (Acta Neuropathol Commun 2019;7:69)
- 26 year old man presented with decreased libido, hypopituitarism and visual disturbances (Neurosurgery 2004;54:753)
- 41 year old woman with nausea, headache and fatigue (Endocr Pathol 2014;25:436)
- 47 year old woman with a history of Cushing disease and a left sided intrasellar lesion (J Surg Case Rep 2020;2020:rjaa104)
- 61 year old man presented with progressively worsening visual acuity, normal pituitary lab values and a mixed solid cystic, intra and suprasellar lesion (Surg Neurol Int 2018;9:145)
- 71 year old man with a previous diagnosis of sellar meningioma and a recurrent lesion on surveillance imaging after 16 years (Ann R Coll Surg Engl 2020;102:e87)
- Complete surgical excision is curative
- Radiotherapy administered in a few patients with unclear benefits
- References: World Neurosurg 2021;145:148, Pituitary 2012;15:227
- Usually white, gray or light yellow soft tissue fragments; hemorrhagic changes can be seen
- Fascicles of spindle cells in fibrillary background reminiscent of glioma
- Possible ill defined whorls reminiscent of sellar meningioma
- Bipolar cells similar to schwannoma
- Sheets and short fascicles of spindle, bipolar tumor cells; storiform pattern can be present
- Variants distinguished based on cytologic features
- Oncocytic tumors have epithelioid cells with oncocytic cytoplasm
- Granular cell variants have large cells with granular cytoplasm
- Ependymal variants have elongated cells with ependymal cell-like features
- Mixed morphologies are not uncommon
- Rarely, focal chronic inflammation
- Absent pericellular reticulin fibers
- Absent Rosenthal fibers, granular bodies and hyalinized blood vessel walls
- Lack of Herring bodies
- Usually negligible mitotic activity
- Rarely, increased cellularity, severe nuclear atypia and high mitotic count
- References: Am J Surg Pathol 2000;24:362, Am J Surg Pathol 2013;37:1694
Contributed by Federico Roncaroli, M.D. and Sylvia L. Asa, M.D., Ph.D.
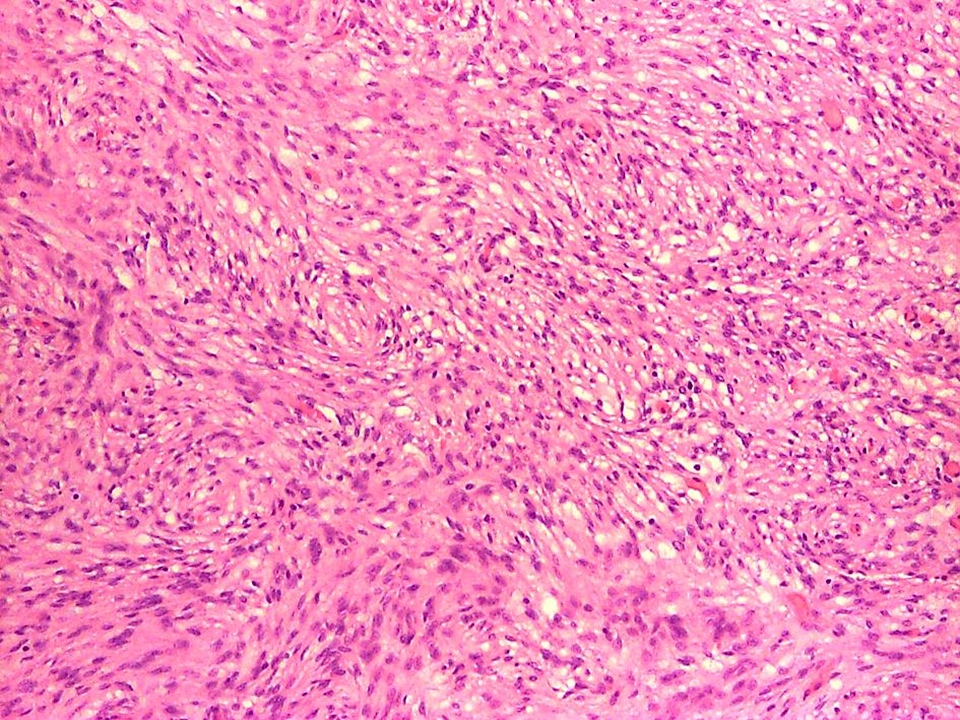
Fascicles of spindle cells

Ill defined whorls
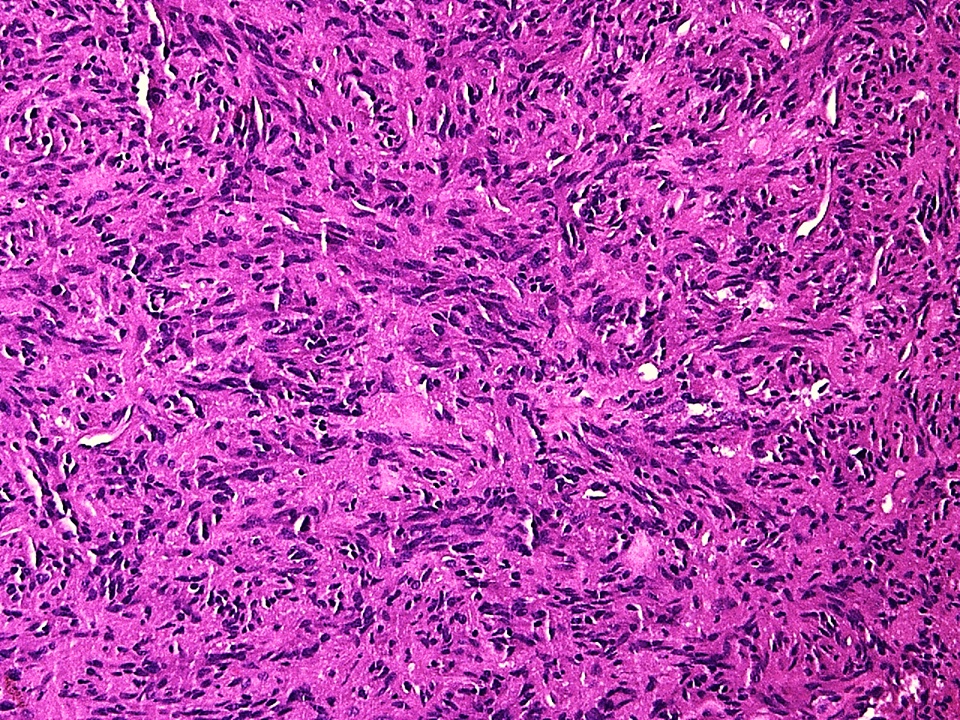
Schwannoma-like features
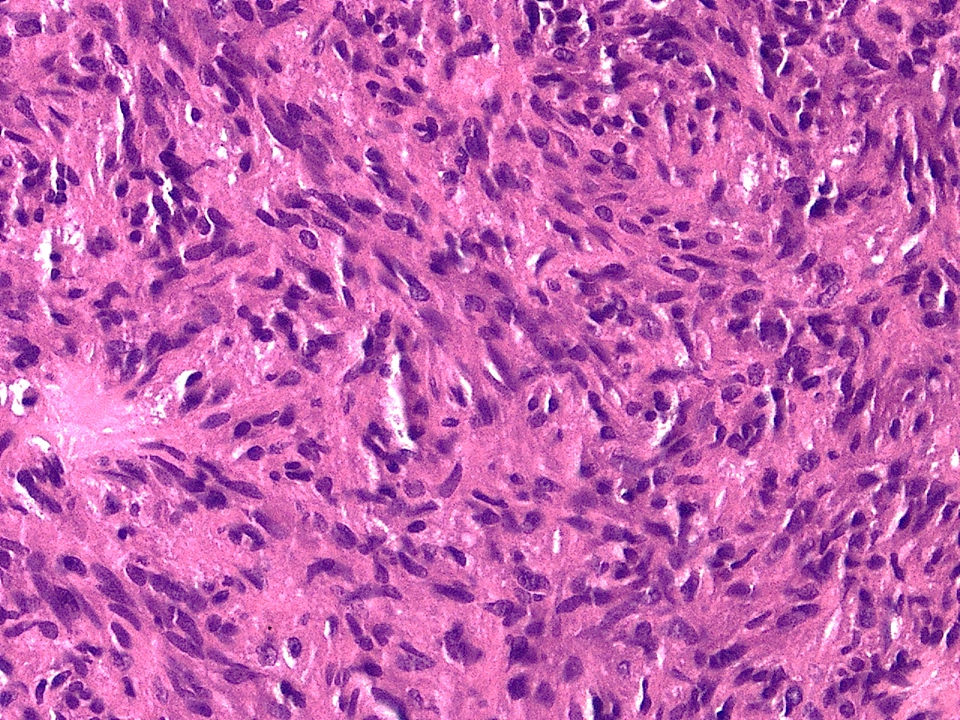
Mild nuclear atypia
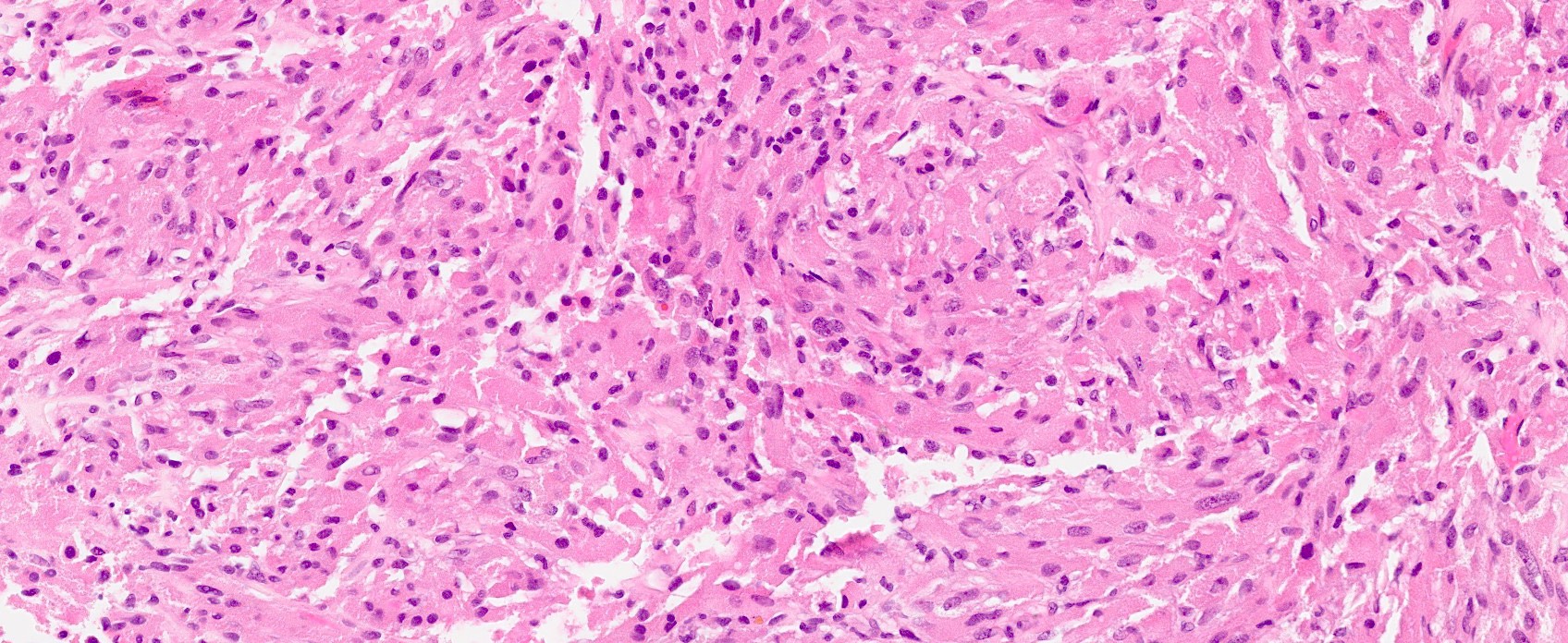
Oncocytic pituicytoma
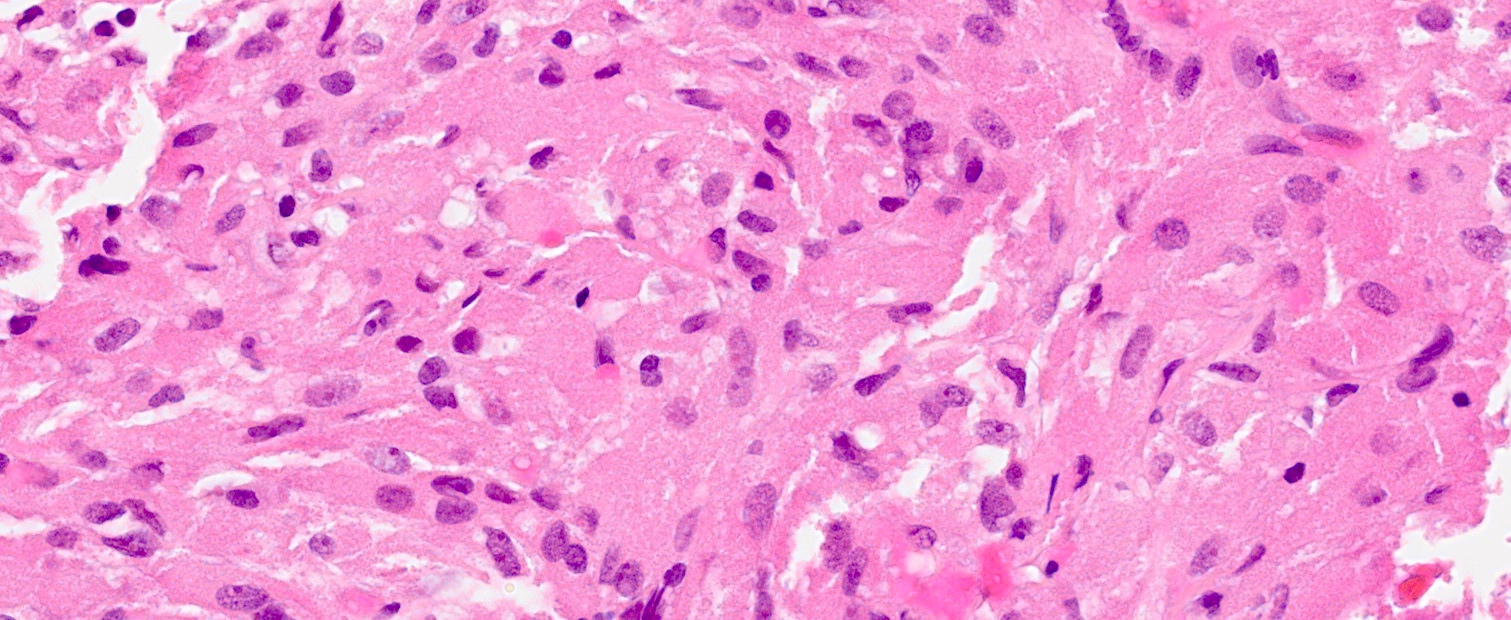
Oncocytic pituicytoma
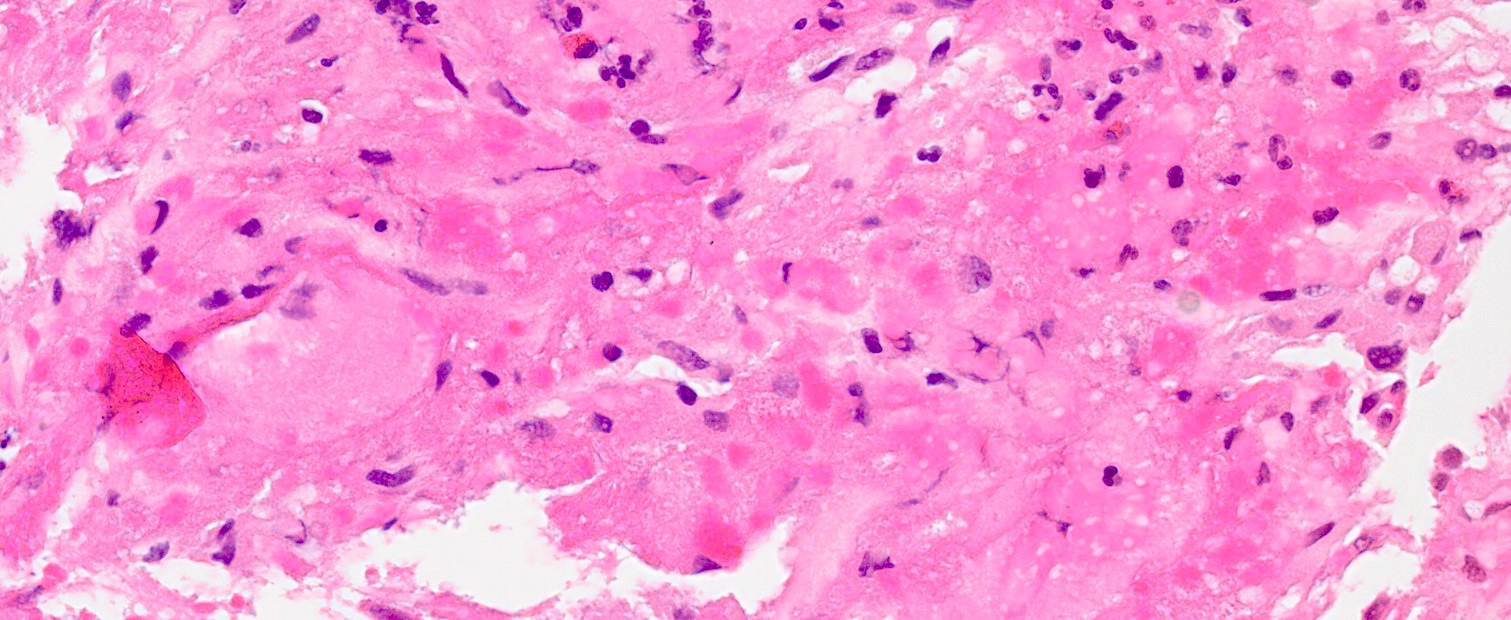
Granular cell pituicytoma
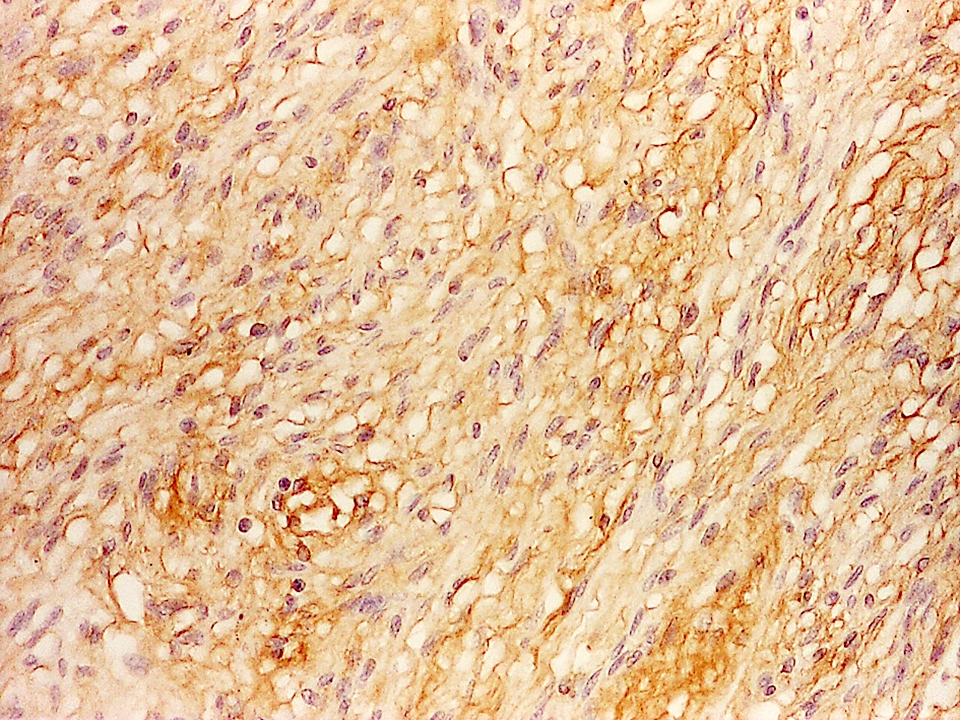
GFAP expression
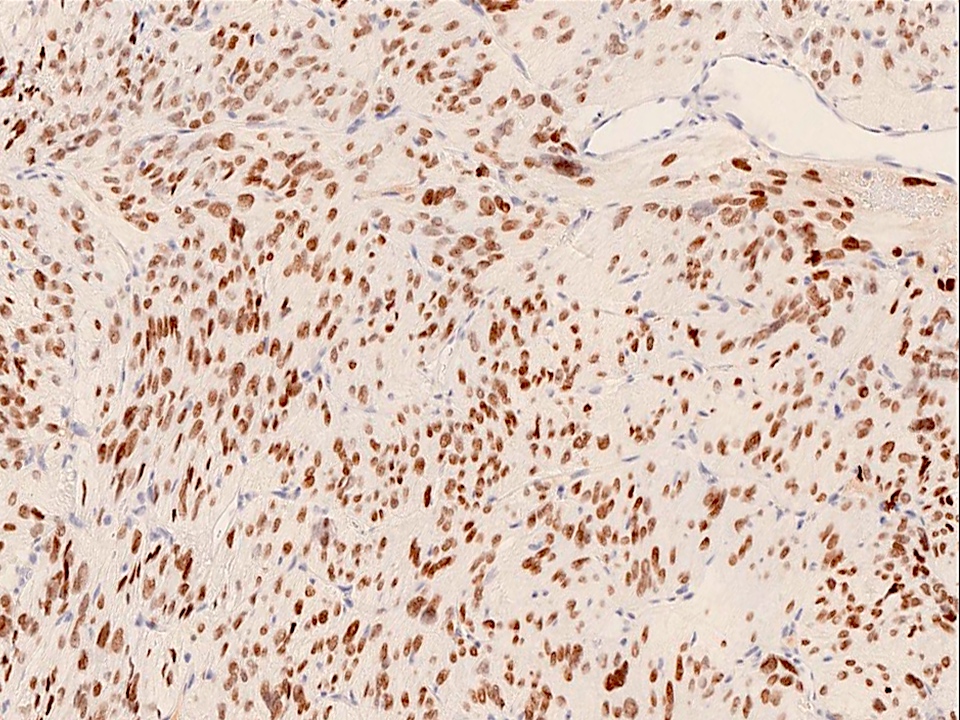
TTF1 nuclear expression
- Aggregates of spindle cells with fibrillary to fine cytoplasm, usually thin walled, delicate vessels and neuropil-like background; features can be reminiscent of meningioma (Diagn Cytopathol 2020;48:342)
- TTF1
- Vimentin, S100
- Variable GFAP (expressed in all cases)
- CD56 (NCAM), galectin3, BCL2, focal SSTR2A (20% of cases)
- Low Ki67 labeling index
- References: Am J Surg Pathol 2000;24:362, Am J Surg Pathol 2013;37:1694
- Pituitary hormones (GH, PRL, ACTH, TSH beta subunit, FSH beta subunit, LH beta subunit and common alpha subunit)
- Pituitary transcription factors (Pit1, SF1 and Tpit)
- Synaptophysin, neurofilament proteins, collagen type IV (absent around single tumor cells)
- EMA
- Cytokeratins
- Smooth muscle actin, CD34, CD38
- Vasopressin
- Reference: Am J Surg Pathol 2013;37:1694
- Elongated or oval tumor cells
- Focal accumulations of intermediate filaments
- No interdigitating cell membranes
- Oncocytic variant has abundant mitochondria rich in lamellar cristae
- No secretory granules
- References: Neurosurgery 2004;54:753, Am J Surg Pathol 2013;37:1694
- Molecular genetics studies have been restricted to the 3 most common subtypes (pituicytomas, granular cell and oncocytic pituicytomas)
- Close clustering on methylation studies indicates a single spectrum (Nature 2018;555:469, Acta Neuropathol 2021;142:1025)
- 2 epigenomic DNA methylation subgroups with only subtle overall methylation differences (Acta Neuropathol 2021;142:1025)
- Variable somatic alterations causing MAPK pathway activation in pituicytomas and oncocytic tumors
- Pathogenic FGFR1, HRAS, BRAF, NF1, CBL, MAP2K2, PTEN and TSC1 variants (Acta Neuropathol 2021;142:1025)
- Rare tumors had accompanying TERT promoter mutation
- SND1 and FAT1 gene mutations also identified (Oncotarget 2016;7:37054)
- Close clustering on methylation studies indicates a single spectrum (Nature 2018;555:469, Acta Neuropathol 2021;142:1025)
- Whole miRNA signature shows global downregulation of miRNA expression (Neurochem Res 2019;44:2360)
- Chromosome 1 alterations in oncocytic tumors including 1p deletion (4/5 cases) and 1p gain (1/5 cases) (J Neuropathol Exp Neurol 2021;80:45)
- Copy number alterations identify a less favorable group with reduced progression free survival (Acta Neuropathol 2021;142:1025)
- IDH1 p.R132H mutation and KIAA1549::BRAF oncogene fusion are absent (Am J Surg Pathol 2013;37:1694)
- Pituitary gland / sellar region, surgical excision:
- Pituicytoma (subtype if relevant) (WHO grade 1) (see comment)
- Comment: Pituicytoma belongs to the group of TTF1 expressing tumors of the posterior hypophysis and infundibulum. It rarely recurs when incompletely removed; follow up is recommended.
- Immunohistochemically, the tumor cells show TTF1 nuclear expression, diffuse vimentin and S100 expression. Immunostains for keratins, synaptophysin, chromogranin and biomarkers of adenohypophyseal neuroendocrine cells are negative.
- Normal neurohypophysis and infundibulum:
- Vasopressin positive
- Herring bodies and axons absent in pituicytoma
- Sellar schwannoma:
- TTF1 negative
- Pericellular collagen IV
- Pilocytic astrocytoma:
- Rosenthal fibers and granular bodies
- Often vascular with features of endothelial proliferation
- Meningioma:
- Pituitary neuroendocrine tumor:
- Rarely, spindle cells
- Chromogranin and synaptophysin positive, as well as pituitary transcription factors and hormones
- TTF1 negative
A middle aged man presented with hypopituitarism. Imaging showed a sellar and suprasellar enhancing lesion. The microscopic image above is from the surgical specimen. Which of the following immunostains is typically positive in these tumor cells?
- Chromogranin
- Cytokeratin CAM5.2
- Neurofilament
- TTF1
- Vasopressin
Comment Here
Reference: Pituicytoma
- Patients present with headaches, visual field disturbances and hypopituitarism
- Patients present with syndrome of antidiuresis (SIAD)
- Surgery is not curative
- These tumors are not responsive to radiotherapy
Comment Here
Reference: Pituicytoma
- Nonneoplastic absolute increase in the number of 1 or more adenohypophyseal cell subtypes seen radiologically as enlargement of the pituitary gland (Pituitary 1999;1:169)
- Relatively common due to physiological and pathological conditions (Pituitary 1999;1:169)
- Remains unrecognized and occurs in heterogenous setting (Endocrinol Diabetes Metab Case Rep 2015;2015:150017)
- Nodular or diffuse hyperplasia of polymorphic acini with intact reticulin, without effacement of gland architecture
- MRI essentially shows symmetrical and diffuse enlargement, similar intensity to gray matter (Pituitary 1999;1:169)
- ICD-10: E23.7 - disorder of the pituitary gland, unspecified
- Very rare, accounting for < 1% of sellar surgical specimens
- Autopsy prevalence 26% (Pituitary 1999;1:169)
- Higher incidence in primary hypothyroidism (Clin Case Rep 2020;9:629)
- Prolactin (PRL) cell hyperplasia in pregnancy (Endocrinol Diabetes Metab Case Rep 2015;2015:150017)
- Age: youth and menopause (AJNR Am J Neuroradiol 1997;18:551)
- Adenohypophysis
- Physiologic response
- Most common: prolactin cell hyperplasia in pregnancy and lactation, peaks immediately postpartum
- Puberty: pituitary height peaks in the 20 - 29 year age group (F > M)
- Elderly: nonfunctional hyperplasia due to basophil cells
- Pars intermedia derived proopiomelanocortin (POMC) cells invasion into the neurohypophysis, women in the 50 - 59 year age group (Hormones (Athens) 2003;2:149)
- Pathologic hyperplasia due to end organ insufficiency
- Longstanding primary hypothyroidism causes TSH cell hyperplasia, young females
- Gonadotroph hyperplasia and sellar expansion in primary hypogonadism due to Klinefelter or Turner syndrome
- Polycystic ovary syndrome: hyperprolactinemia and lactotroph hyperplasia
- ACTH cell hyperplasia due to hypocortisolism in Addison disease
- Rare: pituitary transcription factor gene, PROP1 mutations
- Pathologic hyperplasia associated with ectopic excess of releasing hormones
- GH releasing hormone (GHRH) or corticotropin releasing hormone (CRH) ectopic release causing somatotroph or corticotroph hyperplasia secreted by pancreatic islet cell tumor, pheochromocytoma, bronchial and thymic carcinoid tumors
- ACTH cell hyperplasia due to corticotropin releasing hormone secretion from hypothalamic hamartoma or neuroendocrine tumors
- Iatrogenic: treatment with antipsychotics and excess estrogen in transgender women are associated with increased secretion of prolactin (Int Clin Psychopharmacol 2019;34:89, Int J Mol Sci 2020;21:2024, BMJ Case Rep 2009;2009:bcr02.2009.1589)
- Syndromic: mammosomatotroph hyperplasia in McCune-Albright syndrome, Carney complex, MEN1 related GHRH associated, X linked acrogigantism syndrome (XLAG)
- Idiopathic
- Hereditary (J Clin Endocrinol Metab 2011;96:E2078)
- Mass effect
- Visual disturbance, bitemporal hemianopia, diplopia
- Headaches
- Hormone related hyperplasia
- GH cell: gigantism or acromegaly
- Prolactin cell: hyperprolactinemia
- ACTH cell: Cushing disease
- TSH cell:
- Longstanding primary hypothyroidism results in TSH hyperplasia
- Hyperprolactinemia
- Elevated levels of TRH that stimulates both pituitary TSH and PRL cells due to lack of negative feedback of thyroxin (T4) on the hypothalamus (Pituitary 1999;1:169, Endocrinol Diabetes Metab Case Rep 2015;2015:150056)
- LH / FSH: result of early onset hypogonadism
- History and physical
- Increase or decrease corresponding serum hormone levels
- Imaging
- Biopsy / excision
- Rarely removed surgically
- Most likely identified at autopsy
- References: StatPears: Pituitary Hyperplasia In Primary Hypothyroidism [Accessed 8 April 2021], Case Rep Endocrinol 2019;2019:2012546
- Hormonal levels as indicated
- Symmetric 2 - 3x enlargement of pituitary gland on CT / MRI
- Homogeneously enhancing gland with convex superior margin
- No sellar destruction
- Intensity similar to gray matter
- Homogenous gadolinium uptake
- Reference: StatPearls: Pituitary Gland Imaging [Accessed 13 May 2021]
Images hosted on other servers:

Sagittal and coronal MRI
- Excellent with medical treatment
- Rarely associated with adenoma (Pituitary 1999;1:169)
- 10 year old girl with growth retardation and obesity (Medicine (Baltimore) 2018;97:e12703)
- 12 year old girl with headaches and fatigue (Clin Case Rep 2020;9:629)
- 19 year old woman with abdominal pain and diarrhea and 35 year old woman with secondary amenorrhea and headaches (Endocrinol Diabetes Metab Case Rep 2015;2015:150017)
- 35 year old man with bitemporal hemianopsia (Clin Pract 2015;5:733)
- Usually correct underlying endocrinologic disturbance
- Surgery rarely indicated
- Visual field defects
- Progression in size to establish diagnosis by pathology
- GH and ACTH hyperplasia
- References: StatPears: Pituitary Hyperplasia In Primary Hypothyroidism [Accessed 8 April 2021], AJNR Am J Neuroradiol 1997;18:551
- At autopsy or in the rare event the entire gland is removed, it is diffusely enlarged and tan-white with no discernible nodules
- Lack of well defined lesion
- Specimen may be fragmented
- Diffuse or nodular hyperplasia
- Unevenly enlarged acini
- Usually single cell type
- Relative cellular monomorphism within affected acinus
- Noncompressive; indistinct demarcation
- Difficult to diagnose in fragmented specimen
- Numerical increase in pituitary cells without alteration in architecture
- Rare mitotic activity
- GH cell hyperplasia: chromophobe to pale eosinophilic polygonal cells
- PRL cell hyperplasia: chromophobe with rare microcalcifications
- ACTH cell hyperplasia: amphophilic with large vacuoles (lysosomes) and Crooke cell change
- TSH cell hyperplasia: chromophobe, occasional spindle cells and multiple large PAS+ lysosomes
- LH / FSH hyperplasia: hypervacuolization (castration cells) (Endocrinol Diabetes Metab Case Rep 2015;2015:150017)
Images hosted on other servers:
Pituitary hyperplasia microscopic
- Hypercellular smear with heterogeneous cell populations
- Reticulin is essential for the diagnosis, highlights retention of acinar architecture and may show some expanded acini
- Synaptophysin
- Cell type routine cytoplasmic stains prolactin, GH, TSH, LH, FSH, ACTH
- PAS highlights lysosomes in TSH hyperplasia
- Ki67 labeling may be mildly increased (normal gland is completely negative)
- GFAP
- Neurofilament (2F11)
- Same as normal pituitary:
- GH producing somatotrophs:
- Rough endoplasmic reticulum, well formed Golgi complexes and numerous large, dense secretory granules 100 - 250 nm
- PRL producing lactotrophs:
- Elaborate rough endoplasmic reticulum arranged in parallel arrays, occasionally forming concentric structures known as nebenkern formations
- Prominent Golgi complexes and extrusion of secretory granules at the lateral cell borders known as misplaced exocytosis up to 700 nm
- GH and PRL producing mammosomatotroph:
- Pleomorphic heterogenous granules 150 - 1,000 nm and misplaced exocytosis
- TSH producing thyrotrophs:
- Short dilated rough endoplasmic reticulum and small secretory granules that align along the plasma membrane
- ACTH producing corticotrophs:
- Secretory granules are pleomorphic in shape and electron density with indentations and evaginations of granule membranes, resulting in heart and teardrop shapes
- Also small bundles of intermediate (keratin) filaments throughout the cytoplasm
- GH producing somatotrophs:
- Reference: Ann Clin Lab Sci 1979;9:275, Microsc Res Tech 1992;20:107
- No specific molecular / cytogenetics available
- Reported germline mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene in familiar cases
- Some syndromic association: Carney complex, McCune-Albright syndrome, X linked acrogigantism (XLAG) and less likely, MEN1 related GHRH associated
- Reference: J Clin Endocrinol Metab 2011;96:E2078
- Pituitary, transsphenoidal resection:
- Consistent with pituitary hyperplasia (see comment)
- Comment: Histological sections of the specimen show pituitary tissue composed of unevenly enlarged acini comprised of sheets of monomorphic cells with small round nuclei and moderate amounts of granular eosinophilic cytoplasm. Immunostains were performed that show the lesional tissue cells are strongly positive for synaptophysin, chromogranin, EMA. Ki67 stain labels a rare cell. Reticulin stain highlights retention of acinar architecture and expansion of acini. Prolactin hormone is positive. Other hormonal stains (ACTH, LH, FSH, GH) are negative. This immune profile is consistent with benign pituitary hyperplasia, prolactin hormone producing. Clinical and radiological correlation are recommended.
- Normal adenohypophysis:
- Normal size pituitary gland with heterogeneous cell populations within normal acini
- ACTH cells normally aggregate at the lateral wings of anterior pituitary
- Pituitary adenoma:
- Enlarged mass, well defined from normal pituitary gland
- Homogeneous cell populations with splayed and disrupted reticulin network
- Compresses adjacent normal acini
- Hyperplasia more likely to show strong cytoplasmic immunoreactivity to respective hormones throughout the fragments when compared with adenoma
- Corticotrophin releasing hormone ectopic release
- Longstanding primary hypothyroidism
- Polycystic ovary syndrome
- Prolactin cell hyperplasia in pregnancy and lactation
Comment Here
Reference: Pituitary hyperplasia
- Compresses adjacent normal acini
- Homogeneous cell populations with splayed and disrupted reticulin network
- Microcalcifications are never identified
- Mild acinar expansion which is noncompressive
- Neuroendocrine tumors of the anterior pituitary gland that are composed of secretory cells with pituitary hormone production
- Main tumor types include somatotroph, lactotroph, mammosomatotroph, thyrotroph, corticotroph, gonadotroph, null cell and plurihormonal tumors
- Routine assessment of histology includes determination of:
- Architecture: solid growth, nests, trabeculae or perivascular rosettes
- Cytoplasmic features: acidophilia, basophilia, inclusions indicative of fibrous bodies
- Nuclear features: mitoses, pleomorphism, giant cells
- Other features: inflammatory changes, stroma, hemorrhage, vascular features
- Determination of cell lineage and hormonal activity using stains for transcription factors, hormones and cytokeratins (Endocr Pathol 2022;33:6)
- Evaluation of tumor proliferation potential by mitotic count and Ki67 labeling index
- Evaluation of tumor invasion if possible to identify clinically aggressive tumors
- Ultrastructural features of tumor cells may be important for classification of unusual tumors
- Pituitary neuroendocrine tumor (preferred nomenclature)
- Pituitary adenoma (still acceptable but not preferred terminology)
- Metastatic pituitary neuroendocrine tumor (formerly known as pituitary carcinoma)
- ICD-O: 8272/3 - pituitary carcinoma, NOS
- ICD-11: 2F9A & XH94U0 - neoplasms of unknown behavior of endocrine glands & pituitary adenoma, NOS
- 10 - 15% of intracranial neoplasms
- Mainly occurs in fourth to seventh decade
- F > M
- Incidental tumors are seen in ~14.4% of autopsies and 22.5% of radiologic studies (Cancer 2004;101:613)
- Majority occur in the sella turcica, originating within the adenohypophysis / anterior pituitary lobe, with variable extension upwards into suprasellar region and into adjacent structures, such as cavernous sinuses, sphenoid sinus and sinonasal mucosa
- Ectopic sites include primary location in the sphenoid sinus; rare examples occur in the suprasellar pituitary stalk and clivus (Neurol Med Chir (Tokyo) 2004;44:380, Arq Neuropsiquiatr 2012;70:744, BMC Res Notes 2013;6:411, Pituitary 2020;23:457)
- Rare tumors are seen as a part of ovarian teratoma (Endocr Pathol 2014;25:321, Eur J Intern Med 2005;16:359, Case Rep Womens Health 2020;29:e00279, Endocr Pathol 2020;31:315)
- Cell differentiation is driven by transcription factors leading to 3 main cell lineages
- Transcription factors are (Endocr Pathol 2022;33:6):
- Pit1 (pituitary specific POU class homeodomain transcription factor), which leads to differentiation of somatotrophs, mammosomatotrophs, lactotrophs and thyrotrophs
- SF1 (steroidogenic factor 1), which regulates gonadotroph cell differentiation
- Tpit (T box family member TBX19), which has a transcription factor driving proopiomelanocortin (POMC) lineage with differentiation of corticotrophs
- Most tumors are of unknown etiology and lack mutations that are considered causative; epigenetic alterations are common (Endocr Pathol 2021;32:3)
- Inherited syndromes:
- MEN1 with involvement of the MEN1 gene and loss of menin
- MEN4 with mutation of CDKN1B and loss of p27
- MEN5 with mutation of MAX
- Carney complex with mutation of PRKR1ɑ
- Familial acromegaly with mutation of AIP
- SDHx related disease due to mutation in any of the genes encoding the SDH complex
- Hyperparathyroidism jaw tumor syndrome due to mutation of CDC73 and loss of parafibromin
- Lynch syndrome due to mutation in MLH1 / MSH2 / MSH6 / PMS2
- Noninherited germline predisposition syndromes include:
- X linked acrogigantism due to Xq26 microduplication and GPR101 alterations
- McCune-Albright syndrome with GNAS activating mutations
| Tumor type | Transcription factor(s) | Hormone(s) | Other biomarkers | Clinical features |
| Tpit family | ||||
| Densely granulated corticotroph tumor | Tpit | ACTH | PAS; intense keratins | Florid Cushing disease or silent |
| Sparsely granulated corticotroph tumor | Tpit | ACTH (weak) | PAS (weak); strong keratins | Cushing disease or silent |
| Crooke cell tumor | Tpit | ACTH | PAS; ring-like keratins | Cushing disease or silent |
| Pit1 family | ||||
| Densely granulated somatotroph tumor | Pit1 | GH | Perinuclear keratins | Acromegaly |
| Sparsely granulated somatotroph tumor | Pit1 | GH (weak) | Keratin fibrous bodies (> 70%) | Acromegaly or silent |
| Mammosomatotroph tumor | Pit1, ER | GH, PRL | Perinuclear keratins | Acromegaly and hyperprolactinemia |
| Sparsely granulated lactotroph tumor | Pit1, ER | PRL (juxtanuclear) | Variable keratins | Hyperprolactinemia (tumor size correlates with PRL level) |
| Densely granulated lactotroph tumor | Pit1, ER | PRL | Variable keratins | Hyperprolactinemia |
| Acidophil stem cell tumor | Pit1, ER | PRL with or without GH (weak, focal) | Variable keratins with or without fibrous bodies | Hyperprolactinemia |
| Thyrotroph tumor | Pit1, GATA3 | TSH | Variable keratins | Elevated TSH with or without hyperthyroidism |
| Mature plurihormonal Pit1 lineage tumor | Pit1, GATA3, ER | Variable, often multiple | Perinuclear keratins | Acromegaly, hyperprolactinemia and hyperthyroidism |
| Immature Pit1 lineage tumor | Pit1 with or without GATA3 with or without ER | Variable, often limited / focal | Variable keratins with or without fibrous bodies | Silent or acromegaly or hyperprolactinemia or hyperthyroidism |
| SF1 family | ||||
| Gonadotroph tumor | SF1, GATA3, ER | FSH, LH | Variable keratins, follicular cells | Silent with or without elevated gonadotropins |
| No lineage fidelity | ||||
| Null cell tumor | None | None | Variable | Silent |
| Unclassified plurihormonal tumor | Multiple combinations | Multiple combinations | Variable | Variable |
- Clinical features of hormone excess: acromegaly, gigantism, Cushing disease, sequelae of hyperprolactinemia, hyperthyroidism, rarely gonadotropin excess (Endocr Pathol 2022;33:6)
- Larger tumors (> 1 cm) can be associated with mass effects such as headache, visual disturbance and hypopituitarism
- Hemorrhagic necrosis of large tumors (pituitary apoplexy) may be a surgical emergency
- Current consensus classifies tumors according to pituitary cell lineage and cell differentiation (see Diagrams / tables)
- Classification requires immunohistochemical staining for pituitary transcription factors including the 3 main ones (Pit1, Tpit and SF1) as well as ER and GATA3
- Main pituitary hormones (growth hormone, prolactin, adrenocorticotropic hormone [ACTH], beta thyroid stimulating hormone [TSH], beta luteinizing hormone [LH], beta follicle stimulating hormone [FSH] and alpha subunit of glycoproteins) and keratins (usually CAM 5.2 but also AE1 / AE3 or CK18) (Hum Pathol 2021;107:87)
- Biochemical assessment should be performed in all patients with pituitary tumors (Endocr Pathol 2022;33:6)
- Serum prolactin can be elevated in any patient with a sellar lesion since it is under tonic inhibition and interruption of hypothalamic signaling can result in hyperprolactinemia
- However, this so called stalk effect does not usually result in a level > 200 mcg/L; levels above this usually indicate a tumor producing prolactin
- In patients with true lactotroph tumors, prolactin levels are usually proportional to tumor size
- Patients with large tumors and prolactin levels of 500 mcg/L or less usually do not have a lactotroph tumor
- Growth hormone and IGF1 are biomarkers of acromegaly or gigantism; since growth hormone secretion is pulsatile, IGF1 provides an integrated measure that is more reliable
- Oral glucose tolerance test is a definitive confirmatory test for the diagnosis of acromegaly
- Cortisol and ACTH are elevated in Cushing disease
- The relationship between these is important; a high normal ACTH in a patient with high normal or elevated cortisol should be assessed for Cushing disease
- High dose dexamethasone suppression testing can be useful
- TSH secreting tumors cause increased TSH levels and are usually associated with elevated T3 and T4 levels, hyperthyroidism and goiter
- Gonadotropin excess can be physiological in postmenopausal women; serum levels of FSH and LH as well as testosterone levels in men should be screened
- Cerebrospinal fluid may be xanthochromic with crenated red blood cells and high protein levels in pituitary apoplexy
- Method of classification is based on tumor size and degree of invasion (Endocr Pathol 2022;33:6)
- Important for planning of surgical resection
- Microtumors measure < 1 cm in diameter; macrotumors are > 1 cm
- Identification of tumor extension is important: suprasellar, lateral (cavernous sinus), inferior (sphenoid sinus), posterior fossa
- Density on T2 weighted imaging distinguishes sparsely from densely granulated tumors
Images hosted on other servers:
Sellar and suprasellar masses
- Tumor subtypes are predictive of responsiveness to medical therapies
- For example, densely granulated somatotroph tumors are usually responsive to long acting somatostatin analogues whereas sparsely granulated somatotroph tumors are not; densely granulated corticotroph tumors tend to respond to pasireotide therapy
- Invasive, aggressive tumors adenomas often recur over several years; presurgical growth rates can predict temporal pattern of regrowth
- Immature Pit1 lineage tumors tend to be aggressive (Mod Pathol 2016;29:131)
- Crooke cell tumors are aggressive (Endocr Pathol 2022;33:6)
- ATRX loss has been described in some aggressive corticotroph tumors (J Clin Endocrinol Metab 2021;106:1183, Endocrine 2022;76:228)
- 11 year old boy with a silent corticotroph tumor with adrenocortical choristoma (J Clin Res Pediatr Endocrinol 2022;14:126)
- 17 year old boy with an aggressive prolactinoma treated with temozolomide (Medicine (Baltimore) 2017;96:e8733)
- 19 year old woman with an ACTH producing tumor with signet ring cells (Appl Immunohistochem Mol Morphol 2020;28:e13)
- 33 year old man with Cushing disease and pituitary corticotroph tumor with adrenocortical cells (Neuropathol Appl Neurobiol 2022;48:e12754)
- 37 year old man with Cushing disease and co-occurring infundibular pituicytoma and pituitary adenoma (Endocr Regul 2019;53:263)
- 41 year old woman with plurihormonal tumor of Pit1 and SF1 lineages and collision with corticotroph tumor (Endocr Pathol 2019;30:74)
- 45 year old man with a multihormonal tumor with Pit1 and Tpit lineage cells (BMC Endocr Disord 2017;17:54)
- 47 year old man with a lactotroph PitNET / adenoma associated to granulomatous hypophysitis (Neuropathology 2022 Aug 10 [Epub ahead of print])
- 56 year old woman with a xanthogranulomatous tumor (Mol Clin Oncol 2018;8:445)
- 58 year old women with meningioma pituitary adenoma collision tumors (Medicine (Baltimore) 2017;96:e9139)
- 60 year old woman with a plurihormonal ACTH growth hormone tumor (World Neurosurg 2018;114:e158)
- Surgical approaches
- Commonly via transsphenoidal route (minimally invasive)
- Transfrontal approach for larger invasive tumors
- Partial versus gross total resection
- Dopamine agonists can reduce prolactin levels and the size of sparsely granulated lactotroph tumors
- Somatostatin analogs can reduce hormone (growth hormone and TSH) secretion but may cause little reduction in tumor size
- Clinically nonfunctioning tumors may show little response to medical therapy
- Radiation therapy may be used for tumors resistant to medical therapy or for surgical remnants from subtotal resections
- Therapy may include linear accelerator radiotherapy, stereotactic radiotherapy or radiosurgery
- Single fraction versus fractionated radiosurgery are options (Pituitary 2017;20:489)
- Several studies have evaluated tumor response to temozolomide therapy, particularly in aggressive pituitary tumors (Front Neurol 2021;12:700007, Rev Endocr Metab Disord 2020;21:263)
- Peptide receptor radionuclide therapy with lutetium oxodotreotide has been reported (Endocr Pathol 2019;30:118, Endocr Connect 2019;8:528, J Endocr Soc 2021;5:bvab133)
- These tumors are usually resected as small fragments; however, occasionally they can be removed in a single piece that allows assessment of margins
- Frozen sections are discouraged
- Most cases do not require intraoperative consultation but in unusual situations (e.g., where the differential diagnosis is inflammation), touch preps can be used for cytologic assessment without compromising residual tissue that is retained for proper immunohistochemical analysis
- Architecture varies from solid sheets to nests within a fibrovascular stroma (Endocr Pathol 2022;33:6)
- Some tumors, usually gonadotroph tumors, form pseudorosettes around vascular channels
- Dyscohesive growth is characteristic of sparsely granulated somatotroph tumors (it is associated with loss of E-cadherin expression)
- Most tumor cells are epithelioid but some can be spindle shaped; this is a feature of some immature Pit1 lineage tumors
- Most tumors have a uniform nuclear morphology with stippled chromatin, inconspicuous nucleoli and moderately abundant cytoplasm
- Cells may be classified as acidophilic, basophilic or chromophobic based on tinctorial differences; this usually correlates with content of hormone containing secretory cells (i.e., densely versus sparsely granulated)
- Crooke hyaline change is characterized by large chromophobic or eosinophilic cells with a glassy hyaline appearance (due to accumulation of keratin filaments); it can be seen in neoplastic and nonneoplastic corticotrophs
- Oncocytic change is a feature of some gonadotroph tumors where it tends to be variable; diffuse oncocytosis is a feature of acidophil stem cell tumors
Contributed by Sylvia L. Asa, M.D., Ph.D.
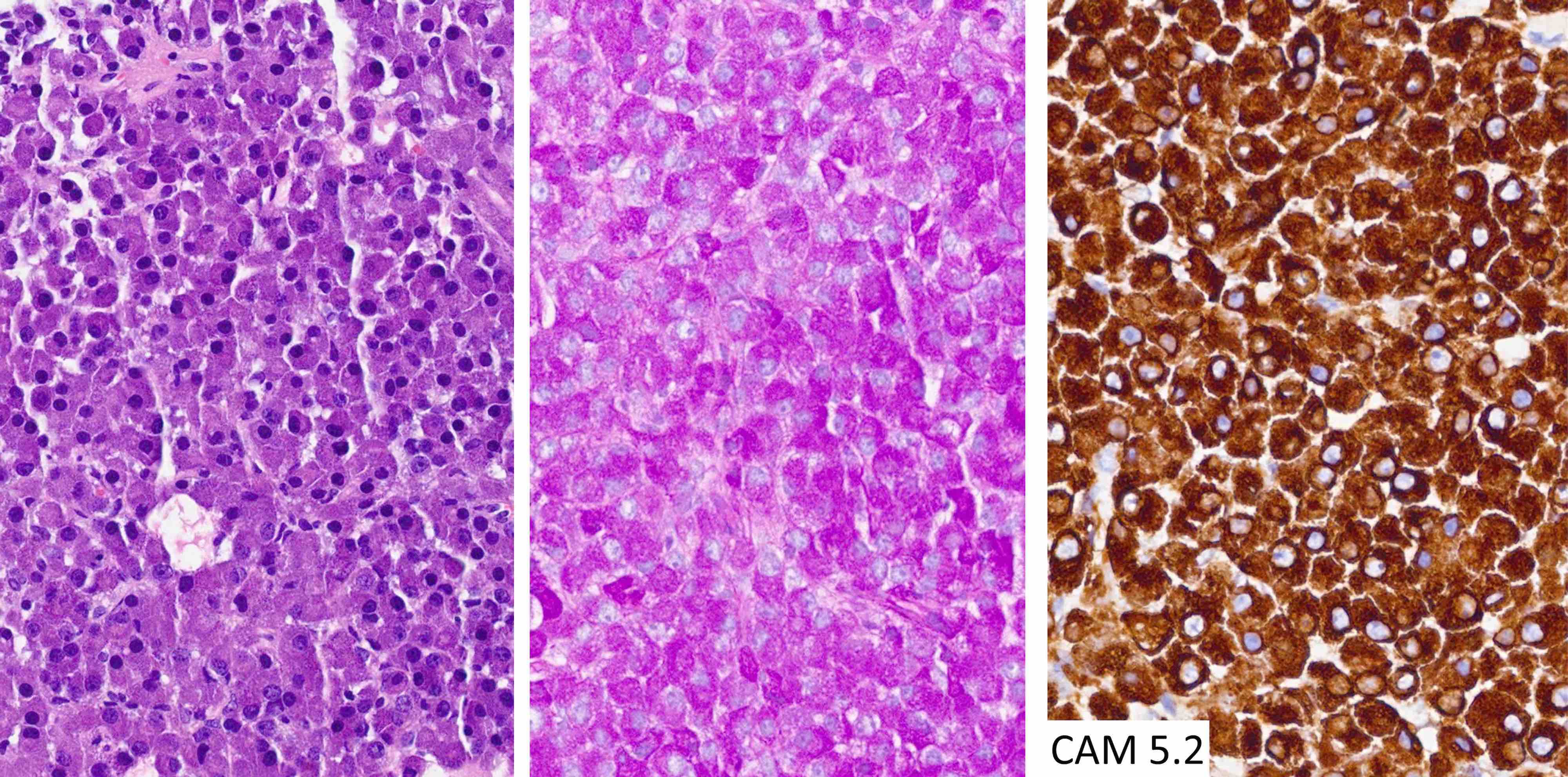
Densely granulated corticotroph tumor
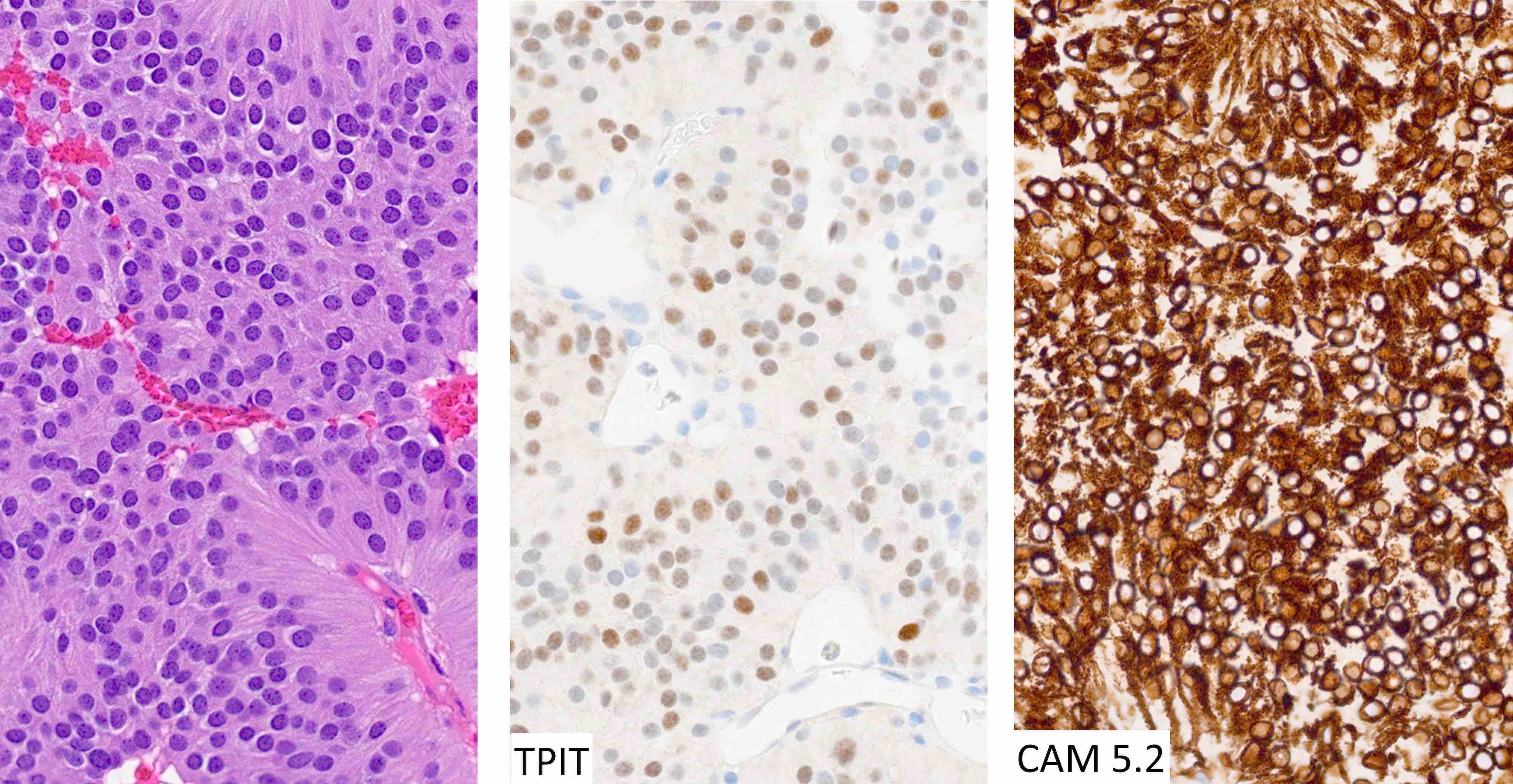
Sparsely granulated corticotroph tumor
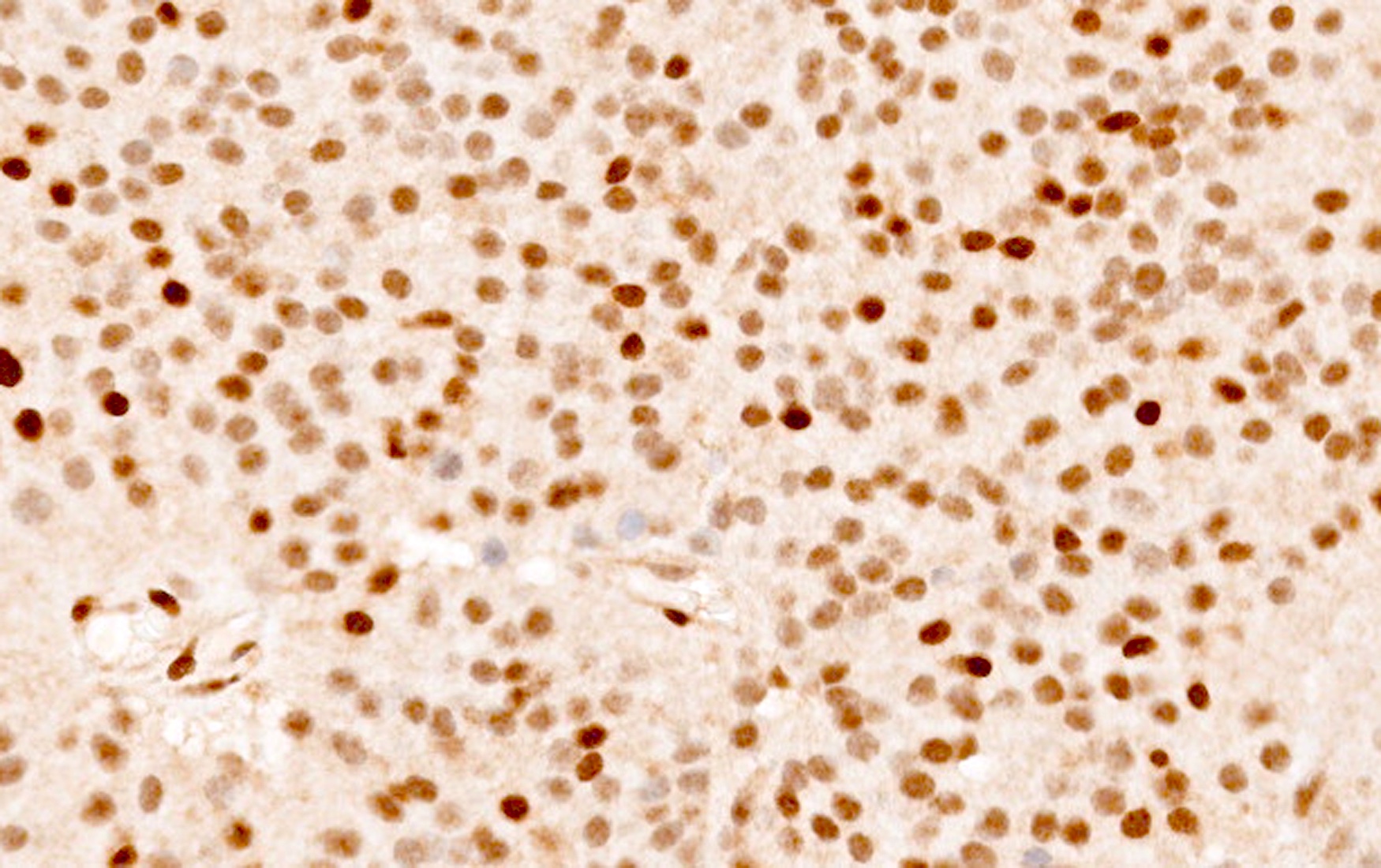
p27 in silent corticotroph tumor
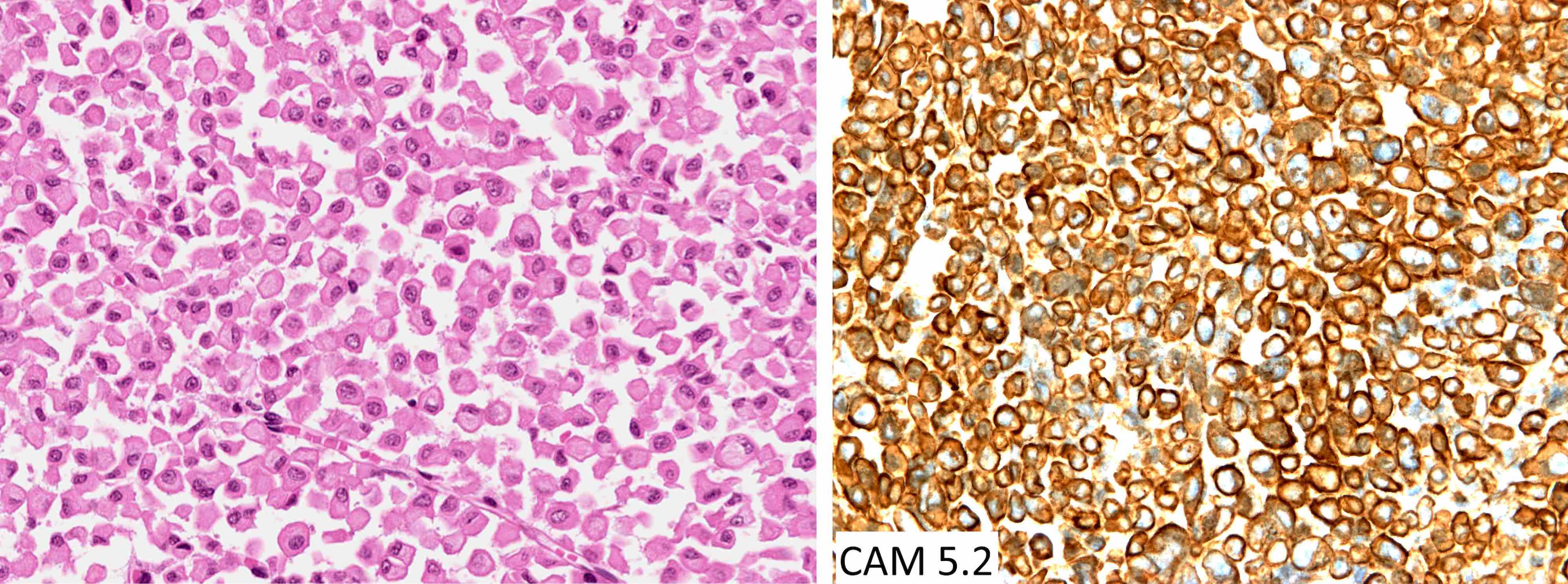
Crooke cell tumor
Densely granulated somatotroph tumor
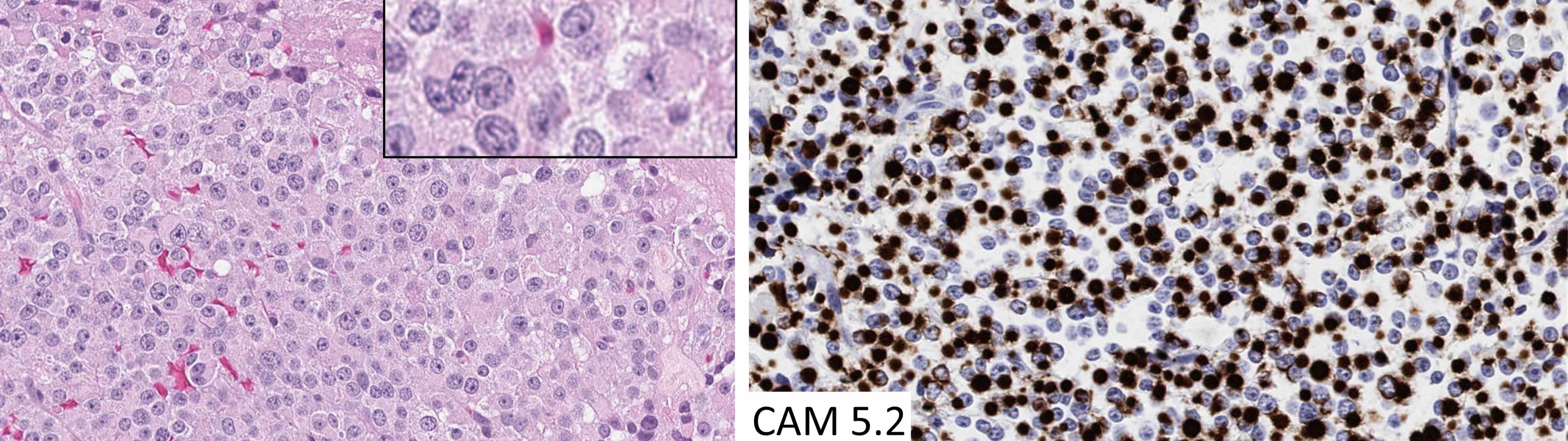
Sparsely granulated somatotroph tumor
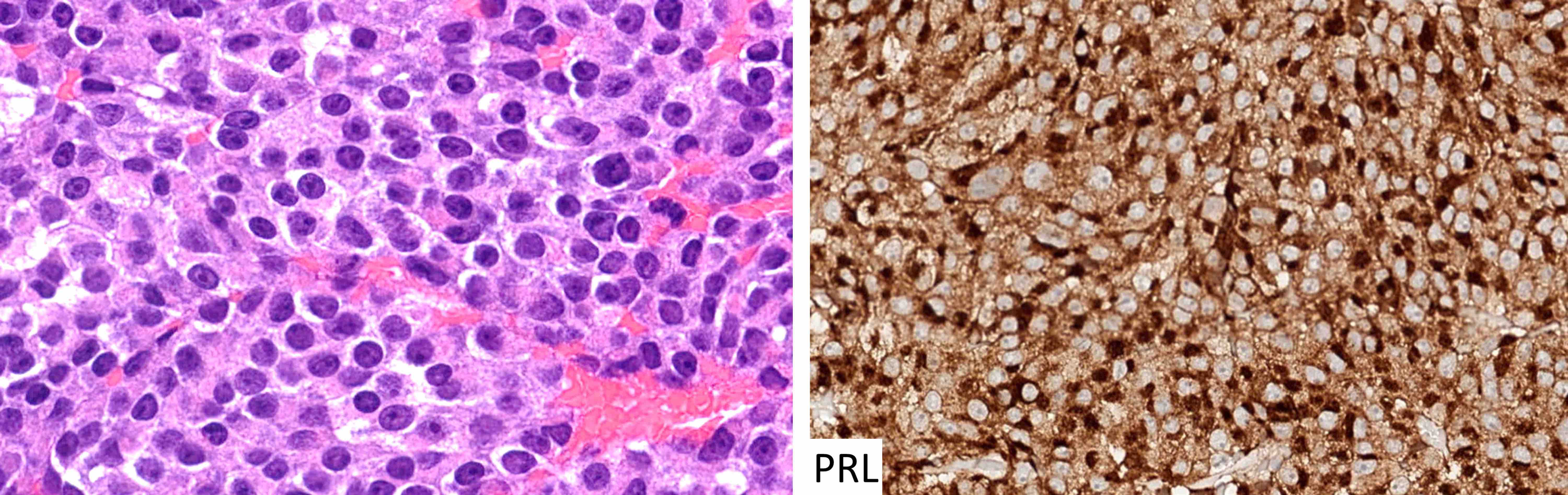
Sparsely granulated lactotroph tumor
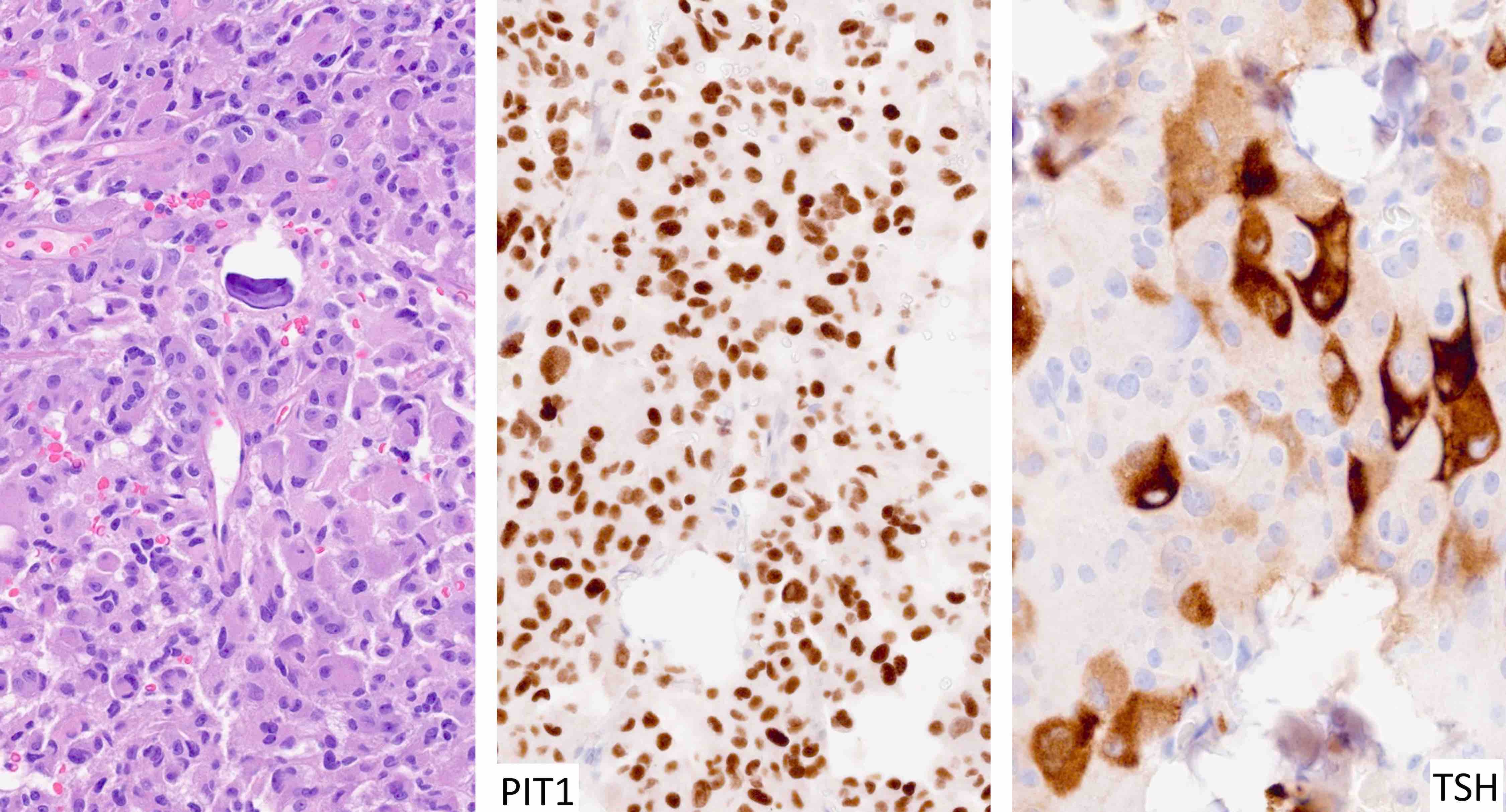
Thyrotroph tumor
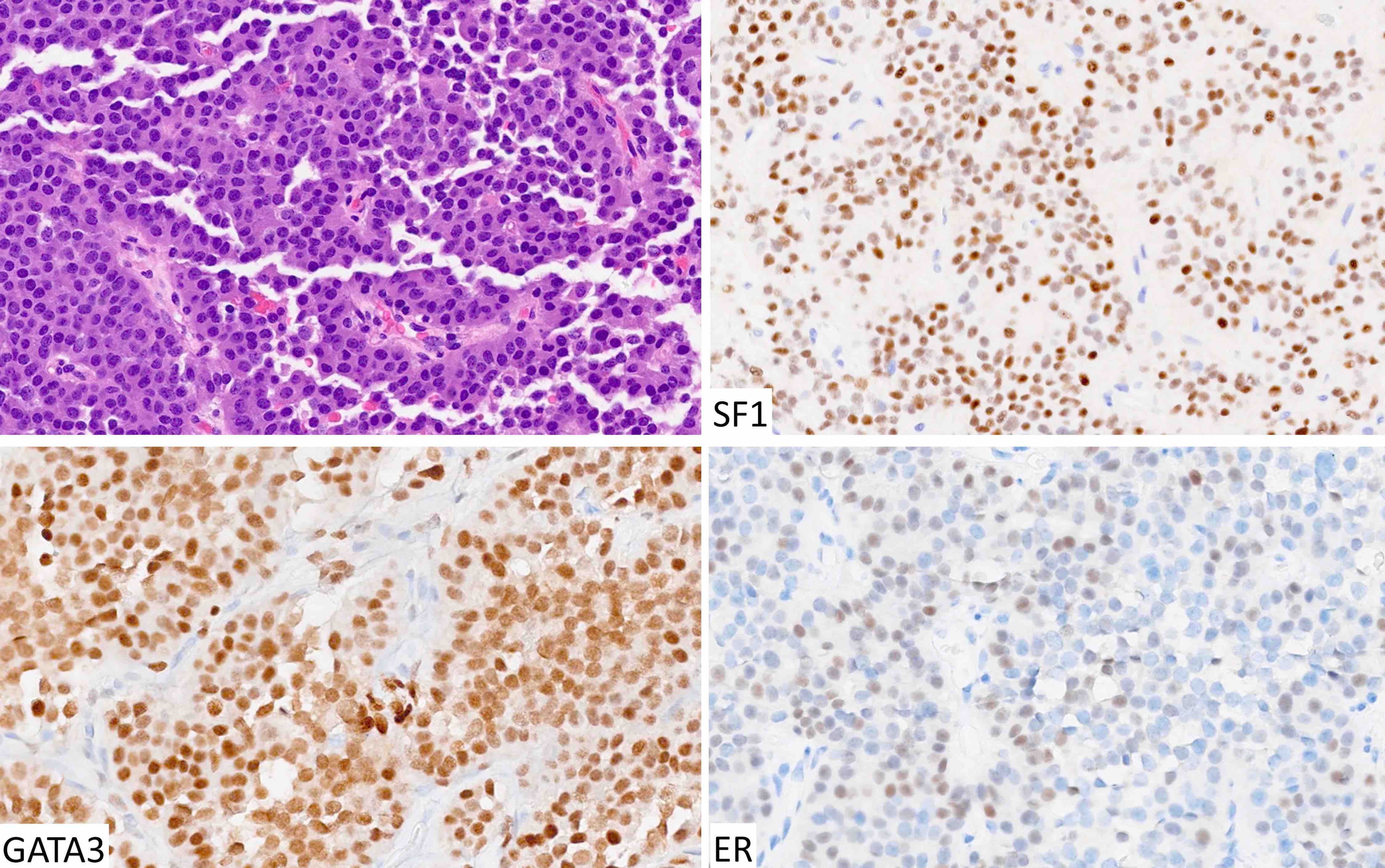
Gonadotroph tumor
- Normal pituitary has mixed cell types on smear preparation whereas tumors show uniform morphology and cell type
- Tumors produce cellular smear with discohesive small round blue cells
- Some tumors have specific cytologic atypia (e.g., fibrous bodies of sparsely granulated somatotroph tumors, Crooke hyaline of Crooke cell tumors)
Contributed by Michael Punsoni, M.D.
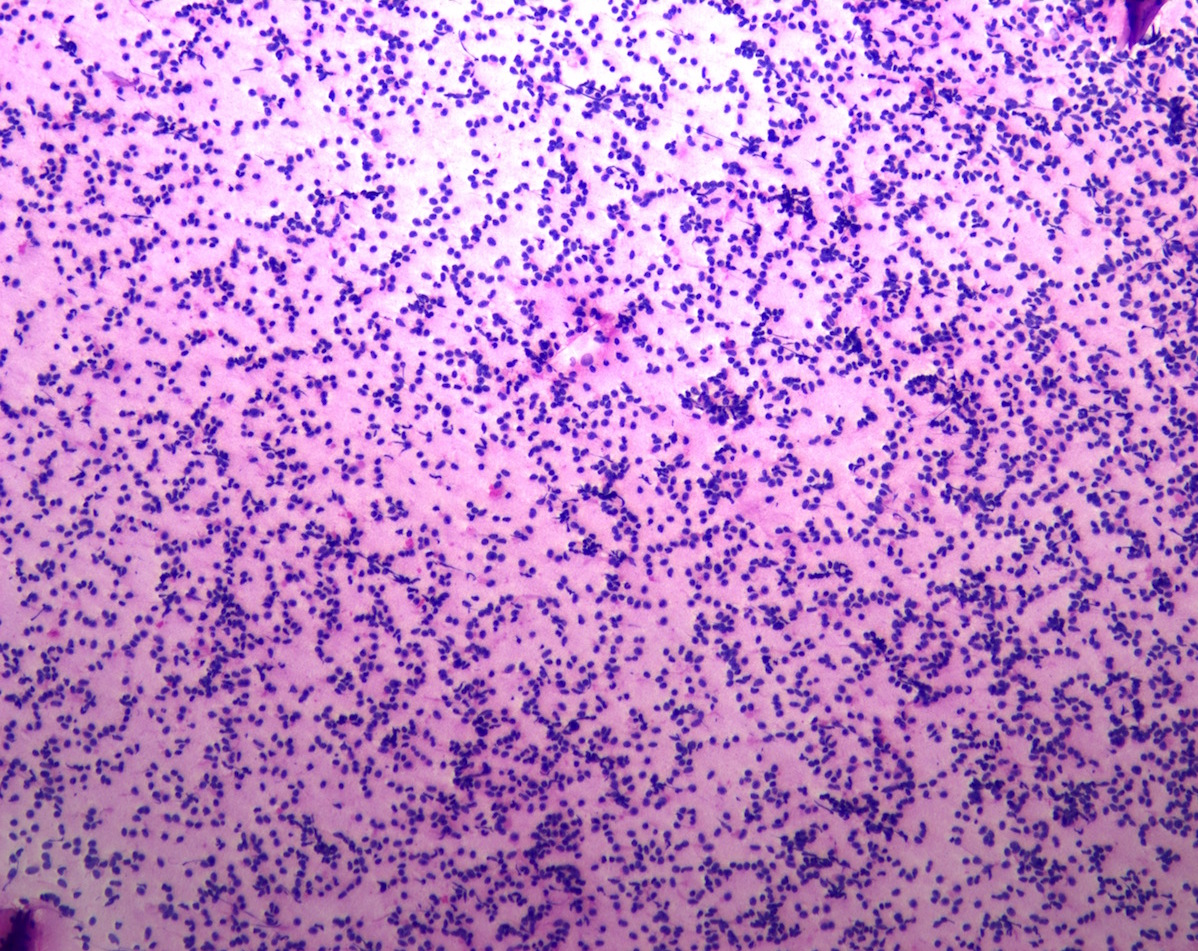
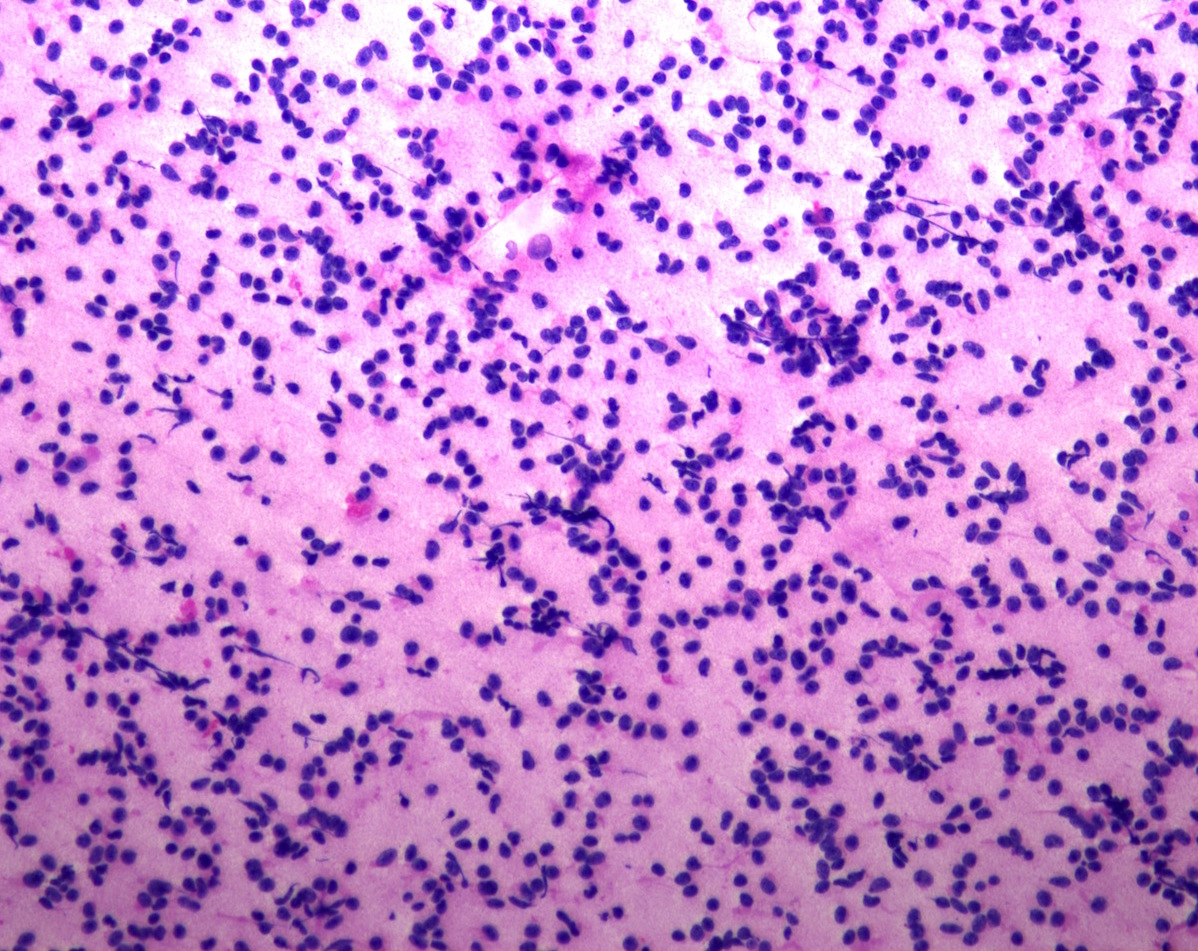
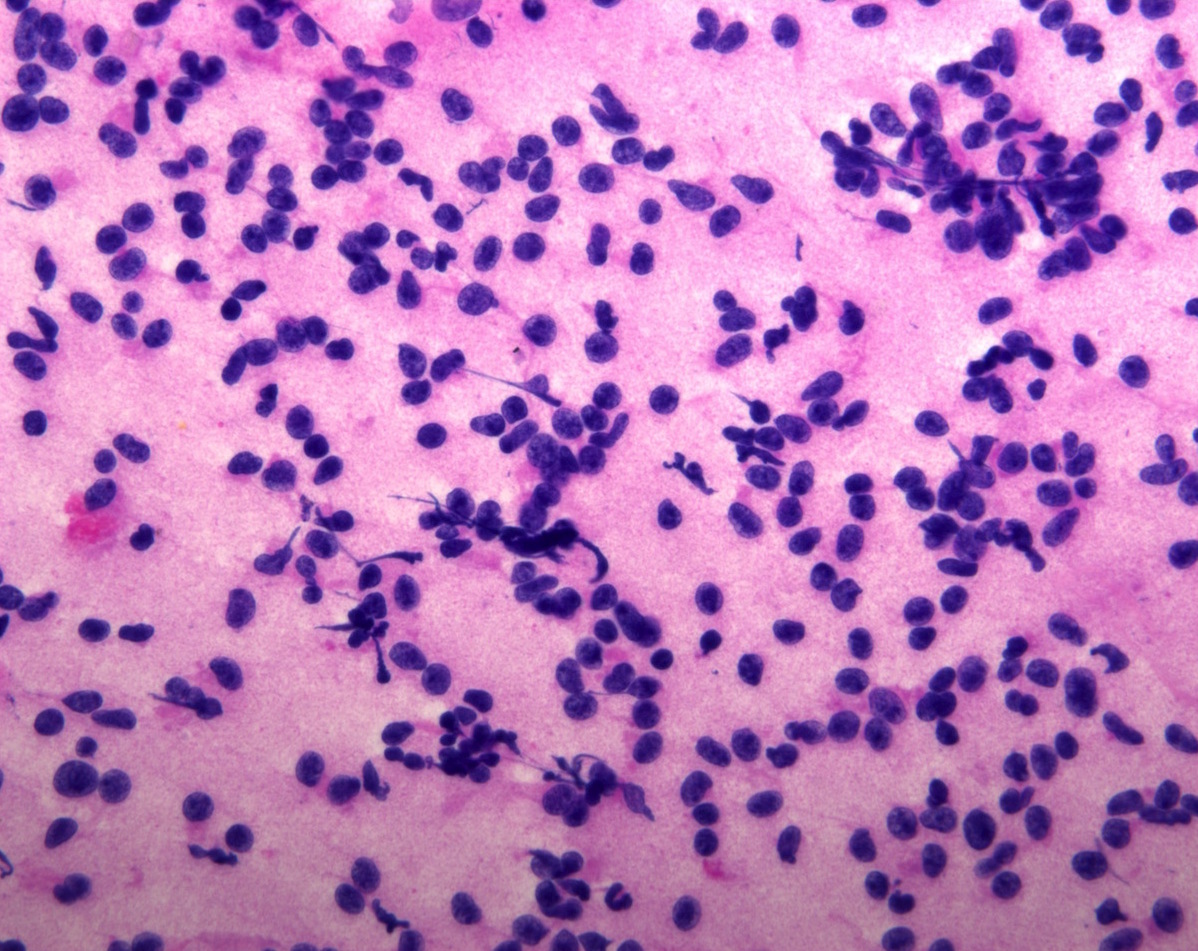
Touch prep showing a cellular population of discohesive small round blue cells
- All pituitary neuroendocrine tumors express INSM1, synaptophysin and chromogranin; positivity for chromogranin A is variable
- Stains for pituitary transcription factors (Pit1, Tpit, SF1, ER and GATA3) and hormones provide the basis for tumor classification (see Diagrams / tables)
- Reticulin staining is useful to distinguish normal acinar architecture, enlarged acini of hyperplasia versus disrupted staining pattern in neoplasms
- Perinuclear CAM 5.2 staining pattern is a feature of densely granulated somatotroph tumors, mammosomatotroph tumors and mature plurihormonal Pit1 lineage tumors
- Paranuclear CAM 5.2 positive fibrous bodies are a conspicuous feature present in > 70% of tumor cells of sparsely granulated somatotroph tumors
- Occasional fibrous bodies can be seen in poorly differentiated Pit1 lineage tumors and acidophil stem cell tumors
- Diffuse and strong cytoplasmic CAM 5.2 staining is characteristic of corticotroph tumors
- Ring-like CAM 5.2 positivity is characteristic of Crooke cell tumors (Brain Pathol 2012;22:443)
- Ki67 immunolabeling often used to characterize tumors with elevated proliferative indices
- Antimitochondrial antibody staining is a feature of oncocytic tumors, including some gonadotroph tumors and the characteristically oncocytic acidophil stem cell tumor
- GFAP: positive in ependymomas
- CK7: positive in choroid plexus tumors and metastatic lesions
- CK20: positive in metastatic lesions
- TTF1: positive in tumors of posterior lobe pituicytes including conventional pituicytoma, granular cell pituicytoma, oncocytic pituicytoma (formerly known as spindle cell oncocytoma) and ependymal pituicytoma (formerly known as sellar ependymoma) (Am J Surg Pathol 2013;37:1694, Endocr Pathol 2014;25:436)
- Mostly supplanted by immunohistochemistry; however, electron microscopy may still play a role in characterization of unusual pituitary tumors and identification of giant mitochondria in acidophil stem cell tumor and spheridia in immature Pit1 lineage tumors (formerly known as silent subtype III adenomas) (Brain Pathol 2012;22:443)
Images hosted on other servers:

Ultrastructural features of some rare pituitary adenomas
- No specific molecular characteristics in routine clinical diagnostic workup
- Most pituitary tumors are sporadic; minority are part of hereditary or familial syndromes
- Somatic mutations in GNAS and USP8 genes have been found in ~40% of sporadic somatotroph tumors and 30 - 60% of sporadic corticotroph tumors, respectively (Endocr Pathol 2021;32:3)
- Pituitary, transsphenoidal resection specimen:
- Mature plurihormonal Pit1 lineage tumor (see synoptic report)
- Synoptic report:
- Clinical features: functional
- Hormone excess (specify): acromegaly, hyperthyroidism
- Tumor size (from imaging): greatest dimension was 3.8 cm
- Additional dimensions: 3.5 x 2.4 cm
- Received: in formalin
- Specimen integrity: fragmented
- Specimen size: 2.4 x 2.2 x 0.4 cm
- Histologic features:
- Reticulin: disrupted
- PAS: negative
- Infiltrating tumor: cannot be determined
- Immunohistochemistry:
- Tpit is negative
- Pit1 is positive
- ER is focally positive
- GATA3 is focally positive
- SF1 is negative
- ACTH is negative
- Growth hormone is positive
- Prolactin is focally positive
- Beta TSH is focally positive
- Alpha subunit is diffuse positive
- Beta FSH is negative
- Beta LH is negative
- Keratin (CAM 5.2) is diffuse
- Ki67 labeling index is 5%
- Tumor type: pituitary neuroendocrine tumor
- Subtype: mature plurihormonal Pit1 lineage tumor
- Additional pathologic findings
- Nontumorous adenohypophysis: present
- Neurohypophysis: not identified
- Gross description: Labeled with the patient's name and hospital number and received in formalin as multiple fragments of tan-pink friable tissue that measure 2.4 x 2.2 x 0.4 cm in aggregate. The specimen is submitted in toto in 1 cassette.
- Normal adenohypophysis:
- Normal acini by reticulin staining
- Pituitary nodular hyperplasia:
- Enlarged acini by reticulin staining
- Metastatic neuroendocrine tumor:
- Pituicytoma:
- Ependymoma:
- Positive for GFAP
- Negative for synaptophysin
- Pituitary blastoma:
- Early childhood tumor, small blastema-like cells, true rosettes, large glandular structures, DICER1 mutation
- Diabetes insipidus is often associated with PitNETs
- GNAS mutations have been described in pituitary tumors
- Most PitNETs are negative for pituitary hormones
- Most PitNETs are part of hereditary or familial syndromes
- The diagnosis of metastatic behavior can be predicted by morphological or immunohistochemical features
Comment Here
Reference: Pituitary neuroendocrine tumor (PitNET)
The chromophobic tumor in the image above has the keratin pattern shown on the right. Which of the following is correct?
- This patient had acromegaly
- This patient had Cushing disease
- This patient would not have had hyperprolactinemia
- This tumor is likely to respond to first generation somatostatin analogues
- This tumor stains for Tpit
Comment Here
Reference: Pituitary neuroendocrine tumor (PitNET)
- Pleomorphic xanthoastrocytoma (PXA) is a circumscribed astrocytic glioma with large multinucleated cells, spindle cells and xanthomatous cells with frequent eosinophilic granular bodies and rich reticulin network
- It is characterized by BRAF p.V600E mutation or other MAPK pathway gene alterations combined with homozygous CDKN2A / B deletion
- Can be graded CNS WHO grade 2 or 3
- Pleomorphic xanthoastrocytoma (PXA) is a circumscribed astrocytoma with superficial location often involving leptomeninges, most often located in temporal lobe
- Classic histology with pleomorphic, xanthomatous and multinucleated tumor cells, often with eosinophilic granular bodies and rich reticulin network
- It shows characteristic genetic alterations including BRAF p.V600E mutation and homozygous deletion of CDKN2A / B
- Assigned CNS WHO grade 2 and 3
- Not recommended: pleomorphic xanthoastrocytoma with anaplastic features; anaplastic pleomorphic xanthoastrocytoma for CNS WHO grade 3
- ICD-O: 9424/3 - pleomorphic xanthoastrocytoma
- ICD-11: 2A00.0Y & XH99U2 - other specified gliomas of brain & pleomorphic xanthoastrocytoma
- PXA accounts for < 0.3% of primary central nervous system (CNS) tumors, with an annual incidence of < 0.7 cases per 100,000 population (Neuro Oncol 2019;21:v1)
- M = F; mean age at diagnosis is 26.3 years
- It typically develops in children and young adults; however, older patients may be affected (J Neurooncol 2012;110:99)
- Majority of tumors occur supratentorially, most commonly in temporal lobes
- Often superficially located with involvement of the overlying leptomeninges (Brain Pathol 2015;25:575)
- There are rare case reports of PXAs involving
- Cerebellum (Surg Neurol 2007;68:89, Acta Neurochir (Wien) 2004;146:1241)
- Spinal cord (J Neurosurg Spine 2006;5:72)
- Pituitary stalk (Neurol India 2018;66:1193)
- Pineal gland (Brain Tumor Pathol 2013;30:242)
- Retina (Am J Surg Pathol 1999;23:79)
- Intraventricular location (Cureus 2023;15:e35975, Cureus 2018;10:e2665)
- It is thought to originate from subpial astrocytes, supported by its frequent superficial location in the cerebral hemispheres and by the fact that both share similar ultrastructural features (Radiol Med 2022;127:1134)
- BRAF mutation or other MAPK pathway alterations combined with homozygous deletion of CDKN2A / B (Brain Pathol 2019;29:85, Brain Pathol 2021;31:20)
- PXAs with TERT promoter mutations are associated with shorter overall survival (Acta Neuropathol 2013;126:907, Acta Neuropathol Commun 2022;10:5)
- PXAs rarely harbor TP53 mutations (Neurogenetics 2001;3:159)
- Alterations in other genes (including SMARCB1, BCOR, BCORL1, ARID1A, ATRX, PTEN, FANCA, FANCD2, FANCI, FANCM, PRKDC, NOTCH2, NOTCH3, NOTCH4 and BCL6) have been described with unclear pathogenetic significance in PXAs (Brain Pathol 2019;29:85)
- No known specific etiology
- Rare cases have been reported in patients with
- Neurofibromatosis type 1 (Clin Neuropathol 2012;31:152, Clin Neuropathol 2012;31:54)
- DiGeorge syndrome (J Neurooncol 2011;102:509)
- Familial melanoma astrocytoma syndrome (Clin Neuropathol 2017;36:213)
- Down syndrome (Front Oncol 2019;9:277)
- Sturge-Weber syndrome (Pediatr Radiol 2005;35:910)
- Many patients present with long history of epileptic seizures
- Other symptoms that are related to raised intracranial pressure include headaches, nausea, vomiting, dizziness, diplopia and somnolence (CNS Oncol 2019;8:CNS39, Cureus 2023;15:e35975)
- Magnetic resonance imaging (MRI) is the most effective imaging method
- Diagnosis is confirmed by biopsy or surgical excision; gross total resection may not be achieved in deeply situated or widely infiltrative tumors
- Diagnostic molecular DNA methylome profile
- Next generation sequencing (NGS)
- Chromosome microarray
- Computed tomography (CT) scan
- Variable appearance (hypodense, hyperdense or mixed)
- May be well or poorly demarcated
- With strong and sometimes heterogenous contrast enhancement
- Tumor cysts are usually hypodense
- Calcification is rare
- Due to superficial location, it may cause scalloping of overlying bone
- MRI scan
- Variable / heterogeneous appearance, often with cystic component
- Solid component is either hypointense or isointense to gray matter on T1 weighted images
- Hyperintense or mixed signal on T2 weighted and fluid attenuated inversion recovery (FLAIR) images
- Cystic component is isointense to cerebrospinal fluid
- Postcontrast enhancement is moderate to strong
- Adjacent edema is not pronounced (Sci Rep 2018;8:14275)
Contributed by Shayan Anwar, M.B.B.S.
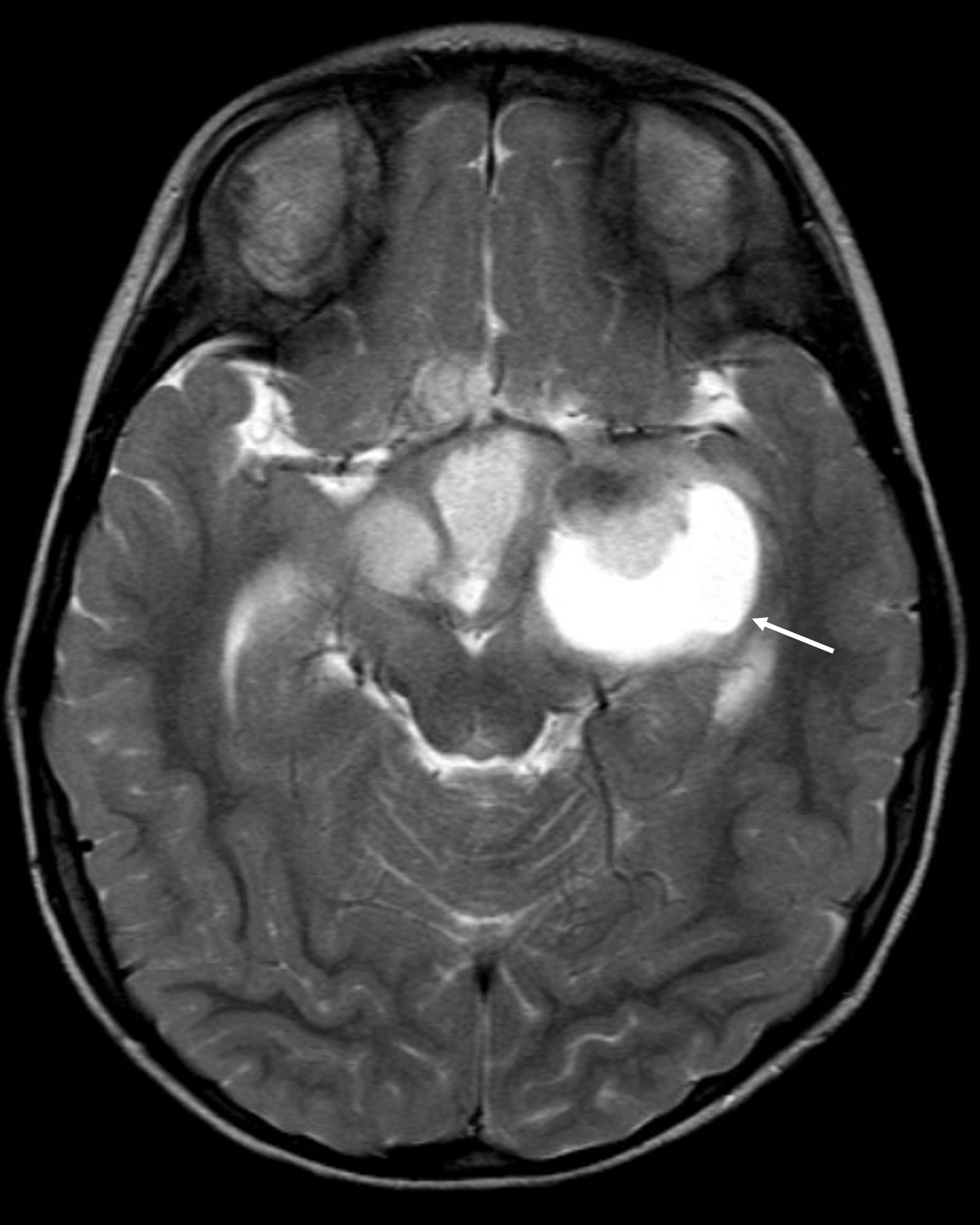
Axial T2 weighted MRI

Coronal FLAIR MRI

Axial T1 weighted precontrast MRI

Axial T1 weighted postcontrast MRI

Axial apparent diffusion coefficient (ADC)
- PXA frequently recurs and is associated with decreased survival compared to other circumscribed low grade gliomas in children and young adults
- Malignant progression is more common in PXA than in other RAS / MAPK driven circumscribed low grade gliomas (Brain Pathol 2015;25:575)
- Extent of surgical excision is the most significant prognostic factor associated with recurrence (Cancer 1999;85:2033)
- Prognostic significance of CNS WHO grading in PXA has been confirmed by 2 large independent studies (Neuro Oncol 2020;22:1474, Brain Pathol 2021;31:20)
- MAPK pathway gene alterations, particularly BRAF p.V600E as well as homozygous deletion of CDKN2A or CDKN2B, are not associated with tumor grade or prognosis (Brain Pathol 2018;28:172, Acta Neuropathol 2011;121:397)
- TERT promoter alteration is associated with more aggressive phenotype and a marker of anaplastic transformation (World Neurosurg 2019;126:624, Acta Neuropathol Commun 2022;10:5)
- 18 year old woman with a pleomorphic xanthoastrocytoma in the left cerebellum (World Neurosurg 2020;139:577)
- 22 and 43 year old men presented with repeated generalized seizures (Medicine (Baltimore) 2020;99:e22478)
- 28 year old man had a sudden limb twitch and visited a local hospital (Int J Gen Med 2020;13:1581)
- 32 year old woman with a mass in her medial right temporal lobe (Diagn Cytopathol 2022;50:E240)
- 33 year old man with an intraventricular anaplastic pleomorphic xanthoastrocytoma (Cureus 2023;15:e35975)
- 48 year old healthy man presented with progressive right upper limb weakness (Cureus 2022;14:e23060)
- For CNS WHO grade 2 PXAs, early intervention with complete surgical excision is treatment of choice and can be followed by wait and watch strategy (Lancet Oncol 2017;18:e315)
- For CNS WHO grade 3 PXAs, postoperative radiotherapy may be offered in adults (Lancet Oncol 2017;18:e315)
- Radiotherapy may play a role for residual or recurrent disease
- Traditional systemic therapies have shown limited benefit in treating PXA
- Targeted BRAF inhibition, alone or in combination with other treatments including MEK inhibition, has been associated with marked radiographic responses in BRAF mutated PXA (CNS Oncol 2019;8:CNS39)
- Variable population of spindled to pleomorphic tumor cells with long and coarse cytoplasmic processes and fibrillary fragments with vessels
- Multinucleated giant cells and tumor cells with microvacuolization resembling xanthomatous cells can be seen (Diagn Cytopathol 2017;45:339)
- Sometimes eosinophilic granular bodies are observed
Contributed by Mohammad Khurram Minhas, M.B.B.S.
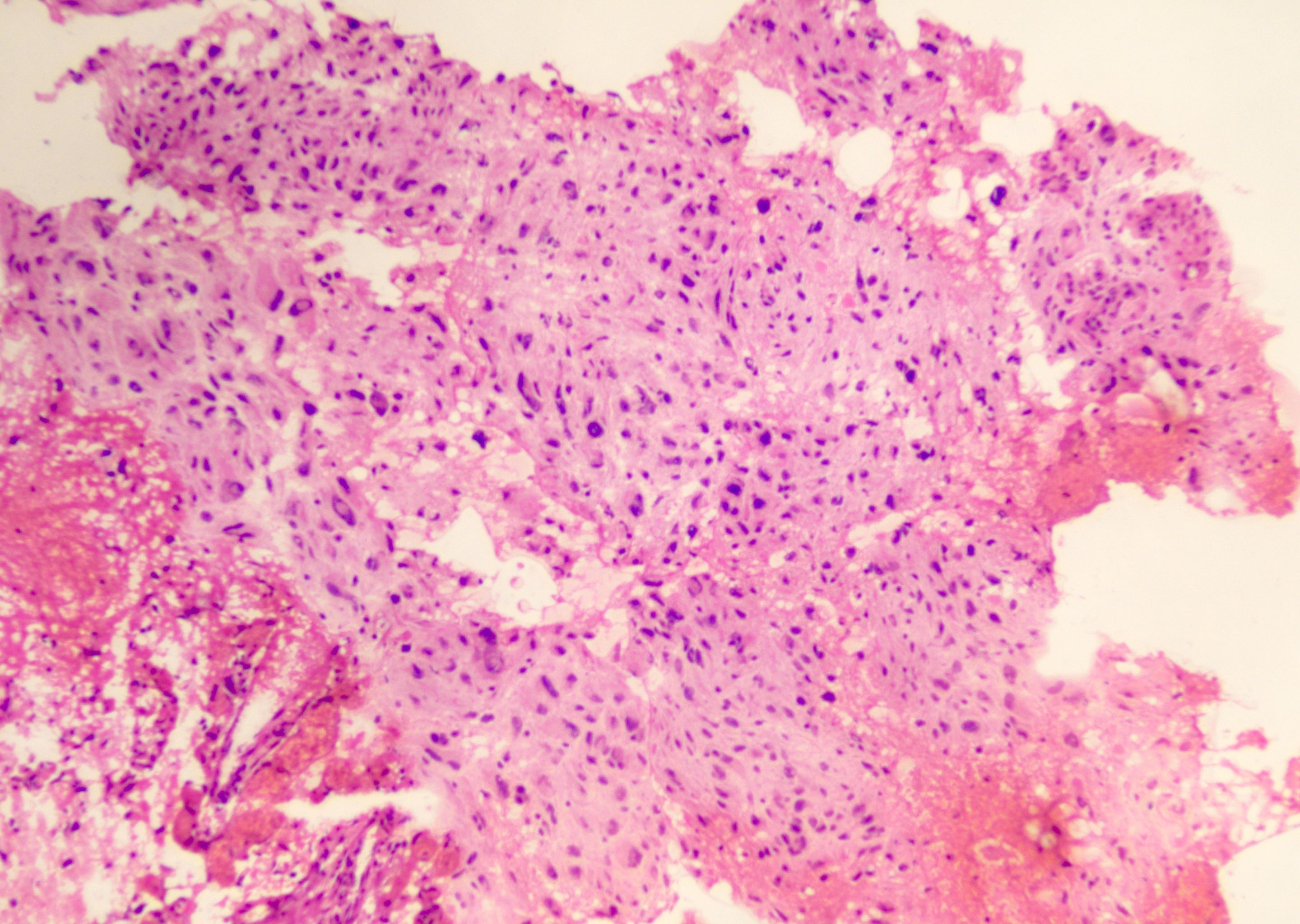
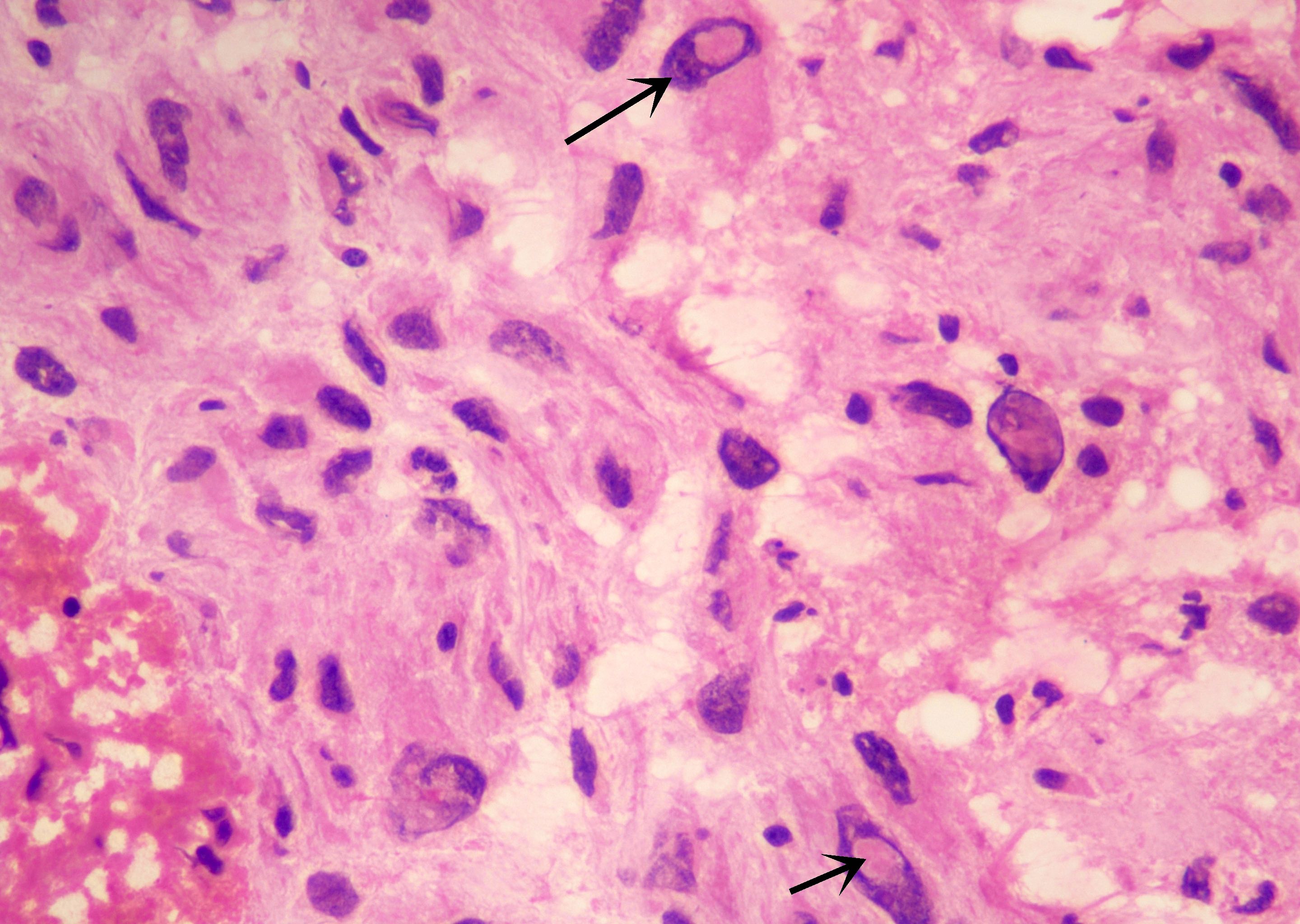
Glial tumor with pleomorphic cells
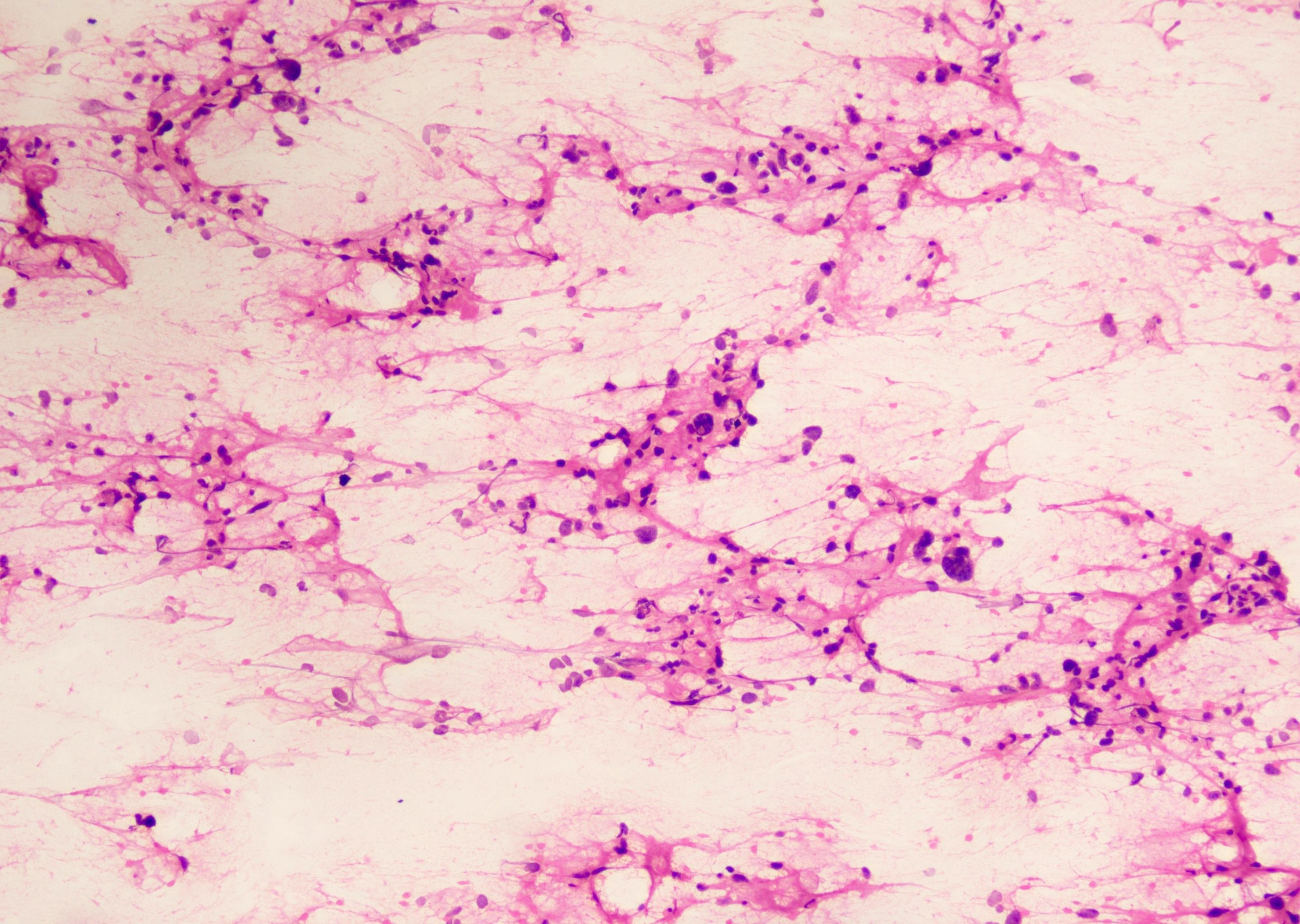

Glial tumor exhibiting fibrillary processes

Intranuclear inclusions
- Mostly solid, noninfiltrative growth pattern; however, microscopic infiltration can be seen at the periphery
- Mixture of spindled, epithelioid, pleomorphic and multinucleated tumor cells, with some showing vacuolated cytoplasm (xanthomatous cells)
- Intranuclear inclusions, prominent nucleoli and perivascular lymphocytic infiltration are common (Cancer 1999;85:2033)
- Frequent eosinophilic granular bodies and dense reticulin deposition
- CNS WHO grading
- CNS WHO grade 2 is assigned to tumors with < 5 mitoses/10 high power fields (HPF)
- Necrosis is commonly seen in tumors with high mitotic activity; however, its significance in isolation is unclear
- Microvascular proliferation is uncommon (Cancer 1999;85:2033)
- CNS WHO grade 3 PXAs may demonstrate less pleomorphism and more diffusely infiltrative pattern
- In CNS WHO grade 3 tumors, a mean Ki67 labeling index of 15% has been reported, whereas it is generally < 1% in CNS WHO grade 2 tumors
- Common cytological patterns of anaplasia include monomorphic small cells, fibrillary morphology, epithelioid and rhabdoid morphology (Brain Pathol 2018;28:172)
Contributed by Aisha Memon, M.B.B.S.

Pleomorphic spindle cells

Epithelioid tumor cells
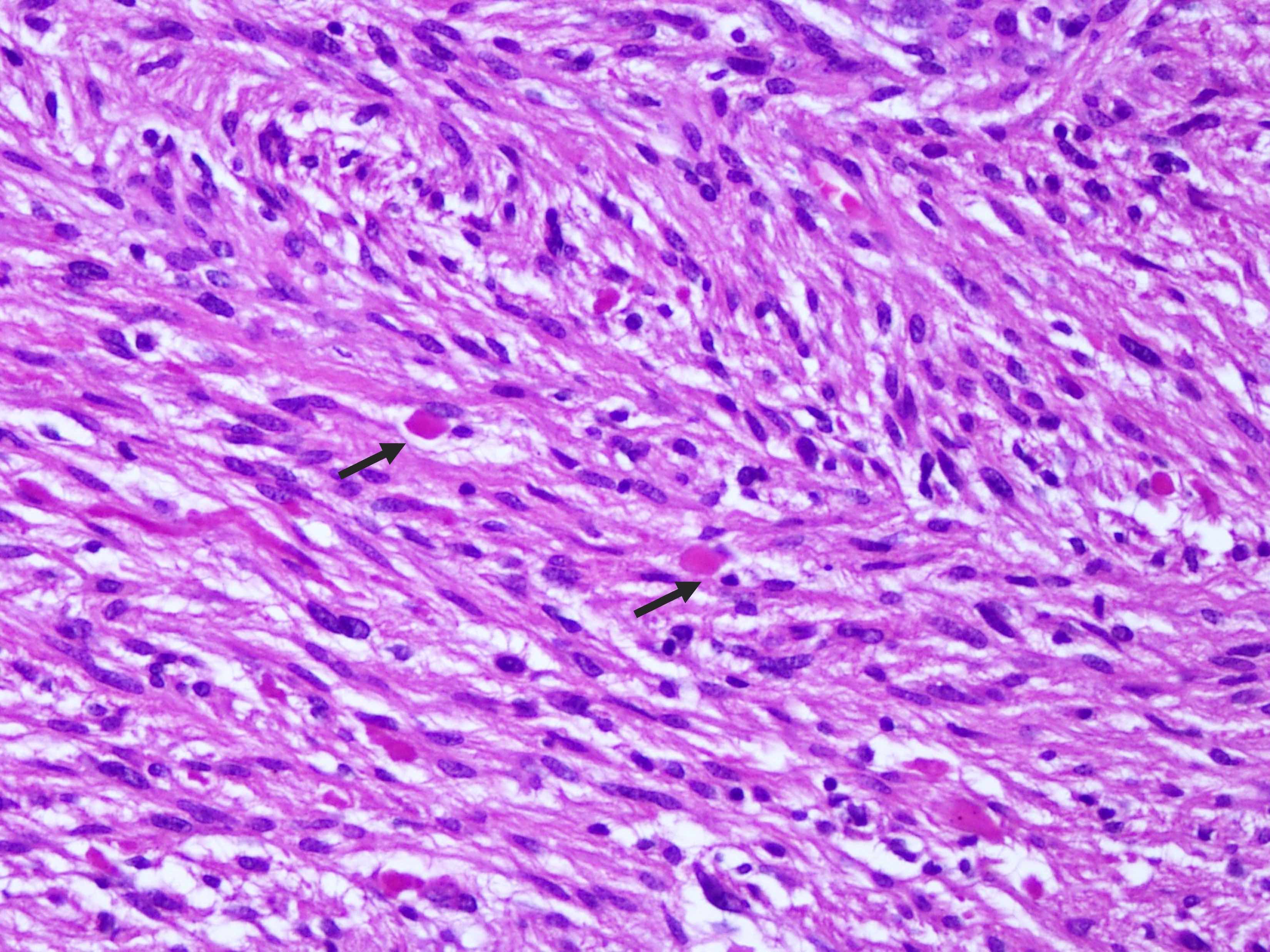
Eosinophilic granular bodies
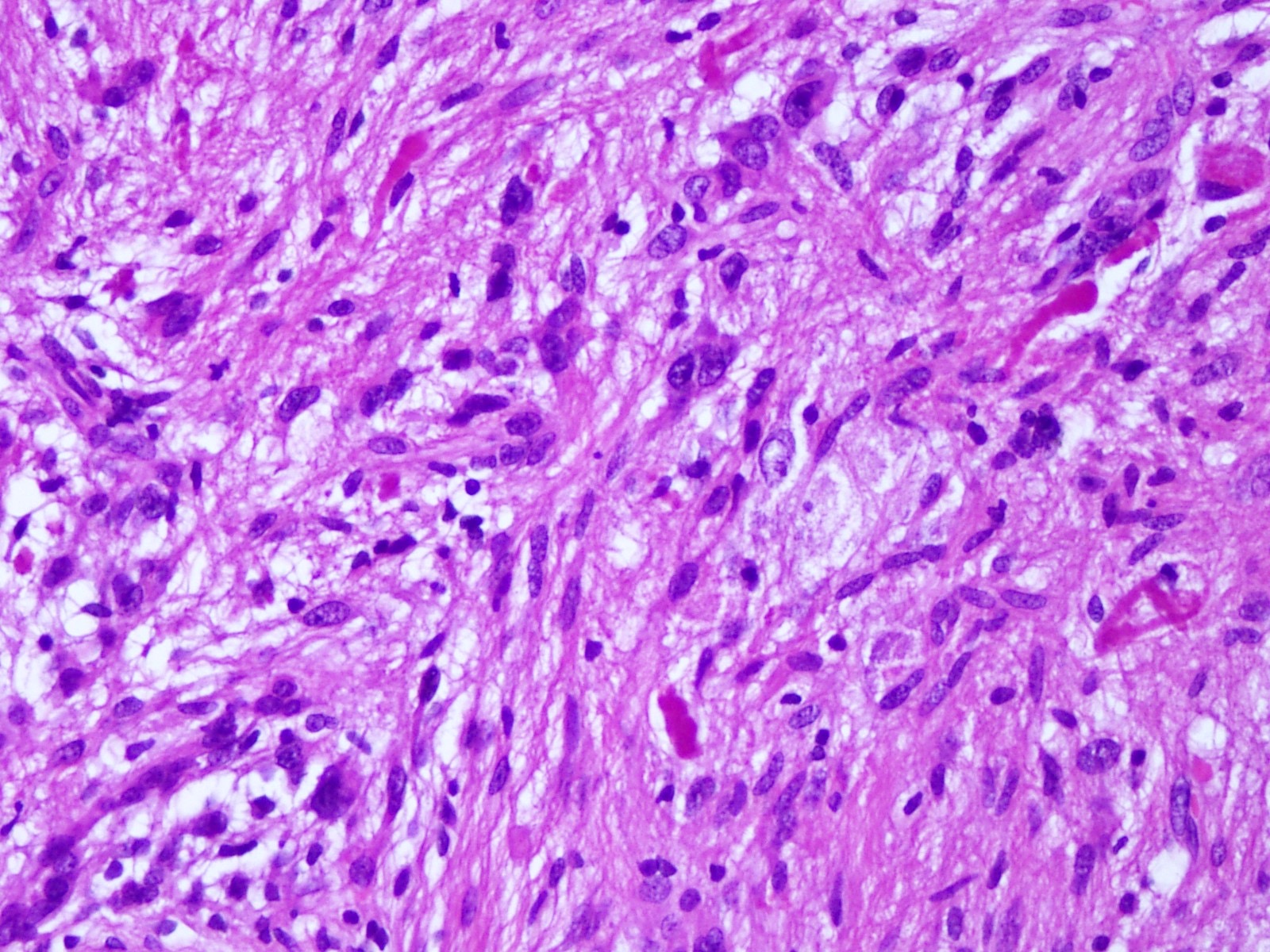
Rosenthal fibers


Xanthomatous cells

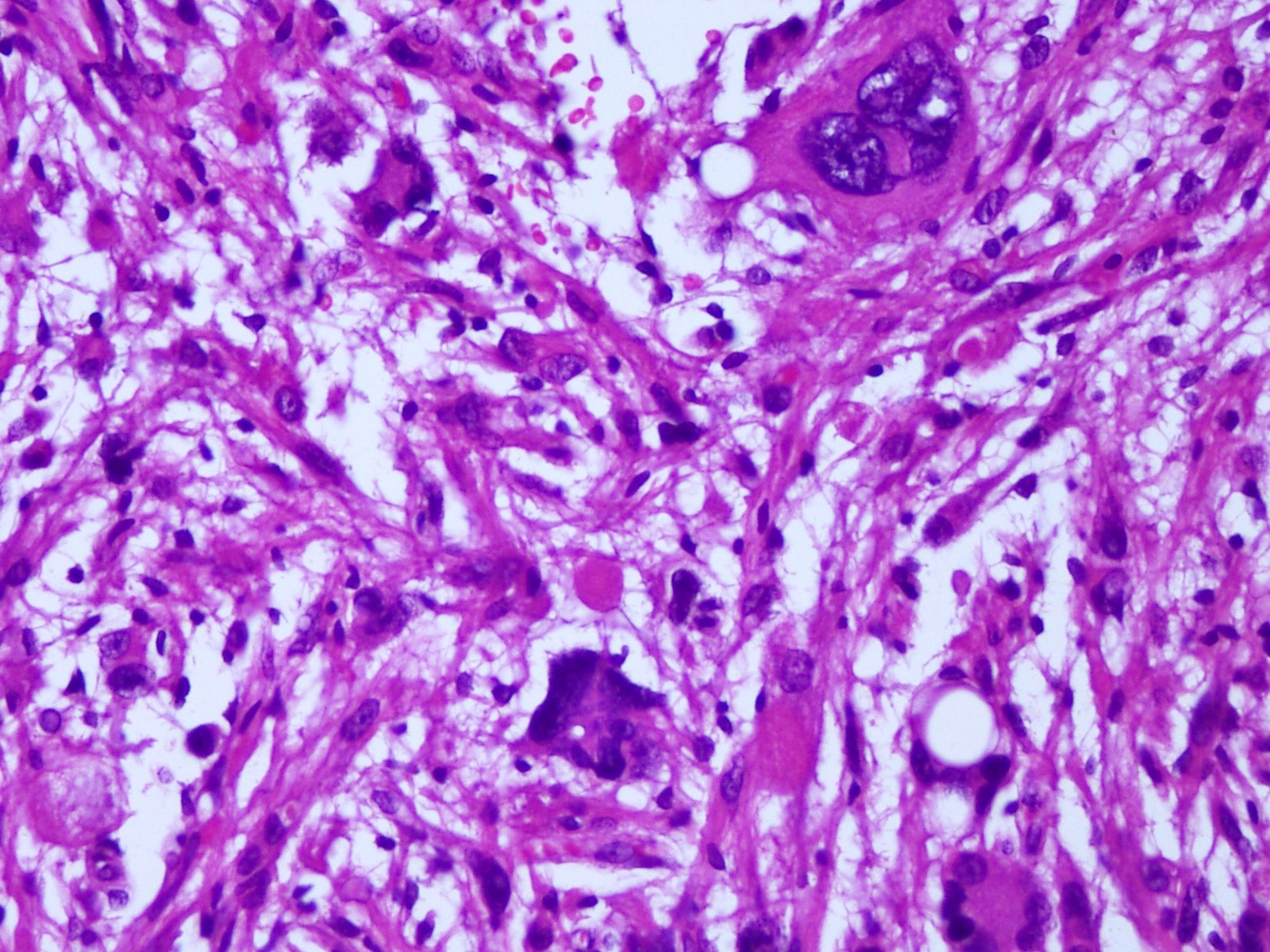
Multinucleated tumor cells

GFAP
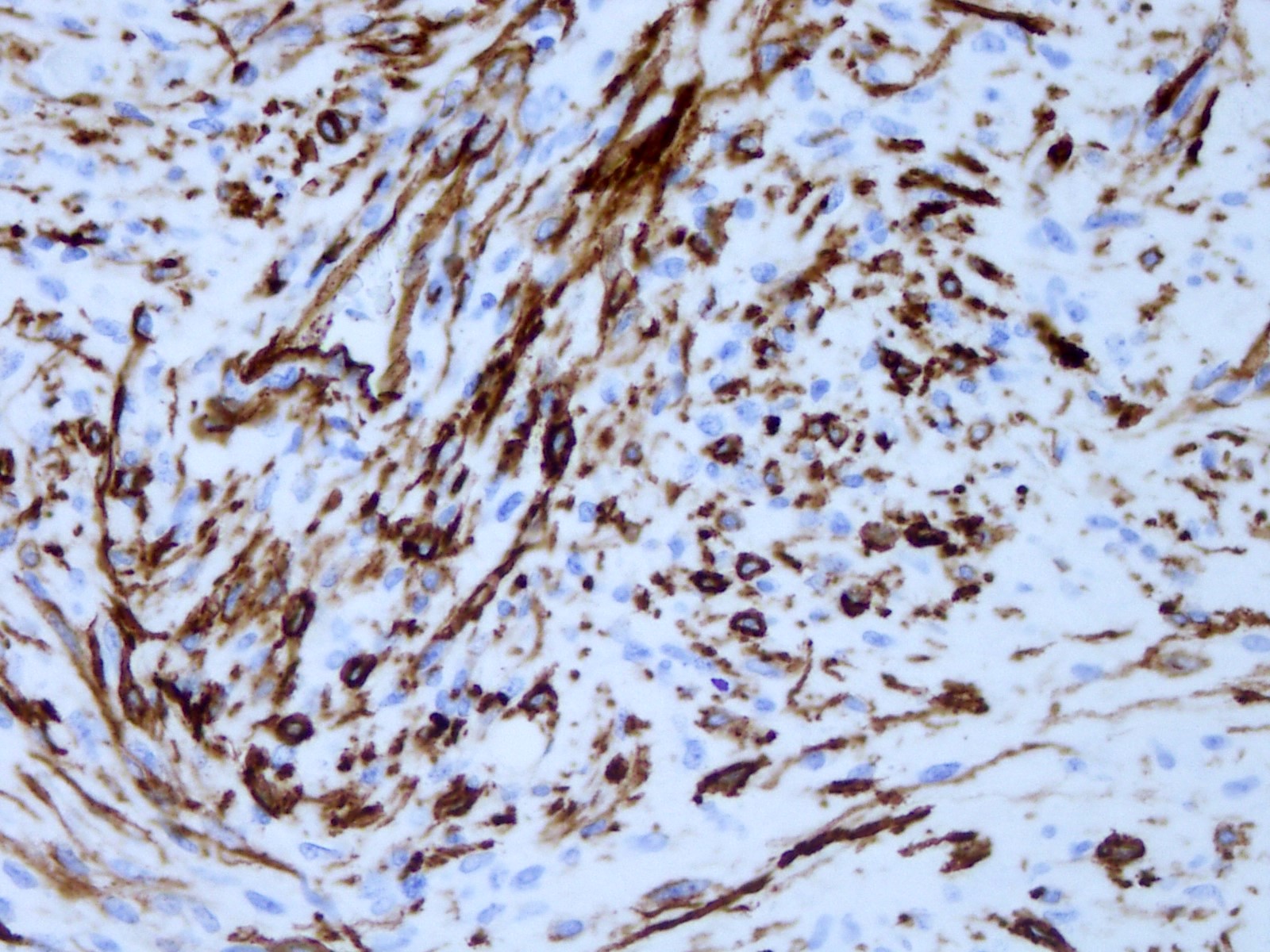
CD34

S100
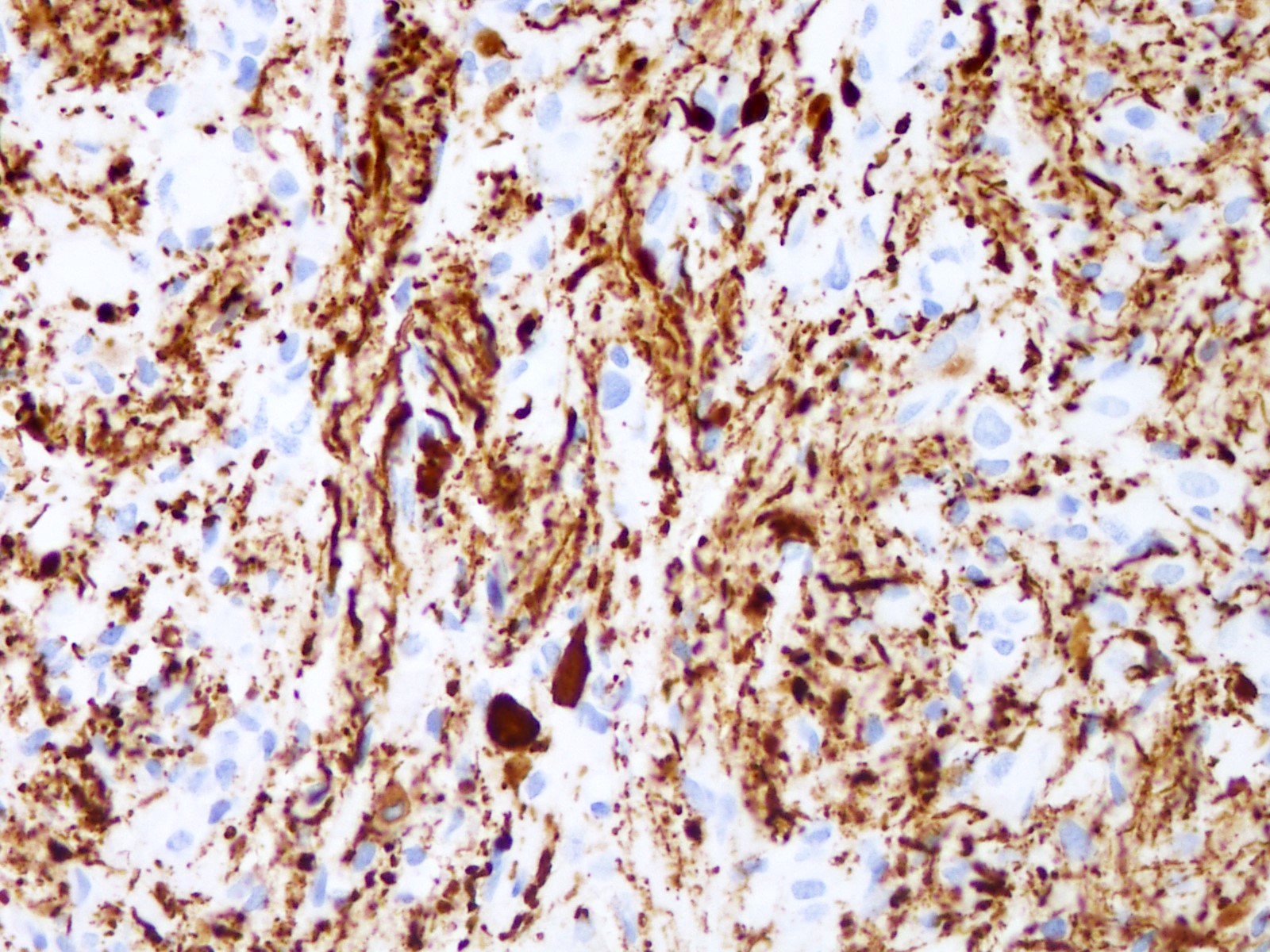
Neurofilament
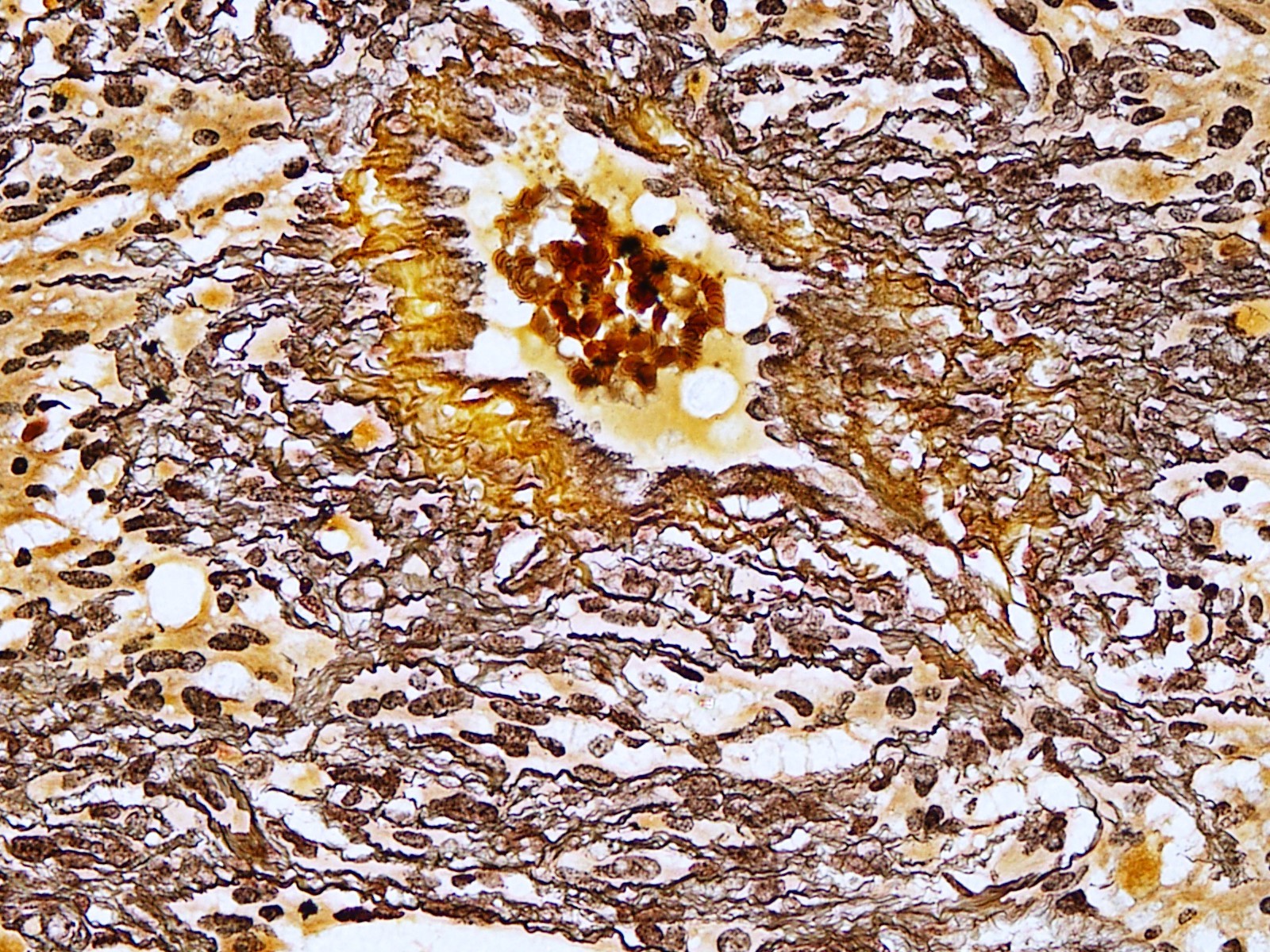
Reticulin stain
Images hosted on other servers:
Pleomorphic xanthoastrocytoma
- GFAP, Olig2 and S100 are usually diffusely positive (Cancer 1999;85:2033)
- Most tumors express CD34 (Acta Neuropathol 2003;105:358)
- Reticulin stain demonstrates rich reticulin network around individual tumor cells
- PAS stain is helpful for identifying eosinophilic granular bodies
- Focal expression of neuronal markers (synaptophysin, neurofilament, class III β tubulin and MAP2) is seen in some PXAs (Am J Surg Pathol 2002;26:479)
- BRAF p.V600E (VE1 antibody) expression is seen in 60 - 80% of cases (Acta Neuropathol Commun 2013;1:20)
- Focal SMARCB1 (INI1) loss has been reported in rare cases that underwent malignant transformation to tumors resembling atypical teratoid / rhabdoid tumors (Brain Pathol 2018;28:172)
- Negative for IDH1 R132H staining
- TP53 usually demonstrates wild type staining pattern (weak expression in rare tumor cells) (Neurogenetics 2001;3:159)
- Presence of relatively few organelles, abundant filaments and microtubules, are characteristic of astrocytes
- Existence of basal laminas in the extracellular space surrounding the surface of the tumor cells and the presence of localized cytoplasmic densities associated with the plasma membrane adjacent to the basal lamina resembling hemidesmosomes (Cancer 1983;52:2055)
- MAPK pathway gene alterations
- Virtually all PXAs harbor alterations in a gene of the MAPK pathway, causing aberrant activation of this pathway
- The most frequent is the missense mutation of BRAF p.V600E occurring in 60 - 80% of PXAs (Acta Neuropathol 2011;121:397, PLoS One 2011;6:e17948, Brain Pathol 2019;29:85, Brain Pathol 2021;31:20)
- In BRAF p.V600E negative tumors, other MAPK pathway gene alterations are found, mostly involving other BRAF alterations (non-p.V600e mutations, non-KIAA1549::BRAF fusions), NTRK1, NTRK2, NTRK3, RAF1 or NF1 (Brain Pathol 2018;28:172)
- CDKN2A or CDKN2B homozygous deletion
- ~94% of PXAs harbor homozygous deletions of CDKN2A / B (Brain Pathol 2019;29:85, Brain Pathol 2021;31:20)
- Combination of BRAF p.V600E mutation and CDKN2A or CDKN2B homozygous deletion is highly suggestive of PXA, although rare cases of ganglioglioma, epithelioid glioblastoma and high grade astrocytoma with piloid features may also have this molecular profile (Acta Neuropathol Commun 2018;6:47, Brain Pathol 2018;28:656, Brain Pathol 2018;28:663, Acta Neuropathol 2018;136:273)
- TERT alterations
- TERT promoter mutations and less commonly, TERT amplification have been found in PXA, more commonly in anaplastic tumors (Acta Neuropathol 2013;126:907)
- Presence of canonical pTERT mutation has been shown to be a robust indicator for poor prognosis in methylation class PXA (Acta Neuropathol Commun 2022;10:5)
- Some PXAs can also harbor gain of chromosome 7 and loss of chromosome 10 (+7 / -10), similar to glioblastomas; however, unlike glioblastomas, PXAs virtually never demonstrate tyrosine kinase receptor gene amplifications (such as EGFR amplification) (Acta Neuropathol 2018;136:793)
Pleomorphic xanthoastrocytoma:
a brief review
- Temporal lobe lesion, gross total resection:
- Pleomorphic xanthoastrocytoma, BRAF V600E mutant, CNS WHO grade 2
- Histological classification: pleomorphic xanthoastrocytoma
- CNS WHO grade: 2
- Molecular information: BRAF V600E mutation, combined with homozygous deletion of CDKN2A / B
- Ganglioglioma:
- Both show presence of eosinophilic granular bodies, lymphocytic infiltration, CD34 expression and BRAF p.V600E mutation
- Presence of true ganglion cells in gangliogliomas and absence of xanthomatous cells
- Absence of reticulin rich network surrounding individual tumor cells in gangliogliomas
- Absence of homozygous deletion of CDKN2A / B in gangliogliomas
- Giant cell glioblastoma:
- Both show gross circumscription, reticulin deposition, marked pleomorphism, multinucleated giant cells and lymphocytic infiltration
- Presence of neuronal antigens in PXAs, which are usually absent in giant cell glioblastoma
- TP53 is mutated in most giant cell glioblastomas (Arch Pathol Lab Med 2003;127:1187)
- Epithelioid glioblastoma:
- Both can show presence of epithelioid cells, frank anaplasia and BRAF p.V600E mutation
- Prior history of a CNS WHO grade 2 PXA can be helpful
- There is a PXA-like epithelioid glioblastoma subset, which clusters by methylation profiling with canonical PXAs with BRAF p.V600E mutation (79%), CDKN2A homozygous deletion (61%) and TERT promoter mutations (30%) (Brain Pathol 2018;28:656)
A 25 year old man presented with history of recurrent headaches and seizures. Magnetic resonance imaging (MRI) scan revealed a solid circumscribed lesion in the temporal cortex with abnormal intensity. Biopsy revealed pleomorphic tumor cells with admixed xanthomatous cells and multinucleated giant cells. Eosinophilic granular bodies were noted along with reticulin rich network. There was no visible mitosis, microvascular proliferation or necrosis. On molecular testing, there was a BRAF p.V600E mutation. What is the most likely diagnosis?
- Epithelioid glioblastoma
- Ganglioglioma
- Giant cell glioblastoma
- Pleomorphic xanthoastrocytoma
- Subependymal giant cell astrocytoma
Comment Here
Reference: Pleomorphic xanthoastrocytoma
- BRAF V600E
- EGFR
- Histone H3
- IDH1
- TERT promoter
Comment Here
Reference: Pleomorphic xanthoastrocytoma
- Indolent cerebral neoplasm characterized by a strong association with seizures in young individuals, diffuse growth patterns, frequent presence of oligodendroglioma-like components, calcifications, CD34 immunoreactivity and MAPK pathway activating genetic abnormalities
- CNS WHO grade 1
- Recently introduced in WHO 2021 classification of CNS tumors
- Recently described, frequently epileptogenic, low grade neuroepithelial tumor (Acta Neuropathol 2017;133:417)
- Typically affects children and young adults
- Characterized by diffuse growth patterns, oligodendroglioma-like cellular components, CD34 immunopositivity and MAPK pathway activating genetic abnormalities
- Generally, exhibits a benign clinical course (Acta Neuropathol 2017;133:417)
- Has been described under the generic designation of long term epilepsy associated tumors (Acta Neuropathol 2014;128:39)
- Not recommended by WHO
- Diffuse glioneuronal tumor
- Diffuse or nonspecific form of dysembryoplastic neuroepithelial tumor
- Massively calcified low grade glioma
- Rare; population based incidence data are unavailable
- Age: mostly occurs in the second and third decades of life (mean age at diagnosis: 22.16 years; range: 4 - 57 years) (Acta Neurol Belg 2023;123:327)
- Slight male predominance (Acta Neurol Belg 2023;123:327)
- Cerebral tumors, usually with cortical and subcortical components (Neuroradiology 2022;64:1255)
- Mostly right sided (64.4%) (Acta Neurol Belg 2023;123:327)
- Majority involve temporal lobes (69%) with frequent involvement of medial / posteroinferior structures (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- Other sites include frontal, parietal and occipital lobes and rarely, third ventricular region (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- Aberrant CD34 expression possibly reflects an origin from developmentally dysregulated neural precursors (Acta Neuropathol 1999;97:481)
- Somatic MAPK pathway activating genetic abnormalities (particularly BRAF mutations or FGFR fusions) definitely drive the pathogenesis of PLNTY; however, the specific mechanisms by which these genetic alterations contribute to the development of PLNTY are not clear (Acta Neuropathol 2017;133:417)
- Unknown predisposing factors
- Germline ATM mutation was identified in a single case, which was associated with progressive disease (J Neuropathol Exp Neurol 2019;78:1100)
Images hosted on other servers:
Prototypical PLNTY
- Typically presents with seizures (intractable epilepsy [47.6%] or a single seizure attack [42.9%]) (Acta Neurol Belg 2023;123:327)
- Less common: headache, dizziness, visual disturbances and mood disorders (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- Neuroimaging: MRI and CT (Acta Neurol Belg 2023;123:327)
- Biopsy
- WHO essential and desirable diagnostic criteria
- Essential diagnostic criteria (presence of the following 6 characteristics)
- Diffuse growth pattern (at least regionally)
- Oligodendroglioma-like components (may be minor)
- Few (if any) mitotic figures
- Regional CD34 expression by tumor cells and by ramified neural cells in associated cerebral cortex
- IDH wild type status
- Unequivocal expression of BRAF p.V600E on immunohistochemical assessment or molecular diagnostic evidence of BRAF V600E mutations, FGFR2 or FGFR3 fusions or potentially other MAPK pathway driving genetic abnormalities
- Desirable diagnostic criteria
- Conspicuous calcification (characteristic, although not constant)
- Absence of 1p / 19q codeletion
- Essential diagnostic criteria (presence of the following 6 characteristics)
- CT
- Often calcified (76.2%) (Neuroradiology 2019;61:1327, Acta Neurol Belg 2023;123:327)
- MRI
- Often superficially situated (Acta Neuropathol 2017;133:417, Neuroradiology 2019;61:1327)
- Circumscribed (76.7%) or blurred outlines (Acta Neurol Belg 2023;123:327)
- Usually cystic and solid (Acta Neurol Belg 2023;123:327, BMC Neurol 2020;20:123)
- Signal heterogeneity with T2 hyperintense or mixed signal, T1 hypointense or mixed signal (Acta Neurol Belg 2023;123:327)
- FLAIR hyperintense
- Majority shows no contrast enhancement (67.5%); patchy or nodular contrast enhancement in 32.5% of cases (Acta Neuropathol 2017;133:417, Acta Neurol Belg 2023;123:327)
- No substantial edema or mass effect (Acta Neuropathol 2017;133:417)
Images hosted on other servers:
FLAIR, CT scan and T1 postcontrast
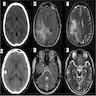
Radiological variability in CT and MRI
- Generally exhibits a benign clinical course (Acta Neuropathol 2017;133:417)
- Amenable to control by gross total resection with relief from seizures or reduced seizure frequency in most cases (Acta Neuropathol 2017;133:417)
- Rarely exhibits local recurrence (11%), tumor progression or malignant transformation (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
- Molecular events associated with less favorable prognosis include germline ATM mutation and somatic mutations involving RB1, TP53, ATRX, PTEN and TEK (J Neuropathol Exp Neurol 2021;80:821, BMC Neurol 2020;20:123, J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
- Radiological contrast enhancement seems to adversely impact prognosis, recurrence and seizure control (Acta Neurol Belg 2023;123:327)
- 13 year old girl with PLNTY mimicking an intracranial granuloma (Indian J Radiol Imaging 2023;33:567)
- 15 year old girl with malignant transformation of PLNTY (Acta Neuropathol 2021;141:123)
- 37 year old man presented with progressive headache (Egyptian Journal of Neurosurgery 2023;38:7)
- 50 year old woman with longstanding epilepsy (Surg Neurol Int 2021;12:470)
- Surgical resection
- Targeted therapies: the MAPK pathway activating genetic alterations are potentially targetable; this provides additional therapeutic options, particularly for tumors that cannot be completely resected or that are located in surgically challenging locations (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821, J Neuropathol Exp Neurol 2019;78:1100)
Images hosted on other servers:
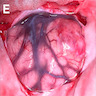
Operative view
- Unencapsulated, indistinctly demarcated from normal brain
- Soft to friable, gray-white masses
- Exhibits both diffuse and compact growth patterns (Acta Neuropathol 2017;133:417)
- Typically shows an oligodendroglioma-like cellular component (Acta Neuropathol 2017;133:417)
- Usually dominant, may be minor
- Ranges from uniform cells with small and round nuclei with perinuclear haloes to cells exhibiting obvious nuclear pleomorphism with more oval or spindled nuclear contours, wrinkled or grooved nuclear membranes and intranuclear pseudoinclusions (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- Astrocytic or morphologically ambiguous elements may also be seen, including fibrillary, spindled and pleomorphic forms (Acta Neuropathol 2017;133:417)
- Subtle perivascular pseudorosettes may be seen
- Calcifications are present in the large majority, ranging from discrete calcospherules to confluent calcific masses with osseous metaplasia (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- Less common / rare: eosinophilic granular bodies, modestly dysmorphic neurons in small numbers and gemistocytic elements (7.7%) (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- Few (if any) mitotic figures; rarely, increased mitotic activity (up to 4 mitoses/10 high power fields) reported (15%) (J Neuropathol Exp Neurol 2021;80:821)
- Generally absent are foci of necrosis, microvascular proliferation, myxoid microcysts, neurocytic or ependymal rosettes, Rosenthal fibers and clearly neoplastic neuronal components (the nosological position of neoplasms otherwise qualifying as PLNTY but having nodular ganglion cell aggregates is unclear) (Acta Neuropathol 2017;133:417)
Contributed by Eman Abdelzaher, M.D., Ph.D. and Jared T. Ahrendsen, M.D., Ph.D.
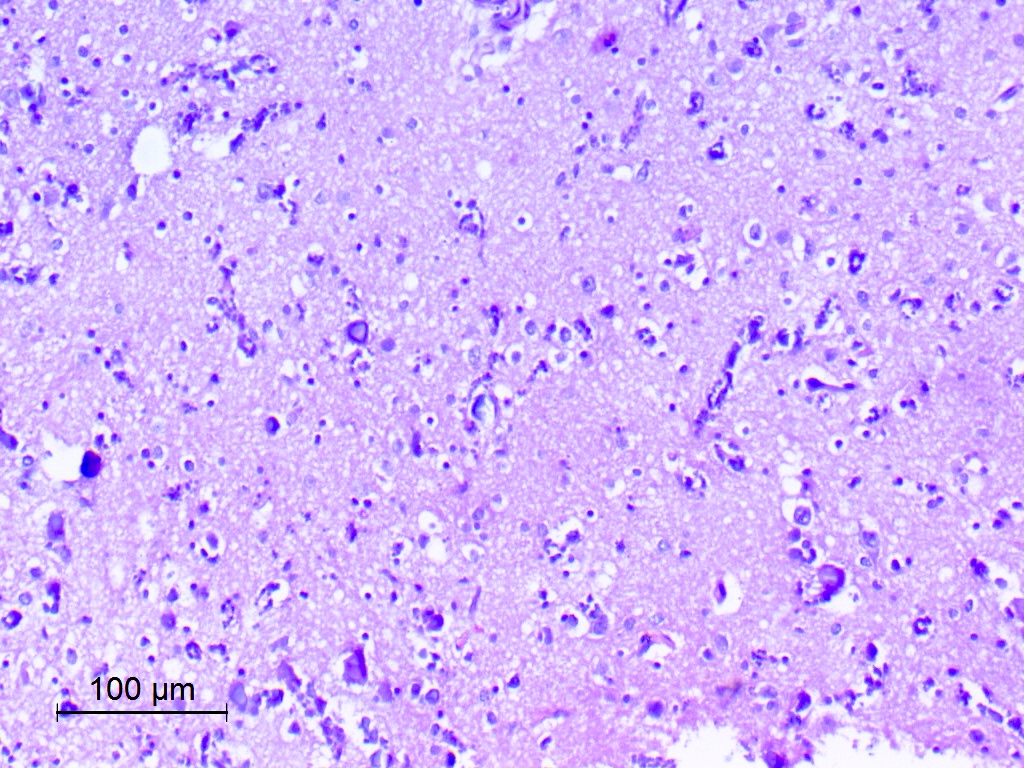
Diffuse growth pattern
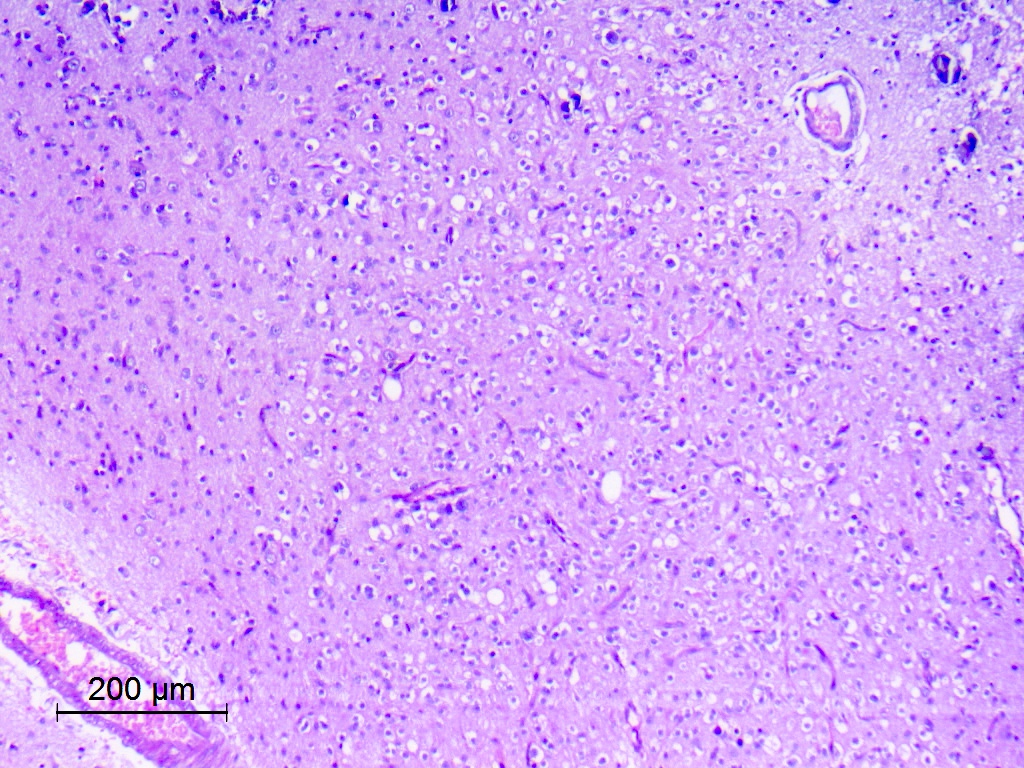
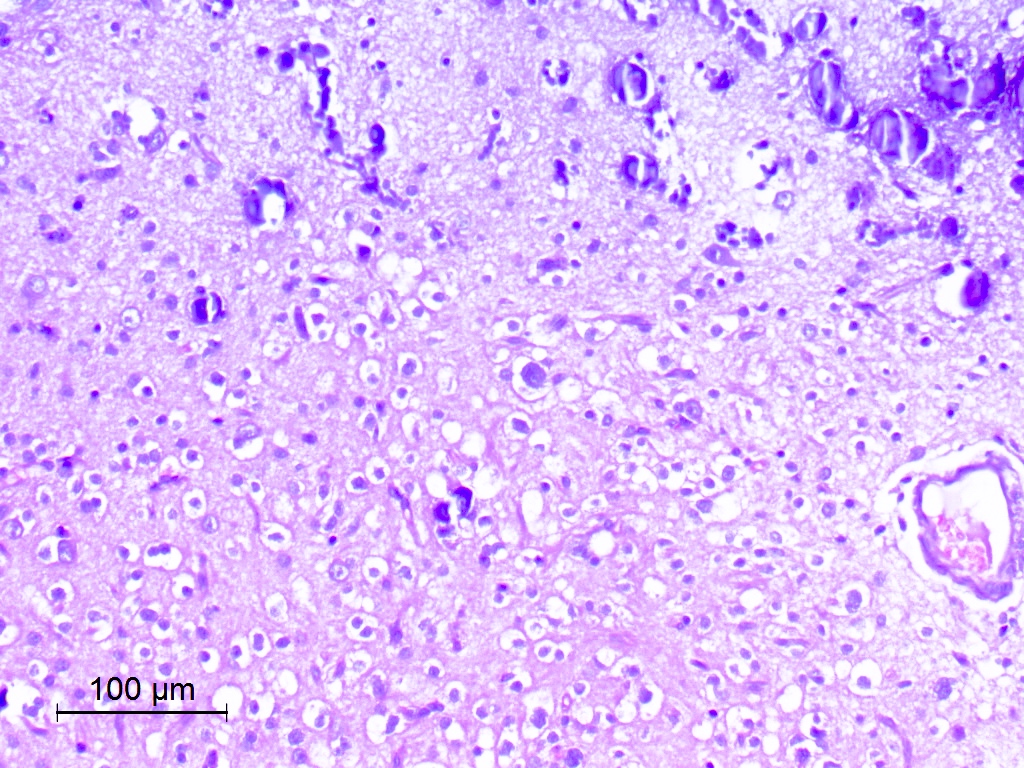
Diffuse growth pattern of oligodendroglioma-like cells
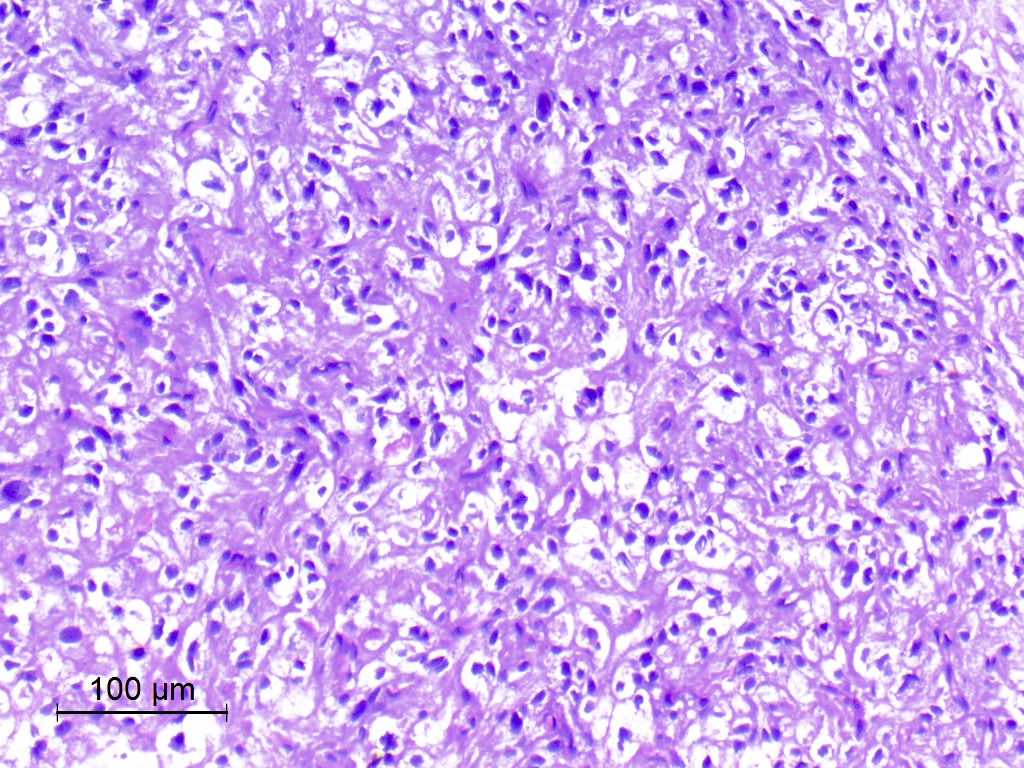
Oligodendroglioma-like cells with mild nuclear pleomorphism
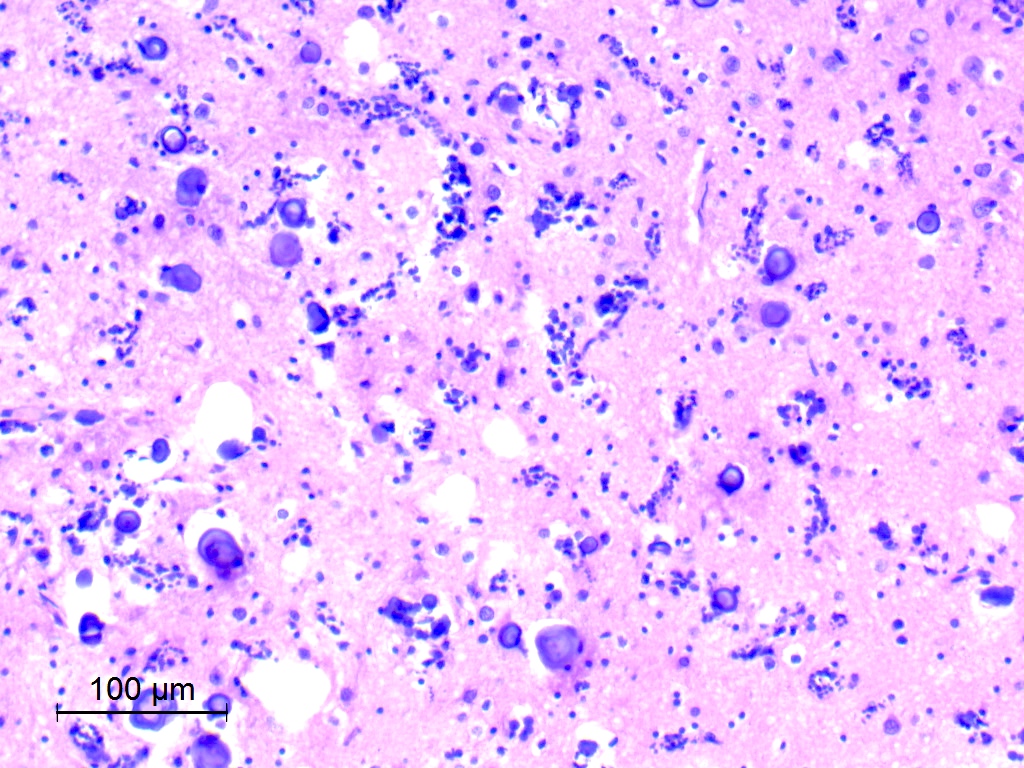
Calcification
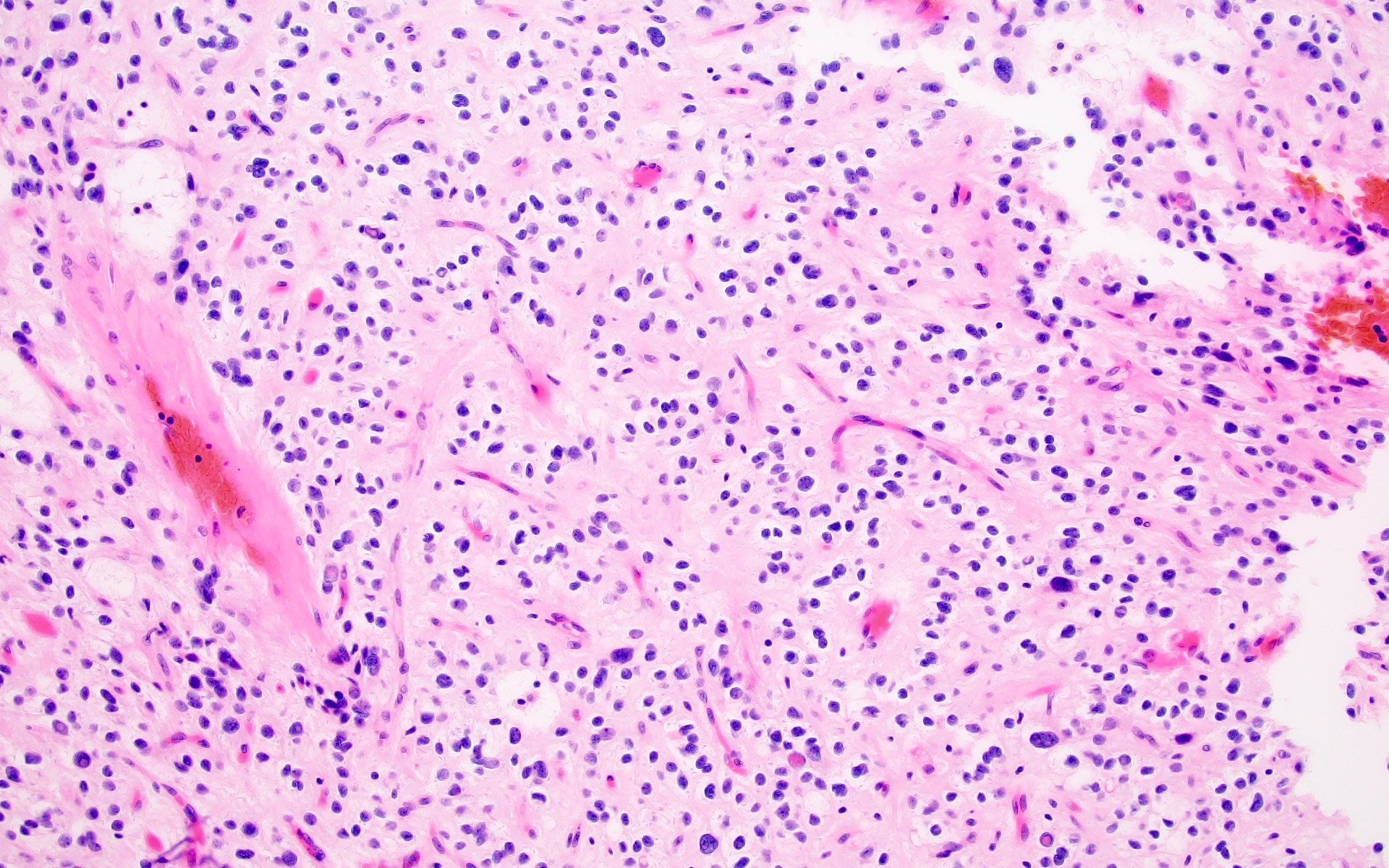
Oligo-like features
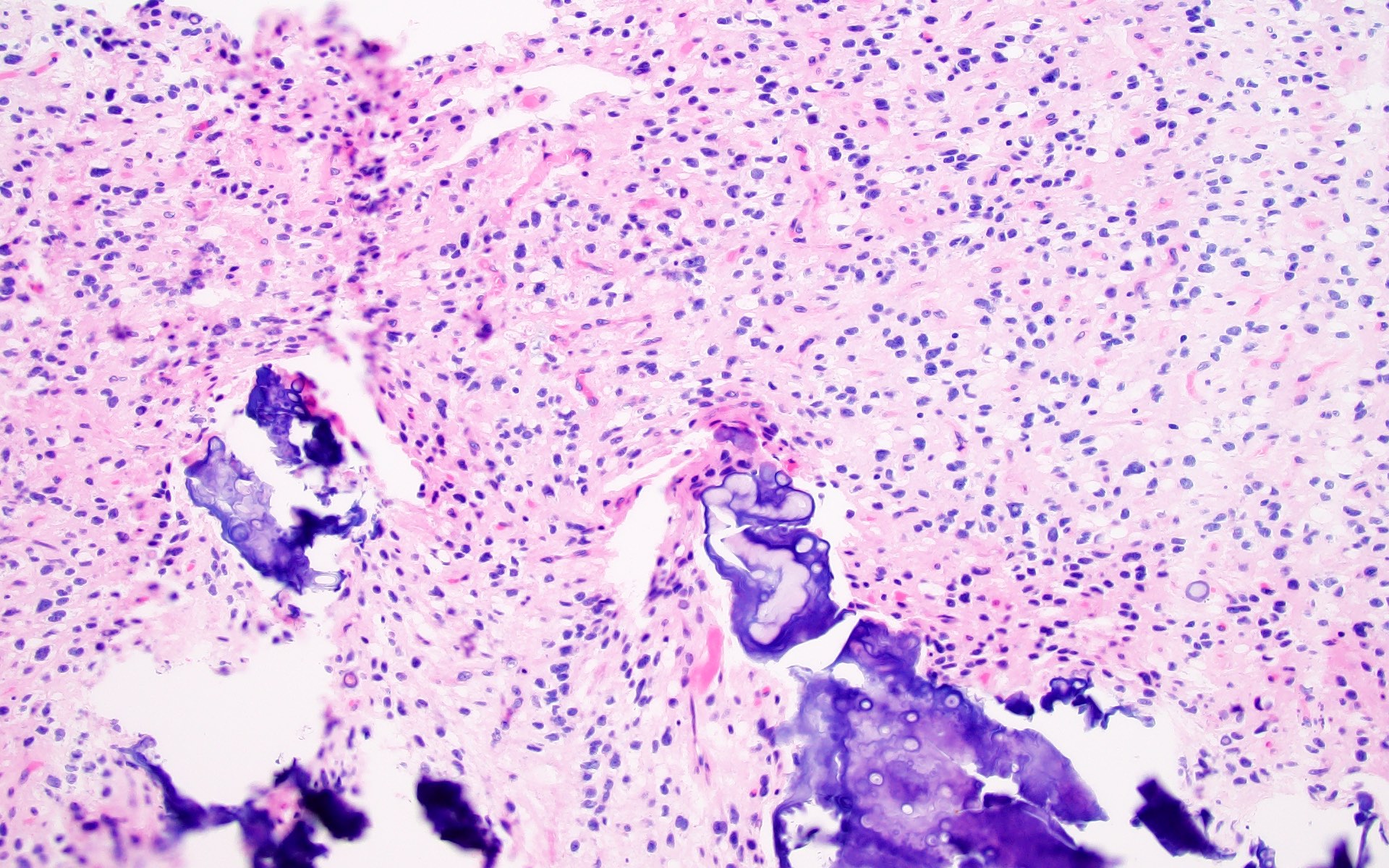
Dystrophic calcifications

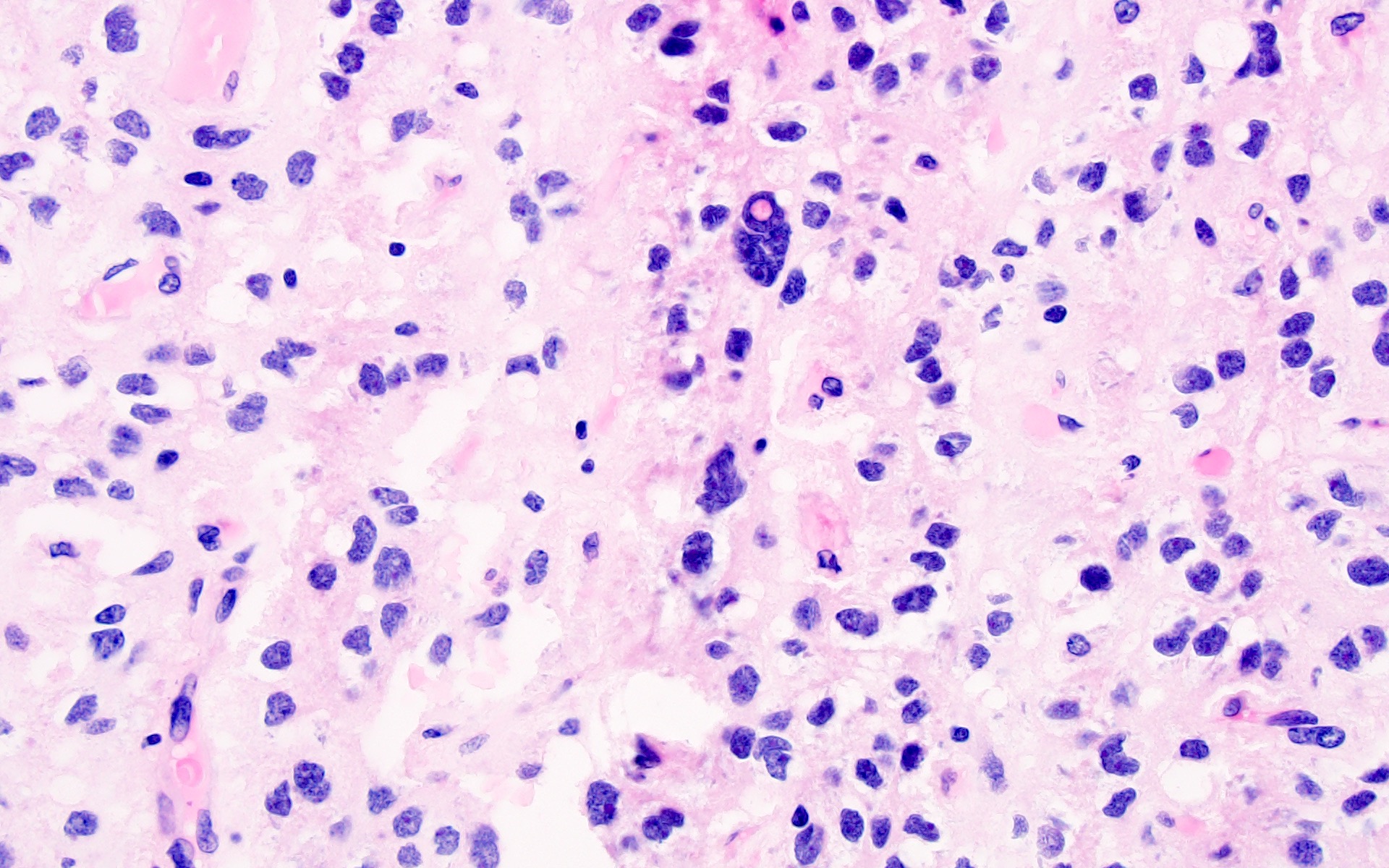
Polymorphic tumor cells
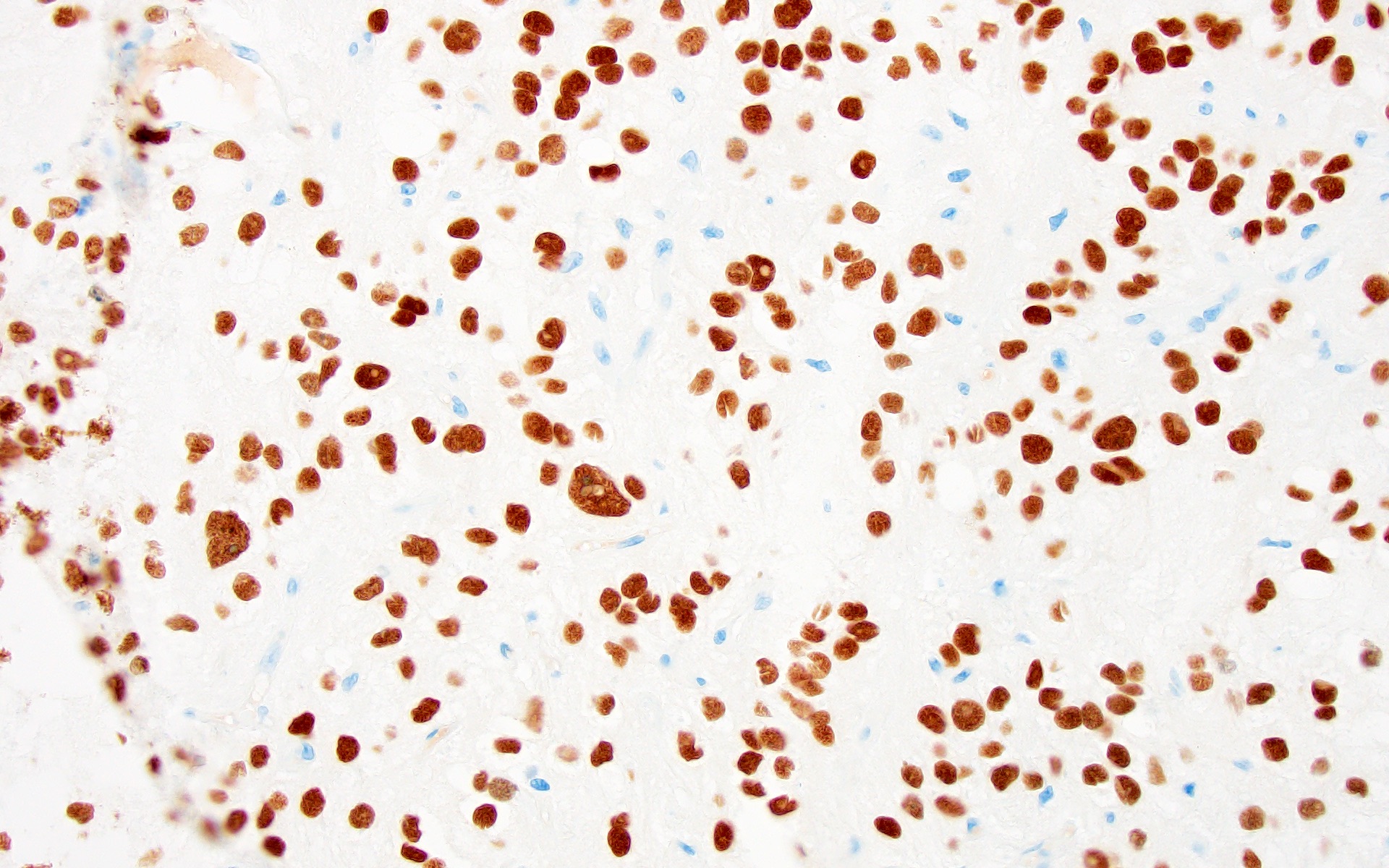
Olig2 diffusely positive
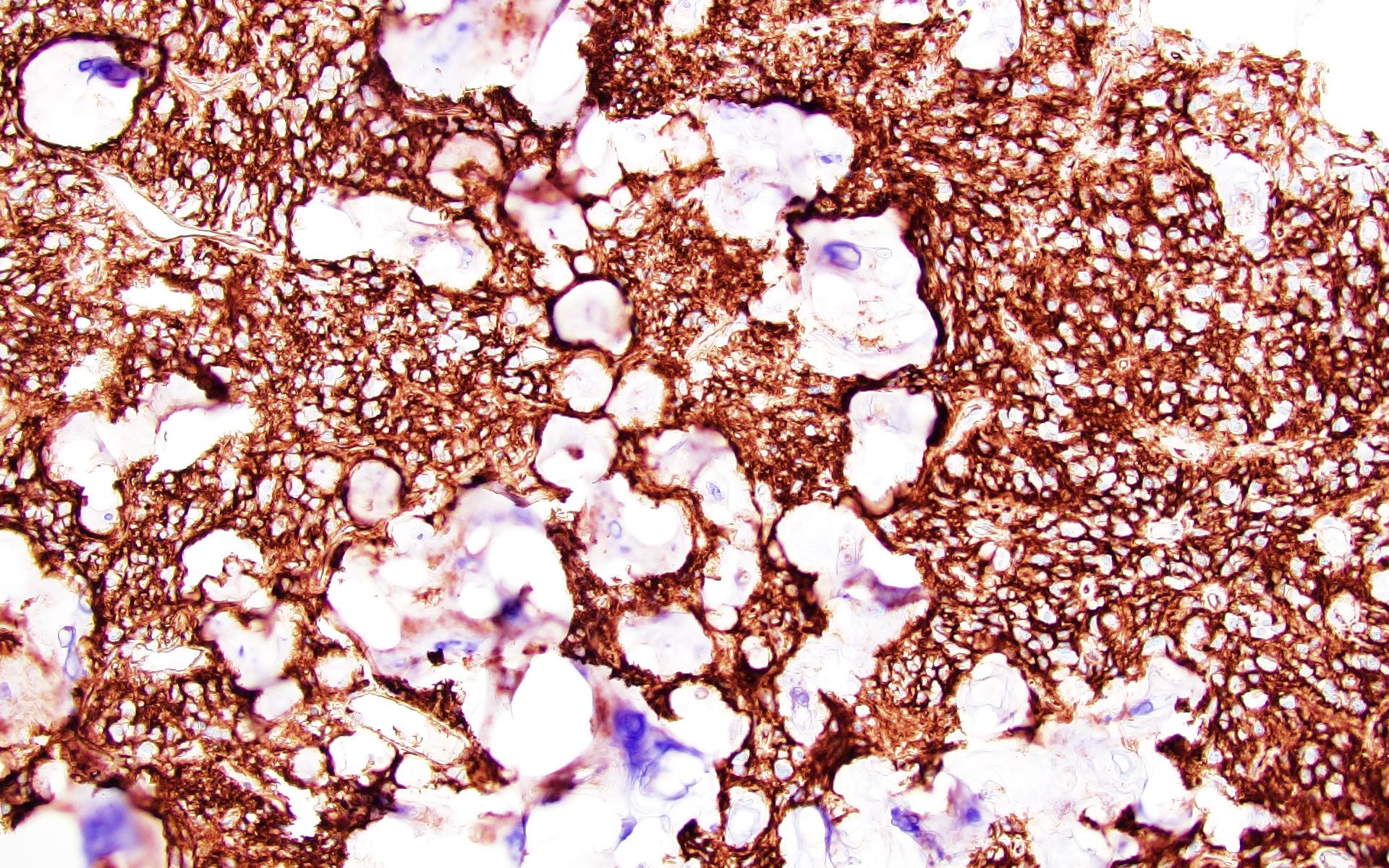
Strong, diffuse CD34 expression
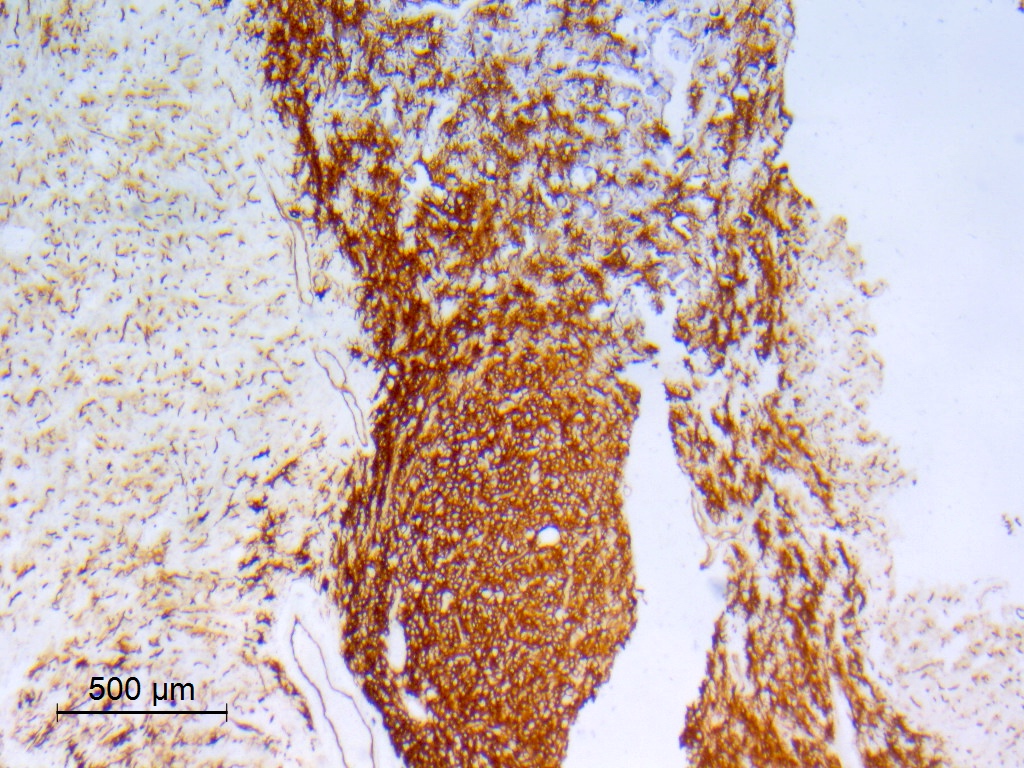
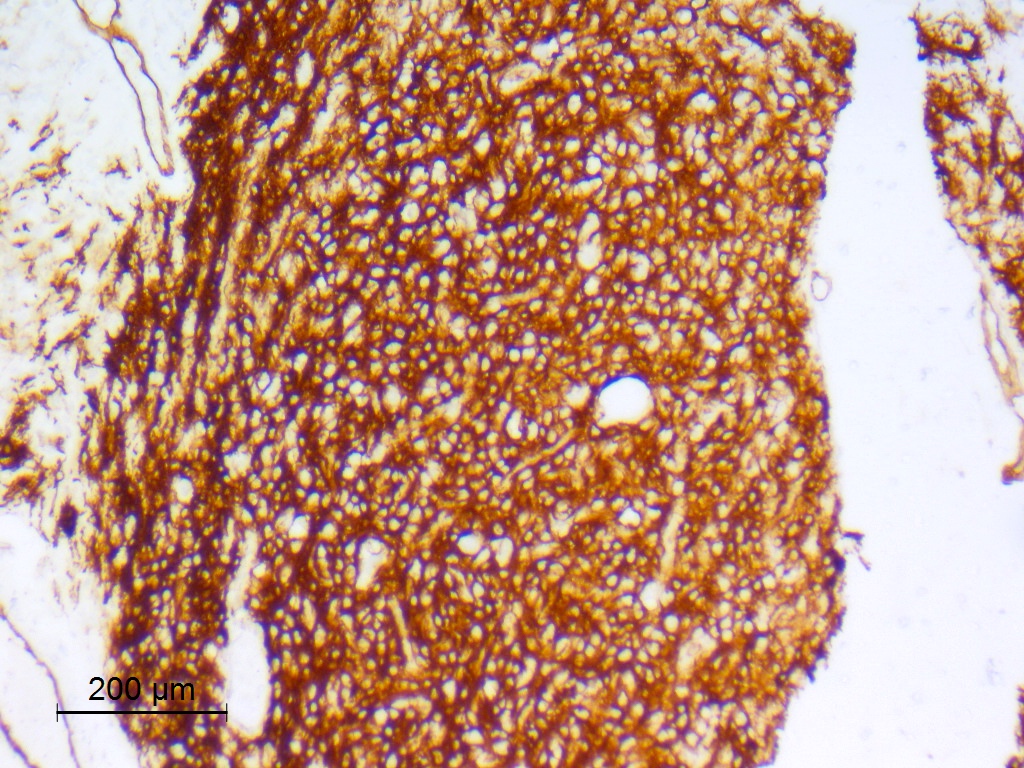
CD34
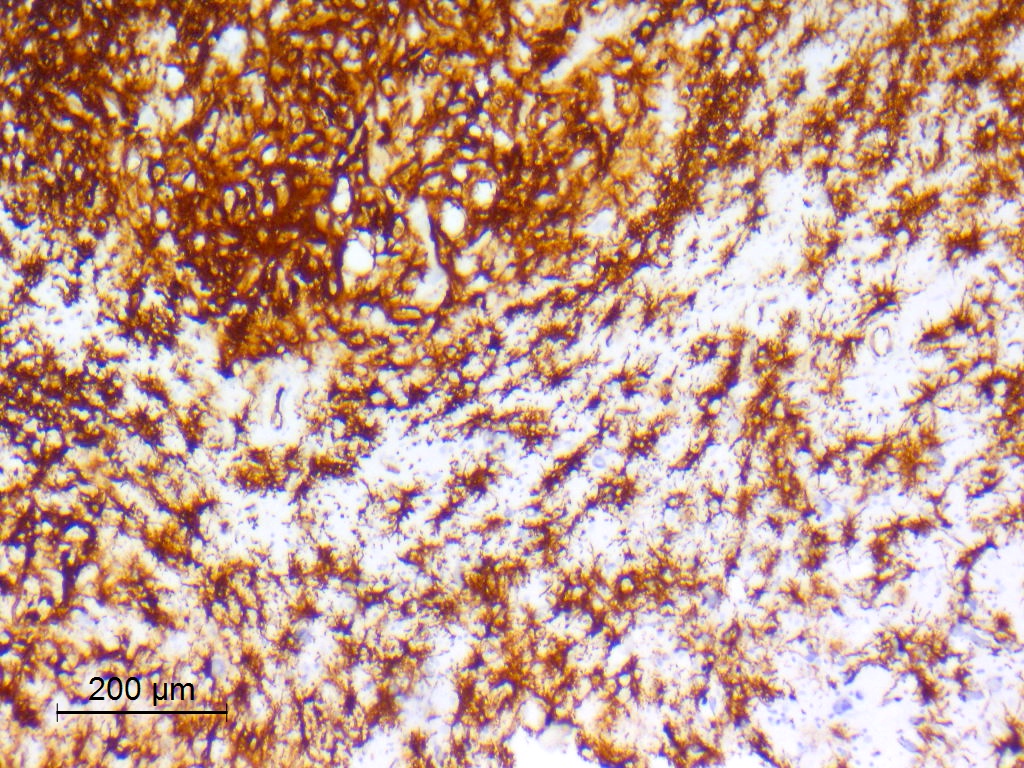
CD34 in tumor cells and ramified neural cells
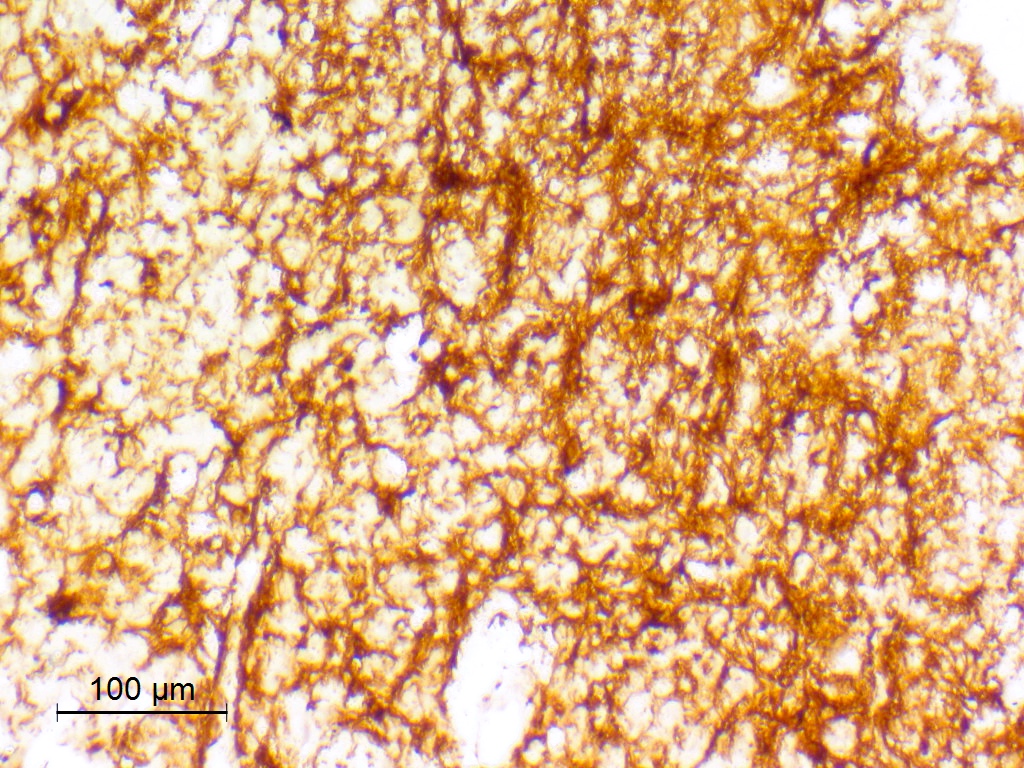
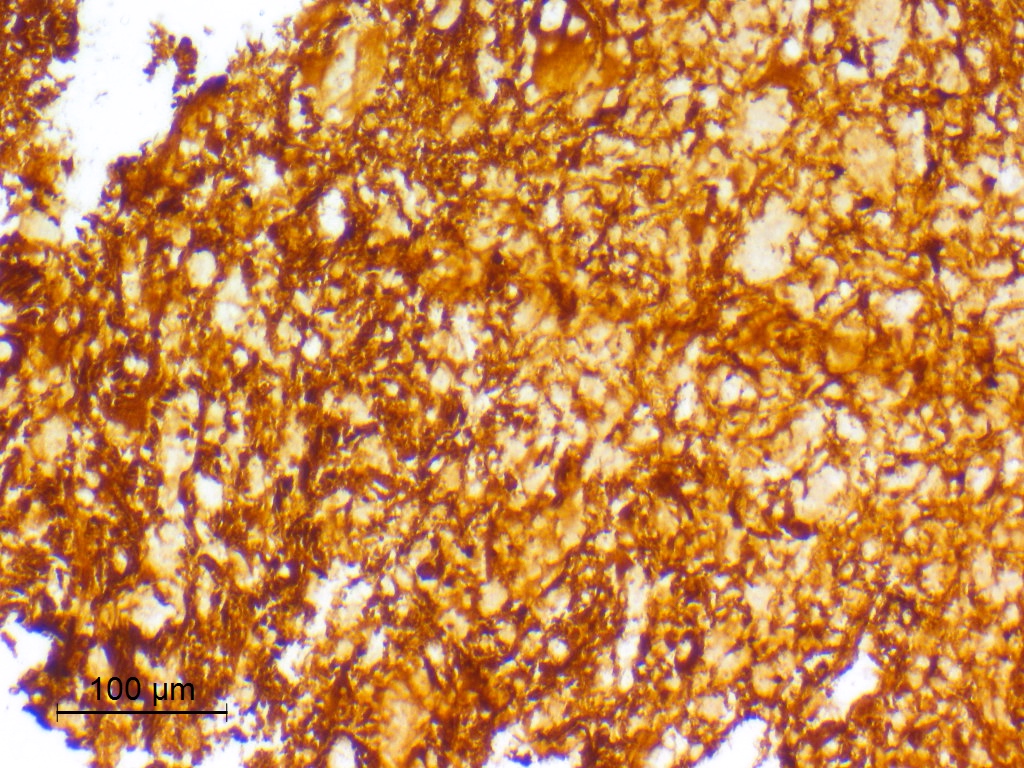
GFAP
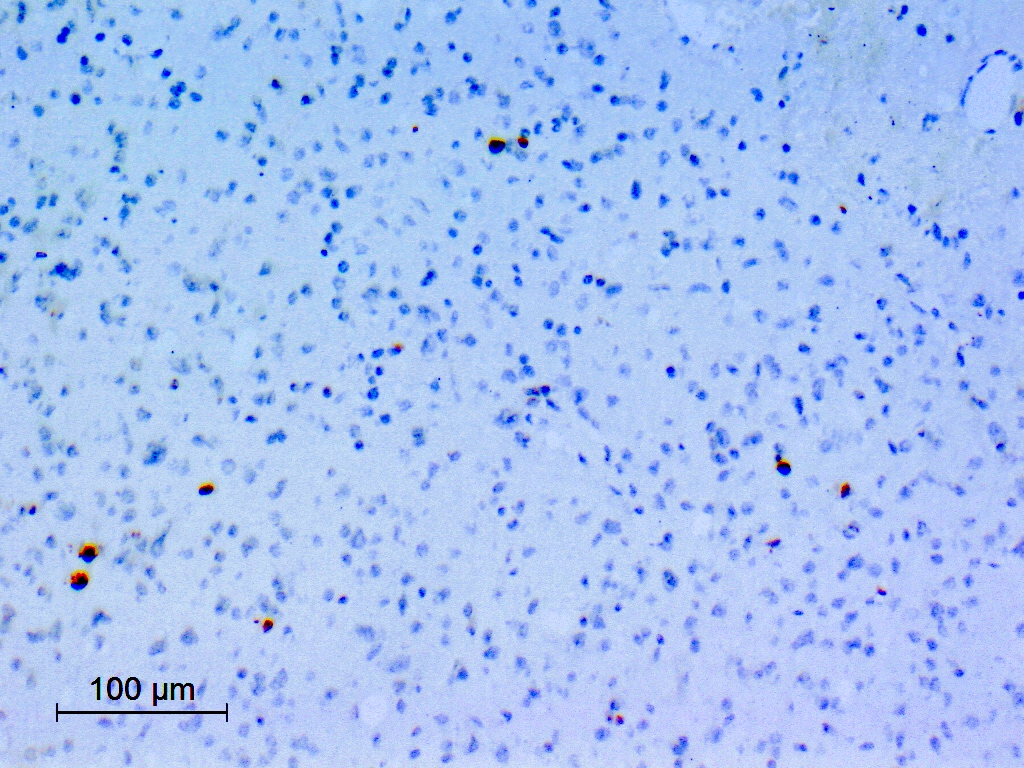
Ki67
- PLNTY cannot be identified on cytological grounds alone
- Intraoperative smear preparations may show
- Tumor cells with rounded nuclei and devoid of cytoplasm
- Tumor cells with irregular nuclear contours and cytoplasmic processes
- Tumor cells attached to blood vessels in a pseudorosette fashion (Acta Neuropathol 2017;133:417)
Images hosted on other servers:
Vague perivascular pseudorosetting
- GFAP: may be regional or diffuse, may be weak (Acta Neuropathol 2017;133:417)
- OLIG2 (Acta Neuropathol 2017;133:417)
- CD34
- May be patchy, or diffuse and intense
- Expressed by both tumor cells and ramified neural elements in the associated cerebral cortex (J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2017;133:417)
- ATRX: retained (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- BRAF p.V600E (30 - 54%) (Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2021;80:821)
- Ki67: generally low (< 1 - 2%) but higher values (up to 5%) have been reported (Acta Neuropathol 2017;133:417)
- Consistently associated with MAPK pathway activation by either BRAF V600E mutations or fusion events involving FGFR2 or FGFR3 in a mutually exclusive fashion; however, these abnormalities are hardly specific to PLNTY as a transforming molecular event (Acta Neuropathol 2017;133:417, Acta Neuropathol 2017;133:417, J Neuropathol Exp Neurol 2019;78:1100, J Neuropathol Exp Neurol 2021;80:821)
- FGFR fusions include FGFR2::CTNNA3, FGFR2:SHTN1 (KIAA1598), FGFR2::INA, FGFR3::TACC3 and FGFR2::MPRIP (J Neuropathol Exp Neurol 2021;80:821, Acta Neurol Belg 2023;123:327)
- Fusion positive tumors occur predominantly in pediatric patients and are more frequently associated with seizures / epilepsy, whereas BRAF V600E mutation positive tumors predominantly affect adult patients (J Neuropathol Exp Neurol 2021;80:821)
- Has a distinct DNA methylation profile most closely aligned to that of ganglioglioma (Acta Neuropathol 2017;133:417)
- Exhibits copy number changes predominantly involving whole chromosomes, suggesting that PLNTY is also characterized by chromosomal instability (J Neuropathol Exp Neurol 2021;80:821)
- PLNTY does not harbor IDH mutations and does not exhibit 1p / 19q codeletion
- Other reported molecular events include NTRK2 alterations, KIAA1549::BRAF fusion and low level FGFR3 amplification; molecular events associated with less favorable prognosis include germline ATM mutation and somatic mutations involving RB1, TP53, ATRX, PTEN and TEK (J Neuropathol Exp Neurol 2021;80:821, BMC Neurol 2020;20:123, J Neuropathol Exp Neurol 2019;78:1100, Acta Neuropathol 2021;141:123)
Images hosted on other servers:

Molecular and clinical findings
FGFR fusions

Distinct methylation signature

Genome wide copy number changes
- Brain mass lesion, temporal lobe, gross total resection:
- Polymorphous low grade neuroepithelial tumor of the young (PLNTY), CNS WHO grade 1
- Molecular genetics: evidence of BRAF V600E mutations, FGFR2 or FGFR3 fusions or potentially other MAPK pathway driving genetic abnormalities
- Adult type diffuse glioma (oligodendroglioma, IDH mutant and 1p / 19q codeleted or low grade diffuse astrocytoma, IDH mutant):
- CD34- (Acta Neuropathol 2017;133:417, World Neurosurg 2019;127:47)
- IDH mutations
- 1p / 19q codeletion in oligodendroglioma
- Lacks MAPK pathway activating genetic abnormalities
- Ependymoma:
- Often OLIG2-
- Dot or ring-like EMA+ (Acta Neuropathol 2017;133:417)
- Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC):
- Characteristic feature is the presence of scattered multinucleated cells (pennies on a plate) and nuclear clusters composed of large pleomorphic nuclei
- Synaptophysin+, GFAP-, CD34- (Neuropathol Appl Neurobiol 2021;47:464)
- Diffuse leptomeningeal glioneuronal tumor:
- Leptomeningeal based
- Synaptophysin+, GFAP-
- Concurrent KIAA1549::BRAF fusion with isolated 1p deletion or 1p / 19q codeletion
- Ganglioglioma:
- Clearly neoplastic neuronal components
- Features that support a diagnosis of PLNTY over ganglioglioma include abundant calcifications, diffuse strong CD34 staining of tumor cells and FGFR2 or FGFR3 fusions (Acta Neuropathol 2017;133:417)
- Nosological position of neoplasms otherwise qualifying as PLNTY but having nodular ganglion cell aggregates is unclear (Acta Neuropathol 2017;133:417)
- Pilocytic astrocytoma:
- Biphasic pattern
- CD34- (Balkan Med J 2019;36:3)
- Diffuse astrocytoma, MYB or MYBL1 altered:
- Diffuse low grade glioma, MAPK pathway altered:
- CD34 is typically limited to a few cells, unlike the widespread expression seen in PLNTY
- PLNTYs likely represent a subset of a larger family of tumors reported as diffuse astrocytomas or oligodendrogliomas of pediatric type having MAPK pathway activating genetic abnormalities (Acta Neuropathol 2017;133:417)
A 14 year old boy presented with refractory epilepsy. MRI showed a right temporal cystic and solid nonenhancing mass lesion. The mass was surgically resected and histopathological examination showed a low grade tumor composed of oligodendroglioma-like tumor cells arranged in a diffuse fashion as shown in the image above. Immunohistochemistry for CD34 showed regional positivity associated with positive ramified neurons in the adjacent cortex. Which of the following molecular events is characteristic of this tumor?
- 1p / 19q codeletion
- IDH mutation
- MAPK pathway activating abnormalities
- PRKCA gene fusion
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
- Absence of calcification
- CD34 negative immunostaining
- Clinical presentation of headache
- Oligodendroglioma-like components
Comment Here
Reference: Polymorphous low grade neuroepithelial tumor of the young (PLNTY)
- Primary central nervous system (CNS) lymphoma occurs first in the brain, eye, spinal cord or leptomeninges, without systemic involvement and not arising in the setting of immunodeficiency
- Majority are high grade diffuse large B cell lymphomas, with only rare cases representing T cell lymphomas, low grade B cell lymphomas or Burkitt lymphoma
- This topic addresses primary diffuse large B cell lymphoma of the CNS and does not include cases that are associated with immunodeficiency (see separate category of immunodeficiency associated lymphoproliferative disorders)
- Rare aggressive large B cell lymphoma occurs predominantly in elderly individuals, with an overall worse prognosis than systemic diffuse large B cell lymphoma
- Composed of large, atypical basophilic lymphocytes with perivascular cuffing and frequent necrosis
- Positive for CD45 and B cell markers and typically positive for BCL6, MUM1 and BCL2
- Unlike CNS lymphomas associated with HIV infection, primary CNS lymphoma is not associated with Epstein-Barr virus infection
- Previously known as reticulum cell sarcoma, diffuse histiocytic lymphoma or lymphomatosis cerebri (no longer recommended)
- Primary CNS lymphoma is used interchangeably with primary diffuse large B cell lymphoma of the CNS
- Excluded from this category are immunodeficiency associated lymphomas (see separate immunodeficiency associated topic)
- Accounts for < 1% of all non-Hodgkin lymphoma cases and 2 - 3% of all CNS tumors (Cancer 2017;123:4314)
- Overall incidence rate is < 1 cases per 100,000 population
- Rates among men and women aged 65+ years have increased in recent decades (1992 - 2011), although the reason for the increase is unclear
- Among immunocompetent individuals (HIV uninfected, nontransplant recipients), the median age at diagnosis among a U.S. population is 67 (range 52 - 77 years) with a similar male to female ratio (Br J Haematol 2016;174:417)
- Majority involve the supratentorial space, with the posterior fossa and spinal cord less frequently involved (Leukemia 2011;25:1797)
- Most patients present with a single tumor but can present with multiple (Isr J Med Sci 1992;28:268)
- 10 - 20% show concurrent intraocular involvement (Cancer 2017;123:4314)
- Extraneural dissemination is very rare but shows a propensity for the testis (J Neurooncol 2006;80:159)
- Not definitively understood
- Central nervous system is an immune privileged site and the lymphoma itself may have genetic and phenotypic features that protect the tumor from ongoing immune surveillance (Virchows Arch 2020;476:647)
- Neoplastic cells may arise from lymphocytes normally residing in the immune privileged milieu of the CNS or from lymphocytes that transform to a malignant clone outside the CNS and then home to the CNS and escape normal immune regulation in the immune privileged site (Virchows Arch 2020;476:647, Curr Oncol Rep 2015;17:60)
- Unknown in immunocompetent patients
- Viruses, including Epstein-Barr virus, do not play a role
- Rising rates in elderly individuals (HIV uninfected, nontransplant recipients) may be related to increasing use of immunosuppressive medications to treat autoimmune conditions in the elderly (Br J Cancer 2011;105:1414)
- Symptoms vary depending on the CNS site involved and commonly include cognitive or behavioral changes, focal neurologic deficits and symptoms of increased intracranial pressure (Cancer 2017;123:4314)
- Seizures are relatively uncommon and patients typically do not present with B symptoms (fever, night sweats, weight loss)
- Ocular manifestations may present as decreased acuity, blurry vision or floaters (Hematol Oncol Clin North Am 2019;33:597)
- Contrast enhanced MRI of the brain is the neuroimaging modality of choice for diagnosis; however, the diagnosis must be confirmed by review of sampled tumor tissue or fluid (Br J Haematol 2019;184:348)
- Stereotactic needle biopsy is the gold standard for diagnosis and classification of CNS lymphoma (Neurooncol Pract 2019;6:415)
- Cerebrospinal fluid (CSF) cytology and flow cytometry may be used when biopsy is not possible or to evaluate for leptomeningeal involvement
- On MRI, there is an isointense to hypointense lesion on T1 weighted images with homogenous gadolinium contrast enhancement
- Lesions are usually solitary, involving the deep white matter and periventricular in location
- In immunocompetent individuals, the lesions typically do not show hemorrhage, calcifications, necrosis or ring enhancement (Hematol Oncol Clin North Am 2019;33:597)
Images hosted on other servers:

Brain MRI with contrast enhancing lesion
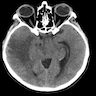
Solid and enhancing nodules on MRI
- Overall poor prognosis with considerably worse prognosis than systemic diffuse large B cell lymphoma
- Age and performance status are the most important prognostic factors
- In the Memorial Sloan Kettering Cancer Center prognostic model, only age and performance status were independent prognostic factors
- The prognostic model from the International Extranodal Lymphoma Study Group includes age and performance status as well as serum lactate dehydrogenase level, CSF protein concentration and involvement of deep structures of the brain (Cancer 2017;123:4314)
- In patients treated with methotrexate based regimens, BCL6 expression may be a predictor of improved progression free survival (Br J Haematol 2014;165:640)
- 25 year old man with 1 week history of severe headaches and changes in vision (Can J Ophthalmol 2019;54:e134)
- 54 year old woman with symptoms of numbness and weakness in the right leg (BMC Neurol 2019;19:90)
- 71 year old man presented with cerebellar symptoms (dizziness, nausea, vomiting and gait imbalance) (J Med Case Rep 2018;12:341)
- 75 year old woman presented with subacute progressive confusion and unusual behavior (Neurooncol Pract 2017;4:46)
- 3 patients presenting with primary CNS diffuse large B cell lymphoma with anaplastic morphology (Diagn Pathol 2019;14:45)
- High dose intravenous methotrexate based regimen is the first line induction therapy
- Consolidation therapy may include conventional chemotherapy, radiotherapy, high dose chemotherapy with autologous stem cell transplantation or their combinations (Transl Oncol 2019;12:523)
- From postmortem examination, tumors have a variable macroscopic appearance, including variable demarcation from the surrounding parenchyma
- Lesions may be firm, homogenous, centrally necrotic, brownish, gray-tan or yellow with areas of hemorrhage (Clin Neuropathol 2008;27:13)
Images hosted on other servers:
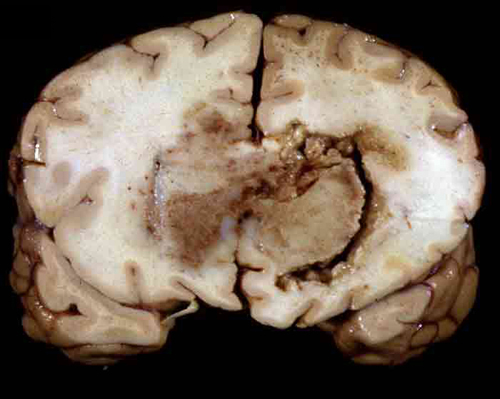
Cerebral lymphoma
spreading across
the corpus callosum
- Histologic features seen on frozen section are similar to those seen on permanent
- Intraoperative cytology is an important component of intraoperative evaluation (Neuropathology 2016;36:313)
Contributed by Genevieve M. Crane, M.D., Ph.D. and Lena Young, D.O.
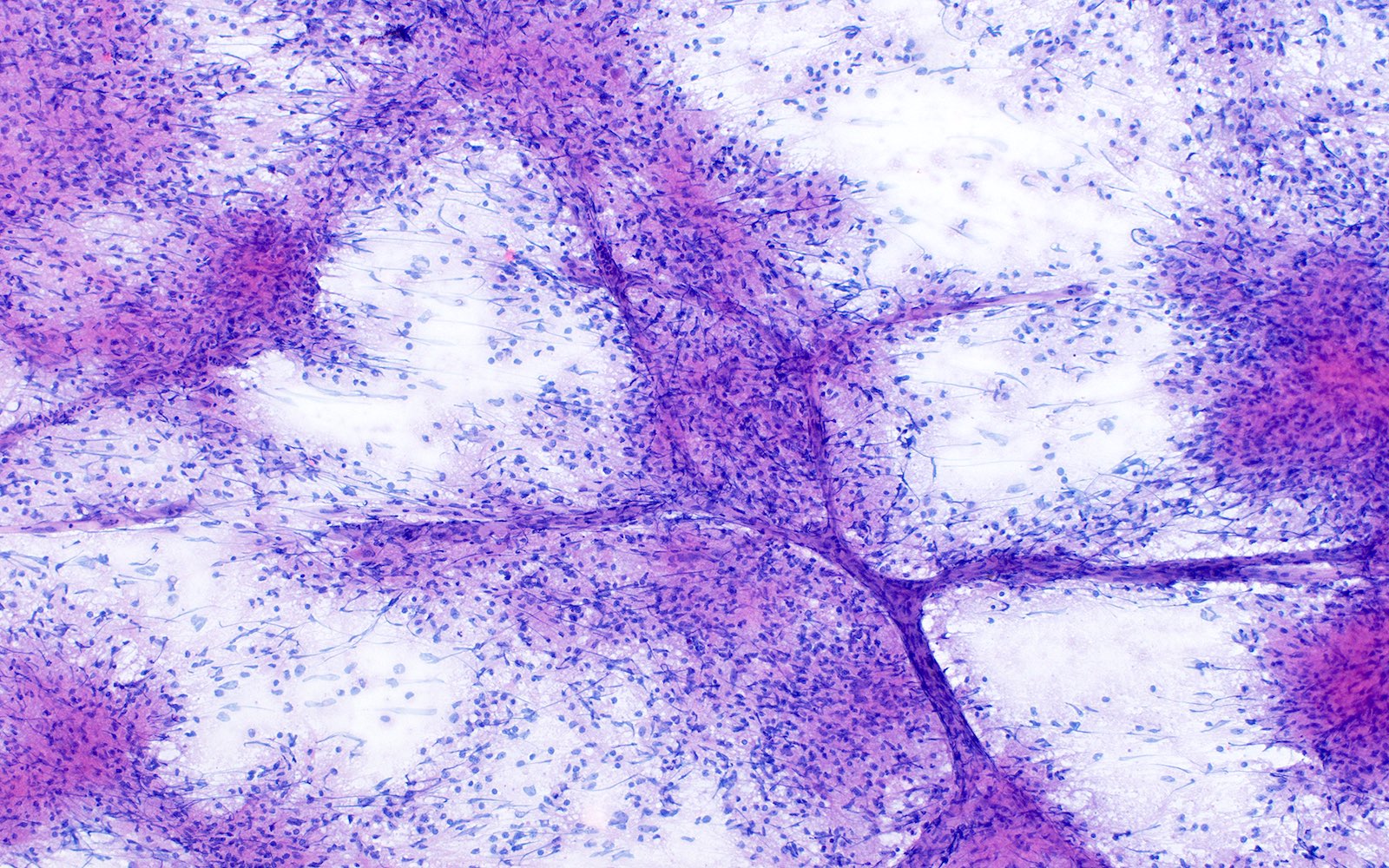
Squash preparation

Frozen section
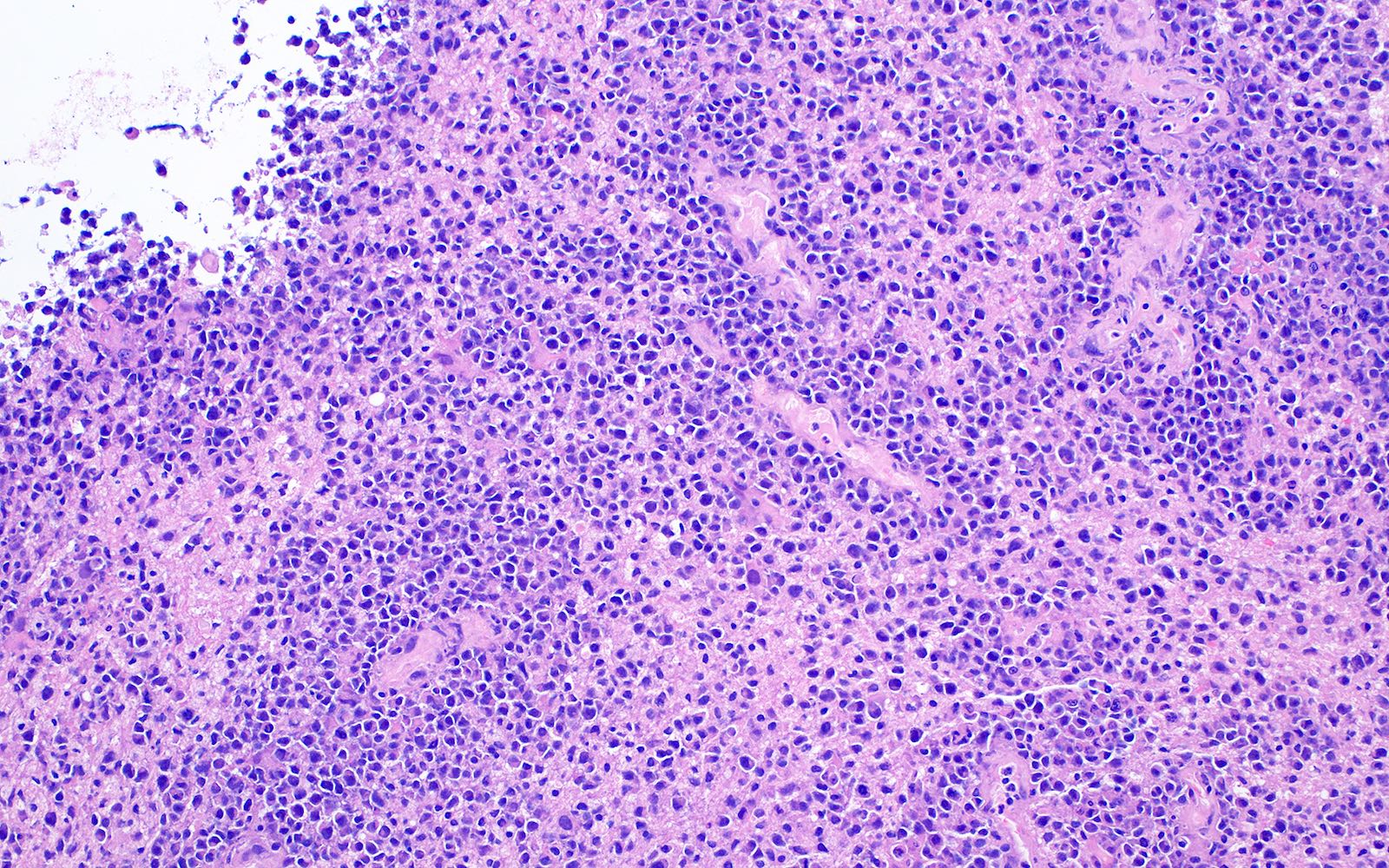
Frozen section control
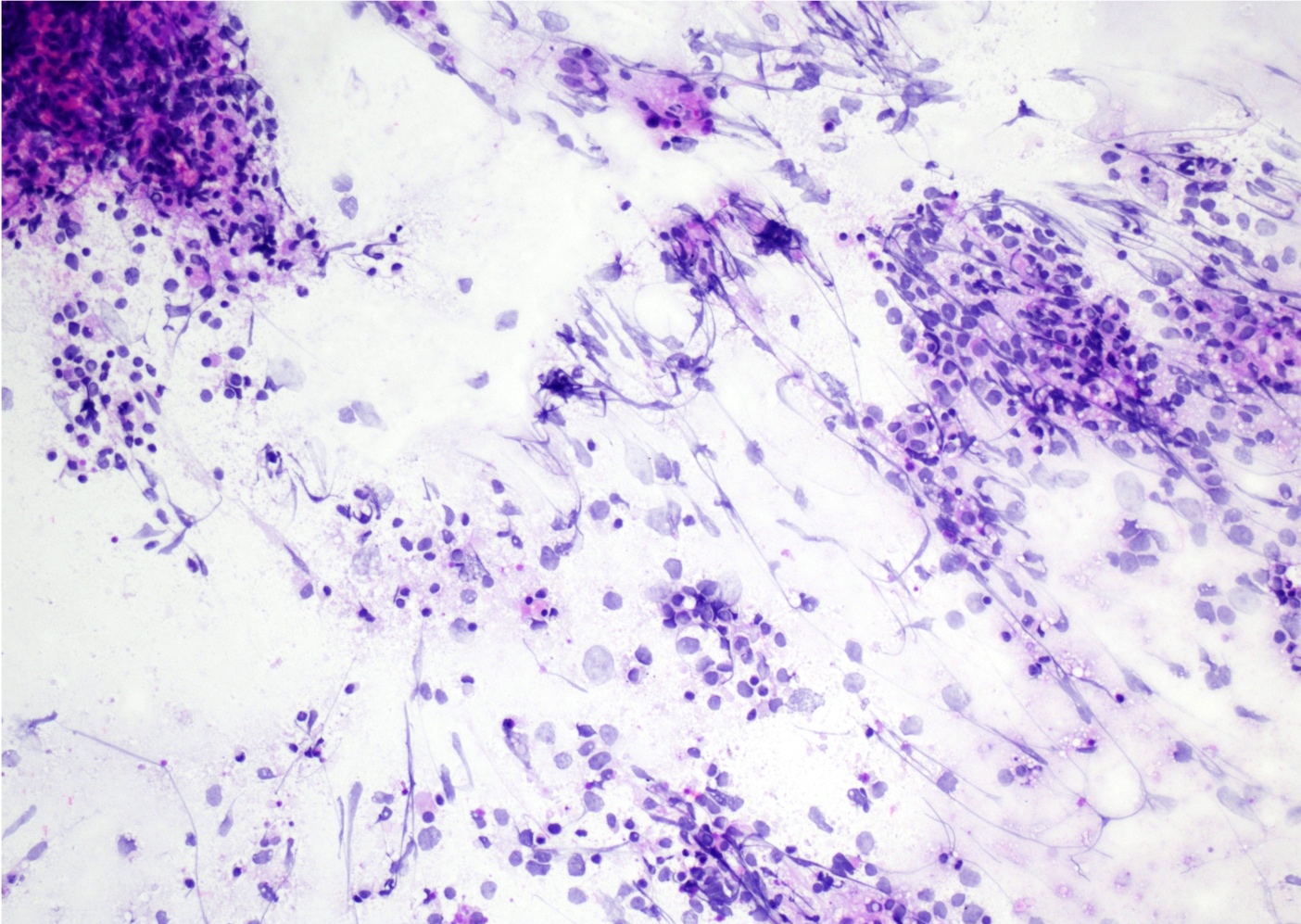
Touch prep of
monomorphic
basophilic cells with
crush artifact
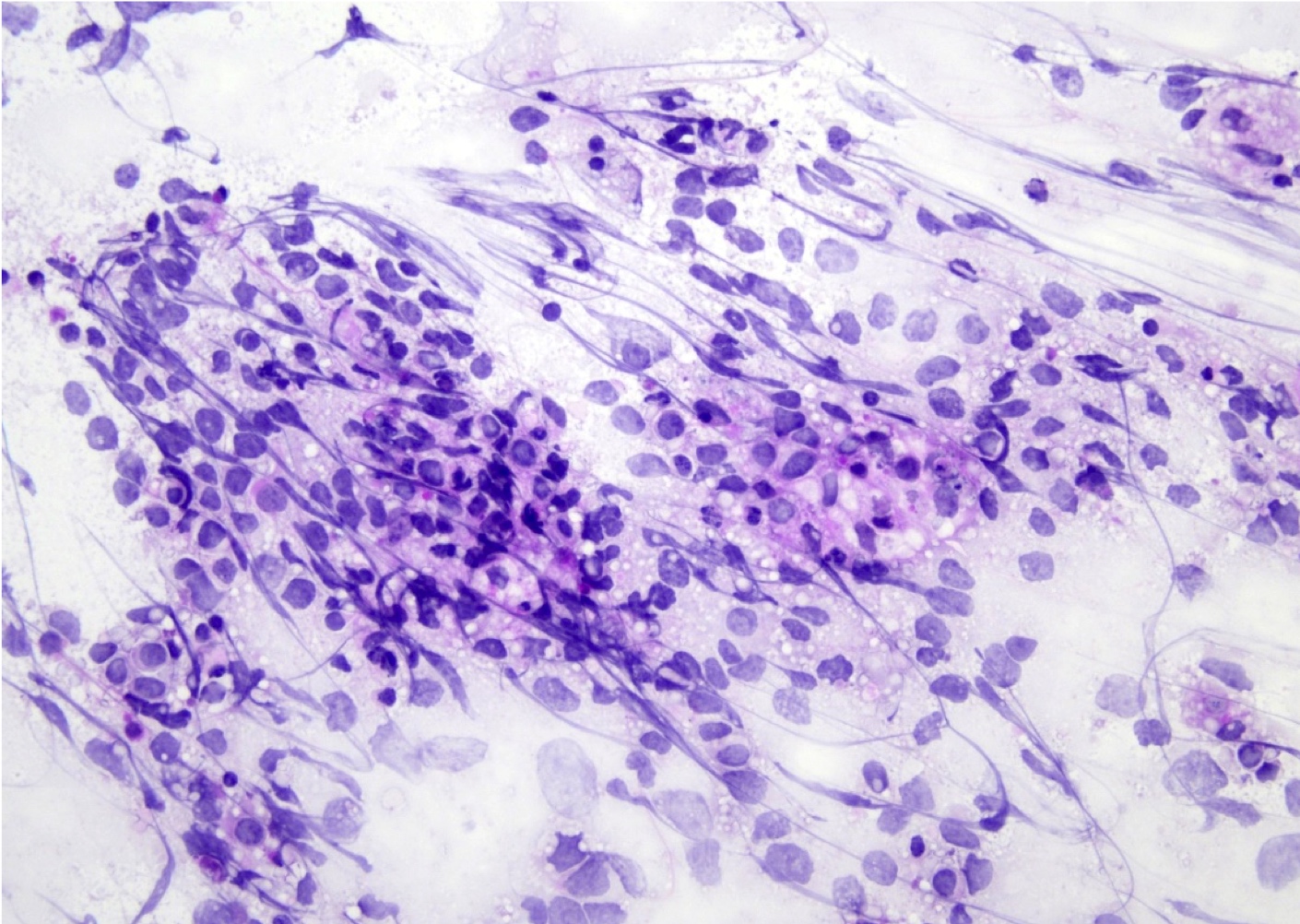
Touch prep
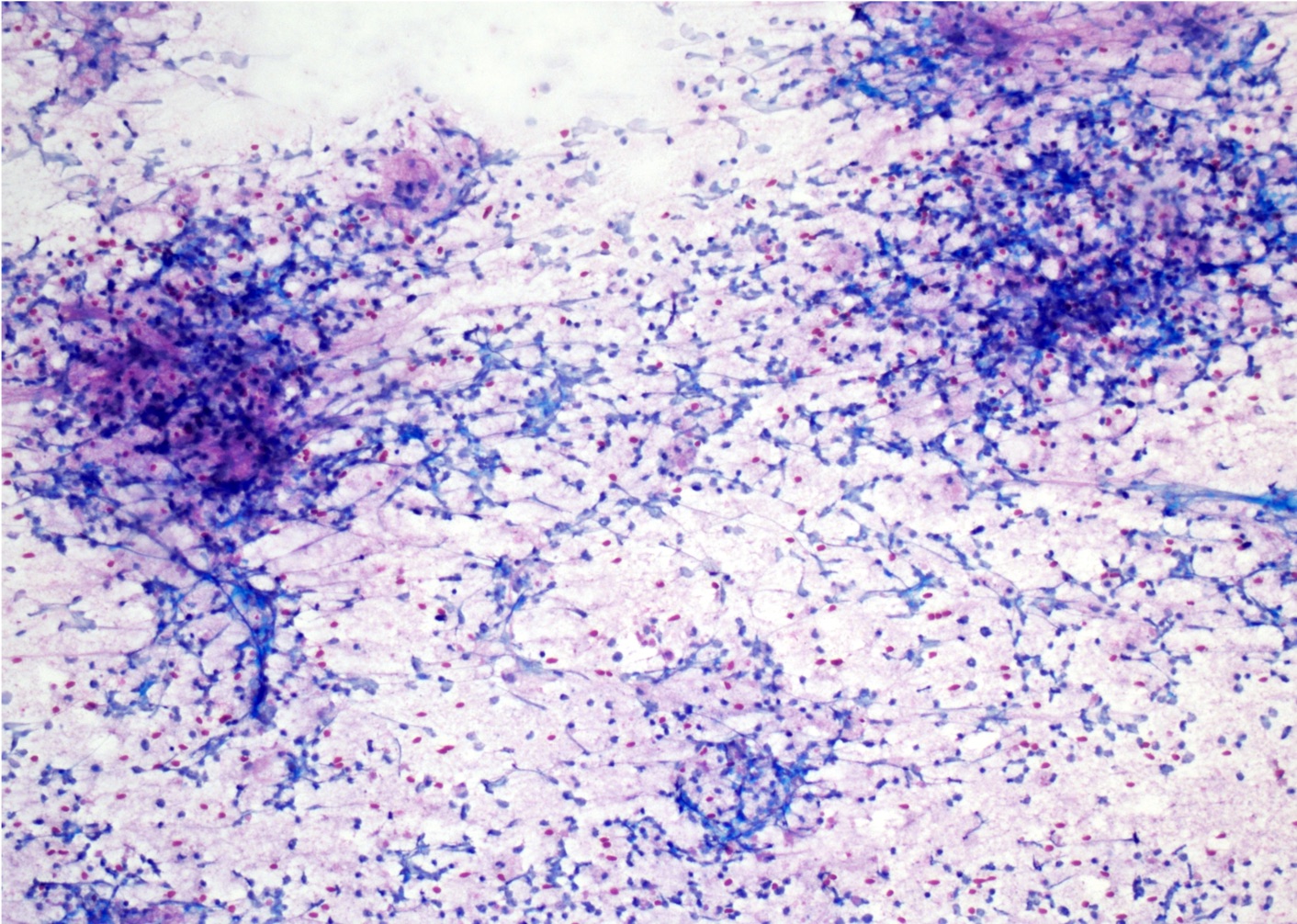
Intraoperative squash preparation
- Characteristic angiocentric growth pattern with cuffs of neoplastic lymphocytes within and around blood vessels (Clin Neuropathol 2008;27:13)
- Diffuse infiltration of brain parenchyma by small clusters or individual cells; necrosis is common
- Accompanied by astrocytic and microglial activation and may be accompanied by a reactive inflammatory infiltrate
- Neoplastic B cells are intermediate to large and frequently have vesicular chromatin and prominent nucleoli
- Corticosteroid administration prior to biopsy may induce dramatic tumor shrinkage with increased macrophages and cellular debris
Contributed by Elizabeth Courville, M.D. and Lena Young, D.O.
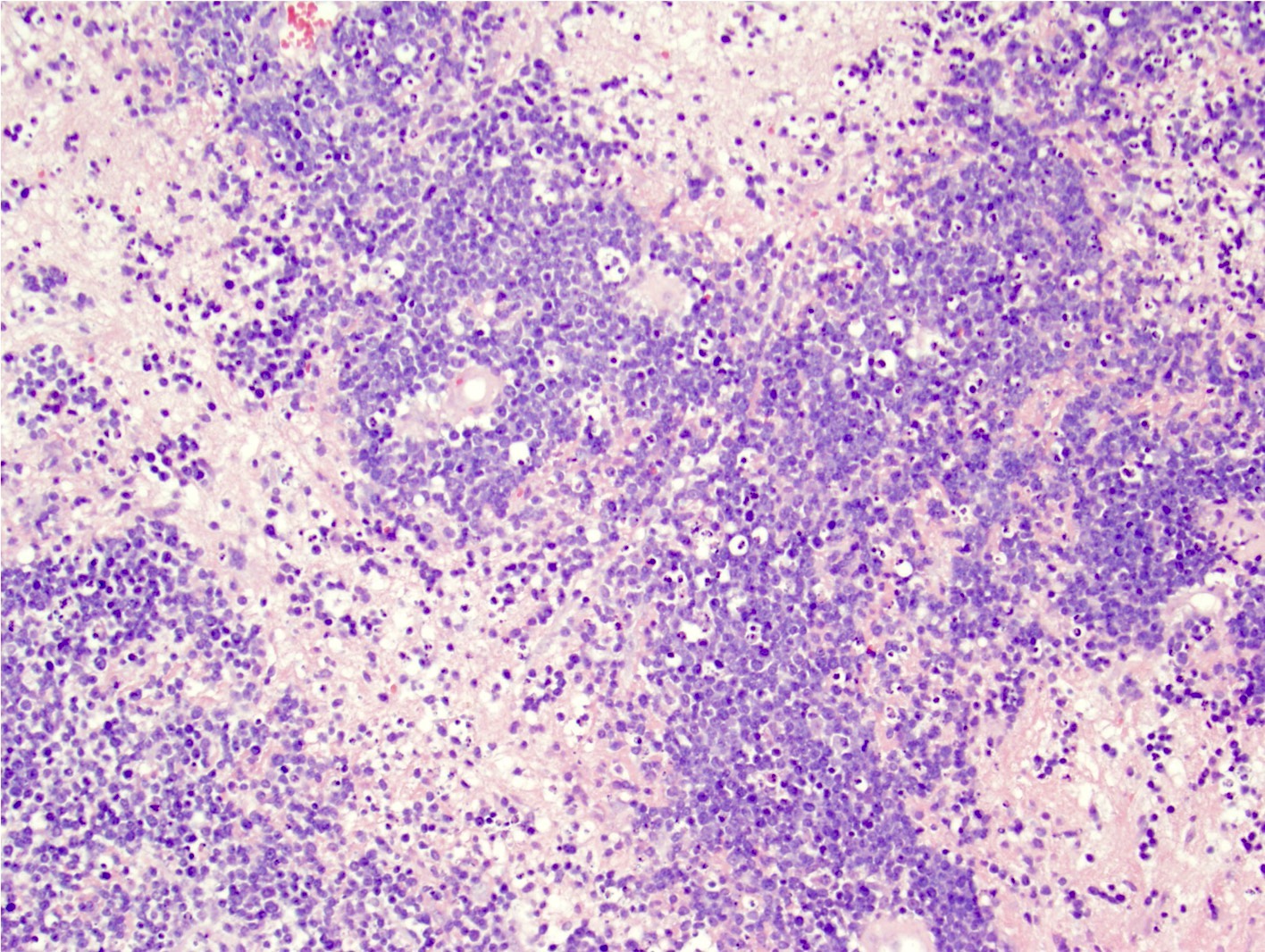
Perivascular infiltrate of atypical lymphoid cells
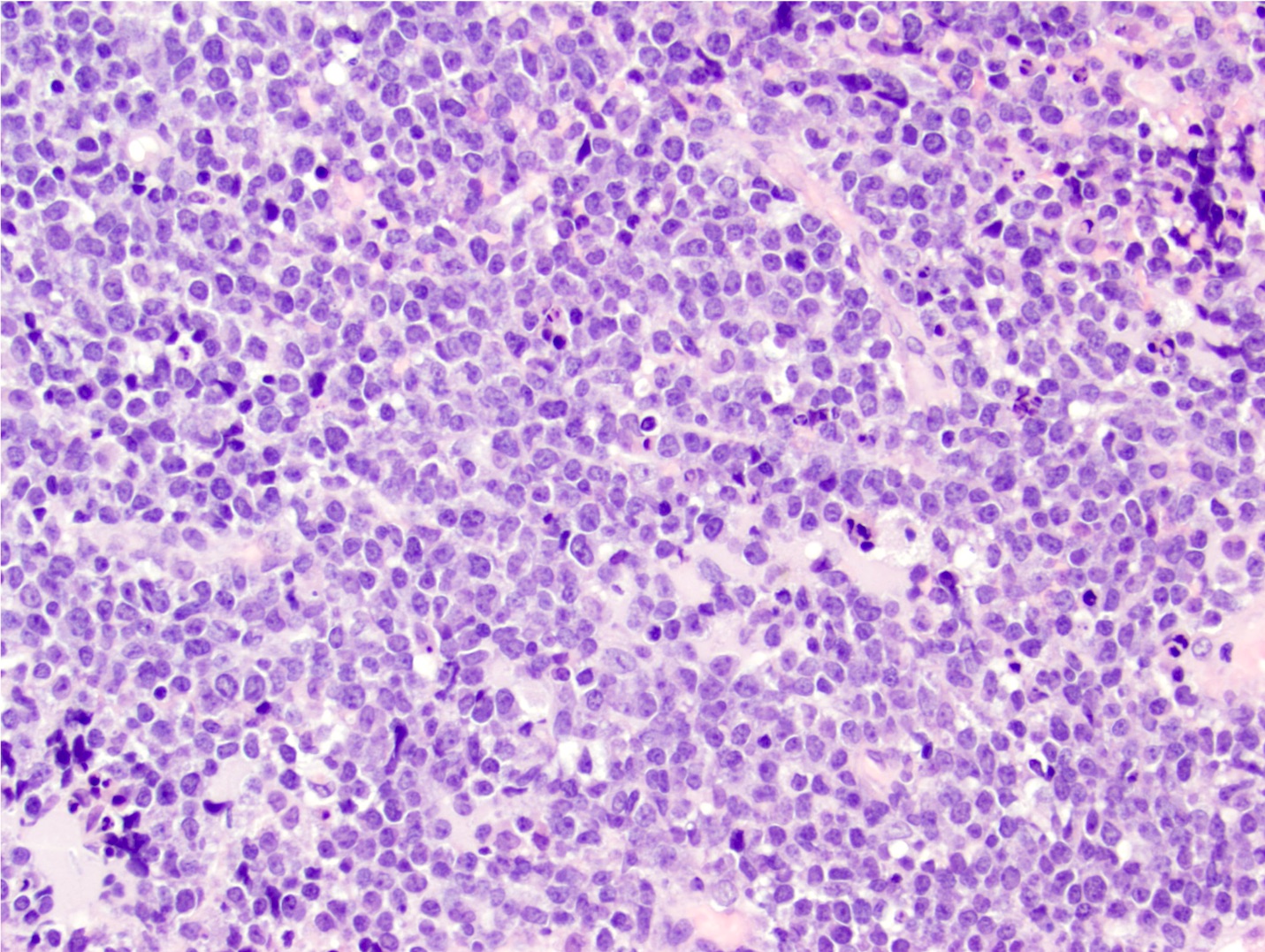
Diffuse infiltrate
of intermediate
to large
mononuclear cells
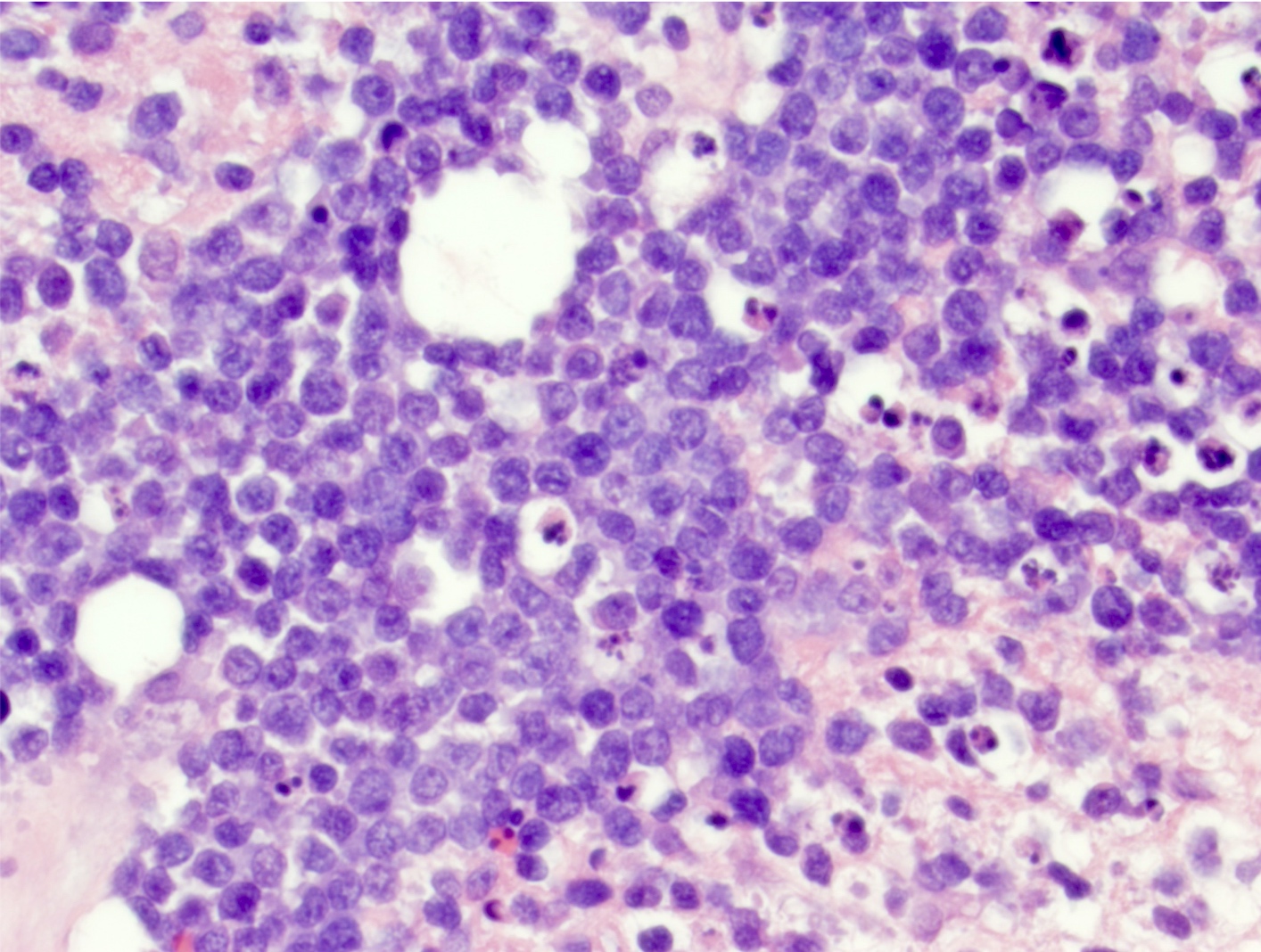
Cytology of mononuclear cells
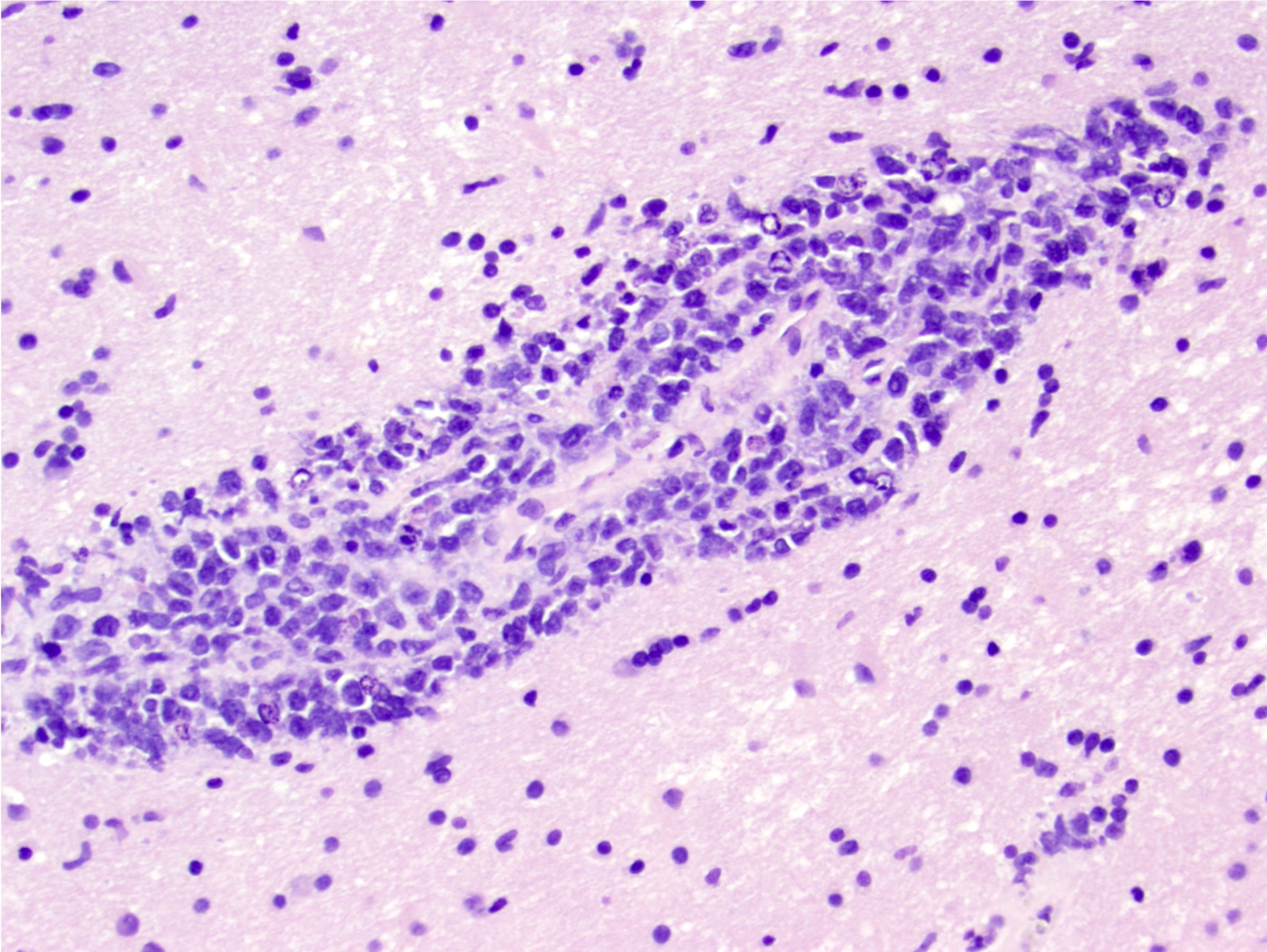
Perivascular cuffing by neoplastic lymphocytes
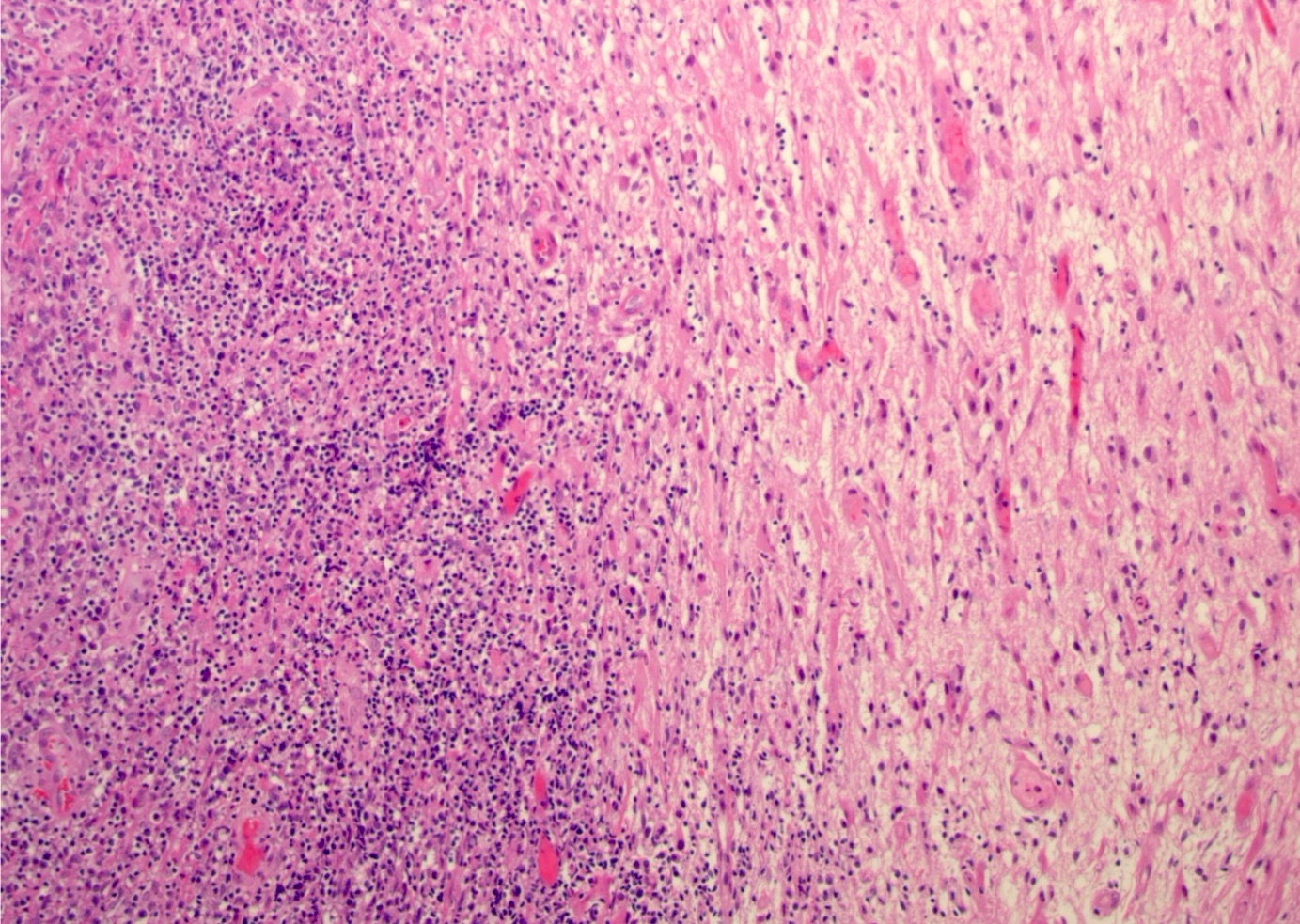
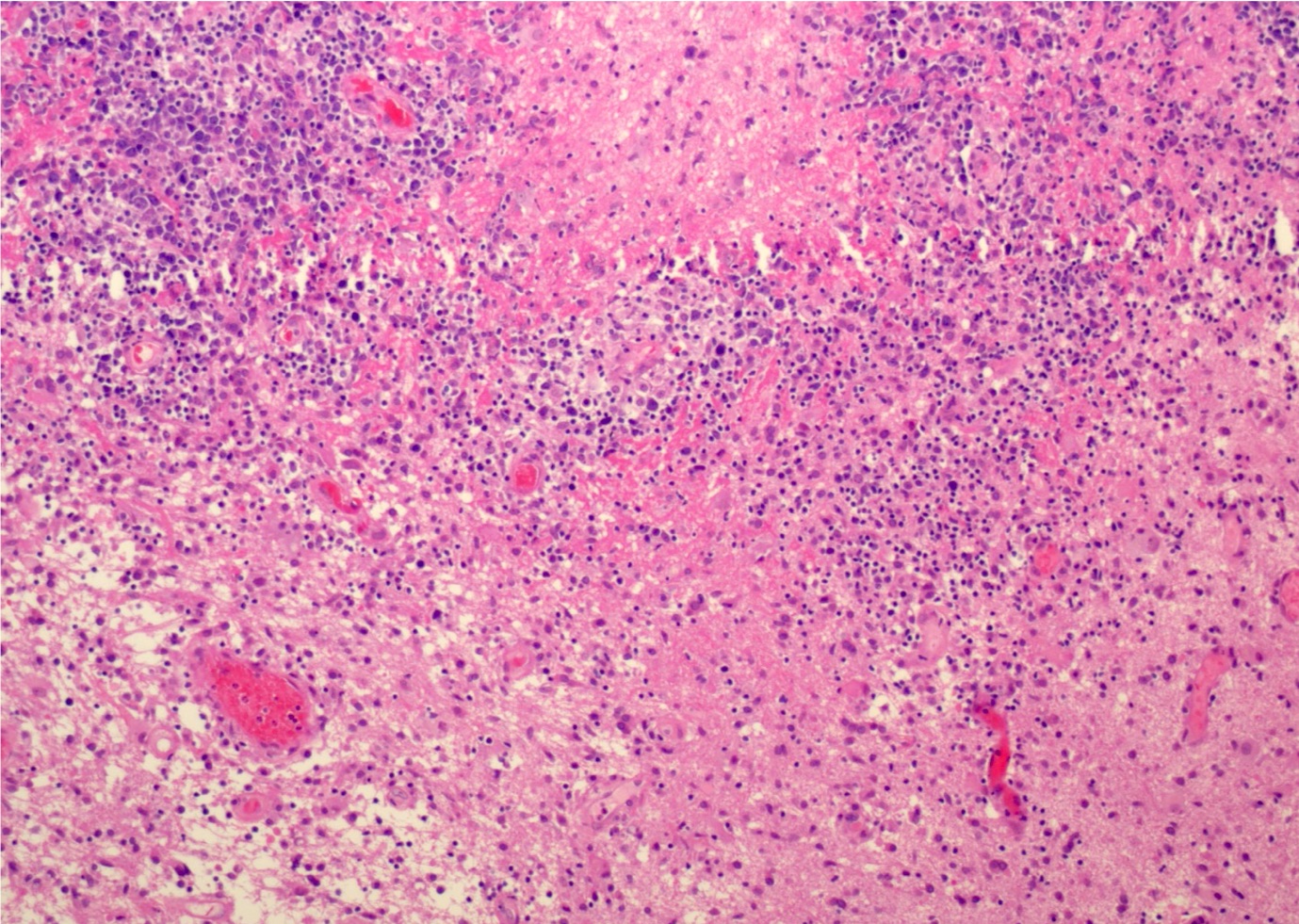
Infiltrate of neoplastic intermediate to large lymphocytes with background inflammation and necrosis
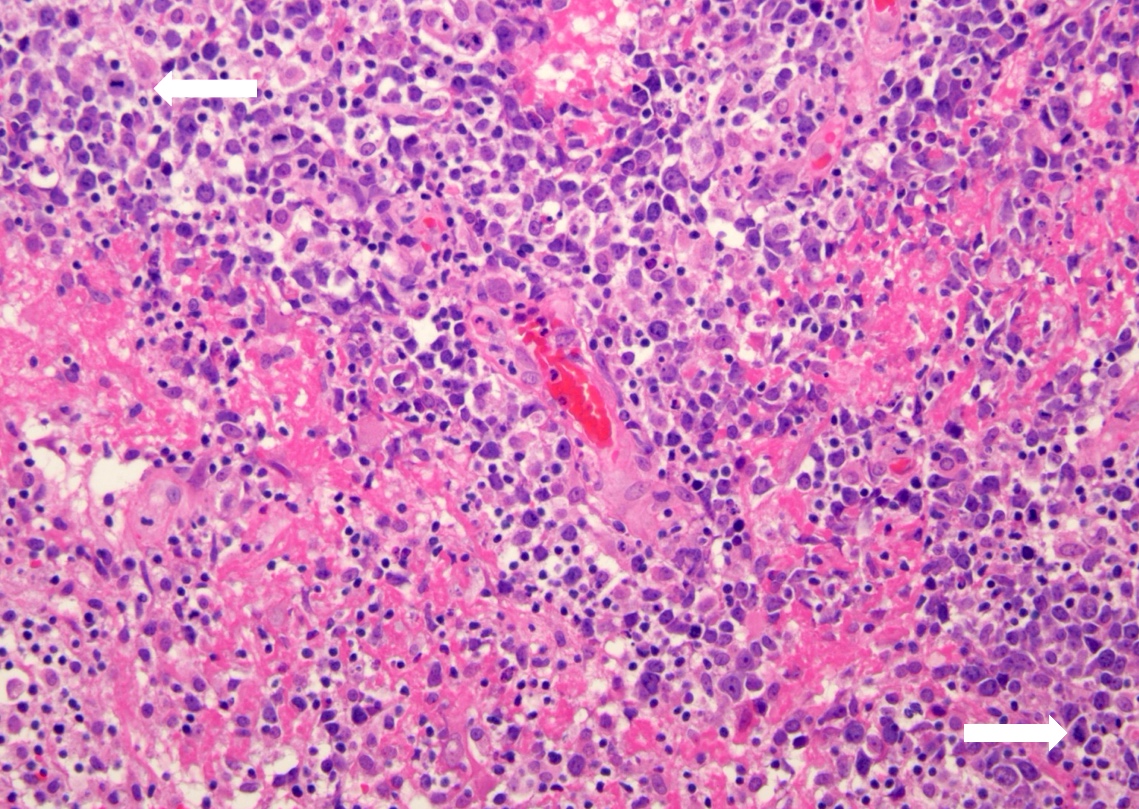
Pleomorphic lymphocytes infiltrating brain

Immunohisto-
chemistry for B and T cells
ISH shows lambda restriction
- Neoplastic lymphocytes in the cerebrospinal fluid are dispersed cells, increased in size compared to background reactive lymphocytes and frequently show irregular nuclear contours, abnormal chromatin and prominent nucleoli
- Mostly discohesive groups of monomorphic, medium to large basophilic lymphoid cells with prominent smearing artifact and background reactive glial cells
- Touch preparations show discohesive, large, atypical basophilic cells with a high nucleus to cytoplasm ratio and open chromatin with immature (lymphoblast) features; in post treatment tumors, only necrotic cells or reactive T cells may remain
- Reference: Asian J Neurosurg 2013;8:195
- PAX5, CD19, CD20, CD22, CD79a (B cell markers)
- BCL6 (60 - 80%)
- MUM1 / IRF4 (90%) (Cibas: Cytology: Diagnostic Principles and Clinical Correlates, 2nd Edition, 2003)
- BCL2 (90%)
- MYC (92%) (Acta Neuropathol 2013;126:603)
- CD38, CD138 (plasma cell markers)
- CD10 (< 10% are positive); CD10 positivity is seen more commonly in systemic diffuse large B cell lymphoma so should prompt careful search for systemic disease (Hematol Oncol 2014;32:57)
- EBV positivity suggests underlying immunodeficiency
- Pertinent negative stains include CD30, ALK1, HHV8, as they are helpful in the evaluation of mimicker neoplasms including systemic large B cell lymphoma, anaplastic large cell lymphoma and HHV8 associated lymphoproliferative disorders respectively
- Specimen is not always sent for flow cytometry from stereotactic needle core biopsies due to limited material; flow cytometry of cerebrospinal fluid can aid in the diagnosis when needle core biopsy is not possible or to evaluate for leptomeningeal involvement
- Flow cytometric immunophenotyping can identify abnormal B cell populations showing light chain restriction
- References: J Exp Clin Cancer Res 2016;35:128, Cytometry B Clin Cytom 2018;94:928
- MYC or BCL2 gene rearrangements are rare; BCL6 gene rearrangements are frequent (Blood Adv 2019;3:3953)
- Oncogenic gain of function mutations in MYD88 (MYD88 L265P) are frequent (Blood 2016;127:869)
- Brain, biopsy:
- Diffuse large B cell lymphoma (see comment)
- Comment: In the absence of systemic disease, the morphologic and immunophenotypic findings are consistent with a diagnosis of primary diffuse large B cell lymphoma of the central nervous system. Clinical correlation is required, such as excluding an immunodeficiency associated lymphoproliferative disorder.
- Systemic malignant lymphoma (diffuse large B cell lymphoma or other) involving the CNS:
- CD10 expression by the lymphoma should strongly raise the possibility
- Prior history and clinical workup is required to exclude systemic lymphoma involving the CNS
- Lymphoma associated with HIV infection:
- EBV positivity suggests this
- Clinical workup is required
- Posttransplant lymphoproliferative disorder (PTLD):
- EBV positive
- History of immunosuppression after allograft
- High grade glioma:
- Lacks the diffuse monomorphic lymphocyte infiltrate, neuronal morphology and IHC staining
- CD45 negative
- Infection including toxoplasma or cerebral abscess:
- Occasionally multiloculate enhancement, generally encapsulated
- Suppurative with mixed acute and chronic inflammation
- Metastatic melanoma or carcinoma:
- Demyelinating lesions:
- May not enhance on imaging
- Generally demarcated
- Histiocytic as opposed to lymphoid infiltrate
- Cerebral infarct:
- Ischemic, "red dead" neurons
- Slight to no lymphoid infiltrate
- Histiocytes predominate

A 73 year old woman presents with progressive confusion and lethargy. Imaging shows a solitary lesion in the parietal lobe. A brain biopsy is performed and intraoperative touch preparations show discohesive, monomorphic, basophilic cells with crush artifact. Permanent sections show perivascular cuffing by intermediate to large mononuclear cells with scant cytoplasm, vesicular chromatin and prominent nucleoli. By immunohistochemical stains, the mononuclear cells are positive for CD20, BCL6 and MUM1 and negative for CD10 and EBV. What is the most likely diagnosis?
- Glioblastoma multiforme (GBM)
- Metastatic melanoma
- Metastatic neuroendocrine carcinoma
- Primary central nervous system lymphoma
Comment Here
Reference: Primary CNS lymphoma
- BCL6
- CD10
- CD20
- IRF4 / MUM1
Comment Here
Reference: Primary CNS lymphoma
- Mucus containing cyst arising from Rathke cleft remnants
- Intrasellar or suprasellar
- Incidental postmortem finding
- Larger cysts, usually in adults, may be symptomatic
- Thin walled, mucinous contents
- Lined by columnar ciliated epithelium with goblet cells
- Variable fragments of pituitary tissue, squamous metaplasia, xanthogranulomatous reaction
- Craniopharyngioma, papillary
- Normal intermediate lobe cysts: small, asymptomatic, lack goblet cells and cilia
- WHO grade 3 due to aggressive behavior and metastatic potential
- High rate of recurrence
- Rhabdoid morphology may increase with recurrences
- May be supratentorial, infratentorial or spinal
- MRI may show irregular tumor borders, cystic component, heterogenous tumoral enhancement and peritumoral edema suggesting a high grade meningioma (AJNR Am J Neuroradiol 2007;28:1462)
Images hosted on other servers:
Marked enhancing mass at left frontal lobe

Hyperostosis, bony erosion in right anterior clinoid process
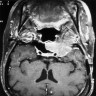
Tumor in right infratemporal fossa
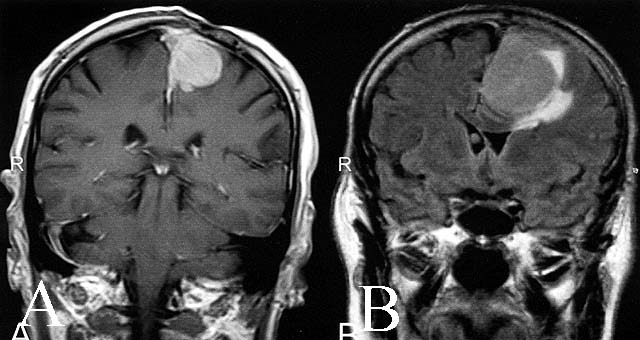
Lesion was isointense on T1 and T2 weighted images
Avidly enhancing, complex falcine meningioma
- 8 year old girl (Med Arh 2010;64:123) and 15 year old boy (Am J Surg Pathol 2001;25:964)
- 47 year old woman with recurrent tumor (Skull Base 2003;13:51)
- 55 year old woman with intraventricular tumor (J Clin Neurosci 2007;14:672)
- 60 year old man with recurrent extracerebral tumor (University of Pittsburgh: A Male in His Late 60's with Recurrent Extracerebral Tumor)
- Lacking malignant features (Clin Neuropathol 2004;23:16)
- Intraspinal tumor with metastasis to liver (J Clin Neurosci 2011;18:714)
- Gross total resection
- Postsurgical radiotherapy (Front Oncol 2013;3:227)
- Radiosurgery (Radiat Oncol 2014;9:38)
- Dural based
- May be well circumscribed or readily adherent to brain parenchyma
- Size can vary
- Sheets of loosely cohesive cells with eccentric nuclei and hyaline, paranuclear inclusions
- Nuclei may show pleomorphism, with vesicular chromatin and prominent nucleoli
- May retain meningothelial features including whorl formation and nuclear pseudoinclusions
- May have brain invasion
- Often increased mitotic activity (greater than 4/10 HP)
- May have papillary features
- Rhabdoid cells with abundant eosinophilic cytoplasm, cytoplasmic hyaline inclusions, eccentric nuclei
- Stout processes consistent with meningioma; however distinct cell borders
- Often vesicular nuclei with prominent nucleoli
- May see mitoses on smear
- May see pseudonuclear inclusions (Diagn Cytopathol 2010;38:594)
Images hosted on other servers:
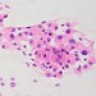
Squash prep 40x
- EMA (variable), PR (in a subset; helpful if positive, not ruled out if negative), vimentin
- INI1 (Mod Pathol 2005;18:951)
- Cytokeratins may pick up cytoplasmic inclusions
- Ki67 (often 10% or more)
- Cytoplasmic whorls of intermediate filaments often entrapping lysosomes or other organelles
- Interdigitating cell membranes and intercellular junctions
- Low grade glioneuronal neoplasm with neurocytic and glial components
- CNS WHO grade 1
- Established as a distinct disease entity by Komori et al. (2002) and later endorsed by WHO classification (2007) (Am J Surg Pathol 2002;26:582, Acta Neuropathol 2007;114:97)
- Rare, slow growing glioneuronal neoplasm (Neurooncol Adv 2020;2:vdaa116)
- Preferentially affects young adults, adolescents and children
- Occurs throughout CNS with predilection for fourth ventricle region
- 2 distinct histological components: neurocytic with distinctive rosettes and glial areas
- Recently, rosette forming glioneuronal tumors (RGNTs) with malignant behavior have been reported (Oncotarget 2017;8:109175)
- Rosette forming glioneuronal tumor of the fourth ventricle (Acta Neuropathol 2007;114:97)
- The 2016 WHO classification modified its description of RGNT and omitted the location qualifier "of the fourth ventricle" (Acta Neuropathol 2016;131:803)
- ICD-O: 9509/1 - rosette forming glioneuronal tumor
- ICD-11: 2A00.21 & XH2JU8 - mixed neuronal glial tumors & rosette forming glioneuronal tumor
- Rare; specific population based incidence rates are not available
- Preferentially affects young adults, adolescents and children; mean age at diagnosis is 28 years (range: 2 - 81 years) (Oncotarget 2017;8:109175)
- No gender predilection (Oncotarget 2017;8:109175)
- Typically arises in midline and involves posterior fossa with predilection for fourth ventricle (38.3%) and cerebella (31.4%) (Oncotarget 2017;8:109175)
- Recently, it was reported to be present nearly throughout the CNS at both supratentorial and infratentorial sites (Oncotarget 2017;8:109175)
- Sites reported include pineal region / tectum, third ventricle, aqueduct, lateral ventricle, spinal cord, thalamus, suprasellar region, basal ganglia, brain stem, septum pellucidum, frontal lobe, temporal lobe and optic chiasm (Quant Imaging Med Surg 2012;2:227, Cureus 2016;8:e857, Interdiscip Neurosurg 2020;19:1, Surg Neurol Int 2020;11:138, Radiol Case Rep 2021;16:3982, Neurochirurgie 2016;62:60, Oncotarget 2017;8:109175, Childs Nerv Syst 2020;36:2867, Neurooncol Adv 2020;2:vdaa116, Neurosurgery 2009;64:E771)
- Histogenesis is unclear; it has been postulated to arise from pluripotent cells of the subependymal plate or the internal granule cell layer of cerebellum or from periventricular stem cell origin with biphenotypic differentiation (J Pediatr Neurosci 2017;12:168, Neurosurgery 2008;62:E1162, Clin Neuropathol 2013;32:370, Virchows Arch 2012;461:581)
- Synergistic interaction between the MAPK and PI3K signaling pathways seem to be involved in the formation of RGNTs; the constitutive activation of the FGFR signaling likely plays a role in a large portion of these tumors together with frequent PIK3CA mutation (Acta Neuropathol 2019;138:497, Cold Spring Harb Mol Case Stud 2016;2:a001057, Neurooncol Adv 2020;2:vdaa116)
- A few cases have been reported in association with neurofibromatosis type 1 and Noonan syndrome, suggesting a genetic susceptibility (Neurosurgery 2009;64:E771, Clin Neuropathol 2011;30:297)
- Nonspecific and site related
- Headache is the most common symptom (60.6%), followed by nausea / vomiting (29%), ataxia (27%) and vertigo / dizziness (20%) (Oncotarget 2017;8:109175)
- Rarely, asymptomatic (7.7%) (Oncotarget 2017;8:109175)
- Neuroimaging: magnetic resonance imaging (MRI) is the preferred modality, computed tomography (CT) (Br J Neurosurg 2012;26:668)
- Biopsy
- WHO essential and desirable diagnostic criteria
- Essential diagnostic criteria (presence of the following 3 characteristics)
- Biphasic histomorphology with a neurocytic component and a glial component
- Uniform neurocytes forming rosettes or perivascular pseudorosettes associated with synaptophysin expression
- (For unresolved lesions) small biopsies showing only 1 tumor component (neurocytic or glial) and a methylation profile of rosette forming glioneuronal tumor
- Desirable diagnostic criteria
- FGFR1 mutation with co-occurring PIK3CA or NF1 mutation
- Essential diagnostic criteria (presence of the following 3 characteristics)
- CT (Oncotarget 2017;8:109175)
- Hypodense midline mass
- Calcification (< 25%)
- MRI (Oncotarget 2017;8:109175)
- Relatively circumscribed midline mass, solid or cystic - solid pattern (47%), cystic pattern (35%) and mixed cystic solid pattern (18%)
- T1 hypointense, T2 hyperintense
- Variable contrast enhancement
- Heterogeneous (44.7%), rim (23.7%) or focal (7.9%) enhancement
- No enhancement (25%)
Contributed by Eman Abdelzaher, M.D., Ph.D.

CT without contrast

CT with contrast
Images hosted on other servers:
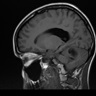
Cerebellar and fourth ventricular RGNT

MRI of cerebellar vermis RGNT
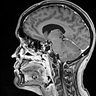
Third ventricle RGNT

MRI of aqueduct and thalamic RGNT
Postcontrast T1 weighted MRI
MRI of pineal region and third ventricle RGNT
- Mostly indolent course with stable disease in 88% of cases (Clin Case Rep 2021;9:e04355, Oncotarget 2017;8:109175)
- Favorable prognosis in terms of survival but location may complicate surgical removal with a significant risk of neurologic injury
- RGNTs have the potential for aggressive behavior with tumor dissemination, recurrence, progression or death reported in 12% of cases (Neurooncol Adv 2020;2:vdaa116, Oncotarget 2017;8:109175
- Risk factors for progression: pediatric age, purely solid tumors and inadequate resection (Oncotarget 2017;8:109175)
- 4 year old girl with spinal RGNT (Radiol Case Rep 2021;16:3982)
- 23 year old woman with diffusely invasive supratentorial RGNT (J Neurosurg Case Lessons 2023;6:CASE23435)
- 24 year old man with highly aggressive RGNT (Brain Tumor Pathol 2015;32:124)
- 41 year old woman with neurofibromatosis type 1 (NF1) and tectal plate RGNT (Cureus 2016;8:e857)
- 49 year old man with disseminated aggressive RGNT and spinal drop metastasis (World Neurosurg 2019;132:7)
- 75 year old woman with a thalamic RGNT and third ventricle dissemination (Neurochirurgie 2016;62:60)
- Surgery is the mainstay of treatment (Neurooncol Adv 2020;2:vdaa116)
- Long term, close follow up is needed
- Effectiveness of chemotherapy and radiotherapy remains unclear (Neurooncol Adv 2020;2:vdaa116, Oncotarget 2017;8:109175)
- Future perspective: targeted therapy with inhibitors of FGFR1, PI3K and MAPK pathways (Acta Neuropathol 2019;138:497)
Images hosted on other servers:
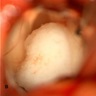
Right frontal exophytic lesion
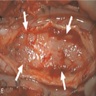
Spinal RGNT, intraoperative picture
- Well demarcated, soft, gelatinous
- Variable cellularity
- Small, bland and uniform nuclei with circle‐like arrangement (Clin Case Rep 2021;9:e04355)
Images hosted on other servers:
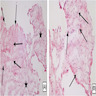
Moderately cellular and hypocellular areas
- Circumscribed, may show limited infiltration of surrounding parenchyma
- Biphasic with 2 distinct histological components: neurocytic and glial
- Neurocytic component
- Consists of uniform neurocytes forming neurocytic rosettes or perivascular pseudorosettes (Clin Case Rep 2021;9:e04355)
- Neurocytes have small round nuclei with fine stippled chromatin and scant cytoplasm
- Neurocytic rosettes are small with ring-like arrangement of neurocytes around delicate eosinophilic neuropil cores; may be arrayed in a cribriform pattern (Brain Pathol 2007;17:308)
- Perivascular pseudorosettes are narrow and formed of neurocytes with delicate cell processes radiating towards central vessels
- Neurocytic elements may lie unanchored in partly microcystic, mucinous matrix and, when sectioned longitudinally, may show a linear arrangement (Brain Pathol 2007;17:308)
- Glial component
- Typically, dominant component and low grade
- Resembles pilocytic astrocytoma with piloid and oligodendroglia-like cells
- Astrocytic tumor cells are spindle to stellate in shape, with elongated or oval nuclei and cytoplasmic processes forming a compact to loosely textured fibrillary background (Acta Neuropathol 2019;138:497)
- May show oligodendroglia-like cells with perinuclear haloes arranged in microcystic pattern or diffuse sheets (Brain Pathol 2007;17:308)
- Mitoses and necrosis absent or rare, atypia absent or minimal (Oncotarget 2017;8:109175, Acta Neuropathol 2019;138:497)
- Rosenthal fibers, eosinophilic granular bodies (EGBs), microcalcifications and hemosiderin deposits may be seen (Brain Pathol 2007;17:308, Acta Neuropathol 2019;138:497)
- Blood vessels may be ectatic, hyalinized, thrombosed or show glomeruloid microvascular proliferation (Oncotarget 2017;8:109175)
- Rarely, dysmorphic neurons may be seen in the glial regions (Brain Pathol 2007;17:308)
Contributed by Eman Abdelzaher, M.D., Ph.D.
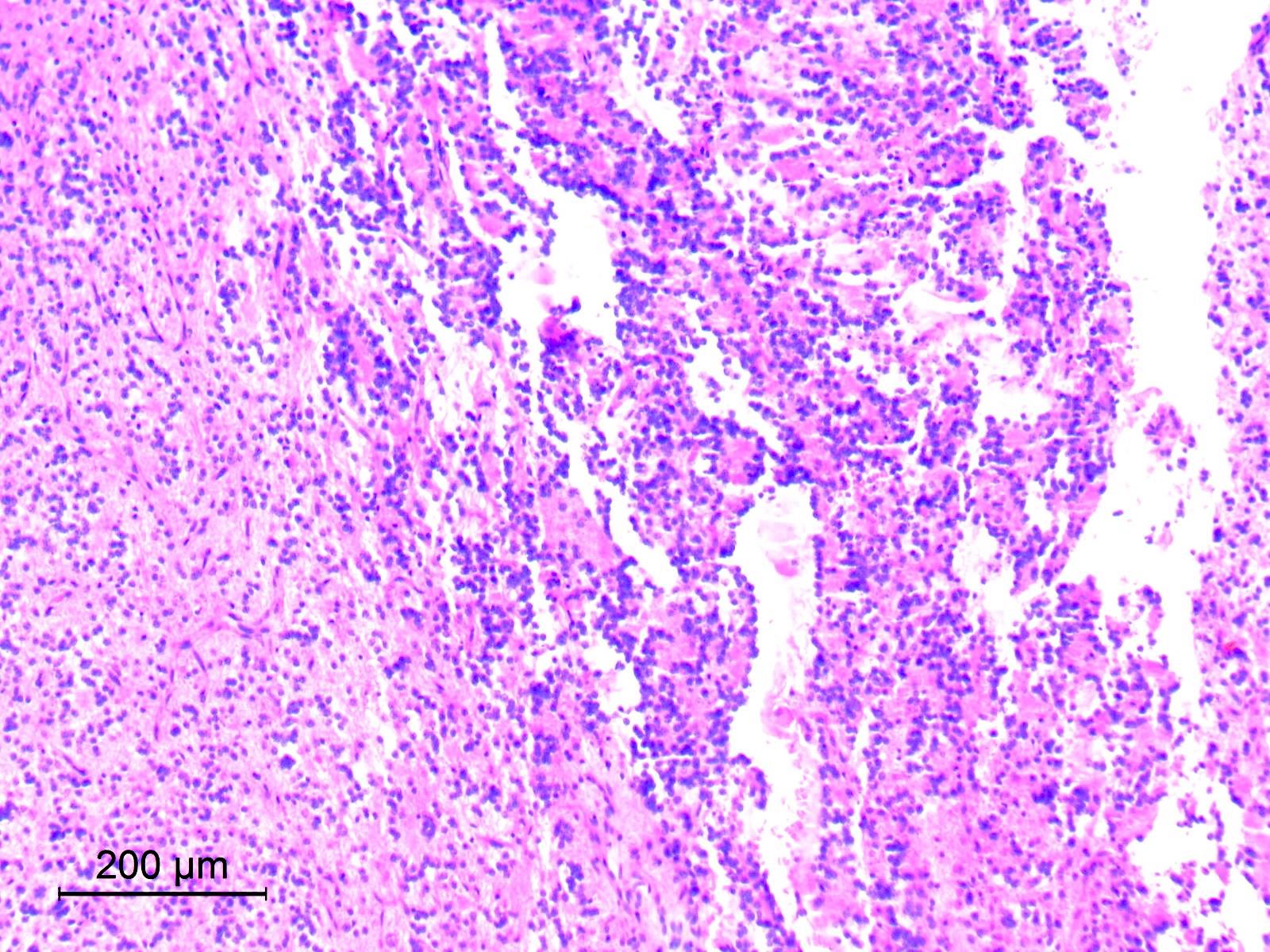
Biphasic neurocytic and glial components
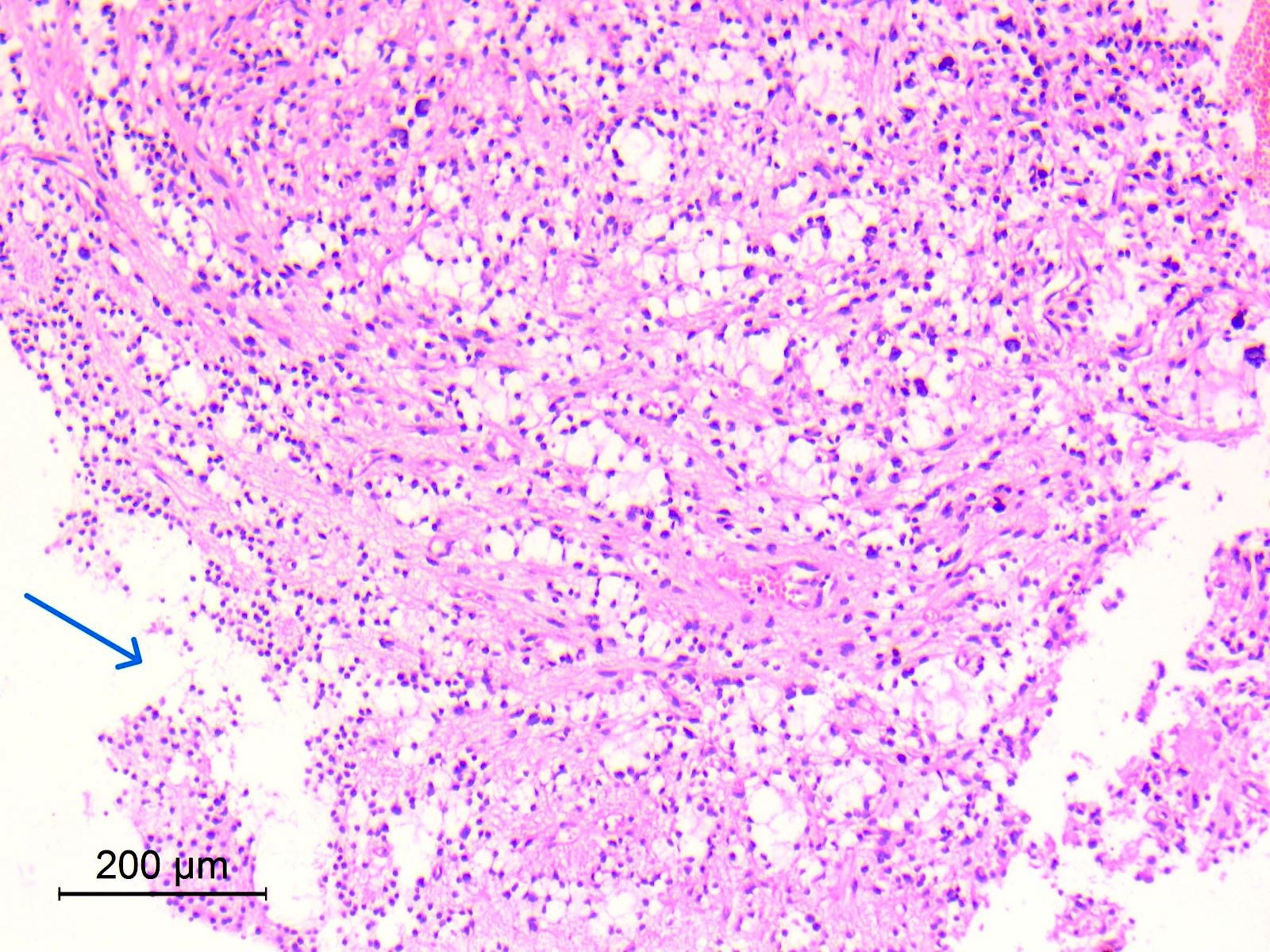
Biphasic neurocytic and pilocytic components
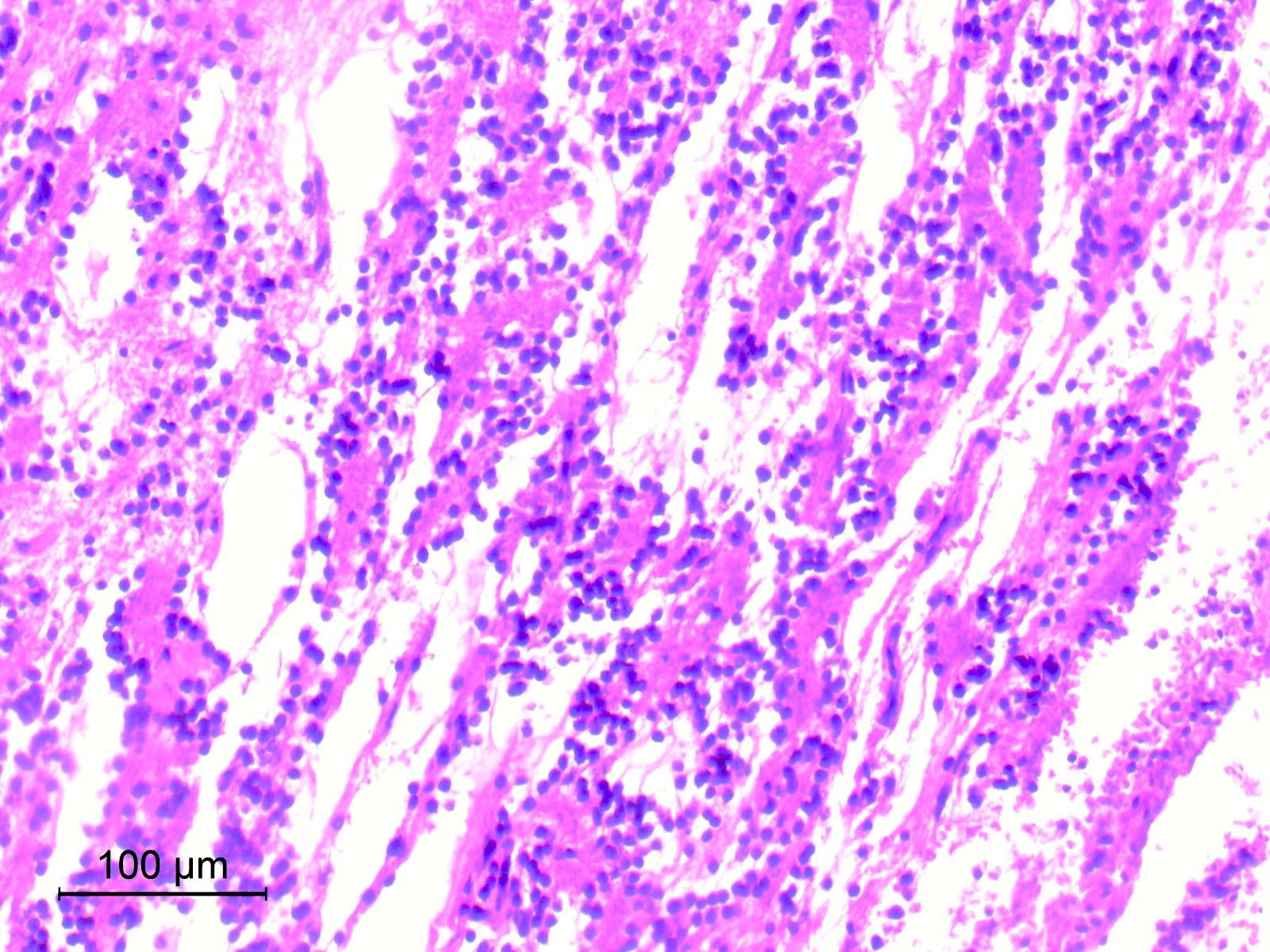

Neurocytic rosettes
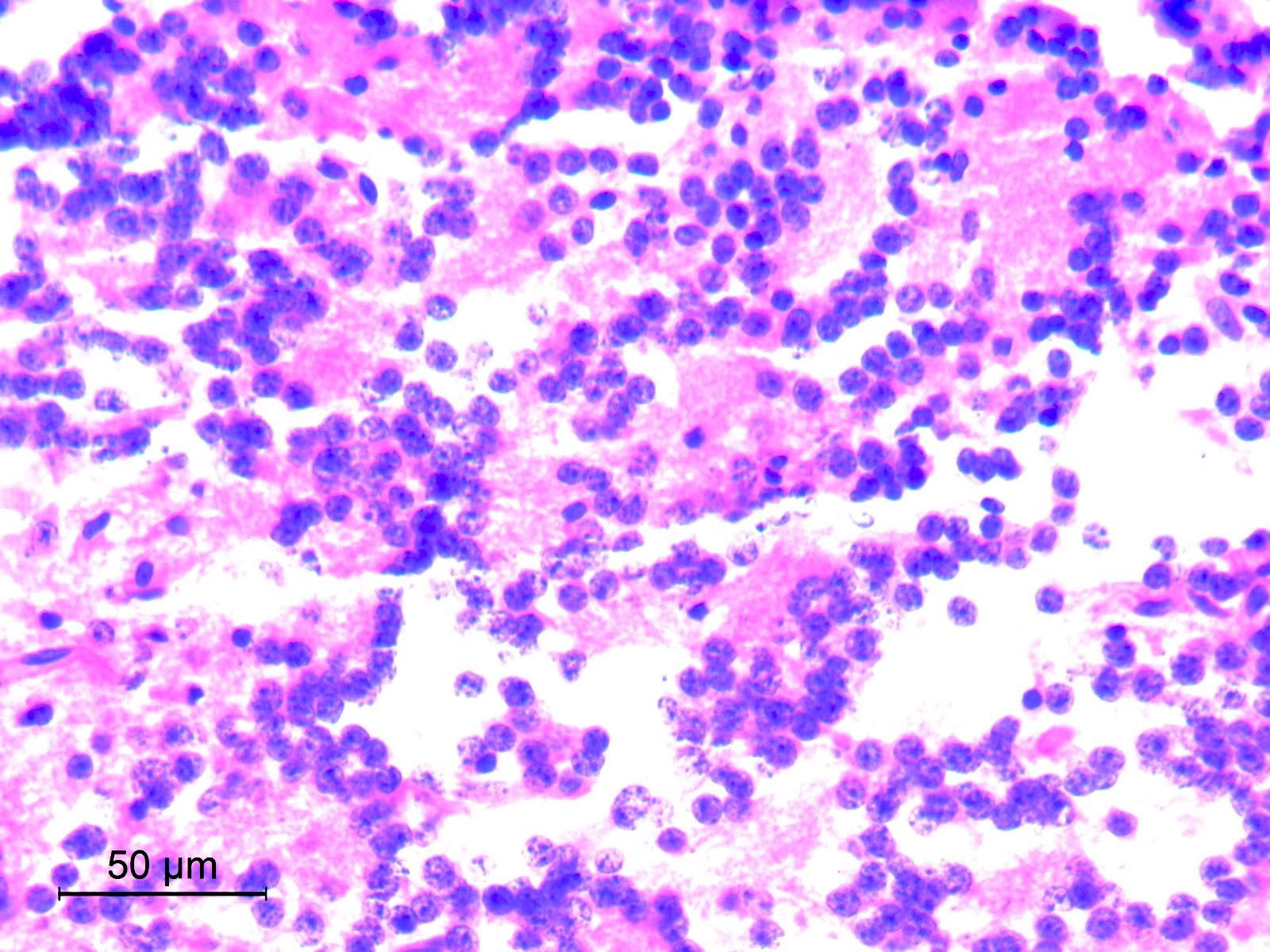
Neurocytes with stippled chromatin
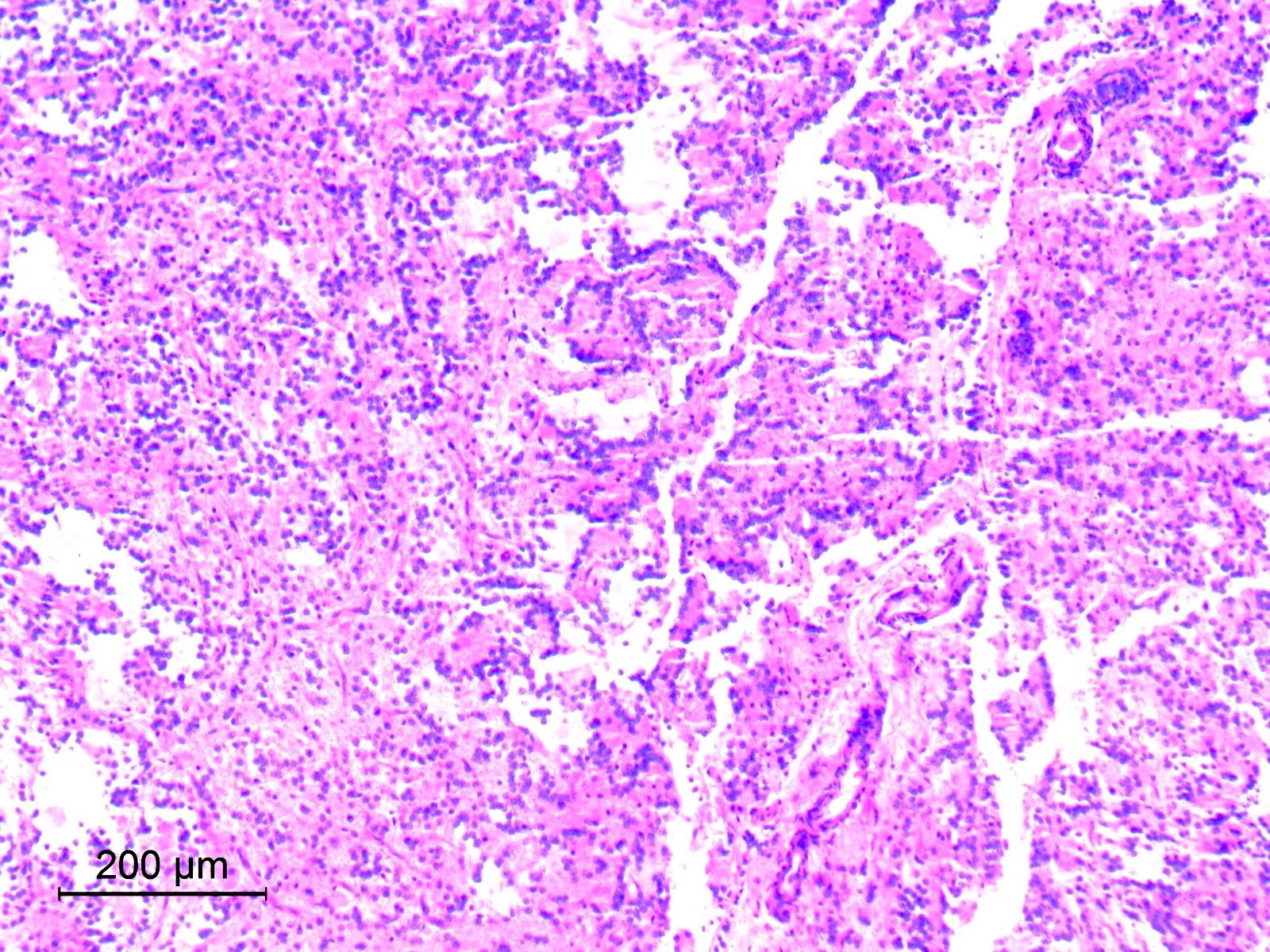

Neurocytic rosettes and pseudorosettes
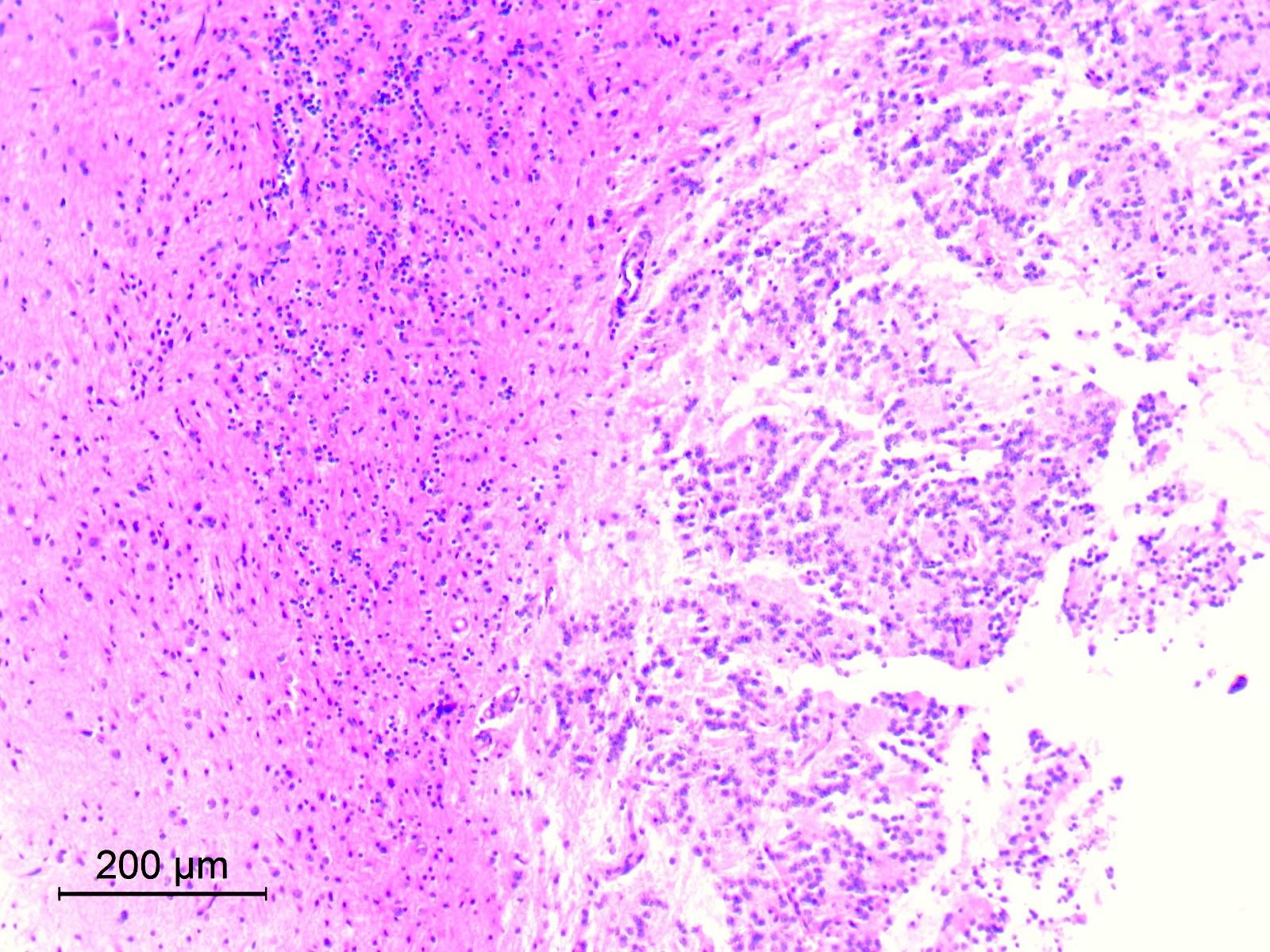
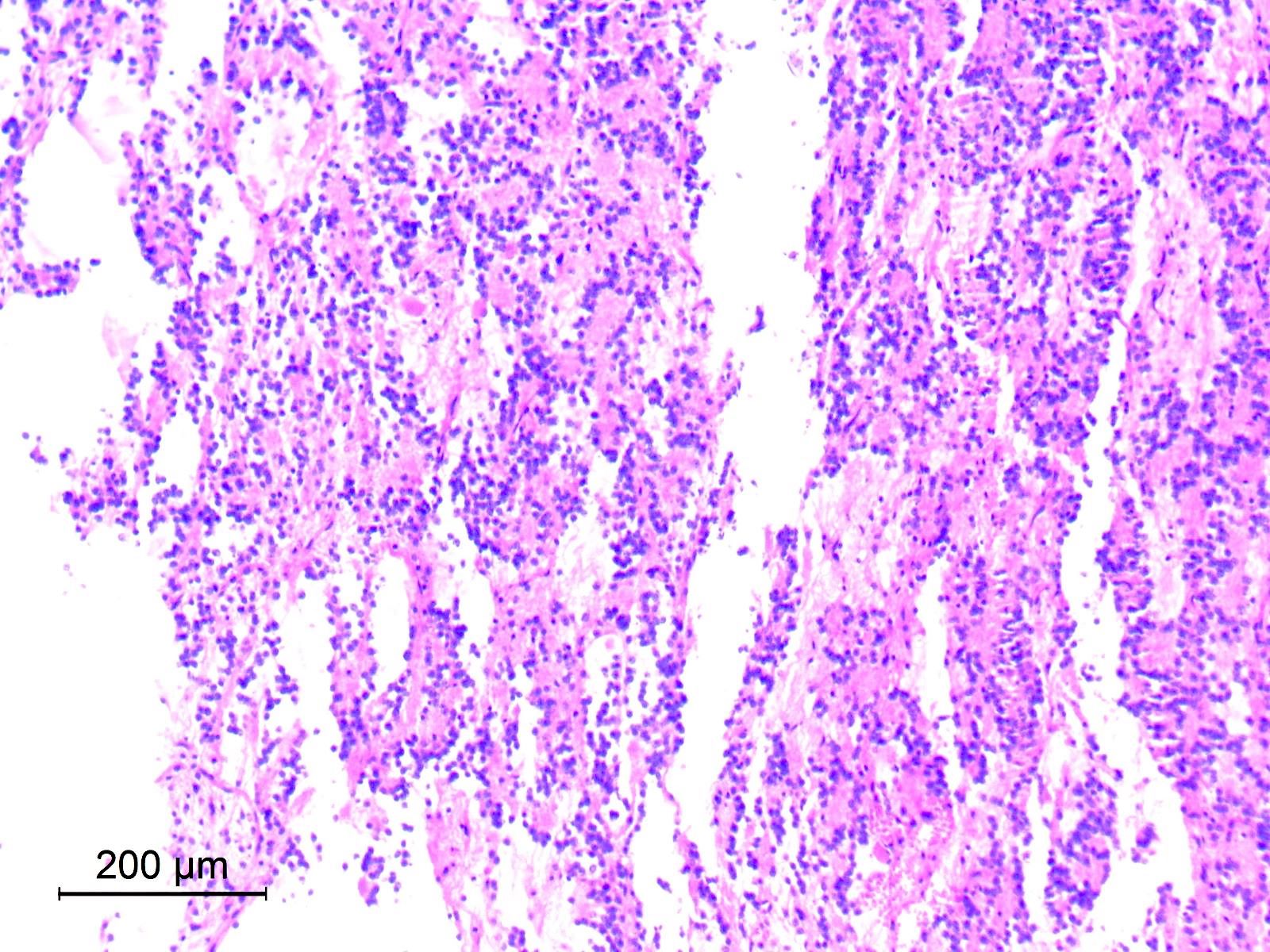
Cribriform pattern
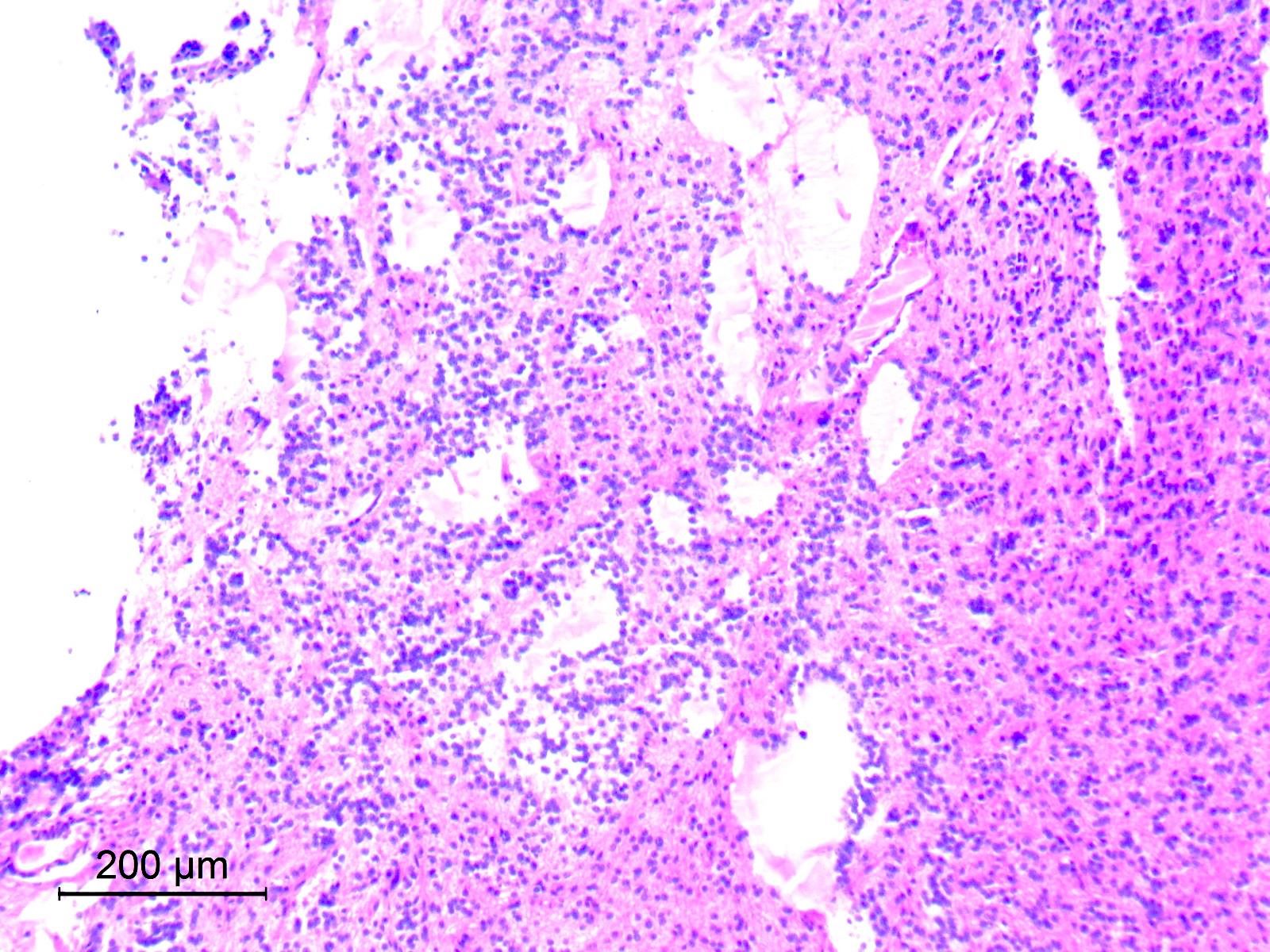
Microcystic pattern
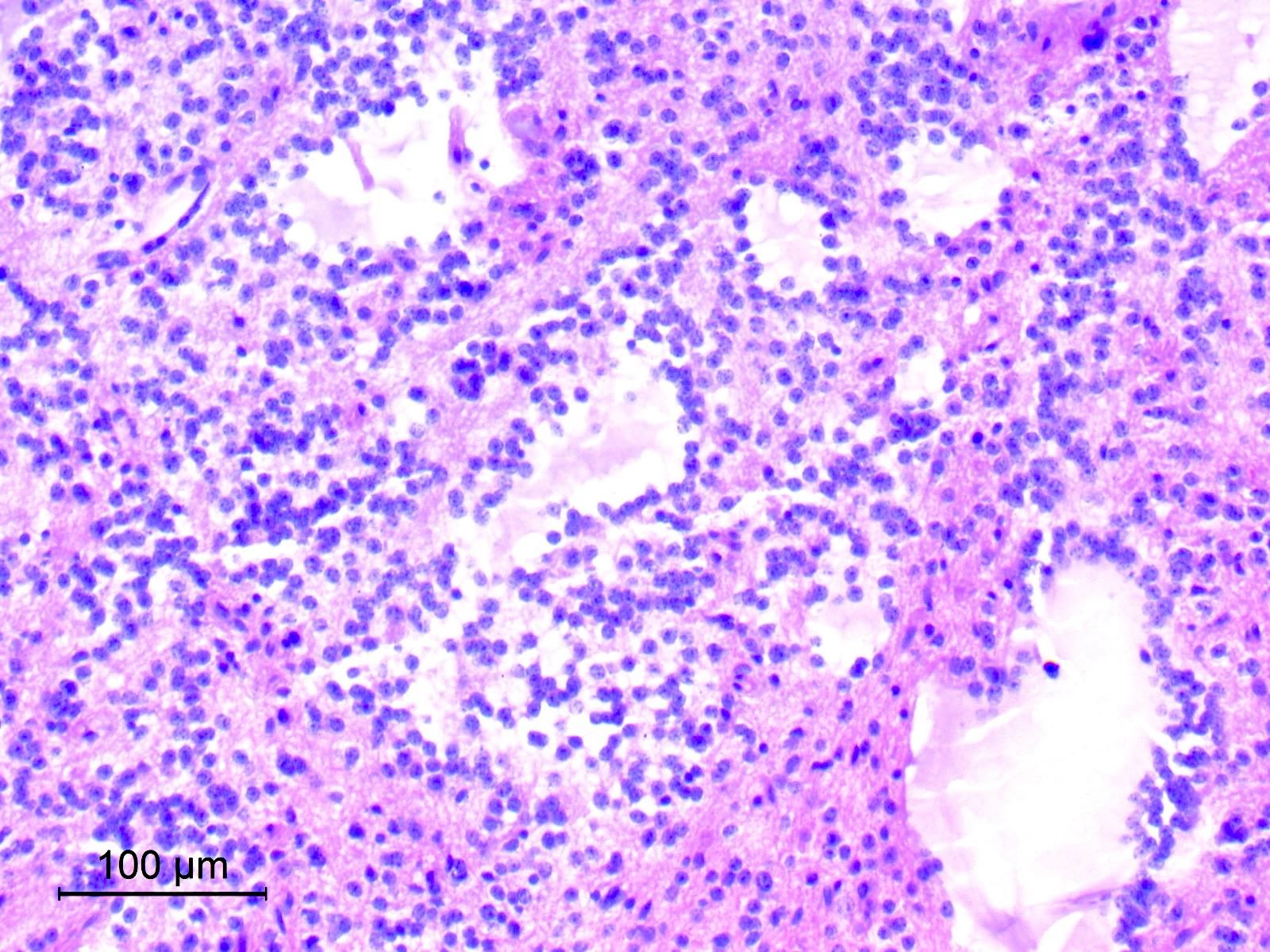
Microcystic pattern
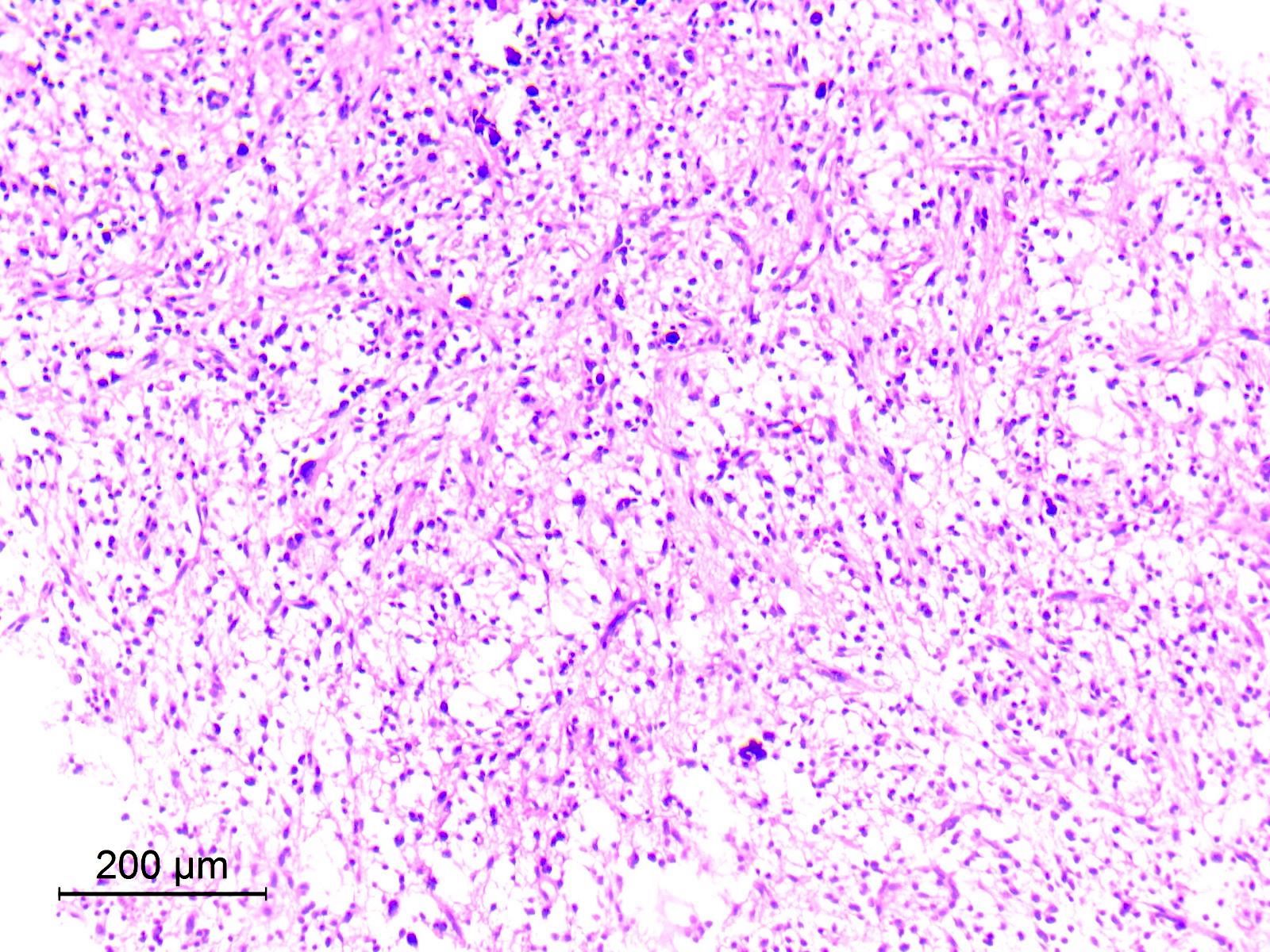
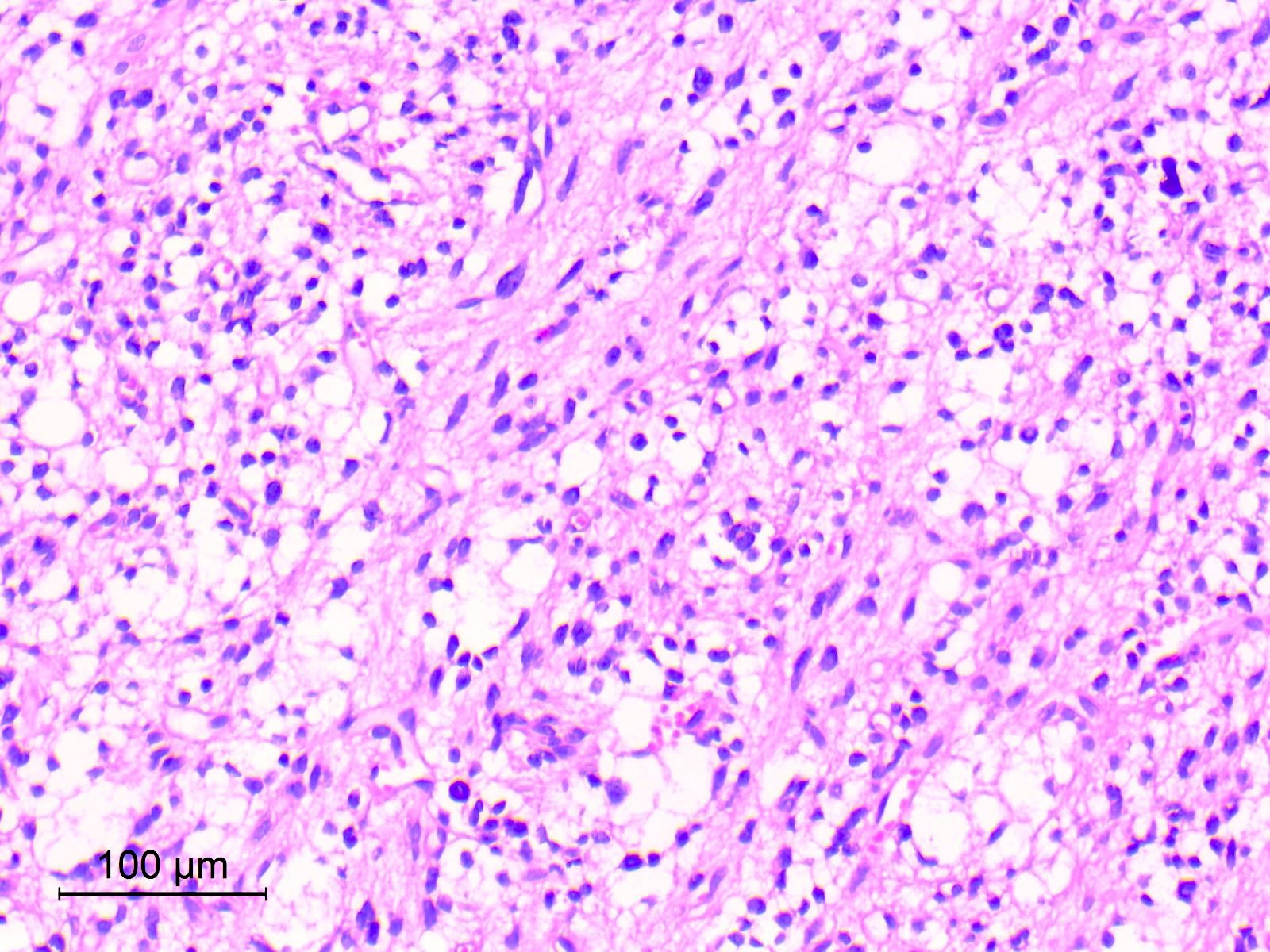
Pilocytic-like area
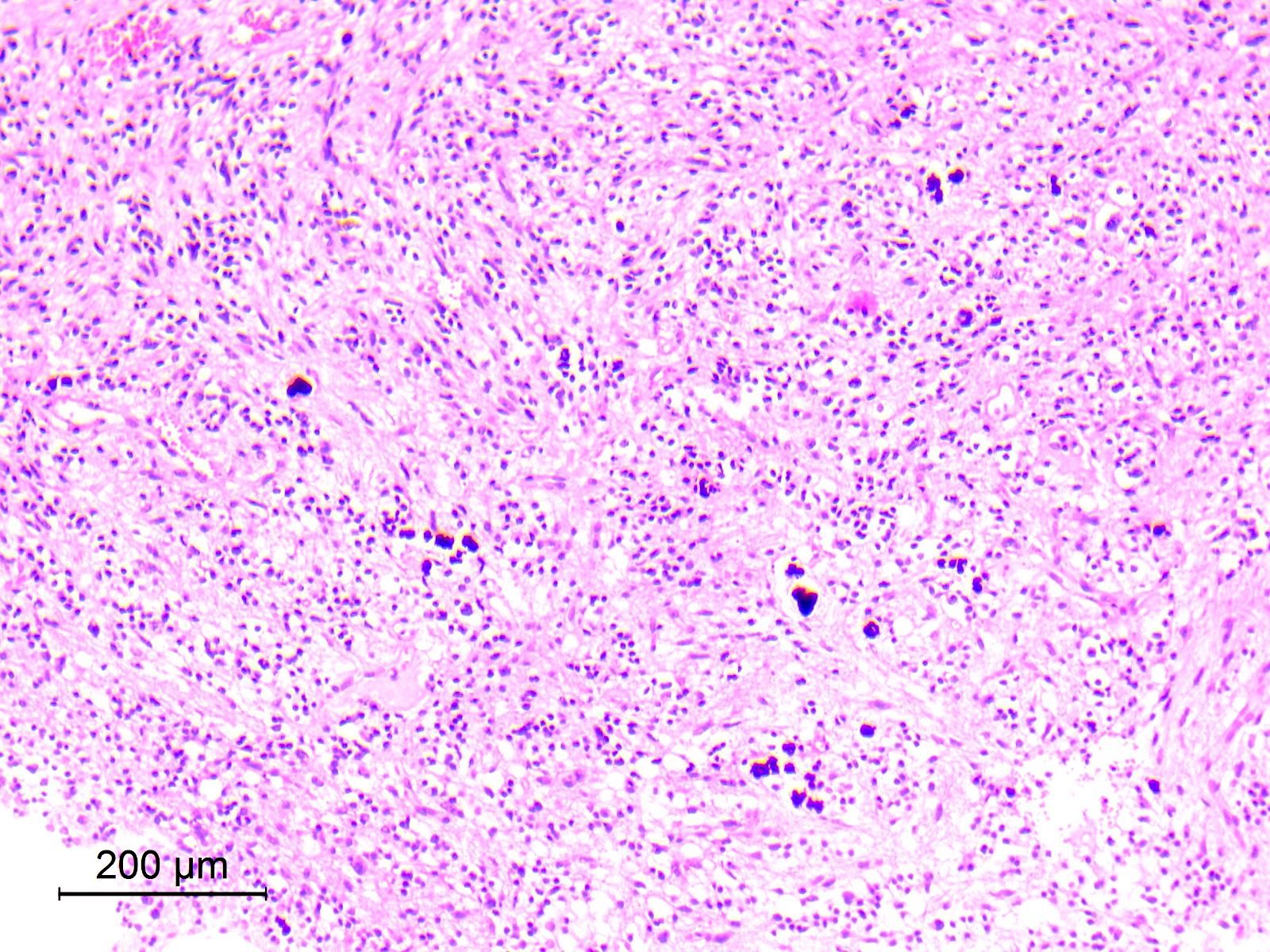
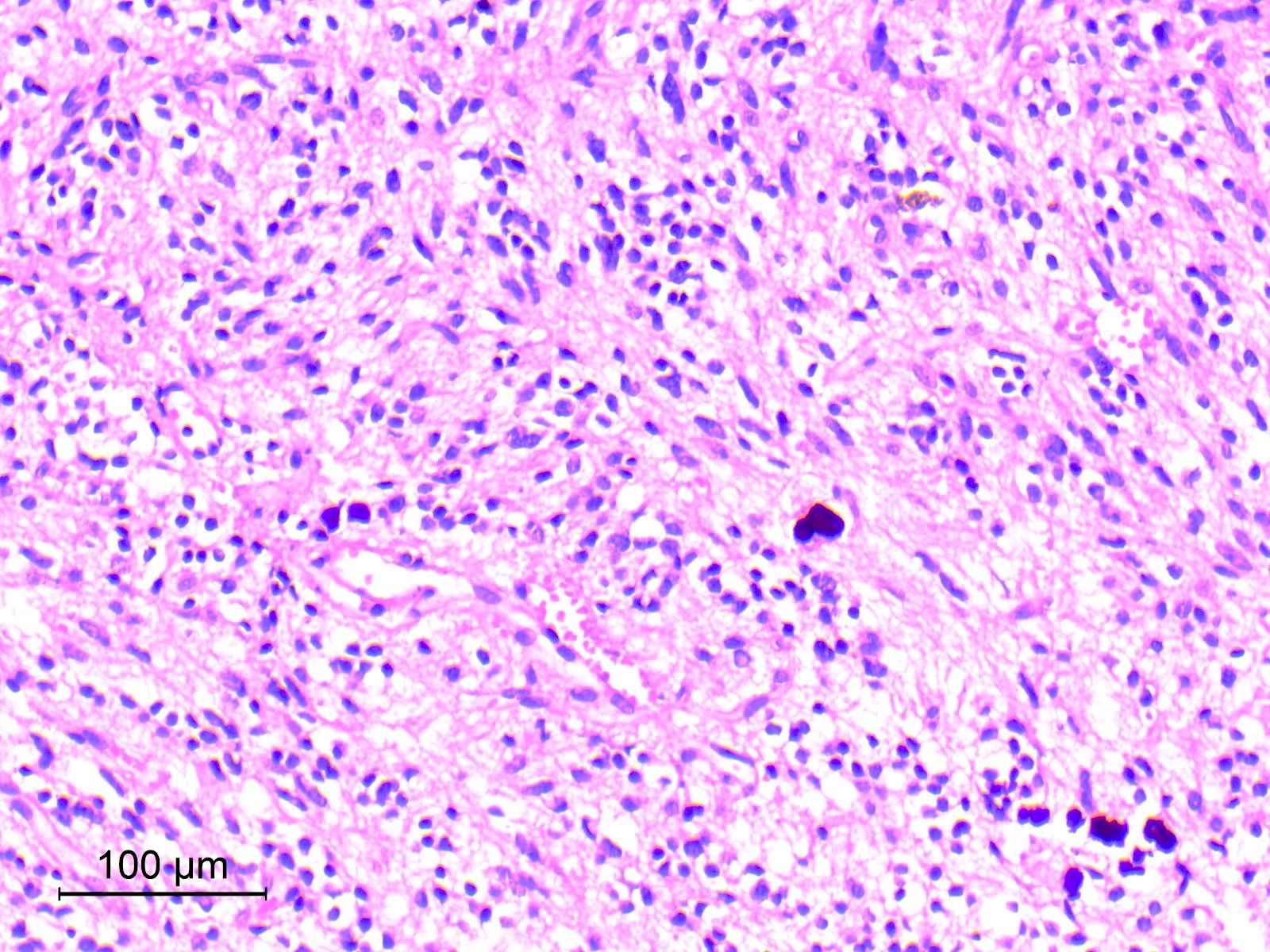
Piloid and oligodendroglia-like cells
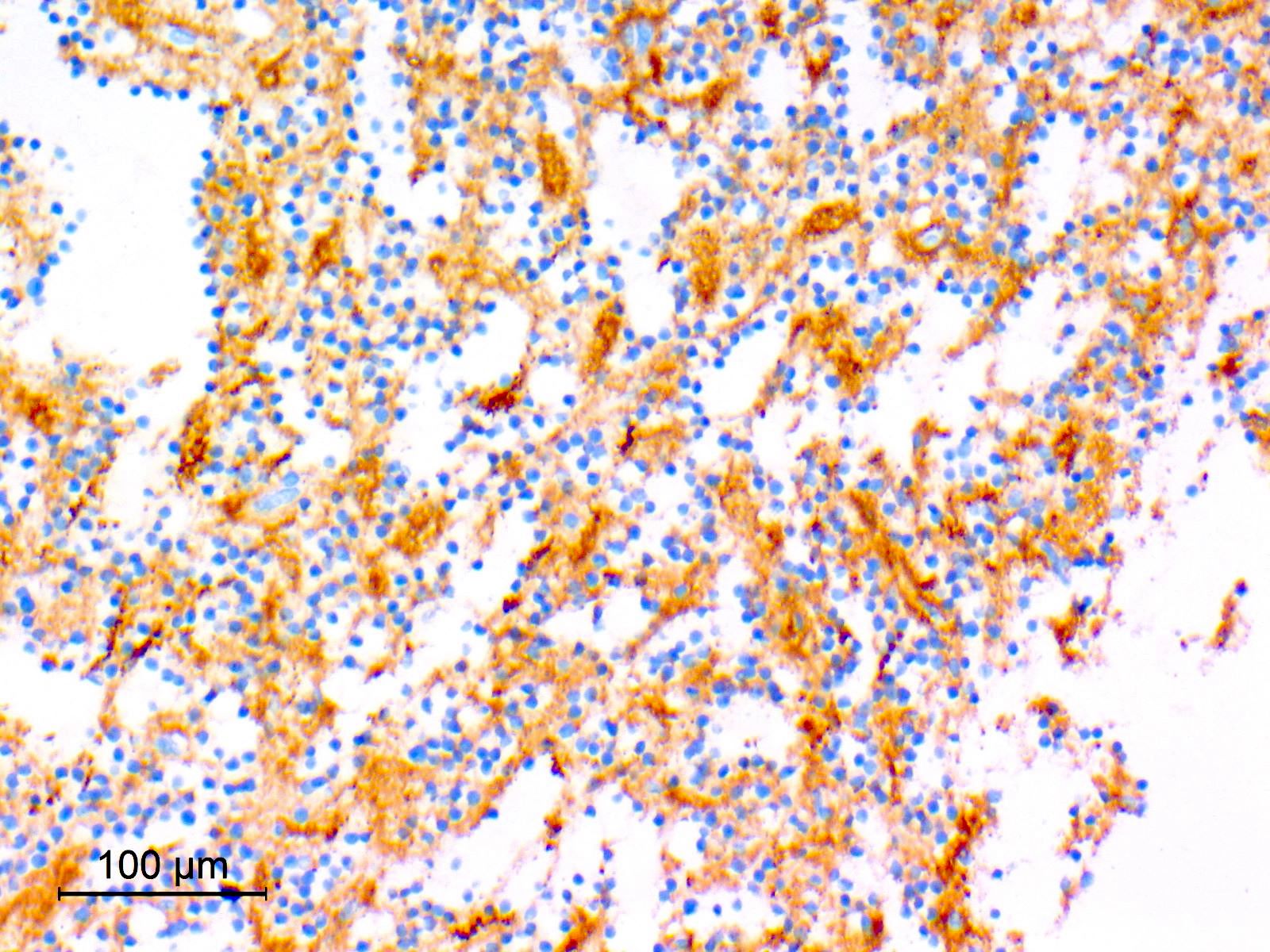
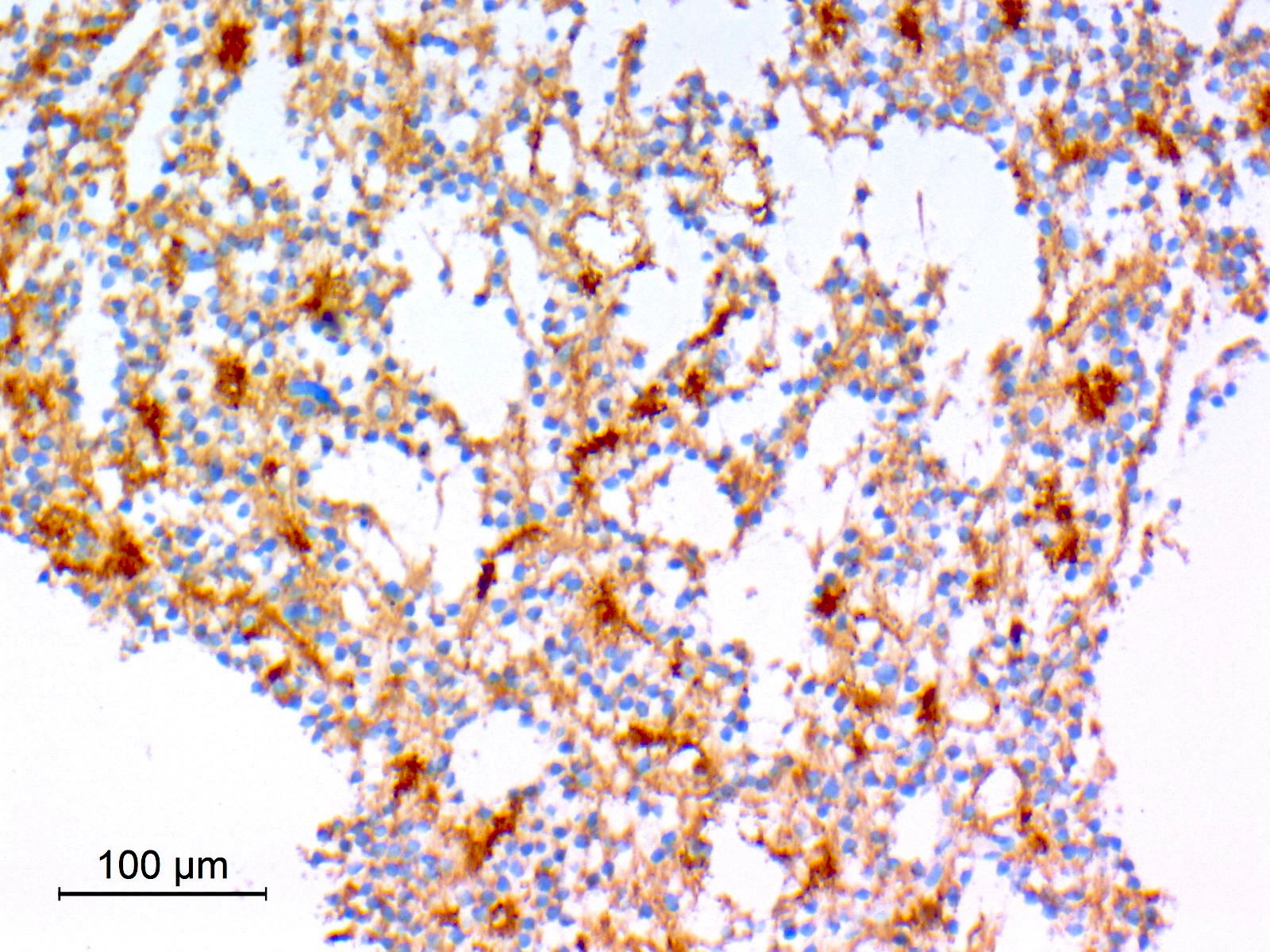
Synaptophysin
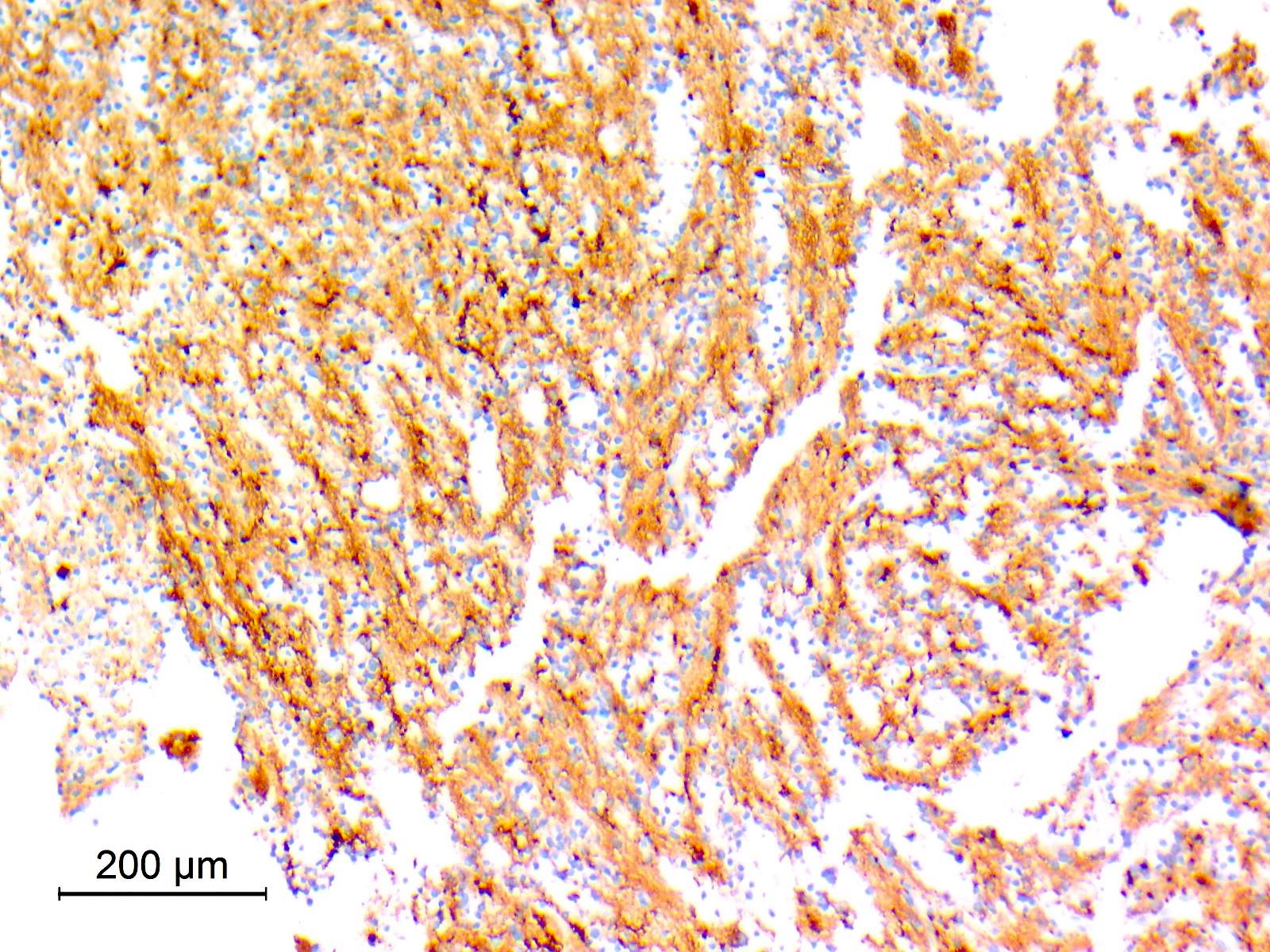

Synaptophysin
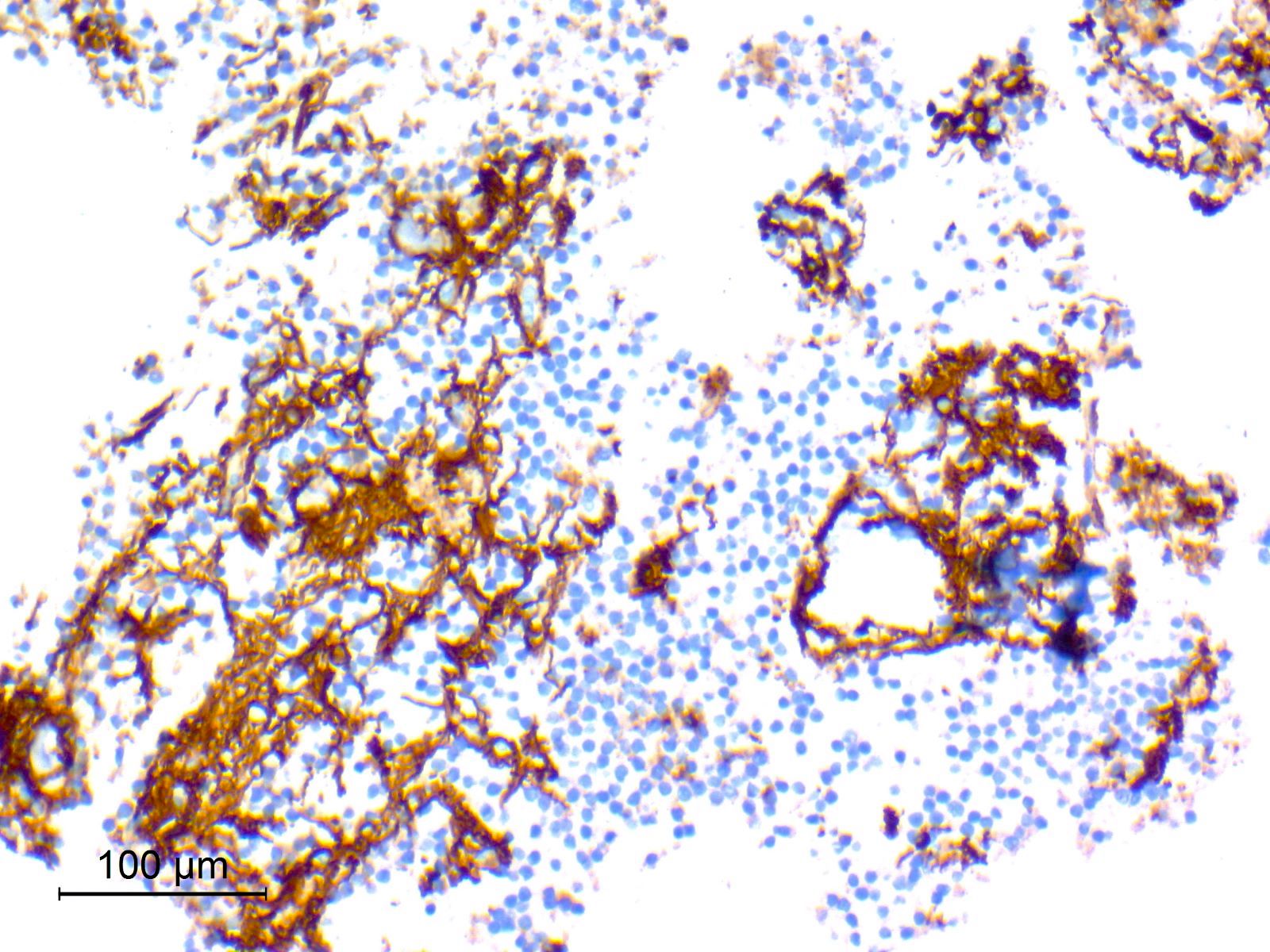
Negative neurocytes by GFAP
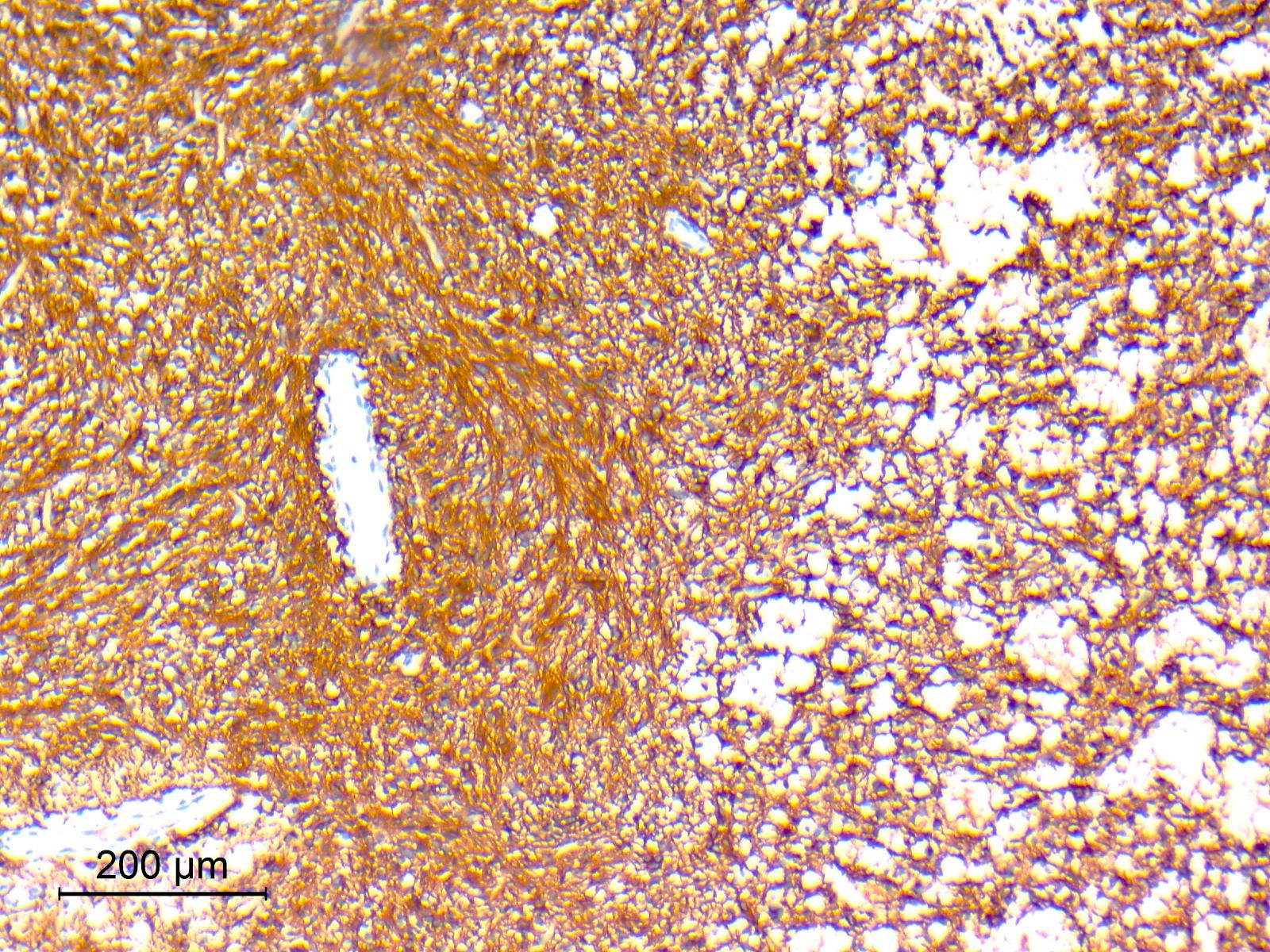
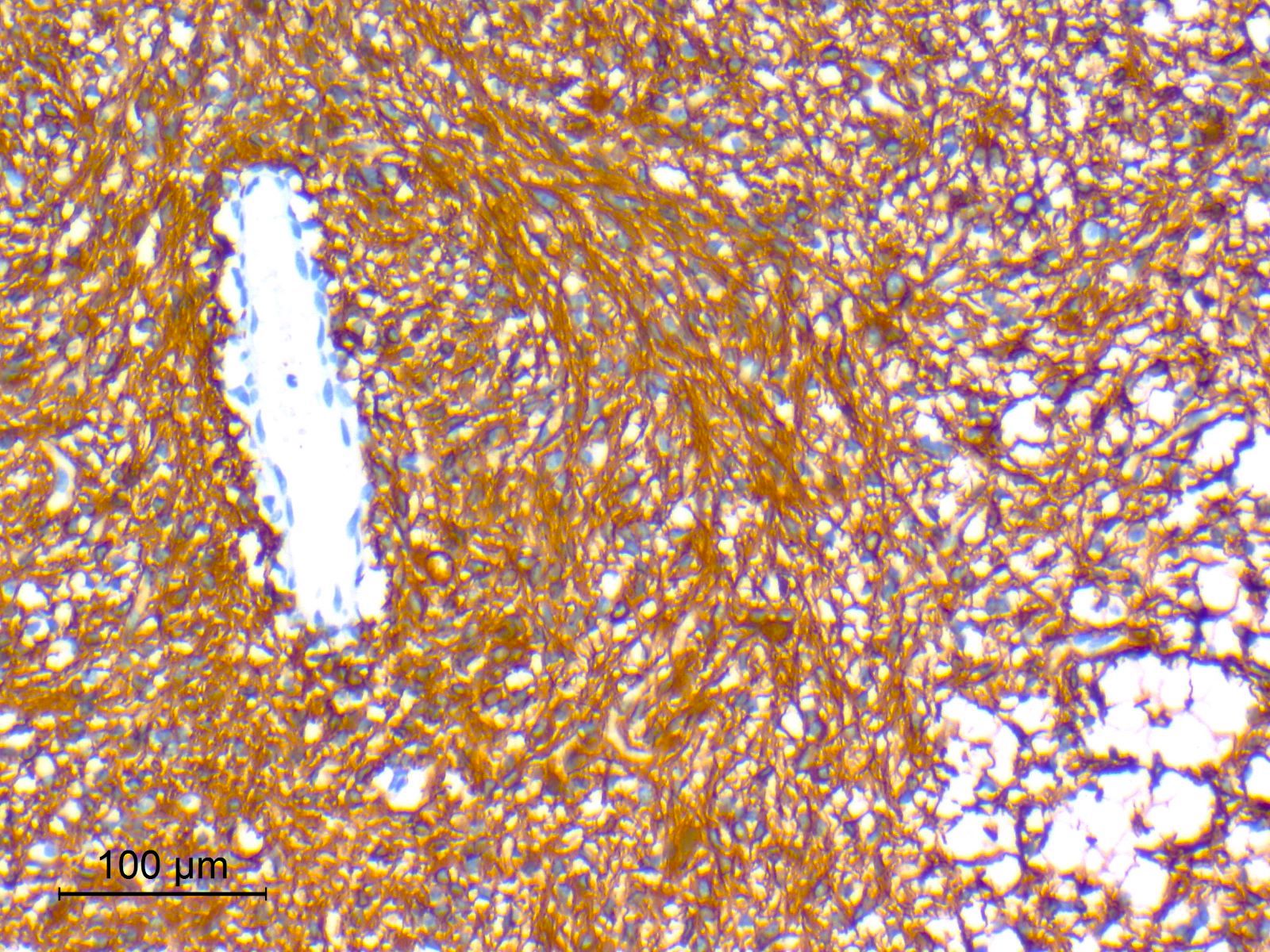
Glial fibrillary acidic protein (GFAP)
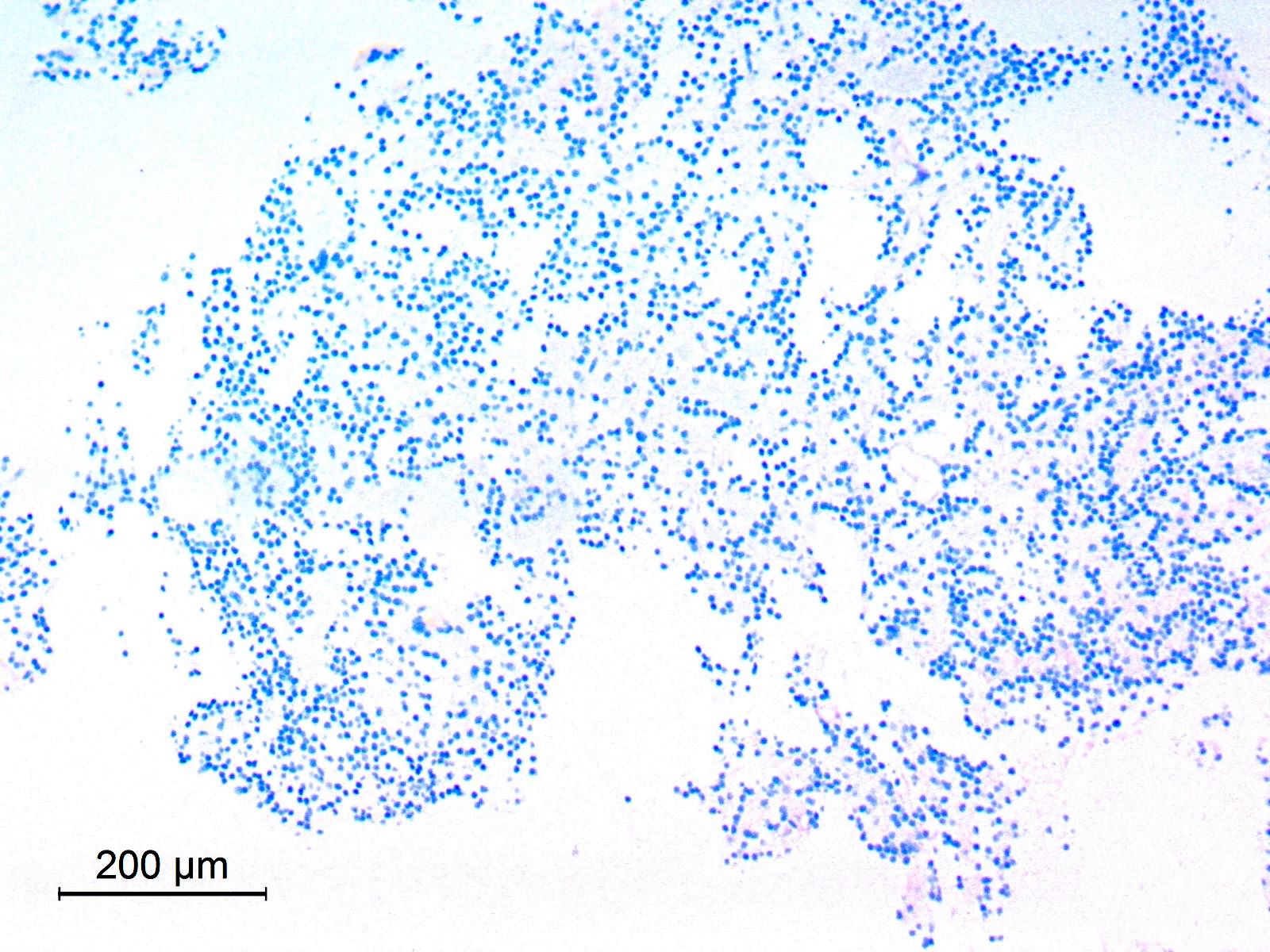
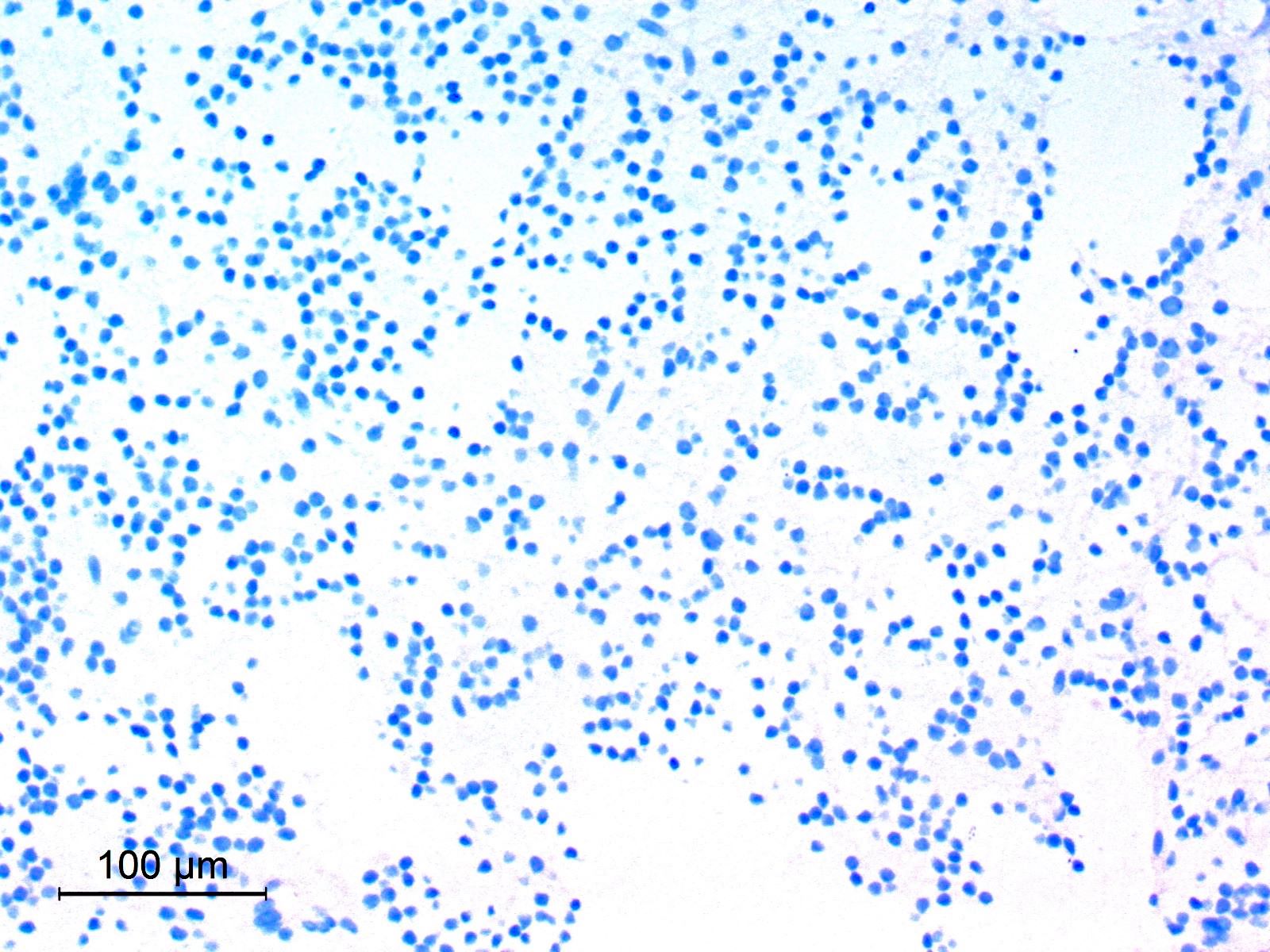
Epithelial membrane antigen (EMA)
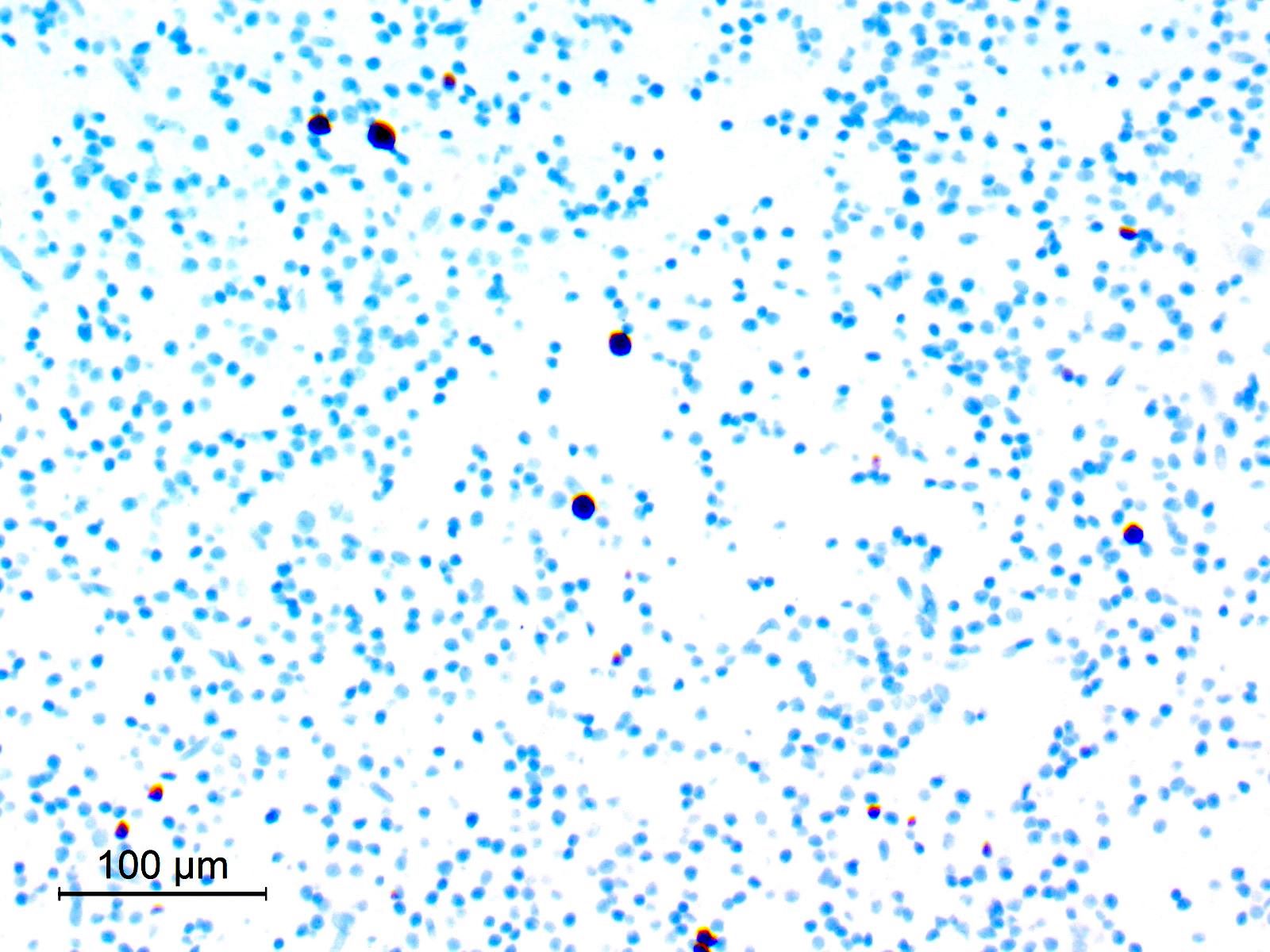
Ki67
- Cellular smears
- Neurocytic cells have uniform round nuclei with delicate chromatin, inconspicuous nucleoli and scant cytoplasm; neurocytic rosettes with fibrillar cores may be seen (Acta Cytol 2021;65:111)
- Glial, fibrillary fragments showing piloid astrocytes with elongated nuclei and coarse bipolar processes
- Dense extracellular mucoid substance
- No atypia
Images hosted on other servers:
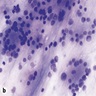
Neurocytic rosettes
- Synaptophysin: highlights neuropil cores of neurocytic rosettes and neuropil of perivascular pseudorosettes (Oncotarget 2017;8:109175, Cancers (Basel) 2023;15:1120)
- MAP2: in neurocytic tumor cells
- Neuron specific enolase (NSE): in neurocytic tumor cells (92%) (Oncotarget 2017;8:109175)
- Neurofilament protein (NFP): in neurocytic tumor cells (60.5%) (Oncotarget 2017;8:109175)
- Glial fibrillary acidic protein (GFAP): in glial component (Oncotarget 2017;8:109175)
- S100: in glial component (Cancers (Basel) 2023;15:1120)
- Olig2: in both neurocytic and pilocytic-like components (Oncotarget 2017;8:109175, Cancers (Basel) 2023;15:1120)
- Low Ki67 proliferation index: generally < 3% (ranging from 0 - 5%), rarely reaching up to 10% (Oncotarget 2017;8:109175, Acta Neuropathol 2019;138:497)
- Epithelial membrane antigen (EMA) (Oncotarget 2017;8:109175)
- CD34 (Acta Neuropathol 2019;138:497)
- NeuN (34%) positive: in neurocytic tumor cells (Oncotarget 2017;8:109175)
- Neurocytic component: neurocytic cells exhibit processes containing aligned microtubules, occasional dense core granules and rare mature synapses (Am J Surg Pathol 2002;26:582)
- Glial component: dense bundles of glial filaments (Neuropathology 2009;29:309)
- Defined by a distinct DNA methylation profile (Acta Neuropathol 2019;138:497)
- FGFR1 hotspot mutations (FGFR1 p.N546 or p.K656) are typical, with co-occurrence of PIK3CA mutations in the majority of cases (Acta Neuropathol 2019;138:497, Acta Neuropathol 2012;123:285)
- Loss of function mutation in NF1 in a subset of cases
- Lack of BRAF alterations
Images hosted on other servers:

RGNT, DNA methylation profiling

RGNT, recurrent genetic alterations
WHO classification of CNS tumors: glioneuronal tumors
- Brain mass lesion, fourth ventricle, gross total resection:
- Rosette forming glioneuronal tumor, CNS WHO grade 1
- Molecular genetics: methylation profile of rosette forming glioneuronal tumor
- Pilocytic astrocytoma:
- No neurocytic component (Pathol Res Pract 2007;203:613)
- Dysembryoplastic neuroepithelial tumor:
- Almost always supratentorial (Pathol Res Pract 2007;203:613)
- Floating neurons
- Lacks well formed rosettes
- Features overlap in some cases with RGNT
- Oligodendroglioma:
- Rare in posterior fossa
- Diffusely infiltrative growth pattern
- No rosettes
- 1p / 19q codeletion in adults (Pathol Res Pract 2007;203:613)
- Ependymoma:
- Perivascular pseudorosettes are GFAP+ and often Olig2- and synaptophysin- (Pathol Res Pract 2007;203:613)
- Cerebellar liponeurocytoma:
- Diffusely synaptophysin+
- Lipidized tumor cells (Can J Surg 2009;52:E117)
- No rosettes
- Papillary glioneuronal tumor:
- Supratentorial
- Perivascular GFAP+ glial cells (Pathol Res Pract 2007;203:613)
- Diffuse leptomeningeal glioneuronal tumor:
- Commonly shows diffuse involvement of the leptomeninges (AJNR Am J Neuroradiol 2020;41:2155)
- Absent or only focal GFAP immunoreactivity
- Characteristic molecular profile of KIAA1549::BRAF fusion with isolated 1p deletion or 1p / 19q codeletion
A 15 year old girl presented with a headache. MRI showed a posterior fossa mass lesion epicentered on the cerebellar vermis. Resection of the tumor was done and the histopathological features of the tumor are shown above. Which of the following is a typical genetic finding in this tumor?
- 1p / 19q codeletion
- BRAF alterations
- FGFR1 mutations
- PRKCA gene fusion
Comment Here
Reference: Rosette forming glioneuronal tumor
- Diffuse growth pattern
- Exclusive supratentorial location
- Neurocytic rosettes with synaptophysin positive neuropil cores
- Papillary architecture
Comment Here
Reference: Rosette forming glioneuronal tumor
- An embryonal tumor of the cerebellum with molecular evidence of SHH pathway activation (2016 WHO definition)
- WHO grade 4 tumors
- Medulloblastomas which fall into the desmoplastic / nodular or medulloblastoma with extensive nodularity histologic categories almost invariably demonstrate SHH activation
- Typically occur in children under 4 years of age, adolescents or young adults
- Those with concurrent TP53 mutation have much worse clinical outcomes and often display large cell / anaplastic histology
- SHH activation can be determined by molecular studies demonstrating mutations in this pathway, genome expression analysis, methylation classification or by immunohistochemistry demonstrating GAB1 expression
- ICD-10: C71.6 - malignant neoplasm of the cerebellum
- TP53 wildtype tumors largely show a bimodal age distribution with cases most common in infants and young adults
- TP53 mutant cases tend to arise in children aged 4 - 17
- M:F = 1.5:1
- References: J Pediatr Hematol Oncol 2009;31:970, J Clin Neurosci 2012;19:1541, Nat Rev Cancer 2012;12:818, Cancer Cell 2014;25:393
- Cerebellum; can involve both the hemispheres or vermis with site of origin possibly age dependent, with 1 study showing hemispheric tumors are more common in older children and young adults and those centered in the vermis more often occurring in infants (Acta Neuropathol 2014;127:931)
- Cerebellar granule cell neuron precursors are thought to give rise to SHH activated medulloblastoma (Cancer Cell 2008;14:123, Cancer Cell 2008;14:135)
- High proportion of TP53 mutant cases contain germline TP53 mutations (Li-Fraumeni syndrome) suggestive of a causal role (J Clin Oncol 2013;31:2927)
- SHH activated and TP53 mutant cases associated with Li-Fraumeni syndrome (Cell 2012;148:59, J Clin Oncol 2013;31:2927)
- SHH activated medulloblastoma is also associated with nevoid basal cell carcinoma syndrome (Gorlin syndrome) (J Clin Oncol 2014;32:4155)
- Patients often present with signs and symptoms of increased cranial pressure (headaches, nausea, vomiting) secondary to obstruction of cerebrospinal fluid (CSF) flow from a large mass impinging on the fourth ventricle
- Signs of cerebellar ataxia are common
- Cases presenting with CSF dissemination may show more specific neurologic signs
- Imaging of the head (CT, MRI) will show a large mass in the lateral hemisphere or involving midline structures
- Diagnosis determined by biopsy or resection, with ancillary studies necessary for medulloblastoma molecular subtyping (immunohistochemistry or methylation profiling or sequencing)
- References: Nature 2010;468:1095, Pediatr Blood Cancer 2013;60:1408
- On CT or MRI: solid, intensely contrast enhancing masses, which are often located in the lateral cerebellar hemisphere but also can be found arising in the midline
- Grape-like pattern may be seen in cases shown to have histology with extensive nodularity (J Neurosurg 1999;91:971, AJNR Am J Neuroradiol 2002;23:1564)
- Edema may be a common imaging feature (Neuroradiology 2011;53:387)
Contributed by John DeWitt, M.D., Ph.D.
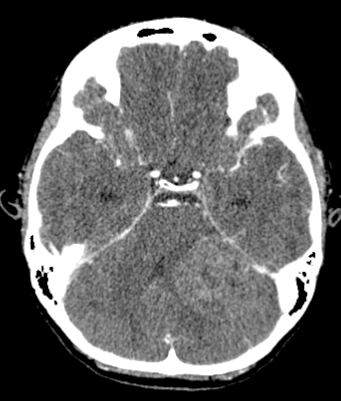
CT scan with contrast
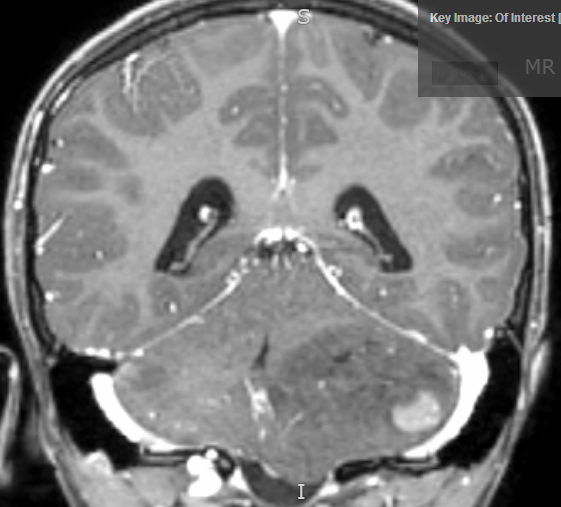
Coronal T1 postcontrast
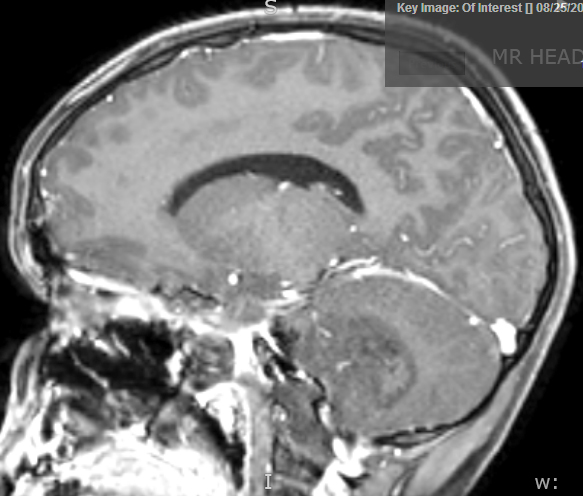
Sagittal T1 postcontrast
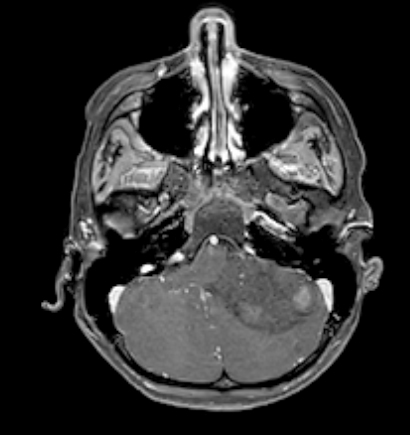
Axial T1 postcontrast
- TP53 mutated tumors typically have a much worse outcome
- 5 year survival of 81% in TP53 wildtype tumors versus 41% in those harboring TP53 mutation (J Clin Oncol 2013;31:2927)
- Presentation with CSF spread is poor prognostic factor
- Histologic subtype can be important in predicting prognosis with those with desmoplastic histology (desmoplastic / nodular and medulloblastoma with extensive nodularity) having a relatively good prognosis versus those with large cell / anaplastic morphology performing poorly
- Tumors with alterations in SMO or PTCH1 are sensitive to SMO inhibitors, while those with downstream alterations (SUFU, GLI2, MYCN) are refractory (Cancer Cell 2014;25:393, J Clin Oncol 2015;33:2646)
- 15 month old girl with Fanconi anemia and a novel mutation in SHH activated medulloblastoma (Biomark Res 2015;3:13)
- 18 month old boy with a novel germline GNAS mutation in SHH activated medulloblastoma (Pediatr Blood Cancer 2020;67:e28103)
- 19 year old woman with headache and vomiting (Surg Neurol Int 2018;9:34)
- SMO inhibitors can be used in SHH activated medulloblastomas that are susceptible (those harboring SMO or PTCH1 mutations) (Cancer Cell 2014;25:393, J Clin Oncol 2015;33:2646)
- Standard treatment includes surgery, radiation, and chemotherapy
- Whole cranial - spinal radiation required in cases with CSF dissemination or drop metastasis
- In general, medulloblastomas present grossly as pink to gray, soft and friable masses arising in cerebellar hemisphere or vermis (with expansion and filling of the fourth ventricle)
- SHH activated medulloblastomas can be more firm and nodular due to increased desmoplastic stroma in many SHH activated tumors
- Areas of gross necrosis may be seen but tend to be less than non-SHH activated tumors
- In disseminated disease, nodules may be grossly evident throughout areas of CSF flow along the neuroaxis
- On frozen section, a dominant population of undifferentiated cells with a high N/C ratio and frequent mitotic figures is often seen
- In cases with desmoplastic / nodular or extensive nodularity histology, these features may be seen at frozen section as well
Contributed by John DeWitt, M.D., Ph.D.
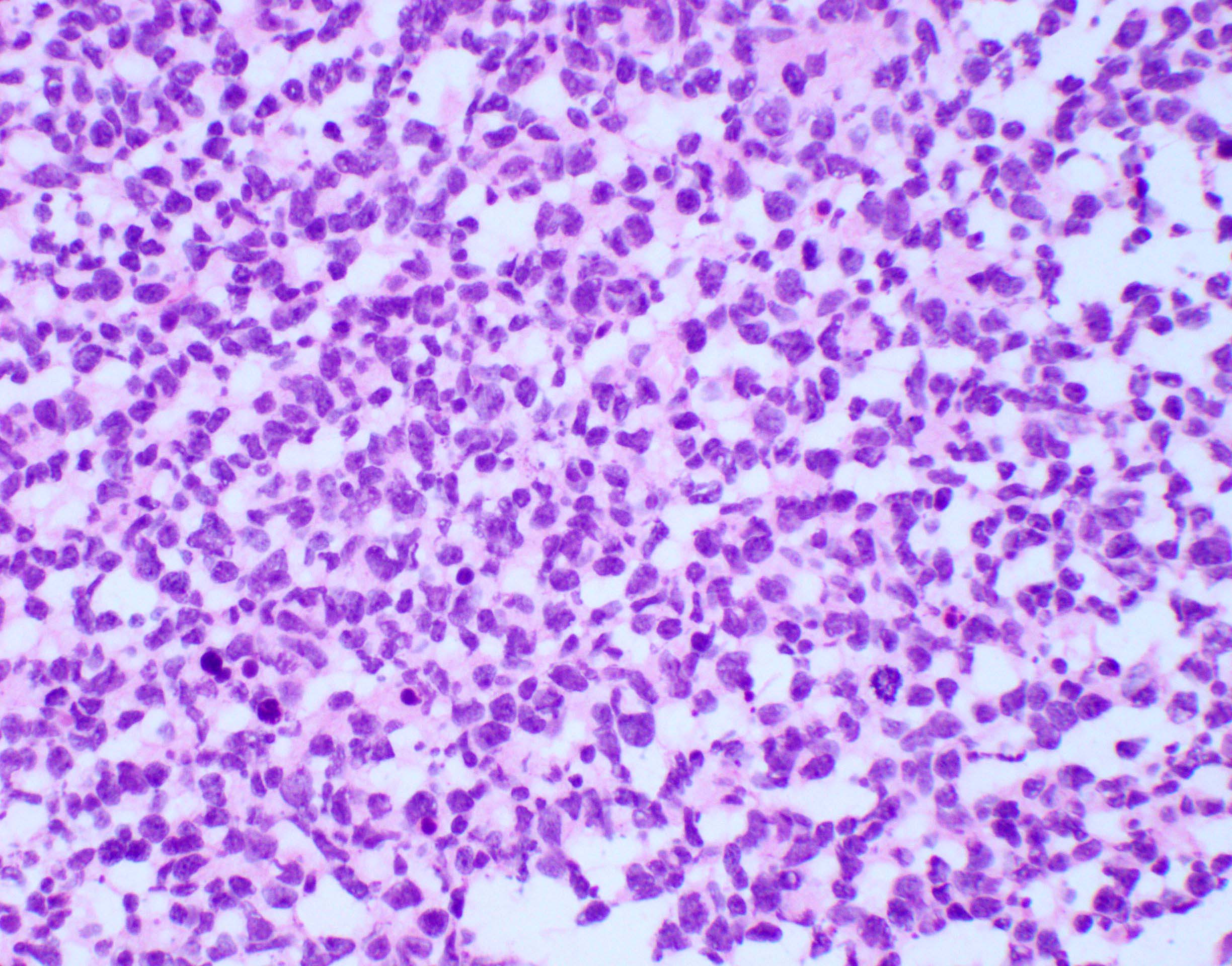
Pleomorphic undifferentiated cells
- Medulloblastomas in general have 4 distinct histologic patterns: classical, desmoplastic / nodular, medulloblastoma with extensive nodularity and large cell / anaplastic
- SHH activated medulloblastomas can present with any of these histologic phenotypes; however, desmoplastic / nodular and extensive nodularity histologic patterns invariably fall into this molecular subtype
- Classic medulloblastoma:
- Small blue round cell tumor
- Syncytial arrangement of densely packed undifferentiated cells (embryonal cells)
- Mitosis with apoptotic bodies
- Homer Wright rosettes
- Desmoplastic / nodular medulloblastoma:
- Densely packed, undifferentiated cells with hyperchromatic and pleomorphic nuclei, which produce dense intercellular reticulin fiber network with nodular reticulin free zones
- Medulloblastoma with extensive nodularity:
- Expanded lobular architecture as reticulin free nodular zones are enlarged and rich in neuropil-like tissue
- Large cell / anaplastic medulloblastoma:
- Anaplasia with marked nuclear pleomorphism, high mitotic count and apoptotic counts
- Nuclear molding and cell wrapping
Contributed by John DeWitt, M.D., Ph.D.
Sheets of undifferentiated cells
Vague nodular formation
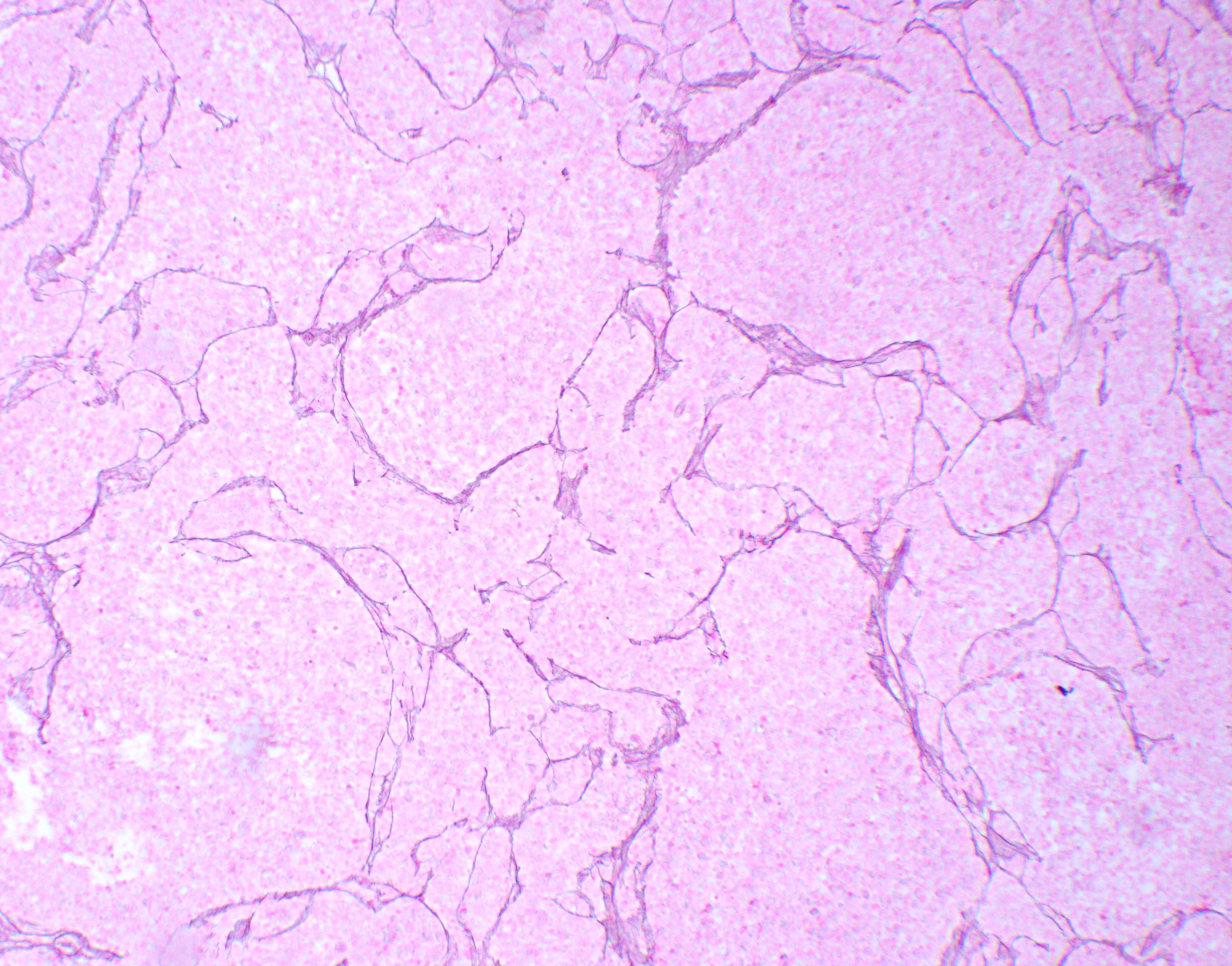
Vague nodular formation with reticulin
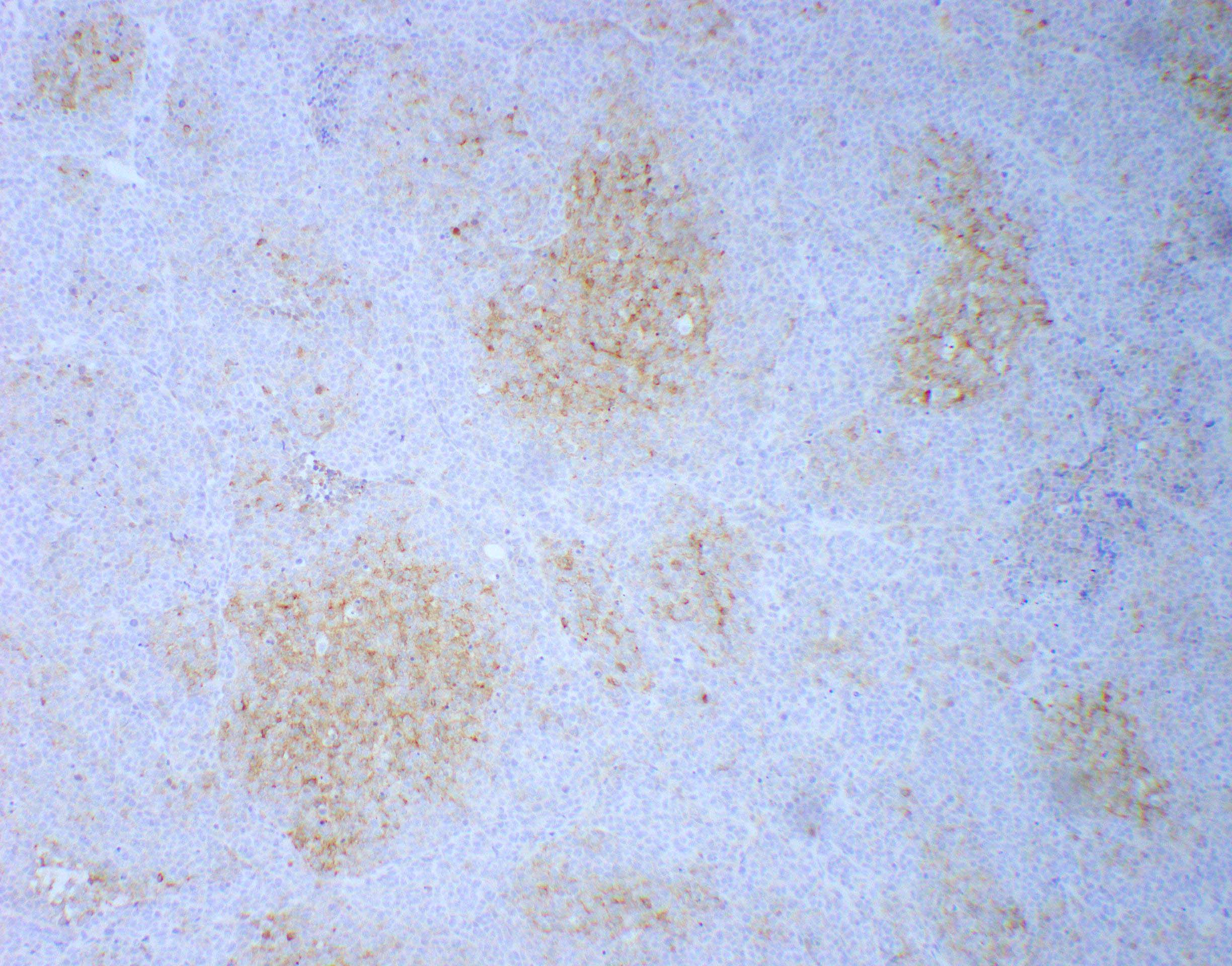
Neuron specific enolase (NSE)
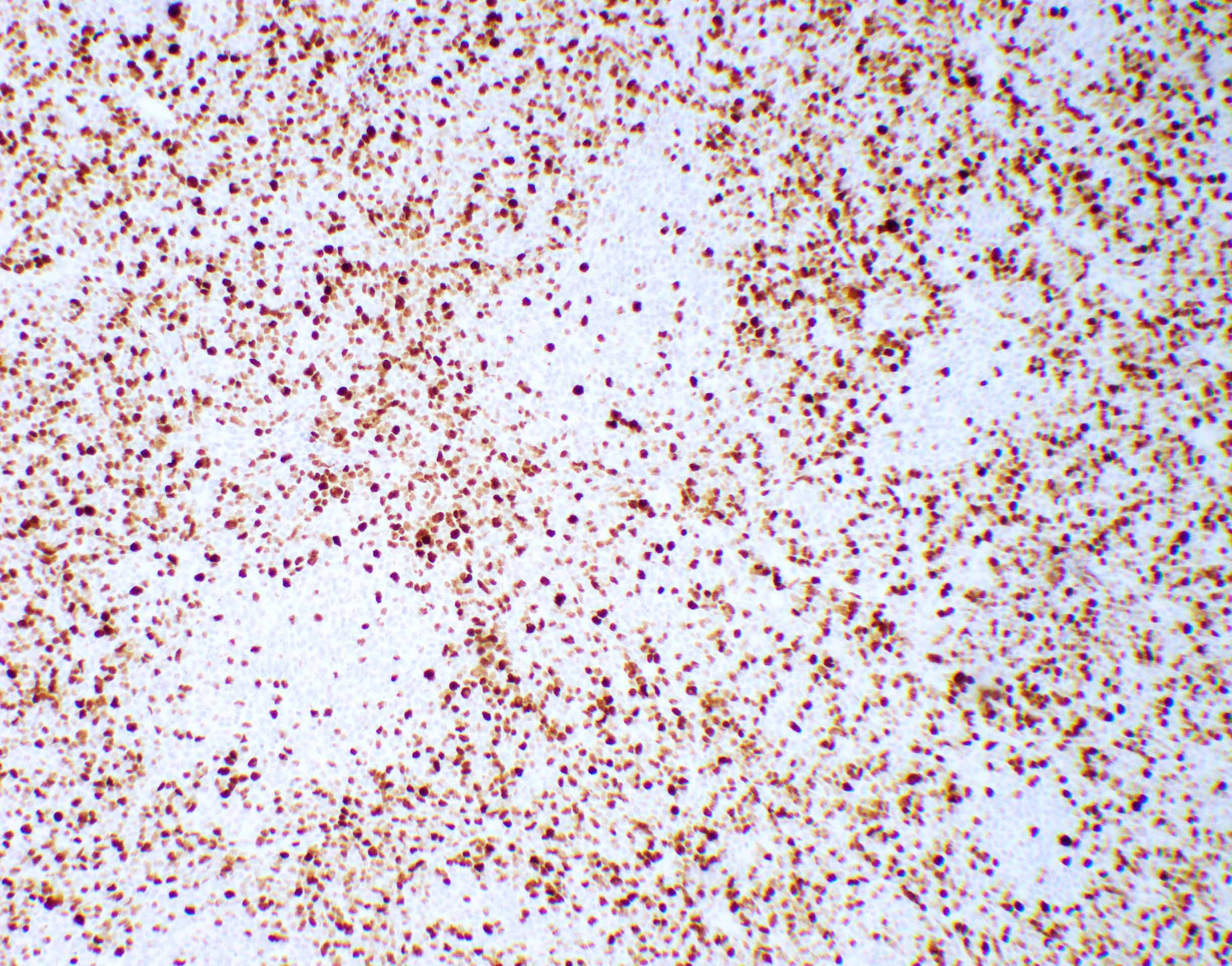
Ki67
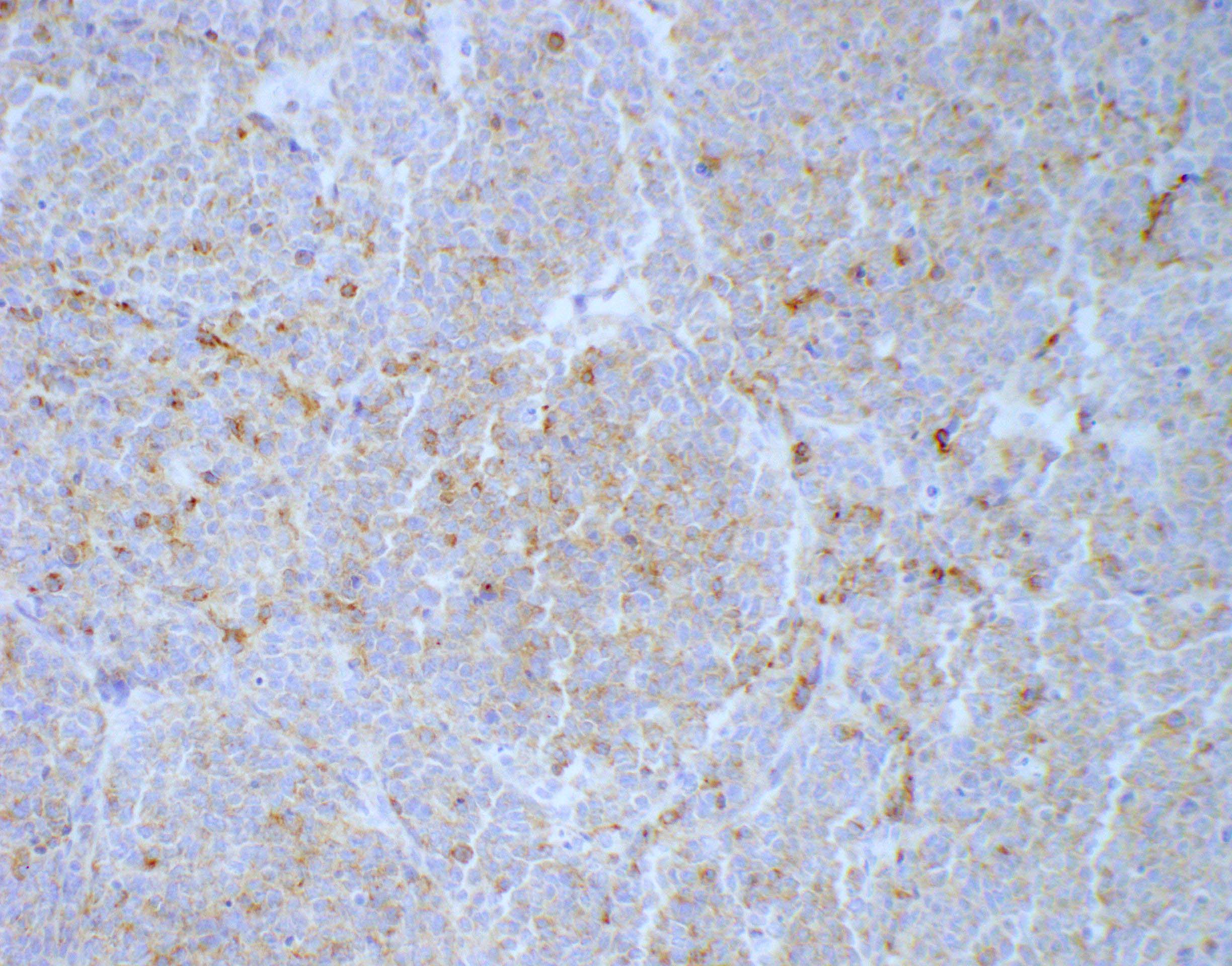
Synaptophysin
Beta catenin
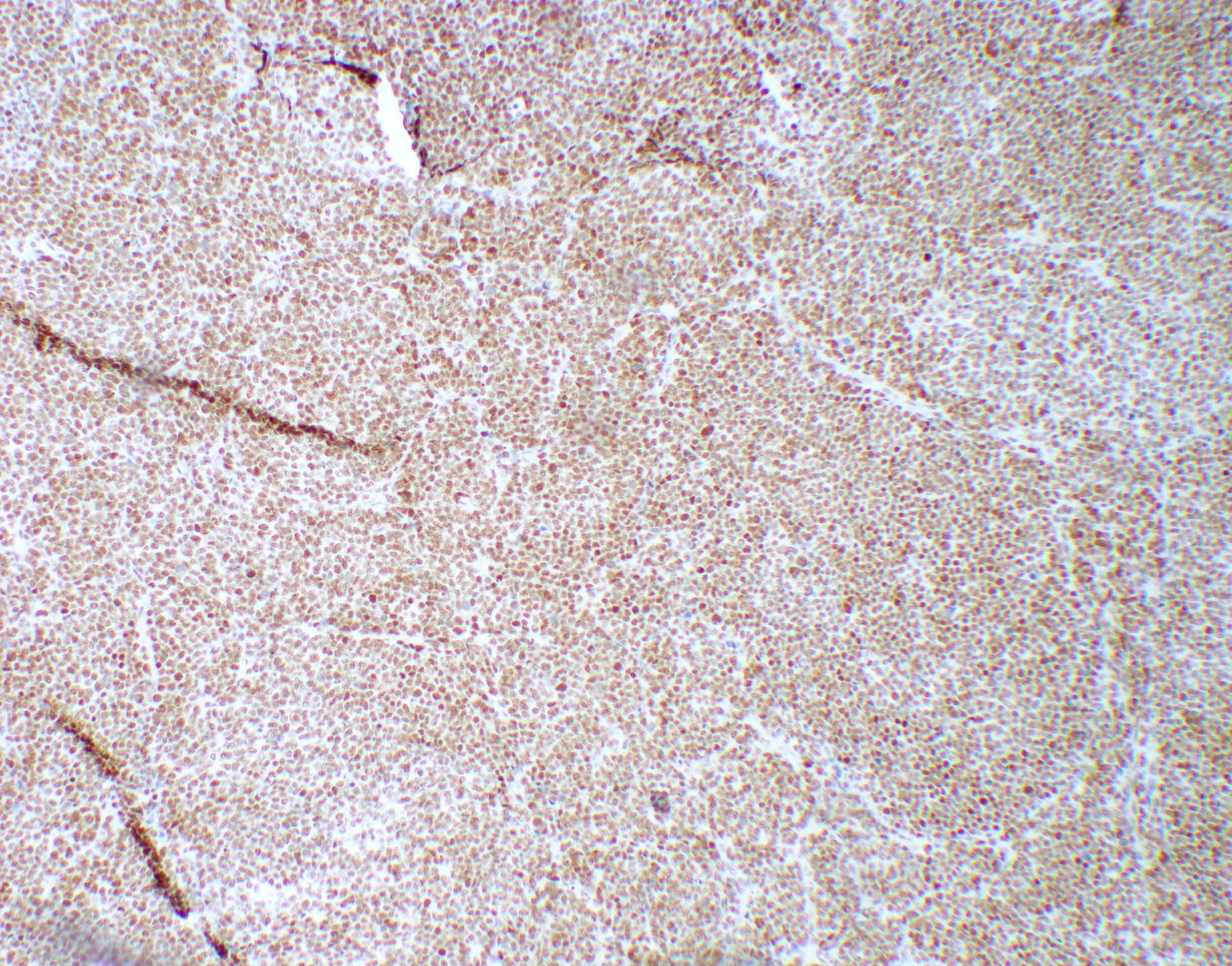
INI1
Contributed by Meaghan Morris, M.D., Ph.D.
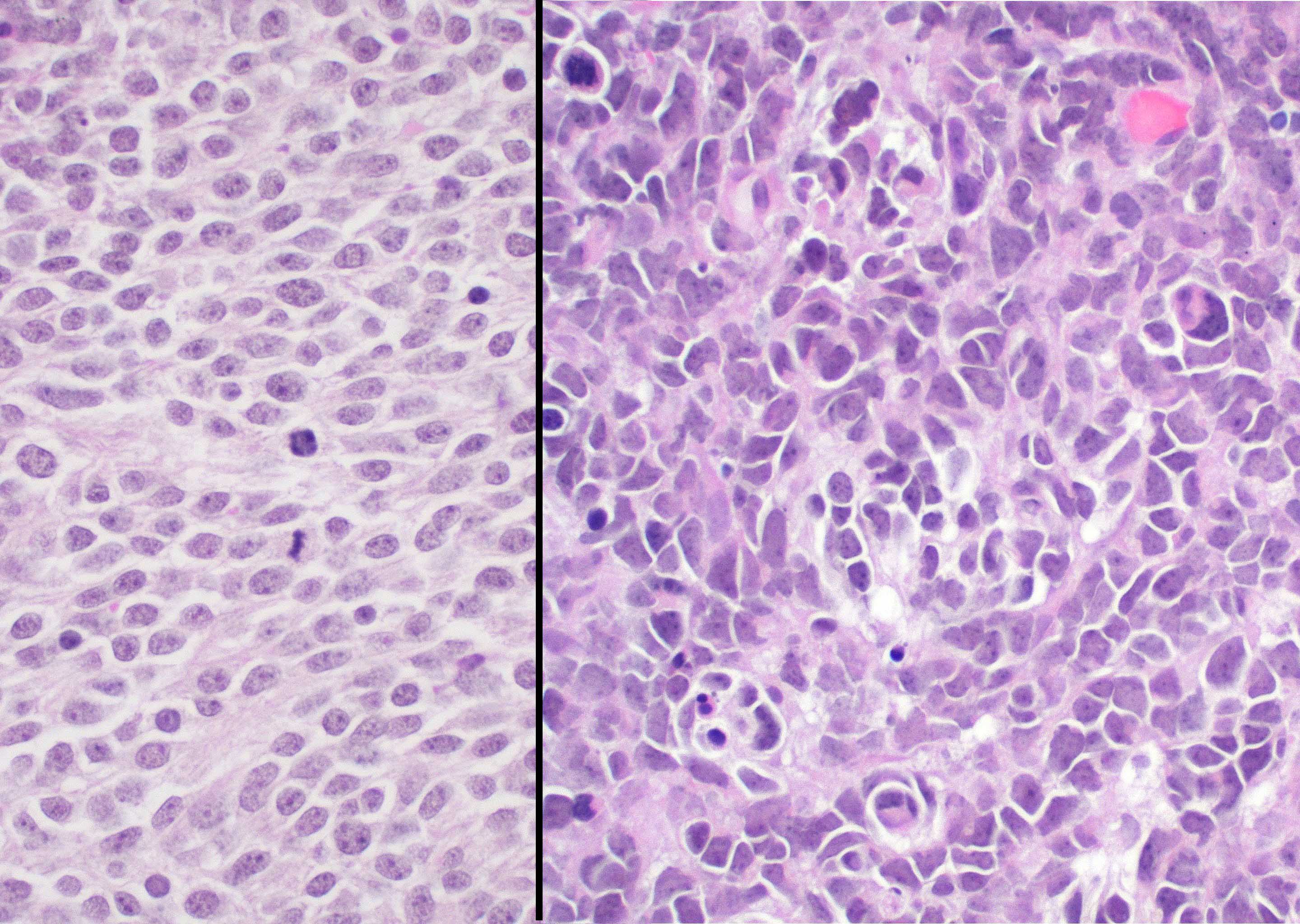
Anaplasia in medulloblastoma
Images hosted on other servers

Medulloblastoma,
desmoplastic / nodular
histologic subtype
- Synaptophysin: positive at least focally in the majority of medulloblastomas
- NeuN: positivity may be seen in nodules of neurocytic differentiation, particularly in cases with nodular / desmoplastic or extensive nodularity histology
- p53: strong pathologic p53 positivity may be seen in some cases and is often associated with anaplasia; often correlated with underlying somatic or germline (Li-Fraumeni) TP53 mutation (J Clin Oncol 2010;28:1345, Cell 2012;148:59)
- Ki67: many positive cells will be seen due to the highly proliferative nature of the tumor; in desmoplastic / nodular histology, Ki67 is higher in the internodular areas and lower in the pale islands
- GAB1: positive in SHH activated medulloblastoma
- YAP1: positive in SHH and WNT activated medulloblastoma
- SMARCB1 / INI1 and SMARCA4 / BRG1: expression is maintained; loss of expression of either of these markers would be consistent with a diagnosis of atypical teratoid / rhabdoid tumor and not medulloblastoma
- Beta catenin: should show cytoplasmic expression; nuclear expression would be consistent with medulloblastoma, WNT activated
- By definition, molecular studies will show activating mutations in the SHH signaling pathway, with a recent study identifying mutations in known pathway genes in 87% of cases (Cancer Cell 2014;25:393)
- Associated with high level amplifications in other genes, including MYCL, GLI2, PPM1D, YAP1 and MDM4 (Nature 2012;488:49)
- Homozygous deletions of PTCH1 and PTEN appear to occur overwhelmingly in the SHH activated subtype of medulloblastoma compared to other groups (Nature 2012;488:49)
- Point mutations in TP53 are associated with chromothripsis and a very poor outcome (Cell 2012;148:59, J Clin Oncol 2013;31:2927)
- Brain, cerebellum, resection:
- Medulloblastoma, classical type, SHH activated and TP53 wildtype, WHO grade 4 (see comment)
- Comment:
- Integrated diagnosis: medulloblastoma, classical type, SHH activated and TP53 wildtype
- Histological diagnosis: medulloblastoma, classical type
- WHO (histological) grade: 4 of 4
- Molecular information:
- P53: scattered positive cells (immunohistochemistry consistent with wildtype)
- INI1: retained (immunohistochemistry consistent with wildtype)
- Beta catenin: diffuse cytoplasmic staining (immunohistochemistry consistent with nonactivated)
- YAP1: positive (immunohistochemistry)
- GAB1: positive (immunohistochemistry)
- Histological features in the tumor include high cell density, hyperchromasia and mitoses. Prominent anaplastic features, such as pleomorphism, prominent nucleoli or cell wrapping, are not appreciated. Desmoplastic or nodular areas are not present. Overall, the histologic and molecular characteristics are consistent with the classical histologic subtype and the SHH activated molecular group.
- Other molecular subtypes of medulloblastoma:
- WNT activated:
- Will show strong nuclear positivity with beta catenin staining
- Group 3 / 4:
- Will not subtype into WNT activated or SHH activated by immunohistochemistry
- Further expression profiling or methylation profiling can identify these specific subtypes
- WNT activated:
- Atypical teratoid / rhabdoid tumor:
- Will show loss of INI1 staining by immunohistochemistry
- Other embryonal tumors:
- CNS neuroblastoma:
- Embryonal tumor with multilayered rosettes
- C19MC altered:
- These tumors will typically be found in the supratentorial region and will lack histologic and molecular features of medulloblastoma
- CNS neuroblastoma:
A histologic section of a tumor resected from the posterior fossa of a young man is shown. What is the most likely diagnosis?
- Metastatic lung adenocarcinoma
- Medulloblastoma, desmoplastic / nodular, SHH activated
- Choroid plexus papilloma
- Pilocytic astrocytoma
- Hemangioblastoma
This is the best answer as the image shows sheets of undifferentiated cells consistent with medulloblastoma. Metastatic lung adenocarcinoma would commonly show gland formation and is unlikely in a young man. Choroid plexus papilloma would show well differentiated cells with a prominent papillary growth pattern. Pilocytic astrocytoma would show prominent glial cells with long thin processes. Hemangioblastoma would show a prominent vascular pattern with interspersed tumor cells and hemorrhage.
Comment Here
Reference: SHH activated
- TP53 mutation
- EGFR amplification
- IDH mutation
- TERT promoter mutation
- 1p19q codeletion
This is the best answer as the presence of TP53 mutation in medulloblastoma, SHH activated is associated with a poor prognosis. EGFR amplification is often present in glioblastoma, IDH wildtype. IDH mutation is associated with improved prognosis in infiltrating gliomas. TERT promoter mutation is seen in oligodendroglioma, IDH mutant, 1p19q codeleted and often present in glioblastoma, IDH wildtype. 1p19q codeletion is seen in oligodendroglioma, IDH mutant, 1p19q codeleted.
Comment Here
Reference: SHH activated
- Schwannoma is a benign nerve sheath tumor composed of differentiated neoplastic Schwann cells
- Classified as CNS WHO grade 1
- CNS primary tumor mostly occurring in the spine / spinal nerve roots (intradural, extramedullary) and vestibular division of the eighth cranial nerve within the cerebellopontine angle
- Multiple tumors in patients with neurofibromatosis type 2 (NF2, bilateral vestibular schwannomas) or schwannomatosis
- Excellent prognosis with nearly absent recurrence after gross total resection
- Malignant transformation is exceptionally rare
- Biphasic, encapsulated tumor composed of compact areas with spindle cells (Antoni A) and palisading nuclei (Verocay bodies) and loose microcystic areas with lipid laden macrophages (Antoni B)
- Neurilemmoma (not recommended)
- Acoustic neuroma (not recommended)
- ICD-O: 9560/0 - schwannoma, NOS
- ICD-11: 2A02.3 & XH98Z3 - benign neoplasm of cranial nerves & schwannoma, NOS
- Majority (90%) are solitary and sporadic
- May occur at any age but the peak incidence is between
- 55 and 75 years for vestibular schwannoma (Neurooncol Adv 2020;2:vdaa135)
- 65 and 75 years for spinal schwannomas (J Neurosurg Spine 2019 Dec 27 [Epub ahead of print])
- Younger (third decade of life) age at onset in patients with neurofibromatosis type 2 (Q J Med 1992;84:603)
- Vestibular schwannoma: most common nonmalignant nerve sheath tumor (75.7%) and third most common nonmalignant primary brain tumor (Neuro Oncol 2022;24:v1)
- Multiple schwannomas in patients with neurofibromatosis type 2 or schwannomatosis (Acta Neuropathol 2020;139:643, Genet Med 2022;24:1967)
- Bilateral vestibular schwannomas are typically seen in patients with neurofibromatosis type 2 (Genet Med 2022;24:1967)
- Most common sites in the CNS:
- Spine, where it is typically intradural and extramedullary
- Vestibular division of the eighth cranial nerve (Neuro Oncol 2022;24:v1)
- NF2 inactivating mutation (frameshift and nonsense) and loss of the remaining wild type allele on chromosome 22, in 50 - 75% of sporadic schwannomas (J Neurosurg 2018;128:911)
- Germline pathogenic NF2 mutation in schwannomas associated with neurofibromatosis type 2 (Genet Med 2022;24:1967)
- Germline mutations of either SMARCB1 or LZTR1 on chromosome 22, followed by a somatic mutation in NF2 along with the deletion of the other chromosome 22 in schwannomas associated with schwannomatosis (Neurology 2015;84:141, Clin Genet 2020;97:376, Genet Med 2022;24:1967)
- Mostly unknown
- Previous irradiation (J Natl Cancer Inst 2002;94:1555)
- Association with neurofibromatosis type 2 and schwannomatosis (Genet Med 2022;24:1967)
- Symptoms depend on the anatomical site of the tumor:
- Tinnitus, unilateral sensorineural hearing loss, vertigo or disequilibrium in patients with vestibular schwannoma (J Neurosurg 2022 Dec 2 [Epub ahead of print])
- Sensory or motor symptoms in patients with spinal schwannoma
- Based on imaging (CT, MRI) / biopsy / resection specimen
- Well circumscribed masses that displace adjacent structures without direct invasion
- On MRI, it may be
- Iso or hyperintense in T2 weighted images (Spine (Phila Pa 1976) 2017;42:E150)
- Hyperintense in fluid attenuated inversion recovery (FLAIR) images
- Heterogeneous or homogeneous enhancement in T1 weighted sequences (J Clin Imaging Sci 2017;7:38)
Contributed by Valeria Barresi, M.D., Ph.D.
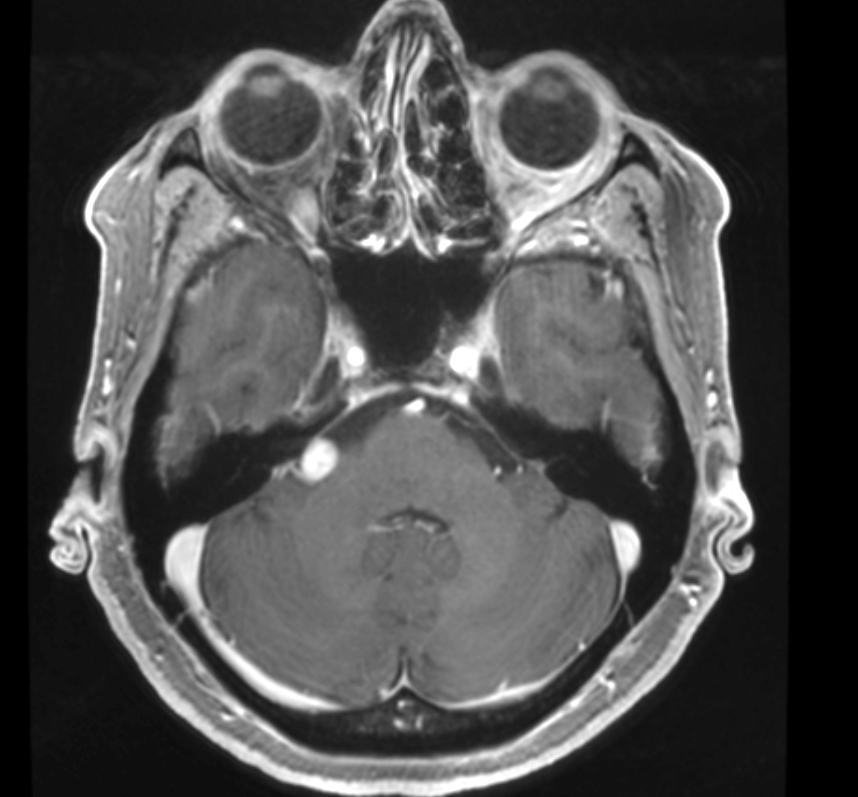
T1 weighted MRI
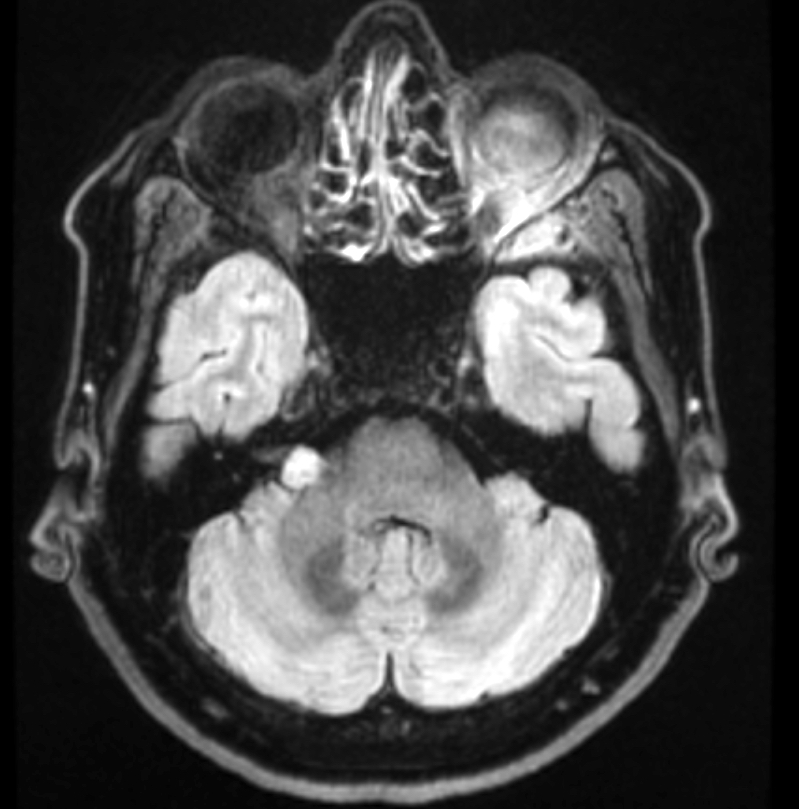
FLAIR MRI
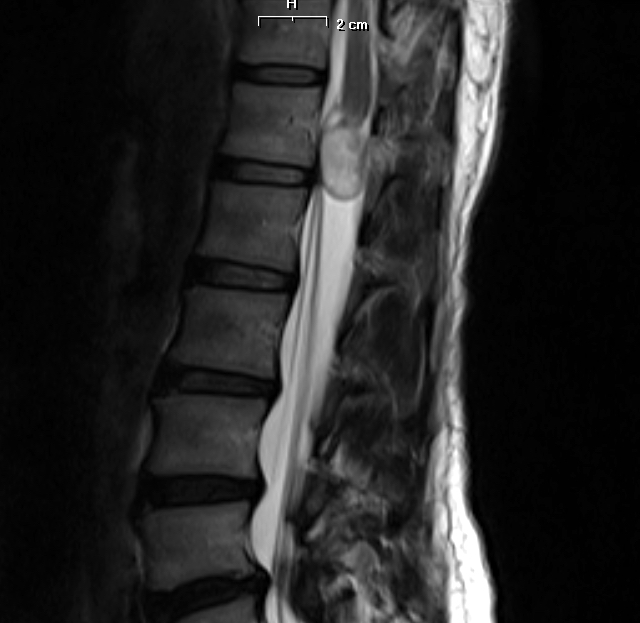
T2 weighted MRI
- Excellent prognosis with nearly absent recurrence after gross total resection (Otol Neurotol 2022;43:702)
- Regrowth after incomplete surgery is correlated with the volume of residual tumor (Laryngoscope 2016;126:1877)
- Malignant transformation is exceptionally rare and has been associated with irradiation and epithelioid subtype (Acta Neurochir (Wien) 2022;164:343, Skull Base 2010;20:381, Neurosurg Rev 2022;45:915, APMIS 2021;129:524, Am J Surg Pathol 2015;39:673)
- 23 year old man with a large multicystic spinal lesion (BMJ Case Rep 2021;14:e242690)
- 48 year old man with a cervical spinal lesion (Asian J Neurosurg 2015;10:42)
- 53 year old woman with intramedullary lesion (Neurosurg Rev 2021;44:1833)
- 70 year old man with an intramedullary mass (Surg Neurol Int 2020;11:454)
- Observation, radiosurgery, surgery or surgery followed by radiosurgery, depending on symptoms, tumor size and association with neurofibromatosis type 2 (Neurooncol Pract 2016;3:281)
- Bevacizumab is considered as an option in patients with neurofibromatosis type 2 (Neuro Oncol 2020;22:31)
- Usually encapsulated, with a smooth surface
- Cut surfaces are firm, light tan and interrupted by white-yellow or hemorrhagic areas (Asian J Neurosurg 2015;10:42, Neurosurg Rev 2021;44:1833)
Images hosted on other servers:
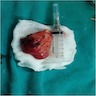
Surgically resected schwannoma
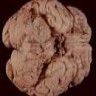
Acoustic schwannoma
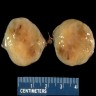
Fish flesh tan cut surface
- Admixture of compact and loose areas, Verocay bodies and wavy / spindled nuclei with pointed ends are useful diagnostic clues in the differential diagnosis versus other spindle cell tumors (e.g., meningioma, ependymoma, sarcoma, solitary fibrous tumor) (Arch Pathol Lab Med 2007;131:1532)
- Additional features: hyalinized blood vessels, chronic inflammatory infiltrates along capsule, hemosiderin
- Additional clues:
- Differential diagnosis versus ependymoma: absence of fibrillary background and perivascular pseudorosettes
- Differential diagnosis versus meningioma: lack of psammoma bodies, whorls or epithelioid syncytia
- Differential diagnosis versus sarcoma: in malignant sarcomas, mitoses are more commonly encountered, nuclear atypia is more uniformly widespread and necrosis may be present (Arch Pathol Lab Med 2007;131:1532)
- Differential diagnosis versus solitary fibrous tumor: lack of staghorn vessels
Contributed by Valeria Barresi, M.D., Ph.D.
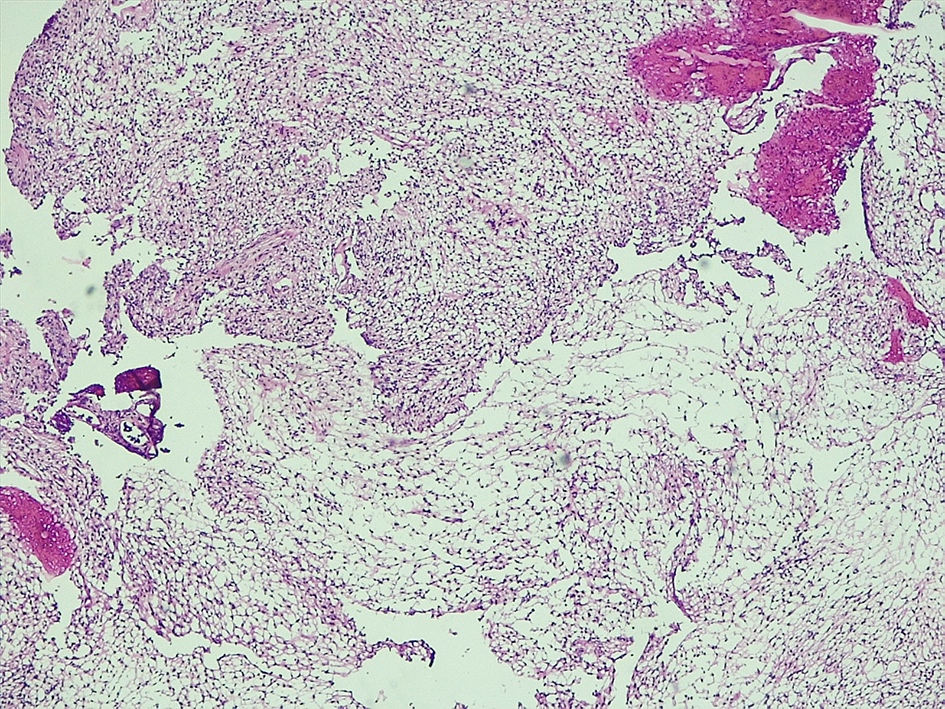
Alternating compact and loose areas
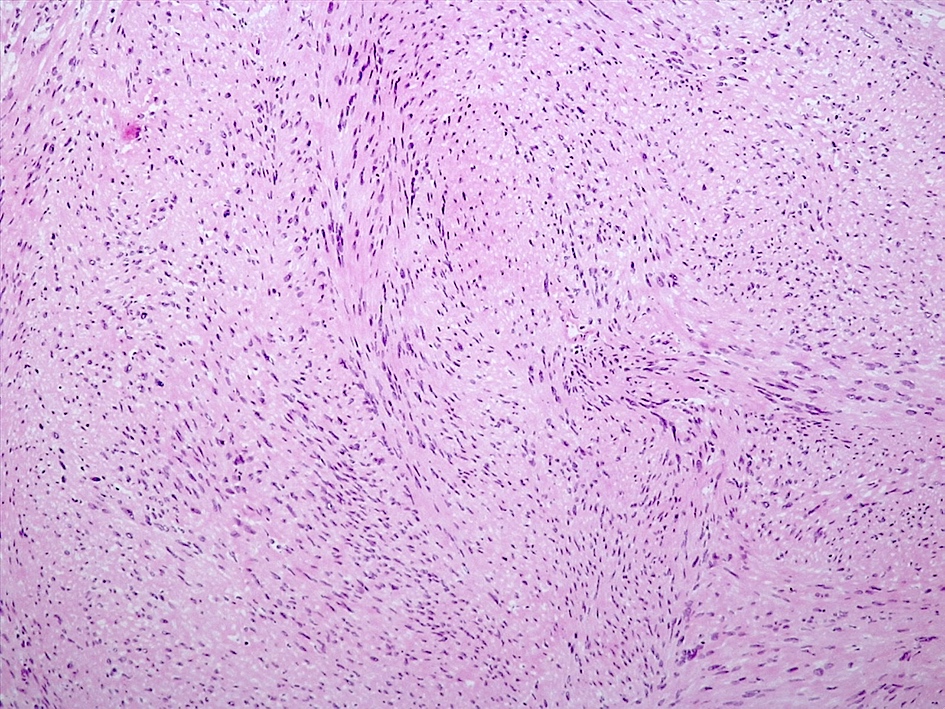
Compact areas composed of spindle cells

Cohesive clusters with sharp borders
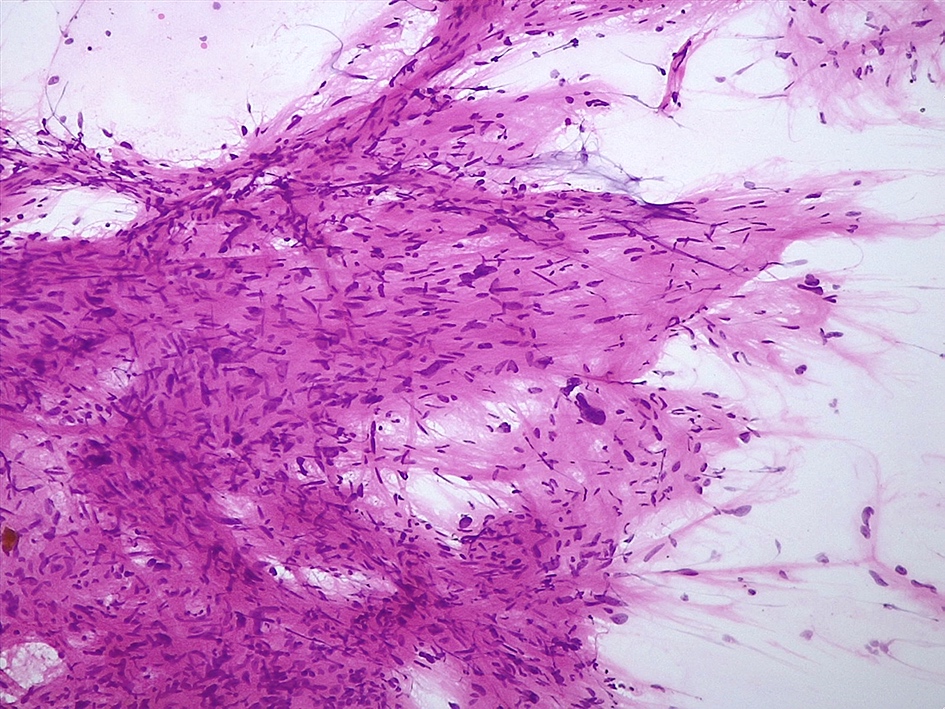
Spindle cell nuclei in fibrillary matrix
- Spindle cell tumo (usually biphasic) and composed of compact hypercellular areas (Antoni A tissue) showing nuclear palisading (Verocay bodies) and loose microcystic areas (Antoni B tissue) with collection of lipid laden histiocytes and thick walled hyalinized blood vessels (Surg Neurol Int 2020;11:454)
- Lymphoid aggregates maybe seen peripherally or in a subcapsular distribution
- Mitoses or focal degenerative atypia may be present
- Ancient subtype: characterized by scattered atypical or bizarre nuclei
- Cellular subtype: composed exclusively or predominantly by Antoni A tissue and devoid of Verocay bodies
- Mitoses may be more conspicuous; uncommon in the CNS (Clin Neurol Neurosurg 2009;111:467)
- Epithelioid subtype: characterized by epithelioid cells with amphophilic or eosinophilic cytoplasm and uniform round nuclei with inconspicuous nucleoli
Contributed by Valeria Barresi, M.D., Ph.D.
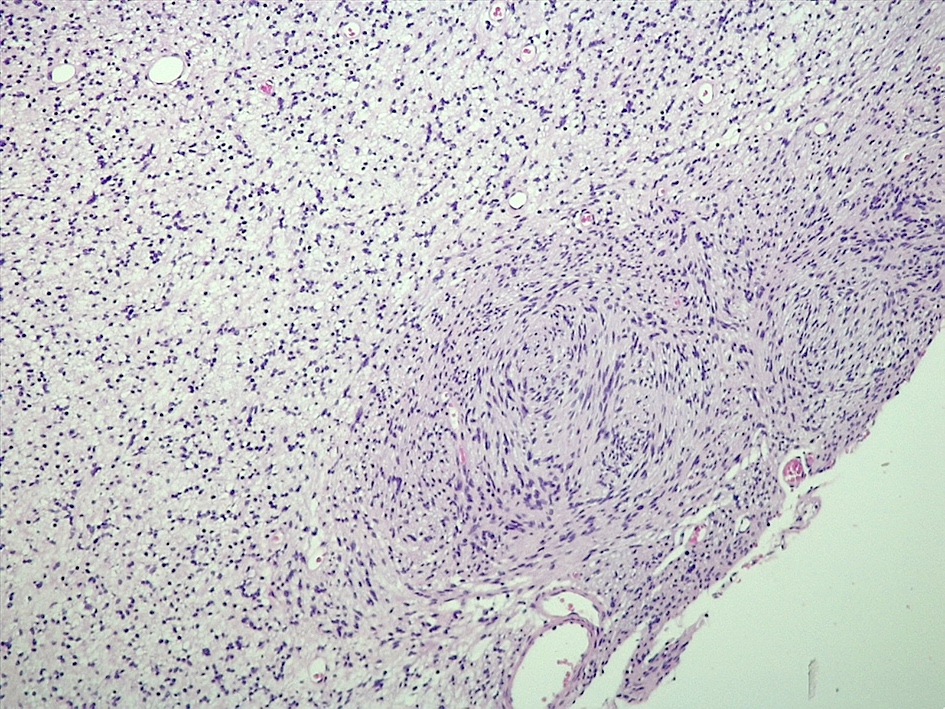
Compact and loose areas
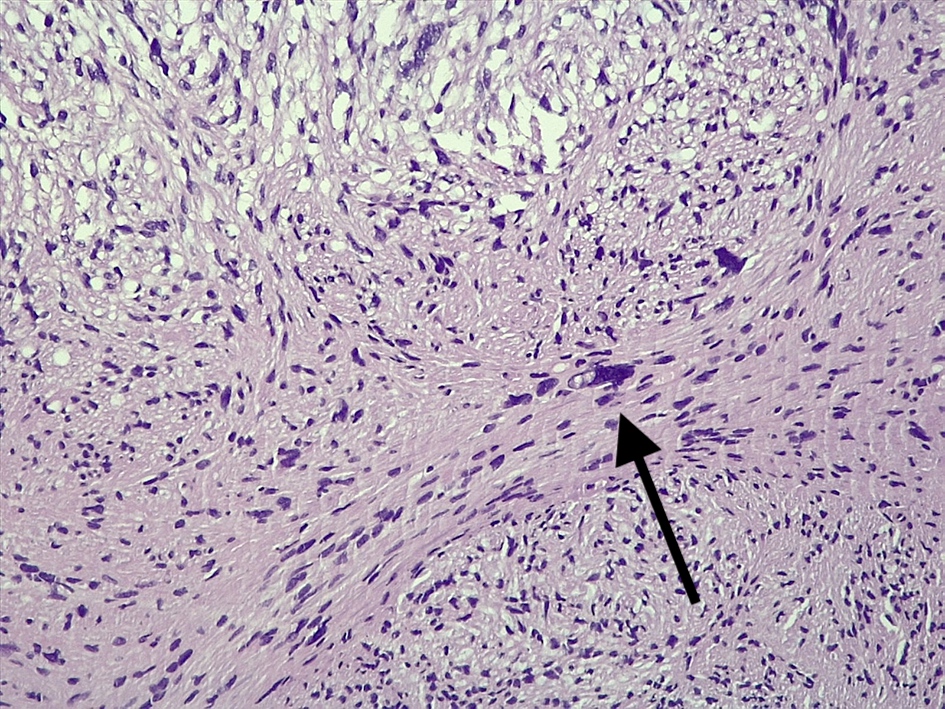
Atypical nuclei
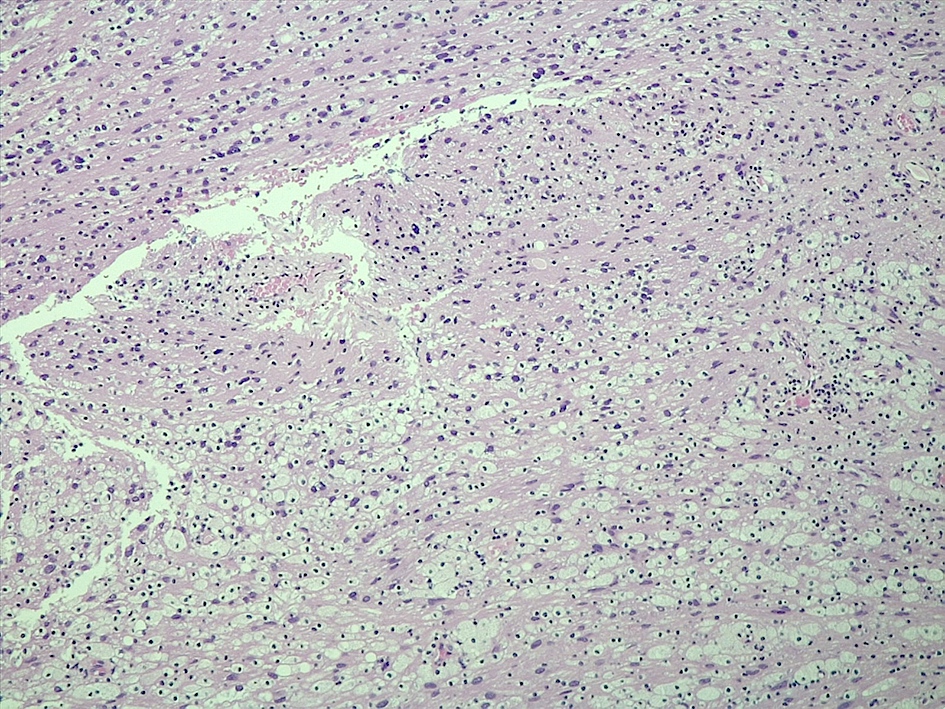
Lipid laden histiocytes
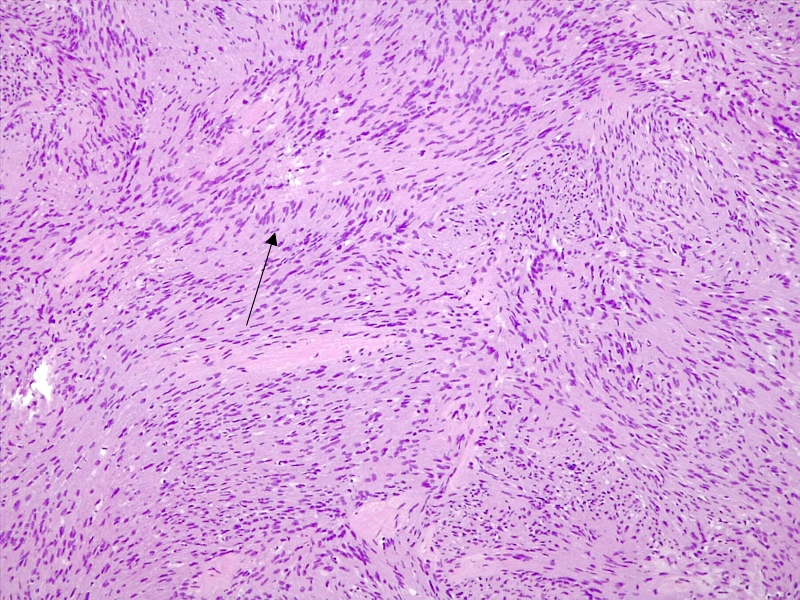
Verocay bodies
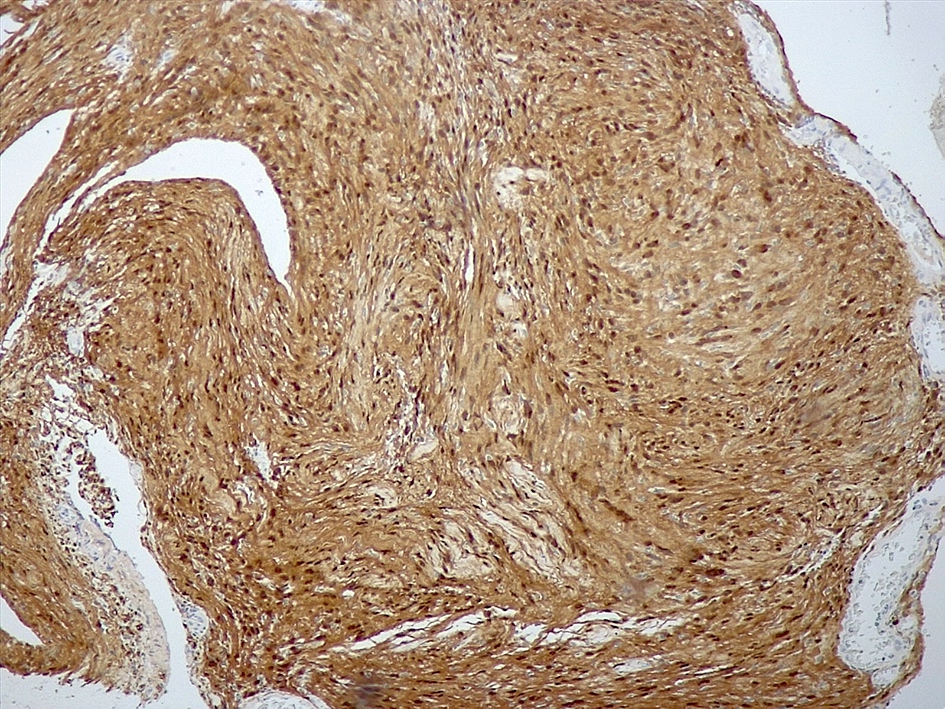
S100
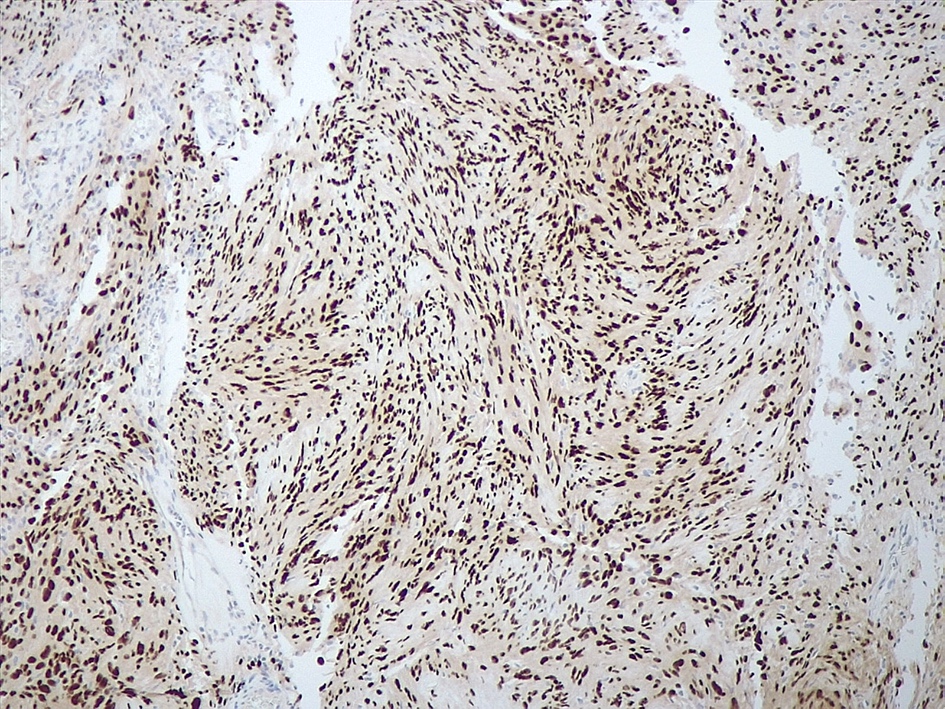
SOX10
- Cohesive lesion with sharp borders in squash preparation or smear and composed of cells with spindle nuclei (Cytopathology 2022;33:196, Cancer Cytopathol 2015;123:171)
- Fibrillary matrix lacking cotton wool look at low magnification
- Hemosiderin deposition may be present (Cytopathology 2022;33:196)
- Nuclear hyperchromasia and pleomorphism may be present
- Strong diffuse SOX10 (Am J Surg Pathol 2008;32:1291)
- Strong diffuse S100 (Appl Immunohistochem Mol Morphol 2012;20:445)
- Neurofilament stain demonstrates axons predominantly pushed to the periphery of the tumor
- EMA (95%) (J Neuropathol Exp Neurol 2017;76:289)
- Progesteron receptor (97%) (J Neuropathol Exp Neurol 2017;76:289)
- CD34 (87%) (J Neuropathol Exp Neurol 2017;76:289)
- STAT6 (J Neuropathol Exp Neurol 2017;76:289, Pathology 2014;46:389)
- SSTR2A (J Neuropathol Exp Neurol 2017;76:289)
- GFAP (J Neuropathol Exp Neurol 2017;76:289)
- CK AE1 / AE3 (J Neuropathol Exp Neurol 2017;76:289)
- MelanA (J Neuropathol Exp Neurol 2017;76:289)
- HMB45 (J Neuropathol Exp Neurol 2017;76:289)
- NF2 inactivating mutation (frameshift and nonsense) and loss of the remaining wild type allele on chromosome 22 is frequent but not specific (J Neurosurg 2018;128:911)
- Distinct methylation pattern (Nat Genet 2016;48:1339, Acta Neuropathol 2016;131:877)
- Spine, tumor, resection:
- Schwannoma, CNS WHO grade 1 (see comment)
- Comment: Spindle cell tumor showing loose areas with lipid laden macrophages and compact areas with nuclear palisading.
- Meningioma, fibrous subtype:
- Solitary fibrous tumor:
- Malignant peripheral nerve sheath tumor:
- Brisk mitotic activity and frequent necrosis
- Frequent necrosis
- Negative or patchy S100 and SOX10 staining
- Complete immunohistochemical loss of H3 p.K27me3 in 50 - 80% of cases
- Neurofibroma:
- Usually not encapsulated
- Presence of shredded carrot collagen bands
- Smaller cells
- Absence of a biphasic pattern
An intradural, extramedullary mass is found in the spine. Histological examination shows a tumor with compact areas composed of spindle cells alternating with loose areas with collection of lipid laden macrophages. What is the most likely diagnosis?
- Ependymoma
- Meningioma, fibrous subtype
- Schwannoma
- Solitary fibrous tumor
Comment Here
Reference: Schwannoma
- Malignant peripheral nerve sheath tumor
- Meningioma, fibrous subtype
- Schwannoma, ancient subtype
- Solitary fibrous tumor
Comment Here
Reference: Schwannoma
- Glial cyst in cerebellum (J Neuroradiol 1995;22:48)
- No communication with ventricles
- 42 year old man with expansive cerebellar lesion (J Neuroradiol 2001;28:209)
- Wall lined by gliosis, Rosenthal fibers present, no epithelial lining
- Solitary fibrous tumors (SFTs) of the CNS are spindle cell neoplasms with a wide range of histopathologic appearances and a pathognomonic NAB2::STAT6 fusion mutation with a characteristic immunophenotype
- Spindle cell neoplasm that ranges in stromal collagenization and cellularity, often with a patternless pattern or dilated staghorn vessels
- Most common in the dura (especially the tentorium) but very rare compared to other primary meningeal tumors (Acta Neuropathol 2019;137:307)
- Pathognomonic NAB2::STAT6 fusion mutation that separates it from other tumors with similar microscopic appearances (Acta Neuropathol 2019;137:307)
- Positive IHC includes STAT6 (nuclear), CD34 and CD99
- Good prognosis but metastasis is possible, even with low grade tumors (Acta Neuropathol 2019;137:307)
- Recent changes to the CNS WHO classification system have made solitary fibrous tumor the sole name for this entity (Neuro Oncol 2021;23:1231)
- Formerly called solitary fibrous tumor / hemangiopericytoma (SFT / HPC) and angioblastic meningioma
- Hemangiopericytoma has been removed from the most recent WHO CNS tumor classification
- ICD-O: 8815/1 - solitary fibrous tumor
- ICD-10 codes no longer listed by the WHO
- ICD-11: 2F7C & XH7E62 - neoplasms of uncertain behavior of connective or other soft tissue & solitary fibrous tumor, NOS
- ICD-11: 2B5Y & XH1HP3 - other specified malignant mesenchymal neoplasms & solitary fibrous tumor, malignant
- < 5% of extra-axial CNS tumors (meningiomas are the most common) (J Clin Neurosci 2014;21:1310)
- Most common in patients in their 40s - 60s (Brain Pathol 2019;29:18)
- M:F = 1.08 (Brain Pathol 2019;29:18)
- Affects the dura most frequently (mainly found in the tentorium)
- Other sites include falx, other dural sites, skull base, cerebellum and pineal gland
- Pathognomonic NAB2::STAT6 fusion mutation on 12q13
- Exact pathogenesis of this neoplasm has not been fully elucidated apart from its spindle cell / fibroblastic nature
- No known familial association
Images hosted on other servers:

Intracranial SFT vascular permeability
- Patients most commonly present with symptoms related to mass effect
- Patients are given a preliminary, often nonspecific diagnosis from CNS imaging
- Found either during a primary workup or incidentally as part of a separate workup
- Typically made by (immuno)histologic examination but can be supplemented with molecular testing for the NAB2::STAT6 mutation
- Minority of cases present with hypoglycemia due to IGF1 production but this is nonspecific (Hum Pathol 1996;27:858)
- Common: dural tail sign (nonspecific) and clear demarcation from surrounding tissue
- Some cases will show a lack of obvious demarcation
- Isodense on noncontrast T1 weighted images but bright on T1 with contrast and T2 weighted images
- Meningiomas may also show a dural tail sign and similar enhancement patterns but are more likely to show intratumoral calcification and spoked wheel vessels (Neuroradiology 2021;63:1215)
Images hosted on other servers:

MRI of spinal SFT
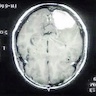
MRI of parieto-occipital SFT

MRI of expansive SFT
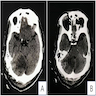
PET / CT after SFT resection
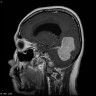
Vividly enhancing mass, various images
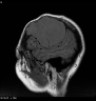
Extra-axial mass, various images
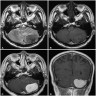
Tumor mass in the left cerebellum
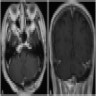
Resection cavity with residual tumor tissue
- > 75% of patients survive 10 years after diagnosis, with shorter survival time associated with higher tumor grade (Neurosurg Rev 2021;44:1299)
- < 50% of tumors recur; the exact rate is cohort dependent (Neurosurg Rev 2021;44:1299)
- Literature is inconclusive as to whether spinal or brain tumors have a better prognosis (Neurosurg Rev 2021;44:1299, J Neurooncol 2019;143:457)
- SFTs can metastasize outside the CNS or to other locations within the CNS (13.3% according to one case review), with metastasis more likely in higher grade and spinal tumors (Neurosurg Rev 2021;44:1299)
- Differences in gene expression associated with increased vascular permeability are thought to increase metastasis risk compared to SFTs outside the CNS (Cancers (Basel) 2021;13:1142)
- 10 year old boy with an intradural extramedullary tumor of the cervical spine (J Neurosurg Pediatr 2017;19:339)
- 26 year old woman with an extra-axial mass found during pregnancy, causing brain herniation (Case Rep Womens Health 2021;29:e00285)
- 45 year old woman with a tentorial mass in the right parieto-occipital region (Int J Surg Case Rep 2019;54:10)
- 53 year old woman with a spindle shaped extradural lesion in the cervical spine (BMC Surg 2021;21:405)
- All tumors are treated with total resection
- Higher grade tumor treatment is often supplemented with adjuvant radiation
- Patients may be monitored for many years and even decades to evaluate for recurrence and metastasis
Images hosted on other servers:

Spinal SFT during resection
- Discrete mass, often contiguous with dura, white to brown
- Gross appearance varies based on the relative amount of stromal collagen and tumor cellularity
- Some tumors can lose their dural attachment and infiltrate surrounding tissue (Ann Diagn Pathol 2003;7:169)
Images hosted on other servers:

SFT metastatic to liver
- Characteristic feature: spindled cells without a single / discrete architecture (patternless pattern)
- Enlarged vascular spaces (staghorn vessels) can suggest SFT
- Typical frozen diagnosis: spindle cell neoplasm, with differential to include SFT, meningioma and possibly other (schwannoma, etc.) depending on location
Contributed by Brenndan Crumley, M.D., M.P.H. and Katherine Schwetye, M.D., Ph.D.
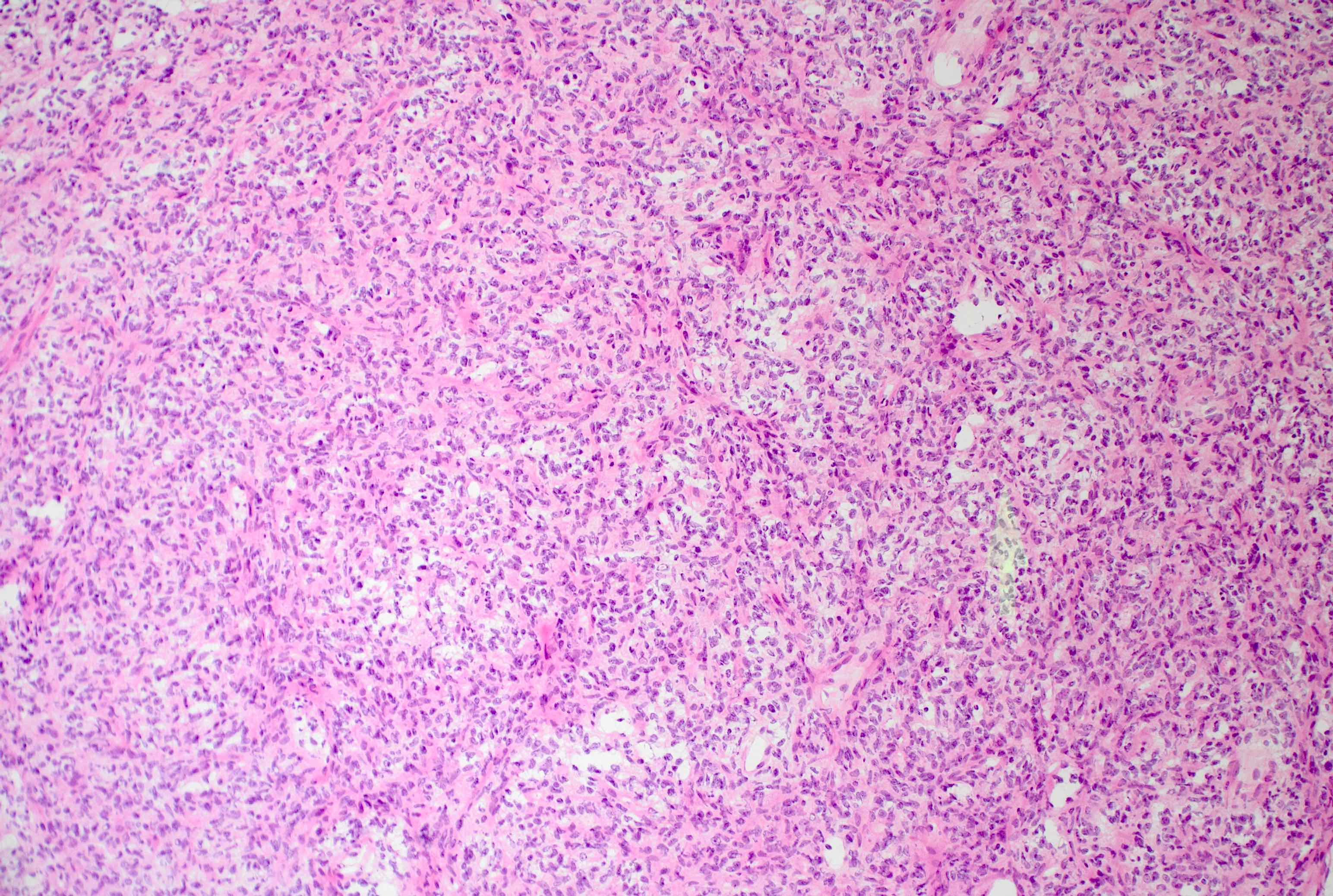
Typical findings
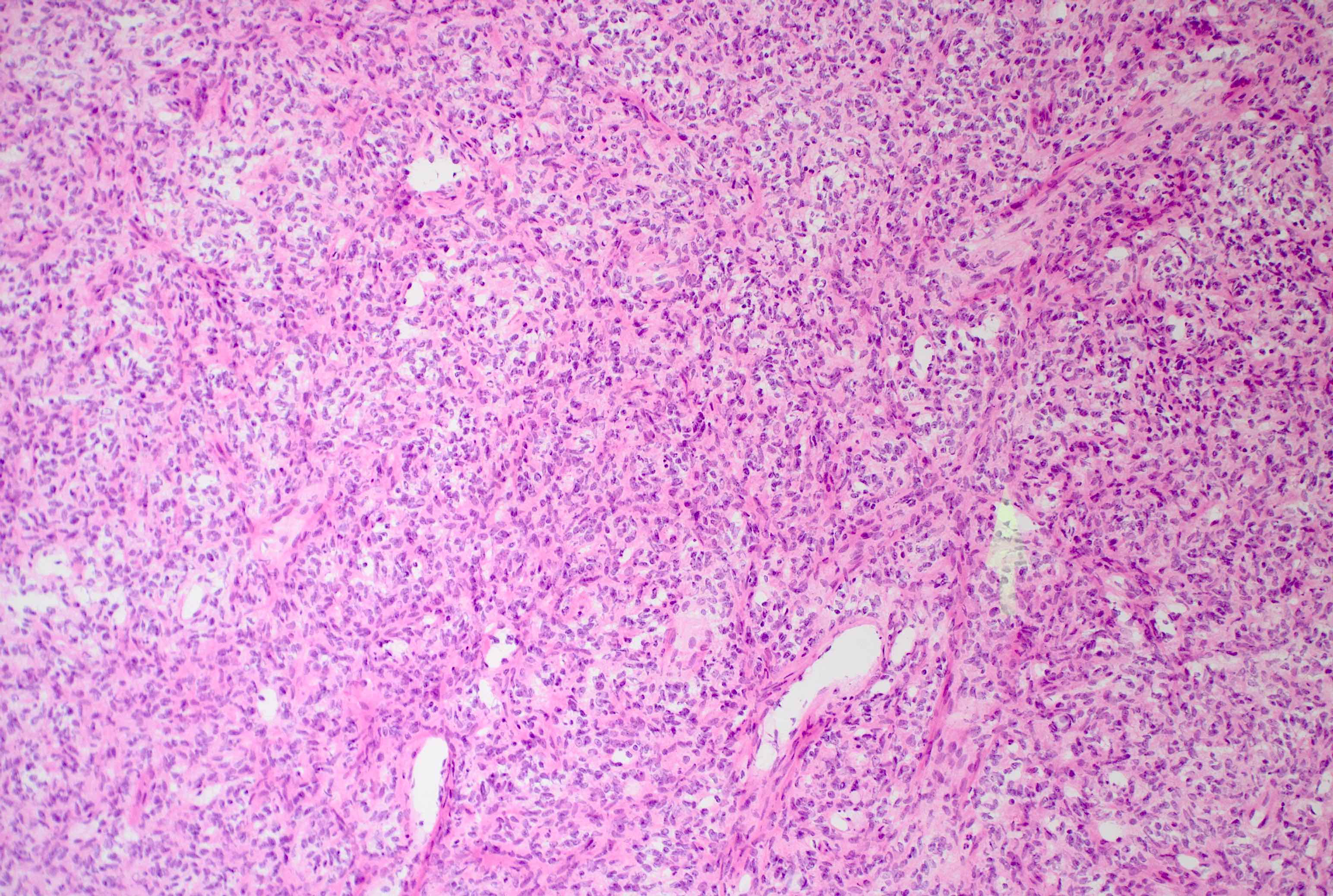
Spindle cells
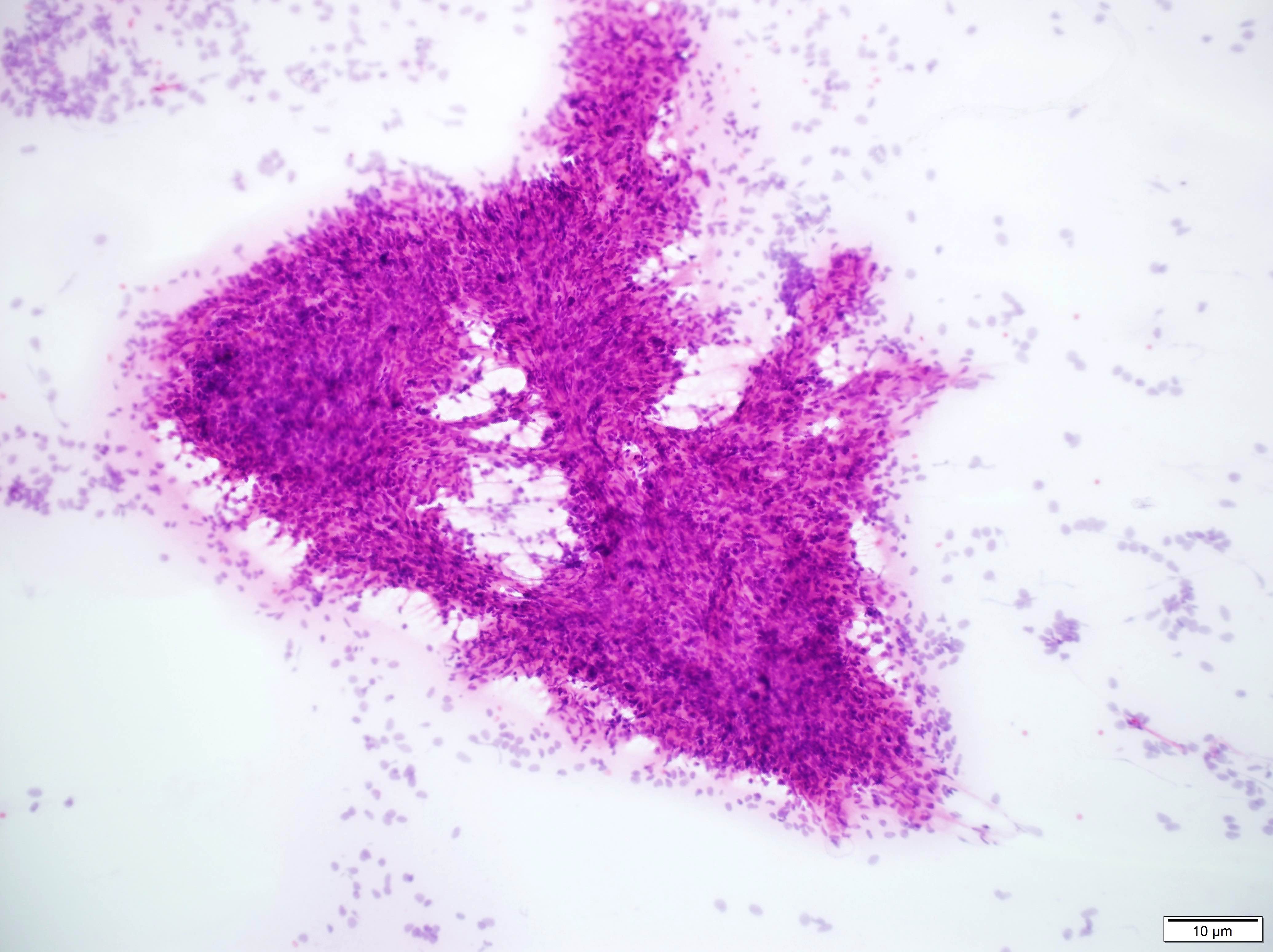
Spindled neoplastic cell aggregate
Images hosted on other servers:
Squash prep
- Essential: patternless pattern of spindle cells, often solid but can also have papillary and other architectures, arranged around branching hyalinized vessels
- Varying amounts of stromal collagen and overall cellularity
- Nuclei are bland, without features seen in mimicking neoplasms
- WHO grading criteria:
- < 5 mitoses/10 high power fields (HPF) = grade 1
- ≥ 5 mitoses/10 HPF = grade 2
- ≥ 5 mitoses/10 HPF with necrosis = grade 3
- Often has staghorn vasculature
Contributed by Brenndan Crumley, M.D., M.P.H. and Katherine Schwetye, M.D., Ph.D.
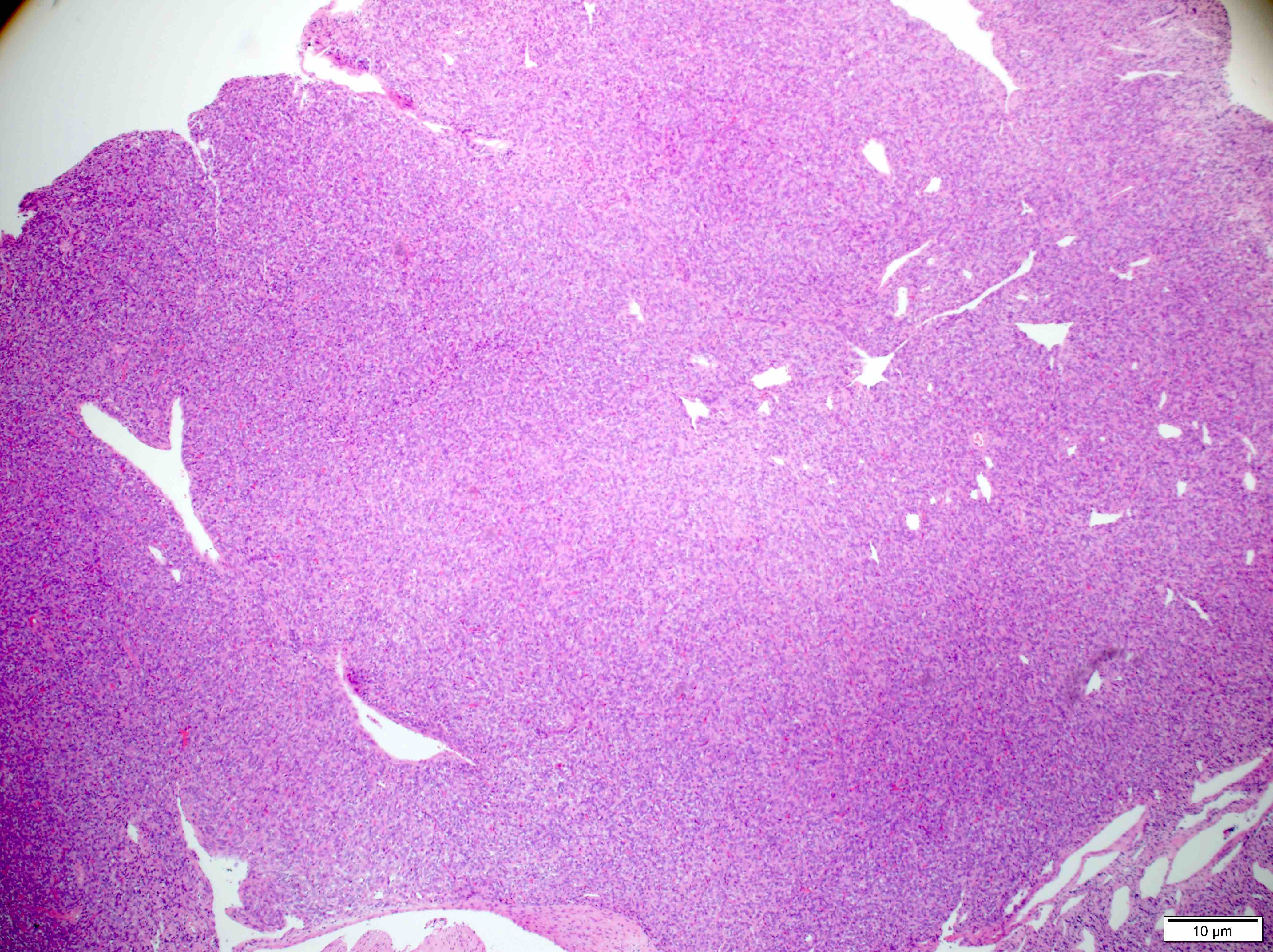
Hypercellular neoplasm, patternless pattern
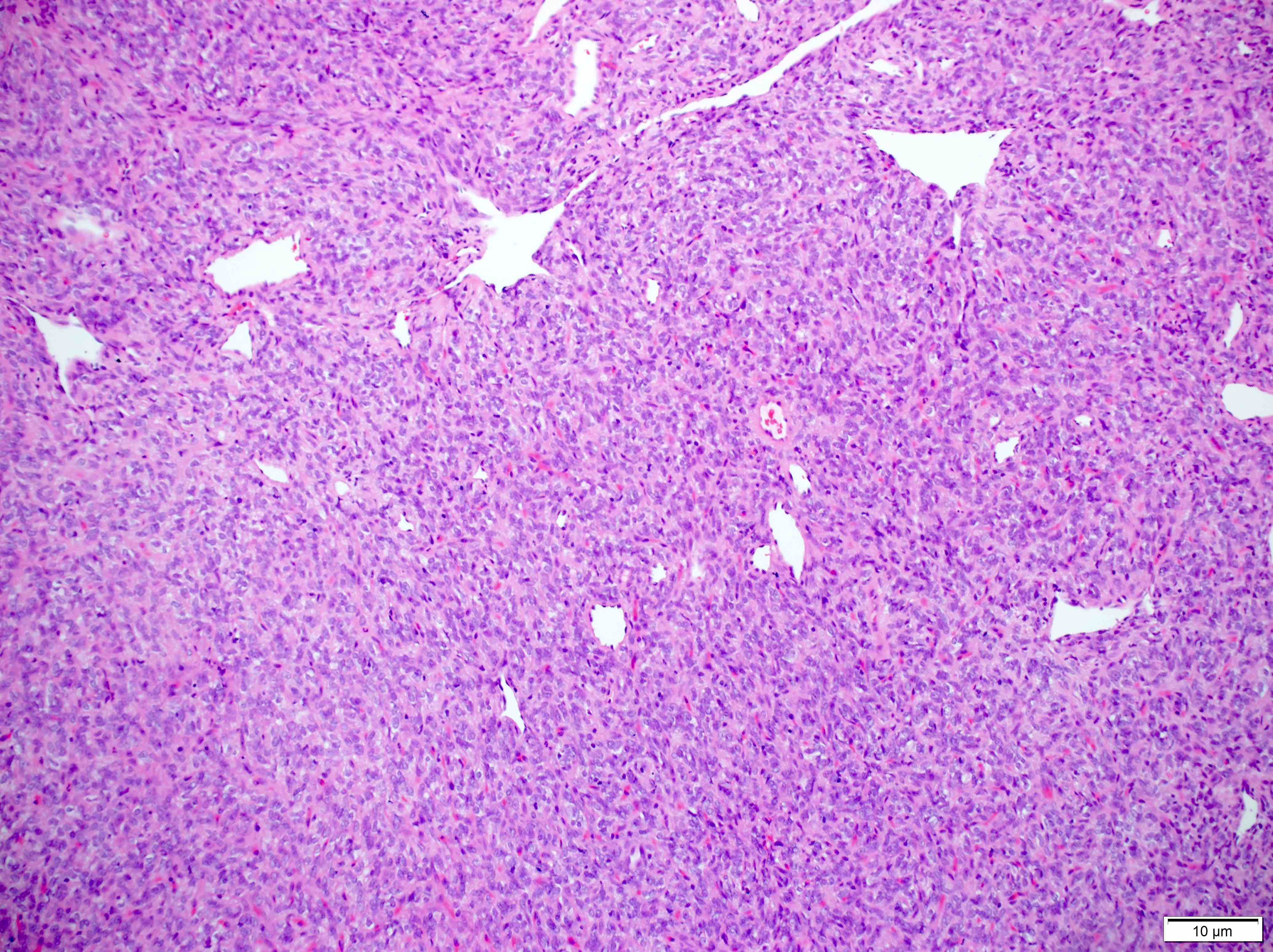
Neoplasm, haphazard spindle cells
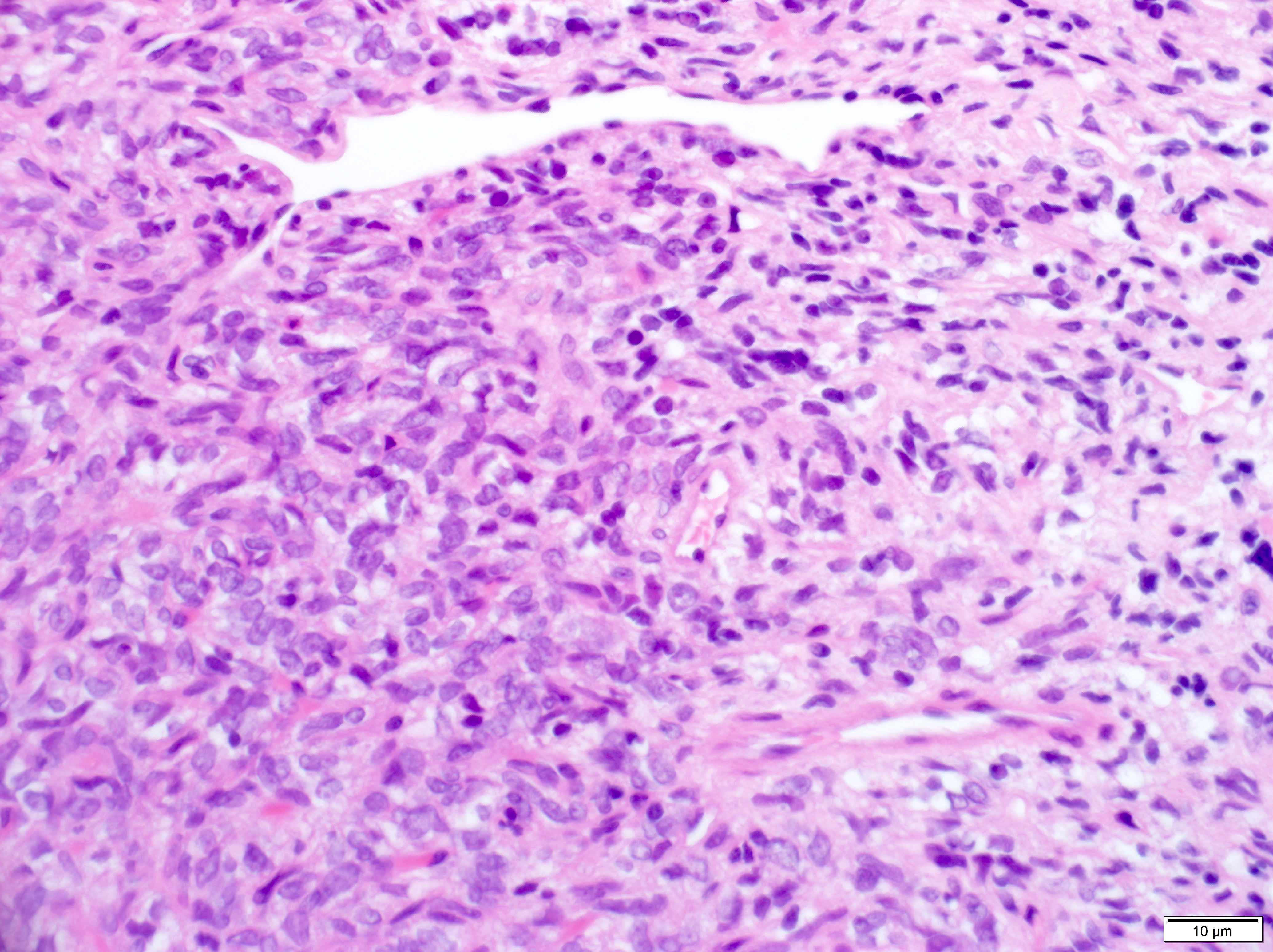
Hypercellularity and staghorn vessels
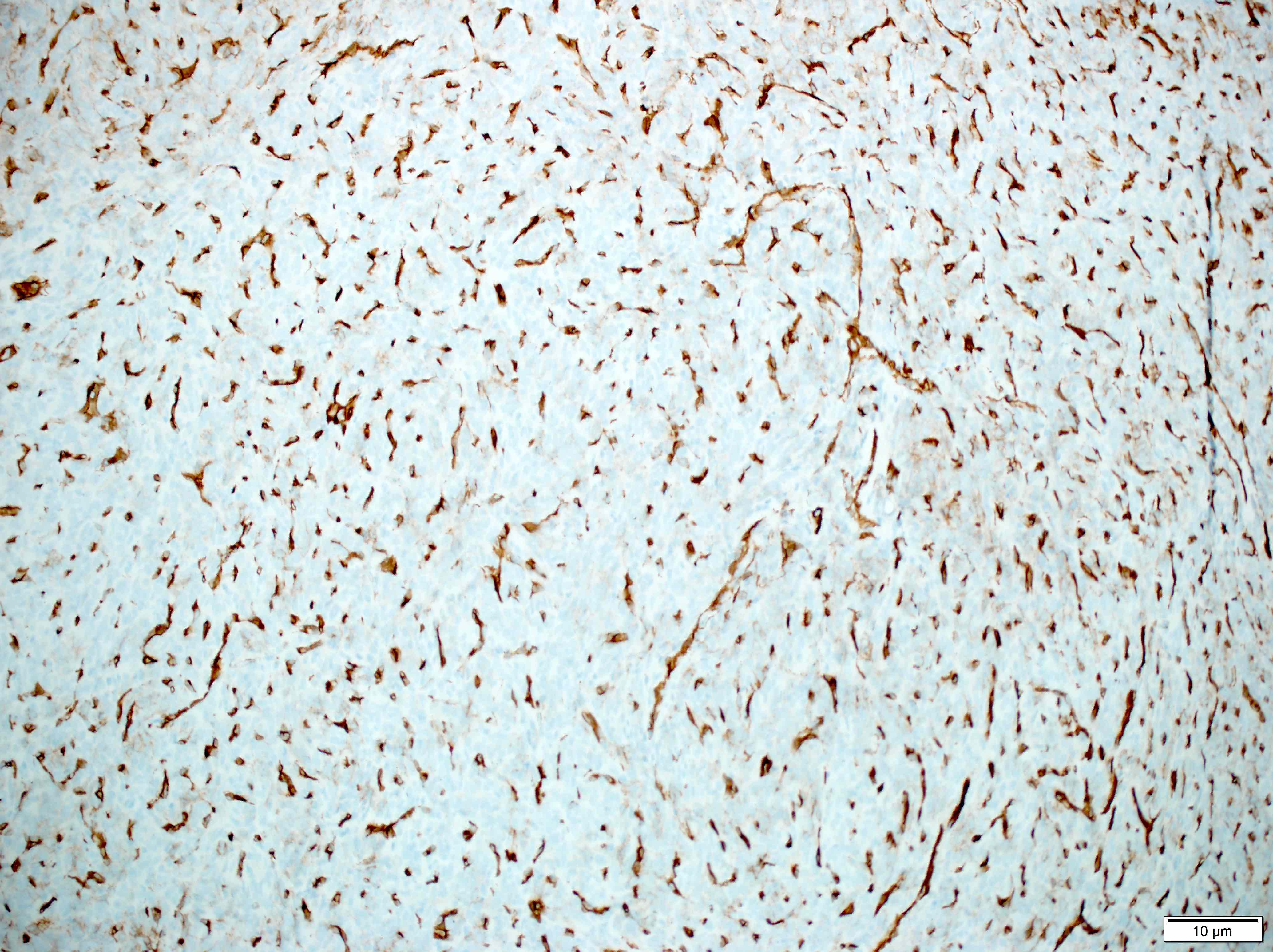
CD34 positivity can be focal
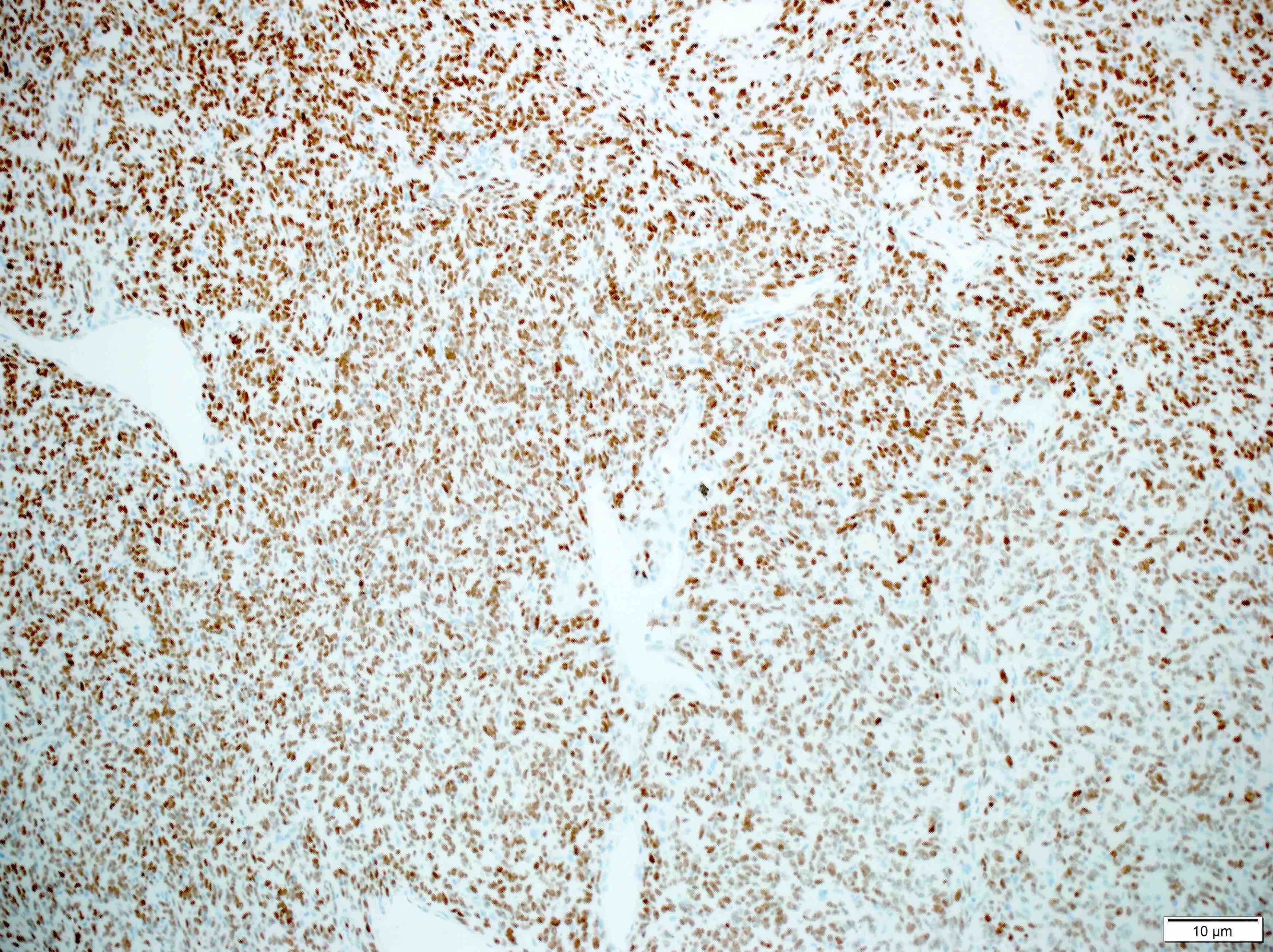
STAT6 nuclear positivity
Images hosted on other servers:

Fibrous dural mass
Occipital dural mass

Occipital dural mass, CD34

Occipital dural mass, reticulin
- Lower grade tumors will show the characteristic patternless pattern but there is not a standardized list of cytology criteria for SFT diagnosis (J Pathol Transl Med 2019;53:192)
- Higher grade tumors can show sheets of monotonous cells with heterochromatic nuclei, often with rhabdoid cells and eosinophilic cytoplasm (J Pathol Transl Med 2019;53:192)
- CD34 (lost in some high grade tumors) (Am J Surg Pathol 1997;21:1354)
- STAT6 (nuclear, lost in some dedifferentiated tumors) (Int J Surg Pathol 2017;25:58)
- Vimentin
- CD99
- SMA (tumor periphery)
- BCL2
- SSTR2A
- S100 (up to 26% some S100 positivity in the CNS) (Iran J Pathol 2016;11:195)
- EMA (up to 29% some EMA positivity in the CNS) (Iran J Pathol 2016;11:195)
- NAB2::STAT6 fusion mutation can be detected by sequencing, RT PCR or a proximity ligation assay
- NAB2 and STAT6 are close to each other on chromosome 12q, which can make genetic analysis difficult and the WHO considers a positive STAT6 immunostain to be sufficient for diagnosis in most cases (Nat Genet 2013;45:131)
Images hosted on other servers:

NAB2::STAT6 mutation analysis results

NAB2::STAT6 RT PCR and sequencing
General overview of solitary fibrous tumors (not specific to CNS tumors) by Dr. Jerad Gardner
- Brain, tentorium, resection:
- Solitary fibrous tumor, CNS WHO grade 1 (see comment)
- Comment: Sections show a neoplasm composed of bland spindle cells within a heavily collagenized stroma, with 3 mitoses in 10 high power fields. A panel of immunohistochemical stains is strongly positive for CD34 and STAT6 (nuclear), with focal positivity for SMA and is negative for EMA and SSTR2A. These findings are consistent with a solitary fibrous tumor.
- (Fibrous) meningioma (closest histologic mimic):
- Dural Ewing sarcoma:
- STAT6-, evidence of EWSR1 rearrangement
- Synovial sarcoma:
- Malignant peripheral nerve sheath tumor:
- Mesenchymal chondrosarcoma:
- Nests of STAT6- small round cells with rare cartilage islands
- Non-SFT metastasis:
- Immunophenotype of the primary tumor, STAT6-
This STAT6 stained slide is taken from the excision of an intracranial neoplasm. Which of the following stains should be negative in this neoplasm?
- ALDH1
- CD34
- SMA
- SSTR2A
Comment Here
Reference: Solitary fibrous tumor
What is the most common location of the extra-axial, STAT6 positive CNS neoplasm seen in this image?
- Cerebellum
- Pineal gland
- Spinal cord
- Tentorium
Comment Here
Reference: Solitary fibrous tumor
- Benign, slowly growing tumor typically arising in wall of lateral ventricles and composed of large ganglioid astrocytes
- Usually associated with tuberous sclerosis, an autosomal dominant syndrome due to mutations in TSC1 gene on #9q34 (hamartin protein) and TSC2 gene on #16p13.3 (tuberin protein)
- WHO grade 1
- Diagnostic criteria for tuberous sclerosis complex (TSC), modified from Roach et al. (J Child Neurol 1999;14:401)
Major features
- Facial angiofibromas or forehead plaque
- Nontraumatic ungual or periungual fibroma
- Hypomelanotic macules (more than 3)
- Shagreen patch (connective tissue nevus)
- Multiple retinal nodular hamartomas
- Cortical tuber
- Subependymal nodule
- Subependymal giant cell astrocytoma
- Cardiac rhabdomyoma, single or multiple
- Lymphangiomatosis
- Renal angiomyolipoma
Minor features
- Multiple randomly distributed pits in dental enamel
- Hamartomatous rectal polyps
- Bone cysts
- Cerebral white matter migration lines
- Gingival fibromas
- Nonrenal hamartomas
- Retinal achromic patch
- "Confetti" skin lesions
- Multiple renal cysts
- Definite TSC: either 2 major features or 1 major feature with 2 minor features
- Probable TSC: 1 major feature and 1 minor feature
- Possible TSC: either 1 major feature or 2 or more minor features
- Tuberous sclerosis also associated with pulmonary and uterine lymphangiomyomatosis, renal angiomyolipoma, cardiac rhabdomyoma
- SEGA present in 6% of tuberous sclerosis patients
Images hosted on other servers:
Enhancing mass in the third ventricle
- 20 year old woman with solitary subependymal giant cell astrocytoma and mutation of TSC2 gene in tumor but not in somatic cells (J Mol Diagn 2005;7:544)
- Circumscribed, often calcified
- Composed mainly of large polygonal to elongate cells resembling astrocytes or ganglion cells with abundant, finely granular eosinophilic cytoplasm, bright pink cellular processes, large round / oval nuclei, prominent nucleoli
- Perivascular pseudorosette formation is common
- Infiltration of mast cells and lymphocytes is common
- No Nissl substance in cytoplasm
- Presence of mitoses, vascular proliferation or necrosis does NOT indicate anaplastic progression
- Mixed glioneuronal phenotype
- GFAP+, S100+, also neurofilament proteins, neuronal associated class III β-tubulin
- HMB45 (unlike other tuberous sclerosis related lesions)
- Ependymoma:
- SEGA cells are larger
- Gemistocytic astrocytoma:
- Intraparenchymal not intraventricular
- Glioma characterized by the clustering of uniform to mildly pleomorphic tumor cell nuclei in an abundant fibrillary matrix prone to microcystic change
- CNS WHO grade 1
- Slow growing tumors that tend to occur within the ventricles of middle aged and elderly adults (Brain Pathol 2008;18:469)
- Histologically characterized by clustered nuclei in an abundant fibrillary background
- Benign biological behavior with excellent prognosis (J Neurosurg 2021;136:736)
- Not recommended by WHO: subependymal glomerate astrocytoma (J Neuropathol Exp Neurol 1954;13:30)
- Rare; < 1% of intracranial neoplasms and ~8% of ependymal tumors (Arch Pathol Lab Med 1999;123:873, J Neurosurg 2021;136:736)
- Age
- Peak incidence in middle aged and elderly adults (mean: 46.7 plus or minus 18.1 years) (J Neurooncol 2007;85:297, Neurol India 2012;60:379, Arch Pathol Lab Med 1999;123:306, J Neurosurg 2021;136:736)
- Rare in children (World Neurosurg 2017;101:599)
- M > F (70.2%) (J Neurosurg 2021;136:736, J Neurooncol 2007;85:297)
- Typically intraventricular, rarely intraparenchymal (1.1%) (J Neurosurg 2021;136:736, Childs Nerv Syst 2021;37:1759)
- Most common
- Lateral ventricles (44.5%): more likely to originate from the walls or horns (51.7%) rather than the septum pellucidum (41.4%) or trigone (10.3%) (J Neurosurg 2021;136:736)
- Fourth ventricle (43.1%): more likely to arise from the floor of the fourth ventricle (53.6%) rather than the roof (10.6%) (J Neurosurg 2021;136:736)
- Less common: third ventricle (3.7%), spinal cord (eccentric intramedullary masses preferentially at cervicothoracic segments) (J Neurooncol 2007;85:297, Arch Pathol Lab Med 1999;123:306, Neurosurgery 1996;38:251, J Neurosurg 2021;136:736, Indian J Neurosurg 2019;8:64)
- Rare: cerebral, cerebellar, brain stem and cerebellopontine angle (Childs Nerv Syst 2021;37:1759, AJNR Am J Neuroradiol 2008;29:190, Surg Neurol Int 2021;12:154, Korean J Radiol 2014;15:151, Brain Pathol 2008;18:469)
- Embryological origin of these tumors remains uncertain; possible precursors include subependymal glia, astrocytes of the subependymal plate, ependymal cells and a mixture of astrocytes and ependymal cells (Brain Pathol 2008;18:469, J Neurosurg 2021;136:736, Arch Pathol Lab Med 1999;123:306)
- Specific mechanisms by which the documented chromosomal or genetic abnormalities contribute to tumorigenesis are currently unknown
- Predisposing factors are unknown
- Rare familial cases are reported (Br J Neurosurg 1987;1:317, J Neurosurg 1994;80:1108, J Neurosurg 2014;121:570)
- TRPS1 gene alterations are reported in sporadic and familial cases (J Neurooncol 2017;134:133, Clin Genet 2023;103:717)
- Losses of chromosomes 19 and 6 appear to play a role in many sporadic cases (Neuro Oncol 2018;20:1616)
- Isolated cases have been described in patients with hereditary aniridia, PAX6 mutation and PTPN11 mutation associated Noonan syndrome (Brain Pathol 2010;20:1033, Case Rep Neurol Med 2019;2019:6091059)
- Rarely associated with craniopharyngiomas (Clin Neuropathol 1996;15:63)
- Often clinically silent and discovered only incidentally on neuroimaging for unrelated reasons or at autopsy (16.1 - 32.5%); asymptomatic cases usually do not exceed 1 cm in diameter (Arch Pathol Lab Med 1999;123:873, J Neurosurg 2021;136:736, J Neurooncol 2007;85:297)
- Clinical presentation is often nonspecific and depends on the location and size of the tumor (Neurol India 2012;60:379, J Neurosurg 1978;49:689, J Neurooncol 2007;85:297, Clin Neurol Neurosurg 1997;99:17)
- Symptomatic tumors usually range from 3 - 5 cm in diameter
- Presenting symptoms can be divided into symptoms caused by ventricular obstruction and intracranial hypertension or those attributable to the compression of neural structures
- Most common: headache (55.5%), followed by nausea or vomiting (20.9%) and dizziness or vertigo (18%) (J Neurooncol 2007;85:297)
- Less common: focal neurologic deficits, seizures (in supratentorial intraparenchymal tumors) and occasionally intratumoral / intraventricular hemorrhage (J Neurooncol 2007;85:297, Childs Nerv Syst 2021;37:1759)
- Subependymomas around the foramen of Monro can cause acute hydrocephalus regardless of tumor size (Neurol India 2012;60:379)
- Neuroimaging: MRI and CT (AJR Am J Roentgenol 1995;165:1245)
- Biopsy
- WHO essential diagnostic criteria
- Circumscribed glioma with clustering of tumor cell nuclei within expansive, focally microcystic fibrillary matrix
- Lack of conspicuous nuclear atypia
- Absent or minimal mitotic activity
- For unresolved lesions: DNA methylation profile aligned with subependymoma
- CT
- Nodular masses with cystic changes (Neurol India 2012;60:379)
- Frequently hypodense (55.6%) rather than isodense (23.6%) or hyperdense masses (20.8%) (J Neurosurg 2021;136:736)
- Absent or minimal enhancement (63.2%); notable contrast enhancement in 36.8% (J Neurosurg 2021;136:736)
- Calcification (23.1%) (J Neurosurg 2021;136:736)
- MRI
- Well circumscribed, sharply demarcated, nodular masses (Neurol India 2012;60:379, J Neurooncol 2007;85:297)
- Hypointense (68%) or isointense (23.8%) on T1 weighted images and hyperintense (> 87%) on T2 weighted images (J Neurosurg 2021;136:736, J Neurooncol 2007;85:297)
- Enhancement is quite variable (J Neurooncol 2007;85:297, Neurol India 2012;60:379)
- Absent or minimal enhancement in the majority
- Notable moderate to marked heterogeneous contrast enhancement (36.7%) (J Neurosurg 2021;136:736)
- Enhancement is more common in posterior fossa subependymomas (AJR Am J Roentgenol 1995;165:1245)
- Hydrocephalus and cystic degeneration are common features (94% and 90.8%, respectively) (J Neurosurg 2021;136:736, J Neurooncol 2007;85:297)
- Peritumoral edema (12.5%) (J Neurooncol 2007;85:297)
- Intraspinal examples appear as eccentric exophytic masses (Indian J Neurosurg 2019;8:64)
Contributed by Eman Abdelzaher, M.D., Ph.D.
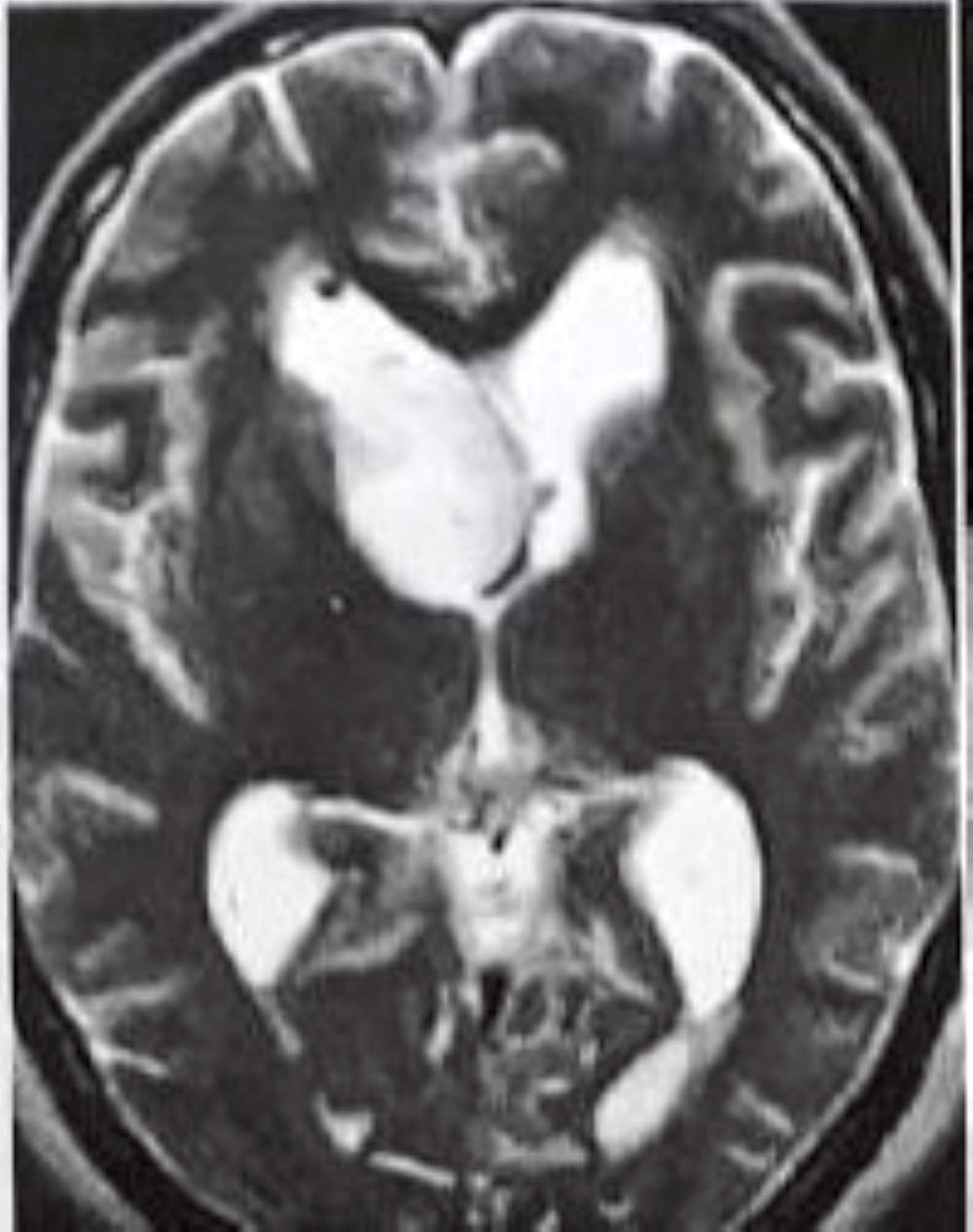
MRI, T2
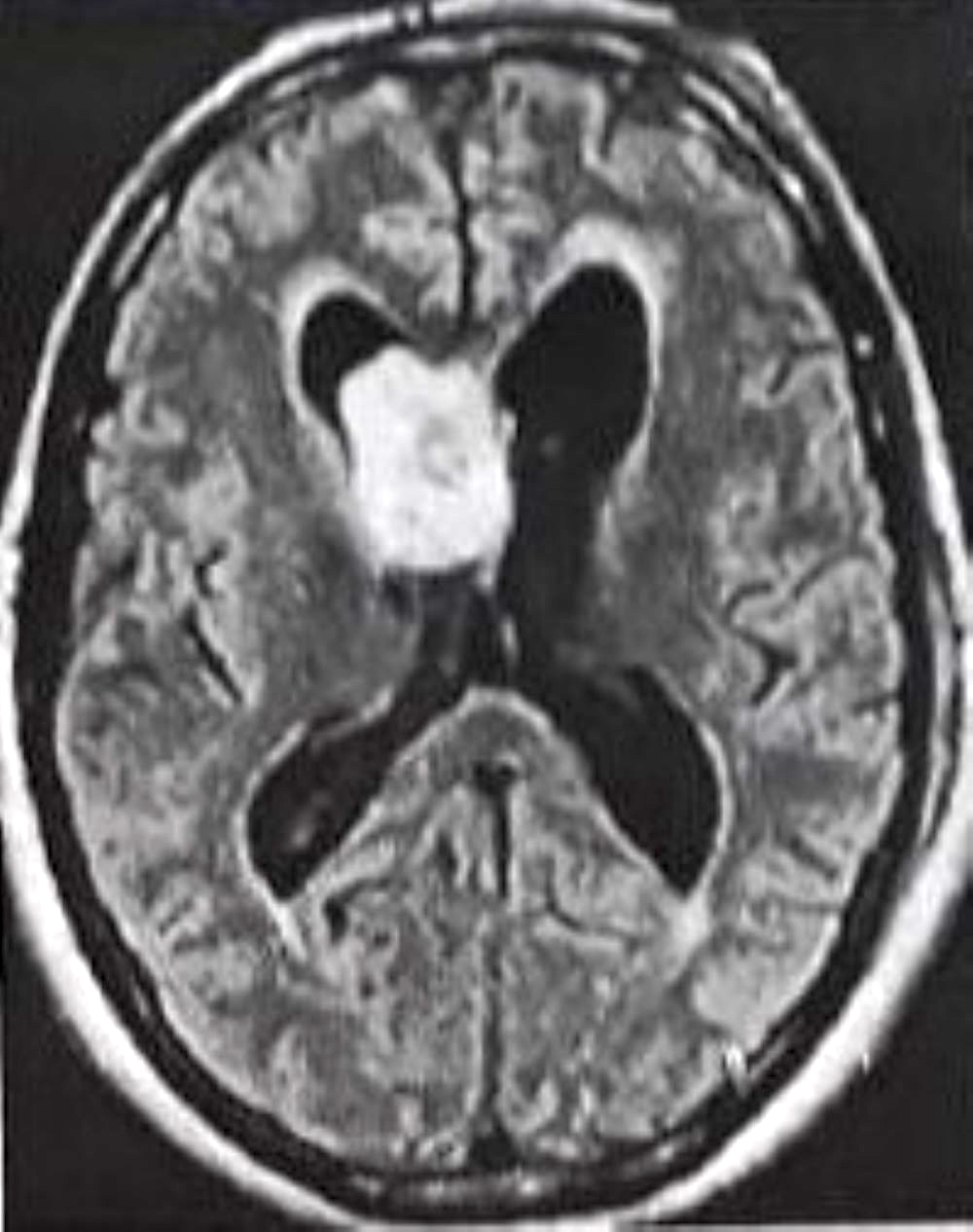
MRI, FLAIR
Images hosted on other servers:
Lateral ventricle
Spinal cord subependymoma, MRI
Intraparenchymal subependymoma, MRI
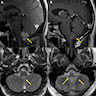
Fourth ventricle subependymoma
- Benign biological behavior with excellent prognosis and excellent postoperative functional outcomes with rare postoperative morbidity (J Neurosurg 2021;136:736, Neurol India 2012;60:379)
- Recurrence is rare; usually related to incomplete excision (1.3 - 11.8%) (J Neurosurg 2021;136:736, J Neurooncol 2007;85:297)
- Elevated proliferation index has been correlated with tumor recurrence and rapid growth (Neurol India 2009;57:191, Spine Surg Relat Res 2018;3:91, Pathol Int 2015;65:438)
- Exceptionally rare: subependymal seeding, anaplastic progression or sudden death due to acute obstructive hydrocephalus or subarachnoid hemorrhage (J Neurosurg 2021;136:736)
- Less favorable prognosis has been associated with older age and poorly defined tumor borders (J Neurooncol 2007;85:297, J Neurosurg 2015;122:49)
- Assessments of chromosome 19 status and DNA methylation profiling may prove useful in the risk stratification of patients with mixed or morphologically ambiguous lesions (Neuro Oncol 2018;20:1616, Cancer Cell 2015;27:728)
- Prognostic significance of mixed ependymoma subependymoma is controversial (J Neurosurg 1978;49:689, J Neurosurg 2015;122:49)
- Brainstem subependymomas can have H3 K27M mutations but this does not carry the rapidly lethal prognosis of diffuse midline gliomas (Hum Pathol 2019;84:262)
- Cytological pleomorphism, occasional mitoses and necrosis have not proved prognostically significant (Arch Pathol Lab Med 1999;123:306)
- Loss of chromosome 6 and TERT mutations are frequent events in ependymoma component of posterior fossa mixed tumors and confer a less favorable prognosis (Acta Neuropathol 2021;141:959)
- 11 year old girl with intraparenchymal subependymoma (Childs Nerv Syst 2021;37:1759)
- 15 year old boy with subependymoma of the cerebellopontine angle and prepontine cistern (AJNR Am J Neuroradiol 2008;29:190)
- 20 year old man with massive subependymoma of the lateral ventricles (Neuroradiology 2005;47:183)
- 25 year old man with mirror image subependymoma (Neurol India 2012;60:684)
- 28 year old man with cerebellar subependymoma (Korean J Radiol 2014;15:151)
- 40 year old man with an intraventricular combined tanycytic ependymoma and subependymoma (Brain Pathol 2013;23:359)
- 57 year old woman with thoracolumbar intramedullary subependymoma with multiple cystic formation (Eur Spine J 2013;22:S317)
- Surgery remains the mainstay of management with an ultimate goal of maximal safe tumor resection (World Neurosurg 2017;101:599, J Neurooncol 2007;85:297)
- Role of radiation therapy is controversial (J Neurooncol 2007;85:297, J Neurosurg 2021;136:736, World Neurosurg 2017;101:599, Arch Pathol Lab Med 1999;123:306)
- Targeted therapy is promising: multikinase inhibitors, topoisomerase inhibitor and pSTAT3 / HIF1α inhibitor could counteract disease progression (J Neuroimmunol 2014;277:168, Cancers (Basel) 2021;13:6218)
Images hosted on other servers:
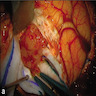
Smooth contoured lobulated mass
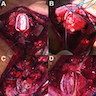
Spinal cord
subependymoma
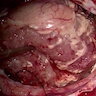
Intraparenchymal
subependymoma
- Generally circumscribed, firm, smooth contoured, lobulated, grayish white masses (J Neurooncol 2007;85:297, Case Rep Neurol 2011;3:227, J Neurosurg 1978;49:689)
- Tumors range in size from 1.5 to 60.0 mm (J Neurosurg 2021;136:736)
- Cystic changes
- Calcification with gritty sensation especially in posterior fossa tumors (J Neurooncol 2007;85:297)
- Focal hemorrhage
Contributed by Eman Abdelzaher, M.D., Ph.D.
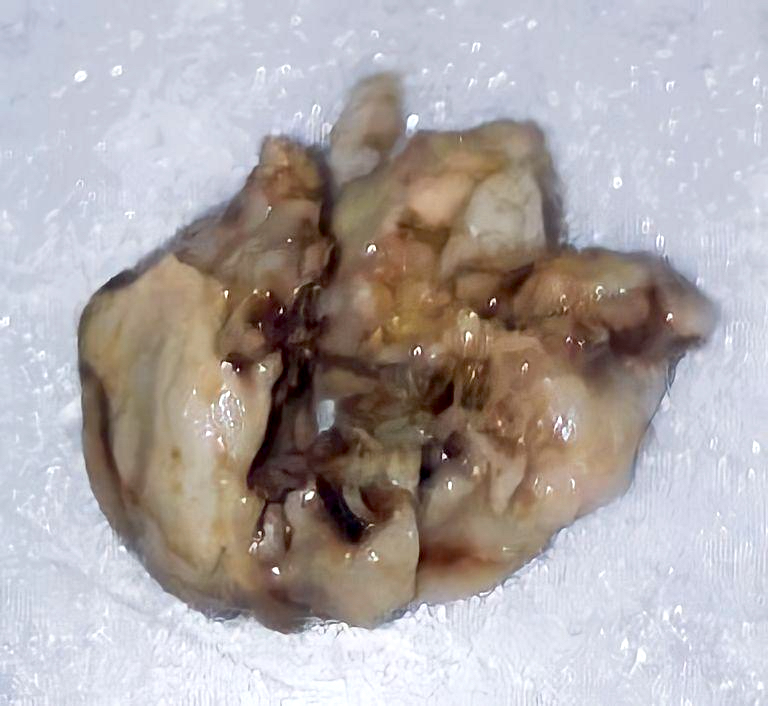
Circumscribed, lobulated mass
- Lobular pattern
- Clusters of monomorphic nuclei with absent or mild pleomorphism; nuclei appear hyperchromatic and pointy with no prominent nucleoli or mitotic figures (Indian J Neurosurg 2019;8:64, OUHSC: Case 501-2 [Accessed 29 January 2024])
- Dense, fine, fibrillary background
- Microcystic formations impart a spongy appearance on low magnification; some bluish mucoid material is identified in some microcysts
Images hosted on other servers:
Monomorphic nuclei in fibrillary pattern
Spongy appearance,
clustering of nuclei,
hyperchromatic /
pointy nuclei
- Nuclear clustering is typical; nuclei are bland, euchromatic, small, round to oval (Neurol India 2012;60:379)
- Abundant densely fibrillar, paucicellular background (Diagn Cytopathol 2018;46:258)
- Common
- Microcysts containing mucoid material, especially near foramen of Monro (Diagn Cytopathol 2018;46:258)
- Calcifications, especially in posterior fossa subependymomas (AJR Am J Roentgenol 1995;165:1245)
- Sclerotic and ectatic blood vessels, hemorrhage and hemosiderin deposits (Arch Pathol Lab Med 1999;123:306)
- Occasional
- Focal perivascular pseudorosettes (J Neurooncol 2007;85:297)
- Nuclear pleomorphism (Arch Pathol Lab Med 1999;123:306)
- Proliferative microvascular abnormalities
- Rare
- Low level mitotic activity
- Necrosis (J Neurooncol 2007;85:297)
- Exceptionally rare
- Melanotic pigmentation (Am J Surg Pathol 1990;14:729)
- Sarcomatous change (Neurosurgery 1991;28:761, Am J Surg Pathol 2008;32:699)
- Mixed histologic pattern (subependymoma together with another glial tumor)
- Mixed ependymoma subependymoma, the most common (13.7%) (J Neurosurg 2021;136:736)
- Subependymoma with elements of fibrillary astroglial or (rarely) gemistocytic morphology (J Neurooncol 2007;85:297)
Contributed by Eman Abdelzaher, M.D., Ph.D., David Taylor, M.D. and Nazila Azordegan, M.D.
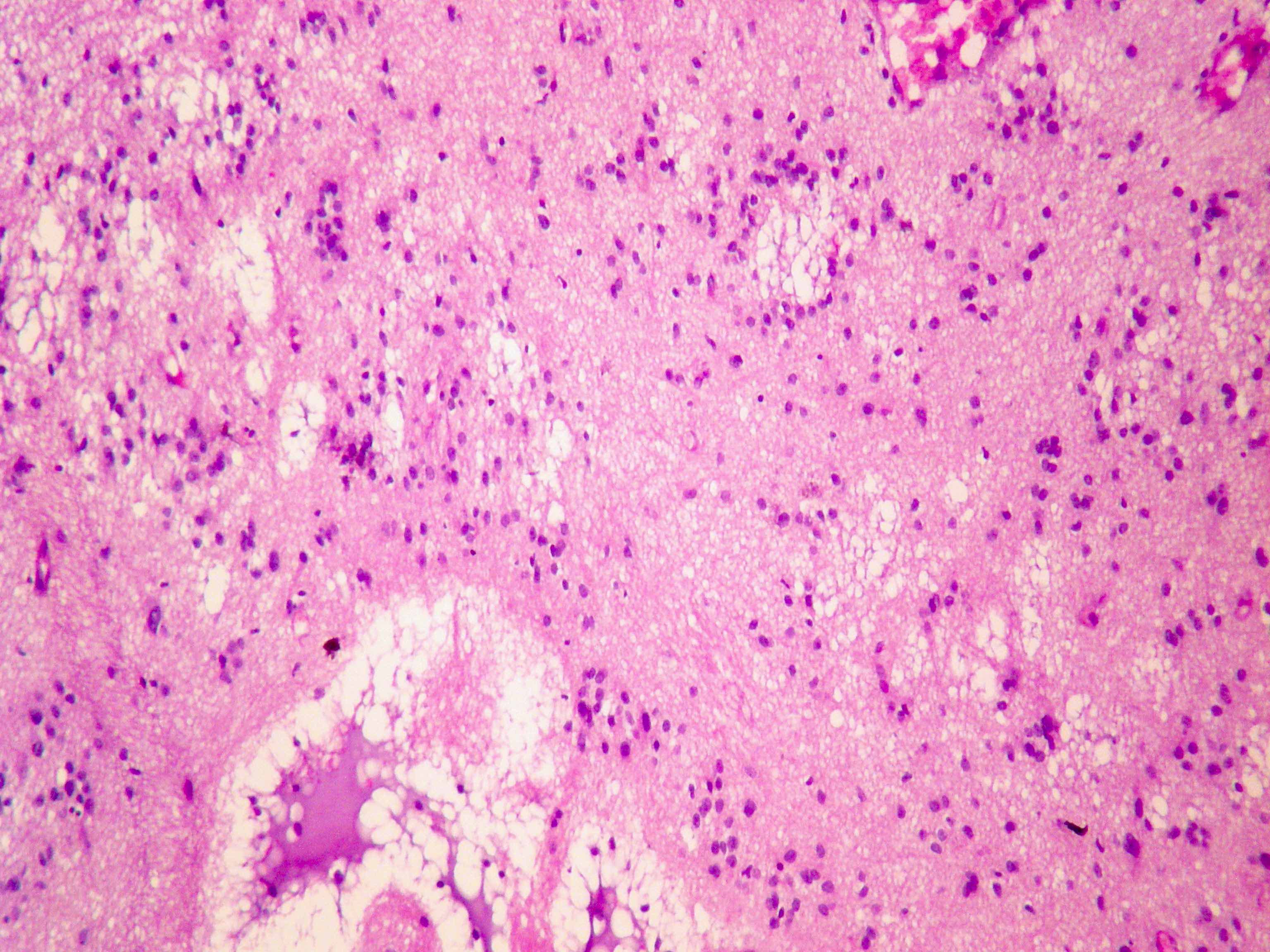
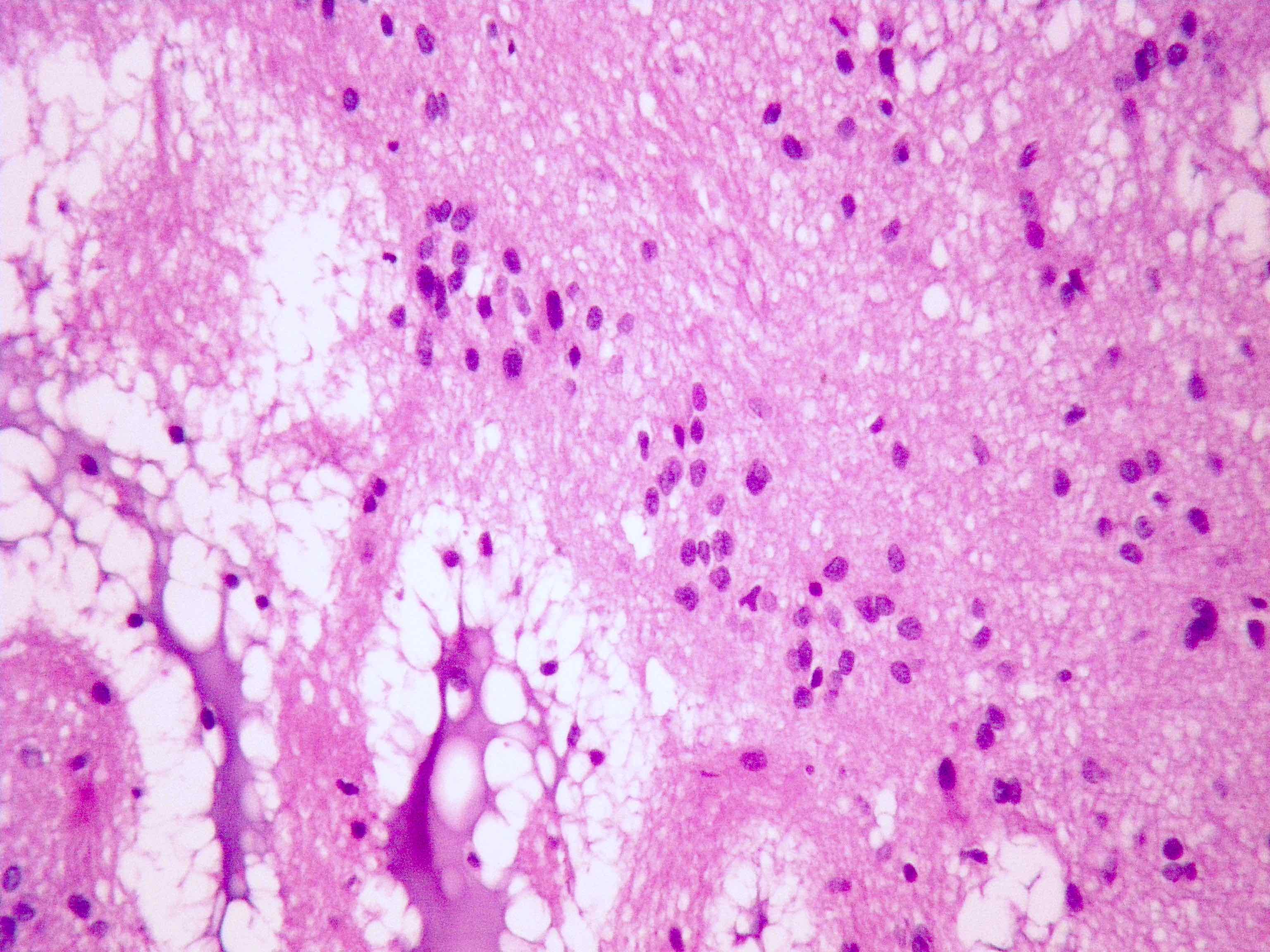
Nuclear clustering and microcystic change
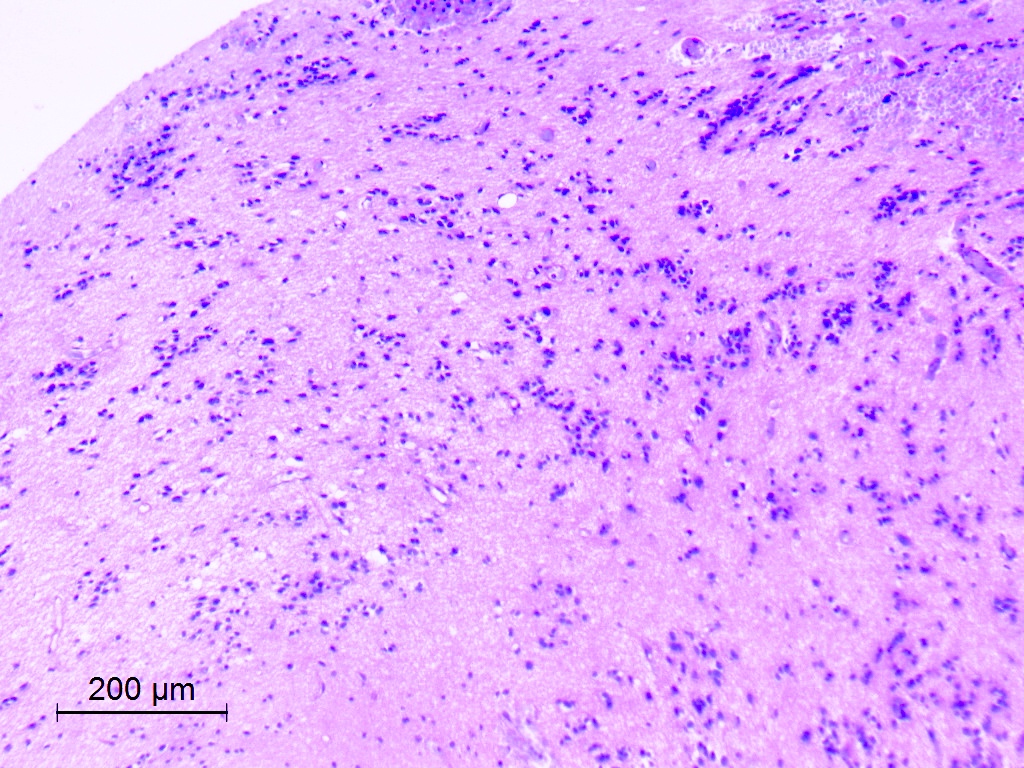
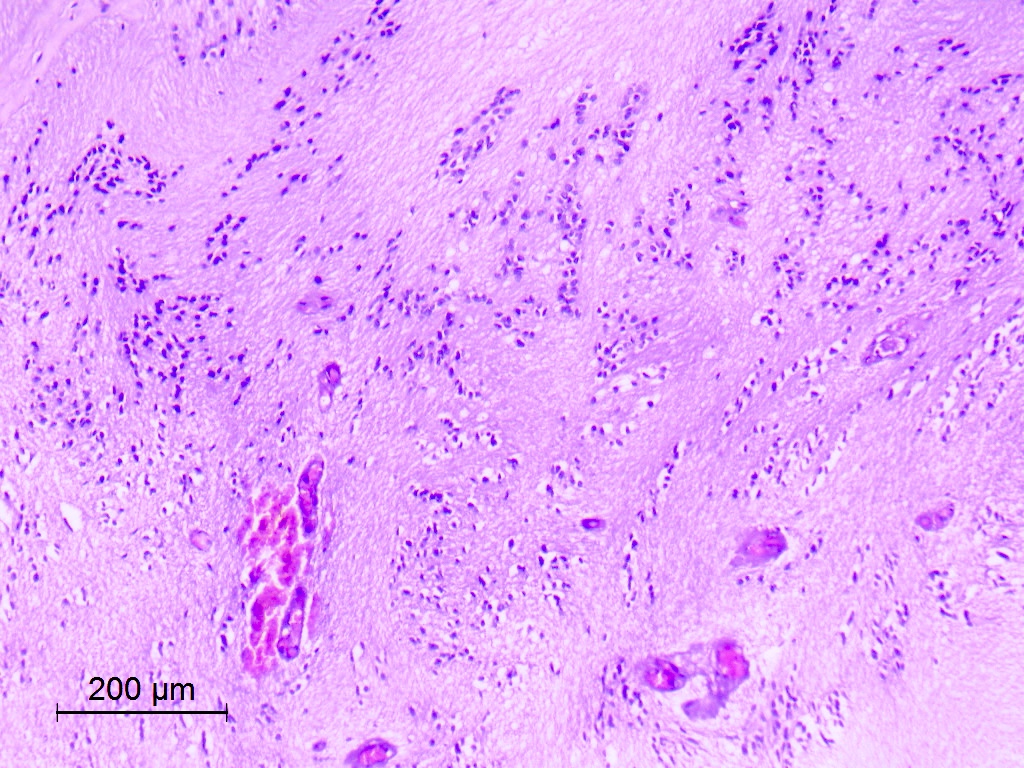
Nuclear clustering
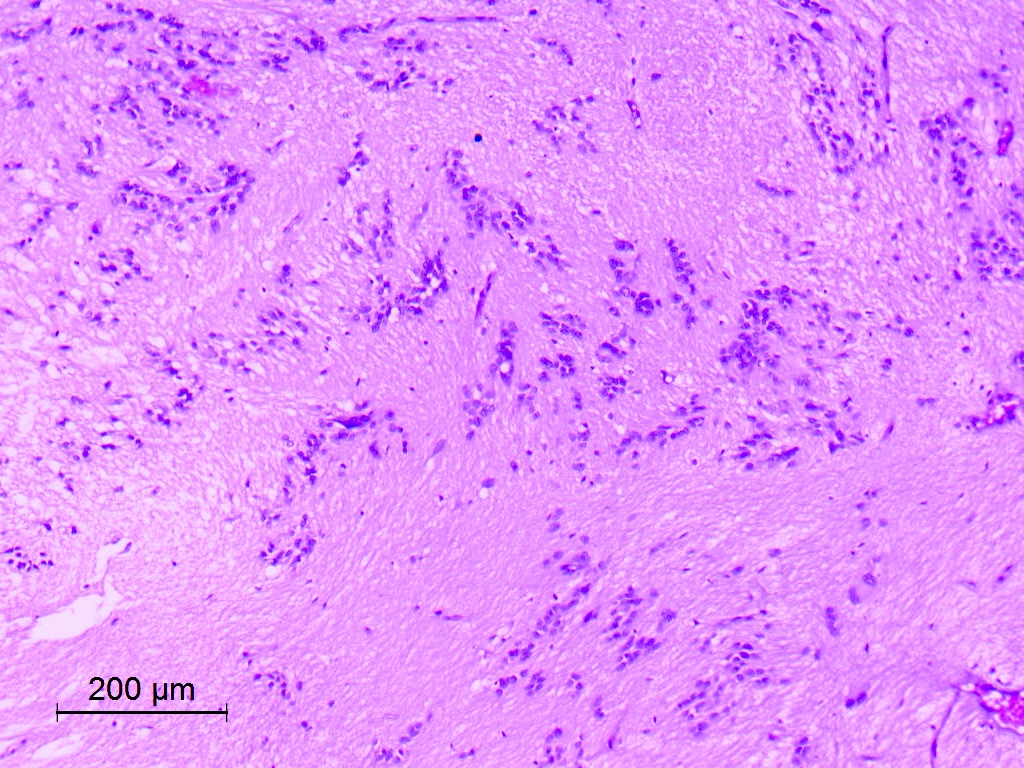
Nuclear clustering
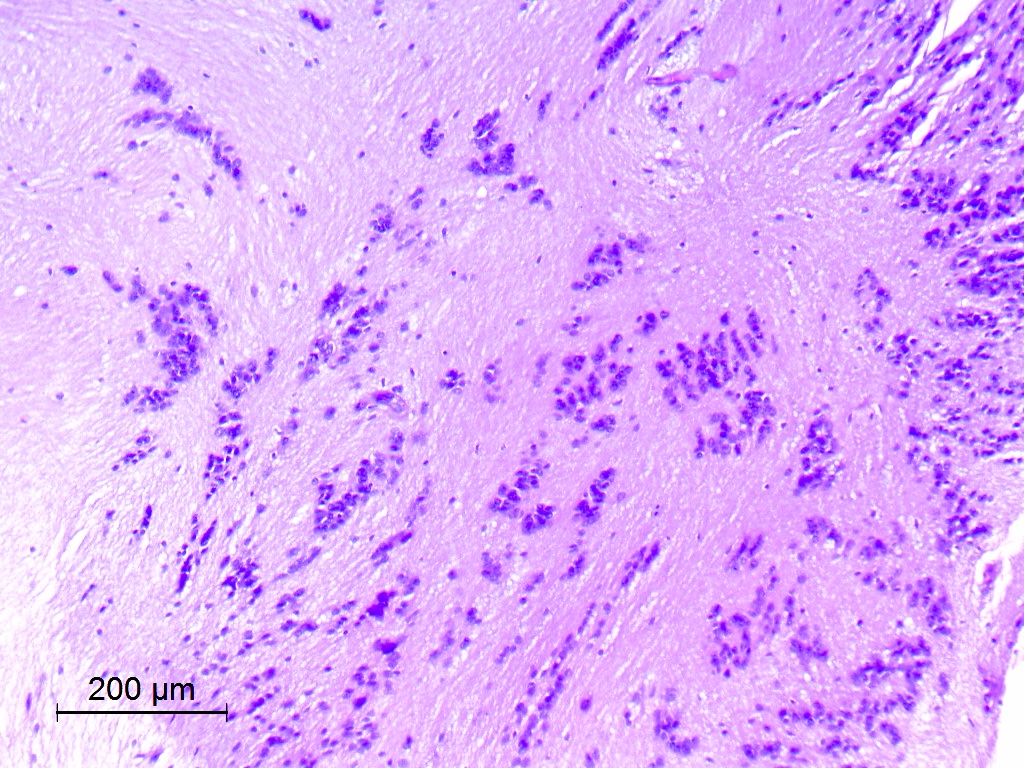
Nuclear clustering

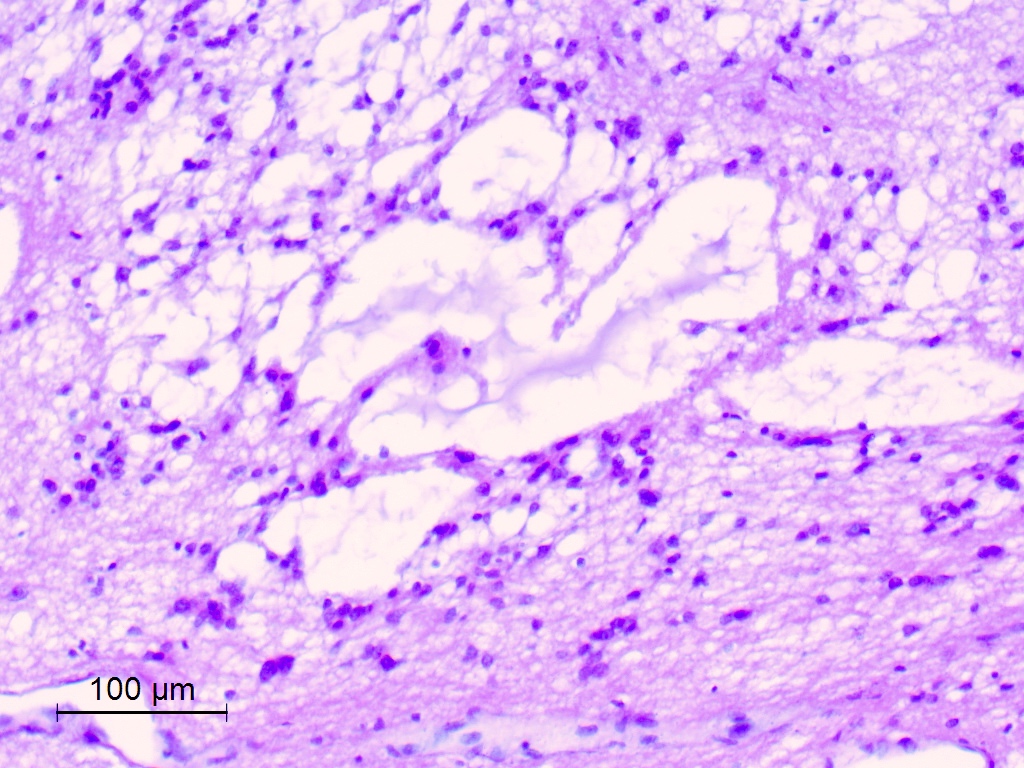
Microcystic change
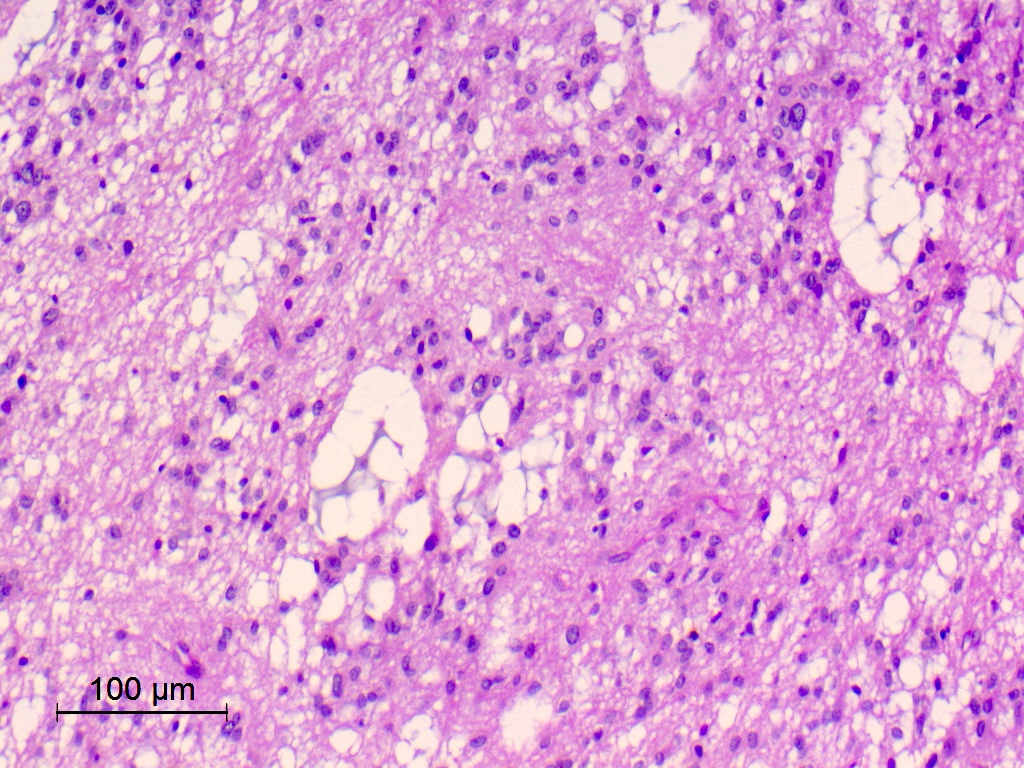
Microcystic change and nuclear pleomorphism
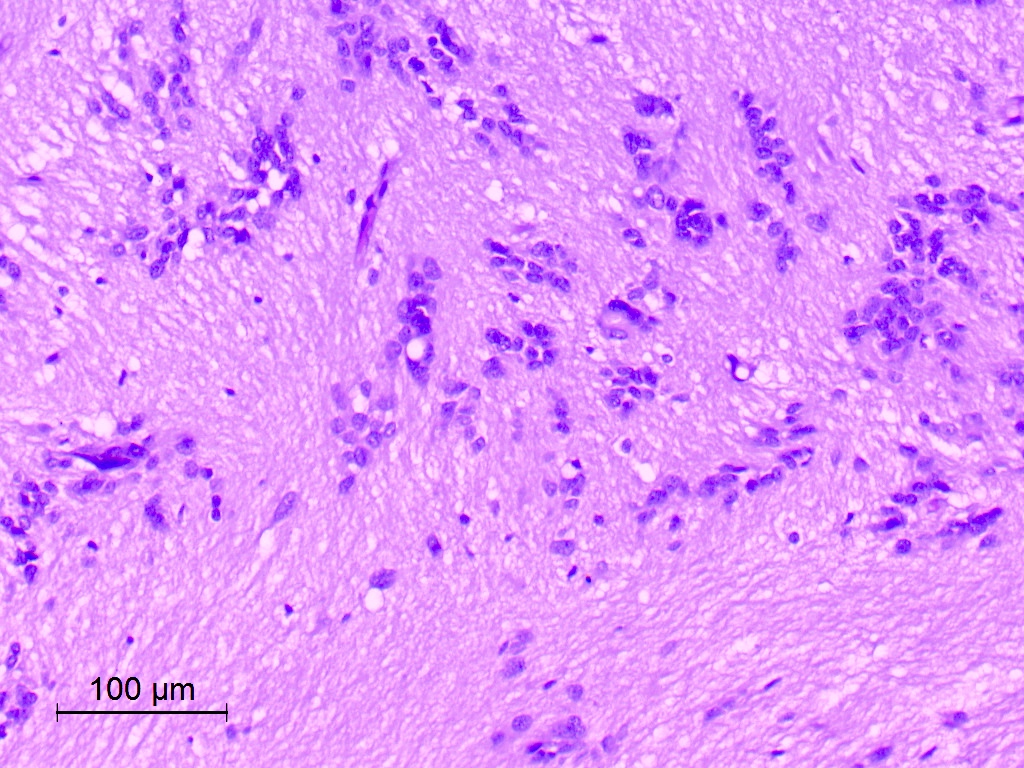
Uniform nuclei
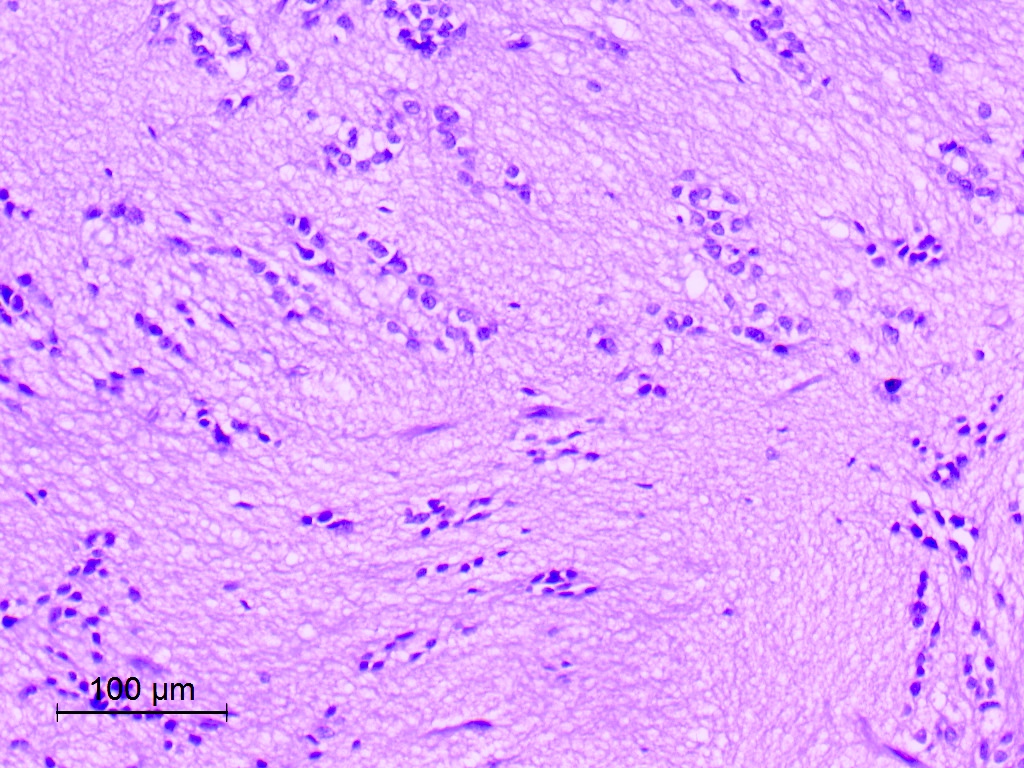
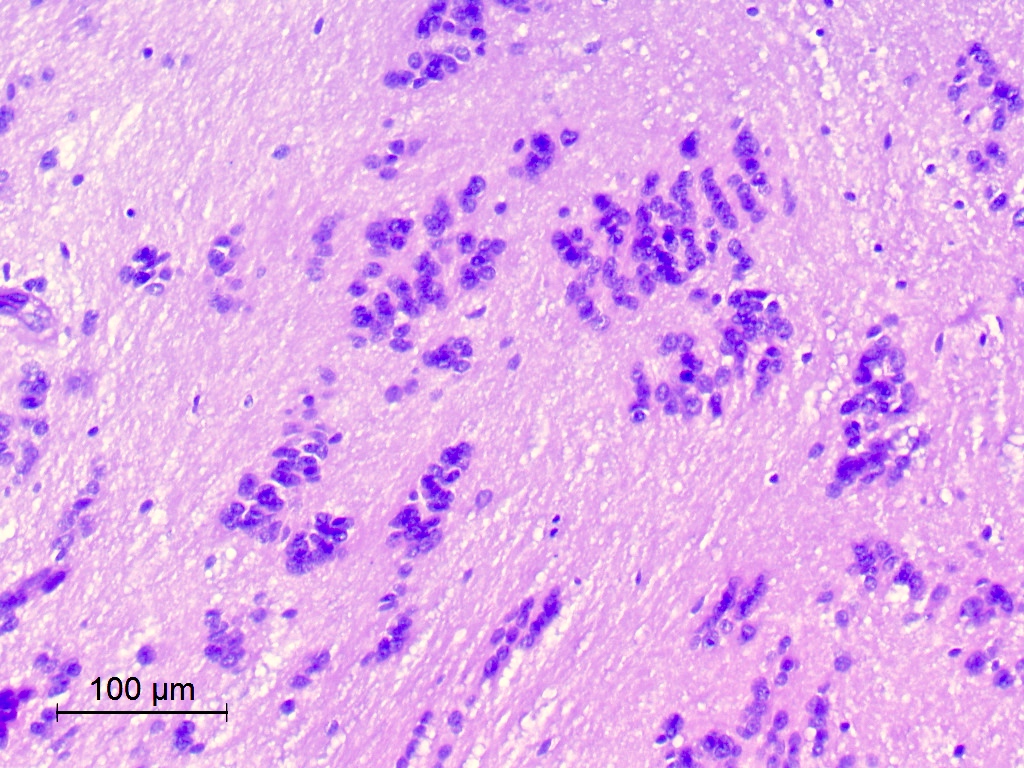
Uniform nuclei
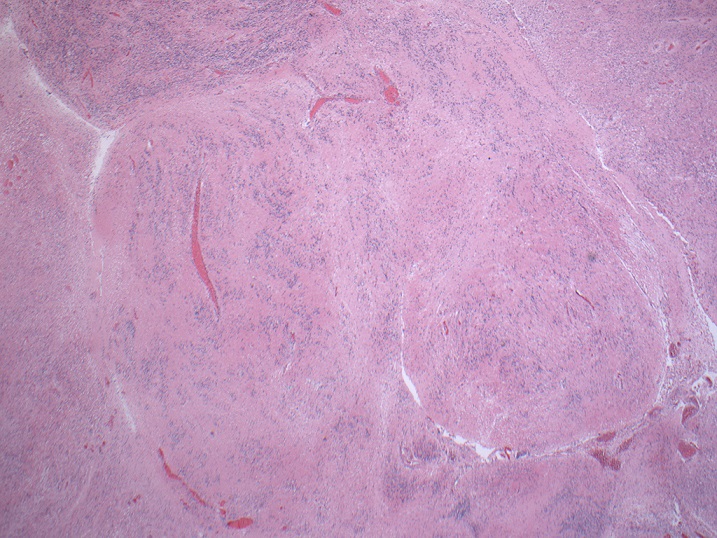
Lobulated tumor
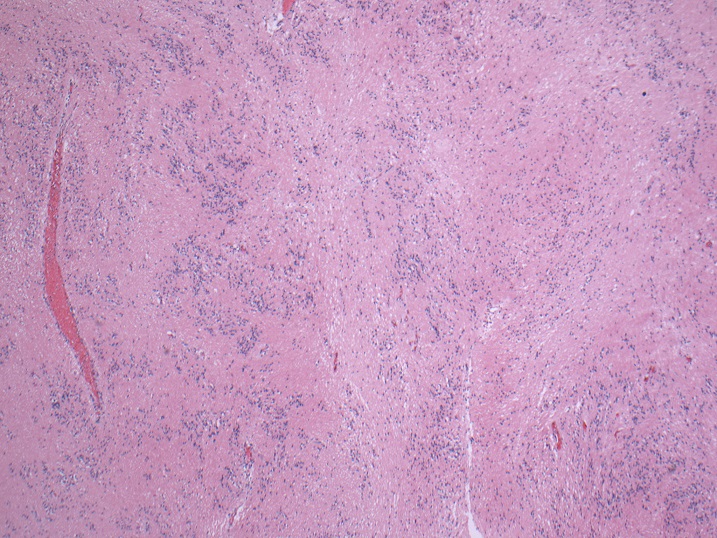
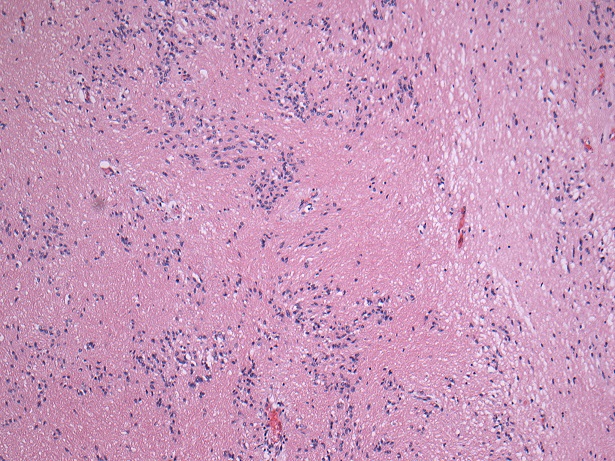
Clustered nuclei in fibrillary background
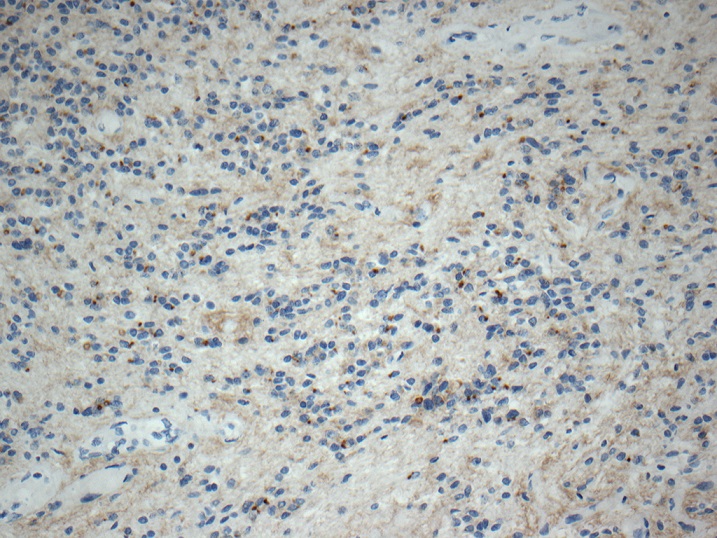
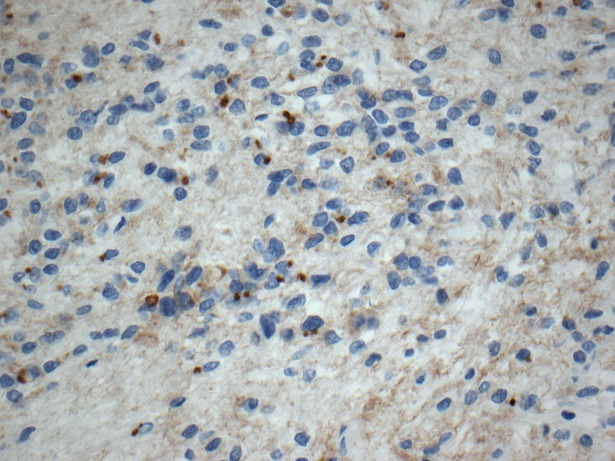
Dot-like perinuclear EMA
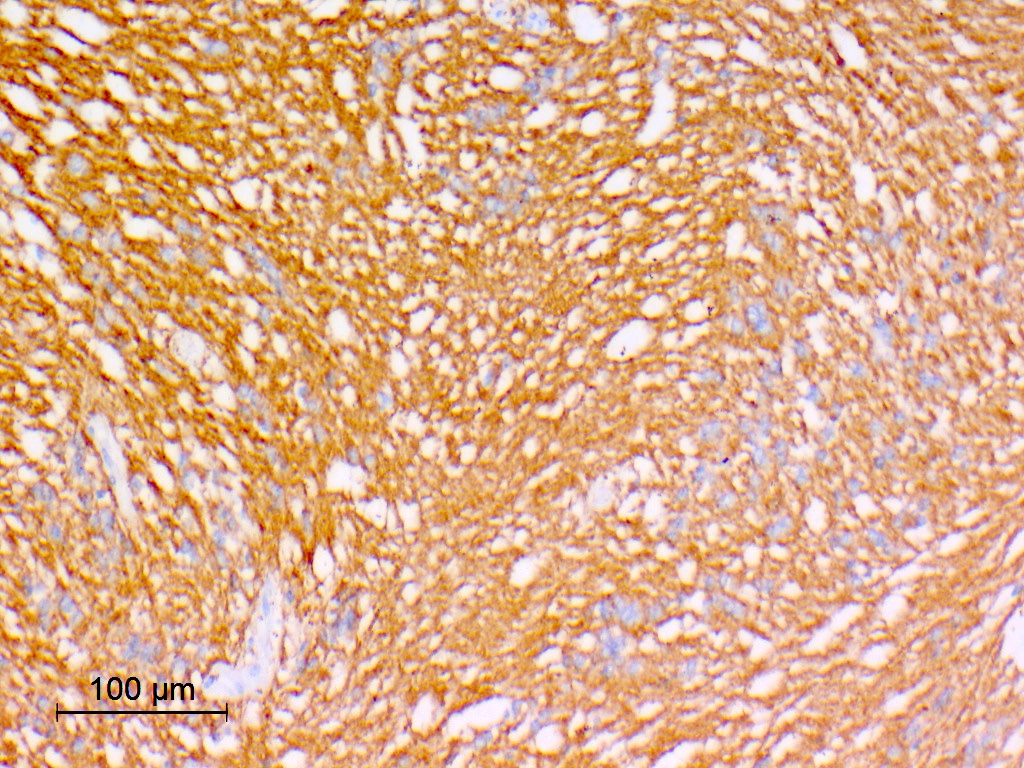
GFAP
Images hosted on other servers:
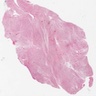
Subependymoma
- Smears poorly; remains as cohesive tissue fragments
- Clusters of bland round or oval nuclei with absent or mild pleomorphism (Diagn Cytopathol 2018;46:258, Indian J Neurosurg 2019;8:64)
- Fine fibrillary matrix of delicate cell processes (Indian J Neurosurg 2019;8:64)
- Microcystic changes (Acta Cytol 2001;45:636)
- Focal mucoid material (Diagn Cytopathol 2018;46:258)
Images hosted on other servers:
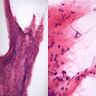
Cohesive tissue
clumps, elongated
cytoplasmic
processes
- GFAP: diffuse (100%) (J Neurooncol 2007;85:297, Cancers (Basel) 2021;13:6218, Hum Pathol 2019;84:262)
- S100 (Diagn Cytopathol 2018;46:258)
- ATRX retained (Surg Neurol Int 2021;12:154, Hum Pathol 2019;84:262)
- H3K27me3: retained (except for rare brainstem lesions) (Hum Pathol 2019;84:262)
- MDM2, HIF1α, topoisomerase IIβ, pSTAT3 and nucleolin (J Neuroimmunol 2014;277:168)
- Aquaporin 1 and aquaporin 4 with different distribution in infratentorial and supratentorial subependymomas (PLoS One 2015;10:e0131367)
- Ki67 typically low but can be variable (J Neurooncol 2007;85:297, Arch Pathol Lab Med 1999;123:306, Diagn Cytopathol 2018;46:258, Neurol India 2009;57:191, Spine Surg Relat Res 2018;3:91, Pathol Int 2015;65:438)
- EMA: may show focal, dot-like positivity (Diagn Cytopathol 2018;46:258, Cancers (Basel) 2021;13:6218, Hum Pathol 2019;84:262)
- Synaptophysin (Diagn Cytopathol 2018;46:258, Pathol Int 2003;53:169)
- Olig2 (Hum Pathol 2019;84:262)
- p53 (Hum Pathol 2019;84:262)
- SOX10: may show limited expression (J Neuropathol Exp Neurol 2016;75:295)
- IDH1 p.R132H (Surg Neurol Int 2021;12:154, Hum Pathol 2019;84:262)
- BRAF p.V600E
- H3K27M: negative (except for rare brainstem lesions) (Hum Pathol 2019;84:262)
- Neurofilament protein: no entrapped axons
- Demonstrate both astrocytic and ependymal characteristics
- Former is evidenced by intermediate filaments within processes and the latter by microlumens, cilia, microvilli and junctional complexes (J Neurosci Rural Pract 2012;3:366, Pathol Int 2003;53:169)
- Microcystic change (Diagn Cytopathol 2018;46:258)
Images hosted on other servers:
Ultrastructural findings in subependymoma
- Distinct DNA methylation profiles of subependymomas at different anatomical locations (supratentorial, posterior fossa and spinal) are documented
- Some histologically defined grade 2 and 3 classic ependymomas may also cluster with molecular subgroups of typical subependymomas (Cancer Cell 2015;27:728, Neuro Oncol 2018;20:1616)
- Chromosomal copy number variations are infrequent in subependymomas at different anatomical locations
- Recurrent copy number abnormalities are loss of chromosome 19 (most frequently within posterior fossa subependymomas [79%] and less frequent within supratentorial [50%] and spinal [40%] subependymomas) and partial chromosome 6 loss in spinal and posterior fossa subependymomas (Neuro Oncol 2018;20:1616, Cancer Cell 2015;27:728, Brain Pathol 2008;18:469)
- TRPS1 and PTPN1 gene mutations have been documented (J Neurosurg 2021;136:736)
- Posterior fossa subependymomas express KIT at high levels (Cancer Cell 2015;27:728)
- Brainstem subependymomas can have H3 K27M mutations but this does not carry the rapidly lethal prognosis of diffuse midline gliomas (Hum Pathol 2019;84:262)
- Do not appear to be associated with NF2 mutations that are found in other ependymal neoplasms (J Neurooncol 2007;85:297)
Images hosted on other servers:

Key molecular and clinical characteristics

Methylation profiling

Copy number variations
Subependymoma
- Lateral ventricular mass lesion, gross total resection:
- Subependymoma, CNS WHO grade 1
- Molecular genetics: DNA methylation profile aligned with subependymoma
- Ependymoma:
- Higher cellularity
- No nuclear clustering
- True rosettes present in some cases (AJNR Am J Neuroradiol 2006;27:488)
- Absent or few microcysts
- Some cases show mixed ependymoma subependymoma features
- Subependymal giant cell astrocytoma:
- Large ganglion-like astrocytes
- Strongly associated with tuberous sclerosis
- Variably immunoreactive for synaptophysin (Neuropathology 2009;29:25)
- Pilocytic astrocytoma:
- Biphasic piloid and spongy areas (Acta Neuropathol 2015;129:775)
- Rosenthal fibers and eosinophilic granular bodies
- Central neurocytoma:
- Cellular
- Uniform oligodendroglioma-like cells
- Neuropil background
- Synaptophysin positive, GFAP negative (Diagn Cytopathol 2018;46:258)
- Adult type diffuse gliomas (oligodendroglioma, IDH mutant and 1p / 19q codeleted or low grade diffuse astrocytomas, IDH mutant):
- Infiltrating growth pattern
- IDH mutations
- 1p / 19q codeletion in oligodendrogliomas (Neuropathol Appl Neurobiol 2022;48:e12790)
66 year old man presented with a headache. MRI of brain showed a nonenhancing mass lesion projecting into the right lateral ventricle. The mass was surgically excised. The image above shows the characteristic morphology of this lesion. What is the typical microscopic finding seen in this entity?
- High mitotic activity
- Nuclear clustering
- Palisading necrosis
- True ependymal rosettes
Comment Here
Reference: Subependymoma
- Intraparenchymal subependymoma
- Posterior fossa subependymomas
- Spinal subependymomas
- Supratentorial subependymomas
Comment Here
Reference: Subependymoma
- Extradural, commonly lumbar, spinal cysts
- Associated with degenerative joint disease
Images hosted on other servers:
Lumbar synovial cyst
- Fibrous wall, mucoid fluid
- Lined by fibrous tissue with or without synovial lining
- Fluid filled cavity within spinal cord (syringomyelia) or brain stem (syringobulbia)
- Hydromyelia: accumulation of cerebrospinal fluid (CSF) causing simple distention of central canal of spinal cord lined by ependymal cells
- Syringomyelia: accumulation of CSF dissecting into surrounding white matter to form a paracentral cavity, not lined by ependyma
- Syringohydromyelia: combination of both, seen in most cases
- Predisposing factors include craniocervical junction abnormalities, spinal cord trauma, spinal cord tumors (Merck Manual: Syrinx of the Spinal Cord or Brain Stem)
Images hosted on other servers:
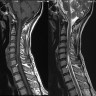
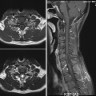
Cervical spine MRI
- Extensively revised in 1993 (Brain Pathol 1993;3:255)
- Third edition in 2000 (J Neuropathol Exp Neurol 2002;61:215)
- Fourth edition in 2007 (Acta Neuropathol 2007;114:97)
- 2016 WHO classification is considered a revision of the fourth edition (Acta Neuropathol 2016;131:803)
- Fifth edition published (online) in 2021 (Neuro Oncol 2021;23:1231)
- 2021 WHO classification, 5th edition (WHO CNS5) summary
- Builds upon the updated fourth edition published in 2016 and the subsequent recommendations of the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) (Brain Pathol 2017;27:851, Acta Neuropathol 2017;133:1, Acta Neuropathol 2018;135:481, Acta Neuropathol 2018;135:639, Acta Neuropathol 2018;136:805, Acta Neuropathol 2019;137:683, Acta Neuropathol 2020;139:603, Brain Pathol 2020;30:844, Brain Pathol 2020;30:863)
- This fifth edition incorporates major changes including further advancing the role of molecular information in the diagnosis of CNS neoplasms, introducing the concept of CNS WHO grade distinct from WHO grade, grading within tumor type, endorsing the use of Arabic numerals for grading instead of Roman numerals and defining new entities based on molecular and histological characteristics (Neuro Oncol 2021;23:1231)
- Taxonomy maintains a hybrid approach including histological and molecular features with some entities requiring specific molecular changes for a diagnosis (e.g., IDH mutant gliomas), while others display a looser association with molecular signatures that are not necessary for a diagnosis (e.g., pleomorphic xanthoastrocytoma and BRAF alteration); for each tumor type, diagnostic criteria are enumerated and listed as essential or desirable
- Grading within tumor type and the use of Arabic (rather than Roman numerals) are both introduced for the first time in CNS classification to move closer to the grading approach of non-CNS neoplasms; however, some idiosyncrasies remain based on historical knowledge and practices, in addition to the use of molecular signatures to determine grade in some cases, which further warrants the introduction and use of CNS WHO grade instead of WHO grade
- Grading within types in gliomas implies the elimination of the term anaplastic, which was previously linked to grade in these tumors
- Anatomic site modifiers that were previously part of a tumor name are removed to make nomenclature simpler and more consistent (e.g., chordoid glioma of the third ventricle is now simply chordoid glioma)
- In addition to NOS (not otherwise specified), NEC (not elsewhere classified) is introduced
- NOS: terminology to be used when molecular information is insufficient, either because testing cannot be fully performed or because the results don't fit within a defined category
- NEC: terminology to be used when necessary testing has been performed and results are available but the results do not fit perfectly in a defined tumor type
- Glial, glioneuronal and neuronal tumors are entirely restructured into 6 different families in a scheme that introduces a distinction between adult type and pediatric type tumors
- Adult type diffuse gliomas
- Pediatric type diffuse low grade gliomas
- Pediatric type diffuse high grade gliomas
- Circumscribed astrocytic gliomas
- Glioneuronal and neuronal tumors
- Ependymomas
- Classification of common adult type diffuse gliomas is drastically simplified to include only 3 types (with grading within tumor for the first 2): astrocytoma, IDH mutant (grade 2, 3 or 4), oligodendroglioma, IDH mutant and 1p / 19q codeleted (grade 2 or 3) and glioblastoma, IDH wild type (grade 4)
- Consequently, glioblastoma is a term exclusively reserved for IDH wild type tumors and the nomenclature for IDH mutant astrocytomas is astrocytoma, IDH mutant, CNS WHO grade 2, 3 or 4
- Classification of medulloblastomas remains largely similar to the 2016 classification, maintaining the incorporation of molecularly defined entities that determine prognostic categories, while adding novel subtypes (4 subgroups of SHH tumors and 8 subgroups of non-WNT / non-SHH medulloblastomas)
- Ependymomas are restructured using a combination of histopathological features, molecular signatures and anatomic site (supratentorial, posterior fossa and spinal)
- New tumor types have been added, such as diffuse hemispheric glioma, H3 G34 mutant or CNS tumor with BCOR internal tandem duplication
- Some newly recognized types are considered provisional (e.g., diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters or intracranial mesenchymal tumor, FET::CREB fusion positive) while some lesions that were introduced in 2016 are no longer provisional (e.g., diffuse leptomeningeal glioneuronal tumor)
- Meningioma is considered a single type with 15 subtypes; grading is significantly changed - while chordoid meningioma and clear cell meningioma are still considered CNS WHO grade 2 based on morphology (pending larger studies), the criteria that define atypical (grade 2) or anaplastic (grade 3) meningiomas should be applied to rhabdoid and papillary meningioma instead of assigning grade 3 based on morphology alone; furthermore, molecular alterations can influence grade, as the presence of TERT promoter mutation or homozygous deletion of CDKN2A/B is considered sufficient for a designation of CNS WHO grade 3 in meningiomas
- New types of mesenchymal neoplasms are included, such as CIC rearranged sarcoma; hemangiopericytoma is now fully retired and solitary fibrous tumor is used instead, as it aligns with the soft tissue nomenclature (the 3 tiered CNS grading scheme remains)
- Paragangliomas are understood as involving cells from the autonomic nervous system, therefore, they are now included in the chapter with nerve tumors and are renamed cauda equina neuroendocrine tumor; melanotic schwannoma is a term that is considered inadequate to reflect the unique features of this lesion, therefore this tumor type is now called malignant melanotic nerve sheath tumor
- Some major changes are introduced to the tumors of the sellar region
- Adamantinomatous craniopharyngioma and papillary craniopharyngioma are now considered 2 different tumor types (rather than subtypes) given their distinct clinicopathological and molecular characteristics
- Tumors of the neurohypophysis are now considered to belong to 1 group, as they may represent morphologic variations of the same tumor
- Pituitary neuroendocrine tumor (PitNET) is a newly introduced term, accompanying pituitary adenoma (pituitary adenoma / pituitary neuroendocrine tumor)
- A new tumor type, characteristic of infancy and associated with DICER1 alterations, is included (pituitary blastoma)
-
Gliomas, glioneuronal tumors and neuronal tumors ICD-O
- Adult type diffuse gliomas
- Pediatric type diffuse low grade gliomas
- Pediatric type diffuse high grade gliomas
- Diffuse midline glioma, H3 K27 altered9385/3
- Diffuse hemispheric glioma, H3 G34 mutant9385/3
- Diffuse pediatric type high grade glioma, H3 wild type and IDH wild type9385/3
- Infant type hemispheric glioma9385/3
- Circumscribed astrocytic gliomas
- Pilocytic astrocytoma9421/1
- High grade astrocytoma with piloid features9421/3
- Pleomorphic xanthoastrocytoma9424/3
- Subependymal giant cell astrocytoma9384/1
- Chordoid glioma9444/1
- Astroblastoma, MN1 altered9430/3
- Glioneuronal and neuronal tumors
- Ganglioglioma9505/1
- Gangliocytoma9492/0
- Desmoplastic infantile ganglioglioma / astrocytoma
- Dysembryoplastic neuroepithelial tumor9413/0
- Diffuse glioneuronal tumor with oligodendroglioma-like features
and nuclear clusters (provisional)- Mixed neuronal - glial tumors2A00.21 (ICD-11)
- Papillary glioneuronal tumor9509/1
- Rosette forming glioneuronal tumor9509/1
- Myxoid glioneuronal tumor9509/1
- Diffuse leptomeningeal glioneuronal tumor9509/3
- Multinodular and vacuolating neuronal tumor9509/0
- Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease)9493/0
- Central neurocytoma9506/1
- Extraventricular neurocytoma9506/1
- Cerebellar liponeurocytoma9506/1
- Ependymal tumors
- Supratentorial ependymoma, NOS9391/3
- Supratentorial ependymoma, ZFTA fusion positive9396/3
- Supratentorial ependymoma, YAP1 fusion positive9396/3
- Posterior fossa ependymoma, NOS9391/3
- Posterior fossa group A (PFA) ependymoma9396/3
- Posterior fossa group B (PFB) ependymoma9396/3
- Spinal ependymoma, NOS9391/3
- Spinal ependymoma, MYCN amplified9396/3
- Myxopapillary ependymoma9394/1
- Subependymoma9383/1
-
Choroid plexus tumors ICD-O
-
Embryonal tumors ICD-O
- Medulloblastoma
- Medulloblastoma, molecularly defined
- Medulloblastoma, histologically defined
- Other CNS embryonal tumors
- Atypical teratoid rhabdoid tumor9508/3
- Cribriform neuroepithelial tumor (provisional)
- Other specified tumors of neuroepithelial tissue of brain2A00.2Y (ICD-11)
- Embryonal tumor with multilayered rosettes9478/3
- CNS neuroblastoma, FOXR2 activated9500/3
- CNS tumor with BCOR internal tandem duplication9500/3
- CNS embryonal tumor, NEC / NOS9473/3
- Pineocytoma9361/1
- Pineal parenchymal tumor of intermediate differentiation9362/3
- Pineoblastoma9362/3
- Papillary tumor of the pineal region9395/3
- Desmoplastic myxoid tumor of the pineal region, SMARCB1 mutant
- Tumors of the pineal gland or pineal region2A00.20 (ICD-11)
- Meningioma9530/0
- Soft tissue tumors
- Fibroblastic and myofibroblastic tumors
- Solitary fibrous tumor8815/1
- Fibroblastic and myofibroblastic tumors
- Vascular tumors
- Hemangiomas and vascular malformations
- Cavernous hemangioma9121/0
- Capillary hemangioma9131/0
- Arteriovenous malformation9123/0
- Hemangioblastoma9161/1
- Hemangiomas and vascular malformations
- Skeletal muscle tumors
- Rhabdomyosarcoma
- Uncertain differentiation
- Intracranial mesenchymal tumor, FET::CREB fusion positive
- Neoplasms of uncertain behavior of connective or other soft tissue2F7C & XH9362 (ICD-11)
- CIC rearranged sarcoma9367/3
- Primary intracranial sarcoma, DICER1 mutant9480/3
- Ewing sarcoma9364/3
- Intracranial mesenchymal tumor, FET::CREB fusion positive
- Chondro-osseous tumors
- Chondrogenic tumors
- Notochordal tumors
-
Pineal tumors ICD-O
-
Cranial and paraspinal nerve tumors ICD-O
-
Meningiomas ICD-O
-
Mesenchymal, nonmeningothelial tumors ICD-O
-
Melanocytic tumors ICD-O
- Diffuse meningeal melanocytic neoplasms
- Circumscribed meningeal melanocytic neoplasms
-
Hematolymphoid tumors ICD-O
- Lymphomas
- CNS lymphomas
- Miscellaneous rare lymphomas in the CNS
- Histiocytic tumors
- Erdheim-Chester disease9749/3
- Rosai-Dorfman disease9749/3
- Juvenile xanthogranuloma9749/1
- Langerhans cell histiocytosis9751/1
- Histiocytic sarcoma9755/3
-
Germ cell tumors ICD-O
- Mature teratoma9080/0
- Immature teratoma9080/3
- Teratoma with somatic type malignancy9084/3
- Germinoma9064/3
- Embryonal carcinoma9070/3
- Yolk sac tumor9071/3
- Choriocarcinoma9100/3
- Mixed germ cell tumor9085/3
-
Tumors of the sellar region ICD-O
- Adamantinomatous craniopharyngioma9351/1
- Papillary craniopharyngioma9352/1
- Pituicytoma, granular cell tumor of the sellar region
and spindle cell oncocytoma- Pituicytoma9432/1
- Granular cell tumor of the sellar region9582/0
- Spindle cell oncocytoma8290/0
- Pituitary adenoma / pituitary neuroendocrine tumor (PitNET)8272/3
- Pituitary blastoma8273/3
-
Metastases to the CNS
Images hosted on other servers:

Key diagnostic genes, molecules, pathways

CNS WHO grades of selected types

Newly recognized WHO 2021 tumor types
- Histological grading is still used based on morphology in many instances; however, in this classification, the presence of certain molecular alterations is incorporated in the grading algorithm
- For example, IDH mutant astrocytomas can be designated as grade 4 based on histology (presence of microvascular proliferation or necrosis) or if there is a homozygous deletion CDKN2A/B independent of histological features
- Similarly, glioblastoma, IDH wild type can now be diagnosed in the absence of the aforementioned microscopic features, if certain molecular alterations are present in a diffuse astrocytic glioma in adults (TERT promoter mutation, EGFR gene amplification or combined gain of entire chromosome 7 and loss of entire chromosome 10 [+7 / -10])
- In the clinical setting, tumor grade remains a key factor influencing choice of therapy
- Anaplastic astrocytoma, IDH mutant
- Anaplastic astrocytoma, IDH wild type
- Glioblastoma, IDH mutant
- Glioblastoma, IDH wild type
Comment Here
Reference: WHO classification of CNS tumors
- CNS WHO grading of gliomas according to the WHO Classification of Central Nervous System Tumors, 5th edition, published in 2021
- Standardization with other fifth edition WHO classification systems (Neuro Oncol 2021;23:1231)
- Switch from Roman to Arabic numeral system
- The term "type" replaces "entity" and "subtype" replaces "variant"
- Shift toward within tumor type grading
- Removal of modifier terms, such as anaplastic
- IDH mutant astrocytic gliomas were 3 separate entities under the 2016 WHO (diffuse astrocytoma, anaplastic astrocytoma and glioblastoma) but now fall under the single tumor type astrocytoma, IDH mutant, with CNS WHO grade ranging from 2 to 4
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted, is assigned a CNS WHO grade of 2 or 3
- Pleomorphic xanthroastrocytoma is assigned a CNS WHO grade of 2 or 3
- Terms that are no longer recommended include diffuse astrocytoma, anaplastic astrocytoma, glioblastoma IDH mutant, anaplastic oligodendroglioma and anaplastic pleomorphic xanthoastrocytoma
- To emphasize difference of grading between CNS and non-CNS tumors, the term CNS WHO grade is endorsed
- Allows designation of not otherwise specified (NOS) or not elsewhere classified (NEC) as modifiers
- Not otherwise specified (NOS): applied when there is lack of diagnostic information (histological or molecular) necessary to classify glioma as a specific WHO diagnostic entity
- Not elsewhere classified (NEC): applied when there is sufficient diagnostic information available (histological and molecular) but the results do not allow for the glioma to be classified as a specific WHO diagnostic entity - often ending up more as a descriptive diagnosis
- New tumor type groupings
- Adult type diffuse gliomas
- Pediatric type diffuse low grade gliomas
- Pediatric type diffuse high grade gliomas
- Circumscribed astrocytic gliomas
- New glioma types
- Diffuse astrocytoma, MYB or MYBL1 altered
- Polymorphous low grade neuroepithelial tumor of the young
- Diffuse low grade glioma, MAPK pathway altered
- Diffuse hemispheric glioma, H3 G34 mutant
- Diffuse pediatric type high grade glioma, H3 wildtype and IDH wildtype
- Infant type hemispheric glioma
- High grade astrocytoma with piloid features
- Removed glioma types
- Diffuse astrocytoma, IDH wildtype
- Anaplastic astrocytoma, IDH wildtype
- Renamed glioma types
- Astrocytoma, IDH mutant
- Eliminates the following previously used terms: diffuse astrocytoma, IDH mutant; anaplastic astrocytoma, IDH mutant; and glioblastoma, IDH mutant
- Diffuse midline glioma, H3 K27 altered
- Terminology replaces the term mutant with altered, to include other mechanisms of histone alteration (e.g., EZHIP overexpression)
- Chordoid glioma
- Omits "of the third ventricle" anatomical designation
- Astroblastoma, MN1 altered
- Adds molecular criterion for diagnosis
- Astrocytoma, IDH mutant
- Changes to existing grading criteria require that either histological or molecular features are present (adult type diffuse gliomas)
- IDH mutant astrocytoma, CNS WHO grade 4 (Neuro Oncol 2021;23:1231)
- IDH mutant diffuse astrocytic glioma with CDKN2A / CDKN2B homozygous deletion is assigned a CNS WHO grade 4, even in the absence of microvascular proliferation or necrosis
- Glioblastoma, IDH wildtype, CNS WHO grade 4 (Acta Neuropathol 2018;136:805)
- Changes require that either histological or molecular criteria are present in order to obtain diagnosis
- Requirements: a diffuse astrocytic glioma with any one or more of the following: microvascular proliferation, necrosis, TERT promoter mutation, EGFR amplification, gain of chromosome 7 with concomitant loss of chromosome 10
- IDH mutant astrocytoma, CNS WHO grade 4 (Neuro Oncol 2021;23:1231)
-
Adult type diffuse gliomas ICD-O codes
- Astrocytoma, IDH mutant
- Astrocytoma, IDH mutant, CNS WHO grade 2 9400/3
- Astrocytoma, IDH mutant, CNS WHO grade 3 9401/3
- Astrocytoma, IDH mutant, CNS WHO grade 4 9445/3
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted, CNS WHO grade 2 9450/3
- Oligodendroglioma, IDH mutant and 1p / 19q codeleted, CNS WHO grade 3 9451/3
- Glioblastoma, IDH wildtype, CNS WHO grade 4 9440/3
-
Pediatric type diffuse low grade gliomas ICD-O codes
- Diffuse astrocytoma, MYB or MYBL1 altered, CNS WHO grade 1 9421/1
- Angiocentric glioma, CNS WHO grade 1 9431/1
- Polymorphous low grade neuroepithelial tumor of the young, CNS WHO grade 1 9413/0
- Diffuse low grade glioma, MAPK pathway altered* 9421/1
-
Pediatric type diffuse high grade gliomas ICD-O codes
- Diffuse midline glioma, H3 K27 altered, CNS WHO grade 4 9385/3
- Diffuse hemispheric glioma, H3 G34 mutant, CNS WHO grade 4 9385/3
- Diffuse pediatric type high grade glioma, H3 wildtype and IDH wildtype, CNS WHO grade 4 9385/3
- Infant type hemispheric glioma* 9385/3
-
Circumscribed astrocytic gliomas ICD-O codes
- Pilocytic astrocytoma, CNS WHO grade 1 9421/1
- High grade astrocytoma with piloid features* 9421/3
- Pleomorphic xanthoastrocytoma, CNS WHO grade 2 or 3 9424/3
- Subependymal giant cell astrocytoma, CNS WHO grade 1 9384/1
- Chordoid glioma, CNS WHO grade 2 9444/1
- Astroblastoma, MN1 altered* 9430/3
- *Definitive CNS WHO grade not established
Contributed by P.J. Cimino, M.D., Ph.D.
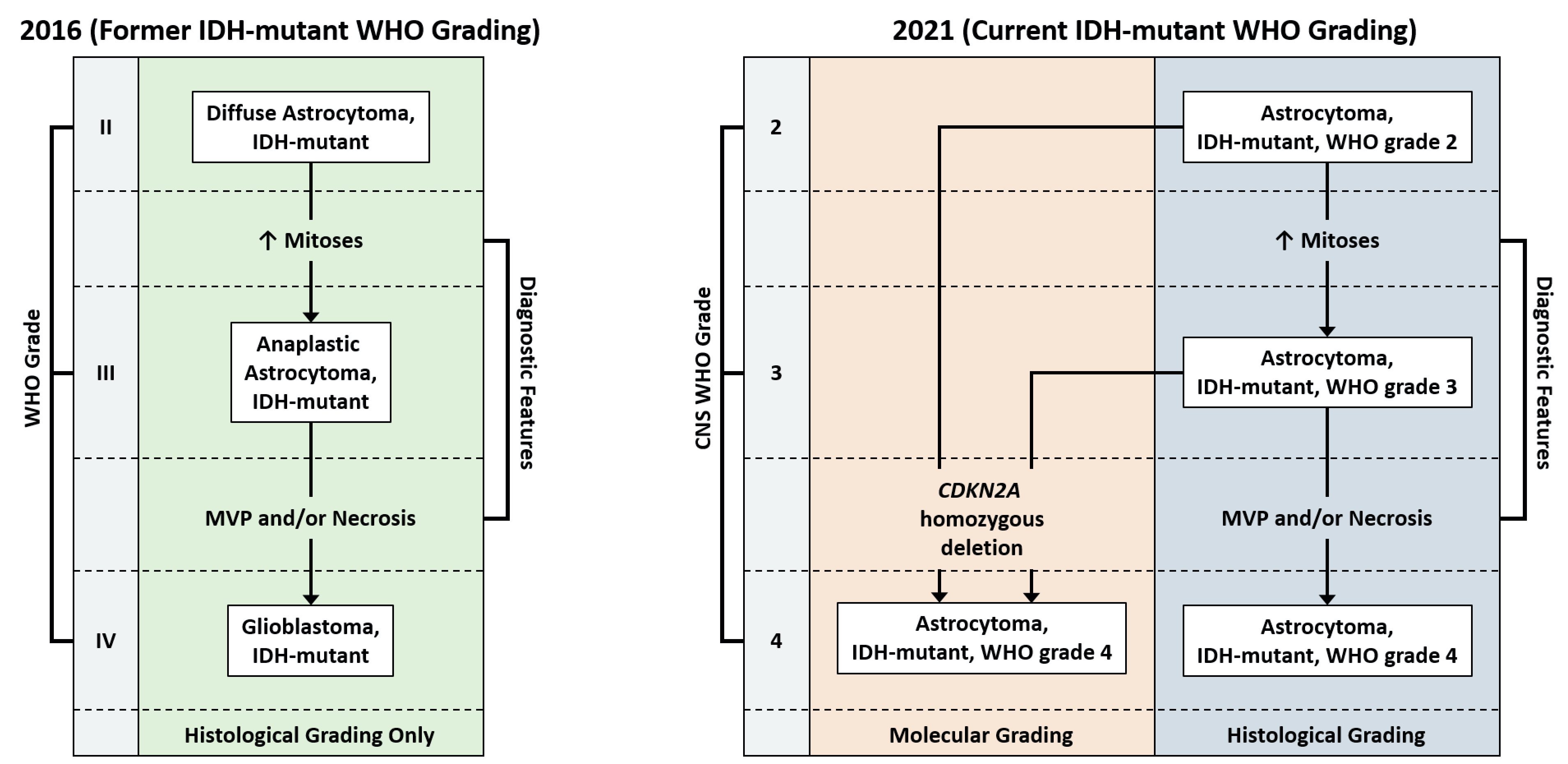
Adult type IDH mutant gliomas
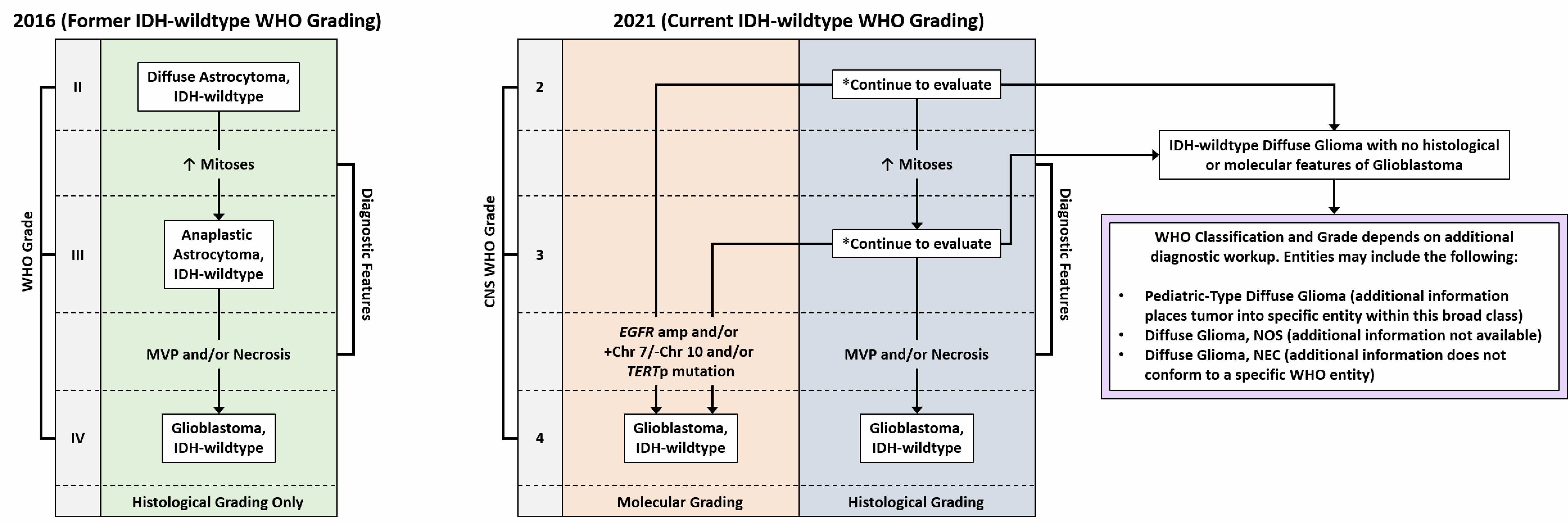
Adult type IDH wildtype gliomas
Contributed by P.J. Cimino, M.D., Ph.D.

Astrocytoma lacking mitoses
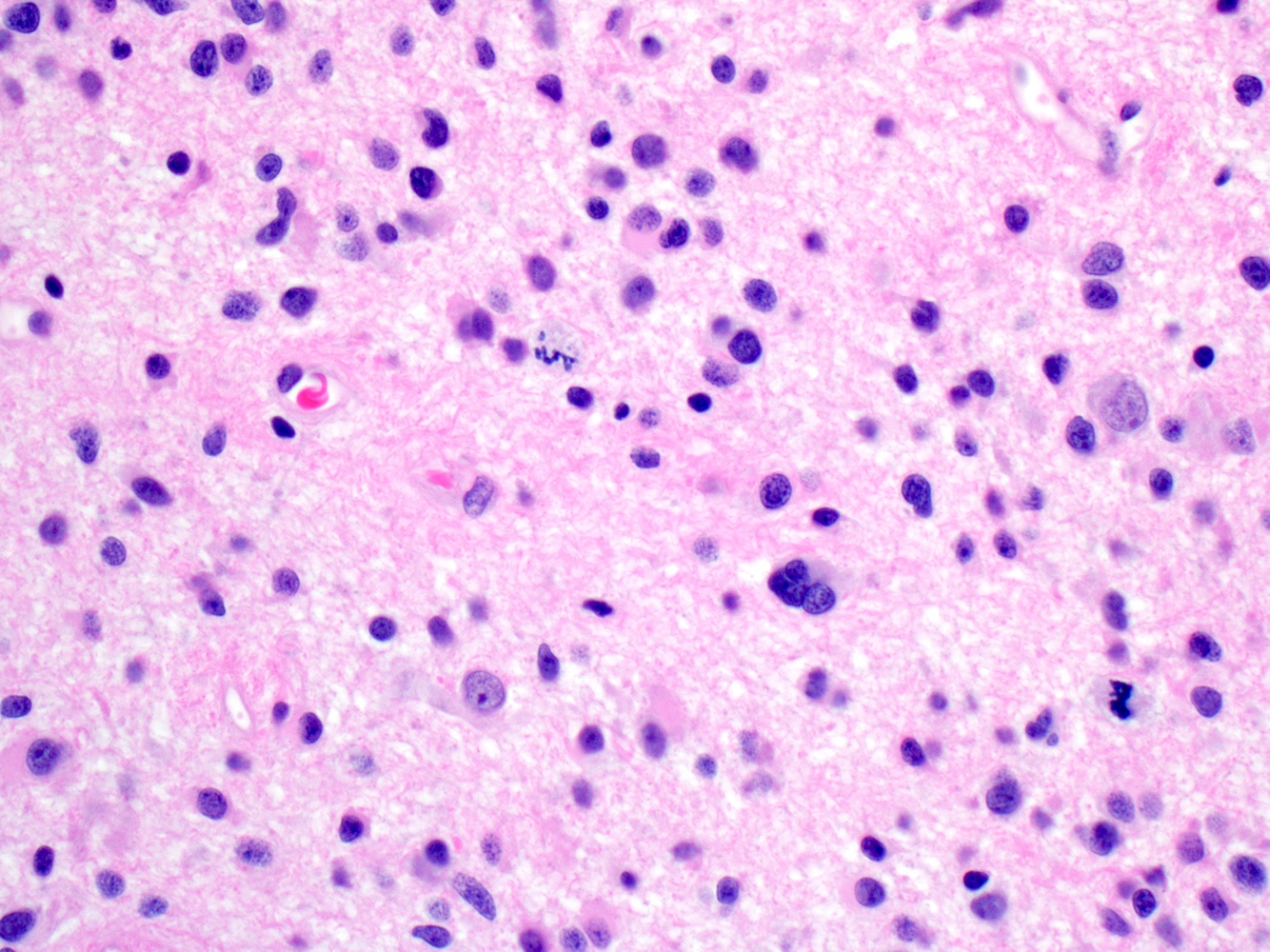
Increased mitotic activity
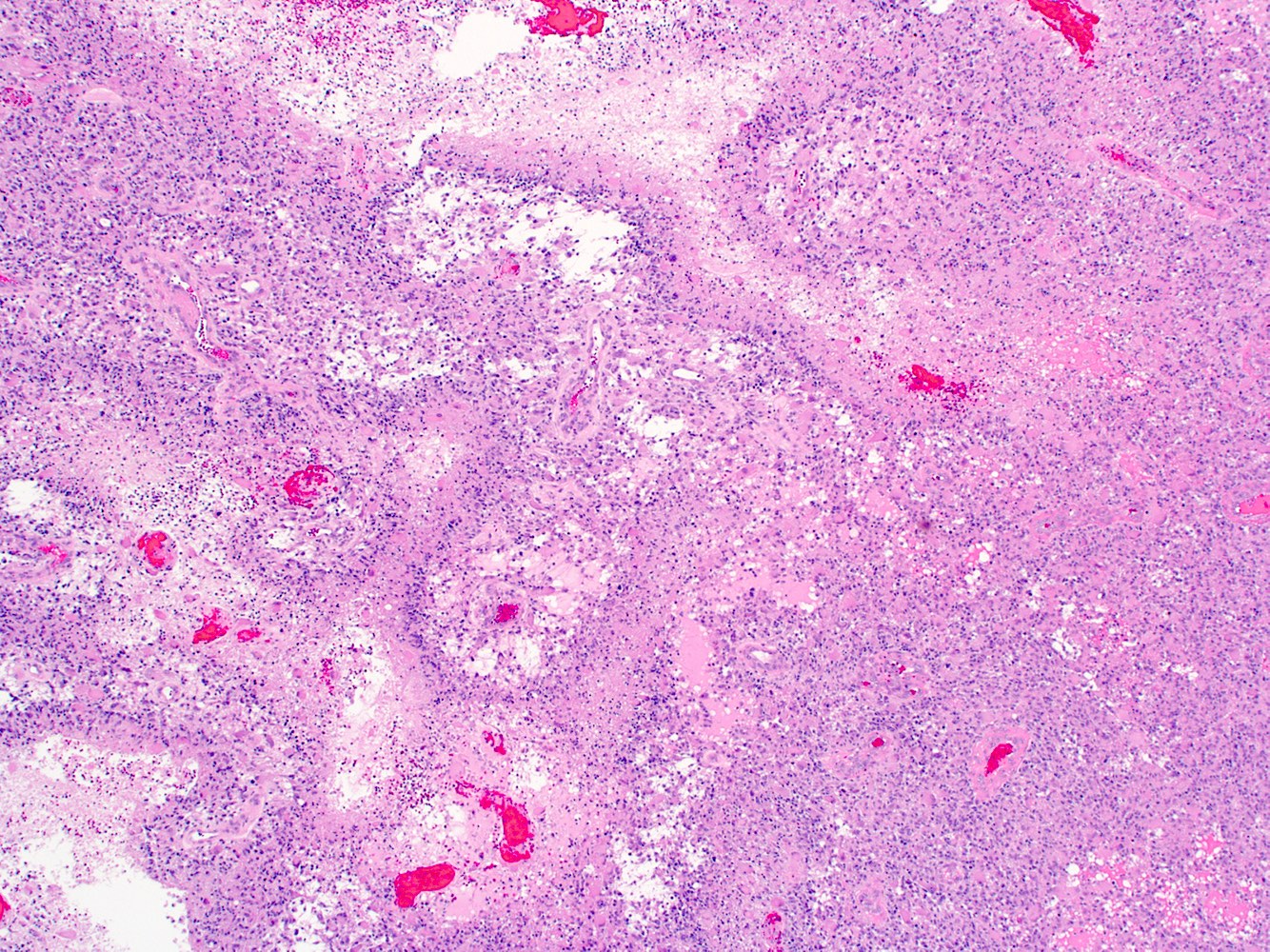
Pseudopalisading necrosis
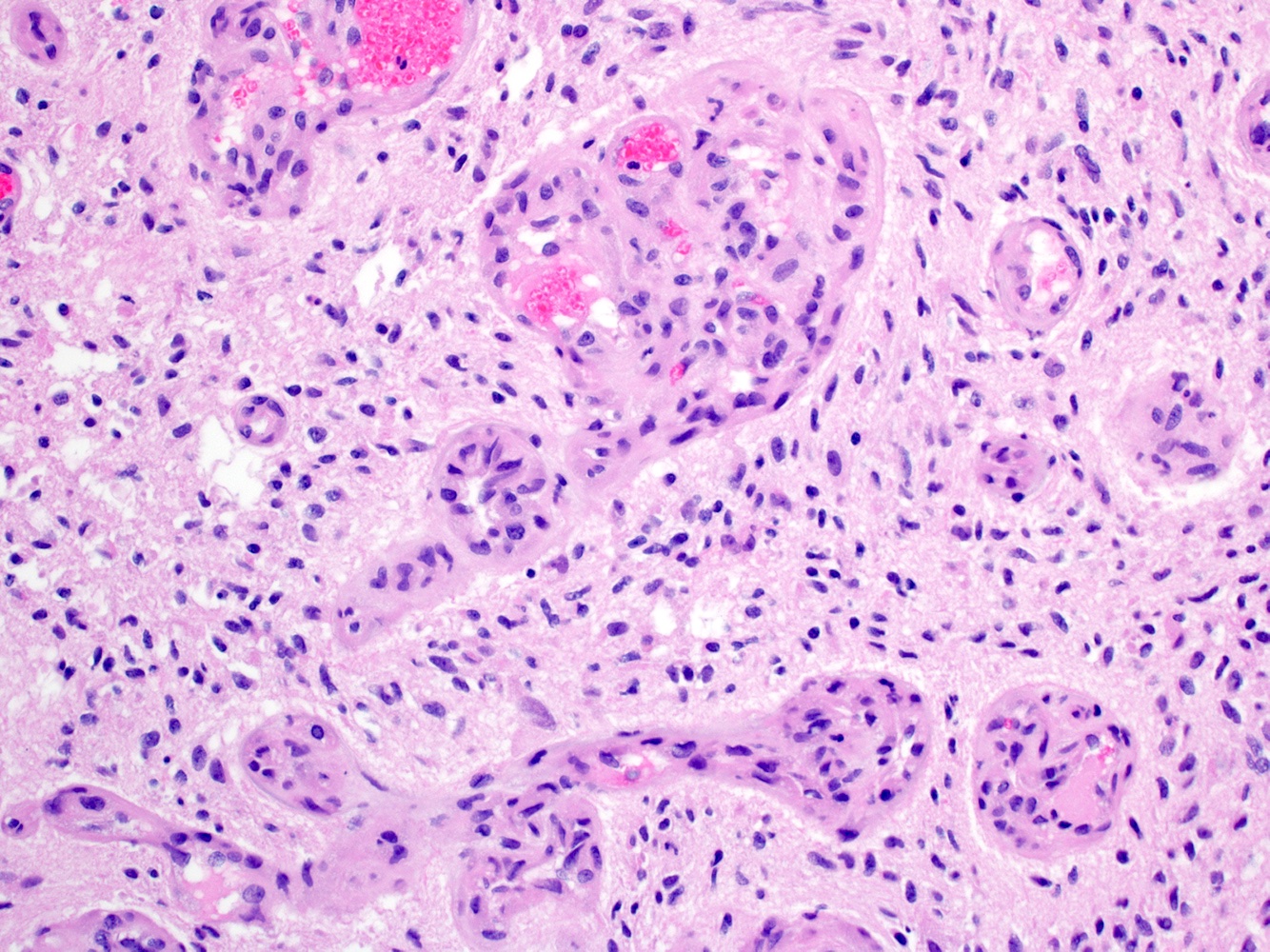
Microvascular proliferation
Contributed by P.J. Cimino, M.D., Ph.D.
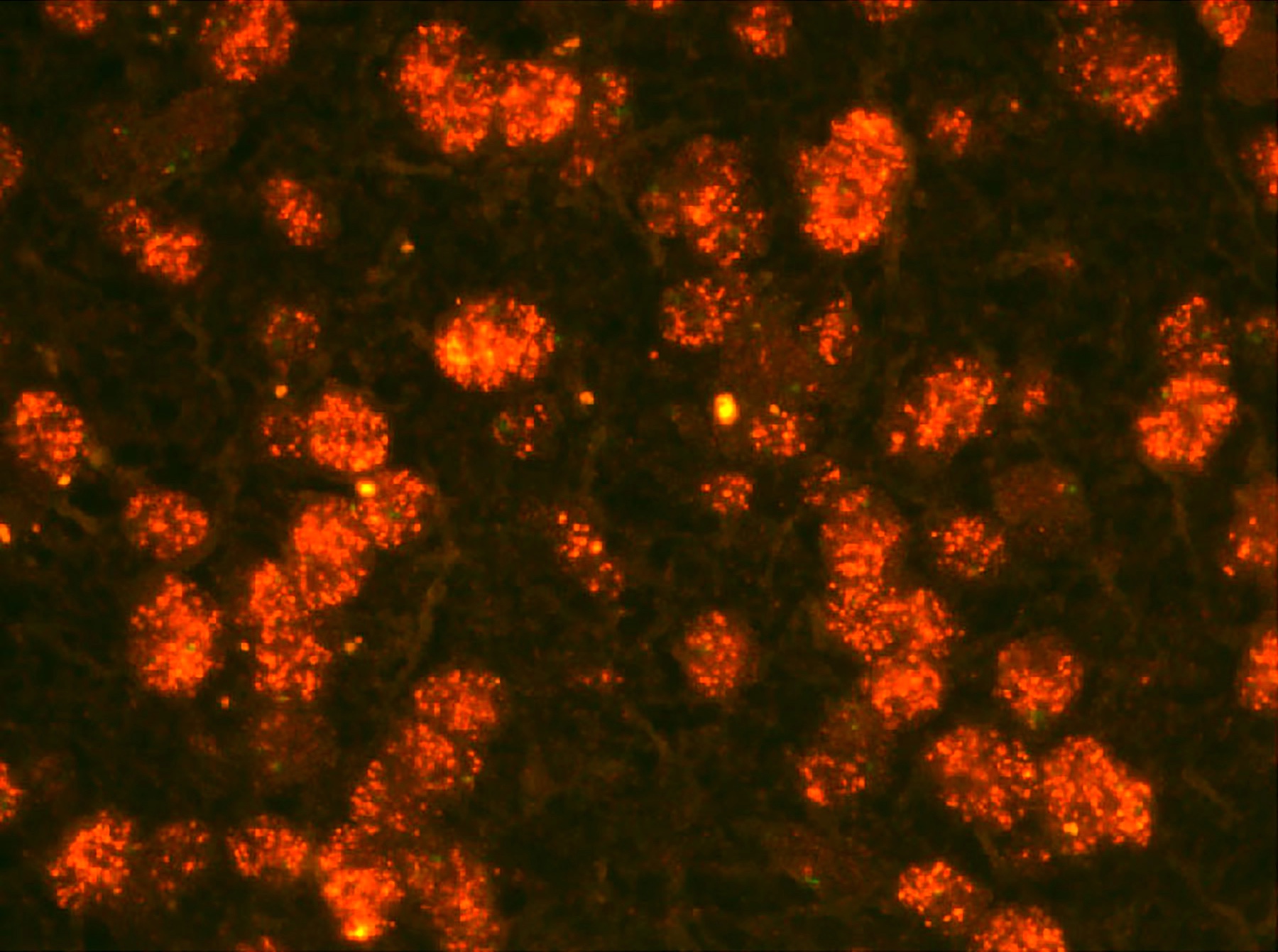
Glioblastoma EGFR amplification
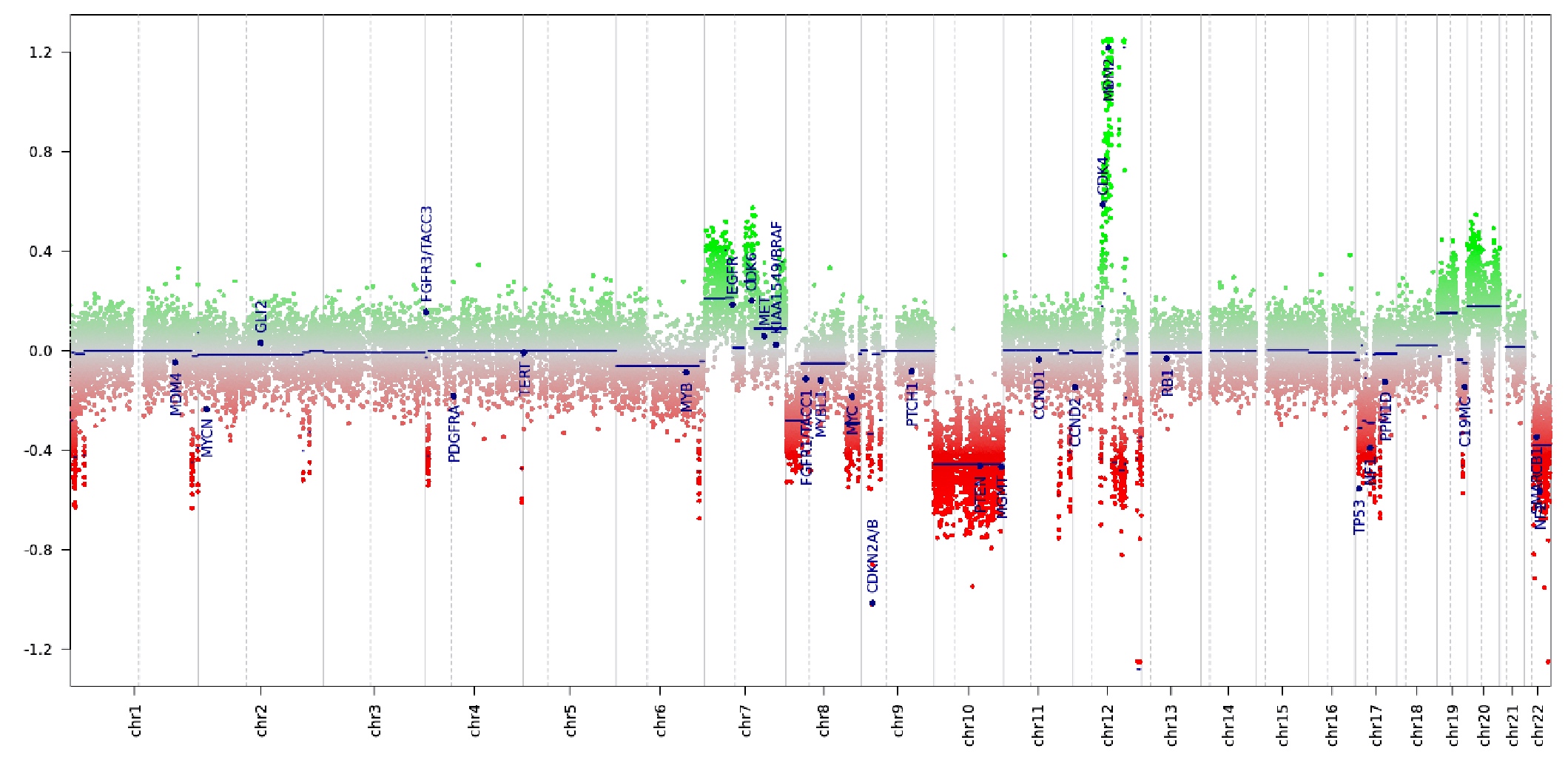
Glioblastoma copy number plot
Images hosted on other servers:

Glioblastoma TERT promoter mutation
2021 WHO classification of CNS tumors: update I - gliomas

This IDH mutant astrocytoma is from a 40 year old woman. There is no microvascular proliferation or necrosis identified. According to the 5th edition of the WHO classification of CNS tumors (2021), a CNS WHO grade 4 should be applied if which of the following genetic alterations is present?
- ATRX loss
- CDKN2A homozygous deletion
- Chromosome 1p / 19q codeletion
- EGFR amplification
- TERT promoter mutation
Comment Here
Reference: WHO grading of gliomas
- Family of neoplasms most likely derived from meningothelial cells of the arachnoid mater
- Arabic numerals (1, 2, 3) now used instead of roman numerals (I, II, III)
- Molecular characteristics now part of the classification for anaplastic (malignant) meningioma, CNS WHO grade 3:
- TERT promotor mutation (Brain Pathol 2014;24:184, Oncotarget 2017;8:109228)
- CDKN2A/B homozygous deletion (Am J Pathol 2001;159:661, Brain Pathol 2002;12:183, Acta Neuropathol 2020;140:409)
-
Meningioma ICD-O
- Meningioma9530/0
Contributed by Jared T. Ahrendsen, M.D., Ph.D.

Criteria for meningioma classification
Contributed by Jared T. Ahrendsen, M.D., Ph.D. and Chunyu Cai, M.D., Ph.D.
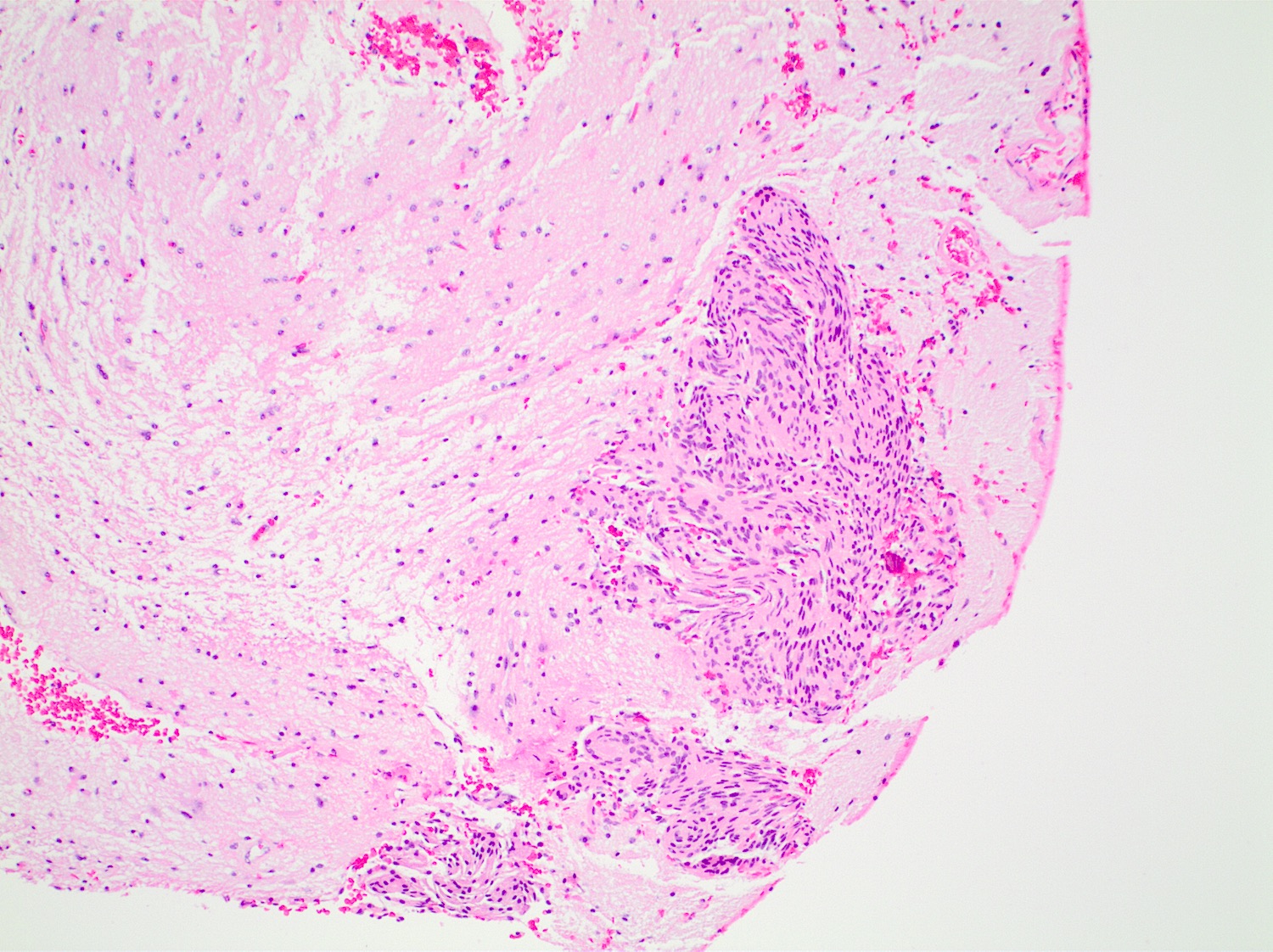
Brain invasion
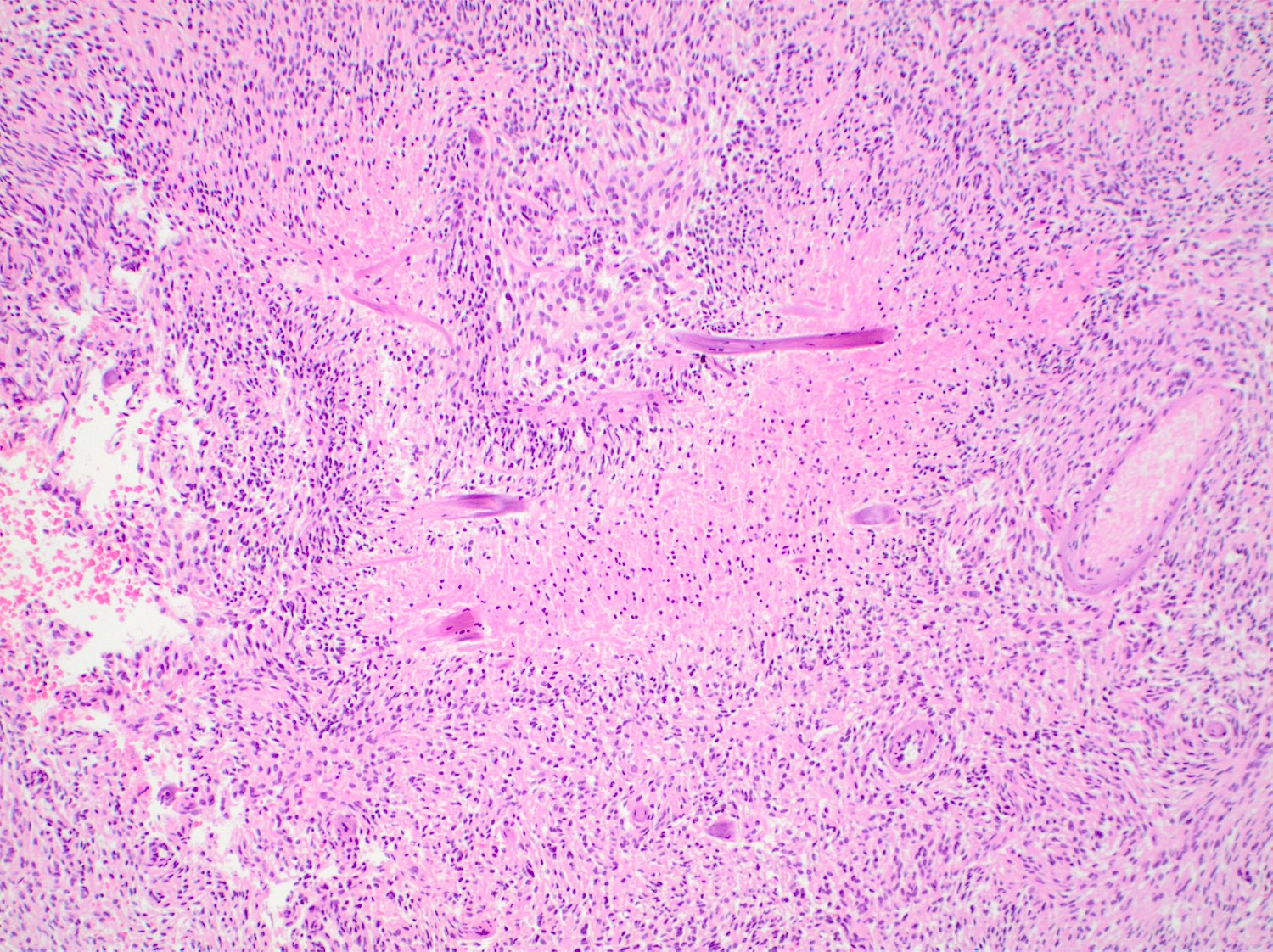
Necrosis
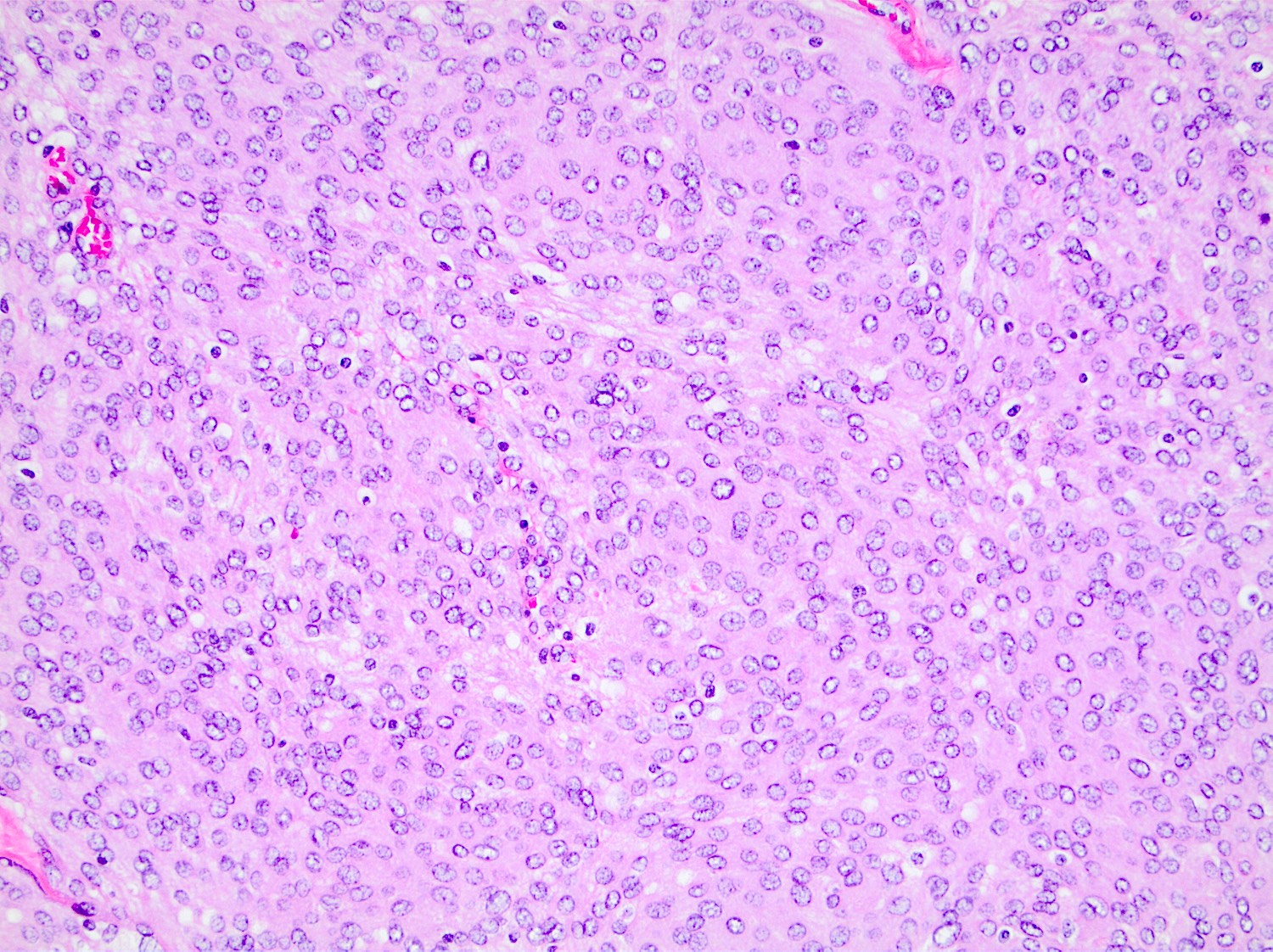
Sheeting
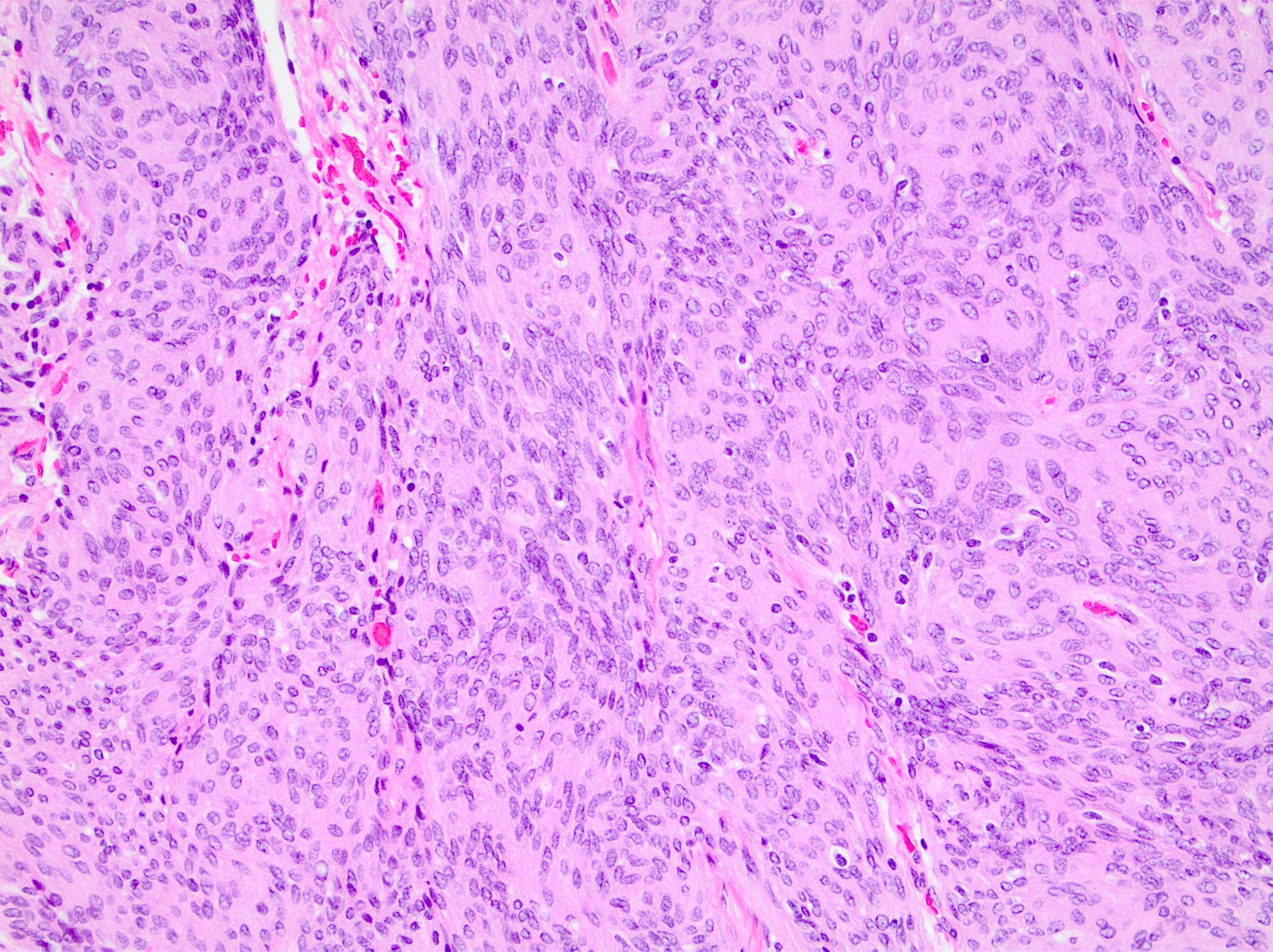
Increased cellularity
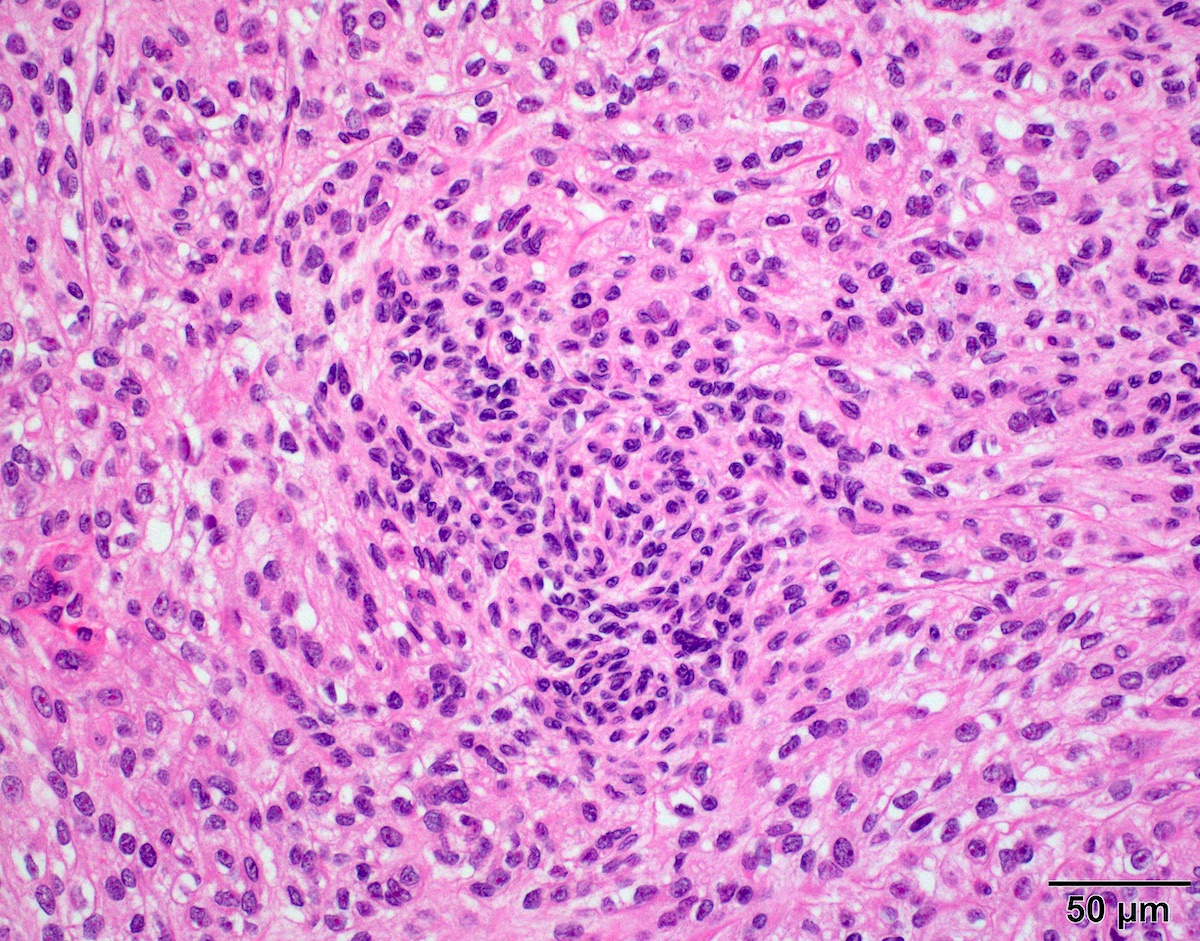
Small cell transformation
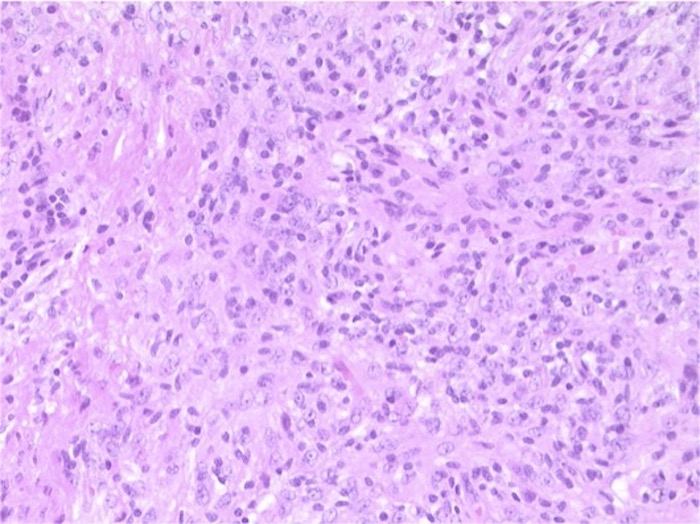
Prominent nucleoli
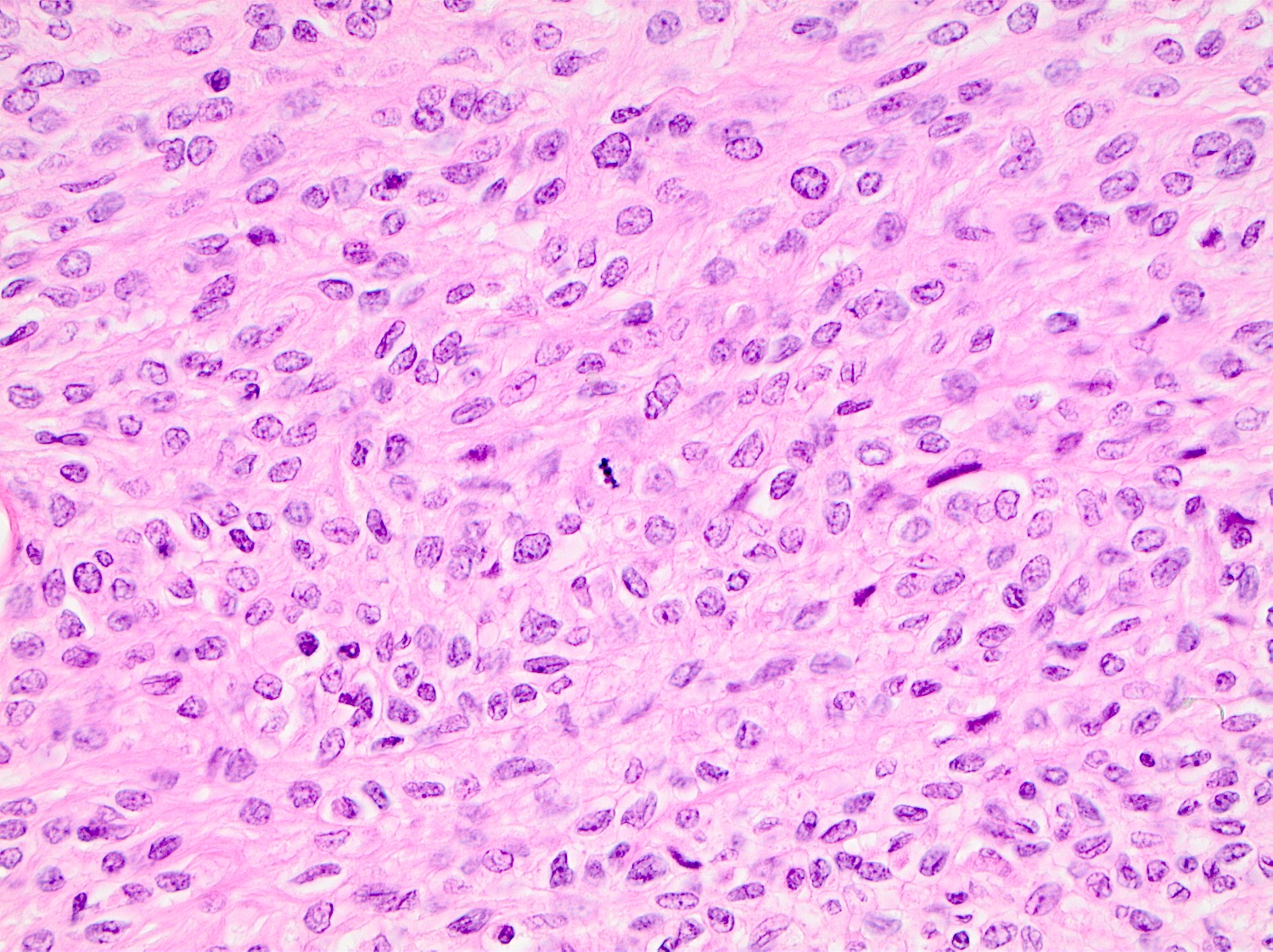
Atypical meningioma

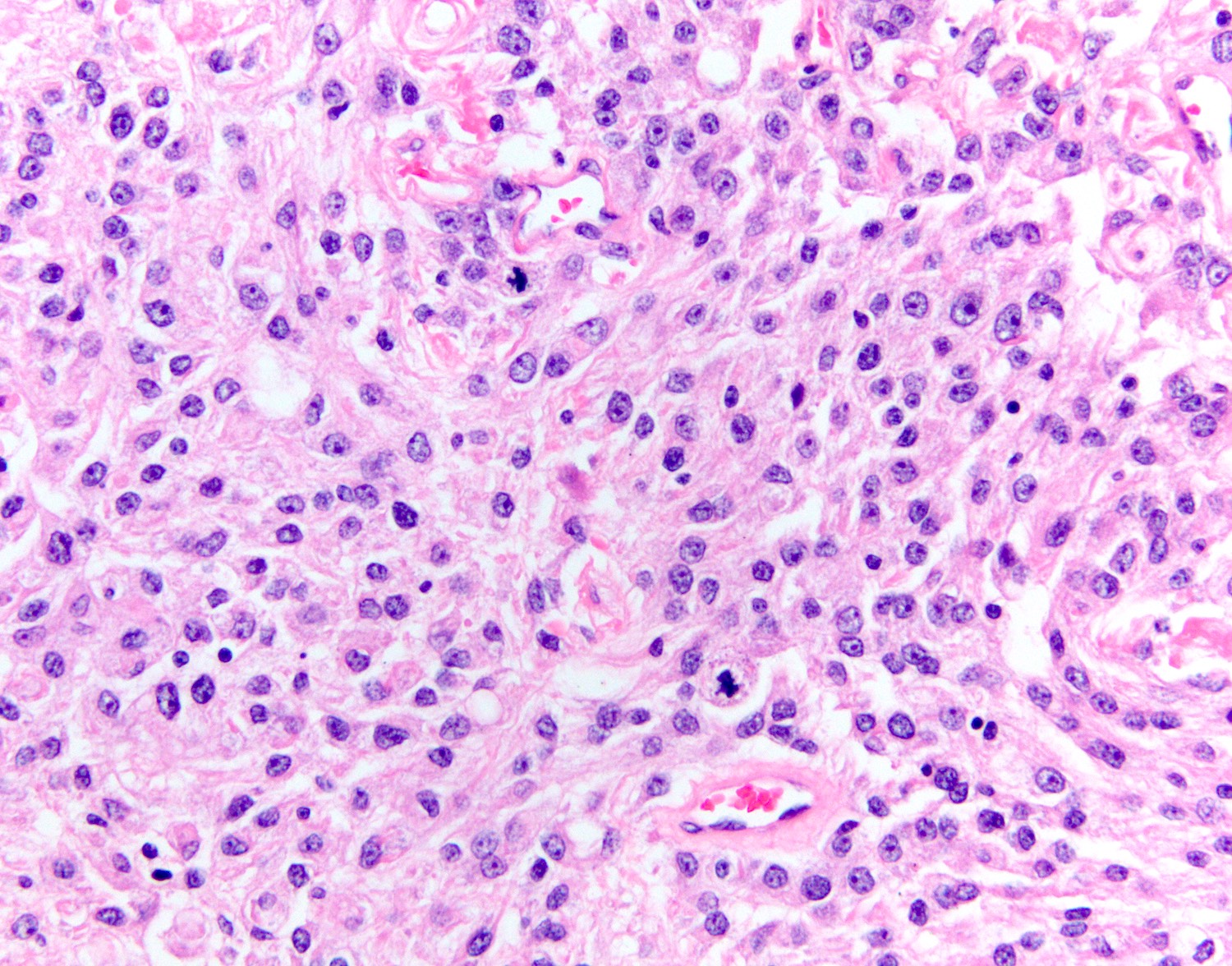
Anaplastic meningioma
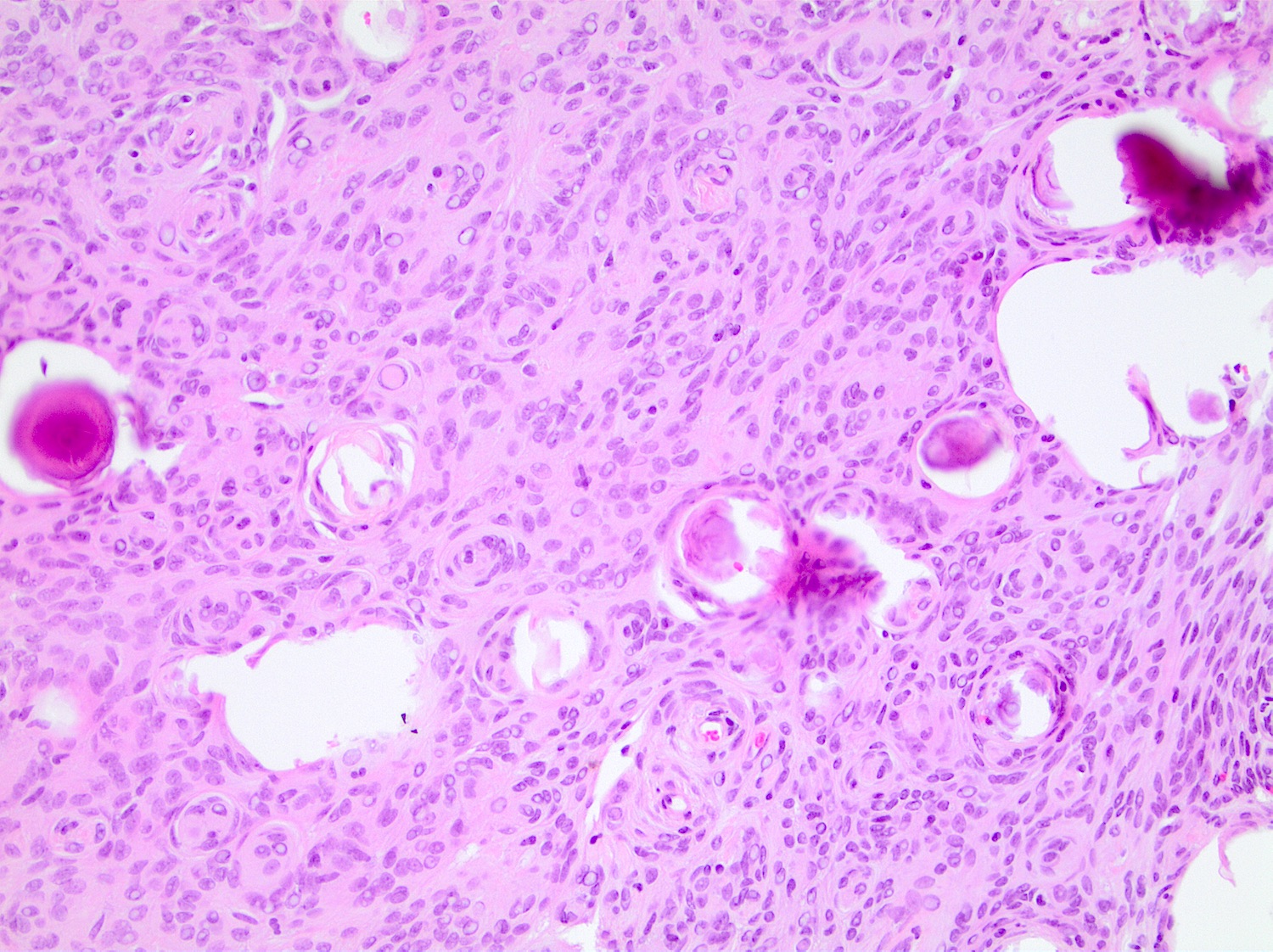
An intracranial neoplasm (histology shown in image) is resected from the lateral convexity of a 65 year old woman. Which of the following molecular alterations, if present, is compatible with a CNS WHO grade 3 designation for this tumor type?
- EGFR amplification
- Germline NF2 mutation
- IDH mutation
- Monosomy 22
- TERT promotor mutation
Comment Here
Reference: WHO grading of meningiomas
Asa: 2020
Ellison: 2013
Gill: 2022
Gray: 2018
Gupta: 2016
Husain: 2021
IARC: 2022
Kleinschmidt-DeMasters: 2022
Love: 2015
Perry: 2017
Rodriguez: 2016
Find related Pathology books: GU/adrenal, head & neck/endocrine, muscle and peripheral nerve nontumor, neuropathology, pediatric
