
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. Linh Dang, Baylor College of Medicine, Texas (USA) for contributing this case.

Orchard Pathology is an all-inclusive CP/AP diagnostic information system designed to accommodate the complex workflow and reporting of clinical, cytology, anatomic, and molecular testing.
Orchard Pathology is used for AP only or as a single system that includes clinical and molecularno more need for two separate systems. The shared database provides access to the patients history and enables the consolidation of clinical results to the pathology report. Unlike legacy text-based AP systems, Orchard Pathology stores report information in structured data fields that enhance EMR integration and simplify data mining for evaluations; correlation studies; quality assurance; and other management reports.
Visit us at
http://www.OrchardSoft.com.
(1) We have extensively updated the CD Markers CD1-CD49 chapter, which now has 68 topics, 479 images and 1092 references.
(2) For a "printer friendly page" of any part of our website, we recommend visiting http://www.printfriendly.com, which has options to print with or without images. As this works better than our "printer friendly" versions of chapters, we will remove the printer friendly versions as we update chapters.
(3) We invite you to join our new PathologyOutlines.com LinkedIn group. To join, you must join LinkedIn (which is free). The purpose of this group is to provide useful professional information to practicing pathologists and laboratory personnel. We invite pathologists and laboratory professionals to share thoughts, ideas and discuss issues related to pathology. This forum is an opportunity to keep up to date with the latest clinical developments and discuss challenges with like-minded colleagues. Just visit http://www.linkedin.com, and search for PathologyOutlines.com in the Groups section.
Visit and follow our Blog to see recent updates to the website.
Case #196
Clinical history:
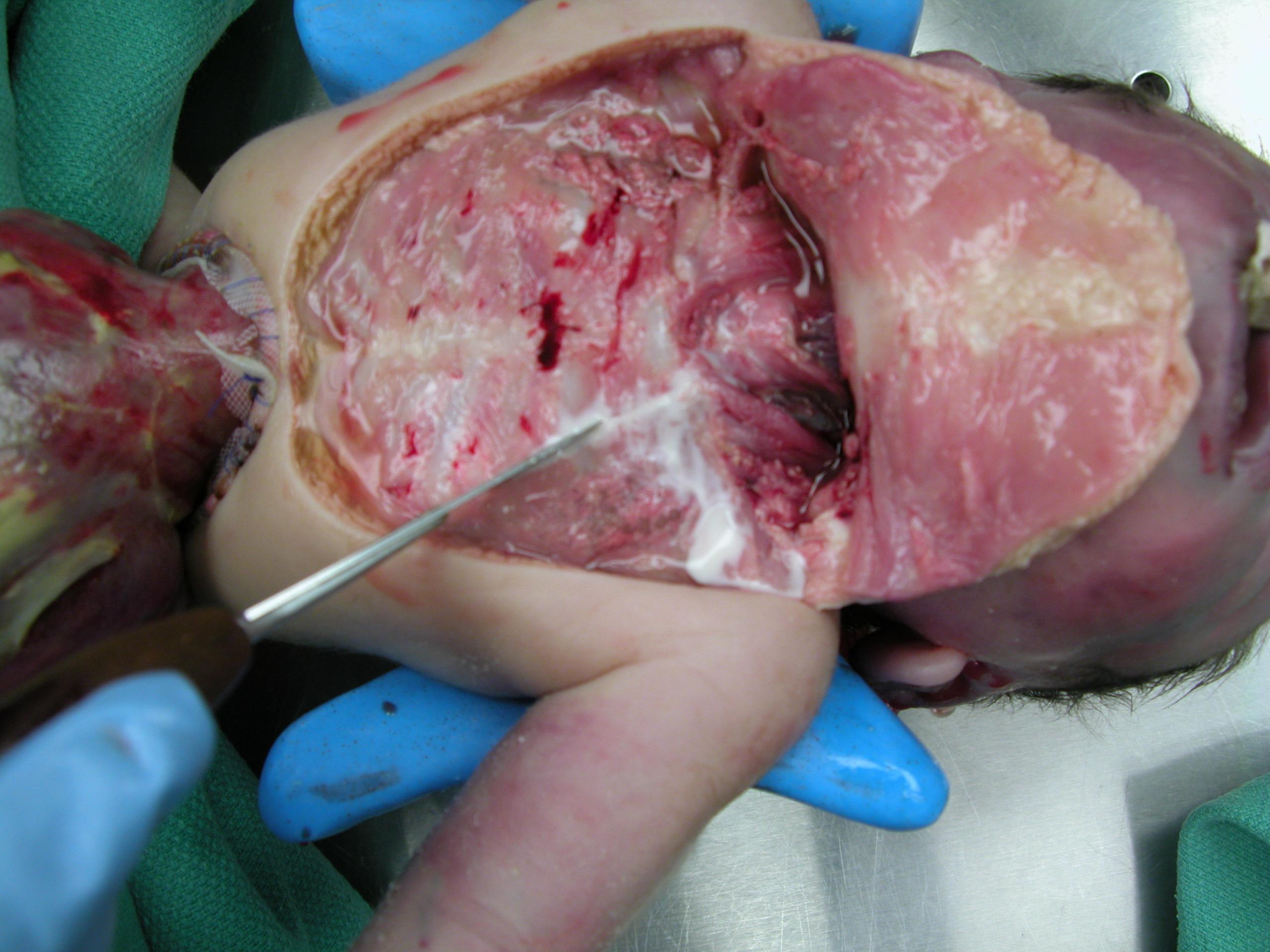
A 12 day old premature male infant was delivered with a ruptured omphalocele and congenital heart defects. On autopsy, bilateral milky white pleural effusions were present. There was 6 ml of fluid on the right and 13 ml on the left. A chemical analysis of the pleural fluid showed 1512 mg/dL of triglycerides, 120 mg/dL of glucose, 125 mmol/L of sodium and 0.5 g/dL of protein.
Clinical image:
Microscopic images:
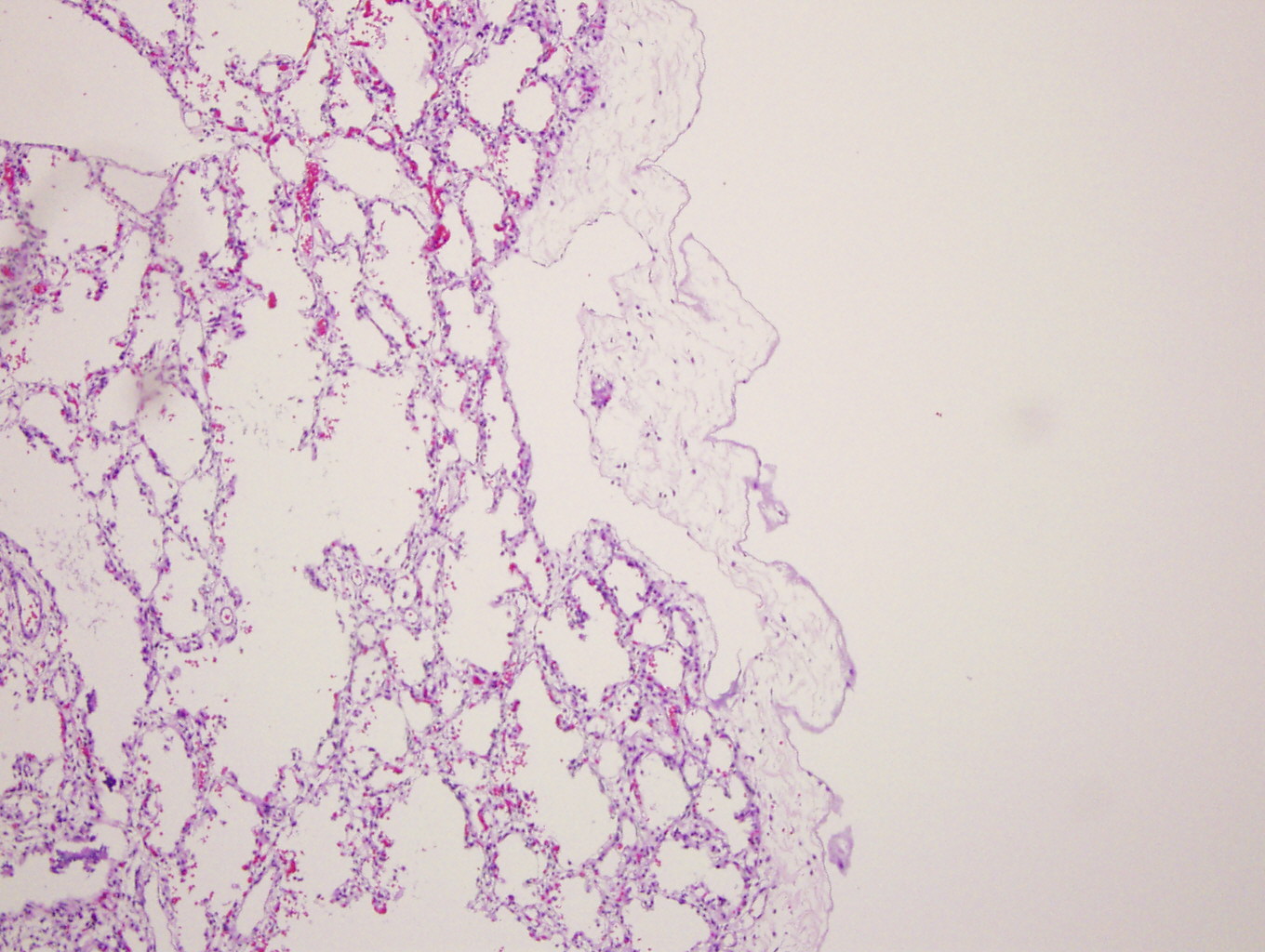
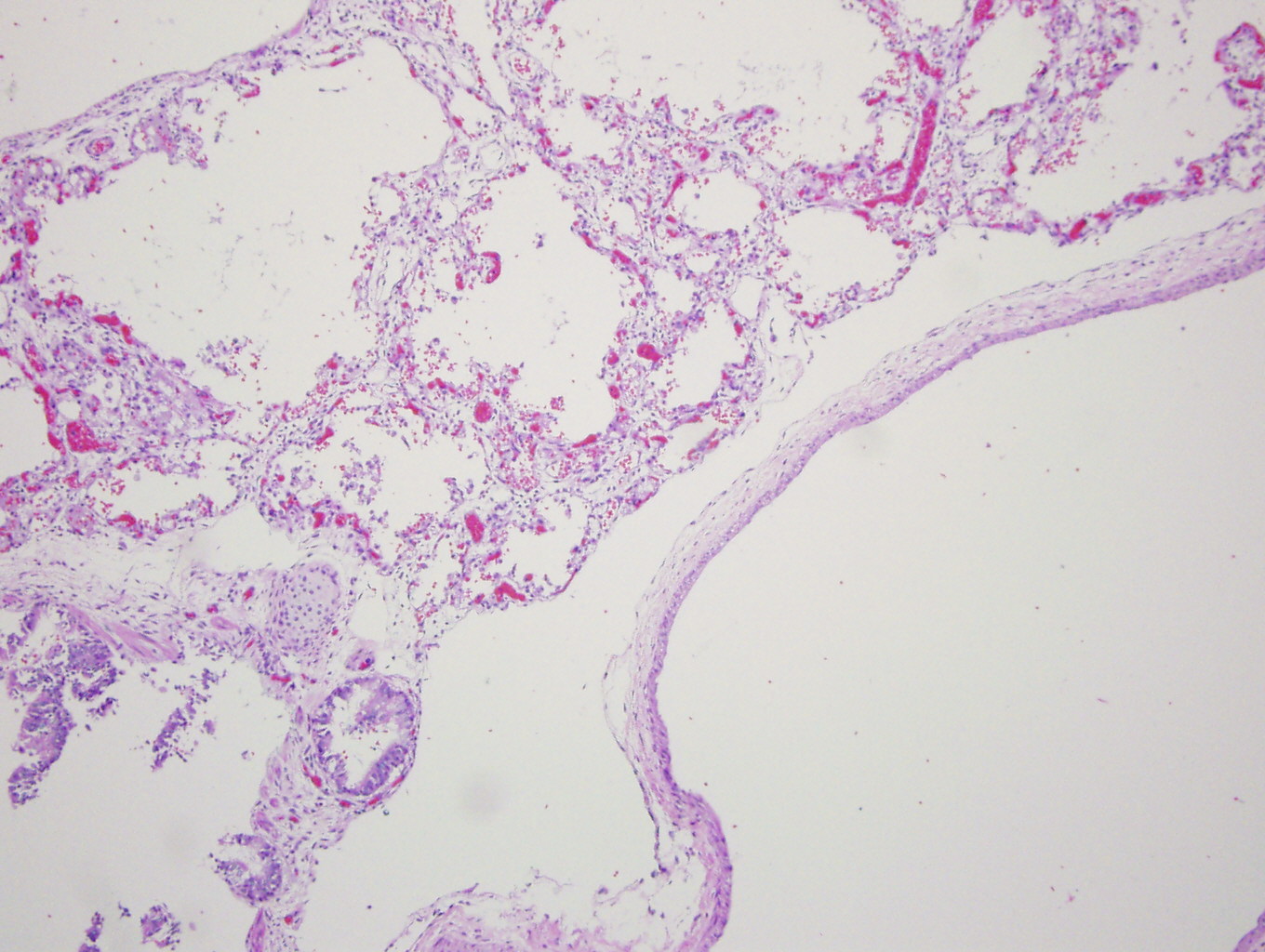
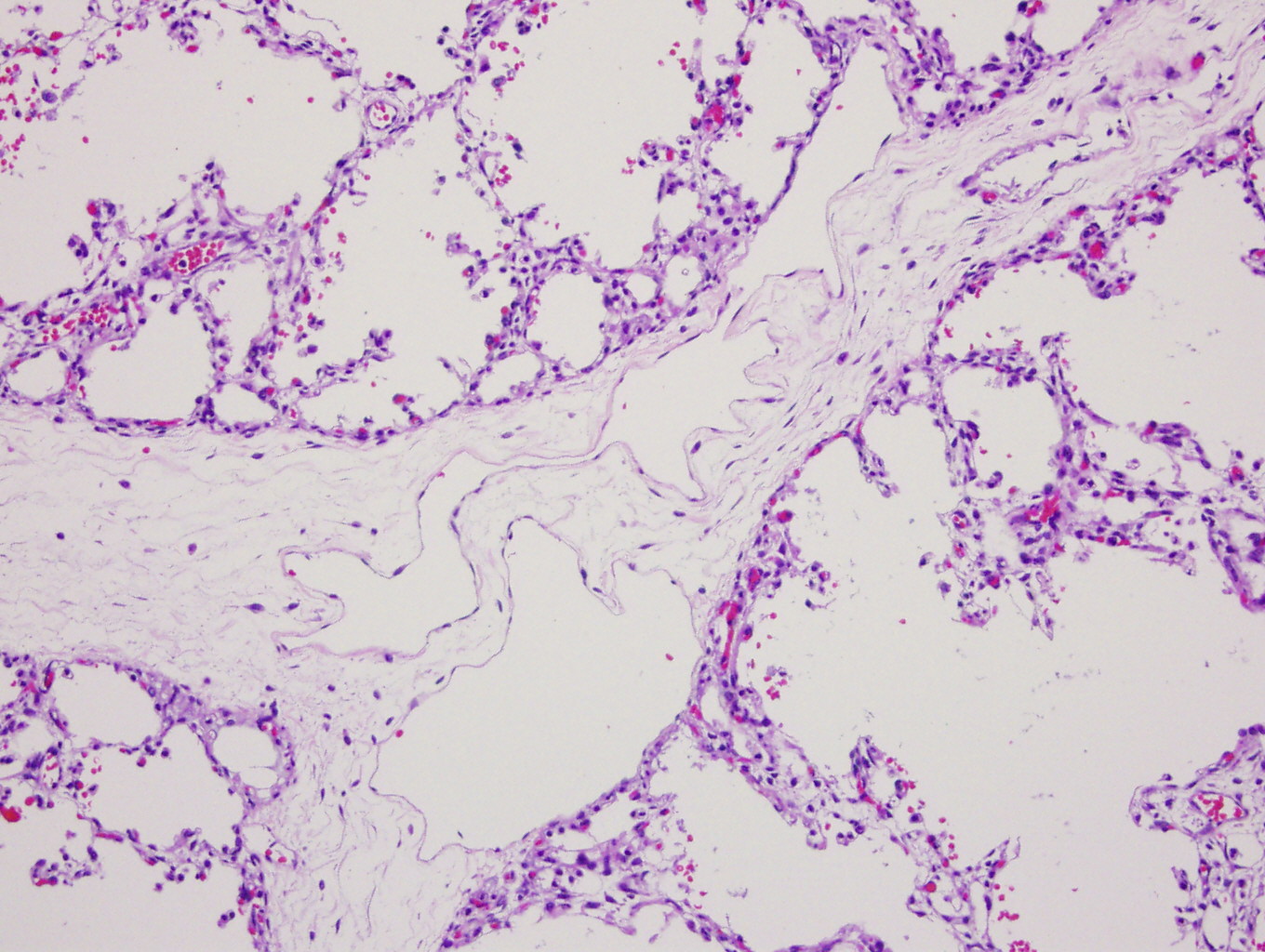

H&E (first 3 images) and D2-40 staining (far right)
What is your diagnosis?
Diagnosis: Primary pulmonary lymphangiectasis
Discussion:
Primary (congenital) pulmonary lymphangiectasis is a rare disease characterized by abnormal dilatation of pulmonary lymphatics, but without lymphatic proliferation (J Korean Med Sci 2007;22:740). Pulmonary lymphangiectasia was classified by Noonan in 1970 into three groups (Am J Dis Child 1970;120:314). Group 1 was generalized lymphangiectasis, including lymphedema with intestinal lymphangiectasis. Group 2 was a secondary disorder due to pulmonary venous hypertension or obstruction associated with cardiovascular anomalies. Group 3 included patients compromised by a primary developmental defect of the pulmonary lymphatics.
In 2000, Faul proposed classifying these disorders as primary or secondary lymphangiectasis (Am J Respir Crit Care Med 2000;161:1037). Primary cases present in neonates with severe respitatory distress with pleural effusion, with generalized edema in some cases. Patients usually require mechanical ventilation. Historically, they were almost always fatal. They are thought to be caused by a failure of pulmonary interstitial connective tissues to regress, leading to the dilation of pulmonary lymphatic capillaries. Secondary cases are due to various disorders that impair lymphatic drainage and increase lymph production, including surgery, radiation, infection, tumor or trauma.
Grossly, the lungs of pulmonary lymphangiectasis are heavy and noncompliant, with numerous dilated lymphatics or cystic spaces in the visceral pleura. Histology shows widened interlobular septa and dilated / cystic lymphatic vessels that stain for D2-40.
The differential diagnosis includes:
- Pulmonary interstitial emphysema: cystic spaces are connected with alveoli and keratin staining shows pneumocytes lining the intact alveolar walls
- Congenital cystic adenomatoid malformation: the entire lung is abormally arranged and immature and cysts are lined by prominent epithelium with goblet cells
- Lymphangiomatosis: an increased number of complex, anastomosing lymphatic channels are present and dilation is secondary, not primary
As in this case, primary pulmonary lymphangiectasis is often fatal in early life. Some authors report a better prognosis more recently due to aggressive neonatal therapies, although in this study, 6 of their 9 cases had an apparently normal neonatal period, suggesting that they were actually secondary disease under the Faul classification (Eur Respir J 2004;24:413).
