
- Cellularity averages 79% at ages 0 - 9 years, 50% at ages 30 - 69 versus 29% at ages 70 - 79
- With aging, hematopoietic tissue is replaced by fat
- Deeper medullary areas are typically more cellular than subcortical areas
- B cell production declines with age (Curr Opin Immunol 2005;17:463) although the relative abundance of pro B, pre B, immature, naive and mature B cells usually does not change appreciably between ages 24 and 88 years
- Occasional patients have exceptionally low numbers of lymphocyte precursors (Blood 2003;101:576)
- Hypocellularity in elderly marrow may be due to increased apoptosis (Mech Ageing Dev 2000;117:57)
- Usually no spicules in bone marrow aspiration from very young children, perhaps due to very low or absent fat content
Contributed by Dragos C. Luca, M.D.
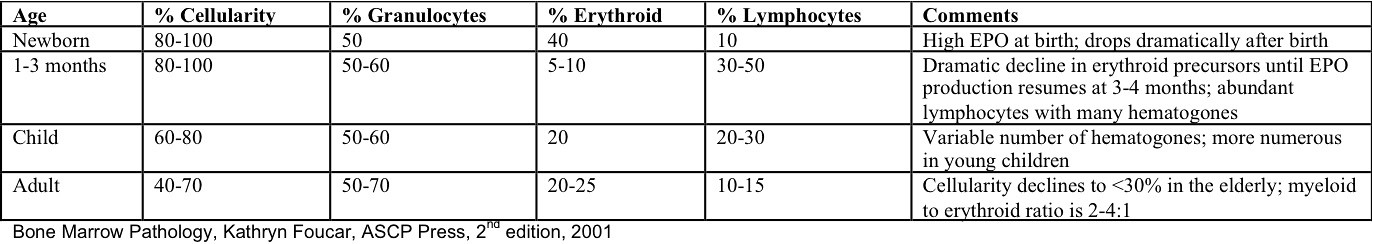
Age related normal values in bone marrow
Images hosted on other servers:
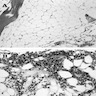
Fatty marrow
AFIP images
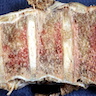
6 month old child has highly cellular marrow (normal for age)


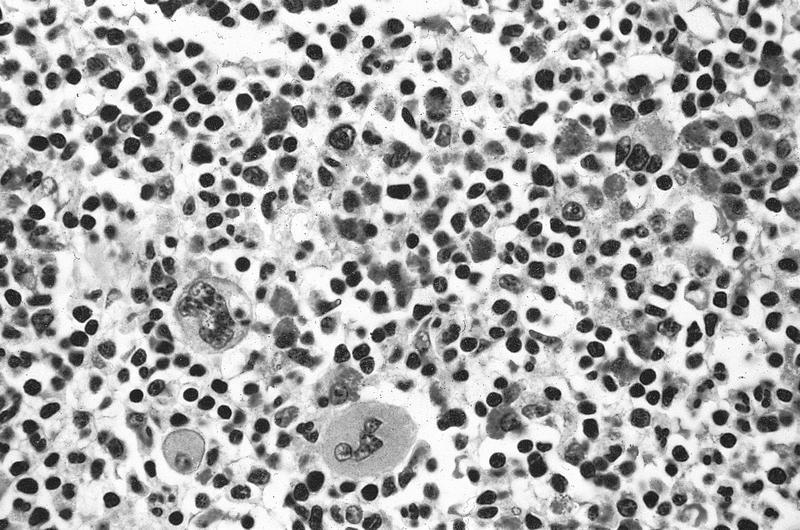
Adult marrow (ages 30 - 69 years)
Images hosted on other servers:
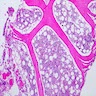
Adult marrow
- Has variable hematological effects (Alcohol Clin Exp Res 2004;28:619)
- May cause megaloblastic anemia due either to folate / B12 deficiency or without these deficiencies
- Cyanamide, used to prevent alcohol intake, may cause aplastic anemia (Eur J Clin Pharmacol 2005;61:467) or granulocytopenia (Intern Med 1997;36:640)
- Alcohol abuse may also cause sideroblastic anemia (Postgrad Med 1992;92:147), severe osteoporosis (Calcif Tissue Int 2005;76:79) or TTP (Eur J Intern Med 2004;15:262)
- 32 year old man with severe pancytopenia (University of Pittsburg Case #428)
- 50 year old man with extensive alcohol and drug abuse, hypocellular marrow and increased blasts (Arch Pathol Lab Med 2005;129:e35)
- 50 year old woman with megaloblastic anemia due to alcohol and mild folate antagonists (Dtsch Med Wochenschr 2005;130:2139)
- May cause hypocellular marrow or reactive myeloblastosis with up to 34% blasts
- Vacuoles in proerythroblasts and other precursors
- Also ringed sideroblasts and iron granules in plasma cells
Images hosted on other servers:

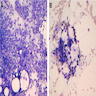
Marked megaloblastic features

Markedly hypercellular
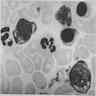
Bone marrow: intracytoplasmic vacuoless
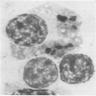
Multiple coarses granules
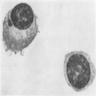
Plasma cell: iron granules forming plaque
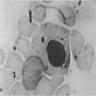
Hemosiderin granules in normoblast
- Leukopenia with vacuoles, hypersegmentation of neutrophils, thrombocytopenia
- Iron granules in plasma cells and other cells
Images hosted on other servers:

Peripheral blood erythrocytes
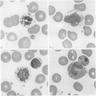
Vacuolation of peripheral blood cells
Images hosted on other servers:
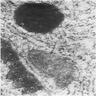
Marrow plasma cell has dense aggregate of iron
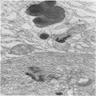
Iron laden organelle
in upper cell may
correspond to
microscopic plaque
- Cytoplasmic vacuoles also due to copper deficiency, hematogones, lipid storage diseases, lipid granulomas, monocytes, Mott cells
- Also called CAMT (congenital amegakaryocytic thrombocytopenia)
- Extremely rare, usually diagnosed in early childhood (Pediatr Blood Cancer 2011;57:199, Hematol Oncol Clin North Am 2009;23:321)
- Initially thought to have variable inheritance (autosomal recessive or X linked) but X linked forms now reinterpreted as mild or attenuated forms of Wiskott-Aldrich syndrome
Etiology
- Most cases caused by homozygous (biallelic) or compound heterozygous mutations in thrombopoietin (TPO) receptor gene c-mpl at 1p34 (Blood 2001;97:139), causing impaired expression of c-mpl gene, causing defective response to TPO (OMIM: Amegakaryocytic Thrombocytopenia, Congenital; CAMT)
- Missense mutations associated with milder or delayed course vs. loss of function mutations (Hum Mutat 2006;27:296)
Clinical features and diagnosis
- Isolated nonimmune thrombocytopenia with decreased marrow megakaryocytes and high serum TPO levels
- Red cell macrocytosis with normal hemoglobin level
- Bone marrow initially normocellular
- Mucocutaneous or GI bleeding, variable physical abnormalities (Br J Haematol 2005;131:636)
Case reports
- Girl with developmental delay, facial malformations and CNS anomalies (Pediatr Blood Cancer 2011;56:452)
- 61 year old woman with systemic sclerosis and anti-c-mpl antibody (Arthritis Rheum 2003;48:1647)
Treatment and prognosis
- Stem cell transplantation
- 50% evolve to aplastic anemia (at mean age 3.5 years); may evolve to myelodysplastic syndrome or leukemia
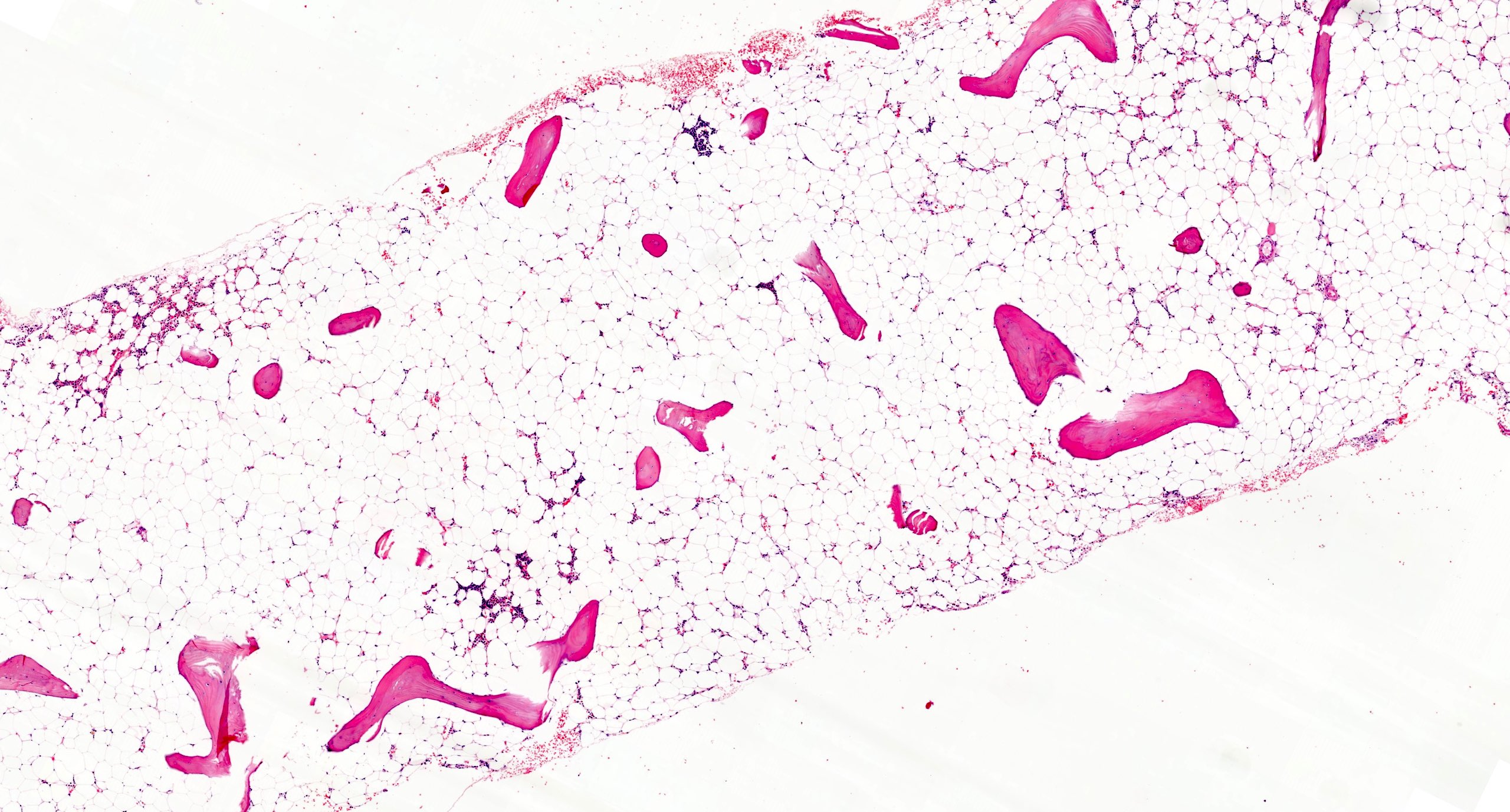
Microscopic (histologic) images
Images hosted on other servers:
Bone marrow aspiration and trephine biopsy
Bone marrow failure in child
- Bone marrow disorder characterized by pancytopenia due to bone marrow trilineage hypoplasia in the absence of any underlying neoplasia or reticulin fibrosis
- Mostly sporadic but can be constitutional (congenital) and occur as a consequence of different germ line genetic defects (Hematology 2015;20:433)
- Acquired disease in the majority of cases characterized by pancytopenia due to ineffective hematopoiesis
- Bone marrow is hypocellular, with lacunar spaces replaced by fatty cells
- Residual nucleated cells include mostly lymphocytes, plasma cells and mast cells with isolated hematopoietic cells
- Sometimes mild atypia but no overt dysplasia
- No increase in blasts; normal CD34 positive count
- No overt reticulin fibrosis
- Careful distinction with other conditions characterized by bone marrow hypocellularity which can mimic aplastic anemia:
- Hypoplastic myelodysplastic syndrome / acute myeloid leukemia
- Subtle infiltration by hairy cell leukemia
- T cell large granular lymphocytic leukemia
- Other possible findings in aplastic anemia, whose detection does not affect diagnosis:
- Cytogenetic or molecular abnormalities (see Molecular / cytogenetics description)
- Small paroxysmal nocturnal hemoglobinuria (PNH) clone
- Bone marrow failure: sometimes used as a synonym of acquired aplastic anemia
- This should be avoided as impaired hematopoiesis due to bone marrow failure can occur in different conditions (including reactive and neoplastic diseases)
- Idiopathic acquired aplastic anemia: should be used only in reference to secondary forms of unknown etiology
- Congenital cases named after the specific disease entity; most common are Fanconi anemia, dyskeratosis congenita and Schwachman-Diamond syndrome
- ICD-10: D61.9 - aplastic anemia, unspecified
- Acquired aplastic anemia:
- Most common form of aplastic anemia
- Rare disease: estimated annual incidence of 2 - 5 cases per million (Eur J Haematol 2018;101:711)
- No sex predilection
- Bimodal age distribution: first peak at 10 - 25 years; second peak at > 60 years
- Geography: lower in Europe and North America; more common in Asia
- Constitutional:
- Rarely encountered in the clinical practice
- Except for X linked disorders, M = F
- Acquired aplastic anemia:
- Most commonly is the result of an autoimmune mediated process
- Likely antigen driven recruitment of cytotoxic and helper T lymphocytes
- Immune mediated destruction of hematopoietic stem cells
- Annihilation of hematopoietic stem cells also occurs as a consequence of both an inflammatory cytokine induced (TNFα and IFNγ) and Fas mediated apoptosis
- Most cases do not have a clear cause and are labeled idiopathic (Eur J Haematol 2018;101:711)
- Constitutional aplastic anemia:
- Results from specific loss of function germ line mutations
- Due to DNA repair defects (Fanconi anemia), telomere defects (dyskeratosis congenita), impairment of differentiation and self renewal pathways (GATA2 deficiency), ribosomopathies (Shwachman-Diamond syndrome and Diamond-Blackfan anemia) (N Engl J Med 2018;379:1643)
- Acquired aplastic anemia (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017, Eur J Haematol 2018;101:711):
- Infectious agents:
- Hepatitis A, B, C viruses
- Other viruses: HIV, EBV, CMV, parvovirus
- Environmental exposures:
- Toxins: benzene, solvents, insecticides
- Drugs: allopurinol, carbamazepine, chloramphenicol, chloroquine, penicillamine, cotrimoxazole, diclofenac, naproxen, valproic acid; numerous other agents less commonly reported
- Others conditions:
- Pregnancy
- Thymoma (J Pediatr Hematol Oncol 2018;40:e464)
- Autoimmune diseases, most commonly systemic lupus erythematosus and rheumatoid arthritis
- Eosinophilic fasciitis
- Infectious agents:
- Constitutional aplastic anemia (see Pathophysiology)
- Signs and symptoms related to presence and severity of cytopenia(s) (i.e. anemia, thrombocytopenia, leukopenia); most common are fatigue, shortness of breath, bleeding, frequent or prolonged infections, bruising (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Constitutional forms clinically manifest at birth or early infancy (Hematology 2015;20:433)
- The following clinical and pathologic findings required for diagnosis:
- Persistent (> 6 months) pancytopenia
- Characteristic features within bone marrow aspirate and biopsy
- Absence of abnormal bone marrow infiltrate and reticulin fibrosis
- Patients exhibit at least 2 of the following:
- Absolute neutrophilic count < 1.5 x 109/L
- Platelet count < 50 x 109/L
- Hemoglobin < 10 g/dL
- Further subclassification based on severity (Br J Haematol 2016;172:187)
- Severe aplastic anemia:
- Bone marrow cellularity < 25% or 25 - 50% with < 30% residual hematopoietic cells
- At least 2 of the following:
- Absolute neutrophilic count < 0.5 x 109/L
- Platelet count < 20 x 109/L
- Reticulocyte count < 0.6 x 109/L
- Very severe aplastic anemia:
- Meets the criteria for severe but absolute neutrophilic count < 0.2 x 109/L
- Nonsevere aplastic anemia:
- Cases not meeting criteria for above groups
- Severe aplastic anemia:
- Constitutional bone marrow failure panels in cases of suspected constitutional etiology (Blood Cells Mol Dis 2015;55:40)
- Main laboratory tests
- Complete blood count, showing pancytopenia (as defined above)
- Low reticulocyte count (< 30 x 109/L)
- Normal vitamin B12, folate and iron (to exclude vitamin deficiency anemias)
- Bone marrow aspirate and biopsy (see Microscopic (histologic) description)
- Additional studies (Br J Haematol 2016;172:187)
- Autoimmune screening (including antinuclear antibody, anti-dsDNA, rheumatoid factor, etc.) to exclude autoimmune etiologies
- Liver function tests and viral studies to rule out posthepatitis aplastic anemia
- Flow cytometry to screen for possible paroxysmal nocturnal hemoglobinuria clones
- Small paroxysmal nocturnal hemoglobinuria clones are detected in up to 60% of aplastic anemia
- They may fluctuate in size or even disappear on follow up studies
- Identification of a small paroxysmal nocturnal hemoglobinuria clone should not discourage a diagnosis of aplastic anemia
- In pediatric cases, additional tests needed (Hematology 2015;20:433, Blood Cells Mol Dis 2015;55:40):
- Chromosomal breakage analysis to exclude Fanconi anemia
- TERC and TERT mutation analysis with consideration of telomere length measurement (to investigate dyskeratosis congenita) (Blood 2009;113:309)
- Nowadays, comprehensive bone marrow failure panels to investigate simultaneously the most common inherited genetic defects
- Higher risk in patients > 45 years old
- Higher responses to immunosuppression in the presence of small paroxysmal nocturnal hemoglobinuria clones (Br J Haematol 2014;164:546)
- Aplastic anemia with somatic mutation(s) more likely to progress into myelodysplastic syndrome / acute leukemia compared to unmutated cases (Blood 2014;124:2698)
- Chromosome 7 abnormalities associated with poorer response to therapy and prognosis
- Cases carrying trisomy 8 or del(13q) show more favorable response to immunosuppressive therapy and better outcome (Blood 2002;99:3129)
- 4 year old girl with quickly progressing peripheral retinal ischemia (Retin Cases Brief Rep 2017;11:108)
- 6 year old South Asian girl diagnosed with celiac disease, presenting with bruises and petechiae (J Med Case Rep 2018;12:16)
- 13 year old girl presenting with opportunistic fungal infection (J Infect Dev Ctries 2015;9:431)
- 14 year old boy with newly diagnosed thymoma (J Pediatr Hematol Oncol 2018;40:e464)
- 25 year old woman with aplastic anemia and delayed response to immunosuppressive therapy (Clin Case Rep 2018;6:1029)
- 32 year old man with acute promyelocytic leukemia in remission (Clin Adv Hematol Oncol 2009;7:670)
- 48 year old woman treated with ipilimumab and nivolumab for metastatic melanoma (Ann Oncol 2017;28:1672)
- 58 year old man with skin induration due to eosinophilic fasciitis (Arthritis Care Res (Hoboken) 2018;70:1095)
- Different options varying by age, comorbidities, availability of a matched donor (Br J Haematol 2016;172:187)
- Bone marrow transplant is the only curative strategy; preferred option for immune aplastic anemia or young patients (Haematologica 2014;99:1784)
- Immunosuppressive agents (i.e. cyclosporine, antithymocyte globulin, corticosteroids) for patients who can't undergo a bone marrow transplant or for clearly immune mediated cases (Blood 2017;129:1428)
- Transfusion support only to relieve symptoms
- Bone marrow stimulating agents (as G-CSF) generally used combined with immunosuppressive agents
- Other agents less commonly used: alemtuzumab, androgens (regarded of poor clinical utility) (N Engl J Med 2018;379:1643, N Engl J Med 2016;374:1922)
- Bone marrow invariably hypocellular for age (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Lacunar spaces replaced by fatty cells
- Trilineage hematopoiesis virtually absent with no / few maturing cells
- Residual nucleated cells include mostly lymphocytes, plasma cells, macrophages, mast cells
- Megaloblastoid erythropoiesis sometimes seen but no overt dysplasia
- Normal blast count
- No evidence of fibrosis
Contributed by Valentina Sangiorgio, M.D.
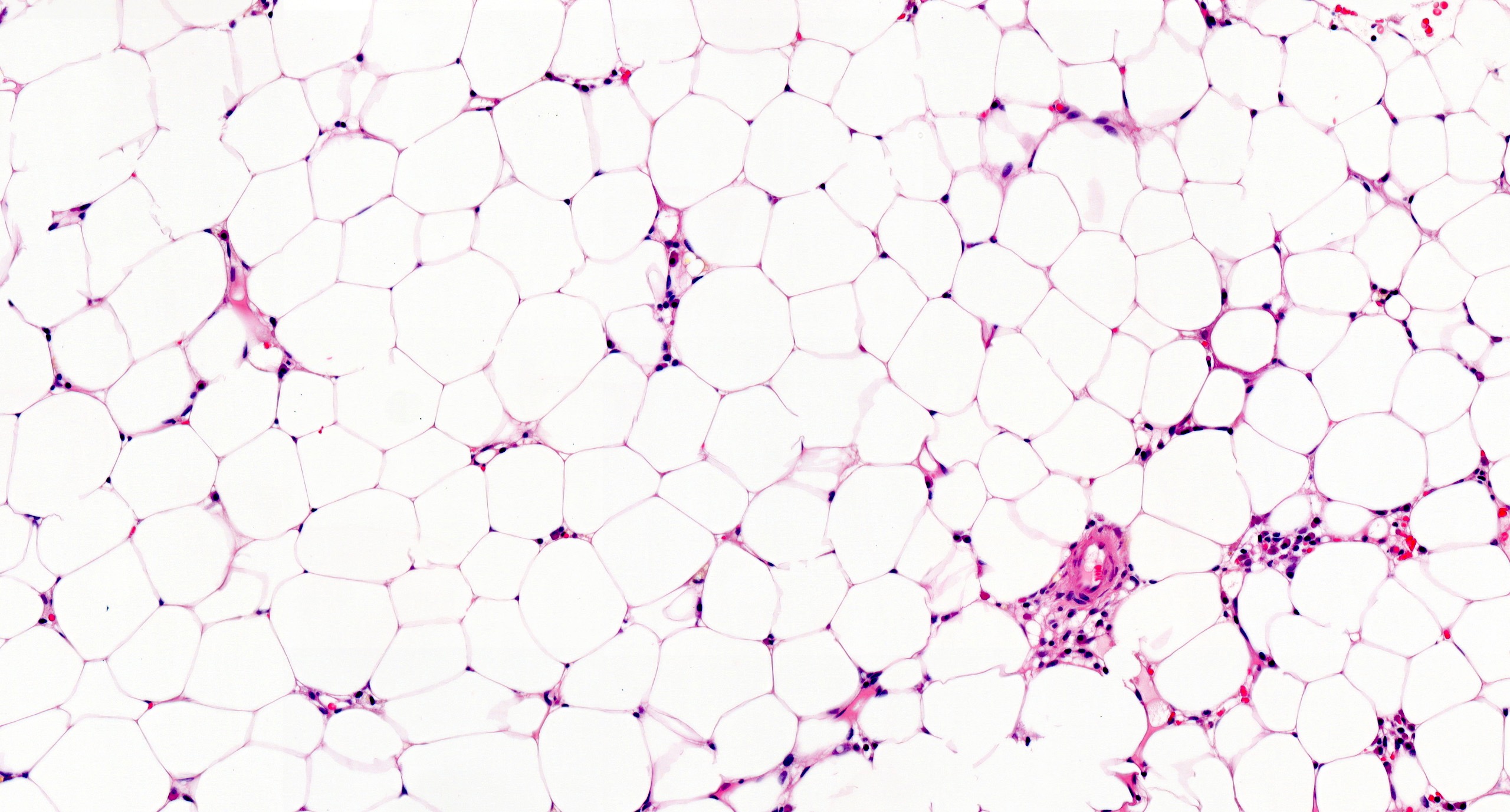
Hypocellular bone marrow
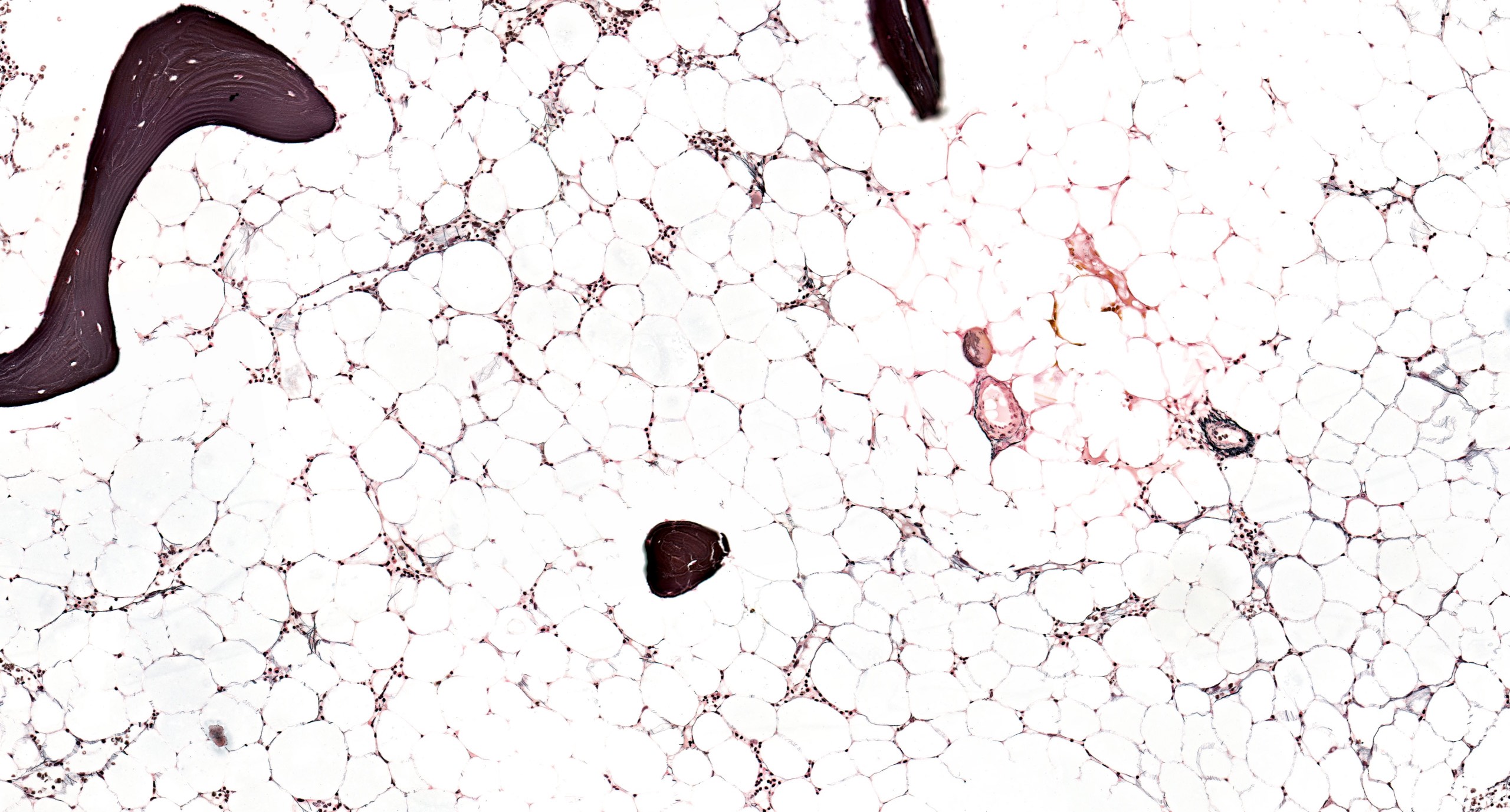
Absence of fibrosis
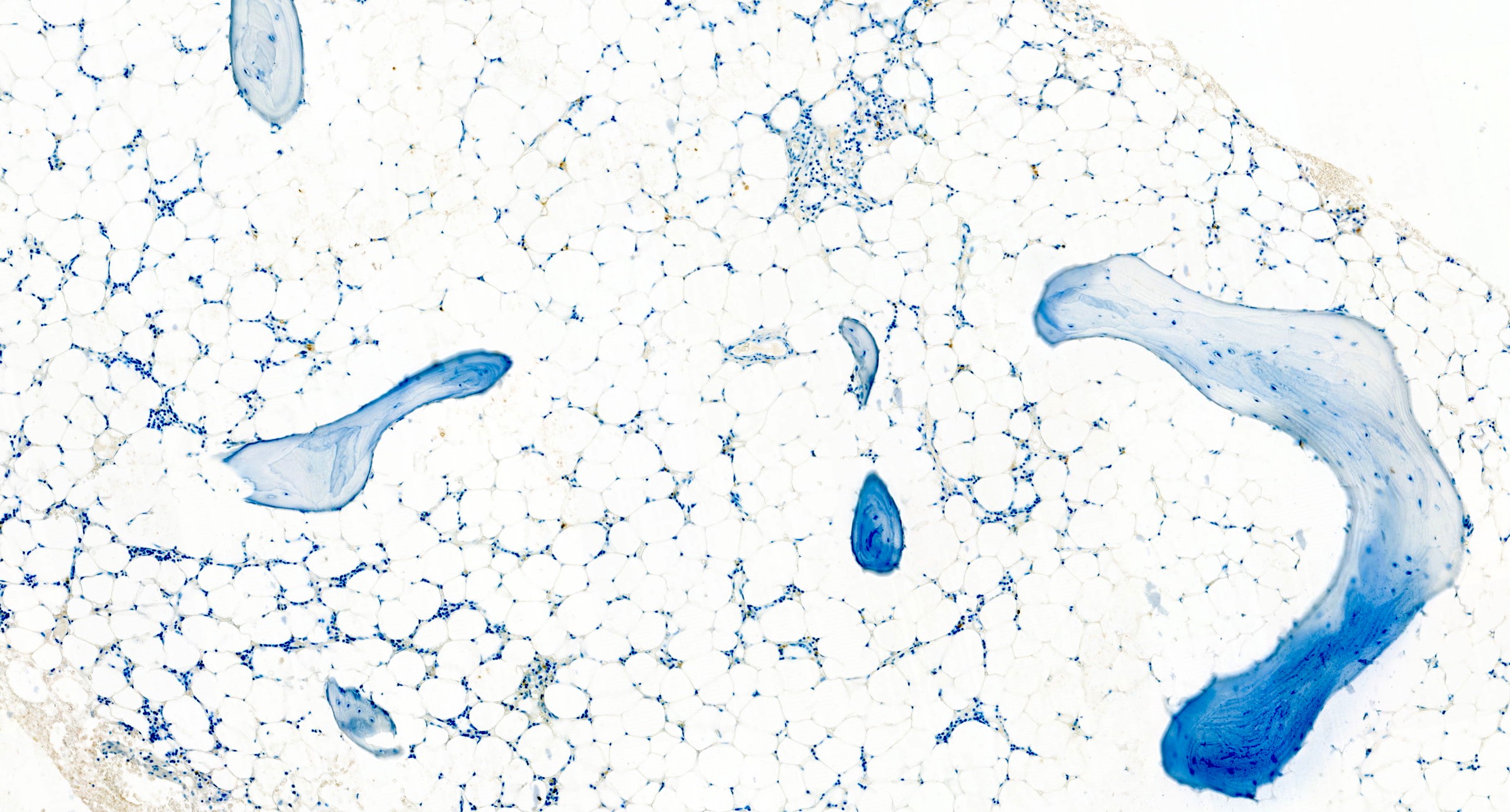
Virtual absence of myeloid cells
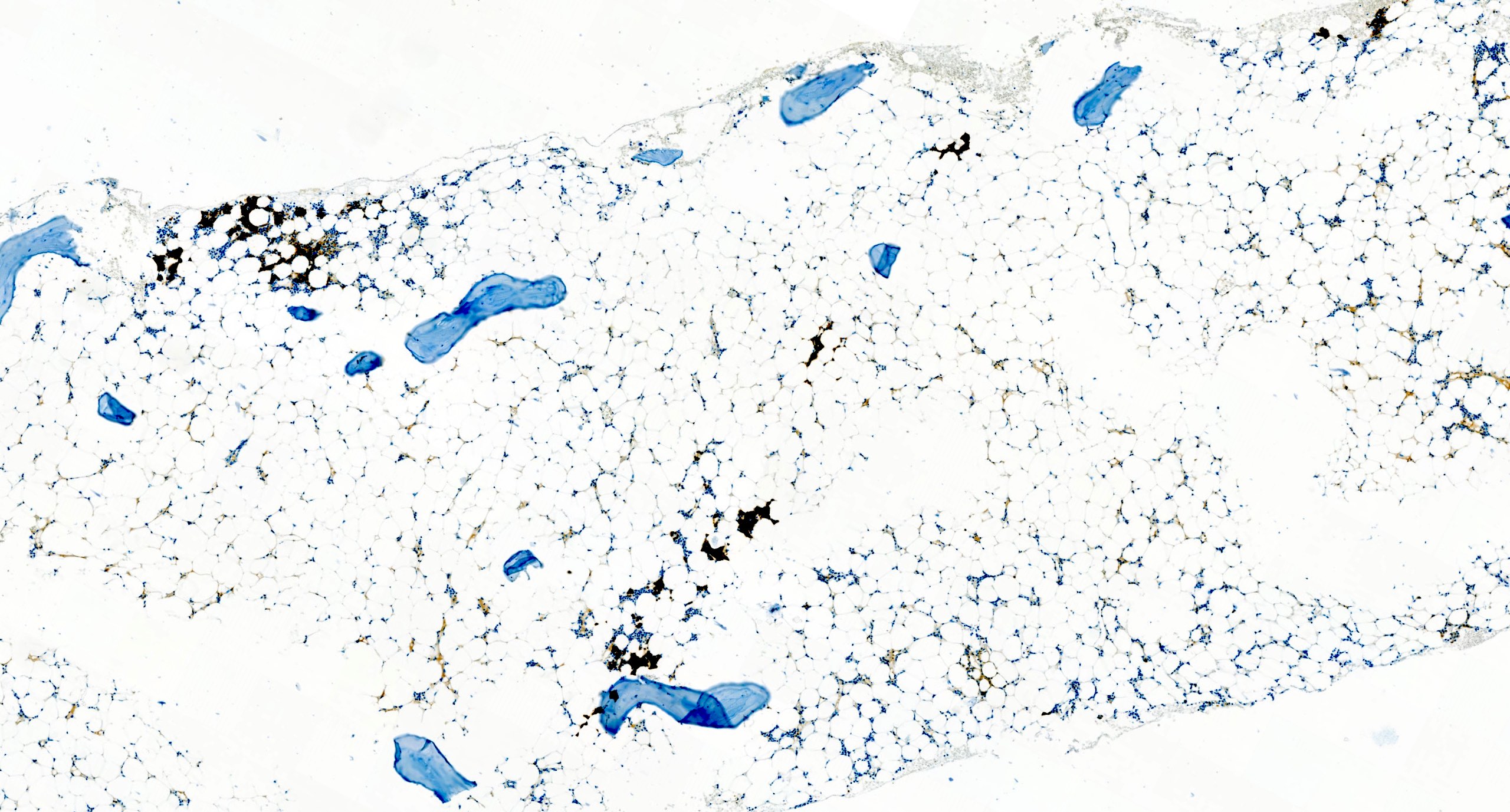
Isolated residual erythroid islands
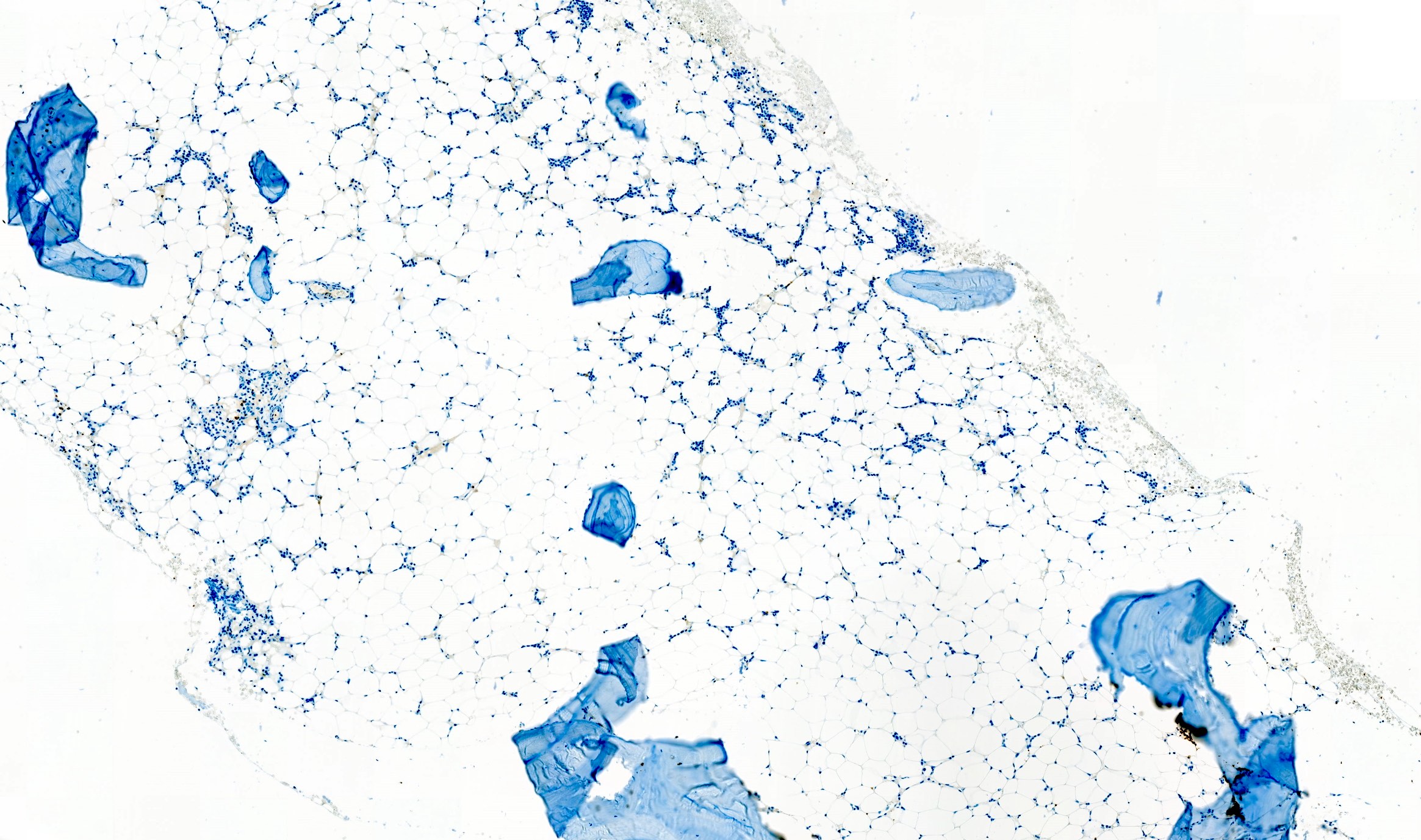
Virtual absence of megakaryocytes

Normal CD34 count

Mast cell hyperplasia
- Severe cytopenia but no overt dysplastic features (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Megaloblastoid changes sometimes seen
- CD34 to assess number and distribution of hematopoietic precursors; an increased CD34 positive count or the presence of abnormal localization of immature precursors (ALIP) suggest hypocellular myelodysplastic syndrome (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- CD20, CD3 to highlight any B and T lymphoid population, respectively; useful for differential diagnosis
- Normal number and phenotype of hematopoietic cells (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- No monoclonal B cell proliferations or atypical T cell populations
- Paroxysmal nocturnal hemoglobinuria clones commonly detected (up to 60% of the cases); do not affect diagnosis if all other diagnostic criteria of aplastic anemia are met
Contributed by Valentina Sangiorgio, M.D.
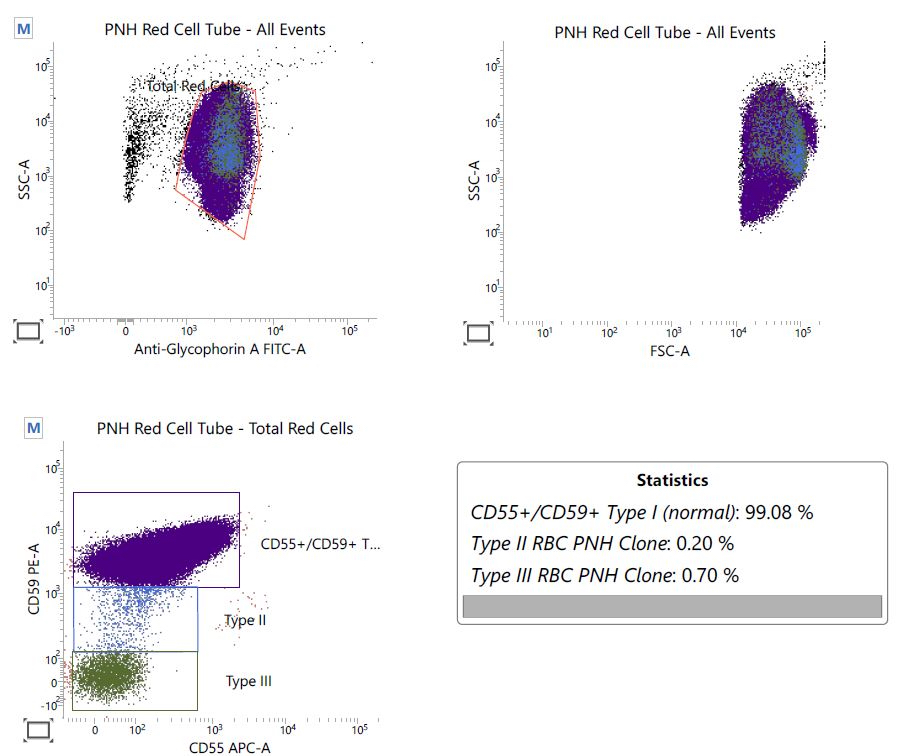
PNH red cell tube
- Cytogenetic findings
- Clonal abnormalities are seen in up to 12% of AA
- Most common abnormalities involve chromosome 7 (around 13% of cases) and trisomy 8 (around 7% of cases) (Am J Hematol 2001;66:167)
- Detection of chromosome 7 abnormalities should prompt further investigations to exclude myelodysplastic syndromes
- Less common alterations: del(13q), trisomy 6, trisomy 15 (< 5% of cases) (N Engl J Med 2015;373:35, Blood 2002;99:3129)
- Unlike in primary myelodysplastic syndrome, aberrancies of chromosome 5 and 20 are infrequent in aplastic anemia
- Detection of any chromosomal abnormality does not suggest per se myelodysplastic syndrome over aplastic anemia
- Karyotyping sometimes problematic in hypocellular marrow; FISH for common myelodysplastic syndrome associated abnormalities should be considered if insufficient metaphases available
- Molecular findings
- Somatic mutations detected in around 20% of aplastic anemia (Blood 2014;124:2698)
- Most common mutations affect ASXL1 and DNMT3A
- Spliceosome gene mutations (SF3B1, SRSF2, U2AF1) and comutations patterns including TET2, DNMT3A and ASXL1 less common in aplastic anemia
- In general, no mutation is disease specific and molecular findings need clinical and pathologic correlation (Blood 2014;124:2698, Blood 2017;129:3371)
- Bone marrow, biopsy and aspirate:
- Hypocellular bone marrow for the patient's age, with features in keeping with aplastic anemia (see comment)
- Comment: The bone marrow is markedly hypocellular for the patient's age (cellularity < 5%). As such, the main histologic differential diagnoses include aplastic anemia and hypoplastic myelodysplastic syndrome. The residual nucleated cells include mostly lymphocytes, plasma cells, mast cells and macrophages. Moreover, overt dysplasia is not observed within any of the three hematopoietic lineages, blast count is within normal limits and there is no evidence of reticulin fibrosis. Given these findings, a diagnosis of aplastic anemia is favored. Correlation with molecular findings (including FISH, conventional cytogenetic and next generation sequencing, whether available) is advised.
- Bone marrow biopsy microscopic description:
- Hypocellular bone marrow for the patient's age (overall cellularity < 5%). Lacunar spaces are extensively replaced by fatty cells; the residual cellularity includes mostly lymphocytes, plasma cells, mast cells and macrophages with only few hematopoietic cells. There is no morphologic increase in blasts. The number of CD34 positive hematopoietic precursors is within normal limits. CD20 and CD3 highlight a population of small B and T lymphocytes, respectively, with a prevalence of the latter, which account for around 25% of the total nucleated cells. Plasma cells are polytypic for kappa and lambda light chains. Reticulin stain reveals a normal network of reticulin fibers.
- Bone marrow aspirate microscopic description:
- Marrow aspirate smear is spicular and shows marked hypocellularity for the patient's age. Myelopoiesis is reduced and left shifted, with only few segmented forms identified. Few normoblastic erythroid precursors are seen. There are scattered megakaryocytes (around 10); however, a reliable assessment of megakaryocytic dysplasia is not possible given the low number of megakaryocytes seen. The residual cellularity includes mostly lymphocytes, plasma cells, mast cells and macrophages. Prussian blue iron stain shows normal storage iron; no ring sideroblasts are noted.
- Bone marrow aspirate cell count (performed on 150 cells) reveals: 1% blasts, 25% promyelocytes / myelocytes, 11% maturing granulocyte forms, 9% erythroid precursors, 31% lymphocytes, 15% plasma cells, 2% eosinophils, 5% mast cells, 1% monocytes. Prussian blue iron stain shows normal storage iron; no ring sideroblasts are noted.
- Paroxysmal nocturnal hemoglobinuria (PNH):
- In classical PNH, there is always evidence of intravascular hemolysis; reticulocyte count is generally high
- Bone marrow is normocellular to hypercellular with erythroid hyperplasia
- PNH clone generally relevant, above 50% and sometimes approaching representation of all circulating cells from the mutated clone (Br J Haematol 2018;182:730)
- Hypoplastic myelodysplastic syndrome:
- Dysplastic hematopoiesis, increased blasts or CD34 positive count and fibrosis suggest hypoplastic myelodysplastic syndrome (Haematologica 2009;94:264, Blood 2017;129:3371)
- Hypoplastic acute myeloid leukemia:
- Acute myeloid leukemia can present with hypocellular bone marrow; diagnosis relies on a proper blast count (≥ 20% defines leukemia) on peripheral blood or bone marrow samples (Haematologica 2009;94:264)
- T cell large granular lymphocytic leukemia:
- Generally presents with isolated anemia; pancytopenia observed only in a minority of the cases
- Lymphocyte count is usually 2 - 20 x 109/L
- Peripheral smears show a characteristic lymphocytosis of large granular lymphocytes
- Bone marrow biopsy shows a cytotoxic T cell infiltrate with a characteristic intrasinusoidal distribution (Blood 2017;129:1082)
- Constitutional bone marrow failure syndrome:
- With multilineage involvement: Fanconi anemia, dyskeratosis congenita
- With single lineage involvement: Diamond-Blackfan anemia, severe congenital neutropenia (Hematology 2015;20:433)
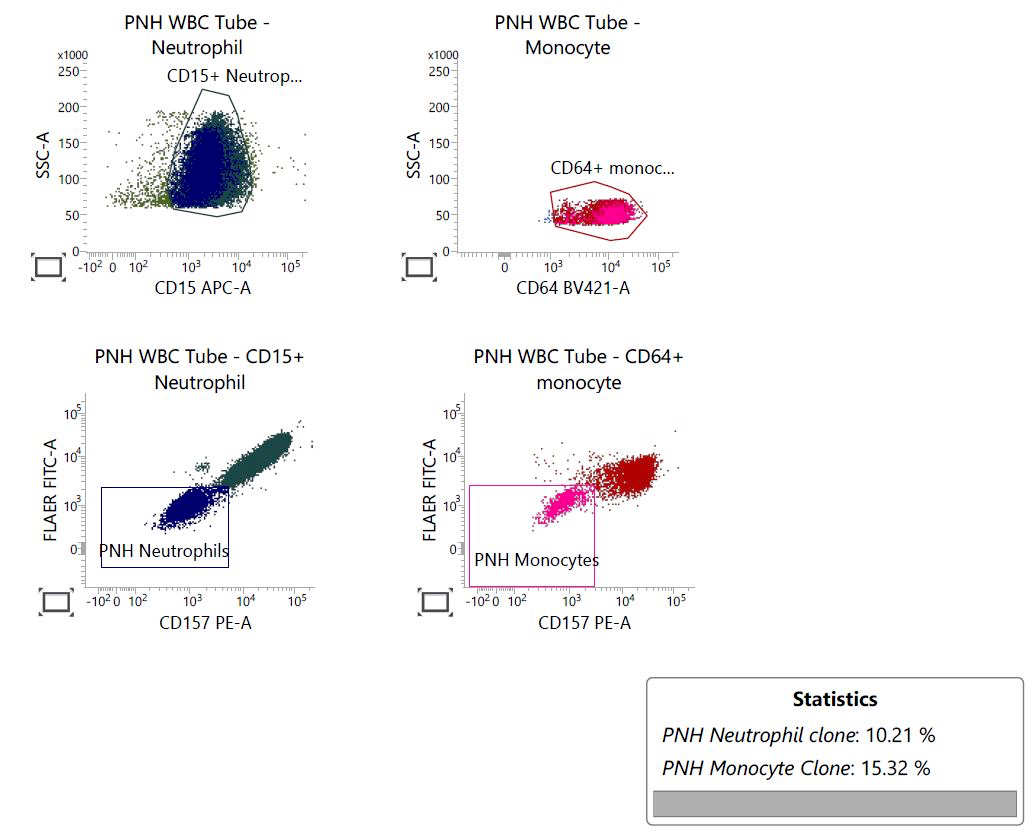
Are these flow cytometry findings compatible with aplastic anemia?
- No, they exclude completely aplastic anemia
- They are not relevant to a diagnostic workup of aplastic anemia
- They require clinical and pathologic correlation
- Yes, they are diagnostic of aplastic anemia
Small paroxysmal nocturnal hemoglobinuria clones are common in aplastic anemia and their detection does not exclude such diagnosis. However, when more relevant clones are detected, as in this case, careful clinical and pathologic investigations are needed to rule out paroxysmal nocturnal hemoglobinuria. True paroxysmal nocturnal hemoglobinuria should not show severe cytopenia(s) and marrow hypocellularity, features most in keeping with aplastic anemia.
Comment Here
Reference: Aplastic anemia (AA)
- 10% blast count
- Bone marrow collagen fibrosis
- Frank multilineage dysplasia
- Trisomy 8
Trisomy 8 is one of the most common cytogenetic abnormalities detected in aplastic anemia. However, this finding is not specific, as it can occur also in other myeloid neoplasms and it has been described in nonneoplastic conditions.
Comment Here
Reference: Aplastic anemia (AA)
- Arsenic intoxication is associated with acute and chronic adverse health effects
- Arsenic is pervasive in water, soil, air; has natural and anthropogenic sources
- Its metabolism involves reduction to a trivalent state and oxidative methylation to a pentavalent state; the trivalent arsenicals, including those methylated, have more potent toxic properties than the pentavalent arsenicals (Toxicol Lett 2002;133:1)
- Arsenic (As) belongs to fifth group of Mendeleyev's periodic table
- Free element of arsenic appears in two allotropic forms: grey and yellow (crystalline)
- Can also exist in various oxidation states, inorganic and organic forms, exposure to inorganic arsenic is associated with most cases of arsenic induced toxicity
- Arsenic contamination in drinking water identified in Russia, New Zealand, Romania, USA
- Inorganic arsenic (e.g. arsenic trioxide) is present in air, usually due to volcanic eruption and purposeful human activities
- Arsenic also found as sulphuric compound in lead, copper, nickel and ferrous ores; also in soil in small quantities
- Also a medicinal used by physicians for 2000 years
- Oral toxicities predominate, cause GI irritation, peripheral neuropathy, vascular lesions, anemia, skin disease; cancer of bladder, kidney, liver, lung (Int J Occup Med Environ Health 2002;15:101)
- Exposure by inhalation is associated with increased risk of lung cancer
- Little information is available on effects of direct dermal contact (Environ Health Perspect 1993;101:79)
- Intracellularly, arsenic accumulates in mitochondria, where it inhibits succinic dehydrogenase and uncouples oxidative phosphorylation, resulting in low ATP levels
- May also down regulate certain cytochrome P450 enzymes and activate c-Jun / AP1 transcription complex
- High doses of inorganic arsenicals induce release of TNF alpha
- Arsenic can induce cross tolerance to cytotoxicity, genotoxicity and apoptosis induced by nickel and other metals (Toxicol Appl Pharmacol 2001;176:127)
- Relative toxicities of arsenicals: As (III) > Mas (III) > DMAs (III) > DMAs (V) > Mas (V) > As (V); trivalent arsenicals increase cell proliferation at low concentrations (0.001 - 0.01 μmol) (Toxicol Appl Pharmacol 2001;172:225)
- Bone marrow depression: due to acute or more commonly chronic arsenic intoxication, may initially manifest as pancytopenia and be reversible; may have nonspecific clinical presentation of pancytopenia, fatigue / weakness (anemia), frequent infections (leukopenia) and easy bruising / petechia (thrombocytopenia)
- Anemia and leukopenia are common in chronic arsenic toxicity, are often accompanied by thrombocytopenia and mild eosinophilia
- Chronic arsenic exposure may be associated with immunosuppression (National Research Council: Arsenic in Drinking Water, 1999)
- Acute intoxication with arsine gas can cause fulminant intravascular hemolysis
- Most sensitive endpoint from arsenic exposure is dermal effects
- Can measure arsenic in blood, urine, hair, fingernails
- Current biological exposure index for U.S. workers of 35 μg/L total urinary arsenic may easily be exceeded by a healthy person eating a seafood meal (The Agency for Toxic Substances and Disease Registry (ATSDR): Arsenic Toxicity)
- Urine test is most reliable method for arsenic exposure within past few days; needs to be done within 24 - 48 hours for accurate analysis of acute exposure
- Hair and fingernail tests measure exposure to high levels over past 6 - 12 months
- Newer microanalytical techniques include synchroton radiation based X ray fluorescence (SXRF) spectroscopy, microparticle induced X ray emission (PIXE) (Biochimie 2009;91:1260)
- Arsenic trioxide approved for treatment of relapsed acute promyelocytic leukemia (APL) for remission induction
- May have activity for various solid tumors, myelodysplastic syndrome, myeloma, autoimmune disease (Ann Hematol 2013;92:719, Chin Med J (Engl) 2012;125:3556, Expert Opin Pharmacother 2012;13:1031, Leukemia 2012;26:1743)
- 35 year old woman with megaloblastic, dyserythropoietic anemia following arsenic ingestion (Ann Clin Lab Sci 1980;10:515)
- 41 year old woman with arsenic intoxication presenting as myelodysplastic syndrome (Am J Hematol 1991;36:291)
- 2 cases of acute arsenic intoxication from contaminated well water (Arch Environ Health 1989;44:385)
- 3 cases of erythroid karyorrhexis in peripheral blood smear in severe arsenic poisoning (Am J Clin Pathol 1984;81:533)
- Chronic arsenic intoxication associated with hemoloytic anemia, megaloblastic anemia and hypoplastic bone marrow (Rinsho Ketsueki 1980;21:409)
- Folic acid deficiency in chronic arsenic poisoning (Lancet 1965;1:784)
- Acute arsenic poisoning: dimercaprol and dimercaptosuccinic acid (DMSA) are chelating agents that sequester arsenic from blood proteins (Biochimie 2009;91:1260)
- Supplemental potassium decreases risk of life threatening arrhythmias from arsenic trioxide
- Sulfur containing substances in garlic may scavenge arsenic from tissue and blood
Images hosted on other servers:
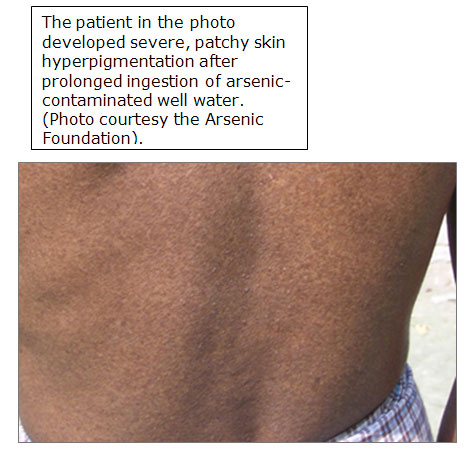
Pigment changes and
palmoplantar hyperkeratoses
are characteristic of
chronic arsenic exposure
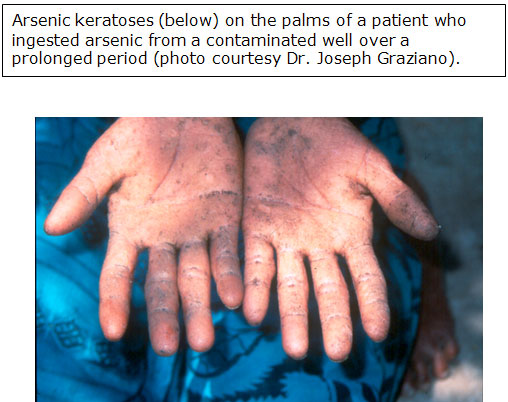
Arsenic keratosis -
so called "raindrops
on a dusty road"
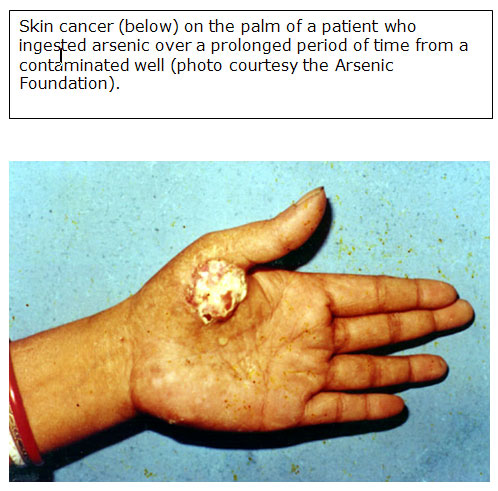
Skin cancer
- Peripheral smear: leukopenia including granulocytopenia and absolute eosinophilia, and profound anemia (normocytic or macrocytic) with coarse basophilic stippling and karyorrhexis (N Engl J Med 1965;273:18, West J Med 1983;139:219, Am J Clin Pathol 1984;81:533, Blut 1985;50:51)
- Dyserythropoiesis including megaloblastic features and typical karyorrhexis in erythropoietic cells, accompanied by basophilic stippling and impairment of mitoses in megakaryocytes and granulopoietic components
- Bone marrow biopsy shows nonspecific changes
AFIP images
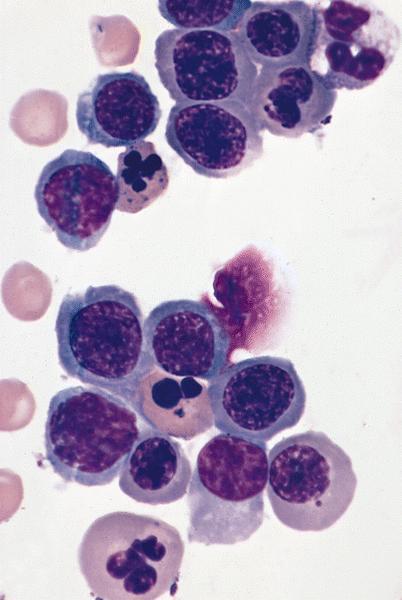
Bone marrow: erythroid
precursors have marked
dysplastic changes including
nuclear lobulation and karyorrhexis
AFIP images

Erythroid precursor with
slightly lobulated
megaloblastoid nucleus
- Arsenic intoxication induces megaloblastic erythropoiesis and other bone marrow changes similar to other dyserythropoietic states, including marked nuclear aberrations involving shape, chromatin distribution and nuclear envelope (Blood 1979;53:820)
Images hosted on other servers:
Abnormal erythroblasts
- No specific cytogenetic abnormalities
- Other heavy metal intoxications, especially those associated with anemia / pancytopenia
- Other conditions associated with megaloblastic changes in erythroid precursors
- Primary autoimmune myelofibrosis (pAIMF): nonneoplastic myelofibrosis with peripheral blood cytopenias and positive autoimmune antibody serologies, typically in the absence of marked splenomegaly, teardrop poikilocytosis, leukoerythroblastosis, cytologic atypia and a defined autoimmune disorder (Am J Hematol 2003;72:8, Hum Pathol 2014;45:2183)
- Secondary autoimmune myelofibrosis (sAIMF): same features of pAIMF but in the setting of a defined autoimmune disorder (Acta Haematol 2017;138:129)
- Nonneoplastic diffuse marrow fibrosis with positive antibody serologies requiring close clinicopathologic correlation for definitive diagnosis
- Usual morphologic features: cytopenia(s) with hypercellular marrow, megakaryocytic and / or erythroid hyperplasias, benign lymphoid aggregates, polytypic plasmacytosis, no or mild hematopoietic dysplasia, mild reticulin fibrosis (MF grade 1)
- No evidence of clonality on ancillary studies (by flow cytometry, chromosomal analysis or next generation sequencing panels for somatic myeloid mutations)
- Usually no or mild splenomegaly
- Typical cases are responsive to glucocorticoids and other immunosuppressive therapy
- Systemic lupus erythematosus (SLE) associated myelofibrosis (sAIMF) (J Clin Pathol 1983;36:1219, Medicine (Baltimore) 1994;73:145, Br J Haematol 2007;139:351)
- ICD-10: D75.81 - myelofibrosis
- Rare
- Incidence and prevalence unknown
- 139 adult cases identified in literature review (Eur J Haematol 2023;111:706)
- Possibly under recognized (Acta Haematol 2017;138:129)
- At least 20 pediatric cases in the literature (Pediatr Blood Cancer 2023;70:e30144, J Clin Rheumatol 2021;27:S378, EJHaem 2020;1:304)
- F:M = ~4:1 (Eur J Haematol 2023;111:706)
- Any age (range: < 1 - 78 years)
- Adult cases (median): 40 - 42 years (Eur J Haematol 2023;111:706)
- Pediatric cases (median): 4.9 - 16.5 years (Pediatr Blood Cancer 2023;70:e30144, J Clin Rheumatol 2021;27:S378)
- ~76% associated with autoimmune syndromes (i.e., sAIMF) (Eur J Haematol 2023;111:706)
- Bone marrow
- Specific pathophysiologic mechanism is not well understood
- Few studies have addressed this question; hypotheses include
- Fibrosis likely driven by overproduction of cytokines
- Predominant source of cytokines is hypothesized to relate to lymphoid aggregates (Clin Adv Hematol Oncol 2018;16:619)
- CD8+ T lymphocytes and IFN gamma have been implicated as having pathogenic roles in a mouse model of primary biliary cholangitis with myelofibrosis (J Autoimmun 2018:89:101)
- IFN gamma signaling through JAK-STAT pathway may play a role
- Overactivation of the JAK-STAT pathway has been associated with multiple autoimmune disorders and malignancies (J Allergy Clin Immunol 2021;147:814)
- Case reports of refractory pAIMF that were responsive to JAK inhibitors (Am J Hematol 2021;96:E283)
- Most hypotheses are extrapolated from studies of other neoplastic myelofibrotic disorders
- Megakaryocytes, platelets and monocytes have been shown to play a role in other etiologies of myelofibrosis (e.g., thrombopoietin associated myelofibrosis, hairy cell leukemia, primary myelofibrosis) via cytokines possibly including PDGF, calmodulin, TGF beta, VEGF and basic fibroblast growth factor (Br J Haematol 2007;139:351, N Engl J Med 2000;342:1255)
- Specific roles of these cytokines in AIMF have not yet been shown
- Increased levels of IFN gamma, TGF beta and IL8 have been associated with cases of primary myelofibrosis (PMF) with autoimmune features (e.g., cases of PMF with positive mitogen stimulated DAT or serologic markers of autoimmunity)
- Unclear if this represents mechanistic overlap between the 2 entities (Leuk Res 2013;37:1509)
- Megakaryocytes, platelets and monocytes have been shown to play a role in other etiologies of myelofibrosis (e.g., thrombopoietin associated myelofibrosis, hairy cell leukemia, primary myelofibrosis) via cytokines possibly including PDGF, calmodulin, TGF beta, VEGF and basic fibroblast growth factor (Br J Haematol 2007;139:351, N Engl J Med 2000;342:1255)
- Fibrosis likely driven by overproduction of cytokines
- Unknown
- Role of potential driver mutations is unknown (Haematologica 2021;106:871)
- Symptomatic
- Anemia (83%)
- Thrombocytopenia (34%)
- Fever (30%)
- Signs / symptoms of autoimmune flare
- Raynaud phenomenon, joint pain, peripheral edema
- Night sweats (6%)
- Rarely splenomegaly, usually mild (1%)
- Asymptomatic (at least 32% of patients)
- sAIMF: may present with clinical features of other autoimmune syndromes (Hum Pathol 2014;45:2183, Eur J Haematol 2023;111:706, Haematologica 2021;106:871)
- Most commonly with systemic lupus erythematosus (SLE)
- Also described in patients with acute inflammatory demyelinating polyneuropathy (AIDP), antiphospholipid syndrome (APLS), autoimmune demyelinating polymyositis, autoimmune hemolytic anemia (AIHA), autoimmune hepatitis, dermatomyositis, diabetes mellitus type I (T1DM), Evans syndrome, Hashimoto thyroiditis, immune thrombocytopenic purpura (ITP), mixed connective tissue disease (MCTD), multiple sclerosis (MS), polymyositis, primary sclerosing cholangitis (PSC), psoriasis, rheumatoid arthritis (RA), Sjögren syndrome (SS), vasculitis, vitiligo
- pAIMF: no established autoimmune disorder but all other features associated with sAIMF (Am J Hematol 2003;72:8)
- Bone marrow biopsy
- May be a dry tap (Clin Adv Hematol Oncol 2018;16:619)
- Peripheral blood smear
- Serologic studies (e.g., autoantibodies)
- Imaging studies (e.g., evaluating spleen)
- Flow cytometry analysis
- Molecular testing (e.g., absent clonal markers of other disorders) (Haematologica 2021;106:871)
- Clinical correlation
- Peripheral cytopenias
- Anemia, thrombocytopenia and / or leukopenia (Eur J Haematol 2023;111:706)
- Serologic autoantibodies
- Cases described with positive antinuclear antibody (ANA), cardiolipin antibodies, cyclic citrullinated peptide antibodies (CCP), direct antiglobulin test (DAT), double stranded DNA antibody (dsDNA), lupus anticoagulant, myeloperoxidase antibodies (MPO), atypical perinuclear antineutrophil cytoplasmic antibody patterns (pANCA), rheumatoid factor (RF), Sjögren syndrome antibodies (SSA / SSB) and / or smooth muscle antibody (Acta Haematol 2017;138:129)
- Elevated inflammatory markers often reported
- E.g., erythrocyte sedimentation rate (ESR) and ferritin (Eur J Haematol 2023;111:706)
- Other abnormal serologic markers
- Hypocomplementemia (Haematologica 2021;106:871)
- Overall good prognosis (Acta Haematol 2017;138:129)
- ~85% of cases with clinical follow up had at least partial symptomatic improvement after treatment (Eur J Haematol 2023;111:706)
- Few reports of long term follow up available (Acta Haematol 2017;138:129, Eur J Haematol 2023;111:706)
- Cases of relapse have been reported (Haematologica 2021;106:871, Pediatr Blood Cancer 2022;69:e29762)
- Factors associated with treatment resistance are unknown
- 15 year old boy with headaches and severe pallor (EJHaem 2020;1:304)
- 44 year old woman with fatigue, joint pains, oral ulcers, sicca syndrome and malar rash (BMJ Case Rep 2019;12:bcr-2018-227520)
- 48 year old woman with anemia and fever (Am J Clin Pathol 2001;116:211)
- 61 year old woman with severe fatigue (Am J Hematol 2021;96:E283)
- Insufficient data on optimal indications and efficacy of different treatment regimens (Haematologica 2021;106:871)
- Adequate prospective trials are difficult due to rarity of AIMF (Eur J Haematol 2023;111:706)
- Usually responds to glucocorticoids
- Most cases with post treatment biopsy data report diminished or reversed marrow fibrosis (Eur J Haematol 2023;111:706)
- Other immunomodulators are sometimes used, alone or with glucocorticoids
- E.g., hydroxychloroquine, mycophenolate, intravenous immunoglobulin, rituximab and others (Eur J Haematol 2023;111:706)
- Ruxolitinib reportedly used in few case reports (Leuk Lymphoma 2023;64:1723, Am J Hematol 2021;96:E283)
- Treatment may aim to control underlying autoimmune disorder (Pediatr Blood Cancer 2023;70:e30144)
- Constellation of morphologic findings may suggest the diagnosis in appropriate clinical context
- Usually mild to moderate reticulin fibrosis of bone marrow
- Mild / delicate fibrosis (MF1; 86%), moderate fibrosis (MF2; 10%), marked fibrosis (MF3; 4%) (Hum Pathol 2014;45:2183)
- Using European consensus classification on grading bone marrow fibrosis (Haematologica 2005;90:1128)
- Mild / delicate fibrosis (MF1; 86%), moderate fibrosis (MF2; 10%), marked fibrosis (MF3; 4%) (Hum Pathol 2014;45:2183)
- Usually hypercellular (72%) or normocellular (24%) bone marrow (Hum Pathol 2014;45:2183)
- Usually with erythroid and megakaryocytic hyperplasia (76% each)
- May show granulocytic hyperplasia (34%)
- Benign lymphoid aggregates (83%) or interstitial infiltrate of lymphocytes (17%) usually present (either / or: 100%; n=29) (Hum Pathol 2014;45:2183)
- Lymphoid aggregates
- Usually small to medium sized
- Nonparatrabecular location
- Well circumscribed
- T and B cell distribution usually in T cell pattern (patterns 1 - 3) (Hum Pathol 2014;45:2183, Hum Pathol 2013;44:512)
- Lymphoid aggregates
- Mild polyclonal / polytypic plasmacytosis may be present (66%), typically < 10% plasma cells (Hum Pathol 2014;45:2183, Am J Hematol 2003;72:8)
- No significant osteosclerosis or bony changes
- No or minimal dysplasia of hematopoietic lineages
- Occasional atypical forms may be present
- Should not meet diagnostic criteria for myeloid neoplasms
- No monoclonal / monotypic population
- No increase in blasts
- Reference: Hum Pathol 2014;45:2183
Contributed by Ria Vergara-Lluri, M.D.
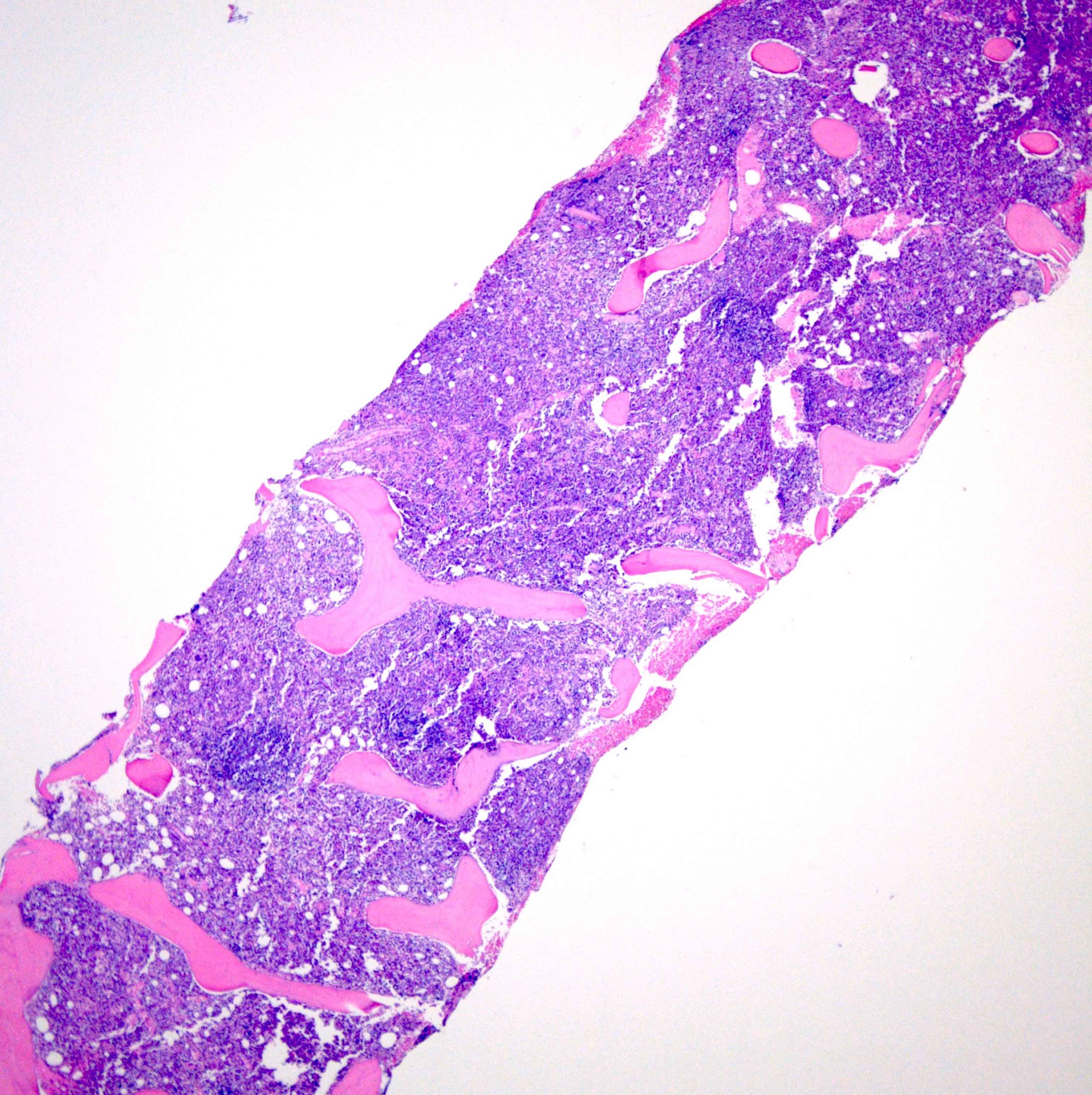
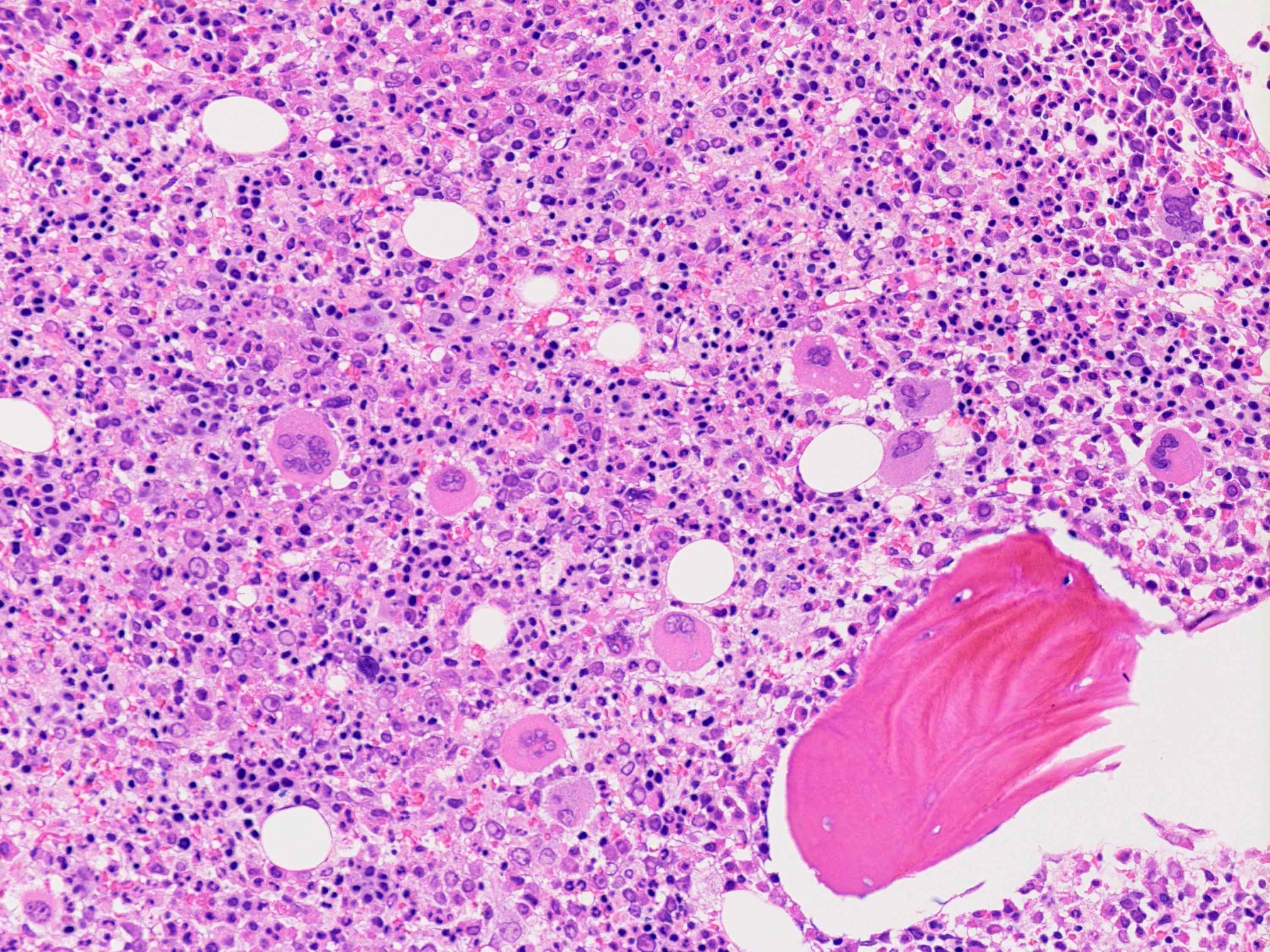
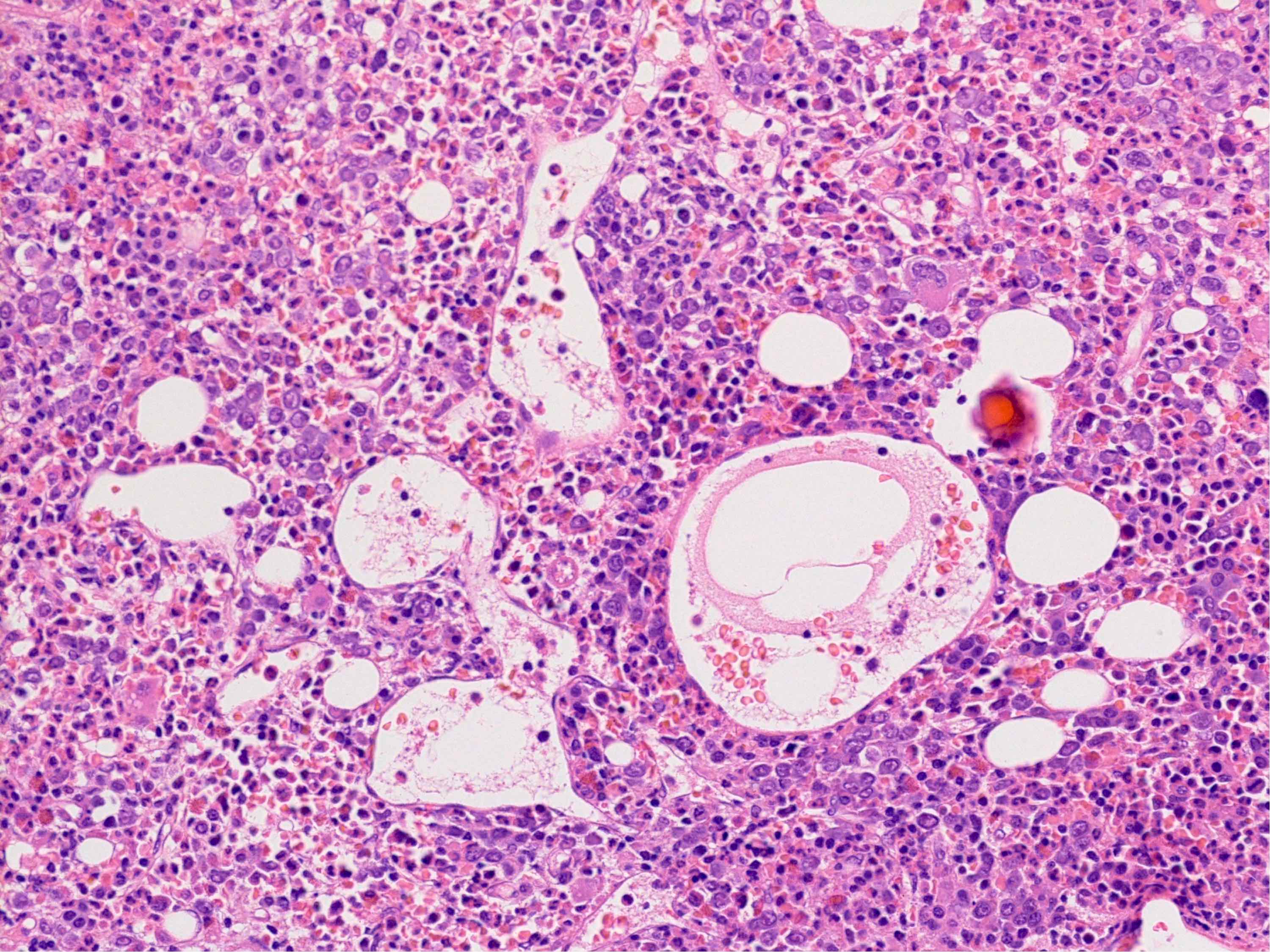
Markedly hypercellular bone marrow
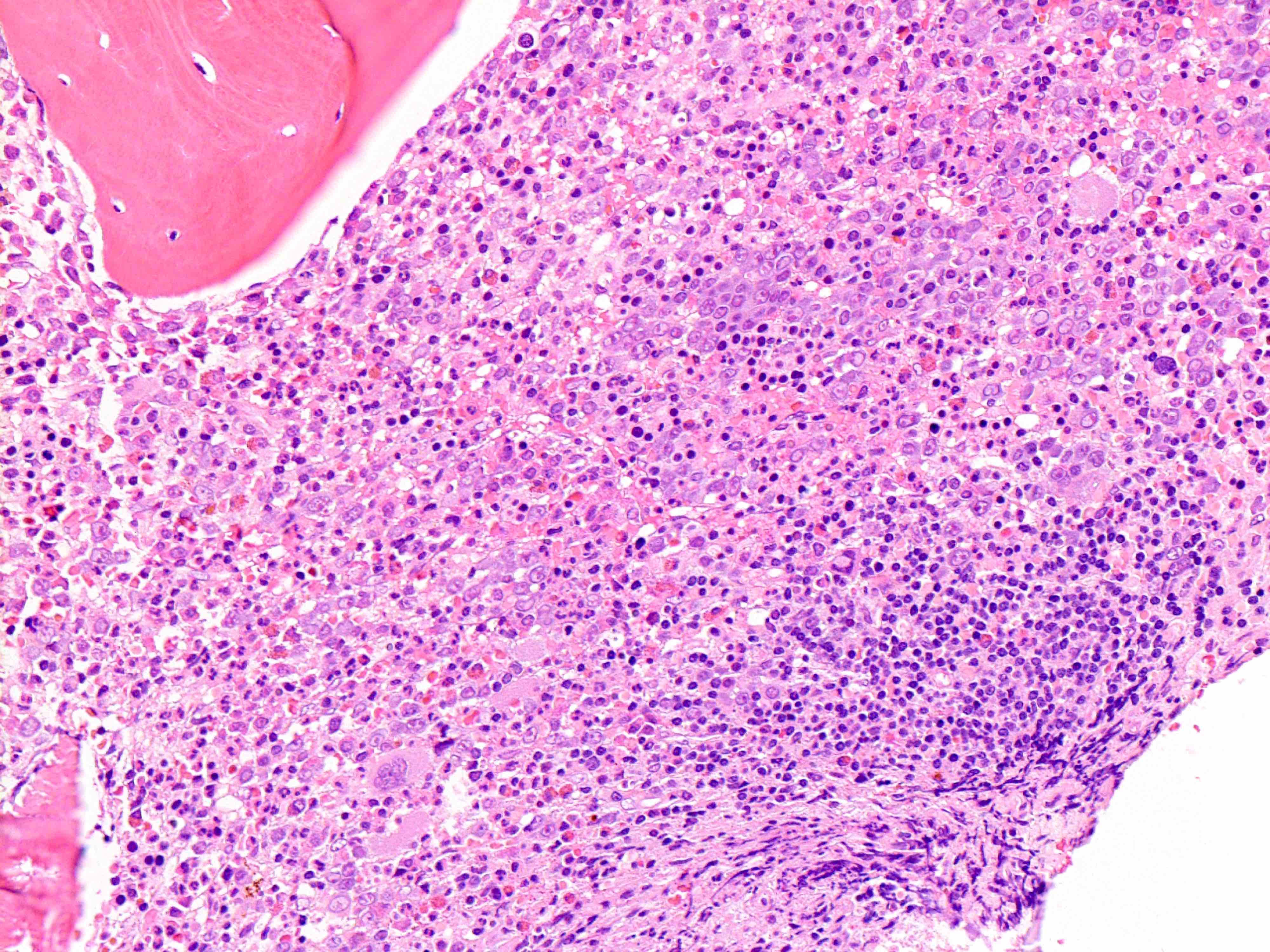
Erythroid hyperplasia
Megakaryocytic hyperplasia without atypia
Sinusoidal dilation
MF grade 1
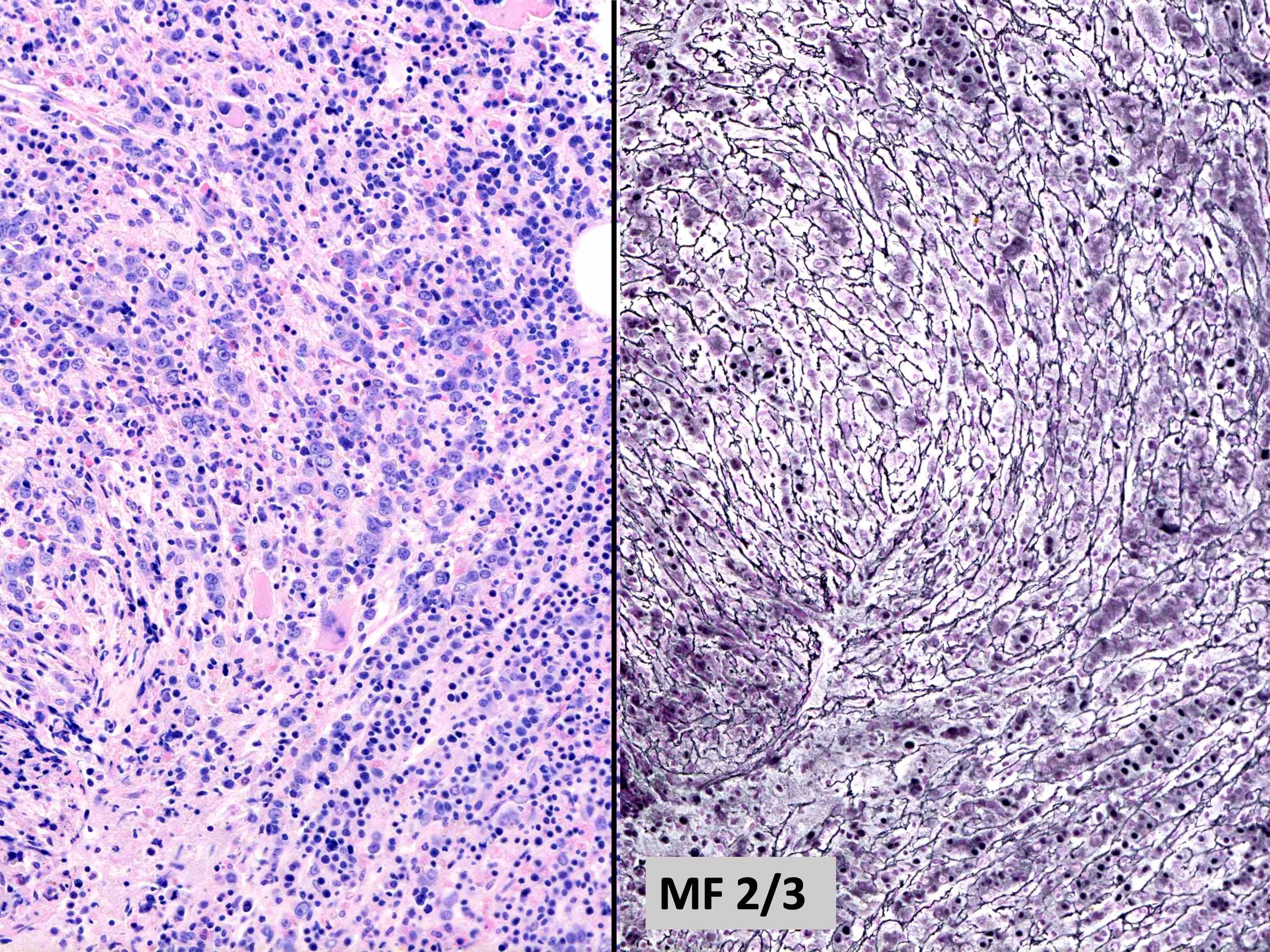
MF grade 2
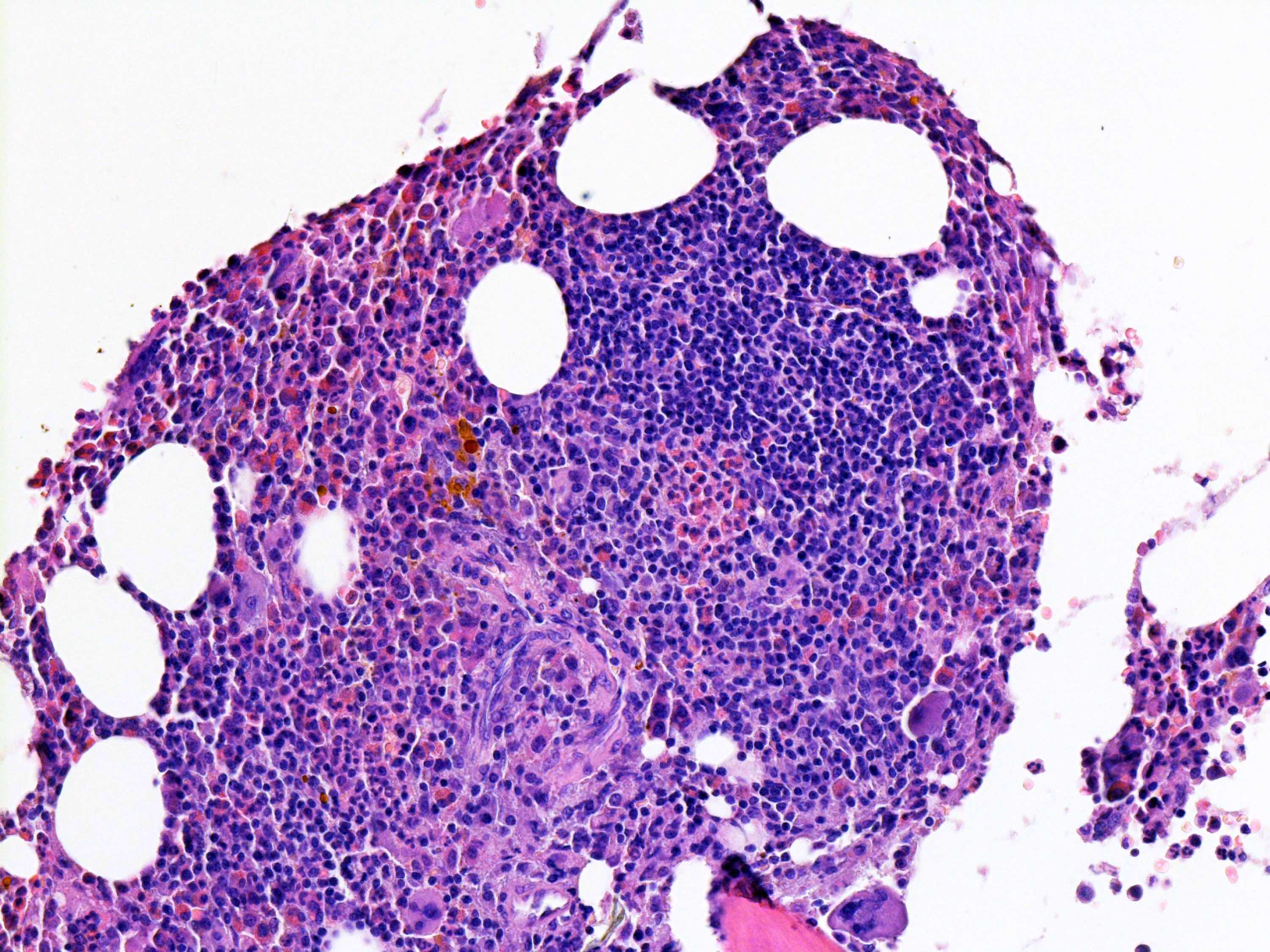
Benign lymphoid aggregate
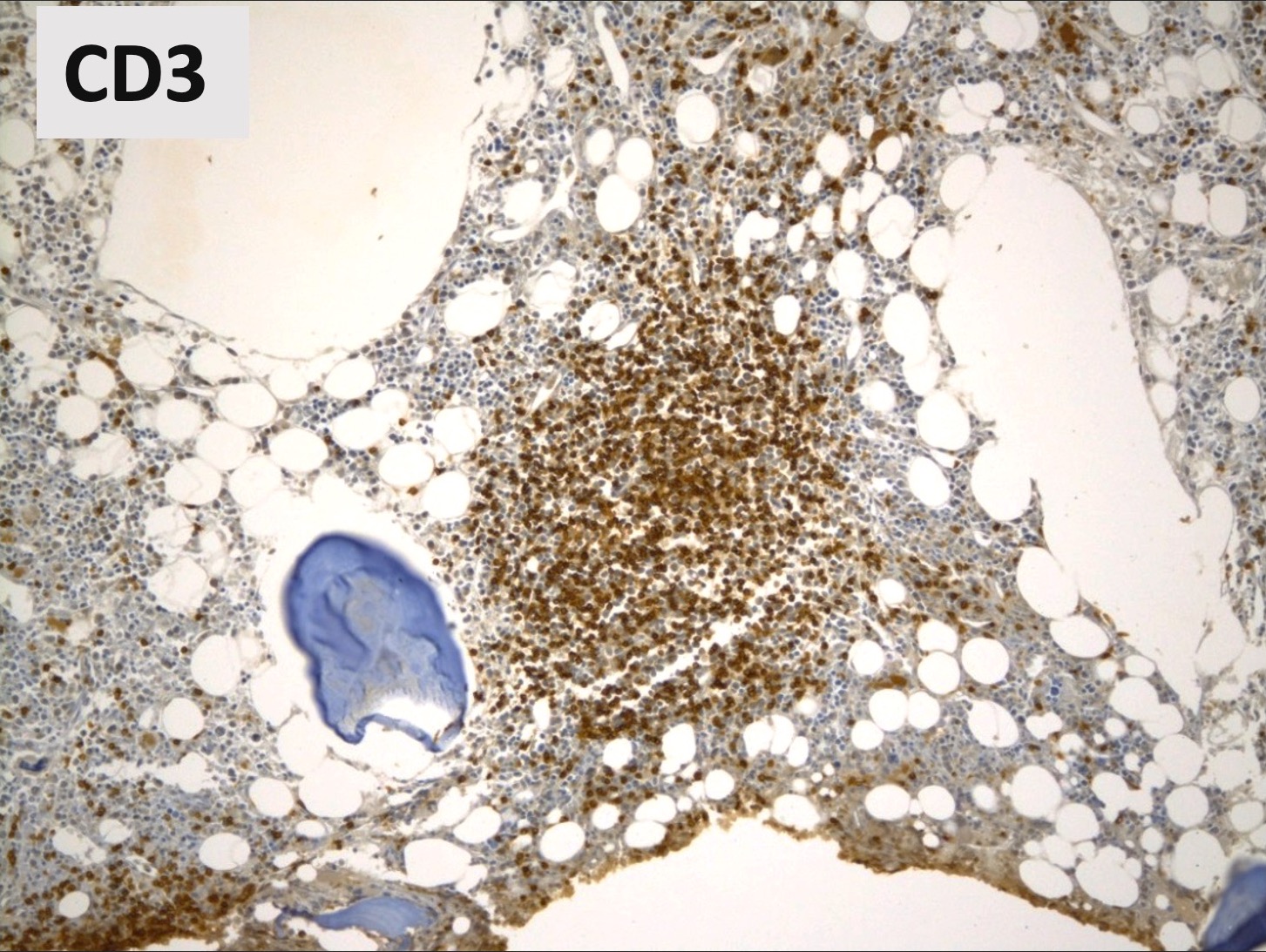
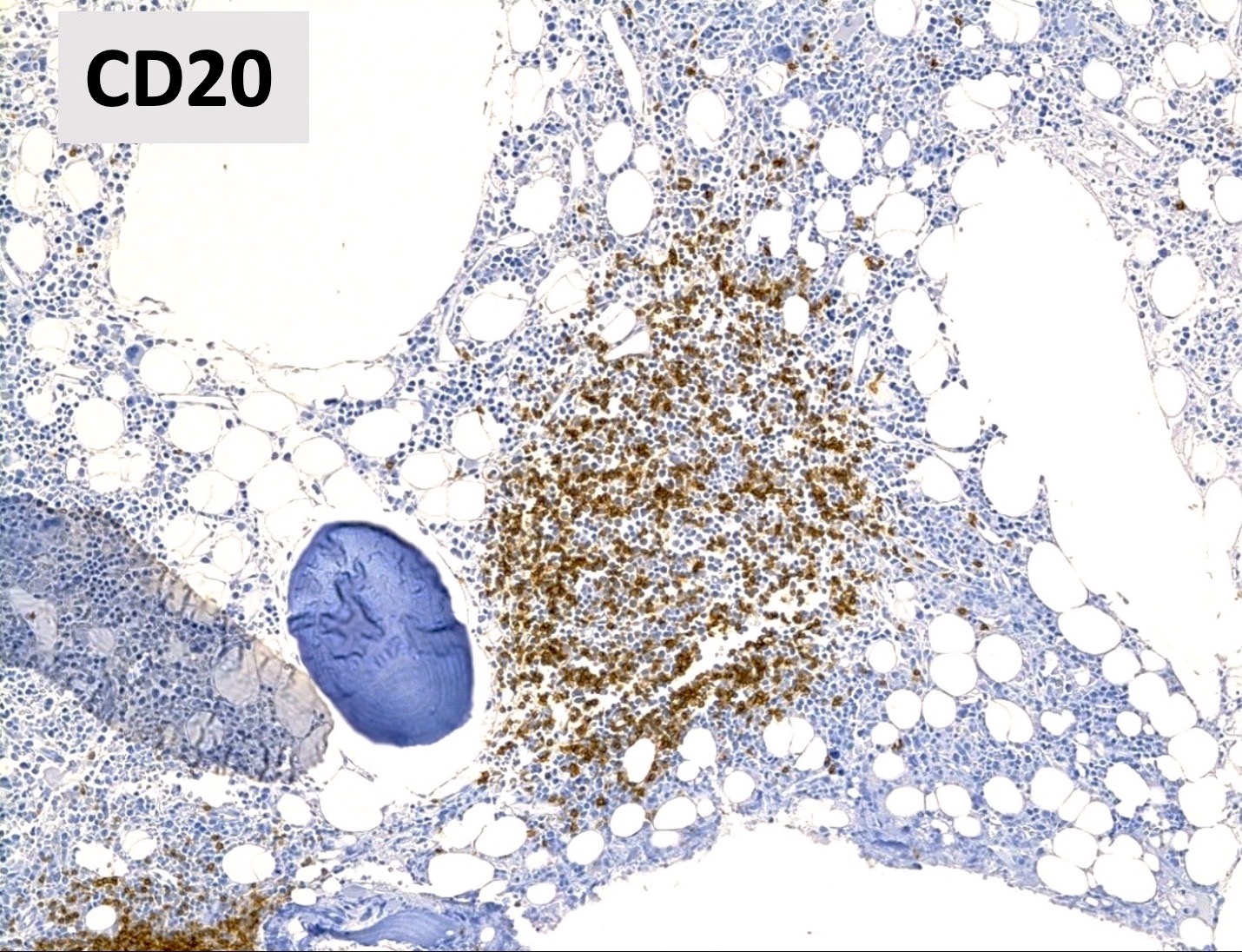
Benign lymphoid aggregate immunostains
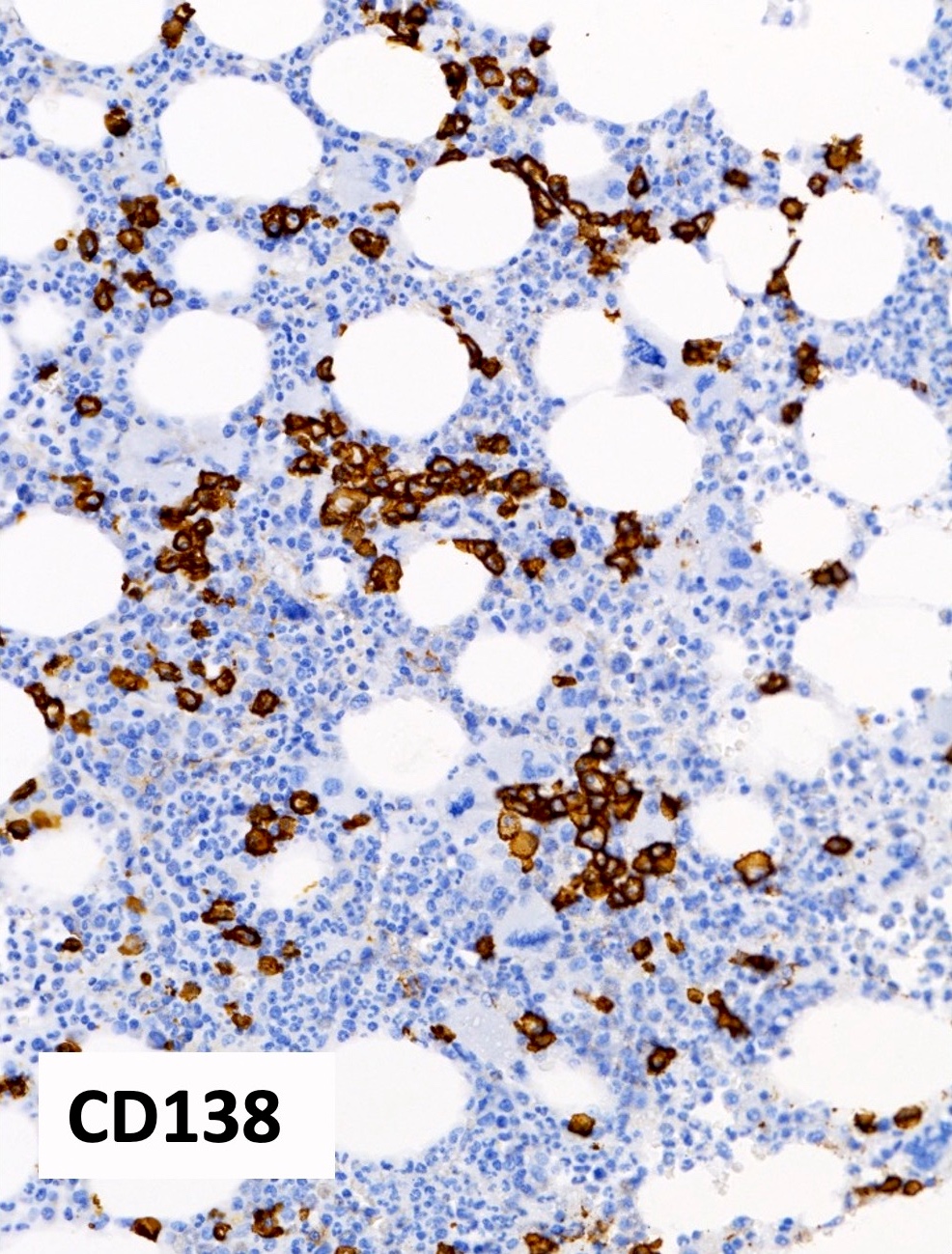

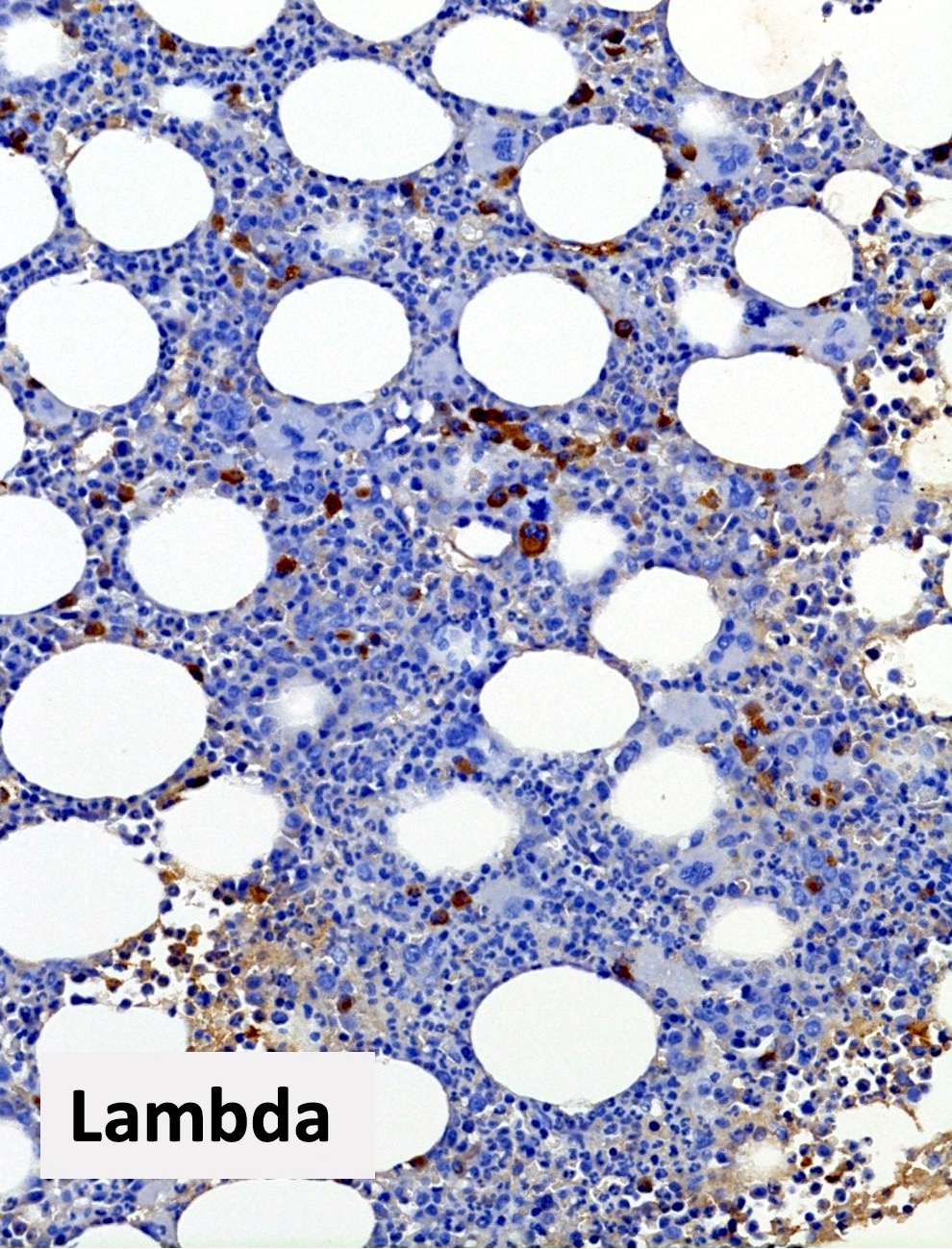
Polytypic plasmacytosis
- Anemia, thrombocytopenia and / or leukopenia
- No or rare dacrocytes (teardrop cells)
- No or rare nucleated red blood cells and blasts
- Usually no eosinophilia or basophilia
- No dysplasia (i.e., no pseudo Pelger-Huët changes, neutrophilic hypogranularity)
- No features worrisome for a myeloid neoplasm (e.g., no significant increase in basophils, eosinophils, neutrophilic left shift, circulating megakaryocytes, etc.)
- Reference: Hum Pathol 2014;45:2183
Contributed by Ria Vergara-Lluri, M.D.
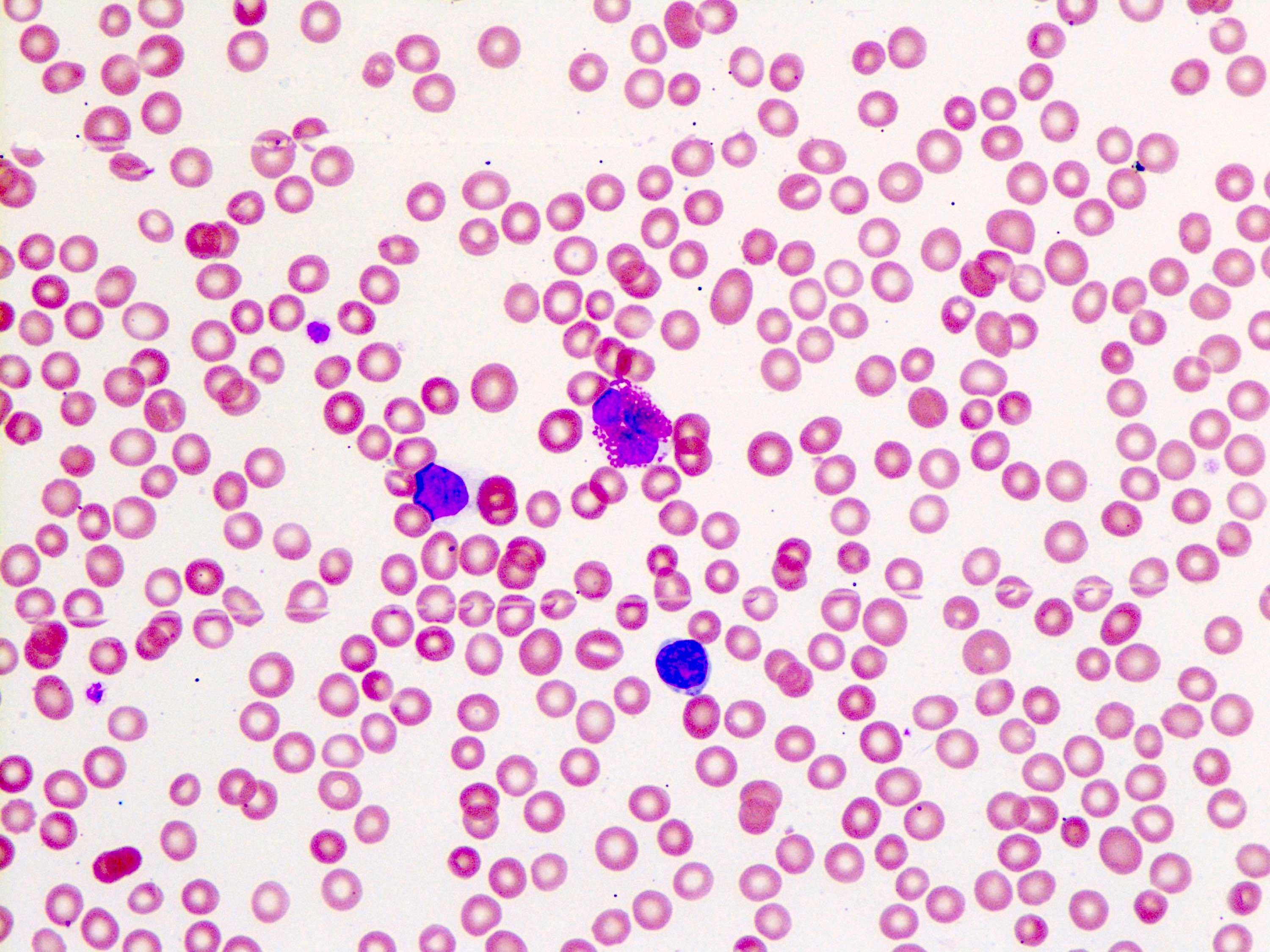
Peripheral blood smear
- Reticulin: highlights bone marrow stromal fibrosis
- CD3 / CD20
- Benign lymphoid aggregates display reactive patterns
- Predominance of T cells over B cells; core of many T cells with peripheral rim of B cells or mixture of T and B cells (Hum Pathol 2013;44:512)
- Interstitial infiltrates show predominance of T over B cells (Pediatr Blood Cancer 2023;70:e30144)
- Benign lymphoid aggregates display reactive patterns
- CD138 / kappa / lambda: show mild polytypic plasmacytosis, if present
- Reference: Hum Pathol 2014;45:2183
- No overt myeloid dysmaturation
- No excess blasts (typically < 3%)
- Polytypic B cells with normal kappa to lambda ratios (Hum Pathol 2014;45:2183)
- No overt pan T cell aberrancies
- Role of potential undiscovered driver mutations is unknown (Haematologica 2021;106:871)
- 1 pediatric AIMF case series found inborn errors of immunity in 50% of cases (n=8) (Pediatr Blood Cancer 2023;70:e30144)
- No JAK2, CALR, MPL mutations
- No molecular or cytogenetic markers of other clonal disease (Eur J Haematol 2023;111:706)
- Negative molecular / cytogenetic testing has been suggested as diagnostic criterion (Haematologica 2021;106:871)
- Peripheral blood:
- Mild normochromic, normocytic anemia
- No circulating blasts
- Mild neutropenia with no overt dysplastic features
- No lymphocytosis nor significant increase in large granular lymphocytes
- Mild thrombocytopenia
- Bone marrow aspirates, imprints, trephine and clot biopsies:
- Moderately hypercellular bone marrow with moderate erythroid and megakaryocytic hyperplasias and adequate numbers of granulocytic precursors
- Mild reticulin fibrosis (MF grade 1; scale 0 - 3)
- Mild lymphocytosis with benign lymphoid aggregates
- Mild polytypic plasmacytosis
- No excess blasts (blasts < 5%)
- No overt dysplastic features
- No diagnostic evidence of lymphoma or plasma cell dyscrasia
- Comment: The constellation of morphologic features is not specific for any one etiology and can also be found in other nonneoplastic settings (such as in other inflammatory disorders, infections, drug exposures, etc.). Thus, these features must be interpreted in the context of pertinent clinical information (e.g., presence of an established rheumatologic disease, etc.), laboratory studies (e.g., rheumatoid factor, antineutrophil antibodies, direct antiglobulin tests, etc.) and imaging findings (e.g., evaluation for splenomegaly, lymphadenopathy, etc.) and may best be undertaken with consultation / communication with hematology and rheumatology clinical services. If other etiologies have been adequately excluded and in the appropriate clinicopathologic context, these morphologic findings could be compatible with autoimmune myelofibrosis (AIMF).
- Nonneoplastic myelofibrosis due to other causes (e.g., idiopathic multicentric Castleman disease, drug induced, infection) (Hematol Oncol 2022;40:191, Bain: Bone Marrow Pathology, 5th Edition, 2019, Br J Haematol 2007;139:351):
- Clinical history or laboratory studies consistent with alternate cause
- For example, idiopathic multicentric Castleman disease (iMCD) can show similar morphologic findings (such as hypercellularity, marrow reticulin fibrosis and megakaryocytic hyperplasia)
- For example, recent romiplostim or eltrombopag administration; evidence of osteomyelitis, tuberculosis or leishmaniasis
- Essential to thoroughly integrate data from clinical history, physical examination, other pathology (e.g., lymph node biopsy results) and imaging findings
- Clinical history or laboratory studies consistent with alternate cause
- Primary myelofibrosis (PMF), prefibrotic and overt fibrotic stages (Acta Haematol 2017;138:129):
- Myeloproliferative neoplasm with poor prognosis
- Palpable splenomegaly (~89 - 100%) and hepatomegaly (~54 - 89%) (Medicine (Baltimore) 1983;62:353)
- May have positive autoimmune antibody serologies (Clin Adv Hematol Oncol 2018;16:619)
- Bone marrow core biopsy
- Prefibrotic stage
- Increased myeloid:erythroid ratio and / or trilineage expansions
- No to minimal reticulin fibrosis (MF0 - MF1)
- Atypical megakaryocyte proliferation
- Often bizarre, hyperchromatic nuclei distributed in loose or dense clusters
- Overt fibrotic stage
- Similar to prefibrotic stage, although cases with more advanced fibrosis may show hypocellularity and / or cellular dropout
- Moderate to severe reticulin fibrosis (MF2 to MF3)
- Varying degrees of osteosclerosis
- Prefibrotic stage
- Driver mutations (JAK2, CALR or MPL) or other clonal markers / cytogenetic abnormalities usually present
- Post myeloproliferative neoplasm myelofibrosis (e.g., post polycythemia vera or post essential thrombocythemia myelofibrosis):
- Prior documented diagnosis of polycythemia vera or essential thrombocythemia
- May present with increasing splenomegaly
- May have positive autoimmune antibody serologies (Acta Haematol 2017;138:129)
- Bone marrow core biopsy
- Bone marrow reticulin fibrosis MF2 or MF3
- Clusters of atypical megakaryocytes
- May show osteosclerosis
- Often has JAK2, CALR or MPL mutation
- Non-Hodgkin lymphomas:
- Chronic myelomonocytic leukemia (CMML) with fibrosis (Oncotarget 2017;8:103274):
- May have splenomegaly (58%, n=45)
- Persistent peripheral blood monocytosis (≥ 1 x 109/L)
- Monocytes > 10% of peripheral blood leukocytes
- Will have morphologic dysplasia in ≥ 1 hematopoietic lineage
- Will have molecular / cytogenetic abnormalities
- Desmoplasia associated with metastatic tumors (Bain: Bone Marrow Pathology, 5th Edition, 2019):
- Normochromic, normocytic anemia is common
- Other cytopenias are less common
- May see leukoerythroblastic anemia (< 50%)
- Aspirate may show tumor cells, necrosis, osteoblasts / osteoclasts
- Trephine may also show osteolysis / osteosclerosis, neoangiogenesis
- Normochromic, normocytic anemia is common
A 47 year old woman presents with fatigue, joint pains and pallor. Laboratory workup is notable for severe anemia, thrombocytopenia and a serum ANA titer of 1:40. A bone marrow biopsy is obtained and shows marked hypercellularity with scattered nonparatrabecular lymphoid aggregates (images above) as well as myelofibrosis highlighted by reticulin staining (not shown). Which of the following findings would be typical for a diagnosis of autoimmune myelofibrosis (AIMF)?
- Increased numbers of megakaryocytes in dense clusters
- MPL mutation on next generation sequencing (NGS) myeloid panel
- Palpable splenomegaly
- Response to glucocorticoids
- Severe reticulin fibrosis (MF grade 3)
Answer E is incorrect because myelofibrosis in AIMF is usually mild to moderate, although rare cases of AIMF have been associated with severe reticulin fibrosis and this finding alone would not be inconsistent with the diagnosis. Answer C is incorrect because palpable splenomegaly is quite uncommon in AIMF, though not exclusionary. Answer B is incorrect because the presence of a pathogenic mutation in MPL ought to prompt assessment favoring neoplasia. The absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL) can be an essential part of the diagnostic work up of AIMF. Answer A is incorrect because while it is typical to see increased numbers of megakaryocytes in AIMF, dense clusters of megakaryocytes (which are very likely to also display cytologic atypia) would be highly unusual for (and morphologically inconsistent with) AIMF. This observation strongly favors a myeloid neoplasm (such as primary myelofibrosis) and should trigger a thorough work up to demonstrate clonality (e.g., karyotype, NGS testing).
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
A 37 year old woman with no significant medical history was admitted to the hospital for acute onset of fevers and severe fatigue. Her vital signs and physical examination were significant only for fever and tachycardia; there is no palpable hepatosplenomegaly.
- Imaging studies showed no organomegaly or lymphadenopathy
- Laboratory studies were remarkable for WBC 3.1 x 109/L, Hb 8.0 g/dL, Plt 39 x 103/uL, ANA titer of 1:80 and a negative DAT
- Peripheral blood smear showed cytopenias without leukoerythroblastosis
- Remainder of the infectious, autoimmune and neoplastic workup was negative
- Bone marrow biopsy displayed a markedly hypercellular marrow with erythroid and megakaryocytic hyperplasias, scattered nonparatrabecular lymphoid aggregates, no overt dysplasia or excess blasts and mild polytypic plasmacytosis (< 10%)
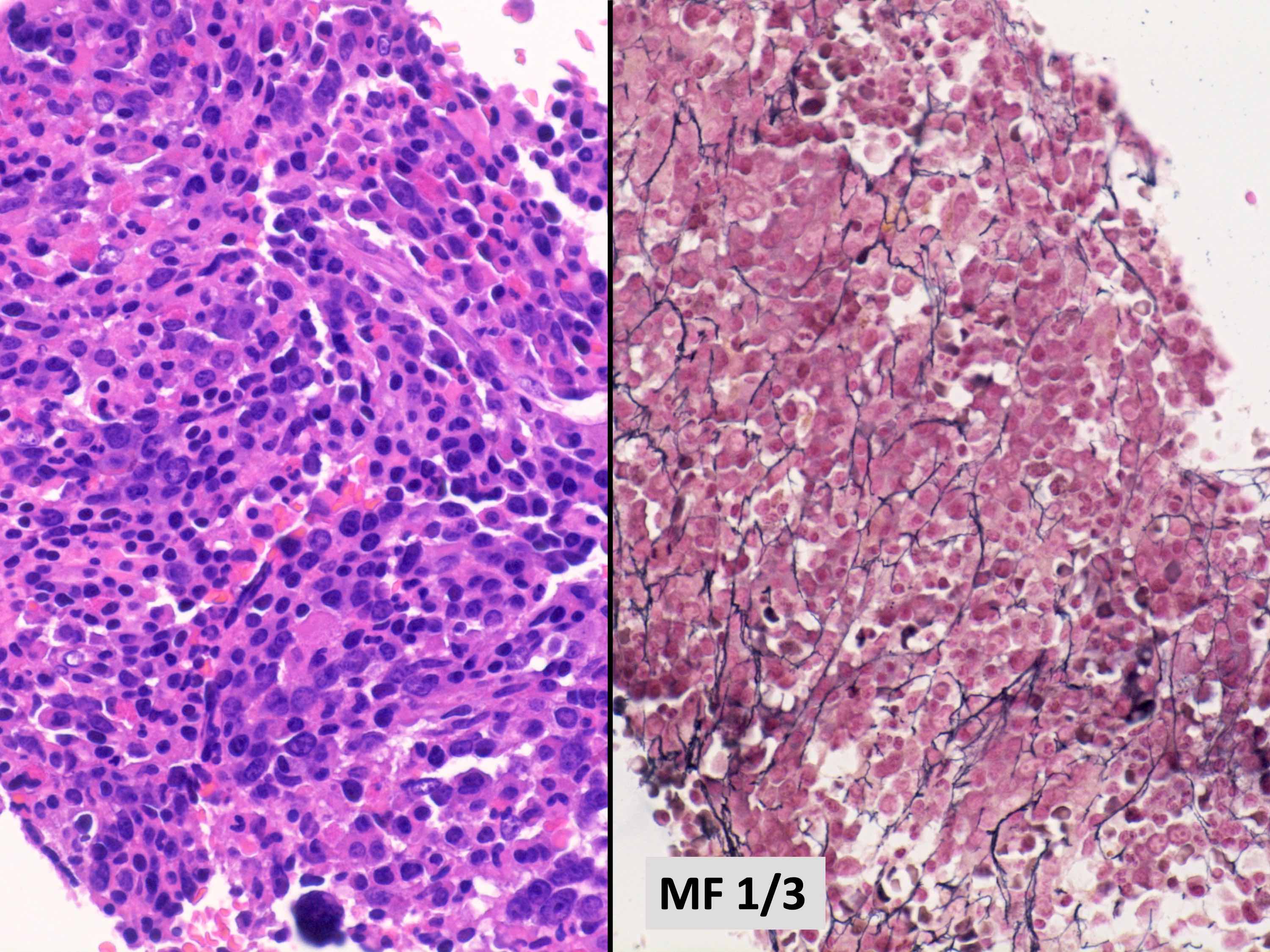
- Reticulin staining revealed moderate reticulin fibrosis (MF2; image shown above)
- Normal karyotype was reported while next generation sequencing (NGS) myeloid panels for somatic mutations were negative
She received a course of glucocorticoids and achieved complete hematologic response and was discharged from the hospital. Rheumatology consultation revealed no established autoimmune disorder. She is being closely monitored by the hematology clinical service. A repeat biopsy performed 1 year after her hospital admission showed similar findings, though decreased reticulin fibrosis is observed (MF1). Which of the following is the most appropriate diagnosis at this time?
- Myelofibrosis, secondary to polycythemia vera
- Primary autoimmune myelofibrosis
- Primary myelofibrosis, overt stage
- Primary myelofibrosis, prefibrotic stage
Secondary AIMF has been described in association with many autoimmune disorders, including systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, autoimmune hemolytic anemia and others. In this vignette, over the course of close clinical follow up, no specific autoimmune, infectious / inflammatory or other hematologic disorder could be established. Furthermore, her favorable response to glucocorticoids and mild decrease in reticulin fibrosis on repeat biopsy is typical of AIMF. Thus, a diagnosis of primary AIMF may be suggested at this time and is the most appropriate choice of those listed here, after careful consideration of the bone marrow findings in the context of clinical information and disease course. Alternate causes of myelofibrosis (including, for example, exposure to drugs, infectious causes, myeloproliferative neoplasm (MPN) or post-MPN myelofibrosis) must be ruled out. Answer A is incorrect because in this case, the patient described has no significant medical history nor evidence of neoplastic progression, making post-MPN myelofibrosis unlikely. Answers C and D are incorrect because the vignette describes a characteristic combination of morphologic and genetic findings that would be seen with AIMF, including the absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL), which can be an essential part of the diagnostic work up.
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
- 0.5% of all white blood cells (least numerous granulated cells in peripheral blood)
- Named because it stains with basic dyes
- Similar to mast cells but apparently generated by different CD34+ precursor cells in bone marrow (basophils mature in marrow, mast cells in connective tissue)
- Progresses from the multipotent myeloid stem cell via a committed progenitor (basophil colony forming unit, CFU-Baso) to basophilic promyelocyte, to basophilic myelocyte, to basophilic metamyelocyte, to basophil
- Leaves bone marrow as terminally differentiated granulocyte
- Basophils and mast cells are effector cells in allergen / IgE mediated immune responses
- They induce type 1 immediate immune response in airways and elsewhere, causing bronchial asthma and other allergic diseases (Allergol Int 2006;55:105)
- Also play a critical role in host defense against helminths (Allergol Int 2006;55:99)
- Factors thought to play key role in basophil production: IL3 (main cytokine), GM-CSF, IL4, IL5 (primary cytokine linked to specific eosinophil and basophil production), stem cell factor (SCF)
- Basophils and their granules contain histamine, sulphated mucopolysaccharides (mostly chondroitin sulfate), peroxidase, low levels of chymase, negligible amount of tryptase, Charcot-Leyden crystal protein, PAF and ECF-A
- Basophil activation test, using CD203c or CD63 as an activation marker, has become a reliable test for in vitro investigation of immediate allergy, complementing other in vitro tests (Clin Mol Allergy 2005;3:9)
- Significant basophilia is more likely to represent neoplasia (chronic myelogenous leukemia, other chronic myeloproliferative disorders, myelodysplastic syndromes, acute leukemias) than reactive processes
- Nonneoplastic basophilia: allergic / hypersensitivity reactions, hypothyroidism, ulcerative colitis, rheumatoid arthritis, renal disease, rare infections (varicella, influenza), irradiation, rare carcinomas and rare drugs (estrogens, antithyroid agents)
- Decreased number of basophils (basophilopenia): certain medications (steroids, epinephrine, thyrotoxicosis therapy), acute stress, acute inflammation
- Basophilic granules are metachromatic (reddish purple) with Toluidine blue and Alcian blue; are also water soluble
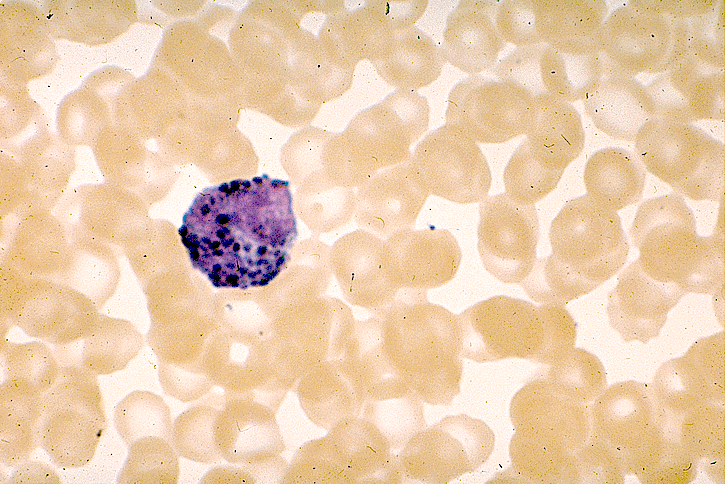
- Basophilic myeloblast: difficult to distinguish from other granulocyte blasts; large round cell with basophilic cytoplasm without granules; N/C ratio is 80%; dispersed chromatin with nucleolus
- Basophilic promyelocyte: intermediate in development between basophilic myeloblast and myelocyte; large round cell with a few undifferentiated cytoplasmic granules; slight chromatin clumping, nucleolus present
- Basophilic myelocyte: round / oval cell; cytoplasm with slight basophilia, moderate cytoplasmic purple black granules of varying size and shape; granules are usually larger than neutrophilic granules; N/C ratio is 50%; chromatin moderately condensed, no distinct nucleolus
- Basophilic metamyelocyte: oval cell with abundant pale cytoplasm, large and fairly uniform specific granules; N/C ratio is 40%; nucleus is small and indented with condensed chromatin, no nucleolus
- Basophil: smaller than other white blood cells (10 - 15 microns); cytoplasm is homogenous pale blue but often obscured by purple blue granules (containing heparin and histamine); N/C is 20%; nucleus is often unsegmented or bilobed, chromatin is coarse
Images hosted on other servers:
Basophilic myelocyte
Basophilic metamyelocyte
Images hosted on other servers:
Basophil
- Commonly used: CD9, CD25, CD38
- Also CD11a, CD11b, CD11c, CD13, CD15u, CD17, CD18, CD26, CD31, CD32, CD33, CD35, CD38, CD43, CD44, CD45, CD46, CD47, CD49d, CD50, CD55, CD58, CD59, CD63, CD68, CD71 (dim by flow cytometry), CD85a, CD85h, CD87, CD88, CD99, CD102, CD116, CD121b, CD123, CD125, CD126, CDw128a (CD181), CD203c, HLA-DR (immature basophils, Allergy 2006;61:1063), histidine decarboxylase, 2D7 (J Clin Pathol 2006;59:396) and basogranulin (Am J Clin Pathol 2006;125:273)
- Allergic subjects: CD32, CD122, CD124, CD130 and CD154 (J Allergy Clin Immunol 2000;106:1190)
- Variable: CD14, CD15, myeloperoxidase (usually negative) and a small proportion are chloroacetate esterase - positive
- Cytochemistry: PAS - positive (not the granules), Sudan black - positive and phosphatase (acid and alkaline) - negative
- Granules ~20nm in diameter with a particulate substructure
Images hosted on other servers:
Basophil
- Trephine:
- A surgical instrument having circular, saw-like edges used to cut out disks of bone, usually from the skull
- Trephine biopsy:
- Biopsy of a portion of bone containing marrow
- Jamshidi type biopsy needle:
- Recommended, usually 11 gauge
- Other needles:
- Single use needle (Biomed Instrum Technol 2005;39:391) for neonates (Br J Haematol 1999;107:458)
- Recommended to use separate needles for trephine biopsy and aspiration (J Clin Pathol 2007;60:212)
- Can use same skin incision but sites a few millimeters apart
- Bilateral biopsies useful if disease is likely focal (lymphoma or metastatic tumors, Cancer 2002;94:1522)
- Ultrasound decalcification may allow more successful FISH, PCR and RT-PCR (Am J Surg Pathol 2006;30:892)
- Note: FISH can be performed on tissue imprints, cytopreps or bone marrow aspirate smears (J Clin Pathol 2005;58:629)
- Note: trephine biopsy may also reveal bone disorders not the reason for the biopsy
- Posterior superior iliac spine
- Alternative site may be necessary in obese patients
Images hosted on other servers:
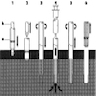
Schematics
- To evaluate:
- Leukemia
- Lymphoma and lymphoproliferative disorders
- Myeloproliferative or myelodysplastic disorders
- Metastatic disease
- Aplastic anemia and other hematologic conditions
- Infectious and metabolic disorders
- Also to evaluate postchemotherapy cellularity and post bone marrow transplant engraftment
- Should be accompanied by aspirated marrow smears and particle crush preparations and by touch imprints from trephine biopsy
- Adverse events in 0.8% (J Clin Pathol 2005;58:406)
- Major complication is hemorrhage / hematoma (apply pressure bandage to biopsy site to prevent; obtain coagulation consultation if patient has bleeding disorder or is on anticoagulants)
- Risk factors for hemorrhage are myeloproliferative disorder, aspirin, other putative platelet dysfunction and thrombocytopenia
- Make imprints from trephine biopsy by gently touching glass slide to specimen
- Possibly freeze part of trephine biopsy for molecular studies (J Clin Pathol 2006;59:1111)
- Fix tissue in formalin, B5 or Zenker
- Decalcify for 45 - 60 minutes
- Embed in paraffin
- Section at 3 - 4 micron intervals, saving tissue for possible special stains and molecular studies
- Some laboratories prefer plastic embedding, which may provide superior cytologic detail (J Clin Pathol 2005;58:897)
- Place some aspirate in EDTA, make smears at bedside with remainder
- Make smears from buffy coat (nucleated cell layer) and particles
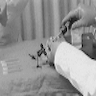
- Position patient properly (MedlinePlus: Bone Marrow Aspiration)
- Prepare and sterilize the site with iodine solution
- Anesthetize skin and bone with lidocaine
- Nick the skin with a blade to facilitate needle insertion
- Insert bone marrow aspiration needle
- Aspiration needle in place with trochar (white) partially withdrawn
- Syringe for aspirating bone marrow is in place
- Part of aspirate is put into EDTA tube to prevent clotting
- Part of aspirate is put on slide to pick particles
- Aspirate smear is made using another slide
- Biopsy needle is inserted at same site
- Feel the give of the needle as it enters the cortex
- Withdraw the trochar from the needle
- After another centimeter push, rotate the needle to cut the end of the specimen
- Withdraw the needle with the specimen inside the needle
- Push the biopsy specimen from the narrow end to the hub end with the trochar
- Place the biopsy specimen in fixative (such as Zenker) for decalcification and processing
Images hosted on other servers:
Blood clot
- Bone marrow biopsies are helpful to determine cellularity and presence of fibrosis
- Purple granular deposits that impair evaluation of touch preparations are due to cartilage in biopsy and are more common in children (J Clin Pathol 2003;56:883)
Images hosted on other servers:


Poor technique

Smear: good and poor technique

Epoxy resin embedded bone marrow biopsies
Images hosted on other servers:
FISH from tissue imprints
- Indications:
- Aplastic anemia, osteopetrosis or other primary / congenital bone marrow disease
- Post high dose chemotherapy for malignancy
- Posttransplant if blood counts do not recover as expected
- Autologous transplantation:
- Graft is patient's own marrow, often after monoclonal antibodies to tumor or cell sorting regimen
- Allogeneic transplantation:
- Graft is from another individual after recipient myeloablative preparatory regimen of high dose chemotherapy, total body radiation or monoclonal antibodies
- Nonmyeloablative allogeneic stem cell transplantation:
- In elderly or those with relatively indolent disease
- Myeloablative steps are reduced or eliminated as curative potential is largely due to graft versus tumor effect
- Similar outcome as traditional approach in patients > age 50 years (Blood 2005;105:1810)
- Examination of bone marrow morphology recommended posttransplant in additional to traditional molecular studies (Arch Pathol Lab Med 2006;130:1479)
- Peripheral blood transplant:
- Uses CD34+ stem cells
- Preparation:
- Chemotherapy, total body irradiation to:
- Immunosuppress patient to prevent rejection
- Eradicate tumor cells (antitumor antibodies also used for this purpose)
- Chemotherapy, total body irradiation to:
- Infection, graft rejection, graft versus host disease, recurrence of malignancy
- Infection:
- Due to immunosuppression
- Less common due to antibiotics, growth factors
- Graft rejection:
- Rare with matched siblings
- Common with unrelated donors
- Characterized by decreasing marrow cellularity and progressive cytopenia
- Decrease in a myeloid cell line may predict impending rejection or be due to drugs or viruses
- Erythroid hypoplasia may be due to parvovirus B19 infection in immunocompromised patients
- Dyserythropoiesis and dysgranulopoiesis may reflect toxic effect of immunosuppressive drugs or antibiotics
- Maturation arrest of granulocytes may occur due to various drugs
- Granulocyte growth factors cause hyperplasia of immature forms and leukemoid peripheral blood reaction with Döhle bodies, abnormally segmented neutrophils and atypical granulation
- Graft versus host disease:
- Associated with increased lymphocytes, plasma cells and eosinophils
- Successful engraftment:
- 0 - 1 week:
- Usually not biopsied
- Marked hypocellularity, hemorrhage, proteinaceous debris, scattered fat cells and macrophages
- 1 - 2 weeks:
- Adipose tissue present
- 2 - 3 weeks:
- Scattered islands of hematopoietic cells
- Often erythroid precursors initially, then promyelocytes and myelocytes
- 5 - 10 weeks:
- Increasing erythroid precursors, granulocytes and megakaryocytes
- Megakaryocyte reconstitution may lag behind other cell lines
- 0 - 1 week:
AFIP images

Promyelocytes and myelocytes in upper field

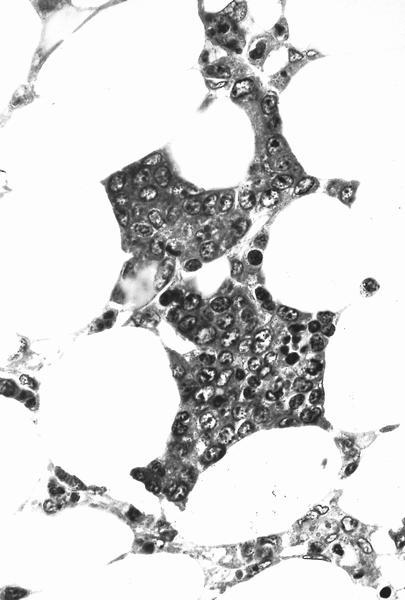
Foci of erythropoiesis
- Rare autosomal recessive immunodeficiency disorder characterized by abnormal intracellular protein transport
- Gene was characterized in 1996 as the LYST or CHS1 gene on 1q42-43 (OMIM: Lysosomal Trafficking Regulator; LYST)
- Mutations cause megagranules in promyelocytes and myeloblasts, which persist in mature forms and are associated with neutropenia and recurrent pyogenic infections
- Patients also have partial albinism (decreased hair and eye pigmentation), photophobia, nystagmus
- Scalp hair analysis may also be useful for diagnosis (Clinics (Sao Paulo) 2006;61:327)
- Bone marrow transplantation for childhood onset cases is curative; disease is otherwise fatal
- Giant inclusion bodies in leukocyte precursor cells (bodies contain lysosomal enzymes)
- Also hemophagocytosis (Hematology Am Soc Hematol Educ Program 2005;1:82)
- Peripheral smear: giant granules in neutrophils, eosinophils and granulocytes
Images hosted on other servers:

Giant inclusion bodies
- Granules: myeloperoxidase
- Suppresses hematopoiesis (Blood 1990;75:1965)
- Monocyte and granulocyte precursors are major site for latent CMV (Intervirology 1999;42:308)
- Rarely CMV can induce clonal T cell proliferations
- Clinical and hematologic features show considerable overlap with EBV infection
- Intrauterine infection is associated with thrombocytopenia and hemolytic anemia in neonates
- CMV pp65 antigenemia assay is more specific in immunocompromised patients than antibody titers
- 57 year old woman with T gamma gene rearrangement and CMV mononucleosis (Am J Hematol 2001;66:64)
- Generally nonspecific findings in bone marrow including myeloid and megakaryocytic suppression
- Infected cells with intranuclear inclusions (not identifiable in many cases); may be highlighted via IHC or ISH
- Nuclear inclusions: single, round or oval, deep blue or amphophilic, surrounding halo, margination of nuclear chromatin
- Cytoplasmic inclusions: multiple, irregular in shape, basophilic
- Occasional hemophagocytosis (Hum Pathol 1998;29:1074), granulomas (Postgrad Med J 1987;63:277), lymphoid aggregates or hematogones (Leuk Res 2003;27:193)
- Atypical lymphocytosis in peripheral blood (mostly increased transformed large granular lymphocytes and NK cells, infrequent infectious mononucleosis-like classic Downey type 2 cells)
- May have circulating CMV infected endothelial cells at feathered edge of peripheral blood smear in immunosuppressed individuals
Images hosted on other servers:
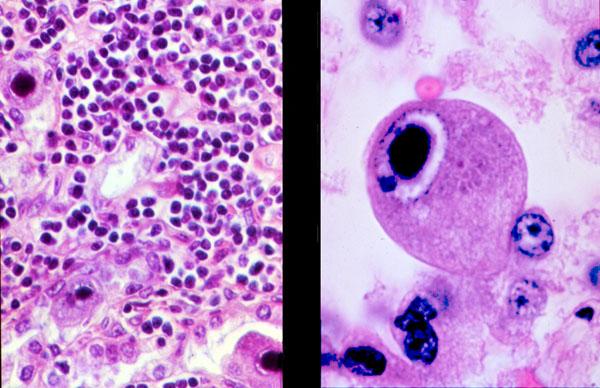
Site unknown (not bone marrow)
- Virus particles with electron dense icosahedral core containing capsomeres with a large pleomorphic lipid envelope
- Particles composed of filamentous or amorphous material detached from nuclear membrane
- Pre B ALL
- T cell lymphoma
- Rare cause of sideroblastic anemia and neutropenia (Am J Hematol 2007;82:625)
- Often due to excess zinc ingestion (CMAJ 2003;169:129, Am J Clin Pathol 2005;123:125)
- Also due to zinc inborn errors of metabolism (J Pediatr Hematol Oncol 2005;27:477, Arch Neurol 2003;60:1303) and total parenteral nutrition (Rinsho Ketsueki 1993;34:171)
- Oral copper gluconate
- Resembles myelodysplasia or sideroblastic anemia
- Presence of cytoplasmic vacuoles is suggestive (Ann Clin Biochem 2005;42:227, Am J Clin Pathol 1992;97:665)
Images hosted on other servers:
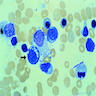
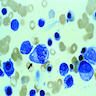
Extensive vacuolation of erythroid precursors
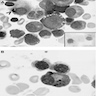
Vacuoles in erythroid precursors

Vacuoles in immature granulocytes

Vacuoles in myelocyte and basophilic normoblast

Cytoplasmic vacuoles in two pronormoblasts


Sideroblasts (Prussian blue)

Pelger-Huët nuclear anomaly
Images hosted on other servers:

Dimorphic RBCs

More pronounced
anisopoikilocytosis
- A member of the inherited bone marrow failure syndromes (BMFS); typically shows an onset in the first year of life (Br J Haematol 2016;172:782)
- Main features include normochromic macrocytic anemia, reticulocytopenia and nearly absent erythroid progenitors in the bone marrow (OMIM: Diamond-Blackfan Anemia 1)
- Predisposition to acute myelogenous leukemia, myelodysplastic syndrome and solid tumors
- Also known as Blackfan-Diamond anemia, congenital hypoplastic anemia and inherited erythroblastopenia (Eur J Hum Genet 2013 Oct;21(10))
- Also known as Blackfan-Diamond anemia, congenital hypoplastic anemia and inherited erythroblastopenia
- A member of the inherited bone marrow failure syndromes (BMFS); typically shows an onset in the first year of life (Br J Haematol 2016;172:782)
- Underlying defect hypothesized to be faulty ribosome biogenesis, resulting in proapoptotic erythropoiesis and erythroid failure (Hematol Oncol Clin North Am 2009;23:261)
- Growth retardation and various congenital abnormalities commonly seen
- Normochromic macrocytic anemia, reticulocytopenia, normocellular for age marrow and nearly absent erythroid progenitors in the bone marrow (OMIM: Diamond-Blackfan Anemia 1)
- Predisposition to acute myelogenous leukemia, myelodysplastic syndrome and solid tumors
- Incidence 1:100,000 ~ 1:200,000 live births and remains consistent across ethnicities (GeneReviews: Diamond-Blackfan Anemia)
- Males and females equally affected
- ~90% diagnosed within first year of life and 35% diagnosed within first month
- ~55% are sporadic cases; ~45% familial with disease inherited mostly in an autosomal dominant pattern
- Incomplete penetrance (GeneReviews: Diamond-Blackfan Anemia)
- Underlying defect hypothesized to be faulty ribosome biogenesis, resulting in proapoptotic erythropoiesis and erythroid failure (Hematol Oncol Clin North Am 2009;23:261)
- Heterozygous mutations in ribosomal proteins (RPs) encoding genes, both small and large ribosomal subunit, have been identified in 60% (Br J Haematol 2016;172:782)
- Mutations identified in 15 RP genes: RPS7, RPS10, RPS17, RPS19, RPS24, RPS26, RPS27, RPS29, RPL5, RPL11, RPL15, RPL26, RPL27, RPL31, RPL35A (Br J Haematol 2016;172:782)
Images hosted on other servers:
Hypothetic model for abortive
ribosome assembly and p53
activation relevant to DBA
- Pallor, weakness, failure to thrive (GeneReviews: Diamond-Blackfan Anemia)
- Growth retardation and various congenital abnormalities seen in 30% - 50%, in particular craniofacial, upper limb, heart and genitourinary malformations
- Higher risk for developing certain malignancies including acute myelogenous leukemia, myelodysplastic syndrome and solid tumors
-
According to 6th Annual Diamond-Blackfan anemia (DBA) International Consensus Conference (Br J Haematol 2008;142:859)
- Classic Diamond-Blackfan anemia is diagnosed if all of the following diagnostic criteria are met:
- Age < 1 year
- Macrocytic anemia with no other significant cytopenias
- Reticulocytopenia
- Normal marrow cellularity with a paucity of erythroid precursors
- A probable diagnosis is made based on the following major and minor supporting criteria, if all of the above diagnostic criteria cannot be met :
- Major supporting criteria:
- The presence of a gene mutation associated with DBA
- Positive family history
- Minor supporting criteria
- Elevated eADA activity
- Congenital anomalies associated with DBA
- Elevated Hemoglobin F
- No evidence of another inherited bone marrow failure syndrome
- A probable diagnosis of Diamond-Blackfan anemia can be made in the following settings:
- Three diagnostic criteria and a positive family history
- Two diagnostic criteria and three minor criteria
- A positive family history and three minor criteria
- Major supporting criteria:
- Nonclassical DBA is diagnosed when there is a DBA associated gene mutation but insufficient diagnostic criteria
- Macrocytic anemia with no other significant cytopenias (GeneReviews: Diamond-Blackfan Anemia)
- Increased red cell mean corpuscular volume (MCV)
- Reticulocytopenia
- Elevated erythrocyte adenosine deaminase activity (eADA) (observed in 80% - 85%)
- Elevated hemoglobin F (HbF) concentration
-
Associated with unfavorable prognosis (Pediatr Res 1999;46:553)
- Young age at presentation
- No family history
- Premature birth
- Hydrops fetalis with de novo mutation in RPS19 gene (Am J Hematol 2013;88:160)
- Infant female with multiple congenital midline defects (J Pediatr Hematol Oncol 2007;29:338)
- 3 month old boy with coexisting Johanson-Blizzard syndrome (J Coll Physicians Surg Pak 2010;20:627)
- 7 year old boy with subsequent Hodgkin lymphoma (J Pediatr Hematol Oncol 2006;28:234)
- 10 year old girl with fatal agranulocytosis after deferiprone therapy (Blood 2007;109:5157)
- 16 year old Japanese boy with colon tumors after hematopoietic stem cell transplantation (Int J Clin Exp Pathol 2015;8:5938)
- 17 year old girl with multihormonal hypopituitarism, hypothyroidism and hypoparathyroidism (Pediatr Endocrinol Diabetes Metab 2013;19:111)
- Child with isolated cleft palate (Cleft Palate Craniofac J 2010 Nov 19 [Epub ahead of print])
- Japanese patient with a novel mutation of ribosomal protein S10 gene (J Pediatr Hematol Oncol 2012;34:293)
- Corticosteroids and red cell transfusions: mainstays of therapy
- Hematopoietic cell transplantation: for steroid refractory patients (Hematol Oncol Clin North Am 2009;23:261)
- Other therapies: androgens, IL3, metoclopramide and immunosuppressive agents (Blood 2007;109:2266)
- Bone marrow examination usually shows normocellular for age, complete loss of erythroid elements or only scattered isolated erythroblasts
- Increased hematogones, especially in infants and young children
- Granulocytic and megakaryocytic lineages are preserved; mild eosinophilia is often present
- Peripheral blood examination usually shows normochromic, macrocytic red blood cells, mild anisopoikilocytosis and the absence of polychromasia (Proytcheva: Diagnostic Pediatric Hematopathology, 1st Edition, 2011)
- Most common genetic causes of Diamond-Blackfan anemia (GeneReviews: Diamond-Blackfan Anemia)
Gene % of DBA Attributed to
Pathogenic Variants in This GeneRPL5 ~6.6% RPL11 ~4.8% RPL35A ~3% RPS10 ~2.6% RPS17 ~1% RPS19 ~25% RPS24 ~2% RPS26 ~6.4%
- Acquired conditions with bone marrow failure: infections (Parvovirus B19 infection, HIV, viral hepatitis, mononucleosis and HTLV1), drugs and toxins, immune mediated diseases (thymoma, myasthenia gravis, systemic lupus erythematosus and multiple endocrinopathies)
- Other genetic conditions with bone marrow failure: Fanconi anemia, Shwachman-Diamond syndrome, Pearson syndrome, dyskeratosis congenita, cartilage hair hypoplasia
- Transient erythroblastopenia of childhood (TEC): acquired anemia caused by decreased production of red blood cell precursors in a previously healthy child; self resolving; 80% of children are age one year or older at diagnosis; normocytic anemia
- Also called Zinsser-Engman-Cole syndrome
- Rare; 90% males; most patients are diagnosed as adults
- X linked recessive inheritance most common; autosomal dominant and recessive forms as well as sporadic occurrences also recognized
- X linked cases are linked to mutations in DKC1 gene encoding dyskerin, a small nucleolar ribonucleoprotein particle, important in ribosomal RNA processing and part of telomerase complex (Int J Hematol 2005;82:184)
- Autosomal dominant cases are linked to mutations in telomerase RNA gene (TERC) that impair telomerase function, leading to critically short telomeres
- Severe variant: Hoyeraal-Hreidarsson syndrome, a multisystem disorder with aplastic anemia, immunodeficiency, microcephaly, cerebellar hypoplasia and growth retardation
Images hosted on other servers:

Defects in cellular proliferation
and differentiation that give rise
to dyskeratosis congenita
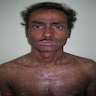
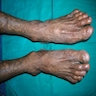
- Clinical triad: cutaneous hyperpigmentation, dystrophic nails, mucosal leukoplakia (may become malignant - squamous cell carcinoma)
- Causes progressive and ultimately fatal loss of hematopoietic renewal (pancytopenia)
- Other common physical findings: ocular abnormalities, dental caries, esophageal webs, genitourinary defects, restrictive pulmonary disorder
- Rarely associated with usual interstitial pneumonia causing death from respiratory failure (Mayo Clin Proc 2005;80:817)
- Although probably an inherited DNA repair defect disorder (similar to Fanconi anemia), no obviously increased sensitivity to mitomycin C and diepoxybutane
- Combined flow cytometry and FISH for very short telomeres in peripheral blood lymphocytes may confirm the diagnosis
- Infant with severe variant of X linked disease (Hoyeraal-Hreidarsson syndrome) and enterocolitis (J Pediatr Gastroenterol Nutr 2009;49:359)
- 38 year old man with dyskeratosis congenita with malignant transformation (BMJ Case Rep 2011 Jan 11;2011)
- Hematologic support with blood products for various cytopenias
- Androgens, G-CSF, erythropoietin, bone marrow transplant, hematopoietic stem cell transplant
- Over two thirds of patients die of bone marrow failure at an early age (20 - 25 years)
- Longer survival associated with increased risk of carcinomas and myelodysplasia (10 - 15%)
Images hosted on other servers:
Reticular skin pigmentation
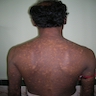
Reticular pigmentation
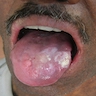
Dorsum of tongue

Ulceroproliferative growth

Clinical data of the DKC proband in the first pedigree
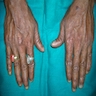
Plantar surface of hands
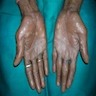
Palmar surface of hands
Hyperpigmentation and dystrophic nails

Epiphora and ectropion
- Bone marrow morphology very similar to Fanconi anemia (initially normo / hypercellular with megaloblastic changes, eventual aplasia in > 50%, possibly late AML)
Images hosted on other servers:

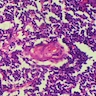
Histopathological features of dyskeratosis congenita
Lymph node: metastatic squamous cell cac
Dorsum of tongue: squamous cell carcinoma
Dorsum of tongue:
richly vascular
with mild chronic
inflammatory infiltrate
- X linked form: DKC1 gene on chromosome Xq28 encodes dyskerin thought to be involved in ribosomal assembly but also associated with the RNA template for telomerase (TERC) and possibly involved in telomere maintenance
- Autosomal dominant form: TERC gene (also known as DKC2) demonstrates "disease anticipation" (earlier onset in successive generations)
- Autosomal recessive form: unknown genetic association
- Normal chromosome breakage studies
- Foucar: Bone Marrow Pathology, 2nd edition, 2001, Foucar: Bone Marrow Pathology, 3rd edition, 2011, Foucar: Non-Neoplastic Disorders of Bone Marrow (Atlas of Non-Tumor Pathology), 1st edition, 2009, Penchansky: Pediatric Bone Marrow, 2004, eMedicine: Dyskeratosis Congenita, OMIM: Dyskeratosis Congenita, X-linked; DKCX, OMIM: Dyskeratosis Congenita, Autosomal Dominant 1; DKCA1, OMIM: Dyskeratosis Congenita, Autosomal Recessive 1; DKCB1
- In embryo, hematopoiesis (other than lymphoid) occurs in yolk sac with formation of mesenchymal derived primitive erythroblasts
- Aorta also contributes lymphomyeloid stem cells, as do embryonic liver and bone marrow (Development 2002;129:4147, Ann N Y Acad Sci 2005;1044:41)
- At weeks 10 to 24, liver is primary hematopoietic organ with production of granulocytes and megakaryocytes in sinusoids
- At months 4 - 5, bone marrow hematopoiesis begins
- By birth, liver and spleen have minimal role in myelopoiesis and bone marrow is major site of hematopoiesis
Images hosted on other servers:
Blood islands in embryo
Images hosted on other servers:
Yolk sac at 18 days
Fetal liver at 17 weeks
Fetal bone marrow at 19 weeks
- Named because granules stain deeply with eosin
- 1 - 4% of all white blood cells (the number of circulating eosinophils is generally low, < 800 / µL)
- Has a role in response to parasitic infections and allergic conditions
- Derived from a common progenitor, CFU-GM, together with granulocytes and monocytes / macrophages
- Progresses from myeloid stem cell to eosinophilic promyelocyte, to eosinophilic myelocyte, to eosinophilic metamyelocyte, then to eosinophil
- IL5 is growth factor that selectively induces eosinophil formation; also induces enhanced eosinophil function and prolonged survival via inhibition of apoptosis; other factors include IL1, IL3
- IL9 enhances IL5 receptor expression
- Eotaxin: eosinophil stimulating chemokine locally produced in tissue (macrophages, eosinophils) but also manifesting systemic effects in bone marrow
- Other basophil and mast cell derived chemoattractants are ECF-A, PAF and LTB4
- Degranulation is strictly controlled, which allows it to differentially release its contents in an ordered manner, which prevents tissue injury during migration (Semin Respir Crit Care Med 2006;27:117)
- Produces IL2, IL3, IL4, IL5, IL7, IL13, IL16, TNF-α, TGF-β, RANTES, eosinophil cationic protein, eosinophil peroxidase (different from myeloperoxidase), eosinophil derived neurotoxin, MBP and Charcot-Leyden crystal lysophospholipase
- Two main functions: modulation of immediate hypersensitivity reactions initiated by basophil / mast cell degranulation and destruction of parasites; key role played by release of eosinophil secondary granule contents
- Eosinophilic promyelocyte: intermediate in development between a myeloblast and myelocyte; 15 microns in diameter with large nucleus and nucleolus; contains a few undifferentiated (primary, coreless) cytoplasmic granules in intensely basophilic cytoplasm
- Eosinophilic myelocyte: round / oval large cells with moderate cytoplasm containing prominent primary purple granules and secondary red orange, refractile granules of similar size; N/C ratio is 50% with moderately condensed chromatin and indistinct nucleolus
- Eosinophilic metamyelocyte: round / oval cells with abundant cytoplasm containing large blue orange granules; N/C ratio is 40%; nucleus is indented with moderately condensed chromatin and no nucleolus
- Eosinophil: 9 - 15 microns with coarsely granular cytoplasm containing refractile orange granules grouped around a single horseshoe shaped nucleus with 2 - 3 lobes (1 lobe - 6%, 2 - 68%, 3 - 22%, 4 - 4%)
Images hosted on other servers:
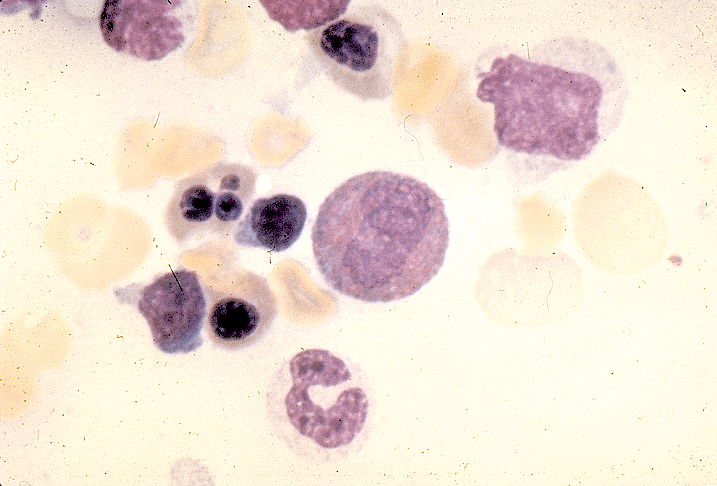
Eosinophilic metamyelocyte

Developing eosinophils
AML-M4eo:
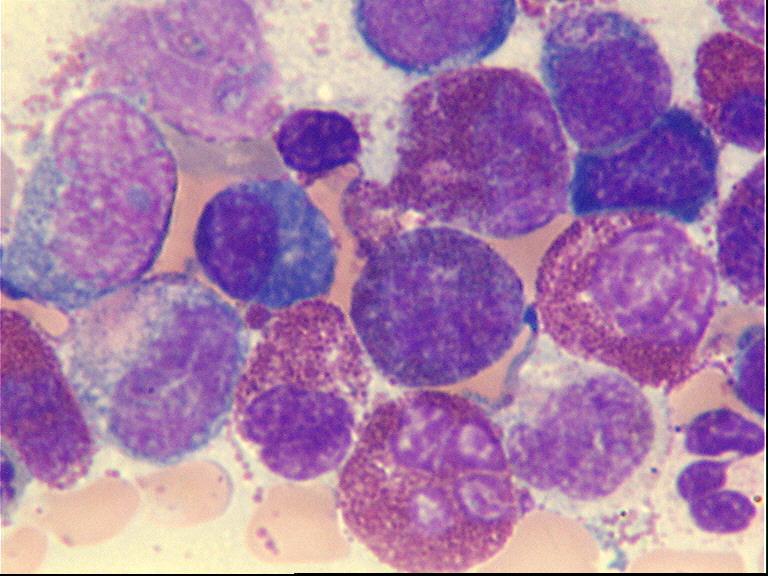
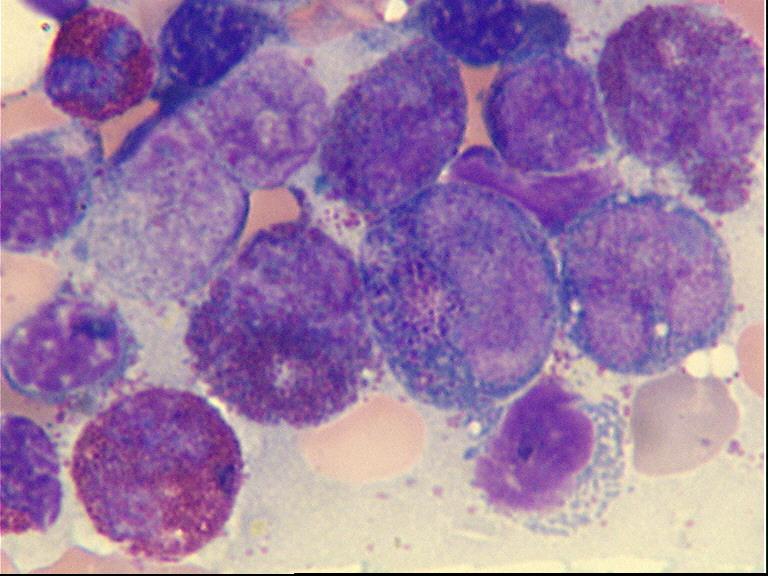


Various eosinophil precursors
Hypereosinophilic syndrome:
Bone marrow aspirate
Bone marrow biopsy
Images hosted on other servers:
Hypereosinophilic syndrome:
Peripheral blood
Peripheral blood:

Eosinophils
- CD9; also CD15, CD16, CD23, CD32, CD35, CD47R (weak), CD49d, CD50, CD52, CD62L, CD69, CD85a, CD85d, CD88, CD89, CD116, CDw125, CD183, myeloperoxidase, Sudan black and PNL2
- Cytochemistry: the naphthol AS-D chloroacetate esterase reaction is normally negative but characteristically faintly positive in the abnormal eosinophils of AML-M4eo
- CD114, tryptase
- Two types of granules: a few rounded homogeneously electron dense granules and many rounded, elongated or oval crystalloid containing ones
Images hosted on other servers:
Normal eosinophil with crystalloid granules
- Erythropoiesis takes place within the bone marrow in units composed of macrophages surrounded by erythroblasts; either erythroid burst forming units (BFU-E, immature) or erythroid colony forming units (CFU-E, more mature)
- Progresses from myeloid stem cell to pronormoblast, to basophilic normoblast, to polychromatophilic normoblast, to orthochromatic normoblast (then extrusion of nucleus), to reticulocyte (young erythrocyte) and then to erythrocyte (red blood cell)
- Characterized by increasing hemoglobin synthesis, decreasing cell size, decreasing cytoplasmic basophilia and progressive chromatin condensation with extrusion of nucleus at the orthochromatic stage of development
- Extruded nuclei bind on macrophage receptors and are subsequently phagocytized; loss of mitotic capability occurs after the early stage of polychromatophilic normoblast
- Average of 4 cell divisions during maturation, so a proerythroblast can give rise to 16 red cells
- Average time in the bone marrow from proerythroblast to reticulocyte is 7 days; average time for reticulocytes to mature into red cells is 1 - 2 days
- Early erythroid precursors cluster in islands randomly distributed throughout marrow but related to vascular structures
- Erythroblast (pronormoblast) islands may be specialized niches where intercellular interconnections and cytokines regulate erythropoiesis (Curr Opin Hematol 2006;13:137)
- Erythropoietin (EPO): lineage specific cytokine (glycoprotein) responsible for erythropoiesis (proliferation, differentiation and survival by delaying apoptosis), produced by renal cells in response to hypoxia and by the bone marrow
- EPO receptor present in highest concentrations on erythroid progenitor cells and proerythroblasts but absent on mature erythrocytes
- Thrombopoietin (TPO): may also have a role in erythroid maturation
- Heme protein and tumor necrosis factor (TNF) exert negative feedback on the production of red blood cells
- Steady state: 2 - 4 X 109 erythrocytes/kg/day; 40,000 - 80,000 reticulocytes/µL/day
- In utero: hypoxic environment → high EPO → high erythroid production; after birth: oxygen → drop in EPO → dramatic drop in the number of erythroid elements in the bone marrow in the first few months of life (as low as < 5%)
- After the initial postnatal period: marked decrease in hematocrit → hypoxia → EPO release by kidney → increase in erythroid elements to the normal 20 - 25% of nucleated cells in bone marrow (sustained hypoxia → up to 5 - 7 times increase in number)
- Sideroblast: normoblast containing small iron granules detectable with Prussian blue (approximately 20% of normoblasts)
- Erythropoiesis stimulators: anemia / hypoxia, thyroid hormones, androgens, growth hormone and corticosteroids
Images hosted on other servers:

Erythropoiesis
- Pronormoblast: 13 - 18 microns, round / ovoid with thin rim of basophilic cytoplasm, large spherical nucleus with fine chromatin and 1 - 2 nucleoli; usually perinuclear halo; N/C ratio is 90%
- Basophilic normoblast: 12 - 17 microns; increase in deeply basophilic cytoplasm compared to pronormoblast and slightly smaller nucleus with slight chromatin condensation; often perinuclear halo; no granules, no nucleolus; N/C ratio is 75 - 85%
- Polychromatophilic normoblast: 12 - 15 microns; round / ovoid with abundant, dull gray to gray green, variegated cytoplasm due to polyribosomes (basophilic) and hemoglobin (eosinophilic); round, condensed and basophilic nucleus has coarse granules that give it a cartwheel appearance; perinuclear halo present; no nucleolus; N/C ratio is 60 - 80%
- Orthochromatophilic normoblast: 8 - 12 microns; round / ovoid cells with pink orange uniformly staining cytoplasm, dark and opaque nucleus that may be pyknotic or in the process of being extruded, no nucleolus
- Reticulocyte: 7 - 10 microns; cannot identify without supravital stain (new methylene blue, brilliant cresyl blue) that colors RNA deep blue and granular; must have at least 2 granules to classify as reticulocyte; cytoplasm is red to pale blue due to RNA, no nucleus is present; larger than mature erythrocyte and lacks central pallor, passes through the bone marrow sinus wall and endothelial cells into the circulating blood where it matures
- Erythrocyte: 7 - 8 microns; round / ovoid biconcave disc with orange red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation
AFIP images
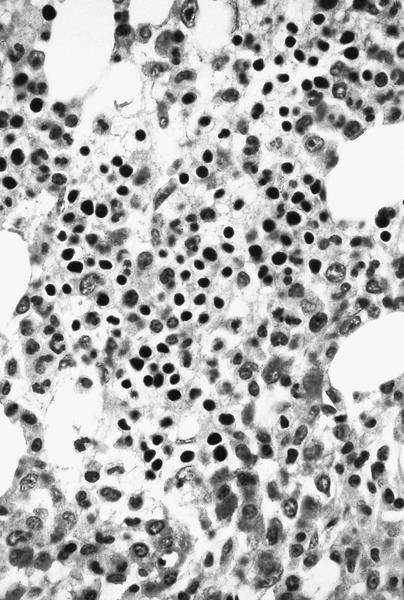
Erythroblast island
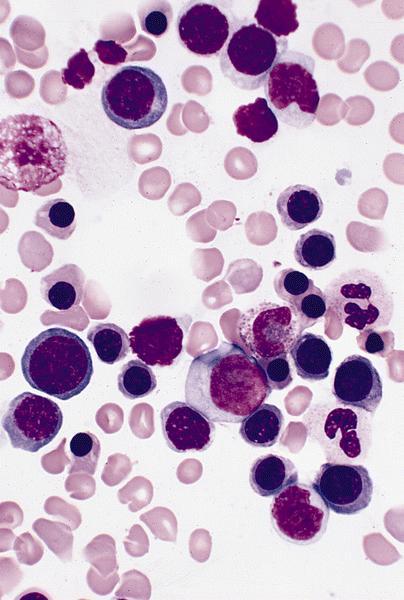
Proerythroblast is large cell at
upper left of center; there are
also basophilic, polychromatophilic
and orthochromatic normoblasts
Images hosted on other servers:
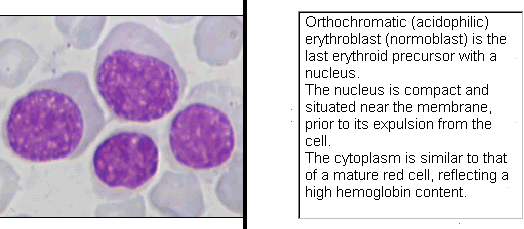
Orthochromatic normoblast

Proerythroblast
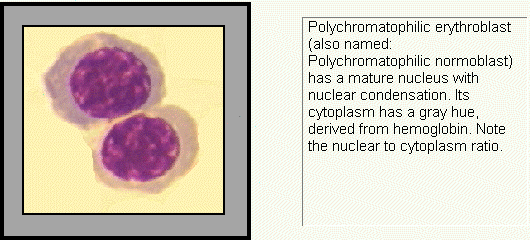
Polychromatophilic normoblast

Basophilic normoblast
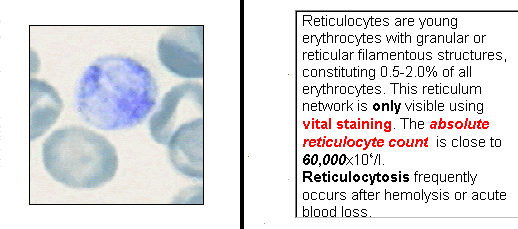
Reticulocyte
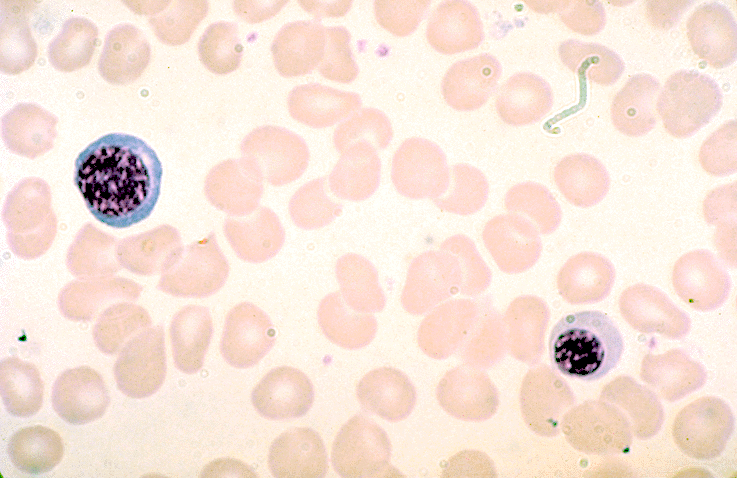
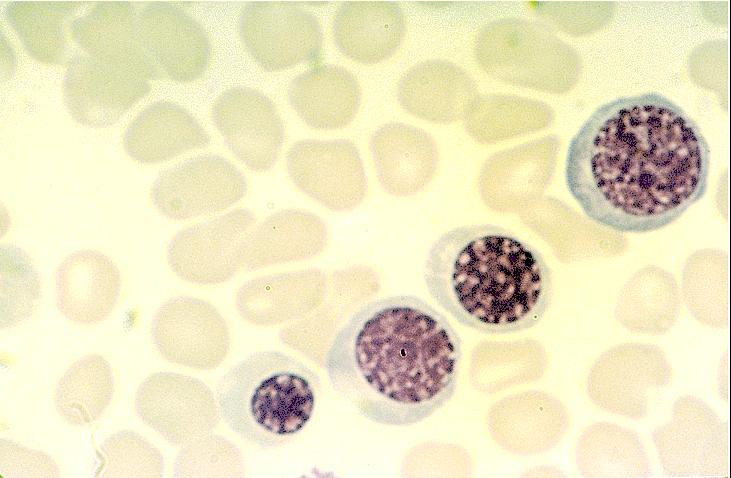
Basophilic and polychromatophilic normoblasts
- GLUT1, CD35, CD36 (early precursors), CD38, CD41, CD43, CD44, CD47, CD49d (erythrocyte precursors only), CD58, CD71 (precursors through reticulocytes), CD75, CD105 (erythrocyte precursors only), CD108, CD111, CD139 (weak), CD147, CD233, CD235a (glycophorin A), CD235b (glycophorin B), CD235ab (glycophorin A/B), CD236 (glycophorin C/D), CD236R (glycophorin C), CD238 (Kell), CD239 (Lutheran), CD240 CE (RhC & RhE), CD240 D (RhD), CD240 DCE (RhD/CE) and CD241 (Rh50, RHAG)
- Expression of both common leukocyte and transferrin receptor antigens progressively declines with maturation while glycophorin expression remains high
- Erythroblasts have characteristic surface invaginations developing into small intracytoplasmic vesicles (rhopheocytotic vesicles)
Images hosted on other servers:

Normal erythrocyte
Reticulocyte
- Originally described in 1927 by Guido Fanconi, who reported 3 brothers with pancytopenia and multiple physical abnormalities (eMedicine: Fanconi Anemia)
- Most common inherited bone marrow failure syndrome; incidence at birth of 1 per 100 - 350K live births (Wikipedia: Fanconi Anemia)
- Typically autosomal recessive with intellectual disability and multiple bone, skin and renal abnormalities
- Associated with MDS / AML (15 - 20% of patients), hepatocellular adenoma
- Presence of significant birth defects correlates with early onset hematologic disease / anemia
- Note: Fanconi syndrome is a generalized dysfunction of proximal renal tubule transport with no primary glomerular involvement
Images hosted on other servers:
Diagnostic algorithm
- Usually diagnosed in infancy; 10% diagnosed at age 16 years or older; rarely diagnosed in 40's or 50's
- Prenatal diagnosis is possible by chromosome breakage analysis of chorionic villus, amniocentesis or cord blood lymphocyte samples
- Highly variable clinical features with occasional patients that are phenotypically normal (up to 30% according to some studies)
- Progressive lethal anemia associated with brown skin pigmentation, hypoplastic bone marrow, renal hypoplasia, absent or hypoplastic thumbs or radii, short stature and other anomalies; some patients have milder phenotype
- Thrombocytopenia: often the first evidence of impending hematopoietic failure
- Variable pancytopenia, macrocytosis, increased fetal hemoglobin and I antigen expression on erythrocytes (evidence of "stressed" erythropoiesis)
- Stimulated peripheral lymphocyte cultures: characteristic pattern of combined G2 phase prolongation and arrested G2 phase, detectable by flow cytometry
- 4 year old boy diagnosed by genetic testing followed by prenatal diagnosis (Ann Lab Med 2012;32:380)
- 4 year old boy with Fanconi anemia and AML (Ann Clin Lab Sci 2011;41:66)
- 13 year old girl with Fanconi anemia and ALL (J Coll Physicians Surg Pak 2012;22:458)
- 50% improve with androgens; also G-CSF; stem cell transplantation
- Late development of myelodysplasia or AML in approximately 15 - 20% of patients
- Median survival is 7 years
Images hosted on other servers:
Clinodactyly with brachymesophalangia
- Early bone marrow specimens may be normocellular with megaloblastic features but gradual aplasia develops with severe pancytopenia by mid to late childhood
- Late stages: decreased / absent megakaryocytes, marked hypocellularity, rare small foci of erythropoiesis / myelopoiesis, fatty replacement
- Rare patients present directly with AML
Images hosted on other servers:
Hypocellular spicule with fatty infiltration
Aspirate smear findings
- At the molecular level, 8 different genetic types of FA complementary groups have been described
- Several genes involved: FAC (9q22.3), FAA (16q24.3), FAG (19p13)
- Chromosomal instability due to increased number of chromosomal breaks, gaps, rearrangements and endoreduplication (OMIM: Fanconi Anemia, Complementation Group A; FANCA)
- 12 mutated genes identified to date; proteins are involved in repair of DNA damage
- Unique cytogenetic finding is increased sensitivity to apoptosis inducing effects of mitomycin C and diepoxybutane
Images hosted on other servers:

Chromosome breakage test

Representative karyotypes
- Indicate hematologic and other diagnoses and indication for bone marrow
- List laboratory data (WBC, Hemoglobin, Hematocrit, MCV, Platelet count, RDW, other)
- Give microscopic description (number, size, shape, other abnormalities) of red blood cells, white blood cells and platelets
- Peripheral smear interpretation
- Biopsy or aspiration site
- Cellularity (% and whether hyper, normo or hypocellular for age)
- Presence or not of trilinear maturation
- Adequacy of specimen
- Indicate myeloid: erythroid ratio (M:E ratio), and whether normal, increased or decreased
- Myeloid cells:
- Normal or abnormal maturation
- Indicate if excess blasts
- Abnormal localization of immature precursors
- Erythroid cell:
- Normal or abnormal maturation
- Provide differential of myeloid and erythroid elements, based on counting 200 - 500 cells in aspirate smear
- Megakaryocytes:
- Normal or abnormal numbers and morphology
- Lymphocytes:
- Lymphoid aggregates
- Hematogones
- Other abnormalities
- Plasma cells:
- Normal or increased
- List abnormalities
- Histiocytes:
- Normal or increased
- List hemophagocytosis or other abnormalities
- Iron stores:
- Quantitate (none, 1+, 2+, 3+ or 4+; indicate if normal, increased or decreased)
- Indicate if ringed sideroblasts are present and how many
- Fibrosis:
- Present or not
- Normal or abnormal
- Bone:
- List any abnormalities present
- Cytogenetic findings
- Molecular findings
- Immunohistochemistry findings
- Other:
- Presence of metastatic tumor or not
- Other findings
- Bone marrow interpretation / diagnosis:
- Correlate with clinical history
- Cytogenetics
- Molecular or immunohistochemistry findings
- Peripheral blood smear
- Specimen type
- Adequacy (adequate, limited, unsatisfactory)
- Results of special stains or other studies, if performed
- Results of cytogenetics, if performed
- Diagnosis (WHO classification)
- Rare disorder of unknown pathogenesis, characterized by fat cell atrophy, focal loss of hematopoietic cells, deposition of extracellular gelatinous substances which are mucopolysacchrides rich in hyaluronic acid and lacking chondrotin sulphate (Am J Surg Pathol 2000;24:56)
- Also referred to as "starvation marrow" or "serous fat atrophy" (Pathology 1983;15:85, Hum Pathol 1978;9:685)
- Almost exclusively adults, 2/3 males
- Higher incidence and severity in young adults (Am J Surg Pathol 2000;24:56, J Assoc Physicians India 2003;51:585)
- Exact pathogenic mechanism unknown
- Deposition of seromucinous, gelatinous, hyaluronic acid rich material in bone marrow stroma
- Associated with hematopoietic atrophy, which may be due to nonsupportive marrow microenvironment and inadequate hematopoietic substrate availability
- May be reversible if underlying disorder is eliminated
- Associated with cancer related cachexia, endstage renal disease, myxedema, anorexia nervosa, infections
- Likely a symptom of severe or chronic illness with malnutrition (Am J Surg Pathol 2000;24:56, Indian J Pathol Microbiol 1997;40:383)
- Common associations by age:
- Age < 40 years: anorexia nervosa, acute febrile states, AIDS
- Ages 50 - 60 years: alcoholism, lymphoma
- Age > 60 years: carcinoma, lymphoma, chronic heart failure (Am J Surg Pathol 2000;24:56)
- Also: acute leukemia (Acta Haematol 1989;82:165), acute myelogenous leukemia postchemotherapy (Am J Hematol 1991;38:220), celiac disease (Pathology 1983;15:85), gastric ulcer, hypothyroidism (Arch Pathol Lab Med 1987;111:375), imatinib mesylate treated chronic myelogenous leukemia (Pathology 2012;44:59, Acta Haematol 2008;119:104), intensive care unit patients (J Clin Pathol 1990;43:850), myelodysplastic syndrome (J Clin Pathol 1996;49:512, Int J Clin Exp Pathol 2013;6:1677), post partial gastrectomy with hemicolectomy (J Ky Med Assoc 1998;96:10), systemic lupus erythematosus (Pathology 1991;23:5)
- Usually malnutrition secondary to anorexia nervosa (Am J Clin Pathol 2002;118:582, Nouv Rev Fr Hematol 1994;36:S85) or starvation but also:
- Infectious: bacterial enteritis, bronchopneumonia, enteric fever, septicemia, toxoplasma, tuberculosis, viral encephalitis or enteritis
- Nutritional: protein calorie malnutrition, iron deficiency, folic / B12 deficiency
- Hematological: aplastic anemia, bone marrow suppression
- Malignancy: leukemia, lymphoma, metastasis
- Miscellaneous: chronic obstructive airway disease, dilated cardiomyopathy, gouty nephropathy, grand mal seizures, Henoch Schönlein purpura, idiopathic thrombocytopenic purpura, ventricular septal defect (J Assoc Physicians India 2003;51:585)
- Bone marrow core biopsy with microscopic examination in correlation with clinical presentations
- Radiologic studies may reveal focal lesions while the findings are usually nonspecific
- Common nonspecific findings include those associated with malnutrition: anemia (Indian J Pathol Microbiol 2005;48:1), deficiency in trace elements or vitamins
- Further laboratory workups are specific to underlying disorders
- May manifest as "hot spot" on 18F-FDG PET / CT scan (Clin Nucl Med 2012;37:798)
- May present as patchy areas of T2 hyperintensity on MRI (Skeletal Radiol 2012;41:357)
- Specific to underlying triggering conditions
- Focal hypoplasia of fat cells and hematopoietic cells
- Accumulation of extracellular gelatinous substances that appear as pink purple material on Romanowsky stained bone marrow preparation
Images hosted on other servers:
Prominent gelatinous transformation
Serous atrophy
Various images
- Alcian blue (pH 2.5), PAS (Periodic Acid Schiff)
- Pink purple on Romanowsky stain
- Randomly aggregated nonamyloid fibrillar and granular material, somewhat similar to amyloid fibrils (Hum Pathol 1978;9:685, Pathology 1983;15:85)
- Amyloidosis: Congo Red+, apple green birefringence on examination with polarized light
- Changes at previous biopsy site: lack of adipose tissue and hematopoietic cells; granulation tissue and new bone formation present, e.g. osteoblasts seen along the endosteal surfaces
- Marrow necrosis: see Cancer 2000;88:1769
- 3 - 6% of total body weight
- Major organ for hematopoiesis at birth; also primary and secondary lymphoid organ
- Hematopoiesis sites change from axial and radial skeleton in newborns to flat bones of central skeleton by mid teens
- Pluripotent stem cells develop into myeloid blasts (myeloblasts, monoblasts, erythroblasts and megakaryoblasts) or lymphoblasts
- Cells are in storage pools for 5 - 7 days, then to blood and then to tissues
- Estimated daily production rates to maintain homeostasis in adults: 50 - 100 x 109 neutrophils, 150 - 200 x 109 platelets and 150 - 200 x 109 erythrocytes
- Cellularity of 90 - 100% in the first years of life, then adult proportions of ~50% reached over a widely variable time interval
- High M:E ratio during the first 3 days of life, progressive decrease by the third week and adult level (3 - 4:1) at 1.5 - 3 years of age
- Lymphocytes predominate in the first 1.5 - 3 years; relatively numerous eosinophils in the first 3 months
- Iron stores are usually absent during the first year of life (exception - first month can have occasional iron stores), then increase progressively and reach adult levels by 5 - 6 years of life
AFIP images


Hematopoietic cell differentiation
Images hosted on other servers:
Hematopoietic cell differentiation

Normal bone marrow differential in adults
- Highly organized meshwork of thin walled capillary - venous sinuses surrounded by extracellular matrix, encased by cortical bone and traversed by trabecular bone (J Clin Pathol 1992;45:645)
- Nutrient (medullary) artery ramifies through marrow space to supply medullary cavity; its arterioles branch into capillaries that are continuous with thin walled sinusoids (capillary - venous sinuses)
- Capillary - venous sinus (CVS, basic bone marrow structural unit): inner endothelial cells and outer adventitial reticular cells (phagocytic, can become lipocytes; also synthesize collagen, laminin, fibronectin and proteoglycans)
- All newly formed mature hematopoietic cells are released into capillary - venous sinus, then pass into the blood: transcellularly (via cytoplasm) for erythrocytes and granulocytes, or via megakaryocyte pseudopod extension and fragmentation for platelets
- Capillary - venous sinus coalesce into venules, then veins, carrying newly formed mature hematopoietic cells to the systemic circulation
- No lymphatic channels
- Topographic distribution: myeloid - peritrabecular, erythroid - central, megakaryocytes associated with sinus walls and lymphocytes throughout (no lymphoid follicles in infancy)
- Also known as "hematopoietic inductive environment"
- Formed by the adventitial (stromal) cells and their products (extracellular matrix proteins, cytokines, inhibitory factors and adhesion molecules)
- Provides support for hematopoietic stem cell renewal and produces factors responsible for osteogenesis
- Heterogeneous cell population: fixed fibroblasts, macrophages and adipose cells
- Extracellular matrix: collagen, fibronectin, laminin, thrombospondin and proteoglycans
- Cells with multilineage hematopoietic differentiation potential and sustained self renewal activity
- Detected by their ability to regenerate long term multilineage hematopoiesis in myeloablated recipients
- Very infrequent in the bone marrow: 0.01 - 0.05% of mononuclear cells
- Defined via functional rather than morphologic characteristics: ability to undergo self renewal and produce multilineage progeny
- Express CD34, various cytokine receptors / adhesion molecules / extracellular matrix protein receptors (SCF, bFGF, IL11 / 3 / 1 / 6, GM-CSF, etc.)
- Endothelial cells likely have major role in regulation of trafficking and homing of hematopoietic stem cells within the bone marrow
- Immunophenotype: CD34+, Thy1+, c-kit+, Rhodamine 123 low, Lin-, CD45RO+, CD38-, flt+ and adhesion molecules
- Spectrum of evolution / maturation: hematopoietic stem cells → progenitor cell with multilineage commitment → progenitor cell with single lineage commitment → blast cell → mature cell
- Developmental continuum: progressive loss of proliferative capacity and gradual acquisition of specific lineage characteristics
- Cytokines produced by T lymphocytes, the monocyte - macrophage lineage and other bone marrow stromal cells (either inducers or inhibitors)
- Stimulatory cytokines (inducers): stem cell factor (SCF), colony stimulating factors (CSFs) and various interleukins (IL)
- Inhibitory cytokines: tumor necrosis factor beta (TNF-β), transforming growth factor alpha (TGF-α) and macrophage inflammatory protein 1alpha (MIP-1α)
- Some factors may be pleiotropic (positive or negative effect): TGF-β
- Myeloid - monocytic: predominant in the bone marrow, progeny includes neutrophils (most numerous), eosinophils, basophils, monocytes / macrophages, dendritic cells and mast cells
- Other: erythroid, megakaryocytic, lymphoid, natural killer and osteoclasts
- Bone marrow biopsy: arterioles, venules, capillaries, sinusoids, adipose tissue, connective tissue and hematopoietic cells; mitotically active cells are usually paratrabecular and perivascular
- Bone marrow aspirate: hematopoietic cells (myeloid, erythroid, megakaryocytic, lymphoid), plasma cells (rare in infants), osteoblasts (most common in infants), osteoclasts, occasional adipocytes, endothelial cells or capillaries
Images hosted on other servers:
Bone marrow biopsy:
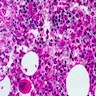
Normal marrow
Bone marrow aspirate smear:
Normal
- Granuloma is formed by aggregates of histiocytes / macrophages, admixed with variable inflammatory cells, with or without giant cells
- An infrequent histopathologic finding, 0.3 - 3% of bone marrow biopsy, related to various conditions, infectious or noninfectious
- Identification of underlying etiologies is the key to treatment and prognosis
- An infrequent histopathologic finding related to various conditions
- Underlying etiologies include infections, sarcoidosis, hematologic and nonhematologic malignancies, drug reaction, autoimmune diseases and other disorders
- 2 common histologic findings: caseating or noncaseating
- Morphologic features only are not reliable to differentiate between causes; it is necessary to combine morphology with microbiologic, serologic, molecular and clinical findings for full interpretation
- Bone marrow granulomas
- Granulomatous bone marrow disease
- Granulomatous lesion in bone marrow
- Granulomatous bone marrow inflammation
- ICD-10: L92.9 - granulomatous disorder of the skin and subcutaneous tissue, unspecified
- Broad age range, mainly aged > 40 years
- Slight male predominance
- Mainly in patients with immune deficiency (HIV), hematologic malignancy and sarcoidosis
- Higher risk in patients with fever of unknown origin (15 times more likely than when biopsied for other reasons) (Hematol Pathol 1988;2:43)
- Bone marrow
- Can be seen in other body sites
- Bone marrow involvement is usually associated with disseminated disease
- Reference: Medicine (Baltimore) 2018;97:e9726
- Unknown at this time
- Infections:
- Bacteria: M. tuberculosis, M. avium, M. pneumoniae, brucellosis, typhoid fever, Q fever, Rocky Mountain spotted fever, Bartonella
- Fungi: histoplasmosis, cryptococcosis, aspergillosis, Candida, Talaromyces marneffei
- Virus: EBV, CMV
- Protozoa: leishmaniasis
- Hematologic disorders:
- Non-Hodgkin lymphoma, mostly T / NK cell origin (e.g., angioimmunoblastic T cell lymphoma)
- Acute lymphoblastic leukemia (ALL)
- Chronic lymphocytic lymphoma (CLL)
- Multiple myeloma (MM)
- Monoclonal gammopathy of undetermined significance (MGUS)
- Acute myeloid leukemia (AML)
- Hodgkin lymphoma
- Myelodysplastic / myeloproliferative neoplasm (MDS / MPN)
- Sarcoidosis
- Drug reaction:
- Anti-inflammatory agents (ibuprofen, indomethacin)
- Antiarrhymthics (amiodarone, procainamide, tocainide)
- Sulfanamide
- Interferon β, interferon α2b
- BCGitis
- Autoimmune disease:
- Lupus
- Sjögren syndrome
- Antineutrophil cytoplasmic antibodies (ANCA) associated vasculitis
- Mixed connective tissue disease
- Rheumatoid arthritis
- Primary biliary cirrhosis
- Behçet disease
- Nonhematologic malignancy:
- Renal cell carcinoma
- Breast cancer
- Ovary cancer
- Bladder cancer
- Hepatic sarcoma
- Other diseases:
- Silicosis
- Various collagen vascular diseases
- Gouty tophi
- Foreign bodies
- Unknown etiology
- References: Medicine (Baltimore) 2018;97:e9726, Int J Lab Hematol 2018;40:123, E J Intern Med 2013;24:468, Hematol Pathol 1988;2:43, Medicine 1983;62:372, J Hematol 2020;9:37, Am J Med Case Rep 2021;9:83
- Fever
- Fatigue and weakness
- Dyspnea and cough
- Lymphadenopathy / hepatosplenomegaly
- Reference: Medicine (Baltimore) 2018;97:e9726
- Diagnosis is based on histological examination of bone marrow biopsy and a series of ancillary tests upon clinical setting
- Anemia, thrombocytopenia, lymphopenia
- Positive or negative for microorganisms
- Serological changes related to infectious or autoimmune etiology
- Reference: Medicine (Baltimore) 2018;97:e9726
- Prognosis varies according to underlying causes
- Good prognosis for those caused by drug reaction (e.g., BCGitis) and for those without definitive underlying causes (West J Med 1996;164:510, E J Intern Med 2013;24:468)
- 9.5 year old Indian girl who presented with bleeding manifestation, skin rash, uveitis and arthritis (Eur J Rheumatol 2020;7:190)
- 46 year old man with fever, shortness of breath, myalgia and general malaise (Am J Med Case Rep 2021;9:83)
- 53 year old woman presented with fever, chills and rigors associated with occipital headache (Blood 2019;133:2730)
- 54 year old woman with history of weight loss and low grade fever (J Ayub Med Coll Abbottabad 2018;30:S671)
- 69 year old woman suffered from cervical pain due to lymphadenopathy (Cancer Radiother 2021;25:51)
- Treatment varies for specific underlying causes
- Steroids for sarcoidosis
- Cytoreductive therapy for hematologic malignancies
- Immunomodulators for autoimmune disorders
- Drug discontinuation for drug reaction
- Reference: Medicine (Baltimore) 2018;97:e9726
- Major types: lipogranuloma (insignificant), epithelioid granuloma (caseating, noncaseating)
- Lipogranuloma: prominent aggregates of histiocytes with variably sized lipid vacuoles; most common type
- Epithelioid granuloma: loose or discrete aggregates of eosinophilic histiocytes / macrophages with variable giant cell formation and variable central caseation / necrosis; usually small, focal and nonnecrotic; often paratrabecularly located; many subtypes; characteristic features for certain causes but not specific
- Caseating / necrotizing granuloma: containing a central area of necrosis (caseation), are significant and usually indicate infection (tuberculosis, Brucella, Rickettsia, fungi)
- Noncaseating granuloma:
- Ring or doughnut granuloma: fibrin and inflammatory cells arranged around a central clear space; associated with Q fever but not specific, can also be seen in Brucella, typhoid, rare viral infections
- Foreign body granuloma: multinucleated giant cell's response to exogenous material, often visible by polarized light
- Sarcoid type granuloma: multiple noncaseating granulomas, occasional fibrinoid necrosis in active cases
- Mycobacterium avium complex related granuloma: clusters of histiocytes with needle-like inclusions in cytoplasm resembling Gaucher cells
- Chronic granulomatous disease: usually nonspecific, presence of active chronic inflammation and pigmented macrophages should raise suspicion (Histopathology 2005;47:508)
- Nonspecific: small and rare epithelioid granulomas may also be clinically insignificant
Contributed by Natasha Savage, M.D. and Yulan Jin, M.D., Ph.D.
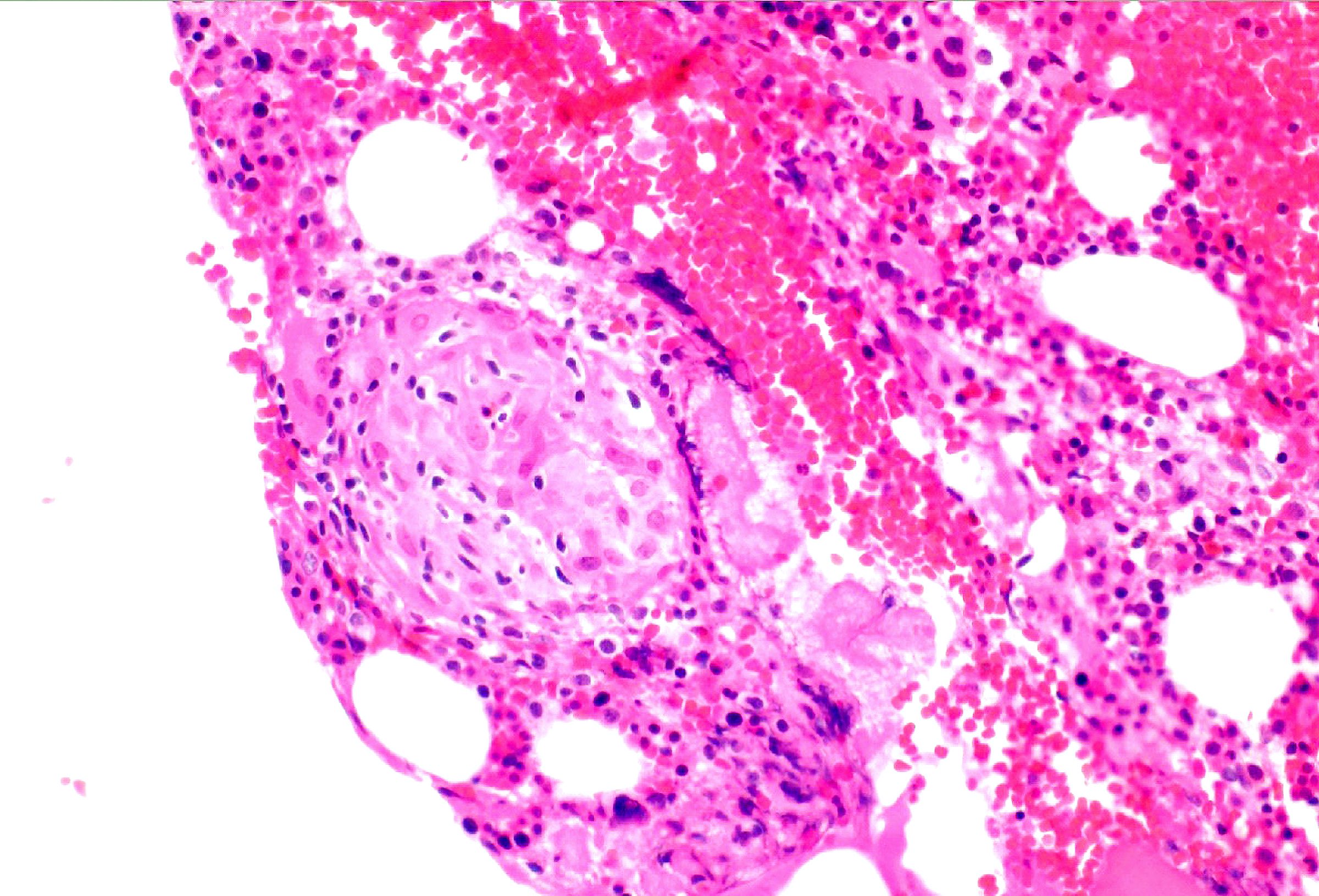
Nonnecrotizing granuloma
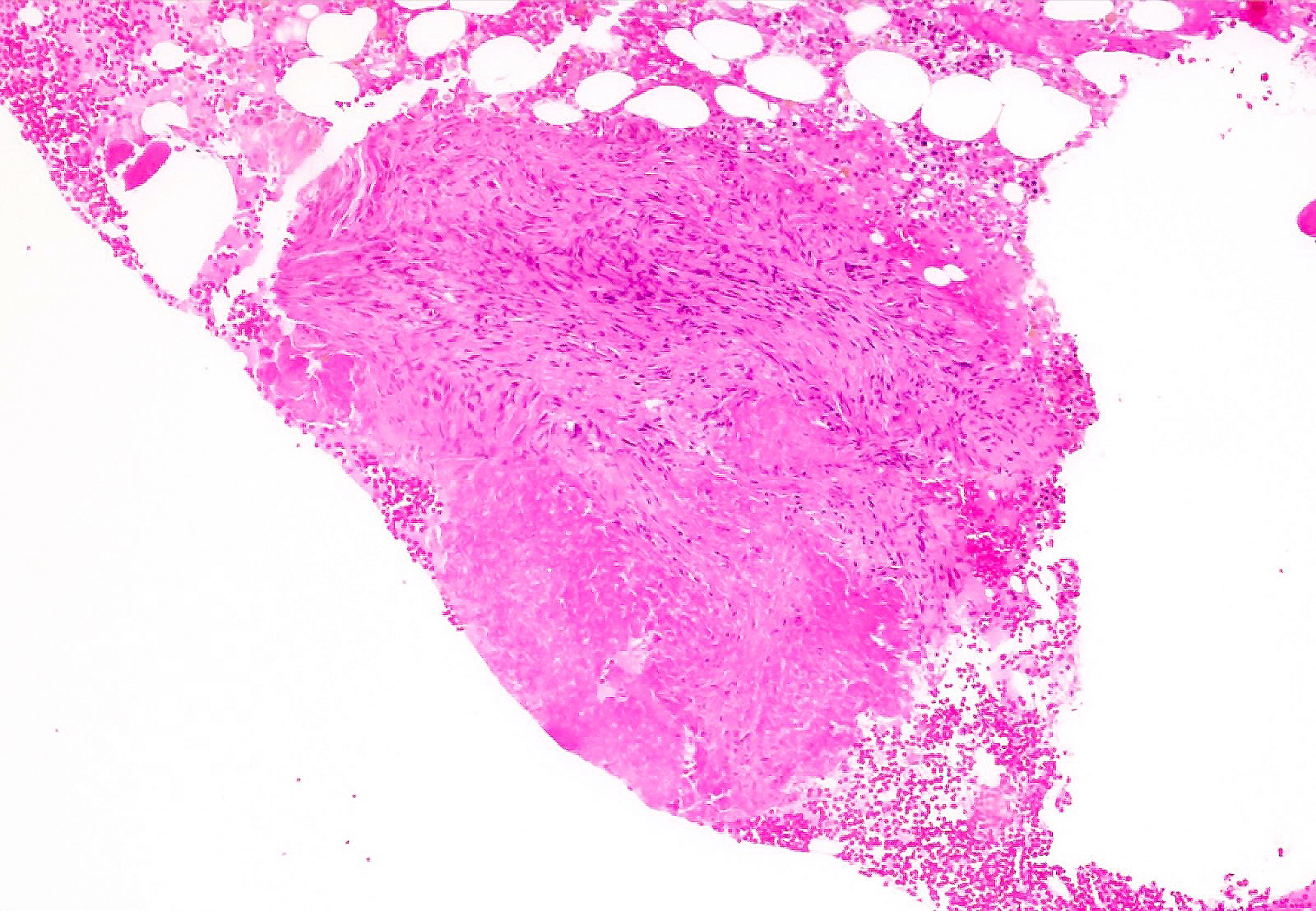
Necrotizing granuloma
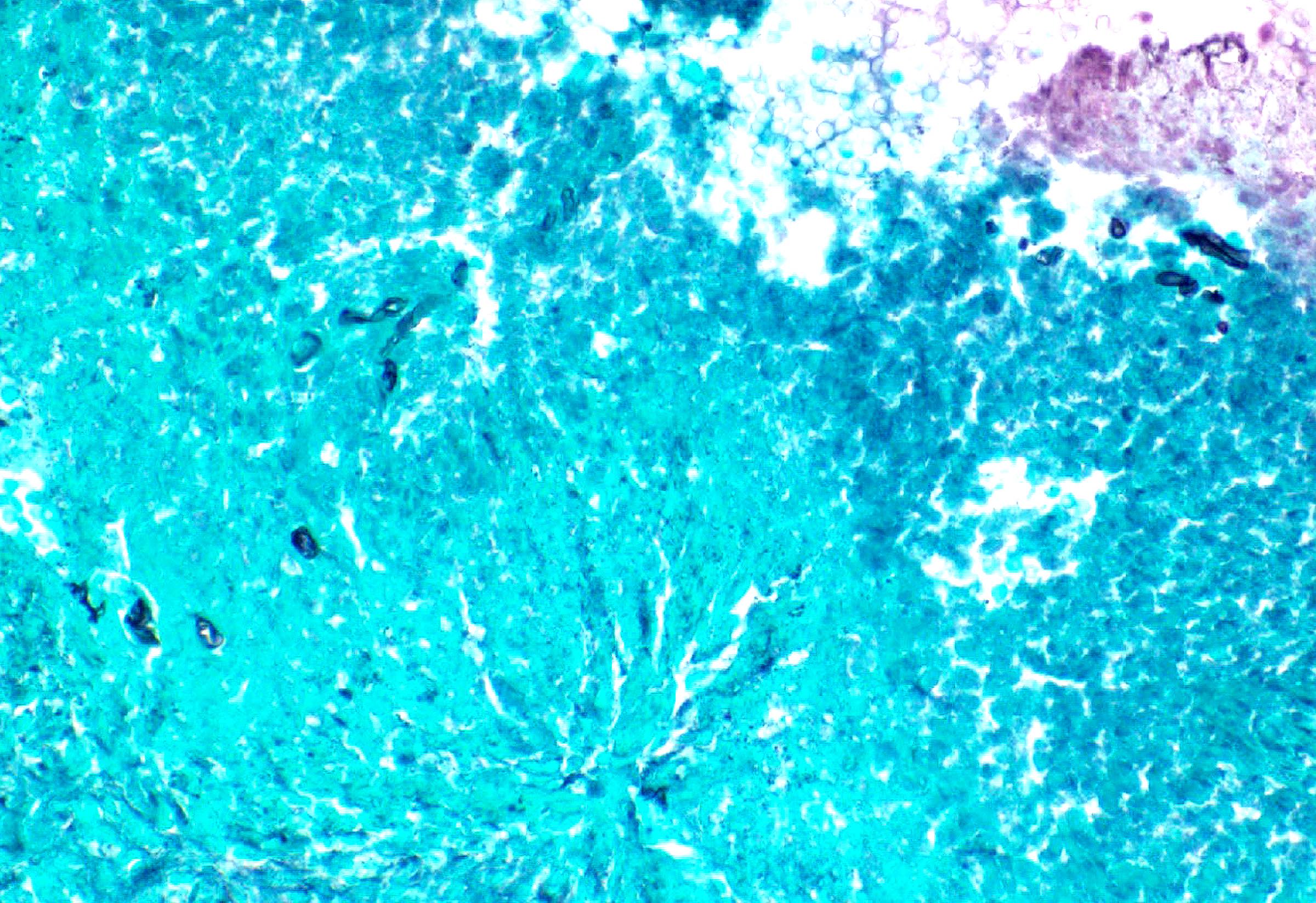
GMS stain
- CD68, CD163: highlight the cluster of histiocytes
- Acid fast bacilli (AFB): positive for mycobacterial infection
- Grocott methenamine silver (GMS): positive for fungal infection (J Clin Tuberc Other Mycobact Dis 2017;7:1)
- Can be applied for diagnosis of non-Hodgkin lymphoma if lymphoma is suspected clinically in a setting of granulomatous inflammation
- Genetic testing for chronic granulomatous disease (CGD): these 5 genes (CYBA, CYBB, NCF1, NCF2 or NCF4) are frequently mutated
- Bone marrow, right posterior iliac crest, core biopsy, clot section:
- Necrotizing granuloma with fungal hyphae (see comment)
- Clinical history: Acute myelogenous leukemia. Status postchemotherapy was in complete remission. Rule out relapse.
- No evidence of residual leukemia
- Comment: The marrow is hypocellular with left shifted granulopoiesis and erythroid predominance. There is no morphologic, immunophenotypic or flow cytometric evidence of residual leukemia. The fungal hyphae identified are poorly preserved, precluding further characterization.
- CBC: WBC: 2.3 k/mm³, Hgb: 10.1 g/dL, MCV: 89.0 fL, Platelets: 56 k/mm³. Differential count (%): Neutrophils: 17, Bands: 40, Lymphocytes: 38, Monocytes: 3, Eosinophils: 1.
- Peripheral smear: Manual review of the peripheral blood shows normocytic / normochromic anemia, thrombocytopenia and moderate leukopenia. RBCs: Normocytic / normochromic red blood cells, mild anisocytosis, mild polychromasia. WBC: Moderate leukopenia. Granulocytes with left shift and toxic changes. Blasts are not increased. Platelets: Moderate thrombocytopenia. Large platelets are noted.
- Bone marrow biopsy: Quality: adequate. Cellularity: hypocellular (5 - 10%). Hematopoiesis: Erythropoiesis is increased. Granulopoiesis is decreased. Megakaryocytes are decreased with normal morphology. No significant changes in medullary bone are noted. Single necrotizing granuloma is identified. Special stains: Reticulin (1+): reticulin fibrosis is present. Iron: stainable iron is increased (3+). PAS: stains appropriately, fungal hyphae are identified. GMS: fungal hyphae are identified. CD34: no increase in blasts. CD117: no increase in mononuclear cells. CD68: positive in histiocytic cells.
- Bone marrow clot section: Quality: adequate. Cellularity: 5 - 10% morphologic features are similar to those observed in the core biopsy.
- Hodgkin lymphoma:
- Histiocytic cell neoplasms (Ann Hematol 2018;97:2117):
- Langerhans cell histiocytosis:
- Often associated with variable numbers of eosinophils
- Expresses CD1a, CD207 (langerin), BCL1 and S100
- Hemophagocytic lymphohistiocytosis:
- Increased histiocytes exhibiting hemophagocytosis of mostly erythrocytes
- Usually presents with very severe clinical symptoms (hepatosplenomegaly) and significant abnormal lab tests (cytopenia, liver function, high ferritin levels, elevated triglyceride level and or soluble IL25 level)
- Histiocytic sarcoma:
- Rosai-Dorfman disease:
- Erdheim-Chester disease:
- Langerhans cell histiocytosis:
- Storage diseases:
- Gaucher disease:
- Niemann-Pick disease:
- Foamy macrophage with round lipid inclusion
- Molecular mutations in SMPD1, NPC1 or NPC2
- Infection:
- Whipple disease:
- Infiltration of foamy macrophage
- Strongly PAS+
- Bacilliform inclusions
- Whipple disease:
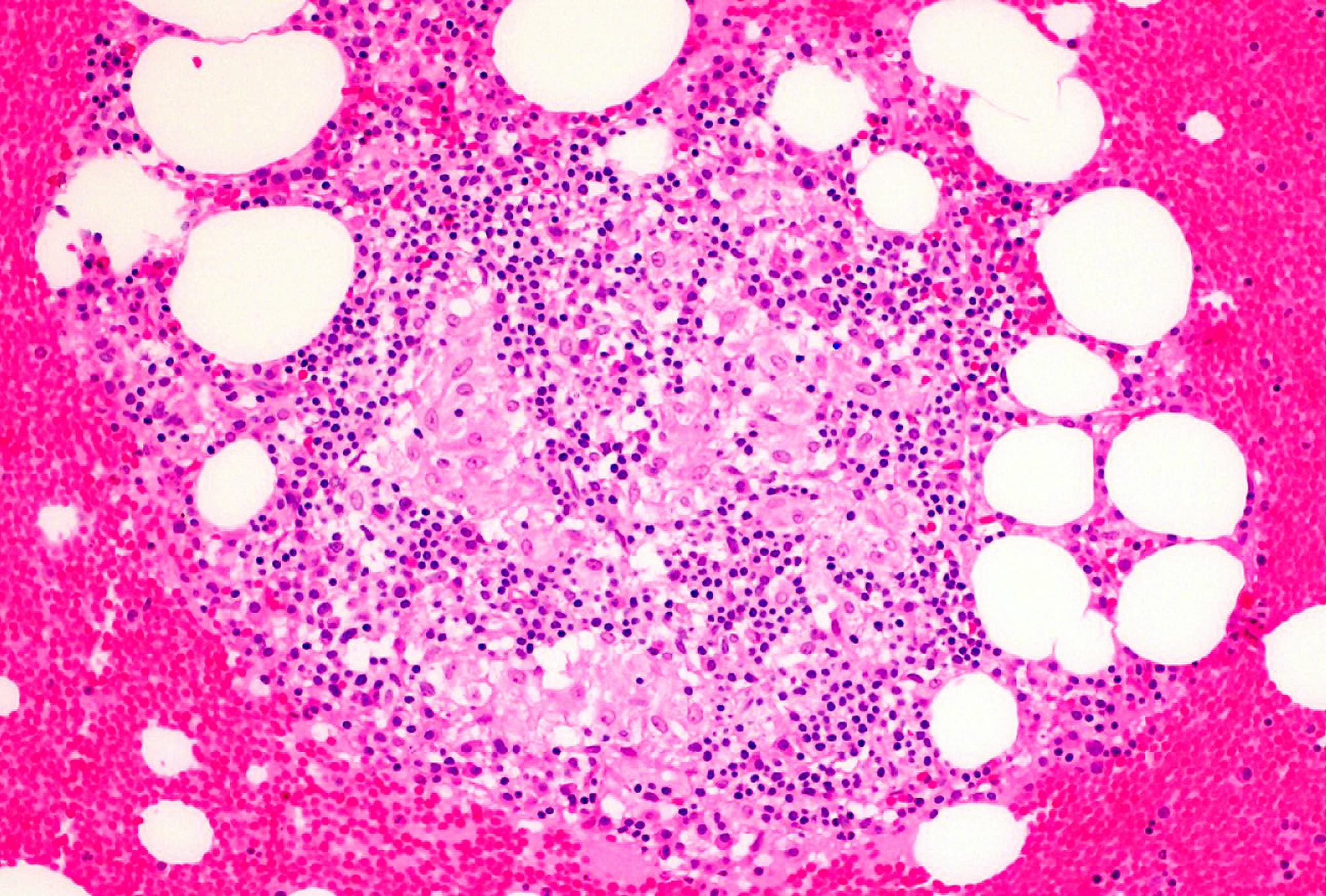
A 64 year old patient with acute myeloid leukemia (AML), status postinduction chemotherapy, had a bone marrow biopsy for disease monitoring. The morphologic findings are shown in the hematoxylin and eosin (H&E) slide (see above image). By immunohistochemistry, the aggregated histiocytic cells are positive for CD64 and negative for CD30, CD1a, langerin, S100, PAS, AFB and GMS. What is the most accurate diagnosis?
- Bone marrow granuloma
- Hodgkin lymphoma
- Langerhans cell histiocytosis
- Whipple Disease
Comment Here
Reference: Granulomatous inflammation
- Detection of bone marrow granulomas can exclude an underlying malignant process
- Different morphologic subtypes of granuloma are diagnostic of specific etiologic conditions
- It is necessary to combine morphology with microbiologic, serologic, molecular and clinical findings to have a full interpretation of underlying etiologies
- They are a frequent histopathologic finding that points to an autoimmune disorder
Comment Here
Reference: Granulomatous inflammation
- Hematogones are normal B lineage progenitors, which normally account for < 1% of the bone marrow cellularity
- Based on sequential antigen expression, they can be divided into 3 stages of maturation: stages 1, 2 and 3 (Leuk Res 2013;37:1404)
- Hematogones are benign B cell precursors and their degree of maturation can be classified by flow cytometry into 3 stages (1, 2 and 3)
- Morphologically, they contain scant cytoplasm and often have round, indented nuclei with absent or inconspicuous nucleoli; the presence of nucleoli generally indicates immaturity
- Increased numbers of hematogones may mimic B lymphoblastic leukemia (B ALL) due to the morphologic and immunophenotypic similarities
- Bone marrow
Images hosted on other servers:

Normal hematogone maturation
- Conditions associated with increased hematogones include (Blood 2001;98:2498)
- Various tumors, including lymphomas and solid tumors
- Nonneoplastic blood cytopenias and autoimmune conditions
- Postchemotherapy, post-bone marrow transplantation
- Viral infections
- Acquired immunodeficiency syndrome (AIDS)
- 7 year old boy with hematogone hyperplasia masquerading as acute lymphoblastic leukemia with concurrent hypercupremia (J Pediatr Hematol Oncol 2020;42:e670)
- 64 year old man with TCL1 positive hematogones in a case of T cell prolymphocytic leukemia after therapy (Hum Pathol 2017;65:175)
- 75 year old man with κ light chain expressing hematogones in a case of λ restricted chronic lymphocytic leukemia (CLL) and multiple myeloma on daratumumab therapy (Blood 2022;140:1917)
- Hematogones are lymphoid appearing cells whose cytomorphology can be best appreciated on bone marrow aspirate smears (typically pediatric)
- Stage 1 hematogones typically have scant to absent cytoplasm (may be moderately to deeply basophilic with no inclusions, granules or vacuoles) and large, round, sometimes indented nuclei with inconspicuous nucleoli
- Generally, the presence of 1 or more nucleoli denotes immaturity; therefore, immature hematogones may resemble lymphoblasts
- Mature hematogones resemble mature lymphocytes with condensed nuclear chromatin; nucleoli are absent
Contributed by Giby V. George, M.B.B.S. and AFIP
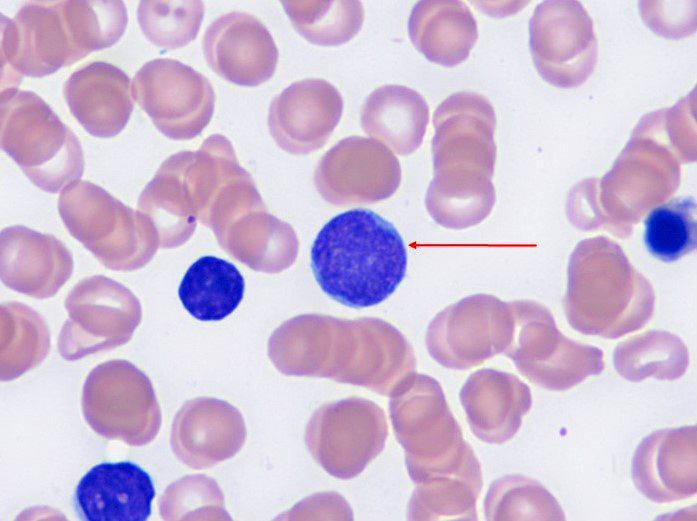
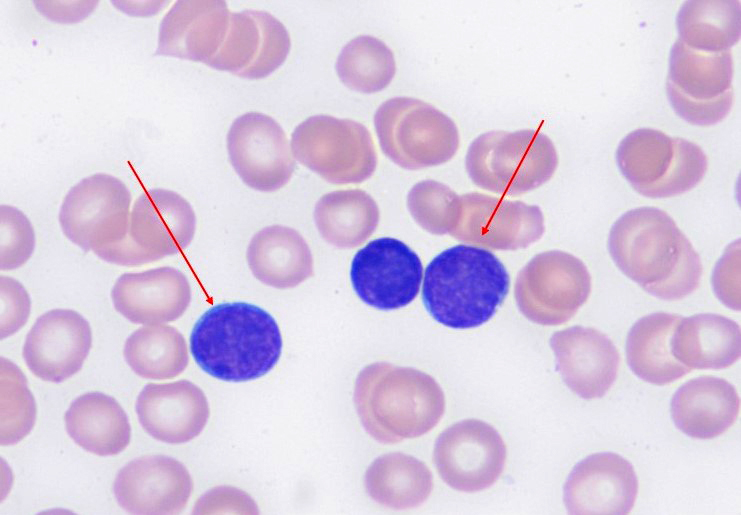
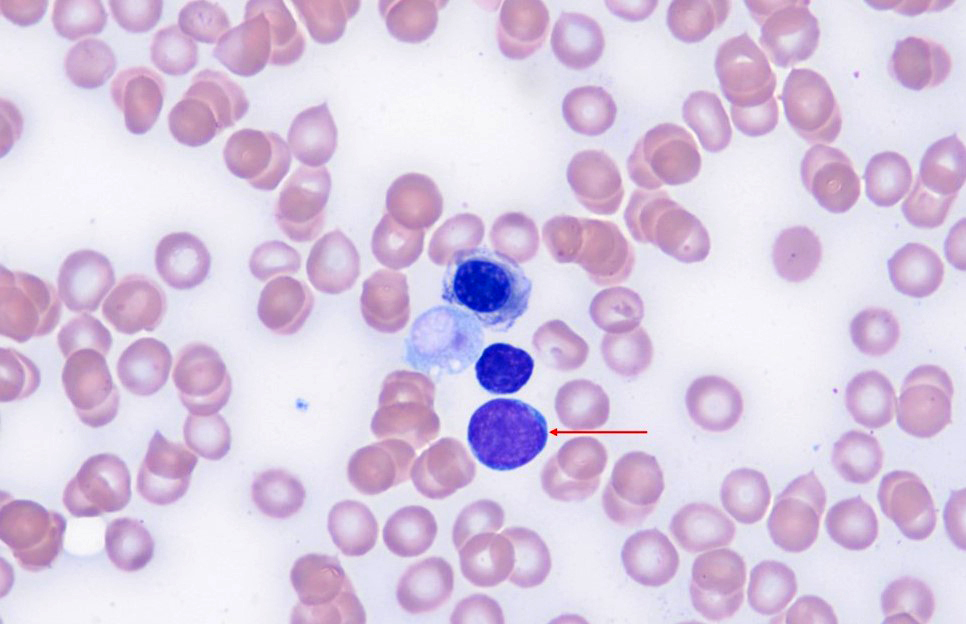
Bone marrow aspirate hematogones
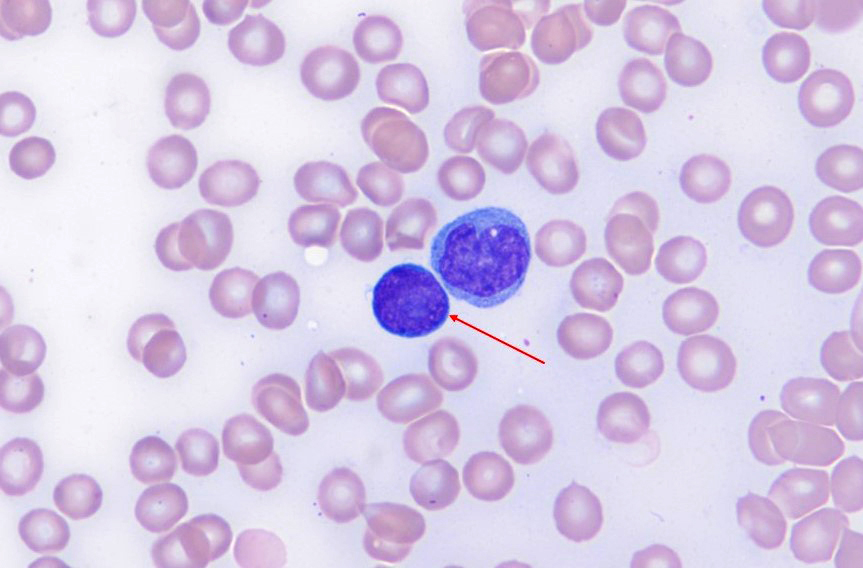
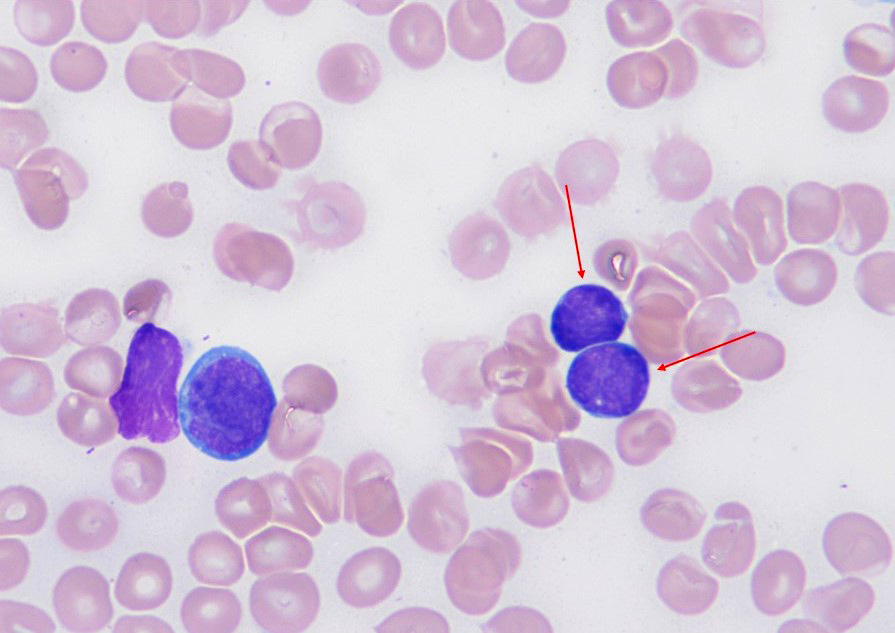
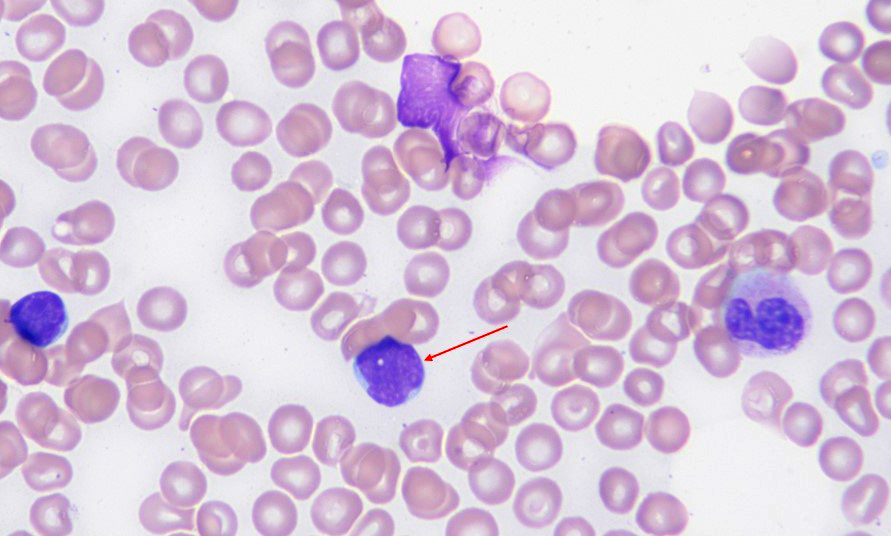
Bone marrow aspirate hematogones
Sparse, lightly basophilic cytoplasm
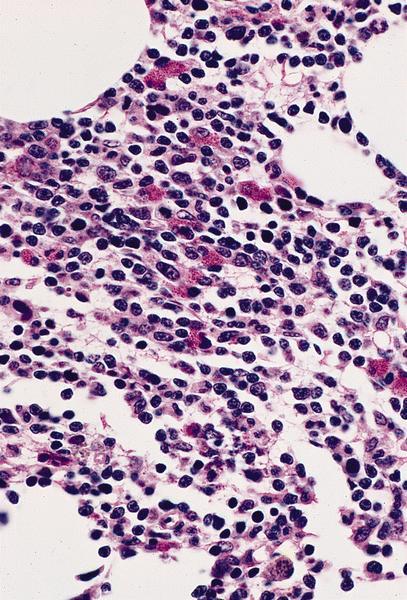
Increased hematogones evenly distributed

Clumped nuclear chromatin
- Hematogones are not usually seen on the peripheral blood smear except in neonates or in umbilical cord blood
- Generally, hematogones demonstrate low side scatter (SSC) by flow cytometry with dimmer expression of CD45 when compared to normal lymphocytes; thus, they lie within the CD45 dim, low SSC gate (Leuk Res 2013;37:1404)
- Stage 1 hematogones exhibit bright expression for TdT, CD34, CD10, CD38 and CD43; they show dim expression of CD19 and CD22 while negative for CD20 and cytoplasmic immunoglobulin (Leuk Res 2013;37:1404)
- Stage 2 hematogones begin to show down regulation / negativity of TdT, CD34 and CD10; they exhibit increased expression of CD19 and CD20 with continued bright expression of CD38 and CD43, with dim expression of CD22 and CD45 and they are generally positive for cytoplasmic immunoglobulin (Leuk Res 2013;37:1404)
- Stage 3 hematogones are negative for TdT and CD34; they demonstrate bright expression of CD19, CD20, CD38, CD43, cytoplasmic and surface immunoglobulin, while dim for CD10, CD22 and CD45 (Leuk Res 2013;37:1404)
- Stage 3 hematogones progress to mature B cells, which are negative for TdT, CD34, CD10 and CD43, while positive for CD19, CD20, CD22, CD45, cytoplasmic / surface immunoglobulin with variable expression of CD38 (Leuk Res 2013;37:1404)
- CD5 expression has also been reported on stage 3 hematogones and mature B cells; in these cases, CD5 expression in conjunction with surface immunoglobulin helps differentiate them from CD5+ neoplastic B cells (Am J Clin Pathol 2009;132:733)
- Rarely, hematogones may exhibit light chain restriction (Leuk Res 2021;111:106704, Leuk Res Rep 2022;17:100316)
- Hematogones may rarely be identified by flow cytometry and IHC in benign lymph nodes (Am J Clin Path 2022;157:202, Hum Pathol 2018;81:131, Int J Lab Hematol 2023;45:592)
- In this case, they should account for 1% or less of the total lymph node population
- Additionally, discrimination from abnormal lymphoblasts is imperative
Contributed by Giby V. George, M.B.B.S.
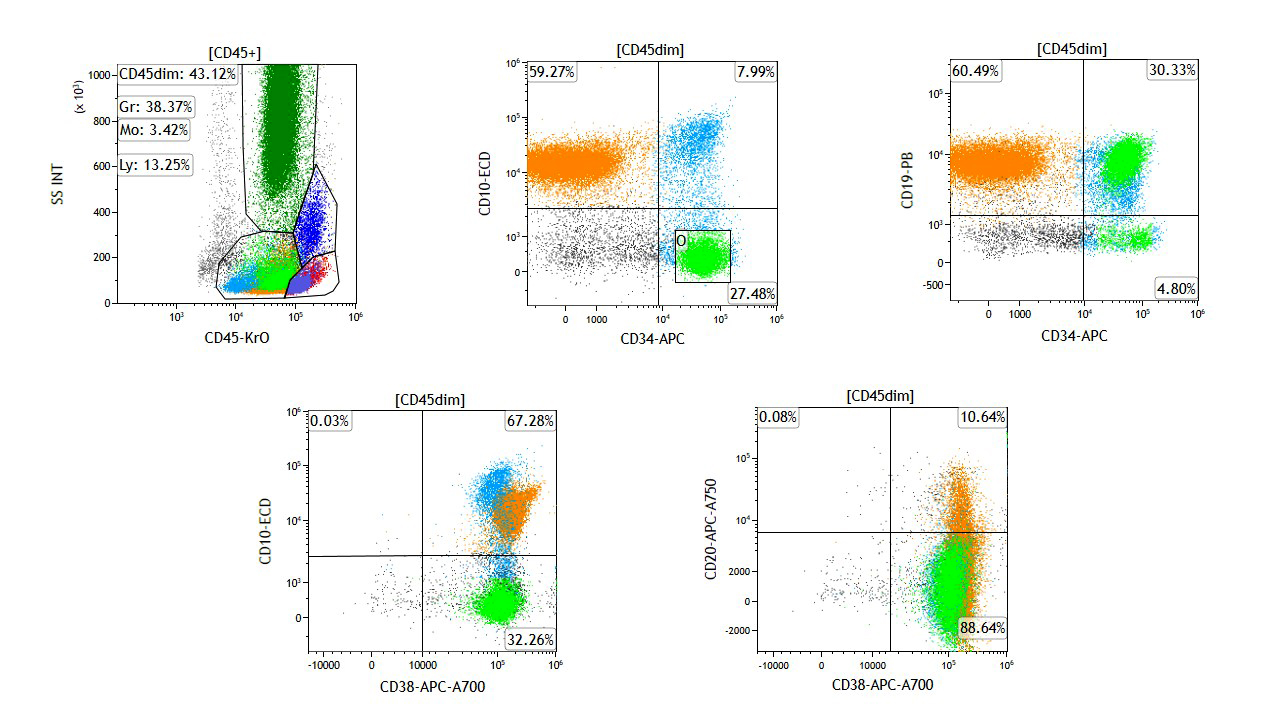
Hematogone maturation in the background of B ALL
Images hosted on other servers:


Normal hematogone maturation by flow cytometry
ICCS: B cells, features of immaturity
Ace My Path: B cell maturation
- Precursor B lymphoblastic leukemia:
- Immature hematogones may resemble lymphoblasts; however, while hematogones usually have inconspicuous nucleoli with scant basophilic cytoplasm (devoid of granules, inclusions or vacuoles), lymphoblasts typically have indented nuclei with fine chromatin and variable nucleoli surrounded by deeply basophilic cytoplasm, which may demonstrate vacuoles
- Flow cytometry will greatly aid in this distinction
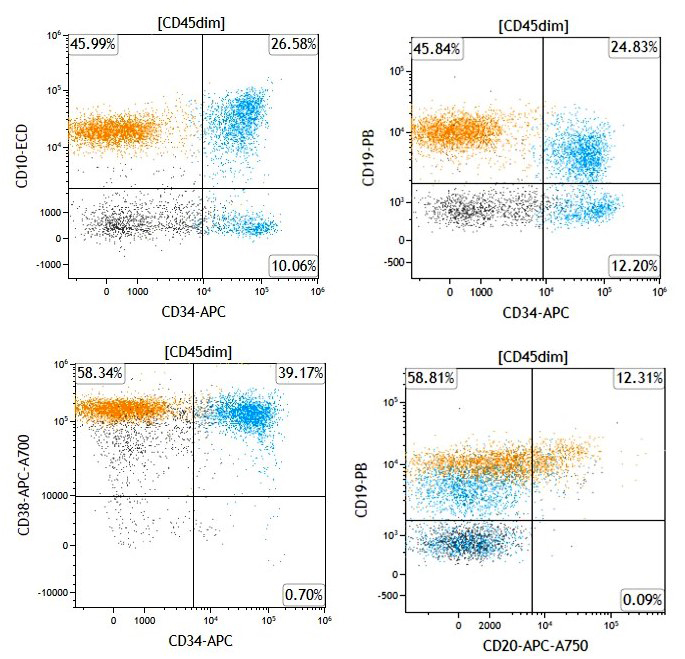
A bone marrow aspirate sample obtained from a 2 year old girl with a history of B lymphoblastic leukemia (B ALL) is submitted for flow cytometric and cytogenetic analysis for minimal residual disease (MRD) assessment. Although cytogenetic workup does not show any abnormalities in the submitted bone marrow specimen, flow cytometry reveals an expanded CD45 dim, low side scatter gate containing 10% of events with distinct subpopulations. One population shows coexpression of CD34, CD10, CD19 and CD38, while another population is positive for CD10 (dimmer), CD19 and CD38 and negative for CD34. What could these events represent?
- Hematogones
- Mature B cell neoplasm
- Normal myeloblasts
- Residual B ALL
Comment Here
Reference: Hematogones
- Pathologic immune activation syndrome
- Congenital or acquired defective natural killer (NK) / T cell function with uncontrolled hypercytokinemia and end organ damage
- Histiocytic infiltration of reticuloendothelial organs with hemophagocytosis of erythrocytes, leukocytes, platelets and their precursor cells
- Divided into genetic (primary) and acquired (secondary) forms
- Sudden onset of a systemic inflammatory response syndrome (SIRS) with symptoms including high unremitting fevers, malaise, rash, jaundice and myalgia, along with accompanying cytopenias, hepatosplenomegaly and generalized lymphadenopathy
- Familial (primary) hemophagocytic lymphohistiocytosis (HLH) is a progressive disease characterized by early age of onset, autosomal recessive inheritance pattern, decreased NK cell function and frequent perforin mutations
- Acquired (secondary) hemophagocytic lymphohistiocytosis is often associated with immunodeficiency, normal or reduced NK cell numbers and normal perforin expression, and may be triggered by infectious agents, malignant neoplasms, graft versus host disease and (rarely) HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome
- Primary and secondary hemophagocytic lymphohistiocytosis are invariably fatal when left untreated but new therapy protocols (including HLH-2004) have reported 5 year survival rates up to 54%
- Familial (primary) hemophagocytic lymphohistiocytosis (FHLH), FHL1 - FHL5 subtypes
- May occur in the setting of Chédiak-Higashi syndrome, Griscelli syndrome type 2, Hermansky-Pudlak syndrome type 2 and X linked lymphoproliferative disorder types 1 and 2 (Hematology Am Soc Hematol Educ Program 2011;2011:178)
- Acquired (secondary) hemophagocytic lymphohistiocytosis
- ICD-10: D76.1 - hemophagocytic lymphohistiocytosis
- Familial hemophagocytic lymphohistiocytosis primarily affects young children with an incidence of approximately 1.5 per 100,000 live births (Pediatr Blood Cancer 2015;62:346)
- Acquired hemophagocytic lymphohistiocytosis is more common in older children and adults; its incidence is unknown but is likely underdiagnosed
- No race or sex predilection
- Involves all reticuloendothelial organs (bone marrow, spleen, liver and lymph nodes)
- Less commonly involves skin, lungs, meninges, cerebrospinal fluid and subcutaneous tissue
- Failure of NK cells to induce apoptosis of the target cell, remove the antigenic stimulus and terminate the inflammatory response
- Accentuated Th1 response leads to high levels of TNF alpha and TNF gamma, causing pancytopenia
- Upregulation of ferritin production secondary to increased heme oxygenase
- Hepatosplenomegaly due to organ infiltration by activated lymphocytes and histiocytes
- Fever induced by interleukin 1 and interleukin 6 (Am J Clin Pathol 2013;139:713)
- Genetic or acquired impaired cytotoxic function of T lymphocytes and NK cells
- Familial hemophagocytic lymphohistiocytosis is an inherited autosomal recessive disease involving mutations in perforin and other genes
- Secondary hemophagocytic lymphohistiocytosis triggers include
- Infection (particularly viruses that include Epstein-Barr virus and cytomegalovirus)
- HLH has also been reported in severe COVID-19 and Mpox infections (Curr Med Sci 2021;41:39, N Engl J Med 2023;388:1246)
- Malignancy (most often NK / T cell neoplasms, such as panniculitis-like T cell lymphoma, nasal type extranodal NK / T cell lymphoma and cutaneous gamma / delta T cell lymphoma)
- Rheumatologic / autoimmune disease
- Immunosuppression
- Pregnancy / HELLP syndrome (rare) (Clin Case Rep 2018;6:2466)
- Infection (particularly viruses that include Epstein-Barr virus and cytomegalovirus)
- Similar triggers can initiate FHLH
- In children, a genetic defect characteristic of FHLH should be ruled out
- In adults, hemophagocytic lymphohistiocytosis should prompt an investigation for underlying malignancy
- Fever
- Malaise
- Myalgia
- Hepatosplenomegaly
- Jaundice
- Generalized lymphadenopathy
- Cytopenias
- Rash
- Neurologic symptoms
- Reference: Inflamm Intest Dis 2020;5:49
- HLH-2004 criteria (updated 2007) include the molecular diagnosis of familial hemophagocytic lymphohistiocytosis or the presence of at least 5 of 8 criteria
- 1. Fever
- 2. Splenomegaly
- 3. Cytopenias (affecting at least 2 lineages in the peripheral blood)
- Hemoglobin levels < 90 g/L
- Platelets < 100 x 10⁹/L
- Neutrophils < 1.0 x 10⁹/L
- 4. Hypertriglyceridemia or hypofibrinogenemia
- Fasting triglycerides ≥ 265 mg/dL
- Fibrinogen ≤ 1.5 g/L
- 5. Documented hemophagocytosis in the bone marrow, spleen or lymph nodes
- 6. Low or absent NK cell activity
- 7. Ferritin ≥ 500 μg/L
- 8. Soluble CD25 ≥ 2,400 U/mL
- Familial hemophagocytic lymphohistiocytosis should have no evidence of malignancy
- Absence of hemophagocytosis during microscopic examination does not exclude the diagnosis of hemophagocytic lymphohistiocytosis (adapted from Pediatr Blood Cancer 2007;48:124)
- Hypofibrinogenemia
- Markedly elevated ferritin
- Hypertriglyceridemia
- Elevated transaminases
- Elevated lactate dehydrogenase
- Elevated serum soluble CD25 (interleukin 2 receptor)
- Reduced or absent NK cell activity by chromium 51 release assay
- Reference: J Blood Med 2014 Jun;5:69
- Hepatosplenomegaly
- Generalized lymphadenopathy
- Alveolar interstitial opacities with pleural effusions
- Gallbladder wall thickening
- Hyperechoic kidneys
- Ascites
- Nonspecific periventricular white matter abnormalities, brain volume loss and enlargement of extra-axial fluid spaces (Pediatr Radiol 2003;33:392)
- Almost invariably fatal if left untreated
- Familial hemophagocytic lymphohistiocytosis tends to recur within the first year of life in 70 - 80% of cases (Am J Clin Pathol 2013;139:713)
- Among the viral associated forms of hemophagocytic lymphohistiocytosis, Epstein-Barr virus carries the worst prognosis
- Delay in diagnosis and multiorgan involvement are associated with an inferior prognosis
- Hemophagocytic lymphohistiocytosis in the setting of HELLP syndrome as well as hemophagocytic lymphohistiocytosis development during the third trimester with associated Epstein-Barr viremia have a poor prognosis (Clin Case Rep 2018;6:2466)
- Newborn twins with thrombocytopenia, coagulation defects and hepatosplenomegaly (N Engl J Med 2004;351:1120)
- 18 month old girl presenting with fluctuating fevers and leishmaniasis (Indian Pediatr 2004;41:605)
- 5 year old girl diagnosed with COVID-19 requiring hematopoietic stem cell transplantation (EJHaem 2022;3:1025)
- 6 year old boy with fever, headache, abdominal pain and typhoid fever (J Emerg Med 2003;25:321)
- 30 year old pregnant woman with HELLP syndrome and persistent fevers upon delivery (Clin Case Rep 2018;6:2466)
- 33 year old HIV positive man with disseminated Mpox infection (N Engl J Med 2023;388:1246)
- Immediate treatment of the underlying disease in secondary hemophagocytic lymphohistiocytosis
- 8 week induction therapy with cyclosporine (HLH-2004 protocol), corticosteroids and etoposide (Am J Clin Pathol 2013;139:713)
- Cyclosporine inhibits T cell activation
- Corticosteroids suppress hypercytokinemia
- Etoposide blocks cell proliferation
- Has demonstrated a 5 year survival rate up to 54%
- Stem cell transplantation is utilized in cases of familial hemophagocytic lymphohistiocytosis
- Hemophagocytosis (histiocyte / macrophage ingestion of erythrocytes, leukocytes, platelets or their precursor cells), including erythrophagocytosis (ingestion of erythrocytes)
- Hemophagocytic cells are best visualized on bone marrow aspirate smear
- Liver sinusoids can demonstrate histiocytes with hemophagocytosis
- Splenic white pulp can be atrophic with extensive infiltration of histiocytes exhibiting hemophagocytosis
- Lymph node involvement in systemic HLH features
- Early disease shows an intense immunoblastic response
- Later stages demonstrate lymphoid depletion with massive sinusoidal infiltration by benign appearing histiocytes and hemophagocytosis
- Enlarged lymph nodes (especially of the axilla) displaying prominent erythrophagocytosis may not be indicative of systemic HLH but rather can be observed in the setting of
- Trauma to lymph drainage sites (i.e., following breast biopsy)
- Renal disease (malignant hypertension, glomerulonephritis, pyelonephritis)
- Infection
- Lymph node hemorrhage or congestion
- Sinus erythrocytosis
- Autoimmune or immunologic conditions
- Idiopathic
- Hemophagocytosis is not a specific finding and can be seen in up to 60% of patients with sepsis (Clin Case Rep 2018;6:2466)
Contributed by Sherif A. Rezk, M.D. and AFIP

Bone marrow
hemophagocytosis
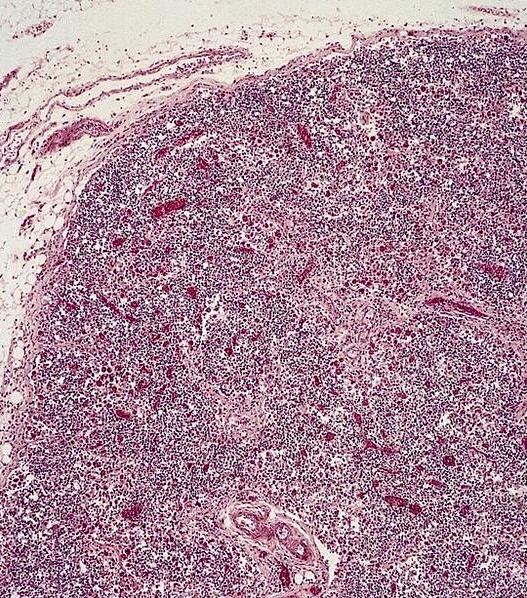
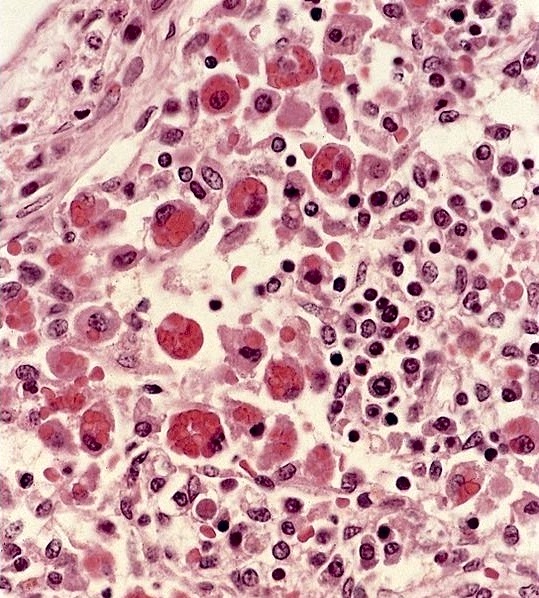
Reactive hemophagocytosis in lymph node
Erythrophagocytosis
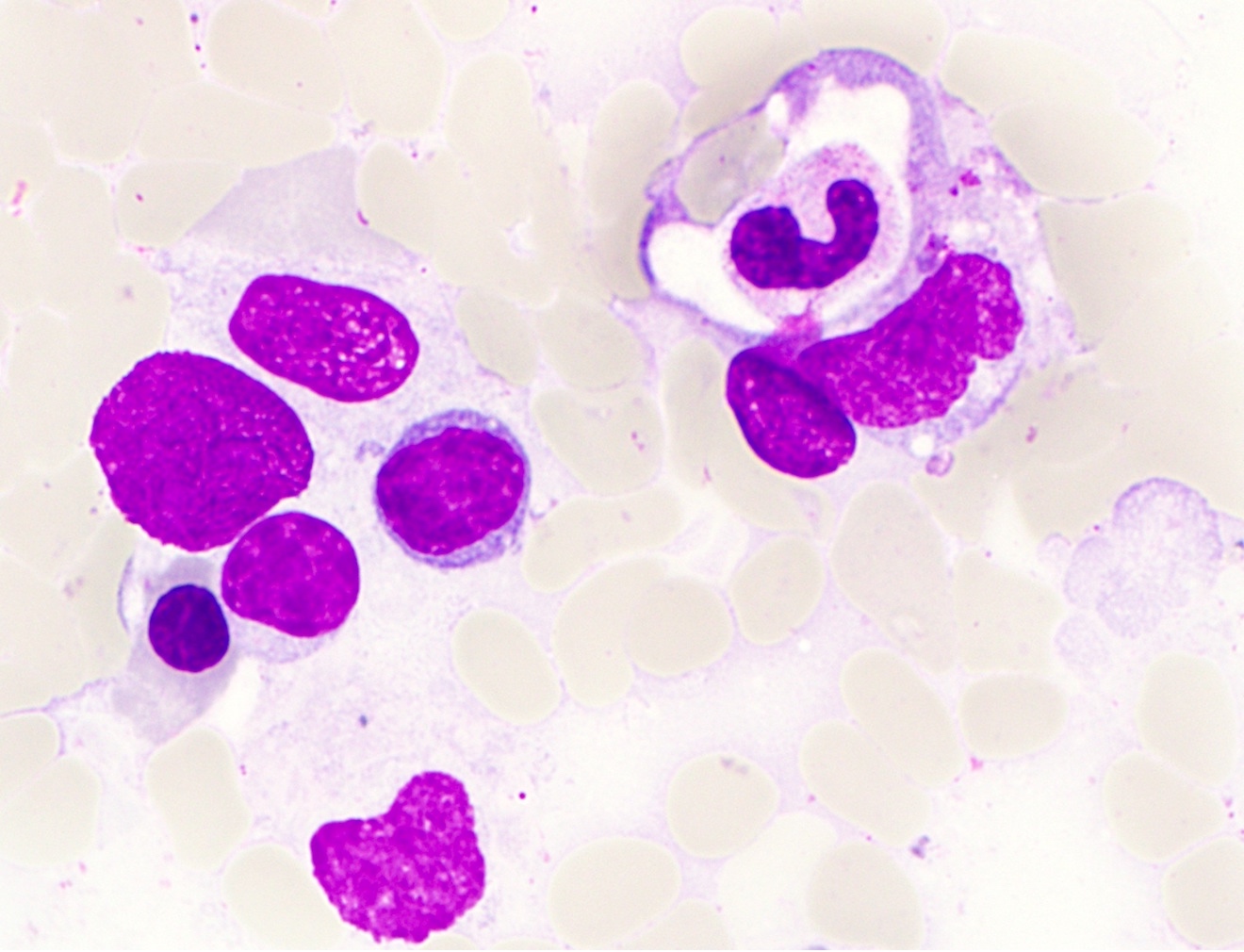
Neutrophil
hemophagocytosis
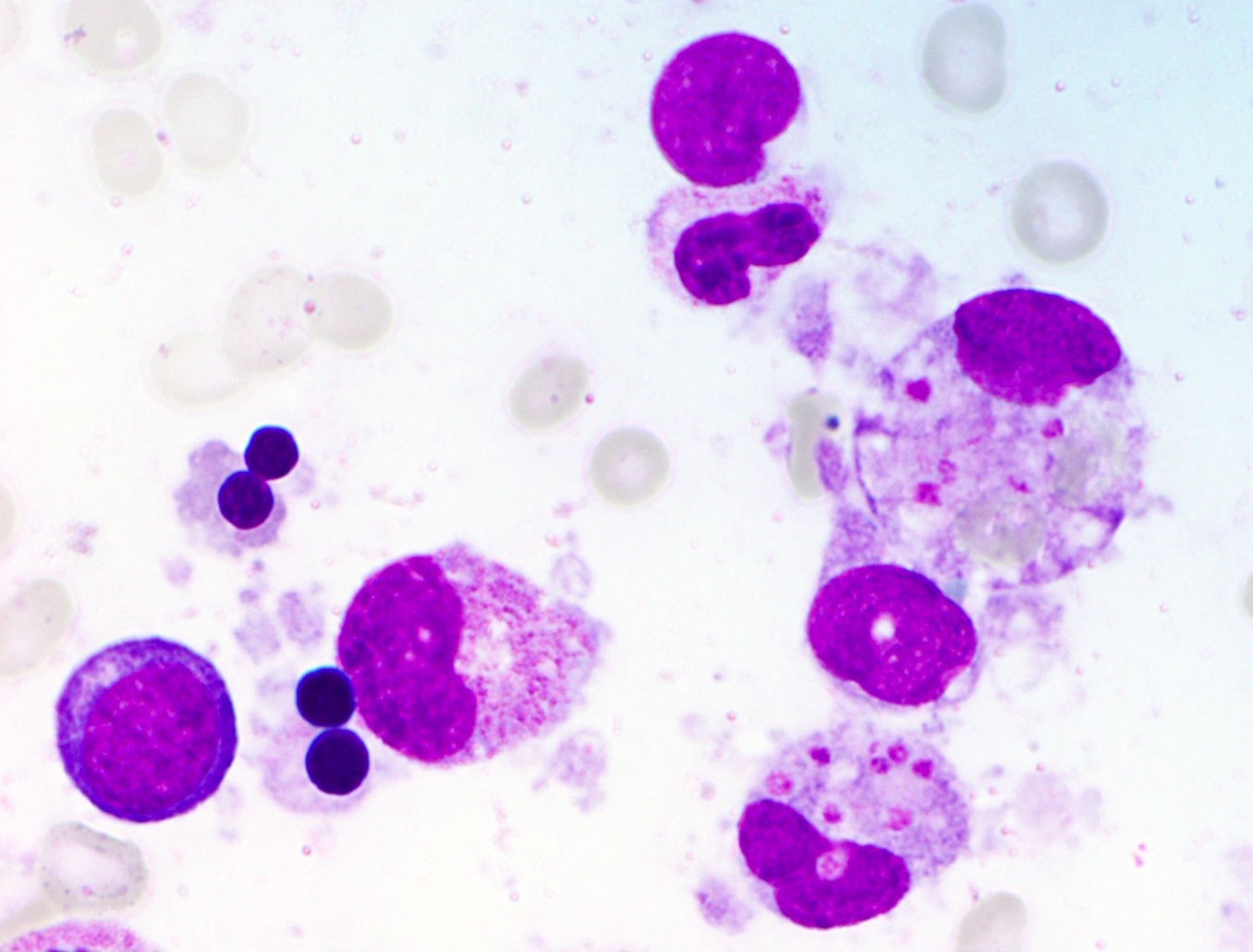
Platelet
hemophagocytosis
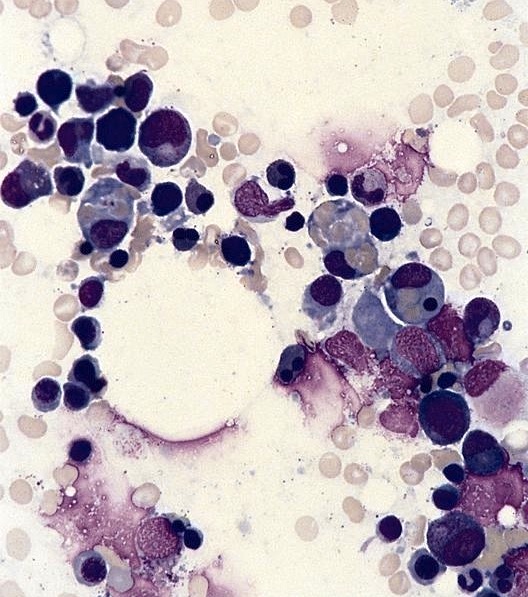
Bone marrow in
reactive
hemophagocytic
syndrome
- Absent or diminished intracellular staining for perforin can indicate mutations in the protein encoding gene
- Flow cytometry may demonstrate decreased expression of CD107a (LAMP1) surface expression on cytotoxic NK / T cells (Am J Clin Pathol 2013;139:713)
- Reduced CD5 and CD7 expression has been observed in circulating and bone marrow CD8 positive T cells in some patients (Blood 2004;104:2007)
- Familial hemophagocytic lymphohistiocytosis is divided into subtypes (FHL1 - FHL5) based on the specific genetic alteration
- Involved genes include PRF1 in 50% of cases (FHL2), UNC13D (FHL3), STX11 (FHL4) and STXBP2 (FHL5)
- Gene responsible for FHL1 has yet to be identified but is located at 9q21_3-22
- Some cases of FHLH can be seen in the setting of macrophage overactivation due to gain of function variants in NLRC4 and deficiency of XIAP (Blood 2020;135:1332)
- Secondary (acquired) hemophagocytic lymphohistiocytosis may demonstrate missense or splice site changes in PRF1, MUNC13-4 and STXBP2 (Blood 2011;118:5794)
- Bone marrow, right iliac crest, aspirate smears, touch imprints, clot and trephine biopsy section:
- Normocellular marrow with trilineage hematopoiesis, marked erythroid hyperplasia and evident hemophagocytosis
- By immunohistochemistry, increased phagocytic histiocytes identified
- Sepsis due to various causes:
- Symptoms of hemophagocytic lymphohistiocytosis (including fever, malaise, myalgia, etc.) can often be seen in acute sepsis of various causes and the diagnosis of hemophagocytic lymphohistiocytosis requires fulfillment of the criteria defined previously
- Myeloproliferative neoplasm / myelodysplastic syndrome (MDS / MPN):
- Hemophagocytic lymphohistiocytosis can also present with cytopenias; careful examination of the bone marrow for dysplasia in various cell lineages will aid in diagnosing MDS / MPN
- Rosai-Dorfman disease:
- Prominent histiocytic infiltration may also be present in hemophagocytic lymphohistiocytosis; however, Rosai-Dorfman disease rarely presents with constitutional symptoms and generally features emperipolesis, as opposed to erythrophagocytosis
- Langerhans cell histiocytosis:
- In spite of the prominent histiocytes sometimes observed in hemophagocytic lymphohistiocytosis, these cells generally lack nuclear grooves and do not demonstrate erythrophagocytosis
- Leishmaniasis / histoplasmosis:
- Platelets phagocytosed by histiocytes in hemophagocytic lymphohistiocytosis can visually mimic intracellular Leishmania donovani or Histoplasma capsulatum organisms
- Clinical history and microbiology analyses, including indirect fluorescent antibody (IFA) testing and polymerase chain reaction (PCR), can confirm the infectious diagnosis
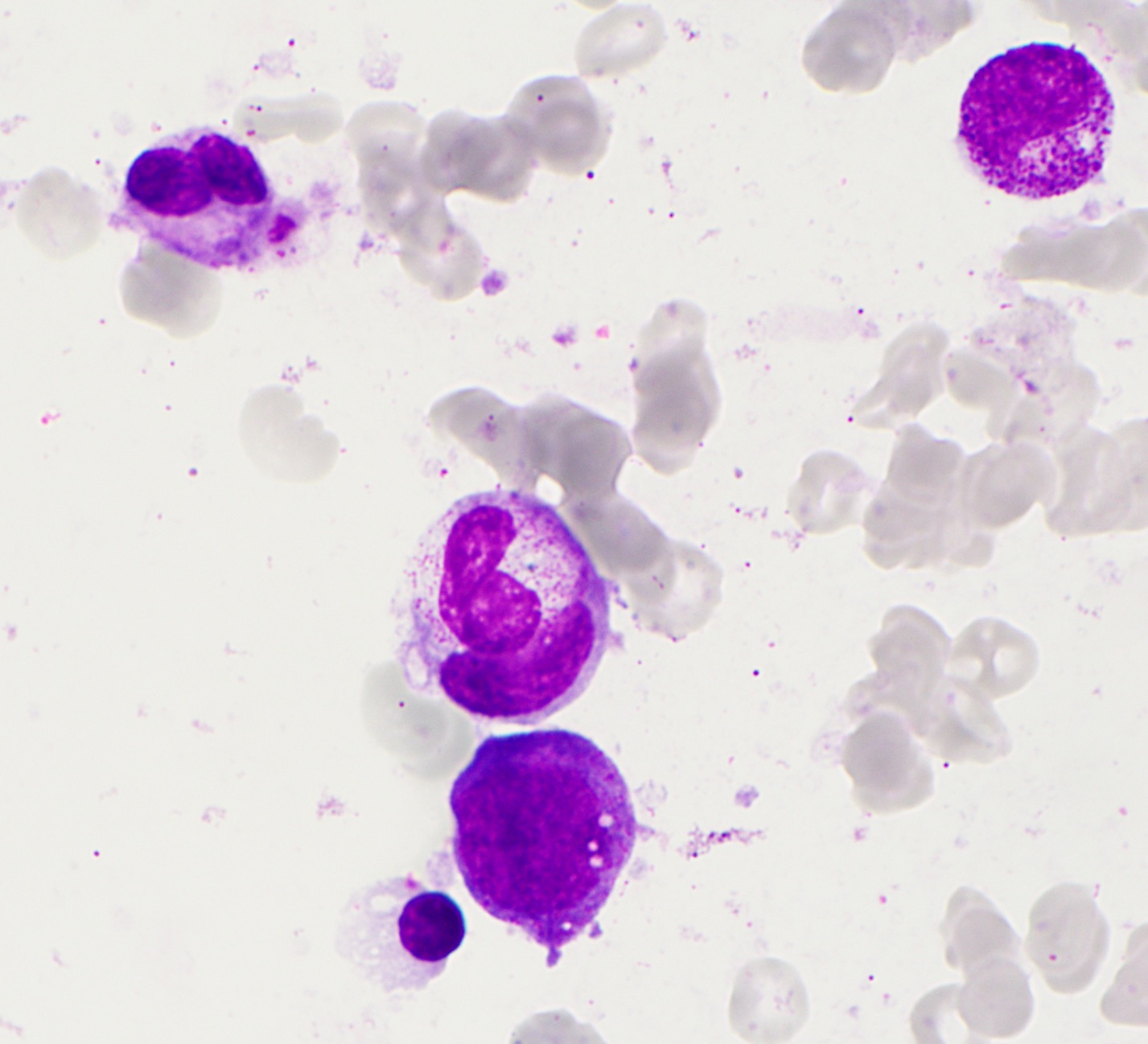
- A 45 year old Latina woman presents with mild normocytic anemia and thrombocytopenia. A representative photograph of her bone marrow aspirate smear is provided. Which of the following diagnoses is most likely in her medical history and could be contributing to the current findings in the bone marrow biopsy?
- Acute human immunodeficiency virus (HIV) infection
- Acute myeloid leukemia
- Panniculitis-like T cell lymphoma
- Triple negative breast cancer
Comment Here
Reference: Hemophagocytic lymphohistiocytosis
- Among the infectious etiologies of hemophagocytic lymphohistiocytosis, which of the following has been most commonly implicated?
- Cytomegalovirus
- Epstein-Barr virus
- Influenza virus
- Parvovirus B19
Comment Here
Reference: Hemophagocytic lymphohistiocytosis
- Must have high index of suspicion for opportunistic infections (acid fast bacilli, Parvovirus B19, Pneumocystis, Histoplasma) in HIV+ patients
- Some recommend GMS and AFB stains on all marrow specimens in AIDS patients, AFB particularly where TB is endemic (Indian J Pathol Microbiol 2005;48:7)
- HIV patients also have increased incidence of lymphoma / other malignancies, reduced iron stores (Am J Clin Pathol 2004;121:393)
- Diagnostic yield for microorganisms is high for bone marrow biopsies (34%) and culture (27%), less (8%) for aspirate smears (J Infect 2007;54:362)
- Acute HIV syndrome (initial infection): sore throat, fever, lymphadenopathy (50 - 70% of patients)
- Opportunistic infections are due to progressive decrease in CD4+ T cells, often below 200/microliter
- Malignancies (secondary and virus associated) may be EBV associated: Burkitt lymphoma, Hodgkin lymphoma, plasmablastic lymphoma or HHV8 associated: Kaposi sarcoma, primary effusion lymphoma
- Neurodegenerative disease: PML / progressive multifocal leukoencephalopathy
- Other: multicentric Castleman disease (HHV8 associated), atherosclerosis
- Hematologic findings: anemia, neutropenia, lymphopenia, thrombocytopenia
- 44 year old AIDS patient presenting with cutaneous Kaposi sarcoma and bacillary angiomatosis (AIDS Patient Care STDS 2002;16:573)
- HIV+ man who died after bleeding from mouth and nose (University of Pittsburgh: HIV Positive Man Died Following Bleeding from Mouth and Nose)
- Usually hypercellular (early in disease course) but interstitium may be loosely structured and hypocellular (Arch Anat Cytol Pathol 1991;39:137)
- Hypocellularity in advanced disease and following potent therapy
- Almost always increased plasma cells
- Often scattered macrophages, dysplastic hematopoietic cells (J Assoc Physicians India 2005;53:705)
- Marrow fibrosis, proportional to number of stained adventitial reticular cells (Arch Pathol Lab Med 2005;129:1137)
- Variable acid fast bacilli without granulomas
- Polymorphous reactive lymphoid hyperplasia
- Proliferation of immunoblasts
- Naked or pyknotic megakaryocyte nuclei (nonspecific, Mod Pathol 1994;7:166)
- Focal fibrinoid necrosis or gelatinous transformation
- Rarely macrophages with PAS+ or GMS+ Pneumocystis jiroveci
- Iron storage abnormalities
- No light chain restriction in lymphocytes / plasma cells (unless malignant)
- Hodgkin lymphoma
- Peripheral T cell lymphoma: rare, T cell receptor rearrangements
- A finding, not a diagnosis
- Usually reactive (infection, blood loss) but also other benign or malignant causes
- Benign causes:
- Affecting predominantly one hematopoietic line:
- Erythroid hyperplasia: often due to hemolytic anemia, high altitude or hypoxia of any other cause, renal disease, hemoglobinopathies, familial polycythemia
- Myeloid hyperplasia: infections, compensatory hyperplasia after cell destruction, chronic blood loss, other stressors, drugs (G-CSF)
- Megakaryocytic hyperplasia: often due to immune thrombocytopenia but also other types of thrombocytopenia; megakaryocytes may have striking nuclear changes
- Affecting predominantly one hematopoietic line:
- Malignant causes:
- Affecting predominantly one hematopoietic line:
- Erythroid: polycythemia vera, dysregulated erythropoietin production by various neoplasms
- Myeloid: myeloid neoplasms (AML, CML, MDS, other MPD besides CML)
- Megakaryocytic: MPD (essential thrombocythemia, CML, primary myelofibrosis), acute megakaryoblastic leukemia
- Affecting predominantly one hematopoietic line:
- Other causes include lymphoma, metastases
- Increased hematopoiesis
- Reduced adipose tissue
Images hosted on other servers:

Megaloblastic anemia
- Normal forms of iron stores in bone marrow:
- Ferritin in erythroblasts
- Hemosiderin in macrophages (normally small and inapparent without special stains)
- Abnormal iron deposits in bone marrow:
- Due to excessive accumulation of hemosiderin within cells of mononuclear phagocyte system, associated with:
- Ringed sideroblasts: seen in nonneoplastic (see below) or neoplastic conditions
- Anemia of chronic disease
- Abnormal iron deposits in other cell types, e.g. plasma cells
- Sideroblasts: normally present; erythroblasts with siderosome, an intracytoplasmic membrane bound ferritin molecule; bone marrow usually has 1 - 2 small siderotic granules in > 10% normoblasts
- Ringed sideroblasts: abnormal finding; 5+ iron granules encircling 1/3 or more of nuclear circumference in erythroid precursors, due to arrangement of iron granules in a ring form around the mitochondria
- Hemosiderin: may be normal or abnormal
- An intracellular iron storage complex which appears to be a complex of ferritin, denatured ferritin and other materials
- The iron within hemosiderin deposits is usually unavailable to supply iron when needed
- Iron is required by many biochemical reactions (i.e. oxidation reduction reactions) but may be toxic if not properly contained
- Approximately 60% of body iron is associated with hemoglobin in circulating red blood cells
- Daily erythropoiesis requires 25 - 30 mg iron per day, provided by macrophages through recycling of heme iron following phagocytosis of senescent red blood cells and heme catabolism
- Intestinal iron absorption (1 - 2 mg per day) only compensates for daily iron losses
- Sideroblastic anemia:
- Bone marrow produces ringed sideroblasts rather than healthy erythrocytes; although body has iron available, it cannot incorporate it into hemoglobin
- Anemia of chronic disease (ACD):
- Hepcidin, a 25 amino acid peptide synthesized in hepatocytes, has central role
- Secreted in plasma and rapidly removed in urine
- Negative regulator of intestinal iron absorption and heme iron recycling by macrophages
- Hepcidin synthesis is stimulated by iron or inflammation (mostly IL6)
- Hepcidin synthesis is repressed by iron deficiency and by all conditions that stimulate bone marrow erythropoiesis such as anemia, bleeding, hemolysis, dyserythropoiesis or erythropoietin injections
- Underlying mechanism for hemochromatosis (adult / child) is a defect in activation of hepcidin normally triggered by iron excess
- A defect in hepcidin repression is responsible for iron refractory iron deficiency anemia (IRIDA)
- Reduced hepcidin filtration in renal insufficiency contributes to associated anemia; stimulation of hepcidin synthesis by inflammation is a major cause of anemia of chronic disorders
- Inflammatory cytokines have important role:
- Decrease ferroportin expression and probably directly blunt erythropoiesis by decreasing the ability of bone marrow to respond to erythropoietin
- Promote production of white blood cells, which causes fewer stem cells to differentiate into red blood cells, even at normal erythropoietin levels and even aside from the effects of hepcidin
- Inhibit erythropoietin release from kidney and survival of circulating red cells is shortened
- Hepcidin, a 25 amino acid peptide synthesized in hepatocytes, has central role
- Sideroblastic anemia:
- Definition: > 5 iron granules, > 1/3 circumference, larger than normal
- Acquired, clonal: refractory anemia with ring sideroblasts (RARS): older adults, ineffective dyserythropoiesis, macrocytic or normocytic hypochromatic anemia, > 15% ringed sideroblasts in bone marrow
- Acquired, reversible: toxins and medications
- Alcohol: most common cause of acquired sideroblastic anemia; has direct toxic effect on erythroid precursors
- Copper deficiency: inadequate copper uptake or secondary to zinc overload; vacuoles in erythroid and myeloid precursors
- Isoniazid: inhibition of pyridoxine metabolism
- Lead: inhibition of δ-ALA dehydratase and heme synthetase
- Congenital: X Linked Sideroblastic Anemia (XLSA): defective aminolevulinic acid synthase (ALAS2) → ↑ affinity to cofactor, pyridoxal-5'-phosphate → ↑ protoporphyrin and heme; microcytic hypochromic anemia, some patients respond to pyridoxine (vitamin B6)
- Congenital: Pearson marrow pancreas syndrome: congenital large deletions or duplications of mitochondrial DNA → sideroblastic anemia and cytoplasmic vacuoles, acidosis and exocrine pancreatic insufficiency
- Anemia of chronic disease (ACD)
- Inflammatory cytokines promote production of white blood cells → fewer stem cells differentiating into red blood cells, even at normal erythropoietin levels and even aside from effects of hepcidin
- In short term, the above effect keeps more iron away from bacterial pathogens in human body (almost all bacteria are iron dependant for metabolism and reproduction) while producing more immune cells in response to infection
- If inflammation continues, the inability of bone marrow to produce red blood cells interferes with normal body functions, as red blood cells require massive amounts of iron for hemoglobin and oxygen transportation
- ACD can also result from Hodgkin lymphoma, lung and breast carcinomas; also noninfectious inflammatory diseases (rheumatoid arthritis, systemic lupus erythematosus)
- Note: all anemia in those with chronic disease is NOT anemia of chronic disease; ACD is often considered separate from anemia of renal failure (due to poor production of erythropoietin) or associated with AZT or other medications
- Laboratory tests (see below)
- Bone marrow biopsy and aspiration
- Cytogenetic and molecular analysis to rule out acquired clonal disorders (RARS in MDS) and congenital disorders
- Reticulocyte count: best test to differentiate abnormalities of production and survival
- Ferritin:
- Storage form of iron; levels directly correlate with total body iron storage
- Most sensitive lab test for iron deficiency anemia
- Also an acute phase reactant, so must obtain CRP (detects inflammation) to rule out elevated ferritin due to inflammation
- Transferrin: iron binding blood plasma proteins for transportation of iron
- Total iron binding capacity (TIBC): blood capacity to bind iron with transferrin
- Percent transferrin saturation: serum iron/TIBC x 100%
- Serum soluble transferrin receptor (STFR):
- Carrier protein for transferrin
- Truncated soluble receptors, shed into blood from erythroblasts in the marrow
- Correlate with iron status of erythroblasts and total mass of erythron; inversely related to iron supply to cells; not altered by inflammation
- STFR is significantly increased in iron deficiency anemia but is normal / only mildly increased in ACD
- Free erythroid protoporphyrin (FEP): increased if decreased iron supply or decreased iron incorporation into heme; normal in thalassemia
- Sideroblastic anemia: ↑ serum iron, ↑ marrow iron, ↑↑ percent saturation, ↑↑ ferritin, ↓ TIBC, ↓ STFR
- ACD: ↓ serum iron, ↓ marrow iron, > 15% percent saturation, ↑ ferritin, ↓ TIBC, normal or ↑ STFR
- 52 year old woman with sideroblastic anemia secondary to zinc toxicity (Blood 2013;122:311)
- Sideroblastic anemia associated with nonneoplastic conditions
- Transfusion may be required in severe cases if patients do not respond to erythropoietin
- Desferrioxamine, a chelating agent, is used for iron overload from transfusions; also therapeutic phlebotomy
- In certain severe cases, bone marrow transplant may be an option although data on success rates are limited
- For isoniazid induced sideroblastic anemia, moderate / high levels of pyrodoxine (vitamin B6) correct the anemia
- Anemia of chronic disease (ACD)
- Treament of underlying chronic disease is optimal although this is only rarely possible with existing knowledge / treatments
- Parenteral iron is increasingly used
- Commercially produced erythropoietin can be helpful in some circumstances but is costly and may be dangerous
- Transfusions if patients are hemodynamically unstable (usually from complications, not the ACD alone)
- New therapeutic perspectives include agonists or antagonists of hepcidin or siRNA to reduce hepcidin synthesis
- Sideroblastic anemia associated with nonneoplastic conditions
- BM biopsy: erythroid hyperplasia with normoblastic to megaloblastic maturation and occasional dysplastic changes
- BM aspirate: Prussian blue stain demonstrates increase in ringed sideroblasts
- Anemia of chronic disease (ACD)
- BM biopsy: may show minimal morphological changes initially; when condition progresses, bone marrow may appear hypocellular with decreases in erythropoietic cells, M:E ratio may be increased with a shift in maturation sequence to later stages
- BM aspirate: Prussian blue stain demonstrates storage iron in macrophages and absence of iron in erythroid precursors
Images hosted on other servers:
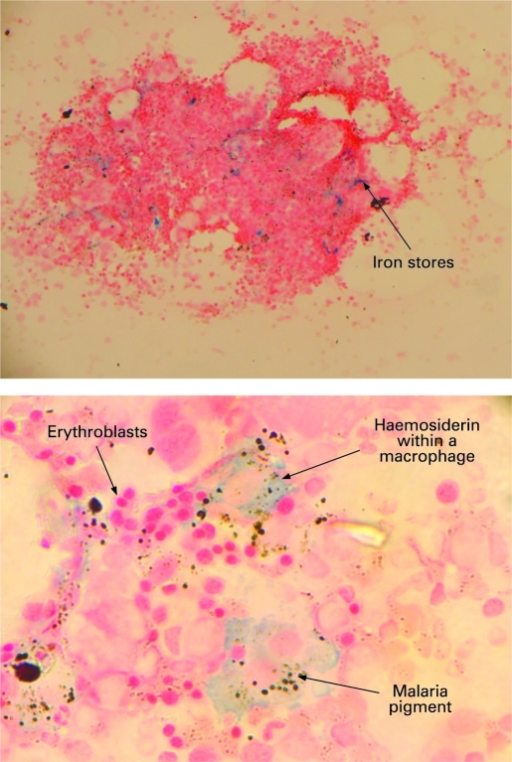
Bone marrow fragment showing iron deposits
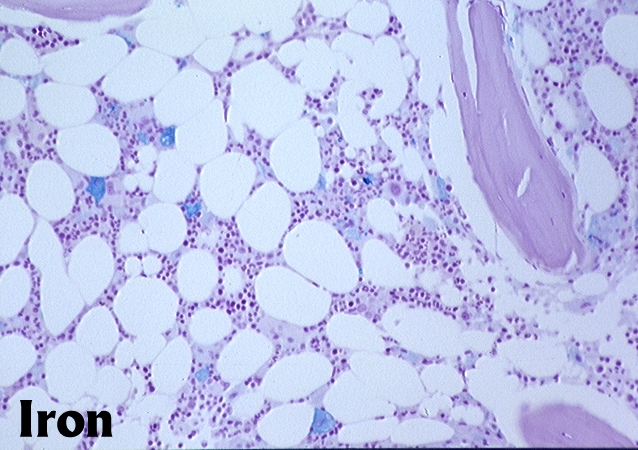
Iron
Normal iron stores
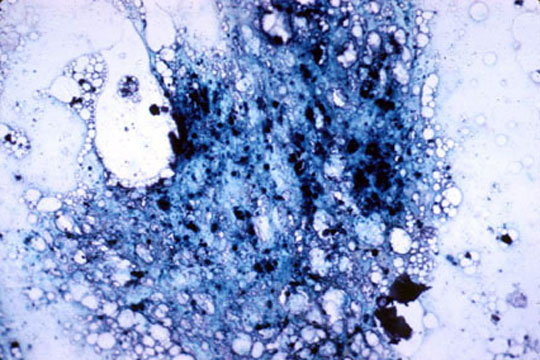
Increased iron stores
- Sideroblastic anemia associated with nonneoplastic conditions:
- Dimorphic (hypochromic and normochromic) red blood cells, basophilic stippling and Pappenheimer bodies (multiple blue dots in inregular sizes and irregularly located or in small clusters)
- Anemia of chronic disease (ACD):
- Often mild normocytic anemia while can sometimes be more severe and can sometimes be microcytic
- Morphologically, ACD often closely resembles iron deficiency anemia and many people with chronic disease are also iron deficient
- The combination of both causes of anemia produces a more severe anemia
- Both ACD and iron deficiency anemia show low reticulocyte production index
- Prussian blue stain (iron stain)
- Sideroblastic anemia: bone marrow EM demonstrates excessive accumulation of iron in macrophages as well as iron granules within erythroblasts and erythrocytes, in particular within mitochondria
Images hosted on other servers:
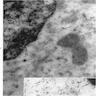
Clumped and free ferritin
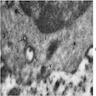
Erythroblast contain ferritin
Macrophage and erythroblast cytoplasm
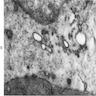
Erythroblasts with ferritin
Clumped, vesticle and free ferritin
Erythroblast with a clump of ferritin

Iron in mitochondria
Erythroblast containing mitochondrial iron
- Lymphocyte precursors originate in bone marrow
- Mature lymphocytes are present in variable proportions in the bone marrow: children < 24 months of age may have up to 60%; adults usually have < 20% but some develop lymphoid aggregates (nonparatrabecular), especially with age
- Unlike other hematopoietic cells, terminal differentiation of mature lymphocytes takes place in secondary lymphoid organs after they leave the bone marrow
- Mature to B cells (humoral immune response) or T/NK cells (cell mediated immune response)
- B cells complete most of their development within the bone marrow, but T cells are generated in the thymus from precursor cells that migrate from the bone marrow
- Recombination assembly of Ig and TCR genes represents the main process in the development of B and T cells (regulated by RAG-1, RAG-2 and TdT)
- Initially (children) have B:T cell ratio ~2:1; after 4 - 5 years of age, proportion of B cells decreases and T cells increases, reaching adult levels during adolescence (1:4 - 5)
- B cell development in marrow is dependent on CD10+ stromal cells (J Pathol 2005;205:311), which form specific, adhesive contacts with developing B lineage cells and also provide growth factors (stem cell factor, IL7, stromal cell derived factor 1)
- Earliest stem cells are in subendosteum, adjacent to inner bone surface; with maturation, B lineage cells move towards central axis of marrow; final stages of development of immature B cells occur in peripheral lymphoid organs (spleen, lymph nodes)
- Germline B cells undergo a series of Ig gene rearrangements to produce a heavy chain (μ) followed by light chains (κ or λ)
- Commitment to B lineage was initially thought to be indicated by CD79; subsequent studies showed that cytoplasmic CD22 appears to be the most specific B cell marker
- A minor subset of B cells also express CD5 (T cell marker)
- Plasma cells are last stage of B cell maturation, express CD38 but lose CD19
- T cells migrate from bone marrow to thymus and mature via TCR gene rearrangements
- Most specific T cell marker appears to be cytoplasmic CD3
- During thymic maturation sequence, T cells are initially double negative for CD4 and CD8 (pro and prethymocytes), then double positive (cortical thymocytes), then express either CD4 or CD8 (medullary thymocytes)
- In NK cells, rearrangements of T cell receptor genes do not occur; proportion of NK cells in bone marrow does not change with age
- Most NK cells (both true and cytotoxic) are morphologically large granular lymphocytes
- Lymphocytes diffusely scattered throughout interstitium; represent 10 - 15% of marrow cells in adults; aggregates often present
- Different subsets (B, T, NK) appear morphologically similar: small to large size, variable amount of cytoplasm, round nuclei, dense chromatin, variable inconspicuous nucleoli
Images hosted on other servers:
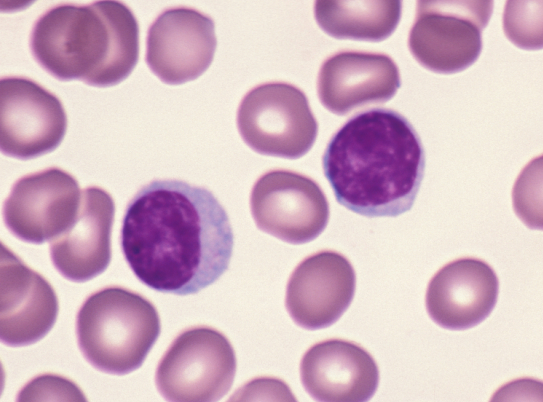
Normal lymphocytes
Images hosted on other servers:
Movement of B lineage cells with maturation
- Earliest immunologically recognizable B cells: TdT, CD34, CD79a, HLA-DR, +/-CD10 and +/-CD45; they subsequently acquire CD19, cCD22, CD22 and CD20
- Pre B cells: +/-TdT, HLA-DR, CD19, CD10, CD20, CD22, CD79a, CD45 and cμ
- Mature B cells: HLA-DR, CD19, CD20, CD22, CD79a, CD79b, CD45 and sIg
- Precursor T cells: TdT, CD34, cCD3, CD2, CD5, CD7, CD38, +/-CD1a and +/-CD45; they are initially double negative for CD4 and CD8 (pro and prethymocytes), then double positive (cortical thymocytes) and then they only express either CD4 or CD8 (medullary thymocytes)
- Mature T cells: CD2, CD3, CD5, CD7; CD45, either CD4 or CD8
- NK cells: CD16, CD56, CD57, +/-sCD3, +/-CD8, +/-CD2, +/-CD7 and +/- cytotoxic proteins (perforin, granzyme, etc.)
Images hosted on other servers:
Small lymphocyte


With cytoplasmic projections
- Benign lymphoid aggregates are composed predominantly of small lymphocytes
- Some benign lymphoid aggregates may have germinal centers made up of centrocytes and centroblasts
- Presence of lymphoid aggregates in the bone marrow biopsy should be evaluated to exclude lymphoproliferative disorders / lymphoma
- Morphology is of paramount importance for the decision to perform ancillary tests
- Small size (< 600 μm), nonparatrabecular location, confined borders, paucity of B cells are usually supportive of benign lymphoid aggregates
- Ancillary modalities such as flow cytometry and molecular studies have their diagnostic limitations and should be taken into consideration when making a diagnosis
- Benign lymphoid aggregates seen in only 1 - 2% of bone marrow biopsy specimens
- More frequently seen in autopsy specimens
- Benign lymphoid aggregates are seen in association with the following (J Clin Pathol 1999;52:294, Virchows Arch A Pathol Anat Histopathol 1991;419:261, Leuk Res 2002;26:525, Am J Clin Pathol 1999;112:844, Eur Respir J 2009;34:405, Blood 2011;117:6438, Mod Pathol 2015;28:367, J Clin Pathol 1993;46:955, Br J Haematol 2016;172:923, Haematologica 2017;102:364)
| Advanced age | |
| Tobacco use | |
| Autoimmune diseases | Rheumatoid arthritis, systemic lupus erythematosus |
| Autoimmune lymphoproliferative syndrome with germline FAS mutation | |
| Inflammatory conditions | |
| Infectious diseases | HIV, hepatitis C virus, hepatitis B virus, mycobacteria, fungal or bacterial and cytomegalovirus infections |
| Myeloproliferative neoplasms | Polycythemia vera, primary myelofibrosis, systemic mastocytosis |
| Myelodysplastic syndromes | |
POEMS syndromes
TEMPI syndromes
| |
| Rituximab and chimeric antigen receptor T cells (CAR T) treatment | |
| Idiopathic hypereosinophilic syndrome |
- Usually, no prior history of lymphoproliferative disorder; however, even if there is a history, that does not necessarily signify the presence of lymphoid aggregates as malignant
- Mean age of presentation is in the sixth decade (Hum Pathol 2013;44:512)
- Incidence increases with age (Hum Pathol 2013;44:512)
- Incidental finding on bone marrow examination
- In all ages, up to 33% of cases may have subsequent malignant lymphoid aggregates in bone marrow or other evidence of lymphoma or lymphoproliferative disorders (Blood 1974;43:389)
- If initial biopsy showed atypical lymphoid aggregates, then follow up with bone marrow biopsy should be suggested
- No treatment for benign lymphoid aggregates
- Lymphoid aggregates are assessed based on the location (intertrabecular / paratrabecular), size and appearance of lymphocyte population
- Characteristics of benign lymphoid aggregates (J Clin Pathol 1999;52:294,
Virchows Arch A Pathol Anat Histopathol 1991;419:261,
J Pathol 1997;181:451,
J Pathol 1996;178:447)
- Small size (< 600 μm)
- Uniform configuration
- Distinct outline
- Nonparatrabecular location
- Becomes smaller or disappears in deeper sections of the specimen
- Lack of cytologic atypia
- Negative for clonality (exception in autoimmune diseases)
- 5 different patterns are described for the T cell and B cell distribution in the lymphoid aggregates (Hum Pathol 2013;44:512)
Patterns CD3+ T cells / CD20+ B cells 1 Predominantly T cells 2 Mixture of T and B cells, haphazard arrangement 3 T cell core surrounded by few B cells 4 B cell core surrounded by T cells 5 Predominantly composed of B cells
- Benign lymphoid aggregates show predominantly patterns 1 - 3 with rare cases of patterns 4 and 5
- Atypical features described in lymphoid aggregates include large size, infiltrative borders, paratrabecular location and predominance of B cells (patterns 4 and 5)
- Patterns 4 and 5 are seen most commonly in lymphomas, with the exception of reactive lymphoid aggregates with BCL6 positive germinal centers (Virchows Arch A Pathol Anat Histopathol 1987;411:543)
- Some of the benign lymphoid aggregates have characteristic morphologic findings based on the etiology (Pathologica 1995;87:640, Eur Respir J 2009;34:405,
Blood 2011;117:6438,
Mod Pathol 2015;28:367,
J Clin Pathol 1993;46:955,
Haematologica 2017;102:364,
Am J Clin Pathol 1999;112:844)
Etiology Lymphoid aggregate morphologic findings POEMS syndrome Composed of mixture of T and B cells,
surrounded by lambda / kappa light chain
restricted or polytypic plasma cellsIdiopathic hypereosinophilic syndrome T cell rich aggregates HIV Large poorly demarcated lymphoid aggregates with cytologic
atypia and histiocytic proliferationsAutoimmune lymphoproliferative
syndrome with germline FAS mutationT cell rich lymphoid aggregates with CD4- / CD8- T cells Rituximab treatment T cell rich lymphoid aggregates CD19 directed CAR T therapy T cell rich lymphoid aggregates with increased CD8+ T cells
Contributed by Barina Aqil, M.D.
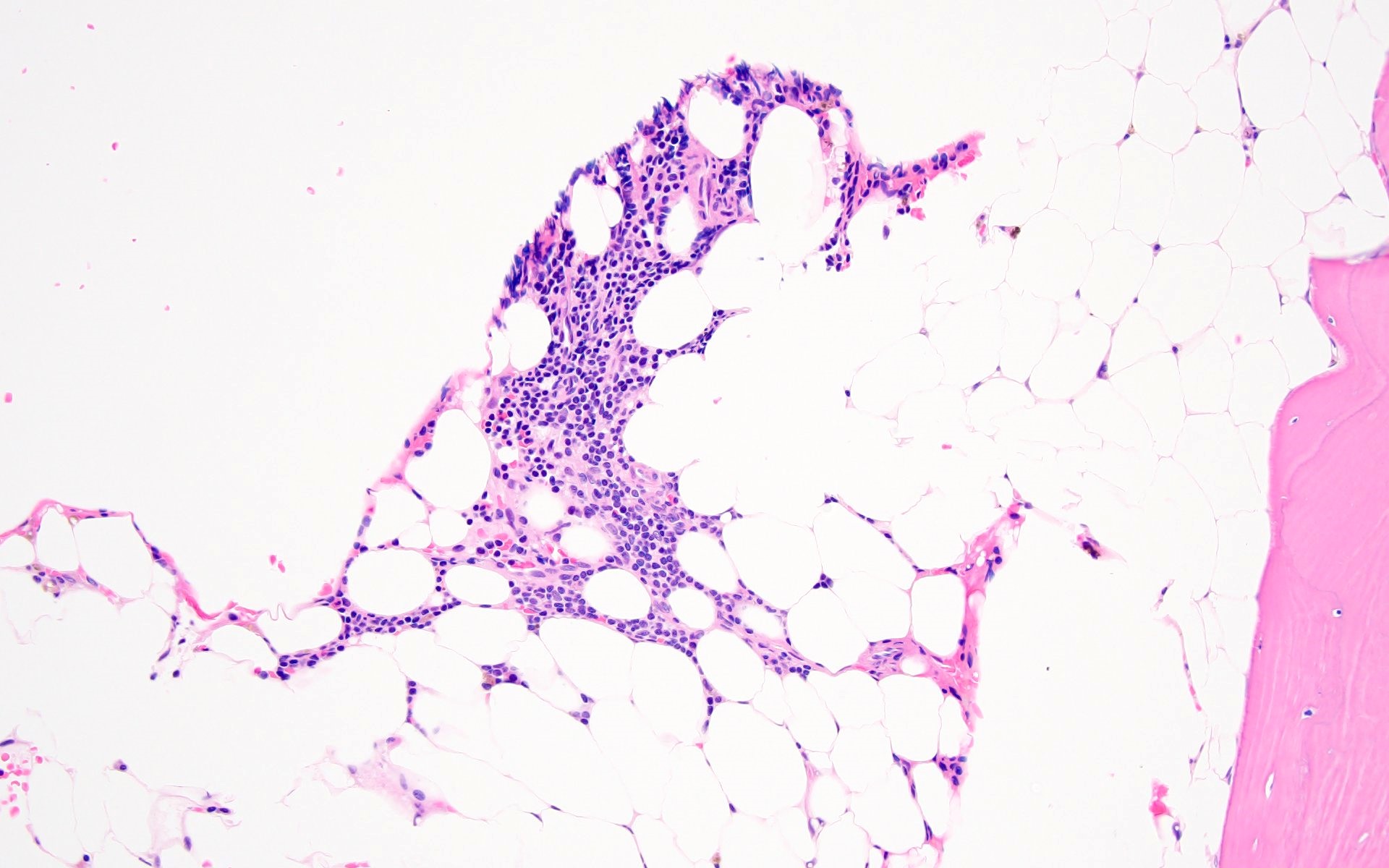
Pattern 1
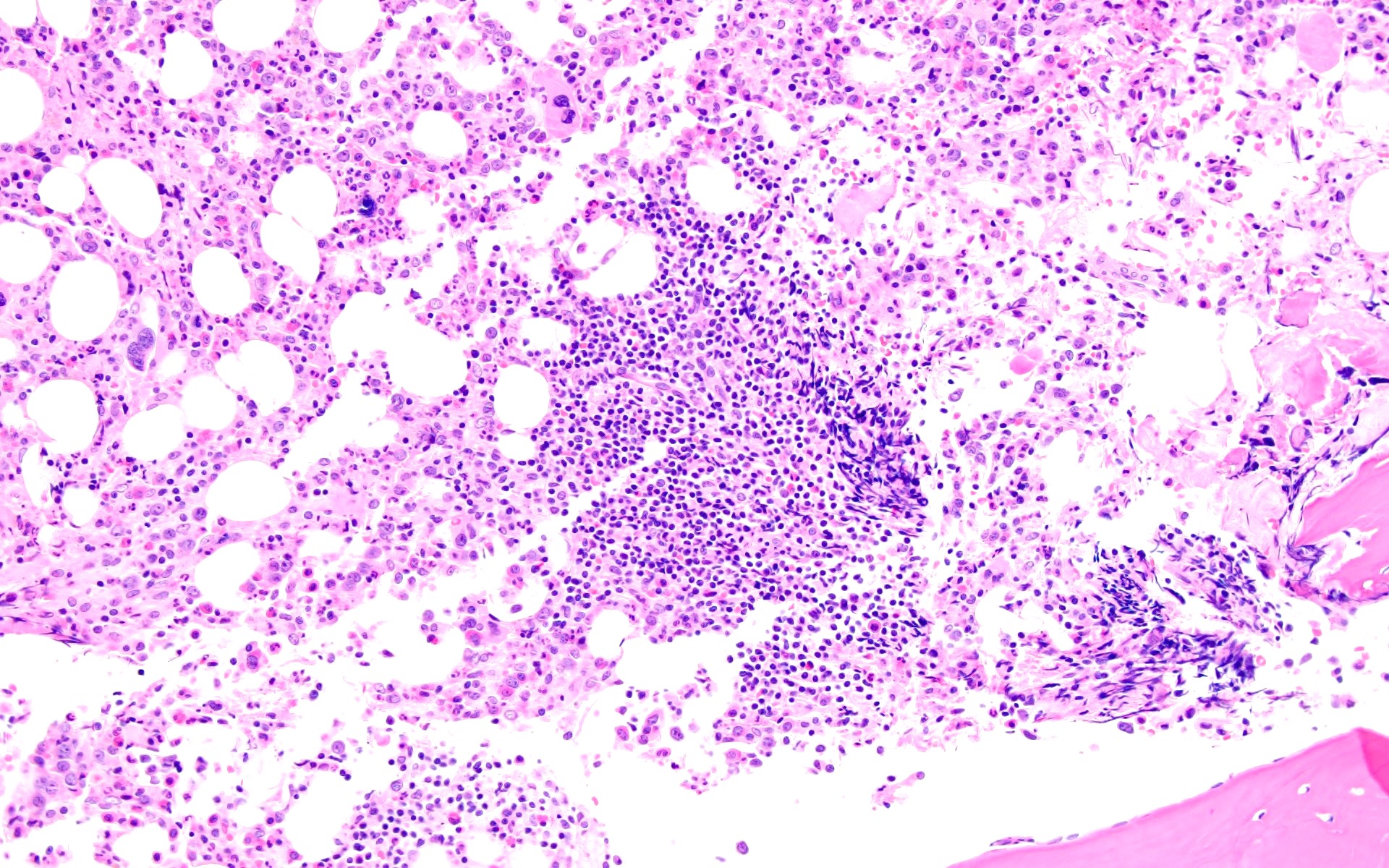
Pattern 2
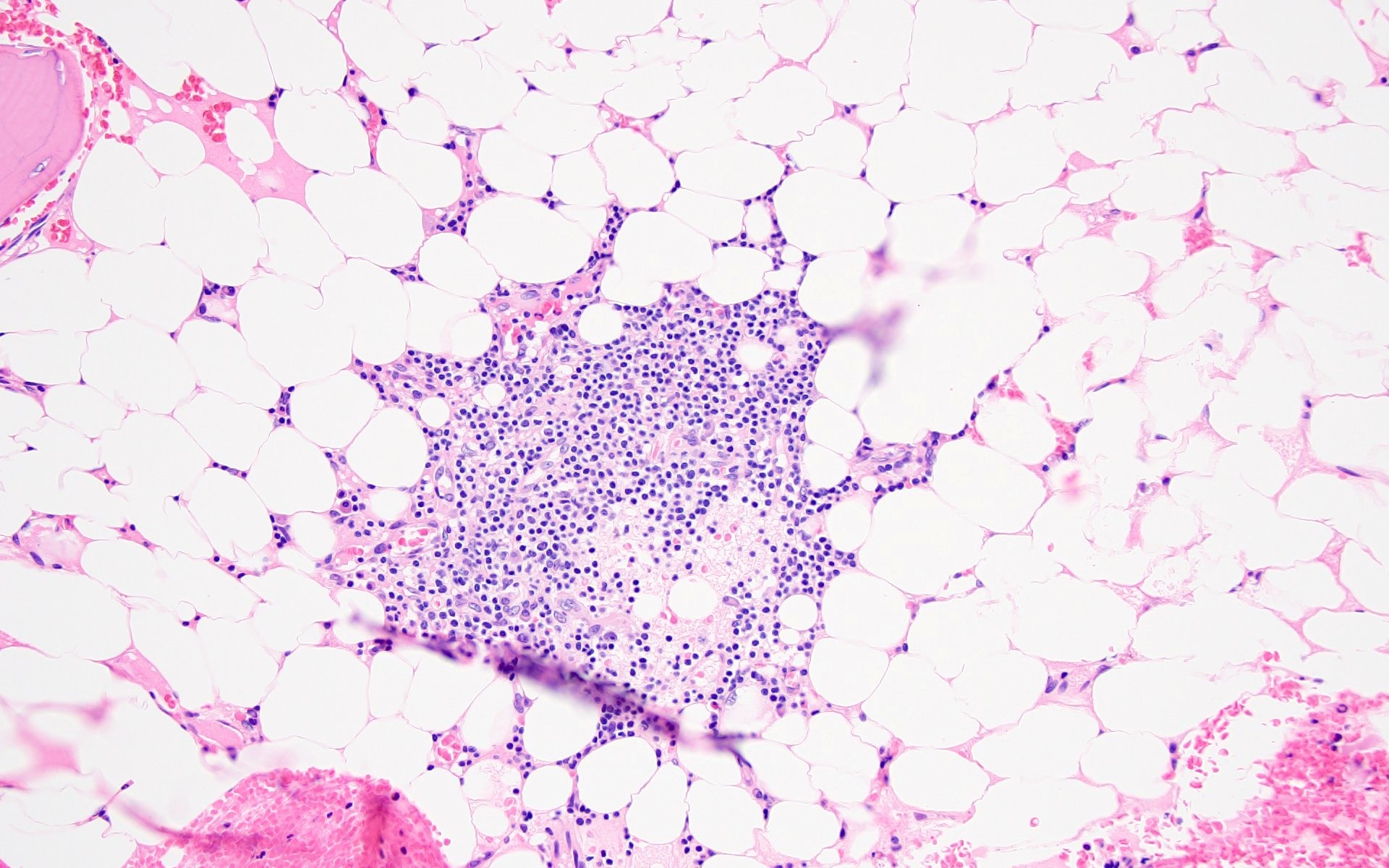
Pattern 3
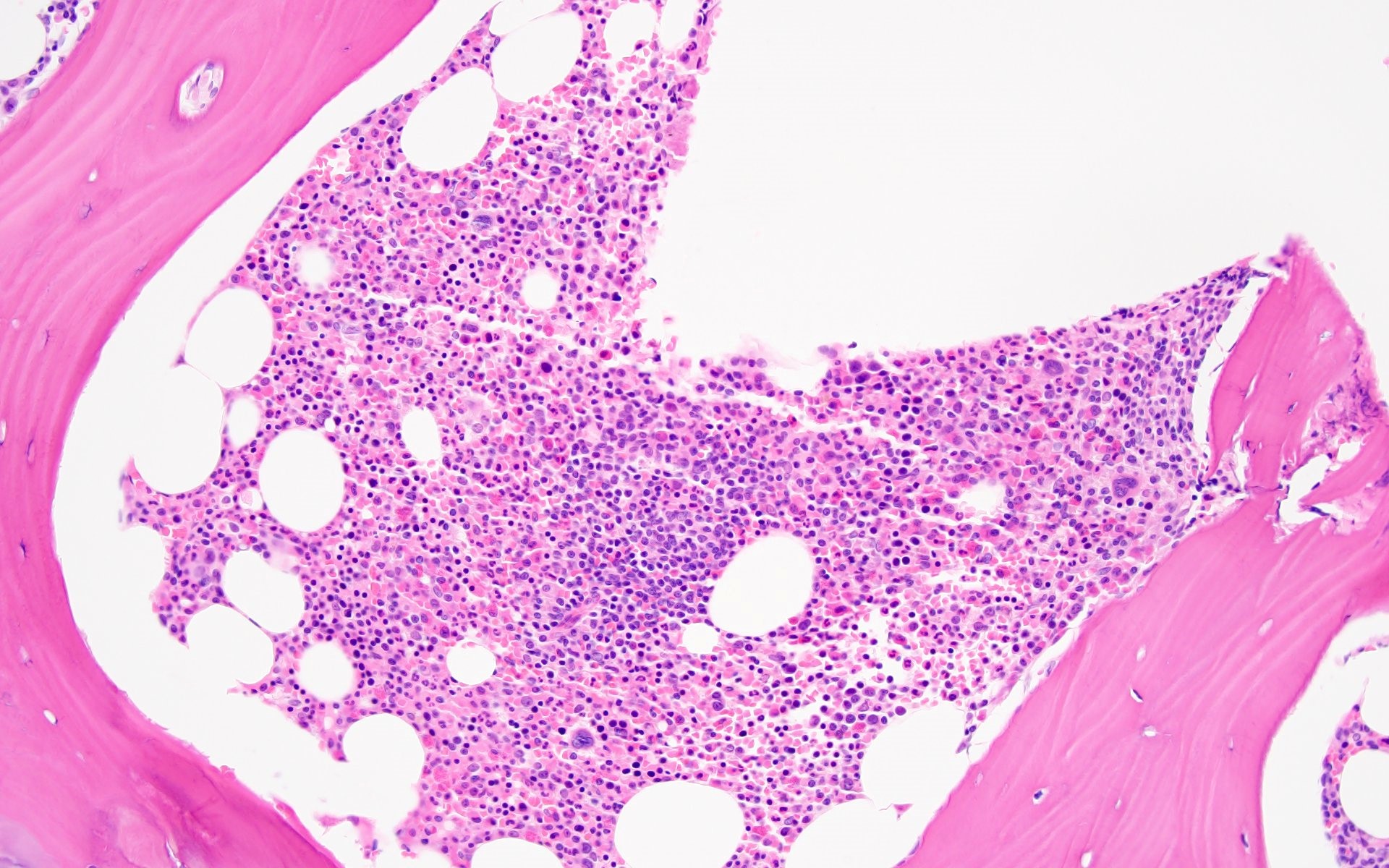
Pattern 4
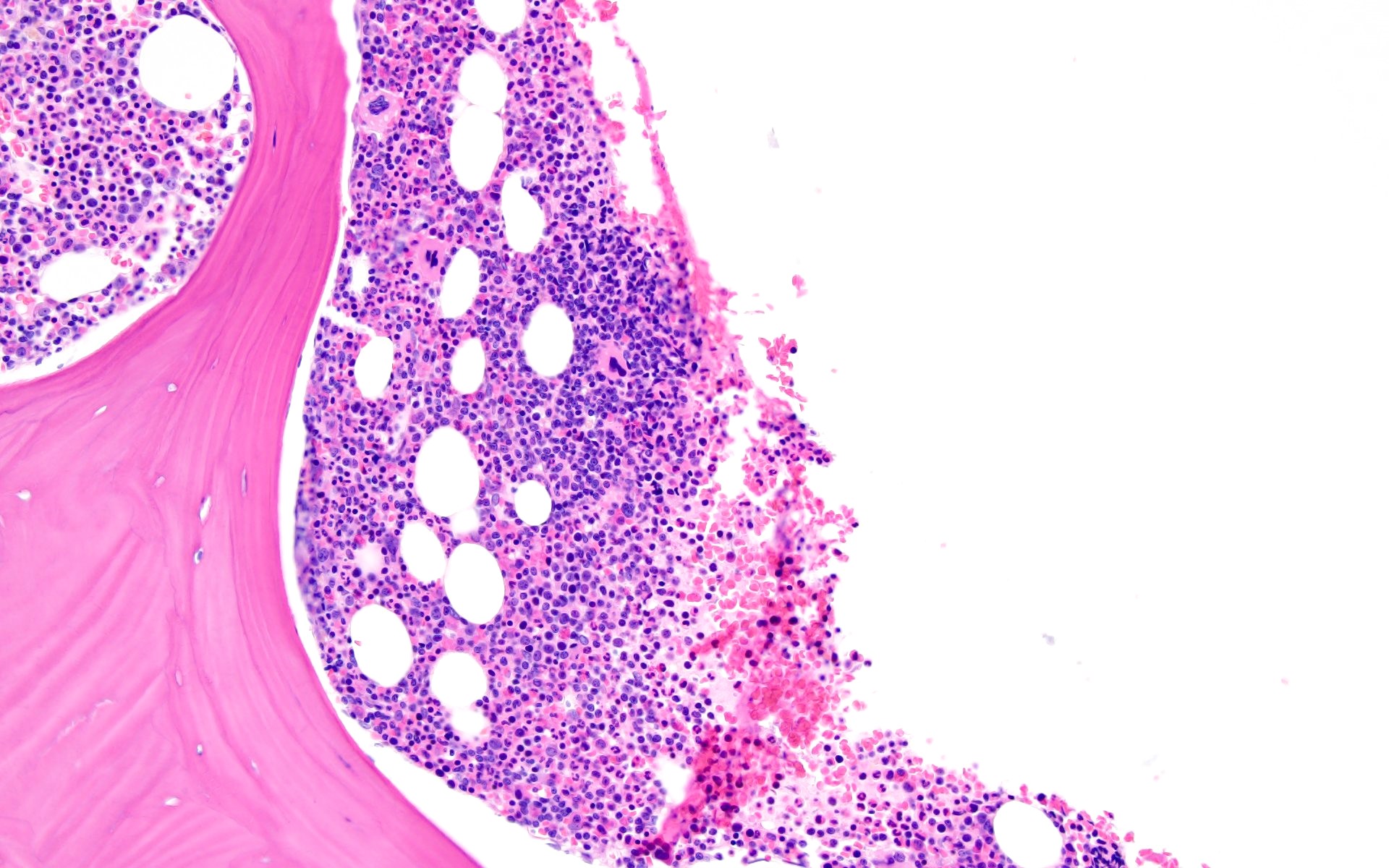
Pattern 5
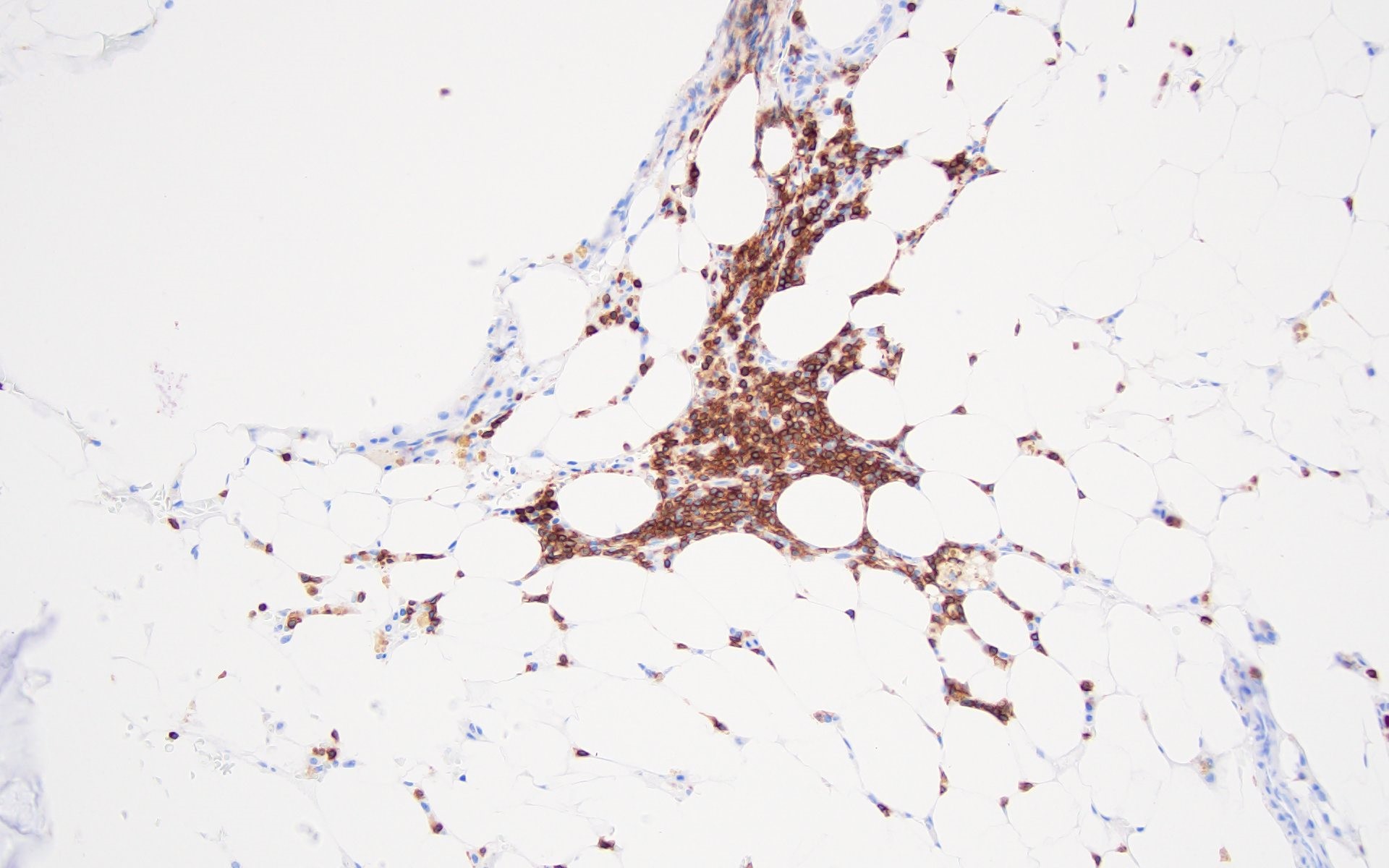
Pattern 1 (CD3)
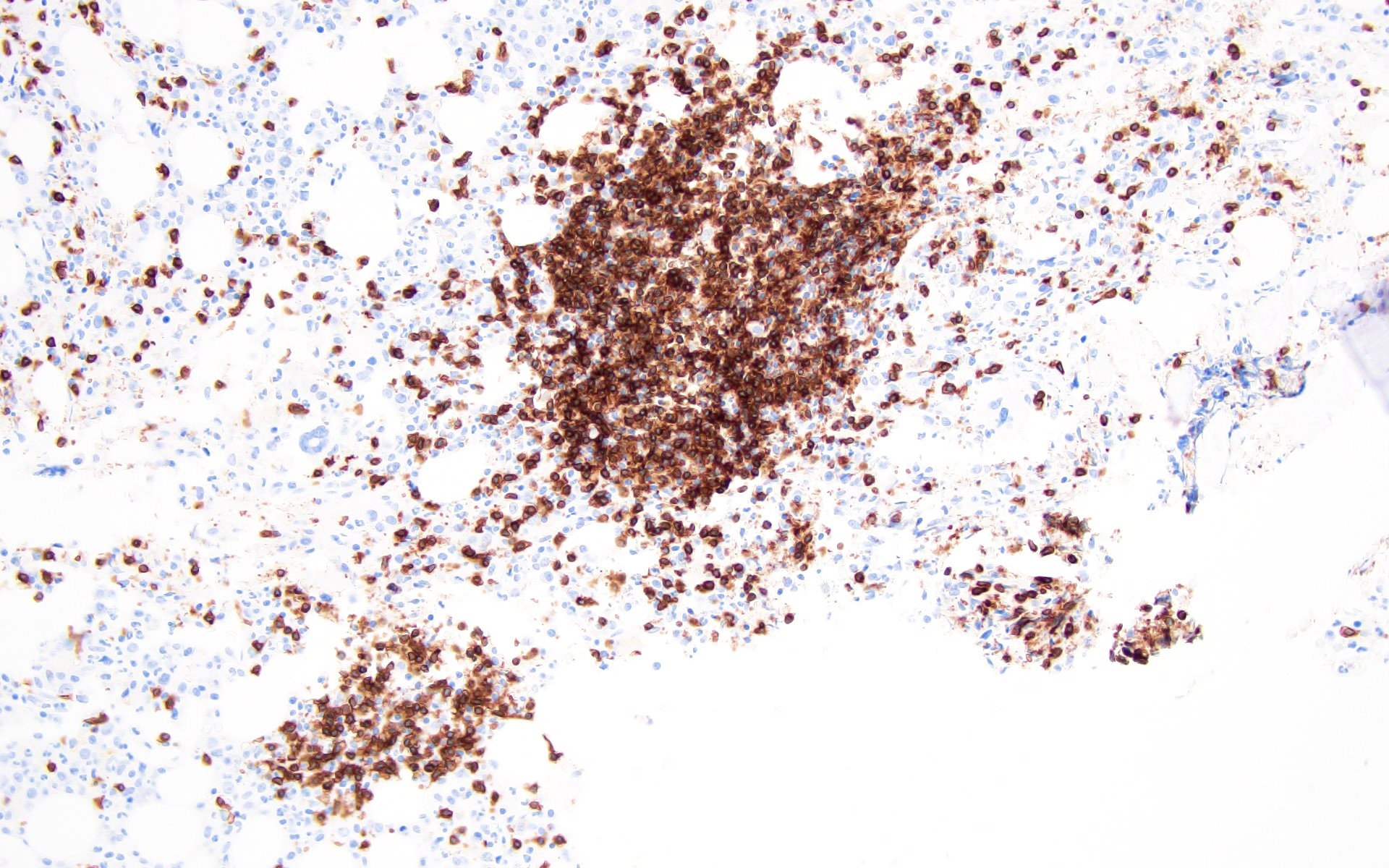
Pattern 2 (CD3)
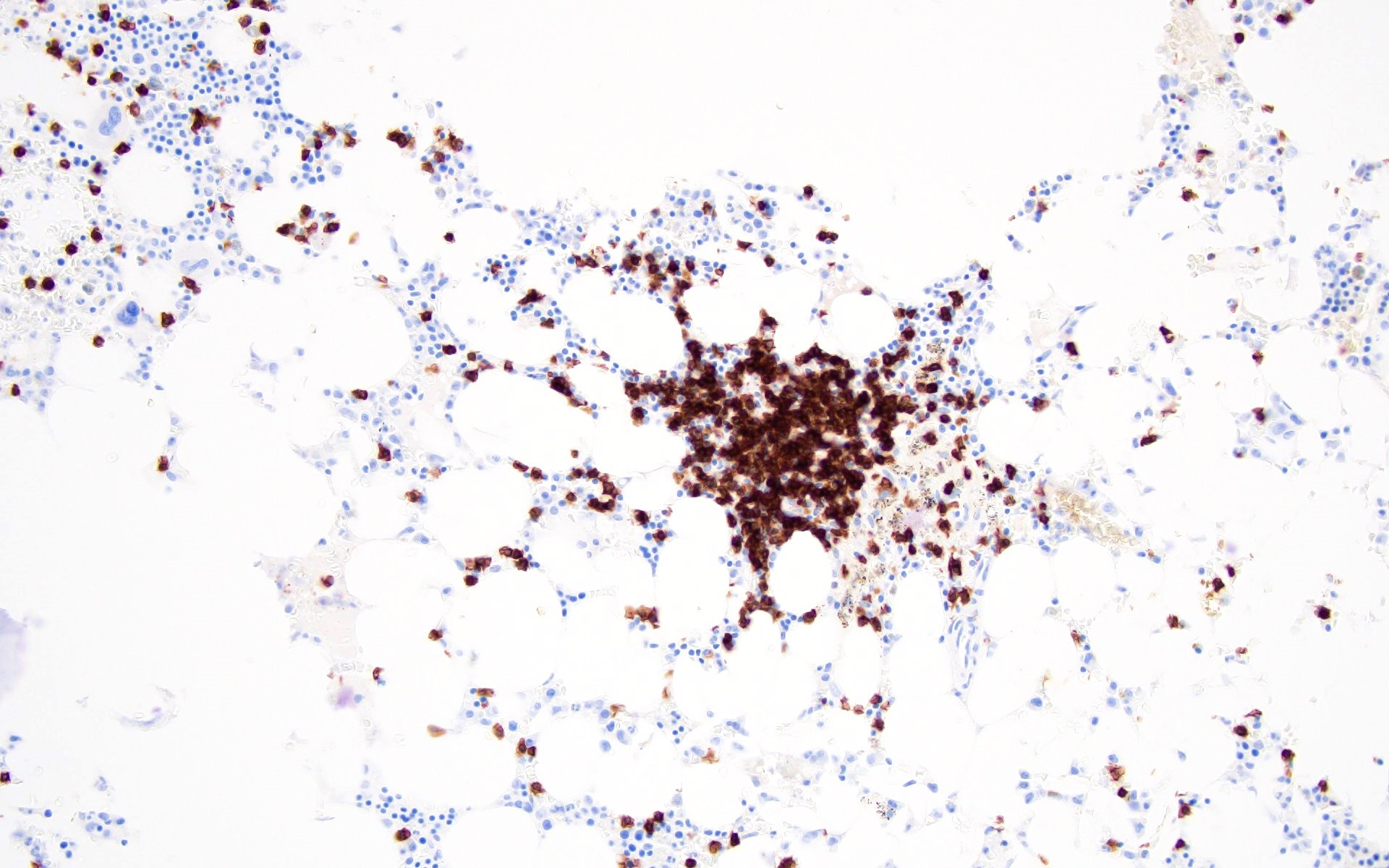
Pattern 3 (CD3)
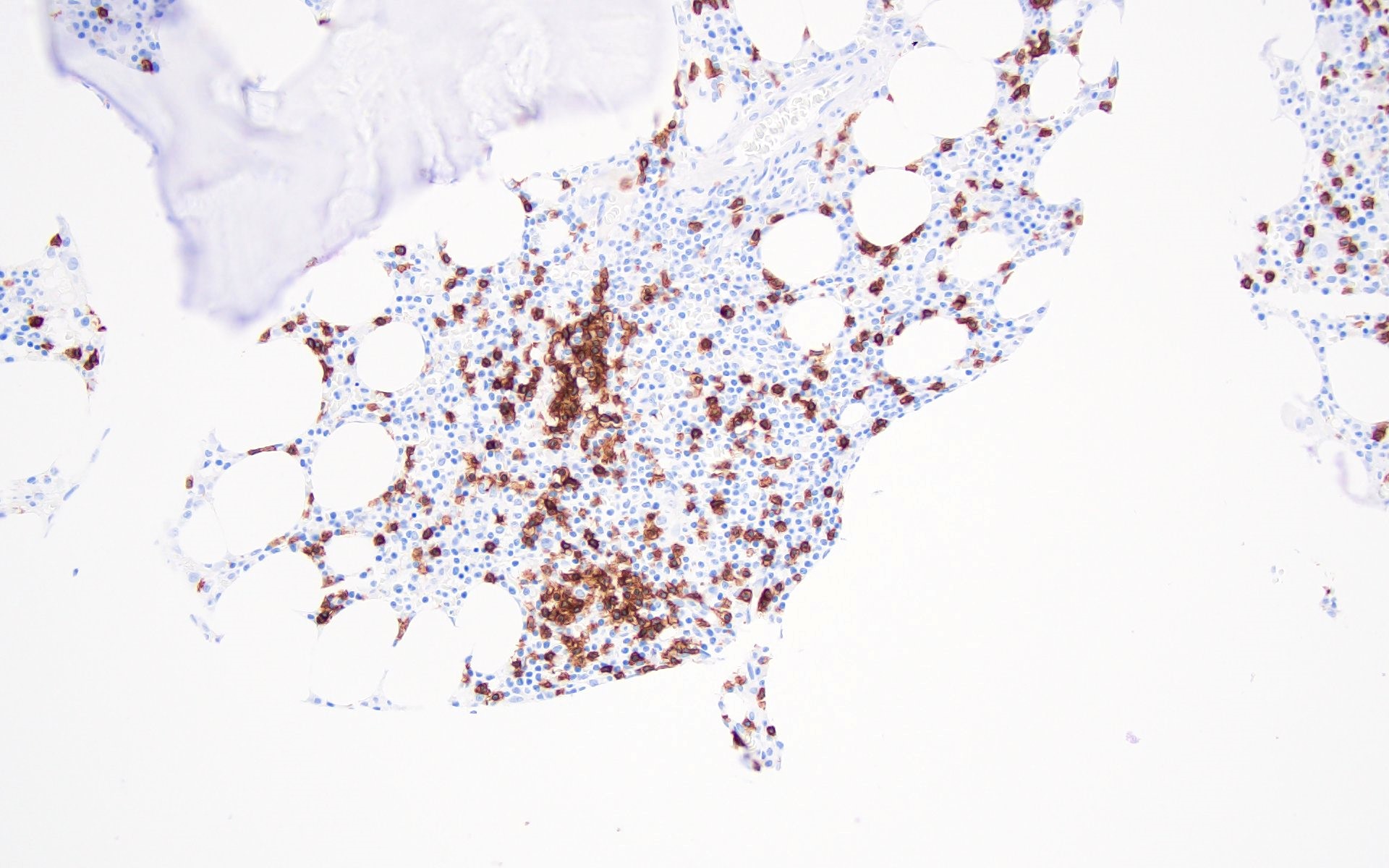
Pattern 4 (CD3)
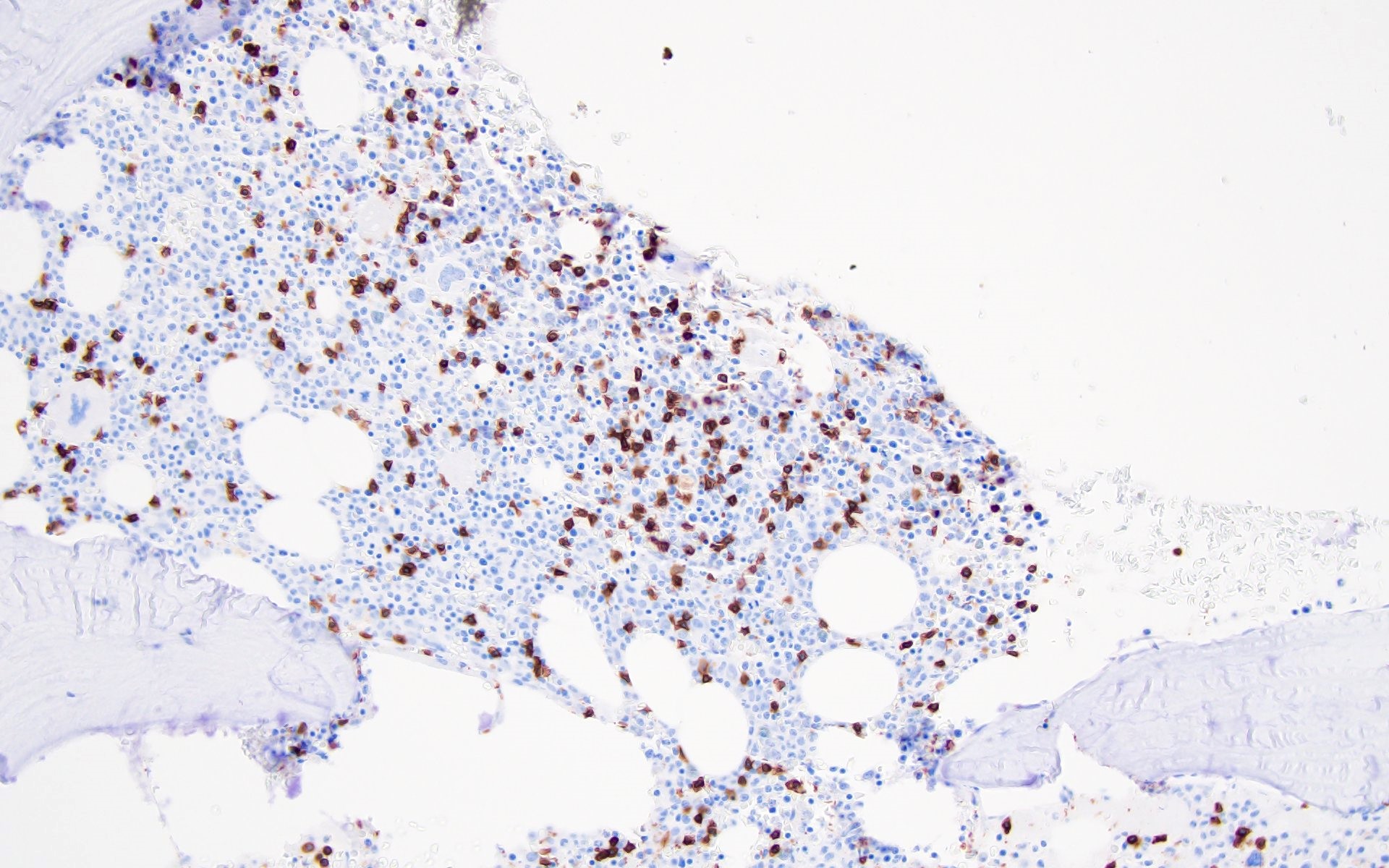
Pattern 5 (CD3)
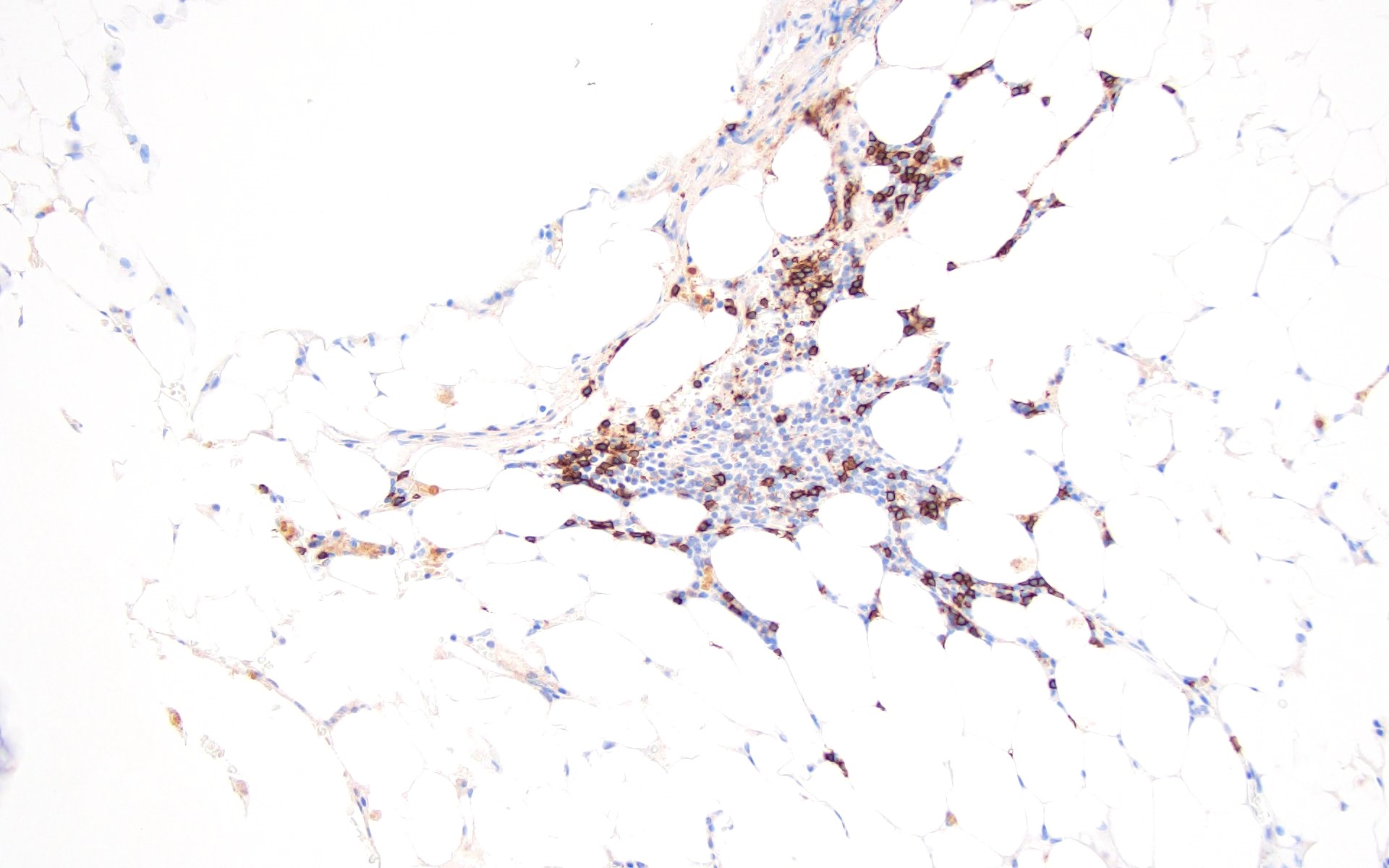
Pattern 1 (CD20)
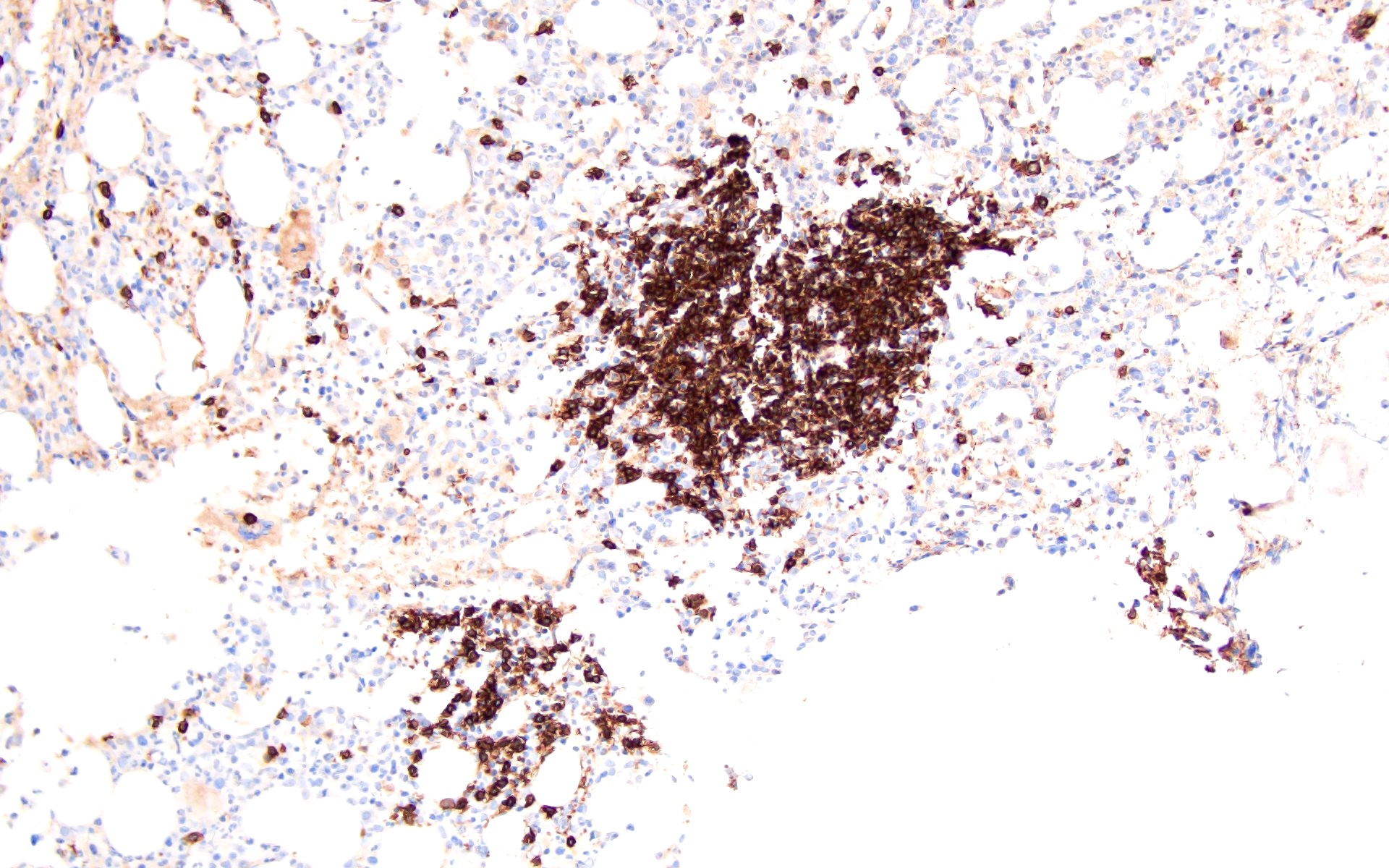
Pattern 2 (CD20)
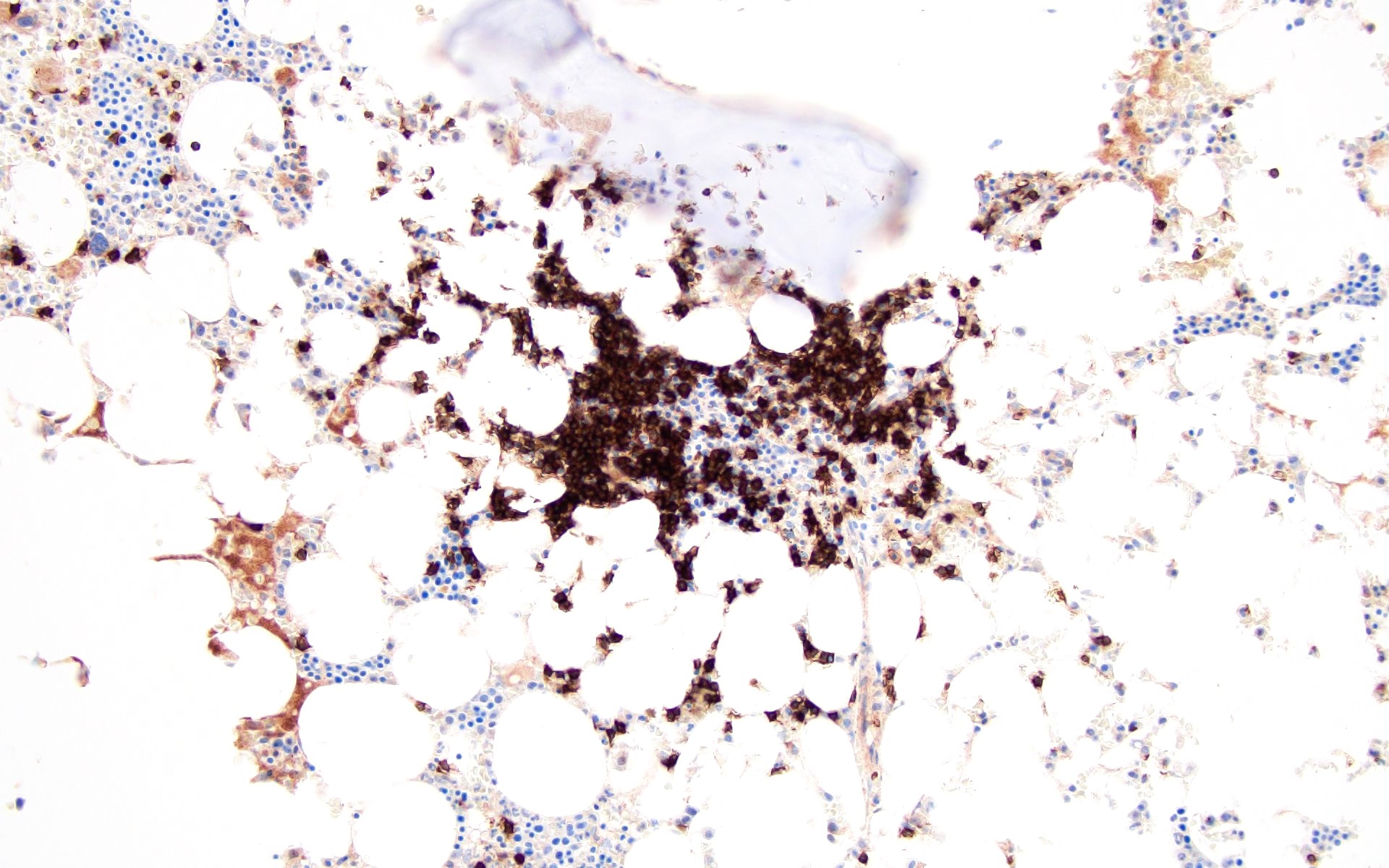
Pattern 3 (CD20)
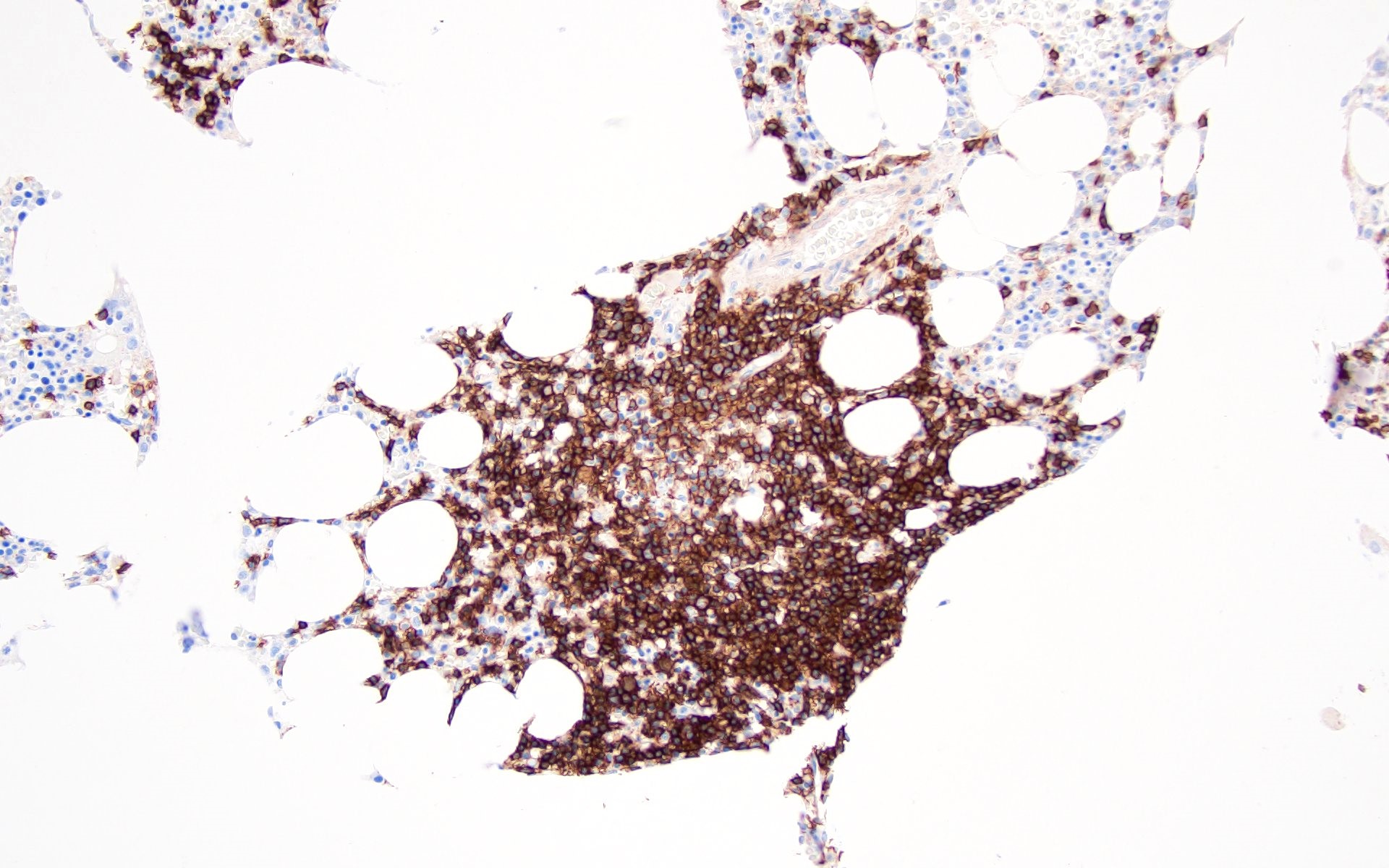
Pattern 4 (CD20)
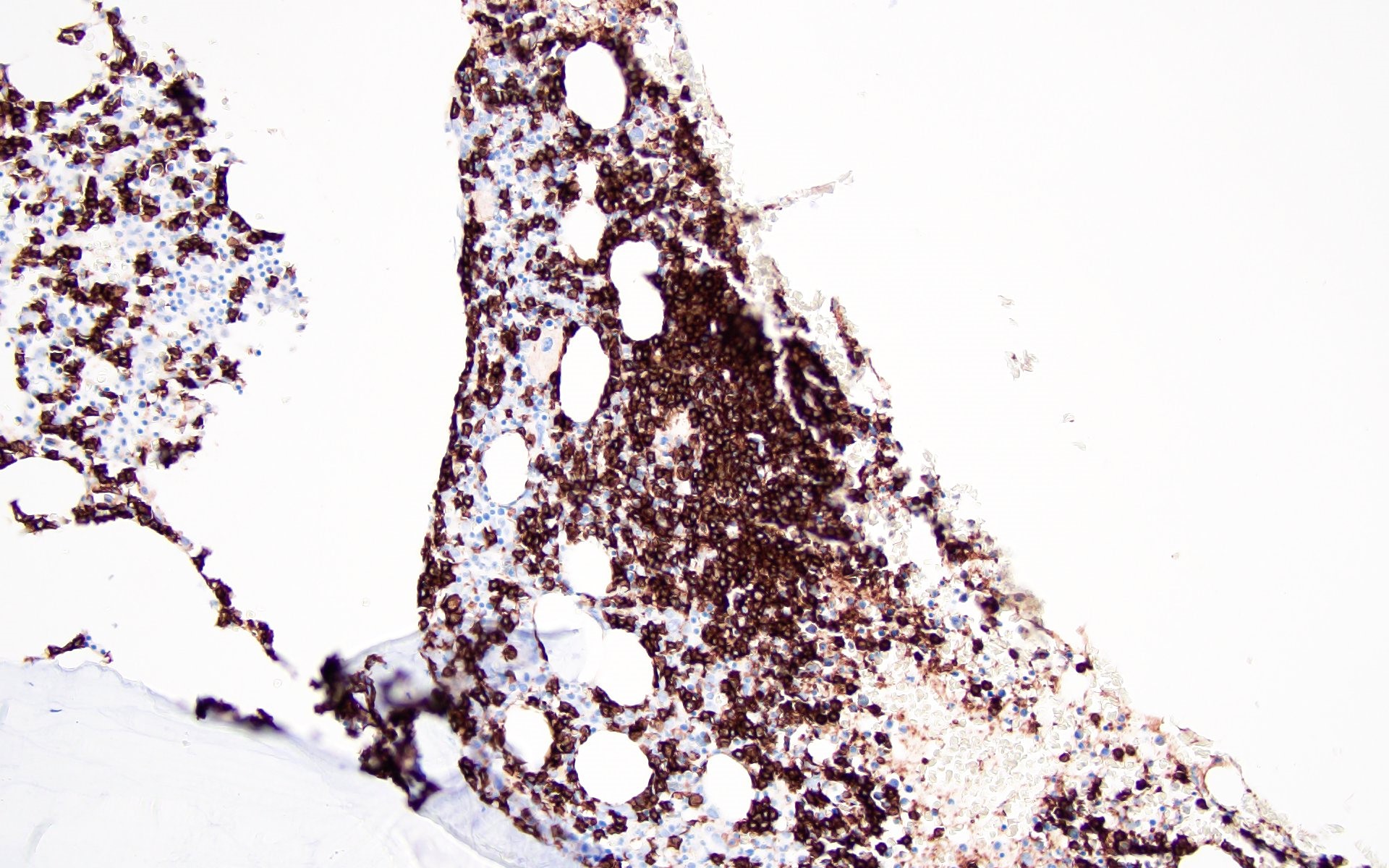
Pattern 5 (CD20)
- Bone marrow aspirate may show lymphocytosis, consisting of small to medium sized, mature appearing lymphocytes without atypia
Contributed by Barina Aqil, M.D.
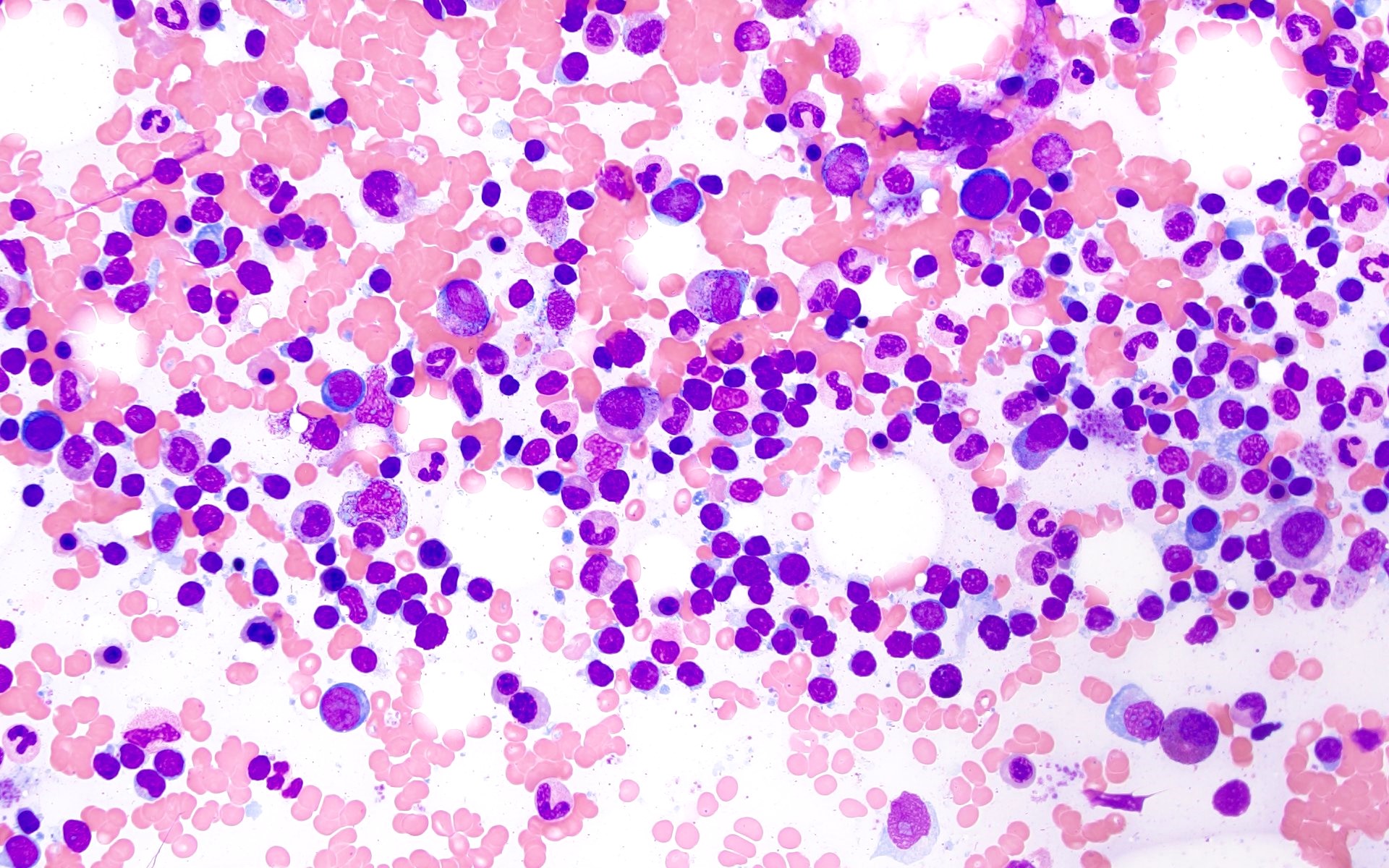
Sheet of small lymphocytes
- No monotypic B cell or phenotypically aberrant T cell populations identified
- Flow cytometry is used to assess the surrogate markers of clonality for B and T cells
- Kappa and lambda immunoglobulin light chains are evaluated for monotypic B cells, which includes kappa:lambda ratio > 3 or < 0.5
- Neoplastic B cells may also show lack of surface immunoglobulin light chains
- Abnormal T cell phenotype is described by abnormal expression patterns of T cell markers; for example, downregulation of surface CD3 expression, loss of CD7 or CD5 as well as a skewed CD4:CD8 ratio or expansion of double positive (CD4+ / CD8+) or double negative (CD4- / CD8-) populations
- Clonal T cell or NK cell populations can be evaluated by assessment of TCR Vβ or KIR repertoire, respectively
- TCR constant β chain 1 (TRBC1) expression is based on the expression of TCR β chain with either a constant region 1 or a constant region 2 (C1 or C2) on mature αβ T cells
- T cell clone is characterized by either a relative increase or a relative decrease in TRBC1+ T cells (Int J Mol Sci 2021;22:1817)
- Molecular tests based on polymerase chain reaction (PCR) are used for detection of immunoglobulin heavy chain (IGH) and light chains or T cell receptor gene rearrangements to identify the clonal nature of lymphoid aggregates and are negative in benign lymphoid aggregates
- Cytogenetic analysis: normal karyotype and no abnormal FISH signals
- Peripheral blood, bone marrow aspirate and left posterior iliac crest, bone marrow core biopsy:
- Normocellular marrow (~30% cellular) with trilineage hematopoiesis and multiple benign lymphoid aggregates (see comment)
- Comment: Flow cytometric immunophenotyping performed on the bone marrow aspirate reveals a polytypic B cell population and a T cell population without evidence of immunophenotypic abnormality based on the markers assayed.
- Microscopic description
- Peripheral blood smear: normocytic normochromic red blood cells with polychromasia and mild anisopoikilocytosis
- Neutrophils are adequate with unremarkable morphology; platelets are adequate with unremarkable morphology
- Bone marrow aspirate smear: the myeloid and erythroid series show progressive maturation with unremarkable morphology
- No overt increase in blasts, lymphocytes or plasma cells noted; megakaryocytes are present with predominantly unremarkable morphology
- Bone marrow core biopsy (decalcified): the unilateral bone marrow core biopsy is normocellular for age (~50% cellular)
- The myeloid and erythroid series show progressive maturation
- Megakaryocytes are adequately present with unremarkable morphology
- Multiple interstitial lymphoid aggregates, which are composed predominantly of small lymphocytes, are noted
- Immunohistochemistry: CD3 and CD20 highlights mixture of interstitially scattered as well as aggregates of CD3+ T cells and CD20+ B cells, with T cell predominance
- Persistent polyclonal B cell lymphocytosis:
- Rare condition of unknown etiology seen in young middle aged women who are usually smokers
- Mostly asymptomatic and are found to have absolute lymphocytosis, elevated serum IgM and binucleated lymphocytes
- Polyclonal B cells detected by flow cytometry
- Rare cases may have lymphadenopathy, hepatomegaly and splenomegaly
- Strong association with human leukocyte antigen DR7 and detection of translocation t(14;18) involving the BCL2 gene has been demonstrated in the literature
- Polymorphous reactive lymphoid hyperplasia:
- Characterized by the presence of interstitially increased lymphocytes or lymphoid aggregates in the bone marrow but it is usually focal, random and poorly circumscribed
- Lymphocytes are polymorphous, polytypic and are seen in the background containing plasma cells, immunoblasts, eosinophils and histiocytes
- Found in any age group and is mostly associated with underlying diseases, such as malignancies, postchemotherapy / stem cell transplant, infections and autoimmune disorders (Leuk Res 2013;37:1404)
- Systemic polyclonal B immunoblastic proliferation:
- Seen in middle aged to elderly population who present with fever, lymphadenopathy, hepatosplenomegaly and absolute lymphocytosis
- Peripheral blood shows circulating immunoblasts and plasmacytoid cells
- Bone marrow is hypercellular with extensive lymphocytic infiltration mimicking lymphoma
- No immunophenotypic abnormality is detected by flow cytometry
- No detection of IGH and TCR gene rearrangements by molecular studies (Cancer 1988;61:1350, Am J Clin Pathol 1992;98:222)
- Malignant lymphoid aggregates:
- Characteristic morphological findings include large / multiple lymphoid aggregates, paratrabecular localization, infiltrative border, distribution around large sinuses with increasing size on deeper sections
- Detection of clonality by flow cytometry and rearrangements by molecular studies
What are the features of malignant lymphoid aggregates?
- Infiltrative border, cytologic atypia, B cell rich pattern
- Large size, nonparatrabecular location, T cell rich pattern
- Large size, uniform configuration, T cell rich pattern
- Small size, intertrabecular location, no cytologic atypia
Comment Here
Reference: Lymphoid aggregates (benign)
- CD10+, BCL6+, BCL2+
- CD10+, BCL6+, BCL2-
- HGAL+, BCL6+, BCL2+
- LMO2+, CD10+, BCL2+
Comment Here
Reference: Lymphoid aggregates (benign)
- Mast cells are important cells of the hematopoietic lineage and play a crucial role in regulating the immune system, causing allergy and autoimmunity
- Mast cells are oval, irregular and sometimes show elongated shapes with cytoplasmic extensions
- Dense, granular cytoplasm packed with basophilic granules is seen
- Neoplastic mast cells may have an atypical spindled shape or decreased granulation
- Mast cell granules show metachromatic staining with Wright, Giemsa and Leishman stains
- Wright stain of bone marrow shows deep pink to dark purple chunky granules that obscure the nucleus
- On H&E they exhibit bland nuclei and pale pink, granular cytoplasm; single cells are often difficult to visualize on H&E
- Also positive for toluidine blue and positive for immunostains tryptase and CD117 (bright expression)
- Reference: Immunol Allergy Clin North Am 2014;34:315, Leuk Res 2001;25:537, Int Arch Allergy Immunol 2018;176:55
- Mastocyte; labrocyte
- Mast cells originate from the same multipotent hematopoietic progenitor cells of the bone marrow as neutrophils, eosinophils, basophils and monocytes (CD34+, KIT+, CD13+) and mature under the influence of the c-kit ligand, thrombopoietin, FLT3 ligand and IL6
- Under normal conditions, mature mast cells do not circulate in the bloodstream
- Mast cells are usually found in mucosal and epithelial tissues, all vascularized tissues except for the central nervous system and retina as well as in normal bone marrow (Blood 2008;112:946, J Histochem Cytochem 2014;62:698)
- Cytoplasm of the mast cell contains numerous large granules that store predominantly histamine, heparin, eicosanoids, cytokines, chondroitin sulfate and neutral proteases (J Histochem Cytochem 2014;62:698)
- Mast cells are known to participate in IgE mediated allergic reactions through FcεRI receptors and activation takes place via cross linking of antigen / IgE / FcεRI (Eur J Immunol 2014;44:2558)
- Mast cells play a key role in the regulation of physiological processes (e.g., vasodilation), enhancement of angiogenesis and elimination of bacteria and parasites (Front Immunol 2016;6:620)
- Reactive mast cell hyperplasia is often associated with chronic inflammatory, fibrogenic conditions and solid tumors (Am J Gastroenterol 2020;115:224)
- Mast cell hyperplasia in bone marrow may be seen in cases of hematologic malignancies such as lymphoplasmacytic lymphoma, chronic lymphocytic leukemia (CLL), myelodysplasia and acute myeloid leukemia (Cytometry B Clin Cytom 2021;100:331)
- Mastocytosis is a hematologic neoplasm defined by expansion and accumulation of neoplastic mast cells (MC) in the skin or in internal organs, such as the bone marrow (BM), spleen, lymph nodes, liver and gastrointestinal tract
- World Health Organization, 5th edition (WHO) and International Consensus classification (ICC) of myeloid neoplasms delineates mastocytosis into cutaneous mastocytosis (CM), systemic mastocytosis (SM) and mast cell sarcoma (MCS) (Leukemia 2022;36:1703, Blood 2022;140:1200)
- Based on disease specific features, systemic mastocytosis is further divided into indolent (ISM), smoldering (SSM), aggressive (ASM), systemic mastocytosis with an associated hematopoietic neoplasm (SM AHN) and mast cell leukemia (MCL) (Hemasphere 2021;5:e646)
- Rare in normal marrow, usually concentrated within particles on aspirate smears and perivascular on biopsy sections
- Mast cells are oval or irregularly shaped cells
- Up to 100% larger than basophils with irregular, elongated shapes and cytoplasmic extensions
- Dense, granular cytoplasm packed with basophilic granules is seen, often obscuring the nucleus and other organelles
- When it can be visualized, the nucleus is round and central (StatPearls: Histology, Mast Cells [Accessed 2 March 2023])
Contributed by Ramya Gadde, M.D. and AFIP
Scattered mast cells
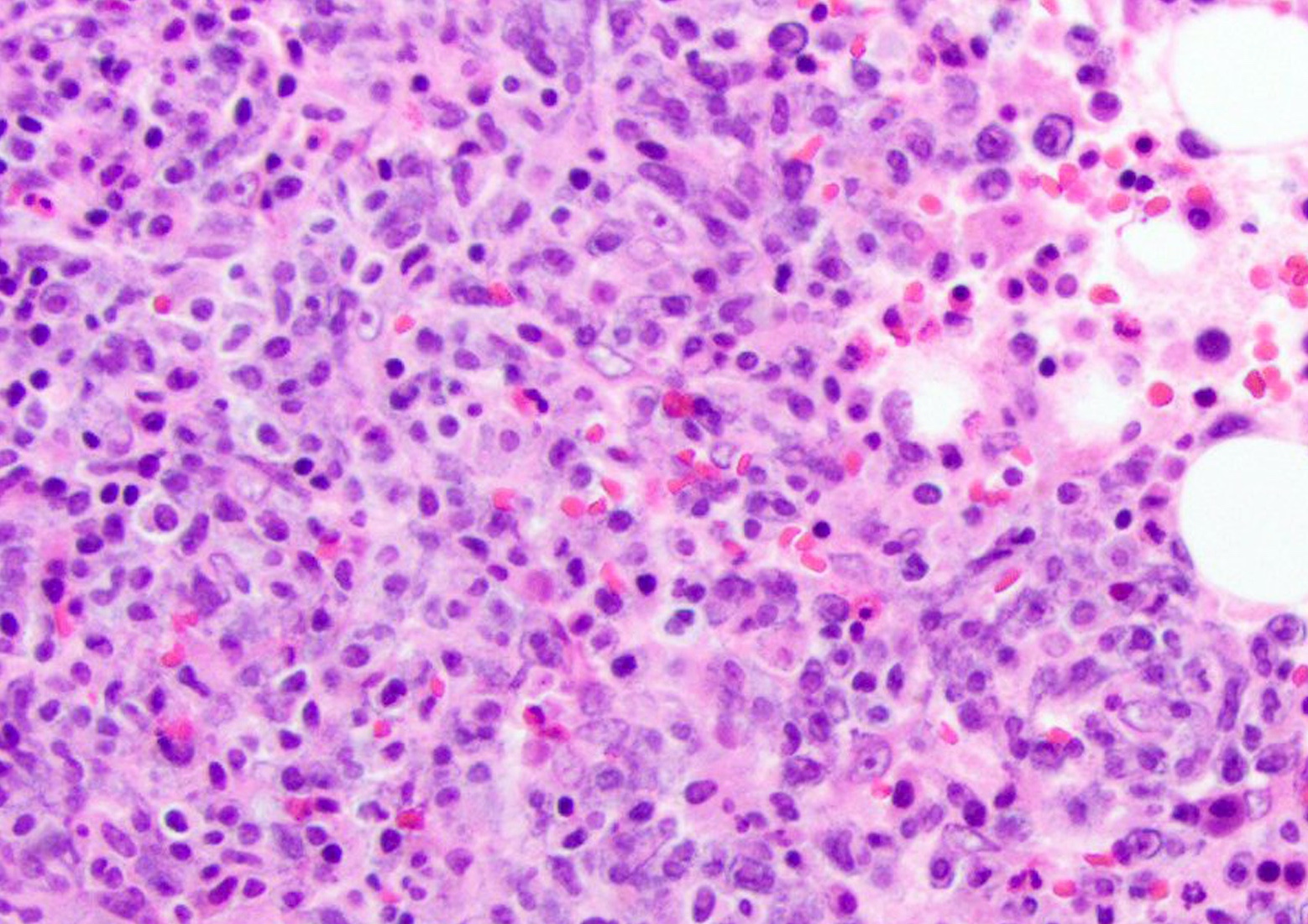
Spindle shaped mast cells
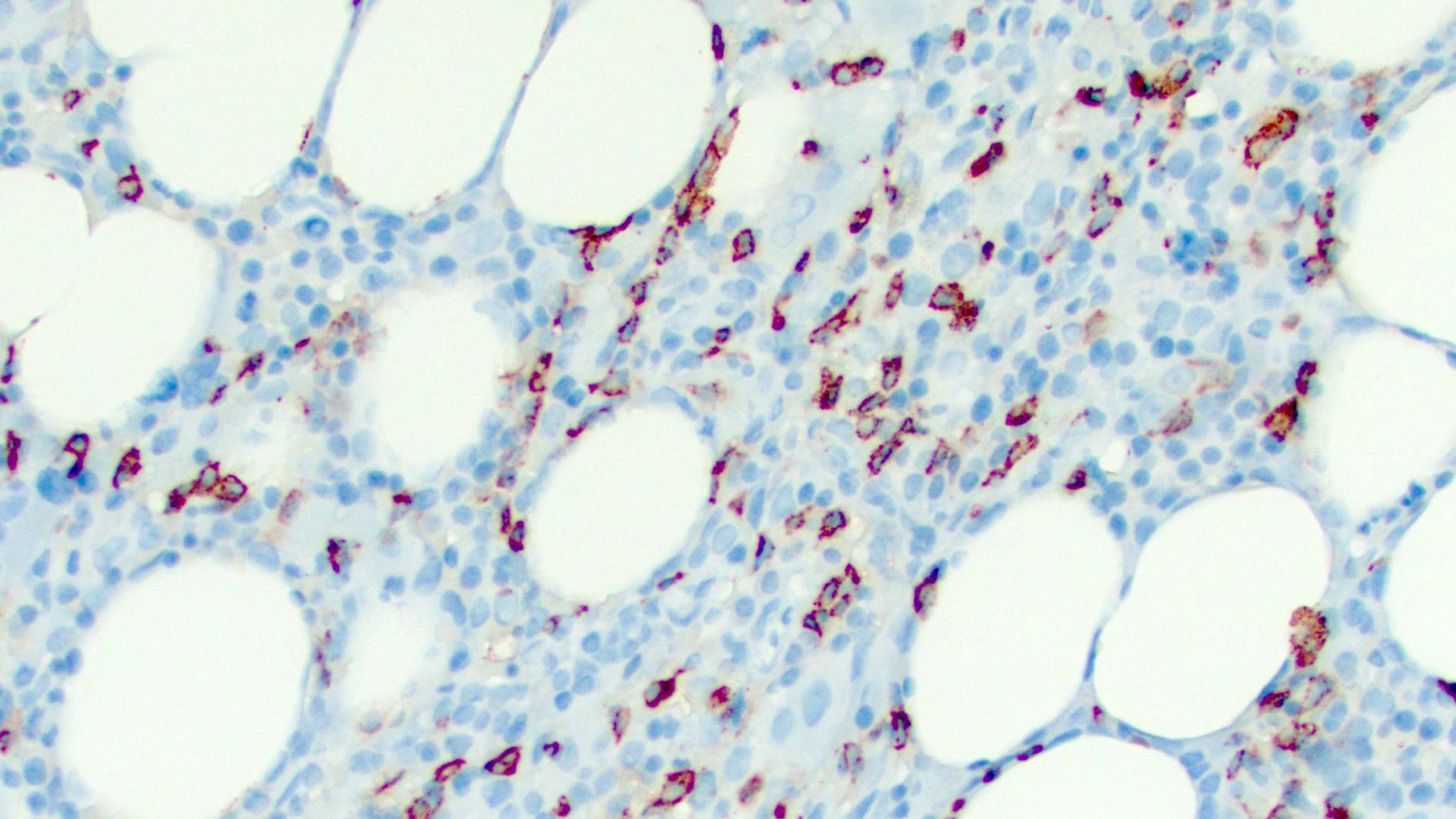
Tryptase
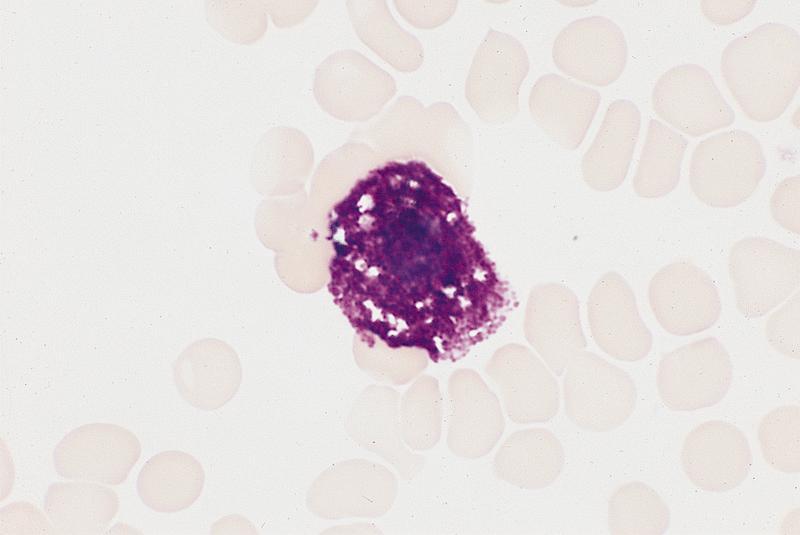
Scattered mast cells with basophilic granules
- Mast cell granules can show metachromatic staining with the Romanowsky combinations (Wright, Giemsa, May-Grünwald Giemsa and Leishman), toluidine blue
- Tryptase, CD117 (Int Arch Allergy Immunol 2018;176:55)
- Aberrant expression of various other hematopoietic antigens by neoplastic mast cells in systemic mastocytosis has been documented; the following antigens may be abnormally expressed, in varying frequency and intensity (Immunol Allergy Clin North Am 2014;34:315):
- Myelomonocytic antigens: CD14, CD33, CD61, CD63, CD68, CD203c, 2D7, myeloperoxidase, naphthol AS-D chloroacetate esterase
- Lymphoid antigens: CD2, CD9, CD25, CD26, CD30, CD52, CD79a
- Stem cell associated antigens: CD44
- Natural killer cell associated antigen: CD57
- Dendritic cell associated antigens: CD35, CD123
- Other antigens: CD99, SDF1 (Immunol Allergy Clin North Am 2014;34:315)
- Electron microscopy demonstrates mast cells, highly granulated cells of the immune system
- Mast cells can be identified by their central nucleus and numerous granules containing histamine and enzymes
- Upon stimulation by an antigen, the cell degranulates
- Reference: Ribatti: Milestones in Immunology - Based on Collected Papers, 1st Edition, 2017
Images hosted on other servers:

Degranulating mast cell
Mast cells with abundant granules
- Systemic mastocytosis:
- Major criteria:
- Multifocal dense infiltrates of mast cells (≥ 15 mast cells in aggregates) in bone marrow biopsies or in sections of other extracutaneous organ(s)
- Minor criteria:
- ≥ 25% of all mast cells are atypical cells or spindle shaped mast cell infiltrates detected in sections of bone marrow or other extracutaneous organs
- KIT activating KIT point mutation(s) at codon 816 or in other critical regions of KIT in bone marrow or another extracutaneous organ
- Mast cells in bone marrow, blood or another extracutaneous organ express 1 or more of CD2, CD30 or CD25
- Baseline serum tryptase concentration > 20 ng/mL (in cases of SM AHN, an elevated tryptase does not count as an SM criterion; in the case of a known hereditary alpha-tryptasaemia, the tryptase level should be adjusted)
- If at least 1 major and 1 minor or 3 minor criteria are fulfilled → the diagnosis is SM can be made (Leukemia 2022;36:1703, Blood 2022;140:1200)
- Major criteria:
- CD2, CD25 and CD30
- CD13 and CD33
- CD34 and CD117
- CD117 and tryptase
Comment Here
Reference: Mast cells
Detection of which of the following activating mutations is used as one of the minor criteria for diagnosis of systemic mastocytosis?
- JAK2
- KIT
- NPM1
- RUNX1
Comment Here
Reference: Mast cells
- Progresses from myeloid stem cell (pluripotential hematopoietic stem cell) to megakaryoblast to promegakaryocyte to megakaryocyte to proplatelets (released into circulation) to platelets (J Thromb Haemost 2003;1:1580, Front Biosci 2007;12:2050)
- Two groups of factors influencing megakaryopoiesis are early acting multilineage cytokines (stem cell factor, GM-CSF, IL3, IL6) and cytokines that are more megakaryocyte specific (thrombopoietin)
- Thrombopoietin: produced by bone marrow stromal cells, hepatocytes and proximal renal tubular epithelial cells; poorly understood regulation
- Maturation is characterized by an increase in size and lobulation of nuclei and is controlled by thrombopoietin (J Clin Invest 2005;115:3339)
- Endomitosis: unique property of megakaryocytes characterized by ongoing DNA synthesis, without mitosis resulting in progressive nuclear lobulation with increasing DNA content (nuclear lobes remain connected)
- Megakaryocytes form demarcation membrane within cytosol, which leads to production of platelets
- Maturation is associated with acquisition of surface GP Ib, GP IIb and von Willebrand factor
- Four types of cytoplasmic granules are alpha, delta, gamma and peroxisomes
- Megakaryocytes reside next to the bone marrow sinuses and project pseudopodia through the sinus walls, releasing platelets directly into the circulation
Images hosted on other servers:
Megakaryopoiesis pathways

Proplatelet formation
- Megakaryocytes are smaller in fetuses and infants and show less nuclear segmentation when compared to older children
- Platelet production appears to be dependent on megakaryocyte ploidy
- Platelet production rate under homeostatic conditions: 2 - 4 x 109/kg/day; may increase 8 - 10x if needed; 10 day survival time in peripheral blood
- Emperipolesis: other cells, especially neutrophils, pass through megakaryoctye tubular system (due to its location) to enter the circulation
- Thrombocytopenia with normal / increased megakaryocytes: congenital (Bernard-Soulier, May-Hegglin, gray platelet syndrome) or acquired (ITP, DIC, TTP, HUS, hypersplenism, megaloblastic anemia)
- Thrombocytopenia with decreased megakaryocytes: congenital (Fanconi, thrombocytopenia with absent radii, X linked amegakaryocytic thrombocytopenia, dyskeratosis congenita, Shwachman-Diamond) or acquired (aplastic anemia, bone marrow infiltration, infections, toxins, immune disorders)
- Thrombocytosis: congenital (familial essential thrombocythemia, other familial myeloproliferative disorders) or acquired (reactive or neoplastic)
- Megakaryoblast: variable size (7 - 35 microns); may be designated micromegakaryoblast if < 15 microns; round / ovoid cells with scanty blue agranular cytoplasm that often forms a rim around nucleus and may have a few small budding protrusions at periphery; nuclei are round / oval with coarse granular chromatin, one or more nucleoli
- Megakaryocyte: randomly disbursed throughout bone marrow; 50 - 150 microns (largest normal nucleated cell in marrow); micromegakaryocytes measure 15 - 30 microns; abundant light blue to pink cytoplasm with numerous purple red or pink granules; nucleus has 8, 16 or 32 overlapping lobes; no nucleolus; megakaryocytes producing platelets may have demarcated granular clumps of platelets streaming from the margins
Contributed by Dragos C. Luca, M.D. and AFIP images
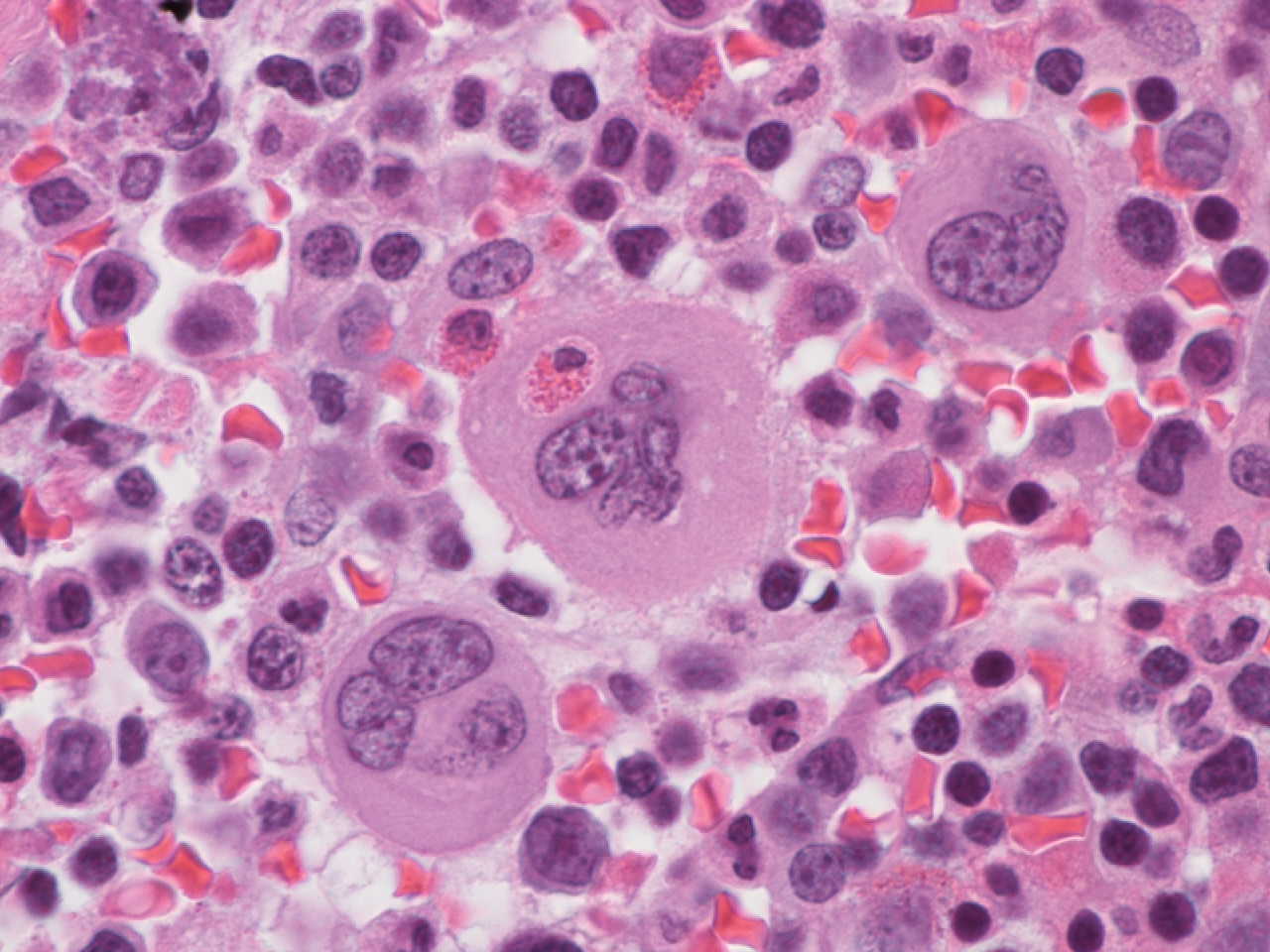
Emperipolesis
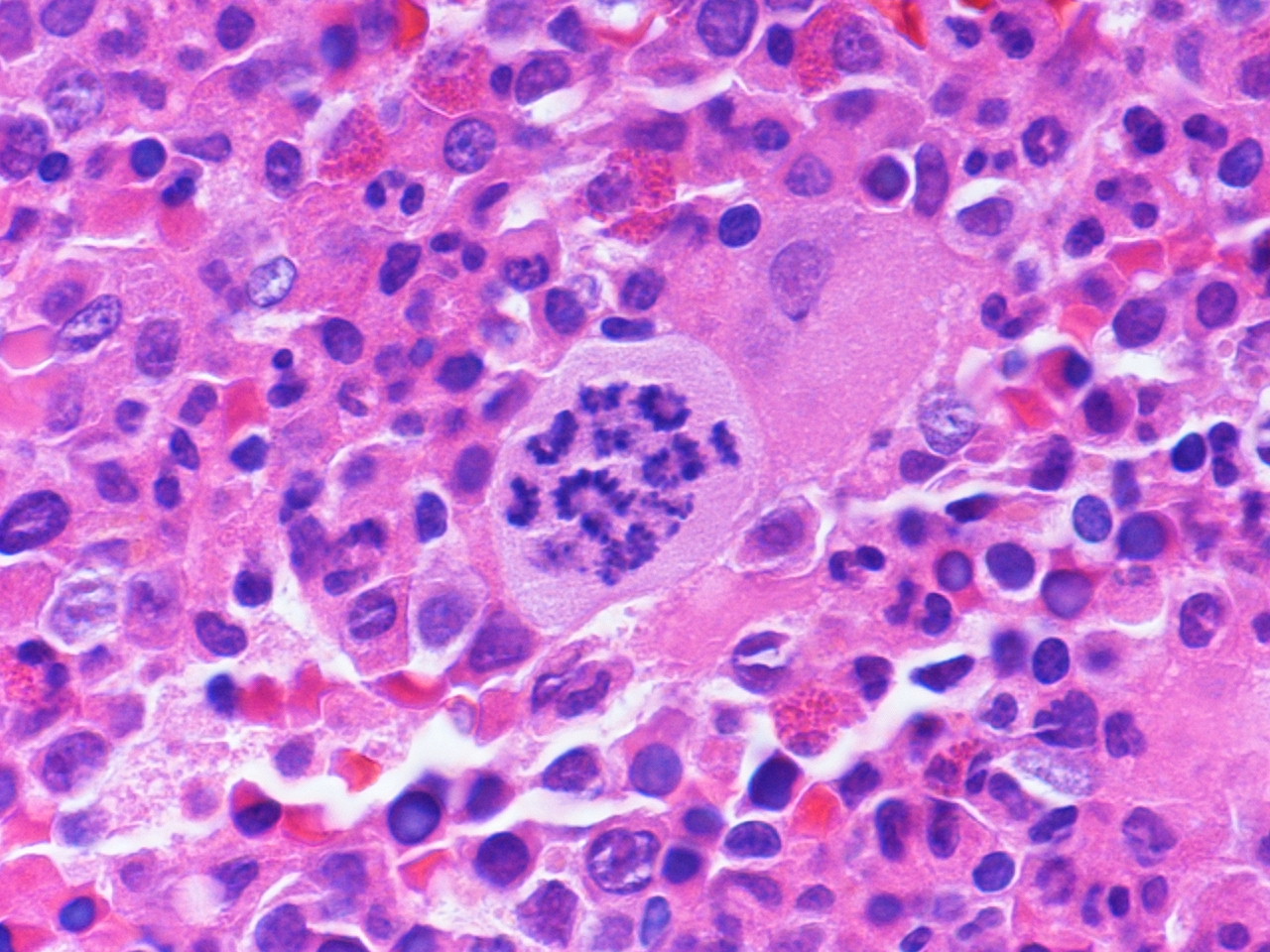
Endomitosis
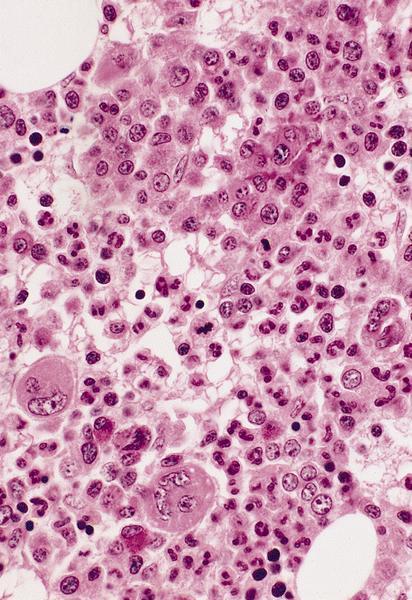
Megakaryocytes
Images hosted on other servers:

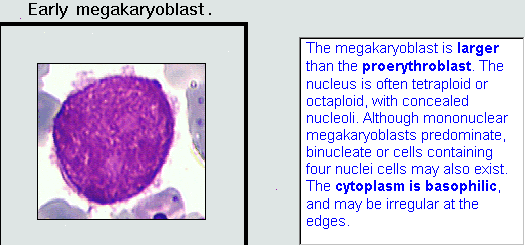
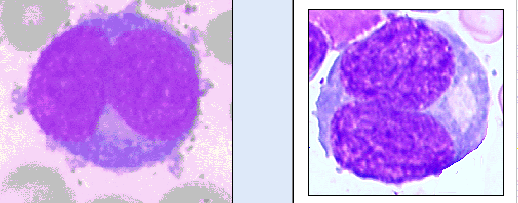
Megakaryoblasts
Megakaryocytes

Emperipolesis

CD62P+ megakaryocyte
Platelet formation
Images hosted on other servers:
Platelets
- Megaloblastic anemia is a disorder of impaired DNA synthesis (with normal RNA synthesis) that manifests with the presence of megaloblasts in the bone marrow resulting in ineffective and abnormal erythropoiesis and macrocytes / macroovalocytes in the peripheral blood
- Common morphologic manifestation of a multitude of entities causing similar impairment in DNA synthesis
- Disorder of impaired DNA synthesis → delayed nuclear maturation → nuclear:cytoplasmic dyssynchrony
- Multitude of etiologies: most commonly vitamin B12 or folate deficiency
- Megaloblasts, macroovalocytes (erythroid lineage) and nuclear hypersegmentation of neutrophils are characteristic morphologic abnormalities
- Megaloblast: abnormal erythroid precursors showing nucleo:cytoplasmic dyssynchrony (more immature nucleus for the degree of maturity of the cytoplasm)
- Macrocyte: mature red blood cell with increased mean corpuscular volume (100 - 110 fL)
- Macroovalocyte: mature red blood cell with macrocytosis and a distinct oval shape with reduced or absent central pallor; commonly seen in megaloblastic anemia but not pathognomic
- ICD-10: D53.1 - other megaloblastic anemias, not elsewhere classified
- Insufficient data regarding incidence and prevalence
- Epidemiology varies depending on the cause of megaloblastic anemia
- Nutritional deficiency of B12 and folate:
- Adults and children in macrobiotic communities and developing countries with rampant malnutrition
- Pregnant women in developing countries with lack of folate supplementation
- Elderly in developed countries
- Pernicious anemia:
- Individuals older than 40 years; incidence of 2% in > 60 years (J Blood Med 2012;3:97)
- More common in individuals of Celtic (e.g. English, Irish, Scottish) or Scandinavian origin
- Patients of any age group on certain chemotherapeutic drugs
- Nutritional deficiency of B12 and folate:
- Bone marrow
- Peripheral blood
- All rapidly dividing epithelia (gut epithelium, uterine cervical epithelium) can show megaloblastic changes due to impaired DNA synthesis: widespread involvement of epithelia is called megaloblastosis
- Reference: StatPearls: Megaloblastic Anemia [Accessed 11 October 2021]
- Normal absorption pathway of cobalamin (B12) (Nat Rev Dis Primers 2017;3:17040):
- Vitamin B12 can be present in diet in free form or bound form
- Free form of B12 / cobalamin binds to protein R binder (TCI or haptocorrin) in the stomach (R binder is produced by salivary glands and gastric mucosal cells)
- This R binder - B12 complex is carried to the duodenum, where R binder is digested and B12 is bound to intrinsic factor (IF) (IF is produced by the parietal cells of the stomach but it binds to B12 in the duodenum)
- B12 - IF complex reaches the terminal ileum, where it is internalized by the cobalamin receptors on the ileal epithelial cells; inside the cytoplasm of ileal epithelial cells, the IF is destroyed
- B12 binds to the protein transcobalamin II (TCII) and is carried bound to TCII to liver (50%) and other organs (50%)
- Normal absorption pathway of folic acid:
- Food folates are present as polyglutamates in diet
- In the GI tract, polyglutamates are broken down to mono and diglutamates; these are absorbed by the proximal jejunum and converted to dihydrofolate (DHF), tetrahydrofolate (THF) and methyl THF
- Folate reaches the plasma as N5 methyl THF; this circulates in the plasma, either loosely bound to albumin or by high affinity binding to folate binding protein
- Inside the cell, N5 methyl THF is utilized for DNA synthesis, with the help of vitamin B12 (see Diagram 1)
- Thus, in the absence of B12 and folate, there is impaired DNA synthesis
- How impaired DNA synthesis leads to megaloblastic anemia (see Diagram 2):
- As the DNA doesn't synthesize normally, nuclear maturation is delayed, while cytoplasmic maturation continues at normal rate, thus forming a cell with mature cytoplasm with relatively immature, large, open nucleus, known as a megaloblast
- Large number of abnormal precursors undergo intramedullary apoptosis leading to anemia
- Additionally, impaired DNA synthesis leads to ineffective hematopoiesis in all 3 cell lines → pancytopenia
- How impaired DNA synthesis leads to megaloblastic anemia (see Diagram 2):
- Pathophysiology of neurological dysfunction in cobalamin deficiency:
- Cobalamin deficiency leads to demyelination with subsequent axonal disruption and gliosis that can affect all parts of the central nervous system
- Peripheral nerves show axonal degeneration without demyelination
- Involvement of the posterior and lateral columns (including the dorsal, pyramidal and spinocerebellar tracts) leads to the classic myelopathic syndrome of B12 deficiency known as subacute combined degeneration
- Biochemical basis:
- Vitamin B12 is a cofactor for the enzyme methylmalonyl CoA mutase, which converts methylmalonyl CoA to succinyl CoA
- With B12 deficiency, methylmalonic acid (MMA) levels accumulate → elevated levels of MMA and homocysteine lead to myelin damage → neurologic deficits, such as neuropathy and ataxia
- Pathophysiology of pernicious anemia:
- Autoimmune disorder with 2 kinds of antibodies: parietal cell antibody (PCA) and intrinsic factor antibody (IFA) → PCA destroys parietal cells by acting parietal cell proton pump ATPase, IFA destroys IF directly → impaired B12 absorption → B12 deficiency → megaloblastic anemia
- In addition, destruction of parietal cells leads to achlorhydria → atrophic gastritis
- Vitamin B12 deficiency
- Dietary deficiency
- Recommended daily intake of 2.4 mcg in ages 14 and older; recent studies recommend a higher level of 4 - 7 mcg (Am J Clin Nutr 2010;91:571)
- Deficiency more common in long term vegans (baseline stores of B12 exceed daily requirement by 1,000 times; thus, it takes years to develop deficiency), breastfed infants of vegan mothers
- Lack of intrinsic factor (IF)
- Chronic gastritis
- Gastric surgery
- Congenital absence of IF
- Pernicious anemia (autoimmune)
- Biologic competition for B12
- Small bowel bacterial overgrowth
- Fish tapeworm disease
- Malabsorption of B12
- Familial: familial selective vitamin B12 malabsorption (Imerslund-Gräsbeck syndrome)
- Drug induced: proton pump inhibitors, H2 receptor antagonists, metformin
- Others: regional enteritis, ileal resection
- Increased requirements
- Pregnancy
- Disseminated cancer
- Dietary deficiency
- Folate deficiency
- Dietary deficiency
- Recommended daily intake of 400 mcg
- Lesser stores than cobalamin, so deficiency develops in 2 - 3 months of dietary restriction
- Uncommon in developed countries due to fortification of foods such as cereals and pasta
- Impaired absorption
- Congenital malabsorption
- Extensive intestinal (jejunal) resection
- Drug interactions: anticonvulsants, trimethoprim, oral contraceptives (J Obstet Gynaecol Can 2015;37:430)
- Increased requirements
- Infancy
- Pregnancy
- Chronic hemolytic anemias
- Myeloproliferative disorders
- Alcoholism (J Nutr 2002;132:2367S)
- Malnutrition
- Direct effects of alcohol on folate metabolism and transport
- Decreased hepatic uptake
- Increased excretion
- Dietary deficiency
- Combined folate and B12 deficiency
- Tropical sprue
- Gluten sensitive enteropathy
- Drug induced suppression of DNA synthesis
- Folate antagonists: methotrexate
- Metabolic inhibitors
- Purine antagonists: 6-mercaptopurine, azathioprine
- Pyrimidine antagonists: 5-fluorouracil, cytosine arabinoside
- Alkylating agents: cyclophosphamide, chlorambucil
- Others: nitrous oxide, arsenic
- Inherited disorders of DNA synthesis
- Orotic aciduria
- Lesch-Nyhan syndrome
- Thiamine responsive megaloblastic anemia
- Transcobalamin II deficiency
- Homocystinuria
- Methylmalonic aciduria
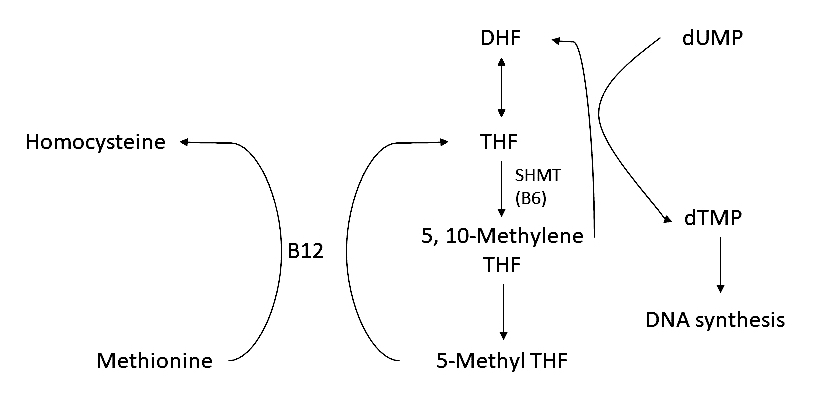
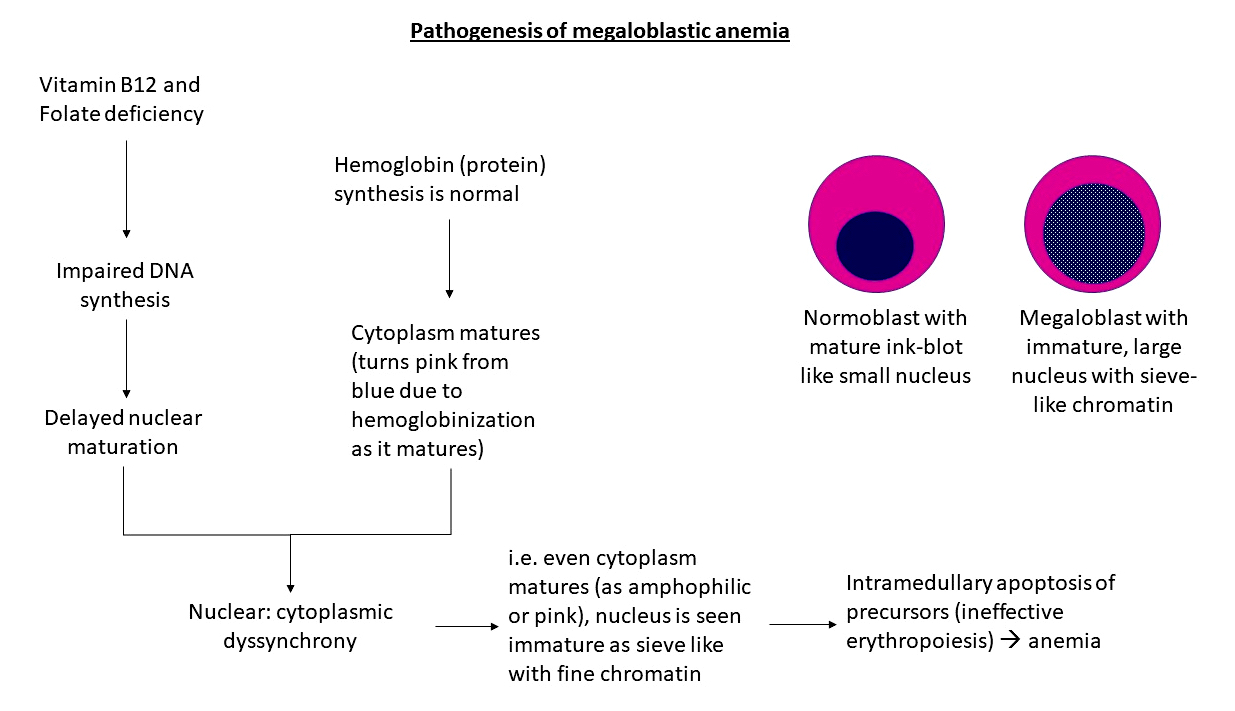
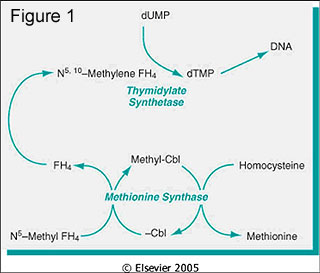

- Depends on the underlying cause of megaloblastic anemia; however, general clinical features resulting from anemia are common to all
- Clinical features resulting from anemia (Med Clin North Am 2017;101:297):
- Weakness, shortness of breath, impaired concentration and exercise ability
- Gradual onset of pallor, tachycardia
- Lemon hue of skin due to increased indirect bilirubin levels due to ineffective erythropoiesis
- Clinical features specific to cobalamin deficiency (Nutrients 2013;5:4521):
- Neurological manifestations, such as paraesthesias, balance disorders, lancinating pains (resulting from peripheral neuropathy); visual disturbances due to optic atrophy, dementia in severe cases
- Beefy red tongue due to atrophy of papillae
- Oral soreness and aphthous stomatitis
- Clinical signs: loss of vibratory sense and proprioception, positive Romberg test, Babinski reflex, hyporeflexia, clonus
- Increased incidence of arterial and venous thrombosis due to raised plasma homocysteine levels
- Clinical features of pernicious anemia:
- Classic triad at presentation: weakness, sore throat, paraesthesiae
- Patients have light blue-gray eyes and premature graying of hair
- Low grade fever, beefy red tongue, tachycardia, blotchy skin pigmentation
- Altered mental status, visual deficits
- Enlarged liver if heart failure is present
- Can be associated with autoimmune diseases, such as type 1 diabetes (3 - 4%), vitiligo (2 - 8%) and autoimmune thyroid disease (3 - 32%) (Nat Commun 2021;12:3761)
- Complete blood count (anemia), peripheral smear, bone marrow aspirate and biopsy, serum vitamin B12 and folate levels, homocysteine levels (hyperhomocysteinemia), formiminoglutamic acid (FIGLU) levels in urine (increased)
- Gastric biopsy in pernicious anemia
- Reference: StatPearls: Megaloblastic Anemia [Accessed 11 October 2021]
- Complete blood count (CBC): anemia, pancytopenia
- Serum B12 levels (isotope dilution or radioimmunoassay, automated chemiluminescence): reduced (normal = 160 - 950 pg/mL)
- Serum homocysteine levels: raised (normal < 15 mcmol/L)
- Serum methylmalonic acid: raised (normal < 0.40 mcmol/mL)
- Combination of serum B12 levels and homocysteine and methylmalonic acid levels is preferred for establishing diagnosis as the measurement of serum B12 alone can miss 10 - 50% of cases (J Am Geriatr Soc 1992;40:1197, J Am Geriatr Soc 1995;43:1290)
- Assessment of folate levels:
- Serum folate level: reduced (< 4 ng/mL suggests deficiency)
- Red cell folate levels: reduced (< 151 ng/mL suggests deficiency)
- Red cell folate levels were considered the true measure of tissue folate levels; however, it is more costly and time consuming and some recent studies suggest serum folate may be superior to red cell folate (Clin Chem Lab Med 2013;51:555)
- 9 week old boy with fever and pancytopenia (J Pak Med Assoc 2020;70:923)
- 7 month old girl with developmental regression (Ital J Pediatr 2020;46:40)
- 39 year old woman with pernicious anemia (Blood 2020;135:1719)
- 69 year old woman with painful tongue (CMAJ 2020;192:E434)
- Supplementation of B12 and folate
- Vitamin B12 changes may be ameliorated by folic acid supplementation alone, because the megaloblastic defects are caused by defect in folate utilization; however, folate does not reverse the neurological symptoms and may aggravate them
- When malabsorption is a cause, parenteral supplementation is needed
- Treat the underlying cause (e.g. bacterial overgrowth, etc.)
- Response to treatment (Klin Lab Diagn 2004;(5):42):
- Reversal of megaloblastic to normoblastic changes: 12 - 48 hours
- Increase in reticulocyte count: within 3 days (starts on second day, peaks at sixth day)
- Normalization of MMA and plasma homocysteine levels: first 5 days
- Rise in white blood cell and platelet counts (if they had been low): 1 week
- Sustained normalization of serum cobalamin: following 2 weeks of therapy
- Neutrophil hypersegmentation persists for 2 weeks or more
- Neurological improvement: first few weeks
- Correction of peripheral blood macrocytosis: first month of treatment
- Normalization of blood counts: before 8 weeks
Images hosted on other servers:
Glossitis, smooth tongue
- Bone marrow is markedly hypercellular with erythroid hyperplasia and reversal of M:E ratio (Greer: Wintrobe's Clinical Hematology, 13th Edition, 2014)
- Hallmark is nuclear:cytoplasmic dyssynchrony / dissociation = megaloblastosis
- Erythroid lineage abnormalities: most apparent
- Megaloblasts: have large, immature nucleus with open, sieve-like chromatin
- Early megaloblasts > intermediate megaloblasts > late megaloblasts
- Some megaloblasts show Howell-Jolly bodies and chromatin stippling
- Frequent normal and abnormal mitoses
- Other features of dyserythropoeisis may also be seen: nuclear budding, irregular nuclear membrane, nuclear fragments, etc.
- Myeloid lineage abnormalities:
- Giant metamyelocytes
- Giant band forms
- Neutrophil hypersegmentation: often precedes anemia
- Misshapen neutrophils → ineffective myelopoiesis → neutropenia
- Megakaryocyte abnormalities: least common
- Nuclei with open chromatin and nuclear lobular hypersegmentation
- Fragmentation → ineffective thrombocytopoiesis
Contributed by Julie Teruya-Feldstein, M.D. and Swati Bhardwaj, M.B.B.S., M.D.
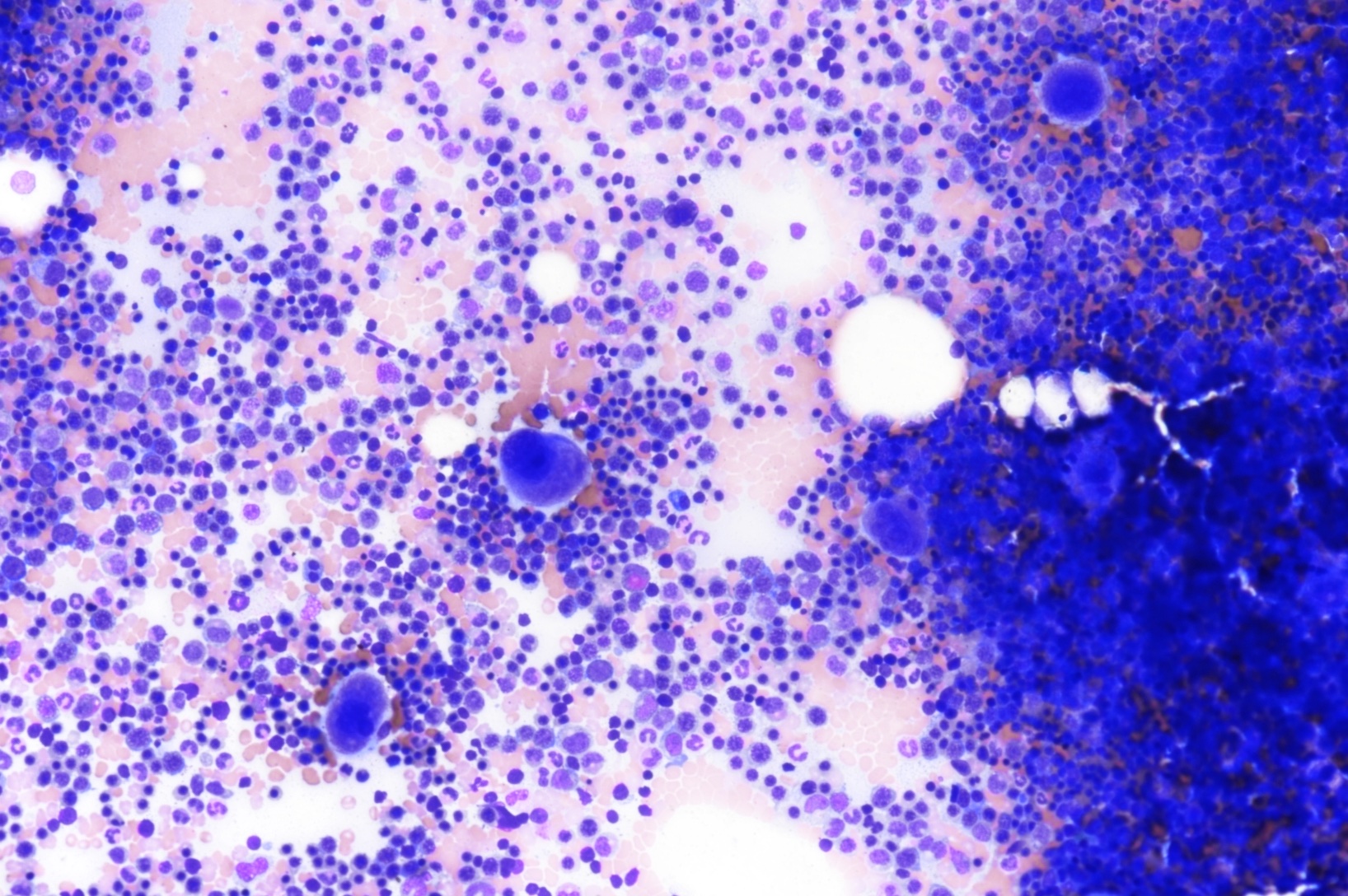
Erythroid hyperplasia
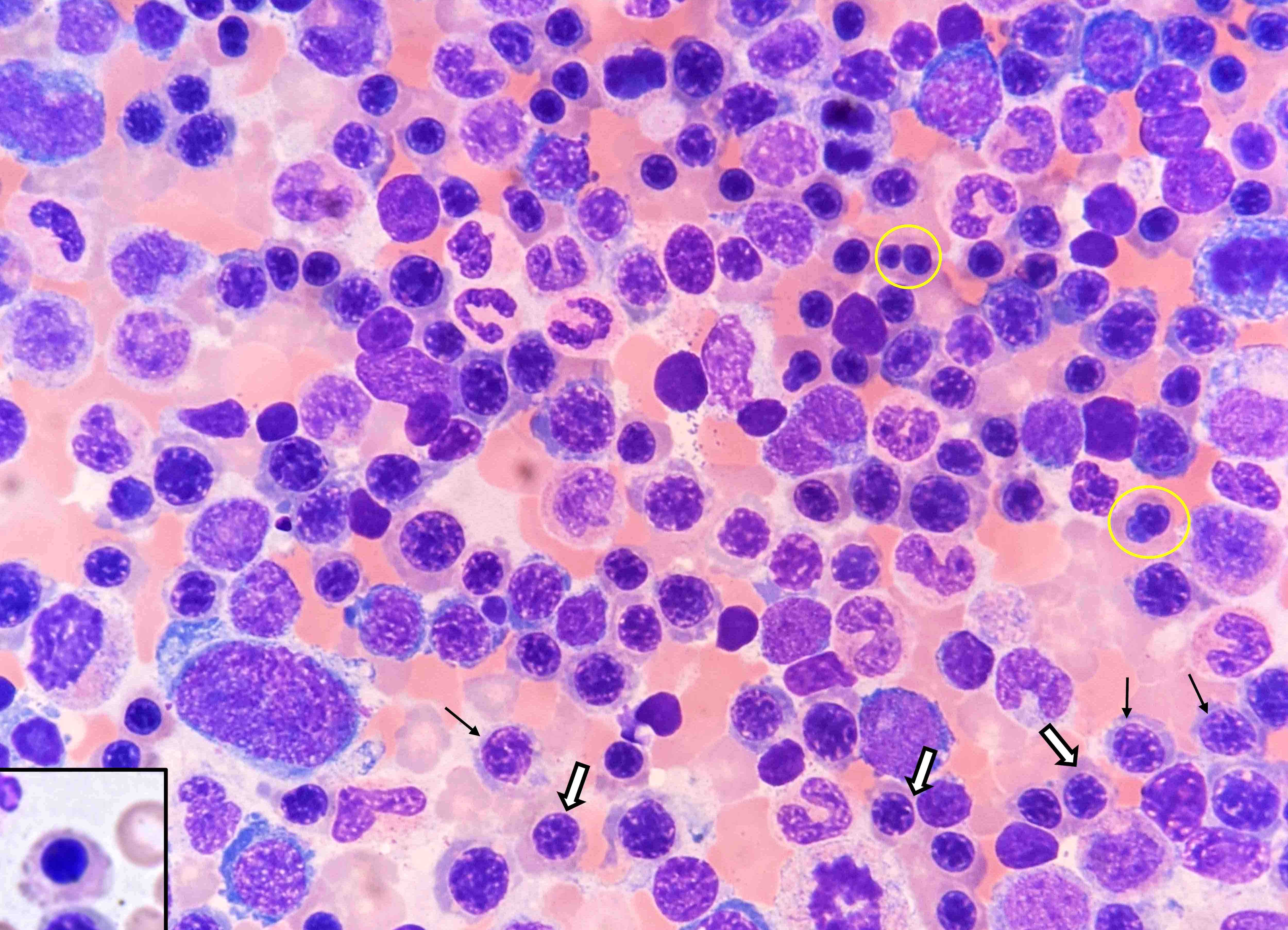
Megaloblasts and erythroid nuclear dysplasia
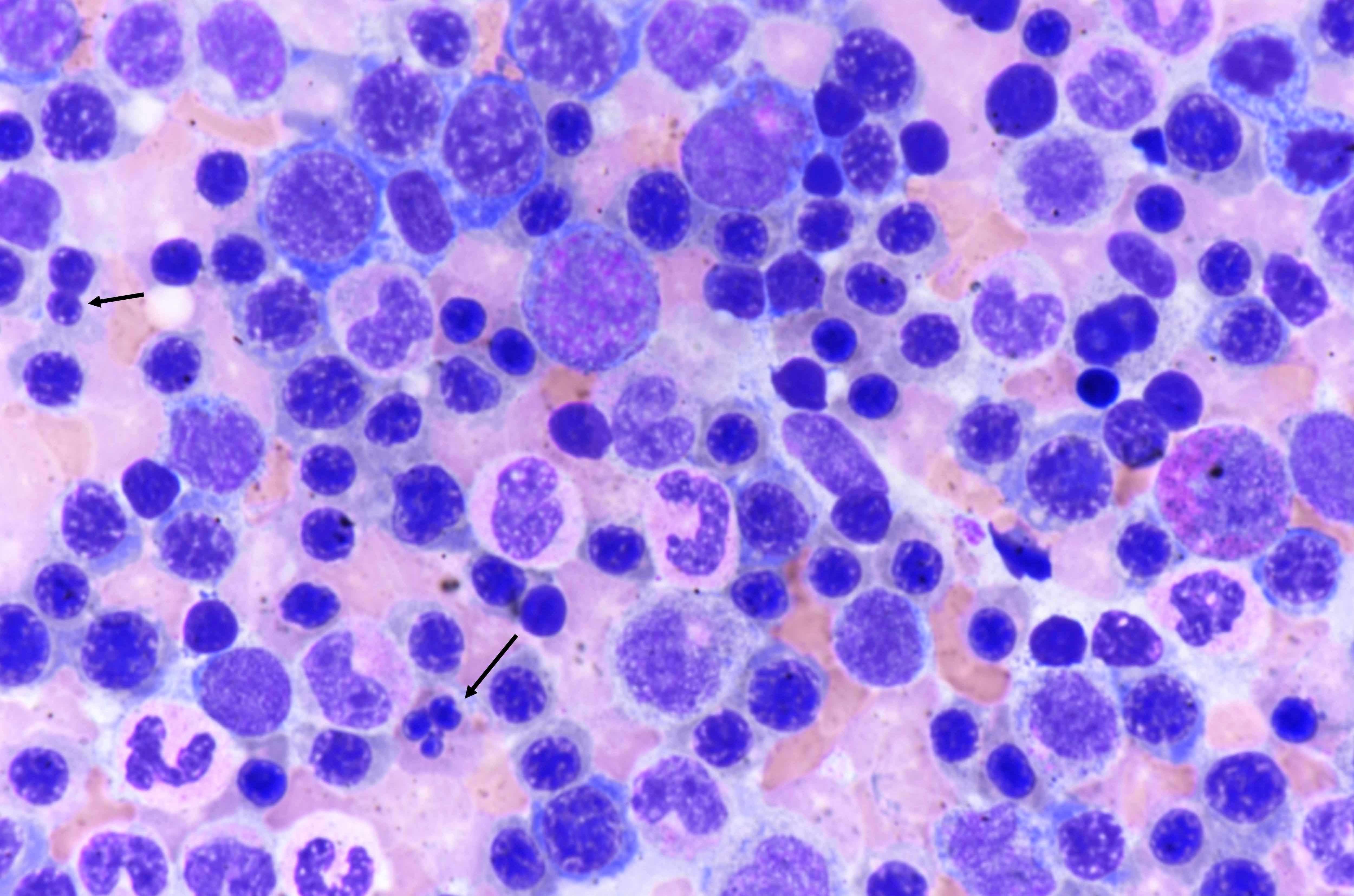
Erythroid hyperplasia and nuclear abnormalities
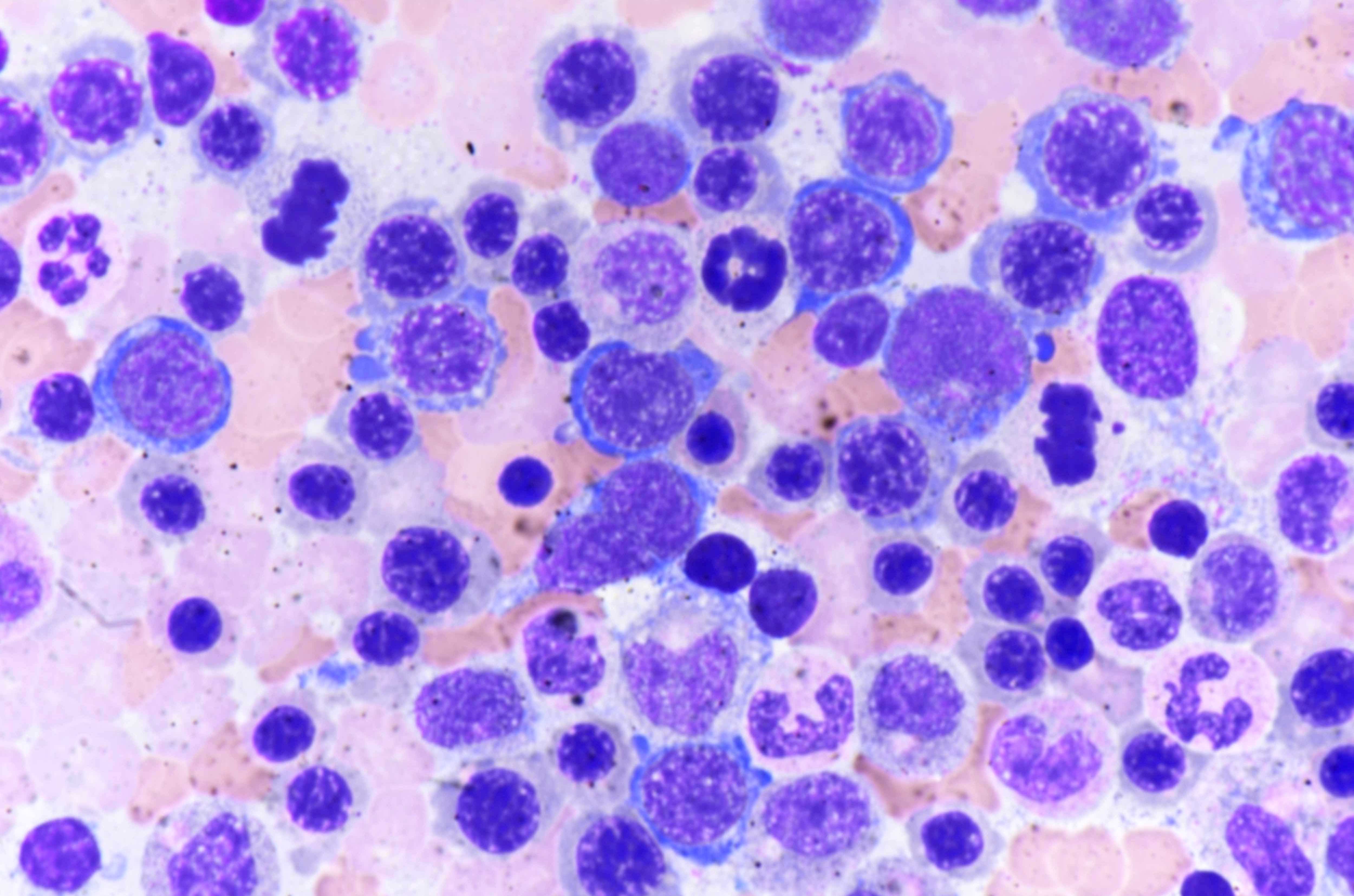
Frequent mitoses and neutrophil hypersegmentation
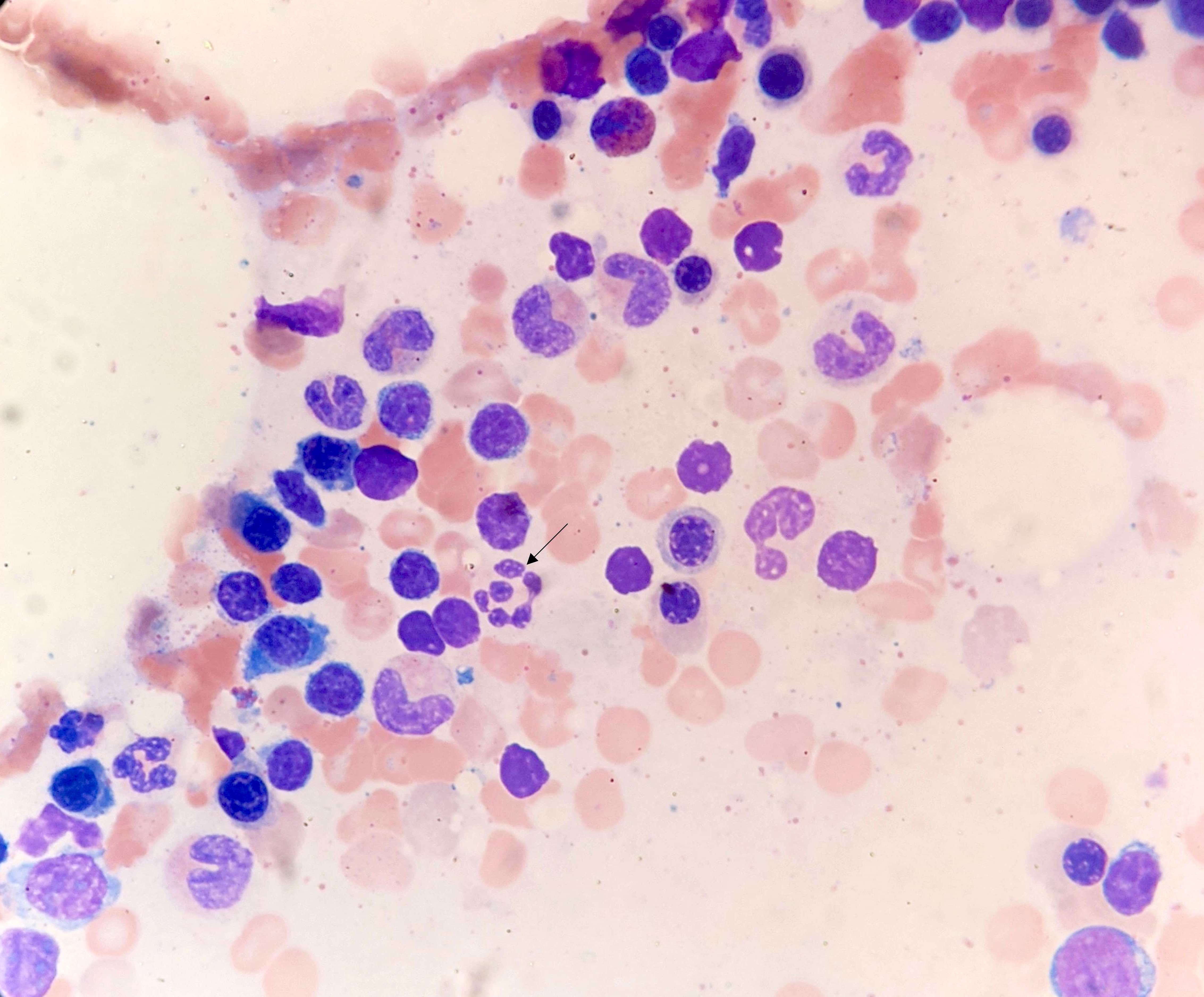
Hypersegmented neutrophil
- Pancytopenia
- Macrocytosis and macroovalocytosis: macroovalocytes are unique to megaloblastic anemia; large, oval, egg shaped cells with diminished / absent central pallor
- Anisopoikilocytosis
- Some red cells show Howell-Jolly bodies, Cabot rings (acidophilic, figure of 8 shape, arginine rich), basophilic stippling
- Reference: Clin Med Res 2006;4:236
Images hosted on other servers:
Macroovalocytosis /
round macrocytes
in liver disease
Macrocytosis with a lymphocyte
Megaloblastoid erythroids, basophils, mast cells in a bone marrow smear
- Bone marrow, right posterior iliac crest, aspirate smears, trephine biopsy:
- Hypercellular marrow (80%) showing erythroid hyperplasia, with reversal of M:E ratio (1:5); erythroid precursors show megaloblastic changes and mild to moderate dyserythropoeisis (see comment)
- Comment: The above findings of bone marrow hypercellularity, erythroid hyperplasia and megaloblastic changes are consistent with megaloblastic anemia in the right clinical context. Advised correlation with serum vitamin B12 and folate levels.
- Peripheral smear: Peripheral smear examination shows pancytopenia. Macrocytes and macroovalocytes are seen along with rare cells with basophilic stippling. Nucleated red cells are seen with immature nucleus (4/100 WBCs). Neutrophils show hypersegmentation.
- Bone marrow aspirate: Adequate for evaluation. Bone marrow aspirate smear is hypercellular with erythroid hyperplasia, reversal of M:E ratio (1:5). Erythroid precursors show megaloblastic changes, nuclear budding and nuclear irregularity. Myeloid lineage cells are reduced in number. Giant metamyelocytes are seen. Neutrophils show hypersegmentation. Megakaryocytes are reduced in number.
- Bone marrow biopsy: Adequate for evaluation. Bone marrow biopsy is hypercellular for age (80%), showing marked erythroid hyperplasia on CD61 staining.
- Myelodysplastic syndrome (MDS):
- Marked dysplasia
- Dysplastic features unique to MDS not seen in megaloblastic anemia include nuclear hyposegmentation and hypogranularity, hypolobation of megakaryocytes, micromonolobated megakaryocytes, hypogranular platelets, increased blasts
- Nonmegaloblastic macrocytic anemias:
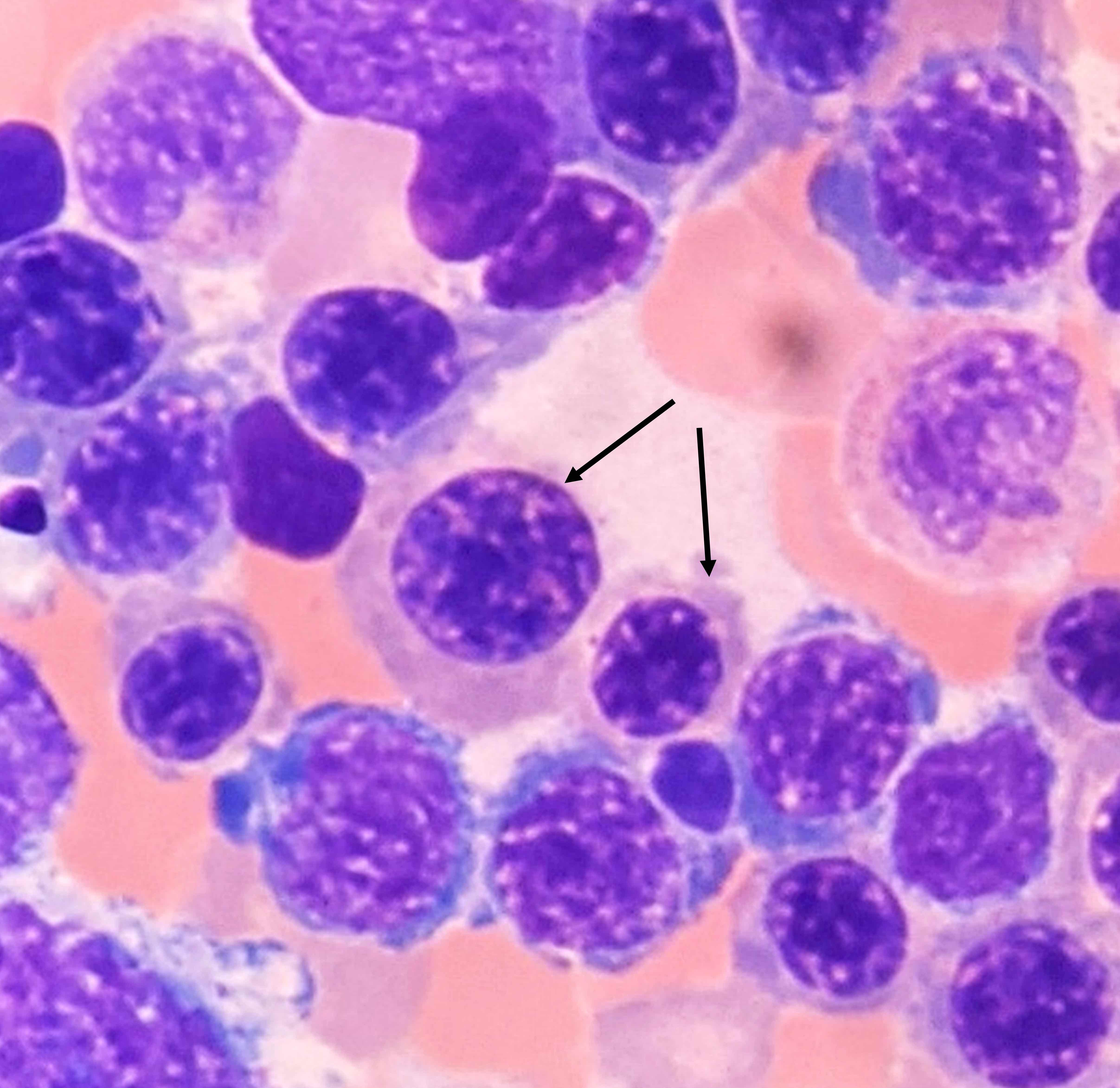
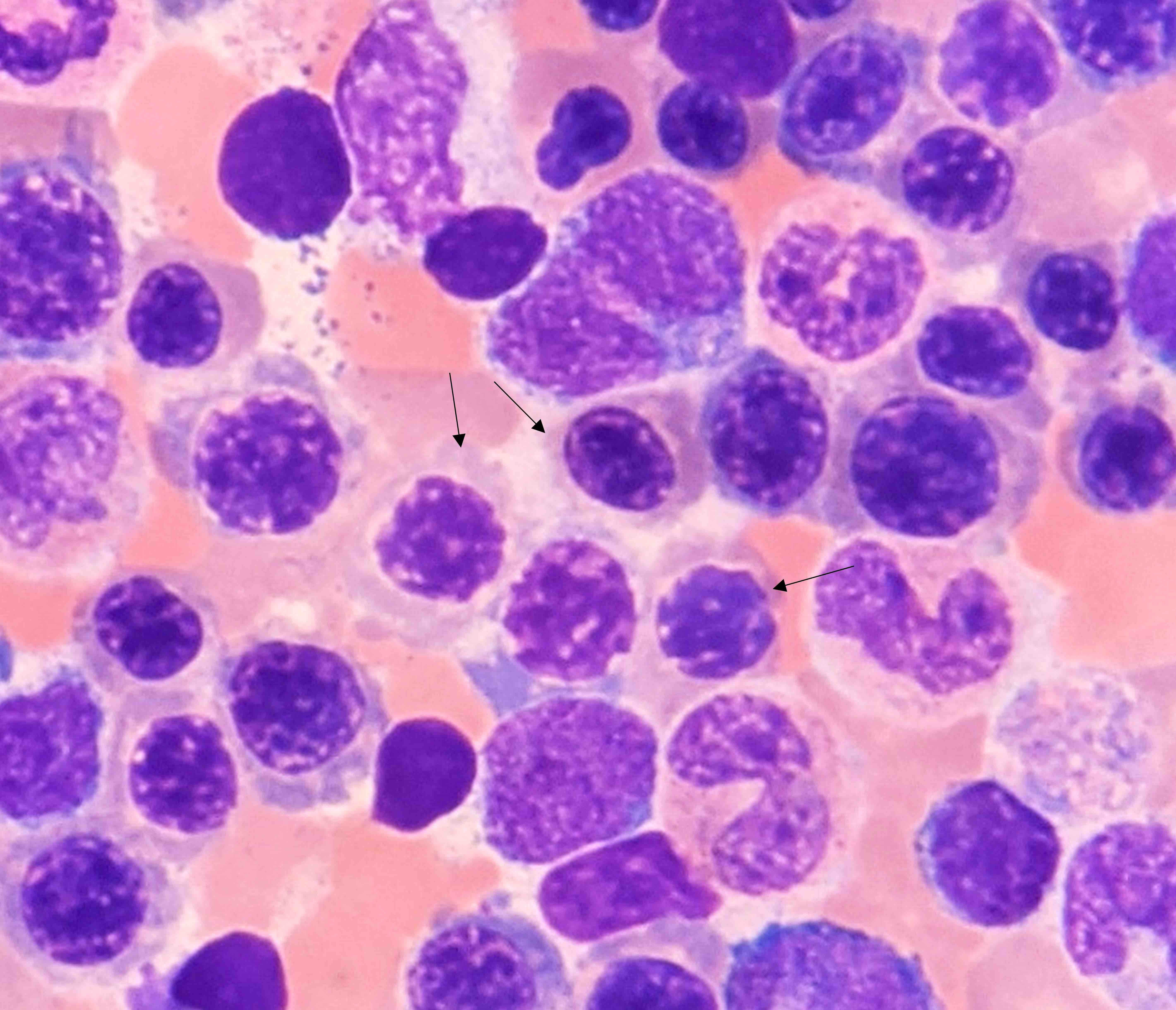
These cells are characteristic of which entity?
- Acute myeloid leukemia
- Iron deficiency anemia
- Megaloblastic anemia
- Sideroblastic anemia
- Duodenum
- Ileum
- Jejunum
- Stomach
Comment Here
Reference: Megaloblastic anemia
- Monocytes usually < 1% of bone marrow cells
- Develop from myeloid stem cell, to monoblast to promonocyte to monocyte (bone marrow) to monocyte (peripheral blood) to macrophage (tissue)
- Same progenitor cell as neutrophils, under the influence of M-CSF
- Difficult to identify monoblasts and promonocytes in normal bone marrow
- Gradual nuclear folding and acquisition of cytoplasmic granules similar to those in neutrophils but fewer and smaller
- Monocytes are also precursors of dendritic cells
- Produce numerous regulatory cytokines in addition to their traditional phagocytic role
- Part of the innate immune system: replenish macrophages and dendritic cells and move quickly to the site of infection (8 - 12 hours)
- A significant proportion (~50%) are stored in the spleen; they spend 1 - 3 days in the bloodstream before moving into tissue
- Causes of monocytosis in bone marrow: reactive / accumulation disorders (high cell turnover states, granulomatous processes, hemophagocytic lymphohistiocytosis, postchemotherapy, fat necrosis, post cytokine therapy, storage diseases, Rosai-Dorfman disease); neoplastic disorders (Langerhans cell histiocytosis, chronic myelomonocytic leukemia, juvenile myelomonocytic leukemia, myeloproliferative diseases, acute myeloid leukemia, malignant histiocytosis, histiocytic sarcoma)
- Causes of monocytopenia in peripheral blood: acute infections, stress, glucocorticoids, aplastic anemia, hairy cell leukemia, acute myeloid leukemia, myelotoxic drugs, genetic syndromes
- Monoblast: 12 - 20 microns, moderate basophilic cytoplasm without granules, may show pseudopod formation, often intense staining on periphery and with perinuclear zone, round / oval nuclei with fine chromatin and 1 - 4 large prominent nucleoli; nucleus may show indentations or folding
- Promonocyte: features intermediate between monoblast and monocyte, more irregular nuclear contour, less basophilic and more granulated cytoplasm
- Monocyte: largest of leukocytes (12 - 20 microns); round with smooth margins or pseudopod-like cytoplasmic extensions; abundant light blue cytoplasm with fine pink azurophilic granules; may have vacuoles or phagocytized material; large bilobed, kidney shaped or U shaped nucleus with moderately clumped chromatin; no nucleolus; N/C ratio is 65 - 80%
AFIP images
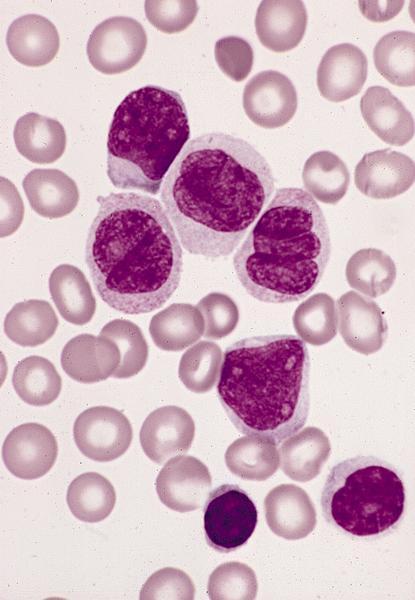
Promonocyte
Bone marrow in acute monocytic leukemia (AML M4):
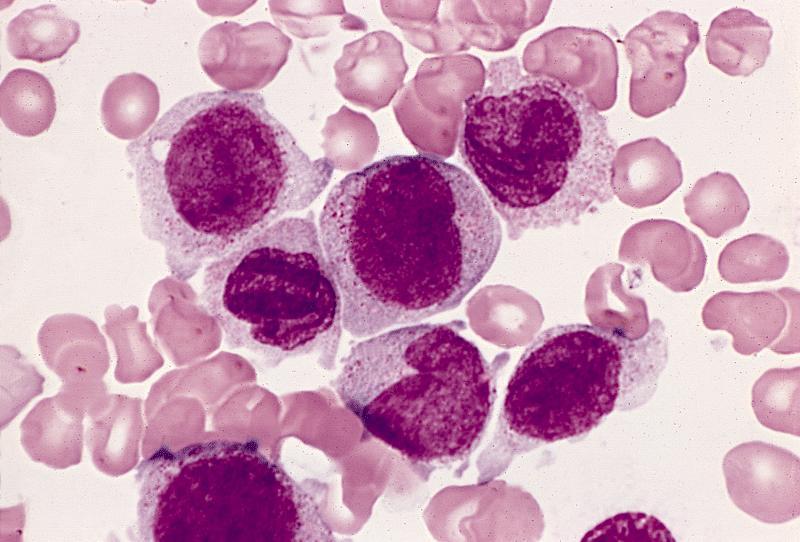
Monoblasts are larger cells with abundant
cytoplasm and round nuclei and
promonocytes are two cells with folded and
creased nuclei next to monoblast in center
Images hosted on other servers:

Monocyte
- Mainly CD14
- Also CD4, CD7, CD11a, CD11b, CD11c, CD11d, CD12, CD13, CD15 (variable), CD15u, CD17, CD18, CD23 (activated), CD29, CD30, CD32, CD33, CD36, CD37, CD38, CD39, CD40, CD43, CD44R, CD45, CD45RB, CD45RC, CD45RO, CD48, CD49a, CD49b, CD49d, CD49e, CD49f, CD51, CD52, CD54, CD61, CD62L, CD64, CD65, CD65s, CD68, CD83 (transient), CD84, CD85A, CD85B, CD85D, CD85E, CD85F, CD85I, CD85J, CD85K, CD85M, CD86, CD87, CD88, CD89, CD91, CD92, CD93, CD97, CD101, CD102, CD105 (activated), CD111, CD112, CD114, CD116, CD122, CD123 (plasmacytoid), CD126, CD127, CD128, CD132, CD137, CD139, CD141, CD148, CD156, CD157, CD163, CD165, CD166 (activated), CD171, CD180, CD210, CD226 and CD227
Images hosted on other servers:

Monocyte - TEM and SEM
- Definition: necrosis of myeloid tissue and medullary stroma in large areas of hematopoietic bone marrow (Cancer 2000;88:1769)
- Repeat biopsy may be needed for diagnosis (another site or after waiting for regeneration)
- Rare, < 1% to 2% of bone marrow biopsies
- Often a severe complication of hematologic malignancies
- Seen in all age groups with no gender predilection
- Compromised microcirculation of oxygen and nutrients to bone marrow cells causes cell death
- Failure of microcirculation is due to inflammation related damage or mechanical obstruction (e.g. DIC, sickle cell disease, tumor emboli)
- Associated with acute lymphoblastic leukemia (ALL) and is often discovered prior to its initial diagnosis or at time of recurrence of ALL
- May be associated with other lymphomas, solid tumor, myeloproliferative disorders or noncancerous conditions (Cancer 2000;88:1769):
- Infection
- Septic shock (Ann Fr Anesth Reanim 2004;23:501)
- Megaloblastic anemia
- Tuberculosis
- Drugs: e.g. fludarabine, imatinib mesylate, interferon alpha
- Sickle cell disease (Am J Med Sci 2000;320:342)
- Disseminated intravascular coagulation
- Hemolytic uremic syndrome
- Antiphospholipid syndrome
- Hyperparathyroidism
- Systemic lupus erythematosus (SLE)
- Bone pain in 75% (Cancer 2000;88:1769); may occur throughout body or localized to lower back / legs
- Fever in 69% (Cancer 2000;88:1769)
- Fatigue
- Jaundice
- Bone marrow biopsy and aspirate demonstrate the histologic and cytologic features of necrosis and underlying diseases
- Should correlate with clinical and laboratory findings
- Anemia (91%), thrombocytopenia (78%), leukoerythroblastic findings (51%)
- Elevated lactic dehydrogenase (LDH) and alkaline phosphatase (ALP) in nearly 50% (Cancer 2000;88:1769)
- Bone marrow scanning
- Technetium 99m sulfur colloid and indium chloride localize selectively to reticuloendothelial elements of bone marrow, corresponding with areas of hematopoiesis
- Hematopoietic areas with changes greater than 2 cm can be visualized
- In bone marrow necrosis, there is little or no isotope uptake throughout the bone marrow cavity; bone marrow scintigraphy confirms the absence of hematopoiesis and also identifies the existence of residual bone marrow activity from which material can be obtained via guided aspiration or biopsy
- In recovery, reappearance of normal hematopoiesis is observed
- Nuclear magnetic resonance
- Noninvasive method to evaluate a large fraction of bone marrow
- Changes in bone marrow signal intensities reflect changes in proportions of fat and water contained in cellular elements
- Bone marrow necrosis is characterized by an increase of water content due to watery changes of bone marrow and replacement of fat by serous material
- While somewhat nondiagnostic, MRI can show the extent of necrosis and can serve as a guide to biopsy sites in which viable hematopoietic bone marrow is suspected; MRI can also document conversion from abnormal to normal bone marrow (Cancer 2000;88:1769)
- Greatly dependent on age of patient and nature of associated disease:
- Children with ALL and bone marrow necrosis appear to have the same prognosis as those without necrosis
- Adults with bone marrow necrosis associated with a noncancerous condition may have a better chance of complete recovery and long term survival than adults with bone marrow necrosis associated with a hematologic malignancy
- May be difficult to determine if necrosis is due to a drug or DIC versus an underlying malignancy; prognosis in these cases is difficult to predict
- Often short survival (Indian J Pathol Microbiol 2000;43:47)
- 14 year old boy with idiopathic bone marrow necrosis (J Clin Diagn Res 2013;7:525)
- 66 year old man with G-CSF associated bone marrow necrosis in AML after induction chemotherapy (Case Rep Hematol 2012;2012:314278)
- Bone marrow necrosis associated with imatinib mesylate in patient with Philadelphia chromosome+ ALL (Ann Hematol 2006;85:542)
- Bone marrow necrosis as presenting feature of childhood ALL (Pediatr Blood Cancer 2007;49:367)
- Treat underlying disease; provide supportive measures for associated anemia, pancytopenia, thrombocytopenia, embolic processes or other complications
- Corticosteroids for CREST syndrome (Eur J Haematol 2005;74:75)
- Gelatinous aspirated specimen
- On trephine biopsy, disruption of normal bone marrow architecture with considerable loss of fat spaces
- Early: nuclear pyknosis and karyorrhexis
- Late: all cell outlines gone, serous transformation of marrow
Images hosted on other servers:
Necrotic and nonnecrotic areas
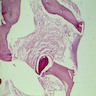
Disruption of normal bone marrow architecture
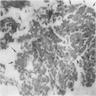
Antiphospholipid syndrome
Nuclear features associated with necrosis
Ghost-like hematopoietic cells
- Amorphous proteinaceous material
- Enmeshed ghost-like hematopoietic cells with irregular or indistinct cell membranes
- Cytoplasm may shrink or be vacuolated
- Nuclear features are indicative of necrosis (pyknosis, karyorrhexis, karyolysis)
- Avascular bone necrosis: may show destruction of spicular architecture
- Marrow aplasia: usually only a loss of myeloid components with no destruction of the reticular architecture
- The myeloid - monocytic lineage predominates in the bone marrow (50 - 70% of nucleated cells), which produces 1 - 2 x 109 granulocytes/kg/day
- Proliferation driven by multiple factors including G-CSF, especially adjacent to bony trabeculae and blood vessels
- Progresses from myeloid stem cell to myeloblast to promyelocyte to neutrophilic myelocyte to neutrophilic metamyelocyte to neutrophilic band to segmented neutrophil (polymorphonuclear neutrophil - PMN)
- Maturation is characterized by decreasing N/C ratio and increasing granule production and nuclear segmentation
- Immature forms are usually paratrabecular or perivascular; exceptions are after bone marrow transplantation, cytokine administration or chemotherapy
- Mature granulocytes pass through the bone marrow sinus wall, through endothelial cell cytoplasm and into the circulation
- Less mature cells are not released into the circulation under homeostatic conditions
- Present in the blood for only a few hours (circulating or marginated pools) before they egress into tissues
- Granulopoiesis is inhibited (negative feedback) by mature granulocytes and the bone marrow microenvironment
- Granulopoiesis can increase tenfold in patients with sustained infections or other inflammatory conditions
- Primary granules: large, round, red pink, electron dense; contain myeloperoxidase, elastase, lysozyme, cathepsin G and acid hydrolases; also called azurophilic (stains sky blue with azure stains used in the past); these granules are formed in promyelocytes (Blood 1979;53:179)
- Secondary (specific) granules: smaller, electron lucent (clear), cause characteristic cytoplasmic color in Wright stained preparations; contain lactoferrin and lysozyme; these granules are formed in myelocytes
- Also tertiary (contain gelatinase) and even quaternary (represented by secretory vesicles probably formed via endocytosis)
- Variations: brisk neutrophilia (30 x 109/L or 30,000/μL) at birth at high altitude, lower counts in black patients than in other populations, age related (newborn: 7,000 - 28,000/μL; infant: 2,500 - 10,000; child: 1,500 - 8,000; adult: 1,500 - 7,000)
- Causes of neutrophilia: constitutional (hereditary neutrophilia, familial cold urticaria, leukocyte adhesion deficiency) and acquired (bacterial infections, chronic Epstein-Barr virus infection, cytokine therapy, eosinophilia myalgia syndrome)
- Causes of neutropenia: constitutional (Fanconi anemia, cyclic neutropenia, Kostmann syndrome, Chédiak-Higashi syndrome) and acquired (drug induced neutropenia, acquired aplastic anemia, megaloblastic anemia)
- Myeloblast: 15 - 20 microns, round / oval; usually scant basophilic cytoplasm with no perinuclear halo; may contain Auer rods (due to fusion of azurophilic granules) or delicate azurophilic granules; round / oval nuclei with occasional indentations or clefts; one or more nucleoli; finely reticulated chromatin; N/C ratio is 80 - 85%
- Type I myeloblast: no granules in cytoplasm
- Type II myeloblast: up to 15 - 20 delicate granules in cytoplasm
- Type III myeloblast: more than 15 - 20 cytoplasmic granules but otherwise has features of a blast cell
- Promyelocyte: 10 - 20 microns; increased basophilic cytoplasm (compared to blasts) with primary coarse red purple, azurophilic granules; large, round / oval nucleus with red blue and fine to slightly condensed chromatin; 1 - 2 nucleoli; N/C ratio is 75 - 85%
- Myelocyte: 10 - 18 microns; round / oval with abundant pink cytoplasm with prominent red purple azurophilic (primary) granules and numerous fine, lilac and specific (neutrophilic) secondary granules; round / oval to slightly indented nucleus with red blue and slightly condensed chromatin; usually no nucleolus; N/C ratio is 50 - 65%
- Metamyelocyte: 10 - 16 microns; moderate pink or colorless cytoplasm with occasional red purple azurophilic (primary) granules and variable fine, lilac and specific (neutrophilic) secondary granules; indented nucleus with light blue purple and granular chromatin; no nucleolus; N/C ratio is 40 - 50%
- Band: 10 - 15 microns; abundant pink cytoplasm with many fine, lilac and neutrophilic (secondary) granules and possibly a few red purple azurophilic (primary) granules; nucleus is indented to more than half the distance from the farthest nuclear margin; elongated and horseshoe shaped nucleus; if lobulated, the bridge or isthmus between the lobes must be wide enough to have two distinct parallel dark margins with light nuclear chromatin in between; has blue purple and clumped granular chromatin; no nucleolus; N/C ratio is 33 - 40%
- Neutrophil: 10 - 15 microns; abundant pink cytoplasm with many fine, lilac and neutrophilic (specific or secondary) granules; lobulated (segmented) nucleus with 2 - 5 lobes, connected by a thin filament of chromatin; the filament is so narrow that there is no visible chromatin between the two sides; filaments may be difficult to visualize due to folding or twisting of nucleus; in other areas, the chromatin is dense with no nucleolus; N/C ratio is 33%
- Classify cell with folded nucleus as neutrophil if:
- Margins of two adjacent lobes are completely separated
- Width of either of the two adjacent lobes markedly narrows or converges towards the junction of the lobes (making it possible for there to be a hidden filament)
- The nucleus is so extensively folded that one cannot determine if a filament is present
- Classify cell with folded nucleus as band if:
- Elongated band form crosses over itself without any evidence of constriction to a filament
- Only the distal tip of the nucleus is slightly bent upon itself
- The hidden area in the fold between two adjacent lobes is so small and the lobe width is so thick that it is unlikely that a thin filament is present
AFIP images
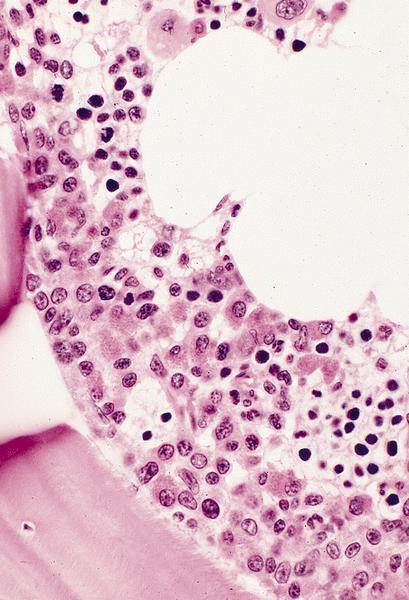
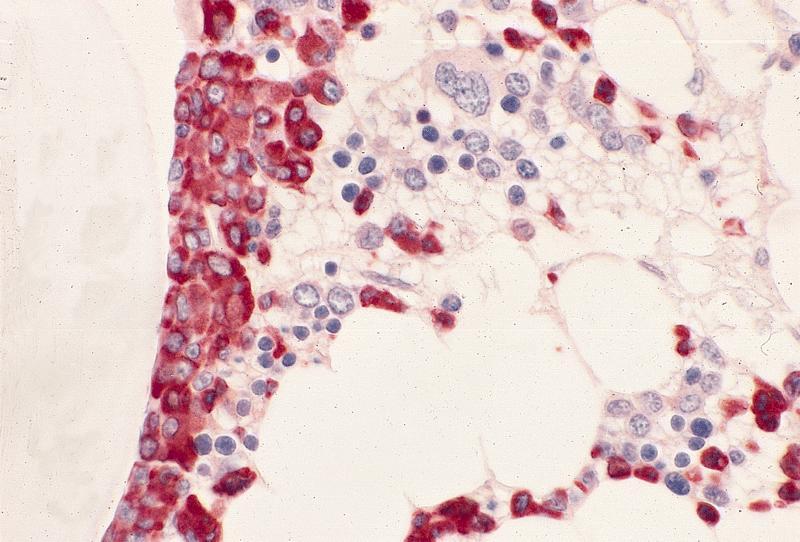
Myelocytes along endosteum of bone trabeculae: H&E and myeloperoxidase stain

Perivascular immature granulocytes
Images hosted on other servers:
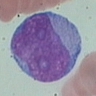
Type I myeloblast
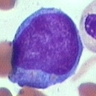
Type II myeloblast
Type III myeloblast
Blast (upper) and promyelocyte (lower)
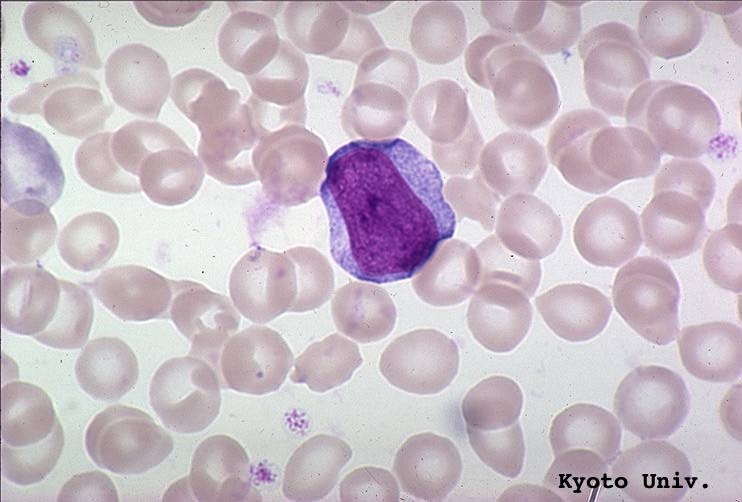
Myeloblast (type not specified)
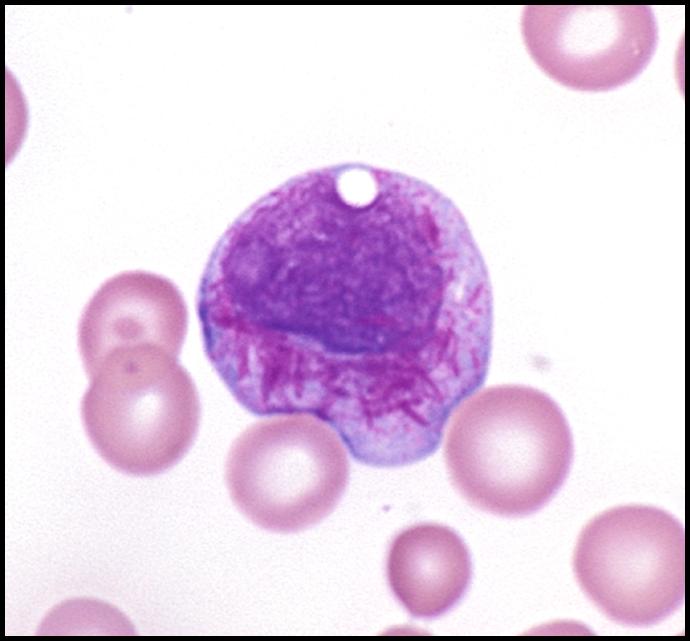
Promyelocytes: with Auer rods
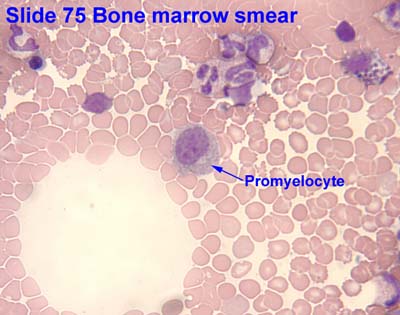
Promyelocytes
3 myelocytes + 2 metamyelocytes (right and lower)
Neutrophilic myelocytes

Myelocyte and bands
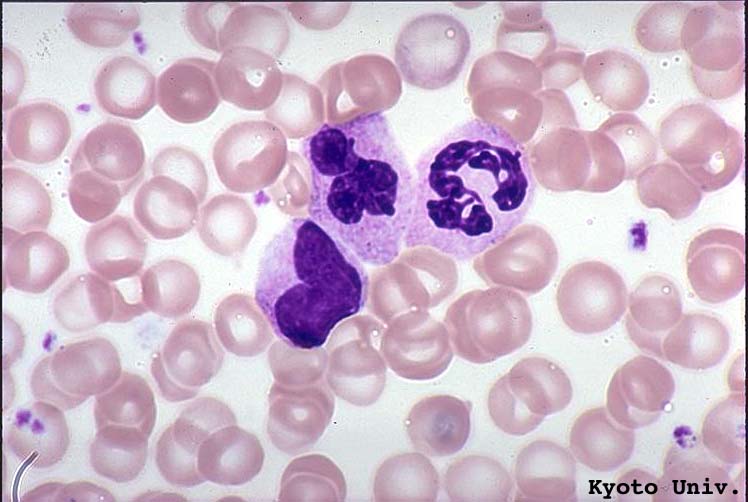
Metamyelocytes
- Neutrophils (may also stain other precursors): CD10, CD11b, CD11c, CD12, CD13, CD14 (weak - 30%), CD15, CD15s, CD15u, CD16a, CD16b, CD17, CD18, CD24, CD29 (low), CD30, CD31, CD32, CD33 (low), CD35, CD37 (low), CD43, CD45RO, CD47, CD47R (weak), CD48 (weak), CD49e, CD62L, CD63 (weak), CD64, CD65s, CD66a, CD66b, CD66c, CD66d, CD66e, CD68, CD69, CD83, CD84, CD85F, CD85M, CD87, CD88, CD89, CD92, CD93, CD97, CD101, CD107a, CD107b (weak), CD114, CD116, CD128a, CD128b, CD132, CD139, CD141, CD148, CD156a, CD157 and CD170
Images hosted on other servers:
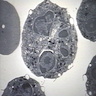
Neutrophil
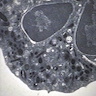
Details of granules
- Autosomal recessive sphingomyelin cholesterol lipidoses
- Types A / B: deficiency of lysosomal acid sphingomyelinase causes accumulation of sphingomyelin and other lipids in macrophages throughout the body
- Bone marrow transplantation (variable improvement) (J Inherit Metab Dis 2003;26:775)
- Clusters and individual foam cells up to 90 microns
- Cytoplasm has clear vacuoles of varying size, may be mulberry or soap bubble-like
- Nuclei are small and round with coarse chromatin
- Similar foam cells in hypercholesterolemia and Tangier disease
- Also disorders with massive cell destruction that overloads body's capacity to digest lipids (thalassemia, sickle cell anemia, ITP, chronic renal failure)
Definition / general:
- Acute neuronopathic form
- Most common type
- Occurs in infants
- Presents with jaundice, hepatomegaly and profound brain damage
- Usually death by age 2 years
- No effective treatment
Molecular description:
- Types A and B due to mutations in SMPD1 gene
Additional references:
Definition / general:
- Chronic form (visceral form) without nervous system involvement
- Highest incidence in those of Turkish, Arabic and North African descent
- Uncommon in Ashkenazi Jews (Am J Hum Genet 2002;71:1413)
- Onset of hepatosplenomegaly in preteen years
- Good prognosis
- May eventually require oxygen therapy due to lung involvement
Treatment:
- Bone marrow transplantation
- Enzyme replacement
Micro description:
- Large macrophages with foamy cytoplasm
- Also sea blue histiocytes (Ann Hematol 2001;80:620)
Molecular description:
- Types A and B due to mutations in SMPD1 gene
Additional references:
Definition / general:
- Chronic neuronopathic form (NPC)
- Rare (1 per 100K live births) disease of newborns
- Autosomal recessive
- May present in first few months of life with cholestatic jaundice or hepatosplenomegaly or develop in adult years
- Fatal in some; others live into adulthood
- 95% have mutations of NPC1 gene
- Involves error of trafficking of cholesterol leading to accumulation of unesterified cholesterol, not sphingomyelin
Micro description:
- Macrophages with abnormal cholesterol storage (foam cells) in 60% (Arch Dis Child 2006;91:841)
- May also have sea blue histiocytes
Micro images:
Images hosted on other servers:
Foamy cells
Molecular description:
- Due to mutations in NPC1 (OMIM: NPC1 Gene; NPC1) or NPC2 (OMIM: NPC2 Gene; NPC2) genes
Additional references:
Definition / general:
- Variable age of presentation
- May have extensive brain damage
- Usually moderate hepatosplenomegaly
- Involves error of trafficking of cholesterol leading to accumulation of unesterified cholesterol, not sphingomyelin
- Associated with family origin in Nova Scotia, Canada
- Bone forming cell that arises from marrow mesenchymal (stromal) cells, unlike osteoclasts which are of hematopoietic origin
- Along endosteal surface of bony trabeculae or along margins in marrow smears
- Common in children; in adults associated with various diseases and healing biopsy sites
- Synthesize angiopoietin and osteopontin, which inhibit hematopoietic stem cell proliferation (Br J Haematol 2006;134:467, J Clin Invest 2006;116:1195)
- Synthesize and transport collagenous matrix, initiate and regulate mineralization, control removal of bone via osteoclasts
- When active are plump and present on bone surface; eventually are encased within the collagen they produce and get flattened (and become osteocytes)
- Activity is promoted by physical activity (Wolff's law of bone adaptation)
- Express parathormone receptors (mediates the activation of osteoclasts) and vitamin D receptor
- Control osteoclast activity via parathyroid hormone (parathormone), PHRP (parathyroid hormone related protein), IL1 and TNF alpha
- Digestion of bone by osteoclasts releases cytokines and growth factors for osteoblasts
Images hosted on other servers:

Interaction of osteoblasts and hematopoietic stem cells
- Large (25 - 50 microns), often oval with abundant blue gray cytoplasm and perinuclear hof separated from the nucleus (unlike plasma cells)
- Nucleus is markedly eccentric and round / ovoid with one or more nucleoli
AFIP images
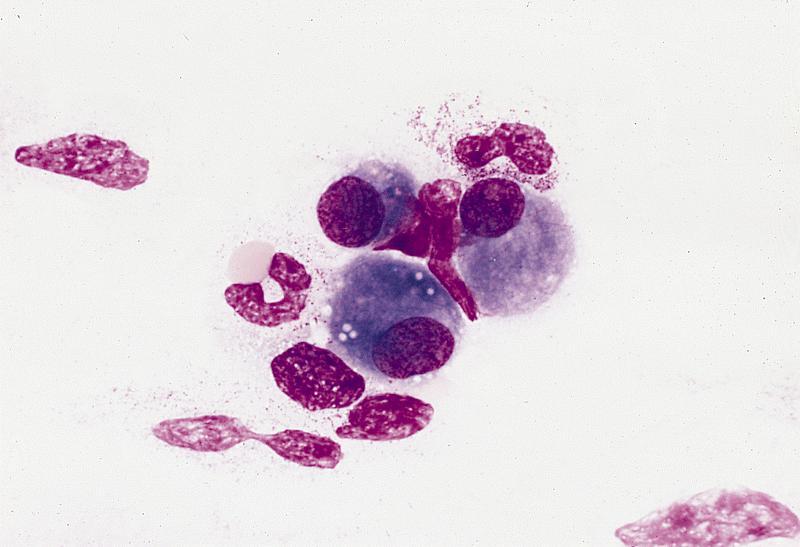
Osteoblasts in marrow smear
Images hosted on other servers:
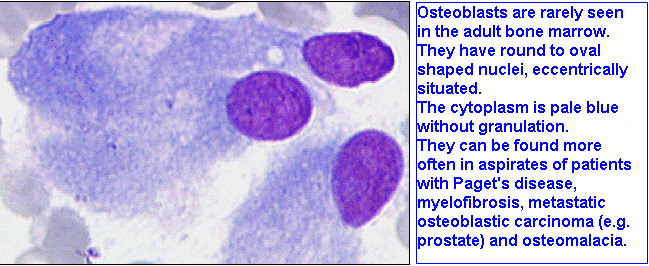


Osteoblasts
- Resemble fibroblasts due to well developed rough endoplasmic reticulum and Golgi zone
Images hosted on other servers:
Colorized SEM
- Cells derived from hematopoietic progenitor cells involved in bone resorption, primarily due to remodeling and not calcium homeostasis
- Along endosteal surface of bony trabeculae (reside in Howship lacunae) or along margins in marrow smears
- Common in children (active bone remodelling); in adults associated with metabolic or neoplastic diseases
- Some overlapping immunophenotypic features with monocytes / macrophages
- Relatively recently identified osteoclast colony stimulating factor and transcription factors involved in osteoclastogenesis
- Activated by parathyroid hormone and by cytokines RANKL and macrophage colony stimulating factor (Arthritis Res Ther 2006;8:201)
- Osteoclasts use their ruffled borders (with villous extensions) to bind to matrix adhesion proteins, produce resorption pits / bays (shallow concavities) called Howship lacunae
- Plasma membrane forms a seal with bone; osteoclast acidifies extracellular area, which solubilizes the mineral and releases enzymes which dissolve the matrix
Images hosted on other servers:

Cross section
Bone resorptive cycle
- Very large (up to 100 microns), multinucleated (2 - 12 nuclei) giant cells associated with bone surface
- Abundant, blue purple pale pink cytoplasm containing many fine, red purple granules
- Also contain bone sand in the cytoplasm
- Multiple, relatively uniform but widely separated nuclei, each with one nucleolus and dense chromatin
- Generally, less mature osteoclasts have basophilic cytoplasm and mature forms have brightly eosinophilic cytoplasm
AFIP images
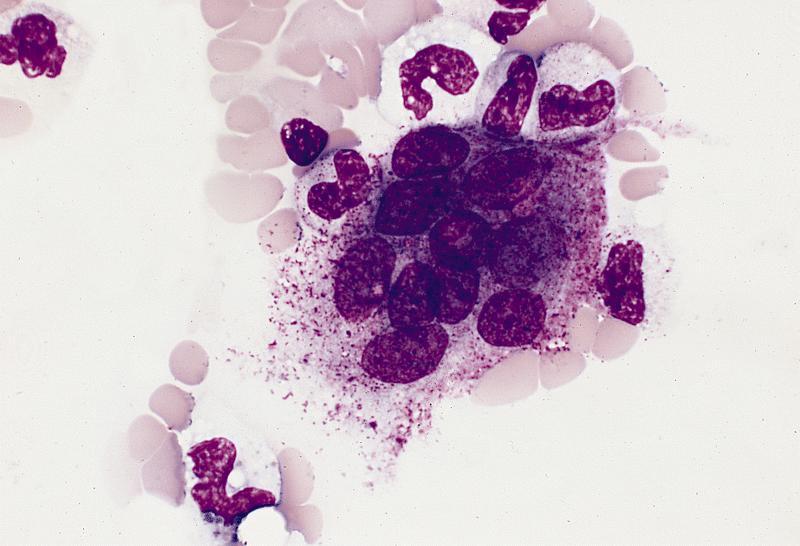
Very large cell
Images hosted on other servers:



Osteoclasts
- Numerous mitochondria, rare lysosomes
- Ruffled edge in area of cell membrane is associated with bone resorption
Images hosted on other servers:

Osteoclast
- Associated with HIV but serology may be negative because patients cannot produce IgG antibodies (Hum Pathol 2000;31:161)
- Affects bone marrow erythroid precursors, causing transient aplastic crisis in patients with hemolytic anemia or chronic anemia
- Affects placenta and other tissues in fetuses, causing fetal hydrops or death
- Also causes erythema infectiosum (Fifth disease), arthropathy (note: B19 DNA is common in rheumatic patients but clinical significance is unclear, J Clin Virol 2005;33:71)
- B19 DNA found in bone marrow of 2% of healthy individuals (J Clin Microbiol 2002;40:933)
- Erythrovirus V9 causes similar pathology (J Clin Microbiol 1999;37:2483)
- Chronic parvovirus infection: associated with immunodeficiency, erythroid hyperplasia, numerous inclusions, particularly in basophilic and polychromatic erythroblasts
- Newborn with leukoerythroblastosis (Haematologica 2005;90:ECR38)
- 7 year old boy with coinfection with falciparum malaria (Haematologica 2005;90:ECR41)
- 22 year old woman with sickle cell-like crisis and bone marrow necrosis associated with parvovirus B19 infection and heterozygosity for haemoglobins S and E (J Intern Med 1999;245:103)
- 29 year old woman with fatal fungal superinfection, sickle cell disease and massive bone marrow necrosis (Haematologica 2006;91:ECR18)
- 41 year old HIV+ man with fulminant parvovirus infection following erythropoietin treatment (Arch Pathol Lab Med 2000;124:441)
- 42 year old woman with systemic lupus erythematosus (Intern Med 2003;42:538)
- 50 year old man with chronic anemia (University of Pittsburgh: Chronic Anemia)
- 63 year old man with hereditary spherocytosis (Postgrad Med J 2003;79:244)
- Marked erythroblast hypoplasia, immature giant erythroblasts
- Occasional erythroblasts may have intranuclear inclusions with surrounding rim of residual chromatin
- Rarely hemophagocytosis (Jpn J Infect Dis 2005;58:149)
Images hosted on other servers:
50 year old man: rare pronormoblast forms
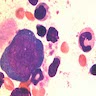
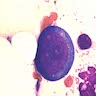
Large and megaloblastic pronormoblast forms
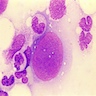
Giant cell with eosinophilic nuclear inclusion
B19+
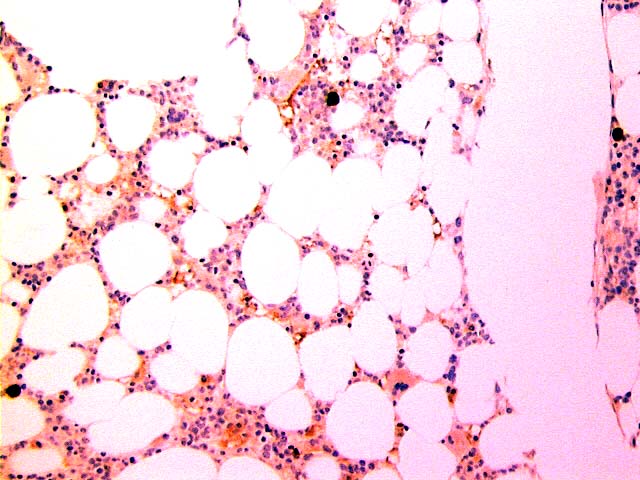
B19+ cells in bone marrow biopsy
- Hemoglobin A, B19
Images hosted on other servers:
Native parvovirus B19
- Congenital dyserythropoietic anemia (J Pediatr Hematol Oncol 2004;26:133)
- Rare childhood disorder with refractory anemia, vacuolization of bone marrow cells, lactic acidosis and pancreas insufficiency
- Those with a mild phenotype or supported through bone marrow failure may develop the encephalopathic features of Kearns-Sayre syndrome (ophthalmoplegia, retinal degeneration, ataxia and endocrine abnormalities)
- Varied organ involvement due to heteroplasmy (mixture of more than one type of mitochondrial DNA; different mixtures cause variable effects), although most die by age 3
- Intramitochondrial iron accumulation causes sideroblastic anemia, also reduction in cytochrome c oxidase activity (Eur J Haematol 2006;77:169)
- Causing nonimmune hydrops fetalis (Chin Med J (Engl) 2003;116:1952)
- 3 month old girl with pancytopenia, failure to thrive, gastroesophageal reflux (University of Pittsburgh: Anemia, Neutropenia, and Lactic Acidosis)
- 6 month old girl without marrow involvement (Arch Dis Child 1997;77:56)
- Very young girl who developed Kearns-Sayre syndrome (Minerva Pediatr 2005;57:143)
- Kearns-Sayre syndrome with features of Pearson marrow pancreas syndrome (Hum Pathol 1999;30:577)
Images hosted on other servers:

Aspirate smear shows few spicules
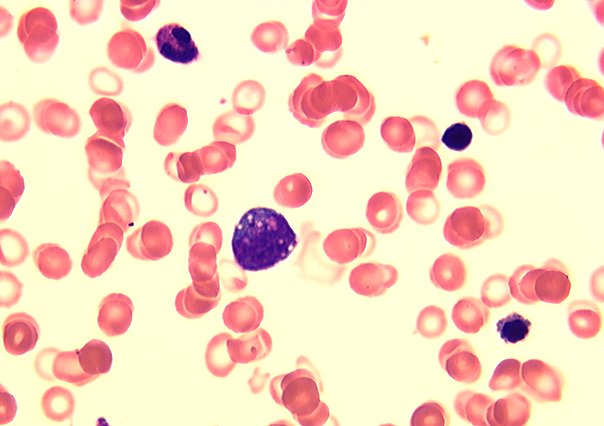
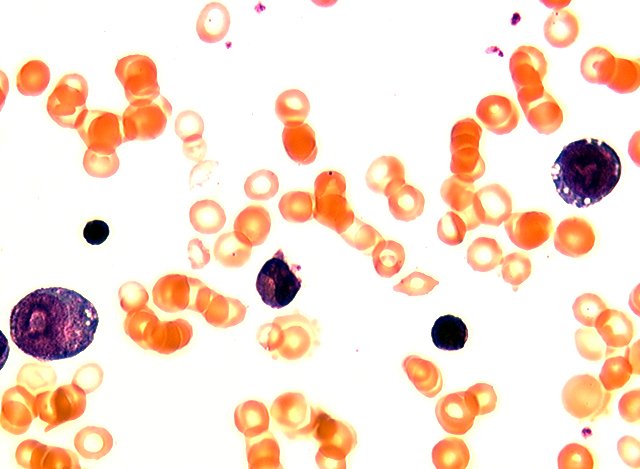

Pronormoblast with vacuoles
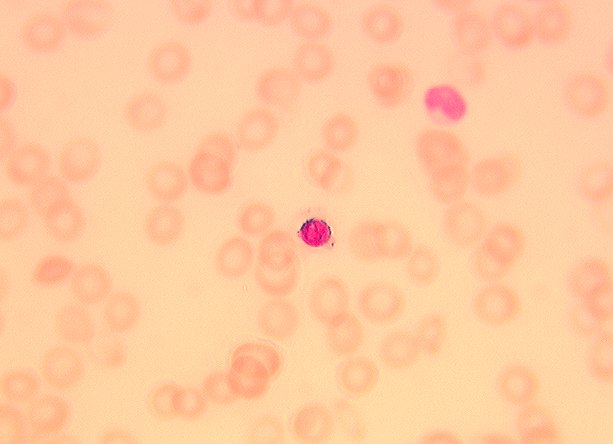
Occasional ringed sideroblasts (Prussian blue)
- Deletion mutations in mitochondrial DNA
- Usually less than 1% of marrow cells; rare in infants
- Often perivascular and in particle crush specimens
- Indeterminate lifespan ranging from days to months
- Produces and secretes antibodies
- Plasmablast: precursor to plasma cell, produces more antibodies than B cells but less than mature plasma cells
- Plasma cells: the last stage of B cell maturation, express CD38 but lose CD19
- Differentiation of mature B cells into plasma cells is dependent upon the transcription factors Blimp1 / PRDM1 and IRF4
- Unlike their precursors, plasma cells cannot act as antigen presenting cells because they no longer display MHC II; they do not take up antigen because they no longer display significant quantities of immunoglobulin on the cell surface
- Plasma cells they cannot switch antibody classes and can only produce a single kind of antibody in a single class of immunoglobulin
- Ovoid cells with abundant deep blue cytoplasm and perinuclear hof, eccentric nucleus with coarse chromatin and clock face (art wheel) pattern
- May have occasional binucleated cells; no nucleoli
Images hosted on other servers:
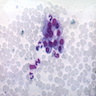
Plasma cells
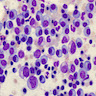

Plasma cells in myeloma
- Prominent Golgi zone and rough endoplasmic reticulum
Images hosted on other servers:
Plasma cell without labels
- Plasmacytosis in bone marrow and peripheral blood can be monoclonal (plasma cell neoplasm) or polyclonal
- See also lymphoid aggregates, polymorphous reactive lymphoid hyperplasia and systemic polyclonal B immunoblastic proliferation
- Reactive plamacytosis in bone marrow can be seen in both neoplastic and nonneoplastic conditions:
- Seen as reactive component in myeloproliferative neoplasms, myelodysplastic syndromes, mastocytosis (Sem Hop 1983;59:2119) or other malignancies
- Seen in polyclonal hypergammaglobulinemia (Int J Hematol 2005;81:62), systemic polyclonal B immunoblastic proliferation (Am J Clin Pathol 1992;98:222, J Forensic Sci 2001;46:156), autoimmune disorders (e.g. SLE), immunocompromise (e.g. HIV infection) and infections (Leuk Lymphoma 2003;44:379)
- Particularly common (86%) in HIV+ patients (Indian J Pathol Microbiol 2005;48:7, Clin Immunol 2001;100:250)
- When strict diagnostic criteria are applied, benign polyclonal plasmacytosis in bone marrow and peripheral blood is uncommon
- Can be associated with fever, leukocytosis, skin rash
- May also see lymphadenopathy, dyspnea, hepatosplenomegaly, jaundice
- Case specific presentation varies by etiology
- Bone marrow biopsy for histomorphologic evaluation and immunophenotypic analysis of plasma cells to demonstrate polyclonality
- When possible, bone marrow aspirate for cytomorphologic evaluation, differential count, flow cytometric analysis (to demonstrate plasma cell polyclonality and confirm lack of aberrant immunophenotype) and cytogenetic / molecular analyses to rule out neoplastic process, if clinically indicated
- CBC and peripheral blood smear to evaluate peripheral blood involvment
- If peripheral blood is involved, use serum protein electrophoresis to assess polyclonality
- 31 year old man with systemic inflammatory syndrome and hepatic inflammatory cell infiltration caused by IL6 producing pheochromocytoma (Endocr J 2005;52:193)
- 41 year old man with polyclonal reactive peripheral blood plasmacytosis mimicking plasma cell leukemia (Leuk Lymphoma 2003;44:379)
- 53 year old woman with methimazole induced aplastic anemia caused by hypocellular bone marrow with plasmacytosis (Thyroid 2004;14:231)
- 72 year old woman with reactive plasmacytosis mimicking multiple myeloma and primary Sjögren syndrome (J Korean Med Sci 2005;20:506)
- 77 year old man with polyclonal plasma cell proliferation with marked hypergammaglobulinemia and multiple autoantibodies (Ann Clin Lab Sci 2006;36:479)
- 89 year old woman with polyclonal proliferation of plasma cells associated with marked hypergammaglobulinemia (Int J Hematol 2005;81:62)
- Systemic polyclonal B immunoblastic proliferation with marked peripheral blood and bone marrow plasmacytosis (Am J Clin Pathol 1992;98:222)
- Mainly symptomatic and supportive
- Treat the underlying condition
- Bone marrow biopsies often show hypercellularity with an increase in plasma cells / plasmacytoid cells, in perivascular or interstitial distribution
- Background trilineage hematopoiesis is present
- Myeloid and erythroid precursors show normal morphology and may be variably decreased in number, depending on the extent of replacement by plasma cells
- Megakaryocytes are often normal in number, morphology and localization
Images hosted on other servers:
Lymph node biopsy
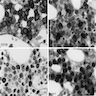
Differentiation of mature plasma cells
- Spectrum of plasma cell differentiation
- Plasmablasts appear large with scant basophilic cytoplasm, prominent nuclei, dispersed fine chromatin, conspicuous nucleoli
- Immature plasma cells show abundant basophilic cytoplasm, round nuclei with condensed chromatin
- With differentiation, plasma cells show more basophilic cytoplasm with perinuclear halos and eccentric nuclei
- Prominent increase of plasma cells / plasmacytoid cells in a variety of sizes and maturation stages
Images hosted on other servers:

Stages of plasma cell differentiation
- CD56 (Am J Pathol 2002;160:1293, Histopathology 2004;44:375) is negative in benign plasma cells (as benign polyclonal plasmacytosis) and ~80% of plasma cell leukemia (Leukemia 1998;12:1977)
- Lack of CD56 expression should not be used solely to rule out plasma cell neoplasm
- Negative for CD20 and surface light chains (as in normal plasma cells)
- Flow cytometric analysis of plasma cells in bone marrow aspirate demonstrates polytypic plasma cells with no immunophenotypic aberration:
- Forward scatter and side scatter larger than those of lymphocytes
- Dimmer CD45, moderate CD19, negative CD20, bright CD38, bright CD138
- Variable intensity of HLA-DR
- Polytypic cytoplasmic kappa and lambda light chains
- Negative for surface kappa and lambda light chains
- Negative for CD56
- A small portion of larger, brighter HLA-DR+ cells can be detected, representing plasmablasts or early plasma cells which are negative for cytoplasmic light chains while the smaller, dim HLA-DR+ cells represent mature plasma cells
Images hosted on other servers:

Plasma cells in bone marrow
- Molecular analysis demonstrates polyclonality of the plasma cells
- Cytogenetic analysis (e.g. karyotyping, FISH) confirms the lack of cytogenetic aberrations
- Castleman disease: see Ann Pathol 1996;16:133
- Myeloma / plasmacytoma: CD56+, clonal, immature and pleomorphic plasma cells
- Plasma cell neoplasms, in particular plasma cell leukemia when peripheral blood is involved
- Plasmacytic SLL / CLL
- Reactive lymphoid hyperplasia of bone marrow is characterized by an increase in benign appearing lymphocytes and can be polymorphous, comprising small to medium sized lymphocytes admixed with plasma cells, histiocytes and sometimes occasional immunoblasts, eosinophils and mast cells:
- Variable peripheral blood lymphocytosis
- May be due to either an interstitial increase in lymphoid cells or an increase in lymphoid aggregates
- A specific example - hematogone hyperplasia:
- Hematogones are precursor B cells with a high nuclear to cytoplasmic ratio, condensed chromatin pattern and no nucleoli
- May be seen in variety of conditions especially in children
- More common after chemotherapy in both adults and children
- Can be difficult to distinguish from lymphoblasts in B cell acute lymphoblastic leukemia
- Associated with immune cytopenia, collagen vascular disease, AIDS (historically) (Arch Anat Cytol Pathol 1991;39:137)
- For these sections - Epidemiology, Pathophysiology, Etiology, Clinical features, Diagnosis, Laboratory, Radiology, Prognostic factors, Individual case reports and Treatment, please review these topics: lymphoid aggregates, persistent polyclonal lymphocytosis, plasmacytosis and systemic polyclonal B immunoblastic proliferation
- Overall, clinical and morphologic features are indicative of a benign reactive process, immunohistochemical and flow cytometric analysis detect no aberrant antigen expression or loss and cytogenetic and molecular findings show no abnormalities or evidence of clonal rearrangement
- Focal, poorly circumscribed, randomly distributed lymphoid aggregates
- Lymphocytes may be irregular and accompanied by plasma cells, immunoblasts, eosinophils, endothelial cells, mast cells, histiocytes and epithelioid histiocytes
- Benign hematogone hyperplasia: described above
- Can be difficult to distinguish from lymphoblasts in B cell acute lymphoblastic leukemia but patient age is important
- Children normally have more lymphocytes (up to 35%) in bone marrow while in adults, the normal value for lymphocytes on marrow aspirate smear is 6 - 25% (Br J Haematol 1996;94:206)
- Can be difficult to distinguish from lymphoblasts in B cell acute lymphoblastic leukemia but patient age is important
- Peripheral T cell lymphoma: clonal; rare in AIDS patients
- Any defect resulting in failure of bone marrow to produce adequate erythroid cells (eMedicine: Pure Red Cell Aplasia)
- Near absence of red blood cell precursors in bone marrow with associated anemia and reticulocytopenia
- Normal numbers of megakaryocytes and white blood cell precursors
- Called either erythroblastopenia or red cell aplasia (RCA)
- Sustained isolated RCA is called pure red cell aplasia (PRCA)
- Incidence of RCA varies by specific subtype, the rarest is Diamond-Blackfan anemia
- Pure RCA predominates in adolescents and adults but can affect any age
- Humoral and cell mediated immunity are likely to be responsible
- Anti-erythroid antibodies may be detected in some patients
- Also due to allogeneic bone marrow transplant from ABO incompatible donor (treat with aggressive plasmapheresis)
- May be due to antibodies to recombinant erythropoietin in those with chronic renal failure
- Primary (idiopathic) or secondary (acquired)
- Associated with at least 40 different drugs; phenytoin is most common
- Also associated with thymoma, hematolymphoid neoplasms (T-LGL leukemia most common), carcinoma, infection (parvovirus B19, mumps, hepatitis, EBV, rarely HIV), immune disorders
- Patients present with severe normochromic, normocytic to slightly macrocytic anemia and marked reticulocytopenia
- Occult parvovirus B19 infection must be excluded by PCR testing in all cases of secondary PRCA
- Evaluation for any underlying condition known to be associated with PRCA is also necessary
- Profound normocytic, normochromic anemia with reticulocytopenia and without polychromasia
- Other blood parameters generally unremarkable
- 19 year old woman with Mycophenolate mofetil treatment for SLE (Lupus 2013;22:213)
- 43 year old man with CD8+ lymphocytosis in bone marrow (J Clin Pathol 2005;58:1118)
- 70 year old woman with associated thymoma and Good syndrome (Ann Thorac Surg 2011;91:1620)
- 73 year old man following erythropoietin treatment (Rinsho Ketsueki 2012;53:110)
- Steroids and immunosuppressants, antilymphocyte globulin, plasmapheresis, IV immunoglobulins
- Bone marrow examination usually reveals complete loss of erythroid lineage
- Sometimes scattered, morphologically unremarkable erythroblasts may be seen especially with IHC
- Other hematopoietic lineages are morphologically normal and show intact maturation
Images hosted on other servers:
No erythroid precursors
- Due to Coxiella burnetii, a rickettsiae usually not identifiable directly (Am J Surg Pathol 1985;9:752)
- Acute primary Q fever is essentially benign but may evolve into subacute Q fever endocarditis or a postinfectious fatigue syndrome
- Organism persists in bone marrow for years (QJM 2005;98:7, Epidemiol Infect 2000;124:543)
- 71 year old man with malaise, fever, sweats (N Engl J Med 2006;354:1937)
- PCR or serology
Images hosted on other servers:
Q fever meningoencephalitis
Q fever pneumonia
- Suggestive finding is a fibrin ring / doughnut granuloma defined as a small, nonnecrotizing granuloma with ring-like structure composed of fibrinoid material, often with a central fat vacuole (Arch Pathol Lab Med 1986;110:1017)
- Rarely hemophagocytosis (Clin Infect Dis 1995;21:1029)
Images hosted on other servers:
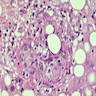
Fibrin ring granuloma with central fatty vacuoles
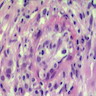
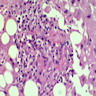
Liver: fibrin ring granuloma
Liver: doughnut granuloma
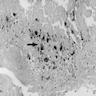
Cardiac valve
Liver: doughnut granuloma (arrow)
Cell lines infected with C. burnetii
Images hosted on other servers:

Within vacuole of host cell
- Note: reactivity varies by type of fixation
- CD45 / LCA: lymphocytes
- Kappa and lambda light chains: to determine clonality or not of lymphocytes and plasma cells
- Myeloperoxidase and lysozyme: for AML subtyping
- B cell markers: CD20, CD79a and possibly PAX5
- T cell marker: CD3
- TdT: for ALL
- Iron stains (Prussian blue): on aspirate smears and trephine biopsies (J Clin Pathol 2005;58:269)
- 0 = Absent
- 1 = Trace
- 2 = Present (sparse)
- 3 = Present (moderate)
- 4 = Abundant (abnormal)
- To assess iron stores, examine 7 - 9 particles (J Clin Pathol 2004;57:1038)
- Absence of staining iron is not diagnostic of iron deficiency anemia (Ann Hematol 2001;80:166)
- Possibly reticulin stain or Masson trichrome stain
- Also CD34 (blasts and blood vessels), Factor VIII (blood vessels), CD61 (megakaryocytes), CD68 (macrophages), Hemoglobin A (for AML-M6) and glycophorin (for erythroid cells)
- Clinical syndrome is associated with splenomegaly, mild thrombocytopenia and bone marrow involvement
- Many reports using the term "syndrome" are actually reporting just a microscopic finding
- Microscopic finding is associated with various hematologic and systemic disorders, including:
- Niemann-Pick disease (Hum Pathol 1982;13:1115)
- Normal marrow
- Ceroid histiocytosis (Blood 1971;37:587)
- Total parenteral nutrition (Leuk Lymphoma 1998;28:523)
- 44 year old man with sea blue histiocytosis secondary to Niemann-Pick disease type B (Ann Hematol 2001;80:620)
- Histiocytes have cytoplasm filled with globules / granules of varying size containing blue or blue green pigment (Romanowsky stain) or yellow tan pigment (H&E)
Images hosted on other servers:
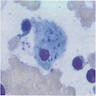
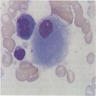
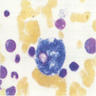
Chronic myelogenous leukemia
Refractory anemia with excess blasts

Refractory anemia with excess blasts in transformation
- Myelin figures
Images hosted on other servers:
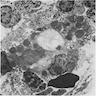
Hyperlipidemia: cytoplasmic electron dense granules
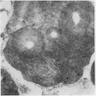
"Finger print" arrangement of membranes
- Also known as Shwachman-Bodian-Diamond syndrome (SBDS)
- Rare autosomal recessive childhood stem cell disorder with peripheral cytopenia (particularly neutropenia), ineffective hematopoiesis, variable marrow cellularity; also pancreatic exocrine insufficiency with pancreatic fatty infiltration
- 25% have bone abnormalities (metaphyseal chondrodysplasia leading to short stature)
- Often considered a congenital neutropenia but 25% develop aplastic anemia; may be a myelodysplastic disorder from inception (Arch Pathol Lab Med 2002;126:1157)
- Additional clinical findings: intellectual disability, facial dysmorphism, dental anomalies, ichthyosis, thoracic deformities
- Often presents with infections at birth due to neutropenia, also malabsorption (steatorrhea)
- Normochromic normocytic anemia, increased HgF levels, thrombocytopenia
- Low serum isoamylase and trypsinogen
- With successful bone marrow transplant (Eur J Pediatr 1999;158:995)
- 25% develop aplastic anemia, 7% develop myelodysplastic syndrome or acute myeloid leukemia
- Supportive treatment (G-CSF, transfusions, pancreatic enzyme replacement)
- Orthopedic management of skeletal deformities
- Hematopoietic stem cell transplant may be curative
- Variable clinical course: pancreatic insufficiency may improve with time but bone marrow failure tends to progress
- Hypocellular marrow; scattered mild dysplastic changes in all cell lines; may have prominent hematogones (Arch Pathol Lab Med 2000;124:1379)
- May demonstrate shift to immaturity in myeloid series
- Variable bone marrow cellularity including normocellular and even occasional hypercellular cases
- Erythroid and megakaryocytic lineages typically preserved initially
Images hosted on other servers:

Bone marrow aspirate

Decreased bone marrow cellularity
- Mutations in Shwachman-Bodian-Diamond syndrome (SBDS) gene (7q11) in 90% (Blood 2004;104:3588)
- Two common gene mutations account for 75% of cases due to gene conversion into a pseudogene that is 97% homologous to the normal SBDS gene
- Genetic testing by sequencing the 5 exons of SBDS: at least one converted allele in 90% of patients, 2 converted alleles in 60% (mainly involving exons 2 and 3)
- Development of MDS / AML mostly associated with chromosome 7 abnormalities
- See also lymphoid aggregates, plasmacytosis and polymorphous reactive lymphoid hyperplasia
- A florid, systemic polyclonal proliferation of B immunoblasts accompanied by polyclonal hypergammaglobulinemia
- Rare, reactive, often associated with an acute immune disorder
- Involves peripheral blood, bone marrow, lymph nodes; often liver and spleen
- In peripheral blood, usually elevated WBC count with high percentage of B immunoblasts
- In middle aged / elderly, may be related to EBV (Pathol Res Pract 2006;202:609) or Pseudomonas (Am J Clin Pathol 1992;98:222)
- > 10% circulating lymphocytes with immunoblastic morphologic features may be seen in hantavirus cardiopulmonary syndrome (Am J Clin Pathol 2001;116:665)
- May mimic a neoplastic process
- CBC: usually leukocytosis with absolute lymphocytosis; neutrophilia with left shift may be present
- Anemia and thrombocytopenia are almost always present
- Positive antiglobulin test, as the anemia is frequently immune mediated
- Hypergammaglobulinemia, polyclonal per immunofixation electrophoresis (IFE)
- Rapid regression of disease process may occur
- Most patients who recover do not relapse but patients may die in acute phase (Cancer 1988;61:1350)
- 43 year old woman (J Forensic Sci 2001;46:156)
- With marked peripheral blood and bone marrow plasmacytosis (Am J Clin Pathol 1992;98:222)
- Supportive care and treatment tailored to associated immune disorder
- Variable response to treatment; many respond to steroids while others require chemotherapy (vincristine and cyclophosphamide) (Cancer 1988;61:1350)
- Hypercellular marrow with extensive infiltration resembling lymphoma or plasmablastic myeloma, due to lymphocytes, immunoblasts, plasma cells, intermediate forms
- Focal lymphocytic aggregates are characteristically present, varying from inconspicuous to large
- Similar infiltrate may be seen in lymph nodes with complete effacement of nodal architecture
- Immunohistochemistry is critical to demonstrate polytypic nature of process
- Bone marrow aspirate smear shows numerous lymphocytes, immunoblasts, plasma cells
- Prominent immunoblastic cytomorphology with intensely basophilic cytoplasm, relatively coarse chromatin pattern, distinct central nucleoli
- Frequently, evidence of maturation to plasma cells can be seen
- Elevated WBC count with absolute lymphocytosis including reactive lymphocytes, immunoblasts, plasma cells
- Neutrophilia with left shift may be present
- Anemia and thrombocytopenia are almost always present
- B cell markers and plasma cell markers in appropriate cellular components
- Plasma cells and immunoblasts are polytypic for immunoglobulin light chain staining pattern
- No aberrant antigen expression
- No light chain restriction
- Polytypic B cells per surface (B cells) or cytoplasmic (plasma cells) light chain expression
- No aberrant antigen loss or expression
- PCR reveals germline immunoglobulin and T cell receptor genes in polyclonal rearrangement pattern
- Rarely, oligoclonal B and T cell populations are demonstrated by PCR
- Clonal cytogenetic abnormalities have been identified in a subset of patients, raising the possibility of a cryptic neoplastic proliferation
- Non-Hodgkin lymphoma with immunoblastic morphologies and some plasma cell dyscrasias (plasmablastic myeloma) have monotypic light chain expression
- Also called selective aplasia, hypoplasia of megakaryocytes with missing radii
- Congenital megakaryocytic disorder with thrombocytopenia and decreased / absent megakaryocytes
- Impaired responsiveness to thrombopoietin (THPO) and high plasma THPO levels
- Rare autosomal recessive disorder of unknown etiology with bilateral absence of radii (thumbs are present) and isolated thrombocytopenia (single line aplasia)
- Lower limbs, GI, cardiovascular and other systems may be involved
- Prenatal diagnosis is possible by detecting bone abnormalities (ultrasound) and thrombocytopenia (cordocentesis)
- Usually severe thrombocytopenia ( < 50,000/μL)
- May have anemia related to extensive bleeding
- Normal or increased WBC
- 50% have leukemoid reaction with WBC > 40,000/μL and left shift
Images hosted on other servers:

Absent radius
- Child with thrombocytopenia absent radius syndrome showing a larger 1q21.1 deletion than his healthy mother has (Eur J Med Genet 2012;55:120)
- Platelet transfusions as needed; rarely stem cell transplantation may be required
- May have increased risk of acute leukemia (Eur J Haematol 2003;70:246)
- Platelet counts spontaneously reach normal levels by age 12 months
- Peripheral blood hematological improvement is associated with normal megakaryocytes in bone marrow
- Megakaryocytes and megakaryocyte precursors are absent or extremely scarce
- Stains for antiplatelet membrane glycoproteins do not detect small immature megakaryocytes
- Bone marrow may show erythroid hyperplasia if significant bleeding has occurred
- Deletions of 1q21.1 (RBM8A)
- Congenital amegakaryocytic thrombocytopenia: biallelic mutations in C-MPL [TPO receptor] at 1p34
- Fanconi anemia: absent thumbs, permanent thrombocytopenia, DEB testing (Am J Med Genet A 2004;124A:35) and mitomycin C testing (C R Biol 2013;336:29) helpful
- Trisomy 18: absent thumbs, permanent thrombocytopenia, karyotyping
- Foucar: Diagnostic Pathology: Blood and Bone Marrow, 1 Har/Psc edition, 2011, Foucar: Non-Neoplastic Disorders of Bone Marrow (Atlas of Nontumor Pathology), 1st edition, 2009, Penachansky: Pediatric Bone Marrow, 2004, OMIM: Thrombocytopenia-Absent Radius Syndrome; TAR, eMedicine: Thrombocytopenia-Absent Radius Syndrome
- Nephrotic cystinosis is a rare autosomal disorder with defective cysteine transport out of lysosomes
- Treated with cysteamine
- Case report: 23 year old man with crystalline histiocytosis in hereditary cysinosis (Arch Pathol Lab Med 2002;126:1135)
- Histology: crystals in marrow histiocytes and giant cells
- Electron microscopy: various abnormal findings (J Clin Pathol 2005;58:939)
- References: eMedicine: Cystinosis
- Also called alpha galactosidase A deficiency, angiokeratoma corporis diffusum universale
- X linked (Xq22.1) recessive lysosomal storage disease that affects 1 per 40,000
- Due to deficiency in lysosomal alpha galactosidase A, which catabolizes neutral glycosphingolipids
- High penetrance in hemizygous males with symptoms at infancy or childhood
- Heterozygous females have later age of presentation, more variable severity due to lyonization of X chromosome
- May have normal leukocyte alpha galactosidase A activity
- Deficiency causes intracellular accumulation of galabiosylceramide (ceramide trihexoside) and digalactosyl ceramide within skin, renal glomeruli and tubular epithelium, blood vessels, corneal epithelium, myocardium and ganglion cells
- Frequently misdiagnosed
- Case reports: 32 year old man with recurrent fevers of unknown origin (University of Pittsburgh: Recurrent Fevers of Unknown Origin); 32 year old man (Arch Pathol Lab Med 1980;104:17)
- Treatment: recombinant human alpha galactosidase A replacement therapy has only a modest effect, perhaps due to heterogeneous distribution (Mol Genet Metab 2007;90:307)
- Histology: cells with small globular, lightly eosinophilic inclusions
- Very rare lysosomal storage disease due to deficiency of beta glucuronidase, which degrades glycosaminoglycans
- Causes progressive accumulation of glycosaminoglycans and subsequent lysosomal distension in multiple tissues
- Histology: Alder-Reilly granules in neutrophils, monocytes, basophils and eosinophils; cytoplasmic inclusions surrounded by clear vacuoles in lymphocytes, plasma cells, osteoblasts, macrophages
- Positive stains: granulocytes, monocytes, lymphocytes - acid phosphatase
- References: Am J Clin Pathol 1982;78:544, eMedicine: Genetics of Mucopolysaccharidosis Type VII, Wikipedia: Sly Syndrome
Images hosted on other servers :
Fabry disease:
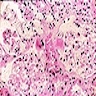
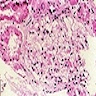
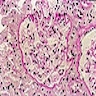
Kidney: podocytes are diffusely distended by intracytoplasmic vacuoles; scattered foam cells in interstitium
Kidney: PAS shows podocytes with foamy cell change
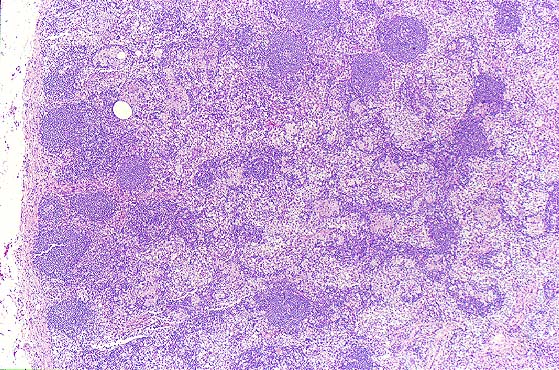
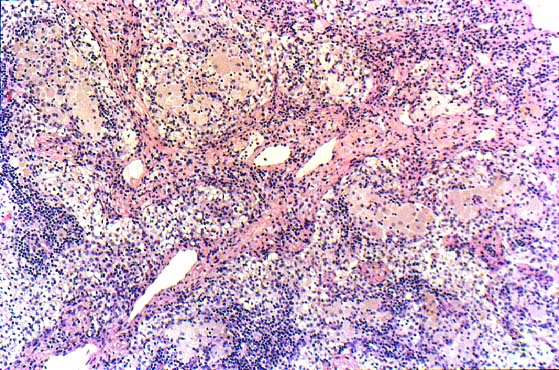

Lymph nodes: reactive hyperplasia
- Fabry disease: PAS, Oil red O, Sudan black, Luxol fast blue (all stain glycolipid and phospholipid-like material)
- Fabry disease:
- Characteristic single membrane bound intracellular inclusions (myelin-like figures, zebra bodies) that are 0.1 to 10 microns in diameter
- Round and lamellated with concentric electron dense layers
- Found in endothelial and smooth muscle cells, myocardium, fibroblasts and glomerular epithelium, urine sediment (Arch Pathol Lab Med 1981;105:361)
Images hosted on other servers - cystinosis:

Normal platelets
Platelets contain variable dense granules

Platelet undergoing spontaneous release
- Fucosidosis
- Gangliosidoses
- Gaucher disease
- Mucopolysaccharidoses: all have different intracellular distribution and ultrastructural features of inclusions, can detect by laboratory assays
- Treatment with chloroquine, amiodarone or aminoglycosides: have similar myelin-like figures (Hum Pathol 2003;34:285)
Cherry: 2022
Foucar: 2009
Foucar: 2017
Foucar: 2023
Hsi: 2017
Hudnall: 2019
IARC: 2017
Jaffe: 2016
Naeim: 2018
Wang: 2020
Find related Pathology books: hematopathology
