
5 October 2016 - Case of the Week #403
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Week page. To subscribe or unsubscribe to Case of the Week or our other email lists, click here.
Thanks to Dr. Avinash Mane, Krishna Institute of Medical Sciences Deemed University (India), for contributing this case. To contribute a Case of the Week, follow the guidelines on our main Case of the Week page.
 Advertisement
Advertisement
Website news:
(1) Add NatPernick@gmail.com to your SafeList / Address book, so our email subscriptions go directly into your inbox.
(2) Please bookmark and use our Amazon.com link to remember to shop at Amazon.com, and at the same time, help PathologyOutlines.com. We receive a commission (~ 5%) from all purchases made through this link, whether pathology related or not, at no cost to you. You get the benefit of wide selection of products and sellers, product reviews, a low price, and the knowledge that you are helping us maintain and expand our free website for the pathology community.
(3) Don't forget to subscribe to our 5 available email newsletters: (a) Case of the Week; (b) Commercial / Promotions email; (c) Jobs, Fellowships and Conferences; (d) Website News / New Books and (e) What's New in Pathology (new). Click here to learn more, and remember, you can unsubscribe at any time.
Visit and follow our Blog to see recent updates to the website.
Case of the Week #403
Clinical history:
A 3 kg boy was born at 38 weeks following spontaneous vaginal delivery. The mother was a healthy 30 year old G3P1 with O+ blood type. Maternal laboratory findings were within normal limits and included a negative antibody screen. There was no significant family medical history, specifically no known history of jaundice, anemia or hematological disorders.
APGARS at birth were normal but within 3 hours of birth, the baby presented in acute respiratory distress with a feeble pulse, marked jaundice, pallor and cyanosis, with a whole body purpuric rash.
Laboratory investigations revealed: Hb - 112 g/L; WBC - 22.7 x 109/L; platelet count - 25 x 109/; reticulocytes - 8.5%. The baby died on the first day of life due to acute bilirubin encephalopathy.
Hematology (blood smear) images:
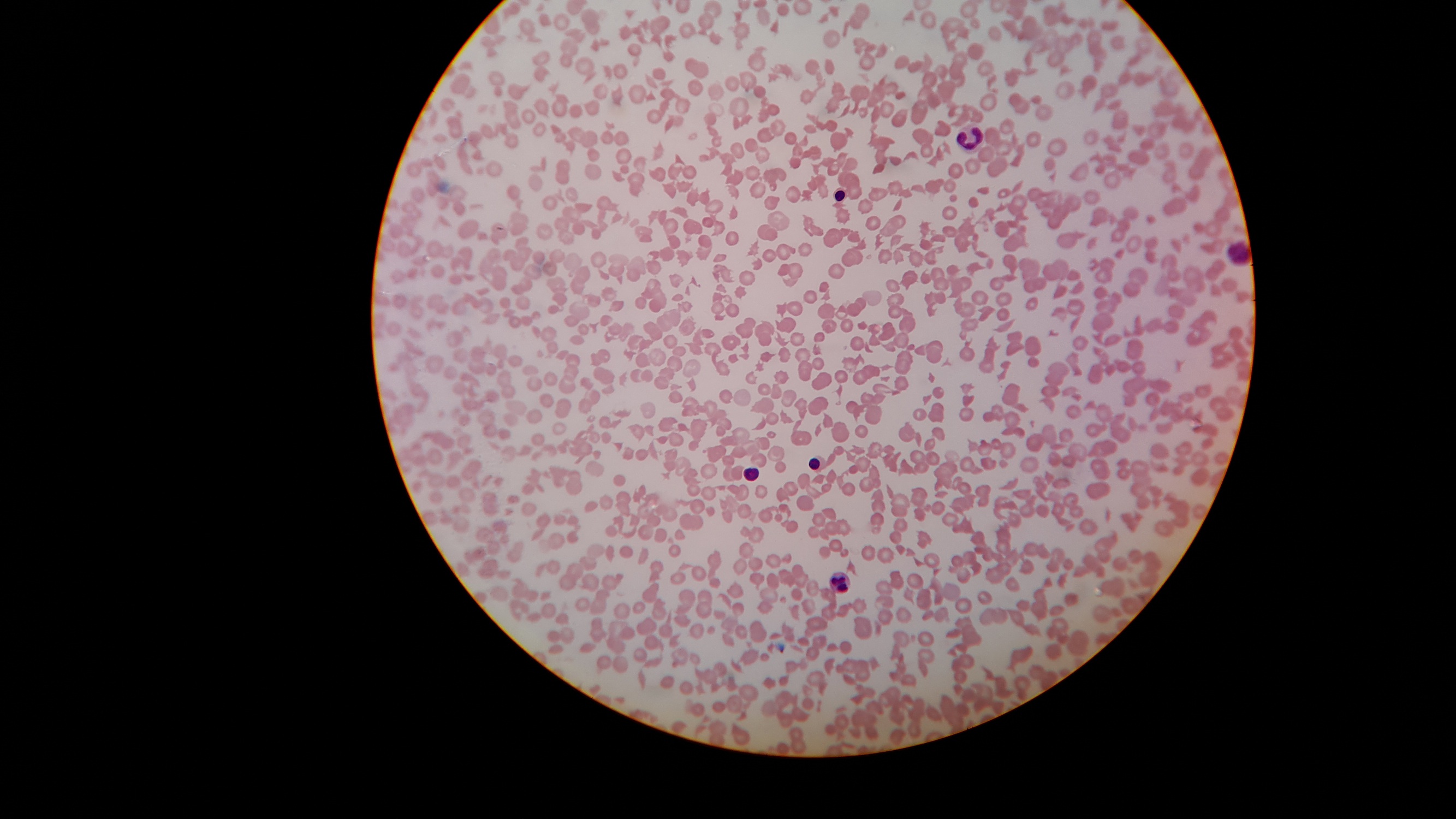
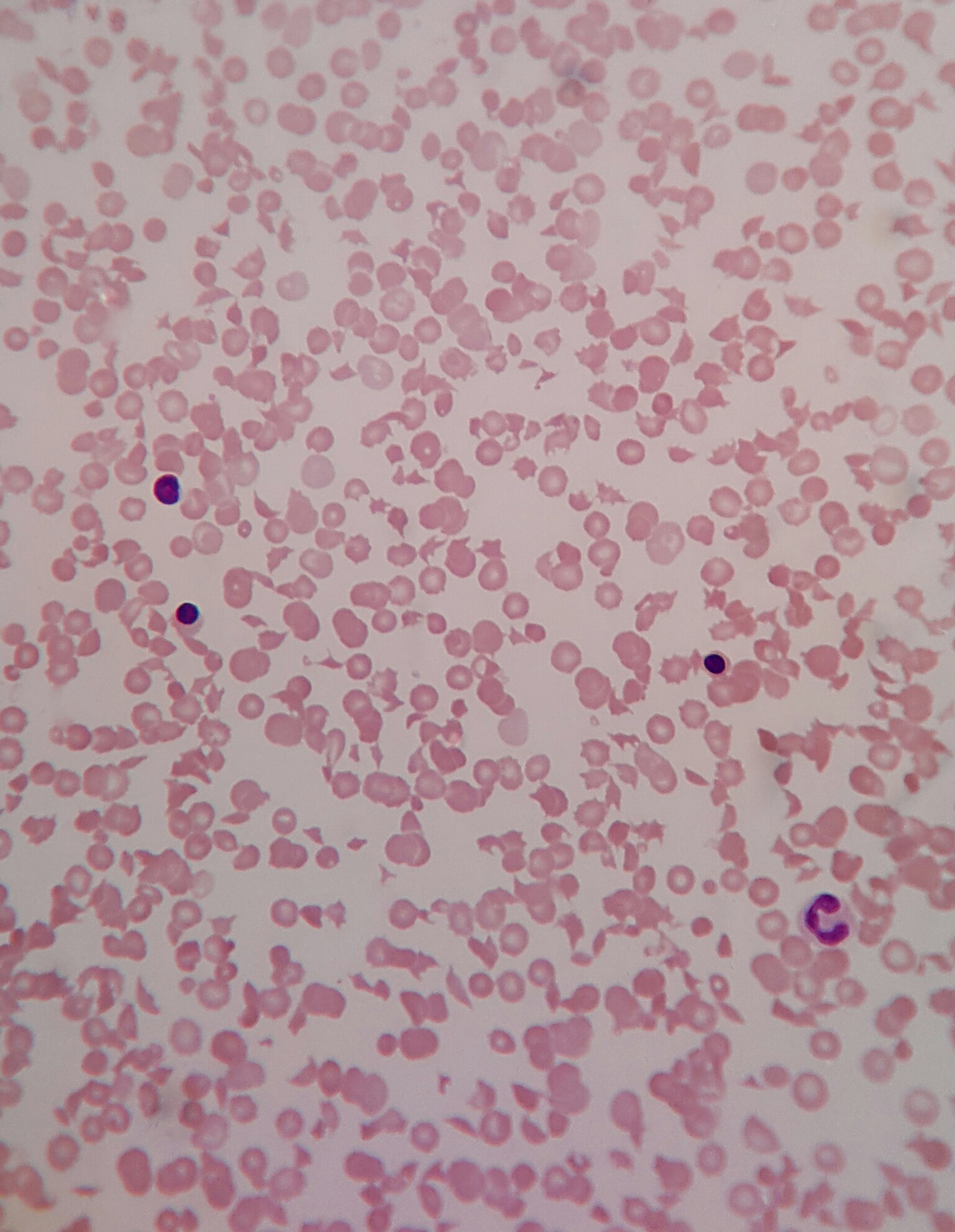
What is your diagnosis?
Diagnosis:
Congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome)
Discussion:
Thrombotic thrombocytopenic purpura (TTP, congenital or acquired) is a rare form of thrombotic microangiopathy (TMA) characterized by thrombocytopenia and microangiopathic hemolytic anemia (MAHA) associated with a range of clinical findings related to end-organ damage. The underlying mechanism is a deficiency in ADAMTS13 (Von Willebrand factor-cleaving protease) enzyme.
In congenital TTP (Upshaw-Schulman syndrome), a genetic defect in the ADAMTS13 gene (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 gene) results in a severe deficiency of ADAMTS13 enzyme.
Acquired TTP is attributed to an auto-immune process (IgG auto-antibody) directed against ADAMTS13.
Clinical presentation:
As described in this case of the week, patients with congenital TTP (Upshaw-Schulman syndrome) typically present shortly after birth or early in the neonatal period with a range of clinical findings, which may include hemolytic anemia with fragmentation of erythrocytes, thrombocytopenia, diffuse and non-focal neurologic findings, decreased renal function and fever.
Pathogenesis and tissue findings:
ADAMTS13 enzyme, also known as Von Willebrand factor-cleaving protease, degrades large Von Willebrand factor (vWF) multimers, decreasing their activity. Deficiency of ADAMTS13 enzyme (either congenital or acquired) results in the persistence of unusually large von Willebrand factor (UL-vWF) multimers. These multimers induce vWF-rich platelet microthromboses. In tissue sections, these microthrombi are found in arterioles and capillaries with little or no associated inflammatory changes (angiopathy). Microthrombi are only seen in small caliber vessels which have high shear stress (arterioles and capillaries) as UL-vWF multimers are elongated and binds platelets to form microthrombi. In vessels without high shear stress, vWF has a globoid form which does not bind platelets in a similar manner.
Laboratory findings:
Findings consistent with TMA and MAHA: schistocytes on peripheral blood smears, low platelet count and reticulocytosis. Increased lactate dehydrogenase (LDH), increased indirect bilirubin and low haptoglobin. Direct antiglobulin (Coombs) test (DAT) is negative.
Diagnosis:
Thrombocytopenia and microangiopathic hemolytic anemia and a severe ADAMTS13 deficiency (< 10% normal values), in the absence of anti-ADAMTS13 antibodies, suggest a diagnosis of congenital TTP. There may be a known family history of jaundice or bleeding disorders, especially in neonates. Diagnosis is confirmed by molecular analysis revealing a double heterozygous or homozygous mutation in the ADAMTS13 gene.
Genetics:
Congenital TTP is transmitted in an autosomal recessive (AR) manner and genetic counseling should be provided to affected families.
Treatment:
Therapeutic plasma exchange transfusion (TPE) using fresh frozen plasma (FFP) to replace ADAMTS13 is the treatment of choice and has reduced mortality from > 90% to less than 20% if initiated in the acute phase (within 4-8 hours) of the suspected diagnosis.
In newborns, exchange transfusion is often required due to the associated anemia and hyperbilirubinemia which may develop in the acute phase. Subsequent acute episodes are treated by therapeutic plasma exchange transfusion and chronic relapsing disease may require prophylactic plasma therapy every 2-3 weeks to maintain ADAMTS13 enzyme levels. There is some controversy regarding the role of platelet transfusions preceding interventional procedures like intravascular catheter placement, which until recently had been considered contraindicated due to significant mortality.
A bio-engineered wild-type recombinant human ADAMTS13 has been successfully investigated in experimental models and human clinical trials are expected soon.
References:
1. Thromb Res 2014;134:1171
2. J Clin Apher 2014;29:148
3. Biorheology 2015;52:319
4. Blood 2012;119:6128
5. Vox Sang 2015;109:168
6. Orphanet
7. OMIM #274150
Discussion by: Dr. Belinda Lategan, St. Boniface Hospital (Canada)
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Week page. To subscribe or unsubscribe to Case of the Week or our other email lists, click here.
Thanks to Dr. Avinash Mane, Krishna Institute of Medical Sciences Deemed University (India), for contributing this case. To contribute a Case of the Week, follow the guidelines on our main Case of the Week page.
AdvertisementWebsite news:
(1) Add NatPernick@gmail.com to your SafeList / Address book, so our email subscriptions go directly into your inbox.
(2) Please bookmark and use our Amazon.com link to remember to shop at Amazon.com, and at the same time, help PathologyOutlines.com. We receive a commission (~ 5%) from all purchases made through this link, whether pathology related or not, at no cost to you. You get the benefit of wide selection of products and sellers, product reviews, a low price, and the knowledge that you are helping us maintain and expand our free website for the pathology community.
(3) Don't forget to subscribe to our 5 available email newsletters: (a) Case of the Week; (b) Commercial / Promotions email; (c) Jobs, Fellowships and Conferences; (d) Website News / New Books and (e) What's New in Pathology (new). Click here to learn more, and remember, you can unsubscribe at any time.
Visit and follow our Blog to see recent updates to the website.
Case of the Week #403
Clinical history:
A 3 kg boy was born at 38 weeks following spontaneous vaginal delivery. The mother was a healthy 30 year old G3P1 with O+ blood type. Maternal laboratory findings were within normal limits and included a negative antibody screen. There was no significant family medical history, specifically no known history of jaundice, anemia or hematological disorders.
APGARS at birth were normal but within 3 hours of birth, the baby presented in acute respiratory distress with a feeble pulse, marked jaundice, pallor and cyanosis, with a whole body purpuric rash.
Laboratory investigations revealed: Hb - 112 g/L; WBC - 22.7 x 109/L; platelet count - 25 x 109/; reticulocytes - 8.5%. The baby died on the first day of life due to acute bilirubin encephalopathy.
Hematology (blood smear) images:
What is your diagnosis?
Diagnosis:
Congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome)
Discussion:
Thrombotic thrombocytopenic purpura (TTP, congenital or acquired) is a rare form of thrombotic microangiopathy (TMA) characterized by thrombocytopenia and microangiopathic hemolytic anemia (MAHA) associated with a range of clinical findings related to end-organ damage. The underlying mechanism is a deficiency in ADAMTS13 (Von Willebrand factor-cleaving protease) enzyme.
In congenital TTP (Upshaw-Schulman syndrome), a genetic defect in the ADAMTS13 gene (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 gene) results in a severe deficiency of ADAMTS13 enzyme.
Acquired TTP is attributed to an auto-immune process (IgG auto-antibody) directed against ADAMTS13.
Clinical presentation:
As described in this case of the week, patients with congenital TTP (Upshaw-Schulman syndrome) typically present shortly after birth or early in the neonatal period with a range of clinical findings, which may include hemolytic anemia with fragmentation of erythrocytes, thrombocytopenia, diffuse and non-focal neurologic findings, decreased renal function and fever.
Pathogenesis and tissue findings:
ADAMTS13 enzyme, also known as Von Willebrand factor-cleaving protease, degrades large Von Willebrand factor (vWF) multimers, decreasing their activity. Deficiency of ADAMTS13 enzyme (either congenital or acquired) results in the persistence of unusually large von Willebrand factor (UL-vWF) multimers. These multimers induce vWF-rich platelet microthromboses. In tissue sections, these microthrombi are found in arterioles and capillaries with little or no associated inflammatory changes (angiopathy). Microthrombi are only seen in small caliber vessels which have high shear stress (arterioles and capillaries) as UL-vWF multimers are elongated and binds platelets to form microthrombi. In vessels without high shear stress, vWF has a globoid form which does not bind platelets in a similar manner.
Laboratory findings:
Findings consistent with TMA and MAHA: schistocytes on peripheral blood smears, low platelet count and reticulocytosis. Increased lactate dehydrogenase (LDH), increased indirect bilirubin and low haptoglobin. Direct antiglobulin (Coombs) test (DAT) is negative.
Diagnosis:
Thrombocytopenia and microangiopathic hemolytic anemia and a severe ADAMTS13 deficiency (< 10% normal values), in the absence of anti-ADAMTS13 antibodies, suggest a diagnosis of congenital TTP. There may be a known family history of jaundice or bleeding disorders, especially in neonates. Diagnosis is confirmed by molecular analysis revealing a double heterozygous or homozygous mutation in the ADAMTS13 gene.
Genetics:
Congenital TTP is transmitted in an autosomal recessive (AR) manner and genetic counseling should be provided to affected families.
Treatment:
Therapeutic plasma exchange transfusion (TPE) using fresh frozen plasma (FFP) to replace ADAMTS13 is the treatment of choice and has reduced mortality from > 90% to less than 20% if initiated in the acute phase (within 4-8 hours) of the suspected diagnosis.
In newborns, exchange transfusion is often required due to the associated anemia and hyperbilirubinemia which may develop in the acute phase. Subsequent acute episodes are treated by therapeutic plasma exchange transfusion and chronic relapsing disease may require prophylactic plasma therapy every 2-3 weeks to maintain ADAMTS13 enzyme levels. There is some controversy regarding the role of platelet transfusions preceding interventional procedures like intravascular catheter placement, which until recently had been considered contraindicated due to significant mortality.
A bio-engineered wild-type recombinant human ADAMTS13 has been successfully investigated in experimental models and human clinical trials are expected soon.
References:
1. Thromb Res 2014;134:1171
2. J Clin Apher 2014;29:148
3. Biorheology 2015;52:319
4. Blood 2012;119:6128
5. Vox Sang 2015;109:168
6. Orphanet
7. OMIM #274150
Discussion by: Dr. Belinda Lategan, St. Boniface Hospital (Canada)
