
15 November 2017 - Case of the Week #443
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Week page. To subscribe or unsubscribe to Case of the Week or our other email lists, click here.
Thanks to Drs. Safa Alshaikh, Maryam Hammad and Zainab Harb, Salmaniya Medical Complex, (Kingdom of Bahrain) for contributing this case and Dr. Hillary Zalaznick, Myriad Genetics, Inc., Utah (USA) for editing the discussion. To contribute a Case of the Week, first make sure that we are currently accepting cases, then follow the guidelines on our main Case of the Week page.

Advertisement
Website news:
(1) Our Industry News page highlights announcements from our Sponsors. It replaces the New Products and Services page and Management page. At the top, it contains links to the Buyers Guide, Promotions Page and Product Questionnaires. Let us know if you have any questions.
(2) Many visitors have indicated that they get better searches when using Google.com to search our website, compared to the Search box in the upper right on our website (which is also from Google). They first visit Google.com, then type PathologyOutlines.com and the search term (for example "Pathologyoutlines.com carcinoid").
(3) See most recent Textbook updates here.
Visit and follow our Blog to see recent updates to the website.
Case of the Week #443
Clinical history:
A 56 year old man with neurofibromatosis type 1 presented with a right axillary swelling, present for many years. Ultrasound showed a 9 x 5 cm well defined heterogeneous soft tissue mass arising from the right axilla. The mass was excised.
Histopathology images:

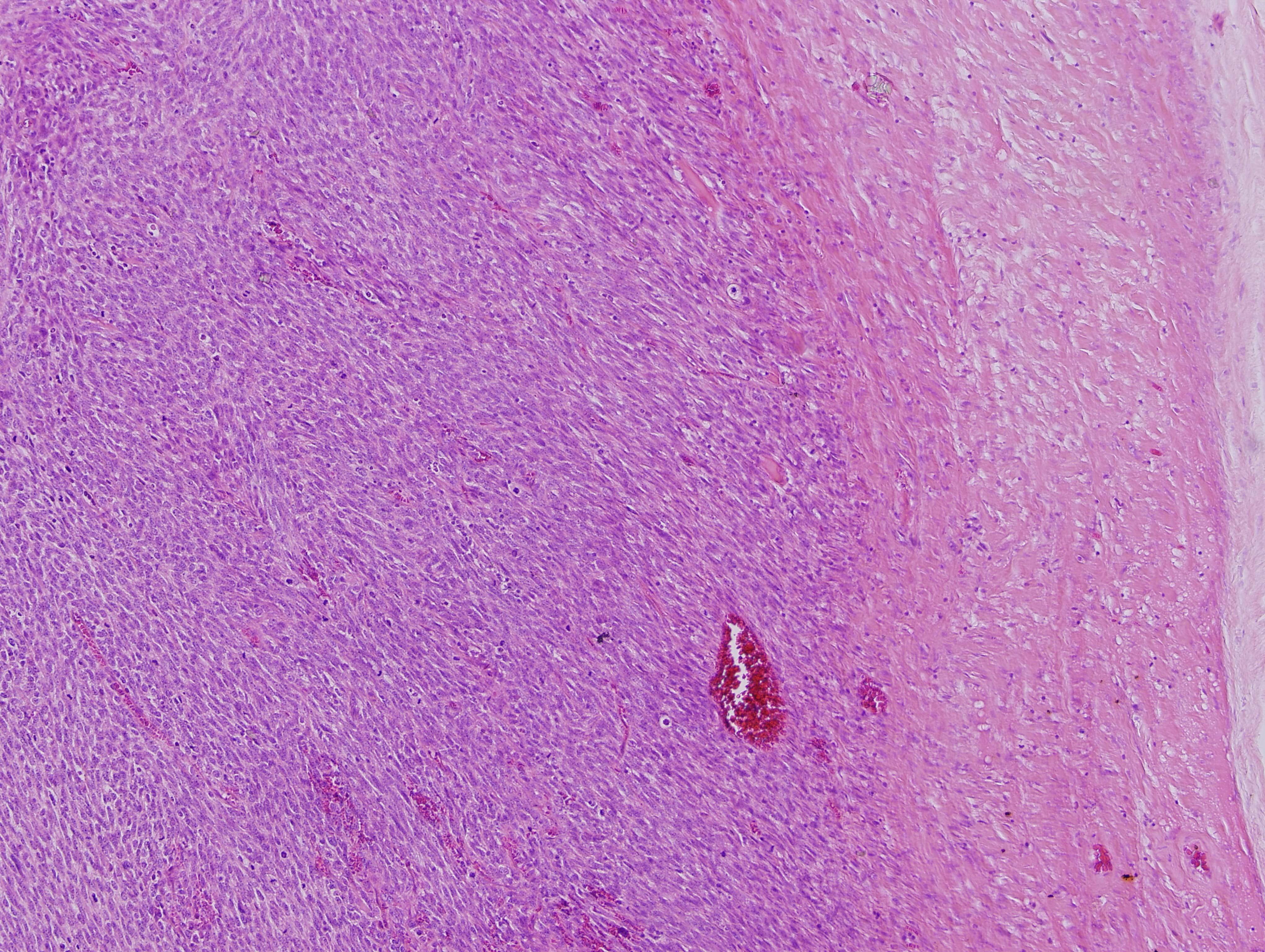
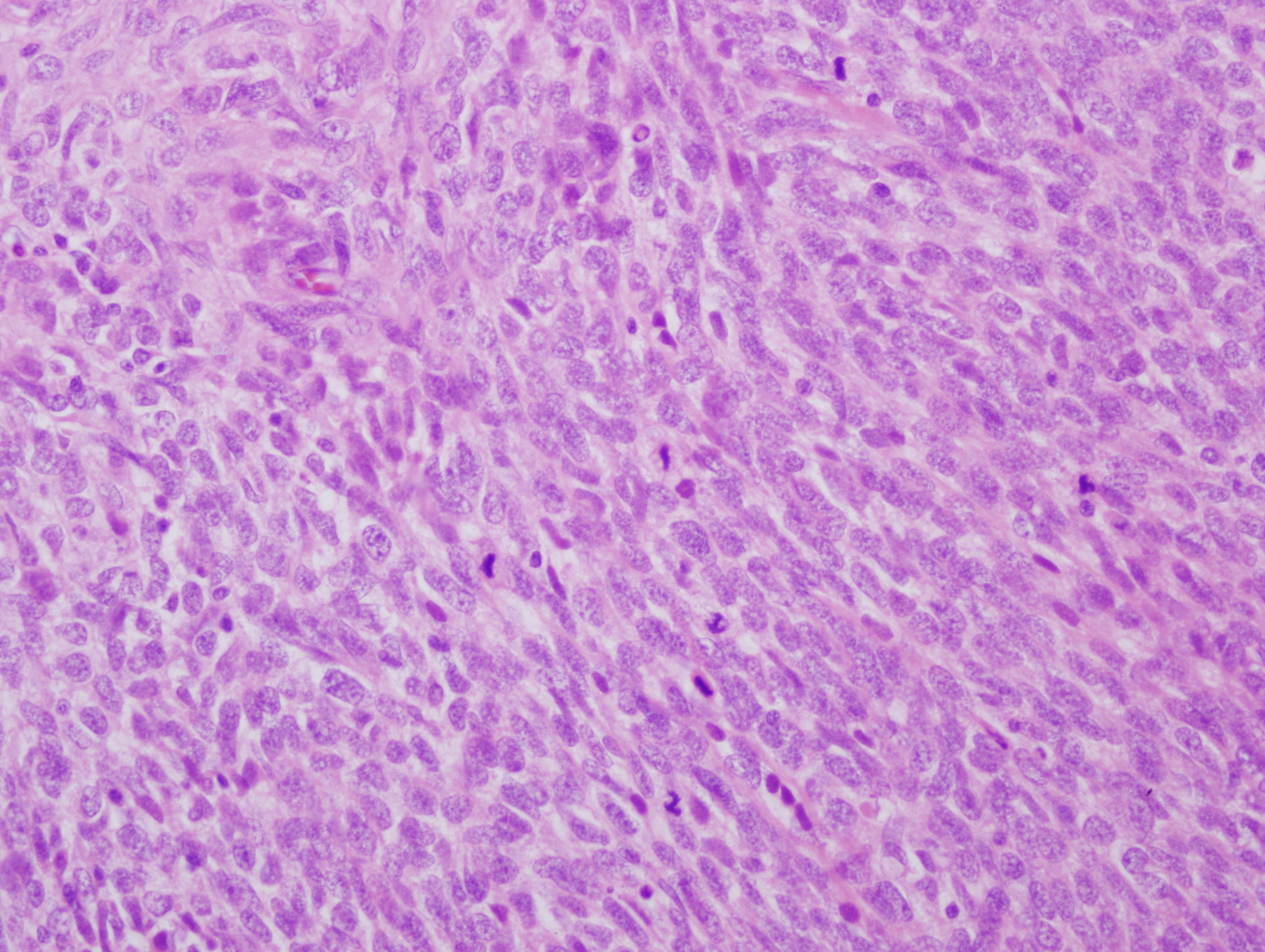
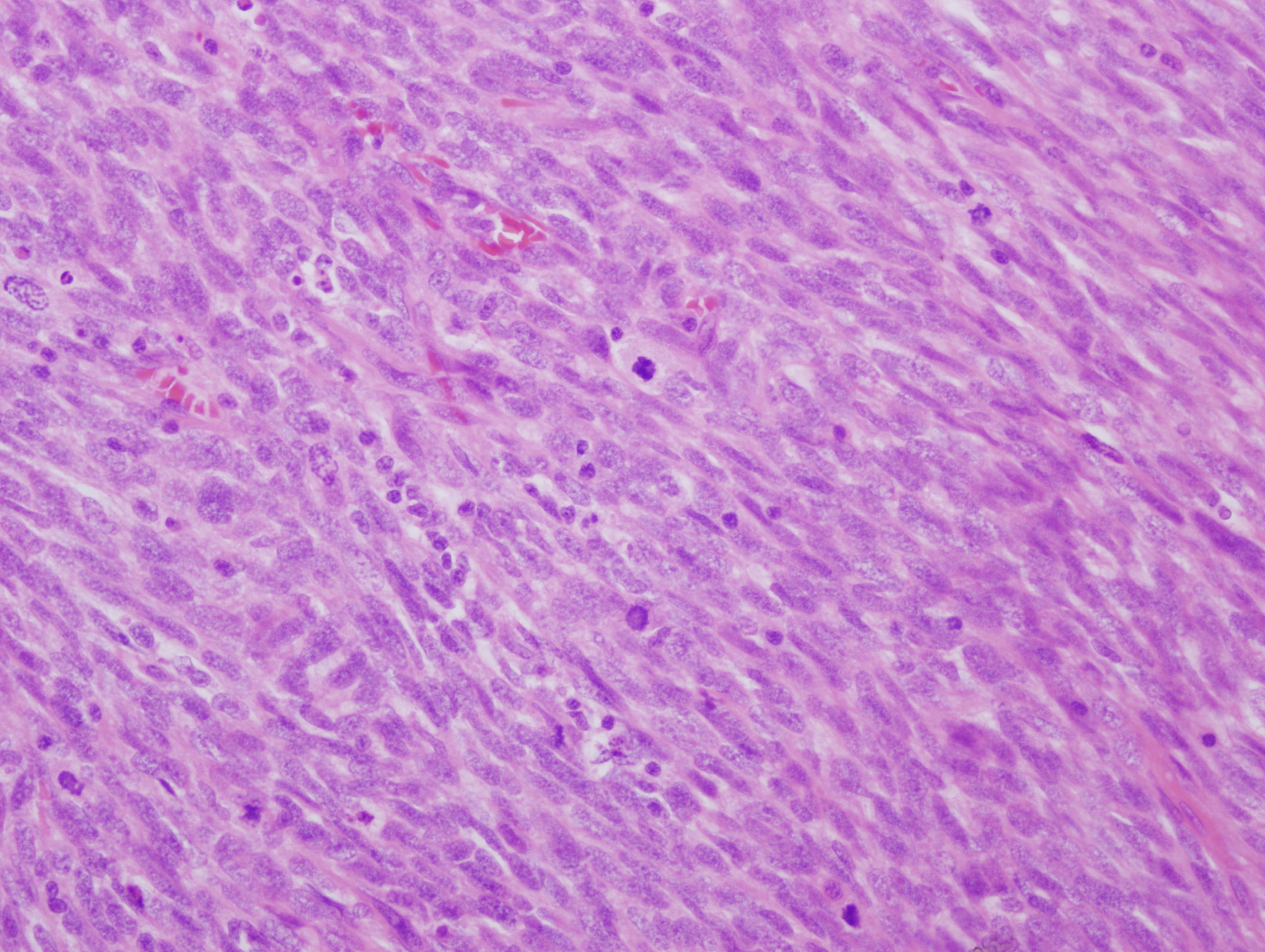
What is your diagnosis?
Diagnosis:
Malignant peripheral nerve sheath tumor
Special stains:
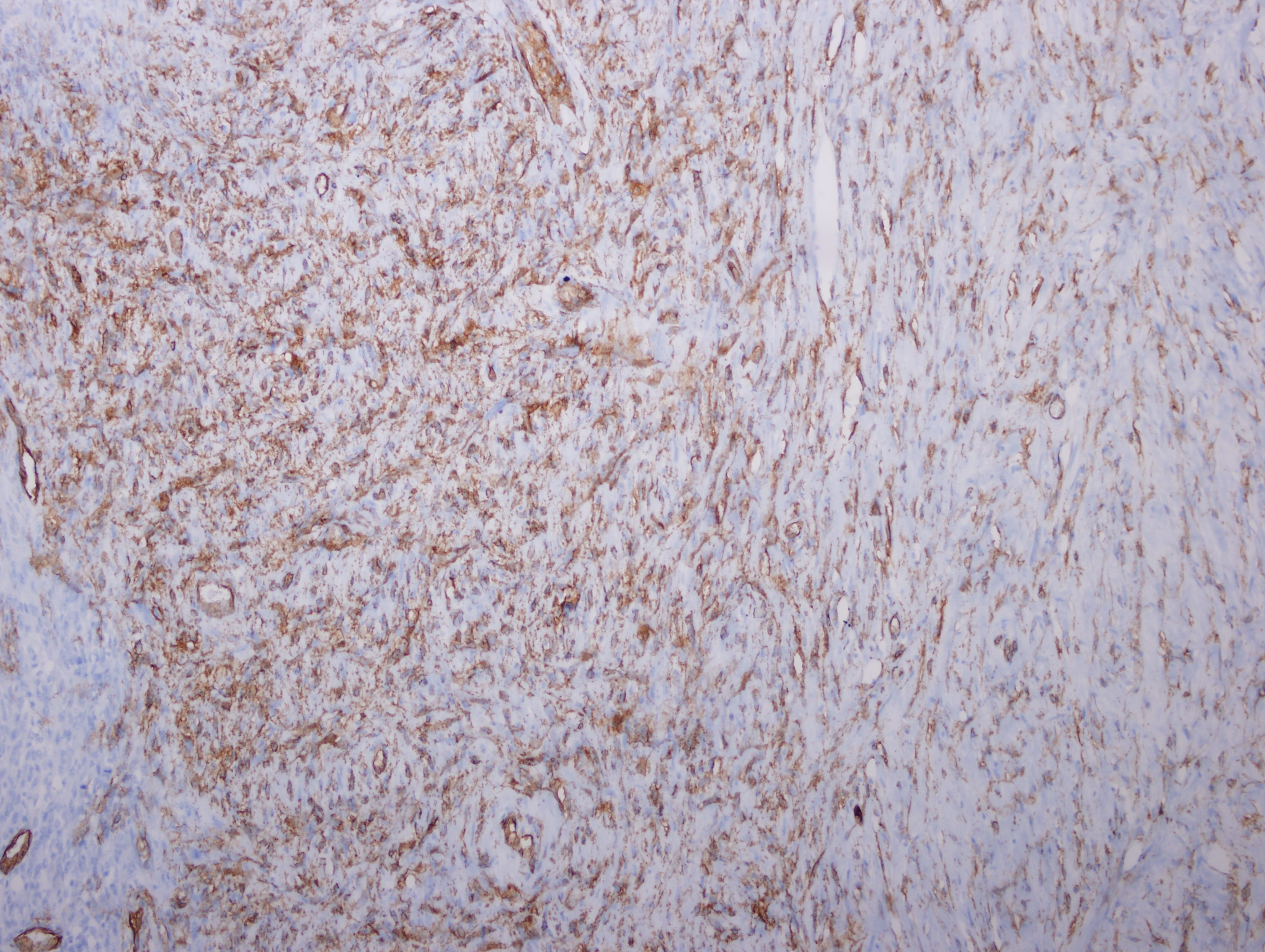
Test question (answer at the end):
A spindle cell tumor with weak positivity for S100 and strong, diffuse nuclear positivity for TLE1 is most likely which of the following?
A. MPNST
B. Cellular schwannoma
C. Fibrosarcoma
D. Synovial sarcoma
Discussion (from author):
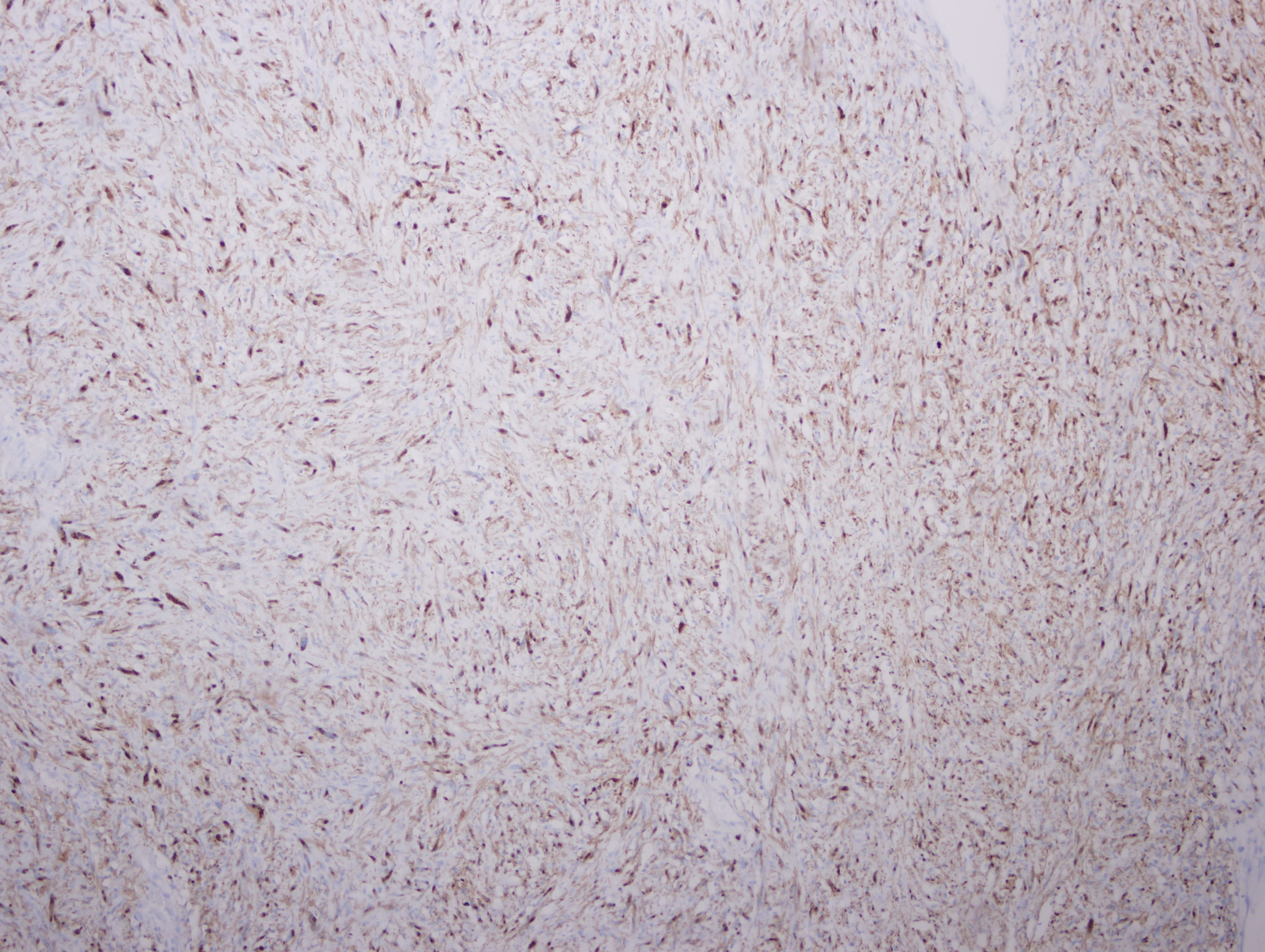
The mass is a spindle cell tumor with hypercellular and occasional myxoid hypocellular areas. The spindle cells are arranged in a somewhat fascicular pattern and occasional whorls are seen. The nuclei are pleomorphic and there are numerous mitotic figures. Some tumor cells are positive for S100 and there is focal positivity for CD34. There was negative staining for EMA, desmin and SMA (not shown). The histologic appearance, immunoprofile, location and patient history of NF1 are consistent with a malignant peripheral nerve sheath tumor (MPNST).
MPNST typically presents as a fusiform mass arising from major nerve trunks, such as the brachial plexus in this patient. MPNST is usually found in adults 25-50 years old and is exceedingly rare in children. Approximately 25-50% of patients with MPNST have NF1 and ~10% of patients with NF1 develop MPNST in their lifetimes. The risk increases to 30% if the patient has a symptomatic plexiform neurofibroma. Exposure to radiation is also a risk factor (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
The diagnosis of MPNST has been inconsistent over the years because of its variable histopathology and lack of specific molecular or immunophenotypic findings. Per Goldblum et al, a diagnosis of MPNST can be made if the tumor fits into one of these three categories:
1. The tumor arises from a peripheral nerve and does not contain heterologous differentiation
2. It arises from a pre-existing benign nerve sheath tumor (typically a neurofibroma), or
3. The histologic features are consistent with a malignant Schwann cell tumor.
Histologic findings associated with malignant Schwann cell tumors, although not entirely specific, include: fascicles of alternating cellularity in a “marbled” pattern, whorled structures reminiscent of tactoid structures and spindled, fusiform or rounded cells with nuclei that are wavy in profile and asymmetrically oval en face (in this particular case the nuclei are less differentiated and do not show these features). MPNSTs can have also show nuclear palisading, rosette-like nodules, perineural / intraneural spread and hyperplastic vascular changes with a subendothelial accentuation of tumor cells. Approximately 10-15% of MPNSTs have heterologous elements, including cartilage and bone, and rarely mucin secreting glands and squamous cells. If rhabdomyoblasts are present, the tumor can be classified as a malignant Triton tumor, which is more aggressive (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
Malignant spindled tumors in NF1 patients should be considered MPNST until proven otherwise. MPNST has both low grade (15% of tumors) and high grade forms (Acta Neuropathol 2012;123:295). Low grade MPNST arising in a neurofibroma can be diagnosed when there is transition to an area with generalized nuclear atypia (enlargement and hyperchromasia), diffuse cellularity, mitotic activity, and an architectural pattern that resembles fibrosarcoma. Rapid enlargement or pain in a pre-existing NF is worrisome for malignant transformation (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014). CDKN2A/B deletions may represent early malignant change, as does loss of p16 or p27 expression (Acta Neuropathol 2012;123:295). Most MPNSTs, however, are high grade and obviously malignant.
The differential diagnosis of MPNST includes cellular schwannoma, monophasic synovial sarcoma, adult type fibrosarcoma and melanoma.
Schwannomas are not typically seen in NF1, and are more common in patients with NF2. Cellular schwannoma resembles MPNST, as it is composed of predominantly Antoni A areas, lacks Verocay bodies, may have focal necrosis and is S100+. However, it has less than 4 mitoses per ten HPF, and the cells surrounding necrotic areas are not atypical. In addition, MPNST staining of S100 is weak and more focal (compared to strong diffuse staining in cellular schwannoma), and cellular schwanoma is typically SOX10+. In the 30% of MPNST that are SOX10+, the staining is weak and focal (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
The cells in synovial sarcoma (SS) are more uniform than those of MPNST, and resemble fibroblasts rather than Schwann cells. The immunoprofiles of SS and MPNST are overlapping: both can be S100 positive and TLE1 positive, but SS shows stronger and more diffuse nuclear positivity for TLE1 (Arch Pathol Lab Med 2015;139:106). SS will stain positive with high molecular weight cytokeratin, which MPNST will not (although some low molecular weight keratin and EMA staining can be present). SS should be negative for CD34. The diagnosis of SS can be confirmed by finding SS18-SSX1 or SS18-SSX2 gene fusions (Acta Neuropathol 2012;123:295).
MPNST can have areas that are indistinguishable from fibrosarcoma, but in general fibrosarcoma has a "herringbone" pattern throughout the lesion and more uniform looking cells than MPNST, which should show areas with nerve sheath differentiation. Fibrosarcoma is S100 and SOX10 negative (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
Spindle cell and desmoplastic melanoma can also share histologic and immunophenotypic features with MPNST. Epithelioid MPNST, a rare variant that has polygonal cells with prominent nucleoli and stronger S100 staining, closely mimics melanoma (Acta Neuropathol 2012;123:295). The diagnosis of MPNST is aided if the tumor arises from a nerve. Melanoma would be favored if there is a dermal location, a junctional melanocytic component, nodular lymphoid aggregates, lymph node metastases and positivity for other melanocytic markers.
MPNST does not respond well to chemotherapy, but radical surgery appears to improve prognosis. Overall five year and ten year survival is approximately 50% and 40%; when treated with radical surgery, the ten year survival rate improves to 80%.
Test Question Answer:
D. Synovial sarcoma. Although MPNST can be positive for TLE1, strong and diffuse nuclear positivity is seen in synovial sarcoma.
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Week page. To subscribe or unsubscribe to Case of the Week or our other email lists, click here.
Thanks to Drs. Safa Alshaikh, Maryam Hammad and Zainab Harb, Salmaniya Medical Complex, (Kingdom of Bahrain) for contributing this case and Dr. Hillary Zalaznick, Myriad Genetics, Inc., Utah (USA) for editing the discussion. To contribute a Case of the Week, first make sure that we are currently accepting cases, then follow the guidelines on our main Case of the Week page.
Advertisement
Website news:
(1) Our Industry News page highlights announcements from our Sponsors. It replaces the New Products and Services page and Management page. At the top, it contains links to the Buyers Guide, Promotions Page and Product Questionnaires. Let us know if you have any questions.
(2) Many visitors have indicated that they get better searches when using Google.com to search our website, compared to the Search box in the upper right on our website (which is also from Google). They first visit Google.com, then type PathologyOutlines.com and the search term (for example "Pathologyoutlines.com carcinoid").
(3) See most recent Textbook updates here.
Visit and follow our Blog to see recent updates to the website.
Case of the Week #443
Clinical history:
A 56 year old man with neurofibromatosis type 1 presented with a right axillary swelling, present for many years. Ultrasound showed a 9 x 5 cm well defined heterogeneous soft tissue mass arising from the right axilla. The mass was excised.
Histopathology images:
What is your diagnosis?
Diagnosis:
Malignant peripheral nerve sheath tumor
Special stains:
CD34
S100
Test question (answer at the end):
A spindle cell tumor with weak positivity for S100 and strong, diffuse nuclear positivity for TLE1 is most likely which of the following?
A. MPNST
B. Cellular schwannoma
C. Fibrosarcoma
D. Synovial sarcoma
Discussion (from author):
The mass is a spindle cell tumor with hypercellular and occasional myxoid hypocellular areas. The spindle cells are arranged in a somewhat fascicular pattern and occasional whorls are seen. The nuclei are pleomorphic and there are numerous mitotic figures. Some tumor cells are positive for S100 and there is focal positivity for CD34. There was negative staining for EMA, desmin and SMA (not shown). The histologic appearance, immunoprofile, location and patient history of NF1 are consistent with a malignant peripheral nerve sheath tumor (MPNST).
MPNST typically presents as a fusiform mass arising from major nerve trunks, such as the brachial plexus in this patient. MPNST is usually found in adults 25-50 years old and is exceedingly rare in children. Approximately 25-50% of patients with MPNST have NF1 and ~10% of patients with NF1 develop MPNST in their lifetimes. The risk increases to 30% if the patient has a symptomatic plexiform neurofibroma. Exposure to radiation is also a risk factor (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
The diagnosis of MPNST has been inconsistent over the years because of its variable histopathology and lack of specific molecular or immunophenotypic findings. Per Goldblum et al, a diagnosis of MPNST can be made if the tumor fits into one of these three categories:
1. The tumor arises from a peripheral nerve and does not contain heterologous differentiation
2. It arises from a pre-existing benign nerve sheath tumor (typically a neurofibroma), or
3. The histologic features are consistent with a malignant Schwann cell tumor.
Histologic findings associated with malignant Schwann cell tumors, although not entirely specific, include: fascicles of alternating cellularity in a “marbled” pattern, whorled structures reminiscent of tactoid structures and spindled, fusiform or rounded cells with nuclei that are wavy in profile and asymmetrically oval en face (in this particular case the nuclei are less differentiated and do not show these features). MPNSTs can have also show nuclear palisading, rosette-like nodules, perineural / intraneural spread and hyperplastic vascular changes with a subendothelial accentuation of tumor cells. Approximately 10-15% of MPNSTs have heterologous elements, including cartilage and bone, and rarely mucin secreting glands and squamous cells. If rhabdomyoblasts are present, the tumor can be classified as a malignant Triton tumor, which is more aggressive (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
Malignant spindled tumors in NF1 patients should be considered MPNST until proven otherwise. MPNST has both low grade (15% of tumors) and high grade forms (Acta Neuropathol 2012;123:295). Low grade MPNST arising in a neurofibroma can be diagnosed when there is transition to an area with generalized nuclear atypia (enlargement and hyperchromasia), diffuse cellularity, mitotic activity, and an architectural pattern that resembles fibrosarcoma. Rapid enlargement or pain in a pre-existing NF is worrisome for malignant transformation (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014). CDKN2A/B deletions may represent early malignant change, as does loss of p16 or p27 expression (Acta Neuropathol 2012;123:295). Most MPNSTs, however, are high grade and obviously malignant.
The differential diagnosis of MPNST includes cellular schwannoma, monophasic synovial sarcoma, adult type fibrosarcoma and melanoma.
Schwannomas are not typically seen in NF1, and are more common in patients with NF2. Cellular schwannoma resembles MPNST, as it is composed of predominantly Antoni A areas, lacks Verocay bodies, may have focal necrosis and is S100+. However, it has less than 4 mitoses per ten HPF, and the cells surrounding necrotic areas are not atypical. In addition, MPNST staining of S100 is weak and more focal (compared to strong diffuse staining in cellular schwannoma), and cellular schwanoma is typically SOX10+. In the 30% of MPNST that are SOX10+, the staining is weak and focal (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
The cells in synovial sarcoma (SS) are more uniform than those of MPNST, and resemble fibroblasts rather than Schwann cells. The immunoprofiles of SS and MPNST are overlapping: both can be S100 positive and TLE1 positive, but SS shows stronger and more diffuse nuclear positivity for TLE1 (Arch Pathol Lab Med 2015;139:106). SS will stain positive with high molecular weight cytokeratin, which MPNST will not (although some low molecular weight keratin and EMA staining can be present). SS should be negative for CD34. The diagnosis of SS can be confirmed by finding SS18-SSX1 or SS18-SSX2 gene fusions (Acta Neuropathol 2012;123:295).
MPNST can have areas that are indistinguishable from fibrosarcoma, but in general fibrosarcoma has a "herringbone" pattern throughout the lesion and more uniform looking cells than MPNST, which should show areas with nerve sheath differentiation. Fibrosarcoma is S100 and SOX10 negative (Goldblum, Folpe and Weiss: Enzinger and Weiss’s Soft Tissue Tumors, 6th ed., 2014).
Spindle cell and desmoplastic melanoma can also share histologic and immunophenotypic features with MPNST. Epithelioid MPNST, a rare variant that has polygonal cells with prominent nucleoli and stronger S100 staining, closely mimics melanoma (Acta Neuropathol 2012;123:295). The diagnosis of MPNST is aided if the tumor arises from a nerve. Melanoma would be favored if there is a dermal location, a junctional melanocytic component, nodular lymphoid aggregates, lymph node metastases and positivity for other melanocytic markers.
MPNST does not respond well to chemotherapy, but radical surgery appears to improve prognosis. Overall five year and ten year survival is approximately 50% and 40%; when treated with radical surgery, the ten year survival rate improves to 80%.
Test Question Answer:
D. Synovial sarcoma. Although MPNST can be positive for TLE1, strong and diffuse nuclear positivity is seen in synovial sarcoma.
